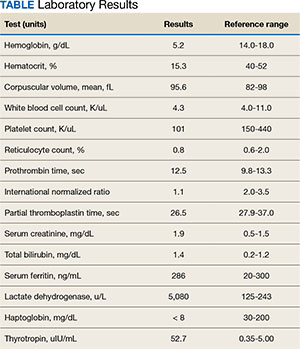
Case Presentation:A 65-year-old male veteran presented to the Veterans Affairs Boston Healthcare System (VABHS) emergency department with progressive fatigue, dyspnea on exertion, lightheadedness, and falls over the last month. New bilateral lower extremity numbness up to his knees developed in the week prior to admission and prompted him to seek care. Additional history included 2 episodes of transient loss of consciousness resulting in falls and a week of diarrhea, which had resolved. His medical history was notable for hypothyroidism secondary to Hashimoto thyroiditis, seizure disorder, vitiligo, treated hepatitis C virus (HCV) infection, alcohol use disorder in remission, diabetes mellitus, posttraumatic stress disorder, and traumatic brain injury. His medications included levothyroxine and carbamazepine. He previously worked as a barber but recently had stopped due to cognitive impairment. On initial evaluation, the patient's vital signs included a temperature of 36.3 °C, heart rate of 77 beats per minute, blood pressure of 139/83 mm Hg, respiratory rate of 18 breaths per minute, and 99% oxygen saturation while breathing ambient air. Physical examination was notable for a frail-appearing man in no acute distress. His conjunctivae were pale, and cardiac auscultation revealed a normal heart rate and irregularly irregular heart rhythm. A neurologic examination revealed decreased vibratory sensation in both feet, delayed and minimal speech, and a blunted affect. His skin was warm and dry with patchy hypopigmentation across the face and forehead. Laboratory results are shown in the Table. Testing 2 years previously found the patient's hemoglobin to be 11.4 g/dL and serum creatinine to be 1.7 mg/dL. A peripheral blood smear showed anisocytosis, hypochromia, decreased platelets, ovalocytes, elliptocytes, and rare teardrop cells, with no schistocytes present. Chest radiography and computed tomography of the head were unremarkable. An abdominal ultrasound revealed a complex hypoechoic mass with peripheral rim vascularity in the right hepatic lobe measuring 3.9 cm × 3.6 cm × 3.9 cm.
Lindsey Ulin, MD, Chief Medical Resident, VABHS and Brigham and Women’s Hospital (BWH):
To build the initial differential diagnosis, we are joined today by 3 internal medicine residents who were not involved in the care of this patient. Dr. Hickey, Dr. Ross and Dr. Manivannan, how did you approach this case?
Meghan Hickey, MD, Senior Internal Medicine Resident, VABHS and Boston Medical Center (BMC):
The constellation of fatigue, weakness, blunted affect, and delayed, minimal speech suggested central nervous system involvement, which I sought to unify with hemolytic anemia and his liver mass. The first diagnosis I considered was Wilson disease; however, this genetic disorder of copper metabolism often presents with liver failure or cirrhosis in young or middle-aged women, so this presentation would be atypical. Next, given the hypopigmentation was reported only on sun-exposed areas of the patient’s face, I considered possibilities other than vitiligo to avoid diagnostic anchoring. One such alternate diagnosis is porphyria cutanea tarda (PCT), which presents in middle-aged and older adults with a photosensitive dermatitis that can include acute sensory deficits. Manifestations of PCT can be triggered by alcohol consumption, though his alcohol use disorder was thought to be in remission, as well as HCV, for which he previously received treatment. However, anemia is uncommon in PCT, so the patient’s low hemoglobin would not be explained by this diagnosis. Lastly, I considered thrombotic thrombocytopenic purpura (TTP) given his anemia, thrombocytopenia, and neurologic symptoms; however, the patient did not have fever or a clear inciting cause, his renal dysfunction was relatively mild, and the peripheral blood smear revealed no schistocytes, which should be present in TTP.
TABLE Laboratory Results
Caroline Ross, MD, and Alan Manivannan, MD; Senior Internal Medicine Residents, VABHS and BMC:
We noted several salient features in the history and physical examination. First, we sought to explain the bilateral lower extremity numbness and decreased vibratory sensation in the feet leading to falls. We also considered his anemia and thrombocytopenia with signs of hemolysis including elevated lactate dehydrogenase (LDH), low haptoglobin, and elevated total bilirubin; however, with normal coagulation parameters. These results initially raised our concern for a thrombotic microangiopathy (TMA) such as TTP. However, the peripheral smear lacked schistocytes, making this less likely. The combination of his neurologic symptoms and TMA-like laboratory findings but without schistocytes raised our concern for vitamin B12 deficiency. Vitamin B12 deficiency can cause a pseudo-TMA picture with laboratory finding similar to TTP; however, schistocytes are typically absent. We also considered the possibility of hepatocellular carcinoma (HCC) with bone marrow infiltration leading to anemia given the finding of a liver mass on his abdominal ultrasound and low reticulocyte index. However, this would not explain his hemolysis. We also considered chronic disseminated intravascular coagulation in the setting of a malignancy as a contributor, but again, the smear lacked schistocytes and his coagulation parameters were normal. Finally, we considered a primary bone marrow process such as myelodysplastic syndrome due to the bicytopenia with poor bone marrow response and smear with tear drop cells and elliptocytes. However, we felt this was less likely as this would not explain his hemolytic anemia.
Dr. Ulin:
To refine the differential diagnosis, we are joined by an expert clinician who was also not involved in the care of this patient to describe his approach to this case. Dr. Orlander, can you walk us through your clinical reasoning?
Jay Orlander, MD, MPH: Professor of Medicine, Section of General Internal Medicine, Boston University Chobanian & Avedisian School of Medicine, Associate Chief, Medical Service, VABHS:
I will first comment on the hepatic mass. The hypoechoic liver mass with peripheral vascularity suggests a growing tumor. The patient has a history of substance use disorder with alcohol and treated HCV. He remains at increased risk for HCC even after prior successful HCV treatment and has 2 of 4 known risk factors for developing HCC— diabetes mellitus and alcohol use—the other 2 being underlying metabolic dysfunctionassociated steatotic liver disease (MASLD) and the presence of hepatic fibrosis, which we have not yet assessed. Worsening liver function can lead to cognitive issues and alcohol to peripheral neuropathy, but his story is not consistent with this. For his liver mass, I recommend a nonurgent magnetic resonance image for further evaluation.
Next, let’s consider his markedly elevated thyrotropin (TSH). Cognitive impairment along with lethargy, fatigue, and decreased exercise tolerance can be prominent features in severe hypothyroidism, but this diagnosis would not explain his hematologic findings.1
I view the principal finding of his laboratory testing as being that his bone marrow is failing to maintain adequate blood elements. He has a markedly low hematocrit along with low platelets and low-normal white blood cell counts. There is an absence of schistocytes on the blood smear, and after correcting his reticulocyte count for his degree of anemia (observed reticulocyte percentage [0.8%] x observed hematocrit [15.3%] / expected hematocrit [40%]), results in a reticulocyte index of 0.12, which is low. This suggests his bone marrow is failing to manufacture red blood cells at an appropriate rate. His haptoglobin is unmeasurable, so there is some free heme circulating. Hence, I infer that hemolysis and ineffective erythropoiesis are both occurring within the bone marrow, which also explains the slight elevation in bilirubin.
Intramedullary hemolysis with a markedly elevated LDH can be seen in severe vitamin B12 deficiency, which has many causes, but one cause in particular warrants consideration in this case: pernicious anemia. Pernicious anemia has an overall prevalence of about 0.1%, but is more common in older adults, and is estimated to be present in 2% to 3% of adults aged > 65 years.2 Prevalence is also increased in patients with other autoimmune diseases such as vitiligo and hypothyroidism, which our patient has.3 The pathophysiology of pernicious anemia relates to either autoimmune gastric parietal cell destruction and/or the development of antibodies against intrinsic factor, which is required for absorption of vitamin B12. Early disease may present with macrocytosis and a normal hemoglobin initially, but anemia develops over time if left untreated. When the primary cause of pernicious anemia is gastric parietal cell destruction, there is also an associated lack of stomach acid production (achlorhydria) with resulting poor micronutrient absorption; specifically, vitamin D, vitamin C, and iron. Hence, 30% of patients diagnosed with pernicious anemia have concurrent iron deficiency, which may counteract macrocytosis and result in a normal mean corpuscular volume. 4 Some medications are also poorly absorbed in achlorhydric states, such as levothyroxine, and treatment doses need to be increased, which could explain his markedly elevated TSH despite presumed medication adherence.
Vitamin B12 is essential for both the peripheral and central nervous systems. Longstanding severe B12 deficiency can explain all of his neurological and neurocognitive changes. The most common neurologic findings in B12 deficiency are symmetric paresthesias or numbness and gait problems. The sensory neuropathy affects the lower extremities more commonly than the upper. Untreated, patients can develop progressive weakness, ataxia, and orthostatic hypotension with syncope, as well as neuropsychiatric changes including depression or mood impairment, cognitive slowing, forgetfulness, and dementia.
Dr. Ulin:
Dr. Orlander, which pieces of objective data are most important in forming your differential diagnosis, and what tests would you obtain next?
Dr. Orlander:
The 3 most salient laboratory tests to me are a complete blood count, with all cell lines impacted but the hemoglobin and hematocrit most dramatically impacted, reticulocyte count of 0.8%, which is inappropriately low and hence suggests a hypoproliferative anemia, and the elevated LDH > 5000 IU/L.
Since my suspected diagnosis is pernicious anemia, I would obtain a blood smear looking for hypersegmented neutrophils, > 1 white blood cells with 5 lobes, or 1 with 6 lobes, which should clinch the diagnosis. Methylmalonic acid (MMA) levels are the most sensitive test for B12 deficiency, so I would also obtain that. Finally, I would check a B12 level, since in a patient with pernicious anemia, I would expect the level to be < 200 pg/mL.
Dr. Ulin:
Before we reveal the results of the patient’s additional workup, how do you approach interpreting B12 levels?
Dr. Orlander:
Measuring B12 can sometimes be problematic: the normal range is considered 200 to 900 pg/mL, but patients with measured low-normal levels in the range of 200 to 400 pg/mL can actually be physiologically deficient. There are also several common causes of falsely low and falsely high B12 levels in the absence of B12 deficiency. Hence, for patients with mild symptoms that could be due to B12 deficiency, many clinicians choose to just treat with B12 supplementation, deeming it safer to treat than miss an early diagnosis. B12 is involved in hydrogen transfer to convert MMA into succinyl-CoA and hence true vitamin B12 deficiency causes an increase in MMA.
Decreased production of vitamin B12 binding proteins, like haptocorrin, has been proposed as the mechanism for spurious low values.5 Certain conditions or medications can also cause spurious low serum vitamin B12 levels and thus might cause the appearance of vitamin B12 deficiency when the patient is not deficient. Examples include multiple myeloma, HIV infection, pregnancy, oral contraceptives, and phenytoin use. An example of spuriously low vitamin B12 level in pregnancy was demonstrated in a series of 50 pregnant individuals with low vitamin B12 levels (45-199 pg/mL), in whom metabolite testing for MMA and homocysteine showed no correlation with vitamin B12 level.6
Further complicating things, some conditions can cause spuriously increased vitamin B12 levels and thus might cause the appearance of normal vitamin B12 levels when the patient is actually deficient.7 Examples include occult malignancy, myeloproliferative neoplasms, alcoholic liver disease, kidney disease, and nitrous oxide exposure (the latter of which is unique in that it can also cause true vitamin B12 deficiency, as evidenced by clinical symptoms and high MMA levels).8,9
Lastly, autoantibodies to intrinsic factor in individuals with pernicious anemia may compete with intrinsic factor in the chemiluminescence assay and result in spuriously normal vitamin B12 levels in the presence of true deficiency.10-12 If the vitamin B12 level is very high (eg, 800 pg/mL), we do not worry about this effect in the absence of clinical features suggesting vitamin B12 deficiency; however, if the vitamin B12 level is borderline or low-normal and/or other clinical features suggest vitamin B12 deficiency, it is prudent to obtain other testing such as an MMA level.
Dr. Ulin:
We are also joined by Dr. Rahul Ganatra, who cared for the patient at the time the diagnosis was made. Dr. Ganatra, can you share the final diagnosis and provide an update on the patient?
Rahul Ganatra, MD, MPH, Director of Continuing Medical Education, VABHS:
The patient’s hemoglobin rose to 6.9 g/dL after transfusion of 2 units of packed red blood cells, and his dyspnea on exertion and fatigue improved. Iron studies, serum thiamine, serum folate, ADAMTS13 activity levels, and AM cortisol level were normal. Upon closer examination of the peripheral blood smear, rare hypersegmented neutrophils were noted. Serum B12 level returned below assay (< 146 pg/mL), and serum MMA was 50,800 nmol/L, confirming the diagnosis of severe vitamin B12 deficiency. Antibodies against intrinsic factor were detected, confirming the diagnosis of pernicious anemia. Treatment was initiated with intramuscular cyanocobalamin every other day and was transitioned to weekly dosing at the time of hospital discharge. After excluding adrenal insufficiency, his levothyroxine dose was increased. Finally, a liver mass biopsy confirmed a concomitant diagnosis of HCC. The patient was discharged home. Five weeks after discharge, his serum B12 level rose to > 1000 pg/mL, and 10 months after discharge, his TSH fell to 0.97 uIU/mL. Several months later, he underwent stereotactic body radiotherapy for the HCC. One year after his initial presentation, he has not resumed work as a barber.
References
Leigh H, Kramer SI. The psychiatric manifestations of endocrine disease. Adv Intern Med. 1984;29:413-445
Lenti MV, Rugge M, Lahner E, et al. Autoimmune gastritis. Nat Rev Dis Primers. 2020;6(1):56.doi:10.1038/s41572-020-0187-8
Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448. doi:10.1056/NEJM199711133372007
. Hershko C, Ronson A, Souroujon M, Maschler I, Heyd J, Patz J. Variable hematologic presentation of autoimmune gastritis: age-related progression from iron deficiency to cobalamin depletion. Blood. 2006;107(4):1673-1679. doi:10.1182/blood-2005-09-3534
Morkbak AL, Hvas AM, Milman N, Nexo E. Holotranscobalamin remains unchanged during pregnancy. Longitudinal changes of cobalamins and their binding proteins during pregnancy and postpartum. Haematologica. 2007;92(12):1711-1712. doi:10.3324/haematol.11636
Metz J, McGrath K, Bennett M, Hyland K, Bottiglieri T. Biochemical indices of vitamin B12 nutrition in pregnant patients with subnormal serum vitamin B12 levels. Am J Hematol. 1995;48(4):251-255. doi:10.1002/ajh.2830480409
Marsden P, Sharma AA, Rotella JA. Review article: clinical manifestations and outcomes of chronic nitrous oxide misuse: a systematic review. Emerg Med Australas. 2022;34(4):492- 503. doi:10.1111/1742-6723.13997
Hamilton MS, Blackmore S, Lee A. Possible cause of false normal B-12 assays. BMJ. 2006;333(7569):654-655. doi:10.1136/bmj.333.7569.654-c
Yang DT, Cook RJ. Spurious elevations of vitamin B12 with pernicious anemia. N Engl J Med. 2012;366(18):1742-1743. doi:10.1056/NEJMc1201655
Carmel R, Agrawal YP. Failures of cobalamin assays in pernicious anemia. N Engl J Med. 2012;367(4):385-386. doi:10.1056/NEJMc1204070
Green R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood. May 11 2017;129(19):2603- 2611. doi:10.1182/blood-2016-10-569186
Miceli E, Lenti MV, Padula D, et al. Common features of patients with autoimmune atrophic gastritis. Clin Gastroenterol Hepatol. 2012;10(7):812-814.doi:10.1016/j.cgh.2012.02.018
Lindsey Ulin, MDa,b; Meghan Hickey, MDb,c; Caroline Ross, MDb,c; Alan Manivannan, MDb,c; Jay Orlander, MD, MPHb,d; Rahul B. Ganatra, MD, MPHb
Author affiliationsa Brigham and Women’s Hospital, Boston, Massachusetts bVeterans Affairs Boston Healthcare System, West Roxbury, Massachusetts c Boston Medical Center, Massachusetts dBoston University Chobanian & Avedisian School of Medicine, Massachusetts
Lindsey Ulin, MDa,b; Meghan Hickey, MDb,c; Caroline Ross, MDb,c; Alan Manivannan, MDb,c; Jay Orlander, MD, MPHb,d; Rahul B. Ganatra, MD, MPHb
Author affiliationsa Brigham and Women’s Hospital, Boston, Massachusetts bVeterans Affairs Boston Healthcare System, West Roxbury, Massachusetts c Boston Medical Center, Massachusetts dBoston University Chobanian & Avedisian School of Medicine, Massachusetts
Author disclosures The authors report no actual or potential conflicts of interest with regard to this article.
