User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
End-of-life dementia care: A palliative perspective
Dr. Casey reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.

Psychiatrists need to be aware of aspects of psychiatric and pain management that are unique to end-of-life care for patients with advanced dementia. The palliative care philosophy presents an opportunity to honestly acknowledge the terminal nature of advanced dementia, limit intrusive and unnecessary care, and thoughtfully address pain and suffering.
Studies suggest that nursing home patients with severe dementia have an average life expectancy of 2 years.1 These patients often have substantial medical comorbidity, frequently with multiple illnesses, each of which has an accepted management that may involve several medications and interventions. Treatment guidelines for individual conditions don’t necessarily take into account multiple interacting illnesses in advanced dementia. Applying recommended treatments for multiple conditions simultaneously may entail prescribing many medications and interventions. Often these techniques are designed to prevent or modify disease over long term, which a patient with advanced dementia is not likely to achieve.2
Such polypharmacy and intensive intervention are not likely to extend life or improve its quality. In fact, the opposite may occur. Dementia patients react poorly to polypharmacy and may require restraint or sedation to accommodate invasive interventions. Feeding tubes are particularly challenging. Studies have revealed that feeding tubes do not extend life in advanced dementia patients.3
Goals for palliative care. The palliative care approach emphasizes relieving suffering in the near term. Applying this philosophy to advanced dementia depends on acknowledging that the patient will not recover from this condition, has a limited life expectancy, and is not likely to benefit from—and in fact may be harmed by—an aggressive approach to comorbid conditions. Instead, these conditions are best managed by controlling pain and suffering in the near term. Hospitalization and invasive interventions are minimized.
Psychiatric management fits well within this approach. Near the end of life, dementia patients often suffer agitation, psychosis, depression, and delirium that may require the expert, judicious use of psychopharmacology. Patients often experience pain, but might not be able to communicate this, except through behavioral changes. Physicians may be overly concerned with possible adverse effects of pain medications, but when appropriately prescribed, these drugs may help relieve suffering. Psychiatrists also have a role in assisting staff and families during an emotionally difficult time.4
1. Mitchell SL, Kiely DK, Hamel DK, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA. 2004;291:2734-2740.
2. Boyd CM, Darer J, Boult C, et al. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases. JAMA. 2005;294:741-743.
3. Li I. Feeding tubes in patients with severe dementia. Am Fam Physician. 2002;65(8):1605-1610,1515.
4. Lyness JM. End of life care: issues relevant to the geriatric psychiatrist. Am J Geriatr Psych. 2004;12(5):457-482.
Dr. Casey reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Psychiatrists need to be aware of aspects of psychiatric and pain management that are unique to end-of-life care for patients with advanced dementia. The palliative care philosophy presents an opportunity to honestly acknowledge the terminal nature of advanced dementia, limit intrusive and unnecessary care, and thoughtfully address pain and suffering.
Studies suggest that nursing home patients with severe dementia have an average life expectancy of 2 years.1 These patients often have substantial medical comorbidity, frequently with multiple illnesses, each of which has an accepted management that may involve several medications and interventions. Treatment guidelines for individual conditions don’t necessarily take into account multiple interacting illnesses in advanced dementia. Applying recommended treatments for multiple conditions simultaneously may entail prescribing many medications and interventions. Often these techniques are designed to prevent or modify disease over long term, which a patient with advanced dementia is not likely to achieve.2
Such polypharmacy and intensive intervention are not likely to extend life or improve its quality. In fact, the opposite may occur. Dementia patients react poorly to polypharmacy and may require restraint or sedation to accommodate invasive interventions. Feeding tubes are particularly challenging. Studies have revealed that feeding tubes do not extend life in advanced dementia patients.3
Goals for palliative care. The palliative care approach emphasizes relieving suffering in the near term. Applying this philosophy to advanced dementia depends on acknowledging that the patient will not recover from this condition, has a limited life expectancy, and is not likely to benefit from—and in fact may be harmed by—an aggressive approach to comorbid conditions. Instead, these conditions are best managed by controlling pain and suffering in the near term. Hospitalization and invasive interventions are minimized.
Psychiatric management fits well within this approach. Near the end of life, dementia patients often suffer agitation, psychosis, depression, and delirium that may require the expert, judicious use of psychopharmacology. Patients often experience pain, but might not be able to communicate this, except through behavioral changes. Physicians may be overly concerned with possible adverse effects of pain medications, but when appropriately prescribed, these drugs may help relieve suffering. Psychiatrists also have a role in assisting staff and families during an emotionally difficult time.4
Dr. Casey reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Psychiatrists need to be aware of aspects of psychiatric and pain management that are unique to end-of-life care for patients with advanced dementia. The palliative care philosophy presents an opportunity to honestly acknowledge the terminal nature of advanced dementia, limit intrusive and unnecessary care, and thoughtfully address pain and suffering.
Studies suggest that nursing home patients with severe dementia have an average life expectancy of 2 years.1 These patients often have substantial medical comorbidity, frequently with multiple illnesses, each of which has an accepted management that may involve several medications and interventions. Treatment guidelines for individual conditions don’t necessarily take into account multiple interacting illnesses in advanced dementia. Applying recommended treatments for multiple conditions simultaneously may entail prescribing many medications and interventions. Often these techniques are designed to prevent or modify disease over long term, which a patient with advanced dementia is not likely to achieve.2
Such polypharmacy and intensive intervention are not likely to extend life or improve its quality. In fact, the opposite may occur. Dementia patients react poorly to polypharmacy and may require restraint or sedation to accommodate invasive interventions. Feeding tubes are particularly challenging. Studies have revealed that feeding tubes do not extend life in advanced dementia patients.3
Goals for palliative care. The palliative care approach emphasizes relieving suffering in the near term. Applying this philosophy to advanced dementia depends on acknowledging that the patient will not recover from this condition, has a limited life expectancy, and is not likely to benefit from—and in fact may be harmed by—an aggressive approach to comorbid conditions. Instead, these conditions are best managed by controlling pain and suffering in the near term. Hospitalization and invasive interventions are minimized.
Psychiatric management fits well within this approach. Near the end of life, dementia patients often suffer agitation, psychosis, depression, and delirium that may require the expert, judicious use of psychopharmacology. Patients often experience pain, but might not be able to communicate this, except through behavioral changes. Physicians may be overly concerned with possible adverse effects of pain medications, but when appropriately prescribed, these drugs may help relieve suffering. Psychiatrists also have a role in assisting staff and families during an emotionally difficult time.4
1. Mitchell SL, Kiely DK, Hamel DK, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA. 2004;291:2734-2740.
2. Boyd CM, Darer J, Boult C, et al. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases. JAMA. 2005;294:741-743.
3. Li I. Feeding tubes in patients with severe dementia. Am Fam Physician. 2002;65(8):1605-1610,1515.
4. Lyness JM. End of life care: issues relevant to the geriatric psychiatrist. Am J Geriatr Psych. 2004;12(5):457-482.
1. Mitchell SL, Kiely DK, Hamel DK, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA. 2004;291:2734-2740.
2. Boyd CM, Darer J, Boult C, et al. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases. JAMA. 2005;294:741-743.
3. Li I. Feeding tubes in patients with severe dementia. Am Fam Physician. 2002;65(8):1605-1610,1515.
4. Lyness JM. End of life care: issues relevant to the geriatric psychiatrist. Am J Geriatr Psych. 2004;12(5):457-482.
Trazodone extended release for major depressive disorder
Extended-release (ER) trazodone—FDA-approved in February 2010—improves symptoms of major depressive disorder (MDD) and allows once-daily dosing (Table 1). Trazodone immediate release (IR) was developed in 1960 and approved by the FDA for treatment of MDD in December 1981. Trazodone IR is now mainly prescribed off-label as a hypnotic at lower-than-antidepressant doses, such as 50 to 100 mg/d at bedtime. The dose needed to achieve antidepressant effect is believed to be ≥300 mg/d. Use of the IR formulation for treating depression has been limited by the need for 3-times-a-day dosing and daytime sedation associated with peaks in serum concentration.
Table 1
Trazodone extended release: Fast facts
| Brand name: Oleptro |
| Class: Triazolopyridine-derived antidepressant |
| Indication: Major depressive disorder |
| Approval date: February 2, 2010 |
| Availability date: August 10, 2010 |
| Manufacturer: Labopharm, Inc. |
| Dosage forms: 150 mg and 300 mg bisectable tablets |
| Starting dose: 150 mg at bedtime |
| Target dose: 300 mg/d; maximum dose 375 mg/d |
Clinical implications
Trazodone ER was designed to eliminate the peaks and troughs in serum concentration seen with trazodone IR. It was hypothesized that by reducing the maximum concentration (Cmax) peaks, trazodone ER would permit higher doses to be better tolerated and help patients to more easily reach target antidepressant doses (≥300 mg/d). Trazodone ER’s once-daily dosing also may increase patient adherence.
How it works
The exact mechanism of action through which trazodone treats depression is not completely understood, but is likely related to enhancing serotonergic activity in the CNS. Trazodone is a triazolopyridine antidepressant, inhibits the serotonin transporter, and is a 5-HT2A and 5-HT2C antagonist. This is why it is sometimes referred as a serotonin antagonist/reuptake inhibitor, but regulatory agencies do not accept this class name. Trazodone is an antagonist at both histamine (H1) and α1-adrenergic receptors, which may mediate trazodone’s sedating properties (H1) and hypotensive (α1-adrenergic) effects.
The ER formulation employs a cross-linked, high-amylose starch excipient that provides controlled release of trazodone over an extended period.
Pharmacokinetics
Trazodone ER has linear pharmacokinetics in doses from 75 to 375 mg. Trazodone ER, 300 mg/d, provides a steady-state exposure equivalent to 100 mg of trazodone IR given 3 times daily, while having a lower Cmax. A high-fat meal can increase Cmax of trazodone ER by 1.9-fold. Trazodone is extensively biotransformed in the liver via the cytochrome P450 (CYP) 3A4 pathway and its metabolites are eliminated within 72 hours. Elimination is predominantly renal, with 70% to 75% of an oral dose being recovered in the urine within 72 hours.1 This formulation maintains its controlled-release properties if bisected.
Because trazodone is a substrate of the CYP3A4 enzyme, its metabolism can be inhibited by CYP3A4 inhibitors. Exercise caution when coadministering medications that cause CYP3A4 inhibition with trazodone ER. The effect of short-term administration of ritonavir (4 doses of 200 mg) on the pharmacokinetics of a single dose of trazodone (50 mg) has been studied in 10 healthy subjects.2 The Cmax of trazodone increased by 34%, area under the curve increased 2.4-fold, half-life increased by 2.2-fold, and clearance decreased by 52%. There is no difference in the half-life between the IR and ER formulations because the ER formulation influences only the release kinetics of the drug, not the half-life of the medication.
Efficacy
Efficacy of trazodone for MDD initially was established in trials conducted with trazodone IR.3-10 The efficacy of the ER formulation was established in a multi-center randomized, double-blind, placebo-controlled trial with 412 patients (age 18 to 80). Patients who met DSM-IV criteria for MDD were randomly assigned to trazodone ER (n=206) or placebo (n=206) for 8 weeks.11 This study showed a statistically significant difference between trazodone ER and placebo after 8 weeks of treatment on the primary outcome measure, which was a change in score on the 17-item Hamilton Depression Rating scale (HAMD-17). HAM-D-17 scores decreased 11.4 points in the trazodone ER group and 9.3 points in the placebo group (P=.012 in the modified intent to treat [ITT] population; P=.009 in the completer analysis). This difference was seen from week 1 and throughout the study. Efficacy of trazodone ER was further supported by statistically significant differences between the drug and placebo in 7 of 13 secondary efficacy endpoints in both the modified ITT and per protocol (PP) populations (HAM-D-17 mood item, mean Montgomery-Åsberg Depression Rating Scale [MADRS] total score, mean Clinical Global Impressions Severity of Illness [CGI-S] score, percentage of HAM-D-17 responders, and 3 quality of sleep items [overall quality of sleep, trouble falling asleep, and awakening during the night]). Overall effect sizes for the HAM-D-17 were -0.26 (modified ITT-last observation carried forward [LOCF] dataset) and -0.33 (PP/observed cases [OC] dataset). The effect sizes in MADRS scores were -0.22 and -0.29 for the modified ITT-LOCF and the PP/OC analyses, respectively.12
Sleep measures. In the study sample >90% of patients had insomnia at baseline (defined as a score ≥2 in any HAM-D-17 sleep item or sum of all 3 sleep items of ≥4). Patients receiving trazodone ER had significant improvement in all 3 HAM-D-17 sleep items. Subjects reported improvement in the overall quality of sleep and awakening during the night after the first week of treatment. Investigators found no significant interaction between improvements in core symptoms of depression and baseline MADRS reduced sleep item or early changes in the HAM-D-17 sleep items. This suggests that the antidepressant effect of trazodone ER was independent of severity of sleep difficulties at baseline and of improvement in insomnia during the study.12
Researchers observed improvement in suicidal ideation on MADRS (item 10) and HAM-D-17 (item 3) after 8 weeks of treatment (effect size -0.2 favoring trazodone ER over placebo).12
In 2 European comparative, randomized, double-blind trials, trazodone prolonged release showed similar antidepressant efficacy as paroxetine4 and setraline5 as measured by HAM-D, MADRS, and CGI-S. This prolonged release formulation made in Europe is not the same technology as the ER formulation recently approved by the FDA.
Tolerability
In the pivotal registration study, trazodone ER was well tolerated at a mean dose of 310 mg/d.11 Twenty-five patients (12.4%) in the trazodone ER group discontinued the drug because of side effects. The most common side effects leading to discontinuation in the active treatment group were dizziness (n=7), sedation (n=5), and somnolence (n=3).11 The most frequent adverse events reported at any study time point were headache (33%), somnolence (31%), dry mouth (25%), dizziness (25%), nausea (21%), sedation (17%), and fatigue (15%) (Table 2).11 In general, these adverse events were mild to moderate and short-lived; most side effects resolved within the first 2 to 3 weeks of treatment with trazodone ER.11
Sexual side effects—delayed ejaculation, delayed time to orgasm, or orgasmic blockade—are common with many anti-depressants. In the pivotal registration study, the incidence of sexual side effects was low (4.9% with trazodone ER vs 2.5% with placebo).11 This is much lower than the rates typically found with selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors, which range from 17% to 41%.13,14 This benefit is thought to be mediated through 5-HT2A and 5-HT2C antagonism. Priapism has been reported in trazodone IR at rates ranging from 1 in 1,000 to 1 in 10,000 and does not appear to be dose-related.15 The rate of priapism in persons using agents for erectile dysfunction ranges from .05% to 6%.15 No case of priapism was seen in the trazodone ER study; however, with its sample size of 412 patients this study was not powered to adequately detect this adverse event.11
There was no significant weight gain difference between the active drug and placebo groups over 8 weeks of treatment.
Safety. Trazodone ER should not be used within 14 days of taking a monoamine oxidase inhibitor.1 Trazodone carries a pregnancy category C, meaning that it should be used only if the potential benefit justifies potential risk to the fetus. In animal studies, trazodone has been shown to cause increased fetal resorption and congenital anomalities with doses up to 50 times the maximum human dose (375 mg/d). Trazo-done may be secreted in breast milk. The drug is best avoided in patients with recent myocardial infarction.
Table 2
Trazodone extended release treatment-emergent adverse events*
| Trazodone ER (n=202) | Placebo (n=204) | |
|---|---|---|
| Headache | 67 (33%) | 55 (27%) |
| Somnolence | 63 (31%) | 32 (16%) |
| Dry mouth | 51 (25%) | 26 (13%) |
| Dizziness | 50 (25%) | 25 (12%) |
| Nausea | 42 (21%) | 26 (13%) |
| Sedation | 34 (17%) | 7 (3%) |
| Fatigue | 30 (15%) | 17 (8%) |
| Diarrhea | 19 (9%) | 23 (11%) |
| Constipation | 16 (8%) | 4 (2%) |
| Back pain | 11 (5%) | 7 (3%) |
| Blurred vision | 11 (5%) | 0 (0%) |
| *Reported by ≥5% of patients Source: Reference 11 | ||
Dosing
The recommended starting dose is 150 mg/d at bedtime. The dose may be increased by 75 mg/d every 3 days, but the maximum dose should not exceed 375 mg/d.1 Trazodone ER is available in 150 mg or 300 mg bisectable tablets. Breaking the tablets in half does not affect the controlled release, but they should not be chewed or crushed.
Related Resource
- Extended-release trazodone (Oleptro) prescribing information. www.oleptro.com/images/9379.pdf.
Drug Brand Names
- Paroxetine • Paxil
- Ritonavir • Norvir
- Sertraline • Zoloft
- Trazodone • Desyrel
- Trazodone extended-release • Oleptro
Disclosures
Dr. Hidalgo receives grant/research support from AstraZeneca, CeNeRx Biopharma, Centers for Disease Control and Prevention, Dainippon Sumitomo Pharma America, Inc., Eli Lilly and Company, Forest Laboratories, Indevus Pharmaceuticals, Janssen Pharmaceuticals, Labopharm, Otsuka, Pfizer, Inc., Repligen Corp., Sanofi-Synthelabo, Sepracor, and the University of South Florida, and is consultant to the MAPI Institute.
Dr. Sheehan has received grant funding support from, been affiliated with, or received honoraria and travel expenses related to lectures/presentations or consultant activities from the following organizations: Abbott Laboratories,1,2,3 Ad Hoc Committee, Treatment Drug and Assessment Research Review,1 Alexza,1 Alza Pharmaceuticals, Palo Alto, CA,1 the American Medical Association,2 American Psychiatric Association Task Force on Benzodiazepine Dependency,1 American Psychiatric Association Task Force on Treatments of Psychiatric Disorders,1 American Psychiatric Association Working Group to Revise DSM III Anxiety Disorders Section,1 Anclote Foundation,2 Anxiety Disorders Resource Center,1 Anxiety Drug Efficacy Case, the FDA,1 Applied Health Outcomes/Xcenda,1 AstraZeneca,1,2,3 Avera Pharmaceuticals,1,2 Boehringer Ingelheim,3 Boots Pharmaceuticals,3 Bristol-Myers Squibb,1,2,3 Burroughs Wellcome,2,3 Cephalon,1 Charter Hospitals,3 Ciba Geigy,3 Committee (RRC) of the National Institute for Mental Health on Anxiety and Phobic Disorder Projects,1 Connecticut and Ohio Academies of Family Physicians,1 Cortex Pharmaceutical,1 Council on Anxiety Disorders,1 CPC Coliseum Medical Center,1 Cypress Bioscience,1 Dista Products Company,3 Division of Drugs and Technology, American Medical Association,1 Eisai,1,2 Eli Lilly and Company,2,3 Excerpta Medica Asia,3 Faxmed, Inc.,1 Forest Laboratories,1,2 Glaxo Pharmaceuticals,3 GlaxoSmithKline,1,2,3 Glaxo-Wellcome,2 Hospital Corporation of America,3 Humana,3 ICI,3 INC Research,1 International Clinical Research (ICR),2 International Society for CNS Drug Development (ISCDD),1 Janssen Pharmaceuticals,1,2,3 Jazz Pharmaceuticals,1,2 Kali-Duphar,2,3 Labopharm,1 Layton Bioscience,1 Lilly Research Laboratories,1 Lundbeck, Denmark,1 Marion Merrell Dow,3 McNeil Pharmaceuticals,3 Mead Johnson,2,3 Medical Outcome Systems,4 MediciNova,1,2 Merck Sharp & Dohme,2,3 National Anxiety Awareness Program,1 National Anxiety Foundation,1 National Depressive and Manic Depressive Association,1 National Institute on Drug Abuse,2 National Institute of Health,2 Neuronetics,1 Novartis Pharmaceuticals Corp.,2 Novo Nordisk,3 Organon,1,3 Orion Pharma,1 Parexel International Corporation,1 Parke-Davis,2,3 Pfizer, Inc.,1,2,3 Pharmacia,1 Pharmacia and Upjohn,1,3 Philadelphia College of Pharmacy and Science,1 Pierre Fabre, France,1 Quintiles,2 Rhone Laboratories,3 Rhone-Poulenc Rorer Pharmaceuticals,3 Roche,1 Roerig,3 Sandoz Pharmaceuticals,2,3 sanofi-aventis,1,2,3 Sanofi-Synthelabo Recherche,1,2 Schering Corporation,3 Sepracor,1 Shire Laboratories, Inc.,1 SmithKline Beecham,1,2,3 Solvay Pharmaceuticals,1,3 Takeda Pharmaceuticals,1 Tampa General Hospital,1 University of South Florida Psychiatry Center,2 University of South Florida College of Medicine, TAP Pharmaceuticals,2,3 Targacept,1 Tampa General Hospital-University Psychiatry Center,3 Tikvah Therapeutics,1 Titan Pharmaceuticals,1 United Bioscience,2 The Upjohn Company,1,2,3 U.S. Congress-House of Representatives Committee,1 University of South Florida Friends of Research in Psychiatry, Board of Trustees,1 Warner Chilcott,2,3 World Health Organization,1 Worldwide Clinical Trials,2 Wyeth-Ayerst,1,2,3 ZARS,1 and Zeneca Pharmaceuticals.1
1: Consultant; 2: Grant/Research Support; 3: Lectures/ Presentations; 4: Stock Holder
1. Oleptro [package insert]. Dublin, Ireland: Labopharm Europe Limited; 2010.
2. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Short-term exposure to low-dose ritonavir impairs clearance and enhances adverse effects of trazodone. J Clin Pharmacol. 2003;43(4):414-422.
3. Beasley CM, Jr, Dornseif BE, Pultz JA, et al. Fluoxetine versus trazodone: efficacy and activating-sedating effects. J Clin Psychiatry. 1991;52:294-299.
4. Kasper S, Olivieri L, Di Loreto G, et al. A comparative, randomized double blind study of trazodone prolonged-release and paroxetine in the treatment of patients with major depressive disorder. Curr Med Res Opin. 2005;21:1139-1146.
5. Munizza C, Olivieri L, Di Loreto G, et al. A comparative, randomized double blind study of trazodone prolonged-release and sertraline in the treatment of patients with major depressive disorder. Curr Med Res Opin. 2006;22:1703-1713.
6. Cunningham LA, Borison RL, Carman JS, et al. A comparison of venlafaxine, trazodone, and placebo in major depression. J Clin Pyschopharmacol. 1994;14:99-106.
7. Weisler RH, Johnston JA, Lineberry CG, et al. Comparison of bupropion and trazodone in the treatment of major depression. J Clin Psychopharmacol. 1994;14:170-179.
8. Feighner JP. Trazodone, a triazolopyridine derivative, in primary depressive disorder. J Clin Psychiatry. 1980;41:250-255.
9. Rickels K, Case WG. Trazodone in depressed outpatients. Am J Psychiatry. 1982;139:803-806.
10. Perry PJ, Garvey MJ, Kelly MW, et al. A comparative trial of fluoxetine versus trazodone in outpatients with major depression. J Clin Psychiatry. 1989;50:290-294.
11. Sheehan DV, Croft HA, Gossen ER, et al. Extended-release trazodone in major depressive disorder: a randomized, double-blind, placebo-controlled study. Psychiatry (Edgmont). 2009;6(5):20-33.
12. Sheehan DV, Rozova A, Gossen ER, et al. The efficacy and tolerability of once-daily controlled-release trazodone for depressed mood, anxiety, insomnia, and suicidality in major depressive disorder. Psychopharmacol Bull. 2009;42(4):5-22.
13. Hu XH, Bull SA, Hunkeler EM, et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry. 2004;65(7):959-965.
14. Landén M, Högberg P, Thase ME. Incidence of sexual side effects in refractory depression during treatment with citalopram or paroxetine. J Clin Psychiatry. 2005;66(1):100-106.
15. Thompson JW, Jr, Ware MR, Blashfield RK. Psychotropic medications and priapism: a comprehensive review. J Clin Psychiatry. 1990;51:430-433.
Extended-release (ER) trazodone—FDA-approved in February 2010—improves symptoms of major depressive disorder (MDD) and allows once-daily dosing (Table 1). Trazodone immediate release (IR) was developed in 1960 and approved by the FDA for treatment of MDD in December 1981. Trazodone IR is now mainly prescribed off-label as a hypnotic at lower-than-antidepressant doses, such as 50 to 100 mg/d at bedtime. The dose needed to achieve antidepressant effect is believed to be ≥300 mg/d. Use of the IR formulation for treating depression has been limited by the need for 3-times-a-day dosing and daytime sedation associated with peaks in serum concentration.
Table 1
Trazodone extended release: Fast facts
| Brand name: Oleptro |
| Class: Triazolopyridine-derived antidepressant |
| Indication: Major depressive disorder |
| Approval date: February 2, 2010 |
| Availability date: August 10, 2010 |
| Manufacturer: Labopharm, Inc. |
| Dosage forms: 150 mg and 300 mg bisectable tablets |
| Starting dose: 150 mg at bedtime |
| Target dose: 300 mg/d; maximum dose 375 mg/d |
Clinical implications
Trazodone ER was designed to eliminate the peaks and troughs in serum concentration seen with trazodone IR. It was hypothesized that by reducing the maximum concentration (Cmax) peaks, trazodone ER would permit higher doses to be better tolerated and help patients to more easily reach target antidepressant doses (≥300 mg/d). Trazodone ER’s once-daily dosing also may increase patient adherence.
How it works
The exact mechanism of action through which trazodone treats depression is not completely understood, but is likely related to enhancing serotonergic activity in the CNS. Trazodone is a triazolopyridine antidepressant, inhibits the serotonin transporter, and is a 5-HT2A and 5-HT2C antagonist. This is why it is sometimes referred as a serotonin antagonist/reuptake inhibitor, but regulatory agencies do not accept this class name. Trazodone is an antagonist at both histamine (H1) and α1-adrenergic receptors, which may mediate trazodone’s sedating properties (H1) and hypotensive (α1-adrenergic) effects.
The ER formulation employs a cross-linked, high-amylose starch excipient that provides controlled release of trazodone over an extended period.
Pharmacokinetics
Trazodone ER has linear pharmacokinetics in doses from 75 to 375 mg. Trazodone ER, 300 mg/d, provides a steady-state exposure equivalent to 100 mg of trazodone IR given 3 times daily, while having a lower Cmax. A high-fat meal can increase Cmax of trazodone ER by 1.9-fold. Trazodone is extensively biotransformed in the liver via the cytochrome P450 (CYP) 3A4 pathway and its metabolites are eliminated within 72 hours. Elimination is predominantly renal, with 70% to 75% of an oral dose being recovered in the urine within 72 hours.1 This formulation maintains its controlled-release properties if bisected.
Because trazodone is a substrate of the CYP3A4 enzyme, its metabolism can be inhibited by CYP3A4 inhibitors. Exercise caution when coadministering medications that cause CYP3A4 inhibition with trazodone ER. The effect of short-term administration of ritonavir (4 doses of 200 mg) on the pharmacokinetics of a single dose of trazodone (50 mg) has been studied in 10 healthy subjects.2 The Cmax of trazodone increased by 34%, area under the curve increased 2.4-fold, half-life increased by 2.2-fold, and clearance decreased by 52%. There is no difference in the half-life between the IR and ER formulations because the ER formulation influences only the release kinetics of the drug, not the half-life of the medication.
Efficacy
Efficacy of trazodone for MDD initially was established in trials conducted with trazodone IR.3-10 The efficacy of the ER formulation was established in a multi-center randomized, double-blind, placebo-controlled trial with 412 patients (age 18 to 80). Patients who met DSM-IV criteria for MDD were randomly assigned to trazodone ER (n=206) or placebo (n=206) for 8 weeks.11 This study showed a statistically significant difference between trazodone ER and placebo after 8 weeks of treatment on the primary outcome measure, which was a change in score on the 17-item Hamilton Depression Rating scale (HAMD-17). HAM-D-17 scores decreased 11.4 points in the trazodone ER group and 9.3 points in the placebo group (P=.012 in the modified intent to treat [ITT] population; P=.009 in the completer analysis). This difference was seen from week 1 and throughout the study. Efficacy of trazodone ER was further supported by statistically significant differences between the drug and placebo in 7 of 13 secondary efficacy endpoints in both the modified ITT and per protocol (PP) populations (HAM-D-17 mood item, mean Montgomery-Åsberg Depression Rating Scale [MADRS] total score, mean Clinical Global Impressions Severity of Illness [CGI-S] score, percentage of HAM-D-17 responders, and 3 quality of sleep items [overall quality of sleep, trouble falling asleep, and awakening during the night]). Overall effect sizes for the HAM-D-17 were -0.26 (modified ITT-last observation carried forward [LOCF] dataset) and -0.33 (PP/observed cases [OC] dataset). The effect sizes in MADRS scores were -0.22 and -0.29 for the modified ITT-LOCF and the PP/OC analyses, respectively.12
Sleep measures. In the study sample >90% of patients had insomnia at baseline (defined as a score ≥2 in any HAM-D-17 sleep item or sum of all 3 sleep items of ≥4). Patients receiving trazodone ER had significant improvement in all 3 HAM-D-17 sleep items. Subjects reported improvement in the overall quality of sleep and awakening during the night after the first week of treatment. Investigators found no significant interaction between improvements in core symptoms of depression and baseline MADRS reduced sleep item or early changes in the HAM-D-17 sleep items. This suggests that the antidepressant effect of trazodone ER was independent of severity of sleep difficulties at baseline and of improvement in insomnia during the study.12
Researchers observed improvement in suicidal ideation on MADRS (item 10) and HAM-D-17 (item 3) after 8 weeks of treatment (effect size -0.2 favoring trazodone ER over placebo).12
In 2 European comparative, randomized, double-blind trials, trazodone prolonged release showed similar antidepressant efficacy as paroxetine4 and setraline5 as measured by HAM-D, MADRS, and CGI-S. This prolonged release formulation made in Europe is not the same technology as the ER formulation recently approved by the FDA.
Tolerability
In the pivotal registration study, trazodone ER was well tolerated at a mean dose of 310 mg/d.11 Twenty-five patients (12.4%) in the trazodone ER group discontinued the drug because of side effects. The most common side effects leading to discontinuation in the active treatment group were dizziness (n=7), sedation (n=5), and somnolence (n=3).11 The most frequent adverse events reported at any study time point were headache (33%), somnolence (31%), dry mouth (25%), dizziness (25%), nausea (21%), sedation (17%), and fatigue (15%) (Table 2).11 In general, these adverse events were mild to moderate and short-lived; most side effects resolved within the first 2 to 3 weeks of treatment with trazodone ER.11
Sexual side effects—delayed ejaculation, delayed time to orgasm, or orgasmic blockade—are common with many anti-depressants. In the pivotal registration study, the incidence of sexual side effects was low (4.9% with trazodone ER vs 2.5% with placebo).11 This is much lower than the rates typically found with selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors, which range from 17% to 41%.13,14 This benefit is thought to be mediated through 5-HT2A and 5-HT2C antagonism. Priapism has been reported in trazodone IR at rates ranging from 1 in 1,000 to 1 in 10,000 and does not appear to be dose-related.15 The rate of priapism in persons using agents for erectile dysfunction ranges from .05% to 6%.15 No case of priapism was seen in the trazodone ER study; however, with its sample size of 412 patients this study was not powered to adequately detect this adverse event.11
There was no significant weight gain difference between the active drug and placebo groups over 8 weeks of treatment.
Safety. Trazodone ER should not be used within 14 days of taking a monoamine oxidase inhibitor.1 Trazodone carries a pregnancy category C, meaning that it should be used only if the potential benefit justifies potential risk to the fetus. In animal studies, trazodone has been shown to cause increased fetal resorption and congenital anomalities with doses up to 50 times the maximum human dose (375 mg/d). Trazo-done may be secreted in breast milk. The drug is best avoided in patients with recent myocardial infarction.
Table 2
Trazodone extended release treatment-emergent adverse events*
| Trazodone ER (n=202) | Placebo (n=204) | |
|---|---|---|
| Headache | 67 (33%) | 55 (27%) |
| Somnolence | 63 (31%) | 32 (16%) |
| Dry mouth | 51 (25%) | 26 (13%) |
| Dizziness | 50 (25%) | 25 (12%) |
| Nausea | 42 (21%) | 26 (13%) |
| Sedation | 34 (17%) | 7 (3%) |
| Fatigue | 30 (15%) | 17 (8%) |
| Diarrhea | 19 (9%) | 23 (11%) |
| Constipation | 16 (8%) | 4 (2%) |
| Back pain | 11 (5%) | 7 (3%) |
| Blurred vision | 11 (5%) | 0 (0%) |
| *Reported by ≥5% of patients Source: Reference 11 | ||
Dosing
The recommended starting dose is 150 mg/d at bedtime. The dose may be increased by 75 mg/d every 3 days, but the maximum dose should not exceed 375 mg/d.1 Trazodone ER is available in 150 mg or 300 mg bisectable tablets. Breaking the tablets in half does not affect the controlled release, but they should not be chewed or crushed.
Related Resource
- Extended-release trazodone (Oleptro) prescribing information. www.oleptro.com/images/9379.pdf.
Drug Brand Names
- Paroxetine • Paxil
- Ritonavir • Norvir
- Sertraline • Zoloft
- Trazodone • Desyrel
- Trazodone extended-release • Oleptro
Disclosures
Dr. Hidalgo receives grant/research support from AstraZeneca, CeNeRx Biopharma, Centers for Disease Control and Prevention, Dainippon Sumitomo Pharma America, Inc., Eli Lilly and Company, Forest Laboratories, Indevus Pharmaceuticals, Janssen Pharmaceuticals, Labopharm, Otsuka, Pfizer, Inc., Repligen Corp., Sanofi-Synthelabo, Sepracor, and the University of South Florida, and is consultant to the MAPI Institute.
Dr. Sheehan has received grant funding support from, been affiliated with, or received honoraria and travel expenses related to lectures/presentations or consultant activities from the following organizations: Abbott Laboratories,1,2,3 Ad Hoc Committee, Treatment Drug and Assessment Research Review,1 Alexza,1 Alza Pharmaceuticals, Palo Alto, CA,1 the American Medical Association,2 American Psychiatric Association Task Force on Benzodiazepine Dependency,1 American Psychiatric Association Task Force on Treatments of Psychiatric Disorders,1 American Psychiatric Association Working Group to Revise DSM III Anxiety Disorders Section,1 Anclote Foundation,2 Anxiety Disorders Resource Center,1 Anxiety Drug Efficacy Case, the FDA,1 Applied Health Outcomes/Xcenda,1 AstraZeneca,1,2,3 Avera Pharmaceuticals,1,2 Boehringer Ingelheim,3 Boots Pharmaceuticals,3 Bristol-Myers Squibb,1,2,3 Burroughs Wellcome,2,3 Cephalon,1 Charter Hospitals,3 Ciba Geigy,3 Committee (RRC) of the National Institute for Mental Health on Anxiety and Phobic Disorder Projects,1 Connecticut and Ohio Academies of Family Physicians,1 Cortex Pharmaceutical,1 Council on Anxiety Disorders,1 CPC Coliseum Medical Center,1 Cypress Bioscience,1 Dista Products Company,3 Division of Drugs and Technology, American Medical Association,1 Eisai,1,2 Eli Lilly and Company,2,3 Excerpta Medica Asia,3 Faxmed, Inc.,1 Forest Laboratories,1,2 Glaxo Pharmaceuticals,3 GlaxoSmithKline,1,2,3 Glaxo-Wellcome,2 Hospital Corporation of America,3 Humana,3 ICI,3 INC Research,1 International Clinical Research (ICR),2 International Society for CNS Drug Development (ISCDD),1 Janssen Pharmaceuticals,1,2,3 Jazz Pharmaceuticals,1,2 Kali-Duphar,2,3 Labopharm,1 Layton Bioscience,1 Lilly Research Laboratories,1 Lundbeck, Denmark,1 Marion Merrell Dow,3 McNeil Pharmaceuticals,3 Mead Johnson,2,3 Medical Outcome Systems,4 MediciNova,1,2 Merck Sharp & Dohme,2,3 National Anxiety Awareness Program,1 National Anxiety Foundation,1 National Depressive and Manic Depressive Association,1 National Institute on Drug Abuse,2 National Institute of Health,2 Neuronetics,1 Novartis Pharmaceuticals Corp.,2 Novo Nordisk,3 Organon,1,3 Orion Pharma,1 Parexel International Corporation,1 Parke-Davis,2,3 Pfizer, Inc.,1,2,3 Pharmacia,1 Pharmacia and Upjohn,1,3 Philadelphia College of Pharmacy and Science,1 Pierre Fabre, France,1 Quintiles,2 Rhone Laboratories,3 Rhone-Poulenc Rorer Pharmaceuticals,3 Roche,1 Roerig,3 Sandoz Pharmaceuticals,2,3 sanofi-aventis,1,2,3 Sanofi-Synthelabo Recherche,1,2 Schering Corporation,3 Sepracor,1 Shire Laboratories, Inc.,1 SmithKline Beecham,1,2,3 Solvay Pharmaceuticals,1,3 Takeda Pharmaceuticals,1 Tampa General Hospital,1 University of South Florida Psychiatry Center,2 University of South Florida College of Medicine, TAP Pharmaceuticals,2,3 Targacept,1 Tampa General Hospital-University Psychiatry Center,3 Tikvah Therapeutics,1 Titan Pharmaceuticals,1 United Bioscience,2 The Upjohn Company,1,2,3 U.S. Congress-House of Representatives Committee,1 University of South Florida Friends of Research in Psychiatry, Board of Trustees,1 Warner Chilcott,2,3 World Health Organization,1 Worldwide Clinical Trials,2 Wyeth-Ayerst,1,2,3 ZARS,1 and Zeneca Pharmaceuticals.1
1: Consultant; 2: Grant/Research Support; 3: Lectures/ Presentations; 4: Stock Holder
Extended-release (ER) trazodone—FDA-approved in February 2010—improves symptoms of major depressive disorder (MDD) and allows once-daily dosing (Table 1). Trazodone immediate release (IR) was developed in 1960 and approved by the FDA for treatment of MDD in December 1981. Trazodone IR is now mainly prescribed off-label as a hypnotic at lower-than-antidepressant doses, such as 50 to 100 mg/d at bedtime. The dose needed to achieve antidepressant effect is believed to be ≥300 mg/d. Use of the IR formulation for treating depression has been limited by the need for 3-times-a-day dosing and daytime sedation associated with peaks in serum concentration.
