User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
A low-frustration strategy for treating somatization
Mrs. M, age 34, was referred for psychiatric evaluation by her primary care physician. She reluctantly agreed to the referral and tells the psychiatrist she “really should be seeing a cardiologist.” Numerous evaluations for chest pain and palpitations—including seven emergency room visits, ECGs and cardiac catheterization—have revealed no medical pathology.
A divorced mother of two children, she says she feels anxious about her “heart condition.” Her father died of a heart attack at age 51. She experiences chest pains at home and at work, particularly when under stress. Sometimes she feels her heart racing and numbness or tingling in her arms.
Although her primary care physician has seen her frequently during the past 6 months, she says the doctor is not taking her complaints seriously. “These chest pains are real,” she says, “so don’t try to tell me they’re all in my head.”
Psychiatrists may be the last doctors patients such as Mrs. M wish to see but the ones best equipped to relieve their suffering. Our experience in treating somatizing patients and the available evidence suggest that cognitive-behavioral therapy (CBT) combined with psychoeducation, reassurance, and sometimes drug therapy is the most effective approach.
Health-related fear—or “illness worry”—is common, occurring in nearly 10% of adults who responded to a recent community survey.2 When this fear drives individuals to their physicians for evaluation, frequently no organic cause is discovered. Full evaluations are expensive and lead to increased use of health care resources, including potentially dangerous invasive testing.3,4
Defining somatization has been a source of confusion.5,6 Some authors consider somatic complaints to be expressions of suppressed psychosocial stressors. Others label them as medically unexplained complaints, although this definition fails to exclude occult medical problems. Kleinman7 defines somatization as “a somatic idiom of psychosocial distress in a setting of medical health-care seeking.” This useful definition links psychosocial problems with somatic complaints and the behavioral drive to obtain a medical evaluation.
In DSM-IV,8 the defining characteristics of somatoform disorders are somatic complaints or disease fears that are out of proportion with any identifiable somatic cause. Entities include somatization disorder, undifferentiated somatization disorder, conversion disorder, pain disorder, hypochondriasis, body dysmorphic disorder, and somatoform disorder–not otherwise specified (NOS).
Subthreshold symptoms. Unfortunately, DSM-IV’s categorization of Axis I somatoform disorders does not capture subthreshold presentations, which are common. Patients with less than the required number of somatic complaints are labeled in a wastebasket fashion with “undifferentiated somatoform disorder.”9
Mrs. M’s persistent chest pain of noncardiac origin is a familiar health anxiety, along with functional GI complaints, headaches, chronic fatigue, and lower back pain. Frustrating to their doctors and frustrated themselves, patients with medically unexplained complaints consume an inordinate amount of physicians’ time.1
Without a clear definition of somatization (Box)2-9 or useful clinical guidelines, psychiatrists must rely on the literature for guidance in managing somatization disorders. This article summarizes the evidence and describes how we apply these findings to practice. And when all else fails, we offer last-ditch advice for managing patients who resist your treatment efforts.
IDENTIFYING COMORBIDITIES
Identifying psychiatric comorbidities is the first step in successfully treating patients with somatoform complaints. In an epidemiologic study, 60% of patients with somatoform complaints also had a mood disorder and 48% had an anxiety disorder.10 In a similar study of patients with hypochondriasis, 88% also had one or more Axis I diagnosis.11
If a patient meets criteria for a comorbid psychiatric disorder and is willing to be treated for it, the somatic complaints may resolve along with the underlying disorder. In fact, the presence of an identifiable Axis I disorder order may predict a more positive prognosis.12
Personality disorders. Somatization in patients with a personality disorder poses unique challenges.13 Granted, when making a diagnosis it is difficult to tease apart somatization from personality disorders because somatization itself may be considered a chronic, maladaptive coping style. However, symptoms such as deception, impulsivity, mood lability, and self-injurious behavior introduce treatment complications that exceed the scope of this article.
Posttraumatic stress disorder (PTSD)—particularly childhood sexual and physical abuse—also predisposes some patients to somatization disorders.14,15 Patients with comorbid PTSD and somatization disorder require highly specialized treatment that is beyond the scope of this review.
COGNITIVE-BEHAVIORAL TREATMENT
Cognitive-behavioral therapy (CBT) is the best-studied and most effective treatment for somatoform disorders.16 CBT for somatization relies on both physiologic and cognitive explanations to account for the patient’s experience, without committing to an “either/or” dichotomy. It offers patients an alternate explanation of what is wrong with them—illness anxiety instead of severe physical illness.
By making patients aware of their automatic thoughts, feelings, behaviors, and underlying beliefs, CBT helps them normalize and cope with their illness anxiety. CBT techniques can be applied in a predetermined course of therapy (such as 12 sessions with a mental health clinician), in a group setting, or piecemeal by any health care provider.
Effective strategies. In a review of 30 controlled trials of CBT for somatoform disorders, Looper17 showed overall effect ranging from 0.38 to 2.5, where 0.2 was defined as a small effect, 0.5 as medium, and 0.8 as large. Hypochondriasis, somatization disorder, body dysmorphic disorder, chronic pain, chronic fatigue, and noncardiac chest pain were included in this review. The most effective strategies:
- included 6 to 16 treatment sessions
- were symptom-focused as opposed to providing general relaxation training
- included maintenance sessions after the initial series.
Four factors of health anxiety. CBT primarily targets the patient’s false beliefs that he or she is physically ill. These beliefs are based on how the patient misinterprets innocuous physical symptoms and responds to them.18 The cognitive theory of health anxiety holds that health anxiety severity is affected by four factors:
- perceived likelihood of illness
- perceived burden of illness
- perceived ability to cope with illness
- perception of the extent to which external factors will help.19
Table 1
Common dysfunctional beliefs of somatizing patients
|
The first two factors worsen and the latter two mitigate health anxiety. An individual patient’s presenting fears often suggest which factors to address. For example, Mrs. M may describe the burden of illness as the focus of her fears (“If I have a heart attack, who will care for my children?”). This information cues you to shift the focus of therapy to helping her cope with child care needs despite her recurring symptoms.
If she focuses on her likelihood of illness, then uncoupling the symptoms from the diagnosis could be more productive. When she reports palpitations, diaphoresis, and dizziness, have her do breathing exercises that induce those symptoms without producing a heart attack.
Table 2
Journaling homework: 5 questions for patients to answer about one symptom each day
|
She might describe feeling unable to cope when she feels symptoms or when cardiologists tell her nothing is wrong with her heart. In that case, focus on relaxation techniques, global stress reduction, and reducing cardiac risk factors to bolster her ability to cope with her illness.
Journaling is a critical component of CBT in treating somatization disorders. Regular journaling by the patient can reveal dysfunctional beliefs that may be driving his or her health anxieties, such as those listed in Table 1. We find it useful to assign patients to answer five questions about one symptom experience each day (Table 2). This self-monitoring provides material to work on with the patient during each session.
Cognitive restructuring. During therapy sessions, we ask patients to suggest alternate explanations for the symptoms recorded in their journals. We then ask them to determine which explanations are more feasible.
For example, if Mrs. M develops palpitations during emotionally charged arguments, we would ask her to develop explanations other than, “I was having a heart attack.” Reality testing includes rhetorical questions such as, “Would you be alive today if you were having a heart attack every time you had palpitations?” Automatic thoughts are successively identified and then tested aloud with the patient:
- “Has every unexplained symptom led to the discovery of a serious illness?”
- “Does every instance of hurt equal harm?”
Eventually, patterns of automatic thoughts emerge, and these reveal the underlying dysfunctional beliefs.
Dysfunctional beliefs are maintained when patients selectively attend to and amplify somatic sensations. Behavioral experiments during sessions can demonstrate to the patient in vivo the process by which they misattribute illness to physical symptoms. For example, overbreathing with a patient during a session may elicit light-headedness, paresthesias, or tachycardia, which can then be linked to overbreathing rather than a chronic or catastrophic illness.
Furthermore, patients can be taught to control the experience. Some patients with headaches or GI pain may be made aware of symptoms by simply asking them to focus their attention on the respective organs. Simply explaining the cycle of misattribution, autonomic activation, and further symptom development with an in vivo demonstration can be illuminating.
Response prevention. Another behavioral technique is to cut back in small increments on actions the patient takes in response to physical symptoms and automatic thoughts. For example, a patient could take medicine and seek reassurance less frequently and avoid rubbing the affected area.
PSYCHOEDUCATION
Two psychoeducation programs for somatization behavior have been formally studied.
The Personal Health Improvement Program20—led by trained facilitators—includes classroom videos, cognitive-behavioral exercises, and home study assignments. After completing the 6-week course, 171 patients with somatization disorders reported reduced physical and psychological distress and increased function. They also visited their primary care physicians less often.
Table 3
How to effectively reassure somatizing patients
| Action | Benefit |
|---|---|
| Review records in front of patients | Demonstrates that you take complaints and histories seriously |
| Acknowledge the severity of patients’ distress | Validates subjective suffering |
| Schedule follow-up visits at regular intervals | Provides access to you and continuity of care; reduces extra phone calls and emergency visits |
| Use clear and simple language | Improves communication |
| Explain that they do not have life-threatening structural disease | Opens door to cognitive restructuring |
| Assign jobs, such as journaling 15 min/day and rounding up medical records | Builds therapeutic alliance, fosters patient responsibility, and restores patients’ sense of control |
| Identify and support the patient’s strengths | Builds self-esteem |
| Use specialty referrals sparingly | Reduces risk of further medical testing and patient anxiety while awaiting results |
Coping with Illness Anxiety21 relies on mini-lectures, demonstrations, videos, and focused group discussions. After six 2-hour sessions, 33 of 43 study patients (78%) used medical services less often and reported reduced disease conviction, consequences of bodily complaints, health anxiety, and checking and avoidance behaviors. Two psychology graduate students taught the course from a manual, with 6 to 9 patients per group.
Psychoeducation in this context relies on didactic presentations, readings, role playing, and videotaped material. The goal is to teach patients to recognize thoughts, emotions, and behaviors that lead to and result from somatic preoccupation. Patients can improve when they recognize dysfunctional behavioral patterns and learn alternate coping strategies.
Somatizing patients—with their aversion to the stigma of mental illness—may find psychoeducation particularly attractive. They can be treated as students who are being educated, rather than as patients who are being treated. Classrooms in both studies cited above were located in medical outpatient offices, not in mental health facilities.
REASSURANCE
Reassurance is a common therapeutic technique in medicine, although it is poorly understood, poorly taught, and not methodically applied. Reassurance alleviates anxiety, enables patients to endure dysphoria, encourages hope, gives insight, and enhances the doctor-patient relationship.22
Table 4
How to avoid becoming frustrated with persistent somatization
| Situation | Response |
|---|---|
| Despite patients’ urgency | Watch and wait, knowing that psychological distress has been chronic |
| Despite patients’ belief that a single pill or procedure will ‘cure’ them | Persist in ‘rehabilitative’ approach |
| Despite patients’ provocations to force you to take a dichotomous approach | Persist in using both physical and psychological explanations |
| Despite your knowledge that patients are actively maintaining their illness beliefs | Try to be patient as they attribute their misfortune to ‘fate,’ ‘bad luck,’ or ‘misfortune’ |
| Despite the fact that you have agreed to treat the patient | Realize that his or her family or culture may reinforce the ‘sick role’ as the only acceptable form of distress |
| Despite patients’ desire to discuss symptoms | Reorient them to sustaining daily function (such as parenting while tolerating fatigue) |
Whereas CBT seeks to challenge patients’ underlying beliefs and restructure their thought processes,23 reassurance can help them tolerate their dysfunctional beliefs and dissuade them from believing their health is dangerously impaired. Reassurance offers a substitute explanation of patients’ dysfunction, although this explanation is not as central or detailed as it is in CBT.
How to reassure. Patients may consider reassurance offered prematurely or by a stranger to be patronizing or dismissive. Reassurance is most effective when:
- given by a trusted person who is reliable, consistent, firm, and empathic
- the patient’s condition has been established as unresponsive to conventional diagnostics or biological therapies.
Patients are most receptive to reassurance when they express distress or frustration with their unexplained symptoms. Affirming that their suffering is legitimate opens the door to further treatment.
Reassurance is least effective when a patient is expressing anger or mistrust, although this is when the physician may feel most pressured to reassure. To successfully reassure a patient, the psychiatrist needs to:
- credibly identify with the patient’s distress
- and listen empathically (such as using body language and facial expressions that convey concern and consideration to the patient).24
Starcevic suggests useful techniques for providing reassurance (Table 3).22
DRUG THERAPIES
Psychotropics are considered a first-line treatment for patients with somatization disorders when:
- the patient spontaneously identifies any discrete, vegetative, or psychological complaints that may respond to drug therapy, such as insomnia, weight loss, sadness, or preoccupation
- the patient meets diagnostic criteria for comorbid anxiety or depressive disorders
- the therapeutic alliance is strong enough to weather the inevitable struggle with side effects and incomplete response to treatment. We do not recommend medication in the first encounter, when it may threaten a nascent alliance.
A common obstacle to prescribing psychotropics to somatizing patients is their sensitivity to suggestions that their complaints are “all in their heads.” To sidestep this resistance, describe the medication as treating the stress caused. by—not causing.—their chronic physical complaints. Proposing antidepressant therapy after—rather than instead of—physical exams and other diagnostics may elicit a more positive response.
Antidepressants. In clinical trials, somatoform complaints show moderate improvement after antidepressant treatment. In a meta-analysis of 6,595 patients with unexplained symptoms treated only with antidepressants, the number needed to treat was 3 to yield a positive response.25 This report of 94 medication trials included patients with headache, fibromyalgia, functional GI syndromes, idiopathic pain, tinnitus, or chronic fatigue.
In other trials:
- Amitriptyline has reduced somatic symptoms in patients labeled as having “masked depression.”26
- Sertraline has reduced disease fear, disease conviction, and bodily preoccupation in patients with hypochondriasis and panic disorder.27
Consider side effects when choosing medication for patients with somatoform disorders. Selective serotonin reuptake inhibitors (SSRIs) in general—and sertraline, citalopram, and escitalopram specifically—have fewer side effects than tricyclics. The adage of “start low, go slow” is appropriate for somatizing patients; we usually start with one-half the dosages recommended for treating depression.
Antipsychotics. In case reports, patients with “atypical psychosis,” “monosymptomatic hypochondriacal psychosis,” or “delusional disorder, somatic type” have responded to antipsychotics. These patients’ somatic beliefs are of delusional intensity, such as the rare fear of being eaten alive by an intestinal parasite (delusional parasitosis). Reported behaviors associated with the delusion include starvation, excessive laxative abuse, ingestion of sharp objects, and self-inflicted stab wounds. Treatments described in the literature include the typical agents pimozide and haloperidol and the atypicals olanzapine and risperidone.
TREATMENT-RESISTANT PATIENTS
Some patients with somatoform disorders will not accept CBT, psychotropics, reassurance, or referrals to group psychoeducation. Despite your best efforts, they may persist in focusing on somatic complaints. If you are willing to maintain a therapeutic relationship with them, be prepared to tolerate several ongoing paradoxes (Table 4).
Behaviorally, you must “listen more and do less.” Emotionally, you must be willing to enter into a long-term relationship with an inherently frustrating patient whose pathologies make you feel therapeutically hopeless and helpless. Understand that their physical symptoms function as a metaphor for psychological distress. You are not required to explore the source, content, or meaning of the metaphor in detail but simply listen to their somatic complaints through that psychological filter.
Related resources
- Starcevic V, Lipsitt D (eds). Hypochondriasis: modern perspectives on an ancient malady. New York: Oxford University Press, 2001.
- Information and support Web site for persons with health anxiety or hypochondria. www.healthanxiety.com
- Anxiety Disorders Association of America. www.adaa.org
Drug brand names
- Amitriptyline • Elavil
- Citalopram • Celexa
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Pimozide • Orap
- Risperidone • Risperdal
- Sertraline • Zoloft
Disclosure
Dr. Isaac reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Wise receives grant support from Eli Lilly & Co. and is a consultant or speaker for Eli Lilly & Co., Pfizer Inc., Bristol-Myers Squibb Co., and GlaxoSmithKline.
1. Katon W, Von Korff M, Lin E, et al. Distressed high utilizers of medical care. DSM-III-R diagnoses and treatment needs. Gen Hosp Psychiatry 1990;12:355-62.
2. Noyes R, Jr, Happel RL, Yagla SJ. Correlates of hypochondriasis in a nonclinical population. Psychosomatics 1999;40:461-9.
3. Mayou R, Sprigings D, Gilbert T. Patients with palpitations referred for 24-hour ECG recording. J Psychosom Res 1999;46:491-6.
4. Mayou RA, Bass C, Hart G, et al. Can clinical assessment of chest pain be made more therapeutic? Q J Med 2000;93:805-11.
5. Lipowski ZJ. Somatization: the experience and communication of psychological distress as somatic symptoms. Psychother Psychosom 1987;47:160-7.
6. Lipowski ZJ. Somatization: medicine’s unsolved problem. Psychosomatics 1987;28(6):294-297.
7. Ware NC, Kleinman A. Culture and somatic experience: the social course of illness in neurasthenia and chronic fatigue syndrome. Psychosom Med 1992;54:546-60.
8. Diagnostic and statistical manual of mental disorders (4th ed., text revision). Washington, DC: American Psychiatric Association, 2000.
9. Bass C, Peveler R, House A. Somatoform disorders: severe psychiatric illnesses neglected by psychiatrists. Br J Psychiatry 2001;179:11-14.
10. Smith GR. The epidemiology and treatment of depression when it coexists with somatoform disorders, somatization, or pain. Gen Hosp Psychiatry 1992;14:265-72.
11. Barsky AJ, Wyshak G, Klerman GL. Psychiatric comorbidity in DSM-III-R hypochondriasis. Arch Gen Psychiatry 1992;49:101-8.
12. Starcevic V. Role of reassurance and psychopathology in hypochondriasis. Psychiatry 1990;53(4):383-95.
13. Rost KM, Akins RN, Brown FW, Smith GR. The comorbidity of DSM-III-R personality disorders in somatization disorder. Gen Hosp Psychiatry 1992;14:322-6.
14. Morrison J. Childhood sexual histories of women with somatization disorder [comment]. Am J Psychiatry 1989;146:239-41.
15. Morse DS, Suchman AL, Frankel RM. The meaning of symptoms in 10 women with somatization disorder and a history of childhood abuse. Arch Fam Med 1997;6:468-76.
16. Kroenke K, Swindle R. Cognitive-behavioral therapy for somatization and symptom syndromes: a critical review of controlled clinical trials. Psychother Psychosom 2000;69:205-15.
17. Looper KJ, Kirmayer LJ. Behavioral medicine approaches to somatoform disorders. J Consult Clin Psychol 2002;70:810-27.
18. Warwick HM, Clark DM, Cobb AM, Salkovskis PM. A controlled trial of cognitive-behavioural treatment of hypochondriasis. Br J Psychiatry 1996;169:189-95.
19. Warwick HM, Salkovskis PM. Cognitive-behavioral treatment of hypochondriasis. In: Lipsitt DR, Starcevic V (eds). Hypochondriasis: Modern perspectives on an ancient malady. New York: Oxford Press, 2001;314-28.
20. McLeod CC, Budd MA. Treatment of somatization in primary care: evaluation of the Personal Health Improvement Program. HMO Pract 1997;11:88-94.
21. Bouman TK, Visser S. Cognitive and behavioural treatment of hypochondriasis. Psychother Psychosom 1998;67:214-21.
22. Starcevic V. Reassurance in the treatment of hypochondriasis. In: Lipsitt DR, Starcevic V (eds). Hypochondriasis: Modern perspectives on an ancient malady. New York: Oxford Press, 2001;291-313.
23. Clark DM, Salkovskis PM, Hackmann A, et al. Two psychological treatments for hypochondriasis. A randomised controlled trial. Br J Psychiatry 1998;173:218-25.
24. Schwartz L. Some notes on reassurance in medical practice. Psychosomatics 1966;7:290-4.
25. O’Malley PG, Jackson JL, Santoro J, et al. Antidepressant therapy for unexplained symptoms and symptom syndromes. J Fam Pract 1999;48:980-90.
26. Kellner R, Fava GA, Lisansky J, et al. Hypochondriacal fears and beliefs in DSM-III melancholia. Changes with amitriptyline. J Affect Disord 1986;10:21-6.
27. Noyes R, Reich J, Clancy J, O’Gorman TW. Reduction in hypochondriasis with treatment of panic disorder. Br J Psychiatry 1986;149:631-5.
Mrs. M, age 34, was referred for psychiatric evaluation by her primary care physician. She reluctantly agreed to the referral and tells the psychiatrist she “really should be seeing a cardiologist.” Numerous evaluations for chest pain and palpitations—including seven emergency room visits, ECGs and cardiac catheterization—have revealed no medical pathology.
A divorced mother of two children, she says she feels anxious about her “heart condition.” Her father died of a heart attack at age 51. She experiences chest pains at home and at work, particularly when under stress. Sometimes she feels her heart racing and numbness or tingling in her arms.
Although her primary care physician has seen her frequently during the past 6 months, she says the doctor is not taking her complaints seriously. “These chest pains are real,” she says, “so don’t try to tell me they’re all in my head.”
Psychiatrists may be the last doctors patients such as Mrs. M wish to see but the ones best equipped to relieve their suffering. Our experience in treating somatizing patients and the available evidence suggest that cognitive-behavioral therapy (CBT) combined with psychoeducation, reassurance, and sometimes drug therapy is the most effective approach.
Health-related fear—or “illness worry”—is common, occurring in nearly 10% of adults who responded to a recent community survey.2 When this fear drives individuals to their physicians for evaluation, frequently no organic cause is discovered. Full evaluations are expensive and lead to increased use of health care resources, including potentially dangerous invasive testing.3,4
Defining somatization has been a source of confusion.5,6 Some authors consider somatic complaints to be expressions of suppressed psychosocial stressors. Others label them as medically unexplained complaints, although this definition fails to exclude occult medical problems. Kleinman7 defines somatization as “a somatic idiom of psychosocial distress in a setting of medical health-care seeking.” This useful definition links psychosocial problems with somatic complaints and the behavioral drive to obtain a medical evaluation.
In DSM-IV,8 the defining characteristics of somatoform disorders are somatic complaints or disease fears that are out of proportion with any identifiable somatic cause. Entities include somatization disorder, undifferentiated somatization disorder, conversion disorder, pain disorder, hypochondriasis, body dysmorphic disorder, and somatoform disorder–not otherwise specified (NOS).
Subthreshold symptoms. Unfortunately, DSM-IV’s categorization of Axis I somatoform disorders does not capture subthreshold presentations, which are common. Patients with less than the required number of somatic complaints are labeled in a wastebasket fashion with “undifferentiated somatoform disorder.”9
Mrs. M’s persistent chest pain of noncardiac origin is a familiar health anxiety, along with functional GI complaints, headaches, chronic fatigue, and lower back pain. Frustrating to their doctors and frustrated themselves, patients with medically unexplained complaints consume an inordinate amount of physicians’ time.1
Without a clear definition of somatization (Box)2-9 or useful clinical guidelines, psychiatrists must rely on the literature for guidance in managing somatization disorders. This article summarizes the evidence and describes how we apply these findings to practice. And when all else fails, we offer last-ditch advice for managing patients who resist your treatment efforts.
IDENTIFYING COMORBIDITIES
Identifying psychiatric comorbidities is the first step in successfully treating patients with somatoform complaints. In an epidemiologic study, 60% of patients with somatoform complaints also had a mood disorder and 48% had an anxiety disorder.10 In a similar study of patients with hypochondriasis, 88% also had one or more Axis I diagnosis.11
If a patient meets criteria for a comorbid psychiatric disorder and is willing to be treated for it, the somatic complaints may resolve along with the underlying disorder. In fact, the presence of an identifiable Axis I disorder order may predict a more positive prognosis.12
Personality disorders. Somatization in patients with a personality disorder poses unique challenges.13 Granted, when making a diagnosis it is difficult to tease apart somatization from personality disorders because somatization itself may be considered a chronic, maladaptive coping style. However, symptoms such as deception, impulsivity, mood lability, and self-injurious behavior introduce treatment complications that exceed the scope of this article.
Posttraumatic stress disorder (PTSD)—particularly childhood sexual and physical abuse—also predisposes some patients to somatization disorders.14,15 Patients with comorbid PTSD and somatization disorder require highly specialized treatment that is beyond the scope of this review.
COGNITIVE-BEHAVIORAL TREATMENT
Cognitive-behavioral therapy (CBT) is the best-studied and most effective treatment for somatoform disorders.16 CBT for somatization relies on both physiologic and cognitive explanations to account for the patient’s experience, without committing to an “either/or” dichotomy. It offers patients an alternate explanation of what is wrong with them—illness anxiety instead of severe physical illness.
By making patients aware of their automatic thoughts, feelings, behaviors, and underlying beliefs, CBT helps them normalize and cope with their illness anxiety. CBT techniques can be applied in a predetermined course of therapy (such as 12 sessions with a mental health clinician), in a group setting, or piecemeal by any health care provider.
Effective strategies. In a review of 30 controlled trials of CBT for somatoform disorders, Looper17 showed overall effect ranging from 0.38 to 2.5, where 0.2 was defined as a small effect, 0.5 as medium, and 0.8 as large. Hypochondriasis, somatization disorder, body dysmorphic disorder, chronic pain, chronic fatigue, and noncardiac chest pain were included in this review. The most effective strategies:
- included 6 to 16 treatment sessions
- were symptom-focused as opposed to providing general relaxation training
- included maintenance sessions after the initial series.
Four factors of health anxiety. CBT primarily targets the patient’s false beliefs that he or she is physically ill. These beliefs are based on how the patient misinterprets innocuous physical symptoms and responds to them.18 The cognitive theory of health anxiety holds that health anxiety severity is affected by four factors:
- perceived likelihood of illness
- perceived burden of illness
- perceived ability to cope with illness
- perception of the extent to which external factors will help.19
Table 1
Common dysfunctional beliefs of somatizing patients
|
The first two factors worsen and the latter two mitigate health anxiety. An individual patient’s presenting fears often suggest which factors to address. For example, Mrs. M may describe the burden of illness as the focus of her fears (“If I have a heart attack, who will care for my children?”). This information cues you to shift the focus of therapy to helping her cope with child care needs despite her recurring symptoms.
If she focuses on her likelihood of illness, then uncoupling the symptoms from the diagnosis could be more productive. When she reports palpitations, diaphoresis, and dizziness, have her do breathing exercises that induce those symptoms without producing a heart attack.
Table 2
Journaling homework: 5 questions for patients to answer about one symptom each day
|
She might describe feeling unable to cope when she feels symptoms or when cardiologists tell her nothing is wrong with her heart. In that case, focus on relaxation techniques, global stress reduction, and reducing cardiac risk factors to bolster her ability to cope with her illness.
Journaling is a critical component of CBT in treating somatization disorders. Regular journaling by the patient can reveal dysfunctional beliefs that may be driving his or her health anxieties, such as those listed in Table 1. We find it useful to assign patients to answer five questions about one symptom experience each day (Table 2). This self-monitoring provides material to work on with the patient during each session.
Cognitive restructuring. During therapy sessions, we ask patients to suggest alternate explanations for the symptoms recorded in their journals. We then ask them to determine which explanations are more feasible.
For example, if Mrs. M develops palpitations during emotionally charged arguments, we would ask her to develop explanations other than, “I was having a heart attack.” Reality testing includes rhetorical questions such as, “Would you be alive today if you were having a heart attack every time you had palpitations?” Automatic thoughts are successively identified and then tested aloud with the patient:
- “Has every unexplained symptom led to the discovery of a serious illness?”
- “Does every instance of hurt equal harm?”
Eventually, patterns of automatic thoughts emerge, and these reveal the underlying dysfunctional beliefs.
Dysfunctional beliefs are maintained when patients selectively attend to and amplify somatic sensations. Behavioral experiments during sessions can demonstrate to the patient in vivo the process by which they misattribute illness to physical symptoms. For example, overbreathing with a patient during a session may elicit light-headedness, paresthesias, or tachycardia, which can then be linked to overbreathing rather than a chronic or catastrophic illness.
Furthermore, patients can be taught to control the experience. Some patients with headaches or GI pain may be made aware of symptoms by simply asking them to focus their attention on the respective organs. Simply explaining the cycle of misattribution, autonomic activation, and further symptom development with an in vivo demonstration can be illuminating.
Response prevention. Another behavioral technique is to cut back in small increments on actions the patient takes in response to physical symptoms and automatic thoughts. For example, a patient could take medicine and seek reassurance less frequently and avoid rubbing the affected area.
PSYCHOEDUCATION
Two psychoeducation programs for somatization behavior have been formally studied.
The Personal Health Improvement Program20—led by trained facilitators—includes classroom videos, cognitive-behavioral exercises, and home study assignments. After completing the 6-week course, 171 patients with somatization disorders reported reduced physical and psychological distress and increased function. They also visited their primary care physicians less often.
Table 3
How to effectively reassure somatizing patients
| Action | Benefit |
|---|---|
| Review records in front of patients | Demonstrates that you take complaints and histories seriously |
| Acknowledge the severity of patients’ distress | Validates subjective suffering |
| Schedule follow-up visits at regular intervals | Provides access to you and continuity of care; reduces extra phone calls and emergency visits |
| Use clear and simple language | Improves communication |
| Explain that they do not have life-threatening structural disease | Opens door to cognitive restructuring |
| Assign jobs, such as journaling 15 min/day and rounding up medical records | Builds therapeutic alliance, fosters patient responsibility, and restores patients’ sense of control |
| Identify and support the patient’s strengths | Builds self-esteem |
| Use specialty referrals sparingly | Reduces risk of further medical testing and patient anxiety while awaiting results |
Coping with Illness Anxiety21 relies on mini-lectures, demonstrations, videos, and focused group discussions. After six 2-hour sessions, 33 of 43 study patients (78%) used medical services less often and reported reduced disease conviction, consequences of bodily complaints, health anxiety, and checking and avoidance behaviors. Two psychology graduate students taught the course from a manual, with 6 to 9 patients per group.
Psychoeducation in this context relies on didactic presentations, readings, role playing, and videotaped material. The goal is to teach patients to recognize thoughts, emotions, and behaviors that lead to and result from somatic preoccupation. Patients can improve when they recognize dysfunctional behavioral patterns and learn alternate coping strategies.
Somatizing patients—with their aversion to the stigma of mental illness—may find psychoeducation particularly attractive. They can be treated as students who are being educated, rather than as patients who are being treated. Classrooms in both studies cited above were located in medical outpatient offices, not in mental health facilities.
REASSURANCE
Reassurance is a common therapeutic technique in medicine, although it is poorly understood, poorly taught, and not methodically applied. Reassurance alleviates anxiety, enables patients to endure dysphoria, encourages hope, gives insight, and enhances the doctor-patient relationship.22
Table 4
How to avoid becoming frustrated with persistent somatization
| Situation | Response |
|---|---|
| Despite patients’ urgency | Watch and wait, knowing that psychological distress has been chronic |
| Despite patients’ belief that a single pill or procedure will ‘cure’ them | Persist in ‘rehabilitative’ approach |
| Despite patients’ provocations to force you to take a dichotomous approach | Persist in using both physical and psychological explanations |
| Despite your knowledge that patients are actively maintaining their illness beliefs | Try to be patient as they attribute their misfortune to ‘fate,’ ‘bad luck,’ or ‘misfortune’ |
| Despite the fact that you have agreed to treat the patient | Realize that his or her family or culture may reinforce the ‘sick role’ as the only acceptable form of distress |
| Despite patients’ desire to discuss symptoms | Reorient them to sustaining daily function (such as parenting while tolerating fatigue) |
Whereas CBT seeks to challenge patients’ underlying beliefs and restructure their thought processes,23 reassurance can help them tolerate their dysfunctional beliefs and dissuade them from believing their health is dangerously impaired. Reassurance offers a substitute explanation of patients’ dysfunction, although this explanation is not as central or detailed as it is in CBT.
How to reassure. Patients may consider reassurance offered prematurely or by a stranger to be patronizing or dismissive. Reassurance is most effective when:
- given by a trusted person who is reliable, consistent, firm, and empathic
- the patient’s condition has been established as unresponsive to conventional diagnostics or biological therapies.
Patients are most receptive to reassurance when they express distress or frustration with their unexplained symptoms. Affirming that their suffering is legitimate opens the door to further treatment.
Reassurance is least effective when a patient is expressing anger or mistrust, although this is when the physician may feel most pressured to reassure. To successfully reassure a patient, the psychiatrist needs to:
- credibly identify with the patient’s distress
- and listen empathically (such as using body language and facial expressions that convey concern and consideration to the patient).24
Starcevic suggests useful techniques for providing reassurance (Table 3).22
DRUG THERAPIES
Psychotropics are considered a first-line treatment for patients with somatization disorders when:
- the patient spontaneously identifies any discrete, vegetative, or psychological complaints that may respond to drug therapy, such as insomnia, weight loss, sadness, or preoccupation
- the patient meets diagnostic criteria for comorbid anxiety or depressive disorders
- the therapeutic alliance is strong enough to weather the inevitable struggle with side effects and incomplete response to treatment. We do not recommend medication in the first encounter, when it may threaten a nascent alliance.
A common obstacle to prescribing psychotropics to somatizing patients is their sensitivity to suggestions that their complaints are “all in their heads.” To sidestep this resistance, describe the medication as treating the stress caused. by—not causing.—their chronic physical complaints. Proposing antidepressant therapy after—rather than instead of—physical exams and other diagnostics may elicit a more positive response.
Antidepressants. In clinical trials, somatoform complaints show moderate improvement after antidepressant treatment. In a meta-analysis of 6,595 patients with unexplained symptoms treated only with antidepressants, the number needed to treat was 3 to yield a positive response.25 This report of 94 medication trials included patients with headache, fibromyalgia, functional GI syndromes, idiopathic pain, tinnitus, or chronic fatigue.
In other trials:
- Amitriptyline has reduced somatic symptoms in patients labeled as having “masked depression.”26
- Sertraline has reduced disease fear, disease conviction, and bodily preoccupation in patients with hypochondriasis and panic disorder.27
Consider side effects when choosing medication for patients with somatoform disorders. Selective serotonin reuptake inhibitors (SSRIs) in general—and sertraline, citalopram, and escitalopram specifically—have fewer side effects than tricyclics. The adage of “start low, go slow” is appropriate for somatizing patients; we usually start with one-half the dosages recommended for treating depression.
Antipsychotics. In case reports, patients with “atypical psychosis,” “monosymptomatic hypochondriacal psychosis,” or “delusional disorder, somatic type” have responded to antipsychotics. These patients’ somatic beliefs are of delusional intensity, such as the rare fear of being eaten alive by an intestinal parasite (delusional parasitosis). Reported behaviors associated with the delusion include starvation, excessive laxative abuse, ingestion of sharp objects, and self-inflicted stab wounds. Treatments described in the literature include the typical agents pimozide and haloperidol and the atypicals olanzapine and risperidone.
TREATMENT-RESISTANT PATIENTS
Some patients with somatoform disorders will not accept CBT, psychotropics, reassurance, or referrals to group psychoeducation. Despite your best efforts, they may persist in focusing on somatic complaints. If you are willing to maintain a therapeutic relationship with them, be prepared to tolerate several ongoing paradoxes (Table 4).
Behaviorally, you must “listen more and do less.” Emotionally, you must be willing to enter into a long-term relationship with an inherently frustrating patient whose pathologies make you feel therapeutically hopeless and helpless. Understand that their physical symptoms function as a metaphor for psychological distress. You are not required to explore the source, content, or meaning of the metaphor in detail but simply listen to their somatic complaints through that psychological filter.
Related resources
- Starcevic V, Lipsitt D (eds). Hypochondriasis: modern perspectives on an ancient malady. New York: Oxford University Press, 2001.
- Information and support Web site for persons with health anxiety or hypochondria. www.healthanxiety.com
- Anxiety Disorders Association of America. www.adaa.org
Drug brand names
- Amitriptyline • Elavil
- Citalopram • Celexa
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Pimozide • Orap
- Risperidone • Risperdal
- Sertraline • Zoloft
Disclosure
Dr. Isaac reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Wise receives grant support from Eli Lilly & Co. and is a consultant or speaker for Eli Lilly & Co., Pfizer Inc., Bristol-Myers Squibb Co., and GlaxoSmithKline.
Mrs. M, age 34, was referred for psychiatric evaluation by her primary care physician. She reluctantly agreed to the referral and tells the psychiatrist she “really should be seeing a cardiologist.” Numerous evaluations for chest pain and palpitations—including seven emergency room visits, ECGs and cardiac catheterization—have revealed no medical pathology.
A divorced mother of two children, she says she feels anxious about her “heart condition.” Her father died of a heart attack at age 51. She experiences chest pains at home and at work, particularly when under stress. Sometimes she feels her heart racing and numbness or tingling in her arms.
Although her primary care physician has seen her frequently during the past 6 months, she says the doctor is not taking her complaints seriously. “These chest pains are real,” she says, “so don’t try to tell me they’re all in my head.”
Psychiatrists may be the last doctors patients such as Mrs. M wish to see but the ones best equipped to relieve their suffering. Our experience in treating somatizing patients and the available evidence suggest that cognitive-behavioral therapy (CBT) combined with psychoeducation, reassurance, and sometimes drug therapy is the most effective approach.
Health-related fear—or “illness worry”—is common, occurring in nearly 10% of adults who responded to a recent community survey.2 When this fear drives individuals to their physicians for evaluation, frequently no organic cause is discovered. Full evaluations are expensive and lead to increased use of health care resources, including potentially dangerous invasive testing.3,4
Defining somatization has been a source of confusion.5,6 Some authors consider somatic complaints to be expressions of suppressed psychosocial stressors. Others label them as medically unexplained complaints, although this definition fails to exclude occult medical problems. Kleinman7 defines somatization as “a somatic idiom of psychosocial distress in a setting of medical health-care seeking.” This useful definition links psychosocial problems with somatic complaints and the behavioral drive to obtain a medical evaluation.
In DSM-IV,8 the defining characteristics of somatoform disorders are somatic complaints or disease fears that are out of proportion with any identifiable somatic cause. Entities include somatization disorder, undifferentiated somatization disorder, conversion disorder, pain disorder, hypochondriasis, body dysmorphic disorder, and somatoform disorder–not otherwise specified (NOS).
Subthreshold symptoms. Unfortunately, DSM-IV’s categorization of Axis I somatoform disorders does not capture subthreshold presentations, which are common. Patients with less than the required number of somatic complaints are labeled in a wastebasket fashion with “undifferentiated somatoform disorder.”9
Mrs. M’s persistent chest pain of noncardiac origin is a familiar health anxiety, along with functional GI complaints, headaches, chronic fatigue, and lower back pain. Frustrating to their doctors and frustrated themselves, patients with medically unexplained complaints consume an inordinate amount of physicians’ time.1
Without a clear definition of somatization (Box)2-9 or useful clinical guidelines, psychiatrists must rely on the literature for guidance in managing somatization disorders. This article summarizes the evidence and describes how we apply these findings to practice. And when all else fails, we offer last-ditch advice for managing patients who resist your treatment efforts.
IDENTIFYING COMORBIDITIES
Identifying psychiatric comorbidities is the first step in successfully treating patients with somatoform complaints. In an epidemiologic study, 60% of patients with somatoform complaints also had a mood disorder and 48% had an anxiety disorder.10 In a similar study of patients with hypochondriasis, 88% also had one or more Axis I diagnosis.11
If a patient meets criteria for a comorbid psychiatric disorder and is willing to be treated for it, the somatic complaints may resolve along with the underlying disorder. In fact, the presence of an identifiable Axis I disorder order may predict a more positive prognosis.12
Personality disorders. Somatization in patients with a personality disorder poses unique challenges.13 Granted, when making a diagnosis it is difficult to tease apart somatization from personality disorders because somatization itself may be considered a chronic, maladaptive coping style. However, symptoms such as deception, impulsivity, mood lability, and self-injurious behavior introduce treatment complications that exceed the scope of this article.
Posttraumatic stress disorder (PTSD)—particularly childhood sexual and physical abuse—also predisposes some patients to somatization disorders.14,15 Patients with comorbid PTSD and somatization disorder require highly specialized treatment that is beyond the scope of this review.
COGNITIVE-BEHAVIORAL TREATMENT
Cognitive-behavioral therapy (CBT) is the best-studied and most effective treatment for somatoform disorders.16 CBT for somatization relies on both physiologic and cognitive explanations to account for the patient’s experience, without committing to an “either/or” dichotomy. It offers patients an alternate explanation of what is wrong with them—illness anxiety instead of severe physical illness.
By making patients aware of their automatic thoughts, feelings, behaviors, and underlying beliefs, CBT helps them normalize and cope with their illness anxiety. CBT techniques can be applied in a predetermined course of therapy (such as 12 sessions with a mental health clinician), in a group setting, or piecemeal by any health care provider.
Effective strategies. In a review of 30 controlled trials of CBT for somatoform disorders, Looper17 showed overall effect ranging from 0.38 to 2.5, where 0.2 was defined as a small effect, 0.5 as medium, and 0.8 as large. Hypochondriasis, somatization disorder, body dysmorphic disorder, chronic pain, chronic fatigue, and noncardiac chest pain were included in this review. The most effective strategies:
- included 6 to 16 treatment sessions
- were symptom-focused as opposed to providing general relaxation training
- included maintenance sessions after the initial series.
Four factors of health anxiety. CBT primarily targets the patient’s false beliefs that he or she is physically ill. These beliefs are based on how the patient misinterprets innocuous physical symptoms and responds to them.18 The cognitive theory of health anxiety holds that health anxiety severity is affected by four factors:
- perceived likelihood of illness
- perceived burden of illness
- perceived ability to cope with illness
- perception of the extent to which external factors will help.19
Table 1
Common dysfunctional beliefs of somatizing patients
|
The first two factors worsen and the latter two mitigate health anxiety. An individual patient’s presenting fears often suggest which factors to address. For example, Mrs. M may describe the burden of illness as the focus of her fears (“If I have a heart attack, who will care for my children?”). This information cues you to shift the focus of therapy to helping her cope with child care needs despite her recurring symptoms.
If she focuses on her likelihood of illness, then uncoupling the symptoms from the diagnosis could be more productive. When she reports palpitations, diaphoresis, and dizziness, have her do breathing exercises that induce those symptoms without producing a heart attack.
Table 2
Journaling homework: 5 questions for patients to answer about one symptom each day
|
She might describe feeling unable to cope when she feels symptoms or when cardiologists tell her nothing is wrong with her heart. In that case, focus on relaxation techniques, global stress reduction, and reducing cardiac risk factors to bolster her ability to cope with her illness.
Journaling is a critical component of CBT in treating somatization disorders. Regular journaling by the patient can reveal dysfunctional beliefs that may be driving his or her health anxieties, such as those listed in Table 1. We find it useful to assign patients to answer five questions about one symptom experience each day (Table 2). This self-monitoring provides material to work on with the patient during each session.
Cognitive restructuring. During therapy sessions, we ask patients to suggest alternate explanations for the symptoms recorded in their journals. We then ask them to determine which explanations are more feasible.
For example, if Mrs. M develops palpitations during emotionally charged arguments, we would ask her to develop explanations other than, “I was having a heart attack.” Reality testing includes rhetorical questions such as, “Would you be alive today if you were having a heart attack every time you had palpitations?” Automatic thoughts are successively identified and then tested aloud with the patient:
- “Has every unexplained symptom led to the discovery of a serious illness?”
- “Does every instance of hurt equal harm?”
Eventually, patterns of automatic thoughts emerge, and these reveal the underlying dysfunctional beliefs.
Dysfunctional beliefs are maintained when patients selectively attend to and amplify somatic sensations. Behavioral experiments during sessions can demonstrate to the patient in vivo the process by which they misattribute illness to physical symptoms. For example, overbreathing with a patient during a session may elicit light-headedness, paresthesias, or tachycardia, which can then be linked to overbreathing rather than a chronic or catastrophic illness.
Furthermore, patients can be taught to control the experience. Some patients with headaches or GI pain may be made aware of symptoms by simply asking them to focus their attention on the respective organs. Simply explaining the cycle of misattribution, autonomic activation, and further symptom development with an in vivo demonstration can be illuminating.
Response prevention. Another behavioral technique is to cut back in small increments on actions the patient takes in response to physical symptoms and automatic thoughts. For example, a patient could take medicine and seek reassurance less frequently and avoid rubbing the affected area.
PSYCHOEDUCATION
Two psychoeducation programs for somatization behavior have been formally studied.
The Personal Health Improvement Program20—led by trained facilitators—includes classroom videos, cognitive-behavioral exercises, and home study assignments. After completing the 6-week course, 171 patients with somatization disorders reported reduced physical and psychological distress and increased function. They also visited their primary care physicians less often.
Table 3
How to effectively reassure somatizing patients
| Action | Benefit |
|---|---|
| Review records in front of patients | Demonstrates that you take complaints and histories seriously |
| Acknowledge the severity of patients’ distress | Validates subjective suffering |
| Schedule follow-up visits at regular intervals | Provides access to you and continuity of care; reduces extra phone calls and emergency visits |
| Use clear and simple language | Improves communication |
| Explain that they do not have life-threatening structural disease | Opens door to cognitive restructuring |
| Assign jobs, such as journaling 15 min/day and rounding up medical records | Builds therapeutic alliance, fosters patient responsibility, and restores patients’ sense of control |
| Identify and support the patient’s strengths | Builds self-esteem |
| Use specialty referrals sparingly | Reduces risk of further medical testing and patient anxiety while awaiting results |
Coping with Illness Anxiety21 relies on mini-lectures, demonstrations, videos, and focused group discussions. After six 2-hour sessions, 33 of 43 study patients (78%) used medical services less often and reported reduced disease conviction, consequences of bodily complaints, health anxiety, and checking and avoidance behaviors. Two psychology graduate students taught the course from a manual, with 6 to 9 patients per group.
