User login
Lichenoid Drug Eruption Secondary to Apalutamide Treatment
To the Editor:
Lichenoid drug eruptions are lichen planus–like hypersensitivity reactions induced by medications. These reactions are rare but cause irritation to the skin, as extreme pruritus is common. One review of 300 consecutive cases of drug eruptions submitted to dermatopathology revealed that 12% of cases were classified as lichenoid drug reactions.1 Lichenoid dermatitis is characterized by extremely pruritic, scaly, eczematous or psoriasiform papules, often along the extensor surfaces and trunk.2 The pruritic nature of the rash can negatively impact quality of life. Treatment typically involves discontinuation of the offending medication, although complete resolution can take months, even after the drug is stopped. Although there have been some data suggesting that topical and/or oral corticosteroids can help with resolution, the rash can persist even with steroid treatment.2
The histopathologic findings of lichenoid drug eruptions show lichen planus–like changes such as hyperkeratosis, irregular acanthosis, and lichenoid interface dermatitis. Accordingly, idiopathic lichen planus is an important differential diagnosis for lichenoid drug eruptions; however, compared to idiopathic lichen planus, lichenoid drug eruptions are more likely to be associated with eosinophils and parakeratosis.1,3 In some cases, the histopathologic distinction between the 2 conditions is impossible, and clinical history needs to be considered to make a diagnosis.1 Drugs known to cause lichenoid drug reactions more commonly include angiotensin-converting enzyme inhibitors, beta blockers, thiazides, gold, penicillamine, and antimalarials.2 Lichenoid drug eruptions also have been documented in patients taking the second-generation nonsteroidal androgen receptor antagonist enzalutamide, which is used for the treatment of prostate cancer.4 More recently, the newer second-generation nonsteroidal androgen receptor antagonist apalutamide has been implicated in several cases of lichenoid drug eruptions.5,6
We present a case of an apalutamide-induced lichenoid drug eruption that was resistant to dose reduction and required discontinuation of treatment due to the negative impact on the patient’s quality of life. Once the rash resolved, the patient transitioned to enzalutamide without any adverse events (AEs).
A 72-year-old man with a history of metastatic prostate cancer (stage IVB) presented to the dermatology clinic with a 4-month history of a dry itchy rash on the face, chest, back, and legs that had developed 2 to 3 months after oncology started him on apalutamide. The patient initially received apalutamide 240 mg/d, which was reduced by his oncologist 3 months later to 180 mg/d following the appearance of the rash. Then apalutamide was held as he awaited improvement of the rash.
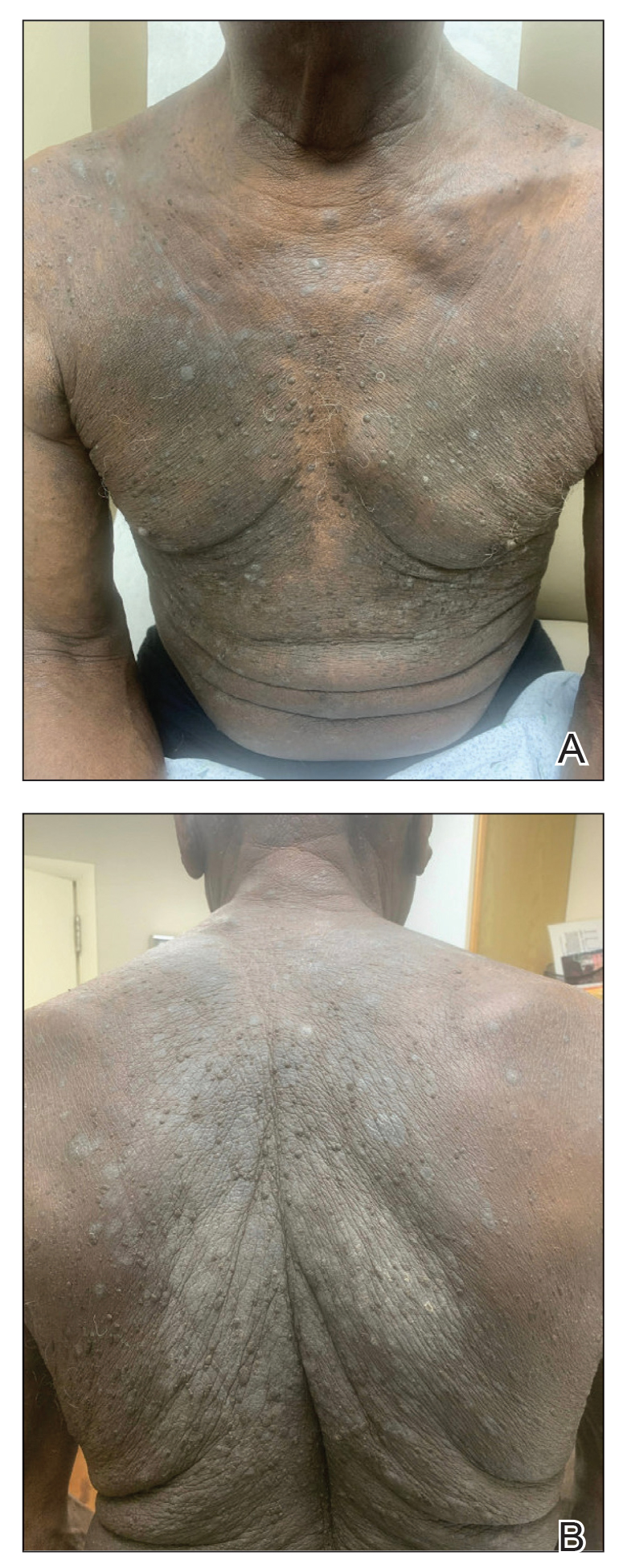
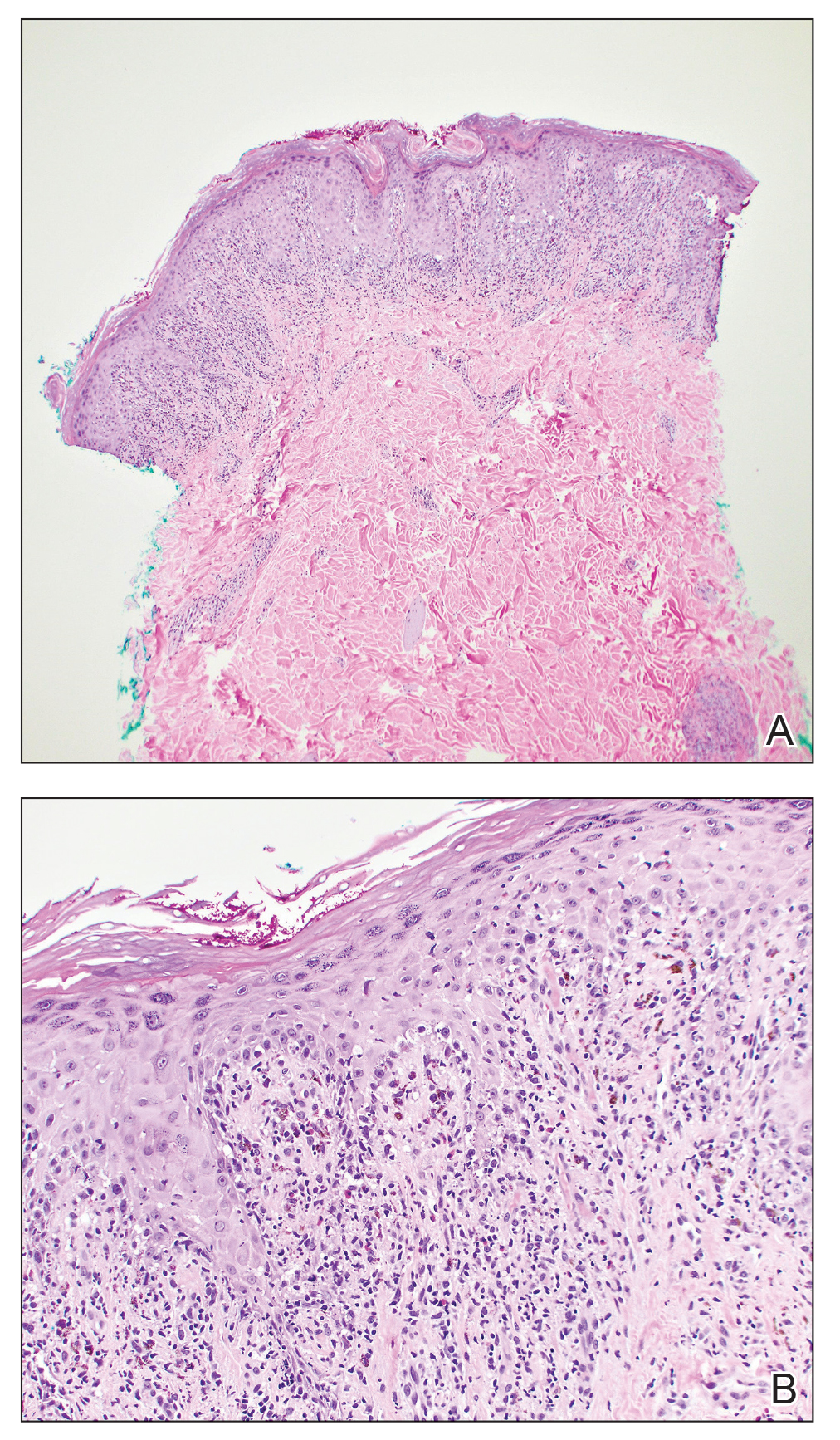
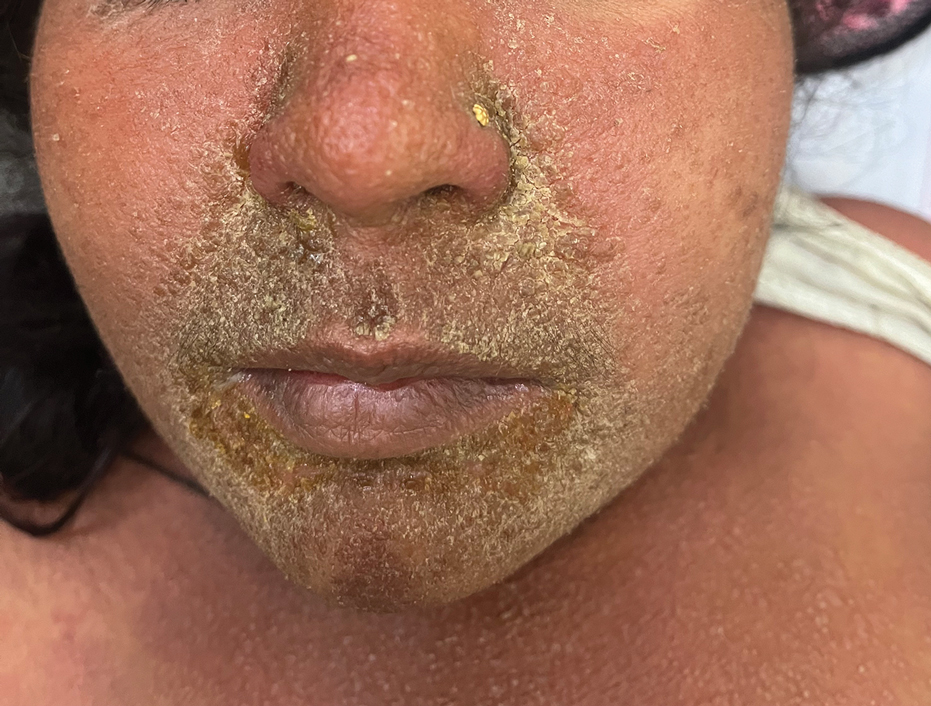
One week after the apalutamide was held, the patient presented to dermatology. He reported that he had tried over-the-counter ammonium lactate 12% lotion twice daily when the rash first developed without improvement. When the apalutamide was held, oncology prescribed mupirocin ointment 2% 3 times daily which yielded minimal relief. On physical examination, widespread lichenified papules and plaques were noted on the face, chest, back, and legs (Figure 1). Dermatology initially prescribed triamcinolone ointment 0.1% twice daily. A 4-mm punch biopsy specimen of the upper back revealed a lichenoid interface dermatitis with numerous eosinophils compatible with a lichenoid hypersensitivity reaction (Figure 2). Considering the clinical and histologic findings, a diagnosis of lichenoid drug eruption secondary to apalutamide treatment was made.
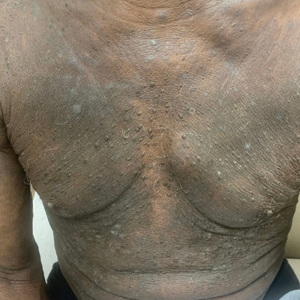
Two weeks after discontinuation of the medication, the rash improved, and the patient restarted apalutamide at a dosage of 120 mg/d; however, the rash re-emerged within 1 month and was resistant to the triamcinolone ointment 0.1%. Apalutamide was again discontinued, and oncology switched the patient to enzalutamide 160 mg/d in an effort to find a medication the patient could better tolerate. Two months after starting enzalutamide, the patient had resolution of the rash and no further dermatologic complications.
Apalutamide is a second-generation nonsteroidal androgen receptor antagonist used in the treatment of nonmetastatic castration-resistant prostate cancer (CRPC) and metastatic castration-sensitive prostate cancer (CSPC).7 It stops the spread and growth of prostate cancer cells by several different mechanisms, including competitively binding androgen receptors, preventing 5α-dihydrotestosterone from binding to androgen receptors, blocking androgen receptor nuclear translocation, impairing co-activator recruitment, and restraining androgen receptor DNA binding.7 The SPARTAN and TITAN phase 3 clinical trials demonstrated increased overall survival and time to progression with apalutamide in both nonmetastatic CRPC and metastatic CSPC. In both trials, the rash was shown to be an AE more commonly associated with apalutamide than placebo.8,9
Until recently, the characteristics of apalutamide-induced drug rashes have not been well described. One literature review reported 6 cases of cutaneous apalutamide-induced drug eruptions.5 Four (66.7%) of these eruptions were maculopapular rashes, only 2 of which were histologically classified as lichenoid in nature. The other 2 eruptions were classified as toxic epidermal necrosis.5 Another study of 303 patients with prostate cancer who were treated with apalutamide recorded the frequency and time to onset of dermatologic AEs.6 Seventy-one (23.4%) of the patients had dermatologic AEs, and of those, only 20 (28.2%) had AEs that resulted in interruptions in apalutamide therapy (with only 5 [25.0%] requiring medication discontinuation). Thirty-two (45.1%) patients were managed with topical or oral corticosteroids or dose modification. In this study, histopathology was examined in 8 cases (one of which had 2 biopsies for a total of 9 biopsies), 7 of which were consistent with lichenoid interface dermatitis.6
Lichenoid interface dermatitis is a rare manifestation of an apalutamide-induced drug eruption and also has been reported secondary to treatment with enzalutamide, another second-generation nonsteroidal androgen receptor antagonist.4 Enzalutamide was the first second-generation nonsteroidal androgen receptor antagonist approved for the treatment of prostate cancer. It originally was approved only for metastatic CRPC after docetaxel therapy in 2012, then later was expanded to metastatic and nonmetastatic CRPC in 2012 and 2018, respectively, as well as metastatic CSPC in 2019.7 Because enzalutamide is from the same medication class as apalutamide and has been on the market longer for the treatment of nonmetastatic CRPC and metastatic CSPC, it is not surprising that similar drug eruptions now are being reported secondary to apalutamide use as well.
It is important for providers to consider lichenoid drug eruptions in the differential diagnosis of pruritic rashes in patients taking second-generation nonsteroidal androgen receptor antagonists such as apalutamide or enzalutamide. Although dose reduction or treatment discontinuation have been the standard of care for patients with extremely pruritic lichenoid drug eruptions secondary to these medications, these are not ideal because they are important for cancer treatment. Interestingly, after our patient’s apalutamide-induced rash resolved and he was switched to enzalutamide, he did not develop any AEs. Based on our patient’s experience, physicians could consider switching their patients to another drug of the same class, as they may be able tolerate that medication. More research is needed to determine how commonly patients tolerate a different second-generation nonsteroidal androgen receptor antagonist after not tolerating another medication from the same class.
- Weyers W, Metze D. Histopathology of drug eruptions—general criteria, common patterns, and differential diagnosis. Dermatol Pract Concept. 2011;1:33-47. doi:10.5826/dpc.0101a09
- Cheraghlou S, Levy LL. Fixed drug eruption, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Thompson DF, Skaehill PA. Drug-induced lichen planus. Pharmacotherapy. 1994;14:561-571.
- Khan S, Saizan AL, O’Brien K, et al. Diffuse hyperpigmented lichenoid drug eruption secondary to enzalutamide. Curr Probl Cancer Case Rep. 2022;5:100135. doi:10.1016/j.cpccr.2021.100135
- Katayama H, Saeki H, Osada S-I. Maculopapular drug eruption caused by apalutamide: case report and review of the literature. J Nippon Med Sch. 2022;89:550-554. doi:10.1272/jnms.JNMS.2022_89-503
- Pan A, Reingold RE, Zhao JL, et al. Dermatologic adverse events in prostate cancer patients treated with the androgen receptor inhibitor apalutamide. J Urol. 2022;207:1010-1019. doi:10.1097/JU.0000000000002425
- Rajaram P, Rivera A, Muthima K, et al. Second-generation androgen receptor antagonists as hormonal therapeutics for three forms of prostate cancer. Molecules. 2020;25:2448. doi:10.3390/molecules25102448
- Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408-1418. doi:10.1056/NEJMoa1715546
- Chi KN, Agarwal N, Bjartell A, et al. Apalutamide for metastatic, castration-sensative prostate cancer. N Engl J Med. 2019;381:13-24. doi:10.1056/NEJMoa1903307
To the Editor:
Lichenoid drug eruptions are lichen planus–like hypersensitivity reactions induced by medications. These reactions are rare but cause irritation to the skin, as extreme pruritus is common. One review of 300 consecutive cases of drug eruptions submitted to dermatopathology revealed that 12% of cases were classified as lichenoid drug reactions.1 Lichenoid dermatitis is characterized by extremely pruritic, scaly, eczematous or psoriasiform papules, often along the extensor surfaces and trunk.2 The pruritic nature of the rash can negatively impact quality of life. Treatment typically involves discontinuation of the offending medication, although complete resolution can take months, even after the drug is stopped. Although there have been some data suggesting that topical and/or oral corticosteroids can help with resolution, the rash can persist even with steroid treatment.2
The histopathologic findings of lichenoid drug eruptions show lichen planus–like changes such as hyperkeratosis, irregular acanthosis, and lichenoid interface dermatitis. Accordingly, idiopathic lichen planus is an important differential diagnosis for lichenoid drug eruptions; however, compared to idiopathic lichen planus, lichenoid drug eruptions are more likely to be associated with eosinophils and parakeratosis.1,3 In some cases, the histopathologic distinction between the 2 conditions is impossible, and clinical history needs to be considered to make a diagnosis.1 Drugs known to cause lichenoid drug reactions more commonly include angiotensin-converting enzyme inhibitors, beta blockers, thiazides, gold, penicillamine, and antimalarials.2 Lichenoid drug eruptions also have been documented in patients taking the second-generation nonsteroidal androgen receptor antagonist enzalutamide, which is used for the treatment of prostate cancer.4 More recently, the newer second-generation nonsteroidal androgen receptor antagonist apalutamide has been implicated in several cases of lichenoid drug eruptions.5,6
We present a case of an apalutamide-induced lichenoid drug eruption that was resistant to dose reduction and required discontinuation of treatment due to the negative impact on the patient’s quality of life. Once the rash resolved, the patient transitioned to enzalutamide without any adverse events (AEs).
A 72-year-old man with a history of metastatic prostate cancer (stage IVB) presented to the dermatology clinic with a 4-month history of a dry itchy rash on the face, chest, back, and legs that had developed 2 to 3 months after oncology started him on apalutamide. The patient initially received apalutamide 240 mg/d, which was reduced by his oncologist 3 months later to 180 mg/d following the appearance of the rash. Then apalutamide was held as he awaited improvement of the rash.
One week after the apalutamide was held, the patient presented to dermatology. He reported that he had tried over-the-counter ammonium lactate 12% lotion twice daily when the rash first developed without improvement. When the apalutamide was held, oncology prescribed mupirocin ointment 2% 3 times daily which yielded minimal relief. On physical examination, widespread lichenified papules and plaques were noted on the face, chest, back, and legs (Figure 1). Dermatology initially prescribed triamcinolone ointment 0.1% twice daily. A 4-mm punch biopsy specimen of the upper back revealed a lichenoid interface dermatitis with numerous eosinophils compatible with a lichenoid hypersensitivity reaction (Figure 2). Considering the clinical and histologic findings, a diagnosis of lichenoid drug eruption secondary to apalutamide treatment was made.
Two weeks after discontinuation of the medication, the rash improved, and the patient restarted apalutamide at a dosage of 120 mg/d; however, the rash re-emerged within 1 month and was resistant to the triamcinolone ointment 0.1%. Apalutamide was again discontinued, and oncology switched the patient to enzalutamide 160 mg/d in an effort to find a medication the patient could better tolerate. Two months after starting enzalutamide, the patient had resolution of the rash and no further dermatologic complications.
Apalutamide is a second-generation nonsteroidal androgen receptor antagonist used in the treatment of nonmetastatic castration-resistant prostate cancer (CRPC) and metastatic castration-sensitive prostate cancer (CSPC).7 It stops the spread and growth of prostate cancer cells by several different mechanisms, including competitively binding androgen receptors, preventing 5α-dihydrotestosterone from binding to androgen receptors, blocking androgen receptor nuclear translocation, impairing co-activator recruitment, and restraining androgen receptor DNA binding.7 The SPARTAN and TITAN phase 3 clinical trials demonstrated increased overall survival and time to progression with apalutamide in both nonmetastatic CRPC and metastatic CSPC. In both trials, the rash was shown to be an AE more commonly associated with apalutamide than placebo.8,9
Until recently, the characteristics of apalutamide-induced drug rashes have not been well described. One literature review reported 6 cases of cutaneous apalutamide-induced drug eruptions.5 Four (66.7%) of these eruptions were maculopapular rashes, only 2 of which were histologically classified as lichenoid in nature. The other 2 eruptions were classified as toxic epidermal necrosis.5 Another study of 303 patients with prostate cancer who were treated with apalutamide recorded the frequency and time to onset of dermatologic AEs.6 Seventy-one (23.4%) of the patients had dermatologic AEs, and of those, only 20 (28.2%) had AEs that resulted in interruptions in apalutamide therapy (with only 5 [25.0%] requiring medication discontinuation). Thirty-two (45.1%) patients were managed with topical or oral corticosteroids or dose modification. In this study, histopathology was examined in 8 cases (one of which had 2 biopsies for a total of 9 biopsies), 7 of which were consistent with lichenoid interface dermatitis.6
Lichenoid interface dermatitis is a rare manifestation of an apalutamide-induced drug eruption and also has been reported secondary to treatment with enzalutamide, another second-generation nonsteroidal androgen receptor antagonist.4 Enzalutamide was the first second-generation nonsteroidal androgen receptor antagonist approved for the treatment of prostate cancer. It originally was approved only for metastatic CRPC after docetaxel therapy in 2012, then later was expanded to metastatic and nonmetastatic CRPC in 2012 and 2018, respectively, as well as metastatic CSPC in 2019.7 Because enzalutamide is from the same medication class as apalutamide and has been on the market longer for the treatment of nonmetastatic CRPC and metastatic CSPC, it is not surprising that similar drug eruptions now are being reported secondary to apalutamide use as well.
It is important for providers to consider lichenoid drug eruptions in the differential diagnosis of pruritic rashes in patients taking second-generation nonsteroidal androgen receptor antagonists such as apalutamide or enzalutamide. Although dose reduction or treatment discontinuation have been the standard of care for patients with extremely pruritic lichenoid drug eruptions secondary to these medications, these are not ideal because they are important for cancer treatment. Interestingly, after our patient’s apalutamide-induced rash resolved and he was switched to enzalutamide, he did not develop any AEs. Based on our patient’s experience, physicians could consider switching their patients to another drug of the same class, as they may be able tolerate that medication. More research is needed to determine how commonly patients tolerate a different second-generation nonsteroidal androgen receptor antagonist after not tolerating another medication from the same class.
To the Editor:
Lichenoid drug eruptions are lichen planus–like hypersensitivity reactions induced by medications. These reactions are rare but cause irritation to the skin, as extreme pruritus is common. One review of 300 consecutive cases of drug eruptions submitted to dermatopathology revealed that 12% of cases were classified as lichenoid drug reactions.1 Lichenoid dermatitis is characterized by extremely pruritic, scaly, eczematous or psoriasiform papules, often along the extensor surfaces and trunk.2 The pruritic nature of the rash can negatively impact quality of life. Treatment typically involves discontinuation of the offending medication, although complete resolution can take months, even after the drug is stopped. Although there have been some data suggesting that topical and/or oral corticosteroids can help with resolution, the rash can persist even with steroid treatment.2
The histopathologic findings of lichenoid drug eruptions show lichen planus–like changes such as hyperkeratosis, irregular acanthosis, and lichenoid interface dermatitis. Accordingly, idiopathic lichen planus is an important differential diagnosis for lichenoid drug eruptions; however, compared to idiopathic lichen planus, lichenoid drug eruptions are more likely to be associated with eosinophils and parakeratosis.1,3 In some cases, the histopathologic distinction between the 2 conditions is impossible, and clinical history needs to be considered to make a diagnosis.1 Drugs known to cause lichenoid drug reactions more commonly include angiotensin-converting enzyme inhibitors, beta blockers, thiazides, gold, penicillamine, and antimalarials.2 Lichenoid drug eruptions also have been documented in patients taking the second-generation nonsteroidal androgen receptor antagonist enzalutamide, which is used for the treatment of prostate cancer.4 More recently, the newer second-generation nonsteroidal androgen receptor antagonist apalutamide has been implicated in several cases of lichenoid drug eruptions.5,6
We present a case of an apalutamide-induced lichenoid drug eruption that was resistant to dose reduction and required discontinuation of treatment due to the negative impact on the patient’s quality of life. Once the rash resolved, the patient transitioned to enzalutamide without any adverse events (AEs).
A 72-year-old man with a history of metastatic prostate cancer (stage IVB) presented to the dermatology clinic with a 4-month history of a dry itchy rash on the face, chest, back, and legs that had developed 2 to 3 months after oncology started him on apalutamide. The patient initially received apalutamide 240 mg/d, which was reduced by his oncologist 3 months later to 180 mg/d following the appearance of the rash. Then apalutamide was held as he awaited improvement of the rash.
One week after the apalutamide was held, the patient presented to dermatology. He reported that he had tried over-the-counter ammonium lactate 12% lotion twice daily when the rash first developed without improvement. When the apalutamide was held, oncology prescribed mupirocin ointment 2% 3 times daily which yielded minimal relief. On physical examination, widespread lichenified papules and plaques were noted on the face, chest, back, and legs (Figure 1). Dermatology initially prescribed triamcinolone ointment 0.1% twice daily. A 4-mm punch biopsy specimen of the upper back revealed a lichenoid interface dermatitis with numerous eosinophils compatible with a lichenoid hypersensitivity reaction (Figure 2). Considering the clinical and histologic findings, a diagnosis of lichenoid drug eruption secondary to apalutamide treatment was made.
Two weeks after discontinuation of the medication, the rash improved, and the patient restarted apalutamide at a dosage of 120 mg/d; however, the rash re-emerged within 1 month and was resistant to the triamcinolone ointment 0.1%. Apalutamide was again discontinued, and oncology switched the patient to enzalutamide 160 mg/d in an effort to find a medication the patient could better tolerate. Two months after starting enzalutamide, the patient had resolution of the rash and no further dermatologic complications.
Apalutamide is a second-generation nonsteroidal androgen receptor antagonist used in the treatment of nonmetastatic castration-resistant prostate cancer (CRPC) and metastatic castration-sensitive prostate cancer (CSPC).7 It stops the spread and growth of prostate cancer cells by several different mechanisms, including competitively binding androgen receptors, preventing 5α-dihydrotestosterone from binding to androgen receptors, blocking androgen receptor nuclear translocation, impairing co-activator recruitment, and restraining androgen receptor DNA binding.7 The SPARTAN and TITAN phase 3 clinical trials demonstrated increased overall survival and time to progression with apalutamide in both nonmetastatic CRPC and metastatic CSPC. In both trials, the rash was shown to be an AE more commonly associated with apalutamide than placebo.8,9
Until recently, the characteristics of apalutamide-induced drug rashes have not been well described. One literature review reported 6 cases of cutaneous apalutamide-induced drug eruptions.5 Four (66.7%) of these eruptions were maculopapular rashes, only 2 of which were histologically classified as lichenoid in nature. The other 2 eruptions were classified as toxic epidermal necrosis.5 Another study of 303 patients with prostate cancer who were treated with apalutamide recorded the frequency and time to onset of dermatologic AEs.6 Seventy-one (23.4%) of the patients had dermatologic AEs, and of those, only 20 (28.2%) had AEs that resulted in interruptions in apalutamide therapy (with only 5 [25.0%] requiring medication discontinuation). Thirty-two (45.1%) patients were managed with topical or oral corticosteroids or dose modification. In this study, histopathology was examined in 8 cases (one of which had 2 biopsies for a total of 9 biopsies), 7 of which were consistent with lichenoid interface dermatitis.6
Lichenoid interface dermatitis is a rare manifestation of an apalutamide-induced drug eruption and also has been reported secondary to treatment with enzalutamide, another second-generation nonsteroidal androgen receptor antagonist.4 Enzalutamide was the first second-generation nonsteroidal androgen receptor antagonist approved for the treatment of prostate cancer. It originally was approved only for metastatic CRPC after docetaxel therapy in 2012, then later was expanded to metastatic and nonmetastatic CRPC in 2012 and 2018, respectively, as well as metastatic CSPC in 2019.7 Because enzalutamide is from the same medication class as apalutamide and has been on the market longer for the treatment of nonmetastatic CRPC and metastatic CSPC, it is not surprising that similar drug eruptions now are being reported secondary to apalutamide use as well.
It is important for providers to consider lichenoid drug eruptions in the differential diagnosis of pruritic rashes in patients taking second-generation nonsteroidal androgen receptor antagonists such as apalutamide or enzalutamide. Although dose reduction or treatment discontinuation have been the standard of care for patients with extremely pruritic lichenoid drug eruptions secondary to these medications, these are not ideal because they are important for cancer treatment. Interestingly, after our patient’s apalutamide-induced rash resolved and he was switched to enzalutamide, he did not develop any AEs. Based on our patient’s experience, physicians could consider switching their patients to another drug of the same class, as they may be able tolerate that medication. More research is needed to determine how commonly patients tolerate a different second-generation nonsteroidal androgen receptor antagonist after not tolerating another medication from the same class.
- Weyers W, Metze D. Histopathology of drug eruptions—general criteria, common patterns, and differential diagnosis. Dermatol Pract Concept. 2011;1:33-47. doi:10.5826/dpc.0101a09
- Cheraghlou S, Levy LL. Fixed drug eruption, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Thompson DF, Skaehill PA. Drug-induced lichen planus. Pharmacotherapy. 1994;14:561-571.
- Khan S, Saizan AL, O’Brien K, et al. Diffuse hyperpigmented lichenoid drug eruption secondary to enzalutamide. Curr Probl Cancer Case Rep. 2022;5:100135. doi:10.1016/j.cpccr.2021.100135
- Katayama H, Saeki H, Osada S-I. Maculopapular drug eruption caused by apalutamide: case report and review of the literature. J Nippon Med Sch. 2022;89:550-554. doi:10.1272/jnms.JNMS.2022_89-503
- Pan A, Reingold RE, Zhao JL, et al. Dermatologic adverse events in prostate cancer patients treated with the androgen receptor inhibitor apalutamide. J Urol. 2022;207:1010-1019. doi:10.1097/JU.0000000000002425
- Rajaram P, Rivera A, Muthima K, et al. Second-generation androgen receptor antagonists as hormonal therapeutics for three forms of prostate cancer. Molecules. 2020;25:2448. doi:10.3390/molecules25102448
- Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408-1418. doi:10.1056/NEJMoa1715546
- Chi KN, Agarwal N, Bjartell A, et al. Apalutamide for metastatic, castration-sensative prostate cancer. N Engl J Med. 2019;381:13-24. doi:10.1056/NEJMoa1903307
- Weyers W, Metze D. Histopathology of drug eruptions—general criteria, common patterns, and differential diagnosis. Dermatol Pract Concept. 2011;1:33-47. doi:10.5826/dpc.0101a09
- Cheraghlou S, Levy LL. Fixed drug eruption, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Thompson DF, Skaehill PA. Drug-induced lichen planus. Pharmacotherapy. 1994;14:561-571.
- Khan S, Saizan AL, O’Brien K, et al. Diffuse hyperpigmented lichenoid drug eruption secondary to enzalutamide. Curr Probl Cancer Case Rep. 2022;5:100135. doi:10.1016/j.cpccr.2021.100135
- Katayama H, Saeki H, Osada S-I. Maculopapular drug eruption caused by apalutamide: case report and review of the literature. J Nippon Med Sch. 2022;89:550-554. doi:10.1272/jnms.JNMS.2022_89-503
- Pan A, Reingold RE, Zhao JL, et al. Dermatologic adverse events in prostate cancer patients treated with the androgen receptor inhibitor apalutamide. J Urol. 2022;207:1010-1019. doi:10.1097/JU.0000000000002425
- Rajaram P, Rivera A, Muthima K, et al. Second-generation androgen receptor antagonists as hormonal therapeutics for three forms of prostate cancer. Molecules. 2020;25:2448. doi:10.3390/molecules25102448
- Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408-1418. doi:10.1056/NEJMoa1715546
- Chi KN, Agarwal N, Bjartell A, et al. Apalutamide for metastatic, castration-sensative prostate cancer. N Engl J Med. 2019;381:13-24. doi:10.1056/NEJMoa1903307
Practice Points
- Although it is rare, patients can develop lichenoid drug eruptions secondary to treatment with second-generation nonsteroidal androgen receptor antagonists such as apalutamide.
- If a patient develops a lichenoid drug eruption while taking a specific second-generation nonsteroidal androgen receptor antagonist, the entire class of medications should not be ruled out, as some patients can tolerate other drugs from that class.
Botulinum Toxin Injection for Treatment of Scleroderma-Related Anterior Neck Sclerosis
To the Editor:
Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body. On its own or in the setting of mixed connective tissue disease, scleroderma can result in systemic or localized symptoms that can limit patients’ functional capabilities, cause pain and discomfort, and reduce self-esteem—all negatively impacting patients’ quality of life.1,2 Neck sclerosis is a common manifestation of scleroderma. There is no curative treatment for scleroderma; thus, therapy is focused on slowing disease progression and improving quality of life. We present a case of neck sclerosis in a 44-year-old woman with scleroderma that was successfully treated with botulinum toxin (BTX) type A injection, resulting in improved skin laxity and appearance with high patient satisfaction. Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region.
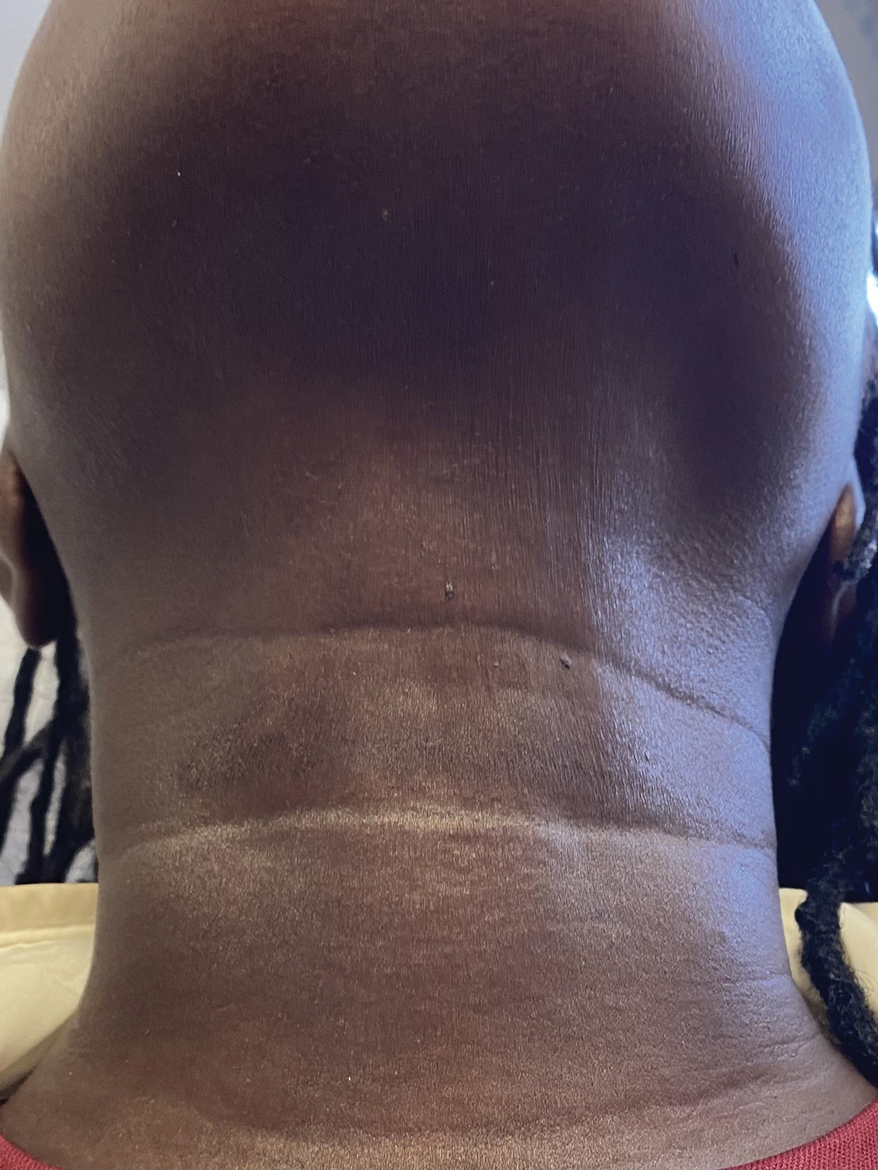
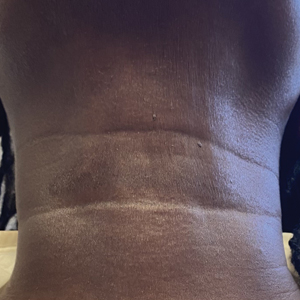
A 44-year-old woman presented to the dermatology clinic for treatment of thickened neck skin with stiffness and tightness that had been present for months to years. She had a history of mixed connective tissue disease (MCTD)(positive anti-ribonucleoprotein, anti–Sjögren syndrome–related antigen, and anti-Smith antibodies) with features of scleroderma and polyarthritis. The patient currently was taking sulfasalazine for the polyarthritis; she previously had taken hydroxychloroquine but discontinued treatment due to ineffectiveness. She was not taking any topical or systemic medications for scleroderma. On physical examination, the skin on the anterior neck appeared thickened with shiny patches (Figure 1). Pinching the skin in the affected area demonstrated sclerosis with high tension.
