User login
The Case
A 52-year-old man presents with abdominal pain. His temperature is 100.8°F, his blood pressure is 170/90 mm/Hg, and his pulse is 110 beats per minute. On exam, he has 2+ lower extremity edema, periorbital edema, and left-sided flank tenderness. His BUN is 42 mg/dL, his creatinine is 2.5 mg/dL, and his albumin is 1.4 g/dL. Urinalysis shows 2+ protein, large blood, and red blood cells (RBCs). What are the next steps in his diagnosis?
Overview
Glomerular diseases involve a wide spectrum of disease processes. They can result from an acute illness, such as an upper respiratory infection that self-resolves, or from chronic disease states, such as HIV. In some instances, such illnesses as systemic lupus erythematosus (SLE) can cause rapidly progressive renal failure, requiring prompt intervention. While glomerular diseases can be daunting, it is essential for hospitalists to be familiar with fundamental concepts and key features unique to each syndrome.
The approach to glomerulonephritis (GN) can be simplified by summarizing various types into the two broad categories of nephrotic and nephritic syndromes, and identifying the key clinical findings (see Table 1, p. below).
The major subtypes of nephrotic syndrome are minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN), and membranoproliferative glomerulonephritis (MPGN). The clinical manifestations of nephrotic syndrome are edema, hyperlipidemia, lipiduria, and hypoalbuminemia.1 The urinalysis is significant for >3.5 g/day of proteinuria showing fatty casts or oval fat bodies.2 The loss of other proteins, such as anti-thrombin III, may put patients at higher risk for developing venous thromboses.1
The major subtypes of nephritic syndrome are post-streptococcal glomerulonephritis (PSGS), IgA nephropathy, Henoch-Schonlein Purpura (HSP), and rapidly progressive GN (RPGN types I, II, and III). The clinical manifestations of nephritic syndrome are hypertension (HTN) and hematuria.1 Nephritic syndromes may present with more rapidly progressive renal failure when compared with nephrotic syndrome.1 The urinalysis is significant for hematuria with RBC casts, and variable levels of proteinuria (typically, less than 3.5 g/day is seen in nephritic syndrome).1
Review of the Data
Nephrotic Syndromes
Minimal change disease. MCD is more common in children than adults, and only accounts for 10% to 15% of glomerular disease cases in adults.3 It is associated with Hodgkin’s lymphoma, NSAID use, and allergic conditions. There usually is an absence of hypertension (HTN). There are no glomerular basement membrane abnormalities seen on light microscopy. Electron microscopy shows effacement of podocytes. On urinalysis, oval fat bodies are seen, which are characteristic of heavy proteinuria. Complement levels are normal. Steroids are first-line treatment, but in adults with relapses or steroid resistance, immunosuppressive agents have also been used.2
Focal segment glomerlosclerosis.
FSGS is the most common primary glomerular disorder in the United States and is the most common cause of nephrotic syndrome among blacks.4,5 It is associated with HIV (collapsing variant), parvovirus B19, heroin use, sickle-cell disease, obesity, chronic vesicoureteral reflux, and HTN.4,6 Sclerosis of segmental glomeruli is seen on light microscopy.
Electron microscopy shows effacement of podocytes. Complement levels are normal. Treatment of primary idiopathic FSGS includes use of renin-angiotensin inhibitors and steroids.4 Immunosuppressives are reserved for relapses. Treatment of secondary FSGS involves identifying the underlying cause.

Membranous nephropathy. MN is twice as common in males as in females and is the most common cause of adult-onset idiopathic nephrotic syndrome, with the average presentation in the fifth or sixth decade of life.7,8 Aside from its idiopathic form, up to 25% of MN cases have an underlying disease process, such as solid organ tumors or hepatitis B.7,9 While nephrotic syndrome overall can increase the risk of thromboembolic complications, MN is the most common nephrotic disorder predisposing the development of renal vein thrombosis.7 Diffuse capillary wall thickening is seen on light microscopy, and electron microscopy shows sub-epithelial immune deposits. Complement levels are normal. Steroids and immunosuppressive agents are used for treatment.10
Membranoproliferative glomerulonephritis. MPGN is a nephrotic syndrome that is more common in children and young adults and can present with features of nephritic syndrome.1,11 It is associated with hepatitis C, SLE, and cryoglobulinemia.11 Light microscopy shows mesangial and endocapillary proliferation, as well as glomerular basement membrane thickening and splitting (“tram track” appearance). Electron microscopy shows subendothelial and dense deposits. It presents with reduced complement levels (C3 and C4).11 Treatment depends on the associated disease.
Nephritic Syndromes
Post-streptococcal glomerulonephritis. PSGN is seen in children and young adults and is associated with skin (impetigo) and throat infections (pharyngitis).12 Hematuria usually presents two to three weeks after a streptococcal infection. The urine is classically dark and smoky-colored. Levels of C3 and CH50 are low, but C4 levels are normal.1 In addition, there are positive antibody titers for ASO and anti-DNase. Light microscopy shows hypercellularity of glomeruli. Electron microscopy shows dome-shaped sub-endothelial deposits. Treatment is usually supportive.
