User login
Cerdulatinib yields ‘encouraging’ results in CTCL, PTCL
LA JOLLA, CALIF. – The spleen tyrosine kinase/Janus kinase inhibitor cerdulatinib has demonstrated activity against relapsed and refractory T-cell lymphomas.

In a phase 2 trial, cerdulatinib produced responses in 34% of patients with peripheral T-cell lymphoma (PTCL) and 26% of those with cutaneous T-cell lymphoma (CTCL).
The best responders were patients with angioimmunoblastic T-cell lymphoma, half of whom achieved a complete response (CR).
The most common grade 3 or higher adverse events (AEs) were amylase increase and lipase increase. However, these increases resolved with dose reduction or interruption, and there were no cases of clinical pancreatitis.
“The data is very encouraging,” said Tatyana Feldman, MD, of the John Theurer Cancer Center in Hackensack, N.J.
Dr. Feldman and her colleagues previously presented results from the phase 2 trial of cerdulatinib (NCT01994382) at the 2018 annual congress of the European Hematology Association.
Dr. Feldman and her colleagues presented data from expansion cohorts of the ongoing trial at the annual T-cell Lymphoma Forum. The cohorts included patients with PTCL or CTCL who had received at least one prior systemic therapy.
PTCL cohort
The 45 PTCL patients had a median age of 65 years (range, 21-84). They had received a median of 3 (range, 1-12) prior therapeutic regimens, 51% were refractory to their last therapy, and 27% had undergone stem cell transplant (SCT).
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 41 patients were evaluable for response.
The overall response rate was 34% (n = 14). Eleven patients had a CR, three had a partial response (PR), and nine had stable disease.
Responses according to subtype were as follows:
- 7 CRs and 1 PR in angioimmunoblastic T-cell lymphoma.
- 2 CRs in PTCL not otherwise specified.
- 1 CR in gamma-delta T-cell lymphoma.
- 1 PR in ALK-negative anaplastic large-cell lymphoma.
- 1 CR and 1 PR in adult T-cell leukemia/lymphoma.
Eight responders have remained on cerdulatinib for anywhere from 3 months to more than 12 months. Five patients have had a response lasting at least 6 months. One patient went on to SCT after achieving a CR.
The most common grade 3 or higher AEs observed in PTCL patients were amylase increase (n = 8), lipase increase (n = 6), pneumonia/lung infection (n = 5), neutropenia (n = 4), diarrhea (n = 4), febrile neutropenia (n = 4), abdominal pain (n = 4), sepsis/bacteremia (n = 3), anemia (n = 3), fatigue (n = 2), and pain (n = 1).
There were two grade 5 AEs – acute respiratory distress syndrome and pneumonia.
CTCL cohort
The 29 CTCL patients had a median age of 62 years (range, 24-79). They had received a median of 4 (range, 1-13) prior therapies, 55% were refractory to their last therapy, and 3% had undergone SCT.
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 27 were evaluable for response.
The overall response rate was 26% (n = 7). Two patients achieved a CR, five achieved a PR, and nine had stable disease. Responses occurred in mycosis fungoides and Sézary syndrome.
Eleven of 23 patients (48%) achieved at least a 50% reduction in skin lesions, and the researchers observed rapid improvements in pruritus.
“I saw patients who would take the first pill, and they would call me and say, ‘I no longer itch,’ ” Dr. Feldman said.
The most common grade 3 or higher AEs in CTCL patients were lipase increase (n = 11), amylase increase (n = 5), sepsis/bacteremia (n = 3), pain (n = 2), fatigue (n = 1), neutropenia (n = 1), and diarrhea (n = 1).
“It’s a very well-tolerated drug,” Dr. Feldman said, adding that there were “really no severe side effects which would prohibit the use of the drug.”
She noted that cerdulatinib’s “favorable” side effect profile might make it a promising candidate for use in combination regimens.
“I think it will be possible to combine it with other drugs in development in T-cell lymphoma. … immunological checkpoint inhibitors, epigenetic modulators such as HDAC [histone deacetylase] inhibitors, methylating agents, and PI3 kinase inhibitors,” Dr. Feldman said.
She reported having no disclosures relevant to this study. The trial is sponsored by Portola Pharmaceuticals.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – The spleen tyrosine kinase/Janus kinase inhibitor cerdulatinib has demonstrated activity against relapsed and refractory T-cell lymphomas.
In a phase 2 trial, cerdulatinib produced responses in 34% of patients with peripheral T-cell lymphoma (PTCL) and 26% of those with cutaneous T-cell lymphoma (CTCL).
The best responders were patients with angioimmunoblastic T-cell lymphoma, half of whom achieved a complete response (CR).
The most common grade 3 or higher adverse events (AEs) were amylase increase and lipase increase. However, these increases resolved with dose reduction or interruption, and there were no cases of clinical pancreatitis.
“The data is very encouraging,” said Tatyana Feldman, MD, of the John Theurer Cancer Center in Hackensack, N.J.
Dr. Feldman and her colleagues previously presented results from the phase 2 trial of cerdulatinib (NCT01994382) at the 2018 annual congress of the European Hematology Association.
Dr. Feldman and her colleagues presented data from expansion cohorts of the ongoing trial at the annual T-cell Lymphoma Forum. The cohorts included patients with PTCL or CTCL who had received at least one prior systemic therapy.
PTCL cohort
The 45 PTCL patients had a median age of 65 years (range, 21-84). They had received a median of 3 (range, 1-12) prior therapeutic regimens, 51% were refractory to their last therapy, and 27% had undergone stem cell transplant (SCT).
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 41 patients were evaluable for response.
The overall response rate was 34% (n = 14). Eleven patients had a CR, three had a partial response (PR), and nine had stable disease.
Responses according to subtype were as follows:
- 7 CRs and 1 PR in angioimmunoblastic T-cell lymphoma.
- 2 CRs in PTCL not otherwise specified.
- 1 CR in gamma-delta T-cell lymphoma.
- 1 PR in ALK-negative anaplastic large-cell lymphoma.
- 1 CR and 1 PR in adult T-cell leukemia/lymphoma.
Eight responders have remained on cerdulatinib for anywhere from 3 months to more than 12 months. Five patients have had a response lasting at least 6 months. One patient went on to SCT after achieving a CR.
The most common grade 3 or higher AEs observed in PTCL patients were amylase increase (n = 8), lipase increase (n = 6), pneumonia/lung infection (n = 5), neutropenia (n = 4), diarrhea (n = 4), febrile neutropenia (n = 4), abdominal pain (n = 4), sepsis/bacteremia (n = 3), anemia (n = 3), fatigue (n = 2), and pain (n = 1).
There were two grade 5 AEs – acute respiratory distress syndrome and pneumonia.
CTCL cohort
The 29 CTCL patients had a median age of 62 years (range, 24-79). They had received a median of 4 (range, 1-13) prior therapies, 55% were refractory to their last therapy, and 3% had undergone SCT.
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 27 were evaluable for response.
The overall response rate was 26% (n = 7). Two patients achieved a CR, five achieved a PR, and nine had stable disease. Responses occurred in mycosis fungoides and Sézary syndrome.
Eleven of 23 patients (48%) achieved at least a 50% reduction in skin lesions, and the researchers observed rapid improvements in pruritus.
“I saw patients who would take the first pill, and they would call me and say, ‘I no longer itch,’ ” Dr. Feldman said.
The most common grade 3 or higher AEs in CTCL patients were lipase increase (n = 11), amylase increase (n = 5), sepsis/bacteremia (n = 3), pain (n = 2), fatigue (n = 1), neutropenia (n = 1), and diarrhea (n = 1).
“It’s a very well-tolerated drug,” Dr. Feldman said, adding that there were “really no severe side effects which would prohibit the use of the drug.”
She noted that cerdulatinib’s “favorable” side effect profile might make it a promising candidate for use in combination regimens.
“I think it will be possible to combine it with other drugs in development in T-cell lymphoma. … immunological checkpoint inhibitors, epigenetic modulators such as HDAC [histone deacetylase] inhibitors, methylating agents, and PI3 kinase inhibitors,” Dr. Feldman said.
She reported having no disclosures relevant to this study. The trial is sponsored by Portola Pharmaceuticals.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – The spleen tyrosine kinase/Janus kinase inhibitor cerdulatinib has demonstrated activity against relapsed and refractory T-cell lymphomas.
In a phase 2 trial, cerdulatinib produced responses in 34% of patients with peripheral T-cell lymphoma (PTCL) and 26% of those with cutaneous T-cell lymphoma (CTCL).
The best responders were patients with angioimmunoblastic T-cell lymphoma, half of whom achieved a complete response (CR).
The most common grade 3 or higher adverse events (AEs) were amylase increase and lipase increase. However, these increases resolved with dose reduction or interruption, and there were no cases of clinical pancreatitis.
“The data is very encouraging,” said Tatyana Feldman, MD, of the John Theurer Cancer Center in Hackensack, N.J.
Dr. Feldman and her colleagues previously presented results from the phase 2 trial of cerdulatinib (NCT01994382) at the 2018 annual congress of the European Hematology Association.
Dr. Feldman and her colleagues presented data from expansion cohorts of the ongoing trial at the annual T-cell Lymphoma Forum. The cohorts included patients with PTCL or CTCL who had received at least one prior systemic therapy.
PTCL cohort
The 45 PTCL patients had a median age of 65 years (range, 21-84). They had received a median of 3 (range, 1-12) prior therapeutic regimens, 51% were refractory to their last therapy, and 27% had undergone stem cell transplant (SCT).
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 41 patients were evaluable for response.
The overall response rate was 34% (n = 14). Eleven patients had a CR, three had a partial response (PR), and nine had stable disease.
