User login
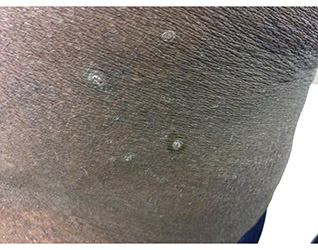
Diagnosis: Perforating disorder
Histology on this patient revealed compact hyperkeratosis and parakeratosis with an entrapped neutrophilic crust and invaginated epidermis. The epidermis showed collagen fibers and the underlying dermis was fibrotic with inflammatory cells. PAS was negative. A Verhoeff-Van Gieson (VVG) stain failed to reveal elastic tissue fibers within the crusted stratum corneum. The results were consistent with a perforating disorder.
Perforating dermatoses result from the extrusion of dermal substances through epidermal channels without any alteration of nearby histologic structures. The substances in the upper dermis induce epidermal hyperplasia that results in the epidermis surrounding the dermal materials, with eventual elimination secondary to keratinocyte maturation and sloughing. Several different forms of perforating dermatoses exist, including reactive perforating collagenosis and the acquired perforating dermatosis diagnostic group (often associated with diabetes mellitus or chronic renal disease), which includes acquired reactive perforating collagenosis (RPC), elastosis perforans serpiginosa, perforating folliculitis, and Kyrle’s disease. Both RPC and perforating folliculitis may demonstrate koebnerization (a phenomenon in which superficial trauma induces lesion formation), which may help a clinician further delineate the specific perforating dermatosis diagnosis.
Reactive perforating collagenosis (RPC) consists of both a familial and an acquired form. The familial form is typically autosomal recessive and presents in childhood but may persist into adulthood. The acquired form develops in adulthood. Clinically, RPC presents as pruritic papules (1-3 mm) with a central dark keratotic plug, which enlarge but often regress after 2 months. Lesions often occur on the extremities and face with onset after superficial trauma (associated with scratching, arthropod bites, etc.). With acquired RPC, underlying systemic diseases are often present, including renal failure, diabetes, and occasionally liver disease or malignant neoplasm. Laboratory tests to screen for these conditions should be ordered as appropriate. In managing RPC, avoiding skin trauma and controlling itching appear to be the most important interventions. Lesions may resolve without treatment, but topical retinoids also may be useful.
Elastosis perforans serpiginosa (EPS) results from changes in both elastic and collagen fibers within the dermis, resulting in a foreign body reaction with transepidermal elimination. Clinically, EPS presents around the second decade of life as erythematous umbilicated papules (2-3 mm) with a central keratin plug arranged in an annular or serpiginous pattern. Lesions usually only involve one anatomic area (frequently the neck and upper extremities). Some of the associated conditions can be remembered with the mnemonic MADPPORES: Marfan syndrome, Acrogeria, Down syndrome, Penicillamine, Pseudoxanthoma elasticum, Osteogenesis imperfecta, Rothmund-Thomson syndrome, Ehlers-Danlos (some forms), and Scleroderma. Regression of lesions often leaves hypopigmented scars. Treatment with cryotherapy may be useful, but electrocautery, dermabrasion, and surgery should be avoided.
Perforating folliculitis is characterized by adult-onset scaly follicular papules with a white central keratotic plug (may contain curled hair) on the extensor surfaces of the extremities and buttocks. Lesions may be pruritic but are more often asymptomatic. The pathophysiology includes the transfollicular extrusion of dermal debris (keratin, cellular debris, degenerated collagen, or degenerated elastic fibers), which may be expressed when the papule is compressed. Perforating folliculitis is often associated with chronic renal failure, diabetes mellitus, and other systemic diseases. The lesions may resolve after stabilization of the underlying disease, including renal transplantation. Management focuses on controlling pruritus with oral antihistamines and topical soothing lotions (menthol, phenol, or camphor), but other unsubstantiated treatments have been tried, including UVB, oral retinoids, and topical retinoids.