Psychoeducation in this context relies on didactic presentations, readings, role playing, and videotaped material. The goal is to teach patients to recognize thoughts, emotions, and behaviors that lead to and result from somatic preoccupation. Patients can improve when they recognize dysfunctional behavioral patterns and learn alternate coping strategies.
Somatizing patients—with their aversion to the stigma of mental illness—may find psychoeducation particularly attractive. They can be treated as students who are being educated, rather than as patients who are being treated. Classrooms in both studies cited above were located in medical outpatient offices, not in mental health facilities.
REASSURANCE
Reassurance is a common therapeutic technique in medicine, although it is poorly understood, poorly taught, and not methodically applied. Reassurance alleviates anxiety, enables patients to endure dysphoria, encourages hope, gives insight, and enhances the doctor-patient relationship.22
Table 4
How to avoid becoming frustrated with persistent somatization
| Situation | Response |
|---|---|
| Despite patients’ urgency | Watch and wait, knowing that psychological distress has been chronic |
| Despite patients’ belief that a single pill or procedure will ‘cure’ them | Persist in ‘rehabilitative’ approach |
| Despite patients’ provocations to force you to take a dichotomous approach | Persist in using both physical and psychological explanations |
| Despite your knowledge that patients are actively maintaining their illness beliefs | Try to be patient as they attribute their misfortune to ‘fate,’ ‘bad luck,’ or ‘misfortune’ |
| Despite the fact that you have agreed to treat the patient | Realize that his or her family or culture may reinforce the ‘sick role’ as the only acceptable form of distress |
| Despite patients’ desire to discuss symptoms | Reorient them to sustaining daily function (such as parenting while tolerating fatigue) |
Whereas CBT seeks to challenge patients’ underlying beliefs and restructure their thought processes,23 reassurance can help them tolerate their dysfunctional beliefs and dissuade them from believing their health is dangerously impaired. Reassurance offers a substitute explanation of patients’ dysfunction, although this explanation is not as central or detailed as it is in CBT.
How to reassure. Patients may consider reassurance offered prematurely or by a stranger to be patronizing or dismissive. Reassurance is most effective when:
- given by a trusted person who is reliable, consistent, firm, and empathic
- the patient’s condition has been established as unresponsive to conventional diagnostics or biological therapies.
Patients are most receptive to reassurance when they express distress or frustration with their unexplained symptoms. Affirming that their suffering is legitimate opens the door to further treatment.
Reassurance is least effective when a patient is expressing anger or mistrust, although this is when the physician may feel most pressured to reassure. To successfully reassure a patient, the psychiatrist needs to:
- credibly identify with the patient’s distress
- and listen empathically (such as using body language and facial expressions that convey concern and consideration to the patient).24
Starcevic suggests useful techniques for providing reassurance (Table 3).22
DRUG THERAPIES
Psychotropics are considered a first-line treatment for patients with somatization disorders when:
- the patient spontaneously identifies any discrete, vegetative, or psychological complaints that may respond to drug therapy, such as insomnia, weight loss, sadness, or preoccupation
- the patient meets diagnostic criteria for comorbid anxiety or depressive disorders
- the therapeutic alliance is strong enough to weather the inevitable struggle with side effects and incomplete response to treatment. We do not recommend medication in the first encounter, when it may threaten a nascent alliance.
A common obstacle to prescribing psychotropics to somatizing patients is their sensitivity to suggestions that their complaints are “all in their heads.” To sidestep this resistance, describe the medication as treating the stress caused. by—not causing.—their chronic physical complaints. Proposing antidepressant therapy after—rather than instead of—physical exams and other diagnostics may elicit a more positive response.
Antidepressants. In clinical trials, somatoform complaints show moderate improvement after antidepressant treatment. In a meta-analysis of 6,595 patients with unexplained symptoms treated only with antidepressants, the number needed to treat was 3 to yield a positive response.25 This report of 94 medication trials included patients with headache, fibromyalgia, functional GI syndromes, idiopathic pain, tinnitus, or chronic fatigue.
In other trials:
- Amitriptyline has reduced somatic symptoms in patients labeled as having “masked depression.”26
- Sertraline has reduced disease fear, disease conviction, and bodily preoccupation in patients with hypochondriasis and panic disorder.27
Consider side effects when choosing medication for patients with somatoform disorders. Selective serotonin reuptake inhibitors (SSRIs) in general—and sertraline, citalopram, and escitalopram specifically—have fewer side effects than tricyclics. The adage of “start low, go slow” is appropriate for somatizing patients; we usually start with one-half the dosages recommended for treating depression.
Antipsychotics. In case reports, patients with “atypical psychosis,” “monosymptomatic hypochondriacal psychosis,” or “delusional disorder, somatic type” have responded to antipsychotics. These patients’ somatic beliefs are of delusional intensity, such as the rare fear of being eaten alive by an intestinal parasite (delusional parasitosis). Reported behaviors associated with the delusion include starvation, excessive laxative abuse, ingestion of sharp objects, and self-inflicted stab wounds. Treatments described in the literature include the typical agents pimozide and haloperidol and the atypicals olanzapine and risperidone.
TREATMENT-RESISTANT PATIENTS
Some patients with somatoform disorders will not accept CBT, psychotropics, reassurance, or referrals to group psychoeducation. Despite your best efforts, they may persist in focusing on somatic complaints. If you are willing to maintain a therapeutic relationship with them, be prepared to tolerate several ongoing paradoxes (Table 4).
Behaviorally, you must “listen more and do less.” Emotionally, you must be willing to enter into a long-term relationship with an inherently frustrating patient whose pathologies make you feel therapeutically hopeless and helpless. Understand that their physical symptoms function as a metaphor for psychological distress. You are not required to explore the source, content, or meaning of the metaphor in detail but simply listen to their somatic complaints through that psychological filter.
Related resources
- Starcevic V, Lipsitt D (eds). Hypochondriasis: modern perspectives on an ancient malady. New York: Oxford University Press, 2001.
- Information and support Web site for persons with health anxiety or hypochondria. www.healthanxiety.com
- Anxiety Disorders Association of America. www.adaa.org
Drug brand names
- Amitriptyline • Elavil
- Citalopram • Celexa
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Pimozide • Orap
- Risperidone • Risperdal
- Sertraline • Zoloft
Disclosure
Dr. Isaac reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Wise receives grant support from Eli Lilly & Co. and is a consultant or speaker for Eli Lilly & Co., Pfizer Inc., Bristol-Myers Squibb Co., and GlaxoSmithKline.
1. Katon W, Von Korff M, Lin E, et al. Distressed high utilizers of medical care. DSM-III-R diagnoses and treatment needs. Gen Hosp Psychiatry 1990;12:355-62.
2. Noyes R, Jr, Happel RL, Yagla SJ. Correlates of hypochondriasis in a nonclinical population. Psychosomatics 1999;40:461-9.
3. Mayou R, Sprigings D, Gilbert T. Patients with palpitations referred for 24-hour ECG recording. J Psychosom Res 1999;46:491-6.
4. Mayou RA, Bass C, Hart G, et al. Can clinical assessment of chest pain be made more therapeutic? Q J Med 2000;93:805-11.
5. Lipowski ZJ. Somatization: the experience and communication of psychological distress as somatic symptoms. Psychother Psychosom 1987;47:160-7.
6. Lipowski ZJ. Somatization: medicine’s unsolved problem. Psychosomatics 1987;28(6):294-297.
7. Ware NC, Kleinman A. Culture and somatic experience: the social course of illness in neurasthenia and chronic fatigue syndrome. Psychosom Med 1992;54:546-60.
8. Diagnostic and statistical manual of mental disorders (4th ed., text revision). Washington, DC: American Psychiatric Association, 2000.
9. Bass C, Peveler R, House A. Somatoform disorders: severe psychiatric illnesses neglected by psychiatrists. Br J Psychiatry 2001;179:11-14.
10. Smith GR. The epidemiology and treatment of depression when it coexists with somatoform disorders, somatization, or pain. Gen Hosp Psychiatry 1992;14:265-72.
11. Barsky AJ, Wyshak G, Klerman GL. Psychiatric comorbidity in DSM-III-R hypochondriasis. Arch Gen Psychiatry 1992;49:101-8.
12. Starcevic V. Role of reassurance and psychopathology in hypochondriasis. Psychiatry 1990;53(4):383-95.
13. Rost KM, Akins RN, Brown FW, Smith GR. The comorbidity of DSM-III-R personality disorders in somatization disorder. Gen Hosp Psychiatry 1992;14:322-6.
14. Morrison J. Childhood sexual histories of women with somatization disorder [comment]. Am J Psychiatry 1989;146:239-41.
15. Morse DS, Suchman AL, Frankel RM. The meaning of symptoms in 10 women with somatization disorder and a history of childhood abuse. Arch Fam Med 1997;6:468-76.
16. Kroenke K, Swindle R. Cognitive-behavioral therapy for somatization and symptom syndromes: a critical review of controlled clinical trials. Psychother Psychosom 2000;69:205-15.
17. Looper KJ, Kirmayer LJ. Behavioral medicine approaches to somatoform disorders. J Consult Clin Psychol 2002;70:810-27.
18. Warwick HM, Clark DM, Cobb AM, Salkovskis PM. A controlled trial of cognitive-behavioural treatment of hypochondriasis. Br J Psychiatry 1996;169:189-95.
19. Warwick HM, Salkovskis PM. Cognitive-behavioral treatment of hypochondriasis. In: Lipsitt DR, Starcevic V (eds). Hypochondriasis: Modern perspectives on an ancient malady. New York: Oxford Press, 2001;314-28.
20. McLeod CC, Budd MA. Treatment of somatization in primary care: evaluation of the Personal Health Improvement Program. HMO Pract 1997;11:88-94.
21. Bouman TK, Visser S. Cognitive and behavioural treatment of hypochondriasis. Psychother Psychosom 1998;67:214-21.
22. Starcevic V. Reassurance in the treatment of hypochondriasis. In: Lipsitt DR, Starcevic V (eds). Hypochondriasis: Modern perspectives on an ancient malady. New York: Oxford Press, 2001;291-313.
23. Clark DM, Salkovskis PM, Hackmann A, et al. Two psychological treatments for hypochondriasis. A randomised controlled trial. Br J Psychiatry 1998;173:218-25.
24. Schwartz L. Some notes on reassurance in medical practice. Psychosomatics 1966;7:290-4.
25. O’Malley PG, Jackson JL, Santoro J, et al. Antidepressant therapy for unexplained symptoms and symptom syndromes. J Fam Pract 1999;48:980-90.
26. Kellner R, Fava GA, Lisansky J, et al. Hypochondriacal fears and beliefs in DSM-III melancholia. Changes with amitriptyline. J Affect Disord 1986;10:21-6.
27. Noyes R, Reich J, Clancy J, O’Gorman TW. Reduction in hypochondriasis with treatment of panic disorder. Br J Psychiatry 1986;149:631-5.
1. Katon W, Von Korff M, Lin E, et al. Distressed high utilizers of medical care. DSM-III-R diagnoses and treatment needs. Gen Hosp Psychiatry 1990;12:355-62.
2. Noyes R, Jr, Happel RL, Yagla SJ. Correlates of hypochondriasis in a nonclinical population. Psychosomatics 1999;40:461-9.
3. Mayou R, Sprigings D, Gilbert T. Patients with palpitations referred for 24-hour ECG recording. J Psychosom Res 1999;46:491-6.
4. Mayou RA, Bass C, Hart G, et al. Can clinical assessment of chest pain be made more therapeutic? Q J Med 2000;93:805-11.
5. Lipowski ZJ. Somatization: the experience and communication of psychological distress as somatic symptoms. Psychother Psychosom 1987;47:160-7.
6. Lipowski ZJ. Somatization: medicine’s unsolved problem. Psychosomatics 1987;28(6):294-297.
7. Ware NC, Kleinman A. Culture and somatic experience: the social course of illness in neurasthenia and chronic fatigue syndrome. Psychosom Med 1992;54:546-60.
8. Diagnostic and statistical manual of mental disorders (4th ed., text revision). Washington, DC: American Psychiatric Association, 2000.
9. Bass C, Peveler R, House A. Somatoform disorders: severe psychiatric illnesses neglected by psychiatrists. Br J Psychiatry 2001;179:11-14.
10. Smith GR. The epidemiology and treatment of depression when it coexists with somatoform disorders, somatization, or pain. Gen Hosp Psychiatry 1992;14:265-72.
11. Barsky AJ, Wyshak G, Klerman GL. Psychiatric comorbidity in DSM-III-R hypochondriasis. Arch Gen Psychiatry 1992;49:101-8.
12. Starcevic V. Role of reassurance and psychopathology in hypochondriasis. Psychiatry 1990;53(4):383-95.
13. Rost KM, Akins RN, Brown FW, Smith GR. The comorbidity of DSM-III-R personality disorders in somatization disorder. Gen Hosp Psychiatry 1992;14:322-6.
14. Morrison J. Childhood sexual histories of women with somatization disorder [comment]. Am J Psychiatry 1989;146:239-41.
15. Morse DS, Suchman AL, Frankel RM. The meaning of symptoms in 10 women with somatization disorder and a history of childhood abuse. Arch Fam Med 1997;6:468-76.
16. Kroenke K, Swindle R. Cognitive-behavioral therapy for somatization and symptom syndromes: a critical review of controlled clinical trials. Psychother Psychosom 2000;69:205-15.
17. Looper KJ, Kirmayer LJ. Behavioral medicine approaches to somatoform disorders. J Consult Clin Psychol 2002;70:810-27.
18. Warwick HM, Clark DM, Cobb AM, Salkovskis PM. A controlled trial of cognitive-behavioural treatment of hypochondriasis. Br J Psychiatry 1996;169:189-95.
19. Warwick HM, Salkovskis PM. Cognitive-behavioral treatment of hypochondriasis. In: Lipsitt DR, Starcevic V (eds). Hypochondriasis: Modern perspectives on an ancient malady. New York: Oxford Press, 2001;314-28.
20. McLeod CC, Budd MA. Treatment of somatization in primary care: evaluation of the Personal Health Improvement Program. HMO Pract 1997;11:88-94.
21. Bouman TK, Visser S. Cognitive and behavioural treatment of hypochondriasis. Psychother Psychosom 1998;67:214-21.
22. Starcevic V. Reassurance in the treatment of hypochondriasis. In: Lipsitt DR, Starcevic V (eds). Hypochondriasis: Modern perspectives on an ancient malady. New York: Oxford Press, 2001;291-313.
23. Clark DM, Salkovskis PM, Hackmann A, et al. Two psychological treatments for hypochondriasis. A randomised controlled trial. Br J Psychiatry 1998;173:218-25.
24. Schwartz L. Some notes on reassurance in medical practice. Psychosomatics 1966;7:290-4.
25. O’Malley PG, Jackson JL, Santoro J, et al. Antidepressant therapy for unexplained symptoms and symptom syndromes. J Fam Pract 1999;48:980-90.
26. Kellner R, Fava GA, Lisansky J, et al. Hypochondriacal fears and beliefs in DSM-III melancholia. Changes with amitriptyline. J Affect Disord 1986;10:21-6.
27. Noyes R, Reich J, Clancy J, O’Gorman TW. Reduction in hypochondriasis with treatment of panic disorder. Br J Psychiatry 1986;149:631-5.
Sodium oxybate: A new way to treat narcolepsy
Existing drug treatments for narcolepsy enhance daytime alertness, and most improve cataplexy, sleep paralysis, and hypnagogic/hypnopompic hallucinations. None of these agents, however, target the nocturnal sleep deficits that lead to daytime symptoms.
Sodium oxybate, one of the most controversial medications to receive FDA approval in recent years (Table 1), has been found to reduce daytime sleepiness and cataplexy by improving nighttime sleep in patients with narcolepsy.
ABOUT SODIUM OXYBATE
Sodium oxybate is also known as gamma-hydroxybutyrate (GHB). An illegal form of GHB—the so-called “date rape drug”—is produced and used illicitly, typically at parties and nightclubs. Some users hide the fast-acting, sedating drug in a cocktail, rendering victims unable to defend against an assault or to recall details leading to the assault.1
Some athletes believe GHB enhances on-field performance by increasing production of growth hormone. Enhanced growth hormone release has no known clinical significance or effect on athletic performance, however.
Table 1
Sodium oxybate: Fast facts
| Drug brand name: Xyrem |
| Class: CNS depressant |
| FDA-approved indications: Treatment of cataplexy |
| Approval date: July 17, 2002 |
| Manufacturer: Orphan Medical |
| Dosing forms: 180 mL oral solution at a concentration of 0.5 grams/mL |
| Recommended dosage: Start at 2.25 grams at bedtime; repeat dose overnight (4.5 grams/d total). Dosage can be increased to 9 grams/d (4.5 grams per dose) by increments of 0.75 grams per dose every 2 weeks. A dropper is supplied to facilitate measurement. |
The U.S. Drug Enforcement Agency (DEA) considers GHB a Schedule 1 (illegal) drug. DEA considers the prescription version a Schedule 3 drug, meaning it can be prescribed with refills as long as a DEA number is listed on the prescription. To prevent misuse, a central pharmacy dispenses sodium oxybate and mandates use of a specific prescription form to verify the physician’s familiarity with the medication. Psychiatrists can call (866) 997-3688 to obtain the form.
Table 2
Sodium oxybate dosing recommendations for patients
|
Sodium oxybate is the only agent FDA-approved for treating cataplexy—muscle weakness common among patients with narcolepsy.
HOW IT WORKS
Developed as an anesthetic, sodium oxybate induces deep sleep and at higher doses causes amnesia.
Derived from gamma-aminobutyric acid (GABA), sodium oxybate’s mechanism of action is unknown. Some believe it binds to the GABA B receptor and partially inhibits the NMDA and AMPA receptor-mediated excitatory neurons in the hippocampus.2
Food alters its bioavailability, so sodium oxybate should be taken several hours after meals to prevent delays in absorption and effect. Patients taking it should not eat at bedtime.
The agent’s pharmacokinetics are nonlinear, meaning that if the dose is doubled, the medication effect is tripled or quadrupled. For this reason, dosage increases must be small (no more than 0.75 grams for each dose) and gradual (at intervals of at least 2 weeks). The medication reaches peak plasma concentration within 30 to 75 minutes, so patients should not take the medication until they are in bed. Its 1-hour half-life explains its brief duration of action and need for repeat dosing overnight (Table 2).
Sodium oxybate does not modify the activity of any cytochrome P-450 enzymes. The medication is high in sodium (0.5 grams in a 3-gram dose) and has a salty taste. Use caution when considering the agent for patients with hypertension or on low-sodium diets.
Sodium oxybate’s safety has not been adequately tested in patients younger than 18 or older than 65 or in those with dementia and other disease processes. Because the drug is metabolized by the liver, the manufacturer recommends prescribing one-half the starting dosage to patients with significant hepatic impairment.
EFFICACY
Sodium oxybate has been shown to indirectly reduce frequency of cataplexy by improving nocturnal sleep:
- In a placebo-controlled, 4-week trial, 136 patients received either placebo or sodium oxybate at bedtime and again overnight in two equally divided doses of 3, 6, or 9 grams each. Patients who received the medication experienced less-frequent cataplexy, reduced daytime sleepiness, and fewer unplanned daytime naps and nocturnal awakenings.3
- A placebo-controlled trial that followed 55 patients for more than 3 years demonstrated long-term efficacy based on the patients’ cataplexy diaries (mean duration of treatment 21 months). Cataplexy returned after abrupt discontinuation.4
Unlike patients with most other disorders, those with narcolepsy generally are willing to repeat a medication overnight. They awaken easily at night—often without an alarm. Patients taking the medication report that they fall asleep again more readily and experience dramatically improved sleep quality and duration.
TOLERABILITY
Sodium oxybate has been well tolerated in relatively small clinical trials.
In the 4-week, placebo-controlled trial,3 nausea, headache, dizziness, and enuresis were most frequently reported. Out of 136 participants, 1 withdrew because of acute confusion and 9 others left because of mild to moderate adverse events. Twelve others experienced one episode of enuresis—probably because they did not fully awaken from deep sleep when developing urinary urgency. Advise patients taking sodium oxybate to urinate before going to bed.
The medication’s propensity to increase slow-wave sleep may cause sleepwalking. Sleepwalking was reported in 32% of patients in one long-term, uncontrolled study.5 If a patient with a history of sleepwalking needs sodium oxybate, advise against sleeping in upper bunks and other dangerous settings, and recommend precautions such as locking doors.
Because of sodium oxybate’s sedating properties, concomitant use of alcohol, barbiturates, and benzodiazepines should be discouraged.
ABUSE POTENTIAL
As discussed, GHB has a high abuse potential with effects such as euphoria, relaxation, and heightened sexual feelings.
Tolerance and dependence has not been reported with sodium oxybate when used as prescribed. A withdrawal state—similar to alcohol and sedative/hypnotic withdrawal and marked by anxiety, tremor, agitation, and delirium—has been reported with GHB abuse (although other chemicals often are used simultaneously in such cases). Narcolepsy patients in clinical trials have abruptly discontinued sodium oxybate after months of use without significant withdrawal.4
Related resources
- Narcolepsy Network Inc. www.narcolepsynetwork.org
Disclosure
Dr. Krahn reports no financial relationship with Orphan Medical or with manufacturers of competing products.
1. Galloway GP, Frederick SL, Staggers FE, Jr, et al. Gamma-hydroxybutyrate: an emerging drug of abuse that causes physical dependence. Addiction 1997;92(1):89-96.
2. Cammalleri M, Brancucci A, Berton F, et al. Gamma-hydroxybutyrate reduces GABA(A)-mediated inhibitory postsynaptic potentials in the CA1 region of hippocampus. Neuropsychopharmacology. 2002;27(9):960-9.
3. U.S. Xyrem Multicenter Study Group. A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep 2002;25(1):42-9.
4. U.S. Xyrem Multi-Center Study Group. The abrupt cessation of therapeutically administered sodium oxybate (GHB) does not cause withdrawal symptoms. J Toxicol Clin Toxicol 2003;41:131-5.
5. Physicians’ Desk Reference (57th ed). Montvale, NJ: Thomson Healthcare, 2003.
6. Mitler MM, Hayduk R. Benefits and risks of pharmacotherapy for narcolepsy. Drug Saf. 2002;25(11):791-809.
Existing drug treatments for narcolepsy enhance daytime alertness, and most improve cataplexy, sleep paralysis, and hypnagogic/hypnopompic hallucinations. None of these agents, however, target the nocturnal sleep deficits that lead to daytime symptoms.
Sodium oxybate, one of the most controversial medications to receive FDA approval in recent years (Table 1), has been found to reduce daytime sleepiness and cataplexy by improving nighttime sleep in patients with narcolepsy.
ABOUT SODIUM OXYBATE
Sodium oxybate is also known as gamma-hydroxybutyrate (GHB). An illegal form of GHB—the so-called “date rape drug”—is produced and used illicitly, typically at parties and nightclubs. Some users hide the fast-acting, sedating drug in a cocktail, rendering victims unable to defend against an assault or to recall details leading to the assault.1
Some athletes believe GHB enhances on-field performance by increasing production of growth hormone. Enhanced growth hormone release has no known clinical significance or effect on athletic performance, however.
Table 1
Sodium oxybate: Fast facts
| Drug brand name: Xyrem |
| Class: CNS depressant |
| FDA-approved indications: Treatment of cataplexy |
| Approval date: July 17, 2002 |
| Manufacturer: Orphan Medical |
| Dosing forms: 180 mL oral solution at a concentration of 0.5 grams/mL |
| Recommended dosage: Start at 2.25 grams at bedtime; repeat dose overnight (4.5 grams/d total). Dosage can be increased to 9 grams/d (4.5 grams per dose) by increments of 0.75 grams per dose every 2 weeks. A dropper is supplied to facilitate measurement. |
The U.S. Drug Enforcement Agency (DEA) considers GHB a Schedule 1 (illegal) drug. DEA considers the prescription version a Schedule 3 drug, meaning it can be prescribed with refills as long as a DEA number is listed on the prescription. To prevent misuse, a central pharmacy dispenses sodium oxybate and mandates use of a specific prescription form to verify the physician’s familiarity with the medication. Psychiatrists can call (866) 997-3688 to obtain the form.
Table 2
Sodium oxybate dosing recommendations for patients
|
Sodium oxybate is the only agent FDA-approved for treating cataplexy—muscle weakness common among patients with narcolepsy.
HOW IT WORKS
Developed as an anesthetic, sodium oxybate induces deep sleep and at higher doses causes amnesia.
Derived from gamma-aminobutyric acid (GABA), sodium oxybate’s mechanism of action is unknown. Some believe it binds to the GABA B receptor and partially inhibits the NMDA and AMPA receptor-mediated excitatory neurons in the hippocampus.2
Food alters its bioavailability, so sodium oxybate should be taken several hours after meals to prevent delays in absorption and effect. Patients taking it should not eat at bedtime.
The agent’s pharmacokinetics are nonlinear, meaning that if the dose is doubled, the medication effect is tripled or quadrupled. For this reason, dosage increases must be small (no more than 0.75 grams for each dose) and gradual (at intervals of at least 2 weeks). The medication reaches peak plasma concentration within 30 to 75 minutes, so patients should not take the medication until they are in bed. Its 1-hour half-life explains its brief duration of action and need for repeat dosing overnight (Table 2).
Sodium oxybate does not modify the activity of any cytochrome P-450 enzymes. The medication is high in sodium (0.5 grams in a 3-gram dose) and has a salty taste. Use caution when considering the agent for patients with hypertension or on low-sodium diets.
Sodium oxybate’s safety has not been adequately tested in patients younger than 18 or older than 65 or in those with dementia and other disease processes. Because the drug is metabolized by the liver, the manufacturer recommends prescribing one-half the starting dosage to patients with significant hepatic impairment.
EFFICACY
Sodium oxybate has been shown to indirectly reduce frequency of cataplexy by improving nocturnal sleep:
- In a placebo-controlled, 4-week trial, 136 patients received either placebo or sodium oxybate at bedtime and again overnight in two equally divided doses of 3, 6, or 9 grams each. Patients who received the medication experienced less-frequent cataplexy, reduced daytime sleepiness, and fewer unplanned daytime naps and nocturnal awakenings.3
- A placebo-controlled trial that followed 55 patients for more than 3 years demonstrated long-term efficacy based on the patients’ cataplexy diaries (mean duration of treatment 21 months). Cataplexy returned after abrupt discontinuation.4
Unlike patients with most other disorders, those with narcolepsy generally are willing to repeat a medication overnight. They awaken easily at night—often without an alarm. Patients taking the medication report that they fall asleep again more readily and experience dramatically improved sleep quality and duration.
TOLERABILITY
Sodium oxybate has been well tolerated in relatively small clinical trials.
In the 4-week, placebo-controlled trial,3 nausea, headache, dizziness, and enuresis were most frequently reported. Out of 136 participants, 1 withdrew because of acute confusion and 9 others left because of mild to moderate adverse events. Twelve others experienced one episode of enuresis—probably because they did not fully awaken from deep sleep when developing urinary urgency. Advise patients taking sodium oxybate to urinate before going to bed.
The medication’s propensity to increase slow-wave sleep may cause sleepwalking. Sleepwalking was reported in 32% of patients in one long-term, uncontrolled study.5 If a patient with a history of sleepwalking needs sodium oxybate, advise against sleeping in upper bunks and other dangerous settings, and recommend precautions such as locking doors.
Because of sodium oxybate’s sedating properties, concomitant use of alcohol, barbiturates, and benzodiazepines should be discouraged.
ABUSE POTENTIAL
As discussed, GHB has a high abuse potential with effects such as euphoria, relaxation, and heightened sexual feelings.
Tolerance and dependence has not been reported with sodium oxybate when used as prescribed. A withdrawal state—similar to alcohol and sedative/hypnotic withdrawal and marked by anxiety, tremor, agitation, and delirium—has been reported with GHB abuse (although other chemicals often are used simultaneously in such cases). Narcolepsy patients in clinical trials have abruptly discontinued sodium oxybate after months of use without significant withdrawal.4
Related resources
- Narcolepsy Network Inc. www.narcolepsynetwork.org
Disclosure
Dr. Krahn reports no financial relationship with Orphan Medical or with manufacturers of competing products.
Existing drug treatments for narcolepsy enhance daytime alertness, and most improve cataplexy, sleep paralysis, and hypnagogic/hypnopompic hallucinations. None of these agents, however, target the nocturnal sleep deficits that lead to daytime symptoms.
Sodium oxybate, one of the most controversial medications to receive FDA approval in recent years (Table 1), has been found to reduce daytime sleepiness and cataplexy by improving nighttime sleep in patients with narcolepsy.
ABOUT SODIUM OXYBATE
Sodium oxybate is also known as gamma-hydroxybutyrate (GHB). An illegal form of GHB—the so-called “date rape drug”—is produced and used illicitly, typically at parties and nightclubs. Some users hide the fast-acting, sedating drug in a cocktail, rendering victims unable to defend against an assault or to recall details leading to the assault.1
Some athletes believe GHB enhances on-field performance by increasing production of growth hormone. Enhanced growth hormone release has no known clinical significance or effect on athletic performance, however.
Table 1
Sodium oxybate: Fast facts
| Drug brand name: Xyrem |
| Class: CNS depressant |
| FDA-approved indications: Treatment of cataplexy |
| Approval date: July 17, 2002 |
| Manufacturer: Orphan Medical |
| Dosing forms: 180 mL oral solution at a concentration of 0.5 grams/mL |
| Recommended dosage: Start at 2.25 grams at bedtime; repeat dose overnight (4.5 grams/d total). Dosage can be increased to 9 grams/d (4.5 grams per dose) by increments of 0.75 grams per dose every 2 weeks. A dropper is supplied to facilitate measurement. |
The U.S. Drug Enforcement Agency (DEA) considers GHB a Schedule 1 (illegal) drug. DEA considers the prescription version a Schedule 3 drug, meaning it can be prescribed with refills as long as a DEA number is listed on the prescription. To prevent misuse, a central pharmacy dispenses sodium oxybate and mandates use of a specific prescription form to verify the physician’s familiarity with the medication. Psychiatrists can call (866) 997-3688 to obtain the form.
Table 2
Sodium oxybate dosing recommendations for patients
|
Sodium oxybate is the only agent FDA-approved for treating cataplexy—muscle weakness common among patients with narcolepsy.
HOW IT WORKS
Developed as an anesthetic, sodium oxybate induces deep sleep and at higher doses causes amnesia.
Derived from gamma-aminobutyric acid (GABA), sodium oxybate’s mechanism of action is unknown. Some believe it binds to the GABA B receptor and partially inhibits the NMDA and AMPA receptor-mediated excitatory neurons in the hippocampus.2
Food alters its bioavailability, so sodium oxybate should be taken several hours after meals to prevent delays in absorption and effect. Patients taking it should not eat at bedtime.
The agent’s pharmacokinetics are nonlinear, meaning that if the dose is doubled, the medication effect is tripled or quadrupled. For this reason, dosage increases must be small (no more than 0.75 grams for each dose) and gradual (at intervals of at least 2 weeks). The medication reaches peak plasma concentration within 30 to 75 minutes, so patients should not take the medication until they are in bed. Its 1-hour half-life explains its brief duration of action and need for repeat dosing overnight (Table 2).
Sodium oxybate does not modify the activity of any cytochrome P-450 enzymes. The medication is high in sodium (0.5 grams in a 3-gram dose) and has a salty taste. Use caution when considering the agent for patients with hypertension or on low-sodium diets.
Sodium oxybate’s safety has not been adequately tested in patients younger than 18 or older than 65 or in those with dementia and other disease processes. Because the drug is metabolized by the liver, the manufacturer recommends prescribing one-half the starting dosage to patients with significant hepatic impairment.
EFFICACY
Sodium oxybate has been shown to indirectly reduce frequency of cataplexy by improving nocturnal sleep:
- In a placebo-controlled, 4-week trial, 136 patients received either placebo or sodium oxybate at bedtime and again overnight in two equally divided doses of 3, 6, or 9 grams each. Patients who received the medication experienced less-frequent cataplexy, reduced daytime sleepiness, and fewer unplanned daytime naps and nocturnal awakenings.3
- A placebo-controlled trial that followed 55 patients for more than 3 years demonstrated long-term efficacy based on the patients’ cataplexy diaries (mean duration of treatment 21 months). Cataplexy returned after abrupt discontinuation.4
Unlike patients with most other disorders, those with narcolepsy generally are willing to repeat a medication overnight. They awaken easily at night—often without an alarm. Patients taking the medication report that they fall asleep again more readily and experience dramatically improved sleep quality and duration.
TOLERABILITY
Sodium oxybate has been well tolerated in relatively small clinical trials.
In the 4-week, placebo-controlled trial,3 nausea, headache, dizziness, and enuresis were most frequently reported. Out of 136 participants, 1 withdrew because of acute confusion and 9 others left because of mild to moderate adverse events. Twelve others experienced one episode of enuresis—probably because they did not fully awaken from deep sleep when developing urinary urgency. Advise patients taking sodium oxybate to urinate before going to bed.
The medication’s propensity to increase slow-wave sleep may cause sleepwalking. Sleepwalking was reported in 32% of patients in one long-term, uncontrolled study.5 If a patient with a history of sleepwalking needs sodium oxybate, advise against sleeping in upper bunks and other dangerous settings, and recommend precautions such as locking doors.
Because of sodium oxybate’s sedating properties, concomitant use of alcohol, barbiturates, and benzodiazepines should be discouraged.
ABUSE POTENTIAL
As discussed, GHB has a high abuse potential with effects such as euphoria, relaxation, and heightened sexual feelings.
Tolerance and dependence has not been reported with sodium oxybate when used as prescribed. A withdrawal state—similar to alcohol and sedative/hypnotic withdrawal and marked by anxiety, tremor, agitation, and delirium—has been reported with GHB abuse (although other chemicals often are used simultaneously in such cases). Narcolepsy patients in clinical trials have abruptly discontinued sodium oxybate after months of use without significant withdrawal.4
Related resources
- Narcolepsy Network Inc. www.narcolepsynetwork.org
Disclosure
Dr. Krahn reports no financial relationship with Orphan Medical or with manufacturers of competing products.
1. Galloway GP, Frederick SL, Staggers FE, Jr, et al. Gamma-hydroxybutyrate: an emerging drug of abuse that causes physical dependence. Addiction 1997;92(1):89-96.
2. Cammalleri M, Brancucci A, Berton F, et al. Gamma-hydroxybutyrate reduces GABA(A)-mediated inhibitory postsynaptic potentials in the CA1 region of hippocampus. Neuropsychopharmacology. 2002;27(9):960-9.
3. U.S. Xyrem Multicenter Study Group. A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep 2002;25(1):42-9.
4. U.S. Xyrem Multi-Center Study Group. The abrupt cessation of therapeutically administered sodium oxybate (GHB) does not cause withdrawal symptoms. J Toxicol Clin Toxicol 2003;41:131-5.
5. Physicians’ Desk Reference (57th ed). Montvale, NJ: Thomson Healthcare, 2003.
6. Mitler MM, Hayduk R. Benefits and risks of pharmacotherapy for narcolepsy. Drug Saf. 2002;25(11):791-809.
1. Galloway GP, Frederick SL, Staggers FE, Jr, et al. Gamma-hydroxybutyrate: an emerging drug of abuse that causes physical dependence. Addiction 1997;92(1):89-96.
2. Cammalleri M, Brancucci A, Berton F, et al. Gamma-hydroxybutyrate reduces GABA(A)-mediated inhibitory postsynaptic potentials in the CA1 region of hippocampus. Neuropsychopharmacology. 2002;27(9):960-9.
3. U.S. Xyrem Multicenter Study Group. A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep 2002;25(1):42-9.
4. U.S. Xyrem Multi-Center Study Group. The abrupt cessation of therapeutically administered sodium oxybate (GHB) does not cause withdrawal symptoms. J Toxicol Clin Toxicol 2003;41:131-5.
5. Physicians’ Desk Reference (57th ed). Montvale, NJ: Thomson Healthcare, 2003.
6. Mitler MM, Hayduk R. Benefits and risks of pharmacotherapy for narcolepsy. Drug Saf. 2002;25(11):791-809.
Clozapine therapy: Timing is everything
HISTORY: Six years of psychosis
Ms. G, age 37, has had paranoid schizophrenia f or 6 years, resulting in numerous hospitalizations and continuous outpatient follow-up. Her family is supportive and supervises her when she’s not hospitalized.
Though fluent in English, Ms. G—a Polish immigrant—speaks primarily in her native tongue during psychotic episodes and becomes increasingly paranoid toward neighbors. As her condition degenerates, she hears her late father’s voice criticizing her. Because of marked social withdrawal and isolation, she cannot maintain basic interpersonal skills or live independently. Her psychosis, apathy, avolition, withdrawal, and lack of focus have persisted despite trials of numerous antipsychotics, including olanzapine, 25 mg nightly for 1 month, and quetiapine, 300 mg bid for 3 weeks.
What are the drug therapy options for this patient?
The authors’ observations
“Treatment-refractory” schizophrenia has numerous definitions. One that is widely accepted but cumbersome—used in the multicenter clozapine trial1 —requires a 5-year absence of periods of good functioning in patients taking an antipsychotic at dosages equivalent to chlorpromazine, 1,000 mg/d. In that time, the patient must have received two or more antipsychotic classes for at least 6 weeks each without achieving significant relief. The Brief Psychiatric Rating Scale (BPRS) score must be at least 45, with item scores of moderate severity for two or more of the following:
- disorganization
- suspiciousness
- hallucinatory behavior
- unusual thought content.
The Clinical Global Impression (CGI) Scale score must be at least 4 (moderately ill). Also, a 6-week trial of haloperidol, with a mean dosage of 60 mg/d:
- must fail to decrease the BPRS score by 20% or to below 35
- or must fail to decrease the CGI severity score to 3 (mildly ill).1
In 1990, an international study group defined treatment-refractory schizophrenia as “the presence of ongoing psychotic symptoms with substantial functional disability and/or behavioral deviances that persist in well-diagnosed persons with schizophrenia despite reasonable and customary pharmacological and psychosocial treatment that has been provided for an adequate period.”2 This definition is far more useful to clinical practice and also considers psychosocial function. Seven levels of treatment response and resistance were suggested, based on presence of positive and negative symptoms, personal and social functioning, and CGI scores.2
Meltzer3 proposed that any person not returning to his or her highest premorbid level of functioning with a tolerable antipsychotic be considered refractory and thus a possible candidate for clozapine therapy.
Ms. G’s illness meets the definition of treatment-refractory schizophrenia. Her CGI score at baseline was 5—severely ill—and several medication trials at sufficient dosages failed to control her positive or negative symptoms. Upon psychotic decompensation, she required prolonged hospitalization and could no longer live independently or work. At this point, she is a possible candidate for clozapine therapy.
TREATMENT: Starting clozapine
Ms. G was started on clozapine, 25 mg at night, titrated to 300 mg at bedtime.
Two weeks later, her paranoia and auditory hallucinations diminished, her interpersonal relationships improved, she was less withdrawn, her thoughts became more organized, and her range of affect expanded. She functioned at her highest level since her initial presentation based on clinical observation and family reports. Her CGI Global Improvement score at this point was 2 (much improved).
Ms. G. continued to take clozapine, 300 mg/d, for 2 years while undergoing weekly blood tests for white blood cell counts (WBC) with differentials. She did not require hospitalization for schizophrenia during this time, and her WBC count averaged between 4,000 and 4,500/mm3, well within the normal range of 3,500 to 12,000/mm3.
Then one day—after maintaining a relatively stable WBC for several weeks—a blood test revealed a WBC of 2,700/mm3. Ms. G exhibited no objective signs of immunosuppression, such as fever or infection. Still, the psychiatrist immediately discontinued clozapine.
Was the treating psychiatrist justified in immediately stopping clozapine after one low WBC reading?
The authors’ observations
Leukopenia, defined as a WBC <3,000/mm3, and agranulocytosis, defined as an absolute neutrophil count <500/mm3, are well-documented adverse reactions to clozapine. Early data on clozapine-associated agranulocytosis cases prior to 1989 suggest that up to 32% were fatal,4 but relatively few cases have occurred since the Clozaril National Registry was instituted in 1977.4,5 Between 1977 and 1997, 585 clozapine-associated agranulocytosis cases were reported in the United States; 19 of these were fatal, suggesting a mortality rate of 3.2% and attesting to the effectiveness of FDA-mandated WBC testing. During this period, 150,409 patients received clozapine.4
The agranulocytosis risk does not appear to be dose-related but declines substantially after the 10th week. Three out of 1,000 patients who take clozapine for 1 year are likely to develop agranulocytosis at the 3- to 6-month mark.4 Although the incidence continues to drop after month 6, it never reaches zero.4-6
Table
Life-threatening effects of clozapine and their reported frequency of occurrence
| Adverse effect | Incidence rate among clozapine users |
|---|---|
| Agranulocytosis | 3/1000 person years* at 6 months |
| Hepatitis | Less than 1% |
| Hyperglycemia with ketoacidosis | Unknown, case reports |
| Myocarditis | 5.0-96.6 cases/100,000 patient years |
| Neuroleptic malignant syndrome | Unknown, several case reports |
| Orthostasis with cardiac collapse | 1/3,000 cases |
| Pulmonary embolism | 1/3,450 person years |
| Seizures | 5% after 1 year of therapy |
| * Person year = 1 person taking medication for 1 year | |
| Source: Clozaril prescribing information. In: Physicians’ Desk Reference (57th ed). Montvale, NJ: Thomson Healthcare, 2003. | |
Other severe adverse effects of clozapine include myocarditis associated with cardiac failure, orthostatic hypotension with circulatory collapse, and rhabdomyolysis (Table).4,7 Leukocytosis and eosinophilia are generally transient and self-limited but may predict agranulocytosis.8,9 The risk of seizure occurs most commonly at dosages greater than 500 mg/d.4
Given these potentially fatal effects, the authors’ treatment guidelines call for potential suspension of clozapine therapy when the WBC is consistently <3,000 mm3 (Algorithm).
CONTINUED TREATMENT: A difficult decision
Ms. G was switched to quetiapine, 100 mg nightly, titrated to 800 mg/d in divided doses.
Approximately 3 weeks later, Ms. G was hospitalized for renewed severe paranoia and command-type auditory hallucinations accompanied by prominent mood lability, avolition, and thought disorganization. During her 7-week hospitalization, she underwent sequential and sometimes overlapping trials of:
- ziprasidone, 160 mg bid
- risperidone, 3 mg bid
- trifluoperazine, 3 mg bid
- haloperidol, 10 mg bid
- divalproex sodium, 500 mg bid
- and carbamazepine, 400 mg bid.
None of these trials significantly improved her psychosis or mood.
At this point, the treating psychiatrists faced a difficult but clear decision: Ms. G was rechallenged on clozapine, 25 mg nightly, titrated again to 300 mg nightly. After she provided informed consent, her WBC was monitored twice daily—morning and evening—for agranulocytosis and to examine WBC patterns. Her average daily WBC counts were 4,200/mm3 in the morning and 5,500/mm3 at night. No physical signs of agranulocytosis emerged.
One week after restarting clozapine, Ms. G became less paranoid and socially more appropriate. Her thought process became increasingly organized, and after 4 weeks she reached her baseline status based upon family reports and the clinician’s CGI Global Improvement rating of 2 (much improved). Her auditory hallucinations resolved, and she was discharged to her family’s care.
Twice-daily blood testing was stopped at discharge. Ms. G continues to take clozapine and receives blood tests every 2 weeks, with no apparent signs of agranulocytosis.
Algorithm Suggested guidelines for managing WBC counts during clozapine therapy
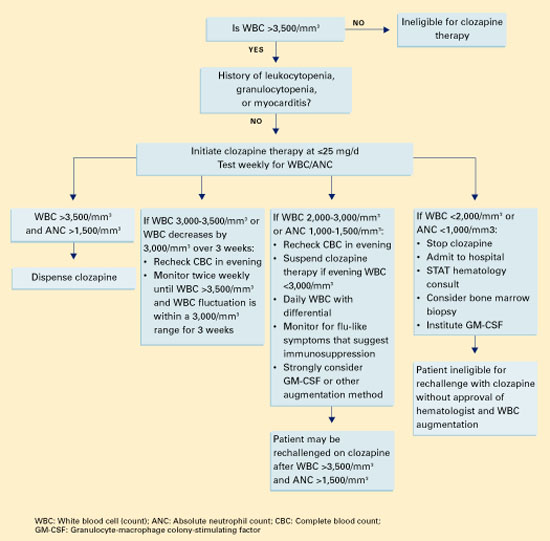
Two treatments for clozapine-dependent agranulocytosis have been described.
- Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a glycoprotein that has been shown to stimulate proliferation of precursor cells in bone marrow and their differentiation into granulocytes and macrophages. Researchers have reported that GM-CSF treatment allows patients to continue taking clozapine after an episode of severe neutropenia.10-12
- Lithium salts have been reported to exploit the natural leukocytosis observed with lithium to counter clozapine-related leukopenia.13 Use of lithium to displace white blood cells has been debated, and anecdotal evidence suggests that combining lithium with clozapine may increase the chance of seizure and neuroleptic malignant syndrome.4 Still, cases reported by Adityanjee and Blier suggest that lithium augmentation is cost-effective and efficacious.14,15
How could Ms. G’s doctors have avoided stopping her clozapine therapy and her subsequent decompensation?