Fed Pract. 2024;41(10). Published online October 15. doi:10.12788/fp.0516
Author and Disclosure Information
Lindsey Ulin, MDa,b; Meghan Hickey, MDb,c; Caroline Ross, MDb,c; Alan Manivannan, MDb,c; Jay Orlander, MD, MPHb,d; Rahul B. Ganatra, MD, MPHb
Author affiliationsa Brigham and Women’s Hospital, Boston, Massachusetts bVeterans Affairs Boston Healthcare System, West Roxbury, Massachusetts c Boston Medical Center, Massachusetts dBoston University Chobanian & Avedisian School of Medicine, Massachusetts
Case Presentation:A 65-year-old male veteran presented to the Veterans Affairs Boston Healthcare System (VABHS) emergency department with progressive fatigue, dyspnea on exertion, lightheadedness, and falls over the last month. New bilateral lower extremity numbness up to his knees developed in the week prior to admission and prompted him to seek care. Additional history included 2 episodes of transient loss of consciousness resulting in falls and a week of diarrhea, which had resolved. His medical history was notable for hypothyroidism secondary to Hashimoto thyroiditis, seizure disorder, vitiligo, treated hepatitis C virus (HCV) infection, alcohol use disorder in remission, diabetes mellitus, posttraumatic stress disorder, and traumatic brain injury. His medications included levothyroxine and carbamazepine. He previously worked as a barber but recently had stopped due to cognitive impairment. On initial evaluation, the patient's vital signs included a temperature of 36.3 °C, heart rate of 77 beats per minute, blood pressure of 139/83 mm Hg, respiratory rate of 18 breaths per minute, and 99% oxygen saturation while breathing ambient air. Physical examination was notable for a frail-appearing man in no acute distress. His conjunctivae were pale, and cardiac auscultation revealed a normal heart rate and irregularly irregular heart rhythm. A neurologic examination revealed decreased vibratory sensation in both feet, delayed and minimal speech, and a blunted affect. His skin was warm and dry with patchy hypopigmentation across the face and forehead. Laboratory results are shown in the Table. Testing 2 years previously found the patient's hemoglobin to be 11.4 g/dL and serum creatinine to be 1.7 mg/dL. A peripheral blood smear showed anisocytosis, hypochromia, decreased platelets, ovalocytes, elliptocytes, and rare teardrop cells, with no schistocytes present. Chest radiography and computed tomography of the head were unremarkable. An abdominal ultrasound revealed a complex hypoechoic mass with peripheral rim vascularity in the right hepatic lobe measuring 3.9 cm × 3.6 cm × 3.9 cm.
Lindsey Ulin, MD, Chief Medical Resident, VABHS and Brigham and Women’s Hospital (BWH):
To build the initial differential diagnosis, we are joined today by 3 internal medicine residents who were not involved in the care of this patient. Dr. Hickey, Dr. Ross and Dr. Manivannan, how did you approach this case?
Meghan Hickey, MD, Senior Internal Medicine Resident, VABHS and Boston Medical Center (BMC):
The constellation of fatigue, weakness, blunted affect, and delayed, minimal speech suggested central nervous system involvement, which I sought to unify with hemolytic anemia and his liver mass. The first diagnosis I considered was Wilson disease; however, this genetic disorder of copper metabolism often presents with liver failure or cirrhosis in young or middle-aged women, so this presentation would be atypical. Next, given the hypopigmentation was reported only on sun-exposed areas of the patient’s face, I considered possibilities other than vitiligo to avoid diagnostic anchoring. One such alternate diagnosis is porphyria cutanea tarda (PCT), which presents in middle-aged and older adults with a photosensitive dermatitis that can include acute sensory deficits. Manifestations of PCT can be triggered by alcohol consumption, though his alcohol use disorder was thought to be in remission, as well as HCV, for which he previously received treatment. However, anemia is uncommon in PCT, so the patient’s low hemoglobin would not be explained by this diagnosis. Lastly, I considered thrombotic thrombocytopenic purpura (TTP) given his anemia, thrombocytopenia, and neurologic symptoms; however, the patient did not have fever or a clear inciting cause, his renal dysfunction was relatively mild, and the peripheral blood smear revealed no schistocytes, which should be present in TTP.
TABLE Laboratory Results
Caroline Ross, MD, and Alan Manivannan, MD; Senior Internal Medicine Residents, VABHS and BMC:
We noted several salient features in the history and physical examination. First, we sought to explain the bilateral lower extremity numbness and decreased vibratory sensation in the feet leading to falls. We also considered his anemia and thrombocytopenia with signs of hemolysis including elevated lactate dehydrogenase (LDH), low haptoglobin, and elevated total bilirubin; however, with normal coagulation parameters. These results initially raised our concern for a thrombotic microangiopathy (TMA) such as TTP. However, the peripheral smear lacked schistocytes, making this less likely. The combination of his neurologic symptoms and TMA-like laboratory findings but without schistocytes raised our concern for vitamin B12 deficiency. Vitamin B12 deficiency can cause a pseudo-TMA picture with laboratory finding similar to TTP; however, schistocytes are typically absent. We also considered the possibility of hepatocellular carcinoma (HCC) with bone marrow infiltration leading to anemia given the finding of a liver mass on his abdominal ultrasound and low reticulocyte index. However, this would not explain his hemolysis. We also considered chronic disseminated intravascular coagulation in the setting of a malignancy as a contributor, but again, the smear lacked schistocytes and his coagulation parameters were normal. Finally, we considered a primary bone marrow process such as myelodysplastic syndrome due to the bicytopenia with poor bone marrow response and smear with tear drop cells and elliptocytes. However, we felt this was less likely as this would not explain his hemolytic anemia.
Dr. Ulin:
To refine the differential diagnosis, we are joined by an expert clinician who was also not involved in the care of this patient to describe his approach to this case. Dr. Orlander, can you walk us through your clinical reasoning?
Jay Orlander, MD, MPH: Professor of Medicine, Section of General Internal Medicine, Boston University Chobanian & Avedisian School of Medicine, Associate Chief, Medical Service, VABHS:
I will first comment on the hepatic mass. The hypoechoic liver mass with peripheral vascularity suggests a growing tumor. The patient has a history of substance use disorder with alcohol and treated HCV. He remains at increased risk for HCC even after prior successful HCV treatment and has 2 of 4 known risk factors for developing HCC— diabetes mellitus and alcohol use—the other 2 being underlying metabolic dysfunctionassociated steatotic liver disease (MASLD) and the presence of hepatic fibrosis, which we have not yet assessed. Worsening liver function can lead to cognitive issues and alcohol to peripheral neuropathy, but his story is not consistent with this. For his liver mass, I recommend a nonurgent magnetic resonance image for further evaluation.
Next, let’s consider his markedly elevated thyrotropin (TSH). Cognitive impairment along with lethargy, fatigue, and decreased exercise tolerance can be prominent features in severe hypothyroidism, but this diagnosis would not explain his hematologic findings.1
I view the principal finding of his laboratory testing as being that his bone marrow is failing to maintain adequate blood elements. He has a markedly low hematocrit along with low platelets and low-normal white blood cell counts. There is an absence of schistocytes on the blood smear, and after correcting his reticulocyte count for his degree of anemia (observed reticulocyte percentage [0.8%] x observed hematocrit [15.3%] / expected hematocrit [40%]), results in a reticulocyte index of 0.12, which is low. This suggests his bone marrow is failing to manufacture red blood cells at an appropriate rate. His haptoglobin is unmeasurable, so there is some free heme circulating. Hence, I infer that hemolysis and ineffective erythropoiesis are both occurring within the bone marrow, which also explains the slight elevation in bilirubin.
Intramedullary hemolysis with a markedly elevated LDH can be seen in severe vitamin B12 deficiency, which has many causes, but one cause in particular warrants consideration in this case: pernicious anemia. Pernicious anemia has an overall prevalence of about 0.1%, but is more common in older adults, and is estimated to be present in 2% to 3% of adults aged > 65 years.2 Prevalence is also increased in patients with other autoimmune diseases such as vitiligo and hypothyroidism, which our patient has.3 The pathophysiology of pernicious anemia relates to either autoimmune gastric parietal cell destruction and/or the development of antibodies against intrinsic factor, which is required for absorption of vitamin B12. Early disease may present with macrocytosis and a normal hemoglobin initially, but anemia develops over time if left untreated. When the primary cause of pernicious anemia is gastric parietal cell destruction, there is also an associated lack of stomach acid production (achlorhydria) with resulting poor micronutrient absorption; specifically, vitamin D, vitamin C, and iron. Hence, 30% of patients diagnosed with pernicious anemia have concurrent iron deficiency, which may counteract macrocytosis and result in a normal mean corpuscular volume. 4 Some medications are also poorly absorbed in achlorhydric states, such as levothyroxine, and treatment doses need to be increased, which could explain his markedly elevated TSH despite presumed medication adherence.
Vitamin B12 is essential for both the peripheral and central nervous systems. Longstanding severe B12 deficiency can explain all of his neurological and neurocognitive changes. The most common neurologic findings in B12 deficiency are symmetric paresthesias or numbness and gait problems. The sensory neuropathy affects the lower extremities more commonly than the upper. Untreated, patients can develop progressive weakness, ataxia, and orthostatic hypotension with syncope, as well as neuropsychiatric changes including depression or mood impairment, cognitive slowing, forgetfulness, and dementia.
Dr. Ulin:
Dr. Orlander, which pieces of objective data are most important in forming your differential diagnosis, and what tests would you obtain next?
Dr. Orlander:
The 3 most salient laboratory tests to me are a complete blood count, with all cell lines impacted but the hemoglobin and hematocrit most dramatically impacted, reticulocyte count of 0.8%, which is inappropriately low and hence suggests a hypoproliferative anemia, and the elevated LDH > 5000 IU/L.
Since my suspected diagnosis is pernicious anemia, I would obtain a blood smear looking for hypersegmented neutrophils, > 1 white blood cells with 5 lobes, or 1 with 6 lobes, which should clinch the diagnosis. Methylmalonic acid (MMA) levels are the most sensitive test for B12 deficiency, so I would also obtain that. Finally, I would check a B12 level, since in a patient with pernicious anemia, I would expect the level to be < 200 pg/mL.
Dr. Ulin:
Before we reveal the results of the patient’s additional workup, how do you approach interpreting B12 levels?
Dr. Orlander:
Measuring B12 can sometimes be problematic: the normal range is considered 200 to 900 pg/mL, but patients with measured low-normal levels in the range of 200 to 400 pg/mL can actually be physiologically deficient. There are also several common causes of falsely low and falsely high B12 levels in the absence of B12 deficiency. Hence, for patients with mild symptoms that could be due to B12 deficiency, many clinicians choose to just treat with B12 supplementation, deeming it safer to treat than miss an early diagnosis. B12 is involved in hydrogen transfer to convert MMA into succinyl-CoA and hence true vitamin B12 deficiency causes an increase in MMA.
Decreased production of vitamin B12 binding proteins, like haptocorrin, has been proposed as the mechanism for spurious low values.5 Certain conditions or medications can also cause spurious low serum vitamin B12 levels and thus might cause the appearance of vitamin B12 deficiency when the patient is not deficient. Examples include multiple myeloma, HIV infection, pregnancy, oral contraceptives, and phenytoin use. An example of spuriously low vitamin B12 level in pregnancy was demonstrated in a series of 50 pregnant individuals with low vitamin B12 levels (45-199 pg/mL), in whom metabolite testing for MMA and homocysteine showed no correlation with vitamin B12 level.6
Further complicating things, some conditions can cause spuriously increased vitamin B12 levels and thus might cause the appearance of normal vitamin B12 levels when the patient is actually deficient.7 Examples include occult malignancy, myeloproliferative neoplasms, alcoholic liver disease, kidney disease, and nitrous oxide exposure (the latter of which is unique in that it can also cause true vitamin B12 deficiency, as evidenced by clinical symptoms and high MMA levels).8,9
Lastly, autoantibodies to intrinsic factor in individuals with pernicious anemia may compete with intrinsic factor in the chemiluminescence assay and result in spuriously normal vitamin B12 levels in the presence of true deficiency.10-12 If the vitamin B12 level is very high (eg, 800 pg/mL), we do not worry about this effect in the absence of clinical features suggesting vitamin B12 deficiency; however, if the vitamin B12 level is borderline or low-normal and/or other clinical features suggest vitamin B12 deficiency, it is prudent to obtain other testing such as an MMA level.
Dr. Ulin:
We are also joined by Dr. Rahul Ganatra, who cared for the patient at the time the diagnosis was made. Dr. Ganatra, can you share the final diagnosis and provide an update on the patient?
Rahul Ganatra, MD, MPH, Director of Continuing Medical Education, VABHS:
The patient’s hemoglobin rose to 6.9 g/dL after transfusion of 2 units of packed red blood cells, and his dyspnea on exertion and fatigue improved. Iron studies, serum thiamine, serum folate, ADAMTS13 activity levels, and AM cortisol level were normal. Upon closer examination of the peripheral blood smear, rare hypersegmented neutrophils were noted. Serum B12 level returned below assay (< 146 pg/mL), and serum MMA was 50,800 nmol/L, confirming the diagnosis of severe vitamin B12 deficiency. Antibodies against intrinsic factor were detected, confirming the diagnosis of pernicious anemia. Treatment was initiated with intramuscular cyanocobalamin every other day and was transitioned to weekly dosing at the time of hospital discharge. After excluding adrenal insufficiency, his levothyroxine dose was increased. Finally, a liver mass biopsy confirmed a concomitant diagnosis of HCC. The patient was discharged home. Five weeks after discharge, his serum B12 level rose to > 1000 pg/mL, and 10 months after discharge, his TSH fell to 0.97 uIU/mL. Several months later, he underwent stereotactic body radiotherapy for the HCC. One year after his initial presentation, he has not resumed work as a barber.
Case Presentation:A 65-year-old male veteran presented to the Veterans Affairs Boston Healthcare System (VABHS) emergency department with progressive fatigue, dyspnea on exertion, lightheadedness, and falls over the last month. New bilateral lower extremity numbness up to his knees developed in the week prior to admission and prompted him to seek care. Additional history included 2 episodes of transient loss of consciousness resulting in falls and a week of diarrhea, which had resolved. His medical history was notable for hypothyroidism secondary to Hashimoto thyroiditis, seizure disorder, vitiligo, treated hepatitis C virus (HCV) infection, alcohol use disorder in remission, diabetes mellitus, posttraumatic stress disorder, and traumatic brain injury. His medications included levothyroxine and carbamazepine. He previously worked as a barber but recently had stopped due to cognitive impairment. On initial evaluation, the patient's vital signs included a temperature of 36.3 °C, heart rate of 77 beats per minute, blood pressure of 139/83 mm Hg, respiratory rate of 18 breaths per minute, and 99% oxygen saturation while breathing ambient air. Physical examination was notable for a frail-appearing man in no acute distress. His conjunctivae were pale, and cardiac auscultation revealed a normal heart rate and irregularly irregular heart rhythm. A neurologic examination revealed decreased vibratory sensation in both feet, delayed and minimal speech, and a blunted affect. His skin was warm and dry with patchy hypopigmentation across the face and forehead. Laboratory results are shown in the Table. Testing 2 years previously found the patient's hemoglobin to be 11.4 g/dL and serum creatinine to be 1.7 mg/dL. A peripheral blood smear showed anisocytosis, hypochromia, decreased platelets, ovalocytes, elliptocytes, and rare teardrop cells, with no schistocytes present. Chest radiography and computed tomography of the head were unremarkable. An abdominal ultrasound revealed a complex hypoechoic mass with peripheral rim vascularity in the right hepatic lobe measuring 3.9 cm × 3.6 cm × 3.9 cm.
Lindsey Ulin, MD, Chief Medical Resident, VABHS and Brigham and Women’s Hospital (BWH):
To build the initial differential diagnosis, we are joined today by 3 internal medicine residents who were not involved in the care of this patient. Dr. Hickey, Dr. Ross and Dr. Manivannan, how did you approach this case?
