User login
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
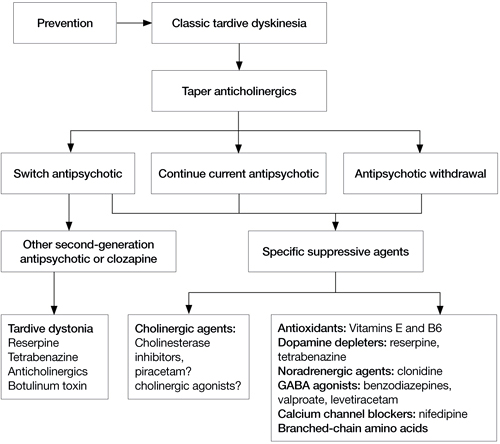
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier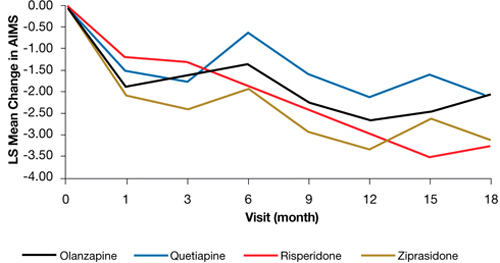
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.