The authors’ observations
Aggressive blood testing and cessation of clozapine therapy are indicated when onset of granulocytopenia and agranulocytosis are suspected. Even with early detection and discontinuation, the chance of infectious disease poses a danger for up to 4 weeks until WBC levels return to normal.4
Given Ms. G’s lack of response to other antipsychotics, however, we had to consider resuming clozapine therapy. Studies have described agranulocytosis management strategies that may allow patients to keep taking clozapine despite low WBC counts (Box).
We also considered the timing of Ms. G’s blood test that showed a WBC count <2,700/mm3. Ahokas16 suggests that evening WBC counts are significantly higher than those taken in the morning and that granulocytes fluctuate in a diurnal pattern. Ms. G’s evening WBC counts were on average 1,300/mm3 higher than morning levels. Allowing for this diurnal variation and comparing evening blood samples could have averted the interruption in Ms. G’s clozapine therapy and prevented relapse in a patient with highly treatment-refractory schizophrenia.
Related resources
- Chong SA, Remington G. Clozapine augmentation: safety and efficacy. Schizophr Bull 2000;26:421-40.
- Emsley R, Oosthuizen P. The new and evolving pharmacotherapy of schizophrenia. Psychiatr Clin North Am 2003;26:141-63.
- Barnas C, Zwierzina H, Hummer M, Sperner-Unterweger B, Stern A, Fleischhacker WW. Granulocyte-macrophage colony-stimulating factor (GM-CSF) treatment of clozapine-induced agranulocytosis: a case report. J Clin Psychiatry 1992;53:245-7.
Drug brand names
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Divalproex sodium • Depakote
- GM-CSF—Filgrastim • Neupogen
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid, others
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trifluoperazine • Stelazine
- Ziprasidone • Geodon
Disclosure
Dr. Rao is a speaker for Pfizer Inc.
Drs. Goforth, Raval, and Sharma report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment resistant schizophrenic: a double blind comparison with chlorpromazine. Arch Gen Psychiatry 1988;45:789-96.
2. Brenner HD, Dencker SJ, Goldstein MJ, et al. Defining treatment refractoriness in schizophrenia. Schizophr Bull 1990;16:551-61.
3. Meltzer HY. Clozapine: is another view valid? Am J Psychiatry 1995;152:821-5.
4. Clozaril prescribing information. In: Physicians’ Desk Reference. (57th ed). Montvale, NJ: Thomson Healthcare, 2003.
5. Alvir JM, Lieberman JA, Safferman AZ, et al. Clozapine-induced agranulocytosis. Incidence and risk factors in the United States. N Engl J Med. 1993;329:162-7.
6. Honigfeld G. Effects of the clozapine national registry system on incidence of deaths related to agranulocytosis. Psychiatr Serv 1996;47:52-6.
7. Scelsa SN, Simpson DM, McQuistion HL, et al. Clozapineinduced myotoxicity in patients with chronic psychotic disorders. Neurology 1996;47:1518-23.
8. Ames D, Wirshing WC, Baker RW, et al. Predictive value of eosinophilia for neutropenia during clozapine treatment. J Clin Psychiatry 1996;57:579-81.
9. Alvir JM, Lieberman JA, Safferman AZ. Do white-cell count spikes predict agranulocytosis in clozapine recipients? Psychopharmacol Bull 1995;31:311-14.
10. Sperner-Unterweger B, Czeipek I, Gaggl S, et al. Treatment of severe clozapine-induced neutropenia with granulocyte colonystimulating factor (G-CSF). Remission despite continuous treatment with clozapine. Br J Psychiatry 1998;172:82-4.
11. Chengappa KN, Gopalani A, Haught MK, et al. The treatment of clozapine-associated agranulocytosis with granulocyte colony-stimulating factor (G-CSF). Psychopharmacol Bull 1996;32:111-21.
12. Lamberti JS, Bellnier TJ, Schwarzkopf SB, Schneider E. Filgrastim treatment of three patients with clozapine-induced agranulocytosis. J Clin Psychiatry 1995;56:256-9.
13. Boshes RA, Manschreck TC, Desrosiers J, et al. Initiation of clozapine therapy in a patient with preexisting leukopenia: a discussion of the rationale of current treatment options. Ann Clin Psychiatry 2001;13:233-7.
14. Adityanjee. Modification of clozapine-induced leukopenia and neutropenia with lithium carbonate. Am J Psychiatry 1995;152:648-9.
15. Blier P, Slater S, Measham T, et al. Lithium and clozapine-induced neutropenia/agranulocytosis. Int Clin Psychopharmacol 1998;13:137-40.
16. Ahokas A, Elonen E. Circadian rhythm of white blood cells during clozapine treatment. Psychopharmacology 1999;144:301-2.
HISTORY: Six years of psychosis
Ms. G, age 37, has had paranoid schizophrenia f or 6 years, resulting in numerous hospitalizations and continuous outpatient follow-up. Her family is supportive and supervises her when she’s not hospitalized.
Though fluent in English, Ms. G—a Polish immigrant—speaks primarily in her native tongue during psychotic episodes and becomes increasingly paranoid toward neighbors. As her condition degenerates, she hears her late father’s voice criticizing her. Because of marked social withdrawal and isolation, she cannot maintain basic interpersonal skills or live independently. Her psychosis, apathy, avolition, withdrawal, and lack of focus have persisted despite trials of numerous antipsychotics, including olanzapine, 25 mg nightly for 1 month, and quetiapine, 300 mg bid for 3 weeks.
What are the drug therapy options for this patient?
The authors’ observations
“Treatment-refractory” schizophrenia has numerous definitions. One that is widely accepted but cumbersome—used in the multicenter clozapine trial1 —requires a 5-year absence of periods of good functioning in patients taking an antipsychotic at dosages equivalent to chlorpromazine, 1,000 mg/d. In that time, the patient must have received two or more antipsychotic classes for at least 6 weeks each without achieving significant relief. The Brief Psychiatric Rating Scale (BPRS) score must be at least 45, with item scores of moderate severity for two or more of the following:
- disorganization
- suspiciousness
- hallucinatory behavior
- unusual thought content.
The Clinical Global Impression (CGI) Scale score must be at least 4 (moderately ill). Also, a 6-week trial of haloperidol, with a mean dosage of 60 mg/d:
- must fail to decrease the BPRS score by 20% or to below 35
- or must fail to decrease the CGI severity score to 3 (mildly ill).1
In 1990, an international study group defined treatment-refractory schizophrenia as “the presence of ongoing psychotic symptoms with substantial functional disability and/or behavioral deviances that persist in well-diagnosed persons with schizophrenia despite reasonable and customary pharmacological and psychosocial treatment that has been provided for an adequate period.”2 This definition is far more useful to clinical practice and also considers psychosocial function. Seven levels of treatment response and resistance were suggested, based on presence of positive and negative symptoms, personal and social functioning, and CGI scores.2
Meltzer3 proposed that any person not returning to his or her highest premorbid level of functioning with a tolerable antipsychotic be considered refractory and thus a possible candidate for clozapine therapy.
Ms. G’s illness meets the definition of treatment-refractory schizophrenia. Her CGI score at baseline was 5—severely ill—and several medication trials at sufficient dosages failed to control her positive or negative symptoms. Upon psychotic decompensation, she required prolonged hospitalization and could no longer live independently or work. At this point, she is a possible candidate for clozapine therapy.
TREATMENT: Starting clozapine
Ms. G was started on clozapine, 25 mg at night, titrated to 300 mg at bedtime.
Two weeks later, her paranoia and auditory hallucinations diminished, her interpersonal relationships improved, she was less withdrawn, her thoughts became more organized, and her range of affect expanded. She functioned at her highest level since her initial presentation based on clinical observation and family reports. Her CGI Global Improvement score at this point was 2 (much improved).
Ms. G. continued to take clozapine, 300 mg/d, for 2 years while undergoing weekly blood tests for white blood cell counts (WBC) with differentials. She did not require hospitalization for schizophrenia during this time, and her WBC count averaged between 4,000 and 4,500/mm3, well within the normal range of 3,500 to 12,000/mm3.
Then one day—after maintaining a relatively stable WBC for several weeks—a blood test revealed a WBC of 2,700/mm3. Ms. G exhibited no objective signs of immunosuppression, such as fever or infection. Still, the psychiatrist immediately discontinued clozapine.
Was the treating psychiatrist justified in immediately stopping clozapine after one low WBC reading?
The authors’ observations
Leukopenia, defined as a WBC <3,000/mm3, and agranulocytosis, defined as an absolute neutrophil count <500/mm3, are well-documented adverse reactions to clozapine. Early data on clozapine-associated agranulocytosis cases prior to 1989 suggest that up to 32% were fatal,4 but relatively few cases have occurred since the Clozaril National Registry was instituted in 1977.4,5 Between 1977 and 1997, 585 clozapine-associated agranulocytosis cases were reported in the United States; 19 of these were fatal, suggesting a mortality rate of 3.2% and attesting to the effectiveness of FDA-mandated WBC testing. During this period, 150,409 patients received clozapine.4
The agranulocytosis risk does not appear to be dose-related but declines substantially after the 10th week. Three out of 1,000 patients who take clozapine for 1 year are likely to develop agranulocytosis at the 3- to 6-month mark.4 Although the incidence continues to drop after month 6, it never reaches zero.4-6
Table
Life-threatening effects of clozapine and their reported frequency of occurrence
| Adverse effect | Incidence rate among clozapine users |
|---|---|
| Agranulocytosis | 3/1000 person years* at 6 months |
| Hepatitis | Less than 1% |
| Hyperglycemia with ketoacidosis | Unknown, case reports |
| Myocarditis | 5.0-96.6 cases/100,000 patient years |
| Neuroleptic malignant syndrome | Unknown, several case reports |
| Orthostasis with cardiac collapse | 1/3,000 cases |
| Pulmonary embolism | 1/3,450 person years |
| Seizures | 5% after 1 year of therapy |
| * Person year = 1 person taking medication for 1 year | |
| Source: Clozaril prescribing information. In: Physicians’ Desk Reference (57th ed). Montvale, NJ: Thomson Healthcare, 2003. | |
Other severe adverse effects of clozapine include myocarditis associated with cardiac failure, orthostatic hypotension with circulatory collapse, and rhabdomyolysis (Table).4,7 Leukocytosis and eosinophilia are generally transient and self-limited but may predict agranulocytosis.8,9 The risk of seizure occurs most commonly at dosages greater than 500 mg/d.4
Given these potentially fatal effects, the authors’ treatment guidelines call for potential suspension of clozapine therapy when the WBC is consistently <3,000 mm3 (Algorithm).
CONTINUED TREATMENT: A difficult decision
Ms. G was switched to quetiapine, 100 mg nightly, titrated to 800 mg/d in divided doses.
Approximately 3 weeks later, Ms. G was hospitalized for renewed severe paranoia and command-type auditory hallucinations accompanied by prominent mood lability, avolition, and thought disorganization. During her 7-week hospitalization, she underwent sequential and sometimes overlapping trials of:
- ziprasidone, 160 mg bid
- risperidone, 3 mg bid
- trifluoperazine, 3 mg bid
- haloperidol, 10 mg bid
- divalproex sodium, 500 mg bid
- and carbamazepine, 400 mg bid.
None of these trials significantly improved her psychosis or mood.
At this point, the treating psychiatrists faced a difficult but clear decision: Ms. G was rechallenged on clozapine, 25 mg nightly, titrated again to 300 mg nightly. After she provided informed consent, her WBC was monitored twice daily—morning and evening—for agranulocytosis and to examine WBC patterns. Her average daily WBC counts were 4,200/mm3 in the morning and 5,500/mm3 at night. No physical signs of agranulocytosis emerged.
One week after restarting clozapine, Ms. G became less paranoid and socially more appropriate. Her thought process became increasingly organized, and after 4 weeks she reached her baseline status based upon family reports and the clinician’s CGI Global Improvement rating of 2 (much improved). Her auditory hallucinations resolved, and she was discharged to her family’s care.
Twice-daily blood testing was stopped at discharge. Ms. G continues to take clozapine and receives blood tests every 2 weeks, with no apparent signs of agranulocytosis.
Algorithm Suggested guidelines for managing WBC counts during clozapine therapy
Two treatments for clozapine-dependent agranulocytosis have been described.
- Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a glycoprotein that has been shown to stimulate proliferation of precursor cells in bone marrow and their differentiation into granulocytes and macrophages. Researchers have reported that GM-CSF treatment allows patients to continue taking clozapine after an episode of severe neutropenia.10-12
- Lithium salts have been reported to exploit the natural leukocytosis observed with lithium to counter clozapine-related leukopenia.13 Use of lithium to displace white blood cells has been debated, and anecdotal evidence suggests that combining lithium with clozapine may increase the chance of seizure and neuroleptic malignant syndrome.4 Still, cases reported by Adityanjee and Blier suggest that lithium augmentation is cost-effective and efficacious.14,15
How could Ms. G’s doctors have avoided stopping her clozapine therapy and her subsequent decompensation?
The authors’ observations
Aggressive blood testing and cessation of clozapine therapy are indicated when onset of granulocytopenia and agranulocytosis are suspected. Even with early detection and discontinuation, the chance of infectious disease poses a danger for up to 4 weeks until WBC levels return to normal.4
Given Ms. G’s lack of response to other antipsychotics, however, we had to consider resuming clozapine therapy. Studies have described agranulocytosis management strategies that may allow patients to keep taking clozapine despite low WBC counts (Box).
We also considered the timing of Ms. G’s blood test that showed a WBC count <2,700/mm3. Ahokas16 suggests that evening WBC counts are significantly higher than those taken in the morning and that granulocytes fluctuate in a diurnal pattern. Ms. G’s evening WBC counts were on average 1,300/mm3 higher than morning levels. Allowing for this diurnal variation and comparing evening blood samples could have averted the interruption in Ms. G’s clozapine therapy and prevented relapse in a patient with highly treatment-refractory schizophrenia.
Related resources
- Chong SA, Remington G. Clozapine augmentation: safety and efficacy. Schizophr Bull 2000;26:421-40.
- Emsley R, Oosthuizen P. The new and evolving pharmacotherapy of schizophrenia. Psychiatr Clin North Am 2003;26:141-63.
- Barnas C, Zwierzina H, Hummer M, Sperner-Unterweger B, Stern A, Fleischhacker WW. Granulocyte-macrophage colony-stimulating factor (GM-CSF) treatment of clozapine-induced agranulocytosis: a case report. J Clin Psychiatry 1992;53:245-7.
Drug brand names
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Divalproex sodium • Depakote
- GM-CSF—Filgrastim • Neupogen
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid, others
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trifluoperazine • Stelazine
- Ziprasidone • Geodon
Disclosure
Dr. Rao is a speaker for Pfizer Inc.
Drs. Goforth, Raval, and Sharma report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
HISTORY: Six years of psychosis
Ms. G, age 37, has had paranoid schizophrenia f or 6 years, resulting in numerous hospitalizations and continuous outpatient follow-up. Her family is supportive and supervises her when she’s not hospitalized.
Though fluent in English, Ms. G—a Polish immigrant—speaks primarily in her native tongue during psychotic episodes and becomes increasingly paranoid toward neighbors. As her condition degenerates, she hears her late father’s voice criticizing her. Because of marked social withdrawal and isolation, she cannot maintain basic interpersonal skills or live independently. Her psychosis, apathy, avolition, withdrawal, and lack of focus have persisted despite trials of numerous antipsychotics, including olanzapine, 25 mg nightly for 1 month, and quetiapine, 300 mg bid for 3 weeks.
What are the drug therapy options for this patient?
The authors’ observations
“Treatment-refractory” schizophrenia has numerous definitions. One that is widely accepted but cumbersome—used in the multicenter clozapine trial1 —requires a 5-year absence of periods of good functioning in patients taking an antipsychotic at dosages equivalent to chlorpromazine, 1,000 mg/d. In that time, the patient must have received two or more antipsychotic classes for at least 6 weeks each without achieving significant relief. The Brief Psychiatric Rating Scale (BPRS) score must be at least 45, with item scores of moderate severity for two or more of the following:
- disorganization
- suspiciousness
- hallucinatory behavior
- unusual thought content.
The Clinical Global Impression (CGI) Scale score must be at least 4 (moderately ill). Also, a 6-week trial of haloperidol, with a mean dosage of 60 mg/d:
- must fail to decrease the BPRS score by 20% or to below 35
- or must fail to decrease the CGI severity score to 3 (mildly ill).1
In 1990, an international study group defined treatment-refractory schizophrenia as “the presence of ongoing psychotic symptoms with substantial functional disability and/or behavioral deviances that persist in well-diagnosed persons with schizophrenia despite reasonable and customary pharmacological and psychosocial treatment that has been provided for an adequate period.”2 This definition is far more useful to clinical practice and also considers psychosocial function. Seven levels of treatment response and resistance were suggested, based on presence of positive and negative symptoms, personal and social functioning, and CGI scores.2
Meltzer3 proposed that any person not returning to his or her highest premorbid level of functioning with a tolerable antipsychotic be considered refractory and thus a possible candidate for clozapine therapy.
Ms. G’s illness meets the definition of treatment-refractory schizophrenia. Her CGI score at baseline was 5—severely ill—and several medication trials at sufficient dosages failed to control her positive or negative symptoms. Upon psychotic decompensation, she required prolonged hospitalization and could no longer live independently or work. At this point, she is a possible candidate for clozapine therapy.
TREATMENT: Starting clozapine
Ms. G was started on clozapine, 25 mg at night, titrated to 300 mg at bedtime.
Two weeks later, her paranoia and auditory hallucinations diminished, her interpersonal relationships improved, she was less withdrawn, her thoughts became more organized, and her range of affect expanded. She functioned at her highest level since her initial presentation based on clinical observation and family reports. Her CGI Global Improvement score at this point was 2 (much improved).
Ms. G. continued to take clozapine, 300 mg/d, for 2 years while undergoing weekly blood tests for white blood cell counts (WBC) with differentials. She did not require hospitalization for schizophrenia during this time, and her WBC count averaged between 4,000 and 4,500/mm3, well within the normal range of 3,500 to 12,000/mm3.
Then one day—after maintaining a relatively stable WBC for several weeks—a blood test revealed a WBC of 2,700/mm3. Ms. G exhibited no objective signs of immunosuppression, such as fever or infection. Still, the psychiatrist immediately discontinued clozapine.
Was the treating psychiatrist justified in immediately stopping clozapine after one low WBC reading?
The authors’ observations
Leukopenia, defined as a WBC <3,000/mm3, and agranulocytosis, defined as an absolute neutrophil count <500/mm3, are well-documented adverse reactions to clozapine. Early data on clozapine-associated agranulocytosis cases prior to 1989 suggest that up to 32% were fatal,4 but relatively few cases have occurred since the Clozaril National Registry was instituted in 1977.4,5 Between 1977 and 1997, 585 clozapine-associated agranulocytosis cases were reported in the United States; 19 of these were fatal, suggesting a mortality rate of 3.2% and attesting to the effectiveness of FDA-mandated WBC testing. During this period, 150,409 patients received clozapine.4
The agranulocytosis risk does not appear to be dose-related but declines substantially after the 10th week. Three out of 1,000 patients who take clozapine for 1 year are likely to develop agranulocytosis at the 3- to 6-month mark.4 Although the incidence continues to drop after month 6, it never reaches zero.4-6
Table
Life-threatening effects of clozapine and their reported frequency of occurrence
| Adverse effect | Incidence rate among clozapine users |
|---|---|
| Agranulocytosis | 3/1000 person years* at 6 months |
| Hepatitis | Less than 1% |
| Hyperglycemia with ketoacidosis | Unknown, case reports |
| Myocarditis | 5.0-96.6 cases/100,000 patient years |
| Neuroleptic malignant syndrome | Unknown, several case reports |
| Orthostasis with cardiac collapse | 1/3,000 cases |
| Pulmonary embolism | 1/3,450 person years |
| Seizures | 5% after 1 year of therapy |
| * Person year = 1 person taking medication for 1 year | |
| Source: Clozaril prescribing information. In: Physicians’ Desk Reference (57th ed). Montvale, NJ: Thomson Healthcare, 2003. | |
Other severe adverse effects of clozapine include myocarditis associated with cardiac failure, orthostatic hypotension with circulatory collapse, and rhabdomyolysis (Table).4,7 Leukocytosis and eosinophilia are generally transient and self-limited but may predict agranulocytosis.8,9 The risk of seizure occurs most commonly at dosages greater than 500 mg/d.4
Given these potentially fatal effects, the authors’ treatment guidelines call for potential suspension of clozapine therapy when the WBC is consistently <3,000 mm3 (Algorithm).
CONTINUED TREATMENT: A difficult decision
Ms. G was switched to quetiapine, 100 mg nightly, titrated to 800 mg/d in divided doses.
Approximately 3 weeks later, Ms. G was hospitalized for renewed severe paranoia and command-type auditory hallucinations accompanied by prominent mood lability, avolition, and thought disorganization. During her 7-week hospitalization, she underwent sequential and sometimes overlapping trials of:
- ziprasidone, 160 mg bid
- risperidone, 3 mg bid
- trifluoperazine, 3 mg bid
- haloperidol, 10 mg bid
- divalproex sodium, 500 mg bid
- and carbamazepine, 400 mg bid.
None of these trials significantly improved her psychosis or mood.
At this point, the treating psychiatrists faced a difficult but clear decision: Ms. G was rechallenged on clozapine, 25 mg nightly, titrated again to 300 mg nightly. After she provided informed consent, her WBC was monitored twice daily—morning and evening—for agranulocytosis and to examine WBC patterns. Her average daily WBC counts were 4,200/mm3 in the morning and 5,500/mm3 at night. No physical signs of agranulocytosis emerged.
One week after restarting clozapine, Ms. G became less paranoid and socially more appropriate. Her thought process became increasingly organized, and after 4 weeks she reached her baseline status based upon family reports and the clinician’s CGI Global Improvement rating of 2 (much improved). Her auditory hallucinations resolved, and she was discharged to her family’s care.
Twice-daily blood testing was stopped at discharge. Ms. G continues to take clozapine and receives blood tests every 2 weeks, with no apparent signs of agranulocytosis.
Algorithm Suggested guidelines for managing WBC counts during clozapine therapy
Two treatments for clozapine-dependent agranulocytosis have been described.
- Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a glycoprotein that has been shown to stimulate proliferation of precursor cells in bone marrow and their differentiation into granulocytes and macrophages. Researchers have reported that GM-CSF treatment allows patients to continue taking clozapine after an episode of severe neutropenia.10-12
- Lithium salts have been reported to exploit the natural leukocytosis observed with lithium to counter clozapine-related leukopenia.13 Use of lithium to displace white blood cells has been debated, and anecdotal evidence suggests that combining lithium with clozapine may increase the chance of seizure and neuroleptic malignant syndrome.4 Still, cases reported by Adityanjee and Blier suggest that lithium augmentation is cost-effective and efficacious.14,15
How could Ms. G’s doctors have avoided stopping her clozapine therapy and her subsequent decompensation?
The authors’ observations
Aggressive blood testing and cessation of clozapine therapy are indicated when onset of granulocytopenia and agranulocytosis are suspected. Even with early detection and discontinuation, the chance of infectious disease poses a danger for up to 4 weeks until WBC levels return to normal.4
Given Ms. G’s lack of response to other antipsychotics, however, we had to consider resuming clozapine therapy. Studies have described agranulocytosis management strategies that may allow patients to keep taking clozapine despite low WBC counts (Box).
We also considered the timing of Ms. G’s blood test that showed a WBC count <2,700/mm3. Ahokas16 suggests that evening WBC counts are significantly higher than those taken in the morning and that granulocytes fluctuate in a diurnal pattern. Ms. G’s evening WBC counts were on average 1,300/mm3 higher than morning levels. Allowing for this diurnal variation and comparing evening blood samples could have averted the interruption in Ms. G’s clozapine therapy and prevented relapse in a patient with highly treatment-refractory schizophrenia.
Related resources
- Chong SA, Remington G. Clozapine augmentation: safety and efficacy. Schizophr Bull 2000;26:421-40.
- Emsley R, Oosthuizen P. The new and evolving pharmacotherapy of schizophrenia. Psychiatr Clin North Am 2003;26:141-63.
- Barnas C, Zwierzina H, Hummer M, Sperner-Unterweger B, Stern A, Fleischhacker WW. Granulocyte-macrophage colony-stimulating factor (GM-CSF) treatment of clozapine-induced agranulocytosis: a case report. J Clin Psychiatry 1992;53:245-7.
Drug brand names
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Divalproex sodium • Depakote
- GM-CSF—Filgrastim • Neupogen
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid, others
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trifluoperazine • Stelazine
- Ziprasidone • Geodon
Disclosure
Dr. Rao is a speaker for Pfizer Inc.
Drs. Goforth, Raval, and Sharma report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment resistant schizophrenic: a double blind comparison with chlorpromazine. Arch Gen Psychiatry 1988;45:789-96.
2. Brenner HD, Dencker SJ, Goldstein MJ, et al. Defining treatment refractoriness in schizophrenia. Schizophr Bull 1990;16:551-61.
3. Meltzer HY. Clozapine: is another view valid? Am J Psychiatry 1995;152:821-5.
4. Clozaril prescribing information. In: Physicians’ Desk Reference. (57th ed). Montvale, NJ: Thomson Healthcare, 2003.
5. Alvir JM, Lieberman JA, Safferman AZ, et al. Clozapine-induced agranulocytosis. Incidence and risk factors in the United States. N Engl J Med. 1993;329:162-7.
6. Honigfeld G. Effects of the clozapine national registry system on incidence of deaths related to agranulocytosis. Psychiatr Serv 1996;47:52-6.
7. Scelsa SN, Simpson DM, McQuistion HL, et al. Clozapineinduced myotoxicity in patients with chronic psychotic disorders. Neurology 1996;47:1518-23.
8. Ames D, Wirshing WC, Baker RW, et al. Predictive value of eosinophilia for neutropenia during clozapine treatment. J Clin Psychiatry 1996;57:579-81.
9. Alvir JM, Lieberman JA, Safferman AZ. Do white-cell count spikes predict agranulocytosis in clozapine recipients? Psychopharmacol Bull 1995;31:311-14.
10. Sperner-Unterweger B, Czeipek I, Gaggl S, et al. Treatment of severe clozapine-induced neutropenia with granulocyte colonystimulating factor (G-CSF). Remission despite continuous treatment with clozapine. Br J Psychiatry 1998;172:82-4.
11. Chengappa KN, Gopalani A, Haught MK, et al. The treatment of clozapine-associated agranulocytosis with granulocyte colony-stimulating factor (G-CSF). Psychopharmacol Bull 1996;32:111-21.
12. Lamberti JS, Bellnier TJ, Schwarzkopf SB, Schneider E. Filgrastim treatment of three patients with clozapine-induced agranulocytosis. J Clin Psychiatry 1995;56:256-9.
13. Boshes RA, Manschreck TC, Desrosiers J, et al. Initiation of clozapine therapy in a patient with preexisting leukopenia: a discussion of the rationale of current treatment options. Ann Clin Psychiatry 2001;13:233-7.
14. Adityanjee. Modification of clozapine-induced leukopenia and neutropenia with lithium carbonate. Am J Psychiatry 1995;152:648-9.
15. Blier P, Slater S, Measham T, et al. Lithium and clozapine-induced neutropenia/agranulocytosis. Int Clin Psychopharmacol 1998;13:137-40.
16. Ahokas A, Elonen E. Circadian rhythm of white blood cells during clozapine treatment. Psychopharmacology 1999;144:301-2.
1. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment resistant schizophrenic: a double blind comparison with chlorpromazine. Arch Gen Psychiatry 1988;45:789-96.
2. Brenner HD, Dencker SJ, Goldstein MJ, et al. Defining treatment refractoriness in schizophrenia. Schizophr Bull 1990;16:551-61.
3. Meltzer HY. Clozapine: is another view valid? Am J Psychiatry 1995;152:821-5.
4. Clozaril prescribing information. In: Physicians’ Desk Reference. (57th ed). Montvale, NJ: Thomson Healthcare, 2003.
5. Alvir JM, Lieberman JA, Safferman AZ, et al. Clozapine-induced agranulocytosis. Incidence and risk factors in the United States. N Engl J Med. 1993;329:162-7.
6. Honigfeld G. Effects of the clozapine national registry system on incidence of deaths related to agranulocytosis. Psychiatr Serv 1996;47:52-6.
7. Scelsa SN, Simpson DM, McQuistion HL, et al. Clozapineinduced myotoxicity in patients with chronic psychotic disorders. Neurology 1996;47:1518-23.
8. Ames D, Wirshing WC, Baker RW, et al. Predictive value of eosinophilia for neutropenia during clozapine treatment. J Clin Psychiatry 1996;57:579-81.
9. Alvir JM, Lieberman JA, Safferman AZ. Do white-cell count spikes predict agranulocytosis in clozapine recipients? Psychopharmacol Bull 1995;31:311-14.
10. Sperner-Unterweger B, Czeipek I, Gaggl S, et al. Treatment of severe clozapine-induced neutropenia with granulocyte colonystimulating factor (G-CSF). Remission despite continuous treatment with clozapine. Br J Psychiatry 1998;172:82-4.
11. Chengappa KN, Gopalani A, Haught MK, et al. The treatment of clozapine-associated agranulocytosis with granulocyte colony-stimulating factor (G-CSF). Psychopharmacol Bull 1996;32:111-21.
12. Lamberti JS, Bellnier TJ, Schwarzkopf SB, Schneider E. Filgrastim treatment of three patients with clozapine-induced agranulocytosis. J Clin Psychiatry 1995;56:256-9.
13. Boshes RA, Manschreck TC, Desrosiers J, et al. Initiation of clozapine therapy in a patient with preexisting leukopenia: a discussion of the rationale of current treatment options. Ann Clin Psychiatry 2001;13:233-7.
14. Adityanjee. Modification of clozapine-induced leukopenia and neutropenia with lithium carbonate. Am J Psychiatry 1995;152:648-9.
15. Blier P, Slater S, Measham T, et al. Lithium and clozapine-induced neutropenia/agranulocytosis. Int Clin Psychopharmacol 1998;13:137-40.
16. Ahokas A, Elonen E. Circadian rhythm of white blood cells during clozapine treatment. Psychopharmacology 1999;144:301-2.
Switching antipsychotics: A balanced approach to ease the transition
When switching antipsychotics for patients with schizophrenia, you can ease this potentially perilous passage by choosing the right time to switch and preventing psychotic relapse. This article describes four keys to a smooth transition:
- assess response and side effects with the existing medication
- weigh the pros and cons of switching, with input from the patient or caregiver
- select a replacement with characteristics that could improve patient function
- choose a switching strategy while considering safety and efficacy data.
RISKS AND BENEFITS OF SWITCHING
The two most compelling reasons to switch antipsychotics are enhanced clinical response and improved tolerability. Others may include lower medication cost, less-frequent monitoring, fewer drug interactions, or easier administration (such as once-daily versus twice-daily dosing).
Table 1
Sample response criteria to evaluate an antipsychotic switch
Improved functioning
|
Decreased symptoms
|
Improved tolerability
|
The greatest risk in a relatively stable patient is for psychotic symptoms to re-emerge. Be very careful when switching patients who:
- might harm themselves or others if their psychosis re-emerges during the switch
- were recently stabilized after an acute psychotic episode and have been maintained for less than 6 months on the medication that controlled their symptoms
- cannot adhere to oral medications and are being maintained on long-acting depot formulations.1
Other factors to consider include:
- the need for more-frequent patient visits during the transition to monitor for adverse effects
- the patient’s willingness to switch
- influence of external stressors—such as recent bereavement—or aspects of the patient’s workplace or living environment that may interfere with adherence to a new regimen
- medication cost and coverage by third-party payers.
Establishing specific response criteria for each patient (Table 1) will help you know when to continue or terminate a switch.
INADEQUATE RESPONSE
Because antipsychotics do not eliminate all positive and negative symptoms, an adequate response is considered a reasonable therapeutic goal. Many patients with schizophrenia respond adequately to traditional or atypical antipsychotics, but approximately one-third do not.2
What is an “adequate” response? No advisory groups or consensus panels have defined this term or set standards for when to switch. Therefore, psychiatrists must evaluate a patient’s response to an antipsychotic by using clinical judgment and patient/caregiver input. To gather the information you need, it is important to:
- identify target symptoms at baseline
- regularly monitor symptom severity, frequency, and intrusion on activities of daily living and quality of life.
Quantitative measures. In research, response is measured as a percent reduction in scores on standard assessments such as the Positive and Negative Syndrome Scales (PANSS). Using the PANSS, however, is too time-consuming for clinical practice.
The Brief Psychiatric Rating Scale (BPRS) is simpler than PANSS and can be administered more quickly but is less specific for negative symptoms. Even so, using the BPRS can help quantify baseline symptoms and monitor clinical response. It is useful to complete a BPRS rating prior to a switch and at subsequent visits during the transition. A >20% reduction in the total score is considered an adequate response.
Qualitative measures. Rating scales do not measure patients’ and sfamilies’ subjective feelings. One patient may view a 50% decrease in psychotic symptoms as extremely favorable and another as unacceptable. Switching therefore may be reasonable for one patient but not for another with a similar clinical response.
Table 2
Treatment-limiting antipsychotic side effects and options for switching
| Treatment-limiting factor | Atypical antipsychotic options |
|---|---|
| Akathisia/activation | Clozapine, olanzapine, quetiapine |
| Hyperprolactinemia | Any atypical antipsychotic except risperidone |
| Insomnia | Olanzapine, quetiapine |
| Orthostatic hypotension | Ziprasidone |
| Pre-existing cardiac dysfunction | Aripiprazole, olanzapine, quetiapine, risperidone |
| Pre-existing diabetes | Aripiprazole, quetiapine, ziprasidone |
| Sedation | Aripiprazole, risperidone, ziprasidone |
| Excessive weight gain | Aripiprazole, quetiapine, risperidone, ziprasidone |
Underdosing contributes to inadequate response, so assess whether an antipsychotic has been given a sufficient trial. For example, 400 to 800 mg/d of quetiapine is considered a therapeutic dosage, assuming adequate tolerability, but some clinicians stop increasing the dosage below that range. Similarly, the usual antipsychotic trial continues at least 3 to 4 weeks at a therapeutic dosage.
TREATMENT-LIMITING EFFECTS
Antipsychotic side effects that justify switching may be treatment-limiting or simply bothersome. For example, switching is necessary for antipsychotic-induced QTc prolongation in a patient with a history of cardiac dysrhythmias and reasonable for excessive daytime sedation in a patient who is working or attending school.
Extrapyramidal symptoms (EPS) and hyperprolactinemia may limit a patient’s tolerance of an antipsychotic. Patients who are especially sensitive to EPS may not tolerate high-potency agents such an haloperidol or even low to moderate dosages of risperidone. Others may not develop EPS while receiving haloperidol or higher dosages of risperidone.
Switching to an antipsychotic with relatively less histamine or alpha-adrenergic blockade may reduce problematic side effects such as sedation and orthostatic hypotension, respectively (Table 2).
Patients vary in how well they tolerate other side effects, such as weight gain. For example, a 10-lb weight gain may be acceptable to one patient and unacceptable to another. The decision to switch may be more obvious in a patient with diabetes, for whom substantial weight gain is unacceptable.
SWITCHING STRATEGIES
No method is universally accepted for switching from one antipsychotic to another. In clinical practice and research, three common methods (Figure) are used:
- immediately discontinuing drug A while starting drug B at full dosage
- slowly tapering drug A while starting drug B at full dosage
- slowly tapering drug A while slowly increasing drug B to full dosage.
Each method has advantages and disadvantages. Gradual cross-titration and tapering may reduce the risk of relapse but increase the risk of side effects. Elaborate regimens may confuse some patients—especially those with cognitive impairment—and increase the risk for adverse events and nonadherence.
Abruptly discontinuing an agent is less confusing and more convenient than gradual tapering, but patients may experience acute withdrawal (as with clozapine). Finally, no guidelines exist on how quickly to make the transition when one or both medications are cross-titrated and tapered. For inpatients receiving intense monitoring, a transition may be completed in 3 to 7 days, whereas outpatients may require 1 to 3 weeks.
Recommendation. The evidence cited in the next section of this article suggests that any of the three methods can be used when switching antipsychotics, except clozapine. When switching from clozapine, extend the cross-taper period to help minimize or eliminate rebound psychosis and cholinergic symptoms.
SWITCHING FROM DEPOT TO ORAL AGENTS
For patients switching from depot to oral antipsychotics, a 1-month cross-titration taper has been shown to be efficient and safe.
Godleski et al3 randomized 26 patients who had received IM depot antipsychotics (haloperidol or fluphenazine decanoate) for at least 3 years to either continue the IM depot antipsychotic or switch to olanzapine. Although the study was designed to assess the safety and efficacy of the switch, it also provided data on the transition method.
Figure 3 common antipsychotic switching strategies
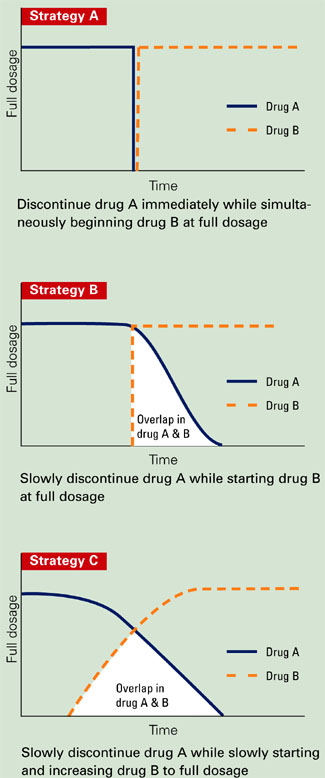
Subjects switching to olanzapine received their routine depot injection plus olanzapine, 10 mg/d for 1 month, followed by olanzapine monotherapy (5 to 20 mg/d) for 2 months. Those who continued IM depot therapy were maintained at a stable dose and dosing interval for 3 months. Safety and efficacy data were collected at baseline and monthly.
Patients receiving olanzapine improved on several efficacy measures, although the clinical relevance was minimal. For example, their mean PANSS total score decreased 3.23 points. One patient—in the control group—was hospitalized. Those who received olanzapine preferred this agent to the IM depot formulations and chose to continue daily olanzapine therapy. Adverse events did not increase significantly while patients received IM depot injections plus olanzapine.
SWITCHING ORAL AGENTS
Clozapine. When risperidone entered the U.S. market in the 1990s, a number of patients who had been treated with clozapine were abruptly switched to risperidone. Many experienced acute symptom exacerbation,4,5 including some whose rebound psychosis was more severe than their original symptoms. Other adverse effects—including nausea, diarrhea, vomiting, headache, restlessness, and sweating— have been attributed to cholinergic rebound caused by abruptly discontinuing clozapine.
To minimize the potential for rebound psychosis and cholinergic symptoms, taper clozapine across a minimum of 1 to 2 weeks. When you need to discontinue clozapine immediately—such as for patients experiencing serious hematologic effects—adding an anticholinergic such as benztropine may minimize the cholinergic rebound.
Risperidone. No studies have formally assessed methods for switching patients to risperidone.
Olanzapine. When switching to olanzapine, a direct switch or cross-titration tapering appear to be viable options.
In one multicenter, open-label study,2 108 patients were randomly assigned to olanzapine, 10 mg/d, after abruptly discontinuing a previous antipsychotic (direct switch) or by cross-titration tapering in a 1:1 fashion. Patients in the crosstitration group started olanzapine and discontinued their original antipsychotics across 2 weeks. Olanzapine dosages were adjusted as needed from 5 to 20 mg/d.
At study entry, approximately 95% of subjects in the direct-switch group and 85% in the crosstitration taper group were taking at least one typical antipsychotic—usually haloperidol. A switch was considered successful if a patient completed the 6-week trial without psychotic symptom worsening or EPS.
The 92 (85%) subjects who completed the study comprised similar percentages from both groups. Their scores on the PANSS total and subscales and Clinical Global Impression (CGI) scale also were similar.
The most common adverse events were somnolence, insomnia, and headache in the directs-witch group (all 11%), and somnolence (15%), headache (9%), insomnia (7%), and increased appetite (7%) in the cross-titration group. Differences in these percentages were not statistically significant.
EPS and akathisia improved significantly in both groups (p< 0.01) after switching to olanzapine. Thirteen (24%) patients in the direct-switch group and 17 (32%) in the cross-titration taper group required at least one dose of benztropine. Use of concomitant medications was similar.
Quetiapine can be abruptly discontinued with minimal risk of adverse events when initiating another antipsychotic.
Cutler et al6 switched 50 stable patients who had been treated with risperidone, thioridazine, haloperidol, or haloperidol plus benztropine. The original antipsychotics were abruptly discontinued, and quetiapine was initiated in a doseescalating fashion and then maintained at 300 mg/d for 12 days. After that, quetiapine was abruptly discontinued and patients were assessed for side effects, including EPS.
Most patients’ BPRS or CGI-Severity of Illness scores did not change significantly. Two patients (4%) experienced psychotic relapse during the switch. The authors speculated that these relapses might have been related to subtherapeutic quetiapine dosing. Transient nausea and vomiting were reported after quetiapine was discontinued.
Ziprasidone. When switching from another antipsychotic to ziprasidone, all three strategies appear well tolerated and maintain symptom control.
Using randomized, open-label trials, Weiden et al7 investigated strategies for switching patients to ziprasidone from olanzapine, risperidone, and traditional antipsychotics. All participants were diagnosed with schizophrenia or schizoaffective disorder and had experienced partial or inadequate response or side effects with their original antipsychotics.
Patients were assigned to one of three switching strategies:
- abruptly discontinue the initial antipsychotic
- decrease the initial antipsychotic’s dosage by 50% for 1 week, then discontinue it
- gradually taper the initial antipsychotic, so that subjects received 100% for 3 days of ziprasidone treatment, 50% for the next 4 days, and none thereafter.
For all three strategies, ziprasidone was started at 80 mg/d for 2 days, with dosing adjusted as needed to 40 to 160 mg/d.
All patients’ total score and positive and negative PANSS subscale scores improved significantly (p<0.01) across 6 weeks, although these data represent symptom changes after switching to ziprasidone. No efficacy or safety data were reported during switching. The authors concluded that patients could switch successfully to ziprasidone over a relatively short period using a variety of methods.
In another study, Stip8 switched 54 patients to ziprasidone from haloperidol. All received ziprasidone, 40 mg bid for 2 days and then 80 mg bid. Haloperidol was discontinued:
- immediately on day 1
- after the dosage was decreased by 50% for 7 days
- or after continuing the full dosage for 2 days, then taking 50% of the initial dosage for 5 days.
All patients maintained symptom control while switching, and 40 of 54 completed the trial. Among responders, BPRS and CGI scores improved significantly across 6 weeks, and EPS improved as expected.
Aripiprazole. Direct-switch and cross-titration tapering methods appear to be effective and welltolerated when switching stable patients to aripiprazole.
In an 8-week, randomized, open-label trial, Casey et al9 switched 311 outpatients with schizophrenia or schizoaffective disorder to aripiprazole. The patients—who had been taking stable dosages of haloperidol, chlorpromazine, risperidone, or olanzapine for at least 1 month—were randomly assigned to three groups:
- group 1 immediately discontinued the previous antipsychotic and started aripiprazole, 30 mg/d
- group 2 started aripiprazole, 30 mg/d, and discontinued the previous antipsychotic across 2 weeks
- group 3 started aripiprazole (10 mg/d in week 1, 20 mg/d in week 2, and 30 mg/d in week 3) and tapered the previous antipsychotic (50% less in week 1, another 50% less in week 2, then discontinued).
Investigators assessed treatment efficacy using the PANSS and CGI at baseline and weeks 4 and 8. They questioned patients about adverse events at each follow-up visit.
Nearly three-fourths (72%) of patients completed the trial. Discontinuation rates were 31% in group 1, 34% in group 2, and 19% in group 3. Efficacy, safety, tolerability, and incidence of discontinuation because of worsening psychosis were comparable across groups.
Similar percentages of patients in each group reported one or more adverse event (89%, 89%, and 81% for groups 1, 2, and 3, respectively). Most adverse events were described as mild to moderate. Insomnia was reported most frequently. Other adverse effects that occurred in >10% of subjects included nausea, akathisia, anxiety, psychosis, headache (groups 2 and 3), somnolence, lightheadedness (groups 1 and 2) vomiting (group 2 only), agitation (group 3 only), and diarrhea (group 2 only).
Seven patients were hospitalized for serious adverse events—usually worsening psychosis. Hospitalization rates were comparable among the three groups.
All groups improved slightly on the Barnes Akathisia Scale, Simpson Angus Rating Scale, and Abnormal Involuntary Movement Scale. Few patients in each group required benztropine (2%, 4%, and 7% for groups 1, 2 and 3, respectively).
Related resources
- Borison RL. Changing antipsychotic medication: guidelines on the transition to treatment with risperidone. The Consensus Study Group on Risperidone Dosing. Clin Ther 1996;18(4):592-607.
Drug brand names
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
Dr. Winans is a consultant for Bristol-Myers Squibb Co. and is a speaker for Pfizer Inc., Abbott Laboratories, and Bristol-Myers Squibb Co.
1. Weiden PJ, Aquila R, Dalheim L, Standard JM. Switching antipsychotic medications. J Clin Psychiatry 1997;58(suppl 10):63-72.
2. Lee CT, Conde BJL, Mazlan M, et al. Switching to olanzapine from previous antipsychotics: a regional collaborative multicenter trial assessing 2 switching techniques in Asia Pacific. J Clin Psychiatry 2002;63:569-76.