Table 1
Trazodone extended release: Fast facts
| Brand name: Oleptro |
| Class: Triazolopyridine-derived antidepressant |
| Indication: Major depressive disorder |
| Approval date: February 2, 2010 |
| Availability date: August 10, 2010 |
| Manufacturer: Labopharm, Inc. |
| Dosage forms: 150 mg and 300 mg bisectable tablets |
| Starting dose: 150 mg at bedtime |
| Target dose: 300 mg/d; maximum dose 375 mg/d |
Clinical implications
Trazodone ER was designed to eliminate the peaks and troughs in serum concentration seen with trazodone IR. It was hypothesized that by reducing the maximum concentration (Cmax) peaks, trazodone ER would permit higher doses to be better tolerated and help patients to more easily reach target antidepressant doses (≥300 mg/d). Trazodone ER’s once-daily dosing also may increase patient adherence.
How it works
The exact mechanism of action through which trazodone treats depression is not completely understood, but is likely related to enhancing serotonergic activity in the CNS. Trazodone is a triazolopyridine antidepressant, inhibits the serotonin transporter, and is a 5-HT2A and 5-HT2C antagonist. This is why it is sometimes referred as a serotonin antagonist/reuptake inhibitor, but regulatory agencies do not accept this class name. Trazodone is an antagonist at both histamine (H1) and α1-adrenergic receptors, which may mediate trazodone’s sedating properties (H1) and hypotensive (α1-adrenergic) effects.
The ER formulation employs a cross-linked, high-amylose starch excipient that provides controlled release of trazodone over an extended period.
Pharmacokinetics
Trazodone ER has linear pharmacokinetics in doses from 75 to 375 mg. Trazodone ER, 300 mg/d, provides a steady-state exposure equivalent to 100 mg of trazodone IR given 3 times daily, while having a lower Cmax. A high-fat meal can increase Cmax of trazodone ER by 1.9-fold. Trazodone is extensively biotransformed in the liver via the cytochrome P450 (CYP) 3A4 pathway and its metabolites are eliminated within 72 hours. Elimination is predominantly renal, with 70% to 75% of an oral dose being recovered in the urine within 72 hours.1 This formulation maintains its controlled-release properties if bisected.
Because trazodone is a substrate of the CYP3A4 enzyme, its metabolism can be inhibited by CYP3A4 inhibitors. Exercise caution when coadministering medications that cause CYP3A4 inhibition with trazodone ER. The effect of short-term administration of ritonavir (4 doses of 200 mg) on the pharmacokinetics of a single dose of trazodone (50 mg) has been studied in 10 healthy subjects.2 The Cmax of trazodone increased by 34%, area under the curve increased 2.4-fold, half-life increased by 2.2-fold, and clearance decreased by 52%. There is no difference in the half-life between the IR and ER formulations because the ER formulation influences only the release kinetics of the drug, not the half-life of the medication.
Efficacy
Efficacy of trazodone for MDD initially was established in trials conducted with trazodone IR.3-10 The efficacy of the ER formulation was established in a multi-center randomized, double-blind, placebo-controlled trial with 412 patients (age 18 to 80). Patients who met DSM-IV criteria for MDD were randomly assigned to trazodone ER (n=206) or placebo (n=206) for 8 weeks.11 This study showed a statistically significant difference between trazodone ER and placebo after 8 weeks of treatment on the primary outcome measure, which was a change in score on the 17-item Hamilton Depression Rating scale (HAMD-17). HAM-D-17 scores decreased 11.4 points in the trazodone ER group and 9.3 points in the placebo group (P=.012 in the modified intent to treat [ITT] population; P=.009 in the completer analysis). This difference was seen from week 1 and throughout the study. Efficacy of trazodone ER was further supported by statistically significant differences between the drug and placebo in 7 of 13 secondary efficacy endpoints in both the modified ITT and per protocol (PP) populations (HAM-D-17 mood item, mean Montgomery-Åsberg Depression Rating Scale [MADRS] total score, mean Clinical Global Impressions Severity of Illness [CGI-S] score, percentage of HAM-D-17 responders, and 3 quality of sleep items [overall quality of sleep, trouble falling asleep, and awakening during the night]). Overall effect sizes for the HAM-D-17 were -0.26 (modified ITT-last observation carried forward [LOCF] dataset) and -0.33 (PP/observed cases [OC] dataset). The effect sizes in MADRS scores were -0.22 and -0.29 for the modified ITT-LOCF and the PP/OC analyses, respectively.12
Sleep measures. In the study sample >90% of patients had insomnia at baseline (defined as a score ≥2 in any HAM-D-17 sleep item or sum of all 3 sleep items of ≥4). Patients receiving trazodone ER had significant improvement in all 3 HAM-D-17 sleep items. Subjects reported improvement in the overall quality of sleep and awakening during the night after the first week of treatment. Investigators found no significant interaction between improvements in core symptoms of depression and baseline MADRS reduced sleep item or early changes in the HAM-D-17 sleep items. This suggests that the antidepressant effect of trazodone ER was independent of severity of sleep difficulties at baseline and of improvement in insomnia during the study.12
Researchers observed improvement in suicidal ideation on MADRS (item 10) and HAM-D-17 (item 3) after 8 weeks of treatment (effect size -0.2 favoring trazodone ER over placebo).12
In 2 European comparative, randomized, double-blind trials, trazodone prolonged release showed similar antidepressant efficacy as paroxetine4 and setraline5 as measured by HAM-D, MADRS, and CGI-S. This prolonged release formulation made in Europe is not the same technology as the ER formulation recently approved by the FDA.
Tolerability
In the pivotal registration study, trazodone ER was well tolerated at a mean dose of 310 mg/d.11 Twenty-five patients (12.4%) in the trazodone ER group discontinued the drug because of side effects. The most common side effects leading to discontinuation in the active treatment group were dizziness (n=7), sedation (n=5), and somnolence (n=3).11 The most frequent adverse events reported at any study time point were headache (33%), somnolence (31%), dry mouth (25%), dizziness (25%), nausea (21%), sedation (17%), and fatigue (15%) (Table 2).11 In general, these adverse events were mild to moderate and short-lived; most side effects resolved within the first 2 to 3 weeks of treatment with trazodone ER.11
Sexual side effects—delayed ejaculation, delayed time to orgasm, or orgasmic blockade—are common with many anti-depressants. In the pivotal registration study, the incidence of sexual side effects was low (4.9% with trazodone ER vs 2.5% with placebo).11 This is much lower than the rates typically found with selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors, which range from 17% to 41%.13,14 This benefit is thought to be mediated through 5-HT2A and 5-HT2C antagonism. Priapism has been reported in trazodone IR at rates ranging from 1 in 1,000 to 1 in 10,000 and does not appear to be dose-related.15 The rate of priapism in persons using agents for erectile dysfunction ranges from .05% to 6%.15 No case of priapism was seen in the trazodone ER study; however, with its sample size of 412 patients this study was not powered to adequately detect this adverse event.11
There was no significant weight gain difference between the active drug and placebo groups over 8 weeks of treatment.
Safety. Trazodone ER should not be used within 14 days of taking a monoamine oxidase inhibitor.1 Trazodone carries a pregnancy category C, meaning that it should be used only if the potential benefit justifies potential risk to the fetus. In animal studies, trazodone has been shown to cause increased fetal resorption and congenital anomalities with doses up to 50 times the maximum human dose (375 mg/d). Trazo-done may be secreted in breast milk. The drug is best avoided in patients with recent myocardial infarction.
Table 2
Trazodone extended release treatment-emergent adverse events*
| Trazodone ER (n=202) | Placebo (n=204) | |
|---|---|---|
| Headache | 67 (33%) | 55 (27%) |
| Somnolence | 63 (31%) | 32 (16%) |
| Dry mouth | 51 (25%) | 26 (13%) |
| Dizziness | 50 (25%) | 25 (12%) |
| Nausea | 42 (21%) | 26 (13%) |
| Sedation | 34 (17%) | 7 (3%) |
| Fatigue | 30 (15%) | 17 (8%) |
| Diarrhea | 19 (9%) | 23 (11%) |
| Constipation | 16 (8%) | 4 (2%) |
| Back pain | 11 (5%) | 7 (3%) |
| Blurred vision | 11 (5%) | 0 (0%) |
| *Reported by ≥5% of patients Source: Reference 11 | ||
Dosing
The recommended starting dose is 150 mg/d at bedtime. The dose may be increased by 75 mg/d every 3 days, but the maximum dose should not exceed 375 mg/d.1 Trazodone ER is available in 150 mg or 300 mg bisectable tablets. Breaking the tablets in half does not affect the controlled release, but they should not be chewed or crushed.
Related Resource
- Extended-release trazodone (Oleptro) prescribing information. www.oleptro.com/images/9379.pdf.
Drug Brand Names
- Paroxetine • Paxil
- Ritonavir • Norvir
- Sertraline • Zoloft
- Trazodone • Desyrel
- Trazodone extended-release • Oleptro
Disclosures
Dr. Hidalgo receives grant/research support from AstraZeneca, CeNeRx Biopharma, Centers for Disease Control and Prevention, Dainippon Sumitomo Pharma America, Inc., Eli Lilly and Company, Forest Laboratories, Indevus Pharmaceuticals, Janssen Pharmaceuticals, Labopharm, Otsuka, Pfizer, Inc., Repligen Corp., Sanofi-Synthelabo, Sepracor, and the University of South Florida, and is consultant to the MAPI Institute.
Dr. Sheehan has received grant funding support from, been affiliated with, or received honoraria and travel expenses related to lectures/presentations or consultant activities from the following organizations: Abbott Laboratories,1,2,3 Ad Hoc Committee, Treatment Drug and Assessment Research Review,1 Alexza,1 Alza Pharmaceuticals, Palo Alto, CA,1 the American Medical Association,2 American Psychiatric Association Task Force on Benzodiazepine Dependency,1 American Psychiatric Association Task Force on Treatments of Psychiatric Disorders,1 American Psychiatric Association Working Group to Revise DSM III Anxiety Disorders Section,1 Anclote Foundation,2 Anxiety Disorders Resource Center,1 Anxiety Drug Efficacy Case, the FDA,1 Applied Health Outcomes/Xcenda,1 AstraZeneca,1,2,3 Avera Pharmaceuticals,1,2 Boehringer Ingelheim,3 Boots Pharmaceuticals,3 Bristol-Myers Squibb,1,2,3 Burroughs Wellcome,2,3 Cephalon,1 Charter Hospitals,3 Ciba Geigy,3 Committee (RRC) of the National Institute for Mental Health on Anxiety and Phobic Disorder Projects,1 Connecticut and Ohio Academies of Family Physicians,1 Cortex Pharmaceutical,1 Council on Anxiety Disorders,1 CPC Coliseum Medical Center,1 Cypress Bioscience,1 Dista Products Company,3 Division of Drugs and Technology, American Medical Association,1 Eisai,1,2 Eli Lilly and Company,2,3 Excerpta Medica Asia,3 Faxmed, Inc.,1 Forest Laboratories,1,2 Glaxo Pharmaceuticals,3 GlaxoSmithKline,1,2,3 Glaxo-Wellcome,2 Hospital Corporation of America,3 Humana,3 ICI,3 INC Research,1 International Clinical Research (ICR),2 International Society for CNS Drug Development (ISCDD),1 Janssen Pharmaceuticals,1,2,3 Jazz Pharmaceuticals,1,2 Kali-Duphar,2,3 Labopharm,1 Layton Bioscience,1 Lilly Research Laboratories,1 Lundbeck, Denmark,1 Marion Merrell Dow,3 McNeil Pharmaceuticals,3 Mead Johnson,2,3 Medical Outcome Systems,4 MediciNova,1,2 Merck Sharp & Dohme,2,3 National Anxiety Awareness Program,1 National Anxiety Foundation,1 National Depressive and Manic Depressive Association,1 National Institute on Drug Abuse,2 National Institute of Health,2 Neuronetics,1 Novartis Pharmaceuticals Corp.,2 Novo Nordisk,3 Organon,1,3 Orion Pharma,1 Parexel International Corporation,1 Parke-Davis,2,3 Pfizer, Inc.,1,2,3 Pharmacia,1 Pharmacia and Upjohn,1,3 Philadelphia College of Pharmacy and Science,1 Pierre Fabre, France,1 Quintiles,2 Rhone Laboratories,3 Rhone-Poulenc Rorer Pharmaceuticals,3 Roche,1 Roerig,3 Sandoz Pharmaceuticals,2,3 sanofi-aventis,1,2,3 Sanofi-Synthelabo Recherche,1,2 Schering Corporation,3 Sepracor,1 Shire Laboratories, Inc.,1 SmithKline Beecham,1,2,3 Solvay Pharmaceuticals,1,3 Takeda Pharmaceuticals,1 Tampa General Hospital,1 University of South Florida Psychiatry Center,2 University of South Florida College of Medicine, TAP Pharmaceuticals,2,3 Targacept,1 Tampa General Hospital-University Psychiatry Center,3 Tikvah Therapeutics,1 Titan Pharmaceuticals,1 United Bioscience,2 The Upjohn Company,1,2,3 U.S. Congress-House of Representatives Committee,1 University of South Florida Friends of Research in Psychiatry, Board of Trustees,1 Warner Chilcott,2,3 World Health Organization,1 Worldwide Clinical Trials,2 Wyeth-Ayerst,1,2,3 ZARS,1 and Zeneca Pharmaceuticals.1
1: Consultant; 2: Grant/Research Support; 3: Lectures/ Presentations; 4: Stock Holder
1. Oleptro [package insert]. Dublin, Ireland: Labopharm Europe Limited; 2010.
2. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Short-term exposure to low-dose ritonavir impairs clearance and enhances adverse effects of trazodone. J Clin Pharmacol. 2003;43(4):414-422.
3. Beasley CM, Jr, Dornseif BE, Pultz JA, et al. Fluoxetine versus trazodone: efficacy and activating-sedating effects. J Clin Psychiatry. 1991;52:294-299.
4. Kasper S, Olivieri L, Di Loreto G, et al. A comparative, randomized double blind study of trazodone prolonged-release and paroxetine in the treatment of patients with major depressive disorder. Curr Med Res Opin. 2005;21:1139-1146.
5. Munizza C, Olivieri L, Di Loreto G, et al. A comparative, randomized double blind study of trazodone prolonged-release and sertraline in the treatment of patients with major depressive disorder. Curr Med Res Opin. 2006;22:1703-1713.
6. Cunningham LA, Borison RL, Carman JS, et al. A comparison of venlafaxine, trazodone, and placebo in major depression. J Clin Pyschopharmacol. 1994;14:99-106.
7. Weisler RH, Johnston JA, Lineberry CG, et al. Comparison of bupropion and trazodone in the treatment of major depression. J Clin Psychopharmacol. 1994;14:170-179.
8. Feighner JP. Trazodone, a triazolopyridine derivative, in primary depressive disorder. J Clin Psychiatry. 1980;41:250-255.
9. Rickels K, Case WG. Trazodone in depressed outpatients. Am J Psychiatry. 1982;139:803-806.
10. Perry PJ, Garvey MJ, Kelly MW, et al. A comparative trial of fluoxetine versus trazodone in outpatients with major depression. J Clin Psychiatry. 1989;50:290-294.
11. Sheehan DV, Croft HA, Gossen ER, et al. Extended-release trazodone in major depressive disorder: a randomized, double-blind, placebo-controlled study. Psychiatry (Edgmont). 2009;6(5):20-33.
12. Sheehan DV, Rozova A, Gossen ER, et al. The efficacy and tolerability of once-daily controlled-release trazodone for depressed mood, anxiety, insomnia, and suicidality in major depressive disorder. Psychopharmacol Bull. 2009;42(4):5-22.
13. Hu XH, Bull SA, Hunkeler EM, et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry. 2004;65(7):959-965.
14. Landén M, Högberg P, Thase ME. Incidence of sexual side effects in refractory depression during treatment with citalopram or paroxetine. J Clin Psychiatry. 2005;66(1):100-106.
15. Thompson JW, Jr, Ware MR, Blashfield RK. Psychotropic medications and priapism: a comprehensive review. J Clin Psychiatry. 1990;51:430-433.
1. Oleptro [package insert]. Dublin, Ireland: Labopharm Europe Limited; 2010.
2. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Short-term exposure to low-dose ritonavir impairs clearance and enhances adverse effects of trazodone. J Clin Pharmacol. 2003;43(4):414-422.
3. Beasley CM, Jr, Dornseif BE, Pultz JA, et al. Fluoxetine versus trazodone: efficacy and activating-sedating effects. J Clin Psychiatry. 1991;52:294-299.
4. Kasper S, Olivieri L, Di Loreto G, et al. A comparative, randomized double blind study of trazodone prolonged-release and paroxetine in the treatment of patients with major depressive disorder. Curr Med Res Opin. 2005;21:1139-1146.
5. Munizza C, Olivieri L, Di Loreto G, et al. A comparative, randomized double blind study of trazodone prolonged-release and sertraline in the treatment of patients with major depressive disorder. Curr Med Res Opin. 2006;22:1703-1713.
6. Cunningham LA, Borison RL, Carman JS, et al. A comparison of venlafaxine, trazodone, and placebo in major depression. J Clin Pyschopharmacol. 1994;14:99-106.
7. Weisler RH, Johnston JA, Lineberry CG, et al. Comparison of bupropion and trazodone in the treatment of major depression. J Clin Psychopharmacol. 1994;14:170-179.
8. Feighner JP. Trazodone, a triazolopyridine derivative, in primary depressive disorder. J Clin Psychiatry. 1980;41:250-255.
9. Rickels K, Case WG. Trazodone in depressed outpatients. Am J Psychiatry. 1982;139:803-806.
10. Perry PJ, Garvey MJ, Kelly MW, et al. A comparative trial of fluoxetine versus trazodone in outpatients with major depression. J Clin Psychiatry. 1989;50:290-294.
11. Sheehan DV, Croft HA, Gossen ER, et al. Extended-release trazodone in major depressive disorder: a randomized, double-blind, placebo-controlled study. Psychiatry (Edgmont). 2009;6(5):20-33.
12. Sheehan DV, Rozova A, Gossen ER, et al. The efficacy and tolerability of once-daily controlled-release trazodone for depressed mood, anxiety, insomnia, and suicidality in major depressive disorder. Psychopharmacol Bull. 2009;42(4):5-22.
13. Hu XH, Bull SA, Hunkeler EM, et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry. 2004;65(7):959-965.
14. Landén M, Högberg P, Thase ME. Incidence of sexual side effects in refractory depression during treatment with citalopram or paroxetine. J Clin Psychiatry. 2005;66(1):100-106.
15. Thompson JW, Jr, Ware MR, Blashfield RK. Psychotropic medications and priapism: a comprehensive review. J Clin Psychiatry. 1990;51:430-433.
‘Firing’ a patient: May a psychiatrist unilaterally terminate care?
Dear Dr. Mossman:
One of my patients, Ms. A, keeps calling in to refill her prescription, but will not come in for an appointment; she needs the medication, but I really shouldn’t keep prescribing it without seeing her. Another patient, Mr. B, has an open chart, but he stopped seeing me last year after I treated him for an acute depressive episode. May I “fire” these patients? If so, what should I do?—Submitted by “Dr. C”
All physicians occasionally encounter patients whom we’d like to stop treating, but because we feel devoted to those we treat, the idea of “firing” a patient makes us uncomfortable. Sometimes, however, ending a treatment relationship is the right choice for the doctor and patient.1
To know why, how, and when you may terminate your professional relationship with a patient, you need to:
- understand the legal and ethical status of a doctor-patient relationship
- know the proper way to end treatment relationships
- decide whether ending your care of the patient is the right medical and ethical choice.
After exploring these points, we’ll return to the cases of Ms. A and Mr. B and consider what Dr. C might do.
- Submit your malpractice-related questions to Dr. Mossman at [email protected].
- Include your name, address, and practice location. If your question is chosen for publication, your name can be withheld by request.
Doctor-patient relationships
Legal and medical authorities characterize the treatment relationship as an implicit contract that imposes certain obligations on the doctor and the patient.2,3 Doctors are compelled to conduct themselves in accordance with the prevailing “standard of care.” Patients’ obligations include being honest and cooperating with care once they have agreed to a treatment plan (Table 1).3
Patients may stop seeing their doctors at any time, but a physician usually must continue to provide all necessary medical attention until either the treatment episode has concluded or both parties agree to end the doctor-patient relationship.2 If a physician wishes to withdraw from a case before the need for services has ended, the physician must either make arrangements for another competent physician to assume care or give the patient ample notice and opportunity to obtain treatment elsewhere.2 If a doctor fails to do this and harm to the patient results, the doctor is guilty of “abandonment,” legally defined as termination of the physician-patient relationship “at an unreasonable time or without affording the patient the opportunity to procure an equally qualified replacement.”2 Physician abandonment can lead to malpractice liability,4 complaints to state licensing authorities,5 and ethical condemnation.6
Table 1
A patient’s responsibilities
| Being truthful |
| Providing a complete medical history |
| Cooperating with agreed-upon treatment and keeping appointments |
| Meeting financial obligations for medical care |
| Health-enhancing behavior |
| Not participating in fraudulent health care |
| Source: Reference 3 |
Terminating without abandoning
Doctors commonly terminate care of their patients when they decide to move or close their practices. Accusations of abandonment may arise if such career decisions are executed improperly, but these matters are not as emotionally troubling for physicians as a decision to “fire” a patient because of the patient’s behavior. Common, legitimate reasons a doctor may consider unilateral termination appear in Table 2.7,8
Certain circumstances are not valid grounds for terminating a doctor-patient relationship. You cannot ethically decline to treat a patient whose problem lies within your areas of clinical competence solely because the patient is seropositive for human immunodeficiency virus,9 nor because of a patient’s race, religion, or other reasons that would constitute illegal discrimination.3 Doctors who practice in rural areas must be especially cautious about terminating care because their patients may have limited access to alternate care sources.10
Meeting with or verbally informing a patient of a termination may be reasonable in some cases, but appropriate unilateral termination of a patient usually requires providing written notification to the patient or person responsible for the patient’s care. Attorneys who specialize in risk management advise doctors to seek legal consultation when preparing a termination-of-care letter and to send it by certified mail. The letter should conform to any applicable rules or regulations where you practice. Typically, required content includes:
- notification that the physician-patient relationship is terminated
- a statement of willingness to provide emergency treatment and access to services for up to 30 days from the mailing date to allow the patient to arrange care from another provider
- an offer to transfer records to the new provider upon receiving the patient’s signed authorization to do so.11
More discussion of the possible contents of termination letters appears in Table 3.7,12-14
Table 2
Common reasons to consider terminating a patient’s care
| Failing to pay bills |
| Repeatedly cancelling or missing appointments |
| Repeatedly failing to follow the agreed-upon treatment plan |
| Overly demanding, rude, disruptive, threatening, or violent behavior toward staff or other patients |
| Patient is very dissatisfied with care |
| Needing specialized services that the physician cannot provide |
| Filing a complaint or legal action against the physician |
| Dishonesty that compromises safety or legality of treatment |
| Physician feels treatment is ineffective |
| Conflict of interest (eg, physician’s religious beliefs preclude providing certain treatments that might be indicated) |
| Developing and acting upon an inappropriate personal interest in the physician |
| Inappropriate response by physician to feelings about the patient (eg, physician feels tempted to act upon an attraction) |
| Source: References 7,8 |
Table 3
Potential elements of termination letters
| Element | Comment | |
|---|---|---|
| Reason for termination | Giving a reason is not required. If an explanation seems necessary, offer a general statement (eg, ‘I have determined it would be best…’) | |
| Adequate time to seek care elsewhere | Typically, at least 30 days. Courts have described appropriate time frames in general terms, such as ‘ample,’ ‘sufficient,’ or ‘reasonable’ | |
| Interim care provisions | Offer interim care for urgent problems until the time limit stated above | |
| Continued care provisions |
| |
| Medical record copies | Offer to provide a summary of treatment or copy of the record to a new provider. Consider enclosing a ‘release of information’ authorization to be returned to the office with the patient’s signature | |
| Sending the letter | Regular and certified mail (return receipt requested). Place a copy of this letter in the patient’s medical record, along with the original certified mail receipt and, if received, the original return receipt | |
| Source: References 7,12-14 | ||
Deciding to ‘fire‘ a patient
Physicians in all specialties encounter patients whose actions generate intensely negative feelings—resentment, anger, even hate.15 But “firing” a patient should be a rare circumstance that’s not undertaken lightly. Many different circumstances can make it reasonable for a physician to consider terminating a patient’s care, so it’s difficult to provide general advice about when firing a patient really is the right thing to do. But 1 “prescription” seems clear: consult a respected colleague first. According to psychiatrist Robert Michels, “Any physician who is thinking of firing a patient should first speak to a colleague… This is an enormous decision and, while it might even be right at times, the physician is probably having a countertransference reaction to his patient and should really understand that before taking action.”1
Having an anonymous consultation with a colleague offers several potential benefits, such as:
- If you’re thinking about firing a patient, you’re probably very upset. A colleague who isn’t emotionally involved can help you assess the matter more dispassionately.
- You may be feeling guilty about disliking the patient. A colleague’s empathy (“Of course you’re angry!”) can help you avoid disowning your feelings, which may make it easier to figure out how to use those feelings to help the patient.15,16
- A colleague may think of solutions that you haven’t considered, which might help you feel less frustrated about how treatment is going.
- A colleague may help you see ways that you’re actually helping the patient, despite feeling that your work is futile.
- If a thoughtful colleague confirms your view that terminating care is appropriate, you’ll feel better about the decision. If you document the anonymous consultation in the patient’s chart, you’ll create a record of your reasonableness and prudence—which will be helpful if you have to defend your action in court.12
Revisiting the case patients
With these thoughts in mind, we return to Dr. C’s clinical dilemmas.
Ms. A. In retrospect, Dr. C might wish he had been clearer with Ms. A about how often she would need to see him for medication monitoring. At this point, however, Dr. C still has options besides firing Ms. A:
- Dr. C can call Ms. A to ask how she’s doing and to explain his medical responsibility to see and reassess her if he is to continue prescribing her medication. He can then follow up with a letter summarizing the conversation.
- Dr. C might ask whether some problem is preventing Ms. A from making an appointment. If, for example, Ms. A has lost her job and health insurance coverage for office visits, Dr. C might suggest options (such as seeing Ms. A once at no charge) or help Ms. A find other ways to obtain follow-up care.
Mr. B. Concerning Mr. B, we wonder, “Why not just leave the chart open?” As is the case with care provided by other specialists—including internists, obstetricians, or dermatologists—psychiatric treatment may occur in discrete episodes over many years. Patients regard a previous care provider as “their doctor” for decades after a treatment episode, and it’s comforting and valuable for former patients to know they can see their “shrink” again if they need to.
Related Resource
- Henderson SM. Advice on abandonment. Oklahoma Board of Medical Licensure and Supervision. www.okmedicalboard.org/download/19980401MD.htm.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Friedman RA. Should a doctor fire a patient? Sometimes it is good medicine. New York Times. September 27, 2005;sect F:1.
2. Dietz LH, Jacobs A, Leming TL, et al. Physicians, surgeons, and other healers, §§130, 216-218. In: American jurisprudence. Vol 61. 2nd ed. New York, NY: Thomson Reuters; 2010.
3. American Medical Association. Code of ethics. Opinions 9.12, 10.02, and 10.015. Available at: http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics.shtml. Accessed October 18, 2010.
4. Lowery v Miller, 157 Wis 2d 503, 460 NW2d 446 (Wis App 1990).
5. Crausman RS. Board of medical licensure and discipline. Available at: http://www.health.ri.gov/hsr/bmld. Accessed October 27, 2010.
6. Pellegrino ED. Nonabandonment: an old obligation revisited. Ann Intern Med. 1995;122:377-378.
7. Harris SM. Take care when firing a patient. American Medical News. Available at: http://www.ama-assn.org/amednews/2008/02/04/bica0204.htm. Accessed October 18, 2010.
8. Gabbard GO. Long-term psychodynamic psychotherapy: a basic text. Arlington, VA: American Psychiatric Publishing, Inc.; 2004.
9. Bragdon v Abbott, 524 U.S. 624 (1998).
10. Henderson SM. Advice on abandonment. Oklahoma Board of Medical Licensure and Supervision. Available at: http://www.okmedicalboard.org/download/19980401MD.htm. Accessed October 18, 2010.
11. Ohio Admin Code Ch, 4731-27(A)(1).
12. Appelbaum PS, Gutheil T. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2006.
13. Appelbaum PS. Law & psychiatry: can a psychiatrist be held responsible when a patient commits murder? Psychiatr Serv. 2002;53:27-29.
14. Tan MW, McDonough WJ. Risk management in psychiatry. Psychiatr Clin North Am. 1990;13:135-147.
15. Groves JE. Taking care of the hateful patient. N Engl J Med. 1978;298:883-887.
16. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17:387-393.

Douglas Mossman, MD
Dr. Mossman is director, Glenn M. Weaver Institute of Law and Psychiatry, University of Cincinnati College of Law, and Adjunct Professor of Clinical Psychiatry and Training Director for the Forensic Psychiatry Fellowship.
Helen M. Farrell, MD
Dr. Farrell, Fellow in Forensic Psychiatry, University of Cincinnati College of Medicine.
Elizabeth Gilday, MD
Dr.Gilday, Fellow in Forensic Psychiatry, University of Cincinnati College of Medicine.
Douglas Mossman, MD
Dr. Mossman is director, Glenn M. Weaver Institute of Law and Psychiatry, University of Cincinnati College of Law, and Adjunct Professor of Clinical Psychiatry and Training Director for the Forensic Psychiatry Fellowship.
Helen M. Farrell, MD
Dr. Farrell, Fellow in Forensic Psychiatry, University of Cincinnati College of Medicine.
Elizabeth Gilday, MD
Dr.Gilday, Fellow in Forensic Psychiatry, University of Cincinnati College of Medicine.
Douglas Mossman, MD
Dr. Mossman is director, Glenn M. Weaver Institute of Law and Psychiatry, University of Cincinnati College of Law, and Adjunct Professor of Clinical Psychiatry and Training Director for the Forensic Psychiatry Fellowship.
Helen M. Farrell, MD
Dr. Farrell, Fellow in Forensic Psychiatry, University of Cincinnati College of Medicine.
Elizabeth Gilday, MD
Dr.Gilday, Fellow in Forensic Psychiatry, University of Cincinnati College of Medicine.
Dear Dr. Mossman:
One of my patients, Ms. A, keeps calling in to refill her prescription, but will not come in for an appointment; she needs the medication, but I really shouldn’t keep prescribing it without seeing her. Another patient, Mr. B, has an open chart, but he stopped seeing me last year after I treated him for an acute depressive episode. May I “fire” these patients? If so, what should I do?—Submitted by “Dr. C”
All physicians occasionally encounter patients whom we’d like to stop treating, but because we feel devoted to those we treat, the idea of “firing” a patient makes us uncomfortable. Sometimes, however, ending a treatment relationship is the right choice for the doctor and patient.1
To know why, how, and when you may terminate your professional relationship with a patient, you need to:
- understand the legal and ethical status of a doctor-patient relationship
- know the proper way to end treatment relationships
- decide whether ending your care of the patient is the right medical and ethical choice.
After exploring these points, we’ll return to the cases of Ms. A and Mr. B and consider what Dr. C might do.
- Submit your malpractice-related questions to Dr. Mossman at [email protected].
- Include your name, address, and practice location. If your question is chosen for publication, your name can be withheld by request.
Doctor-patient relationships
Legal and medical authorities characterize the treatment relationship as an implicit contract that imposes certain obligations on the doctor and the patient.2,3 Doctors are compelled to conduct themselves in accordance with the prevailing “standard of care.” Patients’ obligations include being honest and cooperating with care once they have agreed to a treatment plan (Table 1).3
Patients may stop seeing their doctors at any time, but a physician usually must continue to provide all necessary medical attention until either the treatment episode has concluded or both parties agree to end the doctor-patient relationship.2 If a physician wishes to withdraw from a case before the need for services has ended, the physician must either make arrangements for another competent physician to assume care or give the patient ample notice and opportunity to obtain treatment elsewhere.2 If a doctor fails to do this and harm to the patient results, the doctor is guilty of “abandonment,” legally defined as termination of the physician-patient relationship “at an unreasonable time or without affording the patient the opportunity to procure an equally qualified replacement.”2 Physician abandonment can lead to malpractice liability,4 complaints to state licensing authorities,5 and ethical condemnation.6
Table 1
A patient’s responsibilities
| Being truthful |
| Providing a complete medical history |
| Cooperating with agreed-upon treatment and keeping appointments |
| Meeting financial obligations for medical care |
| Health-enhancing behavior |
| Not participating in fraudulent health care |
| Source: Reference 3 |
Terminating without abandoning
Doctors commonly terminate care of their patients when they decide to move or close their practices. Accusations of abandonment may arise if such career decisions are executed improperly, but these matters are not as emotionally troubling for physicians as a decision to “fire” a patient because of the patient’s behavior. Common, legitimate reasons a doctor may consider unilateral termination appear in Table 2.7,8
Certain circumstances are not valid grounds for terminating a doctor-patient relationship. You cannot ethically decline to treat a patient whose problem lies within your areas of clinical competence solely because the patient is seropositive for human immunodeficiency virus,9 nor because of a patient’s race, religion, or other reasons that would constitute illegal discrimination.3 Doctors who practice in rural areas must be especially cautious about terminating care because their patients may have limited access to alternate care sources.10
Meeting with or verbally informing a patient of a termination may be reasonable in some cases, but appropriate unilateral termination of a patient usually requires providing written notification to the patient or person responsible for the patient’s care. Attorneys who specialize in risk management advise doctors to seek legal consultation when preparing a termination-of-care letter and to send it by certified mail. The letter should conform to any applicable rules or regulations where you practice. Typically, required content includes:
- notification that the physician-patient relationship is terminated
- a statement of willingness to provide emergency treatment and access to services for up to 30 days from the mailing date to allow the patient to arrange care from another provider
- an offer to transfer records to the new provider upon receiving the patient’s signed authorization to do so.11
More discussion of the possible contents of termination letters appears in Table 3.7,12-14
Table 2
Common reasons to consider terminating a patient’s care
| Failing to pay bills |
| Repeatedly cancelling or missing appointments |
| Repeatedly failing to follow the agreed-upon treatment plan |
| Overly demanding, rude, disruptive, threatening, or violent behavior toward staff or other patients |
| Patient is very dissatisfied with care |
| Needing specialized services that the physician cannot provide |
| Filing a complaint or legal action against the physician |
| Dishonesty that compromises safety or legality of treatment |
| Physician feels treatment is ineffective |
| Conflict of interest (eg, physician’s religious beliefs preclude providing certain treatments that might be indicated) |
| Developing and acting upon an inappropriate personal interest in the physician |
| Inappropriate response by physician to feelings about the patient (eg, physician feels tempted to act upon an attraction) |
| Source: References 7,8 |
Table 3
Potential elements of termination letters
| Element | Comment | |
|---|---|---|
| Reason for termination | Giving a reason is not required. If an explanation seems necessary, offer a general statement (eg, ‘I have determined it would be best…’) | |
| Adequate time to seek care elsewhere | Typically, at least 30 days. Courts have described appropriate time frames in general terms, such as ‘ample,’ ‘sufficient,’ or ‘reasonable’ | |
| Interim care provisions | Offer interim care for urgent problems until the time limit stated above | |
| Continued care provisions |
| |
| Medical record copies | Offer to provide a summary of treatment or copy of the record to a new provider. Consider enclosing a ‘release of information’ authorization to be returned to the office with the patient’s signature | |
| Sending the letter | Regular and certified mail (return receipt requested). Place a copy of this letter in the patient’s medical record, along with the original certified mail receipt and, if received, the original return receipt | |
| Source: References 7,12-14 | ||
Deciding to ‘fire‘ a patient
Physicians in all specialties encounter patients whose actions generate intensely negative feelings—resentment, anger, even hate.15 But “firing” a patient should be a rare circumstance that’s not undertaken lightly. Many different circumstances can make it reasonable for a physician to consider terminating a patient’s care, so it’s difficult to provide general advice about when firing a patient really is the right thing to do. But 1 “prescription” seems clear: consult a respected colleague first. According to psychiatrist Robert Michels, “Any physician who is thinking of firing a patient should first speak to a colleague… This is an enormous decision and, while it might even be right at times, the physician is probably having a countertransference reaction to his patient and should really understand that before taking action.”1
Having an anonymous consultation with a colleague offers several potential benefits, such as:
- If you’re thinking about firing a patient, you’re probably very upset. A colleague who isn’t emotionally involved can help you assess the matter more dispassionately.
- You may be feeling guilty about disliking the patient. A colleague’s empathy (“Of course you’re angry!”) can help you avoid disowning your feelings, which may make it easier to figure out how to use those feelings to help the patient.15,16
- A colleague may think of solutions that you haven’t considered, which might help you feel less frustrated about how treatment is going.
- A colleague may help you see ways that you’re actually helping the patient, despite feeling that your work is futile.
- If a thoughtful colleague confirms your view that terminating care is appropriate, you’ll feel better about the decision. If you document the anonymous consultation in the patient’s chart, you’ll create a record of your reasonableness and prudence—which will be helpful if you have to defend your action in court.12
Revisiting the case patients
With these thoughts in mind, we return to Dr. C’s clinical dilemmas.
Ms. A. In retrospect, Dr. C might wish he had been clearer with Ms. A about how often she would need to see him for medication monitoring. At this point, however, Dr. C still has options besides firing Ms. A:
- Dr. C can call Ms. A to ask how she’s doing and to explain his medical responsibility to see and reassess her if he is to continue prescribing her medication. He can then follow up with a letter summarizing the conversation.
- Dr. C might ask whether some problem is preventing Ms. A from making an appointment. If, for example, Ms. A has lost her job and health insurance coverage for office visits, Dr. C might suggest options (such as seeing Ms. A once at no charge) or help Ms. A find other ways to obtain follow-up care.
Mr. B. Concerning Mr. B, we wonder, “Why not just leave the chart open?” As is the case with care provided by other specialists—including internists, obstetricians, or dermatologists—psychiatric treatment may occur in discrete episodes over many years. Patients regard a previous care provider as “their doctor” for decades after a treatment episode, and it’s comforting and valuable for former patients to know they can see their “shrink” again if they need to.
