User login
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported the approval of an inhaled powder formulation of fluticasone furoate and vilanterol as a treatment for chronic obstructive pulmonary disease, based on studies of almost 4,000 patients.
At a meeting on April 17, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted that the results of the studies provided "substantial evidence" to support approval of the fixed-dose combination of 100 mcg of fluticasone furoate (FF) with 25 mcg of vilanterol (VI), administered once a day, for the long-term maintenance treatment of airflow obstruction (in a 9 to 4 vote), and for reducing COPD exacerbations (in an 8 to 5 vote).
The inhaled corticosteroid/long-acting beta-2 agonist (ICS/LABA) combination is formulated in a dry powder, and is administered once a day in a new inhaler that provides 30 doses, according to GlaxoSmithKline, which is developing the product with Theravance.
If the formulation is approved, it would be the first ICS/LABA product that is dosed once a day for COPD, and would be the first such product that would only be available in the combined formulation, since the companies have no plans to pursue approval of the VI component alone. GSK also markets Advair Diskus, the combination of fluticasone propionate and salmeterol, one of the two inhaled ICS/LABA treatments that is FDA approved for the treatment of COPD. The other is the combination of budesonide and formoterol (Symbicort). Both are administered twice a day, and salmeterol and formoterol are both available as separate products for COPD.
The safety and efficacy of three different dosage strengths of FF – 200, 100, and 50 mcg – combined with 25 mcg of VI were evaluated in four pivotal phase III studies of 3,852 mostly Caucasian people (mean age, 62-63 years) with COPD. All patients had a forced expiratory volume in 1 second (FEV1) less than 70% predicted. About half were current smokers.
In two 24-week studies of 2,254 patients evaluating the effect on lung function, the three FF/VI dosage strengths were compared with placebo, FF 100 mcg alone, and VI 25 mcg alone for two primary endpoints: weighted mean FEV1 at 0-4 hours and change from baseline in trough FEV1.
At 24 weeks, there were significant improvements in the weighted mean FEV1 and in trough FEV1 for all three strengths of FF/VI and for VI 25 mcg, compared with placebo. There were also significant improvements in the mean FEV1 (0-4 hours) for all three strengths of FF/VI, compared with FF alone. Improvement in trough FEV1, however, was not significantly better with any of the three doses, when compared with VI 25 mcg alone.
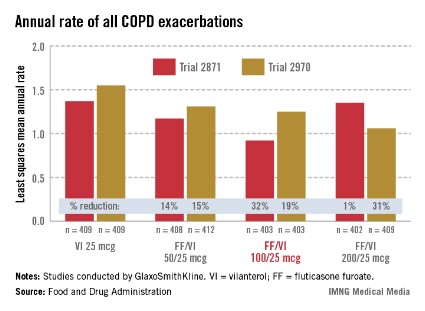
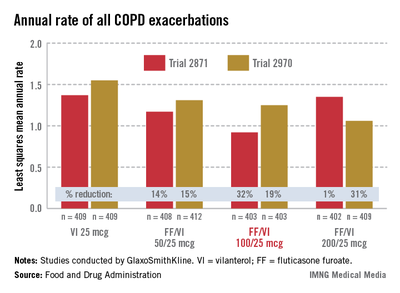
In the two 52-week exacerbation trials of 1,622 patients who had had at least one COPD exacerbation in the previous year, the rate of moderate and severe COPD exacerbations in 1 year was reduced by 21% and 34%, respectively, among those on the FF/VI 100/25 dose over those on VI 25 mcg alone. In one study, the reduction in exacerbations was significant for all three doses, but in the other study, the effect of the proposed 100/25 dose on exacerbations was only numerically, not significantly, improved over the effect of VI 25, the FDA reviewers pointed out. And overall, for the 100/25 dose, the rate of moderate and severe exacerbations was reduced only "by a fraction of an event in 1 year," said Kiya Hamilton, Ph.D., a biostatistician reviewer at the FDA.