Responses according to subtype were as follows:
- 7 CRs and 1 PR in angioimmunoblastic T-cell lymphoma.
- 2 CRs in PTCL not otherwise specified.
- 1 CR in gamma-delta T-cell lymphoma.
- 1 PR in ALK-negative anaplastic large-cell lymphoma.
- 1 CR and 1 PR in adult T-cell leukemia/lymphoma.
Eight responders have remained on cerdulatinib for anywhere from 3 months to more than 12 months. Five patients have had a response lasting at least 6 months. One patient went on to SCT after achieving a CR.
The most common grade 3 or higher AEs observed in PTCL patients were amylase increase (n = 8), lipase increase (n = 6), pneumonia/lung infection (n = 5), neutropenia (n = 4), diarrhea (n = 4), febrile neutropenia (n = 4), abdominal pain (n = 4), sepsis/bacteremia (n = 3), anemia (n = 3), fatigue (n = 2), and pain (n = 1).
There were two grade 5 AEs – acute respiratory distress syndrome and pneumonia.
CTCL cohort
The 29 CTCL patients had a median age of 62 years (range, 24-79). They had received a median of 4 (range, 1-13) prior therapies, 55% were refractory to their last therapy, and 3% had undergone SCT.
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 27 were evaluable for response.
The overall response rate was 26% (n = 7). Two patients achieved a CR, five achieved a PR, and nine had stable disease. Responses occurred in mycosis fungoides and Sézary syndrome.
Eleven of 23 patients (48%) achieved at least a 50% reduction in skin lesions, and the researchers observed rapid improvements in pruritus.
“I saw patients who would take the first pill, and they would call me and say, ‘I no longer itch,’ ” Dr. Feldman said.
The most common grade 3 or higher AEs in CTCL patients were lipase increase (n = 11), amylase increase (n = 5), sepsis/bacteremia (n = 3), pain (n = 2), fatigue (n = 1), neutropenia (n = 1), and diarrhea (n = 1).
“It’s a very well-tolerated drug,” Dr. Feldman said, adding that there were “really no severe side effects which would prohibit the use of the drug.”
She noted that cerdulatinib’s “favorable” side effect profile might make it a promising candidate for use in combination regimens.
“I think it will be possible to combine it with other drugs in development in T-cell lymphoma. … immunological checkpoint inhibitors, epigenetic modulators such as HDAC [histone deacetylase] inhibitors, methylating agents, and PI3 kinase inhibitors,” Dr. Feldman said.
She reported having no disclosures relevant to this study. The trial is sponsored by Portola Pharmaceuticals.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: The overall response rate was 34% in patients with peripheral T-cell lymphoma (PTCL) and 26% in patients with cutaneous T-cell lymphoma (CTCL).
Study details: Expansion cohorts of a phase 2 trial including 45 PTCL patients and 29 CTCL patients
Disclosures: The study was funded by Portola Pharmaceuticals. The investigator reported having no relevant conflicts.

Locally Destructive Metastatic Basal Cell Carcinoma
To the Editor:
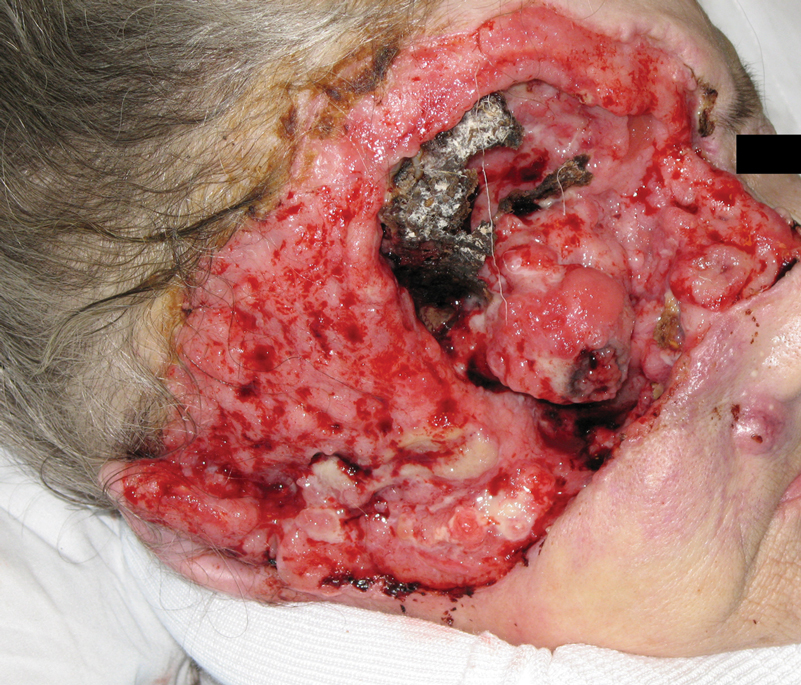
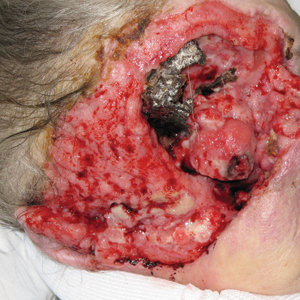
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
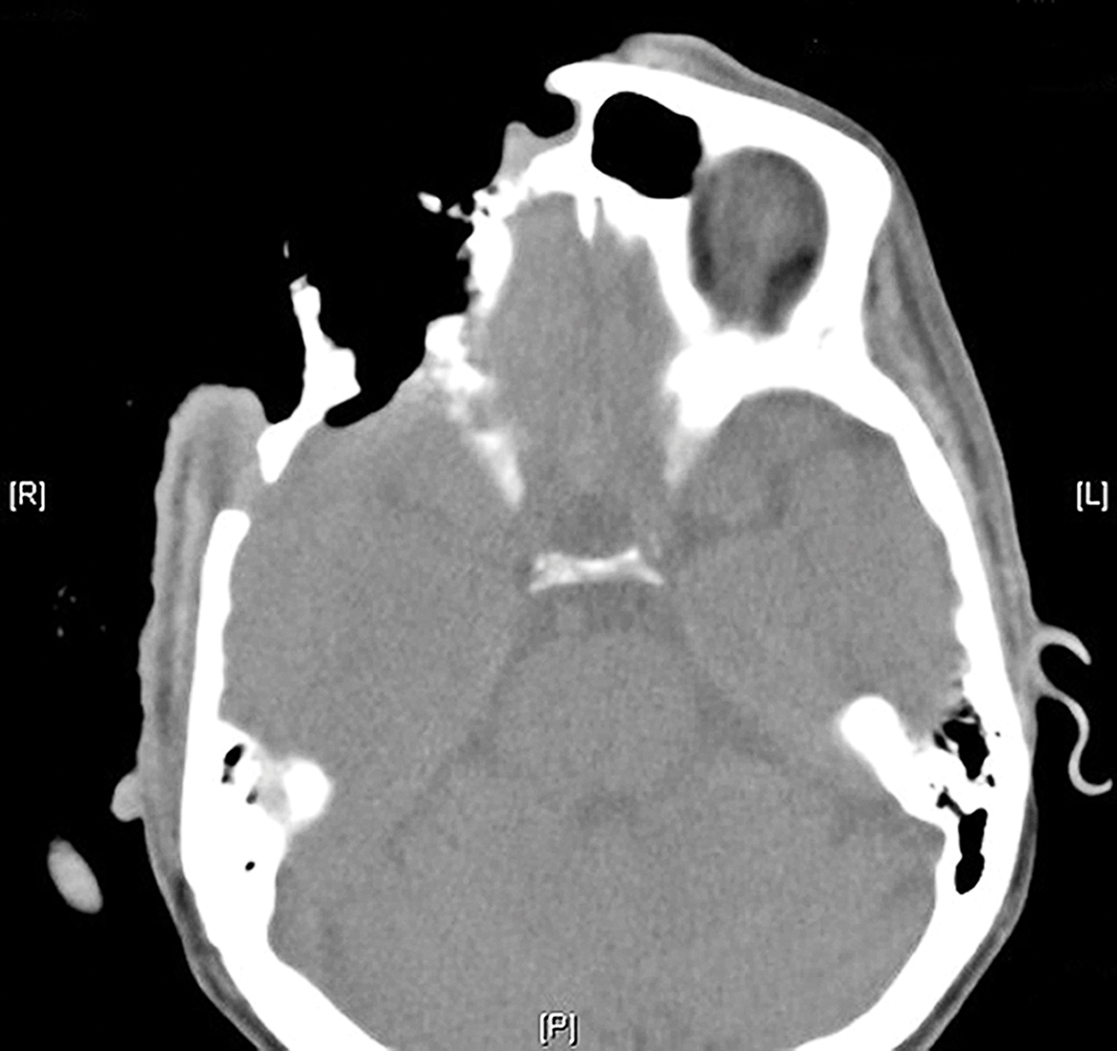
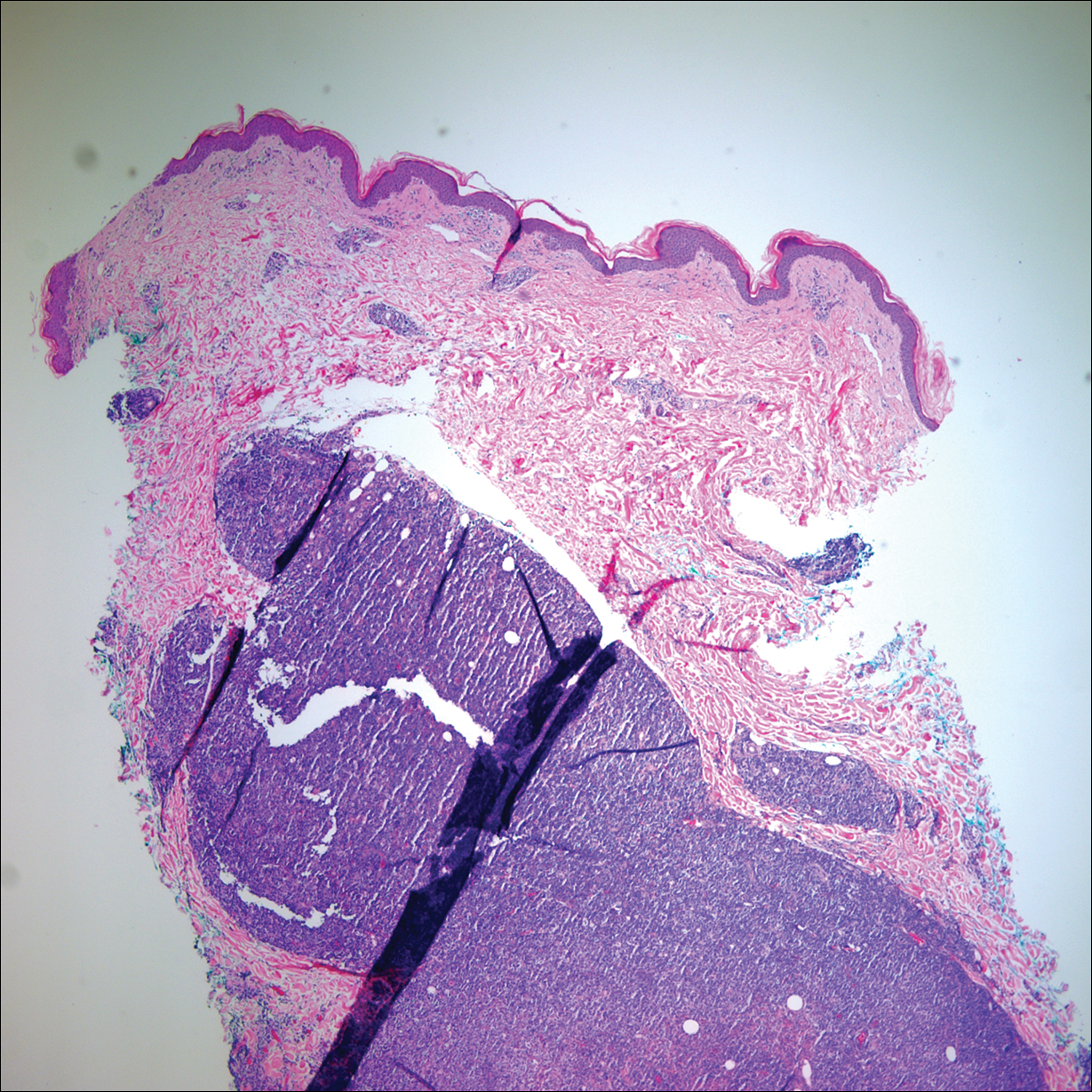
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
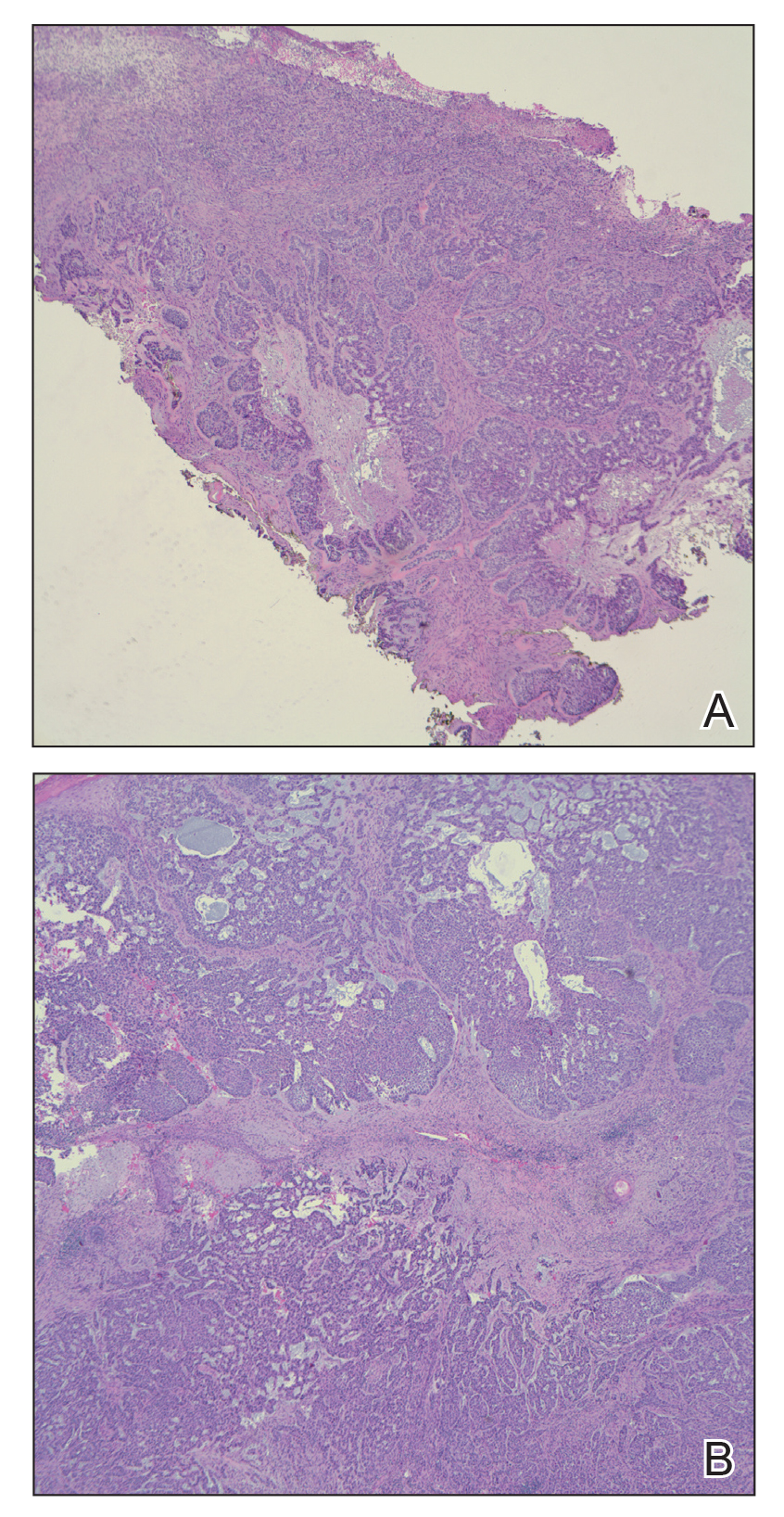