IgA nephropathy. IgA nephropathy is the most common form of glomerular disease worldwide and the most common form of glomerular-related microscopic hematuria in all age groups.2,13 It occurs in all ages but more frequently in males.14 It occurs during or immediately after an upper respiratory infection. Light microscopy shows mesangial cell proliferation and crescentic GN. Electron microscopy shows immune deposits in the mesangium. Complement levels are normal. There has been no proven therapy, but ACE inhibitors (ACE-Is), angiotensin receptor blockers (ARBs), fish oil, steroids, and tonsillectomy have been used with some success.14 The clinical course of IgA nephropathy can be highly variable, with the potential for a benign course to rapidly progressive renal failure, with 15% to 40% of patients developing end-stage renal disease.14
Henoch-Schonlein purpura. HSP affects children more than adults and is the systemic form of IgA nephropathy.14 Most cases are idiopathic. Clinical
manifestations include: HTN; purpuric palpable rash on buttocks, ankles, and legs; bloody diarrhea with abdominal cramps; and pain in wrist, ankle, and knee joints.15 Light microscopy shows mesangial cell proliferation. Immune deposits in the mesangium are seen on electron microscopy. Complement levels are normal. Treatment is supportive.
Rapidly progressive glomerulonephritis types I, II, and III. RPGN represents a wide variety of disease states in which rapid progression to renal failure is seen within days to weeks.16 They are categorized into three sub-categories: I, II, and III.
Type I is an anti-GBM disease, an example being Goodpasture’s syndrome. This condition presents with hemoptysis, pulmonary infiltrates, and hematuria with RBC casts. Anti-GBM antibodies are classically found.1 Complement levels are normal and a linear immunofluorescence pattern is seen. Treatment is steroids, immunosuppressive agents, and plasmapheresis.17
Type II is an immune complex deposition disease, such as HSP, SLE, or post-streptococcal GN, in which granular complex deposits are seen. Treatment is directed toward treating the underlying cause.
Type III is pauci-immune (no immune deposits), showing necrotizing crescentic GN on biopsy, and is associated with a positive ANCA.1,18 They are associated with systemic small-vessel vasculitis, such as granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis), microscopic polyangiitis, and Churg-Strauss syndrome, or can be limited to renal involvement.1,18 Complement levels are normal. Treatment is steroids and immunosuppressive agents, such as cyclophosphamide.
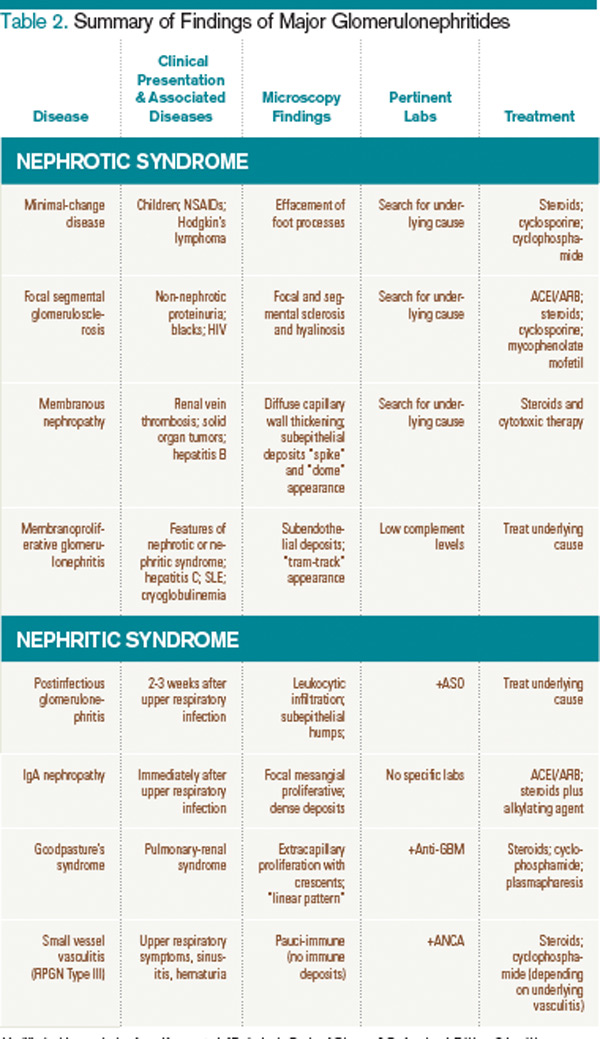
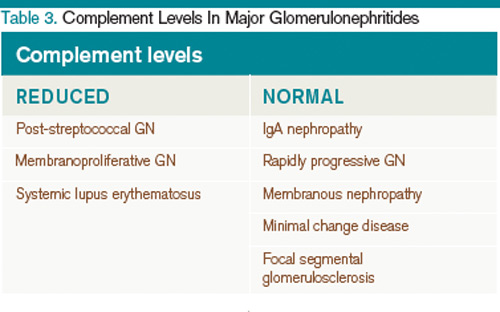
A summary of the findings found in the glomerulonephritides and how complement levels are affected are found in Table 2 and Table 3, respectively.
