User login
Aggressive Merkel Cell Carcinoma in a Liver Transplant Recipient
Merkel cell carcinoma (MCC) is a rare cutaneous neuroendocrine tumor derived from the nerve-associated Merkel cell touch receptors.1 It typically presents as a solitary, rapidly growing, red to violaceous, asymptomatic nodule, though ulcerated, acneform, and cystic lesions also have been described.2 Merkel cell carcinoma follows an aggressive clinical course with a tendency for rapid growth, local recurrence (26%–60% of cases), lymph node invasion, and distant metastases (18%–52% of cases).3
Several risk factors contribute to the development of MCC, including chronic immunosuppression, exposure to UV radiation, and infection with the Merkel cell polyomavirus. Immunosuppression has been shown to increase the risk for MCC and is associated with a worse prognosis independent of stage at diagnosis.4 Organ transplant recipients represent a subset of immunosuppressed patients who are at increased risk for the development of MCC. We report a case of metastatic MCC in a 67-year-old woman 6 years after liver transplantation.
Case Report
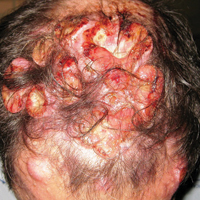
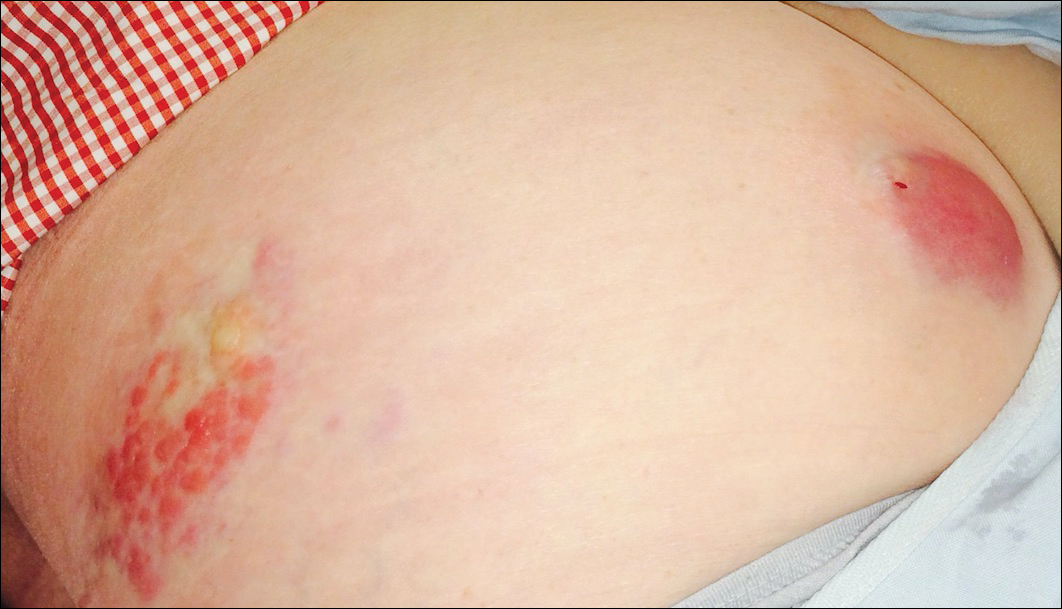
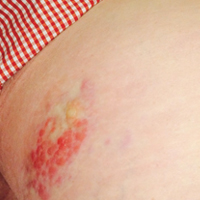
A 67-year-old woman presented to our clinic with 2 masses—1 on the left buttock and 1 on the left hip—of 4 months’ duration. The patient’s medical history was remarkable for autoimmune hepatitis requiring liver transplantation 6 years prior as well as hypertension and thyroid disorder. Her posttransplantation course was unremarkable, and she was maintained on chronic immunosuppression with tacrolimus and mycophenolate mofetil. Six years after transplantation, the patient was observed to have a 4-cm, red-violaceous, painless, dome-shaped tumor on the left buttock (Figure 1). She also was noted to have pink-red papulonodules forming a painless 8-cm plaque on the left hip that was present for 2 weeks prior to presentation (Figure 1). Both lesions were subsequently biopsied.
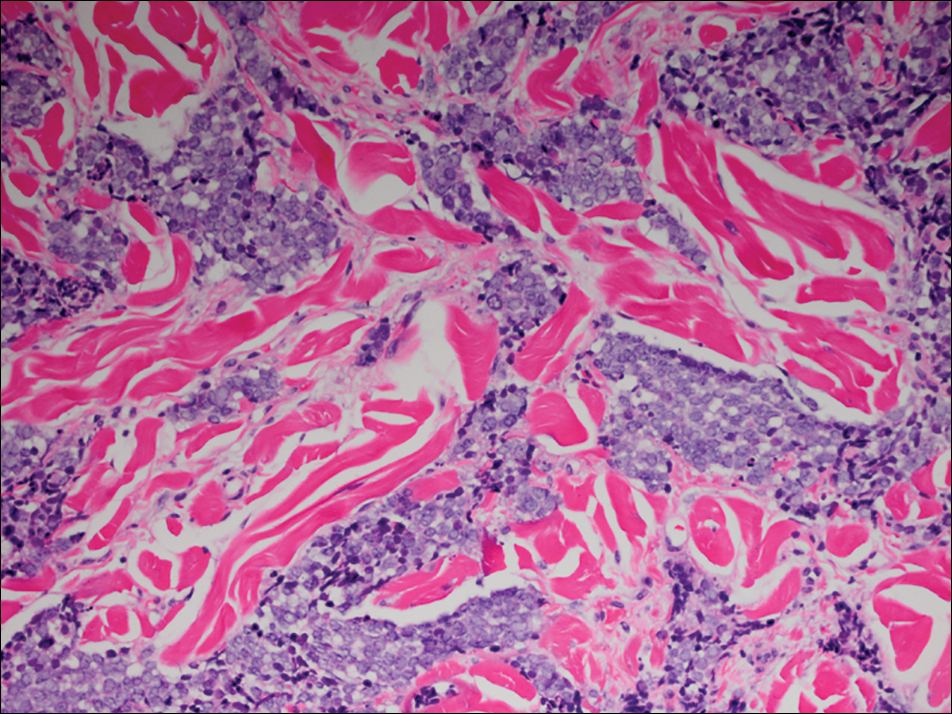
Microscopic examination of both lesions was consistent with the diagnosis of MCC. On histopathology, both samples exhibited a dense cellular dermis composed of atypical basophilic tumor cells with extension into superficial dilated lymphatic channels indicating lymphovascular invasion (Figure 2). Tumor cells were positive for the immunohistochemical markers pankeratin AE1/AE3, CAM 5.2, cytokeratin 20, synaptophysin, chromogranin A, and Merkel cell polyomavirus.
Total-body computed tomography and positron emission tomography revealed a hypermetabolic lobular density in the left gluteal region measuring 3.9×1.1 cm. The mass was associated with avid disease involving the left inguinal, bilateral iliac chain, and retroperitoneal lymph nodes. The patient was determined to have stage IV MCC based on the presence of distant lymph node metastases. The mass on the left hip was identified as an in-transit metastasis from the primary tumor on the left buttock.
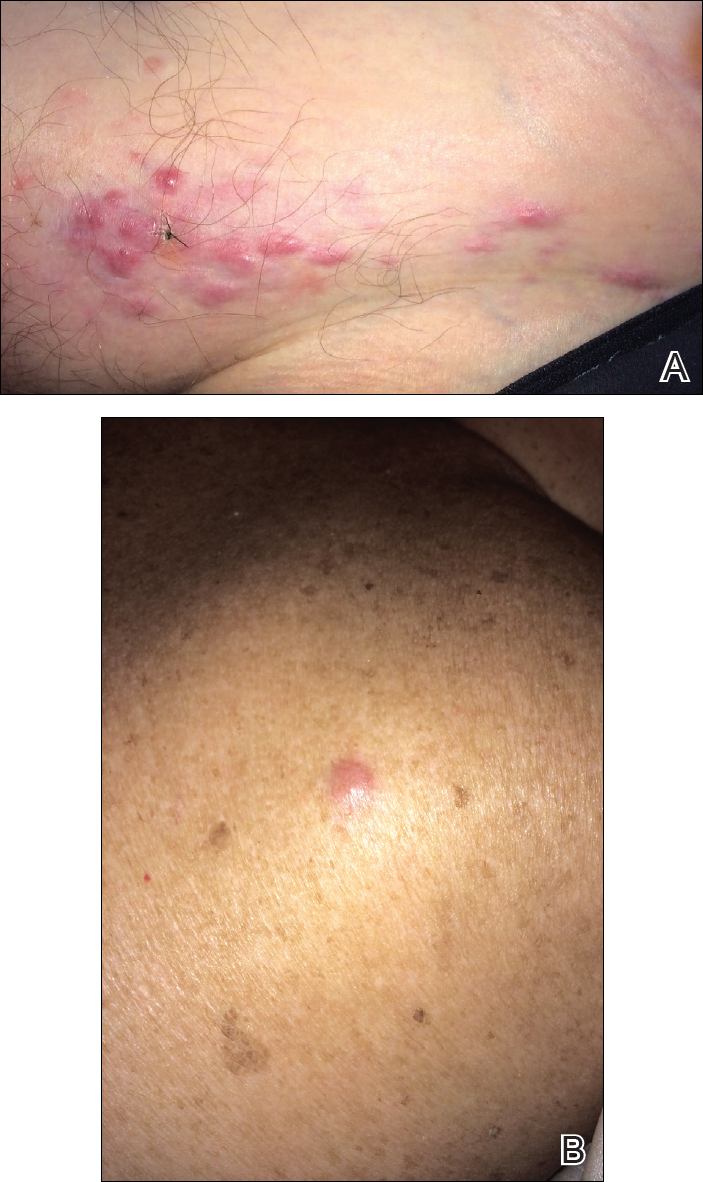
The patient was referred to surgical and medical oncology. The decision was made to start palliative chemotherapy without surgical intervention given the extent of metastases not amenable for resection. The patient was subsequently initiated on chemotherapy with etoposide and carboplatin. After one cycle of chemotherapy, both tumors initially decreased in size; however, 4 months later, despite multiple cycles of chemotherapy, the patient was noted to have growth of existing tumors and interval development of a new 7×5-cm erythematous plaque in the left groin (Figure 3A) and a 1.1×1.0-cm smooth nodule on the right upper back (Figure 3B), both also found to be consistent with distant skin metastases of MCC upon microscopic examination after biopsy. Despite chemotherapy, the patient’s tumor continued to spread and the patient died within 8 months of diagnosis.