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
Practice Points
- Risk factors associated with metastatic spread of basal cell carcinoma (BCC) include larger tumor size, greater depth of invasion, long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.
- The median interval between onset of BCC and metastasis has been shown to be approximately 9 years.
- Vismodegib can be an effective oral therapy for patients with metastatic BCC, locally advanced BCC, recurrence following surgery, or those who are not candidates for surgery or radiation.
IPH4102 on fast track for Sézary syndrome
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
The who have received at least two prior systemic therapies.
IPH4102 is an anti-KIR3DL2 antibody being developed by Innate Pharma as a treatment for T-cell lymphomas.
The FDA’s fast track program is designed to expedite the review of products that are intended to treat serious conditions and have the potential to address unmet medical needs.
The fast track designation for IPH4102 is based on preliminary results from a phase 1 study (NCT02593045) of patients with advanced cutaneous T-cell lymphoma.
Data on 35 Sézary patients in this trial were presented at the 2018 annual meeting of the American Society of Hematology (Blood. 2018;132:684). The patients had a median age of 70 (range, 31-90), and they had received a median of 2 (range, 1-9) prior systemic therapies.
As of Oct. 15, 2018, the overall response rate was 42.9%, with 2 complete responses and 13 partial responses. The median duration of response was 13.8 months, and the median progression-free survival was 11.7 months.
Treatment-related adverse events (AEs) included asthenia (n = 5), lymphopenia (n = 5), fatigue (n = 3), pyrexia (n = 3), arthralgia (n = 2), and diarrhea (n = 1). The only grade 3/4 treatment-related AE was lymphopenia (n = 2).
Four patients experienced six grade 3 or higher AEs that were possibly related to treatment—grade 5 hepatitis (n = 1), grade 4 sepsis (n = 1), grade 3 lymphopenia (n = 3), and grade 3 hypotension (n = 1).
Based on these results, Innate Pharma is planning a phase 2 trial of IPH4102, which is expected to begin in the first half of this year.
Long-term mogamulizumab appears safe, effective in CTCL
LA JOLLA, CALIF. — Prolonged exposure to mogamulizumab can improve responses without compromising safety in patients with cutaneous T-cell lymphoma (CTCL), according to a post hoc analysis of the MAVORIC trial.

Investigators found that exposure to mogamulizumab correlated with response. The highest response rate — 75.6% — was observed in patients exposed to the drug for at least 351 days, and the lowest — 1.9% — was observed in patients exposed to mogamulizumab for less than 72 days.
On the other hand, rates of adverse events (AEs) were similar regardless of how long patients were treated with mogamulizumab.
Youn H. Kim, MD, of Stanford Cancer Institute at Stanford (Calif.) University, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The phase 3 MAVORIC trial (NCT01728805) included 372 adults with CTCL who had failed at least one systemic therapy. The patients were randomized to treatment with mogamulizumab or vorinostat.
Results from this comparison were previously reported at the 10th annual T-cell Lymphoma Forum.