Related Resource
- Henderson SM. Advice on abandonment. Oklahoma Board of Medical Licensure and Supervision. www.okmedicalboard.org/download/19980401MD.htm.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dear Dr. Mossman:
One of my patients, Ms. A, keeps calling in to refill her prescription, but will not come in for an appointment; she needs the medication, but I really shouldn’t keep prescribing it without seeing her. Another patient, Mr. B, has an open chart, but he stopped seeing me last year after I treated him for an acute depressive episode. May I “fire” these patients? If so, what should I do?—Submitted by “Dr. C”
All physicians occasionally encounter patients whom we’d like to stop treating, but because we feel devoted to those we treat, the idea of “firing” a patient makes us uncomfortable. Sometimes, however, ending a treatment relationship is the right choice for the doctor and patient.1
To know why, how, and when you may terminate your professional relationship with a patient, you need to:
- understand the legal and ethical status of a doctor-patient relationship
- know the proper way to end treatment relationships
- decide whether ending your care of the patient is the right medical and ethical choice.
After exploring these points, we’ll return to the cases of Ms. A and Mr. B and consider what Dr. C might do.
- Submit your malpractice-related questions to Dr. Mossman at [email protected].
- Include your name, address, and practice location. If your question is chosen for publication, your name can be withheld by request.
Doctor-patient relationships
Legal and medical authorities characterize the treatment relationship as an implicit contract that imposes certain obligations on the doctor and the patient.2,3 Doctors are compelled to conduct themselves in accordance with the prevailing “standard of care.” Patients’ obligations include being honest and cooperating with care once they have agreed to a treatment plan (Table 1).3
Patients may stop seeing their doctors at any time, but a physician usually must continue to provide all necessary medical attention until either the treatment episode has concluded or both parties agree to end the doctor-patient relationship.2 If a physician wishes to withdraw from a case before the need for services has ended, the physician must either make arrangements for another competent physician to assume care or give the patient ample notice and opportunity to obtain treatment elsewhere.2 If a doctor fails to do this and harm to the patient results, the doctor is guilty of “abandonment,” legally defined as termination of the physician-patient relationship “at an unreasonable time or without affording the patient the opportunity to procure an equally qualified replacement.”2 Physician abandonment can lead to malpractice liability,4 complaints to state licensing authorities,5 and ethical condemnation.6
Table 1
A patient’s responsibilities
| Being truthful |
| Providing a complete medical history |
| Cooperating with agreed-upon treatment and keeping appointments |
| Meeting financial obligations for medical care |
| Health-enhancing behavior |
| Not participating in fraudulent health care |
| Source: Reference 3 |
Terminating without abandoning
Doctors commonly terminate care of their patients when they decide to move or close their practices. Accusations of abandonment may arise if such career decisions are executed improperly, but these matters are not as emotionally troubling for physicians as a decision to “fire” a patient because of the patient’s behavior. Common, legitimate reasons a doctor may consider unilateral termination appear in Table 2.7,8
Certain circumstances are not valid grounds for terminating a doctor-patient relationship. You cannot ethically decline to treat a patient whose problem lies within your areas of clinical competence solely because the patient is seropositive for human immunodeficiency virus,9 nor because of a patient’s race, religion, or other reasons that would constitute illegal discrimination.3 Doctors who practice in rural areas must be especially cautious about terminating care because their patients may have limited access to alternate care sources.10
Meeting with or verbally informing a patient of a termination may be reasonable in some cases, but appropriate unilateral termination of a patient usually requires providing written notification to the patient or person responsible for the patient’s care. Attorneys who specialize in risk management advise doctors to seek legal consultation when preparing a termination-of-care letter and to send it by certified mail. The letter should conform to any applicable rules or regulations where you practice. Typically, required content includes:
- notification that the physician-patient relationship is terminated
- a statement of willingness to provide emergency treatment and access to services for up to 30 days from the mailing date to allow the patient to arrange care from another provider
- an offer to transfer records to the new provider upon receiving the patient’s signed authorization to do so.11
More discussion of the possible contents of termination letters appears in Table 3.7,12-14
Table 2
Common reasons to consider terminating a patient’s care
| Failing to pay bills |
| Repeatedly cancelling or missing appointments |
| Repeatedly failing to follow the agreed-upon treatment plan |
| Overly demanding, rude, disruptive, threatening, or violent behavior toward staff or other patients |
| Patient is very dissatisfied with care |
| Needing specialized services that the physician cannot provide |
| Filing a complaint or legal action against the physician |
| Dishonesty that compromises safety or legality of treatment |
| Physician feels treatment is ineffective |
| Conflict of interest (eg, physician’s religious beliefs preclude providing certain treatments that might be indicated) |
| Developing and acting upon an inappropriate personal interest in the physician |
| Inappropriate response by physician to feelings about the patient (eg, physician feels tempted to act upon an attraction) |
| Source: References 7,8 |
Table 3
Potential elements of termination letters
| Element | Comment | |
|---|---|---|
| Reason for termination | Giving a reason is not required. If an explanation seems necessary, offer a general statement (eg, ‘I have determined it would be best…’) | |
| Adequate time to seek care elsewhere | Typically, at least 30 days. Courts have described appropriate time frames in general terms, such as ‘ample,’ ‘sufficient,’ or ‘reasonable’ | |
| Interim care provisions | Offer interim care for urgent problems until the time limit stated above | |
| Continued care provisions |
| |
| Medical record copies | Offer to provide a summary of treatment or copy of the record to a new provider. Consider enclosing a ‘release of information’ authorization to be returned to the office with the patient’s signature | |
| Sending the letter | Regular and certified mail (return receipt requested). Place a copy of this letter in the patient’s medical record, along with the original certified mail receipt and, if received, the original return receipt | |
| Source: References 7,12-14 | ||
Deciding to ‘fire‘ a patient
Physicians in all specialties encounter patients whose actions generate intensely negative feelings—resentment, anger, even hate.15 But “firing” a patient should be a rare circumstance that’s not undertaken lightly. Many different circumstances can make it reasonable for a physician to consider terminating a patient’s care, so it’s difficult to provide general advice about when firing a patient really is the right thing to do. But 1 “prescription” seems clear: consult a respected colleague first. According to psychiatrist Robert Michels, “Any physician who is thinking of firing a patient should first speak to a colleague… This is an enormous decision and, while it might even be right at times, the physician is probably having a countertransference reaction to his patient and should really understand that before taking action.”1
Having an anonymous consultation with a colleague offers several potential benefits, such as:
- If you’re thinking about firing a patient, you’re probably very upset. A colleague who isn’t emotionally involved can help you assess the matter more dispassionately.
- You may be feeling guilty about disliking the patient. A colleague’s empathy (“Of course you’re angry!”) can help you avoid disowning your feelings, which may make it easier to figure out how to use those feelings to help the patient.15,16
- A colleague may think of solutions that you haven’t considered, which might help you feel less frustrated about how treatment is going.
- A colleague may help you see ways that you’re actually helping the patient, despite feeling that your work is futile.
- If a thoughtful colleague confirms your view that terminating care is appropriate, you’ll feel better about the decision. If you document the anonymous consultation in the patient’s chart, you’ll create a record of your reasonableness and prudence—which will be helpful if you have to defend your action in court.12
Revisiting the case patients
With these thoughts in mind, we return to Dr. C’s clinical dilemmas.
Ms. A. In retrospect, Dr. C might wish he had been clearer with Ms. A about how often she would need to see him for medication monitoring. At this point, however, Dr. C still has options besides firing Ms. A:
- Dr. C can call Ms. A to ask how she’s doing and to explain his medical responsibility to see and reassess her if he is to continue prescribing her medication. He can then follow up with a letter summarizing the conversation.
- Dr. C might ask whether some problem is preventing Ms. A from making an appointment. If, for example, Ms. A has lost her job and health insurance coverage for office visits, Dr. C might suggest options (such as seeing Ms. A once at no charge) or help Ms. A find other ways to obtain follow-up care.
Mr. B. Concerning Mr. B, we wonder, “Why not just leave the chart open?” As is the case with care provided by other specialists—including internists, obstetricians, or dermatologists—psychiatric treatment may occur in discrete episodes over many years. Patients regard a previous care provider as “their doctor” for decades after a treatment episode, and it’s comforting and valuable for former patients to know they can see their “shrink” again if they need to.
Related Resource
- Henderson SM. Advice on abandonment. Oklahoma Board of Medical Licensure and Supervision. www.okmedicalboard.org/download/19980401MD.htm.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Friedman RA. Should a doctor fire a patient? Sometimes it is good medicine. New York Times. September 27, 2005;sect F:1.
2. Dietz LH, Jacobs A, Leming TL, et al. Physicians, surgeons, and other healers, §§130, 216-218. In: American jurisprudence. Vol 61. 2nd ed. New York, NY: Thomson Reuters; 2010.
3. American Medical Association. Code of ethics. Opinions 9.12, 10.02, and 10.015. Available at: http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics.shtml. Accessed October 18, 2010.
4. Lowery v Miller, 157 Wis 2d 503, 460 NW2d 446 (Wis App 1990).
5. Crausman RS. Board of medical licensure and discipline. Available at: http://www.health.ri.gov/hsr/bmld. Accessed October 27, 2010.
6. Pellegrino ED. Nonabandonment: an old obligation revisited. Ann Intern Med. 1995;122:377-378.
7. Harris SM. Take care when firing a patient. American Medical News. Available at: http://www.ama-assn.org/amednews/2008/02/04/bica0204.htm. Accessed October 18, 2010.
8. Gabbard GO. Long-term psychodynamic psychotherapy: a basic text. Arlington, VA: American Psychiatric Publishing, Inc.; 2004.
9. Bragdon v Abbott, 524 U.S. 624 (1998).
10. Henderson SM. Advice on abandonment. Oklahoma Board of Medical Licensure and Supervision. Available at: http://www.okmedicalboard.org/download/19980401MD.htm. Accessed October 18, 2010.
11. Ohio Admin Code Ch, 4731-27(A)(1).
12. Appelbaum PS, Gutheil T. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2006.
13. Appelbaum PS. Law & psychiatry: can a psychiatrist be held responsible when a patient commits murder? Psychiatr Serv. 2002;53:27-29.
14. Tan MW, McDonough WJ. Risk management in psychiatry. Psychiatr Clin North Am. 1990;13:135-147.
15. Groves JE. Taking care of the hateful patient. N Engl J Med. 1978;298:883-887.
16. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17:387-393.
1. Friedman RA. Should a doctor fire a patient? Sometimes it is good medicine. New York Times. September 27, 2005;sect F:1.
2. Dietz LH, Jacobs A, Leming TL, et al. Physicians, surgeons, and other healers, §§130, 216-218. In: American jurisprudence. Vol 61. 2nd ed. New York, NY: Thomson Reuters; 2010.
3. American Medical Association. Code of ethics. Opinions 9.12, 10.02, and 10.015. Available at: http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics.shtml. Accessed October 18, 2010.
4. Lowery v Miller, 157 Wis 2d 503, 460 NW2d 446 (Wis App 1990).
5. Crausman RS. Board of medical licensure and discipline. Available at: http://www.health.ri.gov/hsr/bmld. Accessed October 27, 2010.
6. Pellegrino ED. Nonabandonment: an old obligation revisited. Ann Intern Med. 1995;122:377-378.
7. Harris SM. Take care when firing a patient. American Medical News. Available at: http://www.ama-assn.org/amednews/2008/02/04/bica0204.htm. Accessed October 18, 2010.
8. Gabbard GO. Long-term psychodynamic psychotherapy: a basic text. Arlington, VA: American Psychiatric Publishing, Inc.; 2004.
9. Bragdon v Abbott, 524 U.S. 624 (1998).
10. Henderson SM. Advice on abandonment. Oklahoma Board of Medical Licensure and Supervision. Available at: http://www.okmedicalboard.org/download/19980401MD.htm. Accessed October 18, 2010.
11. Ohio Admin Code Ch, 4731-27(A)(1).
12. Appelbaum PS, Gutheil T. Clinical handbook of psychiatry and the law. 4th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2006.
13. Appelbaum PS. Law & psychiatry: can a psychiatrist be held responsible when a patient commits murder? Psychiatr Serv. 2002;53:27-29.
14. Tan MW, McDonough WJ. Risk management in psychiatry. Psychiatr Clin North Am. 1990;13:135-147.
15. Groves JE. Taking care of the hateful patient. N Engl J Med. 1978;298:883-887.
16. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17:387-393.
Recognizing the unheralded heroes of psychiatry
A large number of individuals contribute in many ways to the process of discovering, applying, and disseminating new psychiatric knowledge. I am, of course, referring to researchers, clinicians, teachers, and advocates who touch the lives of millions of persons who suffer from mental illness every year. This editorial is dedicated to singing the praises of those who quietly contribute to advancing psychiatry.
Research
Patients. Tens of thousands of psychiatric patients sign an informed consent form and volunteer to participate in clinical trials to test new drugs in double-blind, placebo-controlled studies that could lead to FDA approval. Without these volunteers, it would be almost impossible to develop new medications.
Investigators. Psychiatrists and neuroscientists who dedicate their lives to discovering the causes and treatments of psychiatric brain disorders by conducting basic, translational, or clinical research spend countless hours designing, writing, and submitting grant proposals that have a <10% chance of getting funded. They work long hours to discover new knowledge despite limited resources. They move our field forward with cutting-edge discoveries.
Research assistants. They are an army of skilled technical workers who do the heavy lifting in animal or human research and put in long hours to collect data or conduct tests. Yet they are rarely recognized for their critical contributions to science and clinical practice.
Clinicians. Colleagues who refer their patients to research studies are supporting discovery of new knowledge and without them new psychiatric drug development would slow down considerably.
Reviewers of grants and journal articles. It takes a great deal of expertise and time to identify grants worthy of receiving taxpayer support or to review and recommend the best articles for publication in leading psychiatric journals. Many distinguished researchers—who often are clinicians—donate enormous amounts of uncompensated time to these essential tasks. For a list of colleagues who served as Current Psychiatry peer reviewers in 2010.
Interactive clinicians find time to send in letters to the editor about an unusual clinical observation that may generate new research ideas.
Clinical
Psychiatrists and nurse practitioners, who put in far more than the 40-hour work week to meet the need for psychiatric care caused by the severe shortage of psychiatric clinicians in the United States. They sacrifice their personal needs to help others.
Pro bono clinicians, who donate their services without compensation to those who are indigent or uninsured.
Assertive Community Treatment team members, who reach out to the homeless and seriously sick patients who are the most challenging.
Psychiatric nurses, who are the frontline mental health professionals on psychiatric inpatient units and in psychiatric emergency rooms. They deserve our gratitude and thanks.
Families, whose love and dedication to their mentally ill family members can be critical for seeking psychiatric care or adhering to treatment.
Teaching
Teaching faculty, who provide education and training for physicians, psychiatrists, and nurse practitioners of the future. This includes volunteer (unpaid) faculty who donate their time to supervise psychiatric trainees. They prepare the future generations of clinicians, researchers, and teachers.
Mentors, who may be clinicians, researchers, or both, providing personal guidance, coaching, support and role-models for young trainees or early-career psychiatrists, often shaping their lives and careers. Their generosity and generativity are priceless.
Service
Psychiatrists, who actively participate in their local, state, and national professional societies. I estimate that 10% of the members of any association do the bulk of the work to accomplish the stated mission.
Advocacy groups such as the National Alliance on Mental Illness, Mental Health America, and others who do so much to help persons suffering from psychiatric disorders and whose public service advances the cause of our patients and the mental health field.
Colleagues who step forward and do the gut-wrenching but necessary task of turning in colleagues who commit unethical or illegal acts or who come to work intoxicated. These individuals ensure the integrity of the specialty and prompt their colleagues to seek help.
Celebrities and public figures who openly discuss their personal or family struggles with psychiatric illness. They help diminish stigma, enhance the public’s willingness to seek psychiatric care, and reduce discrimination.
I tip my proverbial hat to all the above heroes. Their ongoing contributions are vital for our profession, and they deserve our gratitude.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://CurrentPsychiatry.blogspot.com, or go to our Web site at CurrentPsychiatry.com and click on the “Send Letters” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://CurrentPsychiatry.blogspot.com, or go to our Web site at CurrentPsychiatry.com and click on the “Send Letters” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://CurrentPsychiatry.blogspot.com, or go to our Web site at CurrentPsychiatry.com and click on the “Send Letters” link.
A large number of individuals contribute in many ways to the process of discovering, applying, and disseminating new psychiatric knowledge. I am, of course, referring to researchers, clinicians, teachers, and advocates who touch the lives of millions of persons who suffer from mental illness every year. This editorial is dedicated to singing the praises of those who quietly contribute to advancing psychiatry.
Research
Patients. Tens of thousands of psychiatric patients sign an informed consent form and volunteer to participate in clinical trials to test new drugs in double-blind, placebo-controlled studies that could lead to FDA approval. Without these volunteers, it would be almost impossible to develop new medications.
Investigators. Psychiatrists and neuroscientists who dedicate their lives to discovering the causes and treatments of psychiatric brain disorders by conducting basic, translational, or clinical research spend countless hours designing, writing, and submitting grant proposals that have a <10% chance of getting funded. They work long hours to discover new knowledge despite limited resources. They move our field forward with cutting-edge discoveries.
Research assistants. They are an army of skilled technical workers who do the heavy lifting in animal or human research and put in long hours to collect data or conduct tests. Yet they are rarely recognized for their critical contributions to science and clinical practice.
Clinicians. Colleagues who refer their patients to research studies are supporting discovery of new knowledge and without them new psychiatric drug development would slow down considerably.
Reviewers of grants and journal articles. It takes a great deal of expertise and time to identify grants worthy of receiving taxpayer support or to review and recommend the best articles for publication in leading psychiatric journals. Many distinguished researchers—who often are clinicians—donate enormous amounts of uncompensated time to these essential tasks. For a list of colleagues who served as Current Psychiatry peer reviewers in 2010.
Interactive clinicians find time to send in letters to the editor about an unusual clinical observation that may generate new research ideas.
Clinical
Psychiatrists and nurse practitioners, who put in far more than the 40-hour work week to meet the need for psychiatric care caused by the severe shortage of psychiatric clinicians in the United States. They sacrifice their personal needs to help others.
Pro bono clinicians, who donate their services without compensation to those who are indigent or uninsured.
Assertive Community Treatment team members, who reach out to the homeless and seriously sick patients who are the most challenging.
Psychiatric nurses, who are the frontline mental health professionals on psychiatric inpatient units and in psychiatric emergency rooms. They deserve our gratitude and thanks.
Families, whose love and dedication to their mentally ill family members can be critical for seeking psychiatric care or adhering to treatment.
Teaching
Teaching faculty, who provide education and training for physicians, psychiatrists, and nurse practitioners of the future. This includes volunteer (unpaid) faculty who donate their time to supervise psychiatric trainees. They prepare the future generations of clinicians, researchers, and teachers.
Mentors, who may be clinicians, researchers, or both, providing personal guidance, coaching, support and role-models for young trainees or early-career psychiatrists, often shaping their lives and careers. Their generosity and generativity are priceless.
Service
Psychiatrists, who actively participate in their local, state, and national professional societies. I estimate that 10% of the members of any association do the bulk of the work to accomplish the stated mission.
Advocacy groups such as the National Alliance on Mental Illness, Mental Health America, and others who do so much to help persons suffering from psychiatric disorders and whose public service advances the cause of our patients and the mental health field.
Colleagues who step forward and do the gut-wrenching but necessary task of turning in colleagues who commit unethical or illegal acts or who come to work intoxicated. These individuals ensure the integrity of the specialty and prompt their colleagues to seek help.
Celebrities and public figures who openly discuss their personal or family struggles with psychiatric illness. They help diminish stigma, enhance the public’s willingness to seek psychiatric care, and reduce discrimination.
I tip my proverbial hat to all the above heroes. Their ongoing contributions are vital for our profession, and they deserve our gratitude.
A large number of individuals contribute in many ways to the process of discovering, applying, and disseminating new psychiatric knowledge. I am, of course, referring to researchers, clinicians, teachers, and advocates who touch the lives of millions of persons who suffer from mental illness every year. This editorial is dedicated to singing the praises of those who quietly contribute to advancing psychiatry.
Research
Patients. Tens of thousands of psychiatric patients sign an informed consent form and volunteer to participate in clinical trials to test new drugs in double-blind, placebo-controlled studies that could lead to FDA approval. Without these volunteers, it would be almost impossible to develop new medications.
Investigators. Psychiatrists and neuroscientists who dedicate their lives to discovering the causes and treatments of psychiatric brain disorders by conducting basic, translational, or clinical research spend countless hours designing, writing, and submitting grant proposals that have a <10% chance of getting funded. They work long hours to discover new knowledge despite limited resources. They move our field forward with cutting-edge discoveries.
Research assistants. They are an army of skilled technical workers who do the heavy lifting in animal or human research and put in long hours to collect data or conduct tests. Yet they are rarely recognized for their critical contributions to science and clinical practice.
Clinicians. Colleagues who refer their patients to research studies are supporting discovery of new knowledge and without them new psychiatric drug development would slow down considerably.
Reviewers of grants and journal articles. It takes a great deal of expertise and time to identify grants worthy of receiving taxpayer support or to review and recommend the best articles for publication in leading psychiatric journals. Many distinguished researchers—who often are clinicians—donate enormous amounts of uncompensated time to these essential tasks. For a list of colleagues who served as Current Psychiatry peer reviewers in 2010.
Interactive clinicians find time to send in letters to the editor about an unusual clinical observation that may generate new research ideas.
Clinical
Psychiatrists and nurse practitioners, who put in far more than the 40-hour work week to meet the need for psychiatric care caused by the severe shortage of psychiatric clinicians in the United States. They sacrifice their personal needs to help others.
Pro bono clinicians, who donate their services without compensation to those who are indigent or uninsured.
Assertive Community Treatment team members, who reach out to the homeless and seriously sick patients who are the most challenging.
Psychiatric nurses, who are the frontline mental health professionals on psychiatric inpatient units and in psychiatric emergency rooms. They deserve our gratitude and thanks.
Families, whose love and dedication to their mentally ill family members can be critical for seeking psychiatric care or adhering to treatment.
Teaching
Teaching faculty, who provide education and training for physicians, psychiatrists, and nurse practitioners of the future. This includes volunteer (unpaid) faculty who donate their time to supervise psychiatric trainees. They prepare the future generations of clinicians, researchers, and teachers.
Mentors, who may be clinicians, researchers, or both, providing personal guidance, coaching, support and role-models for young trainees or early-career psychiatrists, often shaping their lives and careers. Their generosity and generativity are priceless.
Service
Psychiatrists, who actively participate in their local, state, and national professional societies. I estimate that 10% of the members of any association do the bulk of the work to accomplish the stated mission.
Advocacy groups such as the National Alliance on Mental Illness, Mental Health America, and others who do so much to help persons suffering from psychiatric disorders and whose public service advances the cause of our patients and the mental health field.
Colleagues who step forward and do the gut-wrenching but necessary task of turning in colleagues who commit unethical or illegal acts or who come to work intoxicated. These individuals ensure the integrity of the specialty and prompt their colleagues to seek help.
Celebrities and public figures who openly discuss their personal or family struggles with psychiatric illness. They help diminish stigma, enhance the public’s willingness to seek psychiatric care, and reduce discrimination.
I tip my proverbial hat to all the above heroes. Their ongoing contributions are vital for our profession, and they deserve our gratitude.
Antiepileptics for psychiatric illness: Find the right match
Discuss this article at http://currentpsychiatry.blogspot.com/2010/12/antiepileptics-for-psychiatric-illness.html#comments
Although antiepileptic drugs (AEDs) are used to treat a spectrum of psychiatric disorders, in some instances they are prescribed without clear evidence of clinical benefit or safety. When considering prescribing an AED, ask yourself:
- Does the evidence show the drug is efficacious for my patient’s disorder or symptoms?
- Which adverse effects are associated with this medication?
- What are the advantages of monitoring the patient’s serum drug concentration?
This review provides an evidence-based framework regarding the safe and effective use of AEDs in psychiatric patients.
For which disorders are AEDs effective?
Bipolar disorder. Multiple studies have found that AEDs are efficacious for treating bipolar disorder. Carbamazepine, valproate (divalproex), and lamotrigine have the most evidence supporting their use (Table 1). For an extensive bibliography of studies supporting AEDs for bipolar disorder and other psychiatric illnesses, see this article at CurrentPsychiatry.com. Carbamazepine and valproate are FDA-approved for treating acute manic or mixed episodes associated with bipolar I disorder in adults, and may be beneficial for maintenance treatment. Lamotrigine is FDA- approved for maintenance treatment of bipolar I disorder in adults; however, it lacks efficacy for mania and acute bipolar depression.1 The use of newer AEDs—including gabapentin, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide—for bipolar disorder is not recommended because evidence is limited or inconclusive.
Major depressive disorder (MDD). Most studies of AEDs in MDD feature open-label designs with small samples. AEDs might have a role as an augmentation strategy, perhaps for patients with agitation or irritability or who partially respond to antidepressants.2
Schizophrenia. Although limited data support the practice, AEDs commonly are combined with antipsychotics to treat patients with schizophrenia.3,4 Clinicians who prescribe carbamazepine should recognize the potential for drug-drug interactions with antipsychotics (ie, increased metabolism of antipsychotics caused by cytochrome P450 [CYP450] 3A4 induction).
Anxiety disorders. AEDs have a limited role in treating anxiety disorders. These agents may be used as augmentation for patients who exhibit partial response or treatment resistance to recommended agents for anxiety disorders, such as selective serotonin reuptake inhibitors (SSRIs) or benzodiazepines. For patients who cannot tolerate SSRIs or benzodiazepines, AEDs may be alternatives.5
Other disorders. AEDs could be used to treat other psychiatric conditions and disorders, including alcohol withdrawal and relapse prevention, benzodiazepine withdrawal, drug dependence and abstinence, obesity, and eating disorders.4,6,7 A list of suggested AEDs for some of these disorders appears in Table 2. However, these recommendations are based on findings from small randomized controlled trials, open-label trials, or case reports.
Table 1
Evidence supporting antiepileptics for mood disorders and schizophrenia
| Medication | Bipolar disorder | Major depressive disorder | Schizophrenia | ||
|---|---|---|---|---|---|
| Mania | Depression | Maintenance | |||
| Carbamazepine | |||||
| Lamotrigine | |||||
| Valproate | |||||
| Gabapentin | |||||
| Levetiracetam | |||||
| Oxcarbazepine | |||||
| Tiagabine | |||||
| Topiramate | |||||
| Zonisamide | |||||
| : strong evidence supporting efficacy; | |||||
| : moderate evidence supporting efficacy; | |||||
| : weak evidence supporting efficacy | |||||
| Source: For an extensive bibliography of studies that support these recommendations, see this article at CurrentPsychiatry.com | |||||
Table 2
Off-label use of antiepileptics for various psychiatric disorders
| Condition/disorder | Possible medication(s)* |
|---|---|
| Alcohol withdrawal/relapse prevention | Carbamazepine, topiramate, valproate |
| Benzodiazepine withdrawal | Carbamazepine, valproate |
| Binge eating disorder | Topiramate, zonisamide |
| Bulimia nervosa | Topiramate |
| Drug dependence/abstinence | Carbamazepine, lamotrigine, topiramate, tiagabine |
| Generalized anxiety disorder | Pregabalin, tiagabine |
| Obesity | Lamotrigine, topiramate, zonisamide |
| Panic disorder | Valproate |
| Posttraumatic stress disorder | Lamotrigine |
| Social phobia | Gabapentin, pregabalin |
| * Based on small randomized controlled trials, open-label trials, or case reports. Further investigation in large systematic trials is needed | |
What about adverse effects?
A thorough understanding of each AED’s adverse effect profile is critical to determine which agent is most suitable for your patient. Factors that may affect the risk of adverse effects include:
- rate of dose escalation
- length of early tolerance development
- rate of increase in and magnitude of peak serum concentrations
- dosing frequency
- pharmacodynamic/pharmacokinetic interactions
- pharmacogenomics.
Cardiovascular effects. Although many AED clinical trials reported “edema” as an adverse effect, peripheral edema specifically has been reported with gabapentin, lamotrigine, tiagabine, and valproate.8 Peripheral edema with these agents generally has not been linked to cardiovascular complications in healthy adults. Carbamazepine and pregabalin may cause conduction abnormalities and should be used with caution in patients with underlying electrocardiogram abnormalities.8
Chronic carbamazepine use results in elevated plasma homocysteine and serum lipoprotein concentrations, which are biomarkers of cardiovascular disease.9 If clinically appropriate, switching from carbamazepine to a non-inducing AED (ie, lamotrigine) may ameliorate such effects. Chronic valproate use has been associated with increased plasma homocysteine levels; increases in serum lipoproteins may parallel valproate-induced weight gain.9
CNS effects. Common acute neurologic effects of AEDs include somnolence, dizziness, and ataxia. The incidence of these effects vary by agent; gabapentin and zonisamide appear to be the most sedating.8 However, in general these effects occur at the start of treatment and abate within a few days with continued treatment or dosage reduction. Starting at a low dose and slowly titrating may help prevent neurologic adverse effects.8 Peripheral neurologic effects—specifically paresthesias—are primarily associated with topiramate and zonisamide and may be attributed to carbonic anhydrase inhibition.8
AEDs’ primary cognitive effects include impaired attention/vigilance, psychomotor speed, and secondary involvement of other cognitive functions (eg, memory). Whereas carbamazepine and valproate have similar cognitive effects (ie, negative effects on attention, learning, memory, and psychomotor speed), newer AEDs except topiramate may produce fewer cognitive adverse effects (Table 3).10 Topiramate is associated with the highest rate of cognitive dysfunction, with frequent complaints of decreased concentration and attention, word-finding problems, and/or impaired memory.8,10
The FDA recently announced a warning of a risk of aseptic meningitis with lamotrigine.11 In 40 reported cases, symptoms—headache, fever, nausea, vomiting, nuchal rigidity, rash, photophobia, and myalgias—occurred between 1 and 42 days of treatment and typically resolved after lamotrigine was withdrawn. In 15 patients in whom lamotrigine was re-initiated, meningitis symptoms returned quickly and with greater severity.11
Dermatologic effects. Skin rashes have been reported with all AEDs; the highest risk is associated with carbamazepine and lamotrigine.12 Predictors of cutaneous reactions to lamotrigine include:
- high initial dose and rapid escalation
- concomitant valproate use without lamotrigine dosage adjustment
- young age.12
A history of AED-induced rash also increases risk. For example, patients with a history of rash with carbamazepine are at risk for rash with oxcarbazepine because of cross-reactivity.
Any AED-induced skin rash may progress to a fatal reaction, such as toxic epidermal necrolysis or Stevens-Johnson syndrome. Carbamazepine and lamotrigine are most strongly associated with these severe reactions.12 Patients who exhibit painful rash, fever, enlarged lymph nodes, malaise, and mucosal involvement may be at risk for a more severe disease course.12 If a patient taking an AED develops a rash, immediately stop the drug and perform a thorough risk-benefit analysis before considering re-initiation.
Hematologic effects. Thrombocytopenia has been reported with carbamazepine, lamotrigine, pregabalin, and valproate. The highest risk is for valproate at doses >50 mg/kg/d or serum concentrations >110 μg/mL in women or >135 μg/mL in men.13,14 Decreased platelet count is common with valproate, but coagulation dysfunction may not be present until counts fall below 50,000/mL. Carbamazepine is associated with leukopenia, which usually occurs in early treatment and resolves without dosage adjustments; however, this agent carries a black-box warning for risks of agranulocytosis and aplastic anemia. Similar postmarketing findings have been reported with lamotrigine.8 Baseline hematologic testing and monitoring is recommended.
Hepatic effects. Transient abnormalities in liver function test (LFT) results often have been reported with carbamazepine, valproate, and zonisamide. Valproate has the highest risk of hepatotoxicity, which generally begins within the first 6 months of therapy and does not correlate with serum concentrations.8 Valproate-induced hepatotoxicity may have acute onset, and hepatic dysfunction may progress despite discontinuing the drug. LFTs are recommended at baseline and regular intervals.8
Metabolic effects. AEDs may increase appetite and body weight. Weight gain is common with valproate and pregabalin, but may occur with carbamazepine and gabapentin as well.8 Weight gain does not appear to be dose-related and may be minimized by diet and exercise. Lamotrigine and levetiracetam do not appear to affect weight, whereas weight loss and anorexia have been reported with topiramate and zonisamide.8
Hyponatremia and syndrome of inappropriate antidiuretic hormone secretion have been reported with both carbamaze-pine and oxcarbazepine; the incidence is higher for oxcarbazepine. For both agents, hyponatremia risk is highest in elderly patients.12 Valproate—alone and concomitant with topiramate—may elevate ammonia levels, but monitoring generally is necessary only in symptomatic patients. Topira-mate and zonisamide increase the risks of hyperchloremic, nonanion gap metabolic acidosis and hypohidrosis; serum bicarbonate should be monitored at baseline and as clinically indicated.12,15
Psychiatric effects. Levetiracetam is associated with aggressive behavior, irritability, and increased anxiety and depression, which usually occur soon after drug initiation.8 Similarly, topiramate use is associated with affective and psychotic symptoms. Carbamazepine, gabapentin, lamotrigine, oxcarbazepine, and valproate have been associated with a decreased risk of psychiatric adverse effects compared with the overall incidence among AEDs.8
An FDA analysis suggested patients receiving AEDs have an elevated risk of suicidal ideation or behaviors, regardless of the indication.16 However, the data for increased suicidality are better supported for epilepsy patients than for those with a psychiatric diagnosis. The increased risk was noted as early as 1 week after initiating an AED and extended up to 6 months. The findings generally were consistent across demographic subgroups and AEDs.16 However, a recent study suggests the risk of suicidal acts or violent death is lowest with topiramate compared with gabapentin, lamotrigine, oxcarbazepine, and tiagabine.17 In patients with bipolar disorder, AEDs might not be associated with increased risk of suicidality and may be protective.18 All patients treated with AEDs should be closely monitored for emergence of or worsening depression, suicidality, and other behavior changes.16
Other effects. Valproate-induced pancreatitis is a rare, life-threatening adverse effect that generally occurs in the first 12 months of treatment and with dose increases.8 Amylase levels are not strong predictors of valproate-induced pancreatitis because elevations occur in asymptomatic users and normal levels have been reported in affected patients. Valproate also is linked to polycystic ovaries; evidence of this association is stronger in women with seizures than in those with mood disorders.19
Secondary to developing metabolic acidosis, both topiramate and zonisamide elevate the risk of developing calcium phosphate kidney stones with long-term use (>1 year).12,20 The risk appears higher in patients who are male, elderly, or have a personal or family history of kidney stones. Encourage patients taking topiramate or zonisamide to increase their fluid intake because this significantly reduces kidney stone risk.
Rare but potentially fatal angioedema has been reported with oxcarbazepine and pregabalin.12 History of angioedema or concurrent use of medications associated with angioedema (eg, angiotensin-converting enzyme inhibitors) may confer additional risk.12
Pregnancy and lactation. Carbamazepine and valproate have been associated with neural tube, craniofacial, and cardiac defects in the developing fetus.21 If possible, these agents should be avoided during pregnancy.21 Despite being teratogenic, carbamaze-pine and valproate are thought to be safe for women who are breast-feeding.8 Lamotrigine is associated with mid-facial clefts with first trimester exposure, but is still believed to be a relatively safe option during pregnancy.2 Because lamotrigine clearance increases as pregnancy progresses, the dosage may need to be increased during pregnancy and decreased after delivery to maintain therapeutic levels. Data are inadequate to assess the safety of gabapentin, levetiracetam, oxcarbaze-pine, tiagabine, topiramate, and zonisamide use during pregnancy and lactation.8,21
Table 422 provides additional clinical pearls regarding AED adverse effects.
Table 3
Comparison of antiepileptics’ effects on cognition
| Medication | Comparative effect on cognition | Compared with |
|---|---|---|
| Carbamazepine | ↑ | Topiramate |
| ↔ | Oxcarbazepine, tiagabine, valproate | |
| ↓ | Gabapentin, lamotrigine, levetiracetam, oxcarbazepine | |
| Lamotrigine | ↑ | Carbamazepine, topiramate |
| ↔ | Gabapentin | |
| Valproate | ↑ | Topiramate |
| ↔ | Carbamazepine, oxcarbazepine | |
| Gabapentin | ↑ | Carbamazepine, topiramate |
| ↔ | Lamotrigine | |
| Levetiracetam | ↑ | Carbamazepine, pregabalin, topiramate |
| Oxcarbazepine | ↔ | Carbamazepine, valproate |
| Pregabalin | ↓ | Levetiracetam |
| Tiagabine | ↑ | Topiramate |
| ↔ | Carbamazepine | |
| Topiramate | ↓ | Carbamazepine, gabapentin, lamotrigine, levetiracetam, tiagabine, valproate |
| ↑: positive profile; ↔: similar profile; ↓: negative profile Source: Reference 10 | ||
Table 4
Managing adverse effects of antiepileptics
| Medication | Comment(s) |
|---|---|
| Carbamazepine | Patients screening positive for the variant HLA-B1502 allele are at an elevated risk of developing Stevens-Johnson syndrome or toxic epidermal necrolysis. All patients of Asian descent should be screened22 |
| Gabapentin | Associated with weight gain, edema, and sedation; no reported effects on liver function tests |
| Lamotrigine | If therapy has been interrupted for ≥5 to 7 days (≥5 half-lives), restart according to initial dosing recommendations to significantly reduce the risk of rash |
| Levetiracetam | Appears to have the highest risk of psychiatric adverse effects |
| Oxcarbazepine | Higher risk of hyponatremia than carbamazepine |
| Pregabalin | Cases of angioedema have been reported (rare); may cause PR prolongation |
| Tiagabine | Elevated risk of seizures and status epilepticus when used in non-seizure patients |
| Topiramate | Increased fluid intake reduces the risk of developing kidney stones |
| Valproate | Tremor, thrombocytopenia, alopecia, and elevated liver enzymes have been associated with higher valproate doses/serum concentrations |
| Zonisamide | Avoid use in patients with severe sulfonamide allergy |
Therapeutic monitoring
Therapeutic serum drug concentration monitoring can help evaluate toxicity, medication adherence, and effects of potential drug-drug interactions. Individual variances in drug metabolism and distribution may affect the correlation between serum concentrations and clinical benefit or toxicity. Therapeutic monitoring can help establish target drug concentrations specific to your patient. The best time to obtain a drug concentration is when your patient is stable or free of most symptoms; this concentration may serve as the patient’s “therapeutic” concentration. Although laboratories have set therapeutic concentration ranges for each medication, treatment should focus on addressing your patient’s clinical presentation, rather than achieving the laboratory-suggested range.