Comment
Transplant recipients represent a well-described cohort of immunosuppressed patients prone to the development of MCC. Merkel cell carcinoma in organ transplant recipients has been most frequently documented to occur after kidney transplantation and less frequently after heart and liver transplantations.5,6 However, the role of organ type and immunosuppressive regimen is not well characterized in the literature. Clarke et al7 investigated the risk for MCC in a large cohort of solid organ transplant recipients based on specific immunosuppression medications. They found a higher risk for MCC in patients who were maintained on cyclosporine, azathioprine, and mTOR (mechanistic target of rapamycin) inhibitors rather than tacrolimus, mycophenolate mofetil, and corticosteroids. In comparison to combination tacrolimus–mycophenolate mofetil, cyclosporine-azathioprine was associated with an increased incidence of MCC; this risk rose remarkably in patients who resided in geographic locations with a higher average of UV exposure. The authors suggested that UV radiation and immunosuppression-induced DNA damage may be synergistic in the development of MCC.7
Merkel cell carcinoma most frequently occurs on sun-exposed sites, including the face, head, and neck (55%); upper and lower extremities (40%); and truncal regions (5%).8 However, case reports highlight MCC arising in atypical locations such as the buttocks and gluteal region in organ transplant recipients.7,9 In the general population, MCC predominantly arises in elderly patients (ie, >70 years), but it is more likely to present at an earlier age in transplant recipients.6,10 In a retrospective analysis of 41 solid organ transplant recipients, 12 were diagnosed before the age of 50 years.6 Data from the US Scientific Registry of Transplant Recipients showed a median age at diagnosis of 62 years, with the highest incidence occurring 10 or more years after transplantation.7
Merkel cell carcinoma behaves aggressively and is the most common cause of skin cancer death after melanoma.11 Organ transplant recipients with MCC have a worse prognosis than MCC patients who are not transplant recipients. In a retrospective registry analysis of 45 de novo cases, Buell at al5 found a 60% mortality rate in transplant recipients, almost double the 33% mortality rate of the general population. Furthermore, Arron et al10 revealed substantially increased rates of disease progression and decreased rates of disease-specific and overall survival in solid organ transplant recipients on immunosuppression compared to immunocompetent controls. The most important factor for poor prognosis is the presence of lymph node invasion, which lowers survival rate.12
Conclusion
Merkel cell carcinoma following liver transplantation is not well described in the literature. We highlight a case of an aggressive MCC arising in a sun-protected site with rapid metastasis 6 years after liver transplantation. This case emphasizes the importance of surveillance for cutaneous malignancy in solid organ transplant recipients.
- Gould VE, Moll R, Moll I, et al. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest. 1985;52:334-353.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29(2, pt 1):143-156.
- Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006;17:1489-1495.
- Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642-646.
- Buell JF, Trofe J, Hanaway MJ, et al. Immunosuppression and Merkel cell cancer. Transplant Proc. 2002;34:1780-1781.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Clarke CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 2015;107. pii:dju382. doi:10.1093/jnci/dju382.
- Rockville Merkel Cell Carcinoma Group. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis and clinical management [published online July 13, 2009]. J Clin Oncol. 2009;27:4021-4026.
- Krejčí K, Tichý T, Horák P, et al. Merkel cell carcinoma of the gluteal region with ipsilateral metastasis into the pancreatic graft of a patient after combined kidney-pancreas transplantation [published online September 20, 2010]. Onkologie. 2010;33:520-524.
- Arron ST, Canavan T, Yu SS. Organ transplant recipients with Merkel cell carcinoma have reduced progression-free, overall, and disease-specific survival independent of stage at presentation [published online July 1, 2014]. J Am Acad Dermatol. 2014;71:684-690.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study [published online July 23, 2009]. J Cutan Pathol. 2010;37:20-27.
- Eng TY, Boersma MG, Fuller CD, et al. Treatment of Merkel cell carcinoma. Am J Clin Oncol. 2004;27:510-515.
Merkel cell carcinoma (MCC) is a rare cutaneous neuroendocrine tumor derived from the nerve-associated Merkel cell touch receptors.1 It typically presents as a solitary, rapidly growing, red to violaceous, asymptomatic nodule, though ulcerated, acneform, and cystic lesions also have been described.2 Merkel cell carcinoma follows an aggressive clinical course with a tendency for rapid growth, local recurrence (26%–60% of cases), lymph node invasion, and distant metastases (18%–52% of cases).3
Several risk factors contribute to the development of MCC, including chronic immunosuppression, exposure to UV radiation, and infection with the Merkel cell polyomavirus. Immunosuppression has been shown to increase the risk for MCC and is associated with a worse prognosis independent of stage at diagnosis.4 Organ transplant recipients represent a subset of immunosuppressed patients who are at increased risk for the development of MCC. We report a case of metastatic MCC in a 67-year-old woman 6 years after liver transplantation.
Case Report
A 67-year-old woman presented to our clinic with 2 masses—1 on the left buttock and 1 on the left hip—of 4 months’ duration. The patient’s medical history was remarkable for autoimmune hepatitis requiring liver transplantation 6 years prior as well as hypertension and thyroid disorder. Her posttransplantation course was unremarkable, and she was maintained on chronic immunosuppression with tacrolimus and mycophenolate mofetil. Six years after transplantation, the patient was observed to have a 4-cm, red-violaceous, painless, dome-shaped tumor on the left buttock (Figure 1). She also was noted to have pink-red papulonodules forming a painless 8-cm plaque on the left hip that was present for 2 weeks prior to presentation (Figure 1). Both lesions were subsequently biopsied.
Microscopic examination of both lesions was consistent with the diagnosis of MCC. On histopathology, both samples exhibited a dense cellular dermis composed of atypical basophilic tumor cells with extension into superficial dilated lymphatic channels indicating lymphovascular invasion (Figure 2). Tumor cells were positive for the immunohistochemical markers pankeratin AE1/AE3, CAM 5.2, cytokeratin 20, synaptophysin, chromogranin A, and Merkel cell polyomavirus.
Total-body computed tomography and positron emission tomography revealed a hypermetabolic lobular density in the left gluteal region measuring 3.9×1.1 cm. The mass was associated with avid disease involving the left inguinal, bilateral iliac chain, and retroperitoneal lymph nodes. The patient was determined to have stage IV MCC based on the presence of distant lymph node metastases. The mass on the left hip was identified as an in-transit metastasis from the primary tumor on the left buttock.
The patient was referred to surgical and medical oncology. The decision was made to start palliative chemotherapy without surgical intervention given the extent of metastases not amenable for resection. The patient was subsequently initiated on chemotherapy with etoposide and carboplatin. After one cycle of chemotherapy, both tumors initially decreased in size; however, 4 months later, despite multiple cycles of chemotherapy, the patient was noted to have growth of existing tumors and interval development of a new 7×5-cm erythematous plaque in the left groin (Figure 3A) and a 1.1×1.0-cm smooth nodule on the right upper back (Figure 3B), both also found to be consistent with distant skin metastases of MCC upon microscopic examination after biopsy. Despite chemotherapy, the patient’s tumor continued to spread and the patient died within 8 months of diagnosis.
Comment
Transplant recipients represent a well-described cohort of immunosuppressed patients prone to the development of MCC. Merkel cell carcinoma in organ transplant recipients has been most frequently documented to occur after kidney transplantation and less frequently after heart and liver transplantations.5,6 However, the role of organ type and immunosuppressive regimen is not well characterized in the literature. Clarke et al7 investigated the risk for MCC in a large cohort of solid organ transplant recipients based on specific immunosuppression medications. They found a higher risk for MCC in patients who were maintained on cyclosporine, azathioprine, and mTOR (mechanistic target of rapamycin) inhibitors rather than tacrolimus, mycophenolate mofetil, and corticosteroids. In comparison to combination tacrolimus–mycophenolate mofetil, cyclosporine-azathioprine was associated with an increased incidence of MCC; this risk rose remarkably in patients who resided in geographic locations with a higher average of UV exposure. The authors suggested that UV radiation and immunosuppression-induced DNA damage may be synergistic in the development of MCC.7
Merkel cell carcinoma most frequently occurs on sun-exposed sites, including the face, head, and neck (55%); upper and lower extremities (40%); and truncal regions (5%).8 However, case reports highlight MCC arising in atypical locations such as the buttocks and gluteal region in organ transplant recipients.7,9 In the general population, MCC predominantly arises in elderly patients (ie, >70 years), but it is more likely to present at an earlier age in transplant recipients.6,10 In a retrospective analysis of 41 solid organ transplant recipients, 12 were diagnosed before the age of 50 years.6 Data from the US Scientific Registry of Transplant Recipients showed a median age at diagnosis of 62 years, with the highest incidence occurring 10 or more years after transplantation.7
Merkel cell carcinoma behaves aggressively and is the most common cause of skin cancer death after melanoma.11 Organ transplant recipients with MCC have a worse prognosis than MCC patients who are not transplant recipients. In a retrospective registry analysis of 45 de novo cases, Buell at al5 found a 60% mortality rate in transplant recipients, almost double the 33% mortality rate of the general population. Furthermore, Arron et al10 revealed substantially increased rates of disease progression and decreased rates of disease-specific and overall survival in solid organ transplant recipients on immunosuppression compared to immunocompetent controls. The most important factor for poor prognosis is the presence of lymph node invasion, which lowers survival rate.12
Conclusion
Merkel cell carcinoma following liver transplantation is not well described in the literature. We highlight a case of an aggressive MCC arising in a sun-protected site with rapid metastasis 6 years after liver transplantation. This case emphasizes the importance of surveillance for cutaneous malignancy in solid organ transplant recipients.