At this year’s meeting, Dr. Kim and her colleagues reported results in 184 patients who were randomized to mogamulizumab — 105 of whom had mycosis fungoides (MF) and 79 of whom had Sézary syndrome (SS).
Patients were exposed to mogamulizumab for a mean of 275.2 days and a median of 170.0 days (range, 1-1,617 days).
The investigators divided patients into the following quartiles according to mogamulizumab exposure:
- Less than 72 days — 52 patients (28%)
- 72-170 days — 40 patients (22%)
- 171-351 days — 47 patients (26%)
- More than 351 days — 45 patients (24%).
Patients exposed to mogamulizumab for longer were more likely to have SS, stage III/IV disease, blood involvement, and a performance status of 0.
Dr. Kim said the SS patients “benefited a lot” from mogamulizumab and therefore remained on treatment longer.
Response
As expected, patients exposed to mogamulizumab for the longest period had the highest global response rates. Confirmed response rates according to drug exposure were as follows:
- Less than 72 days: 1.9% overall, 0% for SS, and 2.9% for MF
- 72-170 days: 10% overall, 18.8% for SS, and 4.2% for MF
- 171-351 days: 29.8% overall, 36.4% for SS, and 24% for MF
- More than 351 days: 75.6% overall, 83.3% for SS, and 66.7% for MF.
In addition, rates of complete response (CR) and partial response (PR) tended to increase with mogamulizumab exposure. Rates of CR, PR, and stable disease (SD) according to exposure time were as follows:
- Less than 72 days: 0% CR, 7.7% PR, and 38.5% SD
- 72-170 days: 2.5% CR, 20% PR, and 62.5% SD
- 171-351 days: 2.1% CR, 34% PR, and 57.4% SD
- More than 351 days: 6.7% CR, 71.1% PR, and 17.8% SD.
Safety
“The percentage of patients reporting adverse events was not different in the long-term treatment-exposure patients, compared to the short-term,” Dr. Kim said.
Percentages of treatment-emergent AEs (TEAEs) and serious AEs (SAEs) according to mogamulizumab exposure were as follows:
- Less than 72 days: 26.6% TEAEs and 6.5% SAEs
- 72-170 days: 18.5% TEAEs and 3.3% SAEs
- 171-351 days: 23.4% TEAEs and 6.0% SAEs
- More than 351 days: 21.7% TEAEs and 4.3% SAEs.
“The majority of the grade 3 events occurred in the first two quartiles, not later, which is important to show,” Dr. Kim said.
Most grade 3 AEs occurred within 170 days of treatment initiation, and the median time to a grade 3 or higher AE was 109 days.
The most common treatment-related AEs in the longest exposure cohort were drug eruption (20.0%), thrombocytopenia (11.1%), stomatitis (8.9%), and anemia (8.9%).
Of all patients in this analysis, 45 experienced drug eruption, which was defined as a skin rash possibly, probably, or definitely related to the study drug.
Nine drug eruption events were grade 3, and the rest were grade 1 or 2. The median time to drug eruption was 107 days.
While drug eruption “didn’t show up early,” there is no cumulative risk with longer exposure to mogamulizumab, Dr. Kim said. Likewise, she said, autoimmune AEs were not dose-cumulative events.
There were two patients with definite autoimmune disease — a 65-year-old man with Miller Fisher syndrome (occurring 199 days after mogamulizumab initiation) and a 40-year-old woman with myositis (151 days) and myocarditis (310 days).
The investigators also identified three patients with possible autoimmune disease, including:
- Pneumonitis (310 days) in a 74-year-old woman
- Polymyalgia rheumatica (209 days) and myopathy (not available) in an 84-year-old man
- Hepatitis (144 days), pneumonitis (about 174 days), and polymyositis (about 174 days) in a 73-year-old man.
Dr. Kim and her colleagues said these data suggest prolonged treatment with mogamulizumab is not associated with an increased safety risk in patients with MF or SS. And the manageable safety profile of mogamulizumab meant that patients who derived a clinical benefit could remain on the drug for an extended period of time.
The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Dr. Kim reported relationships with Merck, Portola Pharmaceuticals, Soligenix, Takeda, TetraLogic Pharmaceuticals, Kyowa Kirin, Seattle Genetics, Medivir, Neumedicines, Eisai, Innate Pharma, Galderma, Miragen Therapeutics, Forty Seven, and Horizon Pharma. Her coinvestigators reported relationships with several companies.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. — Prolonged exposure to mogamulizumab can improve responses without compromising safety in patients with cutaneous T-cell lymphoma (CTCL), according to a post hoc analysis of the MAVORIC trial.
Investigators found that exposure to mogamulizumab correlated with response. The highest response rate — 75.6% — was observed in patients exposed to the drug for at least 351 days, and the lowest — 1.9% — was observed in patients exposed to mogamulizumab for less than 72 days.
On the other hand, rates of adverse events (AEs) were similar regardless of how long patients were treated with mogamulizumab.
Youn H. Kim, MD, of Stanford Cancer Institute at Stanford (Calif.) University, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The phase 3 MAVORIC trial (NCT01728805) included 372 adults with CTCL who had failed at least one systemic therapy. The patients were randomized to treatment with mogamulizumab or vorinostat.
Results from this comparison were previously reported at the 10th annual T-cell Lymphoma Forum.
At this year’s meeting, Dr. Kim and her colleagues reported results in 184 patients who were randomized to mogamulizumab — 105 of whom had mycosis fungoides (MF) and 79 of whom had Sézary syndrome (SS).
Patients were exposed to mogamulizumab for a mean of 275.2 days and a median of 170.0 days (range, 1-1,617 days).
The investigators divided patients into the following quartiles according to mogamulizumab exposure:
- Less than 72 days — 52 patients (28%)
- 72-170 days — 40 patients (22%)
- 171-351 days — 47 patients (26%)
- More than 351 days — 45 patients (24%).
Patients exposed to mogamulizumab for longer were more likely to have SS, stage III/IV disease, blood involvement, and a performance status of 0.
Dr. Kim said the SS patients “benefited a lot” from mogamulizumab and therefore remained on treatment longer.
Response
As expected, patients exposed to mogamulizumab for the longest period had the highest global response rates. Confirmed response rates according to drug exposure were as follows:
- Less than 72 days: 1.9% overall, 0% for SS, and 2.9% for MF
- 72-170 days: 10% overall, 18.8% for SS, and 4.2% for MF
- 171-351 days: 29.8% overall, 36.4% for SS, and 24% for MF
- More than 351 days: 75.6% overall, 83.3% for SS, and 66.7% for MF.
In addition, rates of complete response (CR) and partial response (PR) tended to increase with mogamulizumab exposure. Rates of CR, PR, and stable disease (SD) according to exposure time were as follows:
- Less than 72 days: 0% CR, 7.7% PR, and 38.5% SD
- 72-170 days: 2.5% CR, 20% PR, and 62.5% SD
- 171-351 days: 2.1% CR, 34% PR, and 57.4% SD
- More than 351 days: 6.7% CR, 71.1% PR, and 17.8% SD.
Safety
“The percentage of patients reporting adverse events was not different in the long-term treatment-exposure patients, compared to the short-term,” Dr. Kim said.
Percentages of treatment-emergent AEs (TEAEs) and serious AEs (SAEs) according to mogamulizumab exposure were as follows:
- Less than 72 days: 26.6% TEAEs and 6.5% SAEs
- 72-170 days: 18.5% TEAEs and 3.3% SAEs
- 171-351 days: 23.4% TEAEs and 6.0% SAEs
- More than 351 days: 21.7% TEAEs and 4.3% SAEs.
“The majority of the grade 3 events occurred in the first two quartiles, not later, which is important to show,” Dr. Kim said.
Most grade 3 AEs occurred within 170 days of treatment initiation, and the median time to a grade 3 or higher AE was 109 days.
The most common treatment-related AEs in the longest exposure cohort were drug eruption (20.0%), thrombocytopenia (11.1%), stomatitis (8.9%), and anemia (8.9%).
Of all patients in this analysis, 45 experienced drug eruption, which was defined as a skin rash possibly, probably, or definitely related to the study drug.
Nine drug eruption events were grade 3, and the rest were grade 1 or 2. The median time to drug eruption was 107 days.
While drug eruption “didn’t show up early,” there is no cumulative risk with longer exposure to mogamulizumab, Dr. Kim said. Likewise, she said, autoimmune AEs were not dose-cumulative events.
There were two patients with definite autoimmune disease — a 65-year-old man with Miller Fisher syndrome (occurring 199 days after mogamulizumab initiation) and a 40-year-old woman with myositis (151 days) and myocarditis (310 days).
The investigators also identified three patients with possible autoimmune disease, including:
- Pneumonitis (310 days) in a 74-year-old woman
- Polymyalgia rheumatica (209 days) and myopathy (not available) in an 84-year-old man
- Hepatitis (144 days), pneumonitis (about 174 days), and polymyositis (about 174 days) in a 73-year-old man.
Dr. Kim and her colleagues said these data suggest prolonged treatment with mogamulizumab is not associated with an increased safety risk in patients with MF or SS. And the manageable safety profile of mogamulizumab meant that patients who derived a clinical benefit could remain on the drug for an extended period of time.
The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Dr. Kim reported relationships with Merck, Portola Pharmaceuticals, Soligenix, Takeda, TetraLogic Pharmaceuticals, Kyowa Kirin, Seattle Genetics, Medivir, Neumedicines, Eisai, Innate Pharma, Galderma, Miragen Therapeutics, Forty Seven, and Horizon Pharma. Her coinvestigators reported relationships with several companies.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. — Prolonged exposure to mogamulizumab can improve responses without compromising safety in patients with cutaneous T-cell lymphoma (CTCL), according to a post hoc analysis of the MAVORIC trial.