Secondary Causes of Nephrotic Diseases
Diabetic nephropathy. Diabetic nephropathy is the single most common cause of progressive renal failure in the United States.3 Up to 50% of patients with diabetes present with diabetic nephropathy.19 Current recommendations are to screen yearly for microalbuminuria at the time of diagnosis.3 Treatment involves use of ACE-Is or ARBs to reduce proteinuria and slow the progression of renal disease.
HIV-associated nephropathy. HIV-associated nephropathy commonly presents as the collapsing variant of FSGS. However, it can present as other forms of glomerulopathy, such as MPGN or IgA nephropathy, as well as an immune complex GN with “lupus-like” features without evidence of SLE.19,20 Therefore, HIV nephropathy has now been categorized as a separate entity.3 ACE-Is, HAART therapy, and corticosteroids are the mainstays of treatment.
Amyloidosis. Renal involvement is seen in both primary (AL) and secondary (AA) amyloidosis. Eighty percent of patients with AL have renal disease, and 25% of these patients have nephrotic syndrome.16 Diagnosis is made with Congo Red stain, which shows fibrillary amyloid deposits within the mesangium and capillary walls. Treatment is directed at the underlying process.
Systemic lupus erythematosus. SLE is divided into six classes (I-VI) based on the involvement and severity of renal disease, and steroids and immunosuppressive agents are used for treatment, also based on the severity of the disease.21
Back to the Case
Our patient presented to the hospital with abdominal pain, low-grade fever, HTN, edema, hypoalbuminemia, and new-onset renal failure with gross hematuria and proteinuria. The presence of proteinuria and hypoalbuminemia, combined with peripheral and periorbital edema, suggests glomerular loss of albumin, such as in nephrotic syndrome. His renal failure in the setting of the sudden development of gross hematuria with flank pain is concerning for a renal vein thrombosis, and an abdominal magnetic resonance venography did in fact visualize a renal vein thrombosis.
He was admitted to the hospital and was started on therapeutic intravenous heparin, and bridged to warfarin. Subsequent renal biopsy confirmed the findings of membranous nephropathy, which was suspected due to his renal vein thrombosis. Therapy was initiated with corticosteroids after the biopsy, and he responded well. Because of his risk factors for further thromboembolic events, lifelong anticoagulation therapy was recommended.
Bottom Line
For patients with glomerular disease, differentiating between nephrotic and nephritic syndromes and understanding key clinical and laboratory differences can lead to easier identification and treatment.
Drs. Khan and Smith are assistant professors of medicine, and Dr. Ansari is associate division director, in the Division of Hospital Medicine at Loyola University Medical Center, Maywood, Ill.
References
- Donegio RGB, Salant DJ. Nephrology: Glomerular Diseases. In: ACP Medicine. Dale D, Federman D, eds. Available at: http://www.acpmedicine.com/acpmedicine/institutional/instHtmlReader.action?readerFlag=chapt&chapId=part10_ch05. Accessed Feb. 15, 2012.
- Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. 1998;338:1202-1211.
- Lewis JB, Neilson EG. Chapter 283. Glomerular Diseases. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2012.
- D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398-23411.
- Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis. 2004;44:815-825.
- Balow JE. Nephropathy in the context of HIV infection. Kidney Int. 2005;67:1632-1633.
- Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23:324-332.
- Nickolas TL, Radhakrishnan J, Appel GB. Hyperlipidemia and thrombotic complications in patients with membranous nephropathy. Semin Nephrol. 2003;23:406-411.
- Burstein DM, Korbet SM, Schwartz MM. Membranous glomerulonephritis and malignancy. Am J Kidney Dis. 1993;22:5-10.
- Hofstra JM, Wetzels JF. Management of patients with membranous nephropathy. Nephrol Dial Transplant. 2012;27:6-9.
- Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol. 2010;25:1409-1418.
- Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Pediatr Nephrol. 2011;26:165-180.
- Cohen RA, Brown RS. Clinical practice. Microscopic hematuria. N Engl J Med. 2003;348:2330-2338.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347:738-748.
- McCarthy HJ, Tizard EJ. Clinical practice: Diagnosis and management of henoch-schonlein purpura. Eur J Pediatr. 2010169:643-650.
- Appel GB, Radhakrishnan J. Cecil Medicine: Volume 1: Chapter 123: Glomerular Disorders and Nephrotic Syndromes. MD Consult Preview website. Available at: http://www.mdconsult.com/books/page.do?eid=4-u1.0-B978-1-4377-1604-7..00123-8&isbn=978-1-4377-1604-7&uniqId=313771243-2#4-u1.0-B978-1-4377-1604-7..00123-8. Accessed Feb. 16, 2012.
- Walters G, Willis NS, Craig JC. Interventions for renal vasculitis in adults. Cochrane Database Syst Rev. 2008;(3)(3):CD003232.
- Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164-1177.
- Falk, RJ: Medical Knowledge Self Assessment Program 14. Nephrology: 2006.
- Haas M, Kaul S, Eustace JA. HIV-associated immune complex glomerulonephritis with “lupus-like” features: a clinicopathologic study of 14 cases. Kidney Int. 2005;67:1381.
- Dooley MA, Hogan S, Jennette C, Falk R. Cyclophosphamide therapy for lupus nephritis: poor renal survival in black americans. glomerular disease collaborative network. Kidney Int. 1997;51:1188-1195.