Merkel cell carcinoma (MCC) is a rare cutaneous neuroendocrine tumor derived from the nerve-associated Merkel cell touch receptors.1 It typically presents as a solitary, rapidly growing, red to violaceous, asymptomatic nodule, though ulcerated, acneform, and cystic lesions also have been described.2 Merkel cell carcinoma follows an aggressive clinical course with a tendency for rapid growth, local recurrence (26%–60% of cases), lymph node invasion, and distant metastases (18%–52% of cases).3
Several risk factors contribute to the development of MCC, including chronic immunosuppression, exposure to UV radiation, and infection with the Merkel cell polyomavirus. Immunosuppression has been shown to increase the risk for MCC and is associated with a worse prognosis independent of stage at diagnosis.4 Organ transplant recipients represent a subset of immunosuppressed patients who are at increased risk for the development of MCC. We report a case of metastatic MCC in a 67-year-old woman 6 years after liver transplantation.
Case Report
A 67-year-old woman presented to our clinic with 2 masses—1 on the left buttock and 1 on the left hip—of 4 months’ duration. The patient’s medical history was remarkable for autoimmune hepatitis requiring liver transplantation 6 years prior as well as hypertension and thyroid disorder. Her posttransplantation course was unremarkable, and she was maintained on chronic immunosuppression with tacrolimus and mycophenolate mofetil. Six years after transplantation, the patient was observed to have a 4-cm, red-violaceous, painless, dome-shaped tumor on the left buttock (Figure 1). She also was noted to have pink-red papulonodules forming a painless 8-cm plaque on the left hip that was present for 2 weeks prior to presentation (Figure 1). Both lesions were subsequently biopsied.
Microscopic examination of both lesions was consistent with the diagnosis of MCC. On histopathology, both samples exhibited a dense cellular dermis composed of atypical basophilic tumor cells with extension into superficial dilated lymphatic channels indicating lymphovascular invasion (Figure 2). Tumor cells were positive for the immunohistochemical markers pankeratin AE1/AE3, CAM 5.2, cytokeratin 20, synaptophysin, chromogranin A, and Merkel cell polyomavirus.
Total-body computed tomography and positron emission tomography revealed a hypermetabolic lobular density in the left gluteal region measuring 3.9×1.1 cm. The mass was associated with avid disease involving the left inguinal, bilateral iliac chain, and retroperitoneal lymph nodes. The patient was determined to have stage IV MCC based on the presence of distant lymph node metastases. The mass on the left hip was identified as an in-transit metastasis from the primary tumor on the left buttock.
The patient was referred to surgical and medical oncology. The decision was made to start palliative chemotherapy without surgical intervention given the extent of metastases not amenable for resection. The patient was subsequently initiated on chemotherapy with etoposide and carboplatin. After one cycle of chemotherapy, both tumors initially decreased in size; however, 4 months later, despite multiple cycles of chemotherapy, the patient was noted to have growth of existing tumors and interval development of a new 7×5-cm erythematous plaque in the left groin (Figure 3A) and a 1.1×1.0-cm smooth nodule on the right upper back (Figure 3B), both also found to be consistent with distant skin metastases of MCC upon microscopic examination after biopsy. Despite chemotherapy, the patient’s tumor continued to spread and the patient died within 8 months of diagnosis.
Comment
Transplant recipients represent a well-described cohort of immunosuppressed patients prone to the development of MCC. Merkel cell carcinoma in organ transplant recipients has been most frequently documented to occur after kidney transplantation and less frequently after heart and liver transplantations.5,6 However, the role of organ type and immunosuppressive regimen is not well characterized in the literature. Clarke et al7 investigated the risk for MCC in a large cohort of solid organ transplant recipients based on specific immunosuppression medications. They found a higher risk for MCC in patients who were maintained on cyclosporine, azathioprine, and mTOR (mechanistic target of rapamycin) inhibitors rather than tacrolimus, mycophenolate mofetil, and corticosteroids. In comparison to combination tacrolimus–mycophenolate mofetil, cyclosporine-azathioprine was associated with an increased incidence of MCC; this risk rose remarkably in patients who resided in geographic locations with a higher average of UV exposure. The authors suggested that UV radiation and immunosuppression-induced DNA damage may be synergistic in the development of MCC.7
Merkel cell carcinoma most frequently occurs on sun-exposed sites, including the face, head, and neck (55%); upper and lower extremities (40%); and truncal regions (5%).8 However, case reports highlight MCC arising in atypical locations such as the buttocks and gluteal region in organ transplant recipients.7,9 In the general population, MCC predominantly arises in elderly patients (ie, >70 years), but it is more likely to present at an earlier age in transplant recipients.6,10 In a retrospective analysis of 41 solid organ transplant recipients, 12 were diagnosed before the age of 50 years.6 Data from the US Scientific Registry of Transplant Recipients showed a median age at diagnosis of 62 years, with the highest incidence occurring 10 or more years after transplantation.7
Merkel cell carcinoma behaves aggressively and is the most common cause of skin cancer death after melanoma.11 Organ transplant recipients with MCC have a worse prognosis than MCC patients who are not transplant recipients. In a retrospective registry analysis of 45 de novo cases, Buell at al5 found a 60% mortality rate in transplant recipients, almost double the 33% mortality rate of the general population. Furthermore, Arron et al10 revealed substantially increased rates of disease progression and decreased rates of disease-specific and overall survival in solid organ transplant recipients on immunosuppression compared to immunocompetent controls. The most important factor for poor prognosis is the presence of lymph node invasion, which lowers survival rate.12
Conclusion
Merkel cell carcinoma following liver transplantation is not well described in the literature. We highlight a case of an aggressive MCC arising in a sun-protected site with rapid metastasis 6 years after liver transplantation. This case emphasizes the importance of surveillance for cutaneous malignancy in solid organ transplant recipients.
- Gould VE, Moll R, Moll I, et al. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest. 1985;52:334-353.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29(2, pt 1):143-156.
- Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006;17:1489-1495.
- Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642-646.
- Buell JF, Trofe J, Hanaway MJ, et al. Immunosuppression and Merkel cell cancer. Transplant Proc. 2002;34:1780-1781.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Clarke CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 2015;107. pii:dju382. doi:10.1093/jnci/dju382.
- Rockville Merkel Cell Carcinoma Group. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis and clinical management [published online July 13, 2009]. J Clin Oncol. 2009;27:4021-4026.
- Krejčí K, Tichý T, Horák P, et al. Merkel cell carcinoma of the gluteal region with ipsilateral metastasis into the pancreatic graft of a patient after combined kidney-pancreas transplantation [published online September 20, 2010]. Onkologie. 2010;33:520-524.
- Arron ST, Canavan T, Yu SS. Organ transplant recipients with Merkel cell carcinoma have reduced progression-free, overall, and disease-specific survival independent of stage at presentation [published online July 1, 2014]. J Am Acad Dermatol. 2014;71:684-690.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study [published online July 23, 2009]. J Cutan Pathol. 2010;37:20-27.
- Eng TY, Boersma MG, Fuller CD, et al. Treatment of Merkel cell carcinoma. Am J Clin Oncol. 2004;27:510-515.
- Gould VE, Moll R, Moll I, et al. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest. 1985;52:334-353.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29(2, pt 1):143-156.
- Pectasides D, Pectasides M, Economopoulos T. Merkel cell cancer of the skin. Ann Oncol. 2006;17:1489-1495.
- Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642-646.
- Buell JF, Trofe J, Hanaway MJ, et al. Immunosuppression and Merkel cell cancer. Transplant Proc. 2002;34:1780-1781.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Clarke CA, Robbins HA, Tatalovich Z, et al. Risk of Merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 2015;107. pii:dju382. doi:10.1093/jnci/dju382.
- Rockville Merkel Cell Carcinoma Group. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis and clinical management [published online July 13, 2009]. J Clin Oncol. 2009;27:4021-4026.
- Krejčí K, Tichý T, Horák P, et al. Merkel cell carcinoma of the gluteal region with ipsilateral metastasis into the pancreatic graft of a patient after combined kidney-pancreas transplantation [published online September 20, 2010]. Onkologie. 2010;33:520-524.
- Arron ST, Canavan T, Yu SS. Organ transplant recipients with Merkel cell carcinoma have reduced progression-free, overall, and disease-specific survival independent of stage at presentation [published online July 1, 2014]. J Am Acad Dermatol. 2014;71:684-690.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study [published online July 23, 2009]. J Cutan Pathol. 2010;37:20-27.
- Eng TY, Boersma MG, Fuller CD, et al. Treatment of Merkel cell carcinoma. Am J Clin Oncol. 2004;27:510-515.
Practice Points
- Organ transplant recipients are at an increased risk for Merkel cell carcinoma (MCC).
- Early recognition and diagnosis of MCC is important to improve morbidity and mortality.