Carbamazepine and valproate require therapeutic monitoring to prevent adverse effects from supratherapeutic concentrations (see this article at CurrentPsychiatry.com for a Table listing suggested ranges). The foundation for the therapeutic concentrations of these agents stems from neurology; however, these concentration ranges have been applicable in psychiatry.23
Carbamazepine generally requires more frequent monitoring because it has a narrow therapeutic index and relatively high potential for drug-drug interactions. Compared with lower doses, carbamazepine dosing associated with levels >12 μg/mL is more likely to induce toxicity.23 Carbamazepine autoinduction begins approximately 3 to 5 days after initiation and peaks between 3 to 4 weeks. Therefore, a drop in carbamazepine level from week 1 to week 4 of treatment likely is a pharmacokinetic indicator rather than a sign of nonadherence.
Some acute mania and maintenance bipolar studies have shown a correlation between clinical efficacy and valproate levels.24 A range of 50 to 125 μg/mL is well-accepted in clinical practice.24 For some patients, however, symptoms might not resolve until they are above the therapeutic range, but adverse effects are more likely at higher levels.24
Because concentrations of newer AEDs—including gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide—have not been shown to correlate with therapeutic response, monitoring of serum concentrations is not necessary. However, routine laboratory tests to monitor for adverse effects are recommended.
Table
Therapeutic concentration monitoring for carbamazepine and valproate
| Medication | Suggested therapeutic range (trough level)* | Supratherapeutic presentation |
|---|---|---|
| Carbamazepine | 4 to 12 μg/mL | Ataxia, gastrointestinal upset, drowsiness, dizziness, diplopia, rash |
| Valproate (divalproex) | 50 to 125 μ/mL | Ataxia, nystagmus, tremor, hallucinations |
| *Values may vary among laboratories Source: Reference 23 | ||
Related Resources
- McElroy SL, Keck PE, Post RM, eds. Antiepileptic drugs to treat psychiatric disorders. New York, NY: Informa Health-care USA, Inc.; 2008.
- U.S. Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM100190.
Drug Brand Names
- Carbamazepine • Carbatrol, Equetro, others
- Clozapine • Clozaril
- Gabapentin • Neurontin
- Lamotrigine • Lamictal, Lamictal XR
- Levetiracetam • Keppra, Keppra XR
- Oxcarbazepine • Trileptal
- Pregabalin • Lyrica
- Tiagabine • Gabitril
- Topiramate • Topamax
- Valproate (Divalproex) • Depakote, Depakote ER
- Zonisamide • Zonegram
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bowden CL. Anticonvulsants in bipolar disorders: current research and practice and future directions. Bipolar Disord. 2009;11(suppl 2):20-33.
2. Vigo DV, Baldessarini RJ. Anticonvulsants in the treatment of major depressive disorder: an overview. Harv Rev Psychiatry. 2009;17(4):231-241.
3. Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence? Expert Rev Neurother. 2009;9(1):55-71.
4. Grunze HC. The effectiveness of anticonvulsants in psychiatric disorders. Dialogues Clin Neurosci. 2008;10(1):77-89.
5. Hoffman EJ, Mathew SJ. Anxiety disorders: a comprehensive review of pharmacotherapies. Mt Sinai J Med. 2008;75(3):248-262.
6. Rosenberg JM, Salzman C. Update: new uses for lithium and anticonvulsants. CNS Spectr. 2007;12(11):831-841.
7. McElroy SL, Guerdjikova AI, Martens B, et al. Role of antiepileptic drugs in the management of eating disorders. CNS Drugs. 2009;23(2):139-156.
8. Wilby J, Kainth A, Hawkins N, et al. Clinical effectiveness, tolerability and cost-effectiveness of newer drugs for epilepsy in adults: a systematic review and economic evaluation. Health Technol Assess. 2005;9(15):1-157.
9. Cheng LS, Prasad AN, Rieder MJ. Relationship between antiepileptic drugs and biological markers affecting long-term cardiovascular function in children and adolescents. Can J Clin Pharmacol. 2010;17(1):e5-46.
10. Park SP, Kwon SH. Cognitive effects of antiepileptic drugs. J Clin Neurol. 2008;4(3):99-106.
11. U.S. Food and Drug Administration. FDA drug safety communication: aseptic meningitis associated with use of lamictal (lamotrigine). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm221847.htm. Accessed October 29, 2010.
12. Wade JF, Dang CV, Nelson L, et al. Emergent complications of the newer anticonvulsants. J Emerg Med. 2010;38(2):231-237.
13. Beydoun A, Sackellares JC, Shu V, et al. Safety and efficacy of divalproex sodium monotherapy in partial epilepsy: a double-blind, concentration-response design clinical trial. Neurology. 1997;48(1):182-188.
14. Depakote [package inset]. North Chicago, IL: Abbott Laboratories; 2009.
15. Cerminara C, Seri S, Bombardieri R, et al. Hypohidrosis during topiramate treatment: a rare and reversible side effect. Pediatr Neurol. 2006;34(5):392-394.
16. U.S. Food and Drug Administration. Information for healthcare professionals: suicidal behavior and ideation and antiepileptic drugs. 2008. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100192.htm. Accessed May 10, 2010.
17. Patorno E, Bohn RL, Wahl PM, et al. Anticonvulsant medications and the risk of suicide, attempted suicide, or violent death. JAMA. 2010;303(14):1401-1409.
18. Gibbons RD, Hur K, Brown CH, et al. Relationship between antiepileptic drugs and suicide attempts in patients with bipolar disorder. Arch Gen Psychiatry. 2009;66(12):1354-1360.
19. Bilo L, Meo R. Polycystic ovary syndrome in women using valproate: a review. Gynecol Endocrinol. 2008;24(10):562-570.
20. Welch BJ, Graybeal D, Moe OW, et al. Biochemical and stone-risk profiles with topiramate treatment. Am J Kidney Dis. 2006;48(4):555-563.
21. Harden CL, Meador KJ, Pennell PB, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
22. Chung WH, Hung SI, Chen YT. Genetic predisposition of life-threatening antiepileptic-induced skin reactions. Expert Opin Drug Saf. 2010;9(1):15-21.
23. Warner A, Privitera M, Bates D. Standards of laboratory practice: antiepileptic drug monitoring. Clin Chem. 1998;44(5):1085-1095.
24. Kaneria KM, Patel NC, Keck PE, Jr. Bipolar disorder: new strategy for checking serum valproate. Current Psychiatry. 2005;4(12):31-44.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/12/antiepileptics-for-psychiatric-illness.html#comments
Although antiepileptic drugs (AEDs) are used to treat a spectrum of psychiatric disorders, in some instances they are prescribed without clear evidence of clinical benefit or safety. When considering prescribing an AED, ask yourself:
- Does the evidence show the drug is efficacious for my patient’s disorder or symptoms?
- Which adverse effects are associated with this medication?
- What are the advantages of monitoring the patient’s serum drug concentration?
This review provides an evidence-based framework regarding the safe and effective use of AEDs in psychiatric patients.
For which disorders are AEDs effective?
Bipolar disorder. Multiple studies have found that AEDs are efficacious for treating bipolar disorder. Carbamazepine, valproate (divalproex), and lamotrigine have the most evidence supporting their use (Table 1). For an extensive bibliography of studies supporting AEDs for bipolar disorder and other psychiatric illnesses, see this article at CurrentPsychiatry.com. Carbamazepine and valproate are FDA-approved for treating acute manic or mixed episodes associated with bipolar I disorder in adults, and may be beneficial for maintenance treatment. Lamotrigine is FDA- approved for maintenance treatment of bipolar I disorder in adults; however, it lacks efficacy for mania and acute bipolar depression.1 The use of newer AEDs—including gabapentin, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide—for bipolar disorder is not recommended because evidence is limited or inconclusive.
Major depressive disorder (MDD). Most studies of AEDs in MDD feature open-label designs with small samples. AEDs might have a role as an augmentation strategy, perhaps for patients with agitation or irritability or who partially respond to antidepressants.2
Schizophrenia. Although limited data support the practice, AEDs commonly are combined with antipsychotics to treat patients with schizophrenia.3,4 Clinicians who prescribe carbamazepine should recognize the potential for drug-drug interactions with antipsychotics (ie, increased metabolism of antipsychotics caused by cytochrome P450 [CYP450] 3A4 induction).
Anxiety disorders. AEDs have a limited role in treating anxiety disorders. These agents may be used as augmentation for patients who exhibit partial response or treatment resistance to recommended agents for anxiety disorders, such as selective serotonin reuptake inhibitors (SSRIs) or benzodiazepines. For patients who cannot tolerate SSRIs or benzodiazepines, AEDs may be alternatives.5
Other disorders. AEDs could be used to treat other psychiatric conditions and disorders, including alcohol withdrawal and relapse prevention, benzodiazepine withdrawal, drug dependence and abstinence, obesity, and eating disorders.4,6,7 A list of suggested AEDs for some of these disorders appears in Table 2. However, these recommendations are based on findings from small randomized controlled trials, open-label trials, or case reports.
Table 1
Evidence supporting antiepileptics for mood disorders and schizophrenia
| Medication | Bipolar disorder | Major depressive disorder | Schizophrenia | ||
|---|---|---|---|---|---|
| Mania | Depression | Maintenance | |||
| Carbamazepine | (aggression, impulsivity) | ||||
| Lamotrigine | |||||
| Valproate | (aggression, impulsivity) | ||||
| Gabapentin | |||||
| Levetiracetam | |||||
| Oxcarbazepine | |||||
| Tiagabine | |||||
| Topiramate | |||||
| Zonisamide | |||||
| : strong evidence supporting efficacy; | |||||
| : moderate evidence supporting efficacy; | |||||
| : weak evidence supporting efficacy | |||||
| Source: For an extensive bibliography of studies that support these recommendations, see this article at CurrentPsychiatry.com | |||||
Table 2
Off-label use of antiepileptics for various psychiatric disorders
| Condition/disorder | Possible medication(s)* |
|---|---|
| Alcohol withdrawal/relapse prevention | Carbamazepine, topiramate, valproate |
| Benzodiazepine withdrawal | Carbamazepine, valproate |
| Binge eating disorder | Topiramate, zonisamide |
| Bulimia nervosa | Topiramate |
| Drug dependence/abstinence | Carbamazepine, lamotrigine, topiramate, tiagabine |
| Generalized anxiety disorder | Pregabalin, tiagabine |
| Obesity | Lamotrigine, topiramate, zonisamide |
| Panic disorder | Valproate |
| Posttraumatic stress disorder | Lamotrigine |
| Social phobia | Gabapentin, pregabalin |
| * Based on small randomized controlled trials, open-label trials, or case reports. Further investigation in large systematic trials is needed | |
What about adverse effects?
A thorough understanding of each AED’s adverse effect profile is critical to determine which agent is most suitable for your patient. Factors that may affect the risk of adverse effects include:
- rate of dose escalation
- length of early tolerance development
- rate of increase in and magnitude of peak serum concentrations
- dosing frequency
- pharmacodynamic/pharmacokinetic interactions
- pharmacogenomics.
Cardiovascular effects. Although many AED clinical trials reported “edema” as an adverse effect, peripheral edema specifically has been reported with gabapentin, lamotrigine, tiagabine, and valproate.8 Peripheral edema with these agents generally has not been linked to cardiovascular complications in healthy adults. Carbamazepine and pregabalin may cause conduction abnormalities and should be used with caution in patients with underlying electrocardiogram abnormalities.8
Chronic carbamazepine use results in elevated plasma homocysteine and serum lipoprotein concentrations, which are biomarkers of cardiovascular disease.9 If clinically appropriate, switching from carbamazepine to a non-inducing AED (ie, lamotrigine) may ameliorate such effects. Chronic valproate use has been associated with increased plasma homocysteine levels; increases in serum lipoproteins may parallel valproate-induced weight gain.9
CNS effects. Common acute neurologic effects of AEDs include somnolence, dizziness, and ataxia. The incidence of these effects vary by agent; gabapentin and zonisamide appear to be the most sedating.8 However, in general these effects occur at the start of treatment and abate within a few days with continued treatment or dosage reduction. Starting at a low dose and slowly titrating may help prevent neurologic adverse effects.8 Peripheral neurologic effects—specifically paresthesias—are primarily associated with topiramate and zonisamide and may be attributed to carbonic anhydrase inhibition.8
AEDs’ primary cognitive effects include impaired attention/vigilance, psychomotor speed, and secondary involvement of other cognitive functions (eg, memory). Whereas carbamazepine and valproate have similar cognitive effects (ie, negative effects on attention, learning, memory, and psychomotor speed), newer AEDs except topiramate may produce fewer cognitive adverse effects (Table 3).10 Topiramate is associated with the highest rate of cognitive dysfunction, with frequent complaints of decreased concentration and attention, word-finding problems, and/or impaired memory.8,10
The FDA recently announced a warning of a risk of aseptic meningitis with lamotrigine.11 In 40 reported cases, symptoms—headache, fever, nausea, vomiting, nuchal rigidity, rash, photophobia, and myalgias—occurred between 1 and 42 days of treatment and typically resolved after lamotrigine was withdrawn. In 15 patients in whom lamotrigine was re-initiated, meningitis symptoms returned quickly and with greater severity.11
Dermatologic effects. Skin rashes have been reported with all AEDs; the highest risk is associated with carbamazepine and lamotrigine.12 Predictors of cutaneous reactions to lamotrigine include:
- high initial dose and rapid escalation
- concomitant valproate use without lamotrigine dosage adjustment
- young age.12
A history of AED-induced rash also increases risk. For example, patients with a history of rash with carbamazepine are at risk for rash with oxcarbazepine because of cross-reactivity.
Any AED-induced skin rash may progress to a fatal reaction, such as toxic epidermal necrolysis or Stevens-Johnson syndrome. Carbamazepine and lamotrigine are most strongly associated with these severe reactions.12 Patients who exhibit painful rash, fever, enlarged lymph nodes, malaise, and mucosal involvement may be at risk for a more severe disease course.12 If a patient taking an AED develops a rash, immediately stop the drug and perform a thorough risk-benefit analysis before considering re-initiation.
Hematologic effects. Thrombocytopenia has been reported with carbamazepine, lamotrigine, pregabalin, and valproate. The highest risk is for valproate at doses >50 mg/kg/d or serum concentrations >110 μg/mL in women or >135 μg/mL in men.13,14 Decreased platelet count is common with valproate, but coagulation dysfunction may not be present until counts fall below 50,000/mL. Carbamazepine is associated with leukopenia, which usually occurs in early treatment and resolves without dosage adjustments; however, this agent carries a black-box warning for risks of agranulocytosis and aplastic anemia. Similar postmarketing findings have been reported with lamotrigine.8 Baseline hematologic testing and monitoring is recommended.
Hepatic effects. Transient abnormalities in liver function test (LFT) results often have been reported with carbamazepine, valproate, and zonisamide. Valproate has the highest risk of hepatotoxicity, which generally begins within the first 6 months of therapy and does not correlate with serum concentrations.8 Valproate-induced hepatotoxicity may have acute onset, and hepatic dysfunction may progress despite discontinuing the drug. LFTs are recommended at baseline and regular intervals.8
Metabolic effects. AEDs may increase appetite and body weight. Weight gain is common with valproate and pregabalin, but may occur with carbamazepine and gabapentin as well.8 Weight gain does not appear to be dose-related and may be minimized by diet and exercise. Lamotrigine and levetiracetam do not appear to affect weight, whereas weight loss and anorexia have been reported with topiramate and zonisamide.8
Hyponatremia and syndrome of inappropriate antidiuretic hormone secretion have been reported with both carbamaze-pine and oxcarbazepine; the incidence is higher for oxcarbazepine. For both agents, hyponatremia risk is highest in elderly patients.12 Valproate—alone and concomitant with topiramate—may elevate ammonia levels, but monitoring generally is necessary only in symptomatic patients. Topira-mate and zonisamide increase the risks of hyperchloremic, nonanion gap metabolic acidosis and hypohidrosis; serum bicarbonate should be monitored at baseline and as clinically indicated.12,15
Psychiatric effects. Levetiracetam is associated with aggressive behavior, irritability, and increased anxiety and depression, which usually occur soon after drug initiation.8 Similarly, topiramate use is associated with affective and psychotic symptoms. Carbamazepine, gabapentin, lamotrigine, oxcarbazepine, and valproate have been associated with a decreased risk of psychiatric adverse effects compared with the overall incidence among AEDs.8
An FDA analysis suggested patients receiving AEDs have an elevated risk of suicidal ideation or behaviors, regardless of the indication.16 However, the data for increased suicidality are better supported for epilepsy patients than for those with a psychiatric diagnosis. The increased risk was noted as early as 1 week after initiating an AED and extended up to 6 months. The findings generally were consistent across demographic subgroups and AEDs.16 However, a recent study suggests the risk of suicidal acts or violent death is lowest with topiramate compared with gabapentin, lamotrigine, oxcarbazepine, and tiagabine.17 In patients with bipolar disorder, AEDs might not be associated with increased risk of suicidality and may be protective.18 All patients treated with AEDs should be closely monitored for emergence of or worsening depression, suicidality, and other behavior changes.16
Other effects. Valproate-induced pancreatitis is a rare, life-threatening adverse effect that generally occurs in the first 12 months of treatment and with dose increases.8 Amylase levels are not strong predictors of valproate-induced pancreatitis because elevations occur in asymptomatic users and normal levels have been reported in affected patients. Valproate also is linked to polycystic ovaries; evidence of this association is stronger in women with seizures than in those with mood disorders.19
Secondary to developing metabolic acidosis, both topiramate and zonisamide elevate the risk of developing calcium phosphate kidney stones with long-term use (>1 year).12,20 The risk appears higher in patients who are male, elderly, or have a personal or family history of kidney stones. Encourage patients taking topiramate or zonisamide to increase their fluid intake because this significantly reduces kidney stone risk.
Rare but potentially fatal angioedema has been reported with oxcarbazepine and pregabalin.12 History of angioedema or concurrent use of medications associated with angioedema (eg, angiotensin-converting enzyme inhibitors) may confer additional risk.12
Pregnancy and lactation. Carbamazepine and valproate have been associated with neural tube, craniofacial, and cardiac defects in the developing fetus.21 If possible, these agents should be avoided during pregnancy.21 Despite being teratogenic, carbamaze-pine and valproate are thought to be safe for women who are breast-feeding.8 Lamotrigine is associated with mid-facial clefts with first trimester exposure, but is still believed to be a relatively safe option during pregnancy.2 Because lamotrigine clearance increases as pregnancy progresses, the dosage may need to be increased during pregnancy and decreased after delivery to maintain therapeutic levels. Data are inadequate to assess the safety of gabapentin, levetiracetam, oxcarbaze-pine, tiagabine, topiramate, and zonisamide use during pregnancy and lactation.8,21
Table 422 provides additional clinical pearls regarding AED adverse effects.
Table 3
Comparison of antiepileptics’ effects on cognition
| Medication | Comparative effect on cognition | Compared with |
|---|---|---|
| Carbamazepine | ↑ | Topiramate |
| ↔ | Oxcarbazepine, tiagabine, valproate | |
| ↓ | Gabapentin, lamotrigine, levetiracetam, oxcarbazepine | |
| Lamotrigine | ↑ | Carbamazepine, topiramate |
| ↔ | Gabapentin | |
| Valproate | ↑ | Topiramate |
| ↔ | Carbamazepine, oxcarbazepine | |
| Gabapentin | ↑ | Carbamazepine, topiramate |
| ↔ | Lamotrigine | |
| Levetiracetam | ↑ | Carbamazepine, pregabalin, topiramate |
| Oxcarbazepine | ↔ | Carbamazepine, valproate |
| Pregabalin | ↓ | Levetiracetam |
| Tiagabine | ↑ | Topiramate |
| ↔ | Carbamazepine | |
| Topiramate | ↓ | Carbamazepine, gabapentin, lamotrigine, levetiracetam, tiagabine, valproate |
| ↑: positive profile; ↔: similar profile; ↓: negative profile Source: Reference 10 | ||
Table 4
Managing adverse effects of antiepileptics
| Medication | Comment(s) |
|---|---|
| Carbamazepine | Patients screening positive for the variant HLA-B1502 allele are at an elevated risk of developing Stevens-Johnson syndrome or toxic epidermal necrolysis. All patients of Asian descent should be screened22 |
| Gabapentin | Associated with weight gain, edema, and sedation; no reported effects on liver function tests |
| Lamotrigine | If therapy has been interrupted for ≥5 to 7 days (≥5 half-lives), restart according to initial dosing recommendations to significantly reduce the risk of rash |
| Levetiracetam | Appears to have the highest risk of psychiatric adverse effects |
| Oxcarbazepine | Higher risk of hyponatremia than carbamazepine |
| Pregabalin | Cases of angioedema have been reported (rare); may cause PR prolongation |
| Tiagabine | Elevated risk of seizures and status epilepticus when used in non-seizure patients |
| Topiramate | Increased fluid intake reduces the risk of developing kidney stones |
| Valproate | Tremor, thrombocytopenia, alopecia, and elevated liver enzymes have been associated with higher valproate doses/serum concentrations |
| Zonisamide | Avoid use in patients with severe sulfonamide allergy |
Therapeutic monitoring
Therapeutic serum drug concentration monitoring can help evaluate toxicity, medication adherence, and effects of potential drug-drug interactions. Individual variances in drug metabolism and distribution may affect the correlation between serum concentrations and clinical benefit or toxicity. Therapeutic monitoring can help establish target drug concentrations specific to your patient. The best time to obtain a drug concentration is when your patient is stable or free of most symptoms; this concentration may serve as the patient’s “therapeutic” concentration. Although laboratories have set therapeutic concentration ranges for each medication, treatment should focus on addressing your patient’s clinical presentation, rather than achieving the laboratory-suggested range.
Carbamazepine and valproate require therapeutic monitoring to prevent adverse effects from supratherapeutic concentrations (see this article at CurrentPsychiatry.com for a Table listing suggested ranges). The foundation for the therapeutic concentrations of these agents stems from neurology; however, these concentration ranges have been applicable in psychiatry.23
Carbamazepine generally requires more frequent monitoring because it has a narrow therapeutic index and relatively high potential for drug-drug interactions. Compared with lower doses, carbamazepine dosing associated with levels >12 μg/mL is more likely to induce toxicity.23 Carbamazepine autoinduction begins approximately 3 to 5 days after initiation and peaks between 3 to 4 weeks. Therefore, a drop in carbamazepine level from week 1 to week 4 of treatment likely is a pharmacokinetic indicator rather than a sign of nonadherence.
Some acute mania and maintenance bipolar studies have shown a correlation between clinical efficacy and valproate levels.24 A range of 50 to 125 μg/mL is well-accepted in clinical practice.24 For some patients, however, symptoms might not resolve until they are above the therapeutic range, but adverse effects are more likely at higher levels.24
Because concentrations of newer AEDs—including gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide—have not been shown to correlate with therapeutic response, monitoring of serum concentrations is not necessary. However, routine laboratory tests to monitor for adverse effects are recommended.
Table
Therapeutic concentration monitoring for carbamazepine and valproate
| Medication | Suggested therapeutic range (trough level)* | Supratherapeutic presentation |
|---|---|---|
| Carbamazepine | 4 to 12 μg/mL | Ataxia, gastrointestinal upset, drowsiness, dizziness, diplopia, rash |
| Valproate (divalproex) | 50 to 125 μ/mL | Ataxia, nystagmus, tremor, hallucinations |
| *Values may vary among laboratories Source: Reference 23 | ||
Related Resources
- McElroy SL, Keck PE, Post RM, eds. Antiepileptic drugs to treat psychiatric disorders. New York, NY: Informa Health-care USA, Inc.; 2008.
- U.S. Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM100190.
Drug Brand Names
- Carbamazepine • Carbatrol, Equetro, others
- Clozapine • Clozaril
- Gabapentin • Neurontin
- Lamotrigine • Lamictal, Lamictal XR
- Levetiracetam • Keppra, Keppra XR
- Oxcarbazepine • Trileptal
- Pregabalin • Lyrica
- Tiagabine • Gabitril
- Topiramate • Topamax
- Valproate (Divalproex) • Depakote, Depakote ER
- Zonisamide • Zonegram
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/12/antiepileptics-for-psychiatric-illness.html#comments
Although antiepileptic drugs (AEDs) are used to treat a spectrum of psychiatric disorders, in some instances they are prescribed without clear evidence of clinical benefit or safety. When considering prescribing an AED, ask yourself:
- Does the evidence show the drug is efficacious for my patient’s disorder or symptoms?
- Which adverse effects are associated with this medication?
- What are the advantages of monitoring the patient’s serum drug concentration?
This review provides an evidence-based framework regarding the safe and effective use of AEDs in psychiatric patients.
For which disorders are AEDs effective?
Bipolar disorder. Multiple studies have found that AEDs are efficacious for treating bipolar disorder. Carbamazepine, valproate (divalproex), and lamotrigine have the most evidence supporting their use (Table 1). For an extensive bibliography of studies supporting AEDs for bipolar disorder and other psychiatric illnesses, see this article at CurrentPsychiatry.com. Carbamazepine and valproate are FDA-approved for treating acute manic or mixed episodes associated with bipolar I disorder in adults, and may be beneficial for maintenance treatment. Lamotrigine is FDA- approved for maintenance treatment of bipolar I disorder in adults; however, it lacks efficacy for mania and acute bipolar depression.1 The use of newer AEDs—including gabapentin, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide—for bipolar disorder is not recommended because evidence is limited or inconclusive.
Major depressive disorder (MDD). Most studies of AEDs in MDD feature open-label designs with small samples. AEDs might have a role as an augmentation strategy, perhaps for patients with agitation or irritability or who partially respond to antidepressants.2
Schizophrenia. Although limited data support the practice, AEDs commonly are combined with antipsychotics to treat patients with schizophrenia.3,4 Clinicians who prescribe carbamazepine should recognize the potential for drug-drug interactions with antipsychotics (ie, increased metabolism of antipsychotics caused by cytochrome P450 [CYP450] 3A4 induction).
Anxiety disorders. AEDs have a limited role in treating anxiety disorders. These agents may be used as augmentation for patients who exhibit partial response or treatment resistance to recommended agents for anxiety disorders, such as selective serotonin reuptake inhibitors (SSRIs) or benzodiazepines. For patients who cannot tolerate SSRIs or benzodiazepines, AEDs may be alternatives.5
Other disorders. AEDs could be used to treat other psychiatric conditions and disorders, including alcohol withdrawal and relapse prevention, benzodiazepine withdrawal, drug dependence and abstinence, obesity, and eating disorders.4,6,7 A list of suggested AEDs for some of these disorders appears in Table 2. However, these recommendations are based on findings from small randomized controlled trials, open-label trials, or case reports.
Table 1
Evidence supporting antiepileptics for mood disorders and schizophrenia
| Medication | Bipolar disorder | Major depressive disorder | Schizophrenia | ||
|---|---|---|---|---|---|
| Mania | Depression | Maintenance | |||
| Carbamazepine | (aggression, impulsivity) | ||||
| Lamotrigine | |||||
| Valproate | (aggression, impulsivity) | ||||
| Gabapentin | |||||
| Levetiracetam | |||||
| Oxcarbazepine | |||||
| Tiagabine | |||||
| Topiramate | |||||
| Zonisamide | |||||
| : strong evidence supporting efficacy; | |||||
| : moderate evidence supporting efficacy; | |||||
| : weak evidence supporting efficacy | |||||
| Source: For an extensive bibliography of studies that support these recommendations, see this article at CurrentPsychiatry.com | |||||
Table 2
Off-label use of antiepileptics for various psychiatric disorders
| Condition/disorder | Possible medication(s)* |
|---|---|
| Alcohol withdrawal/relapse prevention | Carbamazepine, topiramate, valproate |
| Benzodiazepine withdrawal | Carbamazepine, valproate |
| Binge eating disorder | Topiramate, zonisamide |
| Bulimia nervosa | Topiramate |
| Drug dependence/abstinence | Carbamazepine, lamotrigine, topiramate, tiagabine |
| Generalized anxiety disorder | Pregabalin, tiagabine |
| Obesity | Lamotrigine, topiramate, zonisamide |
| Panic disorder | Valproate |
| Posttraumatic stress disorder | Lamotrigine |
| Social phobia | Gabapentin, pregabalin |
| * Based on small randomized controlled trials, open-label trials, or case reports. Further investigation in large systematic trials is needed | |
What about adverse effects?
A thorough understanding of each AED’s adverse effect profile is critical to determine which agent is most suitable for your patient. Factors that may affect the risk of adverse effects include:
- rate of dose escalation
- length of early tolerance development
- rate of increase in and magnitude of peak serum concentrations
- dosing frequency
- pharmacodynamic/pharmacokinetic interactions
- pharmacogenomics.
Cardiovascular effects. Although many AED clinical trials reported “edema” as an adverse effect, peripheral edema specifically has been reported with gabapentin, lamotrigine, tiagabine, and valproate.8 Peripheral edema with these agents generally has not been linked to cardiovascular complications in healthy adults. Carbamazepine and pregabalin may cause conduction abnormalities and should be used with caution in patients with underlying electrocardiogram abnormalities.8
Chronic carbamazepine use results in elevated plasma homocysteine and serum lipoprotein concentrations, which are biomarkers of cardiovascular disease.9 If clinically appropriate, switching from carbamazepine to a non-inducing AED (ie, lamotrigine) may ameliorate such effects. Chronic valproate use has been associated with increased plasma homocysteine levels; increases in serum lipoproteins may parallel valproate-induced weight gain.9
CNS effects. Common acute neurologic effects of AEDs include somnolence, dizziness, and ataxia. The incidence of these effects vary by agent; gabapentin and zonisamide appear to be the most sedating.8 However, in general these effects occur at the start of treatment and abate within a few days with continued treatment or dosage reduction. Starting at a low dose and slowly titrating may help prevent neurologic adverse effects.8 Peripheral neurologic effects—specifically paresthesias—are primarily associated with topiramate and zonisamide and may be attributed to carbonic anhydrase inhibition.8
AEDs’ primary cognitive effects include impaired attention/vigilance, psychomotor speed, and secondary involvement of other cognitive functions (eg, memory). Whereas carbamazepine and valproate have similar cognitive effects (ie, negative effects on attention, learning, memory, and psychomotor speed), newer AEDs except topiramate may produce fewer cognitive adverse effects (Table 3).10 Topiramate is associated with the highest rate of cognitive dysfunction, with frequent complaints of decreased concentration and attention, word-finding problems, and/or impaired memory.8,10
The FDA recently announced a warning of a risk of aseptic meningitis with lamotrigine.11 In 40 reported cases, symptoms—headache, fever, nausea, vomiting, nuchal rigidity, rash, photophobia, and myalgias—occurred between 1 and 42 days of treatment and typically resolved after lamotrigine was withdrawn. In 15 patients in whom lamotrigine was re-initiated, meningitis symptoms returned quickly and with greater severity.11
Dermatologic effects. Skin rashes have been reported with all AEDs; the highest risk is associated with carbamazepine and lamotrigine.12 Predictors of cutaneous reactions to lamotrigine include:
- high initial dose and rapid escalation
- concomitant valproate use without lamotrigine dosage adjustment
- young age.12
A history of AED-induced rash also increases risk. For example, patients with a history of rash with carbamazepine are at risk for rash with oxcarbazepine because of cross-reactivity.
Any AED-induced skin rash may progress to a fatal reaction, such as toxic epidermal necrolysis or Stevens-Johnson syndrome. Carbamazepine and lamotrigine are most strongly associated with these severe reactions.12 Patients who exhibit painful rash, fever, enlarged lymph nodes, malaise, and mucosal involvement may be at risk for a more severe disease course.12 If a patient taking an AED develops a rash, immediately stop the drug and perform a thorough risk-benefit analysis before considering re-initiation.
Hematologic effects. Thrombocytopenia has been reported with carbamazepine, lamotrigine, pregabalin, and valproate. The highest risk is for valproate at doses >50 mg/kg/d or serum concentrations >110 μg/mL in women or >135 μg/mL in men.13,14 Decreased platelet count is common with valproate, but coagulation dysfunction may not be present until counts fall below 50,000/mL. Carbamazepine is associated with leukopenia, which usually occurs in early treatment and resolves without dosage adjustments; however, this agent carries a black-box warning for risks of agranulocytosis and aplastic anemia. Similar postmarketing findings have been reported with lamotrigine.8 Baseline hematologic testing and monitoring is recommended.
Hepatic effects. Transient abnormalities in liver function test (LFT) results often have been reported with carbamazepine, valproate, and zonisamide. Valproate has the highest risk of hepatotoxicity, which generally begins within the first 6 months of therapy and does not correlate with serum concentrations.8 Valproate-induced hepatotoxicity may have acute onset, and hepatic dysfunction may progress despite discontinuing the drug. LFTs are recommended at baseline and regular intervals.8
Metabolic effects. AEDs may increase appetite and body weight. Weight gain is common with valproate and pregabalin, but may occur with carbamazepine and gabapentin as well.8 Weight gain does not appear to be dose-related and may be minimized by diet and exercise. Lamotrigine and levetiracetam do not appear to affect weight, whereas weight loss and anorexia have been reported with topiramate and zonisamide.8
Hyponatremia and syndrome of inappropriate antidiuretic hormone secretion have been reported with both carbamaze-pine and oxcarbazepine; the incidence is higher for oxcarbazepine. For both agents, hyponatremia risk is highest in elderly patients.12 Valproate—alone and concomitant with topiramate—may elevate ammonia levels, but monitoring generally is necessary only in symptomatic patients. Topira-mate and zonisamide increase the risks of hyperchloremic, nonanion gap metabolic acidosis and hypohidrosis; serum bicarbonate should be monitored at baseline and as clinically indicated.12,15
Psychiatric effects. Levetiracetam is associated with aggressive behavior, irritability, and increased anxiety and depression, which usually occur soon after drug initiation.8 Similarly, topiramate use is associated with affective and psychotic symptoms. Carbamazepine, gabapentin, lamotrigine, oxcarbazepine, and valproate have been associated with a decreased risk of psychiatric adverse effects compared with the overall incidence among AEDs.8
An FDA analysis suggested patients receiving AEDs have an elevated risk of suicidal ideation or behaviors, regardless of the indication.16 However, the data for increased suicidality are better supported for epilepsy patients than for those with a psychiatric diagnosis. The increased risk was noted as early as 1 week after initiating an AED and extended up to 6 months. The findings generally were consistent across demographic subgroups and AEDs.16 However, a recent study suggests the risk of suicidal acts or violent death is lowest with topiramate compared with gabapentin, lamotrigine, oxcarbazepine, and tiagabine.17 In patients with bipolar disorder, AEDs might not be associated with increased risk of suicidality and may be protective.18 All patients treated with AEDs should be closely monitored for emergence of or worsening depression, suicidality, and other behavior changes.16
Other effects. Valproate-induced pancreatitis is a rare, life-threatening adverse effect that generally occurs in the first 12 months of treatment and with dose increases.8 Amylase levels are not strong predictors of valproate-induced pancreatitis because elevations occur in asymptomatic users and normal levels have been reported in affected patients. Valproate also is linked to polycystic ovaries; evidence of this association is stronger in women with seizures than in those with mood disorders.19
Secondary to developing metabolic acidosis, both topiramate and zonisamide elevate the risk of developing calcium phosphate kidney stones with long-term use (>1 year).12,20 The risk appears higher in patients who are male, elderly, or have a personal or family history of kidney stones. Encourage patients taking topiramate or zonisamide to increase their fluid intake because this significantly reduces kidney stone risk.
Rare but potentially fatal angioedema has been reported with oxcarbazepine and pregabalin.12 History of angioedema or concurrent use of medications associated with angioedema (eg, angiotensin-converting enzyme inhibitors) may confer additional risk.12
Pregnancy and lactation. Carbamazepine and valproate have been associated with neural tube, craniofacial, and cardiac defects in the developing fetus.21 If possible, these agents should be avoided during pregnancy.21 Despite being teratogenic, carbamaze-pine and valproate are thought to be safe for women who are breast-feeding.8 Lamotrigine is associated with mid-facial clefts with first trimester exposure, but is still believed to be a relatively safe option during pregnancy.2 Because lamotrigine clearance increases as pregnancy progresses, the dosage may need to be increased during pregnancy and decreased after delivery to maintain therapeutic levels. Data are inadequate to assess the safety of gabapentin, levetiracetam, oxcarbaze-pine, tiagabine, topiramate, and zonisamide use during pregnancy and lactation.8,21
Table 422 provides additional clinical pearls regarding AED adverse effects.