Investigators found that exposure to mogamulizumab correlated with response. The highest response rate — 75.6% — was observed in patients exposed to the drug for at least 351 days, and the lowest — 1.9% — was observed in patients exposed to mogamulizumab for less than 72 days.
On the other hand, rates of adverse events (AEs) were similar regardless of how long patients were treated with mogamulizumab.
Youn H. Kim, MD, of Stanford Cancer Institute at Stanford (Calif.) University, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The phase 3 MAVORIC trial (NCT01728805) included 372 adults with CTCL who had failed at least one systemic therapy. The patients were randomized to treatment with mogamulizumab or vorinostat.
Results from this comparison were previously reported at the 10th annual T-cell Lymphoma Forum.
At this year’s meeting, Dr. Kim and her colleagues reported results in 184 patients who were randomized to mogamulizumab — 105 of whom had mycosis fungoides (MF) and 79 of whom had Sézary syndrome (SS).
Patients were exposed to mogamulizumab for a mean of 275.2 days and a median of 170.0 days (range, 1-1,617 days).
The investigators divided patients into the following quartiles according to mogamulizumab exposure:
- Less than 72 days — 52 patients (28%)
- 72-170 days — 40 patients (22%)
- 171-351 days — 47 patients (26%)
- More than 351 days — 45 patients (24%).
Patients exposed to mogamulizumab for longer were more likely to have SS, stage III/IV disease, blood involvement, and a performance status of 0.
Dr. Kim said the SS patients “benefited a lot” from mogamulizumab and therefore remained on treatment longer.
Response
As expected, patients exposed to mogamulizumab for the longest period had the highest global response rates. Confirmed response rates according to drug exposure were as follows:
- Less than 72 days: 1.9% overall, 0% for SS, and 2.9% for MF
- 72-170 days: 10% overall, 18.8% for SS, and 4.2% for MF
- 171-351 days: 29.8% overall, 36.4% for SS, and 24% for MF
- More than 351 days: 75.6% overall, 83.3% for SS, and 66.7% for MF.
In addition, rates of complete response (CR) and partial response (PR) tended to increase with mogamulizumab exposure. Rates of CR, PR, and stable disease (SD) according to exposure time were as follows:
- Less than 72 days: 0% CR, 7.7% PR, and 38.5% SD
- 72-170 days: 2.5% CR, 20% PR, and 62.5% SD
- 171-351 days: 2.1% CR, 34% PR, and 57.4% SD
- More than 351 days: 6.7% CR, 71.1% PR, and 17.8% SD.
Safety
“The percentage of patients reporting adverse events was not different in the long-term treatment-exposure patients, compared to the short-term,” Dr. Kim said.
Percentages of treatment-emergent AEs (TEAEs) and serious AEs (SAEs) according to mogamulizumab exposure were as follows:
- Less than 72 days: 26.6% TEAEs and 6.5% SAEs
- 72-170 days: 18.5% TEAEs and 3.3% SAEs
- 171-351 days: 23.4% TEAEs and 6.0% SAEs
- More than 351 days: 21.7% TEAEs and 4.3% SAEs.
“The majority of the grade 3 events occurred in the first two quartiles, not later, which is important to show,” Dr. Kim said.
Most grade 3 AEs occurred within 170 days of treatment initiation, and the median time to a grade 3 or higher AE was 109 days.
The most common treatment-related AEs in the longest exposure cohort were drug eruption (20.0%), thrombocytopenia (11.1%), stomatitis (8.9%), and anemia (8.9%).