Ex Vivo Confocal Microscopy: A Diagnostic Tool for Skin Malignancies
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
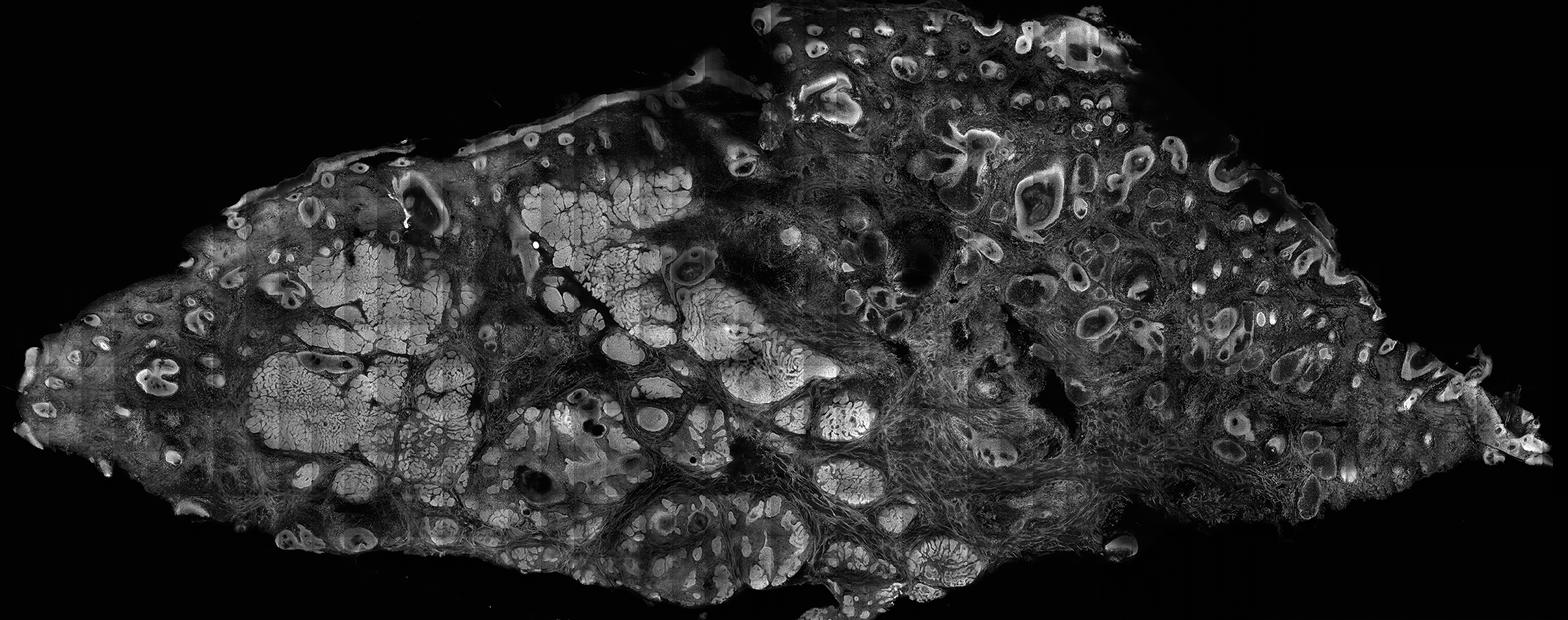
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
Skin cancer is diagnosed in approximately 5.4 million individuals annually in the United States, more than the total number of breast, lung, colon, and prostate cancers diagnosed per year.1 It is estimated that 1 in 5 Americans will develop skin cancer during their lifetime.2 The 2 most common forms of skin cancer are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), accounting for 4 million and 1 million cases diagnosed each year, respectively.3 With the increasing incidence of these skin cancers, the use of noninvasive imaging tools for detection and diagnosis has grown.
Ex vivo confocal microscopy is a diagnostic imaging tool that can be used in real-time at the bedside to assess freshly excised tissue for malignancies. It images tissue samples with cellular resolution and within minutes of biopsy or excision. Ex vivo confocal microscopy is a versatile tool that can assist in the diagnosis and management of skin malignancies such as melanoma, BCC, and SCC.
Reflectance vs Fluorescence Mode
Excised lesions can be examined in reflectance or fluorescence mode in great detail but with slightly varying nuclear-to-dermis contrasts depending on the chromophore that is targeted. In reflectance mode (reflectance confocal microscopy [RCM]), melanin and keratin act as endogenous chromophores because of their high refractive index relative to water,4,5 which allows for the visualization of cellular structures of the skin at low power, as well as microscopic substructures such as melanosomes, cytoplasmic granules, and other cellular organelles at high power. Although an exogenous contrast agent is not required, acetic acid has the capability to highlight nuclei, enhancing the tumor cell-to-dermis contrast in RCM.6 Acetic acid is clinically used as a predictor for certain skin and mucosal membrane neoplasms that blanch when exposed to the solution. In the case of RCM, acetic acid increases the visibility of nuclei by inducing the compaction of chromatin. For the acetowhitening to be effective, the sample must be soaked in the solution for a specific amount of time, depending on the concentration.7 A concentration between 1% and 10% can be used, but the less concentrated the solution, the longer the time of soaking that is required to achieve sufficiently bright nuclei.6
The contrast with acetic acid, however, is quite weak when the tissue is imaged en face, or along the horizontal surface of the sample, due to the collagen in the dermal layer, which has a high reflectance index. This issue is rectified when using the confocal microscope in the fluorescence mode with an exogenous fluorescent dye as a nuclear stain. Fluorescence confocal microscopy (FCM), results in a stronger nuclear-to-dermal contrast because of the role of contrast agents.8 The 1000-fold increase in contrast between nuclei and dermis is the result of dye agents that preferentially bind to nuclear DNA, of which acridine orange is the most commonly used.5,8 Basal cell carcinoma and SCC tumor cells can be visualized with FCM because they appear hyperfluorescent when stained with acridine orange.9 The acridine orange–stained cells display bright nuclei, while the cytoplasm and collagen remains dark. A positive feature of acridine orange is that it does not alter the tissue sample during freezing or formalin fixation and thus has no effect on subsequent histopathology that may need to be performed on the sample.10
High-Resolution Images Aid in Diagnosis
After it is harvested, the tissue sample is soaked in a contrast agent or dye, if needed, depending on the confocal mode to be used. The confocal microscope is then used to take a series of high-resolution individual en face images that are then stitched together to create a final mosaic image that can be up to 12×12 mm.6,11 With a 200-µm depth visibility, confocal microscopy can capture the cellular structures in the epidermis, dermis, and (if compressed enough) subcutaneous fat in just under 3 minutes.12
The images produced through confocal microscopy have an excellent correlation to frozen histological sections and can aid in the diagnosis of many epidermal and dermal malignancies including melanoma, BCC, and SCC. New criteria have been established to aid in the interpretation of the confocal images and identify some of the more common skin cancers.5,12,13 Basal cell carcinoma samples imaged through fluorescence and reflectance in low-power mode display the distinct nodular patterns with well-demarcated edges, as seen on classical histopathology. In the case of FCM, the cells that make up the tumor display hyperfluorescent areas consistent with nucleated cells that are stained with acridine orange. The main features that identify BCC on FCM images include nuclear pleomorphism and crowding, peripheral palisading, clefting of the basaloid islands, increased nucleus-to-cytoplasm ratio, and the presence of a modified dermis surrounding the mass known as the tumoral stroma5,12 (Figure).
In addition to fluorescence and a well-defined tumor silhouette, SCC under FCM displays keratin pearls composed of keratinized squames, nuclear pleomorphism, and fluorescent scales in the stratum corneum that are a result of keratin formation.5,13 The extent of differentiation of the SCC lesion also can be determined by assessing if the silhouette is well defined. A well-defined tumor silhouette is consistent with the diagnosis of a well-differentiated SCC, and vice versa.13 Ex vivo RCM also has been shown to be useful in diagnosing malignant melanomas, with melanin acting as an endogenous chromophore. Some of the features seen on imaging include a disarranged epithelium, hyperreflective roundish and dendritic pagetoid cells, and large hyperreflective polymorphic cells in the superficial chorion.14
Comparison to Conventional Histopathology
Ex vivo confocal microscopy in both the reflectance and fluorescence mode has been shown to perform well compared to conventional histopathology in the diagnosis of biopsy specimens. Ex vivo FCM has been shown to have an overall sensitivity of 88% and specificity of 99% in detecting residual BCC at the margins of excised tissue samples and in the fraction of the time it takes to attain similar results with frozen histopathology.9 Ex vivo RCM has been shown to have a higher prognostic capability, with 100% sensitivity and specificity in identifying BCC when scanning the tissue samples en face.15
Qualitatively, the images produced by RCM and FCM are similar to histopathology in overall architecture. Both techniques enhance the contrast between the epithelium and stroma and create images that can be examined in low as well as high resolution. A substantial difference between confocal microscopy and conventional hematoxylin and eosin–stained histopathology is that the confocal microscope produces images in gray scale. One way to alter the black-and-white images to resemble hematoxylin and eosin–stained slides is through the use of digital staining,16 which could boost clinical acceptance by physicians who are accustomed to the classical pink-purple appearance of pathology slides and could potentially limit the learning curve needed to read the confocal images.
Application in Mohs Micrographic Surgery
An important clinical application of ex vivo FCM imaging that has emerged is the detection of malignant cells at the excision margins during Mohs micrographic surgery. The use of confocal microscopy has the potential to save time by eliminating the need for tissue fixation while still providing good diagnostic accuracy. Implementing FCM as an imaging tool to guide surgical excisions could provide rapid diagnosis of the tissue, expediting excisions and reconstruction or the Mohs procedure while eliminating patient wait time and the need for frozen histopathology. Ex vivo RCM also has been used to establish laser parameters for CO2 laser ablation of superficial and early nodular BCC lesions.17 Other potential uses for ex vivo RCM/FCM could include rapid evaluation of tissue during operating room procedures where rapid frozen sections are currently utilized.