Treatment was associated with an increase in local steroid effects, such as oral candidiasis, as expected. In the exacerbation studies, there were more cases of pneumonia and an increase in pneumonia-related deaths with increasing FF doses, and more fractures among those on the FF/VI combination. Bone loss and pneumonia are recognized as adverse effects of ICS in COPD patients, and with one exception, all the deaths were among those on 200/25 mcg dose, which is not going to be marketed, the FDA said.
In a 12 to 1 vote, the panel agreed that the efficacy data provided substantial evidence that the proposed dose provided "a clinically meaningful benefit" for the long-term maintenance treatment of airflow obstruction in COPD. But they were less united on the impact on exacerbations, voting 8 to 5 that the data provided substantial evidence that the reduction in exacerbations seen in the study was clinically meaningful. The panel also voted 10 to 3 that the safety of the 100/25 mcg dose had been adequately demonstrated for the COPD indication.
Panelists supporting approval cited the totality of the database and the utility of having a once a day dose as among the reasons for their votes. Dr. James Stoller, the Jean Wall Bennett Professor of Medicine at the Cleveland Clinic, supported approval and said that the safety profile was acceptable as long as the formulation was not used to treat patients with mild COPD.
Like other panelists who did not support approval, Dr. Paula Carvalho, professor of medicine in the division of pulmonary and critical care medicine, University of Washington, Seattle, said the benefit-risk profile was not adequate. She added that she was more impressed by the effect that treatment with VI 25 mcg alone had on COPD exacerbations than with the combination. Also voting against approval was Peter Peduzzi, Ph.D., professor of public health at the Yale School of Public Health in New Haven, Conn., who said that the impact on trough FEV1 appeared to be driven by the VI component, and like others voting against approval, he cited safety issues.
If approved, GSK plans to market the product as Breo Ellipta. The FDA’s deadline for making a decision on the approval is May 12. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
The product is under review for both COPD and asthma in the European Union and in Japan. It is not under review for asthma in the United States.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported the approval of an inhaled powder formulation of fluticasone furoate and vilanterol as a treatment for chronic obstructive pulmonary disease, based on studies of almost 4,000 patients.
At a meeting on April 17, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted that the results of the studies provided "substantial evidence" to support approval of the fixed-dose combination of 100 mcg of fluticasone furoate (FF) with 25 mcg of vilanterol (VI), administered once a day, for the long-term maintenance treatment of airflow obstruction (in a 9 to 4 vote), and for reducing COPD exacerbations (in an 8 to 5 vote).
The inhaled corticosteroid/long-acting beta-2 agonist (ICS/LABA) combination is formulated in a dry powder, and is administered once a day in a new inhaler that provides 30 doses, according to GlaxoSmithKline, which is developing the product with Theravance.
If the formulation is approved, it would be the first ICS/LABA product that is dosed once a day for COPD, and would be the first such product that would only be available in the combined formulation, since the companies have no plans to pursue approval of the VI component alone. GSK also markets Advair Diskus, the combination of fluticasone propionate and salmeterol, one of the two inhaled ICS/LABA treatments that is FDA approved for the treatment of COPD. The other is the combination of budesonide and formoterol (Symbicort). Both are administered twice a day, and salmeterol and formoterol are both available as separate products for COPD.
The safety and efficacy of three different dosage strengths of FF – 200, 100, and 50 mcg – combined with 25 mcg of VI were evaluated in four pivotal phase III studies of 3,852 mostly Caucasian people (mean age, 62-63 years) with COPD. All patients had a forced expiratory volume in 1 second (FEV1) less than 70% predicted. About half were current smokers.
In two 24-week studies of 2,254 patients evaluating the effect on lung function, the three FF/VI dosage strengths were compared with placebo, FF 100 mcg alone, and VI 25 mcg alone for two primary endpoints: weighted mean FEV1 at 0-4 hours and change from baseline in trough FEV1.
