User login
Diagnosis: Henoch-Schönlein purpura
Henoch-Schönlein purpura (HSP), also known as immunoglobulin A (IgA) vasculitis, is characterized by palpable purpura, abdominal pain, renal disease, arthritis and/or arthralgia. HSP affects all ages, but is most common in children (the mean age of onset is between six and seven years of age). It also has a slight predilection for males, as well as whites and Asians. Patients are more likely to present during the fall, winter, and spring. It is thought that HSP may be associated with infections that are more prevalent during these times of the year.
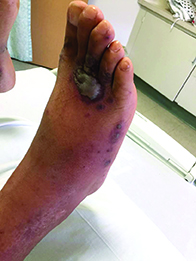
Patients typically have purpura and joint or abdominal pain, although some may present without significant skin manifestations or pain. The rash associated with HSP may appear as erythematous macules or papules that coalesce and evolve into palpable purpura with petechiae, ecchymoses, and/or subcutaneous edema. The skin is typically affected in areas that are more dependent or experience more pressure, such as the lower extremities or buttocks. A more diffuse distribution across the entire body can be seen in those who are nonambulatory.
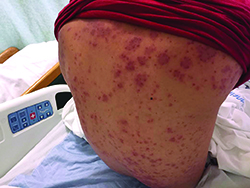
The arthritis associated with HSP usually affects the lower extremities, and it is oligoarticular, nondeforming, and transient, with prominent swelling and tenderness. Gastrointestinal involvement may manifest as nausea, vomiting, abdominal pain, transient paralytic ileus, hemorrhage, bowel ischemia, intussusception (rare in adults), and/or bowel perforation. These symptoms tend to develop 1 week after the rash, but there are reports of gastrointestinal complaints preceding all other manifestations of HSP.
Additionally, there can be renal involvement, which is more common in adults. These patients may experience hematuria with or without red blood cell casts, nephrotic range proteinuria, and/or elevated serum creatinine. Other less common symptoms that have been reported in patients with HSP include scrotal pain and swelling, headaches, seizures, ataxia, central and peripheral neuropathies, impaired lung diffusion capacity, keratitis, and uveitis.
The diagnosis of HSP is classically based upon clinical presentation. The diagnosis may be complicated in cases in which patients do not present with classic signs, such as palpable purpura of the lower extremities and buttocks. In these situations, a biopsy of the skin or kidney may aid in the diagnosis.
The differential diagnosis for HSP includes acute hemorrhagic edema of infancy, coagulopathies, hemolytic uremic syndrome, hypersensitivity vasculitis, IgA nephropathy, immune thrombocytopenia purpura, juvenile idiopathic arthritis, reactive arthritis, rheumatic fever, small vessel vasculitis, and systemic lupus erythematosus.
The etiology of HSP is unclear. Most believe that it is an immune-mediated vasculitis with genetic and environmental influences. In HSP, there is leukocytoclastic vasculitis with deposition of IgA immune complexes, complement component 3 (C3), and fibrin in the affected vessel walls. Small vessels within the papillary dermis are usually present with an inflammatory infiltrate of neutrophils and monocytes.
The treatment of HSP usually consists of supportive care, since most patients will recover spontaneously. Therefore, hydration, rest, and pain relief are essential. Other treatment modalities may be required for more severe or extensive HSP and its complications.
Our patient’s biopsy results were consistent with a leukocytoclastic vasculitis. His biopsy revealed an inflammatory infiltrate of lymphocytes, neutrophils, and nuclear dust with extravasation of erythrocytes and fibrin in the walls of small blood vessels, along with prominent papillary dermal edema and subepidermal vesiculation. Direct immunofluorescence was positive for IgA, C3, and fibrinogen in upper dermal blood vessels. The patient also had renal involvement that was confirmed with a renal biopsy.