Meghan Hickey, MD, Senior Internal Medicine Resident, VABHS and Boston Medical Center (BMC):
The constellation of fatigue, weakness, blunted affect, and delayed, minimal speech suggested central nervous system involvement, which I sought to unify with hemolytic anemia and his liver mass. The first diagnosis I considered was Wilson disease; however, this genetic disorder of copper metabolism often presents with liver failure or cirrhosis in young or middle-aged women, so this presentation would be atypical. Next, given the hypopigmentation was reported only on sun-exposed areas of the patient’s face, I considered possibilities other than vitiligo to avoid diagnostic anchoring. One such alternate diagnosis is porphyria cutanea tarda (PCT), which presents in middle-aged and older adults with a photosensitive dermatitis that can include acute sensory deficits. Manifestations of PCT can be triggered by alcohol consumption, though his alcohol use disorder was thought to be in remission, as well as HCV, for which he previously received treatment. However, anemia is uncommon in PCT, so the patient’s low hemoglobin would not be explained by this diagnosis. Lastly, I considered thrombotic thrombocytopenic purpura (TTP) given his anemia, thrombocytopenia, and neurologic symptoms; however, the patient did not have fever or a clear inciting cause, his renal dysfunction was relatively mild, and the peripheral blood smear revealed no schistocytes, which should be present in TTP.
TABLE Laboratory Results
Caroline Ross, MD, and Alan Manivannan, MD; Senior Internal Medicine Residents, VABHS and BMC:
We noted several salient features in the history and physical examination. First, we sought to explain the bilateral lower extremity numbness and decreased vibratory sensation in the feet leading to falls. We also considered his anemia and thrombocytopenia with signs of hemolysis including elevated lactate dehydrogenase (LDH), low haptoglobin, and elevated total bilirubin; however, with normal coagulation parameters. These results initially raised our concern for a thrombotic microangiopathy (TMA) such as TTP. However, the peripheral smear lacked schistocytes, making this less likely. The combination of his neurologic symptoms and TMA-like laboratory findings but without schistocytes raised our concern for vitamin B12 deficiency. Vitamin B12 deficiency can cause a pseudo-TMA picture with laboratory finding similar to TTP; however, schistocytes are typically absent. We also considered the possibility of hepatocellular carcinoma (HCC) with bone marrow infiltration leading to anemia given the finding of a liver mass on his abdominal ultrasound and low reticulocyte index. However, this would not explain his hemolysis. We also considered chronic disseminated intravascular coagulation in the setting of a malignancy as a contributor, but again, the smear lacked schistocytes and his coagulation parameters were normal. Finally, we considered a primary bone marrow process such as myelodysplastic syndrome due to the bicytopenia with poor bone marrow response and smear with tear drop cells and elliptocytes. However, we felt this was less likely as this would not explain his hemolytic anemia.
Dr. Ulin:
To refine the differential diagnosis, we are joined by an expert clinician who was also not involved in the care of this patient to describe his approach to this case. Dr. Orlander, can you walk us through your clinical reasoning?
Jay Orlander, MD, MPH: Professor of Medicine, Section of General Internal Medicine, Boston University Chobanian & Avedisian School of Medicine, Associate Chief, Medical Service, VABHS:
I will first comment on the hepatic mass. The hypoechoic liver mass with peripheral vascularity suggests a growing tumor. The patient has a history of substance use disorder with alcohol and treated HCV. He remains at increased risk for HCC even after prior successful HCV treatment and has 2 of 4 known risk factors for developing HCC— diabetes mellitus and alcohol use—the other 2 being underlying metabolic dysfunctionassociated steatotic liver disease (MASLD) and the presence of hepatic fibrosis, which we have not yet assessed. Worsening liver function can lead to cognitive issues and alcohol to peripheral neuropathy, but his story is not consistent with this. For his liver mass, I recommend a nonurgent magnetic resonance image for further evaluation.
Next, let’s consider his markedly elevated thyrotropin (TSH). Cognitive impairment along with lethargy, fatigue, and decreased exercise tolerance can be prominent features in severe hypothyroidism, but this diagnosis would not explain his hematologic findings.1
I view the principal finding of his laboratory testing as being that his bone marrow is failing to maintain adequate blood elements. He has a markedly low hematocrit along with low platelets and low-normal white blood cell counts. There is an absence of schistocytes on the blood smear, and after correcting his reticulocyte count for his degree of anemia (observed reticulocyte percentage [0.8%] x observed hematocrit [15.3%] / expected hematocrit [40%]), results in a reticulocyte index of 0.12, which is low. This suggests his bone marrow is failing to manufacture red blood cells at an appropriate rate. His haptoglobin is unmeasurable, so there is some free heme circulating. Hence, I infer that hemolysis and ineffective erythropoiesis are both occurring within the bone marrow, which also explains the slight elevation in bilirubin.
Intramedullary hemolysis with a markedly elevated LDH can be seen in severe vitamin B12 deficiency, which has many causes, but one cause in particular warrants consideration in this case: pernicious anemia. Pernicious anemia has an overall prevalence of about 0.1%, but is more common in older adults, and is estimated to be present in 2% to 3% of adults aged > 65 years.2 Prevalence is also increased in patients with other autoimmune diseases such as vitiligo and hypothyroidism, which our patient has.3 The pathophysiology of pernicious anemia relates to either autoimmune gastric parietal cell destruction and/or the development of antibodies against intrinsic factor, which is required for absorption of vitamin B12. Early disease may present with macrocytosis and a normal hemoglobin initially, but anemia develops over time if left untreated. When the primary cause of pernicious anemia is gastric parietal cell destruction, there is also an associated lack of stomach acid production (achlorhydria) with resulting poor micronutrient absorption; specifically, vitamin D, vitamin C, and iron. Hence, 30% of patients diagnosed with pernicious anemia have concurrent iron deficiency, which may counteract macrocytosis and result in a normal mean corpuscular volume. 4 Some medications are also poorly absorbed in achlorhydric states, such as levothyroxine, and treatment doses need to be increased, which could explain his markedly elevated TSH despite presumed medication adherence.
Vitamin B12 is essential for both the peripheral and central nervous systems. Longstanding severe B12 deficiency can explain all of his neurological and neurocognitive changes. The most common neurologic findings in B12 deficiency are symmetric paresthesias or numbness and gait problems. The sensory neuropathy affects the lower extremities more commonly than the upper. Untreated, patients can develop progressive weakness, ataxia, and orthostatic hypotension with syncope, as well as neuropsychiatric changes including depression or mood impairment, cognitive slowing, forgetfulness, and dementia.
Dr. Ulin:
Dr. Orlander, which pieces of objective data are most important in forming your differential diagnosis, and what tests would you obtain next?
Dr. Orlander:
The 3 most salient laboratory tests to me are a complete blood count, with all cell lines impacted but the hemoglobin and hematocrit most dramatically impacted, reticulocyte count of 0.8%, which is inappropriately low and hence suggests a hypoproliferative anemia, and the elevated LDH > 5000 IU/L.
Since my suspected diagnosis is pernicious anemia, I would obtain a blood smear looking for hypersegmented neutrophils, > 1 white blood cells with 5 lobes, or 1 with 6 lobes, which should clinch the diagnosis. Methylmalonic acid (MMA) levels are the most sensitive test for B12 deficiency, so I would also obtain that. Finally, I would check a B12 level, since in a patient with pernicious anemia, I would expect the level to be < 200 pg/mL.
Dr. Ulin:
Before we reveal the results of the patient’s additional workup, how do you approach interpreting B12 levels?
Dr. Orlander:
Measuring B12 can sometimes be problematic: the normal range is considered 200 to 900 pg/mL, but patients with measured low-normal levels in the range of 200 to 400 pg/mL can actually be physiologically deficient. There are also several common causes of falsely low and falsely high B12 levels in the absence of B12 deficiency. Hence, for patients with mild symptoms that could be due to B12 deficiency, many clinicians choose to just treat with B12 supplementation, deeming it safer to treat than miss an early diagnosis. B12 is involved in hydrogen transfer to convert MMA into succinyl-CoA and hence true vitamin B12 deficiency causes an increase in MMA.
Decreased production of vitamin B12 binding proteins, like haptocorrin, has been proposed as the mechanism for spurious low values.5 Certain conditions or medications can also cause spurious low serum vitamin B12 levels and thus might cause the appearance of vitamin B12 deficiency when the patient is not deficient. Examples include multiple myeloma, HIV infection, pregnancy, oral contraceptives, and phenytoin use. An example of spuriously low vitamin B12 level in pregnancy was demonstrated in a series of 50 pregnant individuals with low vitamin B12 levels (45-199 pg/mL), in whom metabolite testing for MMA and homocysteine showed no correlation with vitamin B12 level.6
Further complicating things, some conditions can cause spuriously increased vitamin B12 levels and thus might cause the appearance of normal vitamin B12 levels when the patient is actually deficient.7 Examples include occult malignancy, myeloproliferative neoplasms, alcoholic liver disease, kidney disease, and nitrous oxide exposure (the latter of which is unique in that it can also cause true vitamin B12 deficiency, as evidenced by clinical symptoms and high MMA levels).8,9
Lastly, autoantibodies to intrinsic factor in individuals with pernicious anemia may compete with intrinsic factor in the chemiluminescence assay and result in spuriously normal vitamin B12 levels in the presence of true deficiency.10-12 If the vitamin B12 level is very high (eg, 800 pg/mL), we do not worry about this effect in the absence of clinical features suggesting vitamin B12 deficiency; however, if the vitamin B12 level is borderline or low-normal and/or other clinical features suggest vitamin B12 deficiency, it is prudent to obtain other testing such as an MMA level.
Dr. Ulin:
We are also joined by Dr. Rahul Ganatra, who cared for the patient at the time the diagnosis was made. Dr. Ganatra, can you share the final diagnosis and provide an update on the patient?
Rahul Ganatra, MD, MPH, Director of Continuing Medical Education, VABHS:
The patient’s hemoglobin rose to 6.9 g/dL after transfusion of 2 units of packed red blood cells, and his dyspnea on exertion and fatigue improved. Iron studies, serum thiamine, serum folate, ADAMTS13 activity levels, and AM cortisol level were normal. Upon closer examination of the peripheral blood smear, rare hypersegmented neutrophils were noted. Serum B12 level returned below assay (< 146 pg/mL), and serum MMA was 50,800 nmol/L, confirming the diagnosis of severe vitamin B12 deficiency. Antibodies against intrinsic factor were detected, confirming the diagnosis of pernicious anemia. Treatment was initiated with intramuscular cyanocobalamin every other day and was transitioned to weekly dosing at the time of hospital discharge. After excluding adrenal insufficiency, his levothyroxine dose was increased. Finally, a liver mass biopsy confirmed a concomitant diagnosis of HCC. The patient was discharged home. Five weeks after discharge, his serum B12 level rose to > 1000 pg/mL, and 10 months after discharge, his TSH fell to 0.97 uIU/mL. Several months later, he underwent stereotactic body radiotherapy for the HCC. One year after his initial presentation, he has not resumed work as a barber.