Kyrle’s disease (hyperkeratosis folliculitis) may have both an acquired and autosomal recessive form. In the inherited form, a genetic disturbance may accelerate epidermal keratinization, resulting in dyskeratotic foci and parakeratosis that penetrate the dermis and induce a granulomatous inflammatory reaction. The acquired form may be associated with diabetic nephropathy, hemodialysis secondary to diabetic nephropathy, congestive heart failure, and liver disease. In contrast to other perforating disorders, the extruded material is predominantly keratin rather than elastic fibers or collagen. Clinically, Kyrle’s disease presents around age 30 years as discrete, pruritic, and scaly folliculocentric papules (1-8 mm) with a hyperkeratotic plug (one-third of diameter). With removal of the plug, a cutaneous depression remains (Kyrle’s phenomenon). The lesions also may coalesce into plaques. Typically, the bilateral lower extremities and extensor surfaces are involved, but lesions may present on any area except for plantar, palmar, and mucosal surfaces. Management should focus on treating the associated medical condition, but UVB and tretinoin also have been used. Other treatment options include allopurinol, psoralen plus UVA, acitretin, phenol, doxycycline, surgical debridement, and topical corticosteroids. Regardless, lesions may resolve and leave atrophic scars.
This case and photo were submitted by Dr. Damon McClain of Three Rivers Dermatology in Coraopolis, Pa., and Mark Ash, a medical student at East Carolina University, Greenville, N.C.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Diagnosis: Perforating disorder
Histology on this patient revealed compact hyperkeratosis and parakeratosis with an entrapped neutrophilic crust and invaginated epidermis. The epidermis showed collagen fibers and the underlying dermis was fibrotic with inflammatory cells. PAS was negative. A Verhoeff-Van Gieson (VVG) stain failed to reveal elastic tissue fibers within the crusted stratum corneum. The results were consistent with a perforating disorder.
Perforating dermatoses result from the extrusion of dermal substances through epidermal channels without any alteration of nearby histologic structures. The substances in the upper dermis induce epidermal hyperplasia that results in the epidermis surrounding the dermal materials, with eventual elimination secondary to keratinocyte maturation and sloughing. Several different forms of perforating dermatoses exist, including reactive perforating collagenosis and the acquired perforating dermatosis diagnostic group (often associated with diabetes mellitus or chronic renal disease), which includes acquired reactive perforating collagenosis (RPC), elastosis perforans serpiginosa, perforating folliculitis, and Kyrle’s disease. Both RPC and perforating folliculitis may demonstrate koebnerization (a phenomenon in which superficial trauma induces lesion formation), which may help a clinician further delineate the specific perforating dermatosis diagnosis.
Reactive perforating collagenosis (RPC) consists of both a familial and an acquired form. The familial form is typically autosomal recessive and presents in childhood but may persist into adulthood. The acquired form develops in adulthood. Clinically, RPC presents as pruritic papules (1-3 mm) with a central dark keratotic plug, which enlarge but often regress after 2 months. Lesions often occur on the extremities and face with onset after superficial trauma (associated with scratching, arthropod bites, etc.). With acquired RPC, underlying systemic diseases are often present, including renal failure, diabetes, and occasionally liver disease or malignant neoplasm. Laboratory tests to screen for these conditions should be ordered as appropriate. In managing RPC, avoiding skin trauma and controlling itching appear to be the most important interventions. Lesions may resolve without treatment, but topical retinoids also may be useful.
Elastosis perforans serpiginosa (EPS) results from changes in both elastic and collagen fibers within the dermis, resulting in a foreign body reaction with transepidermal elimination. Clinically, EPS presents around the second decade of life as erythematous umbilicated papules (2-3 mm) with a central keratin plug arranged in an annular or serpiginous pattern. Lesions usually only involve one anatomic area (frequently the neck and upper extremities). Some of the associated conditions can be remembered with the mnemonic MADPPORES: Marfan syndrome, Acrogeria, Down syndrome, Penicillamine, Pseudoxanthoma elasticum, Osteogenesis imperfecta, Rothmund-Thomson syndrome, Ehlers-Danlos (some forms), and Scleroderma. Regression of lesions often leaves hypopigmented scars. Treatment with cryotherapy may be useful, but electrocautery, dermabrasion, and surgery should be avoided.