Combining In Vivo and Ex Vivo Confocal Microscopy
Many of the diagnostic guidelines created with the use of ex vivo confocal microscopy have been applied to in vivo use, and therefore the use of both modalities is appealing. In vivo confocal microscopy is a noninvasive technique that has been used to map margins of skin tumors such as BCC and lentigo maligna at the bedside.5 It also has been shown to help plan both surgical and nonsurgical treatment modalities and reconstruction before the tumor is excised.18 This technique also can help the patient understand the extent of the excision and any subsequent reconstruction that may be needed.
Limitations
Ex vivo confocal microscopy used as a diagnostic tool does have some limitations. Its novelty may require surgeons and pathologists to be trained to interpret the images properly and correlate them to conventional diagnostic guidelines. The imaging also is limited to a depth of approximately 200 µm; however, the sample may be flipped so that the underside can be imaged as well, which increases the depth to approximately 400 µm. The tissue being imaged must be fixed flat, which may alter its shape. Complex tissue samples may be difficult to flatten out completely and therefore may be difficult to image. A special mount may be required for the sample to be fixed in a proper position for imaging.6
Final Thoughts
Despite some of these limitations, the need for rapid bedside tissue diagnosis makes ex vivo confocal microscopy an attractive device that can be used as an additional diagnostic tool to histopathology and also has been tested in other disciplines, such as breast cancer pathology. In the future, both in vivo and ex vivo confocal microscopy may be utilized to diagnose cutaneous malignancies, guide surgical excisions, and detect lesion progression, and it may become a basis for rapid diagnosis and detection.19
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [published online January 7, 2016]. CA Cancer J Clin. 2016;66:7-30.
- Robinson JK. Sun exposure, sun protection, and vitamin D. JAMA. 2005;294:1541-1543.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Welzel J, Kästle R, Sattler EC. Fluorescence (multiwave) confocal microscopy. Dermatol Clin. 2016;34:527-533.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In vivo and ex vivo confocal microscopy for dermatologic and Mohs surgeons. Dermatol Clin. 2016;34:497-504.
- Patel YG, Nehal KS, Aranda I, et al. Confocal reflectance mosaicing of basal cell carcinomas in Mohs surgical skin excisions. J Biomed Opt. 2007;12:034027.
- Rajadhyaksha M, Gonzalez S, Zavislan JM. Detectability of contrast agents for confocal reflectance imaging of skin and microcirculation. J Biomed Opt. 2004;9:323-331.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of basal cell carcinomas in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol. 2009;160:1242-1250.
- Bennàssar A, Vilata A, Puig S, et al. Ex vivo fluorescence confocal microscopy for fast evaluation of tumour margins during Mohs surgery. Br J Dermatol. 2014;170:360-365.
- Gareau DS, Li Y, Huang B, et al. Confocal mosaicing microscopy in Mohs skin excisions: feasibility of rapid surgical pathology. J Biomed Opt. 2008;13:054001.
- Bini J, Spain J, Nehal K, et al. Confocal mosaicing microscopy of human skin ex vivo: spectral analysis for digital staining to simulate histology-like appearance. J Biomed Opt. 2011;16:076008.
- Bennàssar A, Carrera C, Puig S, et al. Fast evaluation of 69 basal cell carcinomas with ex vivo fluorescence confocal microscopy: criteria description, histopathological correlation, and interobserver agreement. JAMA Dermatol. 2013;149:839-847.
- Longo C, Ragazzi M, Gardini S, et al. Ex vivo fluorescence confocal microscopy in conjunction with Mohs micrographic surgery for cutaneous squamous cell carcinoma. J Am Acad Dermatol. 2015;73:321-322.
- Cinotti E, Haouas M, Grivet D, et al. In vivo and ex vivo confocal microscopy for the management of a melanoma of the eyelid margin. Dermatol Surg. 2015;41:1437-1440.
- , , , ‘En face’ ex vivo reflectance confocal microscopy to help the surgery of basal cell carcinoma of the eyelid [published online December 19, 2016]. Clin Exp Ophthalmol. doi:10.1111/ceo.12904.
- Gareau DS, Jeon H, Nehal KS, et al. Rapid screening of cancer margins in tissue with multimodal confocal microscopy. J Surg Res. 2012;178:533-538.
- Sierra H, Damanpour S, Hibler B, et al. Confocal imaging of carbon dioxide laser-ablated basal cell carcinomas: an ex-vivo study on the uptake of contrast agent and ablation parameters [published online September 22, 2015]. Lasers Surg Med. 2016;48:133-139.
- Hibler BP, Yélamos O, Cordova M, et al. Handheld reflectance confocal microscopy to aid in the management of complex facial lentigo maligna. Cutis. 2017;99:346-352.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside. Lasers Surg Med. 2017;49:7-19.
Practice Points
- Confocal microscopy is an imaging tool that can be used both in vivo and ex vivo to aid in the diagnosis and management of cutaneous neoplasms, including melanoma, basal cell carcinoma, and squamous cell carcinoma, as well as inflammatory dermatoses.
- Ex vivo confocal microscopy can be used in both reflectance and fluorescent modes to render diagnosis in excised tissue or check surgical margins.
- Both in vivo and ex vivo confocal microscopy produces images with cellular resolution with a main limitation being depth of imaging.
What’s on the dermatopathologist’s wish list
NEW YORK – If dermatopathologists had a wish list they could give their dermatologist colleagues, what might it include? High up on the list for many, said Robert Phelps, MD, might be to have them share the clinical picture, treat the specimen gently, and give the best landmarks possible.
Speaking at the summer meeting of the American Academy of Dermatology, Dr. Phelps, director of the dermatopathology service at Mount Sinai Medical Center in New York, led off the dermatopathologist-run session – appropriately titled “Help Me Help You” – by asking, “How can the clinician provide the optimal biopsy?”
It’s always helpful to have as much clinical information as possible, said Dr. Phelps, whose discussion focused on tips for neoplastic lesions. This might include prior history of malignancy, autoimmune disease, pathergy, or other relevant medical history, but clinical pictures can also be a big help, although there can be technical and patient privacy issues to overcome, he noted. If, for example, a larger lesion or rash is being biopsied rather than excised, it can be very helpful to see the larger field and full area of distribution of the lesion in question. Submitting multiple specimens for rashes and larger lesions is always a good idea too, he added.
Although curettage can be a great way to biopsy – and perhaps even definitively treat some lesions – problems can arise on the dermatopathologist’s side when melanocytic lesions are curetted for biopsy, according to Dr. Phelps, a practicing dermatologist and a dermatopathologist. “By virtue of the force of the biopsy, the specimen is often fragmented, and histology can be distorted,” he said. One element of that distortion can be that melanocytes can appear to be free floating, which is a problem. “Dyshesion of melanocytes is usually an indication of atypia … It is an important histologic clue as to the possibility of a malignancy supervening.”
These factors can make it tough for a dermatopathologist to make an accurate call. “If there are free-floating melanocytes from a curetted specimen, I can’t rule out invasive melanoma,” explained Dr. Phelps, since he can’t tell if he is seeing true atypia or disruption that’s an artifact of the collection technique.
In this instance, he said, a dermatopathologist would be “obligated to overcall, because one couldn’t really determine the pathology.” The bottom line? “Don’t curette biopsies of melanocytic lesions.”
Another technique that can interfere with the ability to read a tissue specimen accurately is electrodesiccation. Although it’s often performed in conjunction with curettage, electrodesiccation can cause changes in tissue consistent with thermal injury. “Essentially, the tissue has been burned,” Dr. Phelps pointed out. This can result in a characteristic streaming pattern of nuclei, and the dermis can acquire a “peculiar homogenized appearance,” he said.
Although electrodesiccation can be a useful technique to make sure margins are controlled, “when you do this, just be aware that the interpretation is difficult,” he noted. “It’s difficult to tell where the margins are and if they are the appropriate and correct margins,” he said.
When possible, try to avoid squeezing the tumor, Dr. Phelps advised. Excessive pressure on the specimen can distort cell architecture and make pathological diagnosis really challenging, particularly in lymphoid tumors, he said.
“Often, the tumor is not recognizable,” he added. Crush artifact can result in an appearance of small bluish clumps and smearing of collagen fibers. The effect, he said, can be particularly pronounced with small cell carcinoma and lymphoma, and with rapidly proliferating tumors.
Dr. Phelps said that during his training, he was taught not to use forceps to extract a stubborn punch biopsy specimen; rather, he was trained to use a needle to tease out the specimen. Fear of a self-inflicted needle stick with this technique may be a deterrent, he acknowledged. If forceps are used, he suggested being as gentle as possible and using the finest forceps available.
When pathologists receive an intact excised lesion – one not obtained using a Mohs technique, “delineation of the margin is essential,” Dr. Phelps said. Further, accurate mapping is critical to helping the examiner understand the anatomic orientation of the specimen, a key prerequisite that enables accurate communication from the dermatopathologist back to the clinician if there’s a question regarding the need for retreatment, he added.
For an elliptical excision, ideally, both poles of the ellipse would be suture-tagged, and at least one tag is essential, he said. Then superior and inferior borders can be inked with contrasting colors, and the epidermal borders of the lesion should be marked as well. When the specimen is submitted, it should be accompanied by an accurate map that clearly indicates the coding for medial, lateral, inferior, and superior aspects of the specimen. “Always prepare a specimen diagram for oriented specimens,” Dr. Phelps noted.
Don’t forget to make sure that the left-right orientation on the diagram corresponds to the specimen’s orientation on the patient, he added. Some facilities use a clock face system to indicate orientation and positioning, which may be the clearest method of all.
Sometimes, it’s difficult for the dermatopathologist to visualize whether the specimen is aligned in true medial-lateral fashion, or along skin tension lines, which tend to run diagonally, so “the more clinical information, the better,” he said. “With good mapping, precise retreatment can be optimal,” he said.