3. Godleski LS, Goldsmith J, Vieweg V, et al. Switching from depot antipsychotics to olanzapine in patients with chronic schizophrenia. J Clin Psychiatry 2003;64:119-22.
4. Clinical implications of clozapine discontinuation. Reports of an NIMH workshop. Schizophr Bull 1995;21(2):333-7.
5. Shiovitz TM, Welke TL, Tigel PD, et al. Cholinergic rebound and rapid psychosis following abrupt clozapine withdrawal. Schizophr Bull 1996;22(4):591-5.
6. Cutler AJ, Goldstein JM, Tumas JA. Dosing and switching strategies for quetiapine fumarate. Clin Ther 2002;24(2):209-22.
7. Weiden PJ, Simpson G, Potkin S. Therapeutic response in stable outpatients switched to ziprasidone (poster). Stockholm, Sweden: Association of European Psychiatrists 11th Congress, May 4-8, 2002.
8. Stip E. Haloperidol to ziprasidone switching strategies in schizophrenia (abstract). Eur Neuropsychopharmacol 2001;11(suppl 3):s272.-
9. Casey DE, Carson WH, Saha AR, et al. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology 2003;166(4):391-9.
When switching antipsychotics for patients with schizophrenia, you can ease this potentially perilous passage by choosing the right time to switch and preventing psychotic relapse. This article describes four keys to a smooth transition:
- assess response and side effects with the existing medication
- weigh the pros and cons of switching, with input from the patient or caregiver
- select a replacement with characteristics that could improve patient function
- choose a switching strategy while considering safety and efficacy data.
RISKS AND BENEFITS OF SWITCHING
The two most compelling reasons to switch antipsychotics are enhanced clinical response and improved tolerability. Others may include lower medication cost, less-frequent monitoring, fewer drug interactions, or easier administration (such as once-daily versus twice-daily dosing).
Table 1
Sample response criteria to evaluate an antipsychotic switch
Improved functioning
|
Decreased symptoms
|
Improved tolerability
|
The greatest risk in a relatively stable patient is for psychotic symptoms to re-emerge. Be very careful when switching patients who:
- might harm themselves or others if their psychosis re-emerges during the switch
- were recently stabilized after an acute psychotic episode and have been maintained for less than 6 months on the medication that controlled their symptoms
- cannot adhere to oral medications and are being maintained on long-acting depot formulations.1
Other factors to consider include:
- the need for more-frequent patient visits during the transition to monitor for adverse effects
- the patient’s willingness to switch
- influence of external stressors—such as recent bereavement—or aspects of the patient’s workplace or living environment that may interfere with adherence to a new regimen
- medication cost and coverage by third-party payers.
Establishing specific response criteria for each patient (Table 1) will help you know when to continue or terminate a switch.
INADEQUATE RESPONSE
Because antipsychotics do not eliminate all positive and negative symptoms, an adequate response is considered a reasonable therapeutic goal. Many patients with schizophrenia respond adequately to traditional or atypical antipsychotics, but approximately one-third do not.2
What is an “adequate” response? No advisory groups or consensus panels have defined this term or set standards for when to switch. Therefore, psychiatrists must evaluate a patient’s response to an antipsychotic by using clinical judgment and patient/caregiver input. To gather the information you need, it is important to:
- identify target symptoms at baseline
- regularly monitor symptom severity, frequency, and intrusion on activities of daily living and quality of life.
Quantitative measures. In research, response is measured as a percent reduction in scores on standard assessments such as the Positive and Negative Syndrome Scales (PANSS). Using the PANSS, however, is too time-consuming for clinical practice.
The Brief Psychiatric Rating Scale (BPRS) is simpler than PANSS and can be administered more quickly but is less specific for negative symptoms. Even so, using the BPRS can help quantify baseline symptoms and monitor clinical response. It is useful to complete a BPRS rating prior to a switch and at subsequent visits during the transition. A >20% reduction in the total score is considered an adequate response.
Qualitative measures. Rating scales do not measure patients’ and sfamilies’ subjective feelings. One patient may view a 50% decrease in psychotic symptoms as extremely favorable and another as unacceptable. Switching therefore may be reasonable for one patient but not for another with a similar clinical response.
Table 2
Treatment-limiting antipsychotic side effects and options for switching
| Treatment-limiting factor | Atypical antipsychotic options |
|---|---|
| Akathisia/activation | Clozapine, olanzapine, quetiapine |
| Hyperprolactinemia | Any atypical antipsychotic except risperidone |
| Insomnia | Olanzapine, quetiapine |
| Orthostatic hypotension | Ziprasidone |
| Pre-existing cardiac dysfunction | Aripiprazole, olanzapine, quetiapine, risperidone |
| Pre-existing diabetes | Aripiprazole, quetiapine, ziprasidone |
| Sedation | Aripiprazole, risperidone, ziprasidone |
| Excessive weight gain | Aripiprazole, quetiapine, risperidone, ziprasidone |
Underdosing contributes to inadequate response, so assess whether an antipsychotic has been given a sufficient trial. For example, 400 to 800 mg/d of quetiapine is considered a therapeutic dosage, assuming adequate tolerability, but some clinicians stop increasing the dosage below that range. Similarly, the usual antipsychotic trial continues at least 3 to 4 weeks at a therapeutic dosage.
TREATMENT-LIMITING EFFECTS
Antipsychotic side effects that justify switching may be treatment-limiting or simply bothersome. For example, switching is necessary for antipsychotic-induced QTc prolongation in a patient with a history of cardiac dysrhythmias and reasonable for excessive daytime sedation in a patient who is working or attending school.
Extrapyramidal symptoms (EPS) and hyperprolactinemia may limit a patient’s tolerance of an antipsychotic. Patients who are especially sensitive to EPS may not tolerate high-potency agents such an haloperidol or even low to moderate dosages of risperidone. Others may not develop EPS while receiving haloperidol or higher dosages of risperidone.
Switching to an antipsychotic with relatively less histamine or alpha-adrenergic blockade may reduce problematic side effects such as sedation and orthostatic hypotension, respectively (Table 2).
Patients vary in how well they tolerate other side effects, such as weight gain. For example, a 10-lb weight gain may be acceptable to one patient and unacceptable to another. The decision to switch may be more obvious in a patient with diabetes, for whom substantial weight gain is unacceptable.
SWITCHING STRATEGIES
No method is universally accepted for switching from one antipsychotic to another. In clinical practice and research, three common methods (Figure) are used:
- immediately discontinuing drug A while starting drug B at full dosage
- slowly tapering drug A while starting drug B at full dosage
- slowly tapering drug A while slowly increasing drug B to full dosage.
Each method has advantages and disadvantages. Gradual cross-titration and tapering may reduce the risk of relapse but increase the risk of side effects. Elaborate regimens may confuse some patients—especially those with cognitive impairment—and increase the risk for adverse events and nonadherence.
Abruptly discontinuing an agent is less confusing and more convenient than gradual tapering, but patients may experience acute withdrawal (as with clozapine). Finally, no guidelines exist on how quickly to make the transition when one or both medications are cross-titrated and tapered. For inpatients receiving intense monitoring, a transition may be completed in 3 to 7 days, whereas outpatients may require 1 to 3 weeks.
Recommendation. The evidence cited in the next section of this article suggests that any of the three methods can be used when switching antipsychotics, except clozapine. When switching from clozapine, extend the cross-taper period to help minimize or eliminate rebound psychosis and cholinergic symptoms.
SWITCHING FROM DEPOT TO ORAL AGENTS
For patients switching from depot to oral antipsychotics, a 1-month cross-titration taper has been shown to be efficient and safe.
Godleski et al3 randomized 26 patients who had received IM depot antipsychotics (haloperidol or fluphenazine decanoate) for at least 3 years to either continue the IM depot antipsychotic or switch to olanzapine. Although the study was designed to assess the safety and efficacy of the switch, it also provided data on the transition method.
Figure 3 common antipsychotic switching strategies
Subjects switching to olanzapine received their routine depot injection plus olanzapine, 10 mg/d for 1 month, followed by olanzapine monotherapy (5 to 20 mg/d) for 2 months. Those who continued IM depot therapy were maintained at a stable dose and dosing interval for 3 months. Safety and efficacy data were collected at baseline and monthly.
Patients receiving olanzapine improved on several efficacy measures, although the clinical relevance was minimal. For example, their mean PANSS total score decreased 3.23 points. One patient—in the control group—was hospitalized. Those who received olanzapine preferred this agent to the IM depot formulations and chose to continue daily olanzapine therapy. Adverse events did not increase significantly while patients received IM depot injections plus olanzapine.
SWITCHING ORAL AGENTS
Clozapine. When risperidone entered the U.S. market in the 1990s, a number of patients who had been treated with clozapine were abruptly switched to risperidone. Many experienced acute symptom exacerbation,4,5 including some whose rebound psychosis was more severe than their original symptoms. Other adverse effects—including nausea, diarrhea, vomiting, headache, restlessness, and sweating— have been attributed to cholinergic rebound caused by abruptly discontinuing clozapine.
To minimize the potential for rebound psychosis and cholinergic symptoms, taper clozapine across a minimum of 1 to 2 weeks. When you need to discontinue clozapine immediately—such as for patients experiencing serious hematologic effects—adding an anticholinergic such as benztropine may minimize the cholinergic rebound.
Risperidone. No studies have formally assessed methods for switching patients to risperidone.
Olanzapine. When switching to olanzapine, a direct switch or cross-titration tapering appear to be viable options.
In one multicenter, open-label study,2 108 patients were randomly assigned to olanzapine, 10 mg/d, after abruptly discontinuing a previous antipsychotic (direct switch) or by cross-titration tapering in a 1:1 fashion. Patients in the crosstitration group started olanzapine and discontinued their original antipsychotics across 2 weeks. Olanzapine dosages were adjusted as needed from 5 to 20 mg/d.
At study entry, approximately 95% of subjects in the direct-switch group and 85% in the crosstitration taper group were taking at least one typical antipsychotic—usually haloperidol. A switch was considered successful if a patient completed the 6-week trial without psychotic symptom worsening or EPS.
The 92 (85%) subjects who completed the study comprised similar percentages from both groups. Their scores on the PANSS total and subscales and Clinical Global Impression (CGI) scale also were similar.
The most common adverse events were somnolence, insomnia, and headache in the directs-witch group (all 11%), and somnolence (15%), headache (9%), insomnia (7%), and increased appetite (7%) in the cross-titration group. Differences in these percentages were not statistically significant.
EPS and akathisia improved significantly in both groups (p< 0.01) after switching to olanzapine. Thirteen (24%) patients in the direct-switch group and 17 (32%) in the cross-titration taper group required at least one dose of benztropine. Use of concomitant medications was similar.
Quetiapine can be abruptly discontinued with minimal risk of adverse events when initiating another antipsychotic.
Cutler et al6 switched 50 stable patients who had been treated with risperidone, thioridazine, haloperidol, or haloperidol plus benztropine. The original antipsychotics were abruptly discontinued, and quetiapine was initiated in a doseescalating fashion and then maintained at 300 mg/d for 12 days. After that, quetiapine was abruptly discontinued and patients were assessed for side effects, including EPS.
Most patients’ BPRS or CGI-Severity of Illness scores did not change significantly. Two patients (4%) experienced psychotic relapse during the switch. The authors speculated that these relapses might have been related to subtherapeutic quetiapine dosing. Transient nausea and vomiting were reported after quetiapine was discontinued.
Ziprasidone. When switching from another antipsychotic to ziprasidone, all three strategies appear well tolerated and maintain symptom control.
Using randomized, open-label trials, Weiden et al7 investigated strategies for switching patients to ziprasidone from olanzapine, risperidone, and traditional antipsychotics. All participants were diagnosed with schizophrenia or schizoaffective disorder and had experienced partial or inadequate response or side effects with their original antipsychotics.
Patients were assigned to one of three switching strategies:
- abruptly discontinue the initial antipsychotic
- decrease the initial antipsychotic’s dosage by 50% for 1 week, then discontinue it
- gradually taper the initial antipsychotic, so that subjects received 100% for 3 days of ziprasidone treatment, 50% for the next 4 days, and none thereafter.
For all three strategies, ziprasidone was started at 80 mg/d for 2 days, with dosing adjusted as needed to 40 to 160 mg/d.
All patients’ total score and positive and negative PANSS subscale scores improved significantly (p<0.01) across 6 weeks, although these data represent symptom changes after switching to ziprasidone. No efficacy or safety data were reported during switching. The authors concluded that patients could switch successfully to ziprasidone over a relatively short period using a variety of methods.
In another study, Stip8 switched 54 patients to ziprasidone from haloperidol. All received ziprasidone, 40 mg bid for 2 days and then 80 mg bid. Haloperidol was discontinued:
- immediately on day 1
- after the dosage was decreased by 50% for 7 days
- or after continuing the full dosage for 2 days, then taking 50% of the initial dosage for 5 days.
All patients maintained symptom control while switching, and 40 of 54 completed the trial. Among responders, BPRS and CGI scores improved significantly across 6 weeks, and EPS improved as expected.
Aripiprazole. Direct-switch and cross-titration tapering methods appear to be effective and welltolerated when switching stable patients to aripiprazole.
In an 8-week, randomized, open-label trial, Casey et al9 switched 311 outpatients with schizophrenia or schizoaffective disorder to aripiprazole. The patients—who had been taking stable dosages of haloperidol, chlorpromazine, risperidone, or olanzapine for at least 1 month—were randomly assigned to three groups:
- group 1 immediately discontinued the previous antipsychotic and started aripiprazole, 30 mg/d
- group 2 started aripiprazole, 30 mg/d, and discontinued the previous antipsychotic across 2 weeks
- group 3 started aripiprazole (10 mg/d in week 1, 20 mg/d in week 2, and 30 mg/d in week 3) and tapered the previous antipsychotic (50% less in week 1, another 50% less in week 2, then discontinued).
Investigators assessed treatment efficacy using the PANSS and CGI at baseline and weeks 4 and 8. They questioned patients about adverse events at each follow-up visit.
Nearly three-fourths (72%) of patients completed the trial. Discontinuation rates were 31% in group 1, 34% in group 2, and 19% in group 3. Efficacy, safety, tolerability, and incidence of discontinuation because of worsening psychosis were comparable across groups.
Similar percentages of patients in each group reported one or more adverse event (89%, 89%, and 81% for groups 1, 2, and 3, respectively). Most adverse events were described as mild to moderate. Insomnia was reported most frequently. Other adverse effects that occurred in >10% of subjects included nausea, akathisia, anxiety, psychosis, headache (groups 2 and 3), somnolence, lightheadedness (groups 1 and 2) vomiting (group 2 only), agitation (group 3 only), and diarrhea (group 2 only).
Seven patients were hospitalized for serious adverse events—usually worsening psychosis. Hospitalization rates were comparable among the three groups.
All groups improved slightly on the Barnes Akathisia Scale, Simpson Angus Rating Scale, and Abnormal Involuntary Movement Scale. Few patients in each group required benztropine (2%, 4%, and 7% for groups 1, 2 and 3, respectively).
Related resources
- Borison RL. Changing antipsychotic medication: guidelines on the transition to treatment with risperidone. The Consensus Study Group on Risperidone Dosing. Clin Ther 1996;18(4):592-607.
Drug brand names
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
Dr. Winans is a consultant for Bristol-Myers Squibb Co. and is a speaker for Pfizer Inc., Abbott Laboratories, and Bristol-Myers Squibb Co.
When switching antipsychotics for patients with schizophrenia, you can ease this potentially perilous passage by choosing the right time to switch and preventing psychotic relapse. This article describes four keys to a smooth transition:
- assess response and side effects with the existing medication
- weigh the pros and cons of switching, with input from the patient or caregiver
- select a replacement with characteristics that could improve patient function
- choose a switching strategy while considering safety and efficacy data.
RISKS AND BENEFITS OF SWITCHING
The two most compelling reasons to switch antipsychotics are enhanced clinical response and improved tolerability. Others may include lower medication cost, less-frequent monitoring, fewer drug interactions, or easier administration (such as once-daily versus twice-daily dosing).
Table 1
Sample response criteria to evaluate an antipsychotic switch
Improved functioning
|
Decreased symptoms
|
Improved tolerability
|
The greatest risk in a relatively stable patient is for psychotic symptoms to re-emerge. Be very careful when switching patients who:
- might harm themselves or others if their psychosis re-emerges during the switch
- were recently stabilized after an acute psychotic episode and have been maintained for less than 6 months on the medication that controlled their symptoms
- cannot adhere to oral medications and are being maintained on long-acting depot formulations.1
Other factors to consider include:
- the need for more-frequent patient visits during the transition to monitor for adverse effects
- the patient’s willingness to switch
- influence of external stressors—such as recent bereavement—or aspects of the patient’s workplace or living environment that may interfere with adherence to a new regimen
- medication cost and coverage by third-party payers.
Establishing specific response criteria for each patient (Table 1) will help you know when to continue or terminate a switch.
INADEQUATE RESPONSE
Because antipsychotics do not eliminate all positive and negative symptoms, an adequate response is considered a reasonable therapeutic goal. Many patients with schizophrenia respond adequately to traditional or atypical antipsychotics, but approximately one-third do not.2
What is an “adequate” response? No advisory groups or consensus panels have defined this term or set standards for when to switch. Therefore, psychiatrists must evaluate a patient’s response to an antipsychotic by using clinical judgment and patient/caregiver input. To gather the information you need, it is important to:
- identify target symptoms at baseline
- regularly monitor symptom severity, frequency, and intrusion on activities of daily living and quality of life.
Quantitative measures. In research, response is measured as a percent reduction in scores on standard assessments such as the Positive and Negative Syndrome Scales (PANSS). Using the PANSS, however, is too time-consuming for clinical practice.
The Brief Psychiatric Rating Scale (BPRS) is simpler than PANSS and can be administered more quickly but is less specific for negative symptoms. Even so, using the BPRS can help quantify baseline symptoms and monitor clinical response. It is useful to complete a BPRS rating prior to a switch and at subsequent visits during the transition. A >20% reduction in the total score is considered an adequate response.
Qualitative measures. Rating scales do not measure patients’ and sfamilies’ subjective feelings. One patient may view a 50% decrease in psychotic symptoms as extremely favorable and another as unacceptable. Switching therefore may be reasonable for one patient but not for another with a similar clinical response.
Table 2
Treatment-limiting antipsychotic side effects and options for switching
| Treatment-limiting factor | Atypical antipsychotic options |
|---|---|
| Akathisia/activation | Clozapine, olanzapine, quetiapine |
| Hyperprolactinemia | Any atypical antipsychotic except risperidone |
| Insomnia | Olanzapine, quetiapine |
| Orthostatic hypotension | Ziprasidone |
| Pre-existing cardiac dysfunction | Aripiprazole, olanzapine, quetiapine, risperidone |
| Pre-existing diabetes | Aripiprazole, quetiapine, ziprasidone |
| Sedation | Aripiprazole, risperidone, ziprasidone |
| Excessive weight gain | Aripiprazole, quetiapine, risperidone, ziprasidone |
Underdosing contributes to inadequate response, so assess whether an antipsychotic has been given a sufficient trial. For example, 400 to 800 mg/d of quetiapine is considered a therapeutic dosage, assuming adequate tolerability, but some clinicians stop increasing the dosage below that range. Similarly, the usual antipsychotic trial continues at least 3 to 4 weeks at a therapeutic dosage.
TREATMENT-LIMITING EFFECTS
Antipsychotic side effects that justify switching may be treatment-limiting or simply bothersome. For example, switching is necessary for antipsychotic-induced QTc prolongation in a patient with a history of cardiac dysrhythmias and reasonable for excessive daytime sedation in a patient who is working or attending school.
Extrapyramidal symptoms (EPS) and hyperprolactinemia may limit a patient’s tolerance of an antipsychotic. Patients who are especially sensitive to EPS may not tolerate high-potency agents such an haloperidol or even low to moderate dosages of risperidone. Others may not develop EPS while receiving haloperidol or higher dosages of risperidone.
Switching to an antipsychotic with relatively less histamine or alpha-adrenergic blockade may reduce problematic side effects such as sedation and orthostatic hypotension, respectively (Table 2).
Patients vary in how well they tolerate other side effects, such as weight gain. For example, a 10-lb weight gain may be acceptable to one patient and unacceptable to another. The decision to switch may be more obvious in a patient with diabetes, for whom substantial weight gain is unacceptable.
SWITCHING STRATEGIES
No method is universally accepted for switching from one antipsychotic to another. In clinical practice and research, three common methods (Figure) are used:
- immediately discontinuing drug A while starting drug B at full dosage
- slowly tapering drug A while starting drug B at full dosage
- slowly tapering drug A while slowly increasing drug B to full dosage.
Each method has advantages and disadvantages. Gradual cross-titration and tapering may reduce the risk of relapse but increase the risk of side effects. Elaborate regimens may confuse some patients—especially those with cognitive impairment—and increase the risk for adverse events and nonadherence.
Abruptly discontinuing an agent is less confusing and more convenient than gradual tapering, but patients may experience acute withdrawal (as with clozapine). Finally, no guidelines exist on how quickly to make the transition when one or both medications are cross-titrated and tapered. For inpatients receiving intense monitoring, a transition may be completed in 3 to 7 days, whereas outpatients may require 1 to 3 weeks.
Recommendation. The evidence cited in the next section of this article suggests that any of the three methods can be used when switching antipsychotics, except clozapine. When switching from clozapine, extend the cross-taper period to help minimize or eliminate rebound psychosis and cholinergic symptoms.
SWITCHING FROM DEPOT TO ORAL AGENTS
For patients switching from depot to oral antipsychotics, a 1-month cross-titration taper has been shown to be efficient and safe.
Godleski et al3 randomized 26 patients who had received IM depot antipsychotics (haloperidol or fluphenazine decanoate) for at least 3 years to either continue the IM depot antipsychotic or switch to olanzapine. Although the study was designed to assess the safety and efficacy of the switch, it also provided data on the transition method.
Figure 3 common antipsychotic switching strategies
Subjects switching to olanzapine received their routine depot injection plus olanzapine, 10 mg/d for 1 month, followed by olanzapine monotherapy (5 to 20 mg/d) for 2 months. Those who continued IM depot therapy were maintained at a stable dose and dosing interval for 3 months. Safety and efficacy data were collected at baseline and monthly.
Patients receiving olanzapine improved on several efficacy measures, although the clinical relevance was minimal. For example, their mean PANSS total score decreased 3.23 points. One patient—in the control group—was hospitalized. Those who received olanzapine preferred this agent to the IM depot formulations and chose to continue daily olanzapine therapy. Adverse events did not increase significantly while patients received IM depot injections plus olanzapine.
SWITCHING ORAL AGENTS
Clozapine. When risperidone entered the U.S. market in the 1990s, a number of patients who had been treated with clozapine were abruptly switched to risperidone. Many experienced acute symptom exacerbation,4,5 including some whose rebound psychosis was more severe than their original symptoms. Other adverse effects—including nausea, diarrhea, vomiting, headache, restlessness, and sweating— have been attributed to cholinergic rebound caused by abruptly discontinuing clozapine.
To minimize the potential for rebound psychosis and cholinergic symptoms, taper clozapine across a minimum of 1 to 2 weeks. When you need to discontinue clozapine immediately—such as for patients experiencing serious hematologic effects—adding an anticholinergic such as benztropine may minimize the cholinergic rebound.
Risperidone. No studies have formally assessed methods for switching patients to risperidone.
Olanzapine. When switching to olanzapine, a direct switch or cross-titration tapering appear to be viable options.
In one multicenter, open-label study,2 108 patients were randomly assigned to olanzapine, 10 mg/d, after abruptly discontinuing a previous antipsychotic (direct switch) or by cross-titration tapering in a 1:1 fashion. Patients in the crosstitration group started olanzapine and discontinued their original antipsychotics across 2 weeks. Olanzapine dosages were adjusted as needed from 5 to 20 mg/d.
At study entry, approximately 95% of subjects in the direct-switch group and 85% in the crosstitration taper group were taking at least one typical antipsychotic—usually haloperidol. A switch was considered successful if a patient completed the 6-week trial without psychotic symptom worsening or EPS.
The 92 (85%) subjects who completed the study comprised similar percentages from both groups. Their scores on the PANSS total and subscales and Clinical Global Impression (CGI) scale also were similar.
The most common adverse events were somnolence, insomnia, and headache in the directs-witch group (all 11%), and somnolence (15%), headache (9%), insomnia (7%), and increased appetite (7%) in the cross-titration group. Differences in these percentages were not statistically significant.
EPS and akathisia improved significantly in both groups (p< 0.01) after switching to olanzapine. Thirteen (24%) patients in the direct-switch group and 17 (32%) in the cross-titration taper group required at least one dose of benztropine. Use of concomitant medications was similar.
Quetiapine can be abruptly discontinued with minimal risk of adverse events when initiating another antipsychotic.
Cutler et al6 switched 50 stable patients who had been treated with risperidone, thioridazine, haloperidol, or haloperidol plus benztropine. The original antipsychotics were abruptly discontinued, and quetiapine was initiated in a doseescalating fashion and then maintained at 300 mg/d for 12 days. After that, quetiapine was abruptly discontinued and patients were assessed for side effects, including EPS.
Most patients’ BPRS or CGI-Severity of Illness scores did not change significantly. Two patients (4%) experienced psychotic relapse during the switch. The authors speculated that these relapses might have been related to subtherapeutic quetiapine dosing. Transient nausea and vomiting were reported after quetiapine was discontinued.
Ziprasidone. When switching from another antipsychotic to ziprasidone, all three strategies appear well tolerated and maintain symptom control.
Using randomized, open-label trials, Weiden et al7 investigated strategies for switching patients to ziprasidone from olanzapine, risperidone, and traditional antipsychotics. All participants were diagnosed with schizophrenia or schizoaffective disorder and had experienced partial or inadequate response or side effects with their original antipsychotics.
Patients were assigned to one of three switching strategies:
- abruptly discontinue the initial antipsychotic
- decrease the initial antipsychotic’s dosage by 50% for 1 week, then discontinue it
- gradually taper the initial antipsychotic, so that subjects received 100% for 3 days of ziprasidone treatment, 50% for the next 4 days, and none thereafter.
For all three strategies, ziprasidone was started at 80 mg/d for 2 days, with dosing adjusted as needed to 40 to 160 mg/d.
All patients’ total score and positive and negative PANSS subscale scores improved significantly (p<0.01) across 6 weeks, although these data represent symptom changes after switching to ziprasidone. No efficacy or safety data were reported during switching. The authors concluded that patients could switch successfully to ziprasidone over a relatively short period using a variety of methods.
In another study, Stip8 switched 54 patients to ziprasidone from haloperidol. All received ziprasidone, 40 mg bid for 2 days and then 80 mg bid. Haloperidol was discontinued:
- immediately on day 1
- after the dosage was decreased by 50% for 7 days
- or after continuing the full dosage for 2 days, then taking 50% of the initial dosage for 5 days.
All patients maintained symptom control while switching, and 40 of 54 completed the trial. Among responders, BPRS and CGI scores improved significantly across 6 weeks, and EPS improved as expected.
Aripiprazole. Direct-switch and cross-titration tapering methods appear to be effective and welltolerated when switching stable patients to aripiprazole.
In an 8-week, randomized, open-label trial, Casey et al9 switched 311 outpatients with schizophrenia or schizoaffective disorder to aripiprazole. The patients—who had been taking stable dosages of haloperidol, chlorpromazine, risperidone, or olanzapine for at least 1 month—were randomly assigned to three groups:
- group 1 immediately discontinued the previous antipsychotic and started aripiprazole, 30 mg/d
- group 2 started aripiprazole, 30 mg/d, and discontinued the previous antipsychotic across 2 weeks
- group 3 started aripiprazole (10 mg/d in week 1, 20 mg/d in week 2, and 30 mg/d in week 3) and tapered the previous antipsychotic (50% less in week 1, another 50% less in week 2, then discontinued).
Investigators assessed treatment efficacy using the PANSS and CGI at baseline and weeks 4 and 8. They questioned patients about adverse events at each follow-up visit.
Nearly three-fourths (72%) of patients completed the trial. Discontinuation rates were 31% in group 1, 34% in group 2, and 19% in group 3. Efficacy, safety, tolerability, and incidence of discontinuation because of worsening psychosis were comparable across groups.
Similar percentages of patients in each group reported one or more adverse event (89%, 89%, and 81% for groups 1, 2, and 3, respectively). Most adverse events were described as mild to moderate. Insomnia was reported most frequently. Other adverse effects that occurred in >10% of subjects included nausea, akathisia, anxiety, psychosis, headache (groups 2 and 3), somnolence, lightheadedness (groups 1 and 2) vomiting (group 2 only), agitation (group 3 only), and diarrhea (group 2 only).
Seven patients were hospitalized for serious adverse events—usually worsening psychosis. Hospitalization rates were comparable among the three groups.
All groups improved slightly on the Barnes Akathisia Scale, Simpson Angus Rating Scale, and Abnormal Involuntary Movement Scale. Few patients in each group required benztropine (2%, 4%, and 7% for groups 1, 2 and 3, respectively).
Related resources
- Borison RL. Changing antipsychotic medication: guidelines on the transition to treatment with risperidone. The Consensus Study Group on Risperidone Dosing. Clin Ther 1996;18(4):592-607.
Drug brand names
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
Dr. Winans is a consultant for Bristol-Myers Squibb Co. and is a speaker for Pfizer Inc., Abbott Laboratories, and Bristol-Myers Squibb Co.
1. Weiden PJ, Aquila R, Dalheim L, Standard JM. Switching antipsychotic medications. J Clin Psychiatry 1997;58(suppl 10):63-72.
2. Lee CT, Conde BJL, Mazlan M, et al. Switching to olanzapine from previous antipsychotics: a regional collaborative multicenter trial assessing 2 switching techniques in Asia Pacific. J Clin Psychiatry 2002;63:569-76.
3. Godleski LS, Goldsmith J, Vieweg V, et al. Switching from depot antipsychotics to olanzapine in patients with chronic schizophrenia. J Clin Psychiatry 2003;64:119-22.
4. Clinical implications of clozapine discontinuation. Reports of an NIMH workshop. Schizophr Bull 1995;21(2):333-7.
5. Shiovitz TM, Welke TL, Tigel PD, et al. Cholinergic rebound and rapid psychosis following abrupt clozapine withdrawal. Schizophr Bull 1996;22(4):591-5.
6. Cutler AJ, Goldstein JM, Tumas JA. Dosing and switching strategies for quetiapine fumarate. Clin Ther 2002;24(2):209-22.
7. Weiden PJ, Simpson G, Potkin S. Therapeutic response in stable outpatients switched to ziprasidone (poster). Stockholm, Sweden: Association of European Psychiatrists 11th Congress, May 4-8, 2002.
8. Stip E. Haloperidol to ziprasidone switching strategies in schizophrenia (abstract). Eur Neuropsychopharmacol 2001;11(suppl 3):s272.-
9. Casey DE, Carson WH, Saha AR, et al. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology 2003;166(4):391-9.
1. Weiden PJ, Aquila R, Dalheim L, Standard JM. Switching antipsychotic medications. J Clin Psychiatry 1997;58(suppl 10):63-72.
2. Lee CT, Conde BJL, Mazlan M, et al. Switching to olanzapine from previous antipsychotics: a regional collaborative multicenter trial assessing 2 switching techniques in Asia Pacific. J Clin Psychiatry 2002;63:569-76.
3. Godleski LS, Goldsmith J, Vieweg V, et al. Switching from depot antipsychotics to olanzapine in patients with chronic schizophrenia. J Clin Psychiatry 2003;64:119-22.
4. Clinical implications of clozapine discontinuation. Reports of an NIMH workshop. Schizophr Bull 1995;21(2):333-7.
5. Shiovitz TM, Welke TL, Tigel PD, et al. Cholinergic rebound and rapid psychosis following abrupt clozapine withdrawal. Schizophr Bull 1996;22(4):591-5.
6. Cutler AJ, Goldstein JM, Tumas JA. Dosing and switching strategies for quetiapine fumarate. Clin Ther 2002;24(2):209-22.
7. Weiden PJ, Simpson G, Potkin S. Therapeutic response in stable outpatients switched to ziprasidone (poster). Stockholm, Sweden: Association of European Psychiatrists 11th Congress, May 4-8, 2002.
8. Stip E. Haloperidol to ziprasidone switching strategies in schizophrenia (abstract). Eur Neuropsychopharmacol 2001;11(suppl 3):s272.-
9. Casey DE, Carson WH, Saha AR, et al. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology 2003;166(4):391-9.
Prescribing information: Scroll with the changes
Today’s physician needs to stay abreast of ever-changing prescribing information. Print and CD-ROM drug guides, long regarded as the gold standard in clinical reference, are constrained by press deadlines and start becoming outdated upon publication.
Electronic drug guides, available online or via handheld computers, are updated regularly and provide real-time developments about drug alerts, safety recalls, and changes in prescribing information.
Online drug guides
Benefits. Drug-drug interactions can be checked with a couple of clicks. By contrast, scanning printed lists of interactions for each medication could cost you valuable time.
Some electronic guides list the medication’s cost and formulary availability in addition to dosing, indication(s), drug mechanism, precautions, and adverse reactions.
Drawbacks. Depending on the type of subscription, access to an online drug reference may be restricted. Many institutions purchase access to online references, which are readily available at any terminal or computer in the hospital network. When attempting to access the resource from an outside computer, however, access is denied.
Some facilities circumvent this problem by providing a proxy server to relay access or use a virtual private network to connect to the hospital network. Individual subscriptions, which provide access via password, do not have this limitation.
The table lists benefits and drawbacks of individual programs.
Drug guides for handhelds
Drug guides for personal digital assistants are more accessible, but many handhelds lack sufficient memory for the guides’ databases and programs.
Some programs, such as the American Hospital Formulary Service guide and Physicians’ Desk Reference, can operate from external memory-such as secure digital or compact flash-thus preserving the limited main memory of most handhelds. In working this way, however, these programs’ databases often cannot be updated automatically and require user intervention.
The handheld’s small screen size is another potential drawback. The better handheld drug guides are designed to maximize limited screen space and promote easy navigation by utilizing hyperlinks to jump to related information or pop up windows to provide additional information. Ideally, content should be succinct yet offer sufficient detail.
How to choose a drug reference guide
Electronic drug reference guides vary greatly in their detail, organization of content, and ease of navigation and downloading. To decide which drug reference guide is right for you, try several demonstration versions:
- Compare different features. Look for sufficient content and intuitive navigation.
- Find out if pre-made patient education brochures are available for printing.
- For handheld guides, determine how much memory is required by the program and databases, how often they must be updated, and whether they can be stored on external memory.
- Ask if discounts are offered for purchases during trade shows or for a combination purchase of the guide’s online and handheld versions.
The following table, which lists quick reviews of reference guide demonstration products, can help you explore the options.
If you have any questions about these products or comments about Psyber Psychiatry, click here to contact Dr. Luo or send an e-mail to: [email protected].
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
Today’s physician needs to stay abreast of ever-changing prescribing information. Print and CD-ROM drug guides, long regarded as the gold standard in clinical reference, are constrained by press deadlines and start becoming outdated upon publication.
Electronic drug guides, available online or via handheld computers, are updated regularly and provide real-time developments about drug alerts, safety recalls, and changes in prescribing information.
Online drug guides
Benefits. Drug-drug interactions can be checked with a couple of clicks. By contrast, scanning printed lists of interactions for each medication could cost you valuable time.
Some electronic guides list the medication’s cost and formulary availability in addition to dosing, indication(s), drug mechanism, precautions, and adverse reactions.
Drawbacks. Depending on the type of subscription, access to an online drug reference may be restricted. Many institutions purchase access to online references, which are readily available at any terminal or computer in the hospital network. When attempting to access the resource from an outside computer, however, access is denied.
Some facilities circumvent this problem by providing a proxy server to relay access or use a virtual private network to connect to the hospital network. Individual subscriptions, which provide access via password, do not have this limitation.
The table lists benefits and drawbacks of individual programs.
Drug guides for handhelds
Drug guides for personal digital assistants are more accessible, but many handhelds lack sufficient memory for the guides’ databases and programs.
Some programs, such as the American Hospital Formulary Service guide and Physicians’ Desk Reference, can operate from external memory-such as secure digital or compact flash-thus preserving the limited main memory of most handhelds. In working this way, however, these programs’ databases often cannot be updated automatically and require user intervention.
The handheld’s small screen size is another potential drawback. The better handheld drug guides are designed to maximize limited screen space and promote easy navigation by utilizing hyperlinks to jump to related information or pop up windows to provide additional information. Ideally, content should be succinct yet offer sufficient detail.
How to choose a drug reference guide
Electronic drug reference guides vary greatly in their detail, organization of content, and ease of navigation and downloading. To decide which drug reference guide is right for you, try several demonstration versions:
- Compare different features. Look for sufficient content and intuitive navigation.
- Find out if pre-made patient education brochures are available for printing.
- For handheld guides, determine how much memory is required by the program and databases, how often they must be updated, and whether they can be stored on external memory.
- Ask if discounts are offered for purchases during trade shows or for a combination purchase of the guide’s online and handheld versions.
The following table, which lists quick reviews of reference guide demonstration products, can help you explore the options.
If you have any questions about these products or comments about Psyber Psychiatry, click here to contact Dr. Luo or send an e-mail to: [email protected].
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
Today’s physician needs to stay abreast of ever-changing prescribing information. Print and CD-ROM drug guides, long regarded as the gold standard in clinical reference, are constrained by press deadlines and start becoming outdated upon publication.
Electronic drug guides, available online or via handheld computers, are updated regularly and provide real-time developments about drug alerts, safety recalls, and changes in prescribing information.
Online drug guides
Benefits. Drug-drug interactions can be checked with a couple of clicks. By contrast, scanning printed lists of interactions for each medication could cost you valuable time.
Some electronic guides list the medication’s cost and formulary availability in addition to dosing, indication(s), drug mechanism, precautions, and adverse reactions.
Drawbacks. Depending on the type of subscription, access to an online drug reference may be restricted. Many institutions purchase access to online references, which are readily available at any terminal or computer in the hospital network. When attempting to access the resource from an outside computer, however, access is denied.
Some facilities circumvent this problem by providing a proxy server to relay access or use a virtual private network to connect to the hospital network. Individual subscriptions, which provide access via password, do not have this limitation.
The table lists benefits and drawbacks of individual programs.
Drug guides for handhelds
Drug guides for personal digital assistants are more accessible, but many handhelds lack sufficient memory for the guides’ databases and programs.
Some programs, such as the American Hospital Formulary Service guide and Physicians’ Desk Reference, can operate from external memory-such as secure digital or compact flash-thus preserving the limited main memory of most handhelds. In working this way, however, these programs’ databases often cannot be updated automatically and require user intervention.
The handheld’s small screen size is another potential drawback. The better handheld drug guides are designed to maximize limited screen space and promote easy navigation by utilizing hyperlinks to jump to related information or pop up windows to provide additional information. Ideally, content should be succinct yet offer sufficient detail.
How to choose a drug reference guide
Electronic drug reference guides vary greatly in their detail, organization of content, and ease of navigation and downloading. To decide which drug reference guide is right for you, try several demonstration versions:
- Compare different features. Look for sufficient content and intuitive navigation.
- Find out if pre-made patient education brochures are available for printing.
- For handheld guides, determine how much memory is required by the program and databases, how often they must be updated, and whether they can be stored on external memory.
- Ask if discounts are offered for purchases during trade shows or for a combination purchase of the guide’s online and handheld versions.
The following table, which lists quick reviews of reference guide demonstration products, can help you explore the options.
If you have any questions about these products or comments about Psyber Psychiatry, click here to contact Dr. Luo or send an e-mail to: [email protected].
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
Removing roadblocks to medical care for the severely mentally ill
Severely mentally ill patients often do not get the care they need for chronic medical conditions such as diabetes, hypertension, or hepatitis C. They may lack insurance, forget follow-up appointments, or fail to provide adequate or accurate history because of cognitive deficits. Some fear blood work or other tests, and many adhere poorly to treatment.
12 ways to expedite care
The following tips can help remove some of these roadblocks to proper care:
- Review the patient’s records—psychiatric and medical—to obtain information the patient might not have provided.
- Explain the medical problem in terms the patient understands. Describe why an evaluation or treatment is necessary.
- Ask the patient’s case manager or program nurse to follow up with the patient.
- Refer the patient to a family doctor or internist who participates in the patient’s insurance plan and with whom he or she is comfortable.
- Remind the patient of his or her appointment, preferably that morning. If possible, have the case manager arrange transportation.
- Have a family member or staff member accompany the patient on the visit if the patient expresses fear beforehand. Consider offering an anxiolytic to be taken the day of the visit as needed. Above all, be encouraging.
- Write a note to the doctor about the reason for the referral. Include some history because the patient may not be able to give it, especially within a 15-minute office visit.
- Order specific blood work to be drawn before the visit, if indicated. Have results sent to the doctor’s office.
- Consider screening high-risk patients for hepatitis, including those with a history of IV drug use or multiple sexual partners. Do not automatically attribute liver enzyme elevations to divalproex.
- Review your patient’s diet and exercise habits. Politely discourage excessive sugar and caffeine intake. I always keep some bottled water and raw vegetables on hand to model recommended habits. Many patients have limited incomes, so recommend affordable, healthy foods.
- Monitor for side effects of psychotropics, such as white blood counts for patients on clozapine, liver function and platelet counts with valproate, renal and thyroid function with lithium, etc.
- Don’t give up. If your patient has missed multiple doctors’ appointments, keeps insisting he or she is not sick, or repeatedly fails to comply with diet or medication, continue to be encouraging and advocate the need for proper care.
- Clozapine • Clozaril
- Divalproex • Depakote
- Lithium • Eskalith, Lithobid, others
- Valproate • Depacon
Dr. Szeeley is associate professor, department of psychiatry, University of Medicine and Dentistry of New Jersey, Camden, and heads the department’s division of community psychiatry.
Severely mentally ill patients often do not get the care they need for chronic medical conditions such as diabetes, hypertension, or hepatitis C. They may lack insurance, forget follow-up appointments, or fail to provide adequate or accurate history because of cognitive deficits. Some fear blood work or other tests, and many adhere poorly to treatment.
12 ways to expedite care
The following tips can help remove some of these roadblocks to proper care:
- Review the patient’s records—psychiatric and medical—to obtain information the patient might not have provided.
- Explain the medical problem in terms the patient understands. Describe why an evaluation or treatment is necessary.
- Ask the patient’s case manager or program nurse to follow up with the patient.
- Refer the patient to a family doctor or internist who participates in the patient’s insurance plan and with whom he or she is comfortable.
- Remind the patient of his or her appointment, preferably that morning. If possible, have the case manager arrange transportation.
- Have a family member or staff member accompany the patient on the visit if the patient expresses fear beforehand. Consider offering an anxiolytic to be taken the day of the visit as needed. Above all, be encouraging.
- Write a note to the doctor about the reason for the referral. Include some history because the patient may not be able to give it, especially within a 15-minute office visit.
- Order specific blood work to be drawn before the visit, if indicated. Have results sent to the doctor’s office.
- Consider screening high-risk patients for hepatitis, including those with a history of IV drug use or multiple sexual partners. Do not automatically attribute liver enzyme elevations to divalproex.
- Review your patient’s diet and exercise habits. Politely discourage excessive sugar and caffeine intake. I always keep some bottled water and raw vegetables on hand to model recommended habits. Many patients have limited incomes, so recommend affordable, healthy foods.
- Monitor for side effects of psychotropics, such as white blood counts for patients on clozapine, liver function and platelet counts with valproate, renal and thyroid function with lithium, etc.
- Don’t give up. If your patient has missed multiple doctors’ appointments, keeps insisting he or she is not sick, or repeatedly fails to comply with diet or medication, continue to be encouraging and advocate the need for proper care.
- Clozapine • Clozaril
- Divalproex • Depakote
- Lithium • Eskalith, Lithobid, others
- Valproate • Depacon
Severely mentally ill patients often do not get the care they need for chronic medical conditions such as diabetes, hypertension, or hepatitis C. They may lack insurance, forget follow-up appointments, or fail to provide adequate or accurate history because of cognitive deficits. Some fear blood work or other tests, and many adhere poorly to treatment.
12 ways to expedite care
The following tips can help remove some of these roadblocks to proper care:
- Review the patient’s records—psychiatric and medical—to obtain information the patient might not have provided.
- Explain the medical problem in terms the patient understands. Describe why an evaluation or treatment is necessary.
- Ask the patient’s case manager or program nurse to follow up with the patient.
- Refer the patient to a family doctor or internist who participates in the patient’s insurance plan and with whom he or she is comfortable.
- Remind the patient of his or her appointment, preferably that morning. If possible, have the case manager arrange transportation.
- Have a family member or staff member accompany the patient on the visit if the patient expresses fear beforehand. Consider offering an anxiolytic to be taken the day of the visit as needed. Above all, be encouraging.
- Write a note to the doctor about the reason for the referral. Include some history because the patient may not be able to give it, especially within a 15-minute office visit.
- Order specific blood work to be drawn before the visit, if indicated. Have results sent to the doctor’s office.
- Consider screening high-risk patients for hepatitis, including those with a history of IV drug use or multiple sexual partners. Do not automatically attribute liver enzyme elevations to divalproex.
- Review your patient’s diet and exercise habits. Politely discourage excessive sugar and caffeine intake. I always keep some bottled water and raw vegetables on hand to model recommended habits. Many patients have limited incomes, so recommend affordable, healthy foods.
- Monitor for side effects of psychotropics, such as white blood counts for patients on clozapine, liver function and platelet counts with valproate, renal and thyroid function with lithium, etc.
- Don’t give up. If your patient has missed multiple doctors’ appointments, keeps insisting he or she is not sick, or repeatedly fails to comply with diet or medication, continue to be encouraging and advocate the need for proper care.