References
Leigh H, Kramer SI. The psychiatric manifestations of endocrine disease. Adv Intern Med. 1984;29:413-445
Lenti MV, Rugge M, Lahner E, et al. Autoimmune gastritis. Nat Rev Dis Primers. 2020;6(1):56.doi:10.1038/s41572-020-0187-8
Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448. doi:10.1056/NEJM199711133372007
. Hershko C, Ronson A, Souroujon M, Maschler I, Heyd J, Patz J. Variable hematologic presentation of autoimmune gastritis: age-related progression from iron deficiency to cobalamin depletion. Blood. 2006;107(4):1673-1679. doi:10.1182/blood-2005-09-3534
Morkbak AL, Hvas AM, Milman N, Nexo E. Holotranscobalamin remains unchanged during pregnancy. Longitudinal changes of cobalamins and their binding proteins during pregnancy and postpartum. Haematologica. 2007;92(12):1711-1712. doi:10.3324/haematol.11636
Metz J, McGrath K, Bennett M, Hyland K, Bottiglieri T. Biochemical indices of vitamin B12 nutrition in pregnant patients with subnormal serum vitamin B12 levels. Am J Hematol. 1995;48(4):251-255. doi:10.1002/ajh.2830480409
Marsden P, Sharma AA, Rotella JA. Review article: clinical manifestations and outcomes of chronic nitrous oxide misuse: a systematic review. Emerg Med Australas. 2022;34(4):492- 503. doi:10.1111/1742-6723.13997
Hamilton MS, Blackmore S, Lee A. Possible cause of false normal B-12 assays. BMJ. 2006;333(7569):654-655. doi:10.1136/bmj.333.7569.654-c
Yang DT, Cook RJ. Spurious elevations of vitamin B12 with pernicious anemia. N Engl J Med. 2012;366(18):1742-1743. doi:10.1056/NEJMc1201655
Carmel R, Agrawal YP. Failures of cobalamin assays in pernicious anemia. N Engl J Med. 2012;367(4):385-386. doi:10.1056/NEJMc1204070
Green R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood. May 11 2017;129(19):2603- 2611. doi:10.1182/blood-2016-10-569186
Miceli E, Lenti MV, Padula D, et al. Common features of patients with autoimmune atrophic gastritis. Clin Gastroenterol Hepatol. 2012;10(7):812-814.doi:10.1016/j.cgh.2012.02.018
References
Leigh H, Kramer SI. The psychiatric manifestations of endocrine disease. Adv Intern Med. 1984;29:413-445
Lenti MV, Rugge M, Lahner E, et al. Autoimmune gastritis. Nat Rev Dis Primers. 2020;6(1):56.doi:10.1038/s41572-020-0187-8
Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448. doi:10.1056/NEJM199711133372007
. Hershko C, Ronson A, Souroujon M, Maschler I, Heyd J, Patz J. Variable hematologic presentation of autoimmune gastritis: age-related progression from iron deficiency to cobalamin depletion. Blood. 2006;107(4):1673-1679. doi:10.1182/blood-2005-09-3534
Morkbak AL, Hvas AM, Milman N, Nexo E. Holotranscobalamin remains unchanged during pregnancy. Longitudinal changes of cobalamins and their binding proteins during pregnancy and postpartum. Haematologica. 2007;92(12):1711-1712. doi:10.3324/haematol.11636
Metz J, McGrath K, Bennett M, Hyland K, Bottiglieri T. Biochemical indices of vitamin B12 nutrition in pregnant patients with subnormal serum vitamin B12 levels. Am J Hematol. 1995;48(4):251-255. doi:10.1002/ajh.2830480409
Marsden P, Sharma AA, Rotella JA. Review article: clinical manifestations and outcomes of chronic nitrous oxide misuse: a systematic review. Emerg Med Australas. 2022;34(4):492- 503. doi:10.1111/1742-6723.13997
Hamilton MS, Blackmore S, Lee A. Possible cause of false normal B-12 assays. BMJ. 2006;333(7569):654-655. doi:10.1136/bmj.333.7569.654-c
Yang DT, Cook RJ. Spurious elevations of vitamin B12 with pernicious anemia. N Engl J Med. 2012;366(18):1742-1743. doi:10.1056/NEJMc1201655
Carmel R, Agrawal YP. Failures of cobalamin assays in pernicious anemia. N Engl J Med. 2012;367(4):385-386. doi:10.1056/NEJMc1204070
Green R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood. May 11 2017;129(19):2603- 2611. doi:10.1182/blood-2016-10-569186
Miceli E, Lenti MV, Padula D, et al. Common features of patients with autoimmune atrophic gastritis. Clin Gastroenterol Hepatol. 2012;10(7):812-814.doi:10.1016/j.cgh.2012.02.018
Splenic abscesses are a rare occurrence that represent a marginal proportion of intra-abdominal infections. One study found splenic abscesses in only 0.14% to 0.70% of autopsies and none of the 540 abdominal abscesses they examined originated in the spleen.1 Patients with splenic abscesses tend to present with nonspecific symptoms such as fevers, chills, and abdominal pain.2 Imaging modalities such as abdominal ultrasound and computed tomography (CT) are vital to the workup and diagnosis identification.2 Splenic abscesses are generally associated with another underlying process, as seen in patients who are affected by endocarditis, trauma, metastatic infection, splenic infarction, or neoplasia.2
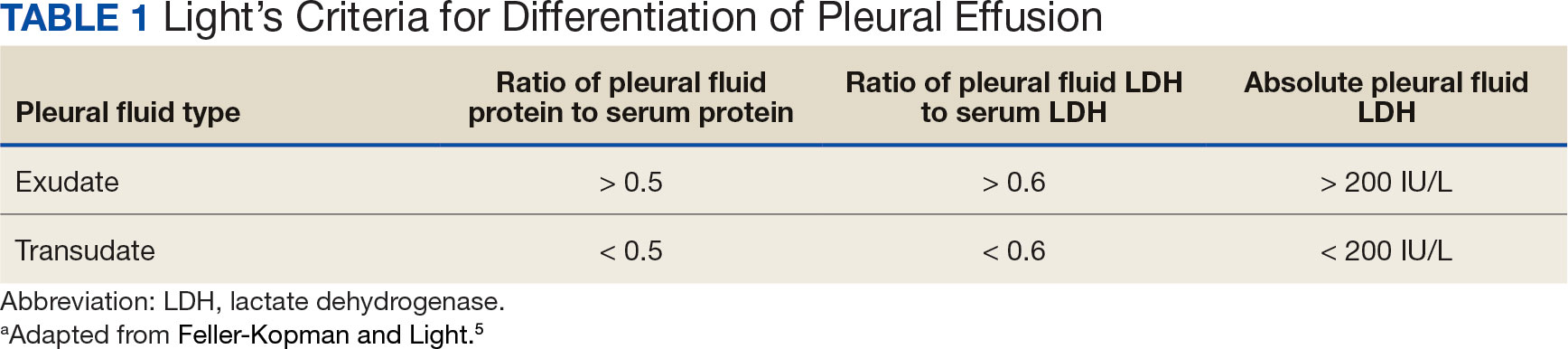
Pleural effusions, or the buildup of fluid within the pleural space, is a common condition typically secondary to another disease.3 Clinical identification of the primary condition may be challenging.3 In the absence of a clear etiology, such as obvious signs of congestive heart failure, further differentiation relies upon pleural fluid analysis, beginning with the distinction between exudate (inflammatory) and transudate (noninflammatory). 3,4 This distinction can be made using Light’s criteria, which relies on protein and lactate dehydrogenase (LDH) ratios between the pleural fluid and serum (Table 1).5 Though rare, half of splenic abscesses are associated with pleural effusion.6 As an inflammatory condition, splenic abscesses have been classically described as a cause of exudative pleural effusions.5,6
A myelodysplastic syndrome is a group of diseases that arise from malignant hematopoietic stem cells, leading to the proliferation of the malignant cells and faulty production of other bone marrow products.7 These disorders can range from single to multilineage dysplasia. Cells are often left in an immature blast form, unable to function appropriately, and vulnerable to destruction. Patients with myeloproliferative disorders frequently suffer from leukopenia and infections attributable to known quantitative and qualitative defects of neutrophils.8
CASE PRESENTATION
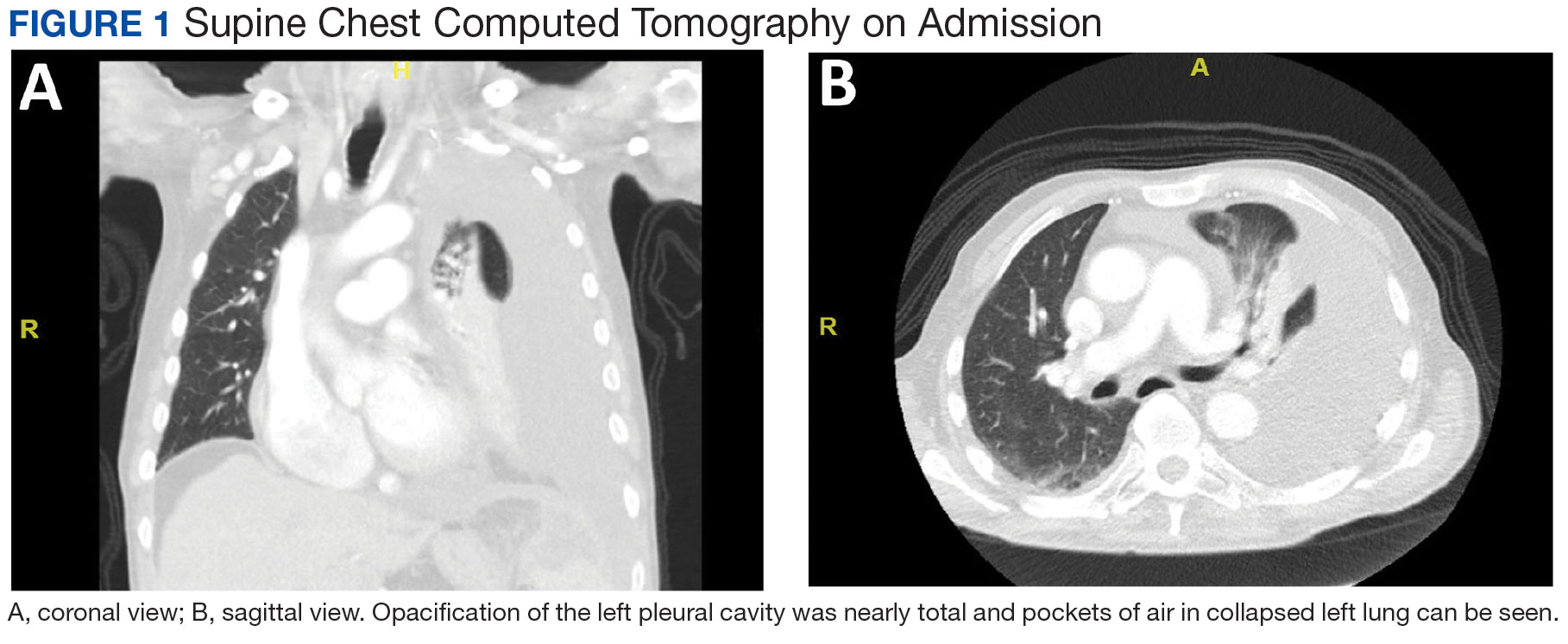
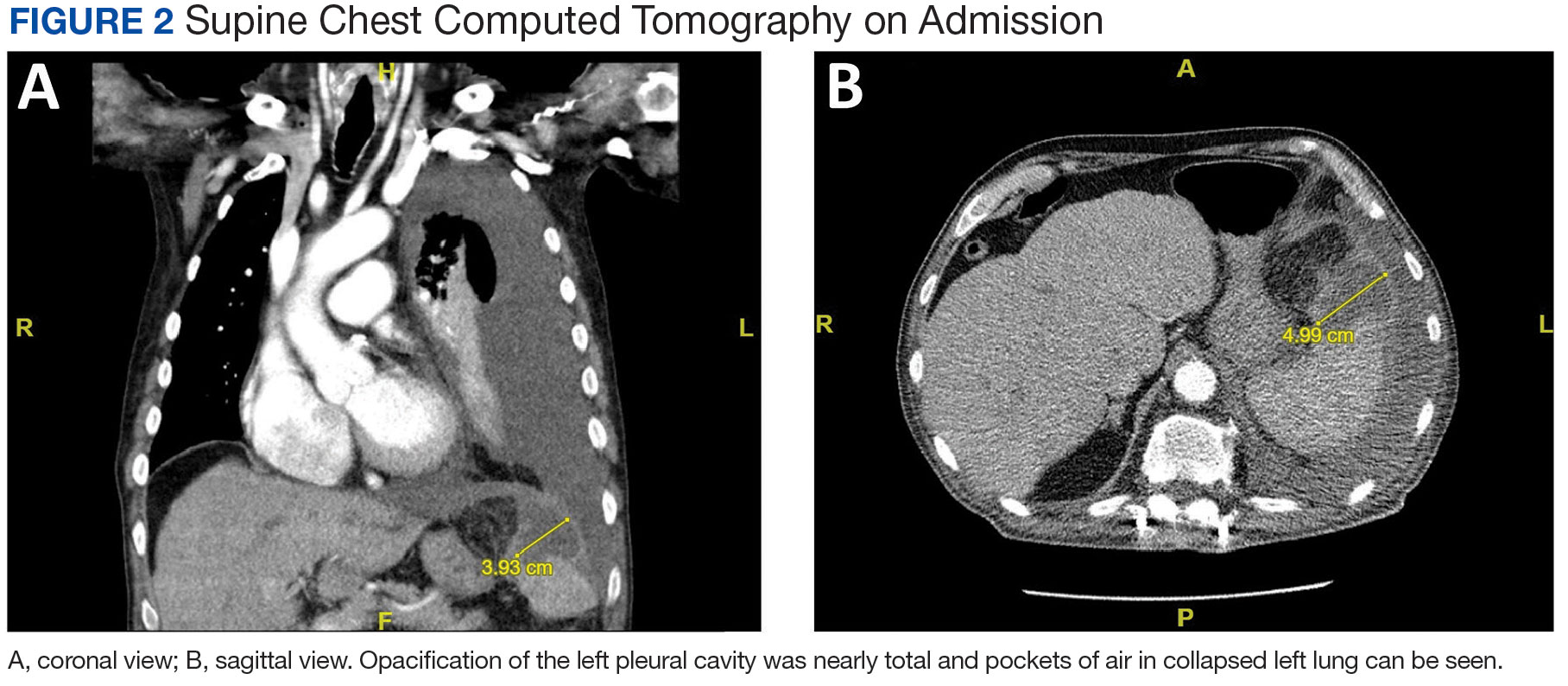
A male aged 80 years presented to the Central Texas Veterans Affairs Hospital (CTVAH) with shortness of breath, weight loss, and fever. On admission, his medical history was notable for atrial fibrillation, myelodysplastic syndrome, hypertension, hyperlipidemia, stable ascending aortic aneurysm, and Vitamin B12 deficiency. A chest CT showed a large left pleural effusion (Figure 1). Additionally, the radiology report noted a nonspecific 4- to 5-cm lobulated subdiaphragmatic mass within the anterior dome of the spleen with surrounding soft tissue swelling and splenomegaly (Figure 2).
A, coronal view; B, sagittal view. Opacification of the left pleural cavity was nearly total and pockets of air in collapsed left lung can be seen.
A, coronal view; B, sagittal view. Opacification of the left pleural cavity was nearly total and pockets of air in collapsed left lung can be seen.
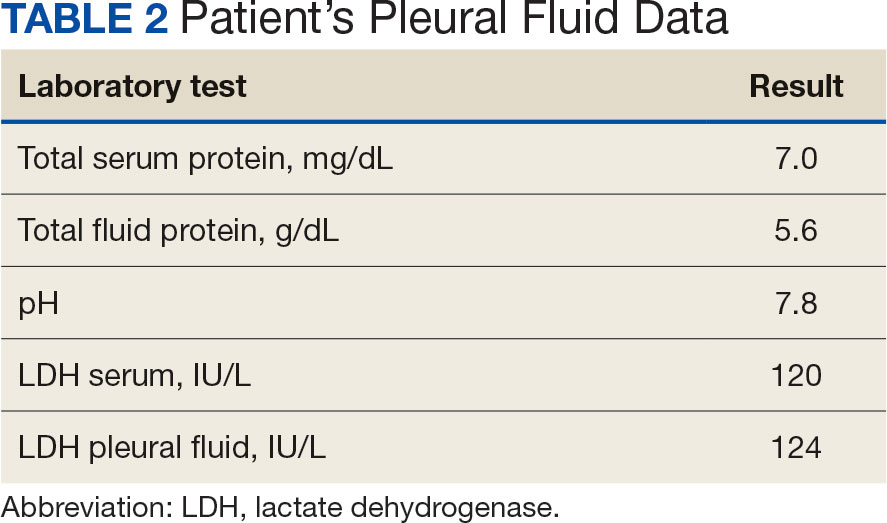
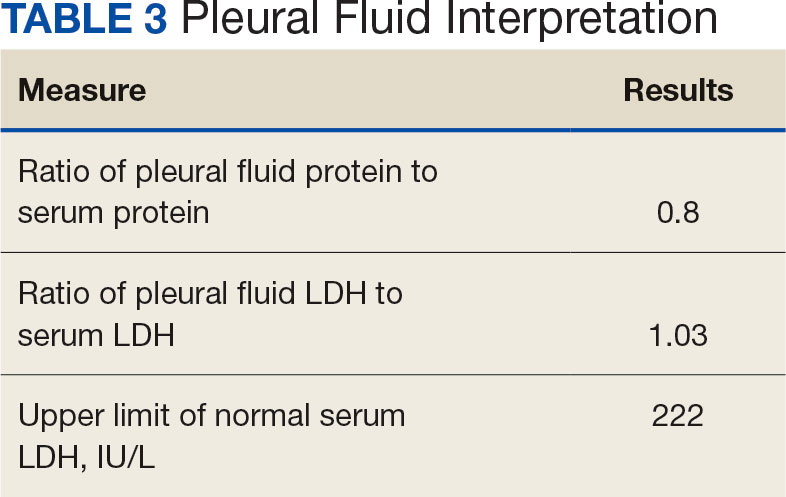
Initial thoracentesis was performed with 1500 mL of straw-colored fluid negative for bacteria, fungi, malignancy, and acid-fast organisms (Tables 2 and 3). The pleural effusion persisted, requiring a second thoracentesis 2 days later that was positive for Escherichia coli (E coli). Given the exudative nature and positive culture, a chest tube was placed, and the pleural effusion was therefore felt to be an empyema, arousing suspicion that the splenic mass seen on CT was an abscess. The site was accessed by interventional radiology, purulent fluid aspirated, and a drain was placed. Cultures grew E coli sensitive to ceftriaxone. Despite receiving intravenous ceftriaxone 2 g daily, the pleural effusion became further complicated due to chest tube obstruction and persistent drainage.
The patient was discharged to Baylor Scott & White Medical Center in Temple, Texas where he underwent decortication with cardiothoracic surgery with several pleural adhesions noted. Following surgery the patient was readmitted to CTVAH and continued ceftriaxone therapy following the infectious disease specialist's recommendation. He was discharged with plans to return to CTVAH for continued care. The patient was readmitted and transitioned to oral levofloxacin 500 mg daily and received physical and occupational therapy. He showed dramatic improvement on this regimen, with a 3-week follow-up CT that indicated only a small left pleural effusion and a 28 mm × 11 mm × 10 mm lesion in the anterior superior spleen. The patient had not returned for further evaluation by thoracic surgery; however, he has continued to see CTVAH primary care without reported recurrence of symptoms.
DISCUSSION
Splenic abscesses are a rare condition typically characterized by hematogenous spread of bacteria from another source, most commonly the endocardium.2 Other differential diagnoses include bacteremia or spread from an intra-abdominal site.2 Staphylococcus aureus and E coli are the most common bacteria seen in splenic abscesses. 2 Treatment includes antibiotics, percutaneous drainage, and, as a last resort, splenectomy.2
Our patient was found to have grown E coli, but no source indicative of spread was identified. He had negative blood cultures, negative findings for intra-abdominal pathologies on CT scans, and a negative echocardiogram for endocarditis. A bronchoscopy showed no evidence of a source from the lungs, and specimens taken from the pleural adhesions were negative for malignancy and bacteria.
This patient had risk factors for the illness, namely his history of being immunocompromised secondary to myelodysplastic syndrome.7 Accordingly, the patient showed persistent leukopenia with neutropenia and lymphocytopenia, which would not be expected for most patients with such an extensive infection. 8 While being immunocompromised undoubtedly contributed to the severity of the patient’s presentation and slow recovery, it does not explain the etiology or origin of his infection. This patient differs from current literature in that his splenic abscess was truly idiopathic rather than resulting from an alternative source.
Complications of splenic abscesses include pleural effusions, as seen with this patient, as well as pneumonia, pneumothorax, hemorrhage, subphrenic abscess, and intraabdominal perforation, among others.2 We determined conclusively that the patient’s pleural effusion was secondary to the splenic abscess, and excluded other bacterial foci strongly suggests that the spleen was the origin of the illness.
CONCLUSIONS
This case suggests splenic abscesses should be considered when evaluating pleural effusion. It further demonstrates that the spleen may be the central source of infection in the absence of iatrogenic inoculation or bacteremia. We hope our findings may lead to earlier identification in similar scenarios and improved patient outcomes in a multidisciplinary approach.
References
Lee WS, Choi ST, Kim KK. Splenic abscess: a single institution study and review of the literature. Yonsei Med J. 2011;52(2):288-292. doi:10.3349/ymj.2011.52.2.288
Lotfollahzadeh S, Mathew G, Zemaitis MR. Splenic Abscess. In: StatPearls. StatPearls Publishing; June 3, 2023.