Perforating folliculitis is characterized by adult-onset scaly follicular papules with a white central keratotic plug (may contain curled hair) on the extensor surfaces of the extremities and buttocks. Lesions may be pruritic but are more often asymptomatic. The pathophysiology includes the transfollicular extrusion of dermal debris (keratin, cellular debris, degenerated collagen, or degenerated elastic fibers), which may be expressed when the papule is compressed. Perforating folliculitis is often associated with chronic renal failure, diabetes mellitus, and other systemic diseases. The lesions may resolve after stabilization of the underlying disease, including renal transplantation. Management focuses on controlling pruritus with oral antihistamines and topical soothing lotions (menthol, phenol, or camphor), but other unsubstantiated treatments have been tried, including UVB, oral retinoids, and topical retinoids.
Kyrle’s disease (hyperkeratosis folliculitis) may have both an acquired and autosomal recessive form. In the inherited form, a genetic disturbance may accelerate epidermal keratinization, resulting in dyskeratotic foci and parakeratosis that penetrate the dermis and induce a granulomatous inflammatory reaction. The acquired form may be associated with diabetic nephropathy, hemodialysis secondary to diabetic nephropathy, congestive heart failure, and liver disease. In contrast to other perforating disorders, the extruded material is predominantly keratin rather than elastic fibers or collagen. Clinically, Kyrle’s disease presents around age 30 years as discrete, pruritic, and scaly folliculocentric papules (1-8 mm) with a hyperkeratotic plug (one-third of diameter). With removal of the plug, a cutaneous depression remains (Kyrle’s phenomenon). The lesions also may coalesce into plaques. Typically, the bilateral lower extremities and extensor surfaces are involved, but lesions may present on any area except for plantar, palmar, and mucosal surfaces. Management should focus on treating the associated medical condition, but UVB and tretinoin also have been used. Other treatment options include allopurinol, psoralen plus UVA, acitretin, phenol, doxycycline, surgical debridement, and topical corticosteroids. Regardless, lesions may resolve and leave atrophic scars.
This case and photo were submitted by Dr. Damon McClain of Three Rivers Dermatology in Coraopolis, Pa., and Mark Ash, a medical student at East Carolina University, Greenville, N.C.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Diagnosis: Perforating disorder
Histology on this patient revealed compact hyperkeratosis and parakeratosis with an entrapped neutrophilic crust and invaginated epidermis. The epidermis showed collagen fibers and the underlying dermis was fibrotic with inflammatory cells. PAS was negative. A Verhoeff-Van Gieson (VVG) stain failed to reveal elastic tissue fibers within the crusted stratum corneum. The results were consistent with a perforating disorder.
Perforating dermatoses result from the extrusion of dermal substances through epidermal channels without any alteration of nearby histologic structures. The substances in the upper dermis induce epidermal hyperplasia that results in the epidermis surrounding the dermal materials, with eventual elimination secondary to keratinocyte maturation and sloughing. Several different forms of perforating dermatoses exist, including reactive perforating collagenosis and the acquired perforating dermatosis diagnostic group (often associated with diabetes mellitus or chronic renal disease), which includes acquired reactive perforating collagenosis (RPC), elastosis perforans serpiginosa, perforating folliculitis, and Kyrle’s disease. Both RPC and perforating folliculitis may demonstrate koebnerization (a phenomenon in which superficial trauma induces lesion formation), which may help a clinician further delineate the specific perforating dermatosis diagnosis.