Dr. Phelps reported that he had no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
NEW YORK – If dermatopathologists had a wish list they could give their dermatologist colleagues, what might it include? High up on the list for many, said Robert Phelps, MD, might be to have them share the clinical picture, treat the specimen gently, and give the best landmarks possible.
Speaking at the summer meeting of the American Academy of Dermatology, Dr. Phelps, director of the dermatopathology service at Mount Sinai Medical Center in New York, led off the dermatopathologist-run session – appropriately titled “Help Me Help You” – by asking, “How can the clinician provide the optimal biopsy?”
It’s always helpful to have as much clinical information as possible, said Dr. Phelps, whose discussion focused on tips for neoplastic lesions. This might include prior history of malignancy, autoimmune disease, pathergy, or other relevant medical history, but clinical pictures can also be a big help, although there can be technical and patient privacy issues to overcome, he noted. If, for example, a larger lesion or rash is being biopsied rather than excised, it can be very helpful to see the larger field and full area of distribution of the lesion in question. Submitting multiple specimens for rashes and larger lesions is always a good idea too, he added.
Although curettage can be a great way to biopsy – and perhaps even definitively treat some lesions – problems can arise on the dermatopathologist’s side when melanocytic lesions are curetted for biopsy, according to Dr. Phelps, a practicing dermatologist and a dermatopathologist. “By virtue of the force of the biopsy, the specimen is often fragmented, and histology can be distorted,” he said. One element of that distortion can be that melanocytes can appear to be free floating, which is a problem. “Dyshesion of melanocytes is usually an indication of atypia … It is an important histologic clue as to the possibility of a malignancy supervening.”
These factors can make it tough for a dermatopathologist to make an accurate call. “If there are free-floating melanocytes from a curetted specimen, I can’t rule out invasive melanoma,” explained Dr. Phelps, since he can’t tell if he is seeing true atypia or disruption that’s an artifact of the collection technique.
In this instance, he said, a dermatopathologist would be “obligated to overcall, because one couldn’t really determine the pathology.” The bottom line? “Don’t curette biopsies of melanocytic lesions.”
Another technique that can interfere with the ability to read a tissue specimen accurately is electrodesiccation. Although it’s often performed in conjunction with curettage, electrodesiccation can cause changes in tissue consistent with thermal injury. “Essentially, the tissue has been burned,” Dr. Phelps pointed out. This can result in a characteristic streaming pattern of nuclei, and the dermis can acquire a “peculiar homogenized appearance,” he said.
Although electrodesiccation can be a useful technique to make sure margins are controlled, “when you do this, just be aware that the interpretation is difficult,” he noted. “It’s difficult to tell where the margins are and if they are the appropriate and correct margins,” he said.
When possible, try to avoid squeezing the tumor, Dr. Phelps advised. Excessive pressure on the specimen can distort cell architecture and make pathological diagnosis really challenging, particularly in lymphoid tumors, he said.
“Often, the tumor is not recognizable,” he added. Crush artifact can result in an appearance of small bluish clumps and smearing of collagen fibers. The effect, he said, can be particularly pronounced with small cell carcinoma and lymphoma, and with rapidly proliferating tumors.
Dr. Phelps said that during his training, he was taught not to use forceps to extract a stubborn punch biopsy specimen; rather, he was trained to use a needle to tease out the specimen. Fear of a self-inflicted needle stick with this technique may be a deterrent, he acknowledged. If forceps are used, he suggested being as gentle as possible and using the finest forceps available.
When pathologists receive an intact excised lesion – one not obtained using a Mohs technique, “delineation of the margin is essential,” Dr. Phelps said. Further, accurate mapping is critical to helping the examiner understand the anatomic orientation of the specimen, a key prerequisite that enables accurate communication from the dermatopathologist back to the clinician if there’s a question regarding the need for retreatment, he added.
For an elliptical excision, ideally, both poles of the ellipse would be suture-tagged, and at least one tag is essential, he said. Then superior and inferior borders can be inked with contrasting colors, and the epidermal borders of the lesion should be marked as well. When the specimen is submitted, it should be accompanied by an accurate map that clearly indicates the coding for medial, lateral, inferior, and superior aspects of the specimen. “Always prepare a specimen diagram for oriented specimens,” Dr. Phelps noted.
Don’t forget to make sure that the left-right orientation on the diagram corresponds to the specimen’s orientation on the patient, he added. Some facilities use a clock face system to indicate orientation and positioning, which may be the clearest method of all.
Sometimes, it’s difficult for the dermatopathologist to visualize whether the specimen is aligned in true medial-lateral fashion, or along skin tension lines, which tend to run diagonally, so “the more clinical information, the better,” he said. “With good mapping, precise retreatment can be optimal,” he said.
Dr. Phelps reported that he had no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
NEW YORK – If dermatopathologists had a wish list they could give their dermatologist colleagues, what might it include? High up on the list for many, said Robert Phelps, MD, might be to have them share the clinical picture, treat the specimen gently, and give the best landmarks possible.
Speaking at the summer meeting of the American Academy of Dermatology, Dr. Phelps, director of the dermatopathology service at Mount Sinai Medical Center in New York, led off the dermatopathologist-run session – appropriately titled “Help Me Help You” – by asking, “How can the clinician provide the optimal biopsy?”
It’s always helpful to have as much clinical information as possible, said Dr. Phelps, whose discussion focused on tips for neoplastic lesions. This might include prior history of malignancy, autoimmune disease, pathergy, or other relevant medical history, but clinical pictures can also be a big help, although there can be technical and patient privacy issues to overcome, he noted. If, for example, a larger lesion or rash is being biopsied rather than excised, it can be very helpful to see the larger field and full area of distribution of the lesion in question. Submitting multiple specimens for rashes and larger lesions is always a good idea too, he added.
Although curettage can be a great way to biopsy – and perhaps even definitively treat some lesions – problems can arise on the dermatopathologist’s side when melanocytic lesions are curetted for biopsy, according to Dr. Phelps, a practicing dermatologist and a dermatopathologist. “By virtue of the force of the biopsy, the specimen is often fragmented, and histology can be distorted,” he said. One element of that distortion can be that melanocytes can appear to be free floating, which is a problem. “Dyshesion of melanocytes is usually an indication of atypia … It is an important histologic clue as to the possibility of a malignancy supervening.”
These factors can make it tough for a dermatopathologist to make an accurate call. “If there are free-floating melanocytes from a curetted specimen, I can’t rule out invasive melanoma,” explained Dr. Phelps, since he can’t tell if he is seeing true atypia or disruption that’s an artifact of the collection technique.
In this instance, he said, a dermatopathologist would be “obligated to overcall, because one couldn’t really determine the pathology.” The bottom line? “Don’t curette biopsies of melanocytic lesions.”
Another technique that can interfere with the ability to read a tissue specimen accurately is electrodesiccation. Although it’s often performed in conjunction with curettage, electrodesiccation can cause changes in tissue consistent with thermal injury. “Essentially, the tissue has been burned,” Dr. Phelps pointed out. This can result in a characteristic streaming pattern of nuclei, and the dermis can acquire a “peculiar homogenized appearance,” he said.
Although electrodesiccation can be a useful technique to make sure margins are controlled, “when you do this, just be aware that the interpretation is difficult,” he noted. “It’s difficult to tell where the margins are and if they are the appropriate and correct margins,” he said.
When possible, try to avoid squeezing the tumor, Dr. Phelps advised. Excessive pressure on the specimen can distort cell architecture and make pathological diagnosis really challenging, particularly in lymphoid tumors, he said.
“Often, the tumor is not recognizable,” he added. Crush artifact can result in an appearance of small bluish clumps and smearing of collagen fibers. The effect, he said, can be particularly pronounced with small cell carcinoma and lymphoma, and with rapidly proliferating tumors.
Dr. Phelps said that during his training, he was taught not to use forceps to extract a stubborn punch biopsy specimen; rather, he was trained to use a needle to tease out the specimen. Fear of a self-inflicted needle stick with this technique may be a deterrent, he acknowledged. If forceps are used, he suggested being as gentle as possible and using the finest forceps available.
When pathologists receive an intact excised lesion – one not obtained using a Mohs technique, “delineation of the margin is essential,” Dr. Phelps said. Further, accurate mapping is critical to helping the examiner understand the anatomic orientation of the specimen, a key prerequisite that enables accurate communication from the dermatopathologist back to the clinician if there’s a question regarding the need for retreatment, he added.
For an elliptical excision, ideally, both poles of the ellipse would be suture-tagged, and at least one tag is essential, he said. Then superior and inferior borders can be inked with contrasting colors, and the epidermal borders of the lesion should be marked as well. When the specimen is submitted, it should be accompanied by an accurate map that clearly indicates the coding for medial, lateral, inferior, and superior aspects of the specimen. “Always prepare a specimen diagram for oriented specimens,” Dr. Phelps noted.
Don’t forget to make sure that the left-right orientation on the diagram corresponds to the specimen’s orientation on the patient, he added. Some facilities use a clock face system to indicate orientation and positioning, which may be the clearest method of all.
Sometimes, it’s difficult for the dermatopathologist to visualize whether the specimen is aligned in true medial-lateral fashion, or along skin tension lines, which tend to run diagonally, so “the more clinical information, the better,” he said. “With good mapping, precise retreatment can be optimal,” he said.