Jany B, Welte T. Pleural effusion in adults-etiology, diagnosis, and treatment. Dtsch Arztebl Int. 2019;116(21):377- 386. doi:10.3238/arztebl.2019.0377
Light RW. Pleural effusions. Med Clin North Am. 2011;95(6):1055-1070. doi:10.1016/j.mcna.2011.08.005
Feller-Kopman D, Light R. Pleural Disease. N Engl J Med. 2018;378(18):1754. doi:10.1056/NEJMc1803858
Ferreiro L, Casal A, Toubes ME, et al. Pleural effusion due to nonmalignant gastrointestinal disease. ERJ Open Res. 2023;9(3):00290-2022. doi:10.1183/23120541.00290-2022
Madison Demmera; Mitchell Clarka; Tayler Acton DOb,c; Nikhil Seth MDa,d
Author affiliations: aTexas A&M School of Medicine, Bryan bCentral Texas Veterans Affairs Hospital, Temple cBaylor College of Medicine, Houston, Texas dBaylor Scott and White Health, Temple, Texas
Author disclosures: The authors report no actual or potential conflicts of interest or outside sources of funding with regard to this article.
Madison Demmera; Mitchell Clarka; Tayler Acton DOb,c; Nikhil Seth MDa,d
Author affiliations: aTexas A&M School of Medicine, Bryan bCentral Texas Veterans Affairs Hospital, Temple cBaylor College of Medicine, Houston, Texas dBaylor Scott and White Health, Temple, Texas
Author disclosures: The authors report no actual or potential conflicts of interest or outside sources of funding with regard to this article.
Fed Pract. 2024;41(9)e509. Published online September 23. doi:10.12788/fp.0509
Author and Disclosure Information
Madison Demmera; Mitchell Clarka; Tayler Acton DOb,c; Nikhil Seth MDa,d
Author affiliations: aTexas A&M School of Medicine, Bryan bCentral Texas Veterans Affairs Hospital, Temple cBaylor College of Medicine, Houston, Texas dBaylor Scott and White Health, Temple, Texas
Author disclosures: The authors report no actual or potential conflicts of interest or outside sources of funding with regard to this article.
Splenic abscesses are a rare occurrence that represent a marginal proportion of intra-abdominal infections. One study found splenic abscesses in only 0.14% to 0.70% of autopsies and none of the 540 abdominal abscesses they examined originated in the spleen.1 Patients with splenic abscesses tend to present with nonspecific symptoms such as fevers, chills, and abdominal pain.2 Imaging modalities such as abdominal ultrasound and computed tomography (CT) are vital to the workup and diagnosis identification.2 Splenic abscesses are generally associated with another underlying process, as seen in patients who are affected by endocarditis, trauma, metastatic infection, splenic infarction, or neoplasia.2
Pleural effusions, or the buildup of fluid within the pleural space, is a common condition typically secondary to another disease.3 Clinical identification of the primary condition may be challenging.3 In the absence of a clear etiology, such as obvious signs of congestive heart failure, further differentiation relies upon pleural fluid analysis, beginning with the distinction between exudate (inflammatory) and transudate (noninflammatory). 3,4 This distinction can be made using Light’s criteria, which relies on protein and lactate dehydrogenase (LDH) ratios between the pleural fluid and serum (Table 1).5 Though rare, half of splenic abscesses are associated with pleural effusion.6 As an inflammatory condition, splenic abscesses have been classically described as a cause of exudative pleural effusions.5,6
A myelodysplastic syndrome is a group of diseases that arise from malignant hematopoietic stem cells, leading to the proliferation of the malignant cells and faulty production of other bone marrow products.7 These disorders can range from single to multilineage dysplasia. Cells are often left in an immature blast form, unable to function appropriately, and vulnerable to destruction. Patients with myeloproliferative disorders frequently suffer from leukopenia and infections attributable to known quantitative and qualitative defects of neutrophils.8
CASE PRESENTATION
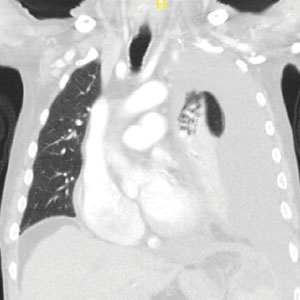
A male aged 80 years presented to the Central Texas Veterans Affairs Hospital (CTVAH) with shortness of breath, weight loss, and fever. On admission, his medical history was notable for atrial fibrillation, myelodysplastic syndrome, hypertension, hyperlipidemia, stable ascending aortic aneurysm, and Vitamin B12 deficiency. A chest CT showed a large left pleural effusion (Figure 1). Additionally, the radiology report noted a nonspecific 4- to 5-cm lobulated subdiaphragmatic mass within the anterior dome of the spleen with surrounding soft tissue swelling and splenomegaly (Figure 2).
A, coronal view; B, sagittal view. Opacification of the left pleural cavity was nearly total and pockets of air in collapsed left lung can be seen.
A, coronal view; B, sagittal view. Opacification of the left pleural cavity was nearly total and pockets of air in collapsed left lung can be seen.
Initial thoracentesis was performed with 1500 mL of straw-colored fluid negative for bacteria, fungi, malignancy, and acid-fast organisms (Tables 2 and 3). The pleural effusion persisted, requiring a second thoracentesis 2 days later that was positive for Escherichia coli (E coli). Given the exudative nature and positive culture, a chest tube was placed, and the pleural effusion was therefore felt to be an empyema, arousing suspicion that the splenic mass seen on CT was an abscess. The site was accessed by interventional radiology, purulent fluid aspirated, and a drain was placed. Cultures grew E coli sensitive to ceftriaxone. Despite receiving intravenous ceftriaxone 2 g daily, the pleural effusion became further complicated due to chest tube obstruction and persistent drainage.
The patient was discharged to Baylor Scott & White Medical Center in Temple, Texas where he underwent decortication with cardiothoracic surgery with several pleural adhesions noted. Following surgery the patient was readmitted to CTVAH and continued ceftriaxone therapy following the infectious disease specialist's recommendation. He was discharged with plans to return to CTVAH for continued care. The patient was readmitted and transitioned to oral levofloxacin 500 mg daily and received physical and occupational therapy. He showed dramatic improvement on this regimen, with a 3-week follow-up CT that indicated only a small left pleural effusion and a 28 mm × 11 mm × 10 mm lesion in the anterior superior spleen. The patient had not returned for further evaluation by thoracic surgery; however, he has continued to see CTVAH primary care without reported recurrence of symptoms.
DISCUSSION
Splenic abscesses are a rare condition typically characterized by hematogenous spread of bacteria from another source, most commonly the endocardium.2 Other differential diagnoses include bacteremia or spread from an intra-abdominal site.2 Staphylococcus aureus and E coli are the most common bacteria seen in splenic abscesses. 2 Treatment includes antibiotics, percutaneous drainage, and, as a last resort, splenectomy.2
Our patient was found to have grown E coli, but no source indicative of spread was identified. He had negative blood cultures, negative findings for intra-abdominal pathologies on CT scans, and a negative echocardiogram for endocarditis. A bronchoscopy showed no evidence of a source from the lungs, and specimens taken from the pleural adhesions were negative for malignancy and bacteria.
This patient had risk factors for the illness, namely his history of being immunocompromised secondary to myelodysplastic syndrome.7 Accordingly, the patient showed persistent leukopenia with neutropenia and lymphocytopenia, which would not be expected for most patients with such an extensive infection. 8 While being immunocompromised undoubtedly contributed to the severity of the patient’s presentation and slow recovery, it does not explain the etiology or origin of his infection. This patient differs from current literature in that his splenic abscess was truly idiopathic rather than resulting from an alternative source.
Complications of splenic abscesses include pleural effusions, as seen with this patient, as well as pneumonia, pneumothorax, hemorrhage, subphrenic abscess, and intraabdominal perforation, among others.2 We determined conclusively that the patient’s pleural effusion was secondary to the splenic abscess, and excluded other bacterial foci strongly suggests that the spleen was the origin of the illness.
CONCLUSIONS
This case suggests splenic abscesses should be considered when evaluating pleural effusion. It further demonstrates that the spleen may be the central source of infection in the absence of iatrogenic inoculation or bacteremia. We hope our findings may lead to earlier identification in similar scenarios and improved patient outcomes in a multidisciplinary approach.
Splenic abscesses are a rare occurrence that represent a marginal proportion of intra-abdominal infections. One study found splenic abscesses in only 0.14% to 0.70% of autopsies and none of the 540 abdominal abscesses they examined originated in the spleen.1 Patients with splenic abscesses tend to present with nonspecific symptoms such as fevers, chills, and abdominal pain.2 Imaging modalities such as abdominal ultrasound and computed tomography (CT) are vital to the workup and diagnosis identification.2 Splenic abscesses are generally associated with another underlying process, as seen in patients who are affected by endocarditis, trauma, metastatic infection, splenic infarction, or neoplasia.2
Pleural effusions, or the buildup of fluid within the pleural space, is a common condition typically secondary to another disease.3 Clinical identification of the primary condition may be challenging.3 In the absence of a clear etiology, such as obvious signs of congestive heart failure, further differentiation relies upon pleural fluid analysis, beginning with the distinction between exudate (inflammatory) and transudate (noninflammatory). 3,4 This distinction can be made using Light’s criteria, which relies on protein and lactate dehydrogenase (LDH) ratios between the pleural fluid and serum (Table 1).5 Though rare, half of splenic abscesses are associated with pleural effusion.6 As an inflammatory condition, splenic abscesses have been classically described as a cause of exudative pleural effusions.5,6
A myelodysplastic syndrome is a group of diseases that arise from malignant hematopoietic stem cells, leading to the proliferation of the malignant cells and faulty production of other bone marrow products.7 These disorders can range from single to multilineage dysplasia. Cells are often left in an immature blast form, unable to function appropriately, and vulnerable to destruction. Patients with myeloproliferative disorders frequently suffer from leukopenia and infections attributable to known quantitative and qualitative defects of neutrophils.8
CASE PRESENTATION
A male aged 80 years presented to the Central Texas Veterans Affairs Hospital (CTVAH) with shortness of breath, weight loss, and fever. On admission, his medical history was notable for atrial fibrillation, myelodysplastic syndrome, hypertension, hyperlipidemia, stable ascending aortic aneurysm, and Vitamin B12 deficiency. A chest CT showed a large left pleural effusion (Figure 1). Additionally, the radiology report noted a nonspecific 4- to 5-cm lobulated subdiaphragmatic mass within the anterior dome of the spleen with surrounding soft tissue swelling and splenomegaly (Figure 2).
A, coronal view; B, sagittal view. Opacification of the left pleural cavity was nearly total and pockets of air in collapsed left lung can be seen.
A, coronal view; B, sagittal view. Opacification of the left pleural cavity was nearly total and pockets of air in collapsed left lung can be seen.
Initial thoracentesis was performed with 1500 mL of straw-colored fluid negative for bacteria, fungi, malignancy, and acid-fast organisms (Tables 2 and 3). The pleural effusion persisted, requiring a second thoracentesis 2 days later that was positive for Escherichia coli (E coli). Given the exudative nature and positive culture, a chest tube was placed, and the pleural effusion was therefore felt to be an empyema, arousing suspicion that the splenic mass seen on CT was an abscess. The site was accessed by interventional radiology, purulent fluid aspirated, and a drain was placed. Cultures grew E coli sensitive to ceftriaxone. Despite receiving intravenous ceftriaxone 2 g daily, the pleural effusion became further complicated due to chest tube obstruction and persistent drainage.
The patient was discharged to Baylor Scott & White Medical Center in Temple, Texas where he underwent decortication with cardiothoracic surgery with several pleural adhesions noted. Following surgery the patient was readmitted to CTVAH and continued ceftriaxone therapy following the infectious disease specialist's recommendation. He was discharged with plans to return to CTVAH for continued care. The patient was readmitted and transitioned to oral levofloxacin 500 mg daily and received physical and occupational therapy. He showed dramatic improvement on this regimen, with a 3-week follow-up CT that indicated only a small left pleural effusion and a 28 mm × 11 mm × 10 mm lesion in the anterior superior spleen. The patient had not returned for further evaluation by thoracic surgery; however, he has continued to see CTVAH primary care without reported recurrence of symptoms.
DISCUSSION
Splenic abscesses are a rare condition typically characterized by hematogenous spread of bacteria from another source, most commonly the endocardium.2 Other differential diagnoses include bacteremia or spread from an intra-abdominal site.2 Staphylococcus aureus and E coli are the most common bacteria seen in splenic abscesses. 2 Treatment includes antibiotics, percutaneous drainage, and, as a last resort, splenectomy.2
Our patient was found to have grown E coli, but no source indicative of spread was identified. He had negative blood cultures, negative findings for intra-abdominal pathologies on CT scans, and a negative echocardiogram for endocarditis. A bronchoscopy showed no evidence of a source from the lungs, and specimens taken from the pleural adhesions were negative for malignancy and bacteria.
This patient had risk factors for the illness, namely his history of being immunocompromised secondary to myelodysplastic syndrome.7 Accordingly, the patient showed persistent leukopenia with neutropenia and lymphocytopenia, which would not be expected for most patients with such an extensive infection. 8 While being immunocompromised undoubtedly contributed to the severity of the patient’s presentation and slow recovery, it does not explain the etiology or origin of his infection. This patient differs from current literature in that his splenic abscess was truly idiopathic rather than resulting from an alternative source.
Complications of splenic abscesses include pleural effusions, as seen with this patient, as well as pneumonia, pneumothorax, hemorrhage, subphrenic abscess, and intraabdominal perforation, among others.2 We determined conclusively that the patient’s pleural effusion was secondary to the splenic abscess, and excluded other bacterial foci strongly suggests that the spleen was the origin of the illness.
CONCLUSIONS
This case suggests splenic abscesses should be considered when evaluating pleural effusion. It further demonstrates that the spleen may be the central source of infection in the absence of iatrogenic inoculation or bacteremia. We hope our findings may lead to earlier identification in similar scenarios and improved patient outcomes in a multidisciplinary approach.
References
Lee WS, Choi ST, Kim KK. Splenic abscess: a single institution study and review of the literature. Yonsei Med J. 2011;52(2):288-292. doi:10.3349/ymj.2011.52.2.288
Lotfollahzadeh S, Mathew G, Zemaitis MR. Splenic Abscess. In: StatPearls. StatPearls Publishing; June 3, 2023.
Jany B, Welte T. Pleural effusion in adults-etiology, diagnosis, and treatment. Dtsch Arztebl Int. 2019;116(21):377- 386. doi:10.3238/arztebl.2019.0377
Light RW. Pleural effusions. Med Clin North Am. 2011;95(6):1055-1070. doi:10.1016/j.mcna.2011.08.005
Feller-Kopman D, Light R. Pleural Disease. N Engl J Med. 2018;378(18):1754. doi:10.1056/NEJMc1803858
Ferreiro L, Casal A, Toubes ME, et al. Pleural effusion due to nonmalignant gastrointestinal disease. ERJ Open Res. 2023;9(3):00290-2022. doi:10.1183/23120541.00290-2022
Toma A, Fenaux P, Dreyfus F, Cordonnier C. Infections in myelodysplastic syndromes. Haematologica. 2012;97(10):1459- 1470. doi:10.3324/haematol2012.063420
References
Lee WS, Choi ST, Kim KK. Splenic abscess: a single institution study and review of the literature. Yonsei Med J. 2011;52(2):288-292. doi:10.3349/ymj.2011.52.2.288
Lotfollahzadeh S, Mathew G, Zemaitis MR. Splenic Abscess. In: StatPearls. StatPearls Publishing; June 3, 2023.
Jany B, Welte T. Pleural effusion in adults-etiology, diagnosis, and treatment. Dtsch Arztebl Int. 2019;116(21):377- 386. doi:10.3238/arztebl.2019.0377
Light RW. Pleural effusions. Med Clin North Am. 2011;95(6):1055-1070. doi:10.1016/j.mcna.2011.08.005
Feller-Kopman D, Light R. Pleural Disease. N Engl J Med. 2018;378(18):1754. doi:10.1056/NEJMc1803858
Ferreiro L, Casal A, Toubes ME, et al. Pleural effusion due to nonmalignant gastrointestinal disease. ERJ Open Res. 2023;9(3):00290-2022. doi:10.1183/23120541.00290-2022
Bevacizumab, an anti-vascular endothelial growth factor monoclonal antibody, is known to inhibit angiogenesis and prevent carcinogenesis. Recent evidence from the IMbrave050 trial indicates that combining bevacizumab with atezolizumab enhances recurrence-free survival (RFS) in high-risk HCC patients undergoing curative treatments. Bevacizumab is notorious for causing endothelial dysfunction that may provoke vasospasm, leading to central hypoperfusion, hypertension, and, albeit rarely, PRES. Similarly, immunotherapy, including atezolizumab, has been implicated in PRES, underscoring a potential risk when these therapies are administered concurrently.