Reactive perforating collagenosis (RPC) consists of both a familial and an acquired form. The familial form is typically autosomal recessive and presents in childhood but may persist into adulthood. The acquired form develops in adulthood. Clinically, RPC presents as pruritic papules (1-3 mm) with a central dark keratotic plug, which enlarge but often regress after 2 months. Lesions often occur on the extremities and face with onset after superficial trauma (associated with scratching, arthropod bites, etc.). With acquired RPC, underlying systemic diseases are often present, including renal failure, diabetes, and occasionally liver disease or malignant neoplasm. Laboratory tests to screen for these conditions should be ordered as appropriate. In managing RPC, avoiding skin trauma and controlling itching appear to be the most important interventions. Lesions may resolve without treatment, but topical retinoids also may be useful.
Elastosis perforans serpiginosa (EPS) results from changes in both elastic and collagen fibers within the dermis, resulting in a foreign body reaction with transepidermal elimination. Clinically, EPS presents around the second decade of life as erythematous umbilicated papules (2-3 mm) with a central keratin plug arranged in an annular or serpiginous pattern. Lesions usually only involve one anatomic area (frequently the neck and upper extremities). Some of the associated conditions can be remembered with the mnemonic MADPPORES: Marfan syndrome, Acrogeria, Down syndrome, Penicillamine, Pseudoxanthoma elasticum, Osteogenesis imperfecta, Rothmund-Thomson syndrome, Ehlers-Danlos (some forms), and Scleroderma. Regression of lesions often leaves hypopigmented scars. Treatment with cryotherapy may be useful, but electrocautery, dermabrasion, and surgery should be avoided.
Perforating folliculitis is characterized by adult-onset scaly follicular papules with a white central keratotic plug (may contain curled hair) on the extensor surfaces of the extremities and buttocks. Lesions may be pruritic but are more often asymptomatic. The pathophysiology includes the transfollicular extrusion of dermal debris (keratin, cellular debris, degenerated collagen, or degenerated elastic fibers), which may be expressed when the papule is compressed. Perforating folliculitis is often associated with chronic renal failure, diabetes mellitus, and other systemic diseases. The lesions may resolve after stabilization of the underlying disease, including renal transplantation. Management focuses on controlling pruritus with oral antihistamines and topical soothing lotions (menthol, phenol, or camphor), but other unsubstantiated treatments have been tried, including UVB, oral retinoids, and topical retinoids.
Kyrle’s disease (hyperkeratosis folliculitis) may have both an acquired and autosomal recessive form. In the inherited form, a genetic disturbance may accelerate epidermal keratinization, resulting in dyskeratotic foci and parakeratosis that penetrate the dermis and induce a granulomatous inflammatory reaction. The acquired form may be associated with diabetic nephropathy, hemodialysis secondary to diabetic nephropathy, congestive heart failure, and liver disease. In contrast to other perforating disorders, the extruded material is predominantly keratin rather than elastic fibers or collagen. Clinically, Kyrle’s disease presents around age 30 years as discrete, pruritic, and scaly folliculocentric papules (1-8 mm) with a hyperkeratotic plug (one-third of diameter). With removal of the plug, a cutaneous depression remains (Kyrle’s phenomenon). The lesions also may coalesce into plaques. Typically, the bilateral lower extremities and extensor surfaces are involved, but lesions may present on any area except for plantar, palmar, and mucosal surfaces. Management should focus on treating the associated medical condition, but UVB and tretinoin also have been used. Other treatment options include allopurinol, psoralen plus UVA, acitretin, phenol, doxycycline, surgical debridement, and topical corticosteroids. Regardless, lesions may resolve and leave atrophic scars.
This case and photo were submitted by Dr. Damon McClain of Three Rivers Dermatology in Coraopolis, Pa., and Mark Ash, a medical student at East Carolina University, Greenville, N.C.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
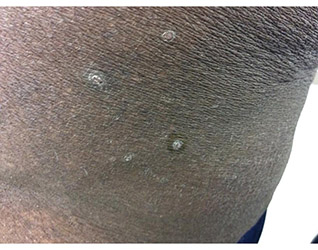
A 41-year-old black male with a past medical history of a kidney transplant, currently on dialysis and waiting for another kidney transplant, presented with a 2-month history of a pruritic rash on the upper body consisting of erythematous papules with central erosions.