The dermatologist (J.J.) discussed potential treatment options to help relax the tension in the skin of the anterior neck, including BTX injections. After receiving counsel on adverse effects, alternative treatments, and postprocedural care, the patient decided to proceed with the procedure. The anterior neck was cleansed with an alcohol swab and 37 units (range, 25–50 units) of incobotulinumtoxinA (reconstituted using 2.5-mL bacteriostatic normal saline per 100 units) was injected transdermally using a 9-point injection technique, with each injection placed approximately 1 cm apart. The approximate treatment area included the space between the sternocleidomastoid anterior edges and below the hyoid bone up to the cricothyroid membrane (anatomic zone II).
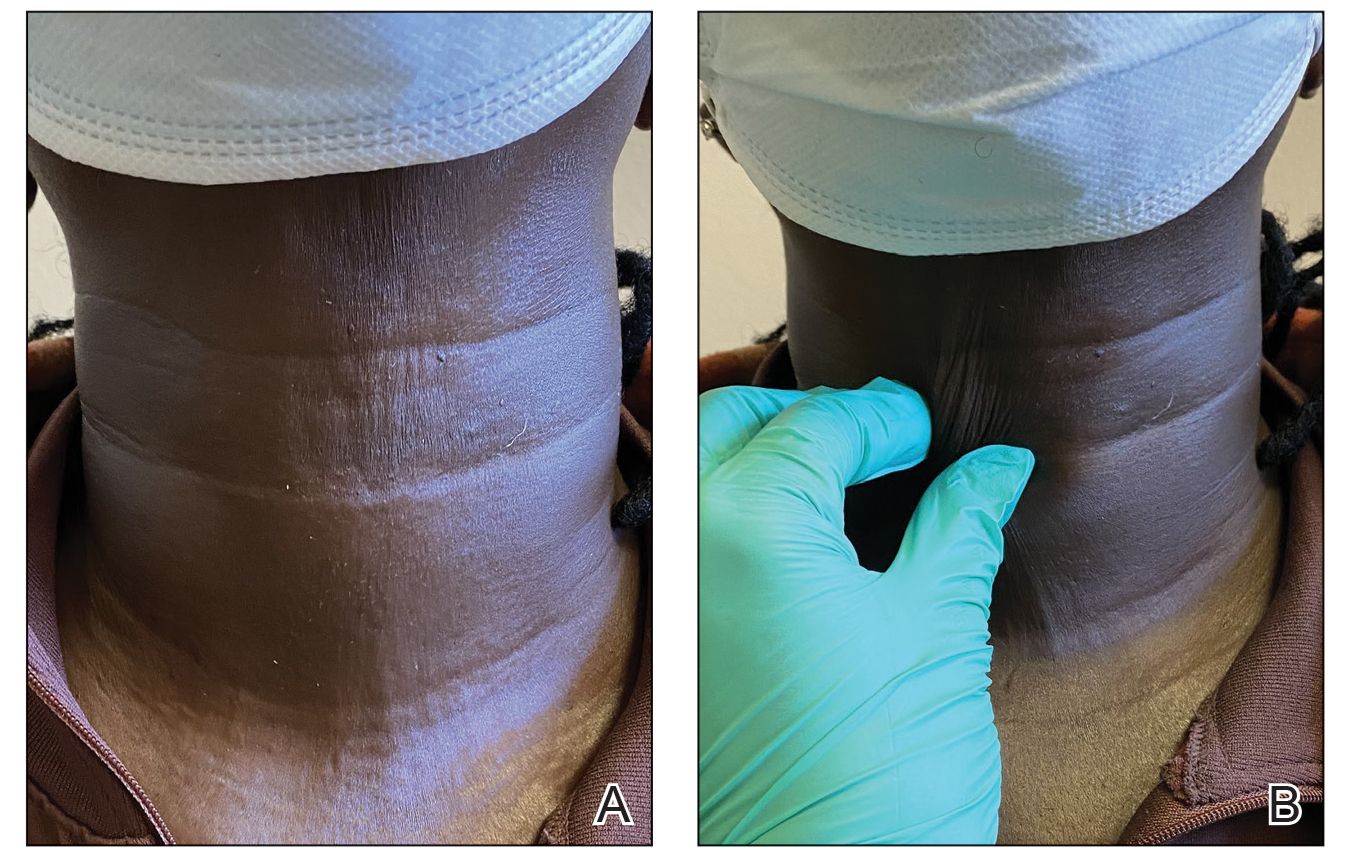
When the patient returned for follow-up 3 weeks later, she reported considerable improvement in the stiffness and appearance of the skin on the anterior neck. On physical examination, the skin of the neck appeared softened, and improved laxity was seen on pinching the skin compared to the initial presentation (Figure 2). The patient expressed satisfaction with the results and denied any adverse events following the procedure.
Mixed connective tissue disease manifests with a combination of features from various disorders—mainly lupus, scleroderma, polymyositis, and rheumatoid arthritis. It is most prevalent in females and often is diagnosed in the third decade of life.3 It is associated with positive antinuclear antibodies and human leukocyte antigen (HLA) II alleles (HLA-DR4, HLA-DR1, and HLA-DR2). Raynaud phenomenon (RP), one of the most common skin manifestations in both scleroderma and MCTD, is present in 75% to 90% of patients with MCTD.3
Scleroderma is a chronic connective tissue disorder that results in excessive collagen deposition in the skin and other organs throughout the body.4 Although the etiology is unknown, scleroderma develops when overactivation of the immune system leads to CD4+ T-lymphocyte infiltration in the skin, along with the release of profibrotic interleukins and growth factors, resulting in fibrosis.4 Subtypes include localized scleroderma (morphea), limited cutaneous systemic sclerosis (formerly known as CREST [calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome), diffuse cutaneous systemic sclerosis, and systemic sclerosis sine scleroderma.5 Scleroderma is associated with positive antinuclear antibodies and HLA II alleles (HLA-DR2 and HLA-DR5).
On its own or in the setting of MCTD, scleroderma can result in systemic or localized symptoms. Overall, the most common symptom is RP.5 Localized scleroderma and limited cutaneous systemic sclerosis manifest with symptoms of the skin and underlying tissues. Diffuse cutaneous systemic sclerosis involves cutaneous and visceral symptoms, including lung, esophageal, and vascular involvement.6 Similar to MCTD, scleroderma is most prevalent in middle-aged females,7 though it occurs at a higher rate and with a more severe disease course in Black patients.8
A highly sensitive and specific test for scleroderma that can aid in diagnosis is the neck sign—tightening of the skin of the neck when the head extends.9,10 In one study, the neck sign was positive in more than 90% of patients with scleroderma and negative for control patients and those with primary RP.9 Thus, neck sclerosis is a common manifestation of scleroderma for which patients may seek treatment.
While there is no curative treatment for scleroderma, skin manifestations can be treated with mycophenolate mofetil or methotrexate.5 Systemic treatments may be recommended if the patient has additional symptoms, such as azathioprine for myositis/arthritis and cyclophosphamide for interstitial lung disease.5 However, it is important to note that these medications are associated with risk for gastrointestinal upset, mouth sores, fatigue, or other complications.
Botulinum toxin is a bacterial protein toxin and neuromodulator that inhibits neurotransmitter release by cleaving SNARE proteins at peripheral nerve terminal junctions.11 It has been used in a variety of dermatologic and nondermatologic conditions, including migraines, hyperhidrosis, contractures, scars, and overactive bladder. It also has been used in aesthetics for facial rejuvenation and minimization of wrinkle appearance. Dermatologists and rheumatologists have successfully used BTX to treat primary and secondary RP—the most common symptom of scleroderma—due to its vasodilatation properties.12 Although our patient did not have RP, use of BTX to treat other features of scleroderma, including en coup de sabre, thoracic outlet syndrome, dyspareunia, gastroparesis, pterygium inversum unguis, and dysphagia has been documented.13-18 An in vivo mouse study that examined the possible mechanism for BTX as a treatment in scleroderma found that BTX injections significantly decreased dermal thickness and inflammation in fibrosis (P<.05). An analysis of oxidative stress and mRNA expression showed that BTX may treat fibrosis by suppressing oxidative stress and inflammatory cells, resulting in decreased apoptosis and oxidant-induced intracellular accumulation of reactive oxygen species.19 Another animal study demonstrated the positive effects of BTX treatment for fibrosis of the bladder in rats.20 In one case report, a female patient with scleroderma and facial fibrosis received perioral BTX injections for cosmetic purposes but also observed improvement in mouth constriction, demonstrating the potential efficacy of BTX for facial fibrosis.21
Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region. We recommend assessing the efficacy of the initial BTX treatment after 2 to 3 weeks, with additional injections as needed to achieve the patient’s desired level of comfort and appearance at approximately 3-month intervals (aligning with the expected duration of efficacy of BTX).22 Our patient experienced considerable relief and high satisfaction with BTX treatment. Given the limitations of sclerosis treatments and the unwanted adverse-effect profile of systemic treatments, BTX injections may be a preferrable treatment option for cutaneous manifestations of scleroderma among patients. Future studies with larger patient populations and a control group are warranted to further explore the use of BTX for the dermatologic treatment of scleroderma.
- Lis-S´wie¸ty A, Skrzypek-Salamon A, Ranosz-Janicka I, et al. Health-related quality of life and its influencing factors in adult patients with localized scleroderma—a cross-sectional study. Health Qual Life Outcomes. 2020;18:133. doi:10.1186/s12955-020-01386-0
- Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. 2015;14:1087-1096. doi:10.1016/j.autrev.2015.07.012
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol. 2012;26:61-72. doi:10.1016/j.berh.2012.01.009
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinico-pathological correlation. G Ital Dermatol Venereol. 2018;153:208-215. doi:10.23736/S0392-0488.18.05922-9
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437. doi:10.1016/j.clindermatol.2013.01.010
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Calderon LM, Pope JE. Scleroderma epidemiology update. Curr Opin Rheumatol. 2021;33:122-127. doi:10.1097/BOR.0000000000000785
- Morgan ND, Gelber AC. African Americans and scleroderma: examining the root cause of the association. Arthritis Care Res (Hoboken). 2019;71:1151-1153. doi:10.1002/acr.23860
- Barnett AJ. The “neck sign” in scleroderma. Arthritis Rheum. 1989;32:209-211. doi:10.1002/anr.1780320215
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J. 1988;64:121-125. doi:10.1136/pgmj.64.748.121
- Rossetto O, Pirazzini M, Fabris F, et al. Botulinum neurotoxins: mechanism of action. Handb Exp Pharmacol. 2021;263:35-47.doi:10.1007/164_2020_355
- Ennis D, Ahmad Z, Anderson MA, et al. Botulinum toxin in the management of primary and secondary Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2021;35:101684. doi:10.1016/j.berh.2021.101684
- Turkmani MG, Alnomair N. Enhancement of the aesthetic outcome of scleroderma en coup de sabre with botulinum toxin injection. JAAD Case Rep. 2018;4:579-581. doi:10.1016/j.jdcr.2018.03.023
- Le EN, Freischlag JA, Christo PJ, et al. Thoracic outlet syndrome secondary to localized scleroderma treated with botulinum toxin injection. Arthritis Care Res (Hoboken). 2010;62:430-433. doi:10.1002/acr.20099
- Mousty E, Rathat G, Rouleau C, et al. Botulinum toxin type A for treatment of dyspareunia caused by localized scleroderma. Acta Obstet Gynecol Scand. 2011;90:926-927. doi:10.1111/j.1600-0412.2011.01183.x
- Tang DM, Friedenberg FK. Gastroparesis: approach, diagnostic evaluation, and management. Dis Mon. 2011;57:74-101. doi:10.1016/j.disamonth.2010.12.007
- Katschinski M. [Diagnosis and treatment of esophageal motility disorders]. Ther Umsch. 2001;58:128-133. doi:10.1024/0040-5930.58.3.128
- Kim DJ, Odell ID. Improvement of pterygium inversum unguis and Raynaud phenomenon with interdigital botulinum toxin injections. JAAD Case Rep. 2022;26:79-81. doi:10.1016/j.jdcr.2022.06.009
- Baral H, Sekiguchi A, Uchiyama A, et al. Inhibition of skin fibrosis in systemic sclerosis by botulinum toxin B via the suppression of oxidative stress. J Dermatol. 2021;48:1052-1061. doi:10.1111/1346-8138.15888
- Jia C, Xing T, Shang Z, et al. Botulinum toxin A improves neurogenic bladder fibrosis by suppressing transforming growth factor β1 expression in rats. Transl Androl Urol. 2021;10:2000-2007. doi:10.21037/tau-21-62
- Hoverson K, Love T, Lam TK, et al. A novel treatment for limited mouth opening due to facial fibrosis: a case series. J Am Acad Dermatol. 2018;78:190-192. doi:10.1016/j.jaad.2017.07.006
- Kollewe K, Mohammadi B, Köhler S, et al. Blepharospasm: long-term treatment with either Botox®, Xeomin® or Dysport®. J Neural Transm (Vienna). 2015;122:427-431. doi:10.1007/s00702-014-1278-z
To the Editor:
Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body. On its own or in the setting of mixed connective tissue disease, scleroderma can result in systemic or localized symptoms that can limit patients’ functional capabilities, cause pain and discomfort, and reduce self-esteem—all negatively impacting patients’ quality of life.1,2 Neck sclerosis is a common manifestation of scleroderma. There is no curative treatment for scleroderma; thus, therapy is focused on slowing disease progression and improving quality of life. We present a case of neck sclerosis in a 44-year-old woman with scleroderma that was successfully treated with botulinum toxin (BTX) type A injection, resulting in improved skin laxity and appearance with high patient satisfaction. Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region.
A 44-year-old woman presented to the dermatology clinic for treatment of thickened neck skin with stiffness and tightness that had been present for months to years. She had a history of mixed connective tissue disease (MCTD)(positive anti-ribonucleoprotein, anti–Sjögren syndrome–related antigen, and anti-Smith antibodies) with features of scleroderma and polyarthritis. The patient currently was taking sulfasalazine for the polyarthritis; she previously had taken hydroxychloroquine but discontinued treatment due to ineffectiveness. She was not taking any topical or systemic medications for scleroderma. On physical examination, the skin on the anterior neck appeared thickened with shiny patches (Figure 1). Pinching the skin in the affected area demonstrated sclerosis with high tension.
The dermatologist (J.J.) discussed potential treatment options to help relax the tension in the skin of the anterior neck, including BTX injections. After receiving counsel on adverse effects, alternative treatments, and postprocedural care, the patient decided to proceed with the procedure. The anterior neck was cleansed with an alcohol swab and 37 units (range, 25–50 units) of incobotulinumtoxinA (reconstituted using 2.5-mL bacteriostatic normal saline per 100 units) was injected transdermally using a 9-point injection technique, with each injection placed approximately 1 cm apart. The approximate treatment area included the space between the sternocleidomastoid anterior edges and below the hyoid bone up to the cricothyroid membrane (anatomic zone II).
When the patient returned for follow-up 3 weeks later, she reported considerable improvement in the stiffness and appearance of the skin on the anterior neck. On physical examination, the skin of the neck appeared softened, and improved laxity was seen on pinching the skin compared to the initial presentation (Figure 2). The patient expressed satisfaction with the results and denied any adverse events following the procedure.
Mixed connective tissue disease manifests with a combination of features from various disorders—mainly lupus, scleroderma, polymyositis, and rheumatoid arthritis. It is most prevalent in females and often is diagnosed in the third decade of life.3 It is associated with positive antinuclear antibodies and human leukocyte antigen (HLA) II alleles (HLA-DR4, HLA-DR1, and HLA-DR2). Raynaud phenomenon (RP), one of the most common skin manifestations in both scleroderma and MCTD, is present in 75% to 90% of patients with MCTD.3
Scleroderma is a chronic connective tissue disorder that results in excessive collagen deposition in the skin and other organs throughout the body.4 Although the etiology is unknown, scleroderma develops when overactivation of the immune system leads to CD4+ T-lymphocyte infiltration in the skin, along with the release of profibrotic interleukins and growth factors, resulting in fibrosis.4 Subtypes include localized scleroderma (morphea), limited cutaneous systemic sclerosis (formerly known as CREST [calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome), diffuse cutaneous systemic sclerosis, and systemic sclerosis sine scleroderma.5 Scleroderma is associated with positive antinuclear antibodies and HLA II alleles (HLA-DR2 and HLA-DR5).
On its own or in the setting of MCTD, scleroderma can result in systemic or localized symptoms. Overall, the most common symptom is RP.5 Localized scleroderma and limited cutaneous systemic sclerosis manifest with symptoms of the skin and underlying tissues. Diffuse cutaneous systemic sclerosis involves cutaneous and visceral symptoms, including lung, esophageal, and vascular involvement.6 Similar to MCTD, scleroderma is most prevalent in middle-aged females,7 though it occurs at a higher rate and with a more severe disease course in Black patients.8
A highly sensitive and specific test for scleroderma that can aid in diagnosis is the neck sign—tightening of the skin of the neck when the head extends.9,10 In one study, the neck sign was positive in more than 90% of patients with scleroderma and negative for control patients and those with primary RP.9 Thus, neck sclerosis is a common manifestation of scleroderma for which patients may seek treatment.
While there is no curative treatment for scleroderma, skin manifestations can be treated with mycophenolate mofetil or methotrexate.5 Systemic treatments may be recommended if the patient has additional symptoms, such as azathioprine for myositis/arthritis and cyclophosphamide for interstitial lung disease.5 However, it is important to note that these medications are associated with risk for gastrointestinal upset, mouth sores, fatigue, or other complications.
Botulinum toxin is a bacterial protein toxin and neuromodulator that inhibits neurotransmitter release by cleaving SNARE proteins at peripheral nerve terminal junctions.11 It has been used in a variety of dermatologic and nondermatologic conditions, including migraines, hyperhidrosis, contractures, scars, and overactive bladder. It also has been used in aesthetics for facial rejuvenation and minimization of wrinkle appearance. Dermatologists and rheumatologists have successfully used BTX to treat primary and secondary RP—the most common symptom of scleroderma—due to its vasodilatation properties.12 Although our patient did not have RP, use of BTX to treat other features of scleroderma, including en coup de sabre, thoracic outlet syndrome, dyspareunia, gastroparesis, pterygium inversum unguis, and dysphagia has been documented.13-18 An in vivo mouse study that examined the possible mechanism for BTX as a treatment in scleroderma found that BTX injections significantly decreased dermal thickness and inflammation in fibrosis (P<.05). An analysis of oxidative stress and mRNA expression showed that BTX may treat fibrosis by suppressing oxidative stress and inflammatory cells, resulting in decreased apoptosis and oxidant-induced intracellular accumulation of reactive oxygen species.19 Another animal study demonstrated the positive effects of BTX treatment for fibrosis of the bladder in rats.20 In one case report, a female patient with scleroderma and facial fibrosis received perioral BTX injections for cosmetic purposes but also observed improvement in mouth constriction, demonstrating the potential efficacy of BTX for facial fibrosis.21
Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region. We recommend assessing the efficacy of the initial BTX treatment after 2 to 3 weeks, with additional injections as needed to achieve the patient’s desired level of comfort and appearance at approximately 3-month intervals (aligning with the expected duration of efficacy of BTX).22 Our patient experienced considerable relief and high satisfaction with BTX treatment. Given the limitations of sclerosis treatments and the unwanted adverse-effect profile of systemic treatments, BTX injections may be a preferrable treatment option for cutaneous manifestations of scleroderma among patients. Future studies with larger patient populations and a control group are warranted to further explore the use of BTX for the dermatologic treatment of scleroderma.
To the Editor:
Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body. On its own or in the setting of mixed connective tissue disease, scleroderma can result in systemic or localized symptoms that can limit patients’ functional capabilities, cause pain and discomfort, and reduce self-esteem—all negatively impacting patients’ quality of life.1,2 Neck sclerosis is a common manifestation of scleroderma. There is no curative treatment for scleroderma; thus, therapy is focused on slowing disease progression and improving quality of life. We present a case of neck sclerosis in a 44-year-old woman with scleroderma that was successfully treated with botulinum toxin (BTX) type A injection, resulting in improved skin laxity and appearance with high patient satisfaction. Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region.
A 44-year-old woman presented to the dermatology clinic for treatment of thickened neck skin with stiffness and tightness that had been present for months to years. She had a history of mixed connective tissue disease (MCTD)(positive anti-ribonucleoprotein, anti–Sjögren syndrome–related antigen, and anti-Smith antibodies) with features of scleroderma and polyarthritis. The patient currently was taking sulfasalazine for the polyarthritis; she previously had taken hydroxychloroquine but discontinued treatment due to ineffectiveness. She was not taking any topical or systemic medications for scleroderma. On physical examination, the skin on the anterior neck appeared thickened with shiny patches (Figure 1). Pinching the skin in the affected area demonstrated sclerosis with high tension.
The dermatologist (J.J.) discussed potential treatment options to help relax the tension in the skin of the anterior neck, including BTX injections. After receiving counsel on adverse effects, alternative treatments, and postprocedural care, the patient decided to proceed with the procedure. The anterior neck was cleansed with an alcohol swab and 37 units (range, 25–50 units) of incobotulinumtoxinA (reconstituted using 2.5-mL bacteriostatic normal saline per 100 units) was injected transdermally using a 9-point injection technique, with each injection placed approximately 1 cm apart. The approximate treatment area included the space between the sternocleidomastoid anterior edges and below the hyoid bone up to the cricothyroid membrane (anatomic zone II).
When the patient returned for follow-up 3 weeks later, she reported considerable improvement in the stiffness and appearance of the skin on the anterior neck. On physical examination, the skin of the neck appeared softened, and improved laxity was seen on pinching the skin compared to the initial presentation (Figure 2). The patient expressed satisfaction with the results and denied any adverse events following the procedure.
Mixed connective tissue disease manifests with a combination of features from various disorders—mainly lupus, scleroderma, polymyositis, and rheumatoid arthritis. It is most prevalent in females and often is diagnosed in the third decade of life.3 It is associated with positive antinuclear antibodies and human leukocyte antigen (HLA) II alleles (HLA-DR4, HLA-DR1, and HLA-DR2). Raynaud phenomenon (RP), one of the most common skin manifestations in both scleroderma and MCTD, is present in 75% to 90% of patients with MCTD.3
Scleroderma is a chronic connective tissue disorder that results in excessive collagen deposition in the skin and other organs throughout the body.4 Although the etiology is unknown, scleroderma develops when overactivation of the immune system leads to CD4+ T-lymphocyte infiltration in the skin, along with the release of profibrotic interleukins and growth factors, resulting in fibrosis.4 Subtypes include localized scleroderma (morphea), limited cutaneous systemic sclerosis (formerly known as CREST [calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome), diffuse cutaneous systemic sclerosis, and systemic sclerosis sine scleroderma.5 Scleroderma is associated with positive antinuclear antibodies and HLA II alleles (HLA-DR2 and HLA-DR5).
On its own or in the setting of MCTD, scleroderma can result in systemic or localized symptoms. Overall, the most common symptom is RP.5 Localized scleroderma and limited cutaneous systemic sclerosis manifest with symptoms of the skin and underlying tissues. Diffuse cutaneous systemic sclerosis involves cutaneous and visceral symptoms, including lung, esophageal, and vascular involvement.6 Similar to MCTD, scleroderma is most prevalent in middle-aged females,7 though it occurs at a higher rate and with a more severe disease course in Black patients.8
A highly sensitive and specific test for scleroderma that can aid in diagnosis is the neck sign—tightening of the skin of the neck when the head extends.9,10 In one study, the neck sign was positive in more than 90% of patients with scleroderma and negative for control patients and those with primary RP.9 Thus, neck sclerosis is a common manifestation of scleroderma for which patients may seek treatment.
While there is no curative treatment for scleroderma, skin manifestations can be treated with mycophenolate mofetil or methotrexate.5 Systemic treatments may be recommended if the patient has additional symptoms, such as azathioprine for myositis/arthritis and cyclophosphamide for interstitial lung disease.5 However, it is important to note that these medications are associated with risk for gastrointestinal upset, mouth sores, fatigue, or other complications.
Botulinum toxin is a bacterial protein toxin and neuromodulator that inhibits neurotransmitter release by cleaving SNARE proteins at peripheral nerve terminal junctions.11 It has been used in a variety of dermatologic and nondermatologic conditions, including migraines, hyperhidrosis, contractures, scars, and overactive bladder. It also has been used in aesthetics for facial rejuvenation and minimization of wrinkle appearance. Dermatologists and rheumatologists have successfully used BTX to treat primary and secondary RP—the most common symptom of scleroderma—due to its vasodilatation properties.12 Although our patient did not have RP, use of BTX to treat other features of scleroderma, including en coup de sabre, thoracic outlet syndrome, dyspareunia, gastroparesis, pterygium inversum unguis, and dysphagia has been documented.13-18 An in vivo mouse study that examined the possible mechanism for BTX as a treatment in scleroderma found that BTX injections significantly decreased dermal thickness and inflammation in fibrosis (P<.05). An analysis of oxidative stress and mRNA expression showed that BTX may treat fibrosis by suppressing oxidative stress and inflammatory cells, resulting in decreased apoptosis and oxidant-induced intracellular accumulation of reactive oxygen species.19 Another animal study demonstrated the positive effects of BTX treatment for fibrosis of the bladder in rats.20 In one case report, a female patient with scleroderma and facial fibrosis received perioral BTX injections for cosmetic purposes but also observed improvement in mouth constriction, demonstrating the potential efficacy of BTX for facial fibrosis.21
Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region. We recommend assessing the efficacy of the initial BTX treatment after 2 to 3 weeks, with additional injections as needed to achieve the patient’s desired level of comfort and appearance at approximately 3-month intervals (aligning with the expected duration of efficacy of BTX).22 Our patient experienced considerable relief and high satisfaction with BTX treatment. Given the limitations of sclerosis treatments and the unwanted adverse-effect profile of systemic treatments, BTX injections may be a preferrable treatment option for cutaneous manifestations of scleroderma among patients. Future studies with larger patient populations and a control group are warranted to further explore the use of BTX for the dermatologic treatment of scleroderma.
- Lis-S´wie¸ty A, Skrzypek-Salamon A, Ranosz-Janicka I, et al. Health-related quality of life and its influencing factors in adult patients with localized scleroderma—a cross-sectional study. Health Qual Life Outcomes. 2020;18:133. doi:10.1186/s12955-020-01386-0
- Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. 2015;14:1087-1096. doi:10.1016/j.autrev.2015.07.012
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol. 2012;26:61-72. doi:10.1016/j.berh.2012.01.009
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinico-pathological correlation. G Ital Dermatol Venereol. 2018;153:208-215. doi:10.23736/S0392-0488.18.05922-9
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437. doi:10.1016/j.clindermatol.2013.01.010
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Calderon LM, Pope JE. Scleroderma epidemiology update. Curr Opin Rheumatol. 2021;33:122-127. doi:10.1097/BOR.0000000000000785
- Morgan ND, Gelber AC. African Americans and scleroderma: examining the root cause of the association. Arthritis Care Res (Hoboken). 2019;71:1151-1153. doi:10.1002/acr.23860
- Barnett AJ. The “neck sign” in scleroderma. Arthritis Rheum. 1989;32:209-211. doi:10.1002/anr.1780320215
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J. 1988;64:121-125. doi:10.1136/pgmj.64.748.121
- Rossetto O, Pirazzini M, Fabris F, et al. Botulinum neurotoxins: mechanism of action. Handb Exp Pharmacol. 2021;263:35-47.doi:10.1007/164_2020_355
- Ennis D, Ahmad Z, Anderson MA, et al. Botulinum toxin in the management of primary and secondary Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2021;35:101684. doi:10.1016/j.berh.2021.101684
- Turkmani MG, Alnomair N. Enhancement of the aesthetic outcome of scleroderma en coup de sabre with botulinum toxin injection. JAAD Case Rep. 2018;4:579-581. doi:10.1016/j.jdcr.2018.03.023
- Le EN, Freischlag JA, Christo PJ, et al. Thoracic outlet syndrome secondary to localized scleroderma treated with botulinum toxin injection. Arthritis Care Res (Hoboken). 2010;62:430-433. doi:10.1002/acr.20099
- Mousty E, Rathat G, Rouleau C, et al. Botulinum toxin type A for treatment of dyspareunia caused by localized scleroderma. Acta Obstet Gynecol Scand. 2011;90:926-927. doi:10.1111/j.1600-0412.2011.01183.x
- Tang DM, Friedenberg FK. Gastroparesis: approach, diagnostic evaluation, and management. Dis Mon. 2011;57:74-101. doi:10.1016/j.disamonth.2010.12.007
- Katschinski M. [Diagnosis and treatment of esophageal motility disorders]. Ther Umsch. 2001;58:128-133. doi:10.1024/0040-5930.58.3.128
- Kim DJ, Odell ID. Improvement of pterygium inversum unguis and Raynaud phenomenon with interdigital botulinum toxin injections. JAAD Case Rep. 2022;26:79-81. doi:10.1016/j.jdcr.2022.06.009
- Baral H, Sekiguchi A, Uchiyama A, et al. Inhibition of skin fibrosis in systemic sclerosis by botulinum toxin B via the suppression of oxidative stress. J Dermatol. 2021;48:1052-1061. doi:10.1111/1346-8138.15888
- Jia C, Xing T, Shang Z, et al. Botulinum toxin A improves neurogenic bladder fibrosis by suppressing transforming growth factor β1 expression in rats. Transl Androl Urol. 2021;10:2000-2007. doi:10.21037/tau-21-62
- Hoverson K, Love T, Lam TK, et al. A novel treatment for limited mouth opening due to facial fibrosis: a case series. J Am Acad Dermatol. 2018;78:190-192. doi:10.1016/j.jaad.2017.07.006
- Kollewe K, Mohammadi B, Köhler S, et al. Blepharospasm: long-term treatment with either Botox®, Xeomin® or Dysport®. J Neural Transm (Vienna). 2015;122:427-431. doi:10.1007/s00702-014-1278-z
- Lis-S´wie¸ty A, Skrzypek-Salamon A, Ranosz-Janicka I, et al. Health-related quality of life and its influencing factors in adult patients with localized scleroderma—a cross-sectional study. Health Qual Life Outcomes. 2020;18:133. doi:10.1186/s12955-020-01386-0
- Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. 2015;14:1087-1096. doi:10.1016/j.autrev.2015.07.012
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol. 2012;26:61-72. doi:10.1016/j.berh.2012.01.009
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinico-pathological correlation. G Ital Dermatol Venereol. 2018;153:208-215. doi:10.23736/S0392-0488.18.05922-9
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437. doi:10.1016/j.clindermatol.2013.01.010
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Calderon LM, Pope JE. Scleroderma epidemiology update. Curr Opin Rheumatol. 2021;33:122-127. doi:10.1097/BOR.0000000000000785
- Morgan ND, Gelber AC. African Americans and scleroderma: examining the root cause of the association. Arthritis Care Res (Hoboken). 2019;71:1151-1153. doi:10.1002/acr.23860
- Barnett AJ. The “neck sign” in scleroderma. Arthritis Rheum. 1989;32:209-211. doi:10.1002/anr.1780320215
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J. 1988;64:121-125. doi:10.1136/pgmj.64.748.121
- Rossetto O, Pirazzini M, Fabris F, et al. Botulinum neurotoxins: mechanism of action. Handb Exp Pharmacol. 2021;263:35-47.doi:10.1007/164_2020_355
- Ennis D, Ahmad Z, Anderson MA, et al. Botulinum toxin in the management of primary and secondary Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2021;35:101684. doi:10.1016/j.berh.2021.101684
- Turkmani MG, Alnomair N. Enhancement of the aesthetic outcome of scleroderma en coup de sabre with botulinum toxin injection. JAAD Case Rep. 2018;4:579-581. doi:10.1016/j.jdcr.2018.03.023
- Le EN, Freischlag JA, Christo PJ, et al. Thoracic outlet syndrome secondary to localized scleroderma treated with botulinum toxin injection. Arthritis Care Res (Hoboken). 2010;62:430-433. doi:10.1002/acr.20099
- Mousty E, Rathat G, Rouleau C, et al. Botulinum toxin type A for treatment of dyspareunia caused by localized scleroderma. Acta Obstet Gynecol Scand. 2011;90:926-927. doi:10.1111/j.1600-0412.2011.01183.x
- Tang DM, Friedenberg FK. Gastroparesis: approach, diagnostic evaluation, and management. Dis Mon. 2011;57:74-101. doi:10.1016/j.disamonth.2010.12.007
- Katschinski M. [Diagnosis and treatment of esophageal motility disorders]. Ther Umsch. 2001;58:128-133. doi:10.1024/0040-5930.58.3.128
- Kim DJ, Odell ID. Improvement of pterygium inversum unguis and Raynaud phenomenon with interdigital botulinum toxin injections. JAAD Case Rep. 2022;26:79-81. doi:10.1016/j.jdcr.2022.06.009
- Baral H, Sekiguchi A, Uchiyama A, et al. Inhibition of skin fibrosis in systemic sclerosis by botulinum toxin B via the suppression of oxidative stress. J Dermatol. 2021;48:1052-1061. doi:10.1111/1346-8138.15888
- Jia C, Xing T, Shang Z, et al. Botulinum toxin A improves neurogenic bladder fibrosis by suppressing transforming growth factor β1 expression in rats. Transl Androl Urol. 2021;10:2000-2007. doi:10.21037/tau-21-62
- Hoverson K, Love T, Lam TK, et al. A novel treatment for limited mouth opening due to facial fibrosis: a case series. J Am Acad Dermatol. 2018;78:190-192. doi:10.1016/j.jaad.2017.07.006
- Kollewe K, Mohammadi B, Köhler S, et al. Blepharospasm: long-term treatment with either Botox®, Xeomin® or Dysport®. J Neural Transm (Vienna). 2015;122:427-431. doi:10.1007/s00702-014-1278-z
Practice Points
- Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body.
- Although there is no curative treatment for scleroderma, there are options to slow disease progression and improve quality of life.
- Botulinum toxin injection may be a preferred treatment option in patients with features of sclerosis or fibrosis related to scleroderma, particularly in the neck region.
Phenytoin-Induced DRESS Syndrome: Clinical and Laboratory Characteristics
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome—a severe cutaneous adverse drug reaction—is characterized by a cutaneous rash and systemic upset in the form of various internal organ and hematologic disturbances. This delayed and idiosyncratic syndrome went by several names, including anticonvulsant hypersensitivity syndrome, before Bocquet et al1 proposed the term DRESS syndrome.
Phenytoin, a hydantoin derivative used in neurology, was implicated in 41% of cases of DRESS syndrome in a study of 100 patients conducted in southern India.2,3 While DRESS syndrome is a newer name, the clinical picture of DRESS secondary to phenytoin use remains similar in that it manifests with a morbilliform rash and systemic upset. We sought to describe the clinical and laboratory characteristics of phenytoin-induced DRESS syndrome in this case series.
The analysis included 23 patients with DRESS syndrome secondary to phenytoin use who presented to a tertiary care institution in North India between July 2021 and December 2022, satisfied the European Registry of Severe Cutaneous Adverse Reaction (RegiSCAR) criteria,4 and achieved a DRESS diagnostic score of more than 1. The mean age of the patients was 44 years (range, 14–74 years). There was a slight female predominance with a male to female ratio of 0.9:1. More than half of the patients (52.2% [12/23]) presented directly to the dermatology outpatient department; the remaining patients were referred from other departments (47.8% [11/23]). Patients primarily were receiving phenytoin for neurologic indications. Specific reasons included antiseizure prophylaxis following a traffic accident (34.8% [8/23]); epilepsy (26.1% [6/23]); and neoplastic (17.4% [4/23]), vascular (17.4% [4/23]), and infectious (4.3% [1/23]) causes. The mean latency period from drug intake to symptom onset was 29 days (range, 6–62 days), and the mean illness duration was 9 days (range, 1–45 days).
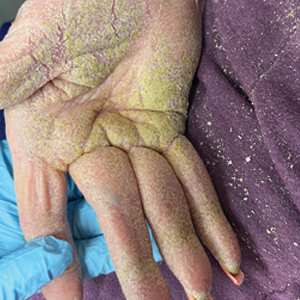
The majority of patients experienced pruritus (91.3% [21/23]) and fever (74.0% [17/23]), and all initially had a rash. Maculopapular morphology was seen in all patients. Erythema multiforme–like (17.4% [4/23]), erythrodermic (17.4% [4/23]), and vesicular (13.0% [3/23]) rashes also were documented (Figure 1). The trunk (100% [23/23]) and extremities (95.7% [22/23]) were involved most often, followed by the palms and soles (56.5% [13/23]). The mean total body surface area affected was 73.65%. Only 7 patients (30.4%) had mucosal involvement; nonhemorrhagic cheilitis was the most common manifestation.