Case Presentation
A 64-year-old woman with a history of hepatitis C and alcoholic cirrhosis was diagnosed with stage II (T2 N0 M0) HCC. Following partial hepatectomy, we proceeded with adjuvant systemic therapy with atezolizumab and bevacizumab (per the IMbrave050 trial). After her 2nd treatment, she developed altered mental status, seizures, and severe hypertension. Labs revealed acute kidney injury and elevated creatinine kinase levels suggesting rhabdomyolysis. Computed tomography head showed no acute findings, but magnetic resonance imaging of the brain identified increased flair attenuated inversion recovery (FLAIR) signal in the brain’s posterior regions, indicating PRES. Symptomatic management with anti-hypertensives and intravenous fluids led to the recovery of mental status to baseline. Further therapy with bevacizumab and atezolizumab was then held off.
Discussion
Therapeutic advances in HCC management through the IMbrave050 trial demonstrate the efficacy of bevacizumab and atezolizumab in reducing RFS, without highlighting the serious side effects like PRES. To our knowledge, this is the first case reported where PRES occurred with the simultaneous use of atezolizumab and bevacizumab. Since both drugs can individually cause PRES, there might be a heightened risk with the co-administration, signaling a critical need for vigilant monitoring and further research into this treatment modality’s long-term safety profile.
Bevacizumab, an anti-vascular endothelial growth factor monoclonal antibody, is known to inhibit angiogenesis and prevent carcinogenesis. Recent evidence from the IMbrave050 trial indicates that combining bevacizumab with atezolizumab enhances recurrence-free survival (RFS) in high-risk HCC patients undergoing curative treatments. Bevacizumab is notorious for causing endothelial dysfunction that may provoke vasospasm, leading to central hypoperfusion, hypertension, and, albeit rarely, PRES. Similarly, immunotherapy, including atezolizumab, has been implicated in PRES, underscoring a potential risk when these therapies are administered concurrently.
Case Presentation
A 64-year-old woman with a history of hepatitis C and alcoholic cirrhosis was diagnosed with stage II (T2 N0 M0) HCC. Following partial hepatectomy, we proceeded with adjuvant systemic therapy with atezolizumab and bevacizumab (per the IMbrave050 trial). After her 2nd treatment, she developed altered mental status, seizures, and severe hypertension. Labs revealed acute kidney injury and elevated creatinine kinase levels suggesting rhabdomyolysis. Computed tomography head showed no acute findings, but magnetic resonance imaging of the brain identified increased flair attenuated inversion recovery (FLAIR) signal in the brain’s posterior regions, indicating PRES. Symptomatic management with anti-hypertensives and intravenous fluids led to the recovery of mental status to baseline. Further therapy with bevacizumab and atezolizumab was then held off.
Discussion
Therapeutic advances in HCC management through the IMbrave050 trial demonstrate the efficacy of bevacizumab and atezolizumab in reducing RFS, without highlighting the serious side effects like PRES. To our knowledge, this is the first case reported where PRES occurred with the simultaneous use of atezolizumab and bevacizumab. Since both drugs can individually cause PRES, there might be a heightened risk with the co-administration, signaling a critical need for vigilant monitoring and further research into this treatment modality’s long-term safety profile.
Background
Bevacizumab, an anti-vascular endothelial growth factor monoclonal antibody, is known to inhibit angiogenesis and prevent carcinogenesis. Recent evidence from the IMbrave050 trial indicates that combining bevacizumab with atezolizumab enhances recurrence-free survival (RFS) in high-risk HCC patients undergoing curative treatments. Bevacizumab is notorious for causing endothelial dysfunction that may provoke vasospasm, leading to central hypoperfusion, hypertension, and, albeit rarely, PRES. Similarly, immunotherapy, including atezolizumab, has been implicated in PRES, underscoring a potential risk when these therapies are administered concurrently.
Case Presentation
A 64-year-old woman with a history of hepatitis C and alcoholic cirrhosis was diagnosed with stage II (T2 N0 M0) HCC. Following partial hepatectomy, we proceeded with adjuvant systemic therapy with atezolizumab and bevacizumab (per the IMbrave050 trial). After her 2nd treatment, she developed altered mental status, seizures, and severe hypertension. Labs revealed acute kidney injury and elevated creatinine kinase levels suggesting rhabdomyolysis. Computed tomography head showed no acute findings, but magnetic resonance imaging of the brain identified increased flair attenuated inversion recovery (FLAIR) signal in the brain’s posterior regions, indicating PRES. Symptomatic management with anti-hypertensives and intravenous fluids led to the recovery of mental status to baseline. Further therapy with bevacizumab and atezolizumab was then held off.
Discussion
Therapeutic advances in HCC management through the IMbrave050 trial demonstrate the efficacy of bevacizumab and atezolizumab in reducing RFS, without highlighting the serious side effects like PRES. To our knowledge, this is the first case reported where PRES occurred with the simultaneous use of atezolizumab and bevacizumab. Since both drugs can individually cause PRES, there might be a heightened risk with the co-administration, signaling a critical need for vigilant monitoring and further research into this treatment modality’s long-term safety profile.
Goblet cell adenocarcinoma (GCA), also known as goblet cell carcinoid, is a rare and distinct type of cancer originating from the appendix. It is characterized by cells that exhibit both mucinous and neuroendocrine differentiation, presenting a more aggressive nature compared to conventional carcinoids and a higher propensity for metastasis.
Case Presentation
A 60-year-old male presented with complaints of abdominal pain, nausea, vomiting, constipation, and weight loss worsening in the last month. He had a history of heavy alcohol intake, smoking, and family history of colon cancer in his grandfather. Initial workup with abdominal CT revealed findings suggestive of early bowel obstruction and possible malignancy. Subsequent EGD showed esophagitis, and colonoscopy identified a cecal mass. Biopsies confirmed malignant cells of enteric type with goblet cell features. Staging CT during hospitalization did not reveal distant metastasis initially. However, diagnostic laparoscopy later identified widespread peritoneal carcinomatosis, precluding surgical intervention. The case was discussed in tumor boards, leading to the initiation of palliative FOLFOX + Bevacizumab chemotherapy. After completing 7 cycles, restaging imaging showed stable disease. Subsequently, the patient experienced worsening obstructive symptoms with CT abdomen and pelvis demonstrating disease progression. Given his condition, decompressive gastrostomy was not feasible. The patient decided to transition to comfort measures only.
Discussion
Goblet cell adenocarcinoma is a rare appendiceal tumor with amphicrine differentiation, occurring at a rate of 0.01–0.05 per 100,000 individuals annually and comprising approximately 15% of all appendiceal neoplasms. These tumors often disseminate within the peritoneum, contributing to their aggressive behavior and challenging management.
Conclusions
Metastatic goblet cell adenocarcinoma presents significant treatment challenges and is associated with a poor prognosis. Tailored treatment strategies, vigilant monitoring, and ongoing research efforts are essential for optimizing patient outcomes and enhancing quality of life in this aggressive cancer
Goblet cell adenocarcinoma (GCA), also known as goblet cell carcinoid, is a rare and distinct type of cancer originating from the appendix. It is characterized by cells that exhibit both mucinous and neuroendocrine differentiation, presenting a more aggressive nature compared to conventional carcinoids and a higher propensity for metastasis.
Case Presentation
A 60-year-old male presented with complaints of abdominal pain, nausea, vomiting, constipation, and weight loss worsening in the last month. He had a history of heavy alcohol intake, smoking, and family history of colon cancer in his grandfather. Initial workup with abdominal CT revealed findings suggestive of early bowel obstruction and possible malignancy. Subsequent EGD showed esophagitis, and colonoscopy identified a cecal mass. Biopsies confirmed malignant cells of enteric type with goblet cell features. Staging CT during hospitalization did not reveal distant metastasis initially. However, diagnostic laparoscopy later identified widespread peritoneal carcinomatosis, precluding surgical intervention. The case was discussed in tumor boards, leading to the initiation of palliative FOLFOX + Bevacizumab chemotherapy. After completing 7 cycles, restaging imaging showed stable disease. Subsequently, the patient experienced worsening obstructive symptoms with CT abdomen and pelvis demonstrating disease progression. Given his condition, decompressive gastrostomy was not feasible. The patient decided to transition to comfort measures only.
Discussion
Goblet cell adenocarcinoma is a rare appendiceal tumor with amphicrine differentiation, occurring at a rate of 0.01–0.05 per 100,000 individuals annually and comprising approximately 15% of all appendiceal neoplasms. These tumors often disseminate within the peritoneum, contributing to their aggressive behavior and challenging management.
Conclusions
Metastatic goblet cell adenocarcinoma presents significant treatment challenges and is associated with a poor prognosis. Tailored treatment strategies, vigilant monitoring, and ongoing research efforts are essential for optimizing patient outcomes and enhancing quality of life in this aggressive cancer
Background
Goblet cell adenocarcinoma (GCA), also known as goblet cell carcinoid, is a rare and distinct type of cancer originating from the appendix. It is characterized by cells that exhibit both mucinous and neuroendocrine differentiation, presenting a more aggressive nature compared to conventional carcinoids and a higher propensity for metastasis.
Case Presentation
A 60-year-old male presented with complaints of abdominal pain, nausea, vomiting, constipation, and weight loss worsening in the last month. He had a history of heavy alcohol intake, smoking, and family history of colon cancer in his grandfather. Initial workup with abdominal CT revealed findings suggestive of early bowel obstruction and possible malignancy. Subsequent EGD showed esophagitis, and colonoscopy identified a cecal mass. Biopsies confirmed malignant cells of enteric type with goblet cell features. Staging CT during hospitalization did not reveal distant metastasis initially. However, diagnostic laparoscopy later identified widespread peritoneal carcinomatosis, precluding surgical intervention. The case was discussed in tumor boards, leading to the initiation of palliative FOLFOX + Bevacizumab chemotherapy. After completing 7 cycles, restaging imaging showed stable disease. Subsequently, the patient experienced worsening obstructive symptoms with CT abdomen and pelvis demonstrating disease progression. Given his condition, decompressive gastrostomy was not feasible. The patient decided to transition to comfort measures only.
Discussion
Goblet cell adenocarcinoma is a rare appendiceal tumor with amphicrine differentiation, occurring at a rate of 0.01–0.05 per 100,000 individuals annually and comprising approximately 15% of all appendiceal neoplasms. These tumors often disseminate within the peritoneum, contributing to their aggressive behavior and challenging management.
Conclusions
Metastatic goblet cell adenocarcinoma presents significant treatment challenges and is associated with a poor prognosis. Tailored treatment strategies, vigilant monitoring, and ongoing research efforts are essential for optimizing patient outcomes and enhancing quality of life in this aggressive cancer
Cholangiocarcinoma (CCA) is a rare and aggressive cancer of the biliary system, accounting for 15% of primary liver cancers. Most CCAs arise spontaneously, with risk factors including primary biliary cirrhosis, liver fluke infection, and biliary malformations. A newly described variant, Inhibin-positive Cholangioblastic (solid-tubulocystic) intrahepatic cholangiocarcinoma (iCCA), mimics neuroendocrine tumors (NET). This report presents a case of this new variant.
Case Presentation
A 53-year-old female with a history of alcohol use disorder and no family history of liver cancer presented with watery diarrhea for a month. Blood tests, including tumor markers, were normal. An ultrasound revealed a large mass in the right hepatic lobe. CT and MRI scans suggested a hemangioma. Due to the mass’s size and spontaneous bleeding risk, she underwent surgical resection. The mass was initially thought to be a hemangioma but was later identified as poorly differentiated intrahepatic CCA with a solid and tubulocystic structure. Pathology showed strong staining for Cytokeratin (CK) 7, CK-19, and Inhibin, and weak staining for synaptophysin, confirming a diagnosis of cholangioblastic iCCA. Genetic testing revealed no actionable variations. She was started on capecitabine for 8 cycles. Follow-up imaging showed no disease recurrence or metastasis.
Discussion
CCA often presents at advanced stages with symptoms like weight loss and jaundice. Diagnosis involves clinical assessment, lab work, and imaging, particularly MRI. Cholangioblastic Intrahepatic CCA (iCCA) is a newly described variant of cholangiocarcinoma. There have been 16 reported cases of the disease. Initially, it was thought to be a NET as it expressed Chromogranin, insulinoma-associated protein-1, and Synaptophysin. Almost half of the reported cases were diagnosed as NET initially. One tool clinicians can use to differentiate them is inhibin. Inhibin has been documented in all of the reported cases of Cholangioblastic iCCA. A novel inhibin-positive cholangioblastic iCCA variant with a Nipped-B-like protein and nucleus accumbens associated-1 (NIPBL-NACC1) fusion transcript has been reported recently, further helping differentiate the two. There is no standard of therapy for this variant. It’s managed similarly to CCAs, relying on surgical resection as the primary treatment. Limited data shows varied responses to neoadjuvant and adjuvant therapy.
Cholangiocarcinoma (CCA) is a rare and aggressive cancer of the biliary system, accounting for 15% of primary liver cancers. Most CCAs arise spontaneously, with risk factors including primary biliary cirrhosis, liver fluke infection, and biliary malformations. A newly described variant, Inhibin-positive Cholangioblastic (solid-tubulocystic) intrahepatic cholangiocarcinoma (iCCA), mimics neuroendocrine tumors (NET). This report presents a case of this new variant.
Case Presentation
A 53-year-old female with a history of alcohol use disorder and no family history of liver cancer presented with watery diarrhea for a month. Blood tests, including tumor markers, were normal. An ultrasound revealed a large mass in the right hepatic lobe. CT and MRI scans suggested a hemangioma. Due to the mass’s size and spontaneous bleeding risk, she underwent surgical resection. The mass was initially thought to be a hemangioma but was later identified as poorly differentiated intrahepatic CCA with a solid and tubulocystic structure. Pathology showed strong staining for Cytokeratin (CK) 7, CK-19, and Inhibin, and weak staining for synaptophysin, confirming a diagnosis of cholangioblastic iCCA. Genetic testing revealed no actionable variations. She was started on capecitabine for 8 cycles. Follow-up imaging showed no disease recurrence or metastasis.
Discussion
CCA often presents at advanced stages with symptoms like weight loss and jaundice. Diagnosis involves clinical assessment, lab work, and imaging, particularly MRI. Cholangioblastic Intrahepatic CCA (iCCA) is a newly described variant of cholangiocarcinoma. There have been 16 reported cases of the disease. Initially, it was thought to be a NET as it expressed Chromogranin, insulinoma-associated protein-1, and Synaptophysin. Almost half of the reported cases were diagnosed as NET initially. One tool clinicians can use to differentiate them is inhibin. Inhibin has been documented in all of the reported cases of Cholangioblastic iCCA. A novel inhibin-positive cholangioblastic iCCA variant with a Nipped-B-like protein and nucleus accumbens associated-1 (NIPBL-NACC1) fusion transcript has been reported recently, further helping differentiate the two. There is no standard of therapy for this variant. It’s managed similarly to CCAs, relying on surgical resection as the primary treatment. Limited data shows varied responses to neoadjuvant and adjuvant therapy.
Background
Cholangiocarcinoma (CCA) is a rare and aggressive cancer of the biliary system, accounting for 15% of primary liver cancers. Most CCAs arise spontaneously, with risk factors including primary biliary cirrhosis, liver fluke infection, and biliary malformations. A newly described variant, Inhibin-positive Cholangioblastic (solid-tubulocystic) intrahepatic cholangiocarcinoma (iCCA), mimics neuroendocrine tumors (NET). This report presents a case of this new variant.
Case Presentation
A 53-year-old female with a history of alcohol use disorder and no family history of liver cancer presented with watery diarrhea for a month. Blood tests, including tumor markers, were normal. An ultrasound revealed a large mass in the right hepatic lobe. CT and MRI scans suggested a hemangioma. Due to the mass’s size and spontaneous bleeding risk, she underwent surgical resection. The mass was initially thought to be a hemangioma but was later identified as poorly differentiated intrahepatic CCA with a solid and tubulocystic structure. Pathology showed strong staining for Cytokeratin (CK) 7, CK-19, and Inhibin, and weak staining for synaptophysin, confirming a diagnosis of cholangioblastic iCCA. Genetic testing revealed no actionable variations. She was started on capecitabine for 8 cycles. Follow-up imaging showed no disease recurrence or metastasis.