The Case
A 52-year-old man presents with abdominal pain. His temperature is 100.8°F, his blood pressure is 170/90 mm/Hg, and his pulse is 110 beats per minute. On exam, he has 2+ lower extremity edema, periorbital edema, and left-sided flank tenderness. His BUN is 42 mg/dL, his creatinine is 2.5 mg/dL, and his albumin is 1.4 g/dL. Urinalysis shows 2+ protein, large blood, and red blood cells (RBCs). What are the next steps in his diagnosis?
Overview
Glomerular diseases involve a wide spectrum of disease processes. They can result from an acute illness, such as an upper respiratory infection that self-resolves, or from chronic disease states, such as HIV. In some instances, such illnesses as systemic lupus erythematosus (SLE) can cause rapidly progressive renal failure, requiring prompt intervention. While glomerular diseases can be daunting, it is essential for hospitalists to be familiar with fundamental concepts and key features unique to each syndrome.
The approach to glomerulonephritis (GN) can be simplified by summarizing various types into the two broad categories of nephrotic and nephritic syndromes, and identifying the key clinical findings (see Table 1, p. below).
The major subtypes of nephrotic syndrome are minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN), and membranoproliferative glomerulonephritis (MPGN). The clinical manifestations of nephrotic syndrome are edema, hyperlipidemia, lipiduria, and hypoalbuminemia.1 The urinalysis is significant for >3.5 g/day of proteinuria showing fatty casts or oval fat bodies.2 The loss of other proteins, such as anti-thrombin III, may put patients at higher risk for developing venous thromboses.1
The major subtypes of nephritic syndrome are post-streptococcal glomerulonephritis (PSGS), IgA nephropathy, Henoch-Schonlein Purpura (HSP), and rapidly progressive GN (RPGN types I, II, and III). The clinical manifestations of nephritic syndrome are hypertension (HTN) and hematuria.1 Nephritic syndromes may present with more rapidly progressive renal failure when compared with nephrotic syndrome.1 The urinalysis is significant for hematuria with RBC casts, and variable levels of proteinuria (typically, less than 3.5 g/day is seen in nephritic syndrome).1
Review of the Data
Nephrotic Syndromes
Minimal change disease. MCD is more common in children than adults, and only accounts for 10% to 15% of glomerular disease cases in adults.3 It is associated with Hodgkin’s lymphoma, NSAID use, and allergic conditions. There usually is an absence of hypertension (HTN). There are no glomerular basement membrane abnormalities seen on light microscopy. Electron microscopy shows effacement of podocytes. On urinalysis, oval fat bodies are seen, which are characteristic of heavy proteinuria. Complement levels are normal. Steroids are first-line treatment, but in adults with relapses or steroid resistance, immunosuppressive agents have also been used.2
Focal segment glomerlosclerosis.
FSGS is the most common primary glomerular disorder in the United States and is the most common cause of nephrotic syndrome among blacks.4,5 It is associated with HIV (collapsing variant), parvovirus B19, heroin use, sickle-cell disease, obesity, chronic vesicoureteral reflux, and HTN.4,6 Sclerosis of segmental glomeruli is seen on light microscopy.
Electron microscopy shows effacement of podocytes. Complement levels are normal. Treatment of primary idiopathic FSGS includes use of renin-angiotensin inhibitors and steroids.4 Immunosuppressives are reserved for relapses. Treatment of secondary FSGS involves identifying the underlying cause.
Membranous nephropathy. MN is twice as common in males as in females and is the most common cause of adult-onset idiopathic nephrotic syndrome, with the average presentation in the fifth or sixth decade of life.7,8 Aside from its idiopathic form, up to 25% of MN cases have an underlying disease process, such as solid organ tumors or hepatitis B.7,9 While nephrotic syndrome overall can increase the risk of thromboembolic complications, MN is the most common nephrotic disorder predisposing the development of renal vein thrombosis.7 Diffuse capillary wall thickening is seen on light microscopy, and electron microscopy shows sub-epithelial immune deposits. Complement levels are normal. Steroids and immunosuppressive agents are used for treatment.10
Membranoproliferative glomerulonephritis. MPGN is a nephrotic syndrome that is more common in children and young adults and can present with features of nephritic syndrome.1,11 It is associated with hepatitis C, SLE, and cryoglobulinemia.11 Light microscopy shows mesangial and endocapillary proliferation, as well as glomerular basement membrane thickening and splitting (“tram track” appearance). Electron microscopy shows subendothelial and dense deposits. It presents with reduced complement levels (C3 and C4).11 Treatment depends on the associated disease.
Nephritic Syndromes
Post-streptococcal glomerulonephritis. PSGN is seen in children and young adults and is associated with skin (impetigo) and throat infections (pharyngitis).12 Hematuria usually presents two to three weeks after a streptococcal infection. The urine is classically dark and smoky-colored. Levels of C3 and CH50 are low, but C4 levels are normal.1 In addition, there are positive antibody titers for ASO and anti-DNase. Light microscopy shows hypercellularity of glomeruli. Electron microscopy shows dome-shaped sub-endothelial deposits. Treatment is usually supportive.