Facial edema, a hallmark feature of DRESS syndrome, was noted in 69.6% (16/23) of patients (Figure 2). Lymphadenopathy was present in 43.5% (10/23) of patients; of those cases, the inguinal (40.0%; n=4) and cervical (30%; n=3) nodes most commonly were involved. Although DRESS syndrome can affect internal organs, this was an issue for only 2 (8.7%) patients who experienced mild hepatomegaly.
Laboratory investigations revealed a mean differential eosinophil percentage of 10.3% (reference range, 1%–4%), while the mean absolute eosinophil count was 1.0634×109/L (reference range, 0.02–0.5×109/L). Other hematologic findings included the mean percentages of neutrophils (60%; reference range, 50%–60%), lymphocytes (19.95%; reference range, 20%–50%), and monocytes (8.70%; reference range, 2%–8%).
Liver function tests revealed transaminitis5 as the most common finding, with mean aspartate aminotransferase levels of 109 U/L (reference range, 8–33 U/L), mean alanine aminotransferase of 97.9 U/L (reference range, 7–56 U/L), and mean alkaline phosphatase levels of 211.35 U/L (reference range, 44–147 U/L). Half of the patients had notable (>2 times the upper limit of normal) transaminitis.
Renal blood workup revealed slightly elevated blood urea nitrogen levels with a mean value of 28.4 mg/dL (reference range, 6–24 mg/dL), and mean serum creatinine was 0.78 mg/dL (reference range for men, 0.7–1.3 mg/dL; for women, 0.6–1.1 mg/dL).
All patients were treated with oral steroids (prednisolone 1 mg/kg/d) before tapering slowly over the following 6 to 8 weeks. The culprit drug (phenytoin) was stopped on the day of presentation. Resolution of rash and itching was seen in all patients by 3 weeks after presentation without any relapse by follow-up at 6 weeks from presentation to the hospital.
Our case series seeks to discuss the clinical and laboratory features of phenytoin-induced DRESS syndrome. Our patients had more erythrodermic and erythema multiforme–like morphologies, less mucosal involvement, more hepatic involvement, and earlier resolution.
- Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg. 1996;15:250-257. doi:10.1016/s1085-5629(96)80038-1
- Patocka J, Wu Q, Nepovimova E, et al. Phenytoin—an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem Toxicol. 2020;142:111393. doi:10.1016/j.fct.2020.111393
- Sasidharanpillai S, Chathoth AT, Khader A, et al. Predictors of disease severity in drug reaction with eosinophilia and systemic symptoms. Indian J Dermatol Venereol Leprol. 2019;85:266-275. doi:10.4103/ijdvl.IJDVL_482_17
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Brit J Dermatol. 2013;169:1071-1080.
- Morán-Mariños C, Alva-Diaz C, De la Cruz Ramirez W, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) induced by phenytoin re-exposure: case report and systematic review. Acta Clin Belg. 2022;77:177-185. doi:10.1080/17843286.2020.1767459
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome—a severe cutaneous adverse drug reaction—is characterized by a cutaneous rash and systemic upset in the form of various internal organ and hematologic disturbances. This delayed and idiosyncratic syndrome went by several names, including anticonvulsant hypersensitivity syndrome, before Bocquet et al1 proposed the term DRESS syndrome.
Phenytoin, a hydantoin derivative used in neurology, was implicated in 41% of cases of DRESS syndrome in a study of 100 patients conducted in southern India.2,3 While DRESS syndrome is a newer name, the clinical picture of DRESS secondary to phenytoin use remains similar in that it manifests with a morbilliform rash and systemic upset. We sought to describe the clinical and laboratory characteristics of phenytoin-induced DRESS syndrome in this case series.
The analysis included 23 patients with DRESS syndrome secondary to phenytoin use who presented to a tertiary care institution in North India between July 2021 and December 2022, satisfied the European Registry of Severe Cutaneous Adverse Reaction (RegiSCAR) criteria,4 and achieved a DRESS diagnostic score of more than 1. The mean age of the patients was 44 years (range, 14–74 years). There was a slight female predominance with a male to female ratio of 0.9:1. More than half of the patients (52.2% [12/23]) presented directly to the dermatology outpatient department; the remaining patients were referred from other departments (47.8% [11/23]). Patients primarily were receiving phenytoin for neurologic indications. Specific reasons included antiseizure prophylaxis following a traffic accident (34.8% [8/23]); epilepsy (26.1% [6/23]); and neoplastic (17.4% [4/23]), vascular (17.4% [4/23]), and infectious (4.3% [1/23]) causes. The mean latency period from drug intake to symptom onset was 29 days (range, 6–62 days), and the mean illness duration was 9 days (range, 1–45 days).
The majority of patients experienced pruritus (91.3% [21/23]) and fever (74.0% [17/23]), and all initially had a rash. Maculopapular morphology was seen in all patients. Erythema multiforme–like (17.4% [4/23]), erythrodermic (17.4% [4/23]), and vesicular (13.0% [3/23]) rashes also were documented (Figure 1). The trunk (100% [23/23]) and extremities (95.7% [22/23]) were involved most often, followed by the palms and soles (56.5% [13/23]). The mean total body surface area affected was 73.65%. Only 7 patients (30.4%) had mucosal involvement; nonhemorrhagic cheilitis was the most common manifestation.
Facial edema, a hallmark feature of DRESS syndrome, was noted in 69.6% (16/23) of patients (Figure 2). Lymphadenopathy was present in 43.5% (10/23) of patients; of those cases, the inguinal (40.0%; n=4) and cervical (30%; n=3) nodes most commonly were involved. Although DRESS syndrome can affect internal organs, this was an issue for only 2 (8.7%) patients who experienced mild hepatomegaly.
Laboratory investigations revealed a mean differential eosinophil percentage of 10.3% (reference range, 1%–4%), while the mean absolute eosinophil count was 1.0634×109/L (reference range, 0.02–0.5×109/L). Other hematologic findings included the mean percentages of neutrophils (60%; reference range, 50%–60%), lymphocytes (19.95%; reference range, 20%–50%), and monocytes (8.70%; reference range, 2%–8%).
Liver function tests revealed transaminitis5 as the most common finding, with mean aspartate aminotransferase levels of 109 U/L (reference range, 8–33 U/L), mean alanine aminotransferase of 97.9 U/L (reference range, 7–56 U/L), and mean alkaline phosphatase levels of 211.35 U/L (reference range, 44–147 U/L). Half of the patients had notable (>2 times the upper limit of normal) transaminitis.
Renal blood workup revealed slightly elevated blood urea nitrogen levels with a mean value of 28.4 mg/dL (reference range, 6–24 mg/dL), and mean serum creatinine was 0.78 mg/dL (reference range for men, 0.7–1.3 mg/dL; for women, 0.6–1.1 mg/dL).
All patients were treated with oral steroids (prednisolone 1 mg/kg/d) before tapering slowly over the following 6 to 8 weeks. The culprit drug (phenytoin) was stopped on the day of presentation. Resolution of rash and itching was seen in all patients by 3 weeks after presentation without any relapse by follow-up at 6 weeks from presentation to the hospital.
Our case series seeks to discuss the clinical and laboratory features of phenytoin-induced DRESS syndrome. Our patients had more erythrodermic and erythema multiforme–like morphologies, less mucosal involvement, more hepatic involvement, and earlier resolution.
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome—a severe cutaneous adverse drug reaction—is characterized by a cutaneous rash and systemic upset in the form of various internal organ and hematologic disturbances. This delayed and idiosyncratic syndrome went by several names, including anticonvulsant hypersensitivity syndrome, before Bocquet et al1 proposed the term DRESS syndrome.
Phenytoin, a hydantoin derivative used in neurology, was implicated in 41% of cases of DRESS syndrome in a study of 100 patients conducted in southern India.2,3 While DRESS syndrome is a newer name, the clinical picture of DRESS secondary to phenytoin use remains similar in that it manifests with a morbilliform rash and systemic upset. We sought to describe the clinical and laboratory characteristics of phenytoin-induced DRESS syndrome in this case series.
The analysis included 23 patients with DRESS syndrome secondary to phenytoin use who presented to a tertiary care institution in North India between July 2021 and December 2022, satisfied the European Registry of Severe Cutaneous Adverse Reaction (RegiSCAR) criteria,4 and achieved a DRESS diagnostic score of more than 1. The mean age of the patients was 44 years (range, 14–74 years). There was a slight female predominance with a male to female ratio of 0.9:1. More than half of the patients (52.2% [12/23]) presented directly to the dermatology outpatient department; the remaining patients were referred from other departments (47.8% [11/23]). Patients primarily were receiving phenytoin for neurologic indications. Specific reasons included antiseizure prophylaxis following a traffic accident (34.8% [8/23]); epilepsy (26.1% [6/23]); and neoplastic (17.4% [4/23]), vascular (17.4% [4/23]), and infectious (4.3% [1/23]) causes. The mean latency period from drug intake to symptom onset was 29 days (range, 6–62 days), and the mean illness duration was 9 days (range, 1–45 days).
The majority of patients experienced pruritus (91.3% [21/23]) and fever (74.0% [17/23]), and all initially had a rash. Maculopapular morphology was seen in all patients. Erythema multiforme–like (17.4% [4/23]), erythrodermic (17.4% [4/23]), and vesicular (13.0% [3/23]) rashes also were documented (Figure 1). The trunk (100% [23/23]) and extremities (95.7% [22/23]) were involved most often, followed by the palms and soles (56.5% [13/23]). The mean total body surface area affected was 73.65%. Only 7 patients (30.4%) had mucosal involvement; nonhemorrhagic cheilitis was the most common manifestation.
Facial edema, a hallmark feature of DRESS syndrome, was noted in 69.6% (16/23) of patients (Figure 2). Lymphadenopathy was present in 43.5% (10/23) of patients; of those cases, the inguinal (40.0%; n=4) and cervical (30%; n=3) nodes most commonly were involved. Although DRESS syndrome can affect internal organs, this was an issue for only 2 (8.7%) patients who experienced mild hepatomegaly.
Laboratory investigations revealed a mean differential eosinophil percentage of 10.3% (reference range, 1%–4%), while the mean absolute eosinophil count was 1.0634×109/L (reference range, 0.02–0.5×109/L). Other hematologic findings included the mean percentages of neutrophils (60%; reference range, 50%–60%), lymphocytes (19.95%; reference range, 20%–50%), and monocytes (8.70%; reference range, 2%–8%).
Liver function tests revealed transaminitis5 as the most common finding, with mean aspartate aminotransferase levels of 109 U/L (reference range, 8–33 U/L), mean alanine aminotransferase of 97.9 U/L (reference range, 7–56 U/L), and mean alkaline phosphatase levels of 211.35 U/L (reference range, 44–147 U/L). Half of the patients had notable (>2 times the upper limit of normal) transaminitis.
Renal blood workup revealed slightly elevated blood urea nitrogen levels with a mean value of 28.4 mg/dL (reference range, 6–24 mg/dL), and mean serum creatinine was 0.78 mg/dL (reference range for men, 0.7–1.3 mg/dL; for women, 0.6–1.1 mg/dL).
All patients were treated with oral steroids (prednisolone 1 mg/kg/d) before tapering slowly over the following 6 to 8 weeks. The culprit drug (phenytoin) was stopped on the day of presentation. Resolution of rash and itching was seen in all patients by 3 weeks after presentation without any relapse by follow-up at 6 weeks from presentation to the hospital.
Our case series seeks to discuss the clinical and laboratory features of phenytoin-induced DRESS syndrome. Our patients had more erythrodermic and erythema multiforme–like morphologies, less mucosal involvement, more hepatic involvement, and earlier resolution.
- Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg. 1996;15:250-257. doi:10.1016/s1085-5629(96)80038-1
- Patocka J, Wu Q, Nepovimova E, et al. Phenytoin—an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem Toxicol. 2020;142:111393. doi:10.1016/j.fct.2020.111393
- Sasidharanpillai S, Chathoth AT, Khader A, et al. Predictors of disease severity in drug reaction with eosinophilia and systemic symptoms. Indian J Dermatol Venereol Leprol. 2019;85:266-275. doi:10.4103/ijdvl.IJDVL_482_17
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Brit J Dermatol. 2013;169:1071-1080.
- Morán-Mariños C, Alva-Diaz C, De la Cruz Ramirez W, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) induced by phenytoin re-exposure: case report and systematic review. Acta Clin Belg. 2022;77:177-185. doi:10.1080/17843286.2020.1767459
- Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg. 1996;15:250-257. doi:10.1016/s1085-5629(96)80038-1
- Patocka J, Wu Q, Nepovimova E, et al. Phenytoin—an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem Toxicol. 2020;142:111393. doi:10.1016/j.fct.2020.111393
- Sasidharanpillai S, Chathoth AT, Khader A, et al. Predictors of disease severity in drug reaction with eosinophilia and systemic symptoms. Indian J Dermatol Venereol Leprol. 2019;85:266-275. doi:10.4103/ijdvl.IJDVL_482_17
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Brit J Dermatol. 2013;169:1071-1080.
- Morán-Mariños C, Alva-Diaz C, De la Cruz Ramirez W, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) induced by phenytoin re-exposure: case report and systematic review. Acta Clin Belg. 2022;77:177-185. doi:10.1080/17843286.2020.1767459
Practice Points
- Phenytoin has been implicated in drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and common symptoms include rash, pruritus, and fever.
- Transaminitis may occur in patients with DRESS syndrome secondary to phenytoin use.
Disseminated Gonococcal Infection of Pharyngeal Origin: Test All Anatomic Sites
To the Editor:
Gonococcal infections, which are caused by the sexually transmitted, gram-negative diplococcus Neisseria gonorrhoeae, are a current and increasing threat to public health. Between 2012 and 2021, the rate of gonococcal infection in the United States increased 137.8% in men and 64.9% in women,1 with an estimated 1.5 million new gonococcal infections occurring each year in the United States as of 2021.2 Neisseria gonorrhoeae is the second most common bacterial sexually transmitted infection (STI), and patients with gonococcal infection frequently are coinfected with Chlamydia trachomatis, which is the most common bacterial STI. Uncomplicated gonococcal infection (also known as gonorrhea) most commonly causes asymptomatic cervicovaginal infection in women and symptomatic urethral infection in men.2 Other uncomplicated manifestations include rectal infection, which can be asymptomatic or manifest with anal pruritus, anal discharge, or tenesmus, and oropharyngeal infection, which can be asymptomatic or manifest with throat pain. If uncomplicated gonococcal infections are left untreated or are incompletely treated, serious complications including septic arthritis, myositis, osteomyelitis, myocarditis, endocarditis, and meningitis might occur.2-5 Ascending, locally invasive infections can cause epididymitis or pelvic inflammatory disease, which is an important cause of infertility in women.2,3 Gonococcal conjunctivitis also can occur, particularly when neonates are exposed to bacteria during vaginal delivery. Although rare, gonococcal bacteria can disseminate widely, with an estimated 0.5% to 3% of uncomplicated gonococcal infections progressing to disseminated gonococcal infection (DGI).3-6 Because DGI can mimic other systemic conditions, including a variety of bacterial and viral infections as well as inflammatory conditions, it can be difficult to diagnose without a high index of clinical suspicion. We present a case of DGI diagnosed based on dermatologic expertise and pharyngeal molecular testing.
A 30-year-old man presented to the emergency department with a rash on the extremeities as well as emesis, fever, sore throat, and severe arthralgia in the wrists, hands, knees, and feet of 2 days’ duration. The patient also had experienced several months of dysuria. He reported daily use of the recreational drug ketamine, multiple new male sexual partners, and unprotected oral and receptive anal sex in recent months. He denied any history of STIs. Physical examination demonstrated tender edematous wrists and fingers, papulovesicles on erythematous bases on the palms, and purpuric macules scattered on the legs (Figure 1). The patient also had tonsillar edema with notable white tonsillar exudate.
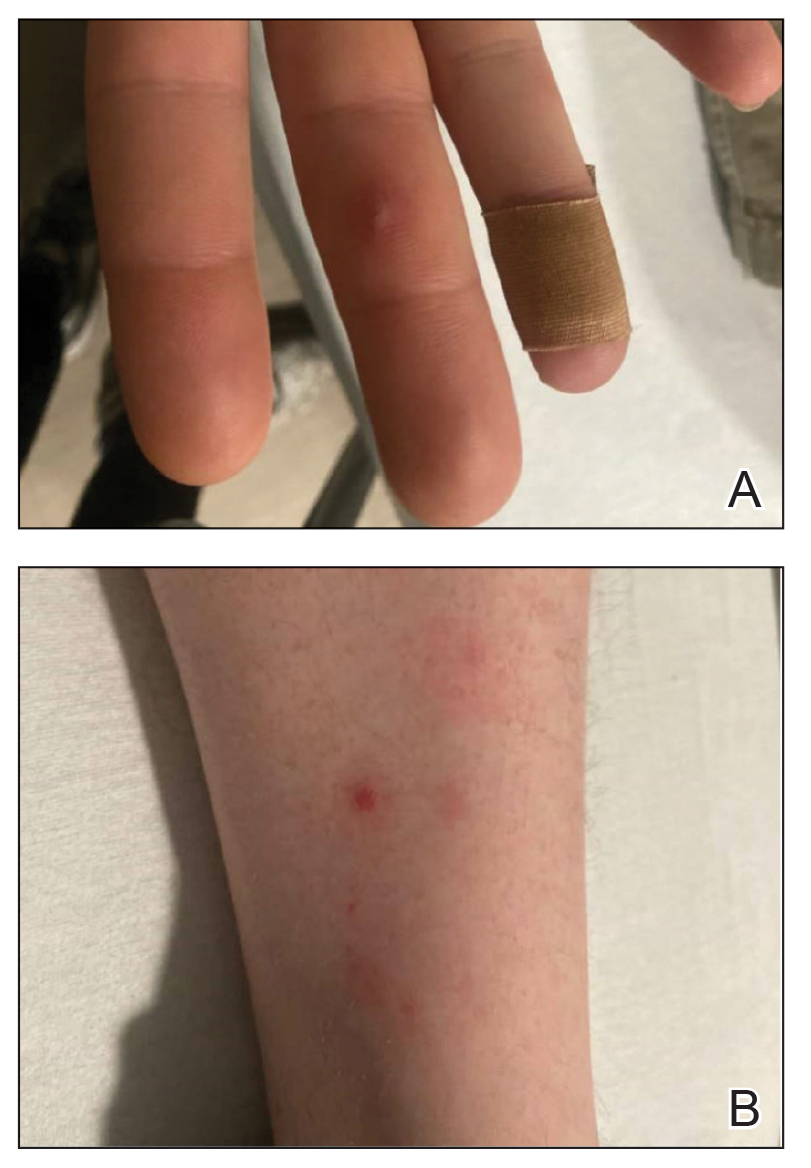
A shave biopsy performed on a papulovesicular lesion on the right thigh showed an intact epidermis with minimal spongiosis and no viral cytopathic changes. There was dermal edema with a moderate superficial and deep neutrophilic infiltrate, mild karyorrhexis, and focal dermal necrosis (Figure 2). Rare acute vasculitis with intravascular fibrin was seen. Periodic acid-Schiff stain for fungi, Gram stain for bacteria, and immunostains for human herpesviruses 1 and 2 were negative.
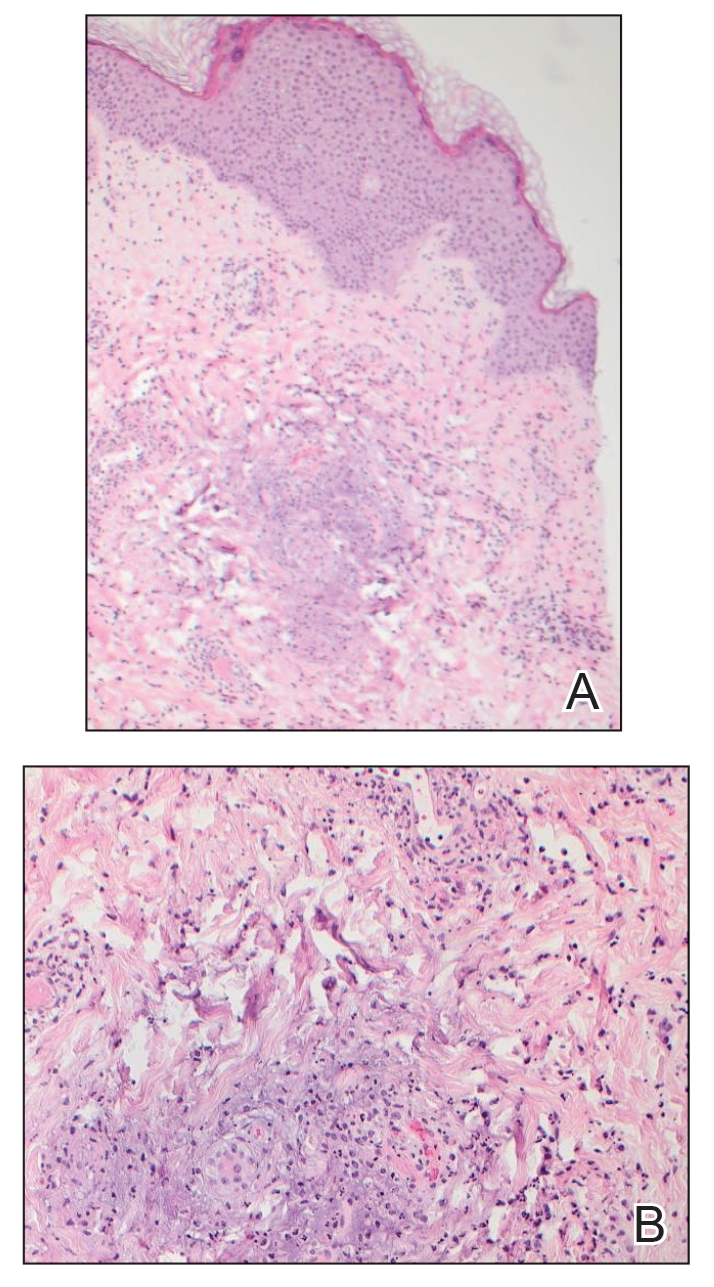
Laboratory studies revealed neutrophil-predominant leukocytosis (white blood cell count, 13.89×109/L [reference range, 4.5–11.0×109/L] with 78.2% neutrophils [reference range, 40.0%–70.0%]) as well as an elevated C-reactive protein level and erythrocyte sedimentation rate (19.98 mg/dL [reference range, <0.05 mg/dL] and 38 mm/h [reference range, 0–15 mm/h], respectively). His liver enzymes, kidney function, prothrombin time, and international normalized ratio were all normal. Urinalysis showed trace amounts of blood and protein, and urine culture was negative for pathogenic bacteria. A rapid plasma reagin test and a fifth-generation HIV antibody test were nonreactive, and bacterial blood cultures were negative for other infectious diseases. Nucleic acid amplification testing (NAAT) performed on a swab from a papulovesicular lesion was negative for human herpesviruses 1 and 2, varicella-zoster virus, orthopoxvirus, and mpox (monkeypox) virus. Based on recommendations from dermatology, NAATs for C trachomatis and N gonorrhoeae were performed on urine and on swabs from the patient’s rectum and pharynx; N gonorrhoeae was detected at the pharynx, but the other sites were negative for both bacteria. A diagnosis of DGI was made based on these results as well as the patient’s clinical presentation of fever, arthralgia, and papulovesicular skin lesions. The patient was treated with 1 g of intravenous ceftriaxone while in the hospital, but unfortunately, he was lost to follow-up and did not complete the full 1-week treatment course.
Disseminated gonococcal infection (also known as arthritis-dermatitis syndrome) is characterized by the abrupt onset of fever, skin lesions, and arthralgia in a symmetric and migratory distribution. Tenosynovitis involving the extensor tendons of the wrists, fingers, knees, and ankles (particularly the Achilles tendon) is characteristic. Skin manifestations usually include hemorrhagic vesicles and papulovesicles limited to the extremities, often with an acral distribution,2-5 though other cutaneous lesions have been described in DGI, including macules, purpura, periurethral abscesses, multifocal cellulitis, and necrotizing fasciitis.7 It is important to consider DGI in a patient who presents with acute systemic symptoms and any of these cutaneous manifestations, even in the absence of joint pain.
Diagnosis of DGI can be difficult, and surveillance is limited in the United States; therefore, the risk factors are somewhat unclear and might be changing. Traditional risk factors for DGI have included immunosuppression due to terminal complement deficiency, female sex, recent menstruation, and pregnancy, but recent data have shown that male sex, HIV infection, use of methamphetamines and other drugs, and use of the monoclonal antibody eculizumab for treatment of complement disorders have been associated with DGI.2,6-8 In the past decade, uncomplicated gonococcal infections have disproportionately affected Black patients, men who have sex with men, adults aged 20 to 25 years, and individuals living in the southern United States.1 It is unclear if the changing demographics of patients with DGI represent true risk factors for dissemination or simply reflect the changing demographics of patients at risk for uncomplicated gonococcal infection.6
Dermatologic expertise in the recognition of cutaneous manifestations of DGI is particularly important due to the limitations of diagnostic tools. The organism is fastidious and difficult to grow in vitro, thus cultures for N gonorrhoeae are not sensitive and require specialized media (eg, Thayer-Martin, modified New York City, or chocolate agar medium with additional antimicrobial agents).3 Molecular assays such as NAATs are more sensitive and specific than culture but are not 100% accurate.2,3,5 Finally, sterile sites such as joints, blood, or cerebrospinal fluid can be difficult to access, and specimens are not always available for specific microbial diagnosis; therefore, even when a gonococcal infection is identified at a mucosal source, physicians must use their clinical judgment to determine whether the mucosal infection is the cause of DGI or if the patient has a separate additional illness.
Once a diagnosis of gonococcal infection is made, any isolated gonococcal bacteria should be tested for antimicrobial susceptibility due to rising rates of drug resistance. Since at least the 1980s, N gonorrhoeae has steadily evolved to have some degree of resistance to most antimicrobials, and epidemiologic evidence indicates that this evolution is continuing.2 Current Centers for Disease Control and Prevention (CDC) recommendations are to treat uncomplicated gonococcal infections with 1 dose of ceftriaxone 500 mg intramuscularly in individuals weighing less than 150 kg (increase to 1 g in those ≥150 kg). Disseminated gonococcal infection requires more aggressive treatment with ceftriaxone 1 g intravenously or intramuscularly every 24 hours for at least 7 days and at a higher dose and for longer duration for patients with endocarditis or meningitis.2 If there is notable clinical improvement after 24 to 48 hours and antimicrobial susceptibility testing confirms an oral agent is appropriate, the patient can be switched to that oral agent to complete treatment. Also, if chlamydia has not been excluded in patients with any type of gonococcal infection, they also should be treated for chlamydia with doxycycline 100 mg twice daily, per CDC guidelines.2 Dermatologists should advocate for patients to be treated for DGI even if the diagnosis is clinical because of the potential for untreated or undertreated patients to progress, to develop additional antimicrobial resistant bacteria, and/or to transmit the infection to others.
This case highlights 2 important points about gonococcal infections and DGI. First, it is important to test and screen patients for gonococcal infection at genitourinary, rectal, and pharyngeal sites. Despite our patient’s report of dysuria, gonococcal infection was only detected via NAAT at the pharynx. As of 2021, CDC guidelines recommend not only testing for gonococcal infection in symptomatic patients at all mucosal sites but also screening all mucosal sites in asymptomatic individuals at high risk.2 Second, dermatologists’ specialized knowledge of cutaneous manifestations provides a valuable tool in the clinical diagnosis of DGI. In this patient, it was the dermatology team’s high index of concern for DGI that led to NAAT testing at all mucosal sites and resulted in an accurate diagnosis. Ultimately, dermatologists play an important role in the diagnosis and management of DGI.
- Centers for Disease Control and Prevention. Sexually transmitted disease surveillance, 2021. Accessed September 9, 2024. https://www.cdc.gov/std/statistics/2022/2021-STD-Surveillance-Report-PDF_ARCHIVED-2-16-24.pdf
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
- Skerlev M, Čulav-Košćak I. Gonorrhea: new challenges. Clin Dermatol. 2014;32:275-281. doi:10.1016/j.clindermatol.2013.08.010
- Mehrany K, Kist JM, O’Connor WJ, et al. Disseminated gonococcemia. Int J Dermatol. 2003;42:208-209. doi:10.1046/j.1365-4362.2003.01720.x
- Sciaudone M, Cope A, Mobley V, et al. Ten years of disseminated gonococcal infections in North Carolina: a review of cases from a large tertiary care hospital. Sex Transm Dis. 2023;50:410-414. doi:10.1097/OLQ.0000000000001794
- Weston EJ, Heidenga BL, Farley MM, et al. Surveillance for disseminated gonococcal infections, Active Bacterial Core surveillance (ABCs)—United States, 2015-2019. Clin Infect Dis. 2022;75:953-958. doi:10.1093/cid/ciac052
- Beatrous SV, Grisoli SB, Riahi RR, et al. Cutaneous manifestations of disseminated gonococcemia. Dermatol Online J. 2017;23:13030/qt33b24006
- Nettleton WD, Kent JB, Macomber K, et al. Notes from the field: ongoing cluster of highly related disseminated gonococcal infections—southwest Michigan, 2019. MMWR Morb Mortal Wkly Rep. 2020;69:353-354. doi:10.15585/mmwr.mm6912az
To the Editor:
Gonococcal infections, which are caused by the sexually transmitted, gram-negative diplococcus Neisseria gonorrhoeae, are a current and increasing threat to public health. Between 2012 and 2021, the rate of gonococcal infection in the United States increased 137.8% in men and 64.9% in women,1 with an estimated 1.5 million new gonococcal infections occurring each year in the United States as of 2021.2 Neisseria gonorrhoeae is the second most common bacterial sexually transmitted infection (STI), and patients with gonococcal infection frequently are coinfected with Chlamydia trachomatis, which is the most common bacterial STI. Uncomplicated gonococcal infection (also known as gonorrhea) most commonly causes asymptomatic cervicovaginal infection in women and symptomatic urethral infection in men.2 Other uncomplicated manifestations include rectal infection, which can be asymptomatic or manifest with anal pruritus, anal discharge, or tenesmus, and oropharyngeal infection, which can be asymptomatic or manifest with throat pain. If uncomplicated gonococcal infections are left untreated or are incompletely treated, serious complications including septic arthritis, myositis, osteomyelitis, myocarditis, endocarditis, and meningitis might occur.2-5 Ascending, locally invasive infections can cause epididymitis or pelvic inflammatory disease, which is an important cause of infertility in women.2,3 Gonococcal conjunctivitis also can occur, particularly when neonates are exposed to bacteria during vaginal delivery. Although rare, gonococcal bacteria can disseminate widely, with an estimated 0.5% to 3% of uncomplicated gonococcal infections progressing to disseminated gonococcal infection (DGI).3-6 Because DGI can mimic other systemic conditions, including a variety of bacterial and viral infections as well as inflammatory conditions, it can be difficult to diagnose without a high index of clinical suspicion. We present a case of DGI diagnosed based on dermatologic expertise and pharyngeal molecular testing.
A 30-year-old man presented to the emergency department with a rash on the extremeities as well as emesis, fever, sore throat, and severe arthralgia in the wrists, hands, knees, and feet of 2 days’ duration. The patient also had experienced several months of dysuria. He reported daily use of the recreational drug ketamine, multiple new male sexual partners, and unprotected oral and receptive anal sex in recent months. He denied any history of STIs. Physical examination demonstrated tender edematous wrists and fingers, papulovesicles on erythematous bases on the palms, and purpuric macules scattered on the legs (Figure 1). The patient also had tonsillar edema with notable white tonsillar exudate.
A shave biopsy performed on a papulovesicular lesion on the right thigh showed an intact epidermis with minimal spongiosis and no viral cytopathic changes. There was dermal edema with a moderate superficial and deep neutrophilic infiltrate, mild karyorrhexis, and focal dermal necrosis (Figure 2). Rare acute vasculitis with intravascular fibrin was seen. Periodic acid-Schiff stain for fungi, Gram stain for bacteria, and immunostains for human herpesviruses 1 and 2 were negative.
Laboratory studies revealed neutrophil-predominant leukocytosis (white blood cell count, 13.89×109/L [reference range, 4.5–11.0×109/L] with 78.2% neutrophils [reference range, 40.0%–70.0%]) as well as an elevated C-reactive protein level and erythrocyte sedimentation rate (19.98 mg/dL [reference range, <0.05 mg/dL] and 38 mm/h [reference range, 0–15 mm/h], respectively). His liver enzymes, kidney function, prothrombin time, and international normalized ratio were all normal. Urinalysis showed trace amounts of blood and protein, and urine culture was negative for pathogenic bacteria. A rapid plasma reagin test and a fifth-generation HIV antibody test were nonreactive, and bacterial blood cultures were negative for other infectious diseases. Nucleic acid amplification testing (NAAT) performed on a swab from a papulovesicular lesion was negative for human herpesviruses 1 and 2, varicella-zoster virus, orthopoxvirus, and mpox (monkeypox) virus. Based on recommendations from dermatology, NAATs for C trachomatis and N gonorrhoeae were performed on urine and on swabs from the patient’s rectum and pharynx; N gonorrhoeae was detected at the pharynx, but the other sites were negative for both bacteria. A diagnosis of DGI was made based on these results as well as the patient’s clinical presentation of fever, arthralgia, and papulovesicular skin lesions. The patient was treated with 1 g of intravenous ceftriaxone while in the hospital, but unfortunately, he was lost to follow-up and did not complete the full 1-week treatment course.
Disseminated gonococcal infection (also known as arthritis-dermatitis syndrome) is characterized by the abrupt onset of fever, skin lesions, and arthralgia in a symmetric and migratory distribution. Tenosynovitis involving the extensor tendons of the wrists, fingers, knees, and ankles (particularly the Achilles tendon) is characteristic. Skin manifestations usually include hemorrhagic vesicles and papulovesicles limited to the extremities, often with an acral distribution,2-5 though other cutaneous lesions have been described in DGI, including macules, purpura, periurethral abscesses, multifocal cellulitis, and necrotizing fasciitis.7 It is important to consider DGI in a patient who presents with acute systemic symptoms and any of these cutaneous manifestations, even in the absence of joint pain.
Diagnosis of DGI can be difficult, and surveillance is limited in the United States; therefore, the risk factors are somewhat unclear and might be changing. Traditional risk factors for DGI have included immunosuppression due to terminal complement deficiency, female sex, recent menstruation, and pregnancy, but recent data have shown that male sex, HIV infection, use of methamphetamines and other drugs, and use of the monoclonal antibody eculizumab for treatment of complement disorders have been associated with DGI.2,6-8 In the past decade, uncomplicated gonococcal infections have disproportionately affected Black patients, men who have sex with men, adults aged 20 to 25 years, and individuals living in the southern United States.1 It is unclear if the changing demographics of patients with DGI represent true risk factors for dissemination or simply reflect the changing demographics of patients at risk for uncomplicated gonococcal infection.6
Dermatologic expertise in the recognition of cutaneous manifestations of DGI is particularly important due to the limitations of diagnostic tools. The organism is fastidious and difficult to grow in vitro, thus cultures for N gonorrhoeae are not sensitive and require specialized media (eg, Thayer-Martin, modified New York City, or chocolate agar medium with additional antimicrobial agents).3 Molecular assays such as NAATs are more sensitive and specific than culture but are not 100% accurate.2,3,5 Finally, sterile sites such as joints, blood, or cerebrospinal fluid can be difficult to access, and specimens are not always available for specific microbial diagnosis; therefore, even when a gonococcal infection is identified at a mucosal source, physicians must use their clinical judgment to determine whether the mucosal infection is the cause of DGI or if the patient has a separate additional illness.
Once a diagnosis of gonococcal infection is made, any isolated gonococcal bacteria should be tested for antimicrobial susceptibility due to rising rates of drug resistance. Since at least the 1980s, N gonorrhoeae has steadily evolved to have some degree of resistance to most antimicrobials, and epidemiologic evidence indicates that this evolution is continuing.2 Current Centers for Disease Control and Prevention (CDC) recommendations are to treat uncomplicated gonococcal infections with 1 dose of ceftriaxone 500 mg intramuscularly in individuals weighing less than 150 kg (increase to 1 g in those ≥150 kg). Disseminated gonococcal infection requires more aggressive treatment with ceftriaxone 1 g intravenously or intramuscularly every 24 hours for at least 7 days and at a higher dose and for longer duration for patients with endocarditis or meningitis.2 If there is notable clinical improvement after 24 to 48 hours and antimicrobial susceptibility testing confirms an oral agent is appropriate, the patient can be switched to that oral agent to complete treatment. Also, if chlamydia has not been excluded in patients with any type of gonococcal infection, they also should be treated for chlamydia with doxycycline 100 mg twice daily, per CDC guidelines.2 Dermatologists should advocate for patients to be treated for DGI even if the diagnosis is clinical because of the potential for untreated or undertreated patients to progress, to develop additional antimicrobial resistant bacteria, and/or to transmit the infection to others.
This case highlights 2 important points about gonococcal infections and DGI. First, it is important to test and screen patients for gonococcal infection at genitourinary, rectal, and pharyngeal sites. Despite our patient’s report of dysuria, gonococcal infection was only detected via NAAT at the pharynx. As of 2021, CDC guidelines recommend not only testing for gonococcal infection in symptomatic patients at all mucosal sites but also screening all mucosal sites in asymptomatic individuals at high risk.2 Second, dermatologists’ specialized knowledge of cutaneous manifestations provides a valuable tool in the clinical diagnosis of DGI. In this patient, it was the dermatology team’s high index of concern for DGI that led to NAAT testing at all mucosal sites and resulted in an accurate diagnosis. Ultimately, dermatologists play an important role in the diagnosis and management of DGI.