Discussion
CCA often presents at advanced stages with symptoms like weight loss and jaundice. Diagnosis involves clinical assessment, lab work, and imaging, particularly MRI. Cholangioblastic Intrahepatic CCA (iCCA) is a newly described variant of cholangiocarcinoma. There have been 16 reported cases of the disease. Initially, it was thought to be a NET as it expressed Chromogranin, insulinoma-associated protein-1, and Synaptophysin. Almost half of the reported cases were diagnosed as NET initially. One tool clinicians can use to differentiate them is inhibin. Inhibin has been documented in all of the reported cases of Cholangioblastic iCCA. A novel inhibin-positive cholangioblastic iCCA variant with a Nipped-B-like protein and nucleus accumbens associated-1 (NIPBL-NACC1) fusion transcript has been reported recently, further helping differentiate the two. There is no standard of therapy for this variant. It’s managed similarly to CCAs, relying on surgical resection as the primary treatment. Limited data shows varied responses to neoadjuvant and adjuvant therapy.
Ampulla of Vater is an extremely rare site for neuroendocrine tumors (NET), accounting for less than 0.3% of gastrointestinal (GI) and 2% of ampullary malignancies. This case report highlights the circuitous diagnosis of this rare tumor in a patient with a history of primary biliary cholangitis presenting with epigastric pain and severe pruritis.
Case Presentation
A 58-year-old female with history of sarcoidosis and primary biliary cholangitis status post sphincterotomy eight months prior, presented with worsening epigastric pain, fatigue, and weight loss over 6 months. Physical exam showed right upper quadrant tenderness. Labs revealed elevated alanine and aspartate aminotransferases at 415 and 195 units/L, with bilirubin of 0.3 mg/dl. Computerized tomography (CT) revealed a 2.3x3.2x4.0 cm peripancreatic hypodensity associated with phlegmon, pancreatic ductal dilation and pneumobilia. Magnetic resonance imaging (MRI) demonstrated a pancreatic head mass. Positron emission tomogram (PET) was negative for distant metastases. After discussion of management options, patient opted for Whipple procedure. The surgical pathology was consistent with invasive ampullary ductal carcinoma of the small intestine, pancreaticobiliary type. However, staining for synaptophysin and chromogranin were positive, with Ki-67 < 55%. Tumor board review confirmed neuroendocrine tumor of the ampulla of Vater. NCCN guidelines recommended active surveillance due to locoregional disease without positive margins or lymph nodes, advising routine follow-up and imaging.
Discussion
Neuroendocrine tumors (NET) at the Ampulla of Vater are exceedingly rare. Often manifesting as obstructive jaundice, they pose diagnostic hurdles, especially in patients with anatomical variations like scarring from primary biliary cholangitis. In a case series of 20 ampullary tumors, only one was neuroendocrine, highlighting their rarity. Accurate diagnosis, achieved through surgical biopsy and immunohistochemical testing, is crucial for appropriate management. Following NCCN guidelines for gastrointestinal NETs, our patient avoided unnecessary systemic treatment meant for adenocarcinoma, preserving her quality of life. Reporting such cases is essential for advancing understanding and refining patient care.
Conclusions
This case had evolving diagnoses, altering both the prognosis and treatment standards. Comorbid primary biliary cholangitis and high-grade tumor complexity posed diagnostic challenges, which was finally confirmed by surgical biopsy. Reporting such cases is vital in aiding tumor management and patient outcomes.
Ampulla of Vater is an extremely rare site for neuroendocrine tumors (NET), accounting for less than 0.3% of gastrointestinal (GI) and 2% of ampullary malignancies. This case report highlights the circuitous diagnosis of this rare tumor in a patient with a history of primary biliary cholangitis presenting with epigastric pain and severe pruritis.
Case Presentation
A 58-year-old female with history of sarcoidosis and primary biliary cholangitis status post sphincterotomy eight months prior, presented with worsening epigastric pain, fatigue, and weight loss over 6 months. Physical exam showed right upper quadrant tenderness. Labs revealed elevated alanine and aspartate aminotransferases at 415 and 195 units/L, with bilirubin of 0.3 mg/dl. Computerized tomography (CT) revealed a 2.3x3.2x4.0 cm peripancreatic hypodensity associated with phlegmon, pancreatic ductal dilation and pneumobilia. Magnetic resonance imaging (MRI) demonstrated a pancreatic head mass. Positron emission tomogram (PET) was negative for distant metastases. After discussion of management options, patient opted for Whipple procedure. The surgical pathology was consistent with invasive ampullary ductal carcinoma of the small intestine, pancreaticobiliary type. However, staining for synaptophysin and chromogranin were positive, with Ki-67 < 55%. Tumor board review confirmed neuroendocrine tumor of the ampulla of Vater. NCCN guidelines recommended active surveillance due to locoregional disease without positive margins or lymph nodes, advising routine follow-up and imaging.
Discussion
Neuroendocrine tumors (NET) at the Ampulla of Vater are exceedingly rare. Often manifesting as obstructive jaundice, they pose diagnostic hurdles, especially in patients with anatomical variations like scarring from primary biliary cholangitis. In a case series of 20 ampullary tumors, only one was neuroendocrine, highlighting their rarity. Accurate diagnosis, achieved through surgical biopsy and immunohistochemical testing, is crucial for appropriate management. Following NCCN guidelines for gastrointestinal NETs, our patient avoided unnecessary systemic treatment meant for adenocarcinoma, preserving her quality of life. Reporting such cases is essential for advancing understanding and refining patient care.
Conclusions
This case had evolving diagnoses, altering both the prognosis and treatment standards. Comorbid primary biliary cholangitis and high-grade tumor complexity posed diagnostic challenges, which was finally confirmed by surgical biopsy. Reporting such cases is vital in aiding tumor management and patient outcomes.
Background
Ampulla of Vater is an extremely rare site for neuroendocrine tumors (NET), accounting for less than 0.3% of gastrointestinal (GI) and 2% of ampullary malignancies. This case report highlights the circuitous diagnosis of this rare tumor in a patient with a history of primary biliary cholangitis presenting with epigastric pain and severe pruritis.
Case Presentation
A 58-year-old female with history of sarcoidosis and primary biliary cholangitis status post sphincterotomy eight months prior, presented with worsening epigastric pain, fatigue, and weight loss over 6 months. Physical exam showed right upper quadrant tenderness. Labs revealed elevated alanine and aspartate aminotransferases at 415 and 195 units/L, with bilirubin of 0.3 mg/dl. Computerized tomography (CT) revealed a 2.3x3.2x4.0 cm peripancreatic hypodensity associated with phlegmon, pancreatic ductal dilation and pneumobilia. Magnetic resonance imaging (MRI) demonstrated a pancreatic head mass. Positron emission tomogram (PET) was negative for distant metastases. After discussion of management options, patient opted for Whipple procedure. The surgical pathology was consistent with invasive ampullary ductal carcinoma of the small intestine, pancreaticobiliary type. However, staining for synaptophysin and chromogranin were positive, with Ki-67 < 55%. Tumor board review confirmed neuroendocrine tumor of the ampulla of Vater. NCCN guidelines recommended active surveillance due to locoregional disease without positive margins or lymph nodes, advising routine follow-up and imaging.
Discussion
Neuroendocrine tumors (NET) at the Ampulla of Vater are exceedingly rare. Often manifesting as obstructive jaundice, they pose diagnostic hurdles, especially in patients with anatomical variations like scarring from primary biliary cholangitis. In a case series of 20 ampullary tumors, only one was neuroendocrine, highlighting their rarity. Accurate diagnosis, achieved through surgical biopsy and immunohistochemical testing, is crucial for appropriate management. Following NCCN guidelines for gastrointestinal NETs, our patient avoided unnecessary systemic treatment meant for adenocarcinoma, preserving her quality of life. Reporting such cases is essential for advancing understanding and refining patient care.
Conclusions
This case had evolving diagnoses, altering both the prognosis and treatment standards. Comorbid primary biliary cholangitis and high-grade tumor complexity posed diagnostic challenges, which was finally confirmed by surgical biopsy. Reporting such cases is vital in aiding tumor management and patient outcomes.
CML is usually classified in chronic phase (CP), accelerated phase (AP) and/or Blast phase (BP). Studies have described another provisional entity as Preleukemic phase of CML which precedes CP and is without leukocytosis and blood features of CP phase of CML. Here we will present a case of metastatic prostate cancer, where next-generation sequencing showed BCR-ABL1 but no overt leukocytosis and then later developed clinical CML after 11 months.
Case Presentation
Our patient was 87-yr-old male with history of metastatic prostate cancer, who presented with elevated PSA levels and new bony metastasis. He underwent next-generation-sequencing (foundation one liquid cdx) in April 2022. It did not show reportable alterations related to prostate cancer but BCR-ABL1 (p210) fusion was detected. It also showed ASXL1-S846fs5 and TET2-Q1548 that are also markers of clonal hematopoiesis. In March 2023 (11 months after the finding of BCR-ABL1), he developed asymptomatic leukocytosis, Workup showed BCR-ABL1(210) of 44% in peripheral blood and bone marrow showed 9% blasts. He was started on Imatinib after shared decision-making and considering the toxicity profile and comorbidities. He followed up regularly with improvement in leukocytosis.
Discussion
The diagnosis of CML is first suspected by typical findings in blood and/or bone marrow and then confirmed by presence of Philadelphia chromosome. Along with chronic and accelerated phases, there is another term described in few cases called “preleukemic or smoldering or aleukemic phase.” This is not a wellestablished term but mostly defined as normal leukocyte count with presence of BCR/ABL1 fusion gene/ ph chromosome. Preleukemic phase of CML is mostly underdiagnosed or misdiagnosed. Our case is unique in that BCR/ABL1 fusion was detected incidentally on next-generation sequencing and patient progressed to chronic phase of CML 11 months later. Upon literature review, few case reports and case series documenting aleukemic/preleukemic phase of CML but timing from the appearance of BCR/ABL1 mutation to actual development to leukocytosis is not well documented. Especially in the era of NGS testing, patients with incidental BCR-ABL1 should be evaluated further irrespective of normal WBC. Further studies need to be done to recognize this early and decrease the delay in treatment.
CML is usually classified in chronic phase (CP), accelerated phase (AP) and/or Blast phase (BP). Studies have described another provisional entity as Preleukemic phase of CML which precedes CP and is without leukocytosis and blood features of CP phase of CML. Here we will present a case of metastatic prostate cancer, where next-generation sequencing showed BCR-ABL1 but no overt leukocytosis and then later developed clinical CML after 11 months.
Case Presentation
Our patient was 87-yr-old male with history of metastatic prostate cancer, who presented with elevated PSA levels and new bony metastasis. He underwent next-generation-sequencing (foundation one liquid cdx) in April 2022. It did not show reportable alterations related to prostate cancer but BCR-ABL1 (p210) fusion was detected. It also showed ASXL1-S846fs5 and TET2-Q1548 that are also markers of clonal hematopoiesis. In March 2023 (11 months after the finding of BCR-ABL1), he developed asymptomatic leukocytosis, Workup showed BCR-ABL1(210) of 44% in peripheral blood and bone marrow showed 9% blasts. He was started on Imatinib after shared decision-making and considering the toxicity profile and comorbidities. He followed up regularly with improvement in leukocytosis.
Discussion
The diagnosis of CML is first suspected by typical findings in blood and/or bone marrow and then confirmed by presence of Philadelphia chromosome. Along with chronic and accelerated phases, there is another term described in few cases called “preleukemic or smoldering or aleukemic phase.” This is not a wellestablished term but mostly defined as normal leukocyte count with presence of BCR/ABL1 fusion gene/ ph chromosome. Preleukemic phase of CML is mostly underdiagnosed or misdiagnosed. Our case is unique in that BCR/ABL1 fusion was detected incidentally on next-generation sequencing and patient progressed to chronic phase of CML 11 months later. Upon literature review, few case reports and case series documenting aleukemic/preleukemic phase of CML but timing from the appearance of BCR/ABL1 mutation to actual development to leukocytosis is not well documented. Especially in the era of NGS testing, patients with incidental BCR-ABL1 should be evaluated further irrespective of normal WBC. Further studies need to be done to recognize this early and decrease the delay in treatment.
Background
CML is usually classified in chronic phase (CP), accelerated phase (AP) and/or Blast phase (BP). Studies have described another provisional entity as Preleukemic phase of CML which precedes CP and is without leukocytosis and blood features of CP phase of CML. Here we will present a case of metastatic prostate cancer, where next-generation sequencing showed BCR-ABL1 but no overt leukocytosis and then later developed clinical CML after 11 months.
Case Presentation
Our patient was 87-yr-old male with history of metastatic prostate cancer, who presented with elevated PSA levels and new bony metastasis. He underwent next-generation-sequencing (foundation one liquid cdx) in April 2022. It did not show reportable alterations related to prostate cancer but BCR-ABL1 (p210) fusion was detected. It also showed ASXL1-S846fs5 and TET2-Q1548 that are also markers of clonal hematopoiesis. In March 2023 (11 months after the finding of BCR-ABL1), he developed asymptomatic leukocytosis, Workup showed BCR-ABL1(210) of 44% in peripheral blood and bone marrow showed 9% blasts. He was started on Imatinib after shared decision-making and considering the toxicity profile and comorbidities. He followed up regularly with improvement in leukocytosis.
Discussion
The diagnosis of CML is first suspected by typical findings in blood and/or bone marrow and then confirmed by presence of Philadelphia chromosome. Along with chronic and accelerated phases, there is another term described in few cases called “preleukemic or smoldering or aleukemic phase.” This is not a wellestablished term but mostly defined as normal leukocyte count with presence of BCR/ABL1 fusion gene/ ph chromosome. Preleukemic phase of CML is mostly underdiagnosed or misdiagnosed. Our case is unique in that BCR/ABL1 fusion was detected incidentally on next-generation sequencing and patient progressed to chronic phase of CML 11 months later. Upon literature review, few case reports and case series documenting aleukemic/preleukemic phase of CML but timing from the appearance of BCR/ABL1 mutation to actual development to leukocytosis is not well documented. Especially in the era of NGS testing, patients with incidental BCR-ABL1 should be evaluated further irrespective of normal WBC. Further studies need to be done to recognize this early and decrease the delay in treatment.
Monoclonal B cell lymphocytosis (MBL) is defined as presence of clonal b cell population that is fewer than 5 × 10(9)/L B-cells in peripheral blood and no other signs of a lymphoproliferative disorder. Patients with MBL are usually monitored with periodic history, physical exam and blood counts. Here we presented a case of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) in breast in a patient with a history of MBL.
Case Presentation
68-year-old male with history of MBL underwent mammogram for breast mass. It showed suspicious 4.4 x 1.6 cm solid and cystic lesion containing a 1.7 x 0.9 x 1.8 cm solid hypervascular mass. Patient underwent left breast mass excision. Histologic sections focus of ADH involving papilloma with uninvolved margins. Lymphoid infiltrates noted had CLL/SLL immunophenotype and that it consists mostly of small B cells positive for CD5, CD20, CD23, CD43, Bcl-2, LEF1. CT CAP and PET/CT were negative for lymphadenopathy. Bone marrow biopsy showed marrow involvement by mature B-cell lymphoproliferative process, immunophenotypically consistent with CLL/SLL. As intra-ductal papilloma completely excised and hemogram was normal tumor board recommended surveillance only for CLL/SLL.
Discussion
MBL can progress to CLL, but it can rarely be presented as an extra-nodal mass in solid organs. We described a case of MBL that progressed to CLL/ SLL in breast mass in a male patient. This is the first reported case in literature where MBL progressed to CLL/ SLL of breast without lymphadenopathy. Upon literature review 8 case reports were found where CLL/SLL were described in breast tissue. 7 of them were in females and 1 one was in male. Two patients had CLL before breast mass but none of them had a history of MBL. 3 described cases in females had CLL/SLL infiltration of breast along with invasive ductal carcinoma. So, a patient with MBL can progress to involve solid organs despite no absolute lymphocytosis and should be considered in differentials of a new mass. Although more common in females, but it can occur in males as well. It’s important to consider the possibility of both CLL/SLL and breast cancer existing simultaneously.
Monoclonal B cell lymphocytosis (MBL) is defined as presence of clonal b cell population that is fewer than 5 × 10(9)/L B-cells in peripheral blood and no other signs of a lymphoproliferative disorder. Patients with MBL are usually monitored with periodic history, physical exam and blood counts. Here we presented a case of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) in breast in a patient with a history of MBL.