IgA nephropathy. IgA nephropathy is the most common form of glomerular disease worldwide and the most common form of glomerular-related microscopic hematuria in all age groups.2,13 It occurs in all ages but more frequently in males.14 It occurs during or immediately after an upper respiratory infection. Light microscopy shows mesangial cell proliferation and crescentic GN. Electron microscopy shows immune deposits in the mesangium. Complement levels are normal. There has been no proven therapy, but ACE inhibitors (ACE-Is), angiotensin receptor blockers (ARBs), fish oil, steroids, and tonsillectomy have been used with some success.14 The clinical course of IgA nephropathy can be highly variable, with the potential for a benign course to rapidly progressive renal failure, with 15% to 40% of patients developing end-stage renal disease.14
Henoch-Schonlein purpura. HSP affects children more than adults and is the systemic form of IgA nephropathy.14 Most cases are idiopathic. Clinical
manifestations include: HTN; purpuric palpable rash on buttocks, ankles, and legs; bloody diarrhea with abdominal cramps; and pain in wrist, ankle, and knee joints.15 Light microscopy shows mesangial cell proliferation. Immune deposits in the mesangium are seen on electron microscopy. Complement levels are normal. Treatment is supportive.
Rapidly progressive glomerulonephritis types I, II, and III. RPGN represents a wide variety of disease states in which rapid progression to renal failure is seen within days to weeks.16 They are categorized into three sub-categories: I, II, and III.
Type I is an anti-GBM disease, an example being Goodpasture’s syndrome. This condition presents with hemoptysis, pulmonary infiltrates, and hematuria with RBC casts. Anti-GBM antibodies are classically found.1 Complement levels are normal and a linear immunofluorescence pattern is seen. Treatment is steroids, immunosuppressive agents, and plasmapheresis.17
Type II is an immune complex deposition disease, such as HSP, SLE, or post-streptococcal GN, in which granular complex deposits are seen. Treatment is directed toward treating the underlying cause.
Type III is pauci-immune (no immune deposits), showing necrotizing crescentic GN on biopsy, and is associated with a positive ANCA.1,18 They are associated with systemic small-vessel vasculitis, such as granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis), microscopic polyangiitis, and Churg-Strauss syndrome, or can be limited to renal involvement.1,18 Complement levels are normal. Treatment is steroids and immunosuppressive agents, such as cyclophosphamide.
A summary of the findings found in the glomerulonephritides and how complement levels are affected are found in Table 2 and Table 3, respectively.
Secondary Causes of Nephrotic Diseases
Diabetic nephropathy. Diabetic nephropathy is the single most common cause of progressive renal failure in the United States.3 Up to 50% of patients with diabetes present with diabetic nephropathy.19 Current recommendations are to screen yearly for microalbuminuria at the time of diagnosis.3 Treatment involves use of ACE-Is or ARBs to reduce proteinuria and slow the progression of renal disease.
HIV-associated nephropathy. HIV-associated nephropathy commonly presents as the collapsing variant of FSGS. However, it can present as other forms of glomerulopathy, such as MPGN or IgA nephropathy, as well as an immune complex GN with “lupus-like” features without evidence of SLE.19,20 Therefore, HIV nephropathy has now been categorized as a separate entity.3 ACE-Is, HAART therapy, and corticosteroids are the mainstays of treatment.
Amyloidosis. Renal involvement is seen in both primary (AL) and secondary (AA) amyloidosis. Eighty percent of patients with AL have renal disease, and 25% of these patients have nephrotic syndrome.16 Diagnosis is made with Congo Red stain, which shows fibrillary amyloid deposits within the mesangium and capillary walls. Treatment is directed at the underlying process.
Systemic lupus erythematosus. SLE is divided into six classes (I-VI) based on the involvement and severity of renal disease, and steroids and immunosuppressive agents are used for treatment, also based on the severity of the disease.21
Back to the Case
Our patient presented to the hospital with abdominal pain, low-grade fever, HTN, edema, hypoalbuminemia, and new-onset renal failure with gross hematuria and proteinuria. The presence of proteinuria and hypoalbuminemia, combined with peripheral and periorbital edema, suggests glomerular loss of albumin, such as in nephrotic syndrome. His renal failure in the setting of the sudden development of gross hematuria with flank pain is concerning for a renal vein thrombosis, and an abdominal magnetic resonance venography did in fact visualize a renal vein thrombosis.
He was admitted to the hospital and was started on therapeutic intravenous heparin, and bridged to warfarin. Subsequent renal biopsy confirmed the findings of membranous nephropathy, which was suspected due to his renal vein thrombosis. Therapy was initiated with corticosteroids after the biopsy, and he responded well. Because of his risk factors for further thromboembolic events, lifelong anticoagulation therapy was recommended.
Bottom Line
For patients with glomerular disease, differentiating between nephrotic and nephritic syndromes and understanding key clinical and laboratory differences can lead to easier identification and treatment.
Drs. Khan and Smith are assistant professors of medicine, and Dr. Ansari is associate division director, in the Division of Hospital Medicine at Loyola University Medical Center, Maywood, Ill.