To the Editor:
Gonococcal infections, which are caused by the sexually transmitted, gram-negative diplococcus Neisseria gonorrhoeae, are a current and increasing threat to public health. Between 2012 and 2021, the rate of gonococcal infection in the United States increased 137.8% in men and 64.9% in women,1 with an estimated 1.5 million new gonococcal infections occurring each year in the United States as of 2021.2 Neisseria gonorrhoeae is the second most common bacterial sexually transmitted infection (STI), and patients with gonococcal infection frequently are coinfected with Chlamydia trachomatis, which is the most common bacterial STI. Uncomplicated gonococcal infection (also known as gonorrhea) most commonly causes asymptomatic cervicovaginal infection in women and symptomatic urethral infection in men.2 Other uncomplicated manifestations include rectal infection, which can be asymptomatic or manifest with anal pruritus, anal discharge, or tenesmus, and oropharyngeal infection, which can be asymptomatic or manifest with throat pain. If uncomplicated gonococcal infections are left untreated or are incompletely treated, serious complications including septic arthritis, myositis, osteomyelitis, myocarditis, endocarditis, and meningitis might occur.2-5 Ascending, locally invasive infections can cause epididymitis or pelvic inflammatory disease, which is an important cause of infertility in women.2,3 Gonococcal conjunctivitis also can occur, particularly when neonates are exposed to bacteria during vaginal delivery. Although rare, gonococcal bacteria can disseminate widely, with an estimated 0.5% to 3% of uncomplicated gonococcal infections progressing to disseminated gonococcal infection (DGI).3-6 Because DGI can mimic other systemic conditions, including a variety of bacterial and viral infections as well as inflammatory conditions, it can be difficult to diagnose without a high index of clinical suspicion. We present a case of DGI diagnosed based on dermatologic expertise and pharyngeal molecular testing.
A 30-year-old man presented to the emergency department with a rash on the extremeities as well as emesis, fever, sore throat, and severe arthralgia in the wrists, hands, knees, and feet of 2 days’ duration. The patient also had experienced several months of dysuria. He reported daily use of the recreational drug ketamine, multiple new male sexual partners, and unprotected oral and receptive anal sex in recent months. He denied any history of STIs. Physical examination demonstrated tender edematous wrists and fingers, papulovesicles on erythematous bases on the palms, and purpuric macules scattered on the legs (Figure 1). The patient also had tonsillar edema with notable white tonsillar exudate.
A shave biopsy performed on a papulovesicular lesion on the right thigh showed an intact epidermis with minimal spongiosis and no viral cytopathic changes. There was dermal edema with a moderate superficial and deep neutrophilic infiltrate, mild karyorrhexis, and focal dermal necrosis (Figure 2). Rare acute vasculitis with intravascular fibrin was seen. Periodic acid-Schiff stain for fungi, Gram stain for bacteria, and immunostains for human herpesviruses 1 and 2 were negative.
Laboratory studies revealed neutrophil-predominant leukocytosis (white blood cell count, 13.89×109/L [reference range, 4.5–11.0×109/L] with 78.2% neutrophils [reference range, 40.0%–70.0%]) as well as an elevated C-reactive protein level and erythrocyte sedimentation rate (19.98 mg/dL [reference range, <0.05 mg/dL] and 38 mm/h [reference range, 0–15 mm/h], respectively). His liver enzymes, kidney function, prothrombin time, and international normalized ratio were all normal. Urinalysis showed trace amounts of blood and protein, and urine culture was negative for pathogenic bacteria. A rapid plasma reagin test and a fifth-generation HIV antibody test were nonreactive, and bacterial blood cultures were negative for other infectious diseases. Nucleic acid amplification testing (NAAT) performed on a swab from a papulovesicular lesion was negative for human herpesviruses 1 and 2, varicella-zoster virus, orthopoxvirus, and mpox (monkeypox) virus. Based on recommendations from dermatology, NAATs for C trachomatis and N gonorrhoeae were performed on urine and on swabs from the patient’s rectum and pharynx; N gonorrhoeae was detected at the pharynx, but the other sites were negative for both bacteria. A diagnosis of DGI was made based on these results as well as the patient’s clinical presentation of fever, arthralgia, and papulovesicular skin lesions. The patient was treated with 1 g of intravenous ceftriaxone while in the hospital, but unfortunately, he was lost to follow-up and did not complete the full 1-week treatment course.
Disseminated gonococcal infection (also known as arthritis-dermatitis syndrome) is characterized by the abrupt onset of fever, skin lesions, and arthralgia in a symmetric and migratory distribution. Tenosynovitis involving the extensor tendons of the wrists, fingers, knees, and ankles (particularly the Achilles tendon) is characteristic. Skin manifestations usually include hemorrhagic vesicles and papulovesicles limited to the extremities, often with an acral distribution,2-5 though other cutaneous lesions have been described in DGI, including macules, purpura, periurethral abscesses, multifocal cellulitis, and necrotizing fasciitis.7 It is important to consider DGI in a patient who presents with acute systemic symptoms and any of these cutaneous manifestations, even in the absence of joint pain.
Diagnosis of DGI can be difficult, and surveillance is limited in the United States; therefore, the risk factors are somewhat unclear and might be changing. Traditional risk factors for DGI have included immunosuppression due to terminal complement deficiency, female sex, recent menstruation, and pregnancy, but recent data have shown that male sex, HIV infection, use of methamphetamines and other drugs, and use of the monoclonal antibody eculizumab for treatment of complement disorders have been associated with DGI.2,6-8 In the past decade, uncomplicated gonococcal infections have disproportionately affected Black patients, men who have sex with men, adults aged 20 to 25 years, and individuals living in the southern United States.1 It is unclear if the changing demographics of patients with DGI represent true risk factors for dissemination or simply reflect the changing demographics of patients at risk for uncomplicated gonococcal infection.6
Dermatologic expertise in the recognition of cutaneous manifestations of DGI is particularly important due to the limitations of diagnostic tools. The organism is fastidious and difficult to grow in vitro, thus cultures for N gonorrhoeae are not sensitive and require specialized media (eg, Thayer-Martin, modified New York City, or chocolate agar medium with additional antimicrobial agents).3 Molecular assays such as NAATs are more sensitive and specific than culture but are not 100% accurate.2,3,5 Finally, sterile sites such as joints, blood, or cerebrospinal fluid can be difficult to access, and specimens are not always available for specific microbial diagnosis; therefore, even when a gonococcal infection is identified at a mucosal source, physicians must use their clinical judgment to determine whether the mucosal infection is the cause of DGI or if the patient has a separate additional illness.
Once a diagnosis of gonococcal infection is made, any isolated gonococcal bacteria should be tested for antimicrobial susceptibility due to rising rates of drug resistance. Since at least the 1980s, N gonorrhoeae has steadily evolved to have some degree of resistance to most antimicrobials, and epidemiologic evidence indicates that this evolution is continuing.2 Current Centers for Disease Control and Prevention (CDC) recommendations are to treat uncomplicated gonococcal infections with 1 dose of ceftriaxone 500 mg intramuscularly in individuals weighing less than 150 kg (increase to 1 g in those ≥150 kg). Disseminated gonococcal infection requires more aggressive treatment with ceftriaxone 1 g intravenously or intramuscularly every 24 hours for at least 7 days and at a higher dose and for longer duration for patients with endocarditis or meningitis.2 If there is notable clinical improvement after 24 to 48 hours and antimicrobial susceptibility testing confirms an oral agent is appropriate, the patient can be switched to that oral agent to complete treatment. Also, if chlamydia has not been excluded in patients with any type of gonococcal infection, they also should be treated for chlamydia with doxycycline 100 mg twice daily, per CDC guidelines.2 Dermatologists should advocate for patients to be treated for DGI even if the diagnosis is clinical because of the potential for untreated or undertreated patients to progress, to develop additional antimicrobial resistant bacteria, and/or to transmit the infection to others.
This case highlights 2 important points about gonococcal infections and DGI. First, it is important to test and screen patients for gonococcal infection at genitourinary, rectal, and pharyngeal sites. Despite our patient’s report of dysuria, gonococcal infection was only detected via NAAT at the pharynx. As of 2021, CDC guidelines recommend not only testing for gonococcal infection in symptomatic patients at all mucosal sites but also screening all mucosal sites in asymptomatic individuals at high risk.2 Second, dermatologists’ specialized knowledge of cutaneous manifestations provides a valuable tool in the clinical diagnosis of DGI. In this patient, it was the dermatology team’s high index of concern for DGI that led to NAAT testing at all mucosal sites and resulted in an accurate diagnosis. Ultimately, dermatologists play an important role in the diagnosis and management of DGI.
- Centers for Disease Control and Prevention. Sexually transmitted disease surveillance, 2021. Accessed September 9, 2024. https://www.cdc.gov/std/statistics/2022/2021-STD-Surveillance-Report-PDF_ARCHIVED-2-16-24.pdf
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
- Skerlev M, Čulav-Košćak I. Gonorrhea: new challenges. Clin Dermatol. 2014;32:275-281. doi:10.1016/j.clindermatol.2013.08.010
- Mehrany K, Kist JM, O’Connor WJ, et al. Disseminated gonococcemia. Int J Dermatol. 2003;42:208-209. doi:10.1046/j.1365-4362.2003.01720.x
- Sciaudone M, Cope A, Mobley V, et al. Ten years of disseminated gonococcal infections in North Carolina: a review of cases from a large tertiary care hospital. Sex Transm Dis. 2023;50:410-414. doi:10.1097/OLQ.0000000000001794
- Weston EJ, Heidenga BL, Farley MM, et al. Surveillance for disseminated gonococcal infections, Active Bacterial Core surveillance (ABCs)—United States, 2015-2019. Clin Infect Dis. 2022;75:953-958. doi:10.1093/cid/ciac052
- Beatrous SV, Grisoli SB, Riahi RR, et al. Cutaneous manifestations of disseminated gonococcemia. Dermatol Online J. 2017;23:13030/qt33b24006
- Nettleton WD, Kent JB, Macomber K, et al. Notes from the field: ongoing cluster of highly related disseminated gonococcal infections—southwest Michigan, 2019. MMWR Morb Mortal Wkly Rep. 2020;69:353-354. doi:10.15585/mmwr.mm6912az
- Centers for Disease Control and Prevention. Sexually transmitted disease surveillance, 2021. Accessed September 9, 2024. https://www.cdc.gov/std/statistics/2022/2021-STD-Surveillance-Report-PDF_ARCHIVED-2-16-24.pdf
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
- Skerlev M, Čulav-Košćak I. Gonorrhea: new challenges. Clin Dermatol. 2014;32:275-281. doi:10.1016/j.clindermatol.2013.08.010
- Mehrany K, Kist JM, O’Connor WJ, et al. Disseminated gonococcemia. Int J Dermatol. 2003;42:208-209. doi:10.1046/j.1365-4362.2003.01720.x
- Sciaudone M, Cope A, Mobley V, et al. Ten years of disseminated gonococcal infections in North Carolina: a review of cases from a large tertiary care hospital. Sex Transm Dis. 2023;50:410-414. doi:10.1097/OLQ.0000000000001794
- Weston EJ, Heidenga BL, Farley MM, et al. Surveillance for disseminated gonococcal infections, Active Bacterial Core surveillance (ABCs)—United States, 2015-2019. Clin Infect Dis. 2022;75:953-958. doi:10.1093/cid/ciac052
- Beatrous SV, Grisoli SB, Riahi RR, et al. Cutaneous manifestations of disseminated gonococcemia. Dermatol Online J. 2017;23:13030/qt33b24006
- Nettleton WD, Kent JB, Macomber K, et al. Notes from the field: ongoing cluster of highly related disseminated gonococcal infections—southwest Michigan, 2019. MMWR Morb Mortal Wkly Rep. 2020;69:353-354. doi:10.15585/mmwr.mm6912az
Practice Points
- Neisseria gonorrhoeae infections of the genitourinary system, rectum, and pharynx can disseminate and cause fever, joint pain, and hemorrhagic papulovesicles that can mimic other serious conditions and require dermatologic expertise to confirm.
- Patients with suspected disseminated gonococcal infection (DGI) as well as patients who are asymptomatic and at increased risk should have all possible anatomic sites of infection—the genitourinary system, rectum, and pharynx—tested with the appropriate molecular assays and culture when appropriate.
- Appropriate recognition and treatment of DGI is vital, as undertreatment can result in serious complications and contribute to the increasing global public health threat of antimicrobial-resistant gonococcal infections.
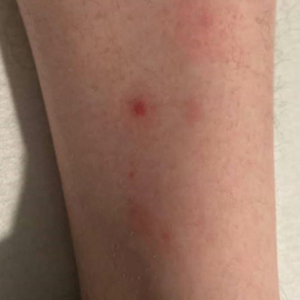
Inspection of Deep Tumor Margins for Accurate Cutaneous Squamous Cell Carcinoma Staging
To the Editor:
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk factors in cutaneous squamous cell carcinoma (cSCC), which may warrant tumor upstaging.1 Intratumor location has not been studied when looking at these high-risk factors. Herein, we report 4 cSCCs initially categorized as well differentiated that were reclassified as moderate to poorly differentiated on analysis of debulk specimens obtained via shave removal.
An 80-year-old man (patient 1) presented with a tender 2-cm erythematous plaque with dried hemorrhagic crusting on the frontal scalp. He had a history of nonmelanoma skin cancers. A biopsy revealed a well-differentiated cSCC, which was upgraded from a T2a tumor to T2b during MMS due to galea involvement. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells at the deep margin (Figure 1A). Given T2b staging, baseline imaging and radiation therapy were recommended.
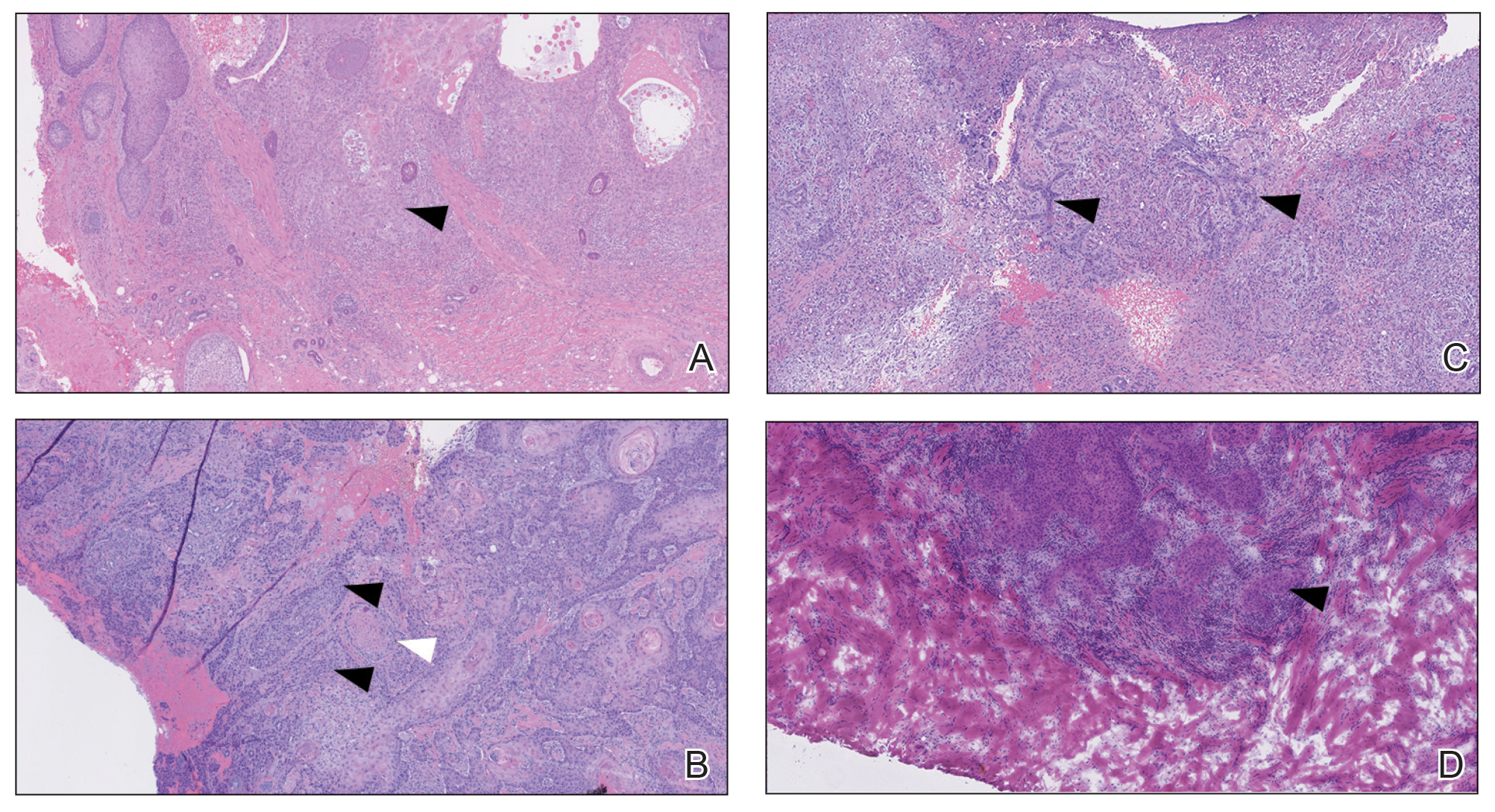
A 75-year-old man (patient 2) presented with a 2-cm erythematous plaque on the left vertex scalp with hemorrhagic crusting, yellow scale, and purulent drainage. He had a history of cSCCs. A biopsy revealed well-differentiated invasive cSCC, which was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Examination of the second Mohs stage revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, infiltration beyond the subcutaneous fat, and perineural invasion (Figure 1B). Given T2b staging, baseline imaging and radiation therapy were recommended.
An 86-year-old woman (patient 3) presented with a tender 2.4-cm plum-colored nodule on the right lower leg. She had a history of basal cell carcinoma. A biopsy revealed a well-differentiated invasive cSCC staged at T2a. Debulk analysis revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, though the staging remained the same (Figure 1C).
An 82-year-old man (patient 4) presented with a 2.7-cm ulcerated nodule with adjacent scaling on the vertex scalp. He had no history of skin cancer. A biopsy revealed a well-differentiated cSCC (Figure 2) that was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells with single-cell extension at the deep margin in the galea (Figure 1D). Given T2b staging, baseline imaging and radiation therapy were recommended.
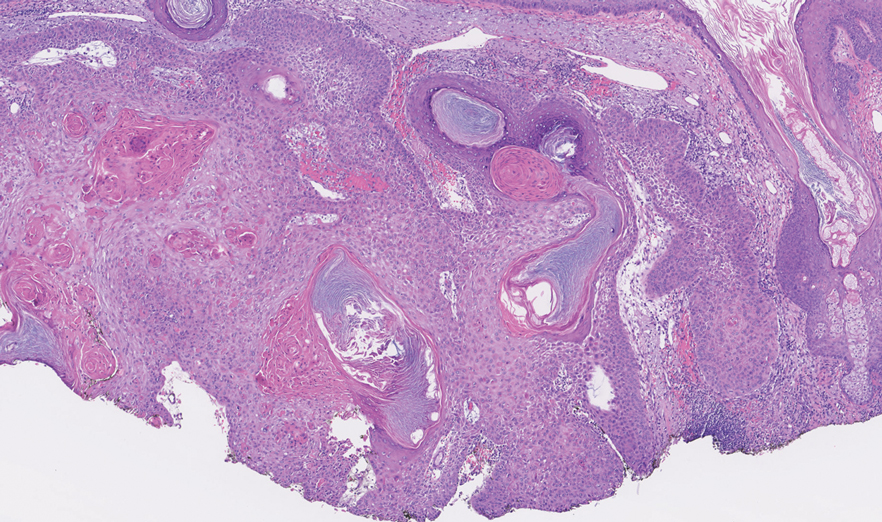
Tumor differentiation is a factor included in the Brigham and Women’s Hospital staging system, and intratumor variability can be clinically relevant for tumor staging.1 Specifically, cSCCs may exhibit intratumor heterogeneity in which predominantly well-differentiated tumors contain focal areas of poorer differentiation.2 This intratumor heterogeneity complicates estimation of tumor risk, as a well-differentiated tumor on biopsy may exhibit poor differentiation at a deeper margin. Our cases highlight that the cells at the deeper margin indeed can show poorer differentiation or other higher-risk tumor features. Thus, the most clinically relevant cells for tumor staging and prognostication may not be visible on initial biopsy, underscoring the utility of close examination of the deep layer of the debulk specimen and Mohs layer for comprehensive staging.
Genetic studies have attempted to identify gene expression patterns in cSCCs that predispose to invasion.3 Three of the top 6 genes in this “invasion signature gene set” were matrix metalloproteases; additionally, IL-24 messenger RNA was upregulated in both the cSCC invasion front and in situ cSCCs. IL-24 has been shown to upregulate the expression of matrix metalloprotease 7 in vitro, suggesting that it may influence tumor progression.3 Although gene expression was not included in this series, the identification of genetic variability in the most poorly differentiated cells residing in the deep margins is of great interest and may reveal mutations contributing to irregular cell morphology and cSCC invasiveness.
Prior studies have indicated that a proportion of cSCCs are histopathologically upgraded from the initial biopsy during MMS due to evidence of perineural invasion, bony invasion, or lesser differentiation noted during MMS stages or debulk analysis.1,4 However, the majority of Mohs surgeons report immediately discarding debulk specimens without further evaluation.5 Herein, we highlight 4 cSCC cases in which the deep margins of the debulk specimen contained the most dedifferentiated cells. Our findings emphasize the importance of thoroughly examining deep tumor margins for complete staging yet also highlight that identifying cells at these margins may not change patient management when high-risk criteria are already met.
- McIlwee BE, Abidi NY, Ravi M, et al. Utility of debulk specimens during Mohs micrographic surgery for cutaneous squamous cell carcinoma. Dermatol Surg. 2021;47:599-604.
- Ramón y Cajal S, Sesé M, Capdevila C, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020;98:161-177.
- Mitsui H, Suárez-Fariñas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014;134:1418-1427.
- Chung E, Hoang S, McEvoy AM, et al. Histopathologic upgrading of cutaneous squamous cell carcinomas during Mohs micrographic surgery: a retrospective cohort study. J Am Acad Dermatol. 2021;85:923-930.
- Alniemi DT, Swanson AM, Lasarev M, et al. Tumor debulking trends for keratinocyte carcinomas among Mohs surgeons. Dermatol Surg. 2021;47:1660-1661.
To the Editor:
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk factors in cutaneous squamous cell carcinoma (cSCC), which may warrant tumor upstaging.1 Intratumor location has not been studied when looking at these high-risk factors. Herein, we report 4 cSCCs initially categorized as well differentiated that were reclassified as moderate to poorly differentiated on analysis of debulk specimens obtained via shave removal.
An 80-year-old man (patient 1) presented with a tender 2-cm erythematous plaque with dried hemorrhagic crusting on the frontal scalp. He had a history of nonmelanoma skin cancers. A biopsy revealed a well-differentiated cSCC, which was upgraded from a T2a tumor to T2b during MMS due to galea involvement. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells at the deep margin (Figure 1A). Given T2b staging, baseline imaging and radiation therapy were recommended.
A 75-year-old man (patient 2) presented with a 2-cm erythematous plaque on the left vertex scalp with hemorrhagic crusting, yellow scale, and purulent drainage. He had a history of cSCCs. A biopsy revealed well-differentiated invasive cSCC, which was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Examination of the second Mohs stage revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, infiltration beyond the subcutaneous fat, and perineural invasion (Figure 1B). Given T2b staging, baseline imaging and radiation therapy were recommended.
An 86-year-old woman (patient 3) presented with a tender 2.4-cm plum-colored nodule on the right lower leg. She had a history of basal cell carcinoma. A biopsy revealed a well-differentiated invasive cSCC staged at T2a. Debulk analysis revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, though the staging remained the same (Figure 1C).
An 82-year-old man (patient 4) presented with a 2.7-cm ulcerated nodule with adjacent scaling on the vertex scalp. He had no history of skin cancer. A biopsy revealed a well-differentiated cSCC (Figure 2) that was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells with single-cell extension at the deep margin in the galea (Figure 1D). Given T2b staging, baseline imaging and radiation therapy were recommended.
Tumor differentiation is a factor included in the Brigham and Women’s Hospital staging system, and intratumor variability can be clinically relevant for tumor staging.1 Specifically, cSCCs may exhibit intratumor heterogeneity in which predominantly well-differentiated tumors contain focal areas of poorer differentiation.2 This intratumor heterogeneity complicates estimation of tumor risk, as a well-differentiated tumor on biopsy may exhibit poor differentiation at a deeper margin. Our cases highlight that the cells at the deeper margin indeed can show poorer differentiation or other higher-risk tumor features. Thus, the most clinically relevant cells for tumor staging and prognostication may not be visible on initial biopsy, underscoring the utility of close examination of the deep layer of the debulk specimen and Mohs layer for comprehensive staging.
Genetic studies have attempted to identify gene expression patterns in cSCCs that predispose to invasion.3 Three of the top 6 genes in this “invasion signature gene set” were matrix metalloproteases; additionally, IL-24 messenger RNA was upregulated in both the cSCC invasion front and in situ cSCCs. IL-24 has been shown to upregulate the expression of matrix metalloprotease 7 in vitro, suggesting that it may influence tumor progression.3 Although gene expression was not included in this series, the identification of genetic variability in the most poorly differentiated cells residing in the deep margins is of great interest and may reveal mutations contributing to irregular cell morphology and cSCC invasiveness.
Prior studies have indicated that a proportion of cSCCs are histopathologically upgraded from the initial biopsy during MMS due to evidence of perineural invasion, bony invasion, or lesser differentiation noted during MMS stages or debulk analysis.1,4 However, the majority of Mohs surgeons report immediately discarding debulk specimens without further evaluation.5 Herein, we highlight 4 cSCC cases in which the deep margins of the debulk specimen contained the most dedifferentiated cells. Our findings emphasize the importance of thoroughly examining deep tumor margins for complete staging yet also highlight that identifying cells at these margins may not change patient management when high-risk criteria are already met.
To the Editor:
Histopathologic analysis of debulk specimens in Mohs micrographic surgery (MMS) may augment identification of high-risk factors in cutaneous squamous cell carcinoma (cSCC), which may warrant tumor upstaging.1 Intratumor location has not been studied when looking at these high-risk factors. Herein, we report 4 cSCCs initially categorized as well differentiated that were reclassified as moderate to poorly differentiated on analysis of debulk specimens obtained via shave removal.
An 80-year-old man (patient 1) presented with a tender 2-cm erythematous plaque with dried hemorrhagic crusting on the frontal scalp. He had a history of nonmelanoma skin cancers. A biopsy revealed a well-differentiated cSCC, which was upgraded from a T2a tumor to T2b during MMS due to galea involvement. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells at the deep margin (Figure 1A). Given T2b staging, baseline imaging and radiation therapy were recommended.
A 75-year-old man (patient 2) presented with a 2-cm erythematous plaque on the left vertex scalp with hemorrhagic crusting, yellow scale, and purulent drainage. He had a history of cSCCs. A biopsy revealed well-differentiated invasive cSCC, which was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Examination of the second Mohs stage revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, infiltration beyond the subcutaneous fat, and perineural invasion (Figure 1B). Given T2b staging, baseline imaging and radiation therapy were recommended.
An 86-year-old woman (patient 3) presented with a tender 2.4-cm plum-colored nodule on the right lower leg. She had a history of basal cell carcinoma. A biopsy revealed a well-differentiated invasive cSCC staged at T2a. Debulk analysis revealed moderately differentiated cSCC, with the least-differentiated cells at the deep margin, though the staging remained the same (Figure 1C).
An 82-year-old man (patient 4) presented with a 2.7-cm ulcerated nodule with adjacent scaling on the vertex scalp. He had no history of skin cancer. A biopsy revealed a well-differentiated cSCC (Figure 2) that was upgraded from a T2a tumor to T2b during MMS due to tumor extension beyond the subcutaneous fat. Debulk analysis revealed moderate to poorly differentiated cSCC, with the least-differentiated cells with single-cell extension at the deep margin in the galea (Figure 1D). Given T2b staging, baseline imaging and radiation therapy were recommended.
Tumor differentiation is a factor included in the Brigham and Women’s Hospital staging system, and intratumor variability can be clinically relevant for tumor staging.1 Specifically, cSCCs may exhibit intratumor heterogeneity in which predominantly well-differentiated tumors contain focal areas of poorer differentiation.2 This intratumor heterogeneity complicates estimation of tumor risk, as a well-differentiated tumor on biopsy may exhibit poor differentiation at a deeper margin. Our cases highlight that the cells at the deeper margin indeed can show poorer differentiation or other higher-risk tumor features. Thus, the most clinically relevant cells for tumor staging and prognostication may not be visible on initial biopsy, underscoring the utility of close examination of the deep layer of the debulk specimen and Mohs layer for comprehensive staging.
Genetic studies have attempted to identify gene expression patterns in cSCCs that predispose to invasion.3 Three of the top 6 genes in this “invasion signature gene set” were matrix metalloproteases; additionally, IL-24 messenger RNA was upregulated in both the cSCC invasion front and in situ cSCCs. IL-24 has been shown to upregulate the expression of matrix metalloprotease 7 in vitro, suggesting that it may influence tumor progression.3 Although gene expression was not included in this series, the identification of genetic variability in the most poorly differentiated cells residing in the deep margins is of great interest and may reveal mutations contributing to irregular cell morphology and cSCC invasiveness.
Prior studies have indicated that a proportion of cSCCs are histopathologically upgraded from the initial biopsy during MMS due to evidence of perineural invasion, bony invasion, or lesser differentiation noted during MMS stages or debulk analysis.1,4 However, the majority of Mohs surgeons report immediately discarding debulk specimens without further evaluation.5 Herein, we highlight 4 cSCC cases in which the deep margins of the debulk specimen contained the most dedifferentiated cells. Our findings emphasize the importance of thoroughly examining deep tumor margins for complete staging yet also highlight that identifying cells at these margins may not change patient management when high-risk criteria are already met.
- McIlwee BE, Abidi NY, Ravi M, et al. Utility of debulk specimens during Mohs micrographic surgery for cutaneous squamous cell carcinoma. Dermatol Surg. 2021;47:599-604.
- Ramón y Cajal S, Sesé M, Capdevila C, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020;98:161-177.
- Mitsui H, Suárez-Fariñas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014;134:1418-1427.
- Chung E, Hoang S, McEvoy AM, et al. Histopathologic upgrading of cutaneous squamous cell carcinomas during Mohs micrographic surgery: a retrospective cohort study. J Am Acad Dermatol. 2021;85:923-930.
- Alniemi DT, Swanson AM, Lasarev M, et al. Tumor debulking trends for keratinocyte carcinomas among Mohs surgeons. Dermatol Surg. 2021;47:1660-1661.
- McIlwee BE, Abidi NY, Ravi M, et al. Utility of debulk specimens during Mohs micrographic surgery for cutaneous squamous cell carcinoma. Dermatol Surg. 2021;47:599-604.
- Ramón y Cajal S, Sesé M, Capdevila C, et al. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020;98:161-177.
- Mitsui H, Suárez-Fariñas M, Gulati N, et al. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014;134:1418-1427.
- Chung E, Hoang S, McEvoy AM, et al. Histopathologic upgrading of cutaneous squamous cell carcinomas during Mohs micrographic surgery: a retrospective cohort study. J Am Acad Dermatol. 2021;85:923-930.
- Alniemi DT, Swanson AM, Lasarev M, et al. Tumor debulking trends for keratinocyte carcinomas among Mohs surgeons. Dermatol Surg. 2021;47:1660-1661.
Practice Points
- A proportion of cutaneous squamous cell carcinomas are upgraded from the initial biopsy during Mohs micrographic surgery due to evidence of perineural invasion, bony invasion, or lesser differentiation noted on Mohs stages or debulk analysis.
- Thorough inspection of the deep tumor margins may be required for accurate tumor staging and evaluation of metastatic risk. Cells at the deep margin of the tumor may demonstrate poorer differentiation and/or other higher-risk tumor features than those closer to the surface.
- Tumor staging may be incomplete until the deep margins are assessed to find the most dysplastic and likely clinically relevant cells, which may be missed without evaluation of the debulked tumor.
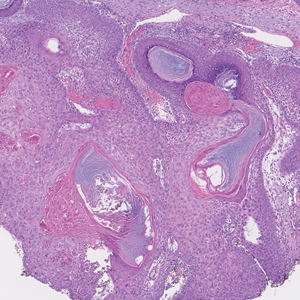
Transient Eruption of Verrucous Keratoses During Encorafenib Therapy: Adverse Event or Paraneoplastic Phenomenon?
To the Editor:
Mutations of the BRAF protein kinase gene are implicated in a variety of malignancies.1 BRAF mutations in malignancies cause the mitogen-activated protein kinase (MAPK) pathway to become constitutively active, which results in unchecked cellular proliferation,2,3 making the BRAF mutation an attractive target for inhibition with pharmacologic agents to potentially halt cancer growth.4 Vemurafenib—the first selective BRAF inhibitor used in clinical practice—initially was approved by the US Food and Drug Administration in 2011. The approval of dabrafenib followed in 2013 and most recently encorafenib in 2018.5
Although targeted treatment of BRAF-mutated malignancies with BRAF inhibitors has become common, it often is associated with cutaneous adverse events (AEs), such as rash, pruritus, photosensitivity, actinic keratosis, and verrucous keratosis. Some reports demonstrate these events in up to 95% of patients undergoing BRAF inhibitor treatment.6 In several cases the eruption of verrucous keratoses is among the most common cutaneous AEs seen among patients receiving BRAF inhibitor treatment.5-7
In general, lesions can appear days to months after therapy is initiated and may resolve after switching to dual therapy with a MEK inhibitor or with complete cessation of BRAF inhibitor therapy.5,7,8 One case of spontaneous resolution of vemurafenib-associated panniculitis during ongoing BRAF inhibitor therapy has been reported9; however, spontaneous resolution of cutaneous AEs is uncommon. Herein, we describe verrucous keratoses in a patient undergoing treatment with encorafenib that resolved spontaneously despite ongoing BRAF inhibitor therapy.
A 61-year-old woman presented to the emergency department with pain in the right lower quadrant. Computed tomography (CT) of the abdomen and pelvis revealed a large ovarian mass. Subsequent bloodwork revealed elevated carcinoembryonic antigen levels. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, right hemicolectomy with ileotransverse side-to-side anastomosis, right pelvic lymph node reduction, and complete cytoreduction. Histopathology revealed an adenocarcinoma of the cecum with tumor invasion into the visceral peritoneum and metastases to the left ovary, fallopian tube, and omentum. A BRAF V600E mutation was detected.
Two months after the initial presentation, the patient started her first cycle of chemotherapy with a combination of folinic acid, fluorouracil, and oxaliplatin. She completed 11 cycles of this regimen, then was switched to capecitabine and oxaliplatin for an additional 2 cycles due to insurance concerns. At the end of treatment, there was no evidence of disease on CT, thus the patient was followed with observation. However, she presented 10 months later to the emergency department with abdominal pain, and CT revealed new lesions in the liver that were concerning for potential metastases. She started oral encorafenib 300 mg/d and intravenous cetuximab 500 mg weekly; after 1 week, encorafenib was reduced to 150 mg/d due to nausea and loss of appetite. Within 2 weeks of starting treatment, the patient reported the relatively abrupt appearance of more than 50 small papules across the shoulders and back (Figure 1A). She was referred to dermatology, and shave biopsies of 2 lesions—one from the left anterior thigh, the other from the right posterior shoulder—revealed verrucous keratosis pathology (Figure 2). At this time, encorafenib was increased again to 300 mg/d as the patient had been tolerating the reduced dose. She continued to report the appearance of new lesions for the next 3 months, after which the lesions were stable for approximately 2 months. By 2.5 months after initiation of therapy, the patient had undergone CT demonstrating resolution of the liver lesions. At 5 months of therapy, the patient reported a stable to slightly reduced number of skin lesions but had begun to experience worsening joint pain, and the dosage of encorafenib was reduced to 225 mg/d. At 7 months of therapy, the dosage was further reduced to 150 mg/d due to persistent arthralgia. A follow-up examination at 10 months of therapy showed improvement in the number and size of the verrucous keratoses, and near resolution was seen by 14 months after the initial onset of the lesions (Figure 1B). At 20 months after initial onset, only 1 remaining verrucous keratosis was identified on physical examination and biopsy. The patient had continued a regimen of encorafenib 150 mg/d and weekly intravenous 500 mg cetuximab up to this point. Over the entire time period that the patient was seen, up to 12 lesions located in high-friction areas had become irritated and were treated with cryotherapy, but this contributed only minorly to the patient’s overall presentation.