Case Presentation
68-year-old male with history of MBL underwent mammogram for breast mass. It showed suspicious 4.4 x 1.6 cm solid and cystic lesion containing a 1.7 x 0.9 x 1.8 cm solid hypervascular mass. Patient underwent left breast mass excision. Histologic sections focus of ADH involving papilloma with uninvolved margins. Lymphoid infiltrates noted had CLL/SLL immunophenotype and that it consists mostly of small B cells positive for CD5, CD20, CD23, CD43, Bcl-2, LEF1. CT CAP and PET/CT were negative for lymphadenopathy. Bone marrow biopsy showed marrow involvement by mature B-cell lymphoproliferative process, immunophenotypically consistent with CLL/SLL. As intra-ductal papilloma completely excised and hemogram was normal tumor board recommended surveillance only for CLL/SLL.
Discussion
MBL can progress to CLL, but it can rarely be presented as an extra-nodal mass in solid organs. We described a case of MBL that progressed to CLL/ SLL in breast mass in a male patient. This is the first reported case in literature where MBL progressed to CLL/ SLL of breast without lymphadenopathy. Upon literature review 8 case reports were found where CLL/SLL were described in breast tissue. 7 of them were in females and 1 one was in male. Two patients had CLL before breast mass but none of them had a history of MBL. 3 described cases in females had CLL/SLL infiltration of breast along with invasive ductal carcinoma. So, a patient with MBL can progress to involve solid organs despite no absolute lymphocytosis and should be considered in differentials of a new mass. Although more common in females, but it can occur in males as well. It’s important to consider the possibility of both CLL/SLL and breast cancer existing simultaneously.
Background
Monoclonal B cell lymphocytosis (MBL) is defined as presence of clonal b cell population that is fewer than 5 × 10(9)/L B-cells in peripheral blood and no other signs of a lymphoproliferative disorder. Patients with MBL are usually monitored with periodic history, physical exam and blood counts. Here we presented a case of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) in breast in a patient with a history of MBL.
Case Presentation
68-year-old male with history of MBL underwent mammogram for breast mass. It showed suspicious 4.4 x 1.6 cm solid and cystic lesion containing a 1.7 x 0.9 x 1.8 cm solid hypervascular mass. Patient underwent left breast mass excision. Histologic sections focus of ADH involving papilloma with uninvolved margins. Lymphoid infiltrates noted had CLL/SLL immunophenotype and that it consists mostly of small B cells positive for CD5, CD20, CD23, CD43, Bcl-2, LEF1. CT CAP and PET/CT were negative for lymphadenopathy. Bone marrow biopsy showed marrow involvement by mature B-cell lymphoproliferative process, immunophenotypically consistent with CLL/SLL. As intra-ductal papilloma completely excised and hemogram was normal tumor board recommended surveillance only for CLL/SLL.
Discussion
MBL can progress to CLL, but it can rarely be presented as an extra-nodal mass in solid organs. We described a case of MBL that progressed to CLL/ SLL in breast mass in a male patient. This is the first reported case in literature where MBL progressed to CLL/ SLL of breast without lymphadenopathy. Upon literature review 8 case reports were found where CLL/SLL were described in breast tissue. 7 of them were in females and 1 one was in male. Two patients had CLL before breast mass but none of them had a history of MBL. 3 described cases in females had CLL/SLL infiltration of breast along with invasive ductal carcinoma. So, a patient with MBL can progress to involve solid organs despite no absolute lymphocytosis and should be considered in differentials of a new mass. Although more common in females, but it can occur in males as well. It’s important to consider the possibility of both CLL/SLL and breast cancer existing simultaneously.
Priapism, defined as a prolonged and often painful penile erection without sexual arousal, constitutes a urological emergency requiring immediate intervention. While commonly associated with conditions like sickle cell anemia and certain medications, malignancy-related priapism is rare and frequently overlooked. Herein, we present a unique case of a 31-year-old male with no significant medical history, who developed persistent priapism as the initial presentation of chronic myeloid leukemia (CML).
Case Presentation
A 31-year-old male without significant medical history, presented to the emergency department with painless priapism, was evaluated by urology and discharged home with precautions. He returned the following day with persistent, now painful priapism. Upon examination, his vital signs were stable. Urology performed aspiration and injection with Sudafed, resulting in mild symptom improvement. Laboratory findings revealed elevated white blood cell count (563.64 k/mcL), anemia (hemoglobin 8.4 g/dL), and a peripheral blood smear showed immature circulating cells with blast forms. He was transferred to a tertiary care center where conservative management addressed bleeding from the penile injection site, with subsequent treatment including leukapheresis and hydroxyurea for cytoreduction. Imaging revealed severe splenomegaly (36 cm) with abdominal mass effect. Peripheral flow cytometry didn’t show malignancy, but cytogenetic analysis showed a BCR/ABL1 fusion gene, confirming chronic myeloid leukemia (CML). Bone marrow biopsy showed hypercellularity without increased blasts. Treatment with dasatinib reduced the white count to 52,000 k/mcL, and was discharged home.
Discussion
Priapism is a urological emergency necessitating immediate intervention to prevent erectile dysfunction and permanent impotence. Management aims to achieve detumescence and typically involves methods such as irrigation or injection of vasoconstrictors into the penis. Malignancy-associated priapism (MAP) often results from venous obstruction due to hyperviscosity. Studies show that CML accounts for approximately 50% cases presenting with MAP, predominantly affecting younger individuals with a mean onset around 27 years of age. Priapism can occur before, during, or after treatment initiation or splenectomy in these patients. Providers should keep a high threshold of suspicion for MAP in patients with no other risk factors as prompt identification and treatment are needed to avoid permanent injury.
Priapism, defined as a prolonged and often painful penile erection without sexual arousal, constitutes a urological emergency requiring immediate intervention. While commonly associated with conditions like sickle cell anemia and certain medications, malignancy-related priapism is rare and frequently overlooked. Herein, we present a unique case of a 31-year-old male with no significant medical history, who developed persistent priapism as the initial presentation of chronic myeloid leukemia (CML).
Case Presentation
A 31-year-old male without significant medical history, presented to the emergency department with painless priapism, was evaluated by urology and discharged home with precautions. He returned the following day with persistent, now painful priapism. Upon examination, his vital signs were stable. Urology performed aspiration and injection with Sudafed, resulting in mild symptom improvement. Laboratory findings revealed elevated white blood cell count (563.64 k/mcL), anemia (hemoglobin 8.4 g/dL), and a peripheral blood smear showed immature circulating cells with blast forms. He was transferred to a tertiary care center where conservative management addressed bleeding from the penile injection site, with subsequent treatment including leukapheresis and hydroxyurea for cytoreduction. Imaging revealed severe splenomegaly (36 cm) with abdominal mass effect. Peripheral flow cytometry didn’t show malignancy, but cytogenetic analysis showed a BCR/ABL1 fusion gene, confirming chronic myeloid leukemia (CML). Bone marrow biopsy showed hypercellularity without increased blasts. Treatment with dasatinib reduced the white count to 52,000 k/mcL, and was discharged home.
Discussion
Priapism is a urological emergency necessitating immediate intervention to prevent erectile dysfunction and permanent impotence. Management aims to achieve detumescence and typically involves methods such as irrigation or injection of vasoconstrictors into the penis. Malignancy-associated priapism (MAP) often results from venous obstruction due to hyperviscosity. Studies show that CML accounts for approximately 50% cases presenting with MAP, predominantly affecting younger individuals with a mean onset around 27 years of age. Priapism can occur before, during, or after treatment initiation or splenectomy in these patients. Providers should keep a high threshold of suspicion for MAP in patients with no other risk factors as prompt identification and treatment are needed to avoid permanent injury.
Introduction
Priapism, defined as a prolonged and often painful penile erection without sexual arousal, constitutes a urological emergency requiring immediate intervention. While commonly associated with conditions like sickle cell anemia and certain medications, malignancy-related priapism is rare and frequently overlooked. Herein, we present a unique case of a 31-year-old male with no significant medical history, who developed persistent priapism as the initial presentation of chronic myeloid leukemia (CML).
Case Presentation
A 31-year-old male without significant medical history, presented to the emergency department with painless priapism, was evaluated by urology and discharged home with precautions. He returned the following day with persistent, now painful priapism. Upon examination, his vital signs were stable. Urology performed aspiration and injection with Sudafed, resulting in mild symptom improvement. Laboratory findings revealed elevated white blood cell count (563.64 k/mcL), anemia (hemoglobin 8.4 g/dL), and a peripheral blood smear showed immature circulating cells with blast forms. He was transferred to a tertiary care center where conservative management addressed bleeding from the penile injection site, with subsequent treatment including leukapheresis and hydroxyurea for cytoreduction. Imaging revealed severe splenomegaly (36 cm) with abdominal mass effect. Peripheral flow cytometry didn’t show malignancy, but cytogenetic analysis showed a BCR/ABL1 fusion gene, confirming chronic myeloid leukemia (CML). Bone marrow biopsy showed hypercellularity without increased blasts. Treatment with dasatinib reduced the white count to 52,000 k/mcL, and was discharged home.
Discussion
Priapism is a urological emergency necessitating immediate intervention to prevent erectile dysfunction and permanent impotence. Management aims to achieve detumescence and typically involves methods such as irrigation or injection of vasoconstrictors into the penis. Malignancy-associated priapism (MAP) often results from venous obstruction due to hyperviscosity. Studies show that CML accounts for approximately 50% cases presenting with MAP, predominantly affecting younger individuals with a mean onset around 27 years of age. Priapism can occur before, during, or after treatment initiation or splenectomy in these patients. Providers should keep a high threshold of suspicion for MAP in patients with no other risk factors as prompt identification and treatment are needed to avoid permanent injury.
Autoimmune hemolytic anemia (AIHA) can rarely be seen as an extra-intestinal manifestation (EIM) of inflammatory bowel disease (IBD), mostly ulcerative colitis (UC). This case report describes the clinical significance of recognizing AIHA in the context of UC.
Case Presentation
A 32-year-old male presented with profound fatigue, pallor, and dyspnea on exertion for one month. He also recalled intermittent bloody diarrhea for two years for which he never sought medical attention. Physical examination was unremarkable except for mid-abdominal tenderness. Labs revealed microcytosis, hemoglobin of 3.8 g/dL, total bilirubin 2.9 mg/ dL, indirect bilirubin of 2.0 mg/dL, LDH 132 U/L alk-p 459 U/L AST 98 U/L ALT 22 U/L. Direct Coombs test was positive suggesting warm AIHA with pan-agglutinin positive on the eluate test. Further testing revealed negative hepatitis and HIV panels and positive fecal calprotectin. CT abdomen and pelvis showed ascites, right pleural effusion and hepatosplenomegaly. Colonoscopy confirmed the diagnosis of ulcerative colitis, with extensive involvement of the colon. Mesalamine was initiated. Hematology was consulted for AIHA, who started the patient on methylprednisone leading to resolution of hemolytic anemia and improvement in gastrointestinal symptoms.
Discussion
IBD typically manifests as colitis, and the incidence of EIM as an initial symptom is observed in less than 10% cases. However, over the course of their lifetime, approximately 25% of patients will experience EIM, underscoring their relevance to clinical outcomes. Anemia is very common in IBD patients, mostly iron deficiency anemia (IDA) or anemia of chronic disease (ACD). However, AIHA can represent a rare but significant EIM of ulcerative colitis (UC), often posing diagnostic challenges. The underlying pathophysiological mechanisms linking UC and AIHA remain incompletely understood, necessitating a multidisciplinary approach to management. Treatment strategies focus on controlling both the hemolysis and the underlying IBD, emphasizing the importance of tailored interventions.
Conclusion
This case underscores the clinical significance of AIHA as an EIM of ulcerative colitis (UC), particularly when presenting as the primary symptom. Timely recognition is paramount to optimizing patient outcomes and preventing disease progression. Further research is warranted to elucidate the underlying mechanisms and therapeutic strategies for AIHA in the context of UC.
Autoimmune hemolytic anemia (AIHA) can rarely be seen as an extra-intestinal manifestation (EIM) of inflammatory bowel disease (IBD), mostly ulcerative colitis (UC). This case report describes the clinical significance of recognizing AIHA in the context of UC.
Case Presentation
A 32-year-old male presented with profound fatigue, pallor, and dyspnea on exertion for one month. He also recalled intermittent bloody diarrhea for two years for which he never sought medical attention. Physical examination was unremarkable except for mid-abdominal tenderness. Labs revealed microcytosis, hemoglobin of 3.8 g/dL, total bilirubin 2.9 mg/ dL, indirect bilirubin of 2.0 mg/dL, LDH 132 U/L alk-p 459 U/L AST 98 U/L ALT 22 U/L. Direct Coombs test was positive suggesting warm AIHA with pan-agglutinin positive on the eluate test. Further testing revealed negative hepatitis and HIV panels and positive fecal calprotectin. CT abdomen and pelvis showed ascites, right pleural effusion and hepatosplenomegaly. Colonoscopy confirmed the diagnosis of ulcerative colitis, with extensive involvement of the colon. Mesalamine was initiated. Hematology was consulted for AIHA, who started the patient on methylprednisone leading to resolution of hemolytic anemia and improvement in gastrointestinal symptoms.
Discussion
IBD typically manifests as colitis, and the incidence of EIM as an initial symptom is observed in less than 10% cases. However, over the course of their lifetime, approximately 25% of patients will experience EIM, underscoring their relevance to clinical outcomes. Anemia is very common in IBD patients, mostly iron deficiency anemia (IDA) or anemia of chronic disease (ACD). However, AIHA can represent a rare but significant EIM of ulcerative colitis (UC), often posing diagnostic challenges. The underlying pathophysiological mechanisms linking UC and AIHA remain incompletely understood, necessitating a multidisciplinary approach to management. Treatment strategies focus on controlling both the hemolysis and the underlying IBD, emphasizing the importance of tailored interventions.
Conclusion
This case underscores the clinical significance of AIHA as an EIM of ulcerative colitis (UC), particularly when presenting as the primary symptom. Timely recognition is paramount to optimizing patient outcomes and preventing disease progression. Further research is warranted to elucidate the underlying mechanisms and therapeutic strategies for AIHA in the context of UC.
Introduction
Autoimmune hemolytic anemia (AIHA) can rarely be seen as an extra-intestinal manifestation (EIM) of inflammatory bowel disease (IBD), mostly ulcerative colitis (UC). This case report describes the clinical significance of recognizing AIHA in the context of UC.
Case Presentation
A 32-year-old male presented with profound fatigue, pallor, and dyspnea on exertion for one month. He also recalled intermittent bloody diarrhea for two years for which he never sought medical attention. Physical examination was unremarkable except for mid-abdominal tenderness. Labs revealed microcytosis, hemoglobin of 3.8 g/dL, total bilirubin 2.9 mg/ dL, indirect bilirubin of 2.0 mg/dL, LDH 132 U/L alk-p 459 U/L AST 98 U/L ALT 22 U/L. Direct Coombs test was positive suggesting warm AIHA with pan-agglutinin positive on the eluate test. Further testing revealed negative hepatitis and HIV panels and positive fecal calprotectin. CT abdomen and pelvis showed ascites, right pleural effusion and hepatosplenomegaly. Colonoscopy confirmed the diagnosis of ulcerative colitis, with extensive involvement of the colon. Mesalamine was initiated. Hematology was consulted for AIHA, who started the patient on methylprednisone leading to resolution of hemolytic anemia and improvement in gastrointestinal symptoms.
Discussion
IBD typically manifests as colitis, and the incidence of EIM as an initial symptom is observed in less than 10% cases. However, over the course of their lifetime, approximately 25% of patients will experience EIM, underscoring their relevance to clinical outcomes. Anemia is very common in IBD patients, mostly iron deficiency anemia (IDA) or anemia of chronic disease (ACD). However, AIHA can represent a rare but significant EIM of ulcerative colitis (UC), often posing diagnostic challenges. The underlying pathophysiological mechanisms linking UC and AIHA remain incompletely understood, necessitating a multidisciplinary approach to management. Treatment strategies focus on controlling both the hemolysis and the underlying IBD, emphasizing the importance of tailored interventions.
Conclusion
This case underscores the clinical significance of AIHA as an EIM of ulcerative colitis (UC), particularly when presenting as the primary symptom. Timely recognition is paramount to optimizing patient outcomes and preventing disease progression. Further research is warranted to elucidate the underlying mechanisms and therapeutic strategies for AIHA in the context of UC.