Dr. Phelps reported that he had no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
Topical Timolol May Improve Overall Scar Cosmesis in Acute Surgical Wounds
Timolol is a nonselective β-adrenergic receptor antagonist indicated for treating glaucoma, heart attacks, hypertension, and migraine headaches. It is made in both an oral and ophthalmic form. In dermatology, the beta-blocker propranolol is approved for the treatment of infantile hemangiomas (IHs). The exact mechanism of action of beta-blockers for the treatment of IHs is not yet completely understood, but it is postulated that they inhibit growth by at least 4 distinct mechanisms: (1) vasoconstriction, (2) inhibition of angiogenesis or vasculogenesis, (3) induction of apoptosis, and (4) recruitment of endothelial progenitor cells to the site of the hemangioma.1
Scar cosmesis can be calculated using the visual analog scale (VAS), which is a subjective scar assessment scored from poor to excellent. The multidimensional VAS is a photograph-based scale derived from evaluating standardized digital photographs in 4 dimensions—pigmentation, vascularity, acceptability, and observer comfort—plus contour. It uses the sum of the individual scores to obtain a single overall score ranging from excellent to poor.2 In this study, we sought to determine if the use of topical timolol after excision or Mohs micrographic surgery (MMS) treatment of nonmelanoma skin cancers improved the overall cosmesis of the scar.
Methods
The study protocol was approved by the institutional review board at Roger Williams Medical Center (Providence, Rhode Island). Eligibility criteria included patients who required excision or MMS for their nonmelanoma skin cancer located below the patella and those who agreed to allow their wounds to heal by secondary intention when given options for closure of their wounds. Patients were randomized to either the timolol (study medication) group or the saline (placebo) group. The initial defects were measured and photographed. Patients were educated on how to apply the study medication. All patients were prescribed 40 mm Hg compression stockings to wear following application of the study medication. Patients were asked to return at 1 and 5 weeks postsurgery and then every 1 to 2 weeks for wound assessment and measurement until their wounds had healed or at 13 weeks, depending on which came first. A healed wound was defined as having no exudate, exhibiting complete reepithelialization, and being stable for 1 week.
Healed wounds were assessed by a blinded outside dermatologist who examined photographs of the wounds and then completed the VAS for each participant’s scar.
Results
A total of 9 participants were enrolled in the study. Three participants were lost to follow-up; 6 completed the study (4 females, 2 males). The mean age was 70 years (age range, 46–89 years). The average wound size was 2×2 cm with a depth of 1 mm. Three participants were in the active medication group and 3 were in the control group.
A VAS was completed for each participant’s scar by an outside blinded dermatologist. Based on the VAS, wounds treated with timolol resulted in more cosmetically favorable scars (scored higher on the VAS) compared to control (mean [SD]: 6.5±0.9 vs 2.5±0.7; P<0.05). See Figures 1 and 2 for representative results.
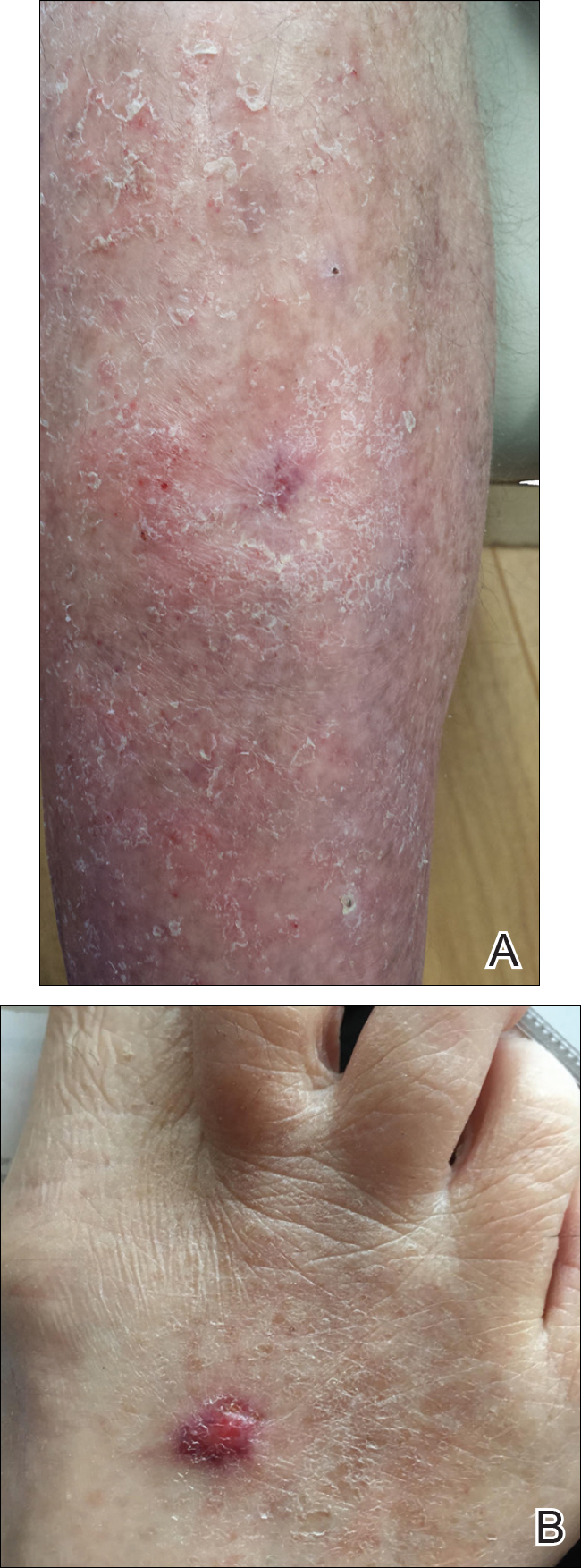
Comment
Dermatologists create acute wounds in patients on a daily basis. Ensuring that patients achieve the most desirable cosmetic outcome is a primary goal for dermatologists and an important component of patient satisfaction. A number of studies have examined patient satisfaction following MMS.3,4 Patient satisfaction is an especially important outcome measure in dermatology, as dermatologic diseases affect cosmetic appearance and are related to quality of life.3,4
Timolol is a nonselective β-adrenergic receptor antagonist that is used in dermatology to treat IHs. In this preliminary study, the authors sought to determine if topical timolol applied to acute wounds following surgical removal of nonmelanoma skin cancers could improve the overall cosmetic outcome of acute surgical scars. The results showed that compared to control, topical timolol resulted in a more cosmetically favorable scar. The results are preliminary, and it would be of future interest to further study the effects of topical timolol on acute surgical wounds from a wound-healing standpoint as well as to further test its effects on the cosmesis of these wounds.
- Chisholm KM, Chang KW, Truong MT, et al. β-Adrenergic receptor expression in vascular tumors [published online June 29, 2012]. Mod Pathol. 2012;25:1446-1451.
- Fearmonti R, Bond J, Erdmann D, et al. A review of scar scales and scar measuring devices. Eplasty. 2010;10:e43.
- Asgari MM, Warton EM, Neugebauer R, et al. Predictors of patient satisfaction with Mohs surgery: analysis of preoperative, intraoperative, and postoperative factors in a prospective cohort. Arch Dermatol. 2011;147:1387-1394.
- Asgari MM, Bertenthal D, Sen S, et al. Patient satisfaction after treatment of nonmelanoma skin cancer. Dermatol Surg. 2009;35:1041-1049.
Timolol is a nonselective β-adrenergic receptor antagonist indicated for treating glaucoma, heart attacks, hypertension, and migraine headaches. It is made in both an oral and ophthalmic form. In dermatology, the beta-blocker propranolol is approved for the treatment of infantile hemangiomas (IHs). The exact mechanism of action of beta-blockers for the treatment of IHs is not yet completely understood, but it is postulated that they inhibit growth by at least 4 distinct mechanisms: (1) vasoconstriction, (2) inhibition of angiogenesis or vasculogenesis, (3) induction of apoptosis, and (4) recruitment of endothelial progenitor cells to the site of the hemangioma.1
Scar cosmesis can be calculated using the visual analog scale (VAS), which is a subjective scar assessment scored from poor to excellent. The multidimensional VAS is a photograph-based scale derived from evaluating standardized digital photographs in 4 dimensions—pigmentation, vascularity, acceptability, and observer comfort—plus contour. It uses the sum of the individual scores to obtain a single overall score ranging from excellent to poor.2 In this study, we sought to determine if the use of topical timolol after excision or Mohs micrographic surgery (MMS) treatment of nonmelanoma skin cancers improved the overall cosmesis of the scar.
Methods
The study protocol was approved by the institutional review board at Roger Williams Medical Center (Providence, Rhode Island). Eligibility criteria included patients who required excision or MMS for their nonmelanoma skin cancer located below the patella and those who agreed to allow their wounds to heal by secondary intention when given options for closure of their wounds. Patients were randomized to either the timolol (study medication) group or the saline (placebo) group. The initial defects were measured and photographed. Patients were educated on how to apply the study medication. All patients were prescribed 40 mm Hg compression stockings to wear following application of the study medication. Patients were asked to return at 1 and 5 weeks postsurgery and then every 1 to 2 weeks for wound assessment and measurement until their wounds had healed or at 13 weeks, depending on which came first. A healed wound was defined as having no exudate, exhibiting complete reepithelialization, and being stable for 1 week.
Healed wounds were assessed by a blinded outside dermatologist who examined photographs of the wounds and then completed the VAS for each participant’s scar.
Results
A total of 9 participants were enrolled in the study. Three participants were lost to follow-up; 6 completed the study (4 females, 2 males). The mean age was 70 years (age range, 46–89 years). The average wound size was 2×2 cm with a depth of 1 mm. Three participants were in the active medication group and 3 were in the control group.
A VAS was completed for each participant’s scar by an outside blinded dermatologist. Based on the VAS, wounds treated with timolol resulted in more cosmetically favorable scars (scored higher on the VAS) compared to control (mean [SD]: 6.5±0.9 vs 2.5±0.7; P<0.05). See Figures 1 and 2 for representative results.