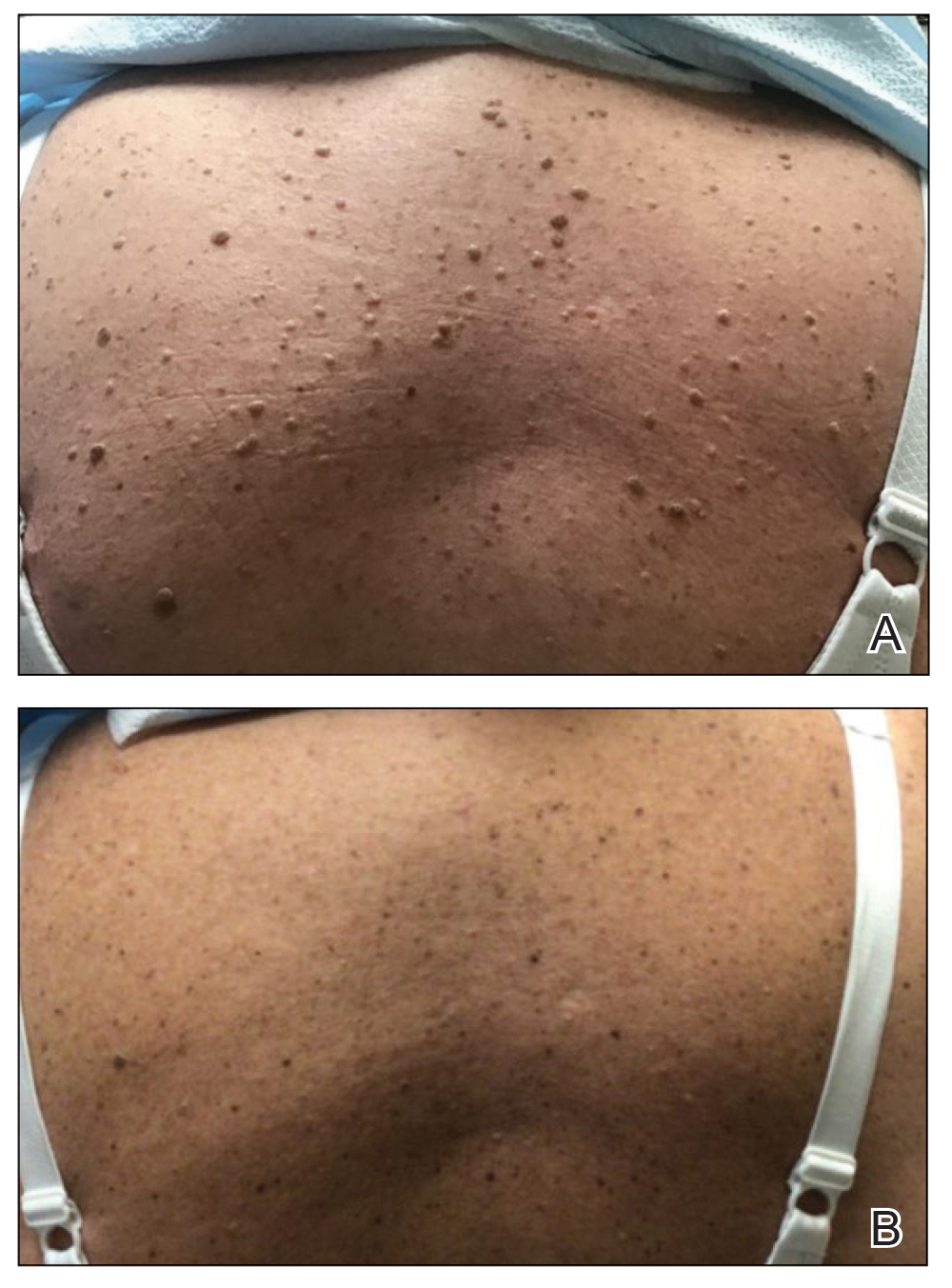
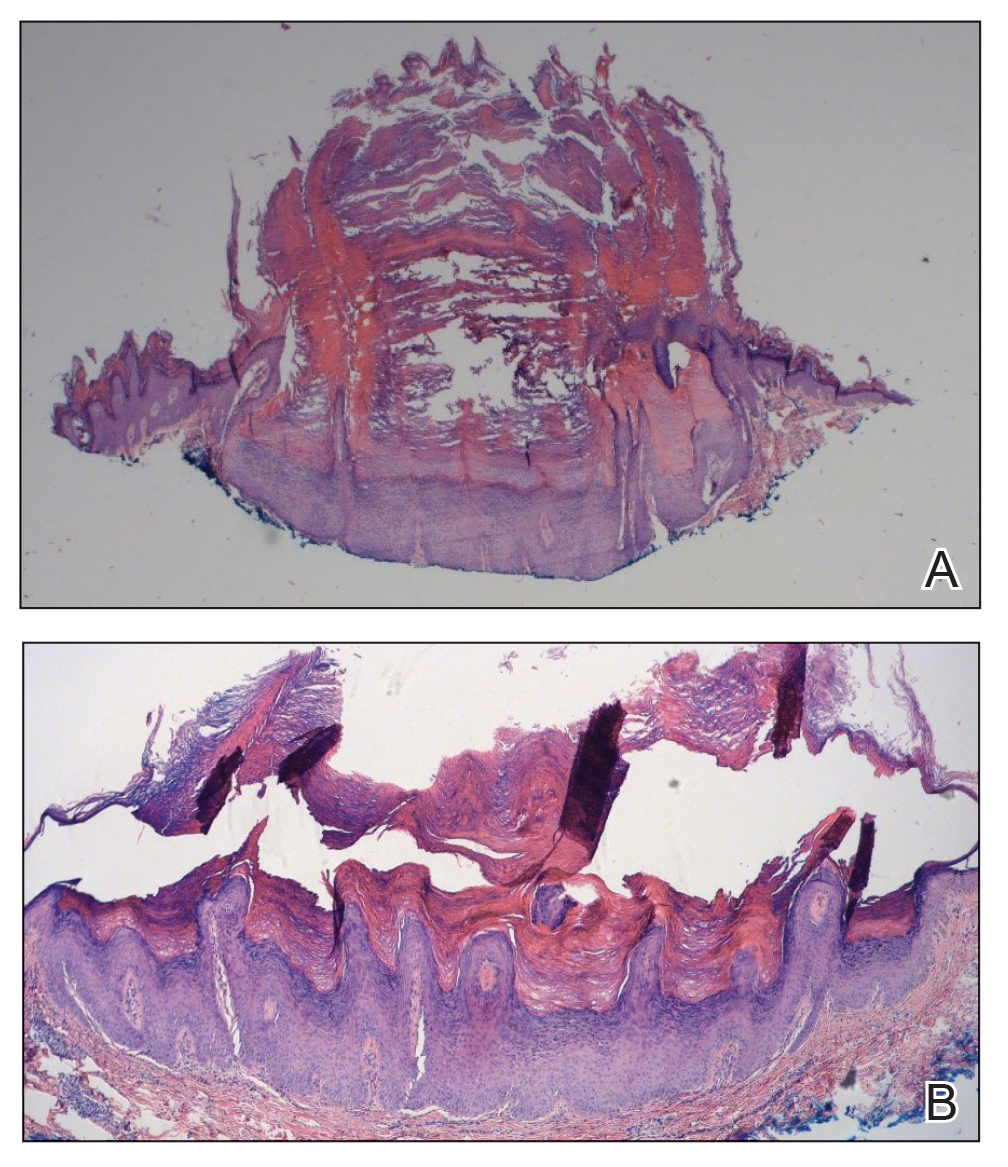
Verrucous keratosis is a known cutaneous AE of BRAF inhibitor treatment with vemurafenib and dabrafenib, with fewer cases attributed to encorafenib.5,6 Within the oncologic setting, the eruption of verrucous papules as a paraneoplastic phenomenon is heavily debated in the literature and is known as the Leser-Trélat sign. This phenomenon is commonly associated with adenocarcinomas of the gastrointestinal tract, as seen in our patient.10 Based on Curth’s postulates—the criteria used to evaluate the relationship between an internal malignancy and a cutaneous disorder—this was unlikely in our patient. The criteria, which do not all need to be met to suggest a paraneoplastic phenomenon, include concurrent onset of the malignancy and the dermatosis, parallel course, association of a specific dermatosis with a specific malignancy, statistical significance of the association, and the presence of a genetic basis for the association.11 Several features favored a drug-related cutaneous eruption vs a paraneoplastic phenomenon: (1) the malignancy was identified months before the cutaneous eruptions manifested; (2) the cutaneous lesions appeared once treatment had already been initiated; and (3) the cutaneous lesions persisted long after the malignancy was no longer identifiable on CT. Indeed, eruption of the papules temporally coincided closely with the initiation of BRAF inhibitor therapy, arguing for correlation.
As a suspected BRAF inhibitor–associated cutaneous AE, the eruption of verrucous keratoses in our patient is remarkable for its spontaneous resolution despite ongoing therapy. It is speculated that keratinocytic proliferation while on BRAF inhibitor therapy may be caused by a paradoxical increase in signaling through CRAF, another Raf isoform that plays a role in the induction of terminal differentiation of keratinocytes, with a subsequent increase in MAPK signaling.12-14 Self-resolution of this cycle despite continuing BRAF inhibitor therapy suggests the possible involvement of balancing and/or alternative mechanistic pathways that may be related to the immune system. Although verrucous keratoses are considered benign proliferations and do not necessarily require any specific treatment or reduction in BRAF inhibitor dosage, they may be treated with cryotherapy, electrocautery, shave removal, or excision,15 which often is done if the lesions become inflamed and cause pain. Additionally, some patients may feel distress from the appearance of the lesions and desire treatment for this reason. Understanding that verrucous keratoses can be a transient cutaneous AE rather than a persistent one may be useful to clinicians as they manage AEs during BRAF inhibitor therapy.
- Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346-356. doi:10.1097/PAT.0b013e328360b61d
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529-545. doi:10.1016/j.hoc.2009.04.001
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246. doi:10.1200/JCO.2010.32.4327
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2012;18:27-35. doi:10.1016/j.molmed.2011.08.001
- Gouda MA, Subbiah V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: from melanoma to tissue-agnostic therapy. ESMO Open. 2023;8:100788. doi:10.1016/j.esmoop.2023.100788
- Gençler B, Gönül M. Cutaneous side effects of BRAF inhibitors in advanced melanoma: review of the literature. Dermatol Res Pract. 2016;2016:5361569. doi:10.1155/2016/5361569.
- Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol. 2012;67:1265-1272. doi:10.1016/j.jaad.2012.04.008
- Naqash AR, File DM, Ziemer CM, et al. Cutaneous adverse reactions in B-RAF positive metastatic melanoma following sequential treatment with B-RAF/MEK inhibitors and immune checkpoint blockade or vice versa. a single-institutional case-series. J Immunother Cancer. 2019;7:4. doi:10.1186/s40425-018-0475-y
- Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J. 2013;19:16. doi:10.5070/d370x41670
- Mirali S, Mufti A, Lansang RP, et al. Eruptive seborrheic keratoses are associated with a co-occurring malignancy in the majority of reported cases: a systematic review. J Cutan Med Surg. 2022;26:57-62. doi:10.1177/12034754211035124
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59:73-98. doi:10.3322/caac.20005
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435. doi:10.1038/nature08833
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-221. doi:10.1016/j.cell.2009.12.040
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 2010;464:427-430. doi:10.1038/nature08902
- Hayat MA. Brain Metastases from Primary Tumors, Volume 3: Epidemiology, Biology, and Therapy of Melanoma and Other Cancers. Academic Press; 2016.
To the Editor:
Mutations of the BRAF protein kinase gene are implicated in a variety of malignancies.1 BRAF mutations in malignancies cause the mitogen-activated protein kinase (MAPK) pathway to become constitutively active, which results in unchecked cellular proliferation,2,3 making the BRAF mutation an attractive target for inhibition with pharmacologic agents to potentially halt cancer growth.4 Vemurafenib—the first selective BRAF inhibitor used in clinical practice—initially was approved by the US Food and Drug Administration in 2011. The approval of dabrafenib followed in 2013 and most recently encorafenib in 2018.5
Although targeted treatment of BRAF-mutated malignancies with BRAF inhibitors has become common, it often is associated with cutaneous adverse events (AEs), such as rash, pruritus, photosensitivity, actinic keratosis, and verrucous keratosis. Some reports demonstrate these events in up to 95% of patients undergoing BRAF inhibitor treatment.6 In several cases the eruption of verrucous keratoses is among the most common cutaneous AEs seen among patients receiving BRAF inhibitor treatment.5-7
In general, lesions can appear days to months after therapy is initiated and may resolve after switching to dual therapy with a MEK inhibitor or with complete cessation of BRAF inhibitor therapy.5,7,8 One case of spontaneous resolution of vemurafenib-associated panniculitis during ongoing BRAF inhibitor therapy has been reported9; however, spontaneous resolution of cutaneous AEs is uncommon. Herein, we describe verrucous keratoses in a patient undergoing treatment with encorafenib that resolved spontaneously despite ongoing BRAF inhibitor therapy.
A 61-year-old woman presented to the emergency department with pain in the right lower quadrant. Computed tomography (CT) of the abdomen and pelvis revealed a large ovarian mass. Subsequent bloodwork revealed elevated carcinoembryonic antigen levels. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, right hemicolectomy with ileotransverse side-to-side anastomosis, right pelvic lymph node reduction, and complete cytoreduction. Histopathology revealed an adenocarcinoma of the cecum with tumor invasion into the visceral peritoneum and metastases to the left ovary, fallopian tube, and omentum. A BRAF V600E mutation was detected.
Two months after the initial presentation, the patient started her first cycle of chemotherapy with a combination of folinic acid, fluorouracil, and oxaliplatin. She completed 11 cycles of this regimen, then was switched to capecitabine and oxaliplatin for an additional 2 cycles due to insurance concerns. At the end of treatment, there was no evidence of disease on CT, thus the patient was followed with observation. However, she presented 10 months later to the emergency department with abdominal pain, and CT revealed new lesions in the liver that were concerning for potential metastases. She started oral encorafenib 300 mg/d and intravenous cetuximab 500 mg weekly; after 1 week, encorafenib was reduced to 150 mg/d due to nausea and loss of appetite. Within 2 weeks of starting treatment, the patient reported the relatively abrupt appearance of more than 50 small papules across the shoulders and back (Figure 1A). She was referred to dermatology, and shave biopsies of 2 lesions—one from the left anterior thigh, the other from the right posterior shoulder—revealed verrucous keratosis pathology (Figure 2). At this time, encorafenib was increased again to 300 mg/d as the patient had been tolerating the reduced dose. She continued to report the appearance of new lesions for the next 3 months, after which the lesions were stable for approximately 2 months. By 2.5 months after initiation of therapy, the patient had undergone CT demonstrating resolution of the liver lesions. At 5 months of therapy, the patient reported a stable to slightly reduced number of skin lesions but had begun to experience worsening joint pain, and the dosage of encorafenib was reduced to 225 mg/d. At 7 months of therapy, the dosage was further reduced to 150 mg/d due to persistent arthralgia. A follow-up examination at 10 months of therapy showed improvement in the number and size of the verrucous keratoses, and near resolution was seen by 14 months after the initial onset of the lesions (Figure 1B). At 20 months after initial onset, only 1 remaining verrucous keratosis was identified on physical examination and biopsy. The patient had continued a regimen of encorafenib 150 mg/d and weekly intravenous 500 mg cetuximab up to this point. Over the entire time period that the patient was seen, up to 12 lesions located in high-friction areas had become irritated and were treated with cryotherapy, but this contributed only minorly to the patient’s overall presentation.
Verrucous keratosis is a known cutaneous AE of BRAF inhibitor treatment with vemurafenib and dabrafenib, with fewer cases attributed to encorafenib.5,6 Within the oncologic setting, the eruption of verrucous papules as a paraneoplastic phenomenon is heavily debated in the literature and is known as the Leser-Trélat sign. This phenomenon is commonly associated with adenocarcinomas of the gastrointestinal tract, as seen in our patient.10 Based on Curth’s postulates—the criteria used to evaluate the relationship between an internal malignancy and a cutaneous disorder—this was unlikely in our patient. The criteria, which do not all need to be met to suggest a paraneoplastic phenomenon, include concurrent onset of the malignancy and the dermatosis, parallel course, association of a specific dermatosis with a specific malignancy, statistical significance of the association, and the presence of a genetic basis for the association.11 Several features favored a drug-related cutaneous eruption vs a paraneoplastic phenomenon: (1) the malignancy was identified months before the cutaneous eruptions manifested; (2) the cutaneous lesions appeared once treatment had already been initiated; and (3) the cutaneous lesions persisted long after the malignancy was no longer identifiable on CT. Indeed, eruption of the papules temporally coincided closely with the initiation of BRAF inhibitor therapy, arguing for correlation.
As a suspected BRAF inhibitor–associated cutaneous AE, the eruption of verrucous keratoses in our patient is remarkable for its spontaneous resolution despite ongoing therapy. It is speculated that keratinocytic proliferation while on BRAF inhibitor therapy may be caused by a paradoxical increase in signaling through CRAF, another Raf isoform that plays a role in the induction of terminal differentiation of keratinocytes, with a subsequent increase in MAPK signaling.12-14 Self-resolution of this cycle despite continuing BRAF inhibitor therapy suggests the possible involvement of balancing and/or alternative mechanistic pathways that may be related to the immune system. Although verrucous keratoses are considered benign proliferations and do not necessarily require any specific treatment or reduction in BRAF inhibitor dosage, they may be treated with cryotherapy, electrocautery, shave removal, or excision,15 which often is done if the lesions become inflamed and cause pain. Additionally, some patients may feel distress from the appearance of the lesions and desire treatment for this reason. Understanding that verrucous keratoses can be a transient cutaneous AE rather than a persistent one may be useful to clinicians as they manage AEs during BRAF inhibitor therapy.
To the Editor:
Mutations of the BRAF protein kinase gene are implicated in a variety of malignancies.1 BRAF mutations in malignancies cause the mitogen-activated protein kinase (MAPK) pathway to become constitutively active, which results in unchecked cellular proliferation,2,3 making the BRAF mutation an attractive target for inhibition with pharmacologic agents to potentially halt cancer growth.4 Vemurafenib—the first selective BRAF inhibitor used in clinical practice—initially was approved by the US Food and Drug Administration in 2011. The approval of dabrafenib followed in 2013 and most recently encorafenib in 2018.5
Although targeted treatment of BRAF-mutated malignancies with BRAF inhibitors has become common, it often is associated with cutaneous adverse events (AEs), such as rash, pruritus, photosensitivity, actinic keratosis, and verrucous keratosis. Some reports demonstrate these events in up to 95% of patients undergoing BRAF inhibitor treatment.6 In several cases the eruption of verrucous keratoses is among the most common cutaneous AEs seen among patients receiving BRAF inhibitor treatment.5-7
In general, lesions can appear days to months after therapy is initiated and may resolve after switching to dual therapy with a MEK inhibitor or with complete cessation of BRAF inhibitor therapy.5,7,8 One case of spontaneous resolution of vemurafenib-associated panniculitis during ongoing BRAF inhibitor therapy has been reported9; however, spontaneous resolution of cutaneous AEs is uncommon. Herein, we describe verrucous keratoses in a patient undergoing treatment with encorafenib that resolved spontaneously despite ongoing BRAF inhibitor therapy.
A 61-year-old woman presented to the emergency department with pain in the right lower quadrant. Computed tomography (CT) of the abdomen and pelvis revealed a large ovarian mass. Subsequent bloodwork revealed elevated carcinoembryonic antigen levels. The patient underwent a hysterectomy, bilateral salpingo-oophorectomy, omentectomy, right hemicolectomy with ileotransverse side-to-side anastomosis, right pelvic lymph node reduction, and complete cytoreduction. Histopathology revealed an adenocarcinoma of the cecum with tumor invasion into the visceral peritoneum and metastases to the left ovary, fallopian tube, and omentum. A BRAF V600E mutation was detected.
Two months after the initial presentation, the patient started her first cycle of chemotherapy with a combination of folinic acid, fluorouracil, and oxaliplatin. She completed 11 cycles of this regimen, then was switched to capecitabine and oxaliplatin for an additional 2 cycles due to insurance concerns. At the end of treatment, there was no evidence of disease on CT, thus the patient was followed with observation. However, she presented 10 months later to the emergency department with abdominal pain, and CT revealed new lesions in the liver that were concerning for potential metastases. She started oral encorafenib 300 mg/d and intravenous cetuximab 500 mg weekly; after 1 week, encorafenib was reduced to 150 mg/d due to nausea and loss of appetite. Within 2 weeks of starting treatment, the patient reported the relatively abrupt appearance of more than 50 small papules across the shoulders and back (Figure 1A). She was referred to dermatology, and shave biopsies of 2 lesions—one from the left anterior thigh, the other from the right posterior shoulder—revealed verrucous keratosis pathology (Figure 2). At this time, encorafenib was increased again to 300 mg/d as the patient had been tolerating the reduced dose. She continued to report the appearance of new lesions for the next 3 months, after which the lesions were stable for approximately 2 months. By 2.5 months after initiation of therapy, the patient had undergone CT demonstrating resolution of the liver lesions. At 5 months of therapy, the patient reported a stable to slightly reduced number of skin lesions but had begun to experience worsening joint pain, and the dosage of encorafenib was reduced to 225 mg/d. At 7 months of therapy, the dosage was further reduced to 150 mg/d due to persistent arthralgia. A follow-up examination at 10 months of therapy showed improvement in the number and size of the verrucous keratoses, and near resolution was seen by 14 months after the initial onset of the lesions (Figure 1B). At 20 months after initial onset, only 1 remaining verrucous keratosis was identified on physical examination and biopsy. The patient had continued a regimen of encorafenib 150 mg/d and weekly intravenous 500 mg cetuximab up to this point. Over the entire time period that the patient was seen, up to 12 lesions located in high-friction areas had become irritated and were treated with cryotherapy, but this contributed only minorly to the patient’s overall presentation.
Verrucous keratosis is a known cutaneous AE of BRAF inhibitor treatment with vemurafenib and dabrafenib, with fewer cases attributed to encorafenib.5,6 Within the oncologic setting, the eruption of verrucous papules as a paraneoplastic phenomenon is heavily debated in the literature and is known as the Leser-Trélat sign. This phenomenon is commonly associated with adenocarcinomas of the gastrointestinal tract, as seen in our patient.10 Based on Curth’s postulates—the criteria used to evaluate the relationship between an internal malignancy and a cutaneous disorder—this was unlikely in our patient. The criteria, which do not all need to be met to suggest a paraneoplastic phenomenon, include concurrent onset of the malignancy and the dermatosis, parallel course, association of a specific dermatosis with a specific malignancy, statistical significance of the association, and the presence of a genetic basis for the association.11 Several features favored a drug-related cutaneous eruption vs a paraneoplastic phenomenon: (1) the malignancy was identified months before the cutaneous eruptions manifested; (2) the cutaneous lesions appeared once treatment had already been initiated; and (3) the cutaneous lesions persisted long after the malignancy was no longer identifiable on CT. Indeed, eruption of the papules temporally coincided closely with the initiation of BRAF inhibitor therapy, arguing for correlation.
As a suspected BRAF inhibitor–associated cutaneous AE, the eruption of verrucous keratoses in our patient is remarkable for its spontaneous resolution despite ongoing therapy. It is speculated that keratinocytic proliferation while on BRAF inhibitor therapy may be caused by a paradoxical increase in signaling through CRAF, another Raf isoform that plays a role in the induction of terminal differentiation of keratinocytes, with a subsequent increase in MAPK signaling.12-14 Self-resolution of this cycle despite continuing BRAF inhibitor therapy suggests the possible involvement of balancing and/or alternative mechanistic pathways that may be related to the immune system. Although verrucous keratoses are considered benign proliferations and do not necessarily require any specific treatment or reduction in BRAF inhibitor dosage, they may be treated with cryotherapy, electrocautery, shave removal, or excision,15 which often is done if the lesions become inflamed and cause pain. Additionally, some patients may feel distress from the appearance of the lesions and desire treatment for this reason. Understanding that verrucous keratoses can be a transient cutaneous AE rather than a persistent one may be useful to clinicians as they manage AEs during BRAF inhibitor therapy.
- Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346-356. doi:10.1097/PAT.0b013e328360b61d
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529-545. doi:10.1016/j.hoc.2009.04.001
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246. doi:10.1200/JCO.2010.32.4327
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2012;18:27-35. doi:10.1016/j.molmed.2011.08.001
- Gouda MA, Subbiah V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: from melanoma to tissue-agnostic therapy. ESMO Open. 2023;8:100788. doi:10.1016/j.esmoop.2023.100788
- Gençler B, Gönül M. Cutaneous side effects of BRAF inhibitors in advanced melanoma: review of the literature. Dermatol Res Pract. 2016;2016:5361569. doi:10.1155/2016/5361569.
- Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol. 2012;67:1265-1272. doi:10.1016/j.jaad.2012.04.008
- Naqash AR, File DM, Ziemer CM, et al. Cutaneous adverse reactions in B-RAF positive metastatic melanoma following sequential treatment with B-RAF/MEK inhibitors and immune checkpoint blockade or vice versa. a single-institutional case-series. J Immunother Cancer. 2019;7:4. doi:10.1186/s40425-018-0475-y
- Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J. 2013;19:16. doi:10.5070/d370x41670
- Mirali S, Mufti A, Lansang RP, et al. Eruptive seborrheic keratoses are associated with a co-occurring malignancy in the majority of reported cases: a systematic review. J Cutan Med Surg. 2022;26:57-62. doi:10.1177/12034754211035124
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59:73-98. doi:10.3322/caac.20005
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435. doi:10.1038/nature08833
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-221. doi:10.1016/j.cell.2009.12.040
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 2010;464:427-430. doi:10.1038/nature08902
- Hayat MA. Brain Metastases from Primary Tumors, Volume 3: Epidemiology, Biology, and Therapy of Melanoma and Other Cancers. Academic Press; 2016.
- Pakneshan S, Salajegheh A, Smith RA, Lam AK. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346-356. doi:10.1097/PAT.0b013e328360b61d
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am. 2009;23:529-545. doi:10.1016/j.hoc.2009.04.001
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246. doi:10.1200/JCO.2010.32.4327
- Ji Z, Flaherty KT, Tsao H. Targeting the RAS pathway in melanoma. Trends Mol Med. 2012;18:27-35. doi:10.1016/j.molmed.2011.08.001
- Gouda MA, Subbiah V. Precision oncology for BRAF-mutant cancers with BRAF and MEK inhibitors: from melanoma to tissue-agnostic therapy. ESMO Open. 2023;8:100788. doi:10.1016/j.esmoop.2023.100788
- Gençler B, Gönül M. Cutaneous side effects of BRAF inhibitors in advanced melanoma: review of the literature. Dermatol Res Pract. 2016;2016:5361569. doi:10.1155/2016/5361569.
- Chu EY, Wanat KA, Miller CJ, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol. 2012;67:1265-1272. doi:10.1016/j.jaad.2012.04.008
- Naqash AR, File DM, Ziemer CM, et al. Cutaneous adverse reactions in B-RAF positive metastatic melanoma following sequential treatment with B-RAF/MEK inhibitors and immune checkpoint blockade or vice versa. a single-institutional case-series. J Immunother Cancer. 2019;7:4. doi:10.1186/s40425-018-0475-y
- Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J. 2013;19:16. doi:10.5070/d370x41670
- Mirali S, Mufti A, Lansang RP, et al. Eruptive seborrheic keratoses are associated with a co-occurring malignancy in the majority of reported cases: a systematic review. J Cutan Med Surg. 2022;26:57-62. doi:10.1177/12034754211035124
- Thiers BH, Sahn RE, Callen JP. Cutaneous manifestations of internal malignancy. CA Cancer J Clin. 2009;59:73-98. doi:10.3322/caac.20005
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-435. doi:10.1038/nature08833
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-221. doi:10.1016/j.cell.2009.12.040
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature. 2010;464:427-430. doi:10.1038/nature08902
- Hayat MA. Brain Metastases from Primary Tumors, Volume 3: Epidemiology, Biology, and Therapy of Melanoma and Other Cancers. Academic Press; 2016.
Practice Points
- Verrucous keratoses are common cutaneous adverse events (AEs) associated with BRAF inhibitor therapy.
- Verrucous papules may be a paraneoplastic phenomenon and can be differentiated from a treatment-related AE based on the timing and progression in relation to tumor burden.
- Although treatment of particularly bothersome lesions with cryotherapy may be warranted, verrucous papules secondary to BRAF inhibitor therapy may resolve spontaneously.
Rare Case of Photodistributed Hyperpigmentation Linked to Kratom Consumption
To the Editor:
Kratom (Mitragyna speciosa) is an evergreen tree native to Southeast Asia.1 Its leaves contain psychoactive compounds including mitragynine and 7-hydroxymitragynine, which exert dose-dependent effects on the central nervous system through opioid and monoaminergic receptors.2,3 At low doses (1–5 g), kratom elicits mild stimulant effects such as increased sociability, alertness, and talkativeness. At high doses (5–15 g), kratom has depressant effects that can provide relief from pain and opioid-withdrawal symptoms.3
Traditionally, kratom has been used in Southeast Asia for recreational and ceremonial purposes, to ease opioid-withdrawal symptoms, and to reduce fatigue from manual labor.4 In the 21st century, availability of kratom expanded to Europe, Australia, and the United States, largely facilitated by widespread dissemination of deceitful marketing and unregulated sales on the internet.1 Although large-scale epidemiologic studies evaluating kratom’s prevalence are scarce, available evidence indicates rising worldwide usage, with a notable increase in kratom-related poison center calls between 2011 and 2017 in the United States.5 In July 2023, kratom made headlines due to the death of a woman in Florida following use of the substance.6
A cross-sectional study revealed that in the United States, kratom typically is used by White individuals for self-treatment of anxiety, depression, pain, and opioid withdrawal.7 However, the potential for severe adverse effects and dependence on kratom can outweigh the benefits.6,8 Reported adverse effects of kratom include tachycardia, hypercholesteremia, liver injury, hallucinations, respiratory depression, seizure, coma, and death.9,10 We present a case of kratom-induced photodistributed hyperpigmentation.
A 63-year-old man presented to the dermatology clinic with diffuse tender, pruritic, hyperpigmented skin lesions that developed over the course of 1 year. The lesions were distributed on sun-exposed areas, including the face, neck, and forearms (Figure 1). The patient reported no other major symptoms, and his health was otherwise unremarkable. He had a medical history of psoriasiform and spongiotic dermatitis consistent with eczema, psoriasis, hypercholesteremia, and hyperlipidemia. The patient was not taking any medications at the time of presentation. He had a family history of plaque psoriasis in his father. Five years prior to the current presentation, the patient was treated with adalimumab for steroid-resistant psoriasis; however, despite initial improvement, he experienced recurrence of scaly erythematous plaques and had discontinued adalimumab the year prior to presentation.
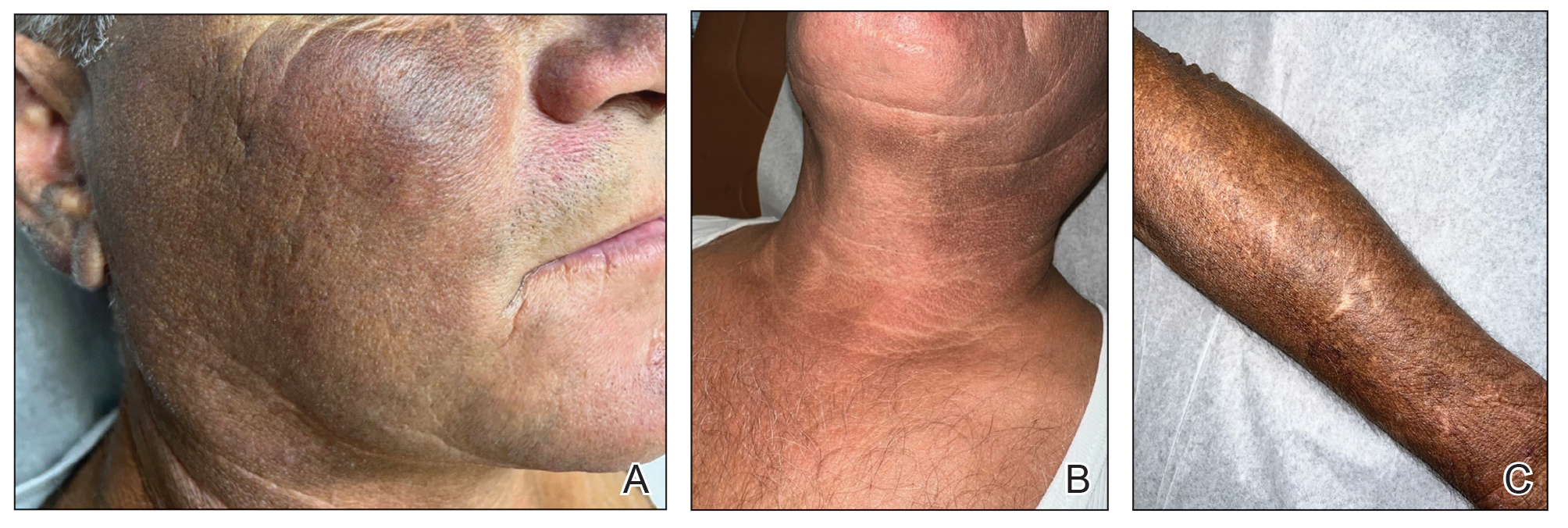
When adalimumab was discontinued, the patient sought alternative treatment for the skin symptoms and began self-administering kratom in an attempt to alleviate associated physical discomfort. He ingested approximately 3 bottles of liquid kratom per day, with each bottle containing 180 mg of mitragynine and less than 8 mg of 7-hydroxymitragynine. Although not scientifically proven, kratom has been colloquially advertised to improve psoriasis.11 The patient reported no other medication use or allergies.
Shave biopsies of hyperpigmented lesions on the right side of the neck, ear, and forearm were performed. Histopathology revealed a sparse superficial, perivascular, lymphocytic infiltrate accompanied by a prominent number of melanophages in the superficial dermis (Figure 2). Special stains further confirmed that the pigment was melanin; the specimens stained positive with Fontana-Masson stain (Figure 3) and negative with an iron stain (Figure 4).
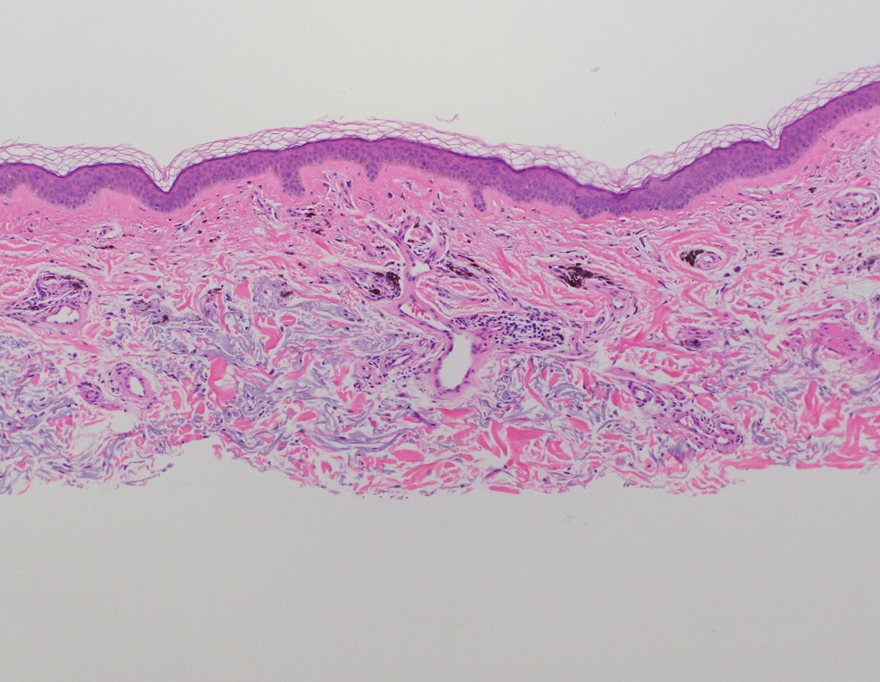
Adalimumab-induced hyperpigmentation was considered. A prior case of adalimumab-induced hyperpigmentation manifested on the face. Histopathology was consistent with a superficial, perivascular, lymphocytic infiltrate with melanophages in the dermis; however, hyperpigmentation was absent in the periorbital area, and affected areas faded 4 months after discontinuation of adalimumab.12 Our patient presented with hyperpigmentation 1 year after adalimumab cessation, and the hyperpigmented areas included the periorbital region. Because of the distinct temporal and clinical features, adalimumab-induced hyperpigmentation was eliminated from the differential diagnosis.
Based on the photodistributed pattern of hyperpigmentation, histopathology, and the temporal relationship between hyperpigmentation onset and kratom usage, a diagnosis of kratom-induced photodistributed hyperpigmentation was made. The patient was advised to discontinue kratom use and use sun protection to prevent further photodamage. The patient subsequently was lost to follow-up.
Kratom alkaloids bind all 3 opioid receptors—μOP, δOP, and κOPs—in a G-protein–biased manner with 7-hydroxymitragynine, the most pharmacologically active alkaloid, exhibiting a higher affinity for μ-opioid receptors.13,14 In human epidermal melanocytes, binding between μ-opioid receptors and β-endorphin, an endogenous opioid, is associated with increased melanin production. This melanogenesis has been linked to hyperpigmentation.15 Given the similarity between kratom alkaloids and β-endorphin in opioid-receptor binding, it is possible that kratom-induced hyperpigmentation may occur through a similar mechanism involving μ-opioid receptors and melanogenesis in epidermal melanocytes. Moreover, some researchers have theorized that sun exposure may result in free radical formation of certain drugs or their metabolites. These free radicals then can interact with cellular DNA, triggering the release of pigmentary mediators and resulting in hyperpigmentation.16 This theory may explain the photodistributed pattern of kratom-induced hyperpigmentation. Further studies are needed to understand the mechanism behind this adverse reaction and its implications for patient treatment.
Literature on kratom-induced hyperpigmentation is limited. Powell et al17 reported a similar case of kratom-induced photodistributed hyperpigmentation—a White man had taken kratom to reduce opioid use and subsequently developed hyperpigmented patches on the arms and face. Moreover, anonymous Reddit users have shared anecdotal reports of hyperpigmentation following kratom use.18
Physicians should be aware of hyperpigmentation as a potential adverse reaction of kratom use as its prevalence increases globally. Further research is warranted to elucidate the mechanism behind this adverse reaction and identify risk factors.