At 24 weeks, there were significant improvements in the weighted mean FEV1 and in trough FEV1 for all three strengths of FF/VI and for VI 25 mcg, compared with placebo. There were also significant improvements in the mean FEV1 (0-4 hours) for all three strengths of FF/VI, compared with FF alone. Improvement in trough FEV1, however, was not significantly better with any of the three doses, when compared with VI 25 mcg alone.
In the two 52-week exacerbation trials of 1,622 patients who had had at least one COPD exacerbation in the previous year, the rate of moderate and severe COPD exacerbations in 1 year was reduced by 21% and 34%, respectively, among those on the FF/VI 100/25 dose over those on VI 25 mcg alone. In one study, the reduction in exacerbations was significant for all three doses, but in the other study, the effect of the proposed 100/25 dose on exacerbations was only numerically, not significantly, improved over the effect of VI 25, the FDA reviewers pointed out. And overall, for the 100/25 dose, the rate of moderate and severe exacerbations was reduced only "by a fraction of an event in 1 year," said Kiya Hamilton, Ph.D., a biostatistician reviewer at the FDA.
Treatment was associated with an increase in local steroid effects, such as oral candidiasis, as expected. In the exacerbation studies, there were more cases of pneumonia and an increase in pneumonia-related deaths with increasing FF doses, and more fractures among those on the FF/VI combination. Bone loss and pneumonia are recognized as adverse effects of ICS in COPD patients, and with one exception, all the deaths were among those on 200/25 mcg dose, which is not going to be marketed, the FDA said.
In a 12 to 1 vote, the panel agreed that the efficacy data provided substantial evidence that the proposed dose provided "a clinically meaningful benefit" for the long-term maintenance treatment of airflow obstruction in COPD. But they were less united on the impact on exacerbations, voting 8 to 5 that the data provided substantial evidence that the reduction in exacerbations seen in the study was clinically meaningful. The panel also voted 10 to 3 that the safety of the 100/25 mcg dose had been adequately demonstrated for the COPD indication.
Panelists supporting approval cited the totality of the database and the utility of having a once a day dose as among the reasons for their votes. Dr. James Stoller, the Jean Wall Bennett Professor of Medicine at the Cleveland Clinic, supported approval and said that the safety profile was acceptable as long as the formulation was not used to treat patients with mild COPD.
Like other panelists who did not support approval, Dr. Paula Carvalho, professor of medicine in the division of pulmonary and critical care medicine, University of Washington, Seattle, said the benefit-risk profile was not adequate. She added that she was more impressed by the effect that treatment with VI 25 mcg alone had on COPD exacerbations than with the combination. Also voting against approval was Peter Peduzzi, Ph.D., professor of public health at the Yale School of Public Health in New Haven, Conn., who said that the impact on trough FEV1 appeared to be driven by the VI component, and like others voting against approval, he cited safety issues.
If approved, GSK plans to market the product as Breo Ellipta. The FDA’s deadline for making a decision on the approval is May 12. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
The product is under review for both COPD and asthma in the European Union and in Japan. It is not under review for asthma in the United States.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported the approval of an inhaled powder formulation of fluticasone furoate and vilanterol as a treatment for chronic obstructive pulmonary disease, based on studies of almost 4,000 patients.
At a meeting on April 17, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted that the results of the studies provided "substantial evidence" to support approval of the fixed-dose combination of 100 mcg of fluticasone furoate (FF) with 25 mcg of vilanterol (VI), administered once a day, for the long-term maintenance treatment of airflow obstruction (in a 9 to 4 vote), and for reducing COPD exacerbations (in an 8 to 5 vote).
The inhaled corticosteroid/long-acting beta-2 agonist (ICS/LABA) combination is formulated in a dry powder, and is administered once a day in a new inhaler that provides 30 doses, according to GlaxoSmithKline, which is developing the product with Theravance.
If the formulation is approved, it would be the first ICS/LABA product that is dosed once a day for COPD, and would be the first such product that would only be available in the combined formulation, since the companies have no plans to pursue approval of the VI component alone. GSK also markets Advair Diskus, the combination of fluticasone propionate and salmeterol, one of the two inhaled ICS/LABA treatments that is FDA approved for the treatment of COPD. The other is the combination of budesonide and formoterol (Symbicort). Both are administered twice a day, and salmeterol and formoterol are both available as separate products for COPD.