Comment
Dermatologists create acute wounds in patients on a daily basis. Ensuring that patients achieve the most desirable cosmetic outcome is a primary goal for dermatologists and an important component of patient satisfaction. A number of studies have examined patient satisfaction following MMS.3,4 Patient satisfaction is an especially important outcome measure in dermatology, as dermatologic diseases affect cosmetic appearance and are related to quality of life.3,4
Timolol is a nonselective β-adrenergic receptor antagonist that is used in dermatology to treat IHs. In this preliminary study, the authors sought to determine if topical timolol applied to acute wounds following surgical removal of nonmelanoma skin cancers could improve the overall cosmetic outcome of acute surgical scars. The results showed that compared to control, topical timolol resulted in a more cosmetically favorable scar. The results are preliminary, and it would be of future interest to further study the effects of topical timolol on acute surgical wounds from a wound-healing standpoint as well as to further test its effects on the cosmesis of these wounds.
Timolol is a nonselective β-adrenergic receptor antagonist indicated for treating glaucoma, heart attacks, hypertension, and migraine headaches. It is made in both an oral and ophthalmic form. In dermatology, the beta-blocker propranolol is approved for the treatment of infantile hemangiomas (IHs). The exact mechanism of action of beta-blockers for the treatment of IHs is not yet completely understood, but it is postulated that they inhibit growth by at least 4 distinct mechanisms: (1) vasoconstriction, (2) inhibition of angiogenesis or vasculogenesis, (3) induction of apoptosis, and (4) recruitment of endothelial progenitor cells to the site of the hemangioma.1
Scar cosmesis can be calculated using the visual analog scale (VAS), which is a subjective scar assessment scored from poor to excellent. The multidimensional VAS is a photograph-based scale derived from evaluating standardized digital photographs in 4 dimensions—pigmentation, vascularity, acceptability, and observer comfort—plus contour. It uses the sum of the individual scores to obtain a single overall score ranging from excellent to poor.2 In this study, we sought to determine if the use of topical timolol after excision or Mohs micrographic surgery (MMS) treatment of nonmelanoma skin cancers improved the overall cosmesis of the scar.
Methods
The study protocol was approved by the institutional review board at Roger Williams Medical Center (Providence, Rhode Island). Eligibility criteria included patients who required excision or MMS for their nonmelanoma skin cancer located below the patella and those who agreed to allow their wounds to heal by secondary intention when given options for closure of their wounds. Patients were randomized to either the timolol (study medication) group or the saline (placebo) group. The initial defects were measured and photographed. Patients were educated on how to apply the study medication. All patients were prescribed 40 mm Hg compression stockings to wear following application of the study medication. Patients were asked to return at 1 and 5 weeks postsurgery and then every 1 to 2 weeks for wound assessment and measurement until their wounds had healed or at 13 weeks, depending on which came first. A healed wound was defined as having no exudate, exhibiting complete reepithelialization, and being stable for 1 week.
Healed wounds were assessed by a blinded outside dermatologist who examined photographs of the wounds and then completed the VAS for each participant’s scar.
Results
A total of 9 participants were enrolled in the study. Three participants were lost to follow-up; 6 completed the study (4 females, 2 males). The mean age was 70 years (age range, 46–89 years). The average wound size was 2×2 cm with a depth of 1 mm. Three participants were in the active medication group and 3 were in the control group.
A VAS was completed for each participant’s scar by an outside blinded dermatologist. Based on the VAS, wounds treated with timolol resulted in more cosmetically favorable scars (scored higher on the VAS) compared to control (mean [SD]: 6.5±0.9 vs 2.5±0.7; P<0.05). See Figures 1 and 2 for representative results.
Comment
Dermatologists create acute wounds in patients on a daily basis. Ensuring that patients achieve the most desirable cosmetic outcome is a primary goal for dermatologists and an important component of patient satisfaction. A number of studies have examined patient satisfaction following MMS.3,4 Patient satisfaction is an especially important outcome measure in dermatology, as dermatologic diseases affect cosmetic appearance and are related to quality of life.3,4
Timolol is a nonselective β-adrenergic receptor antagonist that is used in dermatology to treat IHs. In this preliminary study, the authors sought to determine if topical timolol applied to acute wounds following surgical removal of nonmelanoma skin cancers could improve the overall cosmetic outcome of acute surgical scars. The results showed that compared to control, topical timolol resulted in a more cosmetically favorable scar. The results are preliminary, and it would be of future interest to further study the effects of topical timolol on acute surgical wounds from a wound-healing standpoint as well as to further test its effects on the cosmesis of these wounds.
- Chisholm KM, Chang KW, Truong MT, et al. β-Adrenergic receptor expression in vascular tumors [published online June 29, 2012]. Mod Pathol. 2012;25:1446-1451.
- Fearmonti R, Bond J, Erdmann D, et al. A review of scar scales and scar measuring devices. Eplasty. 2010;10:e43.
- Asgari MM, Warton EM, Neugebauer R, et al. Predictors of patient satisfaction with Mohs surgery: analysis of preoperative, intraoperative, and postoperative factors in a prospective cohort. Arch Dermatol. 2011;147:1387-1394.
- Asgari MM, Bertenthal D, Sen S, et al. Patient satisfaction after treatment of nonmelanoma skin cancer. Dermatol Surg. 2009;35:1041-1049.
- Chisholm KM, Chang KW, Truong MT, et al. β-Adrenergic receptor expression in vascular tumors [published online June 29, 2012]. Mod Pathol. 2012;25:1446-1451.
- Fearmonti R, Bond J, Erdmann D, et al. A review of scar scales and scar measuring devices. Eplasty. 2010;10:e43.
- Asgari MM, Warton EM, Neugebauer R, et al. Predictors of patient satisfaction with Mohs surgery: analysis of preoperative, intraoperative, and postoperative factors in a prospective cohort. Arch Dermatol. 2011;147:1387-1394.
- Asgari MM, Bertenthal D, Sen S, et al. Patient satisfaction after treatment of nonmelanoma skin cancer. Dermatol Surg. 2009;35:1041-1049.
Resident Pearl
- Dermatologists create acute surgical wounds on a daily basis. We should strive for excellent patient outcomes as well as the most desirable cosmetic result. This research article points to a possible new application of a longstanding medication to improve the cosmetic outcome in acute surgical wounds.

Chronic Diffuse Erythematous Papulonodules
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).

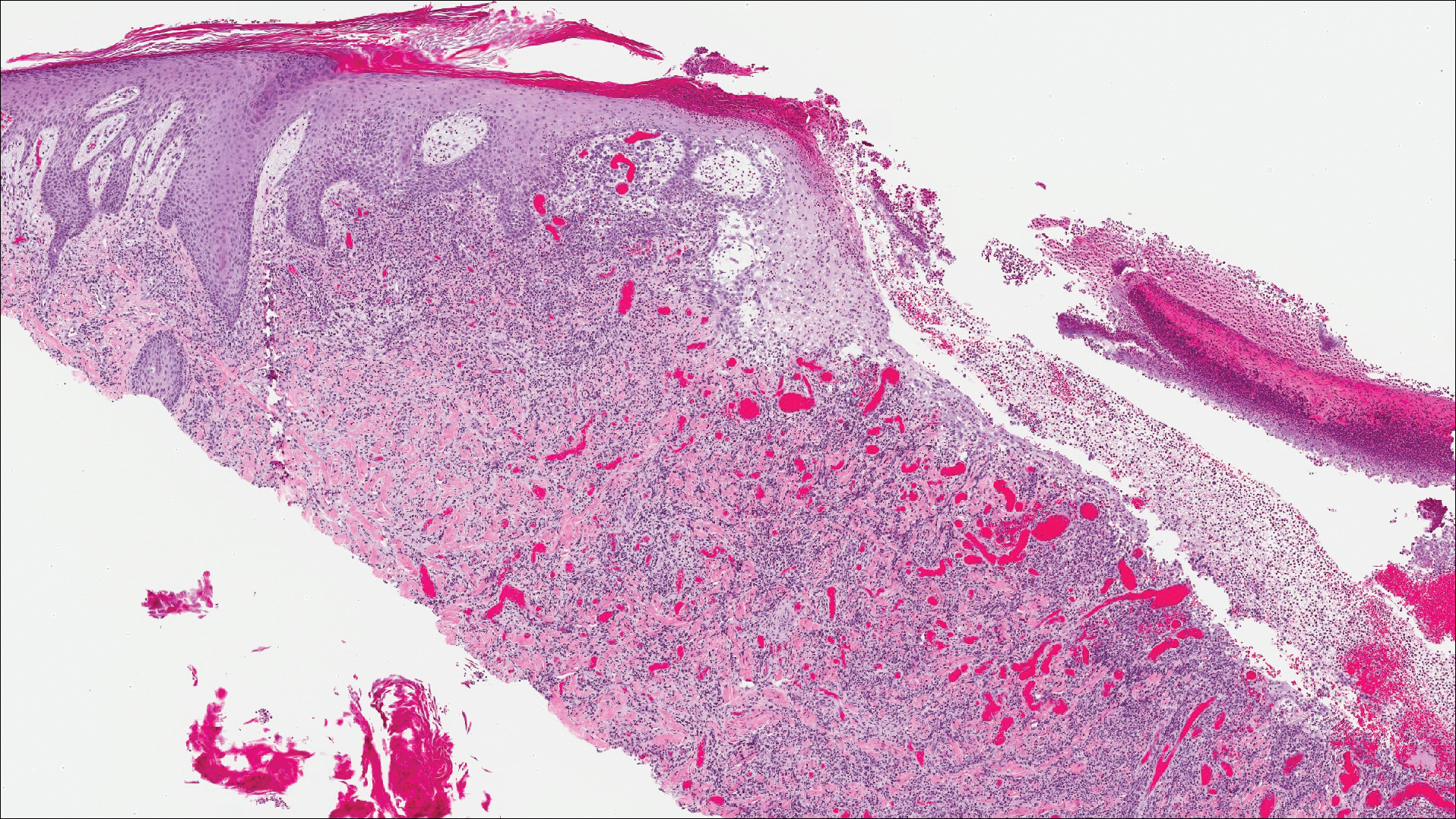