References
- Donegio RGB, Salant DJ. Nephrology: Glomerular Diseases. In: ACP Medicine. Dale D, Federman D, eds. Available at: http://www.acpmedicine.com/acpmedicine/institutional/instHtmlReader.action?readerFlag=chapt&chapId=part10_ch05. Accessed Feb. 15, 2012.
- Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. 1998;338:1202-1211.
- Lewis JB, Neilson EG. Chapter 283. Glomerular Diseases. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2012.
- D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398-23411.
- Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis. 2004;44:815-825.
- Balow JE. Nephropathy in the context of HIV infection. Kidney Int. 2005;67:1632-1633.
- Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23:324-332.
- Nickolas TL, Radhakrishnan J, Appel GB. Hyperlipidemia and thrombotic complications in patients with membranous nephropathy. Semin Nephrol. 2003;23:406-411.
- Burstein DM, Korbet SM, Schwartz MM. Membranous glomerulonephritis and malignancy. Am J Kidney Dis. 1993;22:5-10.
- Hofstra JM, Wetzels JF. Management of patients with membranous nephropathy. Nephrol Dial Transplant. 2012;27:6-9.
- Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol. 2010;25:1409-1418.
- Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Pediatr Nephrol. 2011;26:165-180.
- Cohen RA, Brown RS. Clinical practice. Microscopic hematuria. N Engl J Med. 2003;348:2330-2338.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347:738-748.
- McCarthy HJ, Tizard EJ. Clinical practice: Diagnosis and management of henoch-schonlein purpura. Eur J Pediatr. 2010169:643-650.
- Appel GB, Radhakrishnan J. Cecil Medicine: Volume 1: Chapter 123: Glomerular Disorders and Nephrotic Syndromes. MD Consult Preview website. Available at: http://www.mdconsult.com/books/page.do?eid=4-u1.0-B978-1-4377-1604-7..00123-8&isbn=978-1-4377-1604-7&uniqId=313771243-2#4-u1.0-B978-1-4377-1604-7..00123-8. Accessed Feb. 16, 2012.
- Walters G, Willis NS, Craig JC. Interventions for renal vasculitis in adults. Cochrane Database Syst Rev. 2008;(3)(3):CD003232.
- Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164-1177.
- Falk, RJ: Medical Knowledge Self Assessment Program 14. Nephrology: 2006.
- Haas M, Kaul S, Eustace JA. HIV-associated immune complex glomerulonephritis with “lupus-like” features: a clinicopathologic study of 14 cases. Kidney Int. 2005;67:1381.
- Dooley MA, Hogan S, Jennette C, Falk R. Cyclophosphamide therapy for lupus nephritis: poor renal survival in black americans. glomerular disease collaborative network. Kidney Int. 1997;51:1188-1195.
The Case
A 52-year-old man presents with abdominal pain. His temperature is 100.8°F, his blood pressure is 170/90 mm/Hg, and his pulse is 110 beats per minute. On exam, he has 2+ lower extremity edema, periorbital edema, and left-sided flank tenderness. His BUN is 42 mg/dL, his creatinine is 2.5 mg/dL, and his albumin is 1.4 g/dL. Urinalysis shows 2+ protein, large blood, and red blood cells (RBCs). What are the next steps in his diagnosis?
Overview
Glomerular diseases involve a wide spectrum of disease processes. They can result from an acute illness, such as an upper respiratory infection that self-resolves, or from chronic disease states, such as HIV. In some instances, such illnesses as systemic lupus erythematosus (SLE) can cause rapidly progressive renal failure, requiring prompt intervention. While glomerular diseases can be daunting, it is essential for hospitalists to be familiar with fundamental concepts and key features unique to each syndrome.
The approach to glomerulonephritis (GN) can be simplified by summarizing various types into the two broad categories of nephrotic and nephritic syndromes, and identifying the key clinical findings (see Table 1, p. below).
The major subtypes of nephrotic syndrome are minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN), and membranoproliferative glomerulonephritis (MPGN). The clinical manifestations of nephrotic syndrome are edema, hyperlipidemia, lipiduria, and hypoalbuminemia.1 The urinalysis is significant for >3.5 g/day of proteinuria showing fatty casts or oval fat bodies.2 The loss of other proteins, such as anti-thrombin III, may put patients at higher risk for developing venous thromboses.1
The major subtypes of nephritic syndrome are post-streptococcal glomerulonephritis (PSGS), IgA nephropathy, Henoch-Schonlein Purpura (HSP), and rapidly progressive GN (RPGN types I, II, and III). The clinical manifestations of nephritic syndrome are hypertension (HTN) and hematuria.1 Nephritic syndromes may present with more rapidly progressive renal failure when compared with nephrotic syndrome.1 The urinalysis is significant for hematuria with RBC casts, and variable levels of proteinuria (typically, less than 3.5 g/day is seen in nephritic syndrome).1
Review of the Data
Nephrotic Syndromes
Minimal change disease. MCD is more common in children than adults, and only accounts for 10% to 15% of glomerular disease cases in adults.3 It is associated with Hodgkin’s lymphoma, NSAID use, and allergic conditions. There usually is an absence of hypertension (HTN). There are no glomerular basement membrane abnormalities seen on light microscopy. Electron microscopy shows effacement of podocytes. On urinalysis, oval fat bodies are seen, which are characteristic of heavy proteinuria. Complement levels are normal. Steroids are first-line treatment, but in adults with relapses or steroid resistance, immunosuppressive agents have also been used.2
Focal segment glomerlosclerosis.