This case and photos are courtesy of Tanya Greywal, University of California, San Diego, and Dr. Brooke Resh Sateesh, San Diego Family Dermatology.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Diagnosis: Henoch-Schönlein purpura
Henoch-Schönlein purpura (HSP), also known as immunoglobulin A (IgA) vasculitis, is characterized by palpable purpura, abdominal pain, renal disease, arthritis and/or arthralgia. HSP affects all ages, but is most common in children (the mean age of onset is between six and seven years of age). It also has a slight predilection for males, as well as whites and Asians. Patients are more likely to present during the fall, winter, and spring. It is thought that HSP may be associated with infections that are more prevalent during these times of the year.
Patients typically have purpura and joint or abdominal pain, although some may present without significant skin manifestations or pain. The rash associated with HSP may appear as erythematous macules or papules that coalesce and evolve into palpable purpura with petechiae, ecchymoses, and/or subcutaneous edema. The skin is typically affected in areas that are more dependent or experience more pressure, such as the lower extremities or buttocks. A more diffuse distribution across the entire body can be seen in those who are nonambulatory.
The arthritis associated with HSP usually affects the lower extremities, and it is oligoarticular, nondeforming, and transient, with prominent swelling and tenderness. Gastrointestinal involvement may manifest as nausea, vomiting, abdominal pain, transient paralytic ileus, hemorrhage, bowel ischemia, intussusception (rare in adults), and/or bowel perforation. These symptoms tend to develop 1 week after the rash, but there are reports of gastrointestinal complaints preceding all other manifestations of HSP.
Additionally, there can be renal involvement, which is more common in adults. These patients may experience hematuria with or without red blood cell casts, nephrotic range proteinuria, and/or elevated serum creatinine. Other less common symptoms that have been reported in patients with HSP include scrotal pain and swelling, headaches, seizures, ataxia, central and peripheral neuropathies, impaired lung diffusion capacity, keratitis, and uveitis.
The diagnosis of HSP is classically based upon clinical presentation. The diagnosis may be complicated in cases in which patients do not present with classic signs, such as palpable purpura of the lower extremities and buttocks. In these situations, a biopsy of the skin or kidney may aid in the diagnosis.
The differential diagnosis for HSP includes acute hemorrhagic edema of infancy, coagulopathies, hemolytic uremic syndrome, hypersensitivity vasculitis, IgA nephropathy, immune thrombocytopenia purpura, juvenile idiopathic arthritis, reactive arthritis, rheumatic fever, small vessel vasculitis, and systemic lupus erythematosus.
The etiology of HSP is unclear. Most believe that it is an immune-mediated vasculitis with genetic and environmental influences. In HSP, there is leukocytoclastic vasculitis with deposition of IgA immune complexes, complement component 3 (C3), and fibrin in the affected vessel walls. Small vessels within the papillary dermis are usually present with an inflammatory infiltrate of neutrophils and monocytes.
The treatment of HSP usually consists of supportive care, since most patients will recover spontaneously. Therefore, hydration, rest, and pain relief are essential. Other treatment modalities may be required for more severe or extensive HSP and its complications.
Our patient’s biopsy results were consistent with a leukocytoclastic vasculitis. His biopsy revealed an inflammatory infiltrate of lymphocytes, neutrophils, and nuclear dust with extravasation of erythrocytes and fibrin in the walls of small blood vessels, along with prominent papillary dermal edema and subepidermal vesiculation. Direct immunofluorescence was positive for IgA, C3, and fibrinogen in upper dermal blood vessels. The patient also had renal involvement that was confirmed with a renal biopsy.
This case and photos are courtesy of Tanya Greywal, University of California, San Diego, and Dr. Brooke Resh Sateesh, San Diego Family Dermatology.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Diagnosis: Henoch-Schönlein purpura
Henoch-Schönlein purpura (HSP), also known as immunoglobulin A (IgA) vasculitis, is characterized by palpable purpura, abdominal pain, renal disease, arthritis and/or arthralgia. HSP affects all ages, but is most common in children (the mean age of onset is between six and seven years of age). It also has a slight predilection for males, as well as whites and Asians. Patients are more likely to present during the fall, winter, and spring. It is thought that HSP may be associated with infections that are more prevalent during these times of the year.