Of all patients in this analysis, 45 experienced drug eruption, which was defined as a skin rash possibly, probably, or definitely related to the study drug.
Nine drug eruption events were grade 3, and the rest were grade 1 or 2. The median time to drug eruption was 107 days.
While drug eruption “didn’t show up early,” there is no cumulative risk with longer exposure to mogamulizumab, Dr. Kim said. Likewise, she said, autoimmune AEs were not dose-cumulative events.
There were two patients with definite autoimmune disease — a 65-year-old man with Miller Fisher syndrome (occurring 199 days after mogamulizumab initiation) and a 40-year-old woman with myositis (151 days) and myocarditis (310 days).
The investigators also identified three patients with possible autoimmune disease, including:
- Pneumonitis (310 days) in a 74-year-old woman
- Polymyalgia rheumatica (209 days) and myopathy (not available) in an 84-year-old man
- Hepatitis (144 days), pneumonitis (about 174 days), and polymyositis (about 174 days) in a 73-year-old man.
Dr. Kim and her colleagues said these data suggest prolonged treatment with mogamulizumab is not associated with an increased safety risk in patients with MF or SS. And the manageable safety profile of mogamulizumab meant that patients who derived a clinical benefit could remain on the drug for an extended period of time.
The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Dr. Kim reported relationships with Merck, Portola Pharmaceuticals, Soligenix, Takeda, TetraLogic Pharmaceuticals, Kyowa Kirin, Seattle Genetics, Medivir, Neumedicines, Eisai, Innate Pharma, Galderma, Miragen Therapeutics, Forty Seven, and Horizon Pharma. Her coinvestigators reported relationships with several companies.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: The highest response rate – 75.6% – was observed in patients exposed to mogamulizumab for at least 351 days.
Study details: A post hoc analysis of the MAVORIC trial, including 184 patients treated with mogamulizumab.
Disclosures: The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Investigators disclosed relationships with several companies.
Primary Cutaneous Epstein-Barr Virus–Positive Diffuse Large B-Cell Lymphoma: A Rare and Aggressive Cutaneous Lymphoma
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
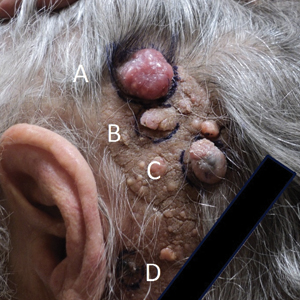
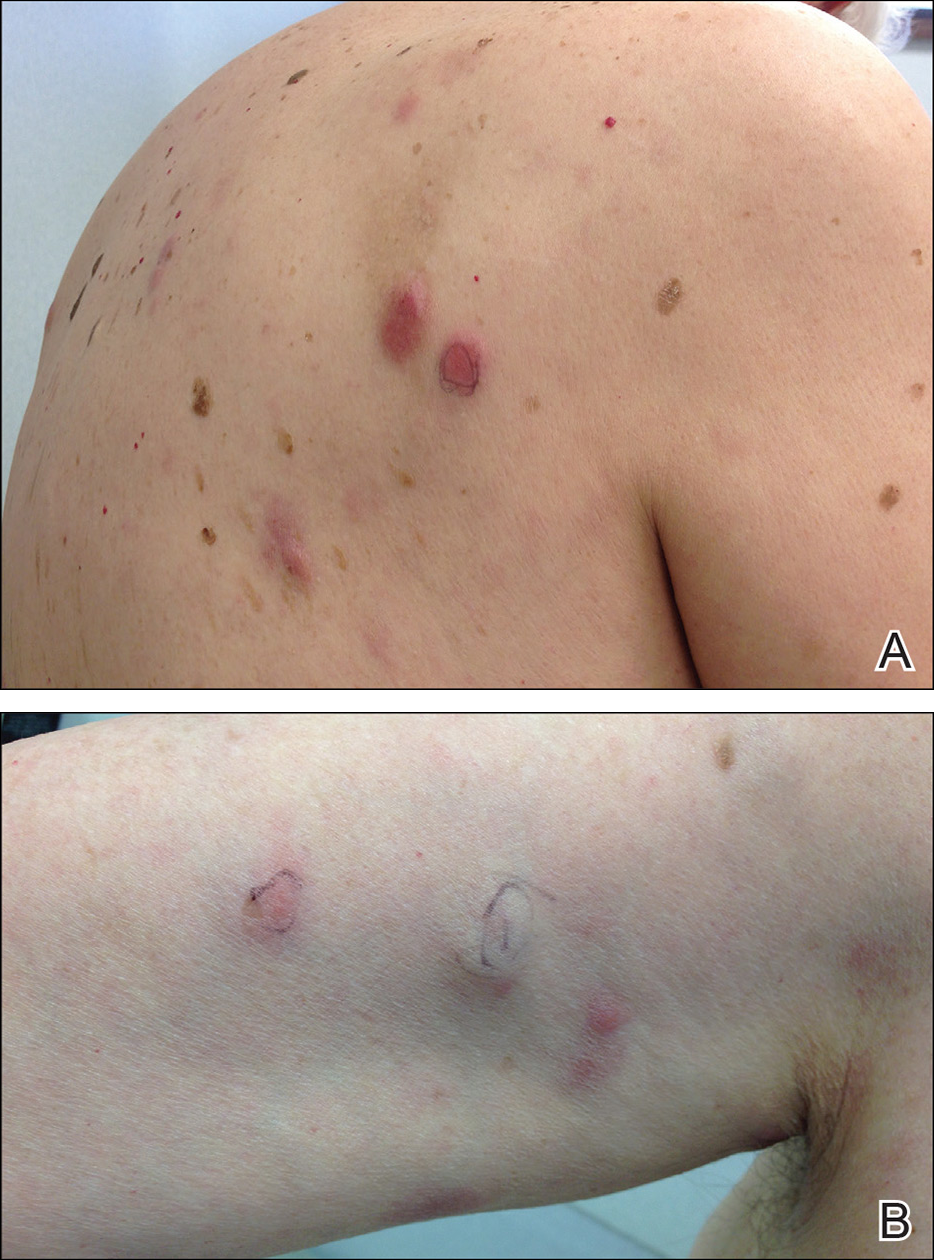
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
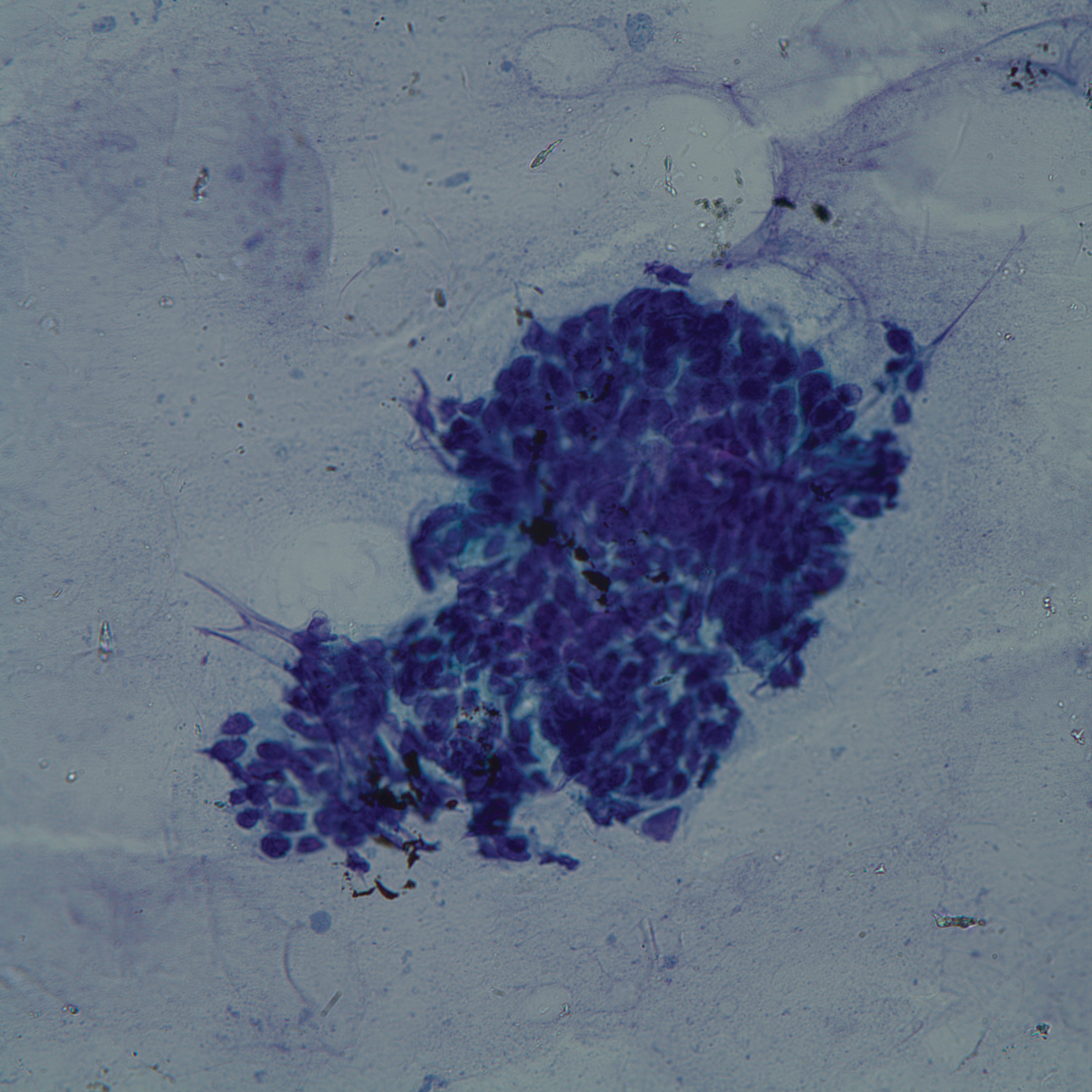
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
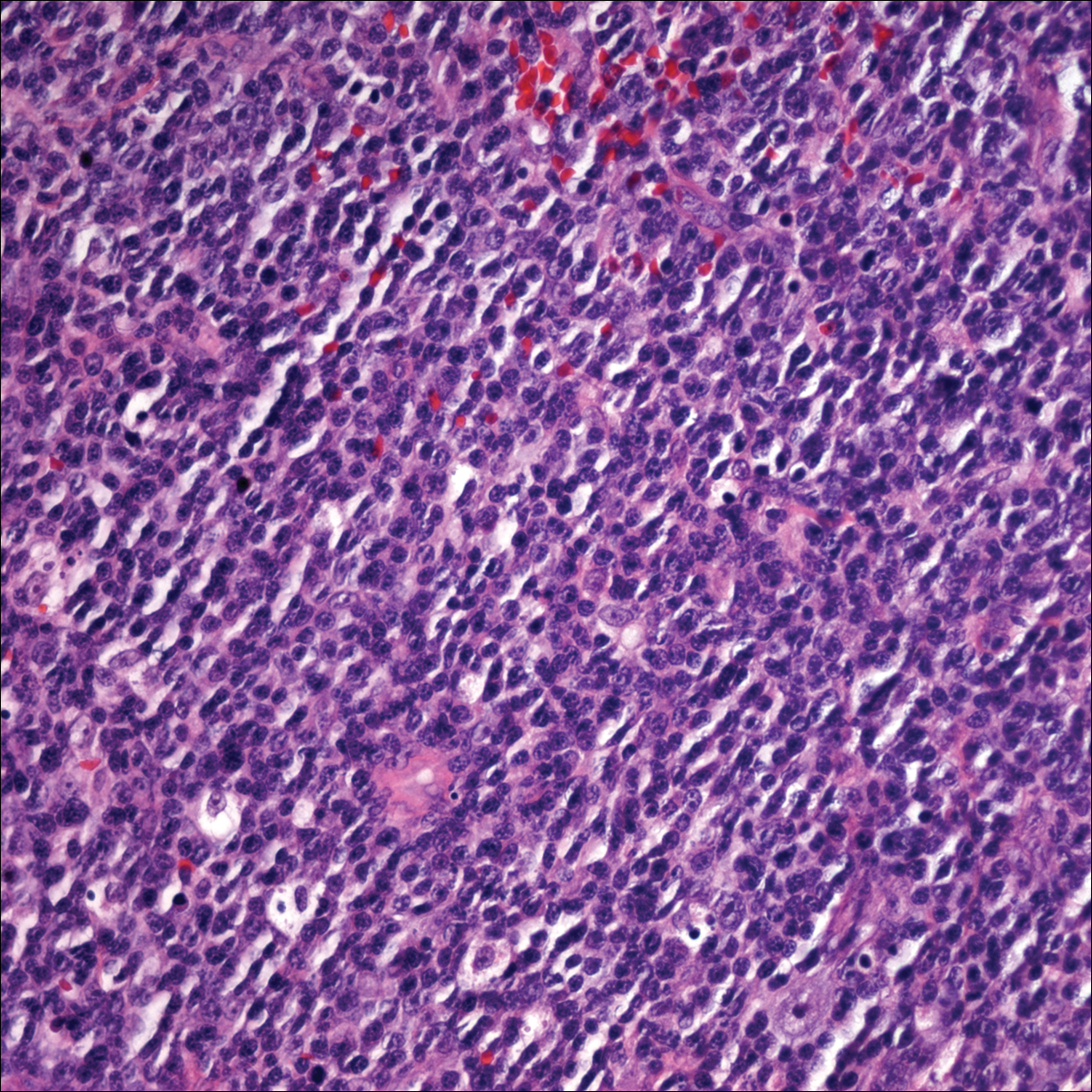
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Practice Points
- Primary cutaneous lymphomas are malignant lymphomas confined to the skin.
- Complete staging workup is necessary to rule out secondary involvement of the skin from a nodal lymphoma.
- Epstein-Barr virus-positive diffuse large B-cell lymphoma is a rare and aggressive primary cutaneous lymphoma.
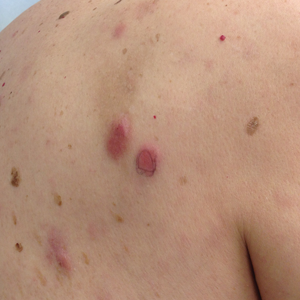
Crizotinib-Induced Lichenoid Drug Eruption in a Patient With Lung Cancer
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
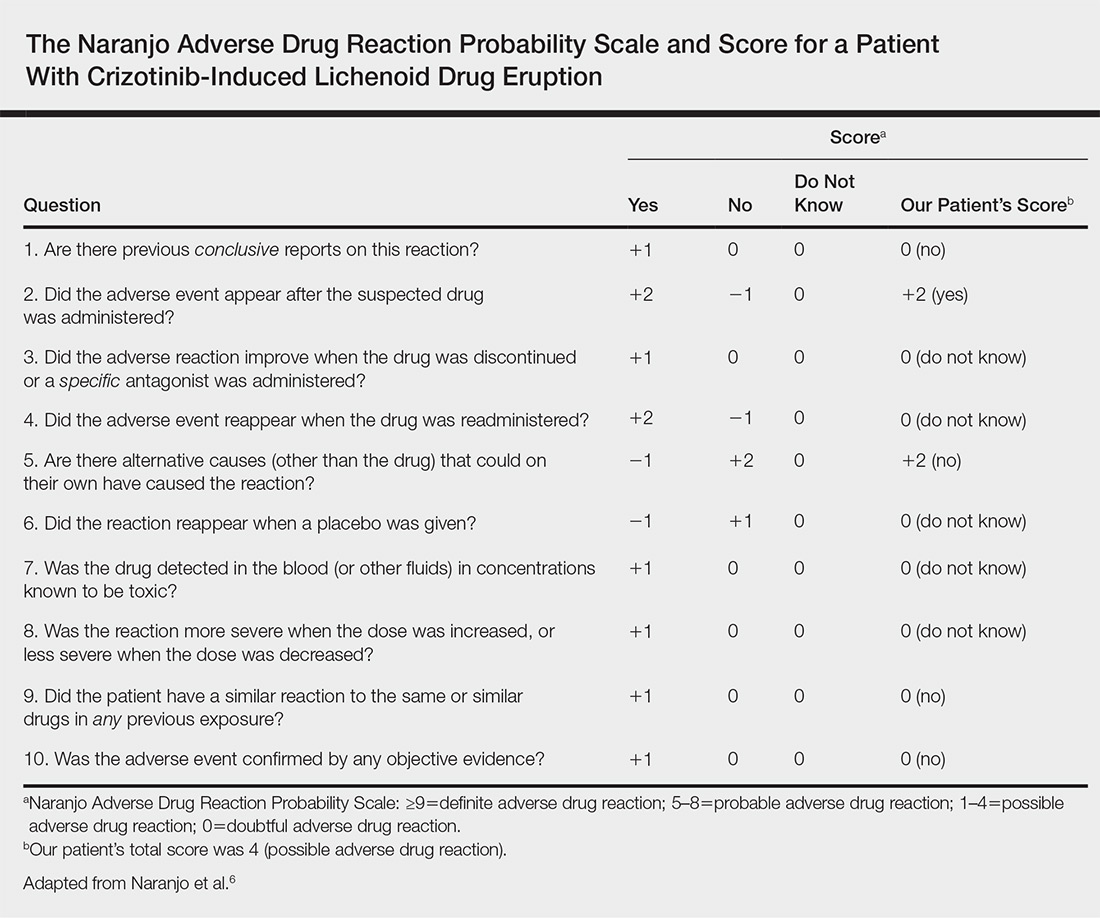
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Practice Points
- Cutaneous lichenoid drug eruptions (LDEs) and photosensitive rash may be caused by crizotinib.