FSGS is the most common primary glomerular disorder in the United States and is the most common cause of nephrotic syndrome among blacks.4,5 It is associated with HIV (collapsing variant), parvovirus B19, heroin use, sickle-cell disease, obesity, chronic vesicoureteral reflux, and HTN.4,6 Sclerosis of segmental glomeruli is seen on light microscopy.
Electron microscopy shows effacement of podocytes. Complement levels are normal. Treatment of primary idiopathic FSGS includes use of renin-angiotensin inhibitors and steroids.4 Immunosuppressives are reserved for relapses. Treatment of secondary FSGS involves identifying the underlying cause.
Membranous nephropathy. MN is twice as common in males as in females and is the most common cause of adult-onset idiopathic nephrotic syndrome, with the average presentation in the fifth or sixth decade of life.7,8 Aside from its idiopathic form, up to 25% of MN cases have an underlying disease process, such as solid organ tumors or hepatitis B.7,9 While nephrotic syndrome overall can increase the risk of thromboembolic complications, MN is the most common nephrotic disorder predisposing the development of renal vein thrombosis.7 Diffuse capillary wall thickening is seen on light microscopy, and electron microscopy shows sub-epithelial immune deposits. Complement levels are normal. Steroids and immunosuppressive agents are used for treatment.10
Membranoproliferative glomerulonephritis. MPGN is a nephrotic syndrome that is more common in children and young adults and can present with features of nephritic syndrome.1,11 It is associated with hepatitis C, SLE, and cryoglobulinemia.11 Light microscopy shows mesangial and endocapillary proliferation, as well as glomerular basement membrane thickening and splitting (“tram track” appearance). Electron microscopy shows subendothelial and dense deposits. It presents with reduced complement levels (C3 and C4).11 Treatment depends on the associated disease.
Nephritic Syndromes
Post-streptococcal glomerulonephritis. PSGN is seen in children and young adults and is associated with skin (impetigo) and throat infections (pharyngitis).12 Hematuria usually presents two to three weeks after a streptococcal infection. The urine is classically dark and smoky-colored. Levels of C3 and CH50 are low, but C4 levels are normal.1 In addition, there are positive antibody titers for ASO and anti-DNase. Light microscopy shows hypercellularity of glomeruli. Electron microscopy shows dome-shaped sub-endothelial deposits. Treatment is usually supportive.
IgA nephropathy. IgA nephropathy is the most common form of glomerular disease worldwide and the most common form of glomerular-related microscopic hematuria in all age groups.2,13 It occurs in all ages but more frequently in males.14 It occurs during or immediately after an upper respiratory infection. Light microscopy shows mesangial cell proliferation and crescentic GN. Electron microscopy shows immune deposits in the mesangium. Complement levels are normal. There has been no proven therapy, but ACE inhibitors (ACE-Is), angiotensin receptor blockers (ARBs), fish oil, steroids, and tonsillectomy have been used with some success.14 The clinical course of IgA nephropathy can be highly variable, with the potential for a benign course to rapidly progressive renal failure, with 15% to 40% of patients developing end-stage renal disease.14
Henoch-Schonlein purpura. HSP affects children more than adults and is the systemic form of IgA nephropathy.14 Most cases are idiopathic. Clinical
manifestations include: HTN; purpuric palpable rash on buttocks, ankles, and legs; bloody diarrhea with abdominal cramps; and pain in wrist, ankle, and knee joints.15 Light microscopy shows mesangial cell proliferation. Immune deposits in the mesangium are seen on electron microscopy. Complement levels are normal. Treatment is supportive.
Rapidly progressive glomerulonephritis types I, II, and III. RPGN represents a wide variety of disease states in which rapid progression to renal failure is seen within days to weeks.16 They are categorized into three sub-categories: I, II, and III.
Type I is an anti-GBM disease, an example being Goodpasture’s syndrome. This condition presents with hemoptysis, pulmonary infiltrates, and hematuria with RBC casts. Anti-GBM antibodies are classically found.1 Complement levels are normal and a linear immunofluorescence pattern is seen. Treatment is steroids, immunosuppressive agents, and plasmapheresis.17
Type II is an immune complex deposition disease, such as HSP, SLE, or post-streptococcal GN, in which granular complex deposits are seen. Treatment is directed toward treating the underlying cause.
Type III is pauci-immune (no immune deposits), showing necrotizing crescentic GN on biopsy, and is associated with a positive ANCA.1,18 They are associated with systemic small-vessel vasculitis, such as granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis), microscopic polyangiitis, and Churg-Strauss syndrome, or can be limited to renal involvement.1,18 Complement levels are normal. Treatment is steroids and immunosuppressive agents, such as cyclophosphamide.
A summary of the findings found in the glomerulonephritides and how complement levels are affected are found in Table 2 and Table 3, respectively.