- Prozialeck WC, Avery BA, Boyer EW, et al. Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy. 2019;70:70-77. doi:10.1016/j.drugpo.2019.05.003
- Bergen-Cico D, MacClurg K. Kratom (Mitragyna speciosa) use, addiction potential, and legal status. In: Preedy VR, ed. Neuropathology of Drug Addictions and Substance Misuse. 2016:903-911. doi:10.1016/B978-0-12-800634-4.00089-5
- Warner ML, Kaufman NC, Grundmann O. The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med. 2016;130:127-138. doi:10.1007/s00414-015-1279-y
- Transnational Institute. Kratom in Thailand: decriminalisation and community control? May 3, 2011. Accessed August 23, 2024. https://www.tni.org/en/publication/kratom-in-thailand-decriminalisation-and-community-control
- Eastlack SC, Cornett EM, Kaye AD. Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020;9:55-69. doi:10.1007/s40122-020-00151-x
- Reyes R. Family of Florida mom who died from herbal substance kratom wins $11M suit. New York Post. July 30, 2023. Updated July 31, 2023. Accessed August 23, 2024. https://nypost.com/2023/07/30/family-of-florida-mom-who-died-from-herbal-substance-kratom-wins-11m-suit/
- Garcia-Romeu A, Cox DJ, Smith KE, et al. Kratom (Mitragyna speciosa): user demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol Depend. 2020;208:107849. doi:10.1016/j.drugalcdep.2020.107849
- Mayo Clinic. Kratom: unsafe and ineffective. Accessed August 23, 2024. https://www.mayoclinic.org/healthy-lifestyle/consumer-health/in-depth/kratom/art-20402171
- Sethi R, Hoang N, Ravishankar DA, et al. Kratom (Mitragyna speciosa): friend or foe? Prim Care Companion CNS Disord. 2020;22:19nr02507.
- Eggleston W, Stoppacher R, Suen K, et al. Kratom use and toxicities in the United States. Pharmacother J Hum Pharmacol Drug Ther. 2019;39:775-777. doi:10.1002/phar.2280
- Qrius. 6 benefits of kratom you should know for healthy skin. March 21, 2023. Accessed August 23, 2024. https://qrius.com/6-benefits-of-kratom-you-should-know-for-healthy-skin/
- Blomberg M, Zachariae COC, Grønhøj F. Hyperpigmentation of the face following adalimumab treatment. Acta Derm Venereol. 2009;89:546-547. doi:10.2340/00015555-0697
- Matsumoto K, Hatori Y, Murayama T, et al. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63-70. doi:10.1016/j.ejphar.2006.08.013
- Jentsch MJ, Pippin MM. Kratom. In: StatPearls. StatPearls Publishing; 2023.
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, et al. Opioids and the skin—where do we stand? Exp Dermatol. 2009;18:424-430.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10. doi:10.5070/D33c97j4z5
- Powell LR, Ryser TJ, Morey GE, et al. Kratom as a novel cause of photodistributed hyperpigmentation. JAAD Case Rep. 2022;28:145-148. doi:10.1016/j.jdcr.2022.07.033
- Haccoon. Skin discoloring? Reddit. June 30, 2019. Accessed August 23, 2024. https://www.reddit.com/r/quittingkratom/comments/c7b1cm/skin_discoloring/
To the Editor:
Kratom (Mitragyna speciosa) is an evergreen tree native to Southeast Asia.1 Its leaves contain psychoactive compounds including mitragynine and 7-hydroxymitragynine, which exert dose-dependent effects on the central nervous system through opioid and monoaminergic receptors.2,3 At low doses (1–5 g), kratom elicits mild stimulant effects such as increased sociability, alertness, and talkativeness. At high doses (5–15 g), kratom has depressant effects that can provide relief from pain and opioid-withdrawal symptoms.3
Traditionally, kratom has been used in Southeast Asia for recreational and ceremonial purposes, to ease opioid-withdrawal symptoms, and to reduce fatigue from manual labor.4 In the 21st century, availability of kratom expanded to Europe, Australia, and the United States, largely facilitated by widespread dissemination of deceitful marketing and unregulated sales on the internet.1 Although large-scale epidemiologic studies evaluating kratom’s prevalence are scarce, available evidence indicates rising worldwide usage, with a notable increase in kratom-related poison center calls between 2011 and 2017 in the United States.5 In July 2023, kratom made headlines due to the death of a woman in Florida following use of the substance.6
A cross-sectional study revealed that in the United States, kratom typically is used by White individuals for self-treatment of anxiety, depression, pain, and opioid withdrawal.7 However, the potential for severe adverse effects and dependence on kratom can outweigh the benefits.6,8 Reported adverse effects of kratom include tachycardia, hypercholesteremia, liver injury, hallucinations, respiratory depression, seizure, coma, and death.9,10 We present a case of kratom-induced photodistributed hyperpigmentation.
A 63-year-old man presented to the dermatology clinic with diffuse tender, pruritic, hyperpigmented skin lesions that developed over the course of 1 year. The lesions were distributed on sun-exposed areas, including the face, neck, and forearms (Figure 1). The patient reported no other major symptoms, and his health was otherwise unremarkable. He had a medical history of psoriasiform and spongiotic dermatitis consistent with eczema, psoriasis, hypercholesteremia, and hyperlipidemia. The patient was not taking any medications at the time of presentation. He had a family history of plaque psoriasis in his father. Five years prior to the current presentation, the patient was treated with adalimumab for steroid-resistant psoriasis; however, despite initial improvement, he experienced recurrence of scaly erythematous plaques and had discontinued adalimumab the year prior to presentation.
When adalimumab was discontinued, the patient sought alternative treatment for the skin symptoms and began self-administering kratom in an attempt to alleviate associated physical discomfort. He ingested approximately 3 bottles of liquid kratom per day, with each bottle containing 180 mg of mitragynine and less than 8 mg of 7-hydroxymitragynine. Although not scientifically proven, kratom has been colloquially advertised to improve psoriasis.11 The patient reported no other medication use or allergies.
Shave biopsies of hyperpigmented lesions on the right side of the neck, ear, and forearm were performed. Histopathology revealed a sparse superficial, perivascular, lymphocytic infiltrate accompanied by a prominent number of melanophages in the superficial dermis (Figure 2). Special stains further confirmed that the pigment was melanin; the specimens stained positive with Fontana-Masson stain (Figure 3) and negative with an iron stain (Figure 4).
Adalimumab-induced hyperpigmentation was considered. A prior case of adalimumab-induced hyperpigmentation manifested on the face. Histopathology was consistent with a superficial, perivascular, lymphocytic infiltrate with melanophages in the dermis; however, hyperpigmentation was absent in the periorbital area, and affected areas faded 4 months after discontinuation of adalimumab.12 Our patient presented with hyperpigmentation 1 year after adalimumab cessation, and the hyperpigmented areas included the periorbital region. Because of the distinct temporal and clinical features, adalimumab-induced hyperpigmentation was eliminated from the differential diagnosis.
Based on the photodistributed pattern of hyperpigmentation, histopathology, and the temporal relationship between hyperpigmentation onset and kratom usage, a diagnosis of kratom-induced photodistributed hyperpigmentation was made. The patient was advised to discontinue kratom use and use sun protection to prevent further photodamage. The patient subsequently was lost to follow-up.
Kratom alkaloids bind all 3 opioid receptors—μOP, δOP, and κOPs—in a G-protein–biased manner with 7-hydroxymitragynine, the most pharmacologically active alkaloid, exhibiting a higher affinity for μ-opioid receptors.13,14 In human epidermal melanocytes, binding between μ-opioid receptors and β-endorphin, an endogenous opioid, is associated with increased melanin production. This melanogenesis has been linked to hyperpigmentation.15 Given the similarity between kratom alkaloids and β-endorphin in opioid-receptor binding, it is possible that kratom-induced hyperpigmentation may occur through a similar mechanism involving μ-opioid receptors and melanogenesis in epidermal melanocytes. Moreover, some researchers have theorized that sun exposure may result in free radical formation of certain drugs or their metabolites. These free radicals then can interact with cellular DNA, triggering the release of pigmentary mediators and resulting in hyperpigmentation.16 This theory may explain the photodistributed pattern of kratom-induced hyperpigmentation. Further studies are needed to understand the mechanism behind this adverse reaction and its implications for patient treatment.
Literature on kratom-induced hyperpigmentation is limited. Powell et al17 reported a similar case of kratom-induced photodistributed hyperpigmentation—a White man had taken kratom to reduce opioid use and subsequently developed hyperpigmented patches on the arms and face. Moreover, anonymous Reddit users have shared anecdotal reports of hyperpigmentation following kratom use.18
Physicians should be aware of hyperpigmentation as a potential adverse reaction of kratom use as its prevalence increases globally. Further research is warranted to elucidate the mechanism behind this adverse reaction and identify risk factors.
To the Editor:
Kratom (Mitragyna speciosa) is an evergreen tree native to Southeast Asia.1 Its leaves contain psychoactive compounds including mitragynine and 7-hydroxymitragynine, which exert dose-dependent effects on the central nervous system through opioid and monoaminergic receptors.2,3 At low doses (1–5 g), kratom elicits mild stimulant effects such as increased sociability, alertness, and talkativeness. At high doses (5–15 g), kratom has depressant effects that can provide relief from pain and opioid-withdrawal symptoms.3
Traditionally, kratom has been used in Southeast Asia for recreational and ceremonial purposes, to ease opioid-withdrawal symptoms, and to reduce fatigue from manual labor.4 In the 21st century, availability of kratom expanded to Europe, Australia, and the United States, largely facilitated by widespread dissemination of deceitful marketing and unregulated sales on the internet.1 Although large-scale epidemiologic studies evaluating kratom’s prevalence are scarce, available evidence indicates rising worldwide usage, with a notable increase in kratom-related poison center calls between 2011 and 2017 in the United States.5 In July 2023, kratom made headlines due to the death of a woman in Florida following use of the substance.6
A cross-sectional study revealed that in the United States, kratom typically is used by White individuals for self-treatment of anxiety, depression, pain, and opioid withdrawal.7 However, the potential for severe adverse effects and dependence on kratom can outweigh the benefits.6,8 Reported adverse effects of kratom include tachycardia, hypercholesteremia, liver injury, hallucinations, respiratory depression, seizure, coma, and death.9,10 We present a case of kratom-induced photodistributed hyperpigmentation.
A 63-year-old man presented to the dermatology clinic with diffuse tender, pruritic, hyperpigmented skin lesions that developed over the course of 1 year. The lesions were distributed on sun-exposed areas, including the face, neck, and forearms (Figure 1). The patient reported no other major symptoms, and his health was otherwise unremarkable. He had a medical history of psoriasiform and spongiotic dermatitis consistent with eczema, psoriasis, hypercholesteremia, and hyperlipidemia. The patient was not taking any medications at the time of presentation. He had a family history of plaque psoriasis in his father. Five years prior to the current presentation, the patient was treated with adalimumab for steroid-resistant psoriasis; however, despite initial improvement, he experienced recurrence of scaly erythematous plaques and had discontinued adalimumab the year prior to presentation.
When adalimumab was discontinued, the patient sought alternative treatment for the skin symptoms and began self-administering kratom in an attempt to alleviate associated physical discomfort. He ingested approximately 3 bottles of liquid kratom per day, with each bottle containing 180 mg of mitragynine and less than 8 mg of 7-hydroxymitragynine. Although not scientifically proven, kratom has been colloquially advertised to improve psoriasis.11 The patient reported no other medication use or allergies.
Shave biopsies of hyperpigmented lesions on the right side of the neck, ear, and forearm were performed. Histopathology revealed a sparse superficial, perivascular, lymphocytic infiltrate accompanied by a prominent number of melanophages in the superficial dermis (Figure 2). Special stains further confirmed that the pigment was melanin; the specimens stained positive with Fontana-Masson stain (Figure 3) and negative with an iron stain (Figure 4).
Adalimumab-induced hyperpigmentation was considered. A prior case of adalimumab-induced hyperpigmentation manifested on the face. Histopathology was consistent with a superficial, perivascular, lymphocytic infiltrate with melanophages in the dermis; however, hyperpigmentation was absent in the periorbital area, and affected areas faded 4 months after discontinuation of adalimumab.12 Our patient presented with hyperpigmentation 1 year after adalimumab cessation, and the hyperpigmented areas included the periorbital region. Because of the distinct temporal and clinical features, adalimumab-induced hyperpigmentation was eliminated from the differential diagnosis.
Based on the photodistributed pattern of hyperpigmentation, histopathology, and the temporal relationship between hyperpigmentation onset and kratom usage, a diagnosis of kratom-induced photodistributed hyperpigmentation was made. The patient was advised to discontinue kratom use and use sun protection to prevent further photodamage. The patient subsequently was lost to follow-up.
Kratom alkaloids bind all 3 opioid receptors—μOP, δOP, and κOPs—in a G-protein–biased manner with 7-hydroxymitragynine, the most pharmacologically active alkaloid, exhibiting a higher affinity for μ-opioid receptors.13,14 In human epidermal melanocytes, binding between μ-opioid receptors and β-endorphin, an endogenous opioid, is associated with increased melanin production. This melanogenesis has been linked to hyperpigmentation.15 Given the similarity between kratom alkaloids and β-endorphin in opioid-receptor binding, it is possible that kratom-induced hyperpigmentation may occur through a similar mechanism involving μ-opioid receptors and melanogenesis in epidermal melanocytes. Moreover, some researchers have theorized that sun exposure may result in free radical formation of certain drugs or their metabolites. These free radicals then can interact with cellular DNA, triggering the release of pigmentary mediators and resulting in hyperpigmentation.16 This theory may explain the photodistributed pattern of kratom-induced hyperpigmentation. Further studies are needed to understand the mechanism behind this adverse reaction and its implications for patient treatment.
Literature on kratom-induced hyperpigmentation is limited. Powell et al17 reported a similar case of kratom-induced photodistributed hyperpigmentation—a White man had taken kratom to reduce opioid use and subsequently developed hyperpigmented patches on the arms and face. Moreover, anonymous Reddit users have shared anecdotal reports of hyperpigmentation following kratom use.18
Physicians should be aware of hyperpigmentation as a potential adverse reaction of kratom use as its prevalence increases globally. Further research is warranted to elucidate the mechanism behind this adverse reaction and identify risk factors.
- Prozialeck WC, Avery BA, Boyer EW, et al. Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy. 2019;70:70-77. doi:10.1016/j.drugpo.2019.05.003
- Bergen-Cico D, MacClurg K. Kratom (Mitragyna speciosa) use, addiction potential, and legal status. In: Preedy VR, ed. Neuropathology of Drug Addictions and Substance Misuse. 2016:903-911. doi:10.1016/B978-0-12-800634-4.00089-5
- Warner ML, Kaufman NC, Grundmann O. The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med. 2016;130:127-138. doi:10.1007/s00414-015-1279-y
- Transnational Institute. Kratom in Thailand: decriminalisation and community control? May 3, 2011. Accessed August 23, 2024. https://www.tni.org/en/publication/kratom-in-thailand-decriminalisation-and-community-control
- Eastlack SC, Cornett EM, Kaye AD. Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020;9:55-69. doi:10.1007/s40122-020-00151-x
- Reyes R. Family of Florida mom who died from herbal substance kratom wins $11M suit. New York Post. July 30, 2023. Updated July 31, 2023. Accessed August 23, 2024. https://nypost.com/2023/07/30/family-of-florida-mom-who-died-from-herbal-substance-kratom-wins-11m-suit/
- Garcia-Romeu A, Cox DJ, Smith KE, et al. Kratom (Mitragyna speciosa): user demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol Depend. 2020;208:107849. doi:10.1016/j.drugalcdep.2020.107849
- Mayo Clinic. Kratom: unsafe and ineffective. Accessed August 23, 2024. https://www.mayoclinic.org/healthy-lifestyle/consumer-health/in-depth/kratom/art-20402171
- Sethi R, Hoang N, Ravishankar DA, et al. Kratom (Mitragyna speciosa): friend or foe? Prim Care Companion CNS Disord. 2020;22:19nr02507.
- Eggleston W, Stoppacher R, Suen K, et al. Kratom use and toxicities in the United States. Pharmacother J Hum Pharmacol Drug Ther. 2019;39:775-777. doi:10.1002/phar.2280
- Qrius. 6 benefits of kratom you should know for healthy skin. March 21, 2023. Accessed August 23, 2024. https://qrius.com/6-benefits-of-kratom-you-should-know-for-healthy-skin/
- Blomberg M, Zachariae COC, Grønhøj F. Hyperpigmentation of the face following adalimumab treatment. Acta Derm Venereol. 2009;89:546-547. doi:10.2340/00015555-0697
- Matsumoto K, Hatori Y, Murayama T, et al. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63-70. doi:10.1016/j.ejphar.2006.08.013
- Jentsch MJ, Pippin MM. Kratom. In: StatPearls. StatPearls Publishing; 2023.
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, et al. Opioids and the skin—where do we stand? Exp Dermatol. 2009;18:424-430.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10. doi:10.5070/D33c97j4z5
- Powell LR, Ryser TJ, Morey GE, et al. Kratom as a novel cause of photodistributed hyperpigmentation. JAAD Case Rep. 2022;28:145-148. doi:10.1016/j.jdcr.2022.07.033
- Haccoon. Skin discoloring? Reddit. June 30, 2019. Accessed August 23, 2024. https://www.reddit.com/r/quittingkratom/comments/c7b1cm/skin_discoloring/
- Prozialeck WC, Avery BA, Boyer EW, et al. Kratom policy: the challenge of balancing therapeutic potential with public safety. Int J Drug Policy. 2019;70:70-77. doi:10.1016/j.drugpo.2019.05.003
- Bergen-Cico D, MacClurg K. Kratom (Mitragyna speciosa) use, addiction potential, and legal status. In: Preedy VR, ed. Neuropathology of Drug Addictions and Substance Misuse. 2016:903-911. doi:10.1016/B978-0-12-800634-4.00089-5
- Warner ML, Kaufman NC, Grundmann O. The pharmacology and toxicology of kratom: from traditional herb to drug of abuse. Int J Legal Med. 2016;130:127-138. doi:10.1007/s00414-015-1279-y
- Transnational Institute. Kratom in Thailand: decriminalisation and community control? May 3, 2011. Accessed August 23, 2024. https://www.tni.org/en/publication/kratom-in-thailand-decriminalisation-and-community-control
- Eastlack SC, Cornett EM, Kaye AD. Kratom—pharmacology, clinical implications, and outlook: a comprehensive review. Pain Ther. 2020;9:55-69. doi:10.1007/s40122-020-00151-x
- Reyes R. Family of Florida mom who died from herbal substance kratom wins $11M suit. New York Post. July 30, 2023. Updated July 31, 2023. Accessed August 23, 2024. https://nypost.com/2023/07/30/family-of-florida-mom-who-died-from-herbal-substance-kratom-wins-11m-suit/
- Garcia-Romeu A, Cox DJ, Smith KE, et al. Kratom (Mitragyna speciosa): user demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol Depend. 2020;208:107849. doi:10.1016/j.drugalcdep.2020.107849
- Mayo Clinic. Kratom: unsafe and ineffective. Accessed August 23, 2024. https://www.mayoclinic.org/healthy-lifestyle/consumer-health/in-depth/kratom/art-20402171
- Sethi R, Hoang N, Ravishankar DA, et al. Kratom (Mitragyna speciosa): friend or foe? Prim Care Companion CNS Disord. 2020;22:19nr02507.
- Eggleston W, Stoppacher R, Suen K, et al. Kratom use and toxicities in the United States. Pharmacother J Hum Pharmacol Drug Ther. 2019;39:775-777. doi:10.1002/phar.2280
- Qrius. 6 benefits of kratom you should know for healthy skin. March 21, 2023. Accessed August 23, 2024. https://qrius.com/6-benefits-of-kratom-you-should-know-for-healthy-skin/
- Blomberg M, Zachariae COC, Grønhøj F. Hyperpigmentation of the face following adalimumab treatment. Acta Derm Venereol. 2009;89:546-547. doi:10.2340/00015555-0697
- Matsumoto K, Hatori Y, Murayama T, et al. Involvement of μ-opioid receptors in antinociception and inhibition of gastrointestinal transit induced by 7-hydroxymitragynine, isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63-70. doi:10.1016/j.ejphar.2006.08.013
- Jentsch MJ, Pippin MM. Kratom. In: StatPearls. StatPearls Publishing; 2023.
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, et al. Opioids and the skin—where do we stand? Exp Dermatol. 2009;18:424-430.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10. doi:10.5070/D33c97j4z5
- Powell LR, Ryser TJ, Morey GE, et al. Kratom as a novel cause of photodistributed hyperpigmentation. JAAD Case Rep. 2022;28:145-148. doi:10.1016/j.jdcr.2022.07.033
- Haccoon. Skin discoloring? Reddit. June 30, 2019. Accessed August 23, 2024. https://www.reddit.com/r/quittingkratom/comments/c7b1cm/skin_discoloring/
Practice Points
- Clinicians should be aware of photodistributed hyperpigmentation as a potential adverse effect of kratom usage.
- Kratom-induced photodistributed hyperpigmentation should be suspected in patients with hyperpigmented lesions in sun-exposed areas of the skin following kratom use. A biopsy of lesions should be obtained to confirm the diagnosis.
- Cessation of kratom should be recommended.
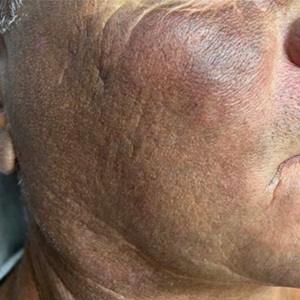
Successful Treatment of Refractory Extensive Pityriasis Rubra Pilaris With Risankizumab and Acitretin
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare papulosquamous condition with an unknown pathogenesis and limited efficacy data, which can make treatment challenging. Some cases of PRP spontaneously resolve in a few months, which is most common in the pediatric population.1 Pityriasis rubra pilaris in adults is likely to persist for years, and spontaneous resolution is unpredictable. Randomized clinical trials are difficult to perform due to the rarity of PRP.
Although there is no cure and no standard protocol for treating PRP, systemic retinoids historically are considered first-line therapy for moderate to severe cases.2 Additional management approaches include symptomatic control with moisturizers and psychological support. Alternative systemic treatments for moderate to severe cases include methotrexate, phototherapy, and cyclosporine.2
Pityriasis rubra pilaris demonstrates a favorable response to methotrexate treatment, especially in type I cases; however, patients on this alternative therapy should be monitored for severe adverse effects (eg, hepatotoxicity, pancytopenia, pneumonitis).2 Phototherapy should be approached with caution. Narrowband UVB, UVA1, and psoralen plus UVA therapy have successfully treated PRP; however, the response is variable. In some cases, the opposite effect can occur, in which the condition is photoaggravated. Phototherapy is a valid alternative form of treatment when used in combination with acitretin, and a phototest should be performed prior to starting this regimen. Cyclosporine is another immunosuppressant that can be considered for PRP treatment, though there are limited data demonstrating its efficacy.2
The introduction of biologic agents has changed the treatment approach for many dermatologic diseases, including PRP. Given the similar features between psoriasis and PRP, the biologics prescribed for psoriasis therapy also are used for patients with PRP that is challenging to treat, such as anti–tumor necrosis factor α inhibitors and IL inhibitors—specifically IL-17 and IL-23. Remission has been achieved with the use of biologics in combination with retinoid therapy.2
Biologic therapies used for PRP effectively inhibit cytokines and reduce the overall inflammatory processes involved in the development of the scaly patches and plaques seen in this condition. However, most reported clinical experiences are case studies, and more research in the form of randomized clinical trials is needed to understand the efficacy and long-term effects of this form of treatment in PRP. We present a case of a patient with refractory adult subtype I PRP that was successfully treated with the IL-23 inhibitor risankizumab.
A 65-year-old man was referred to Florida Academic Dermatology Center (Coral Gables, Florida) with biopsy-proven PRP diagnosed 1 year prior. The patient reported experiencing a debilitating quality of life in the year since diagnosis (Figure 1). Treatment attempts with dupilumab, tralokinumab, intramuscular steroid injections, and topical corticosteroids had failed (Figure 2). Following evaluation at Florida Academic Dermatology Center, the patient was started on acitretin 25 mg every other day and received an initial subcutaneous injection of ixekizumab 160 mg (an IL-17 inhibitor) followed 2 weeks later by a second injection of 80 mg. After the 2 doses of ixekizumab, the patient’s condition worsened with the development of pinpoint hemorrhagic lesions. The medication was discontinued, and he was started on risankizumab 150 mg at the approved dosing regimen for plaque psoriasis in combination with the acitretin therapy. Prior to starting risankizumab, the affected body surface area (BSA) was 80%. At 1-month follow-up, he showed improvement with reduction in scaling and erythema and an affected BSA of 30% (Figure 3). At 4-month follow-up, he continued showing improvement with an affected BSA of 10% (Figure 4). Acitretin was discontinued, and the patient has been successfully maintained on risankizumab 150 mg/mL subcutaneous injections every 12 weeks since.
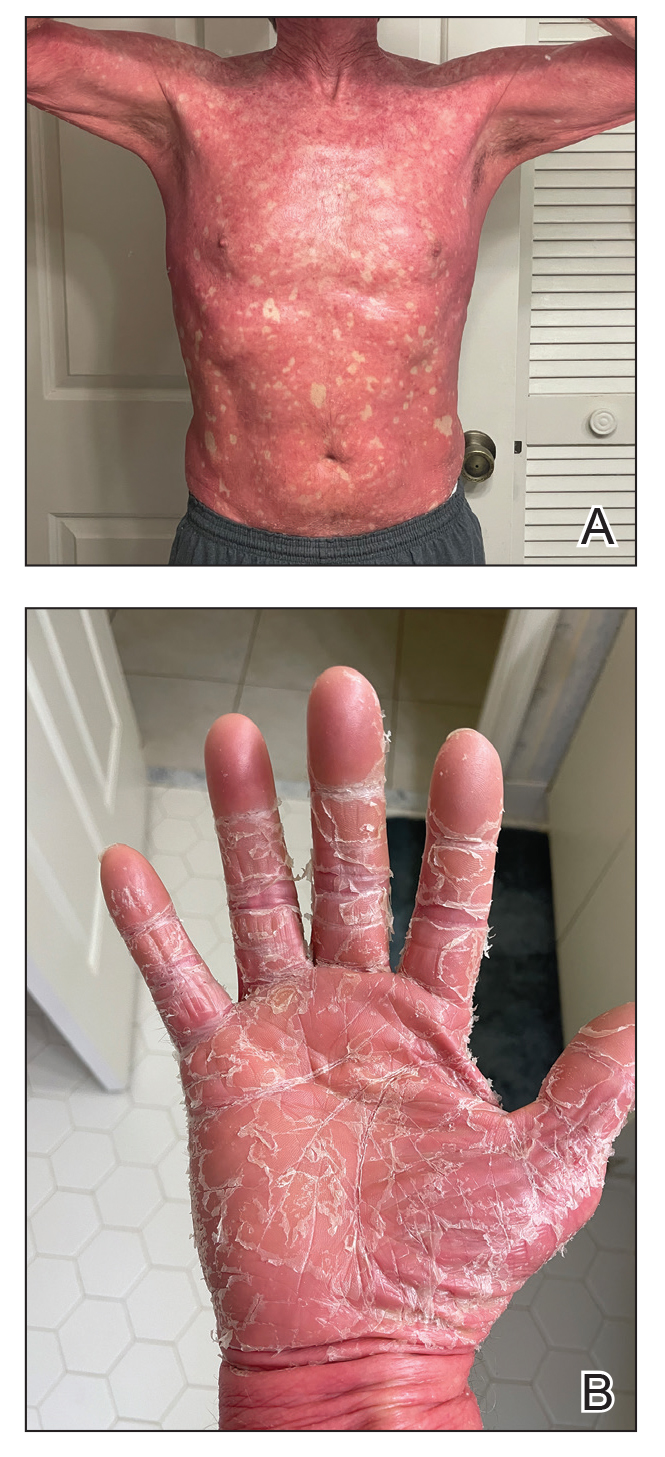
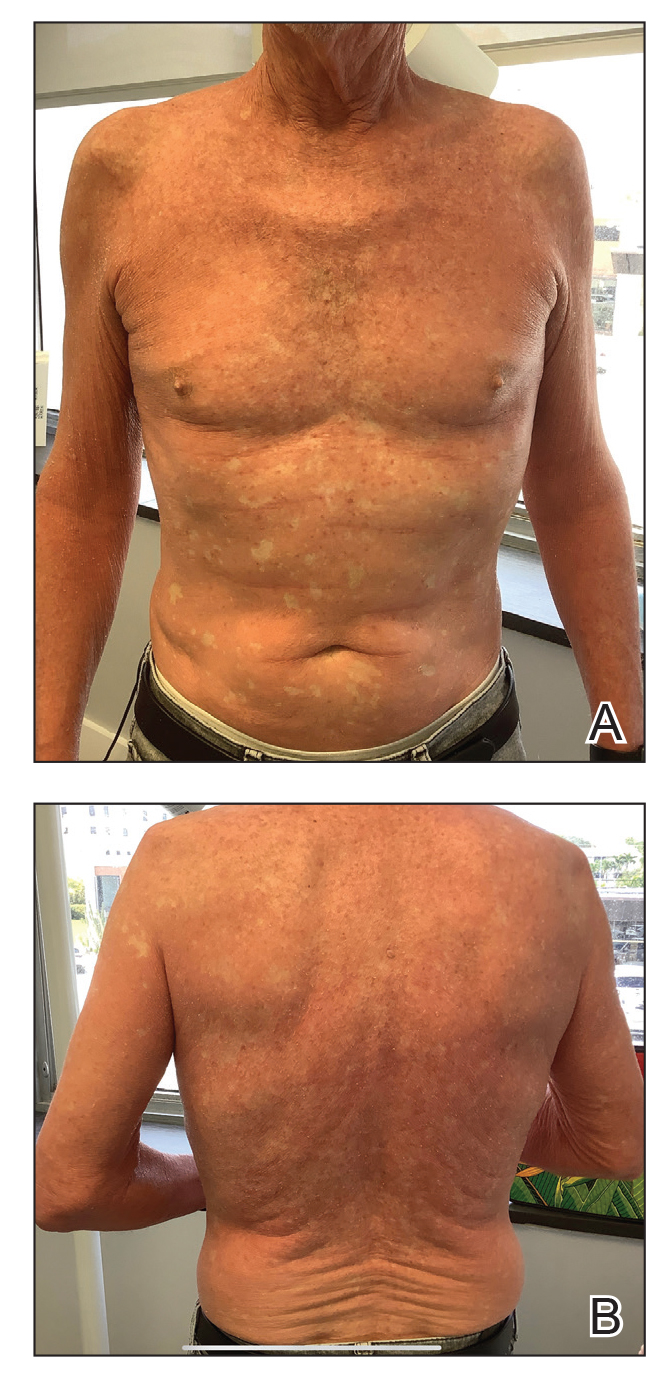
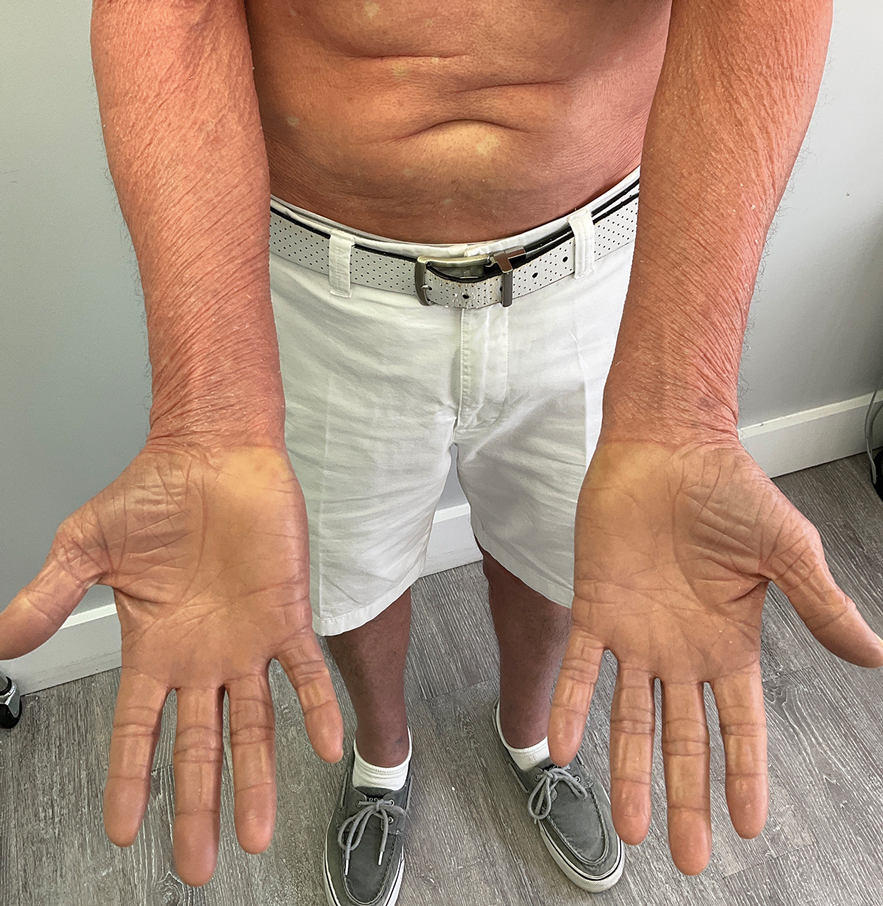
Oral retinoid therapy historically was considered first-line therapy for moderate to severe PRP. A systematic review (N=105) of retinoid therapies showed 83% of patients with PRP who were treated with acitretin plus biologic therapy had a favorable response, whereas only 36% of patients treated with acitretin as monotherapy had the same response, highlighting the importance of dual therapy.3 The use of ustekinumab, ixekizumab, and secukinumab (IL-17 inhibitors) for refractory PRP has been well documented, but a PubMed search of articles indexed for MEDLINE using the search terms risankizumab and pityriasis rubra pilaris yielded only 8 published cases of risankizumab for treatment of PRP.4-8 All patients were diagnosed with refractory PRP, and multiple treatment modalities failed.
Ustekinumab has been shown to create a rapid response and maintain it long term, especially in patients with type 1 PRP who did not respond to systemic therapies or anti–tumor necrosis factor α agents.2 An open-label, single-arm clinical trial found secukinumab was an effective therapy for PRP and demonstrated transcription heterogeneity of this dermatologic condition.9 The researchers proposed that some patients may respond to IL-17 inhibitors but others may not due to the differences in RNA molecules transcribed.9 Our patient demonstrated worsening of his condition with an IL-17 inhibitor but experienced remarkable improvement with risankizumab, an IL-23 inhibitor.
Risankizumab is indicated for the treatment of adults with moderate to severe plaque psoriasis. This humanized IgG1 monoclonal antibody targets the p19 subunit of IL-23, inhibiting its role in the pathogenic helper T cell (TH17) pathway. Research has shown that it is an efficacious and well-tolerated treatment modality for psoriatic conditions.10 It is well known that PRP and psoriasis have similar cytokine activations; therefore, we propose that combination therapy with risankizumab and acitretin may show promise for refractory PRP.
- Gelmetti C, Schiuma AA, Cerri D, et al. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatr Dermatol. 1986;3:446-451. doi:10.1111/j.1525-1470.1986.tb00648.x
- Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2017;10:451-457. doi:10.2147/CCID.S124351
- Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. Eur J Dermatol. 2019;29:524-537. doi:10.1684/ejd.2019.3641
- Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. Br J Dermatol. 2021;184:E148. doi:10.1111/bjd.19681
- Brocco E, Laffitte E. Risankizumab for pityriasis rubra pilaris. Clin Exp Dermatol. 2021;46:1322-1324. doi:10.1111/ced.14715
- Duarte B, Paiva Lopes MJ. Response to: ‘Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab.’ Br J Dermatol. 2021;185:235-236. doi:10.1111/bjd.20061
- Kromer C, Schön MP, Mössner R. Treatment of pityriasis rubra pilaris with risankizumab in two cases. J Dtsch Dermatol Ges. 2021;19:1207-1209. doi:10.1111/ddg.14504
- Kołt-Kamińska M, Osińska A, Kaznowska E, et al. Successful treatment of pityriasis rubra pilaris with risankizumab in children. Dermatol Ther (Heidelb). 2023;13:2431-2441. doi:10.1007/s13555-023-01005-y
- Boudreaux BW, Pincelli TP, Bhullar PK, et al. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. Br J Dermatol. 2022;187:650-658. doi:10.1111/bjd.21708
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156:649-658. doi:10.1001/jamadermatol.2020.0723
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare papulosquamous condition with an unknown pathogenesis and limited efficacy data, which can make treatment challenging. Some cases of PRP spontaneously resolve in a few months, which is most common in the pediatric population.1 Pityriasis rubra pilaris in adults is likely to persist for years, and spontaneous resolution is unpredictable. Randomized clinical trials are difficult to perform due to the rarity of PRP.
Although there is no cure and no standard protocol for treating PRP, systemic retinoids historically are considered first-line therapy for moderate to severe cases.2 Additional management approaches include symptomatic control with moisturizers and psychological support. Alternative systemic treatments for moderate to severe cases include methotrexate, phototherapy, and cyclosporine.2
Pityriasis rubra pilaris demonstrates a favorable response to methotrexate treatment, especially in type I cases; however, patients on this alternative therapy should be monitored for severe adverse effects (eg, hepatotoxicity, pancytopenia, pneumonitis).2 Phototherapy should be approached with caution. Narrowband UVB, UVA1, and psoralen plus UVA therapy have successfully treated PRP; however, the response is variable. In some cases, the opposite effect can occur, in which the condition is photoaggravated. Phototherapy is a valid alternative form of treatment when used in combination with acitretin, and a phototest should be performed prior to starting this regimen. Cyclosporine is another immunosuppressant that can be considered for PRP treatment, though there are limited data demonstrating its efficacy.2
The introduction of biologic agents has changed the treatment approach for many dermatologic diseases, including PRP. Given the similar features between psoriasis and PRP, the biologics prescribed for psoriasis therapy also are used for patients with PRP that is challenging to treat, such as anti–tumor necrosis factor α inhibitors and IL inhibitors—specifically IL-17 and IL-23. Remission has been achieved with the use of biologics in combination with retinoid therapy.2
Biologic therapies used for PRP effectively inhibit cytokines and reduce the overall inflammatory processes involved in the development of the scaly patches and plaques seen in this condition. However, most reported clinical experiences are case studies, and more research in the form of randomized clinical trials is needed to understand the efficacy and long-term effects of this form of treatment in PRP. We present a case of a patient with refractory adult subtype I PRP that was successfully treated with the IL-23 inhibitor risankizumab.