Patients typically have purpura and joint or abdominal pain, although some may present without significant skin manifestations or pain. The rash associated with HSP may appear as erythematous macules or papules that coalesce and evolve into palpable purpura with petechiae, ecchymoses, and/or subcutaneous edema. The skin is typically affected in areas that are more dependent or experience more pressure, such as the lower extremities or buttocks. A more diffuse distribution across the entire body can be seen in those who are nonambulatory.
The arthritis associated with HSP usually affects the lower extremities, and it is oligoarticular, nondeforming, and transient, with prominent swelling and tenderness. Gastrointestinal involvement may manifest as nausea, vomiting, abdominal pain, transient paralytic ileus, hemorrhage, bowel ischemia, intussusception (rare in adults), and/or bowel perforation. These symptoms tend to develop 1 week after the rash, but there are reports of gastrointestinal complaints preceding all other manifestations of HSP.
Additionally, there can be renal involvement, which is more common in adults. These patients may experience hematuria with or without red blood cell casts, nephrotic range proteinuria, and/or elevated serum creatinine. Other less common symptoms that have been reported in patients with HSP include scrotal pain and swelling, headaches, seizures, ataxia, central and peripheral neuropathies, impaired lung diffusion capacity, keratitis, and uveitis.
The diagnosis of HSP is classically based upon clinical presentation. The diagnosis may be complicated in cases in which patients do not present with classic signs, such as palpable purpura of the lower extremities and buttocks. In these situations, a biopsy of the skin or kidney may aid in the diagnosis.
The differential diagnosis for HSP includes acute hemorrhagic edema of infancy, coagulopathies, hemolytic uremic syndrome, hypersensitivity vasculitis, IgA nephropathy, immune thrombocytopenia purpura, juvenile idiopathic arthritis, reactive arthritis, rheumatic fever, small vessel vasculitis, and systemic lupus erythematosus.
The etiology of HSP is unclear. Most believe that it is an immune-mediated vasculitis with genetic and environmental influences. In HSP, there is leukocytoclastic vasculitis with deposition of IgA immune complexes, complement component 3 (C3), and fibrin in the affected vessel walls. Small vessels within the papillary dermis are usually present with an inflammatory infiltrate of neutrophils and monocytes.
The treatment of HSP usually consists of supportive care, since most patients will recover spontaneously. Therefore, hydration, rest, and pain relief are essential. Other treatment modalities may be required for more severe or extensive HSP and its complications.
Our patient’s biopsy results were consistent with a leukocytoclastic vasculitis. His biopsy revealed an inflammatory infiltrate of lymphocytes, neutrophils, and nuclear dust with extravasation of erythrocytes and fibrin in the walls of small blood vessels, along with prominent papillary dermal edema and subepidermal vesiculation. Direct immunofluorescence was positive for IgA, C3, and fibrinogen in upper dermal blood vessels. The patient also had renal involvement that was confirmed with a renal biopsy.
This case and photos are courtesy of Tanya Greywal, University of California, San Diego, and Dr. Brooke Resh Sateesh, San Diego Family Dermatology.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
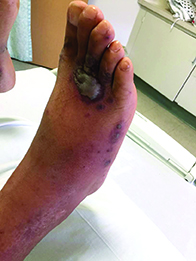
A 40-year-old male with no significant past medical history presented for progressive asymptomatic blisters on the feet, and erythematous lesions on the arms, legs, and trunk. His symptoms began with edema of the feet after a long flight to the Philippines. After a few days, erosions and bullae began forming on his dorsal feet. He was initially evaluated in the Philippines by his primary care physician,who treated him with antibiotics. The patient soon developed pruritic red bumps on his forearms and trunk. The patient was later seen in the emergency department in the United States. At that time, his physical exam was significant for large bullae on a dusky base on the bilateral dorsal feet, and scattered erythematous papules and violaceous macules with dusky centers on the legs, arms, and trunk.