- Clozapine • Clozaril
- Divalproex • Depakote
- Lithium • Eskalith, Lithobid, others
- Valproate • Depacon
Dr. Szeeley is associate professor, department of psychiatry, University of Medicine and Dentistry of New Jersey, Camden, and heads the department’s division of community psychiatry.
Dr. Szeeley is associate professor, department of psychiatry, University of Medicine and Dentistry of New Jersey, Camden, and heads the department’s division of community psychiatry.
Out of the pipeline and into your office
Where do you learn about new psychotropics? One source is the drug reps who come to see us all the time, but their educational materials have a marketing objective.
Current Psychiatry’s “Out of the Pipeline” offers an alernative—a source of early, unbiased information about new medications for psychiatric practice.
This month’s “Out of the Pipeline” examines sodium oxybate (Xyrem), indicated for treating cataplexy in patients with narcolepsy. Sodium oxybate is a legally manufactured form of an illegal “date rape” drug. As an orphan drug, sodium oxybate has undergone less clinical testing than is required for conventional FDA approvals. I want to know as much as possible about this medication before I prescribe it, and the article by Lois E. Krahn, MD—one of Current Psychiatry’s associate editors—is a good start.
Frankly, it is difficult to find qualified “Out of the Pipeline” authors who don’t have apparent conflicts of interest. Almost anyone who could write knowledgeably about premarket experience with a drug has participated in its clinical trials, most of which are supported by pharmaceutical companies. To address this concern, we:
- identify and invite the authors ourselves (without input from pharmaceutical companies)
- emphasize to authors the need for balance and objectivity
- disclose authors’ financial relationships with any companies whose products are mentioned or with manufacturers of competing products
- subject each article to peer review and revision before publication.
I expect more drugs will be developed with novel mechanisms of action and very specific indications. This trend will benefit our patients but increase our need for trusted advice, as in Current Psychiatry.

James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
Where do you learn about new psychotropics? One source is the drug reps who come to see us all the time, but their educational materials have a marketing objective.
Current Psychiatry’s “Out of the Pipeline” offers an alernative—a source of early, unbiased information about new medications for psychiatric practice.
This month’s “Out of the Pipeline” examines sodium oxybate (Xyrem), indicated for treating cataplexy in patients with narcolepsy. Sodium oxybate is a legally manufactured form of an illegal “date rape” drug. As an orphan drug, sodium oxybate has undergone less clinical testing than is required for conventional FDA approvals. I want to know as much as possible about this medication before I prescribe it, and the article by Lois E. Krahn, MD—one of Current Psychiatry’s associate editors—is a good start.
Frankly, it is difficult to find qualified “Out of the Pipeline” authors who don’t have apparent conflicts of interest. Almost anyone who could write knowledgeably about premarket experience with a drug has participated in its clinical trials, most of which are supported by pharmaceutical companies. To address this concern, we:
- identify and invite the authors ourselves (without input from pharmaceutical companies)
- emphasize to authors the need for balance and objectivity
- disclose authors’ financial relationships with any companies whose products are mentioned or with manufacturers of competing products
- subject each article to peer review and revision before publication.
I expect more drugs will be developed with novel mechanisms of action and very specific indications. This trend will benefit our patients but increase our need for trusted advice, as in Current Psychiatry.
Where do you learn about new psychotropics? One source is the drug reps who come to see us all the time, but their educational materials have a marketing objective.
Current Psychiatry’s “Out of the Pipeline” offers an alernative—a source of early, unbiased information about new medications for psychiatric practice.
This month’s “Out of the Pipeline” examines sodium oxybate (Xyrem), indicated for treating cataplexy in patients with narcolepsy. Sodium oxybate is a legally manufactured form of an illegal “date rape” drug. As an orphan drug, sodium oxybate has undergone less clinical testing than is required for conventional FDA approvals. I want to know as much as possible about this medication before I prescribe it, and the article by Lois E. Krahn, MD—one of Current Psychiatry’s associate editors—is a good start.
Frankly, it is difficult to find qualified “Out of the Pipeline” authors who don’t have apparent conflicts of interest. Almost anyone who could write knowledgeably about premarket experience with a drug has participated in its clinical trials, most of which are supported by pharmaceutical companies. To address this concern, we:
- identify and invite the authors ourselves (without input from pharmaceutical companies)
- emphasize to authors the need for balance and objectivity
- disclose authors’ financial relationships with any companies whose products are mentioned or with manufacturers of competing products
- subject each article to peer review and revision before publication.
I expect more drugs will be developed with novel mechanisms of action and very specific indications. This trend will benefit our patients but increase our need for trusted advice, as in Current Psychiatry.
Crossing the line: When does teen substance use become abuse or dependence?
Assessing an adolescent for a possible substance use disorder can be streamlined by choosing age-appropriate screening tools and asking targeted questions. Based on our experience, we offer a 4-step approach to these at-risk patients1 (Box) that focuses on:
- quantifying alcohol or drug abuse and/or dependence
- identifying and treating psychiatric comorbidity
- evaluating and addressing social influences that contribute to substance use
- assessing negative consequences associated with substance abuse.
Adolescent substance use increases the risk of motor vehicle accidents, suicide, transmission of HIV and other sexual diseases, criminal behaviors, and psychological problems. Alcohol and marijuana are the substances most commonly abused by adolescents.
In 2002, the University of Michigan Institute for Social Research’s annual “Monitoring the Future” study reported:
- drunkenness in 7% of 8th graders, 18% of 10th graders, and 30% of 12th graders at least once in the previous 30 days.
- illicit drug use by 18% of 8th graders, 35% of 10th graders, and 41% of 12th graders in the previous 12 months.
Boys used substances more frequently than girls, and boys’ use was more severe. Within the previous month, boys reported greater alcohol use, binge drinking (five or more drinks in one sitting), and heavy drinking, as well as greater illicit drug use in the past year.1
INITIAL EVALUATION
Adolescents generally do not seek substance abuse treatment but are referred because of alcohol- or drug-related legal, school, or family problems. Thus, most present for evaluation with their parents, legal guardians, or officers of the court.
We begin by finding out from parents or guardians the reasons for the evaluation, their perspectives on the adolescent’s behavior, and their expectations of treatment. Then we interview the adolescent alone, assessing for substance use and evaluating peer relationships.
Components. A typical initial evaluation takes 90 minutes to 2 hours and includes:
- psychiatric history and symptoms
- medical history
- previous hospitalizations (medical and psychiatric)
- family history
- social history.
Specifically, the assessment focuses on the reason for the evaluation, with attention to diagnostic criteria for substance use/dependence.
STEP 1: QUANTIFYING DEPENDENCE
As with adults, clinical diagnosis of substance abuse or dependence in adolescents is based on DSM-IV diagnostic criteria (Table 1). Adolescents, however, differ from adults in diagnostic presentation, risk of dependence, and patterns of substance use.
Diagnostic ‘orphans.’ DSM-IV criteria for alcohol use disorders have limitations in adolescents.2 Teens who report one or two dependence symptoms and no abuse symptoms have been described as “diagnostic orphans”3—they fall short of criteria for dependence or abuse but clearly demonstrate substance use patterns. This presentation is common; in a survey of 74,008 high school students, almost 10% of 12th graders reported one or two dependence symptoms and no abuse symptoms.4
Risk of dependence. Adolescents who begin using alcohol or drugs develop dependence more rapidly than adults do.5
Patterns of use. Adolescents are more likely than adults to binge with alcohol and drugs, which may conceal the severity of their abuse. DSM-IV diagnostic criteria for substance abuse or dependence do not consider quantity of use, such as number of drinks or percent of days drinking or using drugs.
Assessment instruments. Many assessment instruments are available to explore adolescent substance use and its associated consequences. Some are described in detail and are available on the Internet. Common screening instruments that can be used for adolescent substance use are compared in Table 2.
DUSI-A and POSIT. Two self-report instruments—Drug Use Screening Inventory-Adolescents (DUSI-A)6 and Problem Oriented Screening Instrument for Teenagers (POSIT)7—can help explore alcohol or drug use in teens who admit to substance use. Anyone who endorses at least one DSM-IV abuse or dependence criterion requires further evaluation. Either test is a good starting point, and both have a built-in “lie” scale.
T-ASI and CASI. The Teen Addiction Severity Index (T-ASI)8 and Comprehensive Adolescent Severity Inventory (CASI)9 are more labor-intensive and require training to administer. These assessments are more appropriate for adolescents with extensive alcohol or drug abuse.
A-OCDS and Deas-MOCS. Our group recently developed the Adolescent Obsessive Compulsive Drinking Scale (A-OCDS)10 and the Deas-Marijuana Obsessive Compulsive Scale (Deas-MOCS).11
These self-report instruments have been validated in treatment- and nontreatment-seeking adolescents and young adults in inpatient and outpatient populations. They are sensitive and specific in identifying problem drinkers and marijuana users, respectively, and are quick, useful screens to determine need for further assessment.
Toxicology is useful for initial assessment and to monitor substance use patterns during treatment.
Urine samples are used to assess marijuana, sedative/hypnotic, amphetamine, cocaine, opiate, and phencyclidine use. Alcohol may be detected in urine, but alcohol levels detected by blood and breath testing are more accurate.
Table 1
Diagnostic criteria for substance abuse and dependence
| Substance abuse | 1 of these 4 symptoms in a 12-month period: Role impairment Hazardous use Legal problems associated with use Social problems |
| Substance dependence | At least 3 of the following: Tolerance Withdrawal Using more or longer than intended Attempting to quit or cut down Much time spent using Activities given up to use Psychological/physical problems resulting from use Subtyped as with or without physiologic features (tolerance or withdrawal symptoms) |
| Source: DSM-IV-TR | |
Marijuana may be detected in the urine for 3 days to 4 weeks, depending on level of use. Cocaine can be detected for 2 to 4 days in urine and longer in hair analysis.
Random screening. Adolescents who use drugs usually know how long substances can be detected, so random urine drug screening is important to treatment progress. We inform adolescents at the beginning of treatment that random screening will be performed to corroborate self-report of substance use. To ensure a reliable urine sample, same-gender staff observe while the adolescent gives the sample.
Table 2
Common screening instruments for alcohol and drug use in adolescents
| Instrument | Items (#) | How administered | Administration time |
|---|---|---|---|
| Drug Use Screening Inventory (DUSI-R), Revised | 159 | Self-report | 20 to 40 minutes |
| Problem-Oriented Screening Instrument for Teenagers (POSIT) | 139 | Self-report | 20 to 30 minutes |
| Teen Addiction Severity Index (T-ASI) | 133 | Clinician | 20 to 45 minutes |
| Comprehensive Adolescent Severity Inventory (CASI) | 245 | Clinician | Varies with experience of administrator |
| Adolescent Obsessive-Compulsive Drinking Scale (A-OCDS) | 14 | Self-report | About 5 minutes |
| Deas-Marijuana Obsessive-Compulsive Scale (Deas-MOCS) | 14 | Self-report | About 5 minutes |
STEP 2: IDENTIFYING PSYCHIATRIC COMORBIDITY
In adolescents, substance use disorder frequently goes hand-in-hand with psychiatric disorders, particularly:
- mood and anxiety disorders
- disruptive disorders (attention-deficit/hyperactivity, oppositional defiant, and conduct disorders)12
- and posttraumatic stress disorder.13
Uncontrolled psychiatric disorders may sabotage substance abuse treatment. Therefore, assess any adolescent presenting with substance use for psychiatric illness.
Did psychiatric symptoms predate or postdate substance use? The answer may suggest self-medication or a substance-induced phenomenon. This assumption does not always apply, however, as many factors affect the relationship between substance use and psychiatric disorders.
Adolescents who meet DSM-IV criteria for conduct disorder—especially those who are highly aggressive—tend to initiate substance use much earlier than adolescents without conduct disorder, and they continue their use longer.
Most adolescents with comorbid psychiatric and substance use disorders develop the psychiatric disorder first. Some report using various substances to medicate their psychiatric symptoms. Early diagnosis and treatment of the psychiatric disorder may prevent or decrease the adolescent’s substance use.
STEPS 3 AND 4: EVALUATING SOCIAL INFLUENCES AND CONSEQUENCES
Social influences that contribute to adolescent alcohol and drug abuse include family dynamics and peer relationships. Consequences include educational and legal problems. We explore these areas with the adolescents and their parents/guardians. In most cases, adolescents are honest when reporting how their alcohol or drug use has affected their lives.
What is his family like? Assess the adolescent’s family, including its structure and history of substance abuse, psychiatric illness, or trauma (Table 3). Adolescents whose parents or siblings use alcohol or drugs are at increased risk for substance use.14 To what extent this association is genetic, environmental, or both is undetermined, but the genetic influence increases as adolescents age.15
Who are her friends? Adolescents who try alcohol or drugs and continue to use them tend to have peers who use these substances.16 Moreover, the severity of adolescents’ substance use is correlated with the number of substance-using peers. To explore peer relationships, ask about:
- peer group composition, including whether peers use alcohol or drugs
- peer interactions, including the adolescent’s ability to assert him- or herself in the peer group
- markers for risky sexual behaviors related to substance use, including infection with HIV and other sexually transmitted diseases.
How is she doing in school? Inquire about the teen’s academic performance, attendance, disciplinary problems, and motivation. Even a small decline in school performance or an increase in disciplinary problems that result in suspension or expulsion can indicate substance use or other at-risk behaviors.
Poor grades or attendance problems suggest but are not the only clues to substance use. Some adolescents with good school performance engage in substance use and may be impaired in other life domains.
Has he been arrested? Substance-abusing adolescents tend to engage in delinquent behaviors, including shoplifting, vandalism, curfew violations, disorderly conduct, and drunken driving. When assessing for delinquency, ask about behaviors that did or did not result in arrest. The teen who avoided arrest for illegal activities may perceive his/her behaviors as less severe than those involving arrest, and it may help to address this denial in individual or group therapy.
Table 3
Questions to assess family influence on an adolescent’s substance use
| Family structure |
|
| Parenting styles |
|
| Substance abuse |
|
| Psychiatric disorders |
|
| Trauma |
|
TREATMENT
We consider any adolescent with dependence symptoms—whether or not the presentation meets full DSM-IV diagnostic criteria—to be a candidate for further assessment and treatment. Early intervention may prevent progression to substance dependence.
Effective treatments:
- are intensive and of sufficient duration to change attitude and behaviors
- are comprehensive and target multiple domains of the adolescent’s life
- are sensitive to cultural and socioeconomic realities
- involve the family
- emphasize pro-social recreational activities, such as playing sports, attending movies, camping, having lunch or dinner with peers, etc.
Inpatient or outpatient? Managed care and insurance restrictions limit many patients’ eligibility for inpatient or residential treatment, so partial hospitalization and outpatient settings have become standard for substance abuse treatment. Partial hospitalization programs vary but may entail several hours, several days per week. Outpatient treatment may encompass individual, group, and family therapy, including after-school programs.
Inpatient treatment is usually reserved for adolescents:
- who need detoxification
- with comorbid psychiatric disorders
- or who may harm themselves or others.
PSYCHOTHERAPIES
Behavioral therapy, family-based therapy, multisystematic therapy (MST), and 12-step approaches have shown efficacy in treating adolescents with substance use disorders.
Behavioral therapy. Behavioral therapy is recommended as initial treatment because substance use plays a functional role in the adolescent’s life and is learned and reinforced in the adolescent’s environment. Homework assignments and role-play are commonly used in therapy.
Three central ingredients are:
- functional analysis (identifying internal and external triggers for starting and continuing substance use)
- skills training (targeting problems identified in the functional analysis)
- and relapse prevention.
Behavioral therapy is more effective than supportive therapy in improving family relationships and school and work attendance and in decreasing substance use, as indicated by fewer positive urine toxicology screens.17
Cognitive-behavioral therapy (CBT) approaches substance use as a maladaptive response to life problems. Its goal is to teach new skills to help the adolescent recognize and avoid high-risk situations and cope with associated problems and behaviors.
In a recent study, Kaminer et al randomly assigned 82 adolescents with psychiatric and substance use disorders to 8 weeks of CBT or psychoeducational therapy (didactic or videotaped presentations of ways to address problems associated with substance use). Substance use was reduced in both treatment groups, but:
- adolescents receiving CBT had significantly fewer positive urine toxicology tests
- adolescents with comorbid conduct disorder were least likely to complete treatment or return for follow-up
- those with depressive and anxiety disorders were most likely to complete treatment.18
Family-based therapy. Two detailed reviews19,20 demonstrate that the adolescent’s family, community, and school relationships affect his or her perceptions and behaviors. Maladaptive relationships in any of these systems may lead to high-risk behaviors. Therefore, family therapy is core to the adolescent’s treatment, regardless of what modality is chosen.
Goals of family therapy may be:
- to help the adolescent abstain from substance use
- to engage in pro-social activities
- to decrease parental denial of the adolescent’s substance use
- to decrease resistance to treatment
- treatment maintenance
- to establish or re-establish structure in the adolescent’s environment
- to improve communication in the family.
Multisystemic therapy is comprehensive and involves all systems that relate to the adolescent’s substance use, including the family, school, community, and legal system. MST requires special training and intensive supervision, so it is usually reserved for adolescents who have not benefited from other forms of treatment.21
12-step approaches. For adolescents, 12-step programs usually augment other treatments and are rarely used alone. Alcoholics Anonymous, Narcotics Anonymous, and other 12-step programs have been studied more extensively in adults than in adolescents.
Adolescents, who often feel invulnerable, may have difficulty accepting the 12-step doctrine of lack of control. A modified 12-step program and workbook for adolescents are available through the American Academy of Child and Adolescent Psychiatry.22
Referral tips. If possible, refer an adolescent to a 12-step group specifically for adolescents. Teens who attend adult groups often perceive their substance use as normal, compared with the more severe and chronic patterns of some adults. Most adolescents relate better to peers with similar problems and may benefit from reminders of the negative consequences of substance use and the benefits of abstinence.
DRUG THERAPY
Drug therapy for adolescents with substance use disorders is usually considered in the context of detoxification, treating withdrawal symptoms, and treating comorbid psychiatric disorders. The same detoxification and withdrawal treatment principles used in adults apply to adolescents.
Clinical withdrawal symptoms are less common in adolescents than adults, probably because of adolescents’ binge patterns of substance use. Even so, some adolescents do experience withdrawal and may be at risk for complications if improperly treated.
Psychiatric comorbidity. To our knowledge, only two double-blind, placebo-controlled studies of drug therapy in treating adolescent substance use disorders have been published.
Depression. Deas et al23 randomized 10 adolescents with alcohol use disorders and depression to 12 weeks of group CBT plus sertraline or placebo. Sertraline was started at 25 mg/d and titrated to a maximum of 100 mg/d. Drinks per drinking day, percent of days drinking, and Hamilton Rating Scale for Depression scores declined similarly in both groups.
Drinking decreased significantly from baseline (by an average 4.7 drinks), and adolescents in both groups no longer met DSM-IV criteria for depression at the end of treatment. CBT’s effectiveness in treating alcohol use disorders and depression might have concealed any difference in effect between sertaline and placebo.
Bipolar disorder. Geller et al24 randomly assigned 25 adolescents with bipolar disorder and substance dependence to lithium or placebo for 6 weeks. Lithium was started as an evening dose of 600 mg and titrated to achieve a lithium blood level of 0.9 to 1.3 mEq/L. Among the 21 adolescents who completed the trial, those receiving lithium had significantly fewer positive urine toxicology screens and higher clinical global assessment of function scores.
Related resources
- National Clearinghouse for Alcohol and Drug Information, Substance Abuse and Mental Health Services Administration, U.S. Department of Health and Human Services. www.health.org/govpubs/bkd306/31k.aspx
- National Institute on Alcohol Abuse and Alcoholism (assessment instruments). www.niaaa.nih.gov/publications/instable.htm
- Deas D, Thomas SE. An overview of controlled studies of adolescent substance abuse treatment. Am J Addict 2001;10:178-89.
Drug brand names
- Sertraline • Zoloft
Disclosure
Dr. Deas receives grant support from Pfizer, Inc. and the National Institute of Alcohol Abuse and Alcoholism.
Dr. Upadhyaya receives grant support from the National Institute of Drug Abuse and GlaxoSmithKline.
Acknowledgment
The authors wish to thank Alva Blair for assistance with preparing this manuscript and Natalie Johnson, MA, Kess Mughelli, BS, and Lakeleia Middleton-Robinson, MA, for technical support.
1. Johnston LD, O’Malley PM, Bachman JG. Monitoring the Future: national results on adolescent drug use. Overview of key findings, 2002. Bethesda, MD: National Institute on Drug Abuse, 2003 (in press).
2. Martin CS, Kaczynski NA, Maisto SA, et al. Patterns of DSM-IV alcohol abuse and dependence symptoms in adolescent drinkers. J Stud Alcohol 1995;56:672-80.
3. Pollock NK, Martin CS. Diagnostic orphans: adolescents with alcohol symptoms who do not qualify for DSM-IV abuse or dependence diagnoses. Am J Psychiatry 1999;156:897-901.
4. Harrison PA, Fulkerson JA, Beebe TJ. DSM-IV substance use disorder criteria for adolescents: a critical examination based on a statewide school survey. Am J Psychiatry 1998;155:486-92.
5. Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse/dependence: results from the National Longitudinal Alcohol Epidemiological Survey. J Subst Abuse 1997;9:103-10.
6. Tarter RE, Laird SB, Bukstein O, Kaminer Y. Validation of the adolescent drug use screening inventory: preliminary findings. Psychol Addict Behav 1992;6:322-6.
7. Rahdert E (ed). The adolescent assessment/referral system manual. DHHS pub. no. (ADM) 91-1735. Rockville, MD: U.S. Department of Health and Human Services, 1991.
8. Kaminer Y, Bukstein OG, Tarter RE. The teen addiction severity index (T-ASI): rationale and reliability. Int J Addict 1991;26:219-26.
9. Meyers K, McLellan AT, Jaeger JL, Pettinati A. The development of the Comprehensive Addiction Severity Index for Adolescents (CASI-A): an interview for assessing multiple problems of adolescents. J Subst Abuse Treat 1995;12:181-93.
10. Deas DV, Roberts JS, Randall CL, Anton RF. Adolescent Obsessive-Compulsive Drinking Scale (A-OCDS): an assessment tool for problem drinking. J Natl Med Assoc 2001;93:92-103.
11. Deas D, Randall CL, Thomas S. The utility of the Deas-Marijuana Obsessive-Compulsive Scale (Deas-MOCS) in an inpatient adolescent substance abusing sample. Drug Alcohol Depend 2002;66(June):S43.-
12. Deas-Nesmith D, Campbell S, Brady KT. Substance use disorders in an adolescent inpatient psychiatric population. JAMA 1998;90:233-8.
13. Clark DB, Lesnick L, Hegedus AM. Traumas and other adverse life events in adolescents with alcohol abuse and dependence. Am Acad Child Adolesc Psychiatry 1997;36:1744-51.
14. Kilpatrick DG, Acierno R, Saunders B, et al. Risk factors for adolescent substance abuse and dependence: data from a national sample. J Consult Clin Psychol 2000;68:19-30.
15. Rose RJ. A developmental behavioral-genetic perspective on alcoholism risk. Alcohol Health Res World 1998;22:131-43.
16. Swadi H. Individual risk factors for adolescent substance use. Drug Alcohol Depend 1999;55:209-24.
17. Azrin N, McMahon P, Donohue B, et al. Behavior therapy for drug abuse: a controlled treatment outcome study. Behav Res Ther 1994;32:857-66.
18. Kaminer Y, Burleson JA, Goldberger R. Cognitive-behavioral coping skills and psychoeducation therapies for adolescent substance abuse. J Nerv Ment Dis 2002;190:737-45.
19. Liddle H, Dakof G. Efficacy of family therapy for drug abuse: promising but not definitive. J Marital Fam Ther 1995;21:511-43.
20. Waldron HB. Adolescent substance abuse and family therapy outcome: a review of randomized trials. Adv Clin Child Psychol 1997;19:199-234.
21. Henggeler SW, Melton LA. Effects of multisystemic therapy on drug use and abuse in serious juvenile offenders: a progress report from two outcome studies. Family Dynamics of Addiction Quarterly 1991;1:40-51.
22. Jaffe S. Step workbook for adolescent chemical dependency recovery. Washington, DC: American Academy of Child and Adolescent Psychiatry, 1990.
23. Deas DV, Randall C, Roberts J, Anton R. A double-blind, placebo-controlled trial of sertraline in depressed adolescent alcoholics: a pilot study. Human Psychopharmacology Clinical and Experimental 2000;15:461-9.
24. Geller B, Cooper T, Sun K, et al. Double-blind and placebo-controlled study of lithium for adolescent bipolar disorders with secondary substance dependency. J Am Acad Child Adolesc Psychiatry 1998;37:171-8.
Assessing an adolescent for a possible substance use disorder can be streamlined by choosing age-appropriate screening tools and asking targeted questions. Based on our experience, we offer a 4-step approach to these at-risk patients1 (Box) that focuses on:
- quantifying alcohol or drug abuse and/or dependence
- identifying and treating psychiatric comorbidity
- evaluating and addressing social influences that contribute to substance use
- assessing negative consequences associated with substance abuse.
Adolescent substance use increases the risk of motor vehicle accidents, suicide, transmission of HIV and other sexual diseases, criminal behaviors, and psychological problems. Alcohol and marijuana are the substances most commonly abused by adolescents.
In 2002, the University of Michigan Institute for Social Research’s annual “Monitoring the Future” study reported:
- drunkenness in 7% of 8th graders, 18% of 10th graders, and 30% of 12th graders at least once in the previous 30 days.
- illicit drug use by 18% of 8th graders, 35% of 10th graders, and 41% of 12th graders in the previous 12 months.
Boys used substances more frequently than girls, and boys’ use was more severe. Within the previous month, boys reported greater alcohol use, binge drinking (five or more drinks in one sitting), and heavy drinking, as well as greater illicit drug use in the past year.1
INITIAL EVALUATION
Adolescents generally do not seek substance abuse treatment but are referred because of alcohol- or drug-related legal, school, or family problems. Thus, most present for evaluation with their parents, legal guardians, or officers of the court.
We begin by finding out from parents or guardians the reasons for the evaluation, their perspectives on the adolescent’s behavior, and their expectations of treatment. Then we interview the adolescent alone, assessing for substance use and evaluating peer relationships.
Components. A typical initial evaluation takes 90 minutes to 2 hours and includes:
- psychiatric history and symptoms
- medical history
- previous hospitalizations (medical and psychiatric)
- family history
- social history.
Specifically, the assessment focuses on the reason for the evaluation, with attention to diagnostic criteria for substance use/dependence.
STEP 1: QUANTIFYING DEPENDENCE
As with adults, clinical diagnosis of substance abuse or dependence in adolescents is based on DSM-IV diagnostic criteria (Table 1). Adolescents, however, differ from adults in diagnostic presentation, risk of dependence, and patterns of substance use.
Diagnostic ‘orphans.’ DSM-IV criteria for alcohol use disorders have limitations in adolescents.2 Teens who report one or two dependence symptoms and no abuse symptoms have been described as “diagnostic orphans”3—they fall short of criteria for dependence or abuse but clearly demonstrate substance use patterns. This presentation is common; in a survey of 74,008 high school students, almost 10% of 12th graders reported one or two dependence symptoms and no abuse symptoms.4
Risk of dependence. Adolescents who begin using alcohol or drugs develop dependence more rapidly than adults do.5
Patterns of use. Adolescents are more likely than adults to binge with alcohol and drugs, which may conceal the severity of their abuse. DSM-IV diagnostic criteria for substance abuse or dependence do not consider quantity of use, such as number of drinks or percent of days drinking or using drugs.
Assessment instruments. Many assessment instruments are available to explore adolescent substance use and its associated consequences. Some are described in detail and are available on the Internet. Common screening instruments that can be used for adolescent substance use are compared in Table 2.
DUSI-A and POSIT. Two self-report instruments—Drug Use Screening Inventory-Adolescents (DUSI-A)6 and Problem Oriented Screening Instrument for Teenagers (POSIT)7—can help explore alcohol or drug use in teens who admit to substance use. Anyone who endorses at least one DSM-IV abuse or dependence criterion requires further evaluation. Either test is a good starting point, and both have a built-in “lie” scale.
T-ASI and CASI. The Teen Addiction Severity Index (T-ASI)8 and Comprehensive Adolescent Severity Inventory (CASI)9 are more labor-intensive and require training to administer. These assessments are more appropriate for adolescents with extensive alcohol or drug abuse.
A-OCDS and Deas-MOCS. Our group recently developed the Adolescent Obsessive Compulsive Drinking Scale (A-OCDS)10 and the Deas-Marijuana Obsessive Compulsive Scale (Deas-MOCS).11
These self-report instruments have been validated in treatment- and nontreatment-seeking adolescents and young adults in inpatient and outpatient populations. They are sensitive and specific in identifying problem drinkers and marijuana users, respectively, and are quick, useful screens to determine need for further assessment.
Toxicology is useful for initial assessment and to monitor substance use patterns during treatment.
Urine samples are used to assess marijuana, sedative/hypnotic, amphetamine, cocaine, opiate, and phencyclidine use. Alcohol may be detected in urine, but alcohol levels detected by blood and breath testing are more accurate.
Table 1
Diagnostic criteria for substance abuse and dependence
| Substance abuse | 1 of these 4 symptoms in a 12-month period: Role impairment Hazardous use Legal problems associated with use Social problems |
| Substance dependence | At least 3 of the following: Tolerance Withdrawal Using more or longer than intended Attempting to quit or cut down Much time spent using Activities given up to use Psychological/physical problems resulting from use Subtyped as with or without physiologic features (tolerance or withdrawal symptoms) |
| Source: DSM-IV-TR | |
Marijuana may be detected in the urine for 3 days to 4 weeks, depending on level of use. Cocaine can be detected for 2 to 4 days in urine and longer in hair analysis.
Random screening. Adolescents who use drugs usually know how long substances can be detected, so random urine drug screening is important to treatment progress. We inform adolescents at the beginning of treatment that random screening will be performed to corroborate self-report of substance use. To ensure a reliable urine sample, same-gender staff observe while the adolescent gives the sample.
Table 2
Common screening instruments for alcohol and drug use in adolescents
| Instrument | Items (#) | How administered | Administration time |
|---|---|---|---|
| Drug Use Screening Inventory (DUSI-R), Revised | 159 | Self-report | 20 to 40 minutes |
| Problem-Oriented Screening Instrument for Teenagers (POSIT) | 139 | Self-report | 20 to 30 minutes |
| Teen Addiction Severity Index (T-ASI) | 133 | Clinician | 20 to 45 minutes |
| Comprehensive Adolescent Severity Inventory (CASI) | 245 | Clinician | Varies with experience of administrator |
| Adolescent Obsessive-Compulsive Drinking Scale (A-OCDS) | 14 | Self-report | About 5 minutes |
| Deas-Marijuana Obsessive-Compulsive Scale (Deas-MOCS) | 14 | Self-report | About 5 minutes |
STEP 2: IDENTIFYING PSYCHIATRIC COMORBIDITY
In adolescents, substance use disorder frequently goes hand-in-hand with psychiatric disorders, particularly:
- mood and anxiety disorders
- disruptive disorders (attention-deficit/hyperactivity, oppositional defiant, and conduct disorders)12
- and posttraumatic stress disorder.13
Uncontrolled psychiatric disorders may sabotage substance abuse treatment. Therefore, assess any adolescent presenting with substance use for psychiatric illness.
Did psychiatric symptoms predate or postdate substance use? The answer may suggest self-medication or a substance-induced phenomenon. This assumption does not always apply, however, as many factors affect the relationship between substance use and psychiatric disorders.
Adolescents who meet DSM-IV criteria for conduct disorder—especially those who are highly aggressive—tend to initiate substance use much earlier than adolescents without conduct disorder, and they continue their use longer.
Most adolescents with comorbid psychiatric and substance use disorders develop the psychiatric disorder first. Some report using various substances to medicate their psychiatric symptoms. Early diagnosis and treatment of the psychiatric disorder may prevent or decrease the adolescent’s substance use.
STEPS 3 AND 4: EVALUATING SOCIAL INFLUENCES AND CONSEQUENCES
Social influences that contribute to adolescent alcohol and drug abuse include family dynamics and peer relationships. Consequences include educational and legal problems. We explore these areas with the adolescents and their parents/guardians. In most cases, adolescents are honest when reporting how their alcohol or drug use has affected their lives.
What is his family like? Assess the adolescent’s family, including its structure and history of substance abuse, psychiatric illness, or trauma (Table 3). Adolescents whose parents or siblings use alcohol or drugs are at increased risk for substance use.14 To what extent this association is genetic, environmental, or both is undetermined, but the genetic influence increases as adolescents age.15
Who are her friends? Adolescents who try alcohol or drugs and continue to use them tend to have peers who use these substances.16 Moreover, the severity of adolescents’ substance use is correlated with the number of substance-using peers. To explore peer relationships, ask about:
- peer group composition, including whether peers use alcohol or drugs
- peer interactions, including the adolescent’s ability to assert him- or herself in the peer group
- markers for risky sexual behaviors related to substance use, including infection with HIV and other sexually transmitted diseases.
How is she doing in school? Inquire about the teen’s academic performance, attendance, disciplinary problems, and motivation. Even a small decline in school performance or an increase in disciplinary problems that result in suspension or expulsion can indicate substance use or other at-risk behaviors.
Poor grades or attendance problems suggest but are not the only clues to substance use. Some adolescents with good school performance engage in substance use and may be impaired in other life domains.
Has he been arrested? Substance-abusing adolescents tend to engage in delinquent behaviors, including shoplifting, vandalism, curfew violations, disorderly conduct, and drunken driving. When assessing for delinquency, ask about behaviors that did or did not result in arrest. The teen who avoided arrest for illegal activities may perceive his/her behaviors as less severe than those involving arrest, and it may help to address this denial in individual or group therapy.
Table 3
Questions to assess family influence on an adolescent’s substance use
| Family structure |
|
| Parenting styles |
|
| Substance abuse |
|
| Psychiatric disorders |
|
| Trauma |
|
TREATMENT
We consider any adolescent with dependence symptoms—whether or not the presentation meets full DSM-IV diagnostic criteria—to be a candidate for further assessment and treatment. Early intervention may prevent progression to substance dependence.
Effective treatments:
- are intensive and of sufficient duration to change attitude and behaviors
- are comprehensive and target multiple domains of the adolescent’s life
- are sensitive to cultural and socioeconomic realities
- involve the family
- emphasize pro-social recreational activities, such as playing sports, attending movies, camping, having lunch or dinner with peers, etc.
Inpatient or outpatient? Managed care and insurance restrictions limit many patients’ eligibility for inpatient or residential treatment, so partial hospitalization and outpatient settings have become standard for substance abuse treatment. Partial hospitalization programs vary but may entail several hours, several days per week. Outpatient treatment may encompass individual, group, and family therapy, including after-school programs.
Inpatient treatment is usually reserved for adolescents:
- who need detoxification
- with comorbid psychiatric disorders
- or who may harm themselves or others.
PSYCHOTHERAPIES
Behavioral therapy, family-based therapy, multisystematic therapy (MST), and 12-step approaches have shown efficacy in treating adolescents with substance use disorders.
Behavioral therapy. Behavioral therapy is recommended as initial treatment because substance use plays a functional role in the adolescent’s life and is learned and reinforced in the adolescent’s environment. Homework assignments and role-play are commonly used in therapy.
Three central ingredients are:
- functional analysis (identifying internal and external triggers for starting and continuing substance use)
- skills training (targeting problems identified in the functional analysis)
- and relapse prevention.
Behavioral therapy is more effective than supportive therapy in improving family relationships and school and work attendance and in decreasing substance use, as indicated by fewer positive urine toxicology screens.17
Cognitive-behavioral therapy (CBT) approaches substance use as a maladaptive response to life problems. Its goal is to teach new skills to help the adolescent recognize and avoid high-risk situations and cope with associated problems and behaviors.
In a recent study, Kaminer et al randomly assigned 82 adolescents with psychiatric and substance use disorders to 8 weeks of CBT or psychoeducational therapy (didactic or videotaped presentations of ways to address problems associated with substance use). Substance use was reduced in both treatment groups, but:
- adolescents receiving CBT had significantly fewer positive urine toxicology tests
- adolescents with comorbid conduct disorder were least likely to complete treatment or return for follow-up
- those with depressive and anxiety disorders were most likely to complete treatment.18
Family-based therapy. Two detailed reviews19,20 demonstrate that the adolescent’s family, community, and school relationships affect his or her perceptions and behaviors. Maladaptive relationships in any of these systems may lead to high-risk behaviors. Therefore, family therapy is core to the adolescent’s treatment, regardless of what modality is chosen.
Goals of family therapy may be:
- to help the adolescent abstain from substance use
- to engage in pro-social activities
- to decrease parental denial of the adolescent’s substance use
- to decrease resistance to treatment
- treatment maintenance
- to establish or re-establish structure in the adolescent’s environment
- to improve communication in the family.
Multisystemic therapy is comprehensive and involves all systems that relate to the adolescent’s substance use, including the family, school, community, and legal system. MST requires special training and intensive supervision, so it is usually reserved for adolescents who have not benefited from other forms of treatment.21
12-step approaches. For adolescents, 12-step programs usually augment other treatments and are rarely used alone. Alcoholics Anonymous, Narcotics Anonymous, and other 12-step programs have been studied more extensively in adults than in adolescents.
Adolescents, who often feel invulnerable, may have difficulty accepting the 12-step doctrine of lack of control. A modified 12-step program and workbook for adolescents are available through the American Academy of Child and Adolescent Psychiatry.22
Referral tips. If possible, refer an adolescent to a 12-step group specifically for adolescents. Teens who attend adult groups often perceive their substance use as normal, compared with the more severe and chronic patterns of some adults. Most adolescents relate better to peers with similar problems and may benefit from reminders of the negative consequences of substance use and the benefits of abstinence.
DRUG THERAPY
Drug therapy for adolescents with substance use disorders is usually considered in the context of detoxification, treating withdrawal symptoms, and treating comorbid psychiatric disorders. The same detoxification and withdrawal treatment principles used in adults apply to adolescents.
Clinical withdrawal symptoms are less common in adolescents than adults, probably because of adolescents’ binge patterns of substance use. Even so, some adolescents do experience withdrawal and may be at risk for complications if improperly treated.
Psychiatric comorbidity. To our knowledge, only two double-blind, placebo-controlled studies of drug therapy in treating adolescent substance use disorders have been published.
Depression. Deas et al23 randomized 10 adolescents with alcohol use disorders and depression to 12 weeks of group CBT plus sertraline or placebo. Sertraline was started at 25 mg/d and titrated to a maximum of 100 mg/d. Drinks per drinking day, percent of days drinking, and Hamilton Rating Scale for Depression scores declined similarly in both groups.
Drinking decreased significantly from baseline (by an average 4.7 drinks), and adolescents in both groups no longer met DSM-IV criteria for depression at the end of treatment. CBT’s effectiveness in treating alcohol use disorders and depression might have concealed any difference in effect between sertaline and placebo.
Bipolar disorder. Geller et al24 randomly assigned 25 adolescents with bipolar disorder and substance dependence to lithium or placebo for 6 weeks. Lithium was started as an evening dose of 600 mg and titrated to achieve a lithium blood level of 0.9 to 1.3 mEq/L. Among the 21 adolescents who completed the trial, those receiving lithium had significantly fewer positive urine toxicology screens and higher clinical global assessment of function scores.
Related resources
- National Clearinghouse for Alcohol and Drug Information, Substance Abuse and Mental Health Services Administration, U.S. Department of Health and Human Services. www.health.org/govpubs/bkd306/31k.aspx
- National Institute on Alcohol Abuse and Alcoholism (assessment instruments). www.niaaa.nih.gov/publications/instable.htm
- Deas D, Thomas SE. An overview of controlled studies of adolescent substance abuse treatment. Am J Addict 2001;10:178-89.
Drug brand names
- Sertraline • Zoloft
Disclosure
Dr. Deas receives grant support from Pfizer, Inc. and the National Institute of Alcohol Abuse and Alcoholism.
Dr. Upadhyaya receives grant support from the National Institute of Drug Abuse and GlaxoSmithKline.
Acknowledgment
The authors wish to thank Alva Blair for assistance with preparing this manuscript and Natalie Johnson, MA, Kess Mughelli, BS, and Lakeleia Middleton-Robinson, MA, for technical support.
Assessing an adolescent for a possible substance use disorder can be streamlined by choosing age-appropriate screening tools and asking targeted questions. Based on our experience, we offer a 4-step approach to these at-risk patients1 (Box) that focuses on:
- quantifying alcohol or drug abuse and/or dependence
- identifying and treating psychiatric comorbidity
- evaluating and addressing social influences that contribute to substance use
- assessing negative consequences associated with substance abuse.
Adolescent substance use increases the risk of motor vehicle accidents, suicide, transmission of HIV and other sexual diseases, criminal behaviors, and psychological problems. Alcohol and marijuana are the substances most commonly abused by adolescents.
In 2002, the University of Michigan Institute for Social Research’s annual “Monitoring the Future” study reported:
- drunkenness in 7% of 8th graders, 18% of 10th graders, and 30% of 12th graders at least once in the previous 30 days.
- illicit drug use by 18% of 8th graders, 35% of 10th graders, and 41% of 12th graders in the previous 12 months.
Boys used substances more frequently than girls, and boys’ use was more severe. Within the previous month, boys reported greater alcohol use, binge drinking (five or more drinks in one sitting), and heavy drinking, as well as greater illicit drug use in the past year.1
INITIAL EVALUATION
Adolescents generally do not seek substance abuse treatment but are referred because of alcohol- or drug-related legal, school, or family problems. Thus, most present for evaluation with their parents, legal guardians, or officers of the court.
We begin by finding out from parents or guardians the reasons for the evaluation, their perspectives on the adolescent’s behavior, and their expectations of treatment. Then we interview the adolescent alone, assessing for substance use and evaluating peer relationships.
Components. A typical initial evaluation takes 90 minutes to 2 hours and includes:
- psychiatric history and symptoms
- medical history
- previous hospitalizations (medical and psychiatric)
- family history
- social history.
Specifically, the assessment focuses on the reason for the evaluation, with attention to diagnostic criteria for substance use/dependence.
STEP 1: QUANTIFYING DEPENDENCE
As with adults, clinical diagnosis of substance abuse or dependence in adolescents is based on DSM-IV diagnostic criteria (Table 1). Adolescents, however, differ from adults in diagnostic presentation, risk of dependence, and patterns of substance use.
Diagnostic ‘orphans.’ DSM-IV criteria for alcohol use disorders have limitations in adolescents.2 Teens who report one or two dependence symptoms and no abuse symptoms have been described as “diagnostic orphans”3—they fall short of criteria for dependence or abuse but clearly demonstrate substance use patterns. This presentation is common; in a survey of 74,008 high school students, almost 10% of 12th graders reported one or two dependence symptoms and no abuse symptoms.4
Risk of dependence. Adolescents who begin using alcohol or drugs develop dependence more rapidly than adults do.5
Patterns of use. Adolescents are more likely than adults to binge with alcohol and drugs, which may conceal the severity of their abuse. DSM-IV diagnostic criteria for substance abuse or dependence do not consider quantity of use, such as number of drinks or percent of days drinking or using drugs.
Assessment instruments. Many assessment instruments are available to explore adolescent substance use and its associated consequences. Some are described in detail and are available on the Internet. Common screening instruments that can be used for adolescent substance use are compared in Table 2.
DUSI-A and POSIT. Two self-report instruments—Drug Use Screening Inventory-Adolescents (DUSI-A)6 and Problem Oriented Screening Instrument for Teenagers (POSIT)7—can help explore alcohol or drug use in teens who admit to substance use. Anyone who endorses at least one DSM-IV abuse or dependence criterion requires further evaluation. Either test is a good starting point, and both have a built-in “lie” scale.
T-ASI and CASI. The Teen Addiction Severity Index (T-ASI)8 and Comprehensive Adolescent Severity Inventory (CASI)9 are more labor-intensive and require training to administer. These assessments are more appropriate for adolescents with extensive alcohol or drug abuse.
A-OCDS and Deas-MOCS. Our group recently developed the Adolescent Obsessive Compulsive Drinking Scale (A-OCDS)10 and the Deas-Marijuana Obsessive Compulsive Scale (Deas-MOCS).11
These self-report instruments have been validated in treatment- and nontreatment-seeking adolescents and young adults in inpatient and outpatient populations. They are sensitive and specific in identifying problem drinkers and marijuana users, respectively, and are quick, useful screens to determine need for further assessment.
Toxicology is useful for initial assessment and to monitor substance use patterns during treatment.
Urine samples are used to assess marijuana, sedative/hypnotic, amphetamine, cocaine, opiate, and phencyclidine use. Alcohol may be detected in urine, but alcohol levels detected by blood and breath testing are more accurate.
Table 1
Diagnostic criteria for substance abuse and dependence
| Substance abuse | 1 of these 4 symptoms in a 12-month period: Role impairment Hazardous use Legal problems associated with use Social problems |
| Substance dependence | At least 3 of the following: Tolerance Withdrawal Using more or longer than intended Attempting to quit or cut down Much time spent using Activities given up to use Psychological/physical problems resulting from use Subtyped as with or without physiologic features (tolerance or withdrawal symptoms) |
| Source: DSM-IV-TR | |
Marijuana may be detected in the urine for 3 days to 4 weeks, depending on level of use. Cocaine can be detected for 2 to 4 days in urine and longer in hair analysis.