A 65-year-old man was referred to Florida Academic Dermatology Center (Coral Gables, Florida) with biopsy-proven PRP diagnosed 1 year prior. The patient reported experiencing a debilitating quality of life in the year since diagnosis (Figure 1). Treatment attempts with dupilumab, tralokinumab, intramuscular steroid injections, and topical corticosteroids had failed (Figure 2). Following evaluation at Florida Academic Dermatology Center, the patient was started on acitretin 25 mg every other day and received an initial subcutaneous injection of ixekizumab 160 mg (an IL-17 inhibitor) followed 2 weeks later by a second injection of 80 mg. After the 2 doses of ixekizumab, the patient’s condition worsened with the development of pinpoint hemorrhagic lesions. The medication was discontinued, and he was started on risankizumab 150 mg at the approved dosing regimen for plaque psoriasis in combination with the acitretin therapy. Prior to starting risankizumab, the affected body surface area (BSA) was 80%. At 1-month follow-up, he showed improvement with reduction in scaling and erythema and an affected BSA of 30% (Figure 3). At 4-month follow-up, he continued showing improvement with an affected BSA of 10% (Figure 4). Acitretin was discontinued, and the patient has been successfully maintained on risankizumab 150 mg/mL subcutaneous injections every 12 weeks since.
Oral retinoid therapy historically was considered first-line therapy for moderate to severe PRP. A systematic review (N=105) of retinoid therapies showed 83% of patients with PRP who were treated with acitretin plus biologic therapy had a favorable response, whereas only 36% of patients treated with acitretin as monotherapy had the same response, highlighting the importance of dual therapy.3 The use of ustekinumab, ixekizumab, and secukinumab (IL-17 inhibitors) for refractory PRP has been well documented, but a PubMed search of articles indexed for MEDLINE using the search terms risankizumab and pityriasis rubra pilaris yielded only 8 published cases of risankizumab for treatment of PRP.4-8 All patients were diagnosed with refractory PRP, and multiple treatment modalities failed.
Ustekinumab has been shown to create a rapid response and maintain it long term, especially in patients with type 1 PRP who did not respond to systemic therapies or anti–tumor necrosis factor α agents.2 An open-label, single-arm clinical trial found secukinumab was an effective therapy for PRP and demonstrated transcription heterogeneity of this dermatologic condition.9 The researchers proposed that some patients may respond to IL-17 inhibitors but others may not due to the differences in RNA molecules transcribed.9 Our patient demonstrated worsening of his condition with an IL-17 inhibitor but experienced remarkable improvement with risankizumab, an IL-23 inhibitor.
Risankizumab is indicated for the treatment of adults with moderate to severe plaque psoriasis. This humanized IgG1 monoclonal antibody targets the p19 subunit of IL-23, inhibiting its role in the pathogenic helper T cell (TH17) pathway. Research has shown that it is an efficacious and well-tolerated treatment modality for psoriatic conditions.10 It is well known that PRP and psoriasis have similar cytokine activations; therefore, we propose that combination therapy with risankizumab and acitretin may show promise for refractory PRP.
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare papulosquamous condition with an unknown pathogenesis and limited efficacy data, which can make treatment challenging. Some cases of PRP spontaneously resolve in a few months, which is most common in the pediatric population.1 Pityriasis rubra pilaris in adults is likely to persist for years, and spontaneous resolution is unpredictable. Randomized clinical trials are difficult to perform due to the rarity of PRP.
Although there is no cure and no standard protocol for treating PRP, systemic retinoids historically are considered first-line therapy for moderate to severe cases.2 Additional management approaches include symptomatic control with moisturizers and psychological support. Alternative systemic treatments for moderate to severe cases include methotrexate, phototherapy, and cyclosporine.2
Pityriasis rubra pilaris demonstrates a favorable response to methotrexate treatment, especially in type I cases; however, patients on this alternative therapy should be monitored for severe adverse effects (eg, hepatotoxicity, pancytopenia, pneumonitis).2 Phototherapy should be approached with caution. Narrowband UVB, UVA1, and psoralen plus UVA therapy have successfully treated PRP; however, the response is variable. In some cases, the opposite effect can occur, in which the condition is photoaggravated. Phototherapy is a valid alternative form of treatment when used in combination with acitretin, and a phototest should be performed prior to starting this regimen. Cyclosporine is another immunosuppressant that can be considered for PRP treatment, though there are limited data demonstrating its efficacy.2
The introduction of biologic agents has changed the treatment approach for many dermatologic diseases, including PRP. Given the similar features between psoriasis and PRP, the biologics prescribed for psoriasis therapy also are used for patients with PRP that is challenging to treat, such as anti–tumor necrosis factor α inhibitors and IL inhibitors—specifically IL-17 and IL-23. Remission has been achieved with the use of biologics in combination with retinoid therapy.2
Biologic therapies used for PRP effectively inhibit cytokines and reduce the overall inflammatory processes involved in the development of the scaly patches and plaques seen in this condition. However, most reported clinical experiences are case studies, and more research in the form of randomized clinical trials is needed to understand the efficacy and long-term effects of this form of treatment in PRP. We present a case of a patient with refractory adult subtype I PRP that was successfully treated with the IL-23 inhibitor risankizumab.
A 65-year-old man was referred to Florida Academic Dermatology Center (Coral Gables, Florida) with biopsy-proven PRP diagnosed 1 year prior. The patient reported experiencing a debilitating quality of life in the year since diagnosis (Figure 1). Treatment attempts with dupilumab, tralokinumab, intramuscular steroid injections, and topical corticosteroids had failed (Figure 2). Following evaluation at Florida Academic Dermatology Center, the patient was started on acitretin 25 mg every other day and received an initial subcutaneous injection of ixekizumab 160 mg (an IL-17 inhibitor) followed 2 weeks later by a second injection of 80 mg. After the 2 doses of ixekizumab, the patient’s condition worsened with the development of pinpoint hemorrhagic lesions. The medication was discontinued, and he was started on risankizumab 150 mg at the approved dosing regimen for plaque psoriasis in combination with the acitretin therapy. Prior to starting risankizumab, the affected body surface area (BSA) was 80%. At 1-month follow-up, he showed improvement with reduction in scaling and erythema and an affected BSA of 30% (Figure 3). At 4-month follow-up, he continued showing improvement with an affected BSA of 10% (Figure 4). Acitretin was discontinued, and the patient has been successfully maintained on risankizumab 150 mg/mL subcutaneous injections every 12 weeks since.
Oral retinoid therapy historically was considered first-line therapy for moderate to severe PRP. A systematic review (N=105) of retinoid therapies showed 83% of patients with PRP who were treated with acitretin plus biologic therapy had a favorable response, whereas only 36% of patients treated with acitretin as monotherapy had the same response, highlighting the importance of dual therapy.3 The use of ustekinumab, ixekizumab, and secukinumab (IL-17 inhibitors) for refractory PRP has been well documented, but a PubMed search of articles indexed for MEDLINE using the search terms risankizumab and pityriasis rubra pilaris yielded only 8 published cases of risankizumab for treatment of PRP.4-8 All patients were diagnosed with refractory PRP, and multiple treatment modalities failed.
Ustekinumab has been shown to create a rapid response and maintain it long term, especially in patients with type 1 PRP who did not respond to systemic therapies or anti–tumor necrosis factor α agents.2 An open-label, single-arm clinical trial found secukinumab was an effective therapy for PRP and demonstrated transcription heterogeneity of this dermatologic condition.9 The researchers proposed that some patients may respond to IL-17 inhibitors but others may not due to the differences in RNA molecules transcribed.9 Our patient demonstrated worsening of his condition with an IL-17 inhibitor but experienced remarkable improvement with risankizumab, an IL-23 inhibitor.
Risankizumab is indicated for the treatment of adults with moderate to severe plaque psoriasis. This humanized IgG1 monoclonal antibody targets the p19 subunit of IL-23, inhibiting its role in the pathogenic helper T cell (TH17) pathway. Research has shown that it is an efficacious and well-tolerated treatment modality for psoriatic conditions.10 It is well known that PRP and psoriasis have similar cytokine activations; therefore, we propose that combination therapy with risankizumab and acitretin may show promise for refractory PRP.
- Gelmetti C, Schiuma AA, Cerri D, et al. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatr Dermatol. 1986;3:446-451. doi:10.1111/j.1525-1470.1986.tb00648.x
- Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2017;10:451-457. doi:10.2147/CCID.S124351
- Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. Eur J Dermatol. 2019;29:524-537. doi:10.1684/ejd.2019.3641
- Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. Br J Dermatol. 2021;184:E148. doi:10.1111/bjd.19681
- Brocco E, Laffitte E. Risankizumab for pityriasis rubra pilaris. Clin Exp Dermatol. 2021;46:1322-1324. doi:10.1111/ced.14715
- Duarte B, Paiva Lopes MJ. Response to: ‘Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab.’ Br J Dermatol. 2021;185:235-236. doi:10.1111/bjd.20061
- Kromer C, Schön MP, Mössner R. Treatment of pityriasis rubra pilaris with risankizumab in two cases. J Dtsch Dermatol Ges. 2021;19:1207-1209. doi:10.1111/ddg.14504
- Kołt-Kamińska M, Osińska A, Kaznowska E, et al. Successful treatment of pityriasis rubra pilaris with risankizumab in children. Dermatol Ther (Heidelb). 2023;13:2431-2441. doi:10.1007/s13555-023-01005-y
- Boudreaux BW, Pincelli TP, Bhullar PK, et al. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. Br J Dermatol. 2022;187:650-658. doi:10.1111/bjd.21708
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156:649-658. doi:10.1001/jamadermatol.2020.0723
- Gelmetti C, Schiuma AA, Cerri D, et al. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatr Dermatol. 1986;3:446-451. doi:10.1111/j.1525-1470.1986.tb00648.x
- Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2017;10:451-457. doi:10.2147/CCID.S124351
- Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. Eur J Dermatol. 2019;29:524-537. doi:10.1684/ejd.2019.3641
- Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. Br J Dermatol. 2021;184:E148. doi:10.1111/bjd.19681
- Brocco E, Laffitte E. Risankizumab for pityriasis rubra pilaris. Clin Exp Dermatol. 2021;46:1322-1324. doi:10.1111/ced.14715
- Duarte B, Paiva Lopes MJ. Response to: ‘Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab.’ Br J Dermatol. 2021;185:235-236. doi:10.1111/bjd.20061
- Kromer C, Schön MP, Mössner R. Treatment of pityriasis rubra pilaris with risankizumab in two cases. J Dtsch Dermatol Ges. 2021;19:1207-1209. doi:10.1111/ddg.14504
- Kołt-Kamińska M, Osińska A, Kaznowska E, et al. Successful treatment of pityriasis rubra pilaris with risankizumab in children. Dermatol Ther (Heidelb). 2023;13:2431-2441. doi:10.1007/s13555-023-01005-y
- Boudreaux BW, Pincelli TP, Bhullar PK, et al. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. Br J Dermatol. 2022;187:650-658. doi:10.1111/bjd.21708
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156:649-658. doi:10.1001/jamadermatol.2020.0723
Practice Points
- Pityriasis rubra pilaris (PRP) is a rare condition that is challenging to treat due to its unknown pathogenesis and limited efficacy data. Systemic retinoids historically were considered first-line therapy for moderate to severe cases of PRP.
- Biologics may be useful for refractory cases of PRP.
- Risankizumab is approved for moderate to severe plaque psoriasis and can be considered off-label for refractory PRP.
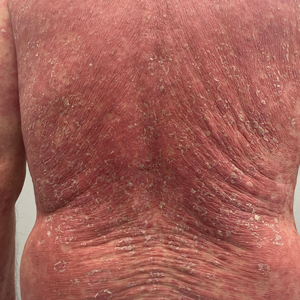
Blaschkolinear Lupus Erythematosus: Strategies for Early Detection and Management
To the Editor:
Chronic cutaneous lupus erythematosus (CCLE) is an inflammatory condition with myriad cutaneous manifestations. Most forms of CCLE have the potential to progress to systemic lupus erythematosus (SLE).1
Blaschkolinear lupus erythematosus (BLE) is an exceedingly rare subtype of cutaneous lupus erythematosus that usually manifests during childhood as linear plaques along the lines of Blaschko.2,3 Under normal conditions, Blaschko lines are not noticeable; they correspond to the direction of ectodermal cell migration during cutaneous embryogenesis.4,5 The embryonic cells travel ventrolaterally, forming a V-shaped pattern on the back, an S-shaped pattern on the trunk, and an hourglass-shaped pattern on the face with several perpendicular intersections near the mouth and nose.6 During their migration, the cells are susceptible to somatic mutations and clonal expansion, resulting in a monoclonal population of genetically heterogenous cells. This phenomenon is known as somatic mosaicism and may lead to an increased susceptibility to an array of congenital and inflammatory dermatoses, such as cutaneous lupus erythematosus.4 Blaschkolinear entities tend to manifest in a unilateral distribution following exposure to a certain environmental trigger, such as trauma, viral illness, or UV radiation, although a trigger is not always present.7 We report a case of BLE manifesting on the head and neck in an adult patient.
A 46-year-old man presented with a pruritic rash of 3 months’ duration on the right cheek that extended inferiorly to the right upper chest. He had a medical history of well-controlled psoriasis, and he denied any antecedent trauma, fevers, chills, arthralgia, or night sweats. There had been no improvement with mometasone ointment 0.1% applied daily for 2 months as prescribed by his primary care provider. Physical examination revealed indurated, red-brown, atrophic plaques in a blaschkolinear distribution around the nose, right upper jaw, right side of the neck, and right upper chest (Figure, A).
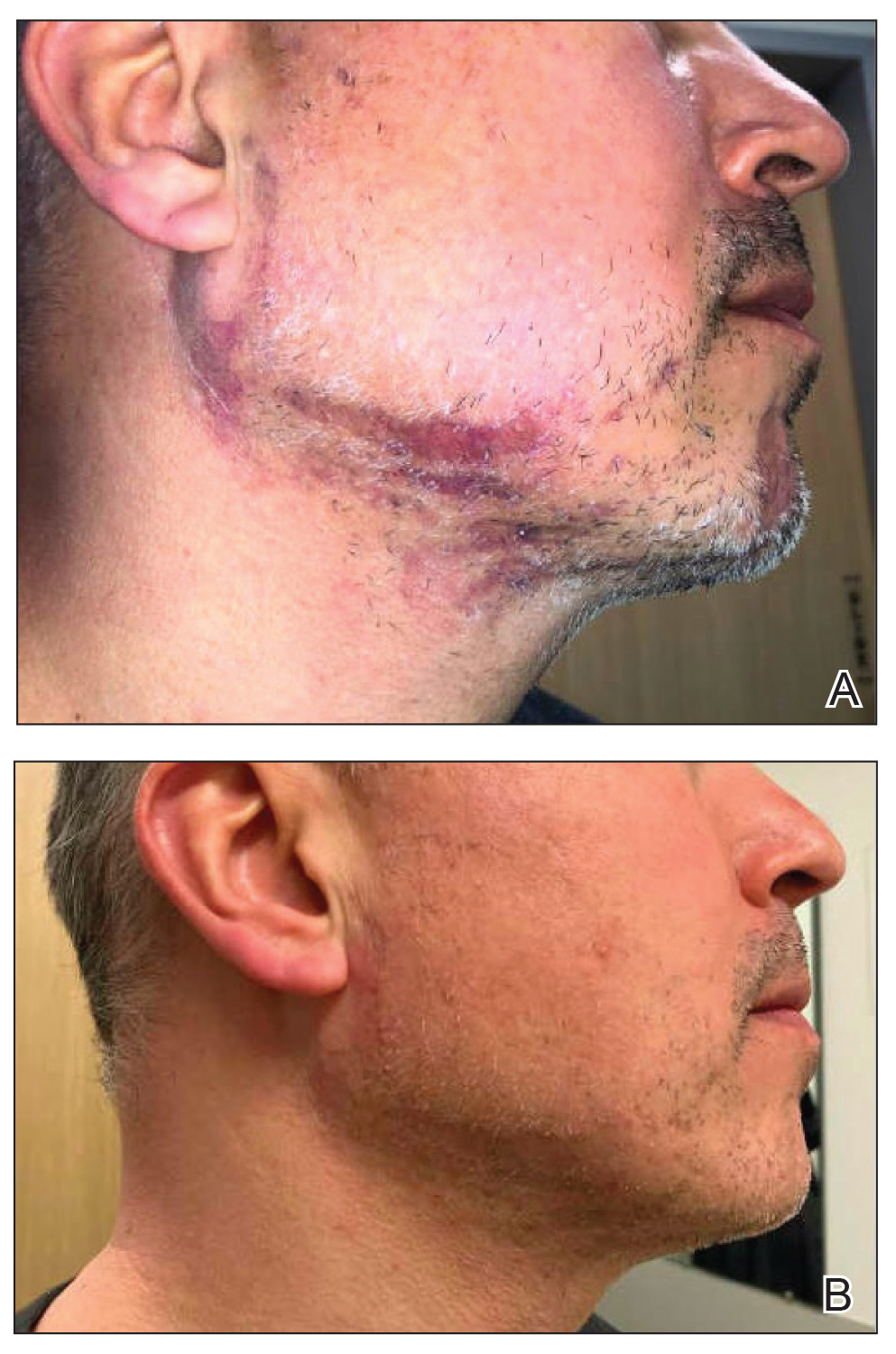
Histopathology of punch biopsies from the right jaw and right upper chest showed an atrophic epidermis with scattered dyskeratotic keratinocytes and vacuolar alteration of the basal cell layer. A superficial and deep perivascular and periadnexal lymphocytic infiltrate was observed in both biopsies. Staining with Verhoeff-van Gieson elastin and periodic acid–Schiff highlighted prominent basement membrane thickening and loss of elastic fibers in the superficial dermis. These findings favored a diagnosis of CCLE, and the clinical blaschkolinear distribution of the rash led to our specific diagnosis of BLE. Laboratory workup for SLE including a complete blood cell count; urine analysis; and testing for liver and kidney function, antinuclearantibodies, complement levels, and erythrocyte sedimentation rate revealed no abnormalities.
The patient started hydroxychloroquine 200 mg twice daily and methotrexate 25 mg weekly along with strict photoprotection measures, including wearing photoprotective clothing and avoiding sunlight during the most intense hours of the day.
Linear lichen planus is an important differential diagnosis to consider in patients with a blaschkolinear eruption.7 Although the clinical manifestations of BLE and linear lichen planus are similar, they differ histopathologically. One study found that only 33.3% of patients (6/18) who clinically presented with blaschkolinear eruptions were correctly diagnosed before histologic examination.7 Visualization of the adnexa as well as the superficial and deep vascular plexuses is paramount in distinguishing between linear lichen planus and BLE; linear lichen planus does not have perivascular and periadnexal infiltration, while BLE does. Thus, in our experience, a punch biopsy—rather than a shave biopsy—should be performed to access the deeper layers of the skin.
Because these 2 entities have noteworthy differences in their management, prognosis, and long-term follow-up, accurate diagnosis is critical. To start, BLE is treated with the use of photoprotection, whereas linear lichen planus is commonly treated with phototherapy. Given the potential for forms of CCLE to progress to SLE, serial monitoring is indicated in patients with BLE. As the risk for progression to SLE is highest in the first 3 years after diagnosis, a review of systems and laboratory testing should occur every 2 to 3 months in the first year after diagnosis (sooner if the disease presentation is more severe).9 Also, treatment with hydroxychloroquine likely delays transformation to SLE and is important in the early management of BLE.10 On the other hand, linear lichen planus tends to self-resolve without progression to systemic involvement, warranting limited follow-up.9
Blaschkolinear lupus erythematosus typically manifests in childhood, but it also can be seen in adults, such as in our patient. Adult-onset BLE is rare but may be underrecognized or underreported in the literature.11 However, dermatologists should consider it in the differential diagnosis for any patient with a blaschkolinear eruption, as establishing the correct diagnosis is key to ensuring prompt and effective treatment for this rare inflammatory condition.
- Grönhagen CM, Fored CM, Granath F, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335-1341. doi:10.1111/j.1365-2133.2011.10272.x
- Requena C, Torrelo A, de Prada I, et al. Linear childhood cutaneous lupus erythematosus following Blaschko lines. J Eur Acad Dermatol Venereol. 2002;16:618-620. doi:10.1046/j.1468-3083.2002.00588.x
- Lim D, Hatami A, Kokta V, et al. Linear cutaneous lupus erythematosus in children-report of two cases and review of the literature: a case report. SAGE Open Med Case Rep. 2020;8:2050313x20979206. doi:10.1177/2050313X20979206
- Jin H, Zhang G, Zhou Y, et al. Old lines tell new tales: Blaschko linear lupus erythematosus. Autoimmun Rev. 2016;15:291-306. doi:10.1016/j.autrev.2015.11.014
- Yu S, Yu H-S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: implications for the role of keratinocytes in lupus erythematosus. Dermatologica Sinica. 2016;34:144-147. doi:10.1016/j.dsi.2015.12.002
- Kouzak SS, Mendes MST, Costa IMC. Cutaneous mosaicisms: concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-517. doi:10.1590/abd1806-4841.20132015
- Liu W, Vano-Galvan S, Liu J-W, et al. Pigmented linear discoid lupus erythematosus following the lines of Blaschko: a retrospective study of a Chinese series. Indian J Dermatol Venereol Leprol. 2020;86:359-365. doi:10.4103/ijdvl.IJDVL_341_19
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Invest Dermatol Symp Proc. 2017;18:S69-S74. doi:10.1016/j.jisp.2016.09.001
- Curtiss P, Walker AM, Chong BF. A systematic review of the progression of cutaneous lupus to systemic lupus erythematosus. Front Immunol. 2022:13:866319. doi:10.3389/fimmu.2022.866319
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404. doi:10.1016/j.berh.2013.07.008
- Milosavljevic K, Fibeger E, Virata AR. A case of linear cutaneous lupus erythematosus in a 55-year-old woman. Am J Case Rep. 2020;21:E921495. doi:10.12659/AJCR.921495
To the Editor:
Chronic cutaneous lupus erythematosus (CCLE) is an inflammatory condition with myriad cutaneous manifestations. Most forms of CCLE have the potential to progress to systemic lupus erythematosus (SLE).1
Blaschkolinear lupus erythematosus (BLE) is an exceedingly rare subtype of cutaneous lupus erythematosus that usually manifests during childhood as linear plaques along the lines of Blaschko.2,3 Under normal conditions, Blaschko lines are not noticeable; they correspond to the direction of ectodermal cell migration during cutaneous embryogenesis.4,5 The embryonic cells travel ventrolaterally, forming a V-shaped pattern on the back, an S-shaped pattern on the trunk, and an hourglass-shaped pattern on the face with several perpendicular intersections near the mouth and nose.6 During their migration, the cells are susceptible to somatic mutations and clonal expansion, resulting in a monoclonal population of genetically heterogenous cells. This phenomenon is known as somatic mosaicism and may lead to an increased susceptibility to an array of congenital and inflammatory dermatoses, such as cutaneous lupus erythematosus.4 Blaschkolinear entities tend to manifest in a unilateral distribution following exposure to a certain environmental trigger, such as trauma, viral illness, or UV radiation, although a trigger is not always present.7 We report a case of BLE manifesting on the head and neck in an adult patient.
A 46-year-old man presented with a pruritic rash of 3 months’ duration on the right cheek that extended inferiorly to the right upper chest. He had a medical history of well-controlled psoriasis, and he denied any antecedent trauma, fevers, chills, arthralgia, or night sweats. There had been no improvement with mometasone ointment 0.1% applied daily for 2 months as prescribed by his primary care provider. Physical examination revealed indurated, red-brown, atrophic plaques in a blaschkolinear distribution around the nose, right upper jaw, right side of the neck, and right upper chest (Figure, A).
Histopathology of punch biopsies from the right jaw and right upper chest showed an atrophic epidermis with scattered dyskeratotic keratinocytes and vacuolar alteration of the basal cell layer. A superficial and deep perivascular and periadnexal lymphocytic infiltrate was observed in both biopsies. Staining with Verhoeff-van Gieson elastin and periodic acid–Schiff highlighted prominent basement membrane thickening and loss of elastic fibers in the superficial dermis. These findings favored a diagnosis of CCLE, and the clinical blaschkolinear distribution of the rash led to our specific diagnosis of BLE. Laboratory workup for SLE including a complete blood cell count; urine analysis; and testing for liver and kidney function, antinuclearantibodies, complement levels, and erythrocyte sedimentation rate revealed no abnormalities.
The patient started hydroxychloroquine 200 mg twice daily and methotrexate 25 mg weekly along with strict photoprotection measures, including wearing photoprotective clothing and avoiding sunlight during the most intense hours of the day.
Linear lichen planus is an important differential diagnosis to consider in patients with a blaschkolinear eruption.7 Although the clinical manifestations of BLE and linear lichen planus are similar, they differ histopathologically. One study found that only 33.3% of patients (6/18) who clinically presented with blaschkolinear eruptions were correctly diagnosed before histologic examination.7 Visualization of the adnexa as well as the superficial and deep vascular plexuses is paramount in distinguishing between linear lichen planus and BLE; linear lichen planus does not have perivascular and periadnexal infiltration, while BLE does. Thus, in our experience, a punch biopsy—rather than a shave biopsy—should be performed to access the deeper layers of the skin.
Because these 2 entities have noteworthy differences in their management, prognosis, and long-term follow-up, accurate diagnosis is critical. To start, BLE is treated with the use of photoprotection, whereas linear lichen planus is commonly treated with phototherapy. Given the potential for forms of CCLE to progress to SLE, serial monitoring is indicated in patients with BLE. As the risk for progression to SLE is highest in the first 3 years after diagnosis, a review of systems and laboratory testing should occur every 2 to 3 months in the first year after diagnosis (sooner if the disease presentation is more severe).9 Also, treatment with hydroxychloroquine likely delays transformation to SLE and is important in the early management of BLE.10 On the other hand, linear lichen planus tends to self-resolve without progression to systemic involvement, warranting limited follow-up.9
Blaschkolinear lupus erythematosus typically manifests in childhood, but it also can be seen in adults, such as in our patient. Adult-onset BLE is rare but may be underrecognized or underreported in the literature.11 However, dermatologists should consider it in the differential diagnosis for any patient with a blaschkolinear eruption, as establishing the correct diagnosis is key to ensuring prompt and effective treatment for this rare inflammatory condition.
To the Editor:
Chronic cutaneous lupus erythematosus (CCLE) is an inflammatory condition with myriad cutaneous manifestations. Most forms of CCLE have the potential to progress to systemic lupus erythematosus (SLE).1
Blaschkolinear lupus erythematosus (BLE) is an exceedingly rare subtype of cutaneous lupus erythematosus that usually manifests during childhood as linear plaques along the lines of Blaschko.2,3 Under normal conditions, Blaschko lines are not noticeable; they correspond to the direction of ectodermal cell migration during cutaneous embryogenesis.4,5 The embryonic cells travel ventrolaterally, forming a V-shaped pattern on the back, an S-shaped pattern on the trunk, and an hourglass-shaped pattern on the face with several perpendicular intersections near the mouth and nose.6 During their migration, the cells are susceptible to somatic mutations and clonal expansion, resulting in a monoclonal population of genetically heterogenous cells. This phenomenon is known as somatic mosaicism and may lead to an increased susceptibility to an array of congenital and inflammatory dermatoses, such as cutaneous lupus erythematosus.4 Blaschkolinear entities tend to manifest in a unilateral distribution following exposure to a certain environmental trigger, such as trauma, viral illness, or UV radiation, although a trigger is not always present.7 We report a case of BLE manifesting on the head and neck in an adult patient.
A 46-year-old man presented with a pruritic rash of 3 months’ duration on the right cheek that extended inferiorly to the right upper chest. He had a medical history of well-controlled psoriasis, and he denied any antecedent trauma, fevers, chills, arthralgia, or night sweats. There had been no improvement with mometasone ointment 0.1% applied daily for 2 months as prescribed by his primary care provider. Physical examination revealed indurated, red-brown, atrophic plaques in a blaschkolinear distribution around the nose, right upper jaw, right side of the neck, and right upper chest (Figure, A).
Histopathology of punch biopsies from the right jaw and right upper chest showed an atrophic epidermis with scattered dyskeratotic keratinocytes and vacuolar alteration of the basal cell layer. A superficial and deep perivascular and periadnexal lymphocytic infiltrate was observed in both biopsies. Staining with Verhoeff-van Gieson elastin and periodic acid–Schiff highlighted prominent basement membrane thickening and loss of elastic fibers in the superficial dermis. These findings favored a diagnosis of CCLE, and the clinical blaschkolinear distribution of the rash led to our specific diagnosis of BLE. Laboratory workup for SLE including a complete blood cell count; urine analysis; and testing for liver and kidney function, antinuclearantibodies, complement levels, and erythrocyte sedimentation rate revealed no abnormalities.
The patient started hydroxychloroquine 200 mg twice daily and methotrexate 25 mg weekly along with strict photoprotection measures, including wearing photoprotective clothing and avoiding sunlight during the most intense hours of the day.
Linear lichen planus is an important differential diagnosis to consider in patients with a blaschkolinear eruption.7 Although the clinical manifestations of BLE and linear lichen planus are similar, they differ histopathologically. One study found that only 33.3% of patients (6/18) who clinically presented with blaschkolinear eruptions were correctly diagnosed before histologic examination.7 Visualization of the adnexa as well as the superficial and deep vascular plexuses is paramount in distinguishing between linear lichen planus and BLE; linear lichen planus does not have perivascular and periadnexal infiltration, while BLE does. Thus, in our experience, a punch biopsy—rather than a shave biopsy—should be performed to access the deeper layers of the skin.
Because these 2 entities have noteworthy differences in their management, prognosis, and long-term follow-up, accurate diagnosis is critical. To start, BLE is treated with the use of photoprotection, whereas linear lichen planus is commonly treated with phototherapy. Given the potential for forms of CCLE to progress to SLE, serial monitoring is indicated in patients with BLE. As the risk for progression to SLE is highest in the first 3 years after diagnosis, a review of systems and laboratory testing should occur every 2 to 3 months in the first year after diagnosis (sooner if the disease presentation is more severe).9 Also, treatment with hydroxychloroquine likely delays transformation to SLE and is important in the early management of BLE.10 On the other hand, linear lichen planus tends to self-resolve without progression to systemic involvement, warranting limited follow-up.9
Blaschkolinear lupus erythematosus typically manifests in childhood, but it also can be seen in adults, such as in our patient. Adult-onset BLE is rare but may be underrecognized or underreported in the literature.11 However, dermatologists should consider it in the differential diagnosis for any patient with a blaschkolinear eruption, as establishing the correct diagnosis is key to ensuring prompt and effective treatment for this rare inflammatory condition.
- Grönhagen CM, Fored CM, Granath F, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335-1341. doi:10.1111/j.1365-2133.2011.10272.x
- Requena C, Torrelo A, de Prada I, et al. Linear childhood cutaneous lupus erythematosus following Blaschko lines. J Eur Acad Dermatol Venereol. 2002;16:618-620. doi:10.1046/j.1468-3083.2002.00588.x
- Lim D, Hatami A, Kokta V, et al. Linear cutaneous lupus erythematosus in children-report of two cases and review of the literature: a case report. SAGE Open Med Case Rep. 2020;8:2050313x20979206. doi:10.1177/2050313X20979206
- Jin H, Zhang G, Zhou Y, et al. Old lines tell new tales: Blaschko linear lupus erythematosus. Autoimmun Rev. 2016;15:291-306. doi:10.1016/j.autrev.2015.11.014
- Yu S, Yu H-S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: implications for the role of keratinocytes in lupus erythematosus. Dermatologica Sinica. 2016;34:144-147. doi:10.1016/j.dsi.2015.12.002
- Kouzak SS, Mendes MST, Costa IMC. Cutaneous mosaicisms: concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-517. doi:10.1590/abd1806-4841.20132015
- Liu W, Vano-Galvan S, Liu J-W, et al. Pigmented linear discoid lupus erythematosus following the lines of Blaschko: a retrospective study of a Chinese series. Indian J Dermatol Venereol Leprol. 2020;86:359-365. doi:10.4103/ijdvl.IJDVL_341_19
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Invest Dermatol Symp Proc. 2017;18:S69-S74. doi:10.1016/j.jisp.2016.09.001
- Curtiss P, Walker AM, Chong BF. A systematic review of the progression of cutaneous lupus to systemic lupus erythematosus. Front Immunol. 2022:13:866319. doi:10.3389/fimmu.2022.866319
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404. doi:10.1016/j.berh.2013.07.008
- Milosavljevic K, Fibeger E, Virata AR. A case of linear cutaneous lupus erythematosus in a 55-year-old woman. Am J Case Rep. 2020;21:E921495. doi:10.12659/AJCR.921495
- Grönhagen CM, Fored CM, Granath F, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335-1341. doi:10.1111/j.1365-2133.2011.10272.x
- Requena C, Torrelo A, de Prada I, et al. Linear childhood cutaneous lupus erythematosus following Blaschko lines. J Eur Acad Dermatol Venereol. 2002;16:618-620. doi:10.1046/j.1468-3083.2002.00588.x
- Lim D, Hatami A, Kokta V, et al. Linear cutaneous lupus erythematosus in children-report of two cases and review of the literature: a case report. SAGE Open Med Case Rep. 2020;8:2050313x20979206. doi:10.1177/2050313X20979206
- Jin H, Zhang G, Zhou Y, et al. Old lines tell new tales: Blaschko linear lupus erythematosus. Autoimmun Rev. 2016;15:291-306. doi:10.1016/j.autrev.2015.11.014
- Yu S, Yu H-S. A patient with subacute cutaneous lupus erythematosus along Blaschko lines: implications for the role of keratinocytes in lupus erythematosus. Dermatologica Sinica. 2016;34:144-147. doi:10.1016/j.dsi.2015.12.002
- Kouzak SS, Mendes MST, Costa IMC. Cutaneous mosaicisms: concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-517. doi:10.1590/abd1806-4841.20132015
- Liu W, Vano-Galvan S, Liu J-W, et al. Pigmented linear discoid lupus erythematosus following the lines of Blaschko: a retrospective study of a Chinese series. Indian J Dermatol Venereol Leprol. 2020;86:359-365. doi:10.4103/ijdvl.IJDVL_341_19
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Invest Dermatol Symp Proc. 2017;18:S69-S74. doi:10.1016/j.jisp.2016.09.001
- Curtiss P, Walker AM, Chong BF. A systematic review of the progression of cutaneous lupus to systemic lupus erythematosus. Front Immunol. 2022:13:866319. doi:10.3389/fimmu.2022.866319
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404. doi:10.1016/j.berh.2013.07.008
- Milosavljevic K, Fibeger E, Virata AR. A case of linear cutaneous lupus erythematosus in a 55-year-old woman. Am J Case Rep. 2020;21:E921495. doi:10.12659/AJCR.921495
Practice Points
- Blaschkolinear lupus erythematosus (BLE), an exceedingly rare subtype of chronic cutaneous lupus erythematosus, usually presents during childhood as linear plaques along the lines of Blaschko.
- It is important to consider linear lichen planus in patients with a blaschkolinear eruption, as the clinical manifestations are similar but there are differences in histopathology, management, prognosis, and long-term follow-up.
- Serial monitoring is indicated in patients with BLE given the potential for progression to systemic lupus erythematosus, which may be delayed with early use of hydroxychloroquine.
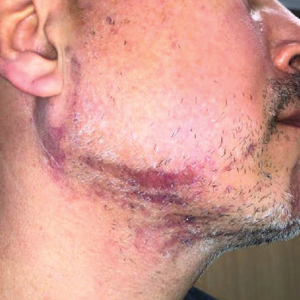
Misdiagnosis of Crusted Scabies: Skin Excoriations Resembling Brown Sugar Are Characteristic
To the Editor:
Crusted scabies (formerly known as Norwegian scabies) is a rare and highly contagious variant of scabies, in which the skin is infested with thousands to millions of Sarcoptes scabiei var hominis mites. We present a case of skin changes that were misdiagnosed as atopic dermatitis, seborrhea, xerosis, and drug eruption on initial presentation, which prompted treatment with a corticosteroid that inadvertently caused progression to crusted scabies.
A 79-year-old woman who uses a wheelchair presented to the clinic with skin changes that consisted of diffuse, severely pruritic, erythematous plaques on the head, neck, trunk, face, and extremities of 2 years’ duration. She had a medical history of hyperlipidemia, hypertension, and hyperglycemia, as well as a stroke that required hospitalization 2 years prior to the onset of the skin changes. She had no history of allergies.
Prior clinical diagnoses by primary care and dermatology included xerosis, atopic dermatitis, seborrhea, and drug eruption. She was treated with a mid-potency topical corticosteroid (triamcinolone acetonide cream 0.1%) twice daily and prednisone 40 mg once daily for 2- to 4-week courses over an 8-month period without reduction in symptoms.
Physical examination at the current presentation revealed golden, crusted, fine, powdery but slightly sticky flakes that spread diffusely across the entire body and came off in crumbles with a simple touch. These widespread crusts were easily visible on clothing. There was underlying diffuse erythema beneath the flaking skin on the trunk and proximal extremities. The scale and shedding skin laid in piles on the patient’s lap and resembled brown sugar (Figure 1). The patient also reported decreased hand function and dexterity due to the yellowbrown, thick, crusty plaques that had developed on both the palmar and dorsal sides of the hands (Figure 2). Erythematous plaques on the scalp, forehead, and inner ears resembled seborrhea (Figure 3). Pruritus severity was rated by the patient as 10 of 10, and she scratched her skin the entire time she was in the clinic. The patient was emotional and stated that she had not been able to sleep due to the discomfort. We suspected scabies, and the patient was reassured to learn that it could be confirmed with a simple skin scrape test.