Table 3
Comparison of antiepileptics’ effects on cognition
| Medication | Comparative effect on cognition | Compared with |
|---|---|---|
| Carbamazepine | ↑ | Topiramate |
| ↔ | Oxcarbazepine, tiagabine, valproate | |
| ↓ | Gabapentin, lamotrigine, levetiracetam, oxcarbazepine | |
| Lamotrigine | ↑ | Carbamazepine, topiramate |
| ↔ | Gabapentin | |
| Valproate | ↑ | Topiramate |
| ↔ | Carbamazepine, oxcarbazepine | |
| Gabapentin | ↑ | Carbamazepine, topiramate |
| ↔ | Lamotrigine | |
| Levetiracetam | ↑ | Carbamazepine, pregabalin, topiramate |
| Oxcarbazepine | ↔ | Carbamazepine, valproate |
| Pregabalin | ↓ | Levetiracetam |
| Tiagabine | ↑ | Topiramate |
| ↔ | Carbamazepine | |
| Topiramate | ↓ | Carbamazepine, gabapentin, lamotrigine, levetiracetam, tiagabine, valproate |
| ↑: positive profile; ↔: similar profile; ↓: negative profile Source: Reference 10 | ||
Table 4
Managing adverse effects of antiepileptics
| Medication | Comment(s) |
|---|---|
| Carbamazepine | Patients screening positive for the variant HLA-B1502 allele are at an elevated risk of developing Stevens-Johnson syndrome or toxic epidermal necrolysis. All patients of Asian descent should be screened22 |
| Gabapentin | Associated with weight gain, edema, and sedation; no reported effects on liver function tests |
| Lamotrigine | If therapy has been interrupted for ≥5 to 7 days (≥5 half-lives), restart according to initial dosing recommendations to significantly reduce the risk of rash |
| Levetiracetam | Appears to have the highest risk of psychiatric adverse effects |
| Oxcarbazepine | Higher risk of hyponatremia than carbamazepine |
| Pregabalin | Cases of angioedema have been reported (rare); may cause PR prolongation |
| Tiagabine | Elevated risk of seizures and status epilepticus when used in non-seizure patients |
| Topiramate | Increased fluid intake reduces the risk of developing kidney stones |
| Valproate | Tremor, thrombocytopenia, alopecia, and elevated liver enzymes have been associated with higher valproate doses/serum concentrations |
| Zonisamide | Avoid use in patients with severe sulfonamide allergy |
Therapeutic monitoring
Therapeutic serum drug concentration monitoring can help evaluate toxicity, medication adherence, and effects of potential drug-drug interactions. Individual variances in drug metabolism and distribution may affect the correlation between serum concentrations and clinical benefit or toxicity. Therapeutic monitoring can help establish target drug concentrations specific to your patient. The best time to obtain a drug concentration is when your patient is stable or free of most symptoms; this concentration may serve as the patient’s “therapeutic” concentration. Although laboratories have set therapeutic concentration ranges for each medication, treatment should focus on addressing your patient’s clinical presentation, rather than achieving the laboratory-suggested range.
Carbamazepine and valproate require therapeutic monitoring to prevent adverse effects from supratherapeutic concentrations (see this article at CurrentPsychiatry.com for a Table listing suggested ranges). The foundation for the therapeutic concentrations of these agents stems from neurology; however, these concentration ranges have been applicable in psychiatry.23
Carbamazepine generally requires more frequent monitoring because it has a narrow therapeutic index and relatively high potential for drug-drug interactions. Compared with lower doses, carbamazepine dosing associated with levels >12 μg/mL is more likely to induce toxicity.23 Carbamazepine autoinduction begins approximately 3 to 5 days after initiation and peaks between 3 to 4 weeks. Therefore, a drop in carbamazepine level from week 1 to week 4 of treatment likely is a pharmacokinetic indicator rather than a sign of nonadherence.
Some acute mania and maintenance bipolar studies have shown a correlation between clinical efficacy and valproate levels.24 A range of 50 to 125 μg/mL is well-accepted in clinical practice.24 For some patients, however, symptoms might not resolve until they are above the therapeutic range, but adverse effects are more likely at higher levels.24
Because concentrations of newer AEDs—including gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide—have not been shown to correlate with therapeutic response, monitoring of serum concentrations is not necessary. However, routine laboratory tests to monitor for adverse effects are recommended.
Table
Therapeutic concentration monitoring for carbamazepine and valproate
| Medication | Suggested therapeutic range (trough level)* | Supratherapeutic presentation |
|---|---|---|
| Carbamazepine | 4 to 12 μg/mL | Ataxia, gastrointestinal upset, drowsiness, dizziness, diplopia, rash |
| Valproate (divalproex) | 50 to 125 μ/mL | Ataxia, nystagmus, tremor, hallucinations |
| *Values may vary among laboratories Source: Reference 23 | ||
Related Resources
- McElroy SL, Keck PE, Post RM, eds. Antiepileptic drugs to treat psychiatric disorders. New York, NY: Informa Health-care USA, Inc.; 2008.
- U.S. Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM100190.
Drug Brand Names
- Carbamazepine • Carbatrol, Equetro, others
- Clozapine • Clozaril
- Gabapentin • Neurontin
- Lamotrigine • Lamictal, Lamictal XR
- Levetiracetam • Keppra, Keppra XR
- Oxcarbazepine • Trileptal
- Pregabalin • Lyrica
- Tiagabine • Gabitril
- Topiramate • Topamax
- Valproate (Divalproex) • Depakote, Depakote ER
- Zonisamide • Zonegram
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bowden CL. Anticonvulsants in bipolar disorders: current research and practice and future directions. Bipolar Disord. 2009;11(suppl 2):20-33.
2. Vigo DV, Baldessarini RJ. Anticonvulsants in the treatment of major depressive disorder: an overview. Harv Rev Psychiatry. 2009;17(4):231-241.
3. Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence? Expert Rev Neurother. 2009;9(1):55-71.
4. Grunze HC. The effectiveness of anticonvulsants in psychiatric disorders. Dialogues Clin Neurosci. 2008;10(1):77-89.
5. Hoffman EJ, Mathew SJ. Anxiety disorders: a comprehensive review of pharmacotherapies. Mt Sinai J Med. 2008;75(3):248-262.
6. Rosenberg JM, Salzman C. Update: new uses for lithium and anticonvulsants. CNS Spectr. 2007;12(11):831-841.
7. McElroy SL, Guerdjikova AI, Martens B, et al. Role of antiepileptic drugs in the management of eating disorders. CNS Drugs. 2009;23(2):139-156.
8. Wilby J, Kainth A, Hawkins N, et al. Clinical effectiveness, tolerability and cost-effectiveness of newer drugs for epilepsy in adults: a systematic review and economic evaluation. Health Technol Assess. 2005;9(15):1-157.
9. Cheng LS, Prasad AN, Rieder MJ. Relationship between antiepileptic drugs and biological markers affecting long-term cardiovascular function in children and adolescents. Can J Clin Pharmacol. 2010;17(1):e5-46.
10. Park SP, Kwon SH. Cognitive effects of antiepileptic drugs. J Clin Neurol. 2008;4(3):99-106.
11. U.S. Food and Drug Administration. FDA drug safety communication: aseptic meningitis associated with use of lamictal (lamotrigine). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm221847.htm. Accessed October 29, 2010.
12. Wade JF, Dang CV, Nelson L, et al. Emergent complications of the newer anticonvulsants. J Emerg Med. 2010;38(2):231-237.
13. Beydoun A, Sackellares JC, Shu V, et al. Safety and efficacy of divalproex sodium monotherapy in partial epilepsy: a double-blind, concentration-response design clinical trial. Neurology. 1997;48(1):182-188.
14. Depakote [package inset]. North Chicago, IL: Abbott Laboratories; 2009.
15. Cerminara C, Seri S, Bombardieri R, et al. Hypohidrosis during topiramate treatment: a rare and reversible side effect. Pediatr Neurol. 2006;34(5):392-394.
16. U.S. Food and Drug Administration. Information for healthcare professionals: suicidal behavior and ideation and antiepileptic drugs. 2008. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100192.htm. Accessed May 10, 2010.
17. Patorno E, Bohn RL, Wahl PM, et al. Anticonvulsant medications and the risk of suicide, attempted suicide, or violent death. JAMA. 2010;303(14):1401-1409.
18. Gibbons RD, Hur K, Brown CH, et al. Relationship between antiepileptic drugs and suicide attempts in patients with bipolar disorder. Arch Gen Psychiatry. 2009;66(12):1354-1360.
19. Bilo L, Meo R. Polycystic ovary syndrome in women using valproate: a review. Gynecol Endocrinol. 2008;24(10):562-570.
20. Welch BJ, Graybeal D, Moe OW, et al. Biochemical and stone-risk profiles with topiramate treatment. Am J Kidney Dis. 2006;48(4):555-563.
21. Harden CL, Meador KJ, Pennell PB, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
22. Chung WH, Hung SI, Chen YT. Genetic predisposition of life-threatening antiepileptic-induced skin reactions. Expert Opin Drug Saf. 2010;9(1):15-21.
23. Warner A, Privitera M, Bates D. Standards of laboratory practice: antiepileptic drug monitoring. Clin Chem. 1998;44(5):1085-1095.
24. Kaneria KM, Patel NC, Keck PE, Jr. Bipolar disorder: new strategy for checking serum valproate. Current Psychiatry. 2005;4(12):31-44.
1. Bowden CL. Anticonvulsants in bipolar disorders: current research and practice and future directions. Bipolar Disord. 2009;11(suppl 2):20-33.
2. Vigo DV, Baldessarini RJ. Anticonvulsants in the treatment of major depressive disorder: an overview. Harv Rev Psychiatry. 2009;17(4):231-241.
3. Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence? Expert Rev Neurother. 2009;9(1):55-71.
4. Grunze HC. The effectiveness of anticonvulsants in psychiatric disorders. Dialogues Clin Neurosci. 2008;10(1):77-89.
5. Hoffman EJ, Mathew SJ. Anxiety disorders: a comprehensive review of pharmacotherapies. Mt Sinai J Med. 2008;75(3):248-262.
6. Rosenberg JM, Salzman C. Update: new uses for lithium and anticonvulsants. CNS Spectr. 2007;12(11):831-841.
7. McElroy SL, Guerdjikova AI, Martens B, et al. Role of antiepileptic drugs in the management of eating disorders. CNS Drugs. 2009;23(2):139-156.
8. Wilby J, Kainth A, Hawkins N, et al. Clinical effectiveness, tolerability and cost-effectiveness of newer drugs for epilepsy in adults: a systematic review and economic evaluation. Health Technol Assess. 2005;9(15):1-157.
9. Cheng LS, Prasad AN, Rieder MJ. Relationship between antiepileptic drugs and biological markers affecting long-term cardiovascular function in children and adolescents. Can J Clin Pharmacol. 2010;17(1):e5-46.
10. Park SP, Kwon SH. Cognitive effects of antiepileptic drugs. J Clin Neurol. 2008;4(3):99-106.
11. U.S. Food and Drug Administration. FDA drug safety communication: aseptic meningitis associated with use of lamictal (lamotrigine). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm221847.htm. Accessed October 29, 2010.
12. Wade JF, Dang CV, Nelson L, et al. Emergent complications of the newer anticonvulsants. J Emerg Med. 2010;38(2):231-237.
13. Beydoun A, Sackellares JC, Shu V, et al. Safety and efficacy of divalproex sodium monotherapy in partial epilepsy: a double-blind, concentration-response design clinical trial. Neurology. 1997;48(1):182-188.
14. Depakote [package inset]. North Chicago, IL: Abbott Laboratories; 2009.
15. Cerminara C, Seri S, Bombardieri R, et al. Hypohidrosis during topiramate treatment: a rare and reversible side effect. Pediatr Neurol. 2006;34(5):392-394.
16. U.S. Food and Drug Administration. Information for healthcare professionals: suicidal behavior and ideation and antiepileptic drugs. 2008. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100192.htm. Accessed May 10, 2010.
17. Patorno E, Bohn RL, Wahl PM, et al. Anticonvulsant medications and the risk of suicide, attempted suicide, or violent death. JAMA. 2010;303(14):1401-1409.
18. Gibbons RD, Hur K, Brown CH, et al. Relationship between antiepileptic drugs and suicide attempts in patients with bipolar disorder. Arch Gen Psychiatry. 2009;66(12):1354-1360.
19. Bilo L, Meo R. Polycystic ovary syndrome in women using valproate: a review. Gynecol Endocrinol. 2008;24(10):562-570.
20. Welch BJ, Graybeal D, Moe OW, et al. Biochemical and stone-risk profiles with topiramate treatment. Am J Kidney Dis. 2006;48(4):555-563.
21. Harden CL, Meador KJ, Pennell PB, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
22. Chung WH, Hung SI, Chen YT. Genetic predisposition of life-threatening antiepileptic-induced skin reactions. Expert Opin Drug Saf. 2010;9(1):15-21.
23. Warner A, Privitera M, Bates D. Standards of laboratory practice: antiepileptic drug monitoring. Clin Chem. 1998;44(5):1085-1095.
24. Kaneria KM, Patel NC, Keck PE, Jr. Bipolar disorder: new strategy for checking serum valproate. Current Psychiatry. 2005;4(12):31-44.
How do SSRIs cause sexual dysfunction?
Although selective serotonin reuptake inhibitors (SSRIs) are frequently prescribed1 and are better tolerated than older antidepressants, side effects such as sexual dysfunction limit patient acceptance of these medications. DSM-IV-TR categorizes medication-induced sexual dysfunction as a type of substance-induced sexual dysfunction.2 These dysfunctions are characterized by impairment of various sexual response phases (Table 1).2,3
Estimating the true incidence and prevalence of SSRI-related sexual dysfunction can be difficult. Zimmerman et al4 compared psychiatrists’ clinical assessments of depressed patients receiving ongoing treatment with results of a standardized side effects questionnaire and found that even though psychiatrists regularly inquired about sexual side effects, on the questionnaire patients reported higher rates of almost all sexual dysfunctions. The incidence of SSRI-induced sexual dysfunction also can be difficult to ascertain because some sexual dysfunctions frequently accompany a primary psychiatric disorder5 or physical illness. Balon6 suggested that the incidence of SSRI-associated sexual dysfunction is 30% to 50%, although others have reported higher incidences.
Few quality studies have focused on identifying the exact nature and causes of SSRI treatment-emergent sexual dysfunction. This article describes mechanisms that may be fundamental to SSRI-associated sexual dysfunction.
Table 1
Sexual dysfunction and the sexual response cycle
| Phase | Description | Dysfunction/disorder |
|---|---|---|
| Desire | Characterized by sexual fantasies and the desire to have sex | Hypoactive sexual desire disorder |
| Sexual aversion disorder | ||
| Hypoactive sexual desire disorder due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired desire | ||
| Excitement | Subjective sense of sexual pleasure and accompanying physiologic changes | Female sexual arousal disorder |
| Erectile disorder | ||
| Erectile disorder due to a general medical condition | ||
| Dyspareunia due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired arousal | ||
| Orgasm | Peaking of sexual pleasure with release of sexual tension | Female orgasmic disorder |
| Male orgasmic disorder | ||
| Premature ejaculation | ||
| Other sexual dysfunction due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired orgasm | ||
| Resolution | A sense of general relaxation, well-being, and muscle relaxation | Postcoital dysphoria |
| Postcoital headache | ||
| Source: References 2,3 | ||
Not just serotonin
Although SSRIs are relatively selective for the serotonergic system, they affect other neurotransmitter systems as well (Table 2).7 For example, at high dosages paroxetine is believed to block norepinephrine reuptake, and it has a clinically significant anticholinergic effect. Also, sertraline is a potent reuptake inhibitor of dopamine.8 Therefore, our discussion will include these neurotransmitters.
In their dual control model of male sexual response, Bancroft et al9 discuss the interplay between excitatory and inhibitory mechanisms at the central and peripheral levels. For example, they describe the role of norepinephrine mediation in the central arousal system via the disinhibition of dopaminergic and a possible testosterone mechanism. They also point to possible inhibition of central sexual arousal by neuropeptidergic and serotonergic mechanisms.
Evidence linking serotonin to sexual dysfunction is inconclusive because there are no exclusively serotonergic agents. Drugs frequently used to test these hypotheses often affect other neurotransmitters, which means conclusions are not specific to serotonin. Animal studies of the impact of serotonin agonist and antagonist agents on mounting and ejaculation have reported inconsistent results.10 Differential roles of 5-HT1 and 5-HT2 receptor activation on sexual behavior may explain some of these inconsistencies.8 However, 1 study found that antiserotonergic pharmacologic agents enhance sexual excitation in laboratory animals,11 and a separate study showed that severing serotonergic axons in the medial forebrain bundle in male rats facilitated ejaculation.12
Monteiro et al13 found a high incidence of anorgasmia in previously orgasmic patients after they received clomipramine, which may be partially attributed to the drug’s serotonergic action. This prompted researchers to hypothesize that central serotonergic tone inhibits sexual behavior. However, based on current evidence, it would be best to consider serotonin as having a modulating effect10—as opposed to a complete inhibitory effect—on human sexual behavior.
Regarding the parasympathetic system, it was long believed that cholinergic innervations mediate penile erection. However, a more plausible hypothesis may be that parasympathetic cholinergic transmission at best has a modulating effect when other neurotransmitters—primarily the adrenergic system—are affected by concomitant pharmacologic interventions. Segraves10 proposed that cholinergic potentiating of adrenergic activity may be primarily responsible for bethanechol-induced reversal of SSRI-induced sexual dysfunction.
The adrenergic system is believed to play a role in penile erection and ejaculation.10 Adrenergic fibers innervate the vas deferens, seminal vesicles, trigone of the urinary bladder, and proximal urethra.14 Penile contractile and erectile tissue is richly innervated by the adrenergic nerve fibers.10 Ejaculation is mediated by α1-adrenergic receptors.10
Table 2
Neurotransmitters affected by SSRIs
| SSRI | Neurotransmitters |
|---|---|
| Citalopram | 5-HT |
| Escitalopram | 5-HT |
| Fluoxetine | 5-HT, NE, DA |
| Fluvoxamine | 5-HT |
| Paroxetine | 5-HT, NE, Ach |
| Sertraline | 5-HT, NE, DA |
| 5-HT: serotonin; Ach: acetylcholine; DA: dopamine; NE: norepinephrine; SSRIs: selective serotonin reuptake inhibitors | |
| Source: Reference 7 | |
The role of nitric oxide synthase
The advent of sildenafil underscored the importance of nitric oxide-mediated relaxation pathways in treating erectile dysfunction. Nitric oxide plays an important role in mediating the penile vasculature changes essential for erection and also is hypothesized to promote penile smooth muscle relaxation via cyclic guanosine monophosphate, there-by contributing to physiologic erection.15 Paroxetine is known to inhibit nitric oxide synthase, which reduces nitric oxide levels. The exact mechanism of this interaction remains unclear; however, it is hypothesized that 3 nitric oxide synthase isoenzymes are structurally similar to cytochrome P450 (CYP450). Paroxetine is a strong CYP2D6 inhibitor, which contributes to low nitric oxide levels in patients taking the drug.16
SSRIs and sexual response
Because decreased libido is part of depressive psychopathology,5 it is difficult to attribute loss of sexual desire directly to SSRIs. Nonetheless, SSRIs are associated with a risk of clinically significant loss of sexual desire that may resemble moderate to severe hypoactive sexual desire disorder.17 Reduced mesolimbic dopaminergic activity as a result of inhibitory serotonergic midbrain raphe nuclei projections is 1 possible cause.18 This hypothesis has lead investigators to examine drug targets in the CNS for hypoactive sexual desire disorder that would inhibit serotonergic tone and lead to brain dopaminergic system stimulation.
Another putative hypothesis for SSRI-induced loss of sexual desire is the role of 5-HT1A receptor-mediated norepinephrine neurotransmission. Because the sympathetic nervous system is believed to be involved in genital arousal in women, a small study analyzed the effect of sympathetic activation on SSRI-induced sexual dysfunction.19 Women who received paroxetine and sertraline—both are highly selective for 5-HT1A—showed improvement in sexual arousal and orgasm after taking ephedrine before sexual activity.19 Women who took fluoxetine, which is less selective for 5-HT1A, show no change or decreased sexual function.
SSRIs are associated with reduced nocturnal penile erections and severe erectile dysfunction, but the relationship is not robust.17 SSRI-induced suppression of rapid eye movement sleep20 may partially explain reduced nocturnal and early morning erections. Supraspinal areas and preganglionic sacral neurons involved in sexual excitement also are reported to have substantial serotonergic activity, which suggests that serotonin has a direct role in erectile dysfunction at a comparative peripheral level.21 However, a recent study17 found no difference in flaccid and peak systolic velocity when comparing patients taking SSRIs with those who do not. This indicates that SSRIs’ affect on spontaneous and sexually aroused erections may be mediated at both central and peripheral levels.
Delayed ejaculation frequently is associated with SSRIs17 and usually is not caused by depressive psychopathology.22 Animal studies show that increased serotonergic tone predicts ejaculatory latency by acting as an inhibitor at the hypothalamus level.23 In contrast, noradrenergic tone enhances ejaculation.24 Antidepressants that increase noradrenaline levels and serotonin levels—such as serotonin-norepinephrine reuptake inhibitors—induce milder ejaculatory delay.17
A recent study17 found impaired climax and reduced libido in partners of patients using SSRIs. Patients receiving SSRIs report less frequent sexual intercourse and heightened guilt associated with masturbation, and SSRIs are associated with psychosocial factors such as higher stress at work and increased risk of conflicts with partners and other family members.17 In addition to biologic mechanisms, these psychosocial and intra-couple factors might contribute to SSRI-associated sexual dysfunction. However, because the temporal association between SSRI use and psychosocial dysfunction is ambiguous, this hypothesis should be interpreted with caution.
SSRIs have been associated with lower serum levels of luteinizing hormone, follicle-stimulating hormone, and testosterone.25 However, these findings need to be replicated before drawing firm conclusions on intermediary role of hormones in SSRI-emergent sexual dysfunction.
Be aware that other medications may contribute to sexual dysfunction experienced by a patient receiving an SSRI (Table 3).26
Table 3
Other than SSRIs, which medications can cause sexual dysfunction?
| Psychotropics |
|---|
| Amphetamines |
| Anticonvulsants |
Antidepressants
|
| Antipsychotics |
| Benzodiazepines |
| Nonpsychotropics |
Antihypertensives
|
| Digoxin |
| Histamine blockers |
| Lipid-lowering agents |
| Narcotics |
| Oral contraceptives |
| SSRIs: selective serotonin reuptake inhibitors |
| Source: Reference 26 |
SSRIs for premature ejaculation?
Because SSRIs can cause delayed ejaculation, they have been used off-label to treat premature ejaculation.27 For this purpose, paroxetine and sertraline have been prescribed with daily or on-demand dosing before sexual intercourse28 and daily fluoxetine has been used.29 However, none of these SSRIs is FDA-approved for treating premature ejaculation, daily dosing of SSRIs exposes patients to undesirable side effects, and inconsistent use of paroxetine can lead to discontinuation syndrome.
These concerns have lead researchers to seek an SSRI that could be used on as-needed basis and would not cause some of the deleterious side effects associated with current SSRIs. The short-acting SSRI dapoxetine is in FDA review for treating premature ejaculation; the drug is approved for this use in several countries outside the United States.30
Sexual health education
Because sexual dysfunction can be caused by underlying psychopathology or physical illness, it is essential to obtain a detailed sexual history at your patient’s initial assessment and at every follow-up visit. Patients and providers may be guarded when discussing sexual health, which can be a barrier to providing comprehensive health care. The organizations listed in Related Resources can provide information and materials to help patients and health care providers better understand sexual health. Addressing the importance of sexual health in a comprehensive, culturally sensitive manner can substantially improve our patients’ medication compliance and prognosis.
- American Association of Sexuality Educators, Counselors, and Therapists. www.aasect.org.
- Sexual Medicine and Wellness Center. www.methodistsexualwellness.com.
- The Sexual Health Network. www.sexualhealth.com.
- International Society for the Study of Women’s Sexual Health. www.isswsh.org.
Drug Brand Names
- Bethanechol • Urecholine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Dapoxetine • Priligy
- Digoxin • Lanoxin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sildenafil • Viagra
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Olfson M, Marcus SC. National patterns in antidepressant medication treatment. Arch Gen Psychiatry. 2009;66(8):848-856.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Sadock BJ, Sadock VA. Abnormal sexuality and sexual dysfunctions. In Sadock BJ, Sadock VA. Kaplan & Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:689-705.
4. Zimmerman M, Galione JN, Attiullah N, et al. Underrecognition of clinically significant side effects in depressed outpatients. J Clin Psychiatry. 2010;71(4):484-490.
5. Casper RC, Redmont DE, Jr, Katz MM, et al. Somatic symptoms in primary affective disorder: presence and relationship to the classification of depression. Arch Gen Psychiatry. 1985;42:1098-1104.
6. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504-1509; quiz 1664.
7. Schatzberg AF, Nemeroff CB. eds. The American Psychiatric Publishing textbook of psychopharmacology. 4th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2009.
8. Schatzberg AF, Cole JO, DeBattista C. Antidepressants. In: Schatzberg AF, Cole JO, DeBattista C. Manual of clinical psychopharmacology. 6th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2007:37-168.
9. Bancroft J, Janssen E. The dual control model of male sexual response: a theoretical approach to centrally mediated erectile dysfunction. Neurosci Biobehav Rev. 2000;24(5):571-579.
10. Segraves RT. Effects of psychotropic drugs on human erection and ejaculation. Arch Gen Psychiatry. 1989;46(3):275-284.
11. Tagliamonte A, Tagliamonte P, Gessa GL, et al. Compulsive sexual activity induced by p-chlorophenylalanine in normal and pinealectomized male rats. Science. 1969;166(911):1433-1435.
12. Rodriguez M, Castro R, Hernandez G, et al. Different roles of catecholaminergic and serotoninergic neurons of the medial forebrain bundle on male rat sexual behavior. Physiol Behav. 1984;33(1):5-11.
13. Monteiro WO, Noshirvani HF, Marks IM, et al. Anorgasmia from clomipramine in obsessive-compulsive disorder. A controlled trial. Br J Psychiatry. 1987;151:107-112.
14. Kleeman FJ. The physiology of the internal urinary sphincter. J Urol. 1970;104(4):549-554.
15. Stahl SM. How psychiatrists can build new therapies for impotence. J Clin Psychiatry. 1998;59(2):47-48.
16. Bredt DS, Hwang PM, Glatt CE, et al. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351(6329):714-718.
17. Corona G, Ricca V, Bandini E, et al. Selective serotonin reuptake inhibitor-induced sexual dysfunction. J Sex Med. 2009;6(5):1259-1269.
18. Pfaus JG. Pathways of sexual desire. J Sex Med. 2009;6(6):1506-1533.
19. Ahrold TK, Meston CM. Effects of SNS activation on SSRI-induced sexual side effects differ by SSRI. J Sex Marital Ther. 2009;35(4):311-319.
20. Hirshkowitz M, Schmidt MH. Sleep-related erections: clinical perspectives and neural mechanisms. Sleep Med Rev. 2005;9(4):311-329.
21. Miner MM, Seftel AD. Centrally acting mechanisms for the treatment of male sexual dysfunction. Urol Clin North Am. 2007;34(4):483-496, v.
22. Labbate LA, Grimes J, Hines A, et al. Sexual dysfunction induced by serotonin reuptake antidepressants. J Sex Marital Ther. 1998;24(1):3-12.
23. Waldinger MD. The neurobiological approach to premature ejaculation. J Urol. 2002;168(6):2359-2367.
24. Meston CM, Frohlich PF. The neurobiology of sexual function. Arch Gen Psychiatry. 2000;57(11):1012-1030.
25. Safarinejad MR. Evaluation of endocrine profile and hypothalamic-pituitary-testis axis in selective serotonin reuptake inhibitor-induced male sexual dysfunction. J Clin Psychopharmacol. 2008;28(4):418-423.
26. Sajith SG, Morgan C, Clarke D. Pharmacological management of inappropriate sexual behaviours: a review of its evidence, rationale and scope in relation to men with intellectual disabilities. J Intellect Disabil Res. 2008;52(12):1078-1090.
27. Giuliano F, Hellstrom WJ. The pharmacological treatment of premature ejaculation. BJU Int. 2008;102(6):668-675.
28. Kim SW, Paick JS. Short-term analysis of the effects of as needed use of sertraline at 5 PM for the treatment of premature ejaculation. Urology. 1999;54(3):544-547.
29. Waldinger MD, Zwinderman AH, Schweitzer DH, et al. Relevance of methodological design for the interpretation of efficacy of drug treatment of premature ejaculation: a systematic review and meta-analysis. Int J Impot Res. 2004;16(4):369-381.
30. Kendirci M, Salem E, Hellstrom WJ. Dapoxetine, a novel selective serotonin transport inhibitor for the treatment of premature ejaculation. Ther Clin Risk Manag. 2007;3(2):277-289.
Although selective serotonin reuptake inhibitors (SSRIs) are frequently prescribed1 and are better tolerated than older antidepressants, side effects such as sexual dysfunction limit patient acceptance of these medications. DSM-IV-TR categorizes medication-induced sexual dysfunction as a type of substance-induced sexual dysfunction.2 These dysfunctions are characterized by impairment of various sexual response phases (Table 1).2,3
Estimating the true incidence and prevalence of SSRI-related sexual dysfunction can be difficult. Zimmerman et al4 compared psychiatrists’ clinical assessments of depressed patients receiving ongoing treatment with results of a standardized side effects questionnaire and found that even though psychiatrists regularly inquired about sexual side effects, on the questionnaire patients reported higher rates of almost all sexual dysfunctions. The incidence of SSRI-induced sexual dysfunction also can be difficult to ascertain because some sexual dysfunctions frequently accompany a primary psychiatric disorder5 or physical illness. Balon6 suggested that the incidence of SSRI-associated sexual dysfunction is 30% to 50%, although others have reported higher incidences.
Few quality studies have focused on identifying the exact nature and causes of SSRI treatment-emergent sexual dysfunction. This article describes mechanisms that may be fundamental to SSRI-associated sexual dysfunction.
Table 1
Sexual dysfunction and the sexual response cycle
| Phase | Description | Dysfunction/disorder |
|---|---|---|
| Desire | Characterized by sexual fantasies and the desire to have sex | Hypoactive sexual desire disorder |
| Sexual aversion disorder | ||
| Hypoactive sexual desire disorder due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired desire | ||
| Excitement | Subjective sense of sexual pleasure and accompanying physiologic changes | Female sexual arousal disorder |
| Erectile disorder | ||
| Erectile disorder due to a general medical condition | ||
| Dyspareunia due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired arousal | ||
| Orgasm | Peaking of sexual pleasure with release of sexual tension | Female orgasmic disorder |
| Male orgasmic disorder | ||
| Premature ejaculation | ||
| Other sexual dysfunction due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired orgasm | ||
| Resolution | A sense of general relaxation, well-being, and muscle relaxation | Postcoital dysphoria |
| Postcoital headache | ||
| Source: References 2,3 | ||
Not just serotonin
Although SSRIs are relatively selective for the serotonergic system, they affect other neurotransmitter systems as well (Table 2).7 For example, at high dosages paroxetine is believed to block norepinephrine reuptake, and it has a clinically significant anticholinergic effect. Also, sertraline is a potent reuptake inhibitor of dopamine.8 Therefore, our discussion will include these neurotransmitters.
In their dual control model of male sexual response, Bancroft et al9 discuss the interplay between excitatory and inhibitory mechanisms at the central and peripheral levels. For example, they describe the role of norepinephrine mediation in the central arousal system via the disinhibition of dopaminergic and a possible testosterone mechanism. They also point to possible inhibition of central sexual arousal by neuropeptidergic and serotonergic mechanisms.
Evidence linking serotonin to sexual dysfunction is inconclusive because there are no exclusively serotonergic agents. Drugs frequently used to test these hypotheses often affect other neurotransmitters, which means conclusions are not specific to serotonin. Animal studies of the impact of serotonin agonist and antagonist agents on mounting and ejaculation have reported inconsistent results.10 Differential roles of 5-HT1 and 5-HT2 receptor activation on sexual behavior may explain some of these inconsistencies.8 However, 1 study found that antiserotonergic pharmacologic agents enhance sexual excitation in laboratory animals,11 and a separate study showed that severing serotonergic axons in the medial forebrain bundle in male rats facilitated ejaculation.12
Monteiro et al13 found a high incidence of anorgasmia in previously orgasmic patients after they received clomipramine, which may be partially attributed to the drug’s serotonergic action. This prompted researchers to hypothesize that central serotonergic tone inhibits sexual behavior. However, based on current evidence, it would be best to consider serotonin as having a modulating effect10—as opposed to a complete inhibitory effect—on human sexual behavior.
Regarding the parasympathetic system, it was long believed that cholinergic innervations mediate penile erection. However, a more plausible hypothesis may be that parasympathetic cholinergic transmission at best has a modulating effect when other neurotransmitters—primarily the adrenergic system—are affected by concomitant pharmacologic interventions. Segraves10 proposed that cholinergic potentiating of adrenergic activity may be primarily responsible for bethanechol-induced reversal of SSRI-induced sexual dysfunction.
The adrenergic system is believed to play a role in penile erection and ejaculation.10 Adrenergic fibers innervate the vas deferens, seminal vesicles, trigone of the urinary bladder, and proximal urethra.14 Penile contractile and erectile tissue is richly innervated by the adrenergic nerve fibers.10 Ejaculation is mediated by α1-adrenergic receptors.10
Table 2
Neurotransmitters affected by SSRIs
| SSRI | Neurotransmitters |
|---|---|
| Citalopram | 5-HT |
| Escitalopram | 5-HT |
| Fluoxetine | 5-HT, NE, DA |
| Fluvoxamine | 5-HT |
| Paroxetine | 5-HT, NE, Ach |
| Sertraline | 5-HT, NE, DA |
| 5-HT: serotonin; Ach: acetylcholine; DA: dopamine; NE: norepinephrine; SSRIs: selective serotonin reuptake inhibitors | |
| Source: Reference 7 | |
The role of nitric oxide synthase
The advent of sildenafil underscored the importance of nitric oxide-mediated relaxation pathways in treating erectile dysfunction. Nitric oxide plays an important role in mediating the penile vasculature changes essential for erection and also is hypothesized to promote penile smooth muscle relaxation via cyclic guanosine monophosphate, there-by contributing to physiologic erection.15 Paroxetine is known to inhibit nitric oxide synthase, which reduces nitric oxide levels. The exact mechanism of this interaction remains unclear; however, it is hypothesized that 3 nitric oxide synthase isoenzymes are structurally similar to cytochrome P450 (CYP450). Paroxetine is a strong CYP2D6 inhibitor, which contributes to low nitric oxide levels in patients taking the drug.16
SSRIs and sexual response
Because decreased libido is part of depressive psychopathology,5 it is difficult to attribute loss of sexual desire directly to SSRIs. Nonetheless, SSRIs are associated with a risk of clinically significant loss of sexual desire that may resemble moderate to severe hypoactive sexual desire disorder.17 Reduced mesolimbic dopaminergic activity as a result of inhibitory serotonergic midbrain raphe nuclei projections is 1 possible cause.18 This hypothesis has lead investigators to examine drug targets in the CNS for hypoactive sexual desire disorder that would inhibit serotonergic tone and lead to brain dopaminergic system stimulation.
Another putative hypothesis for SSRI-induced loss of sexual desire is the role of 5-HT1A receptor-mediated norepinephrine neurotransmission. Because the sympathetic nervous system is believed to be involved in genital arousal in women, a small study analyzed the effect of sympathetic activation on SSRI-induced sexual dysfunction.19 Women who received paroxetine and sertraline—both are highly selective for 5-HT1A—showed improvement in sexual arousal and orgasm after taking ephedrine before sexual activity.19 Women who took fluoxetine, which is less selective for 5-HT1A, show no change or decreased sexual function.
SSRIs are associated with reduced nocturnal penile erections and severe erectile dysfunction, but the relationship is not robust.17 SSRI-induced suppression of rapid eye movement sleep20 may partially explain reduced nocturnal and early morning erections. Supraspinal areas and preganglionic sacral neurons involved in sexual excitement also are reported to have substantial serotonergic activity, which suggests that serotonin has a direct role in erectile dysfunction at a comparative peripheral level.21 However, a recent study17 found no difference in flaccid and peak systolic velocity when comparing patients taking SSRIs with those who do not. This indicates that SSRIs’ affect on spontaneous and sexually aroused erections may be mediated at both central and peripheral levels.
Delayed ejaculation frequently is associated with SSRIs17 and usually is not caused by depressive psychopathology.22 Animal studies show that increased serotonergic tone predicts ejaculatory latency by acting as an inhibitor at the hypothalamus level.23 In contrast, noradrenergic tone enhances ejaculation.24 Antidepressants that increase noradrenaline levels and serotonin levels—such as serotonin-norepinephrine reuptake inhibitors—induce milder ejaculatory delay.17
A recent study17 found impaired climax and reduced libido in partners of patients using SSRIs. Patients receiving SSRIs report less frequent sexual intercourse and heightened guilt associated with masturbation, and SSRIs are associated with psychosocial factors such as higher stress at work and increased risk of conflicts with partners and other family members.17 In addition to biologic mechanisms, these psychosocial and intra-couple factors might contribute to SSRI-associated sexual dysfunction. However, because the temporal association between SSRI use and psychosocial dysfunction is ambiguous, this hypothesis should be interpreted with caution.
SSRIs have been associated with lower serum levels of luteinizing hormone, follicle-stimulating hormone, and testosterone.25 However, these findings need to be replicated before drawing firm conclusions on intermediary role of hormones in SSRI-emergent sexual dysfunction.
Be aware that other medications may contribute to sexual dysfunction experienced by a patient receiving an SSRI (Table 3).26
Table 3
Other than SSRIs, which medications can cause sexual dysfunction?
| Psychotropics |
|---|
| Amphetamines |
| Anticonvulsants |
Antidepressants
|
| Antipsychotics |
| Benzodiazepines |
| Nonpsychotropics |
Antihypertensives
|
| Digoxin |
| Histamine blockers |
| Lipid-lowering agents |
| Narcotics |
| Oral contraceptives |
| SSRIs: selective serotonin reuptake inhibitors |
| Source: Reference 26 |
SSRIs for premature ejaculation?
Because SSRIs can cause delayed ejaculation, they have been used off-label to treat premature ejaculation.27 For this purpose, paroxetine and sertraline have been prescribed with daily or on-demand dosing before sexual intercourse28 and daily fluoxetine has been used.29 However, none of these SSRIs is FDA-approved for treating premature ejaculation, daily dosing of SSRIs exposes patients to undesirable side effects, and inconsistent use of paroxetine can lead to discontinuation syndrome.
These concerns have lead researchers to seek an SSRI that could be used on as-needed basis and would not cause some of the deleterious side effects associated with current SSRIs. The short-acting SSRI dapoxetine is in FDA review for treating premature ejaculation; the drug is approved for this use in several countries outside the United States.30
Sexual health education
Because sexual dysfunction can be caused by underlying psychopathology or physical illness, it is essential to obtain a detailed sexual history at your patient’s initial assessment and at every follow-up visit. Patients and providers may be guarded when discussing sexual health, which can be a barrier to providing comprehensive health care. The organizations listed in Related Resources can provide information and materials to help patients and health care providers better understand sexual health. Addressing the importance of sexual health in a comprehensive, culturally sensitive manner can substantially improve our patients’ medication compliance and prognosis.