- The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.
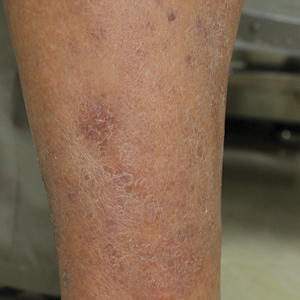
Mohs Micrographic Surgery Overlying a Pacemaker
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.

Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
Practice Points
- Surgical treatment of a cutaneous lesion overlying a cardiac device requires caution, both in avoidance of the device itself as well as electrocautery interference.
- Local anesthesia infiltrates can be used to create a temporary edematous thickening to minimize potential exposure of the device during the procedure.
Cross-contamination of Pathology Specimens: A Cautionary Tale
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
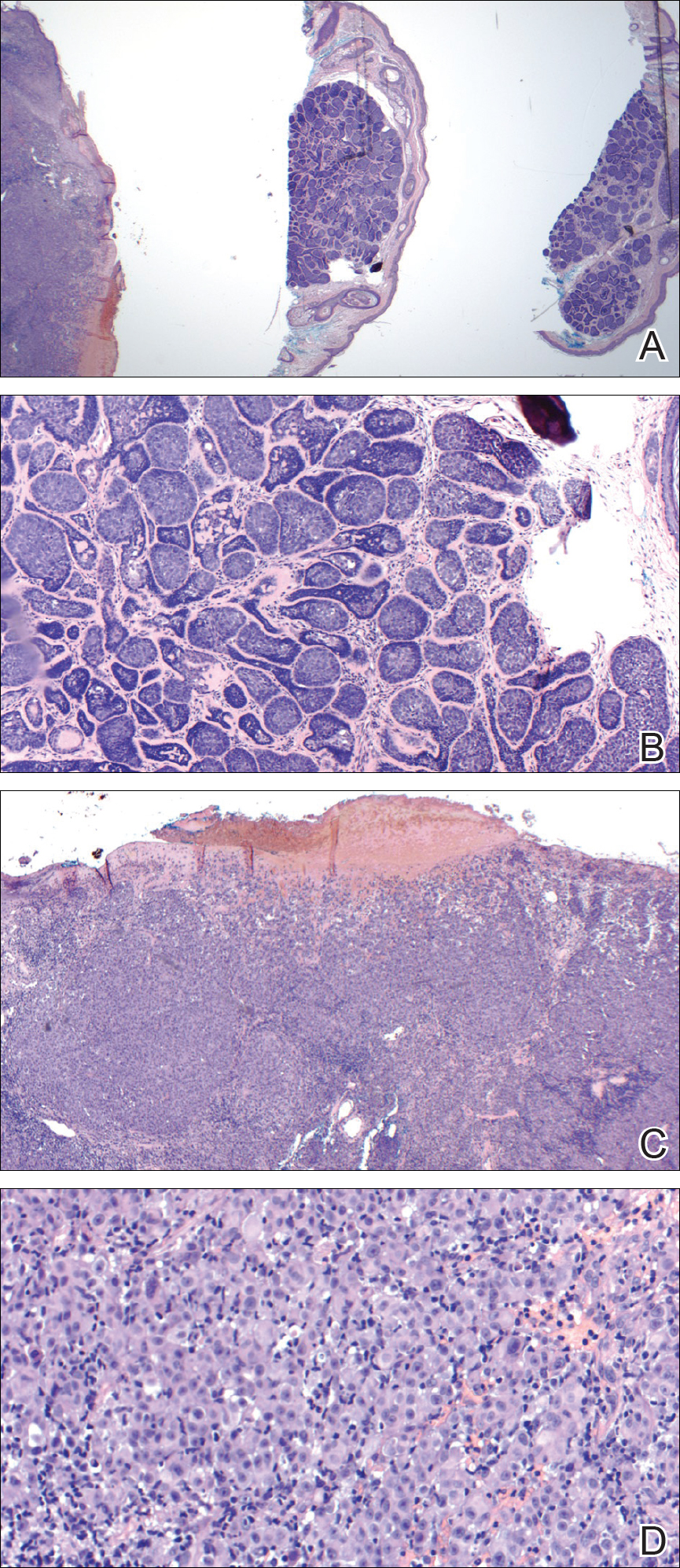
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
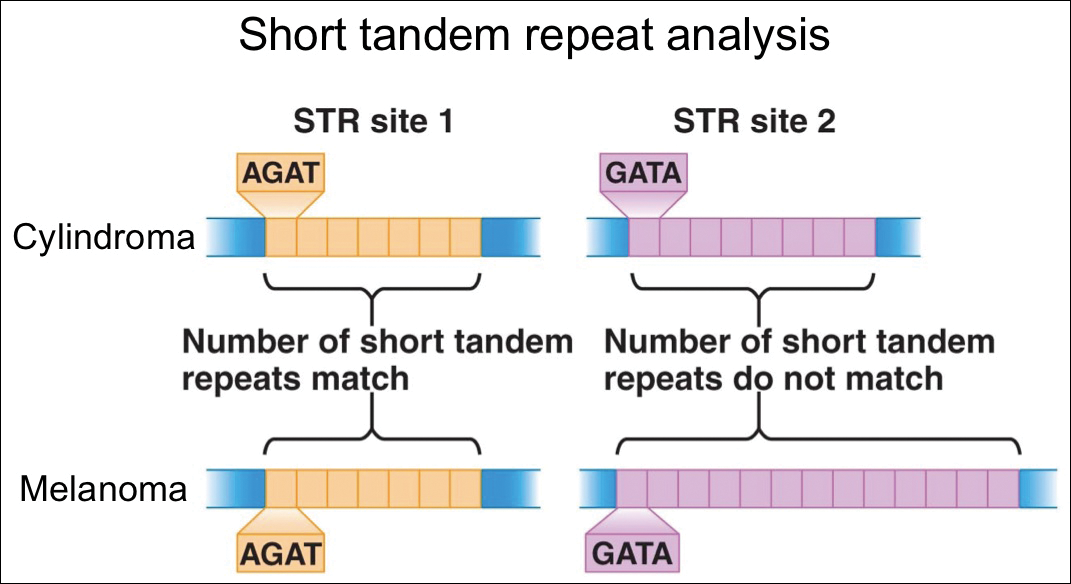
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
Resident Pearl
- Although cross-contamination of pathology specimens is rare, it does occur and can impact diagnosis and management if detected early.
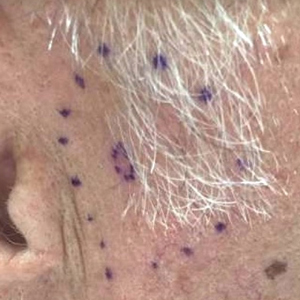
Cutaneous Angiosarcoma of the Lower Leg
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.