Random screening. Adolescents who use drugs usually know how long substances can be detected, so random urine drug screening is important to treatment progress. We inform adolescents at the beginning of treatment that random screening will be performed to corroborate self-report of substance use. To ensure a reliable urine sample, same-gender staff observe while the adolescent gives the sample.
Table 2
Common screening instruments for alcohol and drug use in adolescents
| Instrument | Items (#) | How administered | Administration time |
|---|---|---|---|
| Drug Use Screening Inventory (DUSI-R), Revised | 159 | Self-report | 20 to 40 minutes |
| Problem-Oriented Screening Instrument for Teenagers (POSIT) | 139 | Self-report | 20 to 30 minutes |
| Teen Addiction Severity Index (T-ASI) | 133 | Clinician | 20 to 45 minutes |
| Comprehensive Adolescent Severity Inventory (CASI) | 245 | Clinician | Varies with experience of administrator |
| Adolescent Obsessive-Compulsive Drinking Scale (A-OCDS) | 14 | Self-report | About 5 minutes |
| Deas-Marijuana Obsessive-Compulsive Scale (Deas-MOCS) | 14 | Self-report | About 5 minutes |
STEP 2: IDENTIFYING PSYCHIATRIC COMORBIDITY
In adolescents, substance use disorder frequently goes hand-in-hand with psychiatric disorders, particularly:
- mood and anxiety disorders
- disruptive disorders (attention-deficit/hyperactivity, oppositional defiant, and conduct disorders)12
- and posttraumatic stress disorder.13
Uncontrolled psychiatric disorders may sabotage substance abuse treatment. Therefore, assess any adolescent presenting with substance use for psychiatric illness.
Did psychiatric symptoms predate or postdate substance use? The answer may suggest self-medication or a substance-induced phenomenon. This assumption does not always apply, however, as many factors affect the relationship between substance use and psychiatric disorders.
Adolescents who meet DSM-IV criteria for conduct disorder—especially those who are highly aggressive—tend to initiate substance use much earlier than adolescents without conduct disorder, and they continue their use longer.
Most adolescents with comorbid psychiatric and substance use disorders develop the psychiatric disorder first. Some report using various substances to medicate their psychiatric symptoms. Early diagnosis and treatment of the psychiatric disorder may prevent or decrease the adolescent’s substance use.
STEPS 3 AND 4: EVALUATING SOCIAL INFLUENCES AND CONSEQUENCES
Social influences that contribute to adolescent alcohol and drug abuse include family dynamics and peer relationships. Consequences include educational and legal problems. We explore these areas with the adolescents and their parents/guardians. In most cases, adolescents are honest when reporting how their alcohol or drug use has affected their lives.
What is his family like? Assess the adolescent’s family, including its structure and history of substance abuse, psychiatric illness, or trauma (Table 3). Adolescents whose parents or siblings use alcohol or drugs are at increased risk for substance use.14 To what extent this association is genetic, environmental, or both is undetermined, but the genetic influence increases as adolescents age.15
Who are her friends? Adolescents who try alcohol or drugs and continue to use them tend to have peers who use these substances.16 Moreover, the severity of adolescents’ substance use is correlated with the number of substance-using peers. To explore peer relationships, ask about:
- peer group composition, including whether peers use alcohol or drugs
- peer interactions, including the adolescent’s ability to assert him- or herself in the peer group
- markers for risky sexual behaviors related to substance use, including infection with HIV and other sexually transmitted diseases.
How is she doing in school? Inquire about the teen’s academic performance, attendance, disciplinary problems, and motivation. Even a small decline in school performance or an increase in disciplinary problems that result in suspension or expulsion can indicate substance use or other at-risk behaviors.
Poor grades or attendance problems suggest but are not the only clues to substance use. Some adolescents with good school performance engage in substance use and may be impaired in other life domains.
Has he been arrested? Substance-abusing adolescents tend to engage in delinquent behaviors, including shoplifting, vandalism, curfew violations, disorderly conduct, and drunken driving. When assessing for delinquency, ask about behaviors that did or did not result in arrest. The teen who avoided arrest for illegal activities may perceive his/her behaviors as less severe than those involving arrest, and it may help to address this denial in individual or group therapy.
Table 3
Questions to assess family influence on an adolescent’s substance use
| Family structure |
|
| Parenting styles |
|
| Substance abuse |
|
| Psychiatric disorders |
|
| Trauma |
|
TREATMENT
We consider any adolescent with dependence symptoms—whether or not the presentation meets full DSM-IV diagnostic criteria—to be a candidate for further assessment and treatment. Early intervention may prevent progression to substance dependence.
Effective treatments:
- are intensive and of sufficient duration to change attitude and behaviors
- are comprehensive and target multiple domains of the adolescent’s life
- are sensitive to cultural and socioeconomic realities
- involve the family
- emphasize pro-social recreational activities, such as playing sports, attending movies, camping, having lunch or dinner with peers, etc.
Inpatient or outpatient? Managed care and insurance restrictions limit many patients’ eligibility for inpatient or residential treatment, so partial hospitalization and outpatient settings have become standard for substance abuse treatment. Partial hospitalization programs vary but may entail several hours, several days per week. Outpatient treatment may encompass individual, group, and family therapy, including after-school programs.
Inpatient treatment is usually reserved for adolescents:
- who need detoxification
- with comorbid psychiatric disorders
- or who may harm themselves or others.
PSYCHOTHERAPIES
Behavioral therapy, family-based therapy, multisystematic therapy (MST), and 12-step approaches have shown efficacy in treating adolescents with substance use disorders.
Behavioral therapy. Behavioral therapy is recommended as initial treatment because substance use plays a functional role in the adolescent’s life and is learned and reinforced in the adolescent’s environment. Homework assignments and role-play are commonly used in therapy.
Three central ingredients are:
- functional analysis (identifying internal and external triggers for starting and continuing substance use)
- skills training (targeting problems identified in the functional analysis)
- and relapse prevention.
Behavioral therapy is more effective than supportive therapy in improving family relationships and school and work attendance and in decreasing substance use, as indicated by fewer positive urine toxicology screens.17
Cognitive-behavioral therapy (CBT) approaches substance use as a maladaptive response to life problems. Its goal is to teach new skills to help the adolescent recognize and avoid high-risk situations and cope with associated problems and behaviors.
In a recent study, Kaminer et al randomly assigned 82 adolescents with psychiatric and substance use disorders to 8 weeks of CBT or psychoeducational therapy (didactic or videotaped presentations of ways to address problems associated with substance use). Substance use was reduced in both treatment groups, but:
- adolescents receiving CBT had significantly fewer positive urine toxicology tests
- adolescents with comorbid conduct disorder were least likely to complete treatment or return for follow-up
- those with depressive and anxiety disorders were most likely to complete treatment.18
Family-based therapy. Two detailed reviews19,20 demonstrate that the adolescent’s family, community, and school relationships affect his or her perceptions and behaviors. Maladaptive relationships in any of these systems may lead to high-risk behaviors. Therefore, family therapy is core to the adolescent’s treatment, regardless of what modality is chosen.
Goals of family therapy may be:
- to help the adolescent abstain from substance use
- to engage in pro-social activities
- to decrease parental denial of the adolescent’s substance use
- to decrease resistance to treatment
- treatment maintenance
- to establish or re-establish structure in the adolescent’s environment
- to improve communication in the family.
Multisystemic therapy is comprehensive and involves all systems that relate to the adolescent’s substance use, including the family, school, community, and legal system. MST requires special training and intensive supervision, so it is usually reserved for adolescents who have not benefited from other forms of treatment.21
12-step approaches. For adolescents, 12-step programs usually augment other treatments and are rarely used alone. Alcoholics Anonymous, Narcotics Anonymous, and other 12-step programs have been studied more extensively in adults than in adolescents.
Adolescents, who often feel invulnerable, may have difficulty accepting the 12-step doctrine of lack of control. A modified 12-step program and workbook for adolescents are available through the American Academy of Child and Adolescent Psychiatry.22
Referral tips. If possible, refer an adolescent to a 12-step group specifically for adolescents. Teens who attend adult groups often perceive their substance use as normal, compared with the more severe and chronic patterns of some adults. Most adolescents relate better to peers with similar problems and may benefit from reminders of the negative consequences of substance use and the benefits of abstinence.
DRUG THERAPY
Drug therapy for adolescents with substance use disorders is usually considered in the context of detoxification, treating withdrawal symptoms, and treating comorbid psychiatric disorders. The same detoxification and withdrawal treatment principles used in adults apply to adolescents.
Clinical withdrawal symptoms are less common in adolescents than adults, probably because of adolescents’ binge patterns of substance use. Even so, some adolescents do experience withdrawal and may be at risk for complications if improperly treated.
Psychiatric comorbidity. To our knowledge, only two double-blind, placebo-controlled studies of drug therapy in treating adolescent substance use disorders have been published.
Depression. Deas et al23 randomized 10 adolescents with alcohol use disorders and depression to 12 weeks of group CBT plus sertraline or placebo. Sertraline was started at 25 mg/d and titrated to a maximum of 100 mg/d. Drinks per drinking day, percent of days drinking, and Hamilton Rating Scale for Depression scores declined similarly in both groups.
Drinking decreased significantly from baseline (by an average 4.7 drinks), and adolescents in both groups no longer met DSM-IV criteria for depression at the end of treatment. CBT’s effectiveness in treating alcohol use disorders and depression might have concealed any difference in effect between sertaline and placebo.
Bipolar disorder. Geller et al24 randomly assigned 25 adolescents with bipolar disorder and substance dependence to lithium or placebo for 6 weeks. Lithium was started as an evening dose of 600 mg and titrated to achieve a lithium blood level of 0.9 to 1.3 mEq/L. Among the 21 adolescents who completed the trial, those receiving lithium had significantly fewer positive urine toxicology screens and higher clinical global assessment of function scores.
Related resources
- National Clearinghouse for Alcohol and Drug Information, Substance Abuse and Mental Health Services Administration, U.S. Department of Health and Human Services. www.health.org/govpubs/bkd306/31k.aspx
- National Institute on Alcohol Abuse and Alcoholism (assessment instruments). www.niaaa.nih.gov/publications/instable.htm
- Deas D, Thomas SE. An overview of controlled studies of adolescent substance abuse treatment. Am J Addict 2001;10:178-89.
Drug brand names
- Sertraline • Zoloft
Disclosure
Dr. Deas receives grant support from Pfizer, Inc. and the National Institute of Alcohol Abuse and Alcoholism.
Dr. Upadhyaya receives grant support from the National Institute of Drug Abuse and GlaxoSmithKline.
Acknowledgment
The authors wish to thank Alva Blair for assistance with preparing this manuscript and Natalie Johnson, MA, Kess Mughelli, BS, and Lakeleia Middleton-Robinson, MA, for technical support.
1. Johnston LD, O’Malley PM, Bachman JG. Monitoring the Future: national results on adolescent drug use. Overview of key findings, 2002. Bethesda, MD: National Institute on Drug Abuse, 2003 (in press).
2. Martin CS, Kaczynski NA, Maisto SA, et al. Patterns of DSM-IV alcohol abuse and dependence symptoms in adolescent drinkers. J Stud Alcohol 1995;56:672-80.
3. Pollock NK, Martin CS. Diagnostic orphans: adolescents with alcohol symptoms who do not qualify for DSM-IV abuse or dependence diagnoses. Am J Psychiatry 1999;156:897-901.
4. Harrison PA, Fulkerson JA, Beebe TJ. DSM-IV substance use disorder criteria for adolescents: a critical examination based on a statewide school survey. Am J Psychiatry 1998;155:486-92.
5. Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse/dependence: results from the National Longitudinal Alcohol Epidemiological Survey. J Subst Abuse 1997;9:103-10.
6. Tarter RE, Laird SB, Bukstein O, Kaminer Y. Validation of the adolescent drug use screening inventory: preliminary findings. Psychol Addict Behav 1992;6:322-6.
7. Rahdert E (ed). The adolescent assessment/referral system manual. DHHS pub. no. (ADM) 91-1735. Rockville, MD: U.S. Department of Health and Human Services, 1991.
8. Kaminer Y, Bukstein OG, Tarter RE. The teen addiction severity index (T-ASI): rationale and reliability. Int J Addict 1991;26:219-26.
9. Meyers K, McLellan AT, Jaeger JL, Pettinati A. The development of the Comprehensive Addiction Severity Index for Adolescents (CASI-A): an interview for assessing multiple problems of adolescents. J Subst Abuse Treat 1995;12:181-93.
10. Deas DV, Roberts JS, Randall CL, Anton RF. Adolescent Obsessive-Compulsive Drinking Scale (A-OCDS): an assessment tool for problem drinking. J Natl Med Assoc 2001;93:92-103.
11. Deas D, Randall CL, Thomas S. The utility of the Deas-Marijuana Obsessive-Compulsive Scale (Deas-MOCS) in an inpatient adolescent substance abusing sample. Drug Alcohol Depend 2002;66(June):S43.-
12. Deas-Nesmith D, Campbell S, Brady KT. Substance use disorders in an adolescent inpatient psychiatric population. JAMA 1998;90:233-8.
13. Clark DB, Lesnick L, Hegedus AM. Traumas and other adverse life events in adolescents with alcohol abuse and dependence. Am Acad Child Adolesc Psychiatry 1997;36:1744-51.
14. Kilpatrick DG, Acierno R, Saunders B, et al. Risk factors for adolescent substance abuse and dependence: data from a national sample. J Consult Clin Psychol 2000;68:19-30.
15. Rose RJ. A developmental behavioral-genetic perspective on alcoholism risk. Alcohol Health Res World 1998;22:131-43.
16. Swadi H. Individual risk factors for adolescent substance use. Drug Alcohol Depend 1999;55:209-24.
17. Azrin N, McMahon P, Donohue B, et al. Behavior therapy for drug abuse: a controlled treatment outcome study. Behav Res Ther 1994;32:857-66.
18. Kaminer Y, Burleson JA, Goldberger R. Cognitive-behavioral coping skills and psychoeducation therapies for adolescent substance abuse. J Nerv Ment Dis 2002;190:737-45.
19. Liddle H, Dakof G. Efficacy of family therapy for drug abuse: promising but not definitive. J Marital Fam Ther 1995;21:511-43.
20. Waldron HB. Adolescent substance abuse and family therapy outcome: a review of randomized trials. Adv Clin Child Psychol 1997;19:199-234.
21. Henggeler SW, Melton LA. Effects of multisystemic therapy on drug use and abuse in serious juvenile offenders: a progress report from two outcome studies. Family Dynamics of Addiction Quarterly 1991;1:40-51.
22. Jaffe S. Step workbook for adolescent chemical dependency recovery. Washington, DC: American Academy of Child and Adolescent Psychiatry, 1990.
23. Deas DV, Randall C, Roberts J, Anton R. A double-blind, placebo-controlled trial of sertraline in depressed adolescent alcoholics: a pilot study. Human Psychopharmacology Clinical and Experimental 2000;15:461-9.
24. Geller B, Cooper T, Sun K, et al. Double-blind and placebo-controlled study of lithium for adolescent bipolar disorders with secondary substance dependency. J Am Acad Child Adolesc Psychiatry 1998;37:171-8.
1. Johnston LD, O’Malley PM, Bachman JG. Monitoring the Future: national results on adolescent drug use. Overview of key findings, 2002. Bethesda, MD: National Institute on Drug Abuse, 2003 (in press).
2. Martin CS, Kaczynski NA, Maisto SA, et al. Patterns of DSM-IV alcohol abuse and dependence symptoms in adolescent drinkers. J Stud Alcohol 1995;56:672-80.
3. Pollock NK, Martin CS. Diagnostic orphans: adolescents with alcohol symptoms who do not qualify for DSM-IV abuse or dependence diagnoses. Am J Psychiatry 1999;156:897-901.
4. Harrison PA, Fulkerson JA, Beebe TJ. DSM-IV substance use disorder criteria for adolescents: a critical examination based on a statewide school survey. Am J Psychiatry 1998;155:486-92.
5. Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse/dependence: results from the National Longitudinal Alcohol Epidemiological Survey. J Subst Abuse 1997;9:103-10.
6. Tarter RE, Laird SB, Bukstein O, Kaminer Y. Validation of the adolescent drug use screening inventory: preliminary findings. Psychol Addict Behav 1992;6:322-6.
7. Rahdert E (ed). The adolescent assessment/referral system manual. DHHS pub. no. (ADM) 91-1735. Rockville, MD: U.S. Department of Health and Human Services, 1991.
8. Kaminer Y, Bukstein OG, Tarter RE. The teen addiction severity index (T-ASI): rationale and reliability. Int J Addict 1991;26:219-26.
9. Meyers K, McLellan AT, Jaeger JL, Pettinati A. The development of the Comprehensive Addiction Severity Index for Adolescents (CASI-A): an interview for assessing multiple problems of adolescents. J Subst Abuse Treat 1995;12:181-93.
10. Deas DV, Roberts JS, Randall CL, Anton RF. Adolescent Obsessive-Compulsive Drinking Scale (A-OCDS): an assessment tool for problem drinking. J Natl Med Assoc 2001;93:92-103.
11. Deas D, Randall CL, Thomas S. The utility of the Deas-Marijuana Obsessive-Compulsive Scale (Deas-MOCS) in an inpatient adolescent substance abusing sample. Drug Alcohol Depend 2002;66(June):S43.-
12. Deas-Nesmith D, Campbell S, Brady KT. Substance use disorders in an adolescent inpatient psychiatric population. JAMA 1998;90:233-8.
13. Clark DB, Lesnick L, Hegedus AM. Traumas and other adverse life events in adolescents with alcohol abuse and dependence. Am Acad Child Adolesc Psychiatry 1997;36:1744-51.
14. Kilpatrick DG, Acierno R, Saunders B, et al. Risk factors for adolescent substance abuse and dependence: data from a national sample. J Consult Clin Psychol 2000;68:19-30.
15. Rose RJ. A developmental behavioral-genetic perspective on alcoholism risk. Alcohol Health Res World 1998;22:131-43.
16. Swadi H. Individual risk factors for adolescent substance use. Drug Alcohol Depend 1999;55:209-24.
17. Azrin N, McMahon P, Donohue B, et al. Behavior therapy for drug abuse: a controlled treatment outcome study. Behav Res Ther 1994;32:857-66.
18. Kaminer Y, Burleson JA, Goldberger R. Cognitive-behavioral coping skills and psychoeducation therapies for adolescent substance abuse. J Nerv Ment Dis 2002;190:737-45.
19. Liddle H, Dakof G. Efficacy of family therapy for drug abuse: promising but not definitive. J Marital Fam Ther 1995;21:511-43.
20. Waldron HB. Adolescent substance abuse and family therapy outcome: a review of randomized trials. Adv Clin Child Psychol 1997;19:199-234.
21. Henggeler SW, Melton LA. Effects of multisystemic therapy on drug use and abuse in serious juvenile offenders: a progress report from two outcome studies. Family Dynamics of Addiction Quarterly 1991;1:40-51.
22. Jaffe S. Step workbook for adolescent chemical dependency recovery. Washington, DC: American Academy of Child and Adolescent Psychiatry, 1990.
23. Deas DV, Randall C, Roberts J, Anton R. A double-blind, placebo-controlled trial of sertraline in depressed adolescent alcoholics: a pilot study. Human Psychopharmacology Clinical and Experimental 2000;15:461-9.
24. Geller B, Cooper T, Sun K, et al. Double-blind and placebo-controlled study of lithium for adolescent bipolar disorders with secondary substance dependency. J Am Acad Child Adolesc Psychiatry 1998;37:171-8.
Treating bipolar disorder during pregnancy
Prescribing drug therapy for pregnant bipolar women requires psychiatrists to balance the potential for neonatal malformations against the high risk of relapse when patients discontinue their medications.1 To help you achieve this balance, we offer an evidence-based approach that includes:
- analysis of the FDA’s teratogenicity categories for psychotropics
- review of the safety profiles of drugs used in mood stabilization
- an algorithm for managing patients who are considering conception or are pregnant.
PSYCHOTROPIC RISKS TO OFFSPRING
All psychotropic medications diffuse across the placenta, which exposes the fetus to some degree. Risks include teratogenicity, obstetrical complications, perinatal syndromes, and long-term postnatal behavioral sequelae.
Teratogenicity. A medication is considered teratogenic when prenatal exposure significantly increases the risk of congenital deformities over the baseline risk, which is 2% in the United States.2 The cause of most congenital malformations is unknown. Risk for teratogenicity occurs in the first 12 weeks of gestation, as organs are formed.
Table 1
FDA Use-in-Pregnancy ratings for medications The FDA system weighs the degree to which research findings have ruled out risk to the fetus
| Category | Interpretation |
|---|---|
| A | Controlled studies show no risk |
| B | No evidence of risk in humans |
| C | Risk cannot be ruled out |
| D | Positive evidence of risk |
| X | Contraindicated in pregnancy |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | |
Obstetrical complications include preterm delivery, low birth weight, and delivery complications such as low Apgar scores or behavioral effects requiring intensive care.
Perinatal syndromes include physical and behavioral symptoms noticed shortly after birth (such as jitteriness). These consequences are putatively related to drug use at or near birth and have limited duration.
Postnatal behavioral sequelae include long-term neurobehavioral abnormalities in children who were exposed to psychotropics in utero.
BALANCING RISKS
Risks with medication. The FDA’s “use in pregnancy” rating system (Table 1) uses available data to assess the degree of teratogenic risk. These guidelines can be confusing and are one of many tools to use when considering a possible drug treatment.
Most psychotropics are category “C” or “D,” which imply a chance of harm to the exposed fetus. Category “B” drugs would appear safer, but this rating could simply indicate a lack of adequate human data or that no data have shown harm in animals.
Moreover, a category “D” drug may be chosen more often during pregnancy than a category “C” drug. This may occur when more human data exist on using the category “D” drug in patients with a particular disorder (such as using lithium versus valproate or olanzapine in pregnant bipolar women).
No psychotropics are classified as “A,” meaning either some risks are associated with every psychotropic or the risk of some agents has not been adequately explored. Furthermore, no psychotropics are FDA-approved for use during pregnancy.
Risks without medication. Teratogenicity notwithstanding, psychotropic intervention is the most effective treatment for women with bipolar disorder. Patients who discontinue mood-stabilizing medication after conception increase their risk of relapse into depression or mania,3 either of which could lead to complications and untoward effects on the fetus.
Depression during pregnancy has been linked to low birth weight and preterm delivery.4,5 These effects may be mediated by the illness itself or by other factors that indirectly affect birth outcomes. For example, depression during pregnancy is associated with decreased appetite, substance use and abuse, and lower use of prenatal care.6
Untreated mania may also be associated with perinatal risks, as a pregnant patient in a manic state may engage in impulsive, high-risk behaviors that endanger her and the fetus.7
MOOD STABILIZERS
The FDA categorizes as “D” the three most commonly used mood stabilizers: lithium, valproate, and carbamazepine (Table 2). This rating implies that studies have demonstrated fetal risk but the drug’s potential benefit may still outweigh the risk.
Lithium. The International Registry of Lithium reported increased rates of cardiovascular malformations— such as Ebstein’s anomaly—in children whose mothers took lithium during pregnancy.
Relative risk for Ebstein’s anomaly in children with fetal exposure to lithium may be 20 times higher than the risk in unexposed children, although the absolute risk with lithium exposure remains low (1 in 1,000 births).1,8
No significant neurobehavioral teratogenicity has been reported in infants exposed in utero to lithium, although few cases have been studied. One study reported that 22 lithium-exposed infants attained developmental milestones at a pace comparable to that of unexposed controls.9
“Floppy baby” syndrome, in which infants experience hypotonicity and cyanosis, is the most recognized adverse effect in infants exposed to lithium in utero.10 Its frequency is unknown, but rare. Neonatal hypothyroidism and nephrogenic diabetes insipidus have also been documented.
Anticonvulsants. To date, no studies have examined the outcomes of children whose mothers took anticonvulsants for bipolar disorder during pregnancy, though the research concerning epileptic mothers is extensive.
Neural tube defects. Data associate anticonvulsant exposure with a significantly greater risk for malformations than in the general population. Specifically, anticonvulsants may cause neural tube defects such as spina bifida, ancephaly, and encephaly in 2 to 5% of those exposed, as well as craniofacial anomalies, microcephaly, growth retardation, and heart defects.11-14
Table 2
FDA’s teratogenicity ratings of mood stabilizers and other antimanic agents
| Category | Medication | Teratogenicity |
|---|---|---|
| Mood stabilizers | Lithium Carbamazepine Valproate | Category D Category D Category D |
| Anticonvulsants | Gabapentin Lamotrigine Topiramate | Category C Category C Category C |
| Antipsychotics | Olanzapine Risperidone Chlorpromazine Haloperidol Trifluoperazine | Category C Category C Safety in pregnancy not known Category C Safety in pregnancy not known |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | ||
More minor malformations—such as rotated ears, depressed nasal bridge, short nose, elongated upper lip, and fingernail hypoplasia—have been reported in infants exposed to anticonvulsants in utero.14 These malformations disappear with age.13 Teratogenicity increases with the use of multiple anticonvulsants and possibly with higher maternal plasma levels and toxic metabolites.15
Conclusion. The three most commonly used mood stabilizers are all teratogenic. The least risk may occur with lithium (0.1%) versus valproate (2 to 5%) or carbamazepine (1 to 3%). These risks must be weighed against the up to 50% chance of relapse with medication discontinuation.3
ANTIPSYCHOTICS
Antipsychotics are often used to treat mania because of their rapid effects and sedative properties. Most antipsychotics—specifically, haloperidol, olanzapine, and risperidone—are labeled “C,” specifying that fetal risk cannot be ruled out.
Table 3
FDA’s teratogenicity ratings of common antidepressants
| Category | Medication | Teratogenicity |
|---|---|---|
| Tricyclics | Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline | Category C Category C Safety in pregnancy not known Safety in pregnancy not known Safety in pregnancy not known |
| Selective serotonin reuptake inhibitors | Citalopram Fluoxetine Fluvoxamine Paroxetine Sertraline | Category C Category C Category C Category C Category C |
| Other antidepressants | Bupropion Phenelzine Tranylcypromine | Category B Safety in pregnancy and nursing not known Safety in pregnancy and nursing not known |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | ||
Chlorpromazine and haloperidol have been most studied during pregnancy but in relation to treating hyperemesis gravidarum and psychosis, not bipolar disorder. Results regarding antipsychotics’ teratogenic and behavioral risks are mixed,16-21 probably because the various compounds have different effects on the fetus.
The underlying illness—rather than the medications—may increase the rate of anomalies seen with exposure to antipsychotics:
- Rieder et al22 reported an increased rate of perinatal death in infants of schizophrenic mothers but no significant association between the mothers’ use of antipsychotics and perinatal death.
- Sobel23 compared psychotic women with and without histories of chlorpromazine exposure during pregnancy. Rates of fetal damage were similar and approximately twice that of the general population.
A meta-analysis of 74,337 live births revealed that first-trimester exposure to low-potency antipsychotics increases the relative risk of fetal anomalies in nonpsychotic women. Phenothiazines may increase the 2% baseline incidence of malformations to 2.4%.1 No specific organ malformation following fetal exposure to phenothiazines has been consistently identified.
Olanzapine was recently approved for treating mania. Very little data exist regarding its impact on fetal development when used during pregnancy, although studies on small numbers of women have not revealed teratogenicity.24,25
Conclusion. Psychotic illness itself may increase the risk of poor fetal outcome to a greater extent than does antipsychotic use. Prenatal exposure to low-potency phenothiazines may further increase this risk, although only slightly. The effect of prenatal exposure to atypical antipsychotics requires further study.
BENZODIAZEPINES
Benzodiazepines are rarely a primary treatment for mania or depression. Thus, a comprehensive review of their effect on fetal outcome is beyond the scope of this review. A meta-analysis of exposure in the first trimester suggests a very small but significant increase in risk for cleft palate.1 The absolute risk is <1 in 1,000 cases.
ANTIDEPRESSANTS
Whereas treatment of acute mania is considered a medical emergency, women with bipolar disorder may also relapse into depression during pregnancy. An antidepressant should not be used without a mood stabilizer when treating bipolar I disorder, although a mood stabilizer alone may be inadequate to treat depression. Using tricyclics and selective serotonin reuptake inhibitors (SSRIs) during pregnancy has not been associated with teratogenicity (Table 3),26 although perinatal effects have been reported.1
Tricyclics. In case-control studies involving more than 300,000 live births, 414 incidences of first-trimester exposure to tricyclics were followed. Information from these patients found no significant association between fetal exposure to tricyclics and increased rates of congenital malformations.1 The few studies that have been performed suggest no long-term effects from in utero exposure.26 Although these results suggest that prenatal exposure to tricyclics is relatively safe, more research is needed.
SSRIs. To date, no significant teratogenic effects of SSRIs have been identified in offspring of treated women.
The manufacturer’s register for fluoxetine contains approximately 2,000 cases of treated patients, with no excess cases of congenital anomalies or malformations following prenatal exposure. Citalopram has the next largest database of in utero exposure (n=365), again with no increased risk for teratogenicity. Several smaller systematic reports are available on in utero exposure to sertraline, paroxetine, or escitalopram.26
Most studies of pregnant women taking fluoxetine in the first trimester have found no increased risk of obstetrical complications—including spontaneous pregnancy loss, preterm labor, or low birth weight—compared with women not taking fluoxetine. Taking fluoxetine during the third trimester may increase the risk for perinatal complications,27 although this has been inconsistently reported and requires further study. Effects of other SSRIs in the third trimester have not been systematically explored.
Case reports and one controlled study have addressed possible neonatal perinatal symptoms from in utero exposure to SSRIs.28,29 Preliminary data show no adverse neurobehavioral function in exposed neonates.26
Electroconvulsive therapy (ECT) has been proven effective for acute mania and depression, demonstrating few deleterious effects on neonates. ECT has few side effects and may be safer than drug therapy in this population. Two reviews support the efficacy and relative safety of ECT treatment during pregnancy, although more evidence is needed.30,31
RECOMMENDATIONS
Discuss pregnancy and medication risks with all bipolar women, regardless of proximal plans for pregnancy. If psychotropic medication is used, prescribe carefully during the first trimester, using the minimum number of drugs and the lowest dosages needed to restore or maintain well-being.32
Pros and cons of switching. Some clinicians may encourage a patient to taper a medication during the first trimester because of its unknown or high teratogenicity. Depending on the patient’s illness severity, this might not be the optimal decision. A more conservative option would be to switch to a lower-risk drug during pregnancy.
Lithium has both antidepressant and antimanic properties and is less teratogenic compared with first-trimester exposure to an anticonvulsant. However, if lithium has not been successful for the woman’s mania prophylaxis in the past and she has demonstrated antimanic response to an anticonvulsant, switching to lithium or another anticonvulsant is not recommended.
Algorithm Suggested approach to the bipolar patient who wishes to conceive or is pregnant
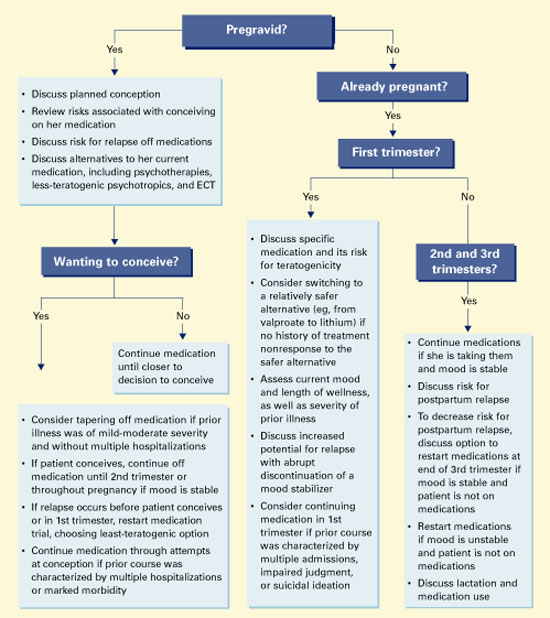
Folate and neural tube defects. As first-trimester exposure to carbamazepine or valproate increases the risk for neural tube defects, using the lowest available dosage may decrease the risk for spina bifida, at least with valproate.
Low maternal folate levels are often associated with neural tube defects from any cause.33 Valproate lowers folate levels by inhibiting one of the enzymes necessary for its formation, which may be a mechanism for the increased risk of spina bifida.34
Folate supplementation. To date, no study has demonstrated that giving folate supplements to women taking anticonvulsants during pregnancy reduces the risk of neural tube defects.35 Nonetheless, we recommend that women who continue to take valproate or carbamazepine during pregnancy receive folate, 3 to 4 mg/d, as a precaution.
Treating manic relapse. Data show high rates of relapse in patients who stop taking lithium, particularly if done abruptly.3 Counsel women taking lithium to plan their pregnancies to allow enough time to taper off the medication prior to conception, if they want to try this. Lithium should be decreased slowly—approximately 50% every 2 weeks—to avoid relapse.
Treat aggressively if relapse occurs during pregnancy. Consider:
- psychiatric hospitalization in case of suicidality or psychosis
- reinstituting drug therapy with a less-teratogenic agent
- ECT for a manic or depressive episode.
As the pregnancy advances and the mother’s volume of distribution increases, dosage increases may be needed to maintain therapeutic drug levels.
Treating depressive relapse. Should depression occur in pregnancy, SSRIs or tricyclics added to mood stabilizer therapy have been shown to be effective, with few teratogenic effects.
Cognitive-behavioral and interpersonal psychotherapies also have shown efficacy in pregnant women with major depressive disorder36 and may be effective for women with bipolar disorder in pregnancy. Cognitive psychotherapies, when used with medication, have been reported effective in preventing relapse in nongravid bipolar patients.36-37
Related resources
Psychiatric disorders during pregnancy. Massachusetts General Hospital Center for Women’s Health. Perinatal Resource Center. www.womensmentalhealth.org/topics/pregnancy_lib.html
Drug brand names
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid et al
- Methylphenidate • Ritalin et al
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Phenelzine • Nardil
- Risperidone • Risperdal
- Sertraline • Zoloft
- Topiramate • Topamax
- Tranylcypromine • Parnate
- Trifluoperazine • Stelazine
- Valproate • Depakote et al
Disclosure
Dr. Altshuler receives research support from Abbott Laboratories, is a consultant to Abbott Laboratories, Forest Laboratories, and Eli Lilly & Co., and is a speaker for GlaxoSmithKline and Janssen Pharmaceutica.
Ms. Richards reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Yonkers receives research support from GlaxoSmithKline and Berlex Laboratories, is a consultant to GlaxoSmithKline, and is a speaker for Eli Lilly and Co., Pfizer Inc., GlaxoSmithKline, and Wyeth Pharmaceuticals.
1. Altshuler L, Cohen L, Szuba, et al. Pharmacologic management of psychiatric illness during pregnancy: dilemmas and guidelines. Am J Psychiatry 1996;153:592-606.
2. Nelson K, Holmes LB. Malformations due to presumed spontaneous mutations in newborn infants. N Engl J Med 1989;320:19-23.
3. Viguera AC, Nonacs R, Cohen LS, et al. Risk of recurrence of bipolar disorder in pregnant and nonpregnant women after discontinuing lithium maintenance. Am J Psychiatry 2000;157:179-84.
4. Steer RA, Scholl TO, Hediger ML, Fischer RL. Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol 1992;45(10):1093-9.
5. Orr ST, Miller CA. Maternal depressive symptoms and the risk of poor pregnancy outcome. Review of the literature and preliminary findings. Epidemiol Rev 1995;17(1):165-71.
6. Zuckerman B, Amaro H, Bauchner H, Cabral H. Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol 1989;160:1107-11.
7. Miller LJ. Psychotic denial of pregnancy: phenomenology and clinical management. Hosp Community Psychiatry 1990;41:1233-7.
8. Cohen LS, Friedman JM, Jefferson JW, et al. A reevaluation of risk of in utero exposure to lithium. JAMA 1994;271(2):146-50correction JAMA 1994;271(19):1485.
9. Schou M. What happened later to the lithium babies? A follow-up study of children born without malformations. Acta Psychiatr Scand 1976;54(3):193-7.
10. Woody JN, London WL, Wilbanks GD. Lithium toxicity in a newborn. Pediatrics 1971;47:94-6.
11. Jones K, Lacro R, Johnson K, Adams J. Patterns of malformations in the children of women treated with carbamazepine during pregnancy. N Engl J Med 1989;320:1661-6.
12. Rosa F. Spina bifida in infants of women treated with carbamazepine during pregnancy. N Engl J Med 1991;324(10):674-7.
13. Koch S, Losche G, Jager-Roman E, et al. Major and minor birth malformations and antiepileptic drugs. Neurology 1992;42:83-8.
14. Jager-Roman E, Deichl A, Jakob S, et al. Fetal growth, major malformations, and minor anomalies in infants born to women receiving valproic acid. J Pediatr 1986;108:997-1004.
15. Nakane Y, Okuma T, Takahashi R, et al. Multi-institutional study on the teratogenicity and fetal toxicity of antiepileptic drugs: a report of a collaborative study group in Japan. Epilepsia 1980;21:663-80.
16. Edlund MJ, Craig TJ. Antipsychotic drug use and birth defects: an epidemiologic reassessment. Compr Psychiatry 1984;25:32-8.
17. Kris EB. Children of mothers maintained on pharmacotherapy during pregnancy and postpartum. Curr Ther Res 1965;7:785-9.
18. Clark CVH, Gorman D, Vernadakis A. Effects of prenatal administration of psychotropic drugs on behavior of developing rats. Dev Psychobiol 1970;3:225-35.
19. Golub M, Kornetsky C. Seizure susceptibility and avoidance conditioning in adult rats treated prenatally with chlorpromazine. Dev Psychobiol 1974;7:79-88.
20. Spear LP, Shalaby IA, Brick J. Chronic administration of haloperidol during development: behavioral and psychopharmacological effects. Psychopharmacology (Berl) 1980;70:47-58.
21. Cagiano R, Barfield RJ, White NR, et al. Subtle behavioral changes produced in rat pups exposed in utero to haloperidol. Eur J Pharmacol 1988;157:45-50.
22. Rieder RO, Rosenthal D, Wender P, Blumenthal H. The offspring of schizophrenics: fetal and neonatal deaths. Arch Gen Psychiatry 1975;32:200-11.
23. Sobel DE. Fetal damage due to ECT, insulin coma, chlorpromazine, or reserpine. Arch Gen Psychiatry 1960;2:606-11.
24. Dickson R. Olanzapine and pregnancy. Can J Psychiatry 1998;43:196-7.
25. Goldstein DJ, Corbin LA, Fung MC. Olanzapine-exposed pregnancies and lactation; early experience. J Clin Psychopharmacol 2000;24(4):399-403.
26. Altshuler LL, Cohen LS, Moline ML, et al. The expert consensus guideline series: treatment of depression in women. Postgrad Med 2001 Mar;(Spec No):1-22.
27. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010-15.
28. Spencer MJ. Fluoxetine hydrochloride (Prozac) toxicity in the neonate. Pediatrics 1993;92:721-2.
29. Cabrera FM, Battaglia G. Delayed decreases in brain 5-HT 2a and 2c receptor density and function in male rat progeny following prenatal fluoxetine. J Pharmacol Exp Ther 1994;269:637-45.
30. Miller LJ. Use of electroconvulsive therapy during pregnancy. Hosp Community Psychiatry 1994;45:444-50.
31. Ferrill MJ, Kehoe WA, Jacisin JJ. ECT during pregnancy: physiologic and pharmacologic considerations. Convuls Ther 1992;8:186-200.
32. Yonkers K, Wisner K, Cohen L, et al. Management of bipolar disorder during pregnancy and the postpartum period. Bipolar Consensus Statement. Submitted for publication.
33. Dansky L, Rosenblatt D, Andermann E. Mechanisms of teratogenesis: folic acid and antiepileptic therapy. Neurology 1992;42(suppl 5):32-42.
34. Wegner C, Nau H. Alteration of embryonic folate metabolism by valproic acid during organogenesis: implications for mechanism of teratogenesis. Neurology 1992;42(suppl 5):17-24.
35. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 1991;338:131-7.
36. Spinelli MG, Endicott J. Controlled clinical trial of interpersonal psychotherapy versus parenting education program for depressed pregnant women. Am J Psychiatry 2003;160:555-62.
37. Lam DH, Watkins ER, Hayward P, et al. A randomized controlled study of cognitive therapy for relapse prevention for bipolar affective disorder: outcome in the first year. Arch Gen Psychiatry 2003;60(2):145-52.
Prescribing drug therapy for pregnant bipolar women requires psychiatrists to balance the potential for neonatal malformations against the high risk of relapse when patients discontinue their medications.1 To help you achieve this balance, we offer an evidence-based approach that includes:
- analysis of the FDA’s teratogenicity categories for psychotropics
- review of the safety profiles of drugs used in mood stabilization
- an algorithm for managing patients who are considering conception or are pregnant.
PSYCHOTROPIC RISKS TO OFFSPRING
All psychotropic medications diffuse across the placenta, which exposes the fetus to some degree. Risks include teratogenicity, obstetrical complications, perinatal syndromes, and long-term postnatal behavioral sequelae.
Teratogenicity. A medication is considered teratogenic when prenatal exposure significantly increases the risk of congenital deformities over the baseline risk, which is 2% in the United States.2 The cause of most congenital malformations is unknown. Risk for teratogenicity occurs in the first 12 weeks of gestation, as organs are formed.
Table 1
FDA Use-in-Pregnancy ratings for medications The FDA system weighs the degree to which research findings have ruled out risk to the fetus
| Category | Interpretation |
|---|---|
| A | Controlled studies show no risk |
| B | No evidence of risk in humans |
| C | Risk cannot be ruled out |
| D | Positive evidence of risk |
| X | Contraindicated in pregnancy |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | |
Obstetrical complications include preterm delivery, low birth weight, and delivery complications such as low Apgar scores or behavioral effects requiring intensive care.
Perinatal syndromes include physical and behavioral symptoms noticed shortly after birth (such as jitteriness). These consequences are putatively related to drug use at or near birth and have limited duration.
Postnatal behavioral sequelae include long-term neurobehavioral abnormalities in children who were exposed to psychotropics in utero.
BALANCING RISKS
Risks with medication. The FDA’s “use in pregnancy” rating system (Table 1) uses available data to assess the degree of teratogenic risk. These guidelines can be confusing and are one of many tools to use when considering a possible drug treatment.
Most psychotropics are category “C” or “D,” which imply a chance of harm to the exposed fetus. Category “B” drugs would appear safer, but this rating could simply indicate a lack of adequate human data or that no data have shown harm in animals.
Moreover, a category “D” drug may be chosen more often during pregnancy than a category “C” drug. This may occur when more human data exist on using the category “D” drug in patients with a particular disorder (such as using lithium versus valproate or olanzapine in pregnant bipolar women).
No psychotropics are classified as “A,” meaning either some risks are associated with every psychotropic or the risk of some agents has not been adequately explored. Furthermore, no psychotropics are FDA-approved for use during pregnancy.
Risks without medication. Teratogenicity notwithstanding, psychotropic intervention is the most effective treatment for women with bipolar disorder. Patients who discontinue mood-stabilizing medication after conception increase their risk of relapse into depression or mania,3 either of which could lead to complications and untoward effects on the fetus.
Depression during pregnancy has been linked to low birth weight and preterm delivery.4,5 These effects may be mediated by the illness itself or by other factors that indirectly affect birth outcomes. For example, depression during pregnancy is associated with decreased appetite, substance use and abuse, and lower use of prenatal care.6
Untreated mania may also be associated with perinatal risks, as a pregnant patient in a manic state may engage in impulsive, high-risk behaviors that endanger her and the fetus.7
MOOD STABILIZERS
The FDA categorizes as “D” the three most commonly used mood stabilizers: lithium, valproate, and carbamazepine (Table 2). This rating implies that studies have demonstrated fetal risk but the drug’s potential benefit may still outweigh the risk.
Lithium. The International Registry of Lithium reported increased rates of cardiovascular malformations— such as Ebstein’s anomaly—in children whose mothers took lithium during pregnancy.
Relative risk for Ebstein’s anomaly in children with fetal exposure to lithium may be 20 times higher than the risk in unexposed children, although the absolute risk with lithium exposure remains low (1 in 1,000 births).1,8
No significant neurobehavioral teratogenicity has been reported in infants exposed in utero to lithium, although few cases have been studied. One study reported that 22 lithium-exposed infants attained developmental milestones at a pace comparable to that of unexposed controls.9
“Floppy baby” syndrome, in which infants experience hypotonicity and cyanosis, is the most recognized adverse effect in infants exposed to lithium in utero.10 Its frequency is unknown, but rare. Neonatal hypothyroidism and nephrogenic diabetes insipidus have also been documented.
Anticonvulsants. To date, no studies have examined the outcomes of children whose mothers took anticonvulsants for bipolar disorder during pregnancy, though the research concerning epileptic mothers is extensive.