- American Association of Sexuality Educators, Counselors, and Therapists. www.aasect.org.
- Sexual Medicine and Wellness Center. www.methodistsexualwellness.com.
- The Sexual Health Network. www.sexualhealth.com.
- International Society for the Study of Women’s Sexual Health. www.isswsh.org.
Drug Brand Names
- Bethanechol • Urecholine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Dapoxetine • Priligy
- Digoxin • Lanoxin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sildenafil • Viagra
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Although selective serotonin reuptake inhibitors (SSRIs) are frequently prescribed1 and are better tolerated than older antidepressants, side effects such as sexual dysfunction limit patient acceptance of these medications. DSM-IV-TR categorizes medication-induced sexual dysfunction as a type of substance-induced sexual dysfunction.2 These dysfunctions are characterized by impairment of various sexual response phases (Table 1).2,3
Estimating the true incidence and prevalence of SSRI-related sexual dysfunction can be difficult. Zimmerman et al4 compared psychiatrists’ clinical assessments of depressed patients receiving ongoing treatment with results of a standardized side effects questionnaire and found that even though psychiatrists regularly inquired about sexual side effects, on the questionnaire patients reported higher rates of almost all sexual dysfunctions. The incidence of SSRI-induced sexual dysfunction also can be difficult to ascertain because some sexual dysfunctions frequently accompany a primary psychiatric disorder5 or physical illness. Balon6 suggested that the incidence of SSRI-associated sexual dysfunction is 30% to 50%, although others have reported higher incidences.
Few quality studies have focused on identifying the exact nature and causes of SSRI treatment-emergent sexual dysfunction. This article describes mechanisms that may be fundamental to SSRI-associated sexual dysfunction.
Table 1
Sexual dysfunction and the sexual response cycle
| Phase | Description | Dysfunction/disorder |
|---|---|---|
| Desire | Characterized by sexual fantasies and the desire to have sex | Hypoactive sexual desire disorder |
| Sexual aversion disorder | ||
| Hypoactive sexual desire disorder due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired desire | ||
| Excitement | Subjective sense of sexual pleasure and accompanying physiologic changes | Female sexual arousal disorder |
| Erectile disorder | ||
| Erectile disorder due to a general medical condition | ||
| Dyspareunia due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired arousal | ||
| Orgasm | Peaking of sexual pleasure with release of sexual tension | Female orgasmic disorder |
| Male orgasmic disorder | ||
| Premature ejaculation | ||
| Other sexual dysfunction due to a general medical condition | ||
| Substance-induced sexual dysfunction with impaired orgasm | ||
| Resolution | A sense of general relaxation, well-being, and muscle relaxation | Postcoital dysphoria |
| Postcoital headache | ||
| Source: References 2,3 | ||
Not just serotonin
Although SSRIs are relatively selective for the serotonergic system, they affect other neurotransmitter systems as well (Table 2).7 For example, at high dosages paroxetine is believed to block norepinephrine reuptake, and it has a clinically significant anticholinergic effect. Also, sertraline is a potent reuptake inhibitor of dopamine.8 Therefore, our discussion will include these neurotransmitters.
In their dual control model of male sexual response, Bancroft et al9 discuss the interplay between excitatory and inhibitory mechanisms at the central and peripheral levels. For example, they describe the role of norepinephrine mediation in the central arousal system via the disinhibition of dopaminergic and a possible testosterone mechanism. They also point to possible inhibition of central sexual arousal by neuropeptidergic and serotonergic mechanisms.
Evidence linking serotonin to sexual dysfunction is inconclusive because there are no exclusively serotonergic agents. Drugs frequently used to test these hypotheses often affect other neurotransmitters, which means conclusions are not specific to serotonin. Animal studies of the impact of serotonin agonist and antagonist agents on mounting and ejaculation have reported inconsistent results.10 Differential roles of 5-HT1 and 5-HT2 receptor activation on sexual behavior may explain some of these inconsistencies.8 However, 1 study found that antiserotonergic pharmacologic agents enhance sexual excitation in laboratory animals,11 and a separate study showed that severing serotonergic axons in the medial forebrain bundle in male rats facilitated ejaculation.12
Monteiro et al13 found a high incidence of anorgasmia in previously orgasmic patients after they received clomipramine, which may be partially attributed to the drug’s serotonergic action. This prompted researchers to hypothesize that central serotonergic tone inhibits sexual behavior. However, based on current evidence, it would be best to consider serotonin as having a modulating effect10—as opposed to a complete inhibitory effect—on human sexual behavior.
Regarding the parasympathetic system, it was long believed that cholinergic innervations mediate penile erection. However, a more plausible hypothesis may be that parasympathetic cholinergic transmission at best has a modulating effect when other neurotransmitters—primarily the adrenergic system—are affected by concomitant pharmacologic interventions. Segraves10 proposed that cholinergic potentiating of adrenergic activity may be primarily responsible for bethanechol-induced reversal of SSRI-induced sexual dysfunction.
The adrenergic system is believed to play a role in penile erection and ejaculation.10 Adrenergic fibers innervate the vas deferens, seminal vesicles, trigone of the urinary bladder, and proximal urethra.14 Penile contractile and erectile tissue is richly innervated by the adrenergic nerve fibers.10 Ejaculation is mediated by α1-adrenergic receptors.10
Table 2
Neurotransmitters affected by SSRIs
| SSRI | Neurotransmitters |
|---|---|
| Citalopram | 5-HT |
| Escitalopram | 5-HT |
| Fluoxetine | 5-HT, NE, DA |
| Fluvoxamine | 5-HT |
| Paroxetine | 5-HT, NE, Ach |
| Sertraline | 5-HT, NE, DA |
| 5-HT: serotonin; Ach: acetylcholine; DA: dopamine; NE: norepinephrine; SSRIs: selective serotonin reuptake inhibitors | |
| Source: Reference 7 | |
The role of nitric oxide synthase
The advent of sildenafil underscored the importance of nitric oxide-mediated relaxation pathways in treating erectile dysfunction. Nitric oxide plays an important role in mediating the penile vasculature changes essential for erection and also is hypothesized to promote penile smooth muscle relaxation via cyclic guanosine monophosphate, there-by contributing to physiologic erection.15 Paroxetine is known to inhibit nitric oxide synthase, which reduces nitric oxide levels. The exact mechanism of this interaction remains unclear; however, it is hypothesized that 3 nitric oxide synthase isoenzymes are structurally similar to cytochrome P450 (CYP450). Paroxetine is a strong CYP2D6 inhibitor, which contributes to low nitric oxide levels in patients taking the drug.16
SSRIs and sexual response
Because decreased libido is part of depressive psychopathology,5 it is difficult to attribute loss of sexual desire directly to SSRIs. Nonetheless, SSRIs are associated with a risk of clinically significant loss of sexual desire that may resemble moderate to severe hypoactive sexual desire disorder.17 Reduced mesolimbic dopaminergic activity as a result of inhibitory serotonergic midbrain raphe nuclei projections is 1 possible cause.18 This hypothesis has lead investigators to examine drug targets in the CNS for hypoactive sexual desire disorder that would inhibit serotonergic tone and lead to brain dopaminergic system stimulation.
Another putative hypothesis for SSRI-induced loss of sexual desire is the role of 5-HT1A receptor-mediated norepinephrine neurotransmission. Because the sympathetic nervous system is believed to be involved in genital arousal in women, a small study analyzed the effect of sympathetic activation on SSRI-induced sexual dysfunction.19 Women who received paroxetine and sertraline—both are highly selective for 5-HT1A—showed improvement in sexual arousal and orgasm after taking ephedrine before sexual activity.19 Women who took fluoxetine, which is less selective for 5-HT1A, show no change or decreased sexual function.
SSRIs are associated with reduced nocturnal penile erections and severe erectile dysfunction, but the relationship is not robust.17 SSRI-induced suppression of rapid eye movement sleep20 may partially explain reduced nocturnal and early morning erections. Supraspinal areas and preganglionic sacral neurons involved in sexual excitement also are reported to have substantial serotonergic activity, which suggests that serotonin has a direct role in erectile dysfunction at a comparative peripheral level.21 However, a recent study17 found no difference in flaccid and peak systolic velocity when comparing patients taking SSRIs with those who do not. This indicates that SSRIs’ affect on spontaneous and sexually aroused erections may be mediated at both central and peripheral levels.
Delayed ejaculation frequently is associated with SSRIs17 and usually is not caused by depressive psychopathology.22 Animal studies show that increased serotonergic tone predicts ejaculatory latency by acting as an inhibitor at the hypothalamus level.23 In contrast, noradrenergic tone enhances ejaculation.24 Antidepressants that increase noradrenaline levels and serotonin levels—such as serotonin-norepinephrine reuptake inhibitors—induce milder ejaculatory delay.17
A recent study17 found impaired climax and reduced libido in partners of patients using SSRIs. Patients receiving SSRIs report less frequent sexual intercourse and heightened guilt associated with masturbation, and SSRIs are associated with psychosocial factors such as higher stress at work and increased risk of conflicts with partners and other family members.17 In addition to biologic mechanisms, these psychosocial and intra-couple factors might contribute to SSRI-associated sexual dysfunction. However, because the temporal association between SSRI use and psychosocial dysfunction is ambiguous, this hypothesis should be interpreted with caution.
SSRIs have been associated with lower serum levels of luteinizing hormone, follicle-stimulating hormone, and testosterone.25 However, these findings need to be replicated before drawing firm conclusions on intermediary role of hormones in SSRI-emergent sexual dysfunction.
Be aware that other medications may contribute to sexual dysfunction experienced by a patient receiving an SSRI (Table 3).26
Table 3
Other than SSRIs, which medications can cause sexual dysfunction?
| Psychotropics |
|---|
| Amphetamines |
| Anticonvulsants |
Antidepressants
|
| Antipsychotics |
| Benzodiazepines |
| Nonpsychotropics |
Antihypertensives
|
| Digoxin |
| Histamine blockers |
| Lipid-lowering agents |
| Narcotics |
| Oral contraceptives |
| SSRIs: selective serotonin reuptake inhibitors |
| Source: Reference 26 |
SSRIs for premature ejaculation?
Because SSRIs can cause delayed ejaculation, they have been used off-label to treat premature ejaculation.27 For this purpose, paroxetine and sertraline have been prescribed with daily or on-demand dosing before sexual intercourse28 and daily fluoxetine has been used.29 However, none of these SSRIs is FDA-approved for treating premature ejaculation, daily dosing of SSRIs exposes patients to undesirable side effects, and inconsistent use of paroxetine can lead to discontinuation syndrome.
These concerns have lead researchers to seek an SSRI that could be used on as-needed basis and would not cause some of the deleterious side effects associated with current SSRIs. The short-acting SSRI dapoxetine is in FDA review for treating premature ejaculation; the drug is approved for this use in several countries outside the United States.30
Sexual health education
Because sexual dysfunction can be caused by underlying psychopathology or physical illness, it is essential to obtain a detailed sexual history at your patient’s initial assessment and at every follow-up visit. Patients and providers may be guarded when discussing sexual health, which can be a barrier to providing comprehensive health care. The organizations listed in Related Resources can provide information and materials to help patients and health care providers better understand sexual health. Addressing the importance of sexual health in a comprehensive, culturally sensitive manner can substantially improve our patients’ medication compliance and prognosis.
- American Association of Sexuality Educators, Counselors, and Therapists. www.aasect.org.
- Sexual Medicine and Wellness Center. www.methodistsexualwellness.com.
- The Sexual Health Network. www.sexualhealth.com.
- International Society for the Study of Women’s Sexual Health. www.isswsh.org.
Drug Brand Names
- Bethanechol • Urecholine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Dapoxetine • Priligy
- Digoxin • Lanoxin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sildenafil • Viagra
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Olfson M, Marcus SC. National patterns in antidepressant medication treatment. Arch Gen Psychiatry. 2009;66(8):848-856.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Sadock BJ, Sadock VA. Abnormal sexuality and sexual dysfunctions. In Sadock BJ, Sadock VA. Kaplan & Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:689-705.
4. Zimmerman M, Galione JN, Attiullah N, et al. Underrecognition of clinically significant side effects in depressed outpatients. J Clin Psychiatry. 2010;71(4):484-490.
5. Casper RC, Redmont DE, Jr, Katz MM, et al. Somatic symptoms in primary affective disorder: presence and relationship to the classification of depression. Arch Gen Psychiatry. 1985;42:1098-1104.
6. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504-1509; quiz 1664.
7. Schatzberg AF, Nemeroff CB. eds. The American Psychiatric Publishing textbook of psychopharmacology. 4th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2009.
8. Schatzberg AF, Cole JO, DeBattista C. Antidepressants. In: Schatzberg AF, Cole JO, DeBattista C. Manual of clinical psychopharmacology. 6th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2007:37-168.
9. Bancroft J, Janssen E. The dual control model of male sexual response: a theoretical approach to centrally mediated erectile dysfunction. Neurosci Biobehav Rev. 2000;24(5):571-579.
10. Segraves RT. Effects of psychotropic drugs on human erection and ejaculation. Arch Gen Psychiatry. 1989;46(3):275-284.
11. Tagliamonte A, Tagliamonte P, Gessa GL, et al. Compulsive sexual activity induced by p-chlorophenylalanine in normal and pinealectomized male rats. Science. 1969;166(911):1433-1435.
12. Rodriguez M, Castro R, Hernandez G, et al. Different roles of catecholaminergic and serotoninergic neurons of the medial forebrain bundle on male rat sexual behavior. Physiol Behav. 1984;33(1):5-11.
13. Monteiro WO, Noshirvani HF, Marks IM, et al. Anorgasmia from clomipramine in obsessive-compulsive disorder. A controlled trial. Br J Psychiatry. 1987;151:107-112.
14. Kleeman FJ. The physiology of the internal urinary sphincter. J Urol. 1970;104(4):549-554.
15. Stahl SM. How psychiatrists can build new therapies for impotence. J Clin Psychiatry. 1998;59(2):47-48.
16. Bredt DS, Hwang PM, Glatt CE, et al. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351(6329):714-718.
17. Corona G, Ricca V, Bandini E, et al. Selective serotonin reuptake inhibitor-induced sexual dysfunction. J Sex Med. 2009;6(5):1259-1269.
18. Pfaus JG. Pathways of sexual desire. J Sex Med. 2009;6(6):1506-1533.
19. Ahrold TK, Meston CM. Effects of SNS activation on SSRI-induced sexual side effects differ by SSRI. J Sex Marital Ther. 2009;35(4):311-319.
20. Hirshkowitz M, Schmidt MH. Sleep-related erections: clinical perspectives and neural mechanisms. Sleep Med Rev. 2005;9(4):311-329.
21. Miner MM, Seftel AD. Centrally acting mechanisms for the treatment of male sexual dysfunction. Urol Clin North Am. 2007;34(4):483-496, v.
22. Labbate LA, Grimes J, Hines A, et al. Sexual dysfunction induced by serotonin reuptake antidepressants. J Sex Marital Ther. 1998;24(1):3-12.
23. Waldinger MD. The neurobiological approach to premature ejaculation. J Urol. 2002;168(6):2359-2367.
24. Meston CM, Frohlich PF. The neurobiology of sexual function. Arch Gen Psychiatry. 2000;57(11):1012-1030.
25. Safarinejad MR. Evaluation of endocrine profile and hypothalamic-pituitary-testis axis in selective serotonin reuptake inhibitor-induced male sexual dysfunction. J Clin Psychopharmacol. 2008;28(4):418-423.
26. Sajith SG, Morgan C, Clarke D. Pharmacological management of inappropriate sexual behaviours: a review of its evidence, rationale and scope in relation to men with intellectual disabilities. J Intellect Disabil Res. 2008;52(12):1078-1090.
27. Giuliano F, Hellstrom WJ. The pharmacological treatment of premature ejaculation. BJU Int. 2008;102(6):668-675.
28. Kim SW, Paick JS. Short-term analysis of the effects of as needed use of sertraline at 5 PM for the treatment of premature ejaculation. Urology. 1999;54(3):544-547.
29. Waldinger MD, Zwinderman AH, Schweitzer DH, et al. Relevance of methodological design for the interpretation of efficacy of drug treatment of premature ejaculation: a systematic review and meta-analysis. Int J Impot Res. 2004;16(4):369-381.
30. Kendirci M, Salem E, Hellstrom WJ. Dapoxetine, a novel selective serotonin transport inhibitor for the treatment of premature ejaculation. Ther Clin Risk Manag. 2007;3(2):277-289.
1. Olfson M, Marcus SC. National patterns in antidepressant medication treatment. Arch Gen Psychiatry. 2009;66(8):848-856.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Sadock BJ, Sadock VA. Abnormal sexuality and sexual dysfunctions. In Sadock BJ, Sadock VA. Kaplan & Sadock’s synopsis of psychiatry: behavioral sciences/clinical psychiatry. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:689-705.
4. Zimmerman M, Galione JN, Attiullah N, et al. Underrecognition of clinically significant side effects in depressed outpatients. J Clin Psychiatry. 2010;71(4):484-490.
5. Casper RC, Redmont DE, Jr, Katz MM, et al. Somatic symptoms in primary affective disorder: presence and relationship to the classification of depression. Arch Gen Psychiatry. 1985;42:1098-1104.
6. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504-1509; quiz 1664.
7. Schatzberg AF, Nemeroff CB. eds. The American Psychiatric Publishing textbook of psychopharmacology. 4th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2009.
8. Schatzberg AF, Cole JO, DeBattista C. Antidepressants. In: Schatzberg AF, Cole JO, DeBattista C. Manual of clinical psychopharmacology. 6th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2007:37-168.
9. Bancroft J, Janssen E. The dual control model of male sexual response: a theoretical approach to centrally mediated erectile dysfunction. Neurosci Biobehav Rev. 2000;24(5):571-579.
10. Segraves RT. Effects of psychotropic drugs on human erection and ejaculation. Arch Gen Psychiatry. 1989;46(3):275-284.
11. Tagliamonte A, Tagliamonte P, Gessa GL, et al. Compulsive sexual activity induced by p-chlorophenylalanine in normal and pinealectomized male rats. Science. 1969;166(911):1433-1435.
12. Rodriguez M, Castro R, Hernandez G, et al. Different roles of catecholaminergic and serotoninergic neurons of the medial forebrain bundle on male rat sexual behavior. Physiol Behav. 1984;33(1):5-11.
13. Monteiro WO, Noshirvani HF, Marks IM, et al. Anorgasmia from clomipramine in obsessive-compulsive disorder. A controlled trial. Br J Psychiatry. 1987;151:107-112.
14. Kleeman FJ. The physiology of the internal urinary sphincter. J Urol. 1970;104(4):549-554.
15. Stahl SM. How psychiatrists can build new therapies for impotence. J Clin Psychiatry. 1998;59(2):47-48.
16. Bredt DS, Hwang PM, Glatt CE, et al. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351(6329):714-718.
17. Corona G, Ricca V, Bandini E, et al. Selective serotonin reuptake inhibitor-induced sexual dysfunction. J Sex Med. 2009;6(5):1259-1269.
18. Pfaus JG. Pathways of sexual desire. J Sex Med. 2009;6(6):1506-1533.
19. Ahrold TK, Meston CM. Effects of SNS activation on SSRI-induced sexual side effects differ by SSRI. J Sex Marital Ther. 2009;35(4):311-319.
20. Hirshkowitz M, Schmidt MH. Sleep-related erections: clinical perspectives and neural mechanisms. Sleep Med Rev. 2005;9(4):311-329.
21. Miner MM, Seftel AD. Centrally acting mechanisms for the treatment of male sexual dysfunction. Urol Clin North Am. 2007;34(4):483-496, v.
22. Labbate LA, Grimes J, Hines A, et al. Sexual dysfunction induced by serotonin reuptake antidepressants. J Sex Marital Ther. 1998;24(1):3-12.
23. Waldinger MD. The neurobiological approach to premature ejaculation. J Urol. 2002;168(6):2359-2367.
24. Meston CM, Frohlich PF. The neurobiology of sexual function. Arch Gen Psychiatry. 2000;57(11):1012-1030.
25. Safarinejad MR. Evaluation of endocrine profile and hypothalamic-pituitary-testis axis in selective serotonin reuptake inhibitor-induced male sexual dysfunction. J Clin Psychopharmacol. 2008;28(4):418-423.
26. Sajith SG, Morgan C, Clarke D. Pharmacological management of inappropriate sexual behaviours: a review of its evidence, rationale and scope in relation to men with intellectual disabilities. J Intellect Disabil Res. 2008;52(12):1078-1090.
27. Giuliano F, Hellstrom WJ. The pharmacological treatment of premature ejaculation. BJU Int. 2008;102(6):668-675.
28. Kim SW, Paick JS. Short-term analysis of the effects of as needed use of sertraline at 5 PM for the treatment of premature ejaculation. Urology. 1999;54(3):544-547.
29. Waldinger MD, Zwinderman AH, Schweitzer DH, et al. Relevance of methodological design for the interpretation of efficacy of drug treatment of premature ejaculation: a systematic review and meta-analysis. Int J Impot Res. 2004;16(4):369-381.
30. Kendirci M, Salem E, Hellstrom WJ. Dapoxetine, a novel selective serotonin transport inhibitor for the treatment of premature ejaculation. Ther Clin Risk Manag. 2007;3(2):277-289.
Treatments for depressed breast cancer patients
Descent after a missed flight
CASE: Psychotic and sleepless
Mr. F, age 30, is referred to our psychiatric outpatient clinic for follow-up care after hospitalization to treat a psychotic episode. His psychotic symptoms started 2 years ago without an identifiable trigger. Mr. F complains of episodic mood symptoms, such as depression, irritability, and angry outbursts; persistent auditory hallucinations (voices calling him names); and persecutory delusions. While in the hospital he was diagnosed with psychotic disorder not otherwise specified and started on olanzapine titrated to 30 mg/d.
During evaluation, Mr. F is depressed and exhibits motor retardation, slow speech, bland affect, impaired short-term memory, and auditory hallucinations. He describes social anxiety and has ideas of reference and problems interpreting facial expressions. He is guarded and suspicious. Although auditory hallucinations and depression affect Mr. F’s daily activities, he is attempting to find a job.
Mr. F has used alcohol since age 16 to escape social difficulties. He says he last used alcohol 1 year ago, but refuses to provide details about how much alcohol he typically consumed. Sporadic cannabis use also started when Mr. F was in his teens.
Mr. F’s symptoms improve with olanzapine, but he complains of weight gain and sedation, so we switch him to aripiprazole, 10 mg/d. Two weeks later he reports feeling jittery and anxious, so we discontinue aripiprazole and start loxapine, 25 mg/d at night, and propranolol, 60 mg/d, for residual akathisia. Despite limited clinical improvement, Mr. F irrationally says he wants to join the Navy. After a week, his psychotic symptoms improve but anxiety persists, so we start clonazepam, 1 mg/d, and oxcarbazepine, 600 mg/d. After 2 weeks he says he feels calmer, but has gained 20 lbs and is constantly tired. Against our advice, Mr. F decides to discontinue loxapine and propranolol, but continues clonazepam and oxcarbazepine.
At his next visit 4 weeks later, Mr. F is in good spirits. He says he is looking for a job as a dental assistant, and shows no apparent signs of psychosis. Mr. F misses his next appointment but returns 3 months later with evident deterioration in his general appearance. He says he is having difficulty sleeping and is depressed, stating “I just lay in bed; I don’t want to deal with life.” He is withdrawn and unwilling to elaborate on his personal problems but asks for a refill of clonazepam and oxcarbazepine, which we provide.
The authors’ observations
Sleep disturbances, including poor sleep efficiency, increased sleep-onset latency, decreased rapid eye movement (REM) sleep latency, and decreased stage 4 of non-REM sleep, occur in 16% to 30% of patients with schizophrenia and are associated with reduced quality of life and poor coping skills.1 Sleep-onset and sleep maintenance problems and sleep-wake reversal generally persist despite antipsychotic treatment.2,3
Slow-wave sleep deficiency can lead to negative symptoms and memory deficits in patients with schizophrenia because4:
- declarative and procedural memory consolidation are associated with slow-wave and stage 2 sleep, respectively
- procedural learning and visual spatial memory are correlated with delta power in slow-wave sleep.3,8
Acute psychosis exacerbations are associated with restless, agitated sleep. Insomnia often is an early warning sign of clinical relapse.5 The etiology of sleep dysfunction in schizophrenia is unknown, but glutamatergic action through N-methyl-d-aspartate receptors, the GABA system,6 and the serotonin system7 have been implicated.
Relapse to alcohol could trigger an exacerbation of Mr. F’s illness; however, he continues to deny alcohol or drug use and we could not identify any evidence of alcohol use at his last visit.
HISTORY: Strange behavior
Mr. F is a first-generation immigrant from Venezuela. He has a general educational development diploma and an associate’s degree. He says he has worked as a dental assistant but lost his job after a driving under the influence charge a year ago. Subsequently, he could not remain employed for long. He lives with his parents.
When Mr. F returns to the clinic 5 months later, he has lost 20 lbs and complains of anxiety and lack of sleep. With stooped posture, slow movements, and a mood-incongruent smile, he admits he ran out of medications and asks for refills, which we provide. He appears somewhat bizarre, wearing a loosely fitting baseball cap that covers his direct field of vision. Mr. F admits that he has been pulling out his hair. His thought process is impoverished and his answers are guarded and evasive. He rejects our recommendation of an antipsychotic; the only medications he is willing to continue are oxcarbazepine and clonazepam.
The authors’ observations
Treatment strategies for sleep disorders in patients with schizophrenia mainly target behavioral aspects of sleep, such as sleep onset and total sleep time, and rarely correct polysomnographic disturbances. Commonly used medications include atypical antipsychotics, benzodiazepines, zolpidem, zopiclone, and antidepressants with sedative properties (Table 1).1 However, new insights on sleep architecture patterns in these individuals have directed focus on other medications. Although antipsychotics, GABAA modulators, and melatonin provide some sleep benefits, none of these agents fully address characteristic sleep disturbances found in patients with schizophrenia.
Recent research has looked at GABAB modulators because of their unique function. GABAB receptors are located on pre-synaptic dopaminergic terminals and inhibit dopamine release and modulate glutamatergic regulation of dopamine. In the glutamate hypofunction model of psychosis, a GABAB agonist would cause disinhibition of glutamate modulation of mesolimbic dopamine and reversal of GABA transmission in the ventral tegmental area.9 Baclofen and γ-hydroxybutyric acid (GHB) currently are the only FDA-approved GABAB receptor agonists. Overall, trials of baclofen have not shown benefit for sleep disturbances in patients with schizophrenia,10,11 perhaps because of the drug’s poor liposolubility and consequent inability to cross the blood-brain barrier. Although hydrophilic like baclofen, GHB, which is also known as sodium oxybate and is FDA-approved for cataplexy due to narcolepsy, might have an advantage because of carrier-mediated transfer across the blood-brain barrier. GHB is thought to act directly as a neurotransmitter but also interacts with dopamine via the GHB receptor and with the GABAB receptor after it is converted to extracellular GABA.
Table 1
Schizophrenia and sleep dysfunction: The effect of psychotropics
| Medication/class | Comments |
|---|---|
| Atypical antipsychotics | In the CATIE study, a large proportion of patients had sleep problems despite antipsychotic treatment Atypicals may improve sleep acutely, but do not normalize it The long-term effects of atypicals on sleep architecture in schizophrenia are unclear; some studies show improved slow-wave sleep but in others slow-wave sleep is reduced |
| GABAA modulators (benzodiazepines, zolpidem, zopiclone) | Decrease sleep latency and nocturnal awakening Do not increase slow-wave sleep and overall sleep quality Decrease slow-wave sleep and REM sleep in rats May impair sleep architecture and cognition |
| Melatonin and modafinil | Melatonin may be useful for improving subjective sleep in patients with schizophrenia, although it does not improve slow-wave sleep parameters Modafinil may enhance cognition |
| GABAB receptor agonists | Few trials in humans but animal studies support a potential therapeutic role Minimal impact on REM sleep Increase slow-wave sleep Human studies with the GABAB agonist GHB show improvement in sleep architecture and subjective sleep |
| CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; GHB: γ-hydroxybutyric acid; REM: rapid eye movement | |
| Source:Reference 1 | |
OUTCOME: A trip cut short
Mr. F does not return to the clinic as scheduled, but 2 months later the U.S. consulate of a Western European country contacts us because Mr. F had a bottle of oxcarbazepine with our contact information. After Mr. F returns to the United States, he tells us his story.
After his last outpatient visit, Mr. F relapsed on alcohol, became despondent over his weakness, and searched for a way to escape his alcohol cravings. He came up with a plan to relocate to an Islamic Middle Eastern country where alcohol is banned and its use heavily punished. Mr. F bought a one-way airplane ticket through a Western Europe connection and departed 7 days later without notifying his family or psychiatrist.
Mr. F’s flight to Western Europe was uneventful. After landing for a connecting flight, his mood improved, his outlook became hopeful, and his auditory hallucinations changed from derogatory to supportive. However, Mr. F became despondent after being barred from his next flight because he did not have a return ticket. He was stranded in the airport with little money and no extra clothing, only his passport and laptop. He slept in the airport and after 3 days set off into the city. Mr. F navigated subway stations, ate at soup kitchens, and sought shelter in hotel lobbies and churches. One week after Mr. F left the airport, the police detained him for disorganized behavior and refusing to vacate a church. He was transported to a hospital, admitted to the psychiatric unit for catatonia, and stabilized on olanzapine, 20 mg/d.
After 1 week, Mr. F was returned to the United States and hospitalized for further evaluation and treatment. On his first day back, Mr. F’s disorganized process appeared to improve. He was euthymic and reported good sleep, tolerable anxiety, and infrequent derogatory auditory hallucinations that were low in volume. On day 3, Mr. F’s mood deteriorated moderately. He became depressed and again experienced derogatory auditory hallucinations. He was internally preoccupied and showed reduced affect and psychomotor activity. Mr. F was discharged the next day to a state-run respite program with a structured plan for psychiatric follow-up, social services, and sobriety maintenance. He remained on olanzapine, 20 mg/d, because we anticipated he would need an adjustment period after his uncommon journey.
The authors’ observations
Psychotic symptoms occurring during long-distance trips have been well described in psychiatric literature. Westbound travel could exacerbate depression. Emerging mania has been documented in eastbound flights, which could be related to sleep deprivation.12,13 The incidence of psychotic exacerbations is correlated with the number of time zones crossed.12
A change in environment, unfamiliar surroundings, presence of strangers, physical inactivity, and a sense of isolation all contribute to jet lag syndrome. Long-distance air travel also disrupts zeitgebers, environmental cues that induce adjustments in the internal body clock.12,14 The body clock is controlled mainly by the SCN in the hypothalamus, which is primarily regulated by the light/dark cycle via melatonin secretion (Figure).
Endogenous changes in circadian rhythms and melatonin secretion abnormalities are present in the pathophysiological mechanism of several psychiatric disorders, including depression, bipolar disorder, and schizophrenia. Trbovic hypothesized that in essence schizophrenia could be a sleep disorder and SCN dysfunction may contribute to the pathogenesis of schizophrenia.15 Several research findings support this hypothesis (Table 2). Recent evidence suggests that abnormal circadian melatonin metabolism may be directly related to the schizophrenia pathophysiology.16 Because melatonin production is regulated by the SCN and jet lag resets the melatonin cycle, a defective SCN may not respond well to such adjustments.
Mr. F’s symptomatology is illustrative of the jet lag scenario. His auditory hallucinations became “more supportive” and helpful during his eastbound flight, whereas after his return to the United States, depression was the predominant mood symptom. Psychotic exacerbation also was noticeable after his return.
There are no recommended treatments for psychosis related to jet lag. Antipsychotics often are used, although there is no accepted agent of choice. Treatment of jet lag includes addressing sleep loss and desynchronization.17 Medications suggested for treatment of sleep loss are antihistamines (H1 receptor antagonists), benzodiazepines, and imidazopyridines (zolpidem, zopiclone). Light therapy or administration of melatonin, ramelteon, or agomelatine can help jet-lagged patients resynchronize with the environment.
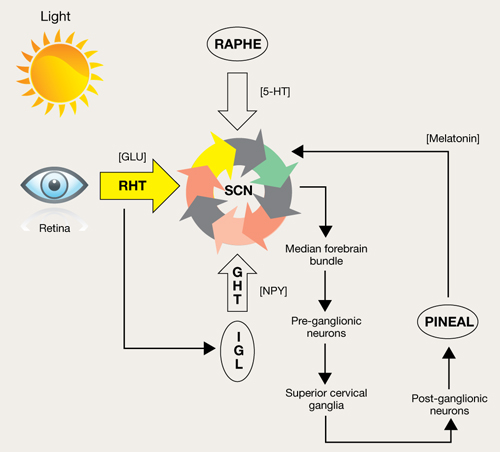
Figure: Pathways for light: Circadian timing system
Photic information reaches the suprachiasmatic nucleus (SCN) through the retinohypothalamic tract (RHT), which uses glutamate (GLU) as a neurotransmitter. A multisynaptic indirect pathway also carries photic information to the SCN. This indirect route arises from the RHT, projects through the intergeniculate leaflet (IGL) of the lateral geniculate nucleus, and finally, the geniculohypothalamic tract (GHT). Neuropeptide Y (NPY) is the neurotransmitter of the GHT. Serotoninergic (5-HT) input to the SCN arrives from the dorsal raphe nuclei. Melatonin, produced in the pineal gland, exerts its effect on circadian timing by feeding back onto the SCN.
Source: Reprinted with permission from reference 14Table 2
Suprachiasmatic nucleus dysfunction may have a role in schizophrenia
| Consequences of SCN dysfunction | Findings relevant to schizophrenia |
|---|---|
| Circadian pattern abnormalities | Individuals with schizophrenia do not have a characteristic circadian pattern of melatonin secretiona Actigraphic studies confirm that patients with schizophrenia have abnormal circadian rhythm activitiesb-d |
| Dopaminergic system abnormalities | The fetal dopaminergic system and D1 dopamine receptors may be involved in the process of synchronizing the SCNe,f |
| Jet lag symptomatology | Jet lag can exacerbate psychiatric disorders,g which suggests that in these patients the SCN is not capable of adjustment |
| Pathologic daytime sleep | Saccadic eye movements in patients with schizophrenia suggest they may be experiencing remnants of REM sleep, supporting the notion that these patients may have dream states during wakefulness |
| REM: rapid eye movement; SCN: suprachiasmatic nucleus | |
| Source: a. Bersani G, Mameli M, Garavini A, et al. Reduction of night/day difference in melatonin blood levels as a possible disease-related index in schizophrenia. Neuro Endocrinol Lett. 2003;24(3-4):181-184. b. Poyurovsky M, Nave R, Epstein R, et al. Actigraphic monitoring (actigraphy) of circadian locomotor activity in schizophrenic patients with acute neuroleptic-induced akathisia. Eur Neuropsychopharmacol. 2000;10(3):171-176. c. Haug HJ, Wirz-Justice A, Rössler W. Actigraphy to measure day structure as a therapeutic variable in the treatment of schizophrenic patients. Acta Psychiatr Scand Suppl. 2000;(407):91-95. d. Martin JL, Jeste DV, Ancoli-Israel S. Older schizophrenia patients have more disrupted sleep and circadian rhythms than age-matched comparison subjects. J Psychiatr Res. 2005;39(3):251-259. e. Strother WN, Norman AB, Lehman MN. D1-dopamine receptor binding and tyrosine hydroxylase-immunoreactivity in the fetal and neonatal hamster suprachiasmatic nucleus. Brain Res Dev Brain Res. 1998;106(1-2):137-144. f. Viswanathan N, Weaver DR, Reppert SM, et al. Entrainment of the fetal hamster circadian pacemaker by prenatal injections of the dopamine agonist SKF 38393. J Neurosci. 1994;14(9):5393-5398. g. Katz G, Durst R, Zislin J, et al. Jet lag causing or exacerbating psychiatric disorders. Harefuah. 2000;138(10):809-812, 912. | |
Related Resources
- Klein DC, Moore R, Reppert SM, eds. Suprachiasmatic nucleus: the mind’s clock. New York, NY: Oxford University Press; 1991.
- Hofstetter JR, Lysaker PH, Mayeda AR. Quality of sleep in patients with schizophrenia is associated with quality of life and coping. BMC Psychiatry. 2005;5:13.
Drug Brand Names
- Agomelatine • Valdoxan
- Aripiprazole • Abilify
- Baclofen • Lioresal
- Clonazepam • Klonopin
- γ-hydroxybutyric acid, sodium oxybate • Xyrem
- Loxapine • Loxitane
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Propranolol • Inderal
- Ramelteon • Rozerem
- Zolpidem • Ambien
- Zoplicone • Lunesta
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kantrowitz J, Citrome L, Javitt D. GABA(B) receptors, schizophrenia and sleep dysfunction: a review of the relationship and its potential clinical and therapeutic implications. CNS Drugs. 2009;23(8):631-669.
2. Poulin J, Daoust AM, Forest G, et al. Sleep architecture and its clinical correlates in first episode and neuroleptic-naïve patients with schizophrenia. Schizophr Res. 2003;62:147-153.
3. Ferrarelli F, Huber R, Peterson MJ. Reduced sleep spindle activity in schizophrenia patients. Am J Psychiatry. 2007;164(3):483-492.
4. Cohrs S. Sleep disturbances in patients with schizophrenia: impact and effect of antipsychotics. CNS Drugs. 2008;22(11):939-962.
5. Haffmans P, Hoencamp E, Knegtering HJ, et al. Sleep disturbance in schizophrenia. Br J Psychiatry. 1994;165(5):697-698.
6. Wisor J, Morairty S, Huynh N, et al. Gene expression in the rat cerebral cortex: comparison of recovery sleep and hypnotic-induced sleep. Neuroscience. 2006;141(1):371-378.
7. Benson KL, Faull KF, Zarcone VP. Evidence for the role of serotonin in the regulation of slow wave sleep in schizophrenia. Sleep. 1991;14(2):133-139.
8. Göder R, Boigs M, Braun S, et al. Impairment of visuospatial memory is associated with decreased slow wave sleep in schizophrenia. J Psychiatr Res. 2004;38:591-599.