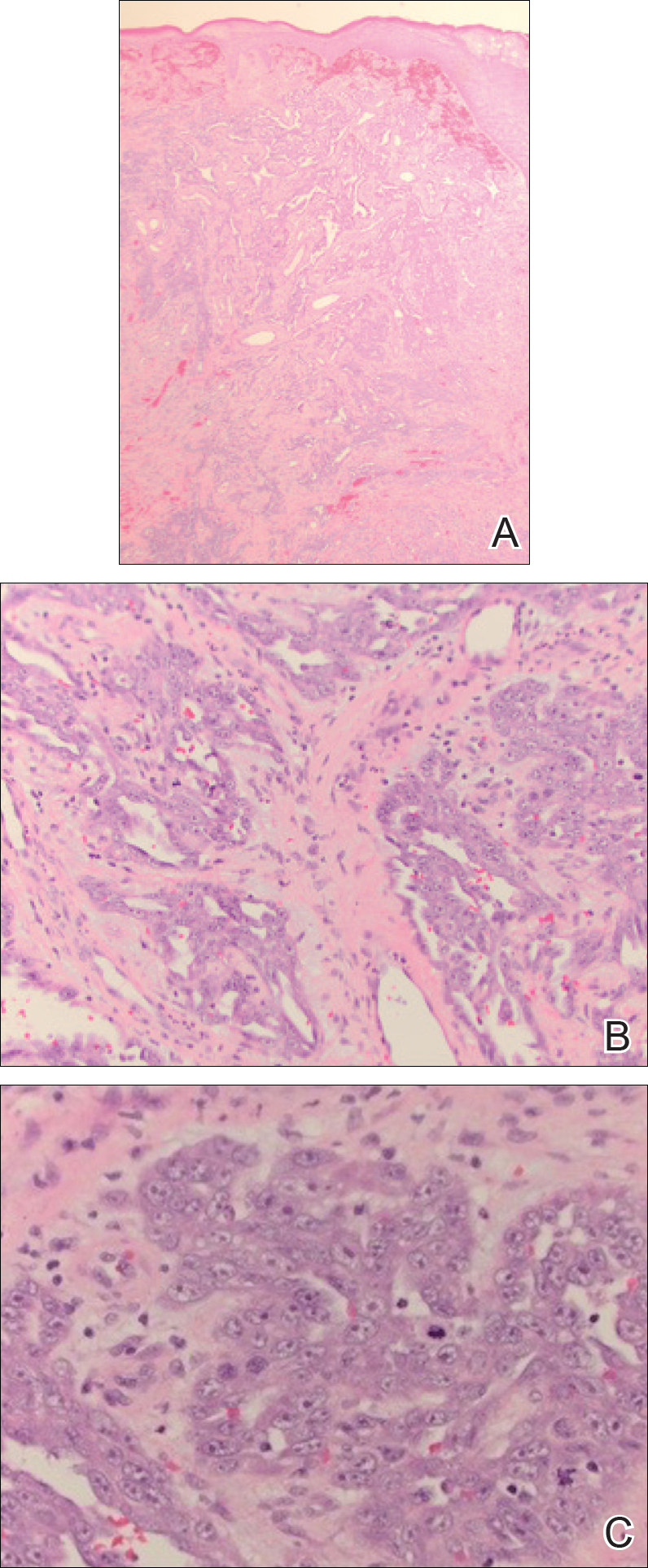
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.

Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.
Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.
Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
Practice Points
- Cutaneous angiosarcoma is a rare malignant vascular neoplasm typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically in the setting of chronic lymphedema following mastectomy (Stewart-Treves syndrome).
- There should be a low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy.
- Histologic analysis of angiosarcoma shows positive staining for CD34 and vimentin with poorly differentiated endothelial atypia with mitotic figures.
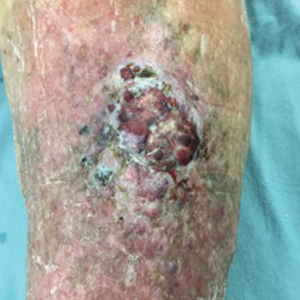
Primary Cutaneous Apocrine Carcinoma Arising Within a Nevus Sebaceus
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
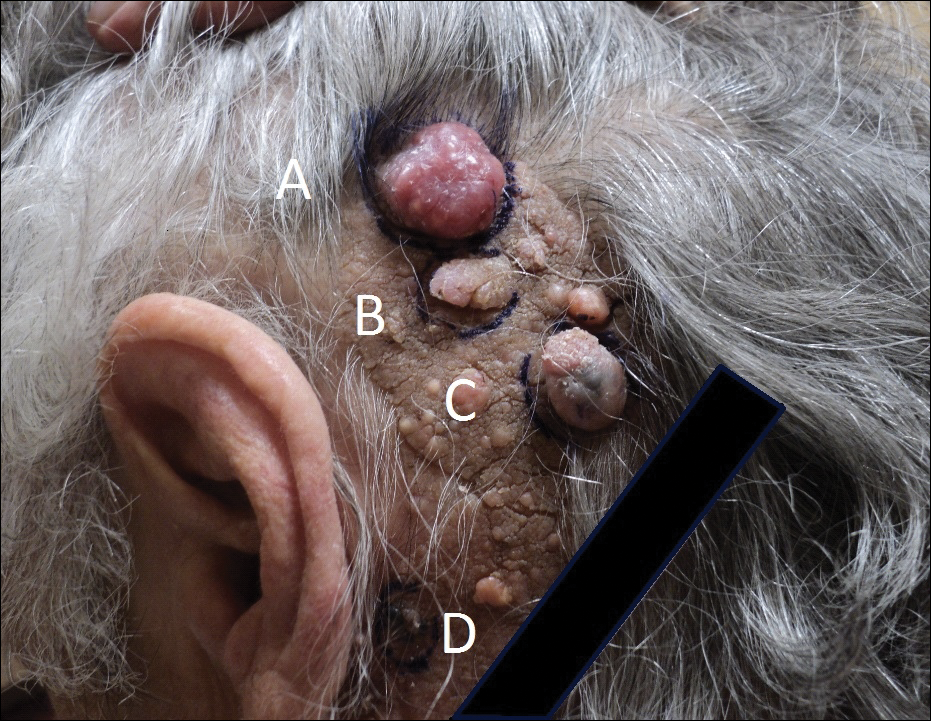

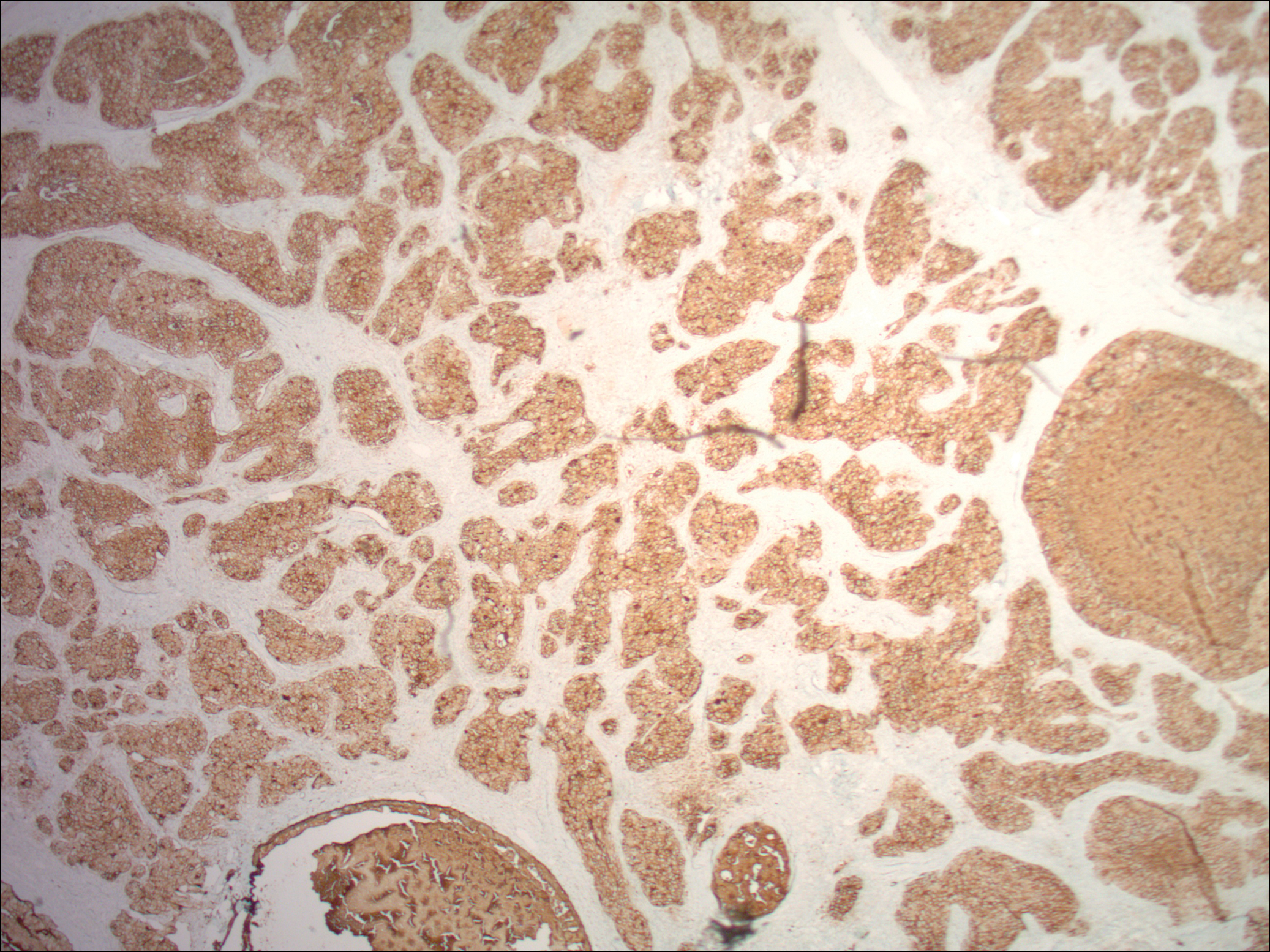
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
Practice Points
- Nevus sebaceus (NS) in the centrofacial region has been correlated with a higher risk for neurological abnormalities, including intellectual disability and seizures.
- Historically, basal cell carcinomas (BCCs) were considered a common occurrence arising from an NS, prompting prophylactic surgical excision of such lesions.
- More recently, it has been recognized that the most common tumor to arise from NS is trichoblastoma rather than BCC; in fact, BCC and other malignancies have been found to be relatively rare compared to their benign counterparts.
- In light of this discovery, observation of NS may be a more prudent course of treatment versus prophylactic surgical excision.