Secondary Causes of Nephrotic Diseases
Diabetic nephropathy. Diabetic nephropathy is the single most common cause of progressive renal failure in the United States.3 Up to 50% of patients with diabetes present with diabetic nephropathy.19 Current recommendations are to screen yearly for microalbuminuria at the time of diagnosis.3 Treatment involves use of ACE-Is or ARBs to reduce proteinuria and slow the progression of renal disease.
HIV-associated nephropathy. HIV-associated nephropathy commonly presents as the collapsing variant of FSGS. However, it can present as other forms of glomerulopathy, such as MPGN or IgA nephropathy, as well as an immune complex GN with “lupus-like” features without evidence of SLE.19,20 Therefore, HIV nephropathy has now been categorized as a separate entity.3 ACE-Is, HAART therapy, and corticosteroids are the mainstays of treatment.
Amyloidosis. Renal involvement is seen in both primary (AL) and secondary (AA) amyloidosis. Eighty percent of patients with AL have renal disease, and 25% of these patients have nephrotic syndrome.16 Diagnosis is made with Congo Red stain, which shows fibrillary amyloid deposits within the mesangium and capillary walls. Treatment is directed at the underlying process.
Systemic lupus erythematosus. SLE is divided into six classes (I-VI) based on the involvement and severity of renal disease, and steroids and immunosuppressive agents are used for treatment, also based on the severity of the disease.21
Back to the Case
Our patient presented to the hospital with abdominal pain, low-grade fever, HTN, edema, hypoalbuminemia, and new-onset renal failure with gross hematuria and proteinuria. The presence of proteinuria and hypoalbuminemia, combined with peripheral and periorbital edema, suggests glomerular loss of albumin, such as in nephrotic syndrome. His renal failure in the setting of the sudden development of gross hematuria with flank pain is concerning for a renal vein thrombosis, and an abdominal magnetic resonance venography did in fact visualize a renal vein thrombosis.
He was admitted to the hospital and was started on therapeutic intravenous heparin, and bridged to warfarin. Subsequent renal biopsy confirmed the findings of membranous nephropathy, which was suspected due to his renal vein thrombosis. Therapy was initiated with corticosteroids after the biopsy, and he responded well. Because of his risk factors for further thromboembolic events, lifelong anticoagulation therapy was recommended.
Bottom Line
For patients with glomerular disease, differentiating between nephrotic and nephritic syndromes and understanding key clinical and laboratory differences can lead to easier identification and treatment.
Drs. Khan and Smith are assistant professors of medicine, and Dr. Ansari is associate division director, in the Division of Hospital Medicine at Loyola University Medical Center, Maywood, Ill.
References
- Donegio RGB, Salant DJ. Nephrology: Glomerular Diseases. In: ACP Medicine. Dale D, Federman D, eds. Available at: http://www.acpmedicine.com/acpmedicine/institutional/instHtmlReader.action?readerFlag=chapt&chapId=part10_ch05. Accessed Feb. 15, 2012.
- Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. 1998;338:1202-1211.
- Lewis JB, Neilson EG. Chapter 283. Glomerular Diseases. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2012.
- D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398-23411.
- Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis. 2004;44:815-825.
- Balow JE. Nephropathy in the context of HIV infection. Kidney Int. 2005;67:1632-1633.
- Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23:324-332.
- Nickolas TL, Radhakrishnan J, Appel GB. Hyperlipidemia and thrombotic complications in patients with membranous nephropathy. Semin Nephrol. 2003;23:406-411.
- Burstein DM, Korbet SM, Schwartz MM. Membranous glomerulonephritis and malignancy. Am J Kidney Dis. 1993;22:5-10.
- Hofstra JM, Wetzels JF. Management of patients with membranous nephropathy. Nephrol Dial Transplant. 2012;27:6-9.
- Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol. 2010;25:1409-1418.
- Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Pediatr Nephrol. 2011;26:165-180.
- Cohen RA, Brown RS. Clinical practice. Microscopic hematuria. N Engl J Med. 2003;348:2330-2338.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347:738-748.
- McCarthy HJ, Tizard EJ. Clinical practice: Diagnosis and management of henoch-schonlein purpura. Eur J Pediatr. 2010169:643-650.
- Appel GB, Radhakrishnan J. Cecil Medicine: Volume 1: Chapter 123: Glomerular Disorders and Nephrotic Syndromes. MD Consult Preview website. Available at: http://www.mdconsult.com/books/page.do?eid=4-u1.0-B978-1-4377-1604-7..00123-8&isbn=978-1-4377-1604-7&uniqId=313771243-2#4-u1.0-B978-1-4377-1604-7..00123-8. Accessed Feb. 16, 2012.
- Walters G, Willis NS, Craig JC. Interventions for renal vasculitis in adults. Cochrane Database Syst Rev. 2008;(3)(3):CD003232.
- Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003;63:1164-1177.
- Falk, RJ: Medical Knowledge Self Assessment Program 14. Nephrology: 2006.
- Haas M, Kaul S, Eustace JA. HIV-associated immune complex glomerulonephritis with “lupus-like” features: a clinicopathologic study of 14 cases. Kidney Int. 2005;67:1381.
- Dooley MA, Hogan S, Jennette C, Falk R. Cyclophosphamide therapy for lupus nephritis: poor renal survival in black americans. glomerular disease collaborative network. Kidney Int. 1997;51:1188-1195.