9. Harte M, O’Connor WT. Evidence for a selective prefrontal cortical GABA(B) receptor mediated inhibition of glutamate release in the ventral tegmental area: a dual probe microdialysis study in the awake rat. Neuroscience. 2005;130(1):215-222.
10. Garbutt JC, van Kammen DP. The interaction between GABA and dopamine: implications for schizophrenia. Schizophr Bull. 1983;9(3):336-353.
11. Finnimore A, Roebuck M, Sajkov D, et al. The effects of the GABA agonist, baclofen, on sleep and breathing. Eur Respir J. 1995;8(2):230-234.
12. Jahuar P, Weller MP. Psychiatric morbidity and time zone changes: a study of patients from Heathrow airport. Br J Psychiatry. 1982;140:231-234.
13. Katz G, Durst R, Zislin Y, et al. Psychiatric aspects of jet lag: review and hypothesis. Med Hypotheses. 2001;56(1):20-23.
14. Waterhouse J, Reilly T, Atkinson G. Jet-lag. Lancet. 1997;350:1611-1616.
15. Trbovic SM. Schizophrenia as a possible dysfunction of the suprachiasmatic nucleus. Med Hypotheses. 2010;74:127-131.
16. Bersani G, Mameli M, Garavini A, et al. Reduction of night/ day difference in melatonin blood levels as a possible disease-related index in schizophrenia. Nuero Endocrinol Lett. 2003;24(3-4):181-184.
17. Brown GM, Pandi-Perumal SR, Trakht I, et al. Melatonin and its relevance to jet lag. Travel Med Infect Dis. 2009;7:69-81.
CASE: Psychotic and sleepless
Mr. F, age 30, is referred to our psychiatric outpatient clinic for follow-up care after hospitalization to treat a psychotic episode. His psychotic symptoms started 2 years ago without an identifiable trigger. Mr. F complains of episodic mood symptoms, such as depression, irritability, and angry outbursts; persistent auditory hallucinations (voices calling him names); and persecutory delusions. While in the hospital he was diagnosed with psychotic disorder not otherwise specified and started on olanzapine titrated to 30 mg/d.
During evaluation, Mr. F is depressed and exhibits motor retardation, slow speech, bland affect, impaired short-term memory, and auditory hallucinations. He describes social anxiety and has ideas of reference and problems interpreting facial expressions. He is guarded and suspicious. Although auditory hallucinations and depression affect Mr. F’s daily activities, he is attempting to find a job.
Mr. F has used alcohol since age 16 to escape social difficulties. He says he last used alcohol 1 year ago, but refuses to provide details about how much alcohol he typically consumed. Sporadic cannabis use also started when Mr. F was in his teens.
Mr. F’s symptoms improve with olanzapine, but he complains of weight gain and sedation, so we switch him to aripiprazole, 10 mg/d. Two weeks later he reports feeling jittery and anxious, so we discontinue aripiprazole and start loxapine, 25 mg/d at night, and propranolol, 60 mg/d, for residual akathisia. Despite limited clinical improvement, Mr. F irrationally says he wants to join the Navy. After a week, his psychotic symptoms improve but anxiety persists, so we start clonazepam, 1 mg/d, and oxcarbazepine, 600 mg/d. After 2 weeks he says he feels calmer, but has gained 20 lbs and is constantly tired. Against our advice, Mr. F decides to discontinue loxapine and propranolol, but continues clonazepam and oxcarbazepine.
At his next visit 4 weeks later, Mr. F is in good spirits. He says he is looking for a job as a dental assistant, and shows no apparent signs of psychosis. Mr. F misses his next appointment but returns 3 months later with evident deterioration in his general appearance. He says he is having difficulty sleeping and is depressed, stating “I just lay in bed; I don’t want to deal with life.” He is withdrawn and unwilling to elaborate on his personal problems but asks for a refill of clonazepam and oxcarbazepine, which we provide.
The authors’ observations
Sleep disturbances, including poor sleep efficiency, increased sleep-onset latency, decreased rapid eye movement (REM) sleep latency, and decreased stage 4 of non-REM sleep, occur in 16% to 30% of patients with schizophrenia and are associated with reduced quality of life and poor coping skills.1 Sleep-onset and sleep maintenance problems and sleep-wake reversal generally persist despite antipsychotic treatment.2,3
Slow-wave sleep deficiency can lead to negative symptoms and memory deficits in patients with schizophrenia because4:
- declarative and procedural memory consolidation are associated with slow-wave and stage 2 sleep, respectively
- procedural learning and visual spatial memory are correlated with delta power in slow-wave sleep.3,8
Acute psychosis exacerbations are associated with restless, agitated sleep. Insomnia often is an early warning sign of clinical relapse.5 The etiology of sleep dysfunction in schizophrenia is unknown, but glutamatergic action through N-methyl-d-aspartate receptors, the GABA system,6 and the serotonin system7 have been implicated.
Relapse to alcohol could trigger an exacerbation of Mr. F’s illness; however, he continues to deny alcohol or drug use and we could not identify any evidence of alcohol use at his last visit.
HISTORY: Strange behavior
Mr. F is a first-generation immigrant from Venezuela. He has a general educational development diploma and an associate’s degree. He says he has worked as a dental assistant but lost his job after a driving under the influence charge a year ago. Subsequently, he could not remain employed for long. He lives with his parents.
When Mr. F returns to the clinic 5 months later, he has lost 20 lbs and complains of anxiety and lack of sleep. With stooped posture, slow movements, and a mood-incongruent smile, he admits he ran out of medications and asks for refills, which we provide. He appears somewhat bizarre, wearing a loosely fitting baseball cap that covers his direct field of vision. Mr. F admits that he has been pulling out his hair. His thought process is impoverished and his answers are guarded and evasive. He rejects our recommendation of an antipsychotic; the only medications he is willing to continue are oxcarbazepine and clonazepam.
The authors’ observations
Treatment strategies for sleep disorders in patients with schizophrenia mainly target behavioral aspects of sleep, such as sleep onset and total sleep time, and rarely correct polysomnographic disturbances. Commonly used medications include atypical antipsychotics, benzodiazepines, zolpidem, zopiclone, and antidepressants with sedative properties (Table 1).1 However, new insights on sleep architecture patterns in these individuals have directed focus on other medications. Although antipsychotics, GABAA modulators, and melatonin provide some sleep benefits, none of these agents fully address characteristic sleep disturbances found in patients with schizophrenia.
Recent research has looked at GABAB modulators because of their unique function. GABAB receptors are located on pre-synaptic dopaminergic terminals and inhibit dopamine release and modulate glutamatergic regulation of dopamine. In the glutamate hypofunction model of psychosis, a GABAB agonist would cause disinhibition of glutamate modulation of mesolimbic dopamine and reversal of GABA transmission in the ventral tegmental area.9 Baclofen and γ-hydroxybutyric acid (GHB) currently are the only FDA-approved GABAB receptor agonists. Overall, trials of baclofen have not shown benefit for sleep disturbances in patients with schizophrenia,10,11 perhaps because of the drug’s poor liposolubility and consequent inability to cross the blood-brain barrier. Although hydrophilic like baclofen, GHB, which is also known as sodium oxybate and is FDA-approved for cataplexy due to narcolepsy, might have an advantage because of carrier-mediated transfer across the blood-brain barrier. GHB is thought to act directly as a neurotransmitter but also interacts with dopamine via the GHB receptor and with the GABAB receptor after it is converted to extracellular GABA.
Table 1
Schizophrenia and sleep dysfunction: The effect of psychotropics
| Medication/class | Comments |
|---|---|
| Atypical antipsychotics | In the CATIE study, a large proportion of patients had sleep problems despite antipsychotic treatment Atypicals may improve sleep acutely, but do not normalize it The long-term effects of atypicals on sleep architecture in schizophrenia are unclear; some studies show improved slow-wave sleep but in others slow-wave sleep is reduced |
| GABAA modulators (benzodiazepines, zolpidem, zopiclone) | Decrease sleep latency and nocturnal awakening Do not increase slow-wave sleep and overall sleep quality Decrease slow-wave sleep and REM sleep in rats May impair sleep architecture and cognition |
| Melatonin and modafinil | Melatonin may be useful for improving subjective sleep in patients with schizophrenia, although it does not improve slow-wave sleep parameters Modafinil may enhance cognition |
| GABAB receptor agonists | Few trials in humans but animal studies support a potential therapeutic role Minimal impact on REM sleep Increase slow-wave sleep Human studies with the GABAB agonist GHB show improvement in sleep architecture and subjective sleep |
| CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; GHB: γ-hydroxybutyric acid; REM: rapid eye movement | |
| Source:Reference 1 | |
OUTCOME: A trip cut short
Mr. F does not return to the clinic as scheduled, but 2 months later the U.S. consulate of a Western European country contacts us because Mr. F had a bottle of oxcarbazepine with our contact information. After Mr. F returns to the United States, he tells us his story.
After his last outpatient visit, Mr. F relapsed on alcohol, became despondent over his weakness, and searched for a way to escape his alcohol cravings. He came up with a plan to relocate to an Islamic Middle Eastern country where alcohol is banned and its use heavily punished. Mr. F bought a one-way airplane ticket through a Western Europe connection and departed 7 days later without notifying his family or psychiatrist.
Mr. F’s flight to Western Europe was uneventful. After landing for a connecting flight, his mood improved, his outlook became hopeful, and his auditory hallucinations changed from derogatory to supportive. However, Mr. F became despondent after being barred from his next flight because he did not have a return ticket. He was stranded in the airport with little money and no extra clothing, only his passport and laptop. He slept in the airport and after 3 days set off into the city. Mr. F navigated subway stations, ate at soup kitchens, and sought shelter in hotel lobbies and churches. One week after Mr. F left the airport, the police detained him for disorganized behavior and refusing to vacate a church. He was transported to a hospital, admitted to the psychiatric unit for catatonia, and stabilized on olanzapine, 20 mg/d.
After 1 week, Mr. F was returned to the United States and hospitalized for further evaluation and treatment. On his first day back, Mr. F’s disorganized process appeared to improve. He was euthymic and reported good sleep, tolerable anxiety, and infrequent derogatory auditory hallucinations that were low in volume. On day 3, Mr. F’s mood deteriorated moderately. He became depressed and again experienced derogatory auditory hallucinations. He was internally preoccupied and showed reduced affect and psychomotor activity. Mr. F was discharged the next day to a state-run respite program with a structured plan for psychiatric follow-up, social services, and sobriety maintenance. He remained on olanzapine, 20 mg/d, because we anticipated he would need an adjustment period after his uncommon journey.
The authors’ observations
Psychotic symptoms occurring during long-distance trips have been well described in psychiatric literature. Westbound travel could exacerbate depression. Emerging mania has been documented in eastbound flights, which could be related to sleep deprivation.12,13 The incidence of psychotic exacerbations is correlated with the number of time zones crossed.12
A change in environment, unfamiliar surroundings, presence of strangers, physical inactivity, and a sense of isolation all contribute to jet lag syndrome. Long-distance air travel also disrupts zeitgebers, environmental cues that induce adjustments in the internal body clock.12,14 The body clock is controlled mainly by the SCN in the hypothalamus, which is primarily regulated by the light/dark cycle via melatonin secretion (Figure).
Endogenous changes in circadian rhythms and melatonin secretion abnormalities are present in the pathophysiological mechanism of several psychiatric disorders, including depression, bipolar disorder, and schizophrenia. Trbovic hypothesized that in essence schizophrenia could be a sleep disorder and SCN dysfunction may contribute to the pathogenesis of schizophrenia.15 Several research findings support this hypothesis (Table 2). Recent evidence suggests that abnormal circadian melatonin metabolism may be directly related to the schizophrenia pathophysiology.16 Because melatonin production is regulated by the SCN and jet lag resets the melatonin cycle, a defective SCN may not respond well to such adjustments.
Mr. F’s symptomatology is illustrative of the jet lag scenario. His auditory hallucinations became “more supportive” and helpful during his eastbound flight, whereas after his return to the United States, depression was the predominant mood symptom. Psychotic exacerbation also was noticeable after his return.
There are no recommended treatments for psychosis related to jet lag. Antipsychotics often are used, although there is no accepted agent of choice. Treatment of jet lag includes addressing sleep loss and desynchronization.17 Medications suggested for treatment of sleep loss are antihistamines (H1 receptor antagonists), benzodiazepines, and imidazopyridines (zolpidem, zopiclone). Light therapy or administration of melatonin, ramelteon, or agomelatine can help jet-lagged patients resynchronize with the environment.
Figure: Pathways for light: Circadian timing system
Photic information reaches the suprachiasmatic nucleus (SCN) through the retinohypothalamic tract (RHT), which uses glutamate (GLU) as a neurotransmitter. A multisynaptic indirect pathway also carries photic information to the SCN. This indirect route arises from the RHT, projects through the intergeniculate leaflet (IGL) of the lateral geniculate nucleus, and finally, the geniculohypothalamic tract (GHT). Neuropeptide Y (NPY) is the neurotransmitter of the GHT. Serotoninergic (5-HT) input to the SCN arrives from the dorsal raphe nuclei. Melatonin, produced in the pineal gland, exerts its effect on circadian timing by feeding back onto the SCN.
Source: Reprinted with permission from reference 14Table 2
Suprachiasmatic nucleus dysfunction may have a role in schizophrenia
| Consequences of SCN dysfunction | Findings relevant to schizophrenia |
|---|---|
| Circadian pattern abnormalities | Individuals with schizophrenia do not have a characteristic circadian pattern of melatonin secretiona Actigraphic studies confirm that patients with schizophrenia have abnormal circadian rhythm activitiesb-d |
| Dopaminergic system abnormalities | The fetal dopaminergic system and D1 dopamine receptors may be involved in the process of synchronizing the SCNe,f |
| Jet lag symptomatology | Jet lag can exacerbate psychiatric disorders,g which suggests that in these patients the SCN is not capable of adjustment |
| Pathologic daytime sleep | Saccadic eye movements in patients with schizophrenia suggest they may be experiencing remnants of REM sleep, supporting the notion that these patients may have dream states during wakefulness |
| REM: rapid eye movement; SCN: suprachiasmatic nucleus | |
| Source: a. Bersani G, Mameli M, Garavini A, et al. Reduction of night/day difference in melatonin blood levels as a possible disease-related index in schizophrenia. Neuro Endocrinol Lett. 2003;24(3-4):181-184. b. Poyurovsky M, Nave R, Epstein R, et al. Actigraphic monitoring (actigraphy) of circadian locomotor activity in schizophrenic patients with acute neuroleptic-induced akathisia. Eur Neuropsychopharmacol. 2000;10(3):171-176. c. Haug HJ, Wirz-Justice A, Rössler W. Actigraphy to measure day structure as a therapeutic variable in the treatment of schizophrenic patients. Acta Psychiatr Scand Suppl. 2000;(407):91-95. d. Martin JL, Jeste DV, Ancoli-Israel S. Older schizophrenia patients have more disrupted sleep and circadian rhythms than age-matched comparison subjects. J Psychiatr Res. 2005;39(3):251-259. e. Strother WN, Norman AB, Lehman MN. D1-dopamine receptor binding and tyrosine hydroxylase-immunoreactivity in the fetal and neonatal hamster suprachiasmatic nucleus. Brain Res Dev Brain Res. 1998;106(1-2):137-144. f. Viswanathan N, Weaver DR, Reppert SM, et al. Entrainment of the fetal hamster circadian pacemaker by prenatal injections of the dopamine agonist SKF 38393. J Neurosci. 1994;14(9):5393-5398. g. Katz G, Durst R, Zislin J, et al. Jet lag causing or exacerbating psychiatric disorders. Harefuah. 2000;138(10):809-812, 912. | |
Related Resources
- Klein DC, Moore R, Reppert SM, eds. Suprachiasmatic nucleus: the mind’s clock. New York, NY: Oxford University Press; 1991.
- Hofstetter JR, Lysaker PH, Mayeda AR. Quality of sleep in patients with schizophrenia is associated with quality of life and coping. BMC Psychiatry. 2005;5:13.
Drug Brand Names
- Agomelatine • Valdoxan
- Aripiprazole • Abilify
- Baclofen • Lioresal
- Clonazepam • Klonopin
- γ-hydroxybutyric acid, sodium oxybate • Xyrem
- Loxapine • Loxitane
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Propranolol • Inderal
- Ramelteon • Rozerem
- Zolpidem • Ambien
- Zoplicone • Lunesta
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Psychotic and sleepless
Mr. F, age 30, is referred to our psychiatric outpatient clinic for follow-up care after hospitalization to treat a psychotic episode. His psychotic symptoms started 2 years ago without an identifiable trigger. Mr. F complains of episodic mood symptoms, such as depression, irritability, and angry outbursts; persistent auditory hallucinations (voices calling him names); and persecutory delusions. While in the hospital he was diagnosed with psychotic disorder not otherwise specified and started on olanzapine titrated to 30 mg/d.
During evaluation, Mr. F is depressed and exhibits motor retardation, slow speech, bland affect, impaired short-term memory, and auditory hallucinations. He describes social anxiety and has ideas of reference and problems interpreting facial expressions. He is guarded and suspicious. Although auditory hallucinations and depression affect Mr. F’s daily activities, he is attempting to find a job.
Mr. F has used alcohol since age 16 to escape social difficulties. He says he last used alcohol 1 year ago, but refuses to provide details about how much alcohol he typically consumed. Sporadic cannabis use also started when Mr. F was in his teens.
Mr. F’s symptoms improve with olanzapine, but he complains of weight gain and sedation, so we switch him to aripiprazole, 10 mg/d. Two weeks later he reports feeling jittery and anxious, so we discontinue aripiprazole and start loxapine, 25 mg/d at night, and propranolol, 60 mg/d, for residual akathisia. Despite limited clinical improvement, Mr. F irrationally says he wants to join the Navy. After a week, his psychotic symptoms improve but anxiety persists, so we start clonazepam, 1 mg/d, and oxcarbazepine, 600 mg/d. After 2 weeks he says he feels calmer, but has gained 20 lbs and is constantly tired. Against our advice, Mr. F decides to discontinue loxapine and propranolol, but continues clonazepam and oxcarbazepine.
At his next visit 4 weeks later, Mr. F is in good spirits. He says he is looking for a job as a dental assistant, and shows no apparent signs of psychosis. Mr. F misses his next appointment but returns 3 months later with evident deterioration in his general appearance. He says he is having difficulty sleeping and is depressed, stating “I just lay in bed; I don’t want to deal with life.” He is withdrawn and unwilling to elaborate on his personal problems but asks for a refill of clonazepam and oxcarbazepine, which we provide.
The authors’ observations
Sleep disturbances, including poor sleep efficiency, increased sleep-onset latency, decreased rapid eye movement (REM) sleep latency, and decreased stage 4 of non-REM sleep, occur in 16% to 30% of patients with schizophrenia and are associated with reduced quality of life and poor coping skills.1 Sleep-onset and sleep maintenance problems and sleep-wake reversal generally persist despite antipsychotic treatment.2,3
Slow-wave sleep deficiency can lead to negative symptoms and memory deficits in patients with schizophrenia because4:
- declarative and procedural memory consolidation are associated with slow-wave and stage 2 sleep, respectively
- procedural learning and visual spatial memory are correlated with delta power in slow-wave sleep.3,8
Acute psychosis exacerbations are associated with restless, agitated sleep. Insomnia often is an early warning sign of clinical relapse.5 The etiology of sleep dysfunction in schizophrenia is unknown, but glutamatergic action through N-methyl-d-aspartate receptors, the GABA system,6 and the serotonin system7 have been implicated.
Relapse to alcohol could trigger an exacerbation of Mr. F’s illness; however, he continues to deny alcohol or drug use and we could not identify any evidence of alcohol use at his last visit.
HISTORY: Strange behavior
Mr. F is a first-generation immigrant from Venezuela. He has a general educational development diploma and an associate’s degree. He says he has worked as a dental assistant but lost his job after a driving under the influence charge a year ago. Subsequently, he could not remain employed for long. He lives with his parents.
When Mr. F returns to the clinic 5 months later, he has lost 20 lbs and complains of anxiety and lack of sleep. With stooped posture, slow movements, and a mood-incongruent smile, he admits he ran out of medications and asks for refills, which we provide. He appears somewhat bizarre, wearing a loosely fitting baseball cap that covers his direct field of vision. Mr. F admits that he has been pulling out his hair. His thought process is impoverished and his answers are guarded and evasive. He rejects our recommendation of an antipsychotic; the only medications he is willing to continue are oxcarbazepine and clonazepam.
The authors’ observations
Treatment strategies for sleep disorders in patients with schizophrenia mainly target behavioral aspects of sleep, such as sleep onset and total sleep time, and rarely correct polysomnographic disturbances. Commonly used medications include atypical antipsychotics, benzodiazepines, zolpidem, zopiclone, and antidepressants with sedative properties (Table 1).1 However, new insights on sleep architecture patterns in these individuals have directed focus on other medications. Although antipsychotics, GABAA modulators, and melatonin provide some sleep benefits, none of these agents fully address characteristic sleep disturbances found in patients with schizophrenia.
Recent research has looked at GABAB modulators because of their unique function. GABAB receptors are located on pre-synaptic dopaminergic terminals and inhibit dopamine release and modulate glutamatergic regulation of dopamine. In the glutamate hypofunction model of psychosis, a GABAB agonist would cause disinhibition of glutamate modulation of mesolimbic dopamine and reversal of GABA transmission in the ventral tegmental area.9 Baclofen and γ-hydroxybutyric acid (GHB) currently are the only FDA-approved GABAB receptor agonists. Overall, trials of baclofen have not shown benefit for sleep disturbances in patients with schizophrenia,10,11 perhaps because of the drug’s poor liposolubility and consequent inability to cross the blood-brain barrier. Although hydrophilic like baclofen, GHB, which is also known as sodium oxybate and is FDA-approved for cataplexy due to narcolepsy, might have an advantage because of carrier-mediated transfer across the blood-brain barrier. GHB is thought to act directly as a neurotransmitter but also interacts with dopamine via the GHB receptor and with the GABAB receptor after it is converted to extracellular GABA.
Table 1
Schizophrenia and sleep dysfunction: The effect of psychotropics
| Medication/class | Comments |
|---|---|
| Atypical antipsychotics | In the CATIE study, a large proportion of patients had sleep problems despite antipsychotic treatment Atypicals may improve sleep acutely, but do not normalize it The long-term effects of atypicals on sleep architecture in schizophrenia are unclear; some studies show improved slow-wave sleep but in others slow-wave sleep is reduced |
| GABAA modulators (benzodiazepines, zolpidem, zopiclone) | Decrease sleep latency and nocturnal awakening Do not increase slow-wave sleep and overall sleep quality Decrease slow-wave sleep and REM sleep in rats May impair sleep architecture and cognition |
| Melatonin and modafinil | Melatonin may be useful for improving subjective sleep in patients with schizophrenia, although it does not improve slow-wave sleep parameters Modafinil may enhance cognition |
| GABAB receptor agonists | Few trials in humans but animal studies support a potential therapeutic role Minimal impact on REM sleep Increase slow-wave sleep Human studies with the GABAB agonist GHB show improvement in sleep architecture and subjective sleep |
| CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; GHB: γ-hydroxybutyric acid; REM: rapid eye movement | |
| Source:Reference 1 | |
OUTCOME: A trip cut short
Mr. F does not return to the clinic as scheduled, but 2 months later the U.S. consulate of a Western European country contacts us because Mr. F had a bottle of oxcarbazepine with our contact information. After Mr. F returns to the United States, he tells us his story.
After his last outpatient visit, Mr. F relapsed on alcohol, became despondent over his weakness, and searched for a way to escape his alcohol cravings. He came up with a plan to relocate to an Islamic Middle Eastern country where alcohol is banned and its use heavily punished. Mr. F bought a one-way airplane ticket through a Western Europe connection and departed 7 days later without notifying his family or psychiatrist.
Mr. F’s flight to Western Europe was uneventful. After landing for a connecting flight, his mood improved, his outlook became hopeful, and his auditory hallucinations changed from derogatory to supportive. However, Mr. F became despondent after being barred from his next flight because he did not have a return ticket. He was stranded in the airport with little money and no extra clothing, only his passport and laptop. He slept in the airport and after 3 days set off into the city. Mr. F navigated subway stations, ate at soup kitchens, and sought shelter in hotel lobbies and churches. One week after Mr. F left the airport, the police detained him for disorganized behavior and refusing to vacate a church. He was transported to a hospital, admitted to the psychiatric unit for catatonia, and stabilized on olanzapine, 20 mg/d.
After 1 week, Mr. F was returned to the United States and hospitalized for further evaluation and treatment. On his first day back, Mr. F’s disorganized process appeared to improve. He was euthymic and reported good sleep, tolerable anxiety, and infrequent derogatory auditory hallucinations that were low in volume. On day 3, Mr. F’s mood deteriorated moderately. He became depressed and again experienced derogatory auditory hallucinations. He was internally preoccupied and showed reduced affect and psychomotor activity. Mr. F was discharged the next day to a state-run respite program with a structured plan for psychiatric follow-up, social services, and sobriety maintenance. He remained on olanzapine, 20 mg/d, because we anticipated he would need an adjustment period after his uncommon journey.
The authors’ observations
Psychotic symptoms occurring during long-distance trips have been well described in psychiatric literature. Westbound travel could exacerbate depression. Emerging mania has been documented in eastbound flights, which could be related to sleep deprivation.12,13 The incidence of psychotic exacerbations is correlated with the number of time zones crossed.12
A change in environment, unfamiliar surroundings, presence of strangers, physical inactivity, and a sense of isolation all contribute to jet lag syndrome. Long-distance air travel also disrupts zeitgebers, environmental cues that induce adjustments in the internal body clock.12,14 The body clock is controlled mainly by the SCN in the hypothalamus, which is primarily regulated by the light/dark cycle via melatonin secretion (Figure).
Endogenous changes in circadian rhythms and melatonin secretion abnormalities are present in the pathophysiological mechanism of several psychiatric disorders, including depression, bipolar disorder, and schizophrenia. Trbovic hypothesized that in essence schizophrenia could be a sleep disorder and SCN dysfunction may contribute to the pathogenesis of schizophrenia.15 Several research findings support this hypothesis (Table 2). Recent evidence suggests that abnormal circadian melatonin metabolism may be directly related to the schizophrenia pathophysiology.16 Because melatonin production is regulated by the SCN and jet lag resets the melatonin cycle, a defective SCN may not respond well to such adjustments.
Mr. F’s symptomatology is illustrative of the jet lag scenario. His auditory hallucinations became “more supportive” and helpful during his eastbound flight, whereas after his return to the United States, depression was the predominant mood symptom. Psychotic exacerbation also was noticeable after his return.
There are no recommended treatments for psychosis related to jet lag. Antipsychotics often are used, although there is no accepted agent of choice. Treatment of jet lag includes addressing sleep loss and desynchronization.17 Medications suggested for treatment of sleep loss are antihistamines (H1 receptor antagonists), benzodiazepines, and imidazopyridines (zolpidem, zopiclone). Light therapy or administration of melatonin, ramelteon, or agomelatine can help jet-lagged patients resynchronize with the environment.
Figure: Pathways for light: Circadian timing system
Photic information reaches the suprachiasmatic nucleus (SCN) through the retinohypothalamic tract (RHT), which uses glutamate (GLU) as a neurotransmitter. A multisynaptic indirect pathway also carries photic information to the SCN. This indirect route arises from the RHT, projects through the intergeniculate leaflet (IGL) of the lateral geniculate nucleus, and finally, the geniculohypothalamic tract (GHT). Neuropeptide Y (NPY) is the neurotransmitter of the GHT. Serotoninergic (5-HT) input to the SCN arrives from the dorsal raphe nuclei. Melatonin, produced in the pineal gland, exerts its effect on circadian timing by feeding back onto the SCN.
Source: Reprinted with permission from reference 14Table 2
Suprachiasmatic nucleus dysfunction may have a role in schizophrenia
| Consequences of SCN dysfunction | Findings relevant to schizophrenia |
|---|---|
| Circadian pattern abnormalities | Individuals with schizophrenia do not have a characteristic circadian pattern of melatonin secretiona Actigraphic studies confirm that patients with schizophrenia have abnormal circadian rhythm activitiesb-d |
| Dopaminergic system abnormalities | The fetal dopaminergic system and D1 dopamine receptors may be involved in the process of synchronizing the SCNe,f |
| Jet lag symptomatology | Jet lag can exacerbate psychiatric disorders,g which suggests that in these patients the SCN is not capable of adjustment |
| Pathologic daytime sleep | Saccadic eye movements in patients with schizophrenia suggest they may be experiencing remnants of REM sleep, supporting the notion that these patients may have dream states during wakefulness |
| REM: rapid eye movement; SCN: suprachiasmatic nucleus | |
| Source: a. Bersani G, Mameli M, Garavini A, et al. Reduction of night/day difference in melatonin blood levels as a possible disease-related index in schizophrenia. Neuro Endocrinol Lett. 2003;24(3-4):181-184. b. Poyurovsky M, Nave R, Epstein R, et al. Actigraphic monitoring (actigraphy) of circadian locomotor activity in schizophrenic patients with acute neuroleptic-induced akathisia. Eur Neuropsychopharmacol. 2000;10(3):171-176. c. Haug HJ, Wirz-Justice A, Rössler W. Actigraphy to measure day structure as a therapeutic variable in the treatment of schizophrenic patients. Acta Psychiatr Scand Suppl. 2000;(407):91-95. d. Martin JL, Jeste DV, Ancoli-Israel S. Older schizophrenia patients have more disrupted sleep and circadian rhythms than age-matched comparison subjects. J Psychiatr Res. 2005;39(3):251-259. e. Strother WN, Norman AB, Lehman MN. D1-dopamine receptor binding and tyrosine hydroxylase-immunoreactivity in the fetal and neonatal hamster suprachiasmatic nucleus. Brain Res Dev Brain Res. 1998;106(1-2):137-144. f. Viswanathan N, Weaver DR, Reppert SM, et al. Entrainment of the fetal hamster circadian pacemaker by prenatal injections of the dopamine agonist SKF 38393. J Neurosci. 1994;14(9):5393-5398. g. Katz G, Durst R, Zislin J, et al. Jet lag causing or exacerbating psychiatric disorders. Harefuah. 2000;138(10):809-812, 912. | |
Related Resources
- Klein DC, Moore R, Reppert SM, eds. Suprachiasmatic nucleus: the mind’s clock. New York, NY: Oxford University Press; 1991.
- Hofstetter JR, Lysaker PH, Mayeda AR. Quality of sleep in patients with schizophrenia is associated with quality of life and coping. BMC Psychiatry. 2005;5:13.
Drug Brand Names
- Agomelatine • Valdoxan
- Aripiprazole • Abilify
- Baclofen • Lioresal
- Clonazepam • Klonopin
- γ-hydroxybutyric acid, sodium oxybate • Xyrem
- Loxapine • Loxitane
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Propranolol • Inderal
- Ramelteon • Rozerem
- Zolpidem • Ambien
- Zoplicone • Lunesta
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kantrowitz J, Citrome L, Javitt D. GABA(B) receptors, schizophrenia and sleep dysfunction: a review of the relationship and its potential clinical and therapeutic implications. CNS Drugs. 2009;23(8):631-669.
2. Poulin J, Daoust AM, Forest G, et al. Sleep architecture and its clinical correlates in first episode and neuroleptic-naïve patients with schizophrenia. Schizophr Res. 2003;62:147-153.
3. Ferrarelli F, Huber R, Peterson MJ. Reduced sleep spindle activity in schizophrenia patients. Am J Psychiatry. 2007;164(3):483-492.
4. Cohrs S. Sleep disturbances in patients with schizophrenia: impact and effect of antipsychotics. CNS Drugs. 2008;22(11):939-962.
5. Haffmans P, Hoencamp E, Knegtering HJ, et al. Sleep disturbance in schizophrenia. Br J Psychiatry. 1994;165(5):697-698.
6. Wisor J, Morairty S, Huynh N, et al. Gene expression in the rat cerebral cortex: comparison of recovery sleep and hypnotic-induced sleep. Neuroscience. 2006;141(1):371-378.
7. Benson KL, Faull KF, Zarcone VP. Evidence for the role of serotonin in the regulation of slow wave sleep in schizophrenia. Sleep. 1991;14(2):133-139.
8. Göder R, Boigs M, Braun S, et al. Impairment of visuospatial memory is associated with decreased slow wave sleep in schizophrenia. J Psychiatr Res. 2004;38:591-599.
9. Harte M, O’Connor WT. Evidence for a selective prefrontal cortical GABA(B) receptor mediated inhibition of glutamate release in the ventral tegmental area: a dual probe microdialysis study in the awake rat. Neuroscience. 2005;130(1):215-222.
10. Garbutt JC, van Kammen DP. The interaction between GABA and dopamine: implications for schizophrenia. Schizophr Bull. 1983;9(3):336-353.
11. Finnimore A, Roebuck M, Sajkov D, et al. The effects of the GABA agonist, baclofen, on sleep and breathing. Eur Respir J. 1995;8(2):230-234.
12. Jahuar P, Weller MP. Psychiatric morbidity and time zone changes: a study of patients from Heathrow airport. Br J Psychiatry. 1982;140:231-234.
13. Katz G, Durst R, Zislin Y, et al. Psychiatric aspects of jet lag: review and hypothesis. Med Hypotheses. 2001;56(1):20-23.
14. Waterhouse J, Reilly T, Atkinson G. Jet-lag. Lancet. 1997;350:1611-1616.
15. Trbovic SM. Schizophrenia as a possible dysfunction of the suprachiasmatic nucleus. Med Hypotheses. 2010;74:127-131.
16. Bersani G, Mameli M, Garavini A, et al. Reduction of night/ day difference in melatonin blood levels as a possible disease-related index in schizophrenia. Nuero Endocrinol Lett. 2003;24(3-4):181-184.
17. Brown GM, Pandi-Perumal SR, Trakht I, et al. Melatonin and its relevance to jet lag. Travel Med Infect Dis. 2009;7:69-81.
1. Kantrowitz J, Citrome L, Javitt D. GABA(B) receptors, schizophrenia and sleep dysfunction: a review of the relationship and its potential clinical and therapeutic implications. CNS Drugs. 2009;23(8):631-669.
2. Poulin J, Daoust AM, Forest G, et al. Sleep architecture and its clinical correlates in first episode and neuroleptic-naïve patients with schizophrenia. Schizophr Res. 2003;62:147-153.
3. Ferrarelli F, Huber R, Peterson MJ. Reduced sleep spindle activity in schizophrenia patients. Am J Psychiatry. 2007;164(3):483-492.
4. Cohrs S. Sleep disturbances in patients with schizophrenia: impact and effect of antipsychotics. CNS Drugs. 2008;22(11):939-962.
5. Haffmans P, Hoencamp E, Knegtering HJ, et al. Sleep disturbance in schizophrenia. Br J Psychiatry. 1994;165(5):697-698.
6. Wisor J, Morairty S, Huynh N, et al. Gene expression in the rat cerebral cortex: comparison of recovery sleep and hypnotic-induced sleep. Neuroscience. 2006;141(1):371-378.
7. Benson KL, Faull KF, Zarcone VP. Evidence for the role of serotonin in the regulation of slow wave sleep in schizophrenia. Sleep. 1991;14(2):133-139.
8. Göder R, Boigs M, Braun S, et al. Impairment of visuospatial memory is associated with decreased slow wave sleep in schizophrenia. J Psychiatr Res. 2004;38:591-599.
9. Harte M, O’Connor WT. Evidence for a selective prefrontal cortical GABA(B) receptor mediated inhibition of glutamate release in the ventral tegmental area: a dual probe microdialysis study in the awake rat. Neuroscience. 2005;130(1):215-222.
10. Garbutt JC, van Kammen DP. The interaction between GABA and dopamine: implications for schizophrenia. Schizophr Bull. 1983;9(3):336-353.
11. Finnimore A, Roebuck M, Sajkov D, et al. The effects of the GABA agonist, baclofen, on sleep and breathing. Eur Respir J. 1995;8(2):230-234.
12. Jahuar P, Weller MP. Psychiatric morbidity and time zone changes: a study of patients from Heathrow airport. Br J Psychiatry. 1982;140:231-234.
13. Katz G, Durst R, Zislin Y, et al. Psychiatric aspects of jet lag: review and hypothesis. Med Hypotheses. 2001;56(1):20-23.
14. Waterhouse J, Reilly T, Atkinson G. Jet-lag. Lancet. 1997;350:1611-1616.
15. Trbovic SM. Schizophrenia as a possible dysfunction of the suprachiasmatic nucleus. Med Hypotheses. 2010;74:127-131.
16. Bersani G, Mameli M, Garavini A, et al. Reduction of night/ day difference in melatonin blood levels as a possible disease-related index in schizophrenia. Nuero Endocrinol Lett. 2003;24(3-4):181-184.
17. Brown GM, Pandi-Perumal SR, Trakht I, et al. Melatonin and its relevance to jet lag. Travel Med Infect Dis. 2009;7:69-81.
Appreciating med checks
I read with great appreciation Dr. Douglas Mossman's column, "Successfully navigating the 15-minute 'med check'" (Malpractice Rx, Current Psychiatry, June 2010, p. 40-43). At first I thought Dr. Mossman believed this practice is inferior care, but I was grateful to see that this is not necessarily so. I have been working to make my med checks "strength-based" and therapeutic despite the brevity, and greatly admire my patients' tenacity despite their suffering. Thank you for legitimizing the work done by community psychiatrists.
Linda J. Griffith, MD
Chief Medical Officer
Consolidated Care, Inc
Assistant Clinical Professor
Department of Psychiatry
Wright State University
Boonshoft School of Medicine
Dayton, OH
I read with great appreciation Dr. Douglas Mossman's column, "Successfully navigating the 15-minute 'med check'" (Malpractice Rx, Current Psychiatry, June 2010, p. 40-43). At first I thought Dr. Mossman believed this practice is inferior care, but I was grateful to see that this is not necessarily so. I have been working to make my med checks "strength-based" and therapeutic despite the brevity, and greatly admire my patients' tenacity despite their suffering. Thank you for legitimizing the work done by community psychiatrists.