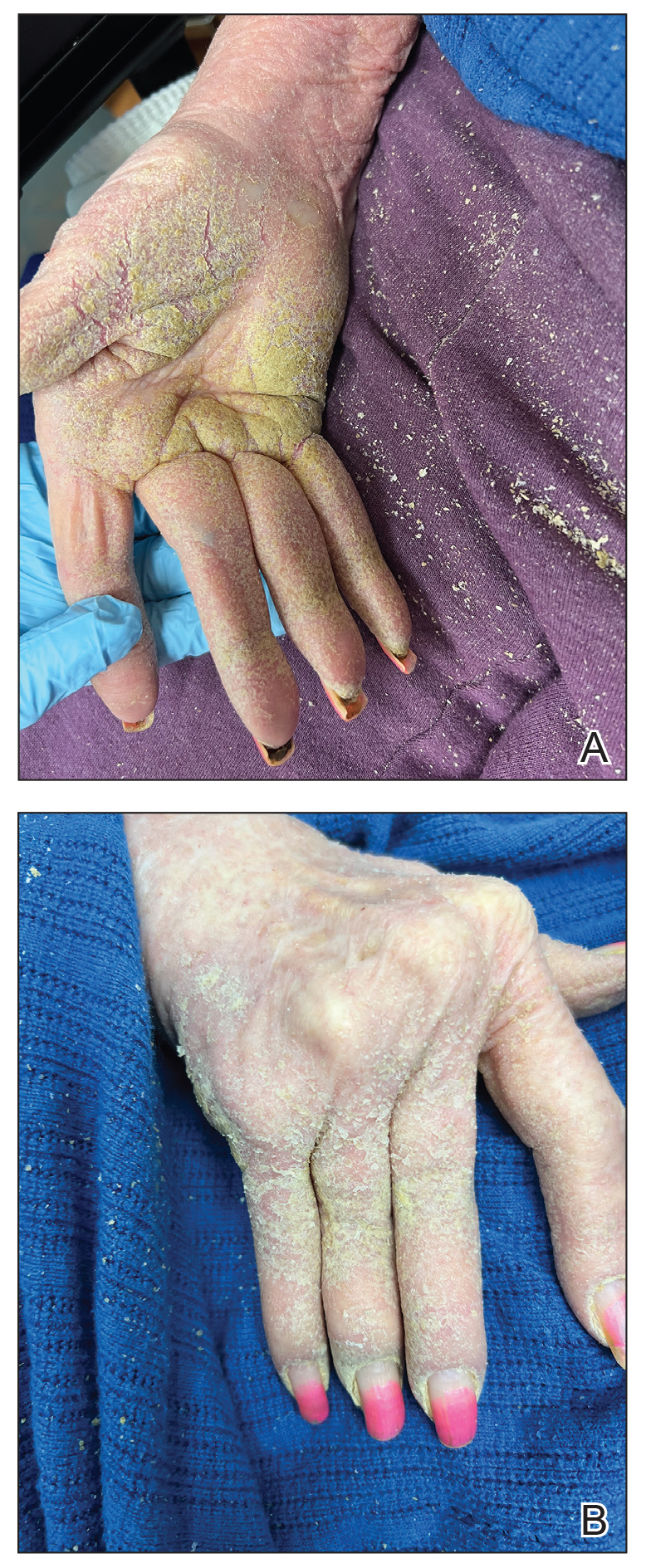
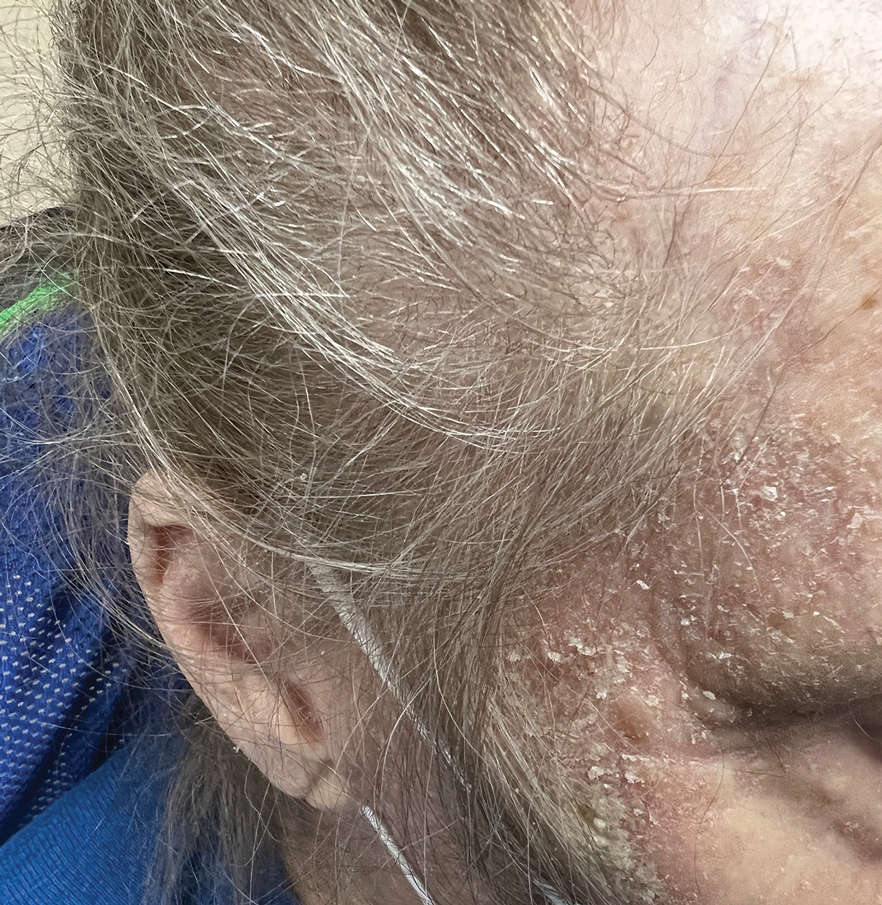
The crusted lesions on the patient's hands were scraped with a #15-blade scalpel, and a routine potassium hydroxide mount was performed. The skin scrapings were placed on a slide with a drop of 10% potassium hydroxide and observed under low-power (×10) and high-power (×40) microscopy, which revealed thousands of mites and eggs (along with previously hatched eggs) (Figure 4) and quickly confirmed a diagnosis of crusted scabies.an extremely contagious form of scabies seen in older patients with compromised immune systems, malnutrition, or disabilities. The patient was prescribed oral ivermectin (3 mg dosed at 200 μg/kg of body weight) and topical permethrin 5%, neither of which she took, as she died of a COVID-19 infection complication 3 days after this diagnostic clinic visit.
Classic and crusted scabies are both caused by infestation of the Sarcoptes scabiei var hominis mite. Classic scabies is a result of an infestation of a small number of mites (commonly 5–15 mites), while crusted scabies is due to hyperinfestation by as many as millions of mites, the latter often requiring more aggressive treatment. The mites are first transmitted to humans by either skin-toskin contact or fomites on bedding and clothing. The scabies mite undergoes 4 life cycle stages: egg, larvae, nymph, and adult. Once female mites are transmitted, they burrow under the skin and lay 2 to 3 eggs per day. The eggs hatch within 3 to 4 days, after which the larvae migrate to the skin surface. The larval stage lasts for 3 to 4 days, during which the larvae burrow into the stratum corneum to create molting pouches, until they molt into slightly larger nymphs. Nymphs can be found in hair follicles or molting pouches until they further molt within 3 to 4 days into adults, which are round, saclike mites. The adult male and female mites then mate, leaving the female fertile for the rest of her 1- to 2-month lifespan. Impregnated female mites traverse the skin surface in search of a burrow site, using the pulvilli on the anterior aspect of 2 legs to hold onto the skin. Once burrowed, the female mite continues to lay eggs for the rest of her life, with approximately 10% of her eggs resulting in adult mites. Male mites feed in shallow pits of the skin until they find a female burrow site for mating.1 This continuous life cycle of the scabies mite gives rise to highly transmissible, pruritic skin excoriations, as demonstrated in our patient.
The skin has a relatively late inflammatory and adaptive immune response to scabies, typically occurring 4 to 6 weeks after the initial infestation.2 This delayed inflammatory response and onset of symptoms may be due to the scabies mite’s ability to alter aspects of the host’s immune response, which differs in classic vs crusted scabies. In classic scabies, there is a predominance of CD4+ T cells in the dermis and minimal CD8+ T cells. The opposite is true in crusted scabies— there is an overwhelming infiltration of CD8+ T cells and minimal CD4+ T cells.3 The CD8+ T-cell predominance in crusted scabies is hypothesized to be the cause of keratinocyte apoptosis, resulting in epidermal hyperproliferation. Keratinocyte apoptosis also secretes cytokines, which may lead to the immunologic targeting of healthy skin cells. The damage of healthy dermal cells contributes to the inability of the skin’s immune system to mount an effective response, allowing the parasite to grow uncontrollably in patients with crusted scabies.4
This ineffective immune response is further exacerbated by corticosteroids, which are commonly prescribed for pruritus experienced by patients with scabies infestations. The mechanism of action of corticosteroids is the production of anti-inflammatory, antimitotic, and immunosuppressive effects.5 Because the integumentary immune system is imbalanced during crusted scabies infestation, the immunosuppressive mechanism of oral and topical corticosteroids further reduces the cellular immune response to scabies. The flourishing of the scabies mites along with keratinocyte apoptosis4 results in the development of hyperkeratotic skin crusting, most frequently on the palms, soles, arms, and legs. Risk factors for crusted scabies include immunosuppression, hospitalization, crowded living conditions, and poor hygiene, though no known risk factors were documented in up to 42% (33/78) of patients with crusted scabies in one study.6
Patients with crusted scabies typically present with generalized, poorly defined, erythematous, fissured plaques covered by scaling and crusts. Plaques on bony prominences such as finger articulations and elbows may have a thick verrucous aspect.1 Skin flaking that resembles brown sugar—a mixture of white sugar and molasses—is a clue to the diagnosis of crusted scabies. Brown sugar has a slightly sandy and sticky texture that ranges in color from very light brown to very dark brown. When present, flakes always appears slightly lighter than the patient’s skin tone. Although skin burrows are pathognomonic and clinically recognizable features of scabies, these burrows can be disguised by lesions, such as the hyperkeratotic plaques seen in our patient. The lesions may or may not be associated with pruritus, which may occur only at night, and bacterial superinfection has been reported in severe cases of crusted scabies,7 as scratching can cause sores, which may lead to infection. In severe cases, the constant scratching could lead to sepsis if the infection enters the bloodstream.8 Another symptom of scabies is a rash that causes small bumps that tend to form in a line, resembling small bites, hives, or pimples, and scaly plaques can lead to misdiagnosis as atopic dermatitis.
Treatment often is delayed due to misdiagnosis, as seen in our patient. Common misdiagnoses include atopic dermatitis, pityriasis rosea, systemic lupus erythematosus, bullous pemphigoid, lichen planus, pediculosis corporis, seborrheic scalp dermatitis, and adverse drug reactions.9 Patients with extensive infestations of crusted scabies should be treated with a 4-week course of permethrin cream 5% daily for 1 week, then twice per week until resolved, and oral ivermectin 200 μg/kg dosed 1 week apart for up to 4 weeks, if needed.1 Topical permethrin works by producing a selective neurotoxic effect on invertebrates such as scabies mites, which disrupts the function of voltage-gated sodium channels, thereby paralyzing the adult mites to halt the spread of infestation. However, treatment with topical medications can be difficult due to the thick crusts that have formed, which make it more challenging for the skin to properly absorb the treatment. Additionally, surgical debridement as an adjunct procedure has been done to improve the effectiveness of topical medications by removing all the mites in skin.10 On the other hand, the mechanism in which ivermectin treats scabies infestations is poorly understood. Current research suggests that ivermectin works by causing persistent opening of pH-gated chloride channels in scabies mites.11 There is emerging concern for drug resistance to these scabicides,12 revealing a need for further research of treatment options.
Patients with crusted scabies can have an extremely large number of mites (up to 2 million), making them more infectious than patients with classic scabies.13 As a result, it is imperative to reduce environmental transmission and risk for reinfection with mites during treatment. Because crusted scabies is transmitted by prolonged skinto- skin contact or by contact with personal items of an infected person (eg, bedding, clothing), treatment guidelines require all clothing, bedding, and towels of a patient with scabies to be machine-washed and dried with hot water and hot dryer cycles. If an item cannot be washed, it should be stored in a sealed plastic bag for 1 week, as scabies mites cannot survive more than 2 to 3 days away from their host of human skin.13 Treatment of close contacts of patients with scabies is recommended, as well as for those in endemic areas or closed communities, such as nursing homes or jails.
- Salavastru CM, Chosidow O, Boffa MJ, et al. European guideline for the management of scabies. J Eur Acad Dermatol Venereol. 2017;31:1248-1253. doi:10.1111/jdv.14351
- Morgan MS, Arlian LG, Markey MP. Sarcoptes scabiei mites modulate gene expression in human skin equivalents. PLoS One. 2013;8:e71143. doi:10.1371/journal.pone.0071143
- Walton SF, Beroukas D, Roberts-Thomson P, et al. New insights into disease pathogenesis in crusted (Norwegian) scabies: the skin immune response in crusted scabies. Br J Dermatol. 2008;158:1247-1255. doi:10.1111/j.1365-2133.2008.08541.x
- Bhat SA, Mounsey KE, Liu X, et al. Host immune responses to the itch mite, Sarcoptes scabiei, in humans. Parasit Vectors. 2017;10:385. doi:10.1186/s13071-017-2320-4
- Binic´ I, Jankovic´ A, Jovanovic´ D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2009;25:188-191. doi:10.3346/jkms.2010.25.1.188
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381. doi:10.1016/j.jinf.2004.08.033
- Yari N, Malone CH, Rivas A. Misdiagnosed crusted scabies in an AIDS patient leads to hyperinfestation. Cutis. 2017;99:202-204.
- American Academy of Dermatology Association. Scabies: signs and symptoms. Accessed July 12, 2024. https://www.aad.org/public/diseases/a-z/scabies-symptoms
- Siegfried EC, Hebert AA. Diagnosis of atopic dermatitis: mimics, overlaps, and complications. J Clin Med. 2015;4:884-917. doi:10.3390/jcm4050884
- Maghrabi MM, Lum S, Joba AT, et al. Norwegian crusted scabies: an unusual case presentation. J Foot Ankle Surg. 2014;53:62-66. doi:10.1053/j.jfas.2013.09.002
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725. doi:10.1056/NEJMct0910329
- Andriantsoanirina V, Izri A, Botterel F, et al. Molecular survey of knockdown resistance to pyrethroids in human scabies mites. Clin Microbiol Infect. 2014;20:O139-O141. doi:10.1111/1469-0691.12334
- Centers for Disease Control and Prevention. Preventing scabies. Published December 18, 2023. Accessed August 9, 2024. https://www.cdc.gov/scabies/prevention/index.html
To the Editor:
Crusted scabies (formerly known as Norwegian scabies) is a rare and highly contagious variant of scabies, in which the skin is infested with thousands to millions of Sarcoptes scabiei var hominis mites. We present a case of skin changes that were misdiagnosed as atopic dermatitis, seborrhea, xerosis, and drug eruption on initial presentation, which prompted treatment with a corticosteroid that inadvertently caused progression to crusted scabies.
A 79-year-old woman who uses a wheelchair presented to the clinic with skin changes that consisted of diffuse, severely pruritic, erythematous plaques on the head, neck, trunk, face, and extremities of 2 years’ duration. She had a medical history of hyperlipidemia, hypertension, and hyperglycemia, as well as a stroke that required hospitalization 2 years prior to the onset of the skin changes. She had no history of allergies.
Prior clinical diagnoses by primary care and dermatology included xerosis, atopic dermatitis, seborrhea, and drug eruption. She was treated with a mid-potency topical corticosteroid (triamcinolone acetonide cream 0.1%) twice daily and prednisone 40 mg once daily for 2- to 4-week courses over an 8-month period without reduction in symptoms.
Physical examination at the current presentation revealed golden, crusted, fine, powdery but slightly sticky flakes that spread diffusely across the entire body and came off in crumbles with a simple touch. These widespread crusts were easily visible on clothing. There was underlying diffuse erythema beneath the flaking skin on the trunk and proximal extremities. The scale and shedding skin laid in piles on the patient’s lap and resembled brown sugar (Figure 1). The patient also reported decreased hand function and dexterity due to the yellowbrown, thick, crusty plaques that had developed on both the palmar and dorsal sides of the hands (Figure 2). Erythematous plaques on the scalp, forehead, and inner ears resembled seborrhea (Figure 3). Pruritus severity was rated by the patient as 10 of 10, and she scratched her skin the entire time she was in the clinic. The patient was emotional and stated that she had not been able to sleep due to the discomfort. We suspected scabies, and the patient was reassured to learn that it could be confirmed with a simple skin scrape test.
The crusted lesions on the patient's hands were scraped with a #15-blade scalpel, and a routine potassium hydroxide mount was performed. The skin scrapings were placed on a slide with a drop of 10% potassium hydroxide and observed under low-power (×10) and high-power (×40) microscopy, which revealed thousands of mites and eggs (along with previously hatched eggs) (Figure 4) and quickly confirmed a diagnosis of crusted scabies.an extremely contagious form of scabies seen in older patients with compromised immune systems, malnutrition, or disabilities. The patient was prescribed oral ivermectin (3 mg dosed at 200 μg/kg of body weight) and topical permethrin 5%, neither of which she took, as she died of a COVID-19 infection complication 3 days after this diagnostic clinic visit.
Classic and crusted scabies are both caused by infestation of the Sarcoptes scabiei var hominis mite. Classic scabies is a result of an infestation of a small number of mites (commonly 5–15 mites), while crusted scabies is due to hyperinfestation by as many as millions of mites, the latter often requiring more aggressive treatment. The mites are first transmitted to humans by either skin-toskin contact or fomites on bedding and clothing. The scabies mite undergoes 4 life cycle stages: egg, larvae, nymph, and adult. Once female mites are transmitted, they burrow under the skin and lay 2 to 3 eggs per day. The eggs hatch within 3 to 4 days, after which the larvae migrate to the skin surface. The larval stage lasts for 3 to 4 days, during which the larvae burrow into the stratum corneum to create molting pouches, until they molt into slightly larger nymphs. Nymphs can be found in hair follicles or molting pouches until they further molt within 3 to 4 days into adults, which are round, saclike mites. The adult male and female mites then mate, leaving the female fertile for the rest of her 1- to 2-month lifespan. Impregnated female mites traverse the skin surface in search of a burrow site, using the pulvilli on the anterior aspect of 2 legs to hold onto the skin. Once burrowed, the female mite continues to lay eggs for the rest of her life, with approximately 10% of her eggs resulting in adult mites. Male mites feed in shallow pits of the skin until they find a female burrow site for mating.1 This continuous life cycle of the scabies mite gives rise to highly transmissible, pruritic skin excoriations, as demonstrated in our patient.
The skin has a relatively late inflammatory and adaptive immune response to scabies, typically occurring 4 to 6 weeks after the initial infestation.2 This delayed inflammatory response and onset of symptoms may be due to the scabies mite’s ability to alter aspects of the host’s immune response, which differs in classic vs crusted scabies. In classic scabies, there is a predominance of CD4+ T cells in the dermis and minimal CD8+ T cells. The opposite is true in crusted scabies— there is an overwhelming infiltration of CD8+ T cells and minimal CD4+ T cells.3 The CD8+ T-cell predominance in crusted scabies is hypothesized to be the cause of keratinocyte apoptosis, resulting in epidermal hyperproliferation. Keratinocyte apoptosis also secretes cytokines, which may lead to the immunologic targeting of healthy skin cells. The damage of healthy dermal cells contributes to the inability of the skin’s immune system to mount an effective response, allowing the parasite to grow uncontrollably in patients with crusted scabies.4
This ineffective immune response is further exacerbated by corticosteroids, which are commonly prescribed for pruritus experienced by patients with scabies infestations. The mechanism of action of corticosteroids is the production of anti-inflammatory, antimitotic, and immunosuppressive effects.5 Because the integumentary immune system is imbalanced during crusted scabies infestation, the immunosuppressive mechanism of oral and topical corticosteroids further reduces the cellular immune response to scabies. The flourishing of the scabies mites along with keratinocyte apoptosis4 results in the development of hyperkeratotic skin crusting, most frequently on the palms, soles, arms, and legs. Risk factors for crusted scabies include immunosuppression, hospitalization, crowded living conditions, and poor hygiene, though no known risk factors were documented in up to 42% (33/78) of patients with crusted scabies in one study.6
Patients with crusted scabies typically present with generalized, poorly defined, erythematous, fissured plaques covered by scaling and crusts. Plaques on bony prominences such as finger articulations and elbows may have a thick verrucous aspect.1 Skin flaking that resembles brown sugar—a mixture of white sugar and molasses—is a clue to the diagnosis of crusted scabies. Brown sugar has a slightly sandy and sticky texture that ranges in color from very light brown to very dark brown. When present, flakes always appears slightly lighter than the patient’s skin tone. Although skin burrows are pathognomonic and clinically recognizable features of scabies, these burrows can be disguised by lesions, such as the hyperkeratotic plaques seen in our patient. The lesions may or may not be associated with pruritus, which may occur only at night, and bacterial superinfection has been reported in severe cases of crusted scabies,7 as scratching can cause sores, which may lead to infection. In severe cases, the constant scratching could lead to sepsis if the infection enters the bloodstream.8 Another symptom of scabies is a rash that causes small bumps that tend to form in a line, resembling small bites, hives, or pimples, and scaly plaques can lead to misdiagnosis as atopic dermatitis.
Treatment often is delayed due to misdiagnosis, as seen in our patient. Common misdiagnoses include atopic dermatitis, pityriasis rosea, systemic lupus erythematosus, bullous pemphigoid, lichen planus, pediculosis corporis, seborrheic scalp dermatitis, and adverse drug reactions.9 Patients with extensive infestations of crusted scabies should be treated with a 4-week course of permethrin cream 5% daily for 1 week, then twice per week until resolved, and oral ivermectin 200 μg/kg dosed 1 week apart for up to 4 weeks, if needed.1 Topical permethrin works by producing a selective neurotoxic effect on invertebrates such as scabies mites, which disrupts the function of voltage-gated sodium channels, thereby paralyzing the adult mites to halt the spread of infestation. However, treatment with topical medications can be difficult due to the thick crusts that have formed, which make it more challenging for the skin to properly absorb the treatment. Additionally, surgical debridement as an adjunct procedure has been done to improve the effectiveness of topical medications by removing all the mites in skin.10 On the other hand, the mechanism in which ivermectin treats scabies infestations is poorly understood. Current research suggests that ivermectin works by causing persistent opening of pH-gated chloride channels in scabies mites.11 There is emerging concern for drug resistance to these scabicides,12 revealing a need for further research of treatment options.
Patients with crusted scabies can have an extremely large number of mites (up to 2 million), making them more infectious than patients with classic scabies.13 As a result, it is imperative to reduce environmental transmission and risk for reinfection with mites during treatment. Because crusted scabies is transmitted by prolonged skinto- skin contact or by contact with personal items of an infected person (eg, bedding, clothing), treatment guidelines require all clothing, bedding, and towels of a patient with scabies to be machine-washed and dried with hot water and hot dryer cycles. If an item cannot be washed, it should be stored in a sealed plastic bag for 1 week, as scabies mites cannot survive more than 2 to 3 days away from their host of human skin.13 Treatment of close contacts of patients with scabies is recommended, as well as for those in endemic areas or closed communities, such as nursing homes or jails.
To the Editor:
Crusted scabies (formerly known as Norwegian scabies) is a rare and highly contagious variant of scabies, in which the skin is infested with thousands to millions of Sarcoptes scabiei var hominis mites. We present a case of skin changes that were misdiagnosed as atopic dermatitis, seborrhea, xerosis, and drug eruption on initial presentation, which prompted treatment with a corticosteroid that inadvertently caused progression to crusted scabies.
A 79-year-old woman who uses a wheelchair presented to the clinic with skin changes that consisted of diffuse, severely pruritic, erythematous plaques on the head, neck, trunk, face, and extremities of 2 years’ duration. She had a medical history of hyperlipidemia, hypertension, and hyperglycemia, as well as a stroke that required hospitalization 2 years prior to the onset of the skin changes. She had no history of allergies.
Prior clinical diagnoses by primary care and dermatology included xerosis, atopic dermatitis, seborrhea, and drug eruption. She was treated with a mid-potency topical corticosteroid (triamcinolone acetonide cream 0.1%) twice daily and prednisone 40 mg once daily for 2- to 4-week courses over an 8-month period without reduction in symptoms.
Physical examination at the current presentation revealed golden, crusted, fine, powdery but slightly sticky flakes that spread diffusely across the entire body and came off in crumbles with a simple touch. These widespread crusts were easily visible on clothing. There was underlying diffuse erythema beneath the flaking skin on the trunk and proximal extremities. The scale and shedding skin laid in piles on the patient’s lap and resembled brown sugar (Figure 1). The patient also reported decreased hand function and dexterity due to the yellowbrown, thick, crusty plaques that had developed on both the palmar and dorsal sides of the hands (Figure 2). Erythematous plaques on the scalp, forehead, and inner ears resembled seborrhea (Figure 3). Pruritus severity was rated by the patient as 10 of 10, and she scratched her skin the entire time she was in the clinic. The patient was emotional and stated that she had not been able to sleep due to the discomfort. We suspected scabies, and the patient was reassured to learn that it could be confirmed with a simple skin scrape test.
The crusted lesions on the patient's hands were scraped with a #15-blade scalpel, and a routine potassium hydroxide mount was performed. The skin scrapings were placed on a slide with a drop of 10% potassium hydroxide and observed under low-power (×10) and high-power (×40) microscopy, which revealed thousands of mites and eggs (along with previously hatched eggs) (Figure 4) and quickly confirmed a diagnosis of crusted scabies.an extremely contagious form of scabies seen in older patients with compromised immune systems, malnutrition, or disabilities. The patient was prescribed oral ivermectin (3 mg dosed at 200 μg/kg of body weight) and topical permethrin 5%, neither of which she took, as she died of a COVID-19 infection complication 3 days after this diagnostic clinic visit.
Classic and crusted scabies are both caused by infestation of the Sarcoptes scabiei var hominis mite. Classic scabies is a result of an infestation of a small number of mites (commonly 5–15 mites), while crusted scabies is due to hyperinfestation by as many as millions of mites, the latter often requiring more aggressive treatment. The mites are first transmitted to humans by either skin-toskin contact or fomites on bedding and clothing. The scabies mite undergoes 4 life cycle stages: egg, larvae, nymph, and adult. Once female mites are transmitted, they burrow under the skin and lay 2 to 3 eggs per day. The eggs hatch within 3 to 4 days, after which the larvae migrate to the skin surface. The larval stage lasts for 3 to 4 days, during which the larvae burrow into the stratum corneum to create molting pouches, until they molt into slightly larger nymphs. Nymphs can be found in hair follicles or molting pouches until they further molt within 3 to 4 days into adults, which are round, saclike mites. The adult male and female mites then mate, leaving the female fertile for the rest of her 1- to 2-month lifespan. Impregnated female mites traverse the skin surface in search of a burrow site, using the pulvilli on the anterior aspect of 2 legs to hold onto the skin. Once burrowed, the female mite continues to lay eggs for the rest of her life, with approximately 10% of her eggs resulting in adult mites. Male mites feed in shallow pits of the skin until they find a female burrow site for mating.1 This continuous life cycle of the scabies mite gives rise to highly transmissible, pruritic skin excoriations, as demonstrated in our patient.
The skin has a relatively late inflammatory and adaptive immune response to scabies, typically occurring 4 to 6 weeks after the initial infestation.2 This delayed inflammatory response and onset of symptoms may be due to the scabies mite’s ability to alter aspects of the host’s immune response, which differs in classic vs crusted scabies. In classic scabies, there is a predominance of CD4+ T cells in the dermis and minimal CD8+ T cells. The opposite is true in crusted scabies— there is an overwhelming infiltration of CD8+ T cells and minimal CD4+ T cells.3 The CD8+ T-cell predominance in crusted scabies is hypothesized to be the cause of keratinocyte apoptosis, resulting in epidermal hyperproliferation. Keratinocyte apoptosis also secretes cytokines, which may lead to the immunologic targeting of healthy skin cells. The damage of healthy dermal cells contributes to the inability of the skin’s immune system to mount an effective response, allowing the parasite to grow uncontrollably in patients with crusted scabies.4
This ineffective immune response is further exacerbated by corticosteroids, which are commonly prescribed for pruritus experienced by patients with scabies infestations. The mechanism of action of corticosteroids is the production of anti-inflammatory, antimitotic, and immunosuppressive effects.5 Because the integumentary immune system is imbalanced during crusted scabies infestation, the immunosuppressive mechanism of oral and topical corticosteroids further reduces the cellular immune response to scabies. The flourishing of the scabies mites along with keratinocyte apoptosis4 results in the development of hyperkeratotic skin crusting, most frequently on the palms, soles, arms, and legs. Risk factors for crusted scabies include immunosuppression, hospitalization, crowded living conditions, and poor hygiene, though no known risk factors were documented in up to 42% (33/78) of patients with crusted scabies in one study.6
Patients with crusted scabies typically present with generalized, poorly defined, erythematous, fissured plaques covered by scaling and crusts. Plaques on bony prominences such as finger articulations and elbows may have a thick verrucous aspect.1 Skin flaking that resembles brown sugar—a mixture of white sugar and molasses—is a clue to the diagnosis of crusted scabies. Brown sugar has a slightly sandy and sticky texture that ranges in color from very light brown to very dark brown. When present, flakes always appears slightly lighter than the patient’s skin tone. Although skin burrows are pathognomonic and clinically recognizable features of scabies, these burrows can be disguised by lesions, such as the hyperkeratotic plaques seen in our patient. The lesions may or may not be associated with pruritus, which may occur only at night, and bacterial superinfection has been reported in severe cases of crusted scabies,7 as scratching can cause sores, which may lead to infection. In severe cases, the constant scratching could lead to sepsis if the infection enters the bloodstream.8 Another symptom of scabies is a rash that causes small bumps that tend to form in a line, resembling small bites, hives, or pimples, and scaly plaques can lead to misdiagnosis as atopic dermatitis.
Treatment often is delayed due to misdiagnosis, as seen in our patient. Common misdiagnoses include atopic dermatitis, pityriasis rosea, systemic lupus erythematosus, bullous pemphigoid, lichen planus, pediculosis corporis, seborrheic scalp dermatitis, and adverse drug reactions.9 Patients with extensive infestations of crusted scabies should be treated with a 4-week course of permethrin cream 5% daily for 1 week, then twice per week until resolved, and oral ivermectin 200 μg/kg dosed 1 week apart for up to 4 weeks, if needed.1 Topical permethrin works by producing a selective neurotoxic effect on invertebrates such as scabies mites, which disrupts the function of voltage-gated sodium channels, thereby paralyzing the adult mites to halt the spread of infestation. However, treatment with topical medications can be difficult due to the thick crusts that have formed, which make it more challenging for the skin to properly absorb the treatment. Additionally, surgical debridement as an adjunct procedure has been done to improve the effectiveness of topical medications by removing all the mites in skin.10 On the other hand, the mechanism in which ivermectin treats scabies infestations is poorly understood. Current research suggests that ivermectin works by causing persistent opening of pH-gated chloride channels in scabies mites.11 There is emerging concern for drug resistance to these scabicides,12 revealing a need for further research of treatment options.
Patients with crusted scabies can have an extremely large number of mites (up to 2 million), making them more infectious than patients with classic scabies.13 As a result, it is imperative to reduce environmental transmission and risk for reinfection with mites during treatment. Because crusted scabies is transmitted by prolonged skinto- skin contact or by contact with personal items of an infected person (eg, bedding, clothing), treatment guidelines require all clothing, bedding, and towels of a patient with scabies to be machine-washed and dried with hot water and hot dryer cycles. If an item cannot be washed, it should be stored in a sealed plastic bag for 1 week, as scabies mites cannot survive more than 2 to 3 days away from their host of human skin.13 Treatment of close contacts of patients with scabies is recommended, as well as for those in endemic areas or closed communities, such as nursing homes or jails.
- Salavastru CM, Chosidow O, Boffa MJ, et al. European guideline for the management of scabies. J Eur Acad Dermatol Venereol. 2017;31:1248-1253. doi:10.1111/jdv.14351
- Morgan MS, Arlian LG, Markey MP. Sarcoptes scabiei mites modulate gene expression in human skin equivalents. PLoS One. 2013;8:e71143. doi:10.1371/journal.pone.0071143
- Walton SF, Beroukas D, Roberts-Thomson P, et al. New insights into disease pathogenesis in crusted (Norwegian) scabies: the skin immune response in crusted scabies. Br J Dermatol. 2008;158:1247-1255. doi:10.1111/j.1365-2133.2008.08541.x
- Bhat SA, Mounsey KE, Liu X, et al. Host immune responses to the itch mite, Sarcoptes scabiei, in humans. Parasit Vectors. 2017;10:385. doi:10.1186/s13071-017-2320-4
- Binic´ I, Jankovic´ A, Jovanovic´ D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2009;25:188-191. doi:10.3346/jkms.2010.25.1.188
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381. doi:10.1016/j.jinf.2004.08.033
- Yari N, Malone CH, Rivas A. Misdiagnosed crusted scabies in an AIDS patient leads to hyperinfestation. Cutis. 2017;99:202-204.
- American Academy of Dermatology Association. Scabies: signs and symptoms. Accessed July 12, 2024. https://www.aad.org/public/diseases/a-z/scabies-symptoms
- Siegfried EC, Hebert AA. Diagnosis of atopic dermatitis: mimics, overlaps, and complications. J Clin Med. 2015;4:884-917. doi:10.3390/jcm4050884
- Maghrabi MM, Lum S, Joba AT, et al. Norwegian crusted scabies: an unusual case presentation. J Foot Ankle Surg. 2014;53:62-66. doi:10.1053/j.jfas.2013.09.002
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725. doi:10.1056/NEJMct0910329
- Andriantsoanirina V, Izri A, Botterel F, et al. Molecular survey of knockdown resistance to pyrethroids in human scabies mites. Clin Microbiol Infect. 2014;20:O139-O141. doi:10.1111/1469-0691.12334
- Centers for Disease Control and Prevention. Preventing scabies. Published December 18, 2023. Accessed August 9, 2024. https://www.cdc.gov/scabies/prevention/index.html
- Salavastru CM, Chosidow O, Boffa MJ, et al. European guideline for the management of scabies. J Eur Acad Dermatol Venereol. 2017;31:1248-1253. doi:10.1111/jdv.14351
- Morgan MS, Arlian LG, Markey MP. Sarcoptes scabiei mites modulate gene expression in human skin equivalents. PLoS One. 2013;8:e71143. doi:10.1371/journal.pone.0071143
- Walton SF, Beroukas D, Roberts-Thomson P, et al. New insights into disease pathogenesis in crusted (Norwegian) scabies: the skin immune response in crusted scabies. Br J Dermatol. 2008;158:1247-1255. doi:10.1111/j.1365-2133.2008.08541.x
- Bhat SA, Mounsey KE, Liu X, et al. Host immune responses to the itch mite, Sarcoptes scabiei, in humans. Parasit Vectors. 2017;10:385. doi:10.1186/s13071-017-2320-4
- Binic´ I, Jankovic´ A, Jovanovic´ D, et al. Crusted (Norwegian) scabies following systemic and topical corticosteroid therapy. J Korean Med Sci. 2009;25:188-191. doi:10.3346/jkms.2010.25.1.188
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381. doi:10.1016/j.jinf.2004.08.033
- Yari N, Malone CH, Rivas A. Misdiagnosed crusted scabies in an AIDS patient leads to hyperinfestation. Cutis. 2017;99:202-204.
- American Academy of Dermatology Association. Scabies: signs and symptoms. Accessed July 12, 2024. https://www.aad.org/public/diseases/a-z/scabies-symptoms
- Siegfried EC, Hebert AA. Diagnosis of atopic dermatitis: mimics, overlaps, and complications. J Clin Med. 2015;4:884-917. doi:10.3390/jcm4050884
- Maghrabi MM, Lum S, Joba AT, et al. Norwegian crusted scabies: an unusual case presentation. J Foot Ankle Surg. 2014;53:62-66. doi:10.1053/j.jfas.2013.09.002
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725. doi:10.1056/NEJMct0910329
- Andriantsoanirina V, Izri A, Botterel F, et al. Molecular survey of knockdown resistance to pyrethroids in human scabies mites. Clin Microbiol Infect. 2014;20:O139-O141. doi:10.1111/1469-0691.12334
- Centers for Disease Control and Prevention. Preventing scabies. Published December 18, 2023. Accessed August 9, 2024. https://www.cdc.gov/scabies/prevention/index.html
PRACTICE POINTS
- Crusted scabies often is misdiagnosed because it mimics common dermatologic conditions, such as atopic dermatitis, psoriasis, drug eruption, and seborrhea. A unique feature of crusted scabies is fine or coarse scaling that resembles brown sugar.
- Immunosuppressants, such as topical corticosteroids, worsen the skin’s immune response to classic scabies infestations, which leads to parasitic overgrowth and the development of crusted scabies.
- Treatment of crusted scabies requires topical and oral scabicide; in addition, all clothing, bedding, and towels should be machine-washed and dried with hot water and hot dryer cycles to prevent environmental transmission and reinfection.