Neural tube defects. Data associate anticonvulsant exposure with a significantly greater risk for malformations than in the general population. Specifically, anticonvulsants may cause neural tube defects such as spina bifida, ancephaly, and encephaly in 2 to 5% of those exposed, as well as craniofacial anomalies, microcephaly, growth retardation, and heart defects.11-14
Table 2
FDA’s teratogenicity ratings of mood stabilizers and other antimanic agents
| Category | Medication | Teratogenicity |
|---|---|---|
| Mood stabilizers | Lithium Carbamazepine Valproate | Category D Category D Category D |
| Anticonvulsants | Gabapentin Lamotrigine Topiramate | Category C Category C Category C |
| Antipsychotics | Olanzapine Risperidone Chlorpromazine Haloperidol Trifluoperazine | Category C Category C Safety in pregnancy not known Category C Safety in pregnancy not known |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | ||
More minor malformations—such as rotated ears, depressed nasal bridge, short nose, elongated upper lip, and fingernail hypoplasia—have been reported in infants exposed to anticonvulsants in utero.14 These malformations disappear with age.13 Teratogenicity increases with the use of multiple anticonvulsants and possibly with higher maternal plasma levels and toxic metabolites.15
Conclusion. The three most commonly used mood stabilizers are all teratogenic. The least risk may occur with lithium (0.1%) versus valproate (2 to 5%) or carbamazepine (1 to 3%). These risks must be weighed against the up to 50% chance of relapse with medication discontinuation.3
ANTIPSYCHOTICS
Antipsychotics are often used to treat mania because of their rapid effects and sedative properties. Most antipsychotics—specifically, haloperidol, olanzapine, and risperidone—are labeled “C,” specifying that fetal risk cannot be ruled out.
Table 3
FDA’s teratogenicity ratings of common antidepressants
| Category | Medication | Teratogenicity |
|---|---|---|
| Tricyclics | Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline | Category C Category C Safety in pregnancy not known Safety in pregnancy not known Safety in pregnancy not known |
| Selective serotonin reuptake inhibitors | Citalopram Fluoxetine Fluvoxamine Paroxetine Sertraline | Category C Category C Category C Category C Category C |
| Other antidepressants | Bupropion Phenelzine Tranylcypromine | Category B Safety in pregnancy and nursing not known Safety in pregnancy and nursing not known |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | ||
Chlorpromazine and haloperidol have been most studied during pregnancy but in relation to treating hyperemesis gravidarum and psychosis, not bipolar disorder. Results regarding antipsychotics’ teratogenic and behavioral risks are mixed,16-21 probably because the various compounds have different effects on the fetus.
The underlying illness—rather than the medications—may increase the rate of anomalies seen with exposure to antipsychotics:
- Rieder et al22 reported an increased rate of perinatal death in infants of schizophrenic mothers but no significant association between the mothers’ use of antipsychotics and perinatal death.
- Sobel23 compared psychotic women with and without histories of chlorpromazine exposure during pregnancy. Rates of fetal damage were similar and approximately twice that of the general population.
A meta-analysis of 74,337 live births revealed that first-trimester exposure to low-potency antipsychotics increases the relative risk of fetal anomalies in nonpsychotic women. Phenothiazines may increase the 2% baseline incidence of malformations to 2.4%.1 No specific organ malformation following fetal exposure to phenothiazines has been consistently identified.
Olanzapine was recently approved for treating mania. Very little data exist regarding its impact on fetal development when used during pregnancy, although studies on small numbers of women have not revealed teratogenicity.24,25
Conclusion. Psychotic illness itself may increase the risk of poor fetal outcome to a greater extent than does antipsychotic use. Prenatal exposure to low-potency phenothiazines may further increase this risk, although only slightly. The effect of prenatal exposure to atypical antipsychotics requires further study.
BENZODIAZEPINES
Benzodiazepines are rarely a primary treatment for mania or depression. Thus, a comprehensive review of their effect on fetal outcome is beyond the scope of this review. A meta-analysis of exposure in the first trimester suggests a very small but significant increase in risk for cleft palate.1 The absolute risk is <1 in 1,000 cases.
ANTIDEPRESSANTS
Whereas treatment of acute mania is considered a medical emergency, women with bipolar disorder may also relapse into depression during pregnancy. An antidepressant should not be used without a mood stabilizer when treating bipolar I disorder, although a mood stabilizer alone may be inadequate to treat depression. Using tricyclics and selective serotonin reuptake inhibitors (SSRIs) during pregnancy has not been associated with teratogenicity (Table 3),26 although perinatal effects have been reported.1
Tricyclics. In case-control studies involving more than 300,000 live births, 414 incidences of first-trimester exposure to tricyclics were followed. Information from these patients found no significant association between fetal exposure to tricyclics and increased rates of congenital malformations.1 The few studies that have been performed suggest no long-term effects from in utero exposure.26 Although these results suggest that prenatal exposure to tricyclics is relatively safe, more research is needed.
SSRIs. To date, no significant teratogenic effects of SSRIs have been identified in offspring of treated women.
The manufacturer’s register for fluoxetine contains approximately 2,000 cases of treated patients, with no excess cases of congenital anomalies or malformations following prenatal exposure. Citalopram has the next largest database of in utero exposure (n=365), again with no increased risk for teratogenicity. Several smaller systematic reports are available on in utero exposure to sertraline, paroxetine, or escitalopram.26
Most studies of pregnant women taking fluoxetine in the first trimester have found no increased risk of obstetrical complications—including spontaneous pregnancy loss, preterm labor, or low birth weight—compared with women not taking fluoxetine. Taking fluoxetine during the third trimester may increase the risk for perinatal complications,27 although this has been inconsistently reported and requires further study. Effects of other SSRIs in the third trimester have not been systematically explored.
Case reports and one controlled study have addressed possible neonatal perinatal symptoms from in utero exposure to SSRIs.28,29 Preliminary data show no adverse neurobehavioral function in exposed neonates.26
Electroconvulsive therapy (ECT) has been proven effective for acute mania and depression, demonstrating few deleterious effects on neonates. ECT has few side effects and may be safer than drug therapy in this population. Two reviews support the efficacy and relative safety of ECT treatment during pregnancy, although more evidence is needed.30,31
RECOMMENDATIONS
Discuss pregnancy and medication risks with all bipolar women, regardless of proximal plans for pregnancy. If psychotropic medication is used, prescribe carefully during the first trimester, using the minimum number of drugs and the lowest dosages needed to restore or maintain well-being.32
Pros and cons of switching. Some clinicians may encourage a patient to taper a medication during the first trimester because of its unknown or high teratogenicity. Depending on the patient’s illness severity, this might not be the optimal decision. A more conservative option would be to switch to a lower-risk drug during pregnancy.
Lithium has both antidepressant and antimanic properties and is less teratogenic compared with first-trimester exposure to an anticonvulsant. However, if lithium has not been successful for the woman’s mania prophylaxis in the past and she has demonstrated antimanic response to an anticonvulsant, switching to lithium or another anticonvulsant is not recommended.
Algorithm Suggested approach to the bipolar patient who wishes to conceive or is pregnant
Folate and neural tube defects. As first-trimester exposure to carbamazepine or valproate increases the risk for neural tube defects, using the lowest available dosage may decrease the risk for spina bifida, at least with valproate.
Low maternal folate levels are often associated with neural tube defects from any cause.33 Valproate lowers folate levels by inhibiting one of the enzymes necessary for its formation, which may be a mechanism for the increased risk of spina bifida.34
Folate supplementation. To date, no study has demonstrated that giving folate supplements to women taking anticonvulsants during pregnancy reduces the risk of neural tube defects.35 Nonetheless, we recommend that women who continue to take valproate or carbamazepine during pregnancy receive folate, 3 to 4 mg/d, as a precaution.
Treating manic relapse. Data show high rates of relapse in patients who stop taking lithium, particularly if done abruptly.3 Counsel women taking lithium to plan their pregnancies to allow enough time to taper off the medication prior to conception, if they want to try this. Lithium should be decreased slowly—approximately 50% every 2 weeks—to avoid relapse.
Treat aggressively if relapse occurs during pregnancy. Consider:
- psychiatric hospitalization in case of suicidality or psychosis
- reinstituting drug therapy with a less-teratogenic agent
- ECT for a manic or depressive episode.
As the pregnancy advances and the mother’s volume of distribution increases, dosage increases may be needed to maintain therapeutic drug levels.
Treating depressive relapse. Should depression occur in pregnancy, SSRIs or tricyclics added to mood stabilizer therapy have been shown to be effective, with few teratogenic effects.
Cognitive-behavioral and interpersonal psychotherapies also have shown efficacy in pregnant women with major depressive disorder36 and may be effective for women with bipolar disorder in pregnancy. Cognitive psychotherapies, when used with medication, have been reported effective in preventing relapse in nongravid bipolar patients.36-37
Related resources
Psychiatric disorders during pregnancy. Massachusetts General Hospital Center for Women’s Health. Perinatal Resource Center. www.womensmentalhealth.org/topics/pregnancy_lib.html
Drug brand names
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid et al
- Methylphenidate • Ritalin et al
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Phenelzine • Nardil
- Risperidone • Risperdal
- Sertraline • Zoloft
- Topiramate • Topamax
- Tranylcypromine • Parnate
- Trifluoperazine • Stelazine
- Valproate • Depakote et al
Disclosure
Dr. Altshuler receives research support from Abbott Laboratories, is a consultant to Abbott Laboratories, Forest Laboratories, and Eli Lilly & Co., and is a speaker for GlaxoSmithKline and Janssen Pharmaceutica.
Ms. Richards reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Yonkers receives research support from GlaxoSmithKline and Berlex Laboratories, is a consultant to GlaxoSmithKline, and is a speaker for Eli Lilly and Co., Pfizer Inc., GlaxoSmithKline, and Wyeth Pharmaceuticals.
Prescribing drug therapy for pregnant bipolar women requires psychiatrists to balance the potential for neonatal malformations against the high risk of relapse when patients discontinue their medications.1 To help you achieve this balance, we offer an evidence-based approach that includes:
- analysis of the FDA’s teratogenicity categories for psychotropics
- review of the safety profiles of drugs used in mood stabilization
- an algorithm for managing patients who are considering conception or are pregnant.
PSYCHOTROPIC RISKS TO OFFSPRING
All psychotropic medications diffuse across the placenta, which exposes the fetus to some degree. Risks include teratogenicity, obstetrical complications, perinatal syndromes, and long-term postnatal behavioral sequelae.
Teratogenicity. A medication is considered teratogenic when prenatal exposure significantly increases the risk of congenital deformities over the baseline risk, which is 2% in the United States.2 The cause of most congenital malformations is unknown. Risk for teratogenicity occurs in the first 12 weeks of gestation, as organs are formed.
Table 1
FDA Use-in-Pregnancy ratings for medications The FDA system weighs the degree to which research findings have ruled out risk to the fetus
| Category | Interpretation |
|---|---|
| A | Controlled studies show no risk |
| B | No evidence of risk in humans |
| C | Risk cannot be ruled out |
| D | Positive evidence of risk |
| X | Contraindicated in pregnancy |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | |
Obstetrical complications include preterm delivery, low birth weight, and delivery complications such as low Apgar scores or behavioral effects requiring intensive care.
Perinatal syndromes include physical and behavioral symptoms noticed shortly after birth (such as jitteriness). These consequences are putatively related to drug use at or near birth and have limited duration.
Postnatal behavioral sequelae include long-term neurobehavioral abnormalities in children who were exposed to psychotropics in utero.
BALANCING RISKS
Risks with medication. The FDA’s “use in pregnancy” rating system (Table 1) uses available data to assess the degree of teratogenic risk. These guidelines can be confusing and are one of many tools to use when considering a possible drug treatment.
Most psychotropics are category “C” or “D,” which imply a chance of harm to the exposed fetus. Category “B” drugs would appear safer, but this rating could simply indicate a lack of adequate human data or that no data have shown harm in animals.
Moreover, a category “D” drug may be chosen more often during pregnancy than a category “C” drug. This may occur when more human data exist on using the category “D” drug in patients with a particular disorder (such as using lithium versus valproate or olanzapine in pregnant bipolar women).
No psychotropics are classified as “A,” meaning either some risks are associated with every psychotropic or the risk of some agents has not been adequately explored. Furthermore, no psychotropics are FDA-approved for use during pregnancy.
Risks without medication. Teratogenicity notwithstanding, psychotropic intervention is the most effective treatment for women with bipolar disorder. Patients who discontinue mood-stabilizing medication after conception increase their risk of relapse into depression or mania,3 either of which could lead to complications and untoward effects on the fetus.
Depression during pregnancy has been linked to low birth weight and preterm delivery.4,5 These effects may be mediated by the illness itself or by other factors that indirectly affect birth outcomes. For example, depression during pregnancy is associated with decreased appetite, substance use and abuse, and lower use of prenatal care.6
Untreated mania may also be associated with perinatal risks, as a pregnant patient in a manic state may engage in impulsive, high-risk behaviors that endanger her and the fetus.7
MOOD STABILIZERS
The FDA categorizes as “D” the three most commonly used mood stabilizers: lithium, valproate, and carbamazepine (Table 2). This rating implies that studies have demonstrated fetal risk but the drug’s potential benefit may still outweigh the risk.
Lithium. The International Registry of Lithium reported increased rates of cardiovascular malformations— such as Ebstein’s anomaly—in children whose mothers took lithium during pregnancy.
Relative risk for Ebstein’s anomaly in children with fetal exposure to lithium may be 20 times higher than the risk in unexposed children, although the absolute risk with lithium exposure remains low (1 in 1,000 births).1,8
No significant neurobehavioral teratogenicity has been reported in infants exposed in utero to lithium, although few cases have been studied. One study reported that 22 lithium-exposed infants attained developmental milestones at a pace comparable to that of unexposed controls.9
“Floppy baby” syndrome, in which infants experience hypotonicity and cyanosis, is the most recognized adverse effect in infants exposed to lithium in utero.10 Its frequency is unknown, but rare. Neonatal hypothyroidism and nephrogenic diabetes insipidus have also been documented.
Anticonvulsants. To date, no studies have examined the outcomes of children whose mothers took anticonvulsants for bipolar disorder during pregnancy, though the research concerning epileptic mothers is extensive.
Neural tube defects. Data associate anticonvulsant exposure with a significantly greater risk for malformations than in the general population. Specifically, anticonvulsants may cause neural tube defects such as spina bifida, ancephaly, and encephaly in 2 to 5% of those exposed, as well as craniofacial anomalies, microcephaly, growth retardation, and heart defects.11-14
Table 2
FDA’s teratogenicity ratings of mood stabilizers and other antimanic agents
| Category | Medication | Teratogenicity |
|---|---|---|
| Mood stabilizers | Lithium Carbamazepine Valproate | Category D Category D Category D |
| Anticonvulsants | Gabapentin Lamotrigine Topiramate | Category C Category C Category C |
| Antipsychotics | Olanzapine Risperidone Chlorpromazine Haloperidol Trifluoperazine | Category C Category C Safety in pregnancy not known Category C Safety in pregnancy not known |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | ||
More minor malformations—such as rotated ears, depressed nasal bridge, short nose, elongated upper lip, and fingernail hypoplasia—have been reported in infants exposed to anticonvulsants in utero.14 These malformations disappear with age.13 Teratogenicity increases with the use of multiple anticonvulsants and possibly with higher maternal plasma levels and toxic metabolites.15
Conclusion. The three most commonly used mood stabilizers are all teratogenic. The least risk may occur with lithium (0.1%) versus valproate (2 to 5%) or carbamazepine (1 to 3%). These risks must be weighed against the up to 50% chance of relapse with medication discontinuation.3
ANTIPSYCHOTICS
Antipsychotics are often used to treat mania because of their rapid effects and sedative properties. Most antipsychotics—specifically, haloperidol, olanzapine, and risperidone—are labeled “C,” specifying that fetal risk cannot be ruled out.
Table 3
FDA’s teratogenicity ratings of common antidepressants
| Category | Medication | Teratogenicity |
|---|---|---|
| Tricyclics | Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline | Category C Category C Safety in pregnancy not known Safety in pregnancy not known Safety in pregnancy not known |
| Selective serotonin reuptake inhibitors | Citalopram Fluoxetine Fluvoxamine Paroxetine Sertraline | Category C Category C Category C Category C Category C |
| Other antidepressants | Bupropion Phenelzine Tranylcypromine | Category B Safety in pregnancy and nursing not known Safety in pregnancy and nursing not known |
| Source: Physicians’ Desk Reference. Montvale, NJ: Medical Economics Co., 2003. | ||
Chlorpromazine and haloperidol have been most studied during pregnancy but in relation to treating hyperemesis gravidarum and psychosis, not bipolar disorder. Results regarding antipsychotics’ teratogenic and behavioral risks are mixed,16-21 probably because the various compounds have different effects on the fetus.
The underlying illness—rather than the medications—may increase the rate of anomalies seen with exposure to antipsychotics:
- Rieder et al22 reported an increased rate of perinatal death in infants of schizophrenic mothers but no significant association between the mothers’ use of antipsychotics and perinatal death.
- Sobel23 compared psychotic women with and without histories of chlorpromazine exposure during pregnancy. Rates of fetal damage were similar and approximately twice that of the general population.
A meta-analysis of 74,337 live births revealed that first-trimester exposure to low-potency antipsychotics increases the relative risk of fetal anomalies in nonpsychotic women. Phenothiazines may increase the 2% baseline incidence of malformations to 2.4%.1 No specific organ malformation following fetal exposure to phenothiazines has been consistently identified.
Olanzapine was recently approved for treating mania. Very little data exist regarding its impact on fetal development when used during pregnancy, although studies on small numbers of women have not revealed teratogenicity.24,25
Conclusion. Psychotic illness itself may increase the risk of poor fetal outcome to a greater extent than does antipsychotic use. Prenatal exposure to low-potency phenothiazines may further increase this risk, although only slightly. The effect of prenatal exposure to atypical antipsychotics requires further study.
BENZODIAZEPINES
Benzodiazepines are rarely a primary treatment for mania or depression. Thus, a comprehensive review of their effect on fetal outcome is beyond the scope of this review. A meta-analysis of exposure in the first trimester suggests a very small but significant increase in risk for cleft palate.1 The absolute risk is <1 in 1,000 cases.
ANTIDEPRESSANTS
Whereas treatment of acute mania is considered a medical emergency, women with bipolar disorder may also relapse into depression during pregnancy. An antidepressant should not be used without a mood stabilizer when treating bipolar I disorder, although a mood stabilizer alone may be inadequate to treat depression. Using tricyclics and selective serotonin reuptake inhibitors (SSRIs) during pregnancy has not been associated with teratogenicity (Table 3),26 although perinatal effects have been reported.1
Tricyclics. In case-control studies involving more than 300,000 live births, 414 incidences of first-trimester exposure to tricyclics were followed. Information from these patients found no significant association between fetal exposure to tricyclics and increased rates of congenital malformations.1 The few studies that have been performed suggest no long-term effects from in utero exposure.26 Although these results suggest that prenatal exposure to tricyclics is relatively safe, more research is needed.
SSRIs. To date, no significant teratogenic effects of SSRIs have been identified in offspring of treated women.
The manufacturer’s register for fluoxetine contains approximately 2,000 cases of treated patients, with no excess cases of congenital anomalies or malformations following prenatal exposure. Citalopram has the next largest database of in utero exposure (n=365), again with no increased risk for teratogenicity. Several smaller systematic reports are available on in utero exposure to sertraline, paroxetine, or escitalopram.26
Most studies of pregnant women taking fluoxetine in the first trimester have found no increased risk of obstetrical complications—including spontaneous pregnancy loss, preterm labor, or low birth weight—compared with women not taking fluoxetine. Taking fluoxetine during the third trimester may increase the risk for perinatal complications,27 although this has been inconsistently reported and requires further study. Effects of other SSRIs in the third trimester have not been systematically explored.
Case reports and one controlled study have addressed possible neonatal perinatal symptoms from in utero exposure to SSRIs.28,29 Preliminary data show no adverse neurobehavioral function in exposed neonates.26
Electroconvulsive therapy (ECT) has been proven effective for acute mania and depression, demonstrating few deleterious effects on neonates. ECT has few side effects and may be safer than drug therapy in this population. Two reviews support the efficacy and relative safety of ECT treatment during pregnancy, although more evidence is needed.30,31
RECOMMENDATIONS
Discuss pregnancy and medication risks with all bipolar women, regardless of proximal plans for pregnancy. If psychotropic medication is used, prescribe carefully during the first trimester, using the minimum number of drugs and the lowest dosages needed to restore or maintain well-being.32
Pros and cons of switching. Some clinicians may encourage a patient to taper a medication during the first trimester because of its unknown or high teratogenicity. Depending on the patient’s illness severity, this might not be the optimal decision. A more conservative option would be to switch to a lower-risk drug during pregnancy.
Lithium has both antidepressant and antimanic properties and is less teratogenic compared with first-trimester exposure to an anticonvulsant. However, if lithium has not been successful for the woman’s mania prophylaxis in the past and she has demonstrated antimanic response to an anticonvulsant, switching to lithium or another anticonvulsant is not recommended.
Algorithm Suggested approach to the bipolar patient who wishes to conceive or is pregnant
Folate and neural tube defects. As first-trimester exposure to carbamazepine or valproate increases the risk for neural tube defects, using the lowest available dosage may decrease the risk for spina bifida, at least with valproate.
Low maternal folate levels are often associated with neural tube defects from any cause.33 Valproate lowers folate levels by inhibiting one of the enzymes necessary for its formation, which may be a mechanism for the increased risk of spina bifida.34
Folate supplementation. To date, no study has demonstrated that giving folate supplements to women taking anticonvulsants during pregnancy reduces the risk of neural tube defects.35 Nonetheless, we recommend that women who continue to take valproate or carbamazepine during pregnancy receive folate, 3 to 4 mg/d, as a precaution.
Treating manic relapse. Data show high rates of relapse in patients who stop taking lithium, particularly if done abruptly.3 Counsel women taking lithium to plan their pregnancies to allow enough time to taper off the medication prior to conception, if they want to try this. Lithium should be decreased slowly—approximately 50% every 2 weeks—to avoid relapse.
Treat aggressively if relapse occurs during pregnancy. Consider:
- psychiatric hospitalization in case of suicidality or psychosis
- reinstituting drug therapy with a less-teratogenic agent
- ECT for a manic or depressive episode.
As the pregnancy advances and the mother’s volume of distribution increases, dosage increases may be needed to maintain therapeutic drug levels.
Treating depressive relapse. Should depression occur in pregnancy, SSRIs or tricyclics added to mood stabilizer therapy have been shown to be effective, with few teratogenic effects.
Cognitive-behavioral and interpersonal psychotherapies also have shown efficacy in pregnant women with major depressive disorder36 and may be effective for women with bipolar disorder in pregnancy. Cognitive psychotherapies, when used with medication, have been reported effective in preventing relapse in nongravid bipolar patients.36-37
Related resources
Psychiatric disorders during pregnancy. Massachusetts General Hospital Center for Women’s Health. Perinatal Resource Center. www.womensmentalhealth.org/topics/pregnancy_lib.html
Drug brand names
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid et al
- Methylphenidate • Ritalin et al
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Phenelzine • Nardil
- Risperidone • Risperdal
- Sertraline • Zoloft
- Topiramate • Topamax
- Tranylcypromine • Parnate
- Trifluoperazine • Stelazine
- Valproate • Depakote et al
Disclosure
Dr. Altshuler receives research support from Abbott Laboratories, is a consultant to Abbott Laboratories, Forest Laboratories, and Eli Lilly & Co., and is a speaker for GlaxoSmithKline and Janssen Pharmaceutica.
Ms. Richards reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Yonkers receives research support from GlaxoSmithKline and Berlex Laboratories, is a consultant to GlaxoSmithKline, and is a speaker for Eli Lilly and Co., Pfizer Inc., GlaxoSmithKline, and Wyeth Pharmaceuticals.
1. Altshuler L, Cohen L, Szuba, et al. Pharmacologic management of psychiatric illness during pregnancy: dilemmas and guidelines. Am J Psychiatry 1996;153:592-606.
2. Nelson K, Holmes LB. Malformations due to presumed spontaneous mutations in newborn infants. N Engl J Med 1989;320:19-23.
3. Viguera AC, Nonacs R, Cohen LS, et al. Risk of recurrence of bipolar disorder in pregnant and nonpregnant women after discontinuing lithium maintenance. Am J Psychiatry 2000;157:179-84.
4. Steer RA, Scholl TO, Hediger ML, Fischer RL. Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol 1992;45(10):1093-9.
5. Orr ST, Miller CA. Maternal depressive symptoms and the risk of poor pregnancy outcome. Review of the literature and preliminary findings. Epidemiol Rev 1995;17(1):165-71.
6. Zuckerman B, Amaro H, Bauchner H, Cabral H. Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol 1989;160:1107-11.
7. Miller LJ. Psychotic denial of pregnancy: phenomenology and clinical management. Hosp Community Psychiatry 1990;41:1233-7.
8. Cohen LS, Friedman JM, Jefferson JW, et al. A reevaluation of risk of in utero exposure to lithium. JAMA 1994;271(2):146-50correction JAMA 1994;271(19):1485.
9. Schou M. What happened later to the lithium babies? A follow-up study of children born without malformations. Acta Psychiatr Scand 1976;54(3):193-7.
10. Woody JN, London WL, Wilbanks GD. Lithium toxicity in a newborn. Pediatrics 1971;47:94-6.
11. Jones K, Lacro R, Johnson K, Adams J. Patterns of malformations in the children of women treated with carbamazepine during pregnancy. N Engl J Med 1989;320:1661-6.
12. Rosa F. Spina bifida in infants of women treated with carbamazepine during pregnancy. N Engl J Med 1991;324(10):674-7.
13. Koch S, Losche G, Jager-Roman E, et al. Major and minor birth malformations and antiepileptic drugs. Neurology 1992;42:83-8.
14. Jager-Roman E, Deichl A, Jakob S, et al. Fetal growth, major malformations, and minor anomalies in infants born to women receiving valproic acid. J Pediatr 1986;108:997-1004.
15. Nakane Y, Okuma T, Takahashi R, et al. Multi-institutional study on the teratogenicity and fetal toxicity of antiepileptic drugs: a report of a collaborative study group in Japan. Epilepsia 1980;21:663-80.
16. Edlund MJ, Craig TJ. Antipsychotic drug use and birth defects: an epidemiologic reassessment. Compr Psychiatry 1984;25:32-8.
17. Kris EB. Children of mothers maintained on pharmacotherapy during pregnancy and postpartum. Curr Ther Res 1965;7:785-9.
18. Clark CVH, Gorman D, Vernadakis A. Effects of prenatal administration of psychotropic drugs on behavior of developing rats. Dev Psychobiol 1970;3:225-35.
19. Golub M, Kornetsky C. Seizure susceptibility and avoidance conditioning in adult rats treated prenatally with chlorpromazine. Dev Psychobiol 1974;7:79-88.
20. Spear LP, Shalaby IA, Brick J. Chronic administration of haloperidol during development: behavioral and psychopharmacological effects. Psychopharmacology (Berl) 1980;70:47-58.
21. Cagiano R, Barfield RJ, White NR, et al. Subtle behavioral changes produced in rat pups exposed in utero to haloperidol. Eur J Pharmacol 1988;157:45-50.
22. Rieder RO, Rosenthal D, Wender P, Blumenthal H. The offspring of schizophrenics: fetal and neonatal deaths. Arch Gen Psychiatry 1975;32:200-11.
23. Sobel DE. Fetal damage due to ECT, insulin coma, chlorpromazine, or reserpine. Arch Gen Psychiatry 1960;2:606-11.
24. Dickson R. Olanzapine and pregnancy. Can J Psychiatry 1998;43:196-7.
25. Goldstein DJ, Corbin LA, Fung MC. Olanzapine-exposed pregnancies and lactation; early experience. J Clin Psychopharmacol 2000;24(4):399-403.
26. Altshuler LL, Cohen LS, Moline ML, et al. The expert consensus guideline series: treatment of depression in women. Postgrad Med 2001 Mar;(Spec No):1-22.
27. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010-15.
28. Spencer MJ. Fluoxetine hydrochloride (Prozac) toxicity in the neonate. Pediatrics 1993;92:721-2.
29. Cabrera FM, Battaglia G. Delayed decreases in brain 5-HT 2a and 2c receptor density and function in male rat progeny following prenatal fluoxetine. J Pharmacol Exp Ther 1994;269:637-45.
30. Miller LJ. Use of electroconvulsive therapy during pregnancy. Hosp Community Psychiatry 1994;45:444-50.
31. Ferrill MJ, Kehoe WA, Jacisin JJ. ECT during pregnancy: physiologic and pharmacologic considerations. Convuls Ther 1992;8:186-200.
32. Yonkers K, Wisner K, Cohen L, et al. Management of bipolar disorder during pregnancy and the postpartum period. Bipolar Consensus Statement. Submitted for publication.
33. Dansky L, Rosenblatt D, Andermann E. Mechanisms of teratogenesis: folic acid and antiepileptic therapy. Neurology 1992;42(suppl 5):32-42.
34. Wegner C, Nau H. Alteration of embryonic folate metabolism by valproic acid during organogenesis: implications for mechanism of teratogenesis. Neurology 1992;42(suppl 5):17-24.
35. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 1991;338:131-7.
36. Spinelli MG, Endicott J. Controlled clinical trial of interpersonal psychotherapy versus parenting education program for depressed pregnant women. Am J Psychiatry 2003;160:555-62.
37. Lam DH, Watkins ER, Hayward P, et al. A randomized controlled study of cognitive therapy for relapse prevention for bipolar affective disorder: outcome in the first year. Arch Gen Psychiatry 2003;60(2):145-52.
1. Altshuler L, Cohen L, Szuba, et al. Pharmacologic management of psychiatric illness during pregnancy: dilemmas and guidelines. Am J Psychiatry 1996;153:592-606.
2. Nelson K, Holmes LB. Malformations due to presumed spontaneous mutations in newborn infants. N Engl J Med 1989;320:19-23.
3. Viguera AC, Nonacs R, Cohen LS, et al. Risk of recurrence of bipolar disorder in pregnant and nonpregnant women after discontinuing lithium maintenance. Am J Psychiatry 2000;157:179-84.
4. Steer RA, Scholl TO, Hediger ML, Fischer RL. Self-reported depression and negative pregnancy outcomes. J Clin Epidemiol 1992;45(10):1093-9.
5. Orr ST, Miller CA. Maternal depressive symptoms and the risk of poor pregnancy outcome. Review of the literature and preliminary findings. Epidemiol Rev 1995;17(1):165-71.
6. Zuckerman B, Amaro H, Bauchner H, Cabral H. Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol 1989;160:1107-11.
7. Miller LJ. Psychotic denial of pregnancy: phenomenology and clinical management. Hosp Community Psychiatry 1990;41:1233-7.
8. Cohen LS, Friedman JM, Jefferson JW, et al. A reevaluation of risk of in utero exposure to lithium. JAMA 1994;271(2):146-50correction JAMA 1994;271(19):1485.
9. Schou M. What happened later to the lithium babies? A follow-up study of children born without malformations. Acta Psychiatr Scand 1976;54(3):193-7.
10. Woody JN, London WL, Wilbanks GD. Lithium toxicity in a newborn. Pediatrics 1971;47:94-6.
11. Jones K, Lacro R, Johnson K, Adams J. Patterns of malformations in the children of women treated with carbamazepine during pregnancy. N Engl J Med 1989;320:1661-6.
12. Rosa F. Spina bifida in infants of women treated with carbamazepine during pregnancy. N Engl J Med 1991;324(10):674-7.
13. Koch S, Losche G, Jager-Roman E, et al. Major and minor birth malformations and antiepileptic drugs. Neurology 1992;42:83-8.
14. Jager-Roman E, Deichl A, Jakob S, et al. Fetal growth, major malformations, and minor anomalies in infants born to women receiving valproic acid. J Pediatr 1986;108:997-1004.
15. Nakane Y, Okuma T, Takahashi R, et al. Multi-institutional study on the teratogenicity and fetal toxicity of antiepileptic drugs: a report of a collaborative study group in Japan. Epilepsia 1980;21:663-80.
16. Edlund MJ, Craig TJ. Antipsychotic drug use and birth defects: an epidemiologic reassessment. Compr Psychiatry 1984;25:32-8.
17. Kris EB. Children of mothers maintained on pharmacotherapy during pregnancy and postpartum. Curr Ther Res 1965;7:785-9.
18. Clark CVH, Gorman D, Vernadakis A. Effects of prenatal administration of psychotropic drugs on behavior of developing rats. Dev Psychobiol 1970;3:225-35.
19. Golub M, Kornetsky C. Seizure susceptibility and avoidance conditioning in adult rats treated prenatally with chlorpromazine. Dev Psychobiol 1974;7:79-88.
20. Spear LP, Shalaby IA, Brick J. Chronic administration of haloperidol during development: behavioral and psychopharmacological effects. Psychopharmacology (Berl) 1980;70:47-58.
21. Cagiano R, Barfield RJ, White NR, et al. Subtle behavioral changes produced in rat pups exposed in utero to haloperidol. Eur J Pharmacol 1988;157:45-50.
22. Rieder RO, Rosenthal D, Wender P, Blumenthal H. The offspring of schizophrenics: fetal and neonatal deaths. Arch Gen Psychiatry 1975;32:200-11.
23. Sobel DE. Fetal damage due to ECT, insulin coma, chlorpromazine, or reserpine. Arch Gen Psychiatry 1960;2:606-11.
24. Dickson R. Olanzapine and pregnancy. Can J Psychiatry 1998;43:196-7.
25. Goldstein DJ, Corbin LA, Fung MC. Olanzapine-exposed pregnancies and lactation; early experience. J Clin Psychopharmacol 2000;24(4):399-403.
26. Altshuler LL, Cohen LS, Moline ML, et al. The expert consensus guideline series: treatment of depression in women. Postgrad Med 2001 Mar;(Spec No):1-22.
27. Chambers CD, Johnson KA, Dick LM, et al. Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010-15.
28. Spencer MJ. Fluoxetine hydrochloride (Prozac) toxicity in the neonate. Pediatrics 1993;92:721-2.
29. Cabrera FM, Battaglia G. Delayed decreases in brain 5-HT 2a and 2c receptor density and function in male rat progeny following prenatal fluoxetine. J Pharmacol Exp Ther 1994;269:637-45.
30. Miller LJ. Use of electroconvulsive therapy during pregnancy. Hosp Community Psychiatry 1994;45:444-50.
31. Ferrill MJ, Kehoe WA, Jacisin JJ. ECT during pregnancy: physiologic and pharmacologic considerations. Convuls Ther 1992;8:186-200.
32. Yonkers K, Wisner K, Cohen L, et al. Management of bipolar disorder during pregnancy and the postpartum period. Bipolar Consensus Statement. Submitted for publication.
33. Dansky L, Rosenblatt D, Andermann E. Mechanisms of teratogenesis: folic acid and antiepileptic therapy. Neurology 1992;42(suppl 5):32-42.
34. Wegner C, Nau H. Alteration of embryonic folate metabolism by valproic acid during organogenesis: implications for mechanism of teratogenesis. Neurology 1992;42(suppl 5):17-24.
35. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 1991;338:131-7.
36. Spinelli MG, Endicott J. Controlled clinical trial of interpersonal psychotherapy versus parenting education program for depressed pregnant women. Am J Psychiatry 2003;160:555-62.
37. Lam DH, Watkins ER, Hayward P, et al. A randomized controlled study of cognitive therapy for relapse prevention for bipolar affective disorder: outcome in the first year. Arch Gen Psychiatry 2003;60(2):145-52.
Intermittent explosive disorder: Taming temper tantrums in the volatile, impulsive adult
Mr. P, age 41, has a “problem with anger.” Since age 17, he has had sudden outbursts of screaming and shouting, with occasional minor damage to objects. These outbursts—including episodes of “road rage”—occur once or more per week and almost daily for months at a time.
Mr. P has also had more violent episodes— sometimes every 2 to 3 months—in which he has punched holes in walls, destroyed a computer with a hammer, and assaulted other people with his fists. These events are not premeditated and are typically triggered by Mr. P’s frustration at not being “perfect” or by others breaking what he considers “general rules of conduct.”
The day before his initial visit, while he was stuck in traffic, Mr. P saw a car speeding down the shoulder. Enraged, he pulled in front of the car so that the driver had to slam on the brakes. He jumped out of his car and approached the other driver, shouting obscenities. The other driver locked her door and tried to ignore Mr. P until he returned to his car. Mr. P noted that this episode “ruined” his day because of his lingering anger and irritability.
Intermittent explosive disorder (IED) is more common and complex than was once thought, based on recent evidence. Recurrent, problematic, impulsive aggression is highly comorbid with other psychiatric conditions—including mood and personality disorders—and undermines social relationships and job performance. Typical characteristics of IED are outlined in Table 1.1-3
Table 1
Typical characteristics of intermittent explosive disorder
| Onset in childhood or adolescence (mean age 15), with average duration ±20 years |
| Aggressive outbursts: |
|
| Some episodes may appear without identifiable provocation |
| Male to female ratio 3:1, although some data suggest gender parity |
| Source: Adapted from references 1-3 |
This article offers updated diagnostic criteria and a two-pronged algorithm that can help you diagnose and treat this aggression disorder.
HOW COMMON IS IED?
DSM-IV states that IED is “apparently rare.” This statement is far from surprising, given the limitations of DSM criteria. Surveys of hospitalized patients in the 1980s found that only 1.1% met DSM-III criteria for IED.4 In another study of more than 400 patients seeking treatment for aggression, only 1.8% met DSM-III criteria for IED (although far more would likely have met DSM-IV criteria).5
A more recent survey of 411 psychiatric outpatients6 found that 3.8% met current and 6.2% met lifetime DSM-IV criteria for IED, using the Structured Clinical Interview for DSM-IV Diagnoses (SCID). Reanalysis of a threefold larger data set from the same study site (Coccaro and Zimmerman, unpublished) yielded the same result.
Far from rare. More recently, our findings from a small sample suggested that the community rate of lifetime IED is about 4% by DSM-IV criteria and 5% by research criteria. In the United States, we estimate that the lifetime rate of IED could be 4.5 to 18 million persons using DSM-IV criteria or 6.7 to 22.2 million using IED research criteria. If so, IED is at least as common as other major psychiatric disorders, including schizophrenia or bipolar illness. The ongoing National Comorbidity Study is expected to produce more definitive community data.
PSYCHIATRIC COMORBIDITY
Axis I disorders. IED is highly comorbid with mood, anxiety, and substance use disorders,3,7,8 although no causal relationship has been shown
Mood and substance abuse disorders. IED’s age of onset may precede that of mood and substance use disorders, according to analysis of our unpublished data. If so, comorbid IED may not occur in the context of mood or substance use disorders.
Anxiety disorders. We have noted a similar pattern with IED and anxiety disorders, although phobic anxiety disorders (simple or social phobia) tend to manifest earlier than IED. This suggests that early-onset phobic anxiety might be associated with an increased risk of IED in adolescence or young adulthood.
Bipolar disorder. McElroy9 has suggested a relationship between IED and bipolar disorder. In some samples, as many as one-half of IED patients (56%) have comorbid bipolar disorder when one includes bipolar II and cyclothymia.3 Moreover, some subjects’ aggressive episodes appear to resemble “microdysphoric manic episodes.”9 Other studies,8 however, find a much lower rate (10% or less) of IED comorbidity with bipolar illness.
Bipolar disorder overall may not be highly comorbid with IED, although rates may be higher in specialty clinic samples. In individuals with any kind of bipolar disorder, mood stabilizers— rather than selective serotonin reuptake inhibitors (SSRIs)—are probably the better choice as first-line treatment of IED.9
Axis II disorders. DSM-IV allows IED diagnosis in individuals with borderline or antisocial personality disorder, as long as these cluster B disorders do not better explain the aggressive behavior. How a clinician makes this distinction is not clear; in fact, most clinicians do not diagnose IED in patients with personality disorders, regardless of the clinical picture.
IED comorbidity with borderline or antisocial personality disorders varies with the sample. Persons with personality disorders who seek treatment of aggressive behavior are more likely to have comorbid IED (90%) than those not seeking treatment who are outpatients (50%) or in the community (25%).1,7
Individuals with personality disorders and IED score higher in aggression and lower in psychosocial function than do similar individuals without IED,7 indicating that the additional diagnosis is relevant.
Case report continued.
Mr. P’s outbursts have cost him several friendships, including romantic relationships. He has never advanced at work because he is seen as too volatile to supervise subordinates. Though some of Mr. P’s aggressive outbursts have occurred under the influence of alcohol, most are not related to alcohol or drug use. He has no medical problems and no other psychiatric history.
A full diagnostic evaluation uncovers a personality disorder, not otherwise specified (eight scattered traits from obsessive-compulsive personality disorder and from each of the cluster B personality disorders), and no Axis I condition other than intermittent explosive disorder.
PROBLEMS DEFINING IED
Intermittent explosive disorder is the only DSM diagnosis that applies to persons with histories of recurrent, problematic aggression not caused by another mental or physical disorder. Even so, little research on IED is available. DSM criteria for IED are poorly operationalized and have improved only modestly since the diagnosis was first included in DSM-III. In that revision, IED had four criteria.
“A” criteria specified recurrent outbursts of “seriously assaultive or destructive behavior,” but left unanswered important questions such as:
- What behavior crosses the threshold for seriously” assaultive or destructive?
- Does any physical assault qualify, or only those that cause physical injury (or stigmata)?
- How often or within what time must the behavior occur?
The phrase “recurrent acts of aggression” suggested that at least three acts of aggression were required to reach the threshold, but DSM-III provided no guidelines.
“B” criteria stated that the aggression should be out of proportion to the provocation. But how should one judge this criterion, when provocative stimuli sometimes are clearly sufficient to prompt a justifiably aggressive act?
“C” criteria excluded persons who are aggressive or impulsive between ill-defined “aggressive episodes.” This exclusion was especially limiting because individuals with recurrent, problematic, aggressive behaviors generally are impulsive and aggressive between more-severe outbursts. Excluding those who otherwise met diagnostic criteria for IED led to a spuriously low prevalence rate and limited the number of research subjects. DSM-IV eliminated this criterion but made no other notable changes in IED criteria.
“D” criteria in DSM-III and III-R further restricted the number of individuals who could meet this diagnosis:
- In DSM-III, antisocial personality disorder excluded the diagnosis of IED.
- In DSM-III-R, borderline personality disorder was added as an exclusionary factor.
Because of these restrictions, very few clinically valid cases of IED (individuals meeting A and B criteria) could receive an IED diagnosis.10
EVOLVING DIAGNOSTIC CRITERIA
By the early 1990s, DSM diagnostic criteria clearly severely restricted the study of recurrent, problematic aggression, even though research since DSM-III had greatly advanced our understanding of human aggression. For example, data linked impulsive aggression to deficits in central serotonergic function and suggested that agents that enhance serotonergic activity could modify this behavior.
Some investigators proposed research criteria for IED (IED-R) so that individuals with recurrent, problematic, impulsive aggression could be identified and studied. Research criteria first published in 19987 proposed six changes/clarifications in IED diagnostic criteria:
Lower-intensity aggression. The scope of aggressive behavior was expanded to include verbal and indirect physical aggression, provided that these behaviors are associated with distress and/or impairment. Data from double-blind, placebo-controlled trials indicated that these lower-intensity (although usually higher frequency) behaviors respond well to treatment with SSRIs.11,12
Impulsivity. The aggression was specified as impulsive. This change identified individuals with greater liability for deficits in central serotonergic function and excluded individuals with premeditated or criminal aggression.
A minimal frequency of aggression over time was proposed to make the IED diagnosis more reliable and to ensure that persons with only occasional impulsive aggressive outbursts (especially of low severity) were given this diagnosis.
Subjective distress (in the individual) and/or social or occupational dysfunction was proposed so that putatively aggressive individuals are not diagnosed for manifesting behaviors that are not functionally severe.
Diagnostic exclusionary criteria were modified so that individuals with:
- antisocial or borderline personality disorder could be diagnosed with IED if otherwise warranted
- aggressive behaviors confined within major depression episodes could not be diagnosed with IED.
This last change recognized that impulsive, aggressive outbursts could point to major depressive disorder.
When the revised criteria were tested in patients seeking treatment for aggression, those who met IED-R criteria were found to exhibit significantly greater aggression and impulsivity (using validated scales) and lower global functioning than those who did not.7 Statistical adjustments made to account for aggression score differences eliminated the difference in global functioning, which suggested a direct link between aggression and global function in individuals with IED-R.
Two patterns. Later research uncovered at least patterns of aggressive outbursts:
- low intensity at high frequency (such as verbal arguments or door slamming approximately twice weekly)
- high intensity at low frequency (such as physical aggression resulting in injury or destruction of nontrivial property at least three times per year).
Data revealed that 69% of individuals with IED-like histories displayed both aggression patterns, 20% displayed only the high-intensity/low-frequency pattern, and 11% displayed only the low-intensity/high-frequency pattern.
Because further analysis revealed no important differences between these groups in measures of aggression and impulsivity, IED-R criteria were revised to include both patterns in the “A” criteria. This revision integrated the essences of IED-R and DSM criteria into one diagnostic set (Table 2).
INFLUENCE OF HEREDITY
No twin or adoption studies of IED have been performed. However, family history data suggest that IED (or IED-type behavior) is familial. I recently conducted a blinded, controlled, family history study using IED-R criteria and found a significantly elevated risk for IED (p < 0.01) in relatives of persons with a history of IED (26%), compared with non-IED controls (8%). Comorbid conditions did not affect the risk among the IED subjects or their relatives, suggesting that IED is familial and independent of other conditions.13
Nearly all studies of aggression’s biology and treatment have measured aggression as a dimensional variable along a continuous scale from low to high.14 Our studies have allowed us to explore biological and treatment response correlates. In preliminary analyses, we have found that the maximal prolactin response to d-fenfluramine challenge and the number of platelet serotonin transporter binding sites are:
- reduced in subjects meeting research criteria for IED
- inversely correlated with dimensional measures of impulsive aggression.