Linda J. Griffith, MD
Chief Medical Officer
Consolidated Care, Inc
Assistant Clinical Professor
Department of Psychiatry
Wright State University
Boonshoft School of Medicine
Dayton, OH
I read with great appreciation Dr. Douglas Mossman's column, "Successfully navigating the 15-minute 'med check'" (Malpractice Rx, Current Psychiatry, June 2010, p. 40-43). At first I thought Dr. Mossman believed this practice is inferior care, but I was grateful to see that this is not necessarily so. I have been working to make my med checks "strength-based" and therapeutic despite the brevity, and greatly admire my patients' tenacity despite their suffering. Thank you for legitimizing the work done by community psychiatrists.
Linda J. Griffith, MD
Chief Medical Officer
Consolidated Care, Inc
Assistant Clinical Professor
Department of Psychiatry
Wright State University
Boonshoft School of Medicine
Dayton, OH
Evaluating for conversion disorder: When to suspect Creutzfeldt-Jakob disease
Consider this rare disorder in patients with rapid-onset neurologic symptoms
Ms. J, age 63, is admitted to Neurology with progressive dizziness and cognitive impairment. She had developed word-finding difficulties, weakness, memory problems, and an episode of arm shaking, which prompted referral for inpatient workup. Ms. J has a history of hypertension, palpitations, and diabetes mellitus.
Her neurologic examination is variable; some examiners find pronounced aphasia and right-sided weakness, whereas others document a nearly normal examination. Lumbar puncture (LP) shows normal cell count, glucose, protein, and negative Gram’s stain; MRI of the brain is normal. Enterovirus polymerase chain reaction, cryptococcal antigen, and Lyme antibody are negative. Electroencephalography (EEG) demonstrates diffuse slowing. The primary team requests psychiatric consultation to assess for conversion disorder.
Ms. J is cooperative with psychiatric evaluation. She denies current or past psychiatric symptomatology and does not meet criteria for major depression, dysthymia, adjustment disorder, anxiety disorder, psychosis, or mania. She denies personal or family history of suicidal or homicidal ideation, intent, or plan. Her youngest son died 5 years earlier; she is financially secure and her 40-year marriage is stable. Ms. J denies having a history of substance use, physical or sexual abuse, or trauma.
In the Cardiology clinic 2 months ago, Ms. J denied having neurologic symptoms and was noted to be doing well. Her neurologic symptoms began shortly after that visit and steadily progressed. She is unable to identify an inciting event or stressor. Ms. J worked until 2 weeks before this admission. Neurologic examination at the time of psychiatric consultation is notable for waxing and waning expressive aphasia, right homonymous hemianopsia, and mildly decreased strength in the left biceps and forearm.
Ms. J presented to her cardiologist reporting dizziness and blurred vision 6 weeks ago, and she was observed in the hospital 3 weeks earlier for further evaluation. Laboratory testing during that hospitalization included blood counts, basic metabolic panel, thyroid function studies, erythrocyte sedimentation rate, thiamine, folic acid and vitamin B12, rapid plasma reagin and human immunodeficiency virus antibody, and LP, all reported as within normal limits.
Thorough review of Ms. J’s medical records reveals abnormalities that would be difficult to ascribe to conversion disorder. Specifically, 6 weeks ago, MRI of the brain demonstrated restricted diffusion in the left occipital lobe, and cerebrospinal fluid (CSF) neuron-specific enolase was moderately elevated at 34 ng/mL. The psychiatric consultant discusses these findings and concern for possible rapidly progressive dementia or Creutzfeldt-Jakob disease (CJD) with the primary team, Ms. J, and her family.
Ms. J is discharged with testing for CSF protein 14-3-3 pending and medical follow-up in 10 days. At follow-up 1 week later, her symptoms are worse; she is completely aphasic and wheelchair-bound. Antithyroglobulin and antimicrosomal thyroid antibodies and paraneoplastic antibody panel return normal. CSF protein 14-3-3 ultimately returns positive, supporting a clinical diagnosis of CJD. Ms. J dies shortly after hospital follow-up, less than 4 months after her first complaint of neurologic symptoms. No autopsy is performed.
Patients with conversion disorder may present with neurologic symptoms such as blindness, seizures, paralysis, or sensory loss with no identifiable anatomical or medical explanation.1 Conversion seizures—also known as pseudoseizures or nonepileptic seizures—may be clinically indistinguishable from generalized tonic-clonic seizures, but no EEG correlate can be identified.1,2 Conversion disorder is conceptualized as an unconscious manifestation of psychological conflict or stress—patients are not aware they are producing symptoms—and has been associated with emotional, sexual, and physical trauma.3,4
Conversion disorder is a diagnosis of exclusion and requires thorough evaluation to rule out neurologic or medical etiologies. The differential diagnosis for conversion disorder includes the broad medical differential diagnosis for the symptom, whether it be paralysis, seizures, sensory loss, or other presenting symptoms. Therefore, when evaluating patients for conversion disorder, be vigilant to the possibility of not only underlying psychological stress or trauma but also undiscovered medical or neurologic illness.
In Ms. J’s case, the primary team began to suspect there was no organic cause of her neurologic symptoms. However, psychiatric evaluation revealed that Ms. J had no history of stress or trauma that typically would be associated with conversion disorder, nor did she manifest other psychiatric symptoms, except waxing and waning mental status, which raised concerns for possible delirium or encephalopathy. Additionally, slowing on EEG was a nonspecific but abnormal finding that made conversion disorder unlikely. The consulting psychiatrist discussed this slowing, in conjunction with the abnormal MRI and elevated CSF neuron-specific enolase, with members of the referring Neurology service, who ordered additional testing of CSF for protein 14-3-3.
Creutzfeldt-Jakob disease
CJD is a rapidly progressive neurodegenerative disorder characterized by cognitive changes, behavioral changes, gait disturbances, akinetic mutism, and myoclonus.5 CJD results from the transition of prion proteins, which are present in the normal human brain, to disease-associated forms that aggregate and propagate and result in neurotoxicity with spongiform changes in neurons.6 The transition of normal prions to disease-associated prions may be hereditary, iatrogenic, infectious, or sporadic. Because the pathologic prion protein can be transmitted and normal sterilization procedures do not prevent the spread of CJD, special precautions should be taken to avoid contact with blood or CSF from patients suspected of having CJD.5
CJD most commonly occurs in the sporadic form, for which there are no identifiable risk factors, with an average age of onset between age 50 and 70. The disease affects women and men equally at a rate of 1 to 2 persons per million per year worldwide.6,7 Most patients with CJD die within 12 months of diagnosis8; median survival is 4 to 5 months.7,9 Although there is no approved or standard effective treatment for this uniformly fatal disease, research into the possibility of genetic or post-translational treatments is ongoing. One group reported inhibition of prion propagation by quinacrine and chlorpromazine in vitro.10 Clinical studies of quinacrine have demonstrated tolerability but no impact on the course of CJD.6
Clues to diagnosis. Although there is no treatment for CJD, early diagnosis can help patients and families understand the relentless progression of symptoms and also permits end-of-life planning and palliative care.11 Diagnosing CJD requires a high level of suspicion and traditionally has required brain biopsy or autopsy for conclusive diagnosis, although in some cases rare EEG findings of periodic sharp wave complexes or generalized periodic epileptiform discharges (GPEDs) have suggested the diagnosis.7,8,12 Recently, specific MRI findings have been described with fluid attenuated inversion recovery (FLAIR) and diffusion sequences.9,13,14
Routine LP for CSF examination (including cell count, protein, and glucose) frequently is normal.8 Specific testing to assess for CJD is required. Elevated levels of CSF neuron-specific enolase (normal <30 ng/mL) and protein 14-3-3 (normal <8 ng/mL) are fairly sensitive and specific for CJD when assessed in patients with the proper clinical history, although normal levels of these proteins have been detected in patients later confirmed to have CJD.7,15 A large multinational collaborative study of confirmed CJD cases that evaluated diagnostic test characteristics recommended that because each test has limitations and can be falsely negative—even in a case of later-confirmed CJD—a rational approach to diagnosis includes brain MRI with diffusion-weighted imaging, CSF analysis for protein 14-3-3, and EEG to assess for periodic sharp wave complexes or GPEDs.16
Because CJD presentation varies widely, most clinicians will not consider the diagnosis until the disease has progressed or the patient has died. Patients who present with psychological symptoms or predominant language disturbances and dysphagia may be referred to a psychiatrist or an ear, nose, and throat specialist before seeing a neurologist.9 Patients may be extensively evaluated and treated for conversion disorder when the correct diagnosis is CJD.17
Sporadic CJD traditionally is associated with neurologic presentations, whereas variant CJD is believed to present with psychiatric symptomatology. However, in a 25-year retrospective review of 126 patients with sporadic CJD, 80% of cases demonstrated psychiatric symptoms within the first 100 days of the disease course.18 Of these, nearly 25% showed psychiatric symptoms at presentation, including sleep disturbances, psychotic symptoms, agitation, and anxiety.
Psychiatrists should be aware of distinguishing features of rapidly progressive dementias and CJD, especially in the setting of psychiatric consultation, to rule out somatic etiologies of unexplained neurologic symptoms. It is important to obtain a history of baseline functioning, duration of decline, and psychiatric symptomatology to differentiate between organic and somatic causes. Differential diagnosis for rapidly progressive cognitive impairment is broad and includes delirium from diverse medical causes; rapidly progressive dementia such as accelerated Alzheimer’s disease, Lewy body disease, or frontotemporal dementia; and psychogenic causes, including conversion disorder (Table 1).7,8,12Table 2 provides distinguishing features of CJD, Alzheimer’s disease, Lewy body disease, and frontotemporal dementia with motor neuron disease.7,8,19
Table 1
Differential diagnosis of rapidly progressive dementia
| Celiac disease |
| Central nervous system vasculitis |
| Creutzfeldt-Jakob disease |
| Delirium (numerous possible etiologies) |
| Focal status epilepticus |
| Hashimoto’s encephalopathy |
Infection
|
Intoxication
|
| Limbic encephalopathy from paraneoplastic antibody syndrome |
| Lymphomatoid granulomatosis |
Malignancy
|
| Porphyria |
| Progressive supranuclear palsy |
Psychiatric disorder
|
| Sarcoidosis |
| Stroke |
| Vitamin deficiency (vitamin E, thiamine) |
| EBV: Epstein-Barr virus; HIV: human immunodeficiency virus; HSV: herpes simplex virus Source: References 7,8,12 |
Table 2
Distinguishing features of Creutzfeldt-Jakob disease
| Sporadic CJD | AD | DLBD | FTD-MND | |
|---|---|---|---|---|
| Time course | Rapid progression (median survival 4 to 5 months) | Insidious onset; progressive decline | Insidious onset; progressive decline | May experience rapid course to death |
| Age at onset | Age 50 to 70 | Incidence increases with age (usual onset age 65 to 85) | Older (age ~80) | Young age at onset |
| EEG findings | Periodic atypical triphasic waves; GPEDs | Normal or diffuse abnormalities | Rarely atypical triphasic waves | Increased slow activity, decreased fast activity |
| MRI findings | Restricted diffusion | Generalized atrophy | Generalized atrophy | Frontal atrophy |
| AD: Alzheimer’s disease; CJD: Creutzfeldt-Jakob disease; DLBD: diffuse Lewy body dementia; EEG: electroencephalography; FTD-MND: frontotemporal dementia with motor neuron disease; GPEDs: generalized periodic epileptiform discharges Source: References 7,8,19 | ||||
Related Resources
National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
Centers for Disease Control and Prevention. About CJD. www.cdc.gov/ncidod/dvrd/cjd.
Drug Brand Names
Chlorpromazine • Thorazine, Largactil
Quinacine • Atabrine
Disclosure
Dr. Gagliardi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry. 2006;163(9):1510-1517.
2. Teo WY, Choong CT. Neurological presentations of conversion disorders in a group of Singapore children. Pediatr Int. 2008;50(4):533-536.
3. Brown RJ, Cardena E, Nuenhuis E, et al. Should conversion disorder be reclassified as a dissociative disorder in DSM-V? Psychosomatics. 2007;48:369-378.
4. Stone J, Carson A, Aditya H, et al. The role of physical injury in motor and sensory conversion symptoms: a systematic and narrative review. J Psychosom Res. 2009;66:383-390.
5. National Institute of Neurological Disorders and Stroke Creutzfeldt-Jakob disease fact sheet. Available at: http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm. Accessed August 7, 2010.
6. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334-344.
7. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:98-108.
8. Josephs KA, Ahlskog E, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201-207.
9. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease.Curr Psychiatry Rep. 2003;5:43-46.
10. Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapies for prion disease. PNAS. 2001;98:9836-9841.
11. Cumbler E, Furfari K, Guerrasio J. Creutzfeldt-Jacob disease presenting as severe depression: a case report. Cases J. 2009;2:122-124.
12. Tan KM, Worrell GA, Parisi JE, et al. Creutzfeldt-Jakob disease with focal electroencephalographic and magnetic resonance imaging findings. Arch Neurol. 2007;64:600-601.
13. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443-449.
14. Manners DN, Parchi P, Tonon C, et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72:1425-1431.
15. Aksamit AJ, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. 2001;57:728-730.
16. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
17. Solvason HB, Harris B, Zeifert P, et al. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002;159(4):528-537.
18. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
19. Liedorp M, van der Flier WM, Hoogervorst EL, et al. Associations between patterns of EEG abnormalities and diagnosis in a large memory clinic cohort. Dement Geriatr Cogn Disord. 2009;27:18-23.
Consider this rare disorder in patients with rapid-onset neurologic symptoms
Ms. J, age 63, is admitted to Neurology with progressive dizziness and cognitive impairment. She had developed word-finding difficulties, weakness, memory problems, and an episode of arm shaking, which prompted referral for inpatient workup. Ms. J has a history of hypertension, palpitations, and diabetes mellitus.
Her neurologic examination is variable; some examiners find pronounced aphasia and right-sided weakness, whereas others document a nearly normal examination. Lumbar puncture (LP) shows normal cell count, glucose, protein, and negative Gram’s stain; MRI of the brain is normal. Enterovirus polymerase chain reaction, cryptococcal antigen, and Lyme antibody are negative. Electroencephalography (EEG) demonstrates diffuse slowing. The primary team requests psychiatric consultation to assess for conversion disorder.
Ms. J is cooperative with psychiatric evaluation. She denies current or past psychiatric symptomatology and does not meet criteria for major depression, dysthymia, adjustment disorder, anxiety disorder, psychosis, or mania. She denies personal or family history of suicidal or homicidal ideation, intent, or plan. Her youngest son died 5 years earlier; she is financially secure and her 40-year marriage is stable. Ms. J denies having a history of substance use, physical or sexual abuse, or trauma.
In the Cardiology clinic 2 months ago, Ms. J denied having neurologic symptoms and was noted to be doing well. Her neurologic symptoms began shortly after that visit and steadily progressed. She is unable to identify an inciting event or stressor. Ms. J worked until 2 weeks before this admission. Neurologic examination at the time of psychiatric consultation is notable for waxing and waning expressive aphasia, right homonymous hemianopsia, and mildly decreased strength in the left biceps and forearm.
Ms. J presented to her cardiologist reporting dizziness and blurred vision 6 weeks ago, and she was observed in the hospital 3 weeks earlier for further evaluation. Laboratory testing during that hospitalization included blood counts, basic metabolic panel, thyroid function studies, erythrocyte sedimentation rate, thiamine, folic acid and vitamin B12, rapid plasma reagin and human immunodeficiency virus antibody, and LP, all reported as within normal limits.
Thorough review of Ms. J’s medical records reveals abnormalities that would be difficult to ascribe to conversion disorder. Specifically, 6 weeks ago, MRI of the brain demonstrated restricted diffusion in the left occipital lobe, and cerebrospinal fluid (CSF) neuron-specific enolase was moderately elevated at 34 ng/mL. The psychiatric consultant discusses these findings and concern for possible rapidly progressive dementia or Creutzfeldt-Jakob disease (CJD) with the primary team, Ms. J, and her family.
Ms. J is discharged with testing for CSF protein 14-3-3 pending and medical follow-up in 10 days. At follow-up 1 week later, her symptoms are worse; she is completely aphasic and wheelchair-bound. Antithyroglobulin and antimicrosomal thyroid antibodies and paraneoplastic antibody panel return normal. CSF protein 14-3-3 ultimately returns positive, supporting a clinical diagnosis of CJD. Ms. J dies shortly after hospital follow-up, less than 4 months after her first complaint of neurologic symptoms. No autopsy is performed.
Patients with conversion disorder may present with neurologic symptoms such as blindness, seizures, paralysis, or sensory loss with no identifiable anatomical or medical explanation.1 Conversion seizures—also known as pseudoseizures or nonepileptic seizures—may be clinically indistinguishable from generalized tonic-clonic seizures, but no EEG correlate can be identified.1,2 Conversion disorder is conceptualized as an unconscious manifestation of psychological conflict or stress—patients are not aware they are producing symptoms—and has been associated with emotional, sexual, and physical trauma.3,4
Conversion disorder is a diagnosis of exclusion and requires thorough evaluation to rule out neurologic or medical etiologies. The differential diagnosis for conversion disorder includes the broad medical differential diagnosis for the symptom, whether it be paralysis, seizures, sensory loss, or other presenting symptoms. Therefore, when evaluating patients for conversion disorder, be vigilant to the possibility of not only underlying psychological stress or trauma but also undiscovered medical or neurologic illness.
In Ms. J’s case, the primary team began to suspect there was no organic cause of her neurologic symptoms. However, psychiatric evaluation revealed that Ms. J had no history of stress or trauma that typically would be associated with conversion disorder, nor did she manifest other psychiatric symptoms, except waxing and waning mental status, which raised concerns for possible delirium or encephalopathy. Additionally, slowing on EEG was a nonspecific but abnormal finding that made conversion disorder unlikely. The consulting psychiatrist discussed this slowing, in conjunction with the abnormal MRI and elevated CSF neuron-specific enolase, with members of the referring Neurology service, who ordered additional testing of CSF for protein 14-3-3.
Creutzfeldt-Jakob disease
CJD is a rapidly progressive neurodegenerative disorder characterized by cognitive changes, behavioral changes, gait disturbances, akinetic mutism, and myoclonus.5 CJD results from the transition of prion proteins, which are present in the normal human brain, to disease-associated forms that aggregate and propagate and result in neurotoxicity with spongiform changes in neurons.6 The transition of normal prions to disease-associated prions may be hereditary, iatrogenic, infectious, or sporadic. Because the pathologic prion protein can be transmitted and normal sterilization procedures do not prevent the spread of CJD, special precautions should be taken to avoid contact with blood or CSF from patients suspected of having CJD.5
CJD most commonly occurs in the sporadic form, for which there are no identifiable risk factors, with an average age of onset between age 50 and 70. The disease affects women and men equally at a rate of 1 to 2 persons per million per year worldwide.6,7 Most patients with CJD die within 12 months of diagnosis8; median survival is 4 to 5 months.7,9 Although there is no approved or standard effective treatment for this uniformly fatal disease, research into the possibility of genetic or post-translational treatments is ongoing. One group reported inhibition of prion propagation by quinacrine and chlorpromazine in vitro.10 Clinical studies of quinacrine have demonstrated tolerability but no impact on the course of CJD.6
Clues to diagnosis. Although there is no treatment for CJD, early diagnosis can help patients and families understand the relentless progression of symptoms and also permits end-of-life planning and palliative care.11 Diagnosing CJD requires a high level of suspicion and traditionally has required brain biopsy or autopsy for conclusive diagnosis, although in some cases rare EEG findings of periodic sharp wave complexes or generalized periodic epileptiform discharges (GPEDs) have suggested the diagnosis.7,8,12 Recently, specific MRI findings have been described with fluid attenuated inversion recovery (FLAIR) and diffusion sequences.9,13,14
Routine LP for CSF examination (including cell count, protein, and glucose) frequently is normal.8 Specific testing to assess for CJD is required. Elevated levels of CSF neuron-specific enolase (normal <30 ng/mL) and protein 14-3-3 (normal <8 ng/mL) are fairly sensitive and specific for CJD when assessed in patients with the proper clinical history, although normal levels of these proteins have been detected in patients later confirmed to have CJD.7,15 A large multinational collaborative study of confirmed CJD cases that evaluated diagnostic test characteristics recommended that because each test has limitations and can be falsely negative—even in a case of later-confirmed CJD—a rational approach to diagnosis includes brain MRI with diffusion-weighted imaging, CSF analysis for protein 14-3-3, and EEG to assess for periodic sharp wave complexes or GPEDs.16
Because CJD presentation varies widely, most clinicians will not consider the diagnosis until the disease has progressed or the patient has died. Patients who present with psychological symptoms or predominant language disturbances and dysphagia may be referred to a psychiatrist or an ear, nose, and throat specialist before seeing a neurologist.9 Patients may be extensively evaluated and treated for conversion disorder when the correct diagnosis is CJD.17
Sporadic CJD traditionally is associated with neurologic presentations, whereas variant CJD is believed to present with psychiatric symptomatology. However, in a 25-year retrospective review of 126 patients with sporadic CJD, 80% of cases demonstrated psychiatric symptoms within the first 100 days of the disease course.18 Of these, nearly 25% showed psychiatric symptoms at presentation, including sleep disturbances, psychotic symptoms, agitation, and anxiety.
Psychiatrists should be aware of distinguishing features of rapidly progressive dementias and CJD, especially in the setting of psychiatric consultation, to rule out somatic etiologies of unexplained neurologic symptoms. It is important to obtain a history of baseline functioning, duration of decline, and psychiatric symptomatology to differentiate between organic and somatic causes. Differential diagnosis for rapidly progressive cognitive impairment is broad and includes delirium from diverse medical causes; rapidly progressive dementia such as accelerated Alzheimer’s disease, Lewy body disease, or frontotemporal dementia; and psychogenic causes, including conversion disorder (Table 1).7,8,12Table 2 provides distinguishing features of CJD, Alzheimer’s disease, Lewy body disease, and frontotemporal dementia with motor neuron disease.7,8,19
Table 1
Differential diagnosis of rapidly progressive dementia
| Celiac disease |
| Central nervous system vasculitis |
| Creutzfeldt-Jakob disease |
| Delirium (numerous possible etiologies) |
| Focal status epilepticus |
| Hashimoto’s encephalopathy |
Infection
|
Intoxication
|
| Limbic encephalopathy from paraneoplastic antibody syndrome |
| Lymphomatoid granulomatosis |
Malignancy
|
| Porphyria |
| Progressive supranuclear palsy |
Psychiatric disorder
|
| Sarcoidosis |
| Stroke |
| Vitamin deficiency (vitamin E, thiamine) |
| EBV: Epstein-Barr virus; HIV: human immunodeficiency virus; HSV: herpes simplex virus Source: References 7,8,12 |
Table 2
Distinguishing features of Creutzfeldt-Jakob disease
| Sporadic CJD | AD | DLBD | FTD-MND | |
|---|---|---|---|---|
| Time course | Rapid progression (median survival 4 to 5 months) | Insidious onset; progressive decline | Insidious onset; progressive decline | May experience rapid course to death |
| Age at onset | Age 50 to 70 | Incidence increases with age (usual onset age 65 to 85) | Older (age ~80) | Young age at onset |
| EEG findings | Periodic atypical triphasic waves; GPEDs | Normal or diffuse abnormalities | Rarely atypical triphasic waves | Increased slow activity, decreased fast activity |
| MRI findings | Restricted diffusion | Generalized atrophy | Generalized atrophy | Frontal atrophy |
| AD: Alzheimer’s disease; CJD: Creutzfeldt-Jakob disease; DLBD: diffuse Lewy body dementia; EEG: electroencephalography; FTD-MND: frontotemporal dementia with motor neuron disease; GPEDs: generalized periodic epileptiform discharges Source: References 7,8,19 | ||||
Related Resources
National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
Centers for Disease Control and Prevention. About CJD. www.cdc.gov/ncidod/dvrd/cjd.
Drug Brand Names
Chlorpromazine • Thorazine, Largactil
Quinacine • Atabrine
Disclosure
Dr. Gagliardi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Consider this rare disorder in patients with rapid-onset neurologic symptoms
Ms. J, age 63, is admitted to Neurology with progressive dizziness and cognitive impairment. She had developed word-finding difficulties, weakness, memory problems, and an episode of arm shaking, which prompted referral for inpatient workup. Ms. J has a history of hypertension, palpitations, and diabetes mellitus.
Her neurologic examination is variable; some examiners find pronounced aphasia and right-sided weakness, whereas others document a nearly normal examination. Lumbar puncture (LP) shows normal cell count, glucose, protein, and negative Gram’s stain; MRI of the brain is normal. Enterovirus polymerase chain reaction, cryptococcal antigen, and Lyme antibody are negative. Electroencephalography (EEG) demonstrates diffuse slowing. The primary team requests psychiatric consultation to assess for conversion disorder.
Ms. J is cooperative with psychiatric evaluation. She denies current or past psychiatric symptomatology and does not meet criteria for major depression, dysthymia, adjustment disorder, anxiety disorder, psychosis, or mania. She denies personal or family history of suicidal or homicidal ideation, intent, or plan. Her youngest son died 5 years earlier; she is financially secure and her 40-year marriage is stable. Ms. J denies having a history of substance use, physical or sexual abuse, or trauma.
In the Cardiology clinic 2 months ago, Ms. J denied having neurologic symptoms and was noted to be doing well. Her neurologic symptoms began shortly after that visit and steadily progressed. She is unable to identify an inciting event or stressor. Ms. J worked until 2 weeks before this admission. Neurologic examination at the time of psychiatric consultation is notable for waxing and waning expressive aphasia, right homonymous hemianopsia, and mildly decreased strength in the left biceps and forearm.
Ms. J presented to her cardiologist reporting dizziness and blurred vision 6 weeks ago, and she was observed in the hospital 3 weeks earlier for further evaluation. Laboratory testing during that hospitalization included blood counts, basic metabolic panel, thyroid function studies, erythrocyte sedimentation rate, thiamine, folic acid and vitamin B12, rapid plasma reagin and human immunodeficiency virus antibody, and LP, all reported as within normal limits.
Thorough review of Ms. J’s medical records reveals abnormalities that would be difficult to ascribe to conversion disorder. Specifically, 6 weeks ago, MRI of the brain demonstrated restricted diffusion in the left occipital lobe, and cerebrospinal fluid (CSF) neuron-specific enolase was moderately elevated at 34 ng/mL. The psychiatric consultant discusses these findings and concern for possible rapidly progressive dementia or Creutzfeldt-Jakob disease (CJD) with the primary team, Ms. J, and her family.
Ms. J is discharged with testing for CSF protein 14-3-3 pending and medical follow-up in 10 days. At follow-up 1 week later, her symptoms are worse; she is completely aphasic and wheelchair-bound. Antithyroglobulin and antimicrosomal thyroid antibodies and paraneoplastic antibody panel return normal. CSF protein 14-3-3 ultimately returns positive, supporting a clinical diagnosis of CJD. Ms. J dies shortly after hospital follow-up, less than 4 months after her first complaint of neurologic symptoms. No autopsy is performed.
Patients with conversion disorder may present with neurologic symptoms such as blindness, seizures, paralysis, or sensory loss with no identifiable anatomical or medical explanation.1 Conversion seizures—also known as pseudoseizures or nonepileptic seizures—may be clinically indistinguishable from generalized tonic-clonic seizures, but no EEG correlate can be identified.1,2 Conversion disorder is conceptualized as an unconscious manifestation of psychological conflict or stress—patients are not aware they are producing symptoms—and has been associated with emotional, sexual, and physical trauma.3,4
Conversion disorder is a diagnosis of exclusion and requires thorough evaluation to rule out neurologic or medical etiologies. The differential diagnosis for conversion disorder includes the broad medical differential diagnosis for the symptom, whether it be paralysis, seizures, sensory loss, or other presenting symptoms. Therefore, when evaluating patients for conversion disorder, be vigilant to the possibility of not only underlying psychological stress or trauma but also undiscovered medical or neurologic illness.
In Ms. J’s case, the primary team began to suspect there was no organic cause of her neurologic symptoms. However, psychiatric evaluation revealed that Ms. J had no history of stress or trauma that typically would be associated with conversion disorder, nor did she manifest other psychiatric symptoms, except waxing and waning mental status, which raised concerns for possible delirium or encephalopathy. Additionally, slowing on EEG was a nonspecific but abnormal finding that made conversion disorder unlikely. The consulting psychiatrist discussed this slowing, in conjunction with the abnormal MRI and elevated CSF neuron-specific enolase, with members of the referring Neurology service, who ordered additional testing of CSF for protein 14-3-3.
Creutzfeldt-Jakob disease
CJD is a rapidly progressive neurodegenerative disorder characterized by cognitive changes, behavioral changes, gait disturbances, akinetic mutism, and myoclonus.5 CJD results from the transition of prion proteins, which are present in the normal human brain, to disease-associated forms that aggregate and propagate and result in neurotoxicity with spongiform changes in neurons.6 The transition of normal prions to disease-associated prions may be hereditary, iatrogenic, infectious, or sporadic. Because the pathologic prion protein can be transmitted and normal sterilization procedures do not prevent the spread of CJD, special precautions should be taken to avoid contact with blood or CSF from patients suspected of having CJD.5
CJD most commonly occurs in the sporadic form, for which there are no identifiable risk factors, with an average age of onset between age 50 and 70. The disease affects women and men equally at a rate of 1 to 2 persons per million per year worldwide.6,7 Most patients with CJD die within 12 months of diagnosis8; median survival is 4 to 5 months.7,9 Although there is no approved or standard effective treatment for this uniformly fatal disease, research into the possibility of genetic or post-translational treatments is ongoing. One group reported inhibition of prion propagation by quinacrine and chlorpromazine in vitro.10 Clinical studies of quinacrine have demonstrated tolerability but no impact on the course of CJD.6
Clues to diagnosis. Although there is no treatment for CJD, early diagnosis can help patients and families understand the relentless progression of symptoms and also permits end-of-life planning and palliative care.11 Diagnosing CJD requires a high level of suspicion and traditionally has required brain biopsy or autopsy for conclusive diagnosis, although in some cases rare EEG findings of periodic sharp wave complexes or generalized periodic epileptiform discharges (GPEDs) have suggested the diagnosis.7,8,12 Recently, specific MRI findings have been described with fluid attenuated inversion recovery (FLAIR) and diffusion sequences.9,13,14
Routine LP for CSF examination (including cell count, protein, and glucose) frequently is normal.8 Specific testing to assess for CJD is required. Elevated levels of CSF neuron-specific enolase (normal <30 ng/mL) and protein 14-3-3 (normal <8 ng/mL) are fairly sensitive and specific for CJD when assessed in patients with the proper clinical history, although normal levels of these proteins have been detected in patients later confirmed to have CJD.7,15 A large multinational collaborative study of confirmed CJD cases that evaluated diagnostic test characteristics recommended that because each test has limitations and can be falsely negative—even in a case of later-confirmed CJD—a rational approach to diagnosis includes brain MRI with diffusion-weighted imaging, CSF analysis for protein 14-3-3, and EEG to assess for periodic sharp wave complexes or GPEDs.16
Because CJD presentation varies widely, most clinicians will not consider the diagnosis until the disease has progressed or the patient has died. Patients who present with psychological symptoms or predominant language disturbances and dysphagia may be referred to a psychiatrist or an ear, nose, and throat specialist before seeing a neurologist.9 Patients may be extensively evaluated and treated for conversion disorder when the correct diagnosis is CJD.17
Sporadic CJD traditionally is associated with neurologic presentations, whereas variant CJD is believed to present with psychiatric symptomatology. However, in a 25-year retrospective review of 126 patients with sporadic CJD, 80% of cases demonstrated psychiatric symptoms within the first 100 days of the disease course.18 Of these, nearly 25% showed psychiatric symptoms at presentation, including sleep disturbances, psychotic symptoms, agitation, and anxiety.
Psychiatrists should be aware of distinguishing features of rapidly progressive dementias and CJD, especially in the setting of psychiatric consultation, to rule out somatic etiologies of unexplained neurologic symptoms. It is important to obtain a history of baseline functioning, duration of decline, and psychiatric symptomatology to differentiate between organic and somatic causes. Differential diagnosis for rapidly progressive cognitive impairment is broad and includes delirium from diverse medical causes; rapidly progressive dementia such as accelerated Alzheimer’s disease, Lewy body disease, or frontotemporal dementia; and psychogenic causes, including conversion disorder (Table 1).7,8,12Table 2 provides distinguishing features of CJD, Alzheimer’s disease, Lewy body disease, and frontotemporal dementia with motor neuron disease.7,8,19
Table 1
Differential diagnosis of rapidly progressive dementia
| Celiac disease |
| Central nervous system vasculitis |
| Creutzfeldt-Jakob disease |
| Delirium (numerous possible etiologies) |
| Focal status epilepticus |
| Hashimoto’s encephalopathy |
Infection
|
Intoxication
|
| Limbic encephalopathy from paraneoplastic antibody syndrome |
| Lymphomatoid granulomatosis |
Malignancy
|
| Porphyria |
| Progressive supranuclear palsy |
Psychiatric disorder
|
| Sarcoidosis |
| Stroke |
| Vitamin deficiency (vitamin E, thiamine) |
| EBV: Epstein-Barr virus; HIV: human immunodeficiency virus; HSV: herpes simplex virus Source: References 7,8,12 |
Table 2
Distinguishing features of Creutzfeldt-Jakob disease
| Sporadic CJD | AD | DLBD | FTD-MND | |
|---|---|---|---|---|
| Time course | Rapid progression (median survival 4 to 5 months) | Insidious onset; progressive decline | Insidious onset; progressive decline | May experience rapid course to death |
| Age at onset | Age 50 to 70 | Incidence increases with age (usual onset age 65 to 85) | Older (age ~80) | Young age at onset |
| EEG findings | Periodic atypical triphasic waves; GPEDs | Normal or diffuse abnormalities | Rarely atypical triphasic waves | Increased slow activity, decreased fast activity |
| MRI findings | Restricted diffusion | Generalized atrophy | Generalized atrophy | Frontal atrophy |
| AD: Alzheimer’s disease; CJD: Creutzfeldt-Jakob disease; DLBD: diffuse Lewy body dementia; EEG: electroencephalography; FTD-MND: frontotemporal dementia with motor neuron disease; GPEDs: generalized periodic epileptiform discharges Source: References 7,8,19 | ||||
Related Resources
National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
Centers for Disease Control and Prevention. About CJD. www.cdc.gov/ncidod/dvrd/cjd.
Drug Brand Names
Chlorpromazine • Thorazine, Largactil
Quinacine • Atabrine
Disclosure
Dr. Gagliardi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry. 2006;163(9):1510-1517.
2. Teo WY, Choong CT. Neurological presentations of conversion disorders in a group of Singapore children. Pediatr Int. 2008;50(4):533-536.
3. Brown RJ, Cardena E, Nuenhuis E, et al. Should conversion disorder be reclassified as a dissociative disorder in DSM-V? Psychosomatics. 2007;48:369-378.
4. Stone J, Carson A, Aditya H, et al. The role of physical injury in motor and sensory conversion symptoms: a systematic and narrative review. J Psychosom Res. 2009;66:383-390.
5. National Institute of Neurological Disorders and Stroke Creutzfeldt-Jakob disease fact sheet. Available at: http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm. Accessed August 7, 2010.
6. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334-344.
7. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:98-108.
8. Josephs KA, Ahlskog E, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201-207.
9. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease.Curr Psychiatry Rep. 2003;5:43-46.
10. Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapies for prion disease. PNAS. 2001;98:9836-9841.
11. Cumbler E, Furfari K, Guerrasio J. Creutzfeldt-Jacob disease presenting as severe depression: a case report. Cases J. 2009;2:122-124.
12. Tan KM, Worrell GA, Parisi JE, et al. Creutzfeldt-Jakob disease with focal electroencephalographic and magnetic resonance imaging findings. Arch Neurol. 2007;64:600-601.
13. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443-449.
14. Manners DN, Parchi P, Tonon C, et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72:1425-1431.
15. Aksamit AJ, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. 2001;57:728-730.
16. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
17. Solvason HB, Harris B, Zeifert P, et al. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002;159(4):528-537.
18. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
19. Liedorp M, van der Flier WM, Hoogervorst EL, et al. Associations between patterns of EEG abnormalities and diagnosis in a large memory clinic cohort. Dement Geriatr Cogn Disord. 2009;27:18-23.
1. Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry. 2006;163(9):1510-1517.
2. Teo WY, Choong CT. Neurological presentations of conversion disorders in a group of Singapore children. Pediatr Int. 2008;50(4):533-536.
3. Brown RJ, Cardena E, Nuenhuis E, et al. Should conversion disorder be reclassified as a dissociative disorder in DSM-V? Psychosomatics. 2007;48:369-378.
4. Stone J, Carson A, Aditya H, et al. The role of physical injury in motor and sensory conversion symptoms: a systematic and narrative review. J Psychosom Res. 2009;66:383-390.
5. National Institute of Neurological Disorders and Stroke Creutzfeldt-Jakob disease fact sheet. Available at: http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm. Accessed August 7, 2010.
6. Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334-344.
7. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64:98-108.
8. Josephs KA, Ahlskog E, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201-207.
9. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease.Curr Psychiatry Rep. 2003;5:43-46.
10. Korth C, May BCH, Cohen FE, et al. Acridine and phenothiazine derivatives as pharmacotherapies for prion disease. PNAS. 2001;98:9836-9841.
11. Cumbler E, Furfari K, Guerrasio J. Creutzfeldt-Jacob disease presenting as severe depression: a case report. Cases J. 2009;2:122-124.
12. Tan KM, Worrell GA, Parisi JE, et al. Creutzfeldt-Jakob disease with focal electroencephalographic and magnetic resonance imaging findings. Arch Neurol. 2007;64:600-601.
13. Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;63:443-449.
14. Manners DN, Parchi P, Tonon C, et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology. 2009;72:1425-1431.
15. Aksamit AJ, Preissner CM, Homburger HA. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology. 2001;57:728-730.
16. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278-2287.
17. Solvason HB, Harris B, Zeifert P, et al. Psychological versus biological clinical interpretation: a patient with prion disease. Am J Psychiatry. 2002;159(4):528-537.
18. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17:489-495.
19. Liedorp M, van der Flier WM, Hoogervorst EL, et al. Associations between patterns of EEG abnormalities and diagnosis in a large memory clinic cohort. Dement Geriatr Cogn Disord. 2009;27:18-23.