The safety and efficacy of three different dosage strengths of FF – 200, 100, and 50 mcg – combined with 25 mcg of VI were evaluated in four pivotal phase III studies of 3,852 mostly Caucasian people (mean age, 62-63 years) with COPD. All patients had a forced expiratory volume in 1 second (FEV1) less than 70% predicted. About half were current smokers.
In two 24-week studies of 2,254 patients evaluating the effect on lung function, the three FF/VI dosage strengths were compared with placebo, FF 100 mcg alone, and VI 25 mcg alone for two primary endpoints: weighted mean FEV1 at 0-4 hours and change from baseline in trough FEV1.
At 24 weeks, there were significant improvements in the weighted mean FEV1 and in trough FEV1 for all three strengths of FF/VI and for VI 25 mcg, compared with placebo. There were also significant improvements in the mean FEV1 (0-4 hours) for all three strengths of FF/VI, compared with FF alone. Improvement in trough FEV1, however, was not significantly better with any of the three doses, when compared with VI 25 mcg alone.
In the two 52-week exacerbation trials of 1,622 patients who had had at least one COPD exacerbation in the previous year, the rate of moderate and severe COPD exacerbations in 1 year was reduced by 21% and 34%, respectively, among those on the FF/VI 100/25 dose over those on VI 25 mcg alone. In one study, the reduction in exacerbations was significant for all three doses, but in the other study, the effect of the proposed 100/25 dose on exacerbations was only numerically, not significantly, improved over the effect of VI 25, the FDA reviewers pointed out. And overall, for the 100/25 dose, the rate of moderate and severe exacerbations was reduced only "by a fraction of an event in 1 year," said Kiya Hamilton, Ph.D., a biostatistician reviewer at the FDA.
Treatment was associated with an increase in local steroid effects, such as oral candidiasis, as expected. In the exacerbation studies, there were more cases of pneumonia and an increase in pneumonia-related deaths with increasing FF doses, and more fractures among those on the FF/VI combination. Bone loss and pneumonia are recognized as adverse effects of ICS in COPD patients, and with one exception, all the deaths were among those on 200/25 mcg dose, which is not going to be marketed, the FDA said.
In a 12 to 1 vote, the panel agreed that the efficacy data provided substantial evidence that the proposed dose provided "a clinically meaningful benefit" for the long-term maintenance treatment of airflow obstruction in COPD. But they were less united on the impact on exacerbations, voting 8 to 5 that the data provided substantial evidence that the reduction in exacerbations seen in the study was clinically meaningful. The panel also voted 10 to 3 that the safety of the 100/25 mcg dose had been adequately demonstrated for the COPD indication.
Panelists supporting approval cited the totality of the database and the utility of having a once a day dose as among the reasons for their votes. Dr. James Stoller, the Jean Wall Bennett Professor of Medicine at the Cleveland Clinic, supported approval and said that the safety profile was acceptable as long as the formulation was not used to treat patients with mild COPD.
Like other panelists who did not support approval, Dr. Paula Carvalho, professor of medicine in the division of pulmonary and critical care medicine, University of Washington, Seattle, said the benefit-risk profile was not adequate. She added that she was more impressed by the effect that treatment with VI 25 mcg alone had on COPD exacerbations than with the combination. Also voting against approval was Peter Peduzzi, Ph.D., professor of public health at the Yale School of Public Health in New Haven, Conn., who said that the impact on trough FEV1 appeared to be driven by the VI component, and like others voting against approval, he cited safety issues.
If approved, GSK plans to market the product as Breo Ellipta. The FDA’s deadline for making a decision on the approval is May 12. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist might be given a waiver, but not at this meeting.
The product is under review for both COPD and asthma in the European Union and in Japan. It is not under review for asthma in the United States.
AT AN FDA ADVISORY PANEL MEETING