Table 2
Updated diagnostic criteria for intermittent explosive disorder
| A. Recurrent incidents of aggression manifest as either: |
| 1. Verbal or physical aggression towards other people, animals, or property occurring twice weekly on average for 1 month |
| OR |
| 2. Three episodes involving physical assault against other people or destruction of property over a 1-year period |
| B. The degree of aggressiveness expressed is grossly out of proportion to the provocation or any precipitating psychosocial stressors |
| C. The aggressive behavior is generally not premeditated (ie, is impulsive) and is not committed to achieve a tangible objective (such as money, power, intimidation, etc.) |
| D. The aggressive behavior causes marked distress in the individual or impairs occupational or interpersonal functioning |
| E. The aggressive behavior is not better explained by another mental disorder (such as a major depressive/manic/psychotic disorder, attention-deficit/hyperactivity disorder, general medical condition [head trauma, Alzheimer’s disease], or due to the direct physiologic effects of a substance) |
| Source: Adapted from reference 7 |
Earlier, Virkkunen et al15 reported reduced cerebrospinal fluid 5-hydroxyindoleacetic acid concentrations in persons diagnosed with IED based on DSM-III criteria, compared with persons who were not diagnosed with IED and those who demonstrated nonimpulsive aggression.
TREATING IED
Cognitive therapy. Few double-blind, randomized, placebo-controlled trials of any treatments for IED have been published. Trials using cognitive-behavioral approaches have reduced self-rated anger and its expression in young adults with anger disorders.16 Although many of these subjects may have had IED, it is not known if this approach works in IED.
Table 3
Characteristic behaviors of aggressive individuals*
| Severity | Behaviors |
|---|---|
| Mildly aggressive | Occasional verbal arguments and/or temper tantrums |
| Moderately aggressive | Frequent verbal arguments and temper tantrums (about twice weekly on average), occasional destruction of property, rare or occasional physical assault against others (usually without injury) |
| Highly aggressive | Frequent verbal arguments and temper tantrums (about twice weekly) and/or more than occasional destruction of property or physical assault against others, sometimes with injury |
| * Characteristics given are descriptive and not based on data. | |
Drug therapy. SSRIs. A trial by this author using fluoxetine showed that impulsive aggressive behavior responds to treatment that targets the central serotonergic system.12 Forty subjects with personality disorders and histories of impulsive aggression received fluoxetine, 20 to 60 mg qd, or placebo for 12 weeks. Fluoxetine reduced overt aggression and irritability about 67% more than placebo, as assessed by the Overt Aggression Scale Modified for Outpatients (OAS-M).
All subjects met research criteria for IED. A reanalysis suggests that SSRIs may be most effective in moderately aggressive patients (Table 3),17 whose serotonergic system may be less impaired than that of highly aggressive patients.18
Mood stabilizers. Impulsively aggressive subjects who do not respond to an SSRI may respond to a mood stabilizer.19 An antiaggressive response in IED-like subjects has been reported for lithium,20 carbamazepine,21 and diphenylhydantoin.22
Recently, Hollander et al23 reported greater reduction in overt aggression scores in IED subjects with a DSM cluster B personality disorder who were treated with divalproex, compared with placebo. This study used the same design and outcome measure as our study12 and included subjects who met both DSM-IV and research criteria for IED.
For unknown reasons, divalproex was no more effective than placebo in IED subjects without cluster B personality disorder. More research is needed to uncover predictors of antiaggressive response in IED subjects.
Unipolar vs. bipolar. McElroy9 has suggested using SSRIs (or other antidepressants) as first-line treatment for IED subjects with unipolar affective symptoms and mood stabilizers for those with bipolar affective symptoms. IED subjects without bipolar affective symptoms should be treated first with SSRIs (Algorithm). Preliminary data suggest a role for atypical antipsychotics to treat aggressive behavior in patients with schizophrenia or bipolar disorder, but no empiric data exist.
Beta blockers such as propranolol also may be considered.2 However, beta blockers are more difficult to dose and are associated with more burdensome side effects, compared with SSRIs.
Algorithm Suggested 2-pronged approach for treating intermittent explosive disorder
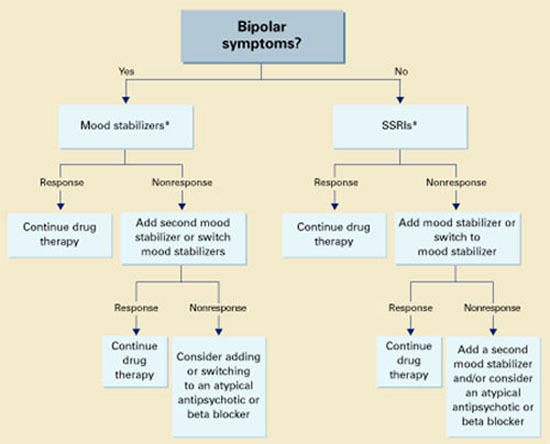
* With or without an anger management program, which may precede drug interventionThe full effects of antiaggressive treatment with an SSRI (E. Coccaro, unpublished observations) or a mood stabilizer19 may take 3 months to observe12,20,22,23 and tend to disappear soon after treatment is discontinued.
Therefore, an adequate trial of SSRIs or mood stabilizers is no less than 3 months. If improvement is seen, continue drug treatment indefinitely.
Case report continued.
Mr. P was started on an SSRI. His aggressive outbursts decreased in intensity and frequency over 3 months but were not eliminated. After 6 months he dropped out of treatment, but returned 5 weeks later because his aggressive outbursts had resumed their pre-treatment level.
SSRI treatment was restarted, and Mr. P began a 12-week anger management course of relaxation training, cognitive restructuring, and coping skills training. He gained greater control over his aggressive outbursts and continues monthly medication checks and anger management “booster sessions.”
Related resources
- Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
- Olvera RL. Intermittent explosive disorder: epidemiology, diagnosis and management. CNS Drugs 2002;16:517-26.
Drug brand names
- Carbamazepine • Tegretol
- Diphenylhydantoin • Dilantin
- Divalproex • Depakote
- Fluoxetine • Prozac
- Lithium • Lithobid
- Propanolol • Inderal
Disclosure
Dr. Coccaro reports that he receives research grants and serves on the speaker’s bureau or as a consultant to Eli Lilly and Co., Abbott Laboratories, GlaxoSmithKline, and Forrest Laboratories.
1. Coccaro EF, Schimdt CA, Samuels JF, et al. Lifetime rates of intermittent explosive disorder in a community sample (abstract). Philadelphia: American Psychiatric Association annual meeting, 2002.
2. Mattes JA. Comparative effectiveness of carbamazepine and propranolol for rage outbursts. J Neuropsychiatry Clin Neurosci 1990;2:159-64.
3. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry 1998;59:203-10.
4. Monopolis S, Lion JR. Problems in the diagnosis of intermittent explosive disorder. Am J Psychiatry 1983;140:1200-2.
5. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (abstract 265). Washington, DC: American Psychiatric Association annual meeting, 1998.
6. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (APA new research abstracts #265). Washington, DC: American Psychiatric Publishing, Inc., 1998.
7. Coccaro EF, Kavoussi RJ, Berman ME, Lish JD. Intermittent explosive disorder-revised: development, reliability and validity of research criteria. Compr Psychiatry 1998;39:368-76.
8. Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
9. McElroy SL. Recognition and treatment of DSM-IV intermittent explosive disorder. J Clin Psychiatry 1999;60(suppl 15):12-16.
10. Felthous AR, Bryant G, Wingerter CB, Barratt E. The diagnosis of intermittent explosive disorder in violent men. Bull Am Acad Psychiatry Law 1991;19:71-9.
11. Salzman C, Wolfson AN, Schatzberg A, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. J Clin Psychopharmacology 1995;15:23-9.
12. Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality disordered subjects. Arch Gen Psychiatry 1997;54:1081-8.
13. Coccaro EF. Family history study of intermittent explosive disorder (abstract). Washington, DC: American Psychiatric Association annual meeting, 1999.
14. Coccaro EF, Siever LJ. Pathophysiology and treatment of aggression. In: Davis KL, Charney D, Coyle JT, Nemeroff D (eds). Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002;1709-24
15. Virkkunen M, Rawlings R, Tokola R, et al. CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry 1994;51:20-7.
16. Deffenbacher JL. Psychosocial interventions: anger disorders. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
17. Lee R, Coccaro EF. Treatment of aggression: serotonergic agents. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
18. Coccaro EF, Kavoussi RJ, Hauger RL. Serotonin function and antiaggressive responses to fluoxetine: a pilot study. Biol Psychiatry 1997;42:546-52.
19. Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. J Clin Psychiatry 1998;59:676-80.
20. Sheard MH, Marini J, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry 1976;133:1409-13.
21. Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder: alprazolam, carbamazepine, trifluroperazine, and tranylcypromine. Arch Gen Psychiatry 1988;45:111-19.
22. Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacology 1997;17:341-9.
23. Hollander E, Tracy KA, Swann AC, et al. Divalproex sodium is superior to placebo for impulsive aggression in Cluster B personality disorders. Neuropsychopharmacology 2003;28:1186-97.
Mr. P, age 41, has a “problem with anger.” Since age 17, he has had sudden outbursts of screaming and shouting, with occasional minor damage to objects. These outbursts—including episodes of “road rage”—occur once or more per week and almost daily for months at a time.
Mr. P has also had more violent episodes— sometimes every 2 to 3 months—in which he has punched holes in walls, destroyed a computer with a hammer, and assaulted other people with his fists. These events are not premeditated and are typically triggered by Mr. P’s frustration at not being “perfect” or by others breaking what he considers “general rules of conduct.”
The day before his initial visit, while he was stuck in traffic, Mr. P saw a car speeding down the shoulder. Enraged, he pulled in front of the car so that the driver had to slam on the brakes. He jumped out of his car and approached the other driver, shouting obscenities. The other driver locked her door and tried to ignore Mr. P until he returned to his car. Mr. P noted that this episode “ruined” his day because of his lingering anger and irritability.
Intermittent explosive disorder (IED) is more common and complex than was once thought, based on recent evidence. Recurrent, problematic, impulsive aggression is highly comorbid with other psychiatric conditions—including mood and personality disorders—and undermines social relationships and job performance. Typical characteristics of IED are outlined in Table 1.1-3
Table 1
Typical characteristics of intermittent explosive disorder
| Onset in childhood or adolescence (mean age 15), with average duration ±20 years |
| Aggressive outbursts: |
|
| Some episodes may appear without identifiable provocation |
| Male to female ratio 3:1, although some data suggest gender parity |
| Source: Adapted from references 1-3 |
This article offers updated diagnostic criteria and a two-pronged algorithm that can help you diagnose and treat this aggression disorder.
HOW COMMON IS IED?
DSM-IV states that IED is “apparently rare.” This statement is far from surprising, given the limitations of DSM criteria. Surveys of hospitalized patients in the 1980s found that only 1.1% met DSM-III criteria for IED.4 In another study of more than 400 patients seeking treatment for aggression, only 1.8% met DSM-III criteria for IED (although far more would likely have met DSM-IV criteria).5
A more recent survey of 411 psychiatric outpatients6 found that 3.8% met current and 6.2% met lifetime DSM-IV criteria for IED, using the Structured Clinical Interview for DSM-IV Diagnoses (SCID). Reanalysis of a threefold larger data set from the same study site (Coccaro and Zimmerman, unpublished) yielded the same result.
Far from rare. More recently, our findings from a small sample suggested that the community rate of lifetime IED is about 4% by DSM-IV criteria and 5% by research criteria. In the United States, we estimate that the lifetime rate of IED could be 4.5 to 18 million persons using DSM-IV criteria or 6.7 to 22.2 million using IED research criteria. If so, IED is at least as common as other major psychiatric disorders, including schizophrenia or bipolar illness. The ongoing National Comorbidity Study is expected to produce more definitive community data.
PSYCHIATRIC COMORBIDITY
Axis I disorders. IED is highly comorbid with mood, anxiety, and substance use disorders,3,7,8 although no causal relationship has been shown
Mood and substance abuse disorders. IED’s age of onset may precede that of mood and substance use disorders, according to analysis of our unpublished data. If so, comorbid IED may not occur in the context of mood or substance use disorders.
Anxiety disorders. We have noted a similar pattern with IED and anxiety disorders, although phobic anxiety disorders (simple or social phobia) tend to manifest earlier than IED. This suggests that early-onset phobic anxiety might be associated with an increased risk of IED in adolescence or young adulthood.
Bipolar disorder. McElroy9 has suggested a relationship between IED and bipolar disorder. In some samples, as many as one-half of IED patients (56%) have comorbid bipolar disorder when one includes bipolar II and cyclothymia.3 Moreover, some subjects’ aggressive episodes appear to resemble “microdysphoric manic episodes.”9 Other studies,8 however, find a much lower rate (10% or less) of IED comorbidity with bipolar illness.
Bipolar disorder overall may not be highly comorbid with IED, although rates may be higher in specialty clinic samples. In individuals with any kind of bipolar disorder, mood stabilizers— rather than selective serotonin reuptake inhibitors (SSRIs)—are probably the better choice as first-line treatment of IED.9
Axis II disorders. DSM-IV allows IED diagnosis in individuals with borderline or antisocial personality disorder, as long as these cluster B disorders do not better explain the aggressive behavior. How a clinician makes this distinction is not clear; in fact, most clinicians do not diagnose IED in patients with personality disorders, regardless of the clinical picture.
IED comorbidity with borderline or antisocial personality disorders varies with the sample. Persons with personality disorders who seek treatment of aggressive behavior are more likely to have comorbid IED (90%) than those not seeking treatment who are outpatients (50%) or in the community (25%).1,7
Individuals with personality disorders and IED score higher in aggression and lower in psychosocial function than do similar individuals without IED,7 indicating that the additional diagnosis is relevant.
Case report continued.
Mr. P’s outbursts have cost him several friendships, including romantic relationships. He has never advanced at work because he is seen as too volatile to supervise subordinates. Though some of Mr. P’s aggressive outbursts have occurred under the influence of alcohol, most are not related to alcohol or drug use. He has no medical problems and no other psychiatric history.
A full diagnostic evaluation uncovers a personality disorder, not otherwise specified (eight scattered traits from obsessive-compulsive personality disorder and from each of the cluster B personality disorders), and no Axis I condition other than intermittent explosive disorder.
PROBLEMS DEFINING IED
Intermittent explosive disorder is the only DSM diagnosis that applies to persons with histories of recurrent, problematic aggression not caused by another mental or physical disorder. Even so, little research on IED is available. DSM criteria for IED are poorly operationalized and have improved only modestly since the diagnosis was first included in DSM-III. In that revision, IED had four criteria.
“A” criteria specified recurrent outbursts of “seriously assaultive or destructive behavior,” but left unanswered important questions such as:
- What behavior crosses the threshold for seriously” assaultive or destructive?
- Does any physical assault qualify, or only those that cause physical injury (or stigmata)?
- How often or within what time must the behavior occur?
The phrase “recurrent acts of aggression” suggested that at least three acts of aggression were required to reach the threshold, but DSM-III provided no guidelines.
“B” criteria stated that the aggression should be out of proportion to the provocation. But how should one judge this criterion, when provocative stimuli sometimes are clearly sufficient to prompt a justifiably aggressive act?
“C” criteria excluded persons who are aggressive or impulsive between ill-defined “aggressive episodes.” This exclusion was especially limiting because individuals with recurrent, problematic, aggressive behaviors generally are impulsive and aggressive between more-severe outbursts. Excluding those who otherwise met diagnostic criteria for IED led to a spuriously low prevalence rate and limited the number of research subjects. DSM-IV eliminated this criterion but made no other notable changes in IED criteria.
“D” criteria in DSM-III and III-R further restricted the number of individuals who could meet this diagnosis:
- In DSM-III, antisocial personality disorder excluded the diagnosis of IED.
- In DSM-III-R, borderline personality disorder was added as an exclusionary factor.
Because of these restrictions, very few clinically valid cases of IED (individuals meeting A and B criteria) could receive an IED diagnosis.10
EVOLVING DIAGNOSTIC CRITERIA
By the early 1990s, DSM diagnostic criteria clearly severely restricted the study of recurrent, problematic aggression, even though research since DSM-III had greatly advanced our understanding of human aggression. For example, data linked impulsive aggression to deficits in central serotonergic function and suggested that agents that enhance serotonergic activity could modify this behavior.
Some investigators proposed research criteria for IED (IED-R) so that individuals with recurrent, problematic, impulsive aggression could be identified and studied. Research criteria first published in 19987 proposed six changes/clarifications in IED diagnostic criteria:
Lower-intensity aggression. The scope of aggressive behavior was expanded to include verbal and indirect physical aggression, provided that these behaviors are associated with distress and/or impairment. Data from double-blind, placebo-controlled trials indicated that these lower-intensity (although usually higher frequency) behaviors respond well to treatment with SSRIs.11,12
Impulsivity. The aggression was specified as impulsive. This change identified individuals with greater liability for deficits in central serotonergic function and excluded individuals with premeditated or criminal aggression.
A minimal frequency of aggression over time was proposed to make the IED diagnosis more reliable and to ensure that persons with only occasional impulsive aggressive outbursts (especially of low severity) were given this diagnosis.
Subjective distress (in the individual) and/or social or occupational dysfunction was proposed so that putatively aggressive individuals are not diagnosed for manifesting behaviors that are not functionally severe.
Diagnostic exclusionary criteria were modified so that individuals with:
- antisocial or borderline personality disorder could be diagnosed with IED if otherwise warranted
- aggressive behaviors confined within major depression episodes could not be diagnosed with IED.
This last change recognized that impulsive, aggressive outbursts could point to major depressive disorder.
When the revised criteria were tested in patients seeking treatment for aggression, those who met IED-R criteria were found to exhibit significantly greater aggression and impulsivity (using validated scales) and lower global functioning than those who did not.7 Statistical adjustments made to account for aggression score differences eliminated the difference in global functioning, which suggested a direct link between aggression and global function in individuals with IED-R.
Two patterns. Later research uncovered at least patterns of aggressive outbursts:
- low intensity at high frequency (such as verbal arguments or door slamming approximately twice weekly)
- high intensity at low frequency (such as physical aggression resulting in injury or destruction of nontrivial property at least three times per year).
Data revealed that 69% of individuals with IED-like histories displayed both aggression patterns, 20% displayed only the high-intensity/low-frequency pattern, and 11% displayed only the low-intensity/high-frequency pattern.
Because further analysis revealed no important differences between these groups in measures of aggression and impulsivity, IED-R criteria were revised to include both patterns in the “A” criteria. This revision integrated the essences of IED-R and DSM criteria into one diagnostic set (Table 2).
INFLUENCE OF HEREDITY
No twin or adoption studies of IED have been performed. However, family history data suggest that IED (or IED-type behavior) is familial. I recently conducted a blinded, controlled, family history study using IED-R criteria and found a significantly elevated risk for IED (p < 0.01) in relatives of persons with a history of IED (26%), compared with non-IED controls (8%). Comorbid conditions did not affect the risk among the IED subjects or their relatives, suggesting that IED is familial and independent of other conditions.13
Nearly all studies of aggression’s biology and treatment have measured aggression as a dimensional variable along a continuous scale from low to high.14 Our studies have allowed us to explore biological and treatment response correlates. In preliminary analyses, we have found that the maximal prolactin response to d-fenfluramine challenge and the number of platelet serotonin transporter binding sites are:
- reduced in subjects meeting research criteria for IED
- inversely correlated with dimensional measures of impulsive aggression.
Table 2
Updated diagnostic criteria for intermittent explosive disorder
| A. Recurrent incidents of aggression manifest as either: |
| 1. Verbal or physical aggression towards other people, animals, or property occurring twice weekly on average for 1 month |
| OR |
| 2. Three episodes involving physical assault against other people or destruction of property over a 1-year period |
| B. The degree of aggressiveness expressed is grossly out of proportion to the provocation or any precipitating psychosocial stressors |
| C. The aggressive behavior is generally not premeditated (ie, is impulsive) and is not committed to achieve a tangible objective (such as money, power, intimidation, etc.) |
| D. The aggressive behavior causes marked distress in the individual or impairs occupational or interpersonal functioning |
| E. The aggressive behavior is not better explained by another mental disorder (such as a major depressive/manic/psychotic disorder, attention-deficit/hyperactivity disorder, general medical condition [head trauma, Alzheimer’s disease], or due to the direct physiologic effects of a substance) |
| Source: Adapted from reference 7 |
Earlier, Virkkunen et al15 reported reduced cerebrospinal fluid 5-hydroxyindoleacetic acid concentrations in persons diagnosed with IED based on DSM-III criteria, compared with persons who were not diagnosed with IED and those who demonstrated nonimpulsive aggression.
TREATING IED
Cognitive therapy. Few double-blind, randomized, placebo-controlled trials of any treatments for IED have been published. Trials using cognitive-behavioral approaches have reduced self-rated anger and its expression in young adults with anger disorders.16 Although many of these subjects may have had IED, it is not known if this approach works in IED.
Table 3
Characteristic behaviors of aggressive individuals*
| Severity | Behaviors |
|---|---|
| Mildly aggressive | Occasional verbal arguments and/or temper tantrums |
| Moderately aggressive | Frequent verbal arguments and temper tantrums (about twice weekly on average), occasional destruction of property, rare or occasional physical assault against others (usually without injury) |
| Highly aggressive | Frequent verbal arguments and temper tantrums (about twice weekly) and/or more than occasional destruction of property or physical assault against others, sometimes with injury |
| * Characteristics given are descriptive and not based on data. | |
Drug therapy. SSRIs. A trial by this author using fluoxetine showed that impulsive aggressive behavior responds to treatment that targets the central serotonergic system.12 Forty subjects with personality disorders and histories of impulsive aggression received fluoxetine, 20 to 60 mg qd, or placebo for 12 weeks. Fluoxetine reduced overt aggression and irritability about 67% more than placebo, as assessed by the Overt Aggression Scale Modified for Outpatients (OAS-M).
All subjects met research criteria for IED. A reanalysis suggests that SSRIs may be most effective in moderately aggressive patients (Table 3),17 whose serotonergic system may be less impaired than that of highly aggressive patients.18
Mood stabilizers. Impulsively aggressive subjects who do not respond to an SSRI may respond to a mood stabilizer.19 An antiaggressive response in IED-like subjects has been reported for lithium,20 carbamazepine,21 and diphenylhydantoin.22
Recently, Hollander et al23 reported greater reduction in overt aggression scores in IED subjects with a DSM cluster B personality disorder who were treated with divalproex, compared with placebo. This study used the same design and outcome measure as our study12 and included subjects who met both DSM-IV and research criteria for IED.
For unknown reasons, divalproex was no more effective than placebo in IED subjects without cluster B personality disorder. More research is needed to uncover predictors of antiaggressive response in IED subjects.
Unipolar vs. bipolar. McElroy9 has suggested using SSRIs (or other antidepressants) as first-line treatment for IED subjects with unipolar affective symptoms and mood stabilizers for those with bipolar affective symptoms. IED subjects without bipolar affective symptoms should be treated first with SSRIs (Algorithm). Preliminary data suggest a role for atypical antipsychotics to treat aggressive behavior in patients with schizophrenia or bipolar disorder, but no empiric data exist.
Beta blockers such as propranolol also may be considered.2 However, beta blockers are more difficult to dose and are associated with more burdensome side effects, compared with SSRIs.
Algorithm Suggested 2-pronged approach for treating intermittent explosive disorder
* With or without an anger management program, which may precede drug interventionThe full effects of antiaggressive treatment with an SSRI (E. Coccaro, unpublished observations) or a mood stabilizer19 may take 3 months to observe12,20,22,23 and tend to disappear soon after treatment is discontinued.
Therefore, an adequate trial of SSRIs or mood stabilizers is no less than 3 months. If improvement is seen, continue drug treatment indefinitely.
Case report continued.
Mr. P was started on an SSRI. His aggressive outbursts decreased in intensity and frequency over 3 months but were not eliminated. After 6 months he dropped out of treatment, but returned 5 weeks later because his aggressive outbursts had resumed their pre-treatment level.
SSRI treatment was restarted, and Mr. P began a 12-week anger management course of relaxation training, cognitive restructuring, and coping skills training. He gained greater control over his aggressive outbursts and continues monthly medication checks and anger management “booster sessions.”
Related resources
- Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
- Olvera RL. Intermittent explosive disorder: epidemiology, diagnosis and management. CNS Drugs 2002;16:517-26.
Drug brand names
- Carbamazepine • Tegretol
- Diphenylhydantoin • Dilantin
- Divalproex • Depakote
- Fluoxetine • Prozac
- Lithium • Lithobid
- Propanolol • Inderal
Disclosure
Dr. Coccaro reports that he receives research grants and serves on the speaker’s bureau or as a consultant to Eli Lilly and Co., Abbott Laboratories, GlaxoSmithKline, and Forrest Laboratories.
Mr. P, age 41, has a “problem with anger.” Since age 17, he has had sudden outbursts of screaming and shouting, with occasional minor damage to objects. These outbursts—including episodes of “road rage”—occur once or more per week and almost daily for months at a time.
Mr. P has also had more violent episodes— sometimes every 2 to 3 months—in which he has punched holes in walls, destroyed a computer with a hammer, and assaulted other people with his fists. These events are not premeditated and are typically triggered by Mr. P’s frustration at not being “perfect” or by others breaking what he considers “general rules of conduct.”
The day before his initial visit, while he was stuck in traffic, Mr. P saw a car speeding down the shoulder. Enraged, he pulled in front of the car so that the driver had to slam on the brakes. He jumped out of his car and approached the other driver, shouting obscenities. The other driver locked her door and tried to ignore Mr. P until he returned to his car. Mr. P noted that this episode “ruined” his day because of his lingering anger and irritability.
Intermittent explosive disorder (IED) is more common and complex than was once thought, based on recent evidence. Recurrent, problematic, impulsive aggression is highly comorbid with other psychiatric conditions—including mood and personality disorders—and undermines social relationships and job performance. Typical characteristics of IED are outlined in Table 1.1-3
Table 1
Typical characteristics of intermittent explosive disorder
| Onset in childhood or adolescence (mean age 15), with average duration ±20 years |
| Aggressive outbursts: |
|
| Some episodes may appear without identifiable provocation |
| Male to female ratio 3:1, although some data suggest gender parity |
| Source: Adapted from references 1-3 |
This article offers updated diagnostic criteria and a two-pronged algorithm that can help you diagnose and treat this aggression disorder.
HOW COMMON IS IED?
DSM-IV states that IED is “apparently rare.” This statement is far from surprising, given the limitations of DSM criteria. Surveys of hospitalized patients in the 1980s found that only 1.1% met DSM-III criteria for IED.4 In another study of more than 400 patients seeking treatment for aggression, only 1.8% met DSM-III criteria for IED (although far more would likely have met DSM-IV criteria).5
A more recent survey of 411 psychiatric outpatients6 found that 3.8% met current and 6.2% met lifetime DSM-IV criteria for IED, using the Structured Clinical Interview for DSM-IV Diagnoses (SCID). Reanalysis of a threefold larger data set from the same study site (Coccaro and Zimmerman, unpublished) yielded the same result.
Far from rare. More recently, our findings from a small sample suggested that the community rate of lifetime IED is about 4% by DSM-IV criteria and 5% by research criteria. In the United States, we estimate that the lifetime rate of IED could be 4.5 to 18 million persons using DSM-IV criteria or 6.7 to 22.2 million using IED research criteria. If so, IED is at least as common as other major psychiatric disorders, including schizophrenia or bipolar illness. The ongoing National Comorbidity Study is expected to produce more definitive community data.
PSYCHIATRIC COMORBIDITY
Axis I disorders. IED is highly comorbid with mood, anxiety, and substance use disorders,3,7,8 although no causal relationship has been shown
Mood and substance abuse disorders. IED’s age of onset may precede that of mood and substance use disorders, according to analysis of our unpublished data. If so, comorbid IED may not occur in the context of mood or substance use disorders.
Anxiety disorders. We have noted a similar pattern with IED and anxiety disorders, although phobic anxiety disorders (simple or social phobia) tend to manifest earlier than IED. This suggests that early-onset phobic anxiety might be associated with an increased risk of IED in adolescence or young adulthood.
Bipolar disorder. McElroy9 has suggested a relationship between IED and bipolar disorder. In some samples, as many as one-half of IED patients (56%) have comorbid bipolar disorder when one includes bipolar II and cyclothymia.3 Moreover, some subjects’ aggressive episodes appear to resemble “microdysphoric manic episodes.”9 Other studies,8 however, find a much lower rate (10% or less) of IED comorbidity with bipolar illness.
Bipolar disorder overall may not be highly comorbid with IED, although rates may be higher in specialty clinic samples. In individuals with any kind of bipolar disorder, mood stabilizers— rather than selective serotonin reuptake inhibitors (SSRIs)—are probably the better choice as first-line treatment of IED.9
Axis II disorders. DSM-IV allows IED diagnosis in individuals with borderline or antisocial personality disorder, as long as these cluster B disorders do not better explain the aggressive behavior. How a clinician makes this distinction is not clear; in fact, most clinicians do not diagnose IED in patients with personality disorders, regardless of the clinical picture.
IED comorbidity with borderline or antisocial personality disorders varies with the sample. Persons with personality disorders who seek treatment of aggressive behavior are more likely to have comorbid IED (90%) than those not seeking treatment who are outpatients (50%) or in the community (25%).1,7
Individuals with personality disorders and IED score higher in aggression and lower in psychosocial function than do similar individuals without IED,7 indicating that the additional diagnosis is relevant.
Case report continued.
Mr. P’s outbursts have cost him several friendships, including romantic relationships. He has never advanced at work because he is seen as too volatile to supervise subordinates. Though some of Mr. P’s aggressive outbursts have occurred under the influence of alcohol, most are not related to alcohol or drug use. He has no medical problems and no other psychiatric history.
A full diagnostic evaluation uncovers a personality disorder, not otherwise specified (eight scattered traits from obsessive-compulsive personality disorder and from each of the cluster B personality disorders), and no Axis I condition other than intermittent explosive disorder.
PROBLEMS DEFINING IED
Intermittent explosive disorder is the only DSM diagnosis that applies to persons with histories of recurrent, problematic aggression not caused by another mental or physical disorder. Even so, little research on IED is available. DSM criteria for IED are poorly operationalized and have improved only modestly since the diagnosis was first included in DSM-III. In that revision, IED had four criteria.
“A” criteria specified recurrent outbursts of “seriously assaultive or destructive behavior,” but left unanswered important questions such as:
- What behavior crosses the threshold for seriously” assaultive or destructive?
- Does any physical assault qualify, or only those that cause physical injury (or stigmata)?
- How often or within what time must the behavior occur?
The phrase “recurrent acts of aggression” suggested that at least three acts of aggression were required to reach the threshold, but DSM-III provided no guidelines.
“B” criteria stated that the aggression should be out of proportion to the provocation. But how should one judge this criterion, when provocative stimuli sometimes are clearly sufficient to prompt a justifiably aggressive act?
“C” criteria excluded persons who are aggressive or impulsive between ill-defined “aggressive episodes.” This exclusion was especially limiting because individuals with recurrent, problematic, aggressive behaviors generally are impulsive and aggressive between more-severe outbursts. Excluding those who otherwise met diagnostic criteria for IED led to a spuriously low prevalence rate and limited the number of research subjects. DSM-IV eliminated this criterion but made no other notable changes in IED criteria.
“D” criteria in DSM-III and III-R further restricted the number of individuals who could meet this diagnosis:
- In DSM-III, antisocial personality disorder excluded the diagnosis of IED.
- In DSM-III-R, borderline personality disorder was added as an exclusionary factor.
Because of these restrictions, very few clinically valid cases of IED (individuals meeting A and B criteria) could receive an IED diagnosis.10
EVOLVING DIAGNOSTIC CRITERIA
By the early 1990s, DSM diagnostic criteria clearly severely restricted the study of recurrent, problematic aggression, even though research since DSM-III had greatly advanced our understanding of human aggression. For example, data linked impulsive aggression to deficits in central serotonergic function and suggested that agents that enhance serotonergic activity could modify this behavior.
Some investigators proposed research criteria for IED (IED-R) so that individuals with recurrent, problematic, impulsive aggression could be identified and studied. Research criteria first published in 19987 proposed six changes/clarifications in IED diagnostic criteria:
Lower-intensity aggression. The scope of aggressive behavior was expanded to include verbal and indirect physical aggression, provided that these behaviors are associated with distress and/or impairment. Data from double-blind, placebo-controlled trials indicated that these lower-intensity (although usually higher frequency) behaviors respond well to treatment with SSRIs.11,12
Impulsivity. The aggression was specified as impulsive. This change identified individuals with greater liability for deficits in central serotonergic function and excluded individuals with premeditated or criminal aggression.
A minimal frequency of aggression over time was proposed to make the IED diagnosis more reliable and to ensure that persons with only occasional impulsive aggressive outbursts (especially of low severity) were given this diagnosis.
Subjective distress (in the individual) and/or social or occupational dysfunction was proposed so that putatively aggressive individuals are not diagnosed for manifesting behaviors that are not functionally severe.
Diagnostic exclusionary criteria were modified so that individuals with:
- antisocial or borderline personality disorder could be diagnosed with IED if otherwise warranted
- aggressive behaviors confined within major depression episodes could not be diagnosed with IED.
This last change recognized that impulsive, aggressive outbursts could point to major depressive disorder.
When the revised criteria were tested in patients seeking treatment for aggression, those who met IED-R criteria were found to exhibit significantly greater aggression and impulsivity (using validated scales) and lower global functioning than those who did not.7 Statistical adjustments made to account for aggression score differences eliminated the difference in global functioning, which suggested a direct link between aggression and global function in individuals with IED-R.
Two patterns. Later research uncovered at least patterns of aggressive outbursts:
- low intensity at high frequency (such as verbal arguments or door slamming approximately twice weekly)
- high intensity at low frequency (such as physical aggression resulting in injury or destruction of nontrivial property at least three times per year).
Data revealed that 69% of individuals with IED-like histories displayed both aggression patterns, 20% displayed only the high-intensity/low-frequency pattern, and 11% displayed only the low-intensity/high-frequency pattern.
Because further analysis revealed no important differences between these groups in measures of aggression and impulsivity, IED-R criteria were revised to include both patterns in the “A” criteria. This revision integrated the essences of IED-R and DSM criteria into one diagnostic set (Table 2).
INFLUENCE OF HEREDITY
No twin or adoption studies of IED have been performed. However, family history data suggest that IED (or IED-type behavior) is familial. I recently conducted a blinded, controlled, family history study using IED-R criteria and found a significantly elevated risk for IED (p < 0.01) in relatives of persons with a history of IED (26%), compared with non-IED controls (8%). Comorbid conditions did not affect the risk among the IED subjects or their relatives, suggesting that IED is familial and independent of other conditions.13
Nearly all studies of aggression’s biology and treatment have measured aggression as a dimensional variable along a continuous scale from low to high.14 Our studies have allowed us to explore biological and treatment response correlates. In preliminary analyses, we have found that the maximal prolactin response to d-fenfluramine challenge and the number of platelet serotonin transporter binding sites are:
- reduced in subjects meeting research criteria for IED
- inversely correlated with dimensional measures of impulsive aggression.
Table 2
Updated diagnostic criteria for intermittent explosive disorder
| A. Recurrent incidents of aggression manifest as either: |
| 1. Verbal or physical aggression towards other people, animals, or property occurring twice weekly on average for 1 month |
| OR |
| 2. Three episodes involving physical assault against other people or destruction of property over a 1-year period |
| B. The degree of aggressiveness expressed is grossly out of proportion to the provocation or any precipitating psychosocial stressors |
| C. The aggressive behavior is generally not premeditated (ie, is impulsive) and is not committed to achieve a tangible objective (such as money, power, intimidation, etc.) |
| D. The aggressive behavior causes marked distress in the individual or impairs occupational or interpersonal functioning |
| E. The aggressive behavior is not better explained by another mental disorder (such as a major depressive/manic/psychotic disorder, attention-deficit/hyperactivity disorder, general medical condition [head trauma, Alzheimer’s disease], or due to the direct physiologic effects of a substance) |
| Source: Adapted from reference 7 |
Earlier, Virkkunen et al15 reported reduced cerebrospinal fluid 5-hydroxyindoleacetic acid concentrations in persons diagnosed with IED based on DSM-III criteria, compared with persons who were not diagnosed with IED and those who demonstrated nonimpulsive aggression.
TREATING IED
Cognitive therapy. Few double-blind, randomized, placebo-controlled trials of any treatments for IED have been published. Trials using cognitive-behavioral approaches have reduced self-rated anger and its expression in young adults with anger disorders.16 Although many of these subjects may have had IED, it is not known if this approach works in IED.
Table 3
Characteristic behaviors of aggressive individuals*
| Severity | Behaviors |
|---|---|
| Mildly aggressive | Occasional verbal arguments and/or temper tantrums |
| Moderately aggressive | Frequent verbal arguments and temper tantrums (about twice weekly on average), occasional destruction of property, rare or occasional physical assault against others (usually without injury) |
| Highly aggressive | Frequent verbal arguments and temper tantrums (about twice weekly) and/or more than occasional destruction of property or physical assault against others, sometimes with injury |
| * Characteristics given are descriptive and not based on data. | |
Drug therapy. SSRIs. A trial by this author using fluoxetine showed that impulsive aggressive behavior responds to treatment that targets the central serotonergic system.12 Forty subjects with personality disorders and histories of impulsive aggression received fluoxetine, 20 to 60 mg qd, or placebo for 12 weeks. Fluoxetine reduced overt aggression and irritability about 67% more than placebo, as assessed by the Overt Aggression Scale Modified for Outpatients (OAS-M).
All subjects met research criteria for IED. A reanalysis suggests that SSRIs may be most effective in moderately aggressive patients (Table 3),17 whose serotonergic system may be less impaired than that of highly aggressive patients.18
Mood stabilizers. Impulsively aggressive subjects who do not respond to an SSRI may respond to a mood stabilizer.19 An antiaggressive response in IED-like subjects has been reported for lithium,20 carbamazepine,21 and diphenylhydantoin.22
Recently, Hollander et al23 reported greater reduction in overt aggression scores in IED subjects with a DSM cluster B personality disorder who were treated with divalproex, compared with placebo. This study used the same design and outcome measure as our study12 and included subjects who met both DSM-IV and research criteria for IED.
For unknown reasons, divalproex was no more effective than placebo in IED subjects without cluster B personality disorder. More research is needed to uncover predictors of antiaggressive response in IED subjects.
Unipolar vs. bipolar. McElroy9 has suggested using SSRIs (or other antidepressants) as first-line treatment for IED subjects with unipolar affective symptoms and mood stabilizers for those with bipolar affective symptoms. IED subjects without bipolar affective symptoms should be treated first with SSRIs (Algorithm). Preliminary data suggest a role for atypical antipsychotics to treat aggressive behavior in patients with schizophrenia or bipolar disorder, but no empiric data exist.
Beta blockers such as propranolol also may be considered.2 However, beta blockers are more difficult to dose and are associated with more burdensome side effects, compared with SSRIs.
Algorithm Suggested 2-pronged approach for treating intermittent explosive disorder
* With or without an anger management program, which may precede drug interventionThe full effects of antiaggressive treatment with an SSRI (E. Coccaro, unpublished observations) or a mood stabilizer19 may take 3 months to observe12,20,22,23 and tend to disappear soon after treatment is discontinued.
Therefore, an adequate trial of SSRIs or mood stabilizers is no less than 3 months. If improvement is seen, continue drug treatment indefinitely.
Case report continued.
Mr. P was started on an SSRI. His aggressive outbursts decreased in intensity and frequency over 3 months but were not eliminated. After 6 months he dropped out of treatment, but returned 5 weeks later because his aggressive outbursts had resumed their pre-treatment level.
SSRI treatment was restarted, and Mr. P began a 12-week anger management course of relaxation training, cognitive restructuring, and coping skills training. He gained greater control over his aggressive outbursts and continues monthly medication checks and anger management “booster sessions.”
Related resources
- Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
- Olvera RL. Intermittent explosive disorder: epidemiology, diagnosis and management. CNS Drugs 2002;16:517-26.
Drug brand names
- Carbamazepine • Tegretol
- Diphenylhydantoin • Dilantin
- Divalproex • Depakote
- Fluoxetine • Prozac
- Lithium • Lithobid
- Propanolol • Inderal
Disclosure
Dr. Coccaro reports that he receives research grants and serves on the speaker’s bureau or as a consultant to Eli Lilly and Co., Abbott Laboratories, GlaxoSmithKline, and Forrest Laboratories.
1. Coccaro EF, Schimdt CA, Samuels JF, et al. Lifetime rates of intermittent explosive disorder in a community sample (abstract). Philadelphia: American Psychiatric Association annual meeting, 2002.
2. Mattes JA. Comparative effectiveness of carbamazepine and propranolol for rage outbursts. J Neuropsychiatry Clin Neurosci 1990;2:159-64.
3. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry 1998;59:203-10.
4. Monopolis S, Lion JR. Problems in the diagnosis of intermittent explosive disorder. Am J Psychiatry 1983;140:1200-2.
5. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (abstract 265). Washington, DC: American Psychiatric Association annual meeting, 1998.
6. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (APA new research abstracts #265). Washington, DC: American Psychiatric Publishing, Inc., 1998.
7. Coccaro EF, Kavoussi RJ, Berman ME, Lish JD. Intermittent explosive disorder-revised: development, reliability and validity of research criteria. Compr Psychiatry 1998;39:368-76.
8. Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
9. McElroy SL. Recognition and treatment of DSM-IV intermittent explosive disorder. J Clin Psychiatry 1999;60(suppl 15):12-16.
10. Felthous AR, Bryant G, Wingerter CB, Barratt E. The diagnosis of intermittent explosive disorder in violent men. Bull Am Acad Psychiatry Law 1991;19:71-9.
11. Salzman C, Wolfson AN, Schatzberg A, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. J Clin Psychopharmacology 1995;15:23-9.
12. Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality disordered subjects. Arch Gen Psychiatry 1997;54:1081-8.
13. Coccaro EF. Family history study of intermittent explosive disorder (abstract). Washington, DC: American Psychiatric Association annual meeting, 1999.
14. Coccaro EF, Siever LJ. Pathophysiology and treatment of aggression. In: Davis KL, Charney D, Coyle JT, Nemeroff D (eds). Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002;1709-24
15. Virkkunen M, Rawlings R, Tokola R, et al. CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry 1994;51:20-7.
16. Deffenbacher JL. Psychosocial interventions: anger disorders. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
17. Lee R, Coccaro EF. Treatment of aggression: serotonergic agents. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
18. Coccaro EF, Kavoussi RJ, Hauger RL. Serotonin function and antiaggressive responses to fluoxetine: a pilot study. Biol Psychiatry 1997;42:546-52.
19. Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. J Clin Psychiatry 1998;59:676-80.
20. Sheard MH, Marini J, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry 1976;133:1409-13.
21. Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder: alprazolam, carbamazepine, trifluroperazine, and tranylcypromine. Arch Gen Psychiatry 1988;45:111-19.
22. Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacology 1997;17:341-9.
23. Hollander E, Tracy KA, Swann AC, et al. Divalproex sodium is superior to placebo for impulsive aggression in Cluster B personality disorders. Neuropsychopharmacology 2003;28:1186-97.
1. Coccaro EF, Schimdt CA, Samuels JF, et al. Lifetime rates of intermittent explosive disorder in a community sample (abstract). Philadelphia: American Psychiatric Association annual meeting, 2002.
2. Mattes JA. Comparative effectiveness of carbamazepine and propranolol for rage outbursts. J Neuropsychiatry Clin Neurosci 1990;2:159-64.
3. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry 1998;59:203-10.
4. Monopolis S, Lion JR. Problems in the diagnosis of intermittent explosive disorder. Am J Psychiatry 1983;140:1200-2.
5. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (abstract 265). Washington, DC: American Psychiatric Association annual meeting, 1998.
6. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (APA new research abstracts #265). Washington, DC: American Psychiatric Publishing, Inc., 1998.
7. Coccaro EF, Kavoussi RJ, Berman ME, Lish JD. Intermittent explosive disorder-revised: development, reliability and validity of research criteria. Compr Psychiatry 1998;39:368-76.
8. Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
9. McElroy SL. Recognition and treatment of DSM-IV intermittent explosive disorder. J Clin Psychiatry 1999;60(suppl 15):12-16.
10. Felthous AR, Bryant G, Wingerter CB, Barratt E. The diagnosis of intermittent explosive disorder in violent men. Bull Am Acad Psychiatry Law 1991;19:71-9.
11. Salzman C, Wolfson AN, Schatzberg A, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. J Clin Psychopharmacology 1995;15:23-9.
12. Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality disordered subjects. Arch Gen Psychiatry 1997;54:1081-8.
13. Coccaro EF. Family history study of intermittent explosive disorder (abstract). Washington, DC: American Psychiatric Association annual meeting, 1999.
14. Coccaro EF, Siever LJ. Pathophysiology and treatment of aggression. In: Davis KL, Charney D, Coyle JT, Nemeroff D (eds). Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002;1709-24
15. Virkkunen M, Rawlings R, Tokola R, et al. CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry 1994;51:20-7.
16. Deffenbacher JL. Psychosocial interventions: anger disorders. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
17. Lee R, Coccaro EF. Treatment of aggression: serotonergic agents. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
18. Coccaro EF, Kavoussi RJ, Hauger RL. Serotonin function and antiaggressive responses to fluoxetine: a pilot study. Biol Psychiatry 1997;42:546-52.
19. Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. J Clin Psychiatry 1998;59:676-80.
20. Sheard MH, Marini J, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry 1976;133:1409-13.
21. Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder: alprazolam, carbamazepine, trifluroperazine, and tranylcypromine. Arch Gen Psychiatry 1988;45:111-19.
22. Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacology 1997;17:341-9.
23. Hollander E, Tracy KA, Swann AC, et al. Divalproex sodium is superior to placebo for impulsive aggression in Cluster B personality disorders. Neuropsychopharmacology 2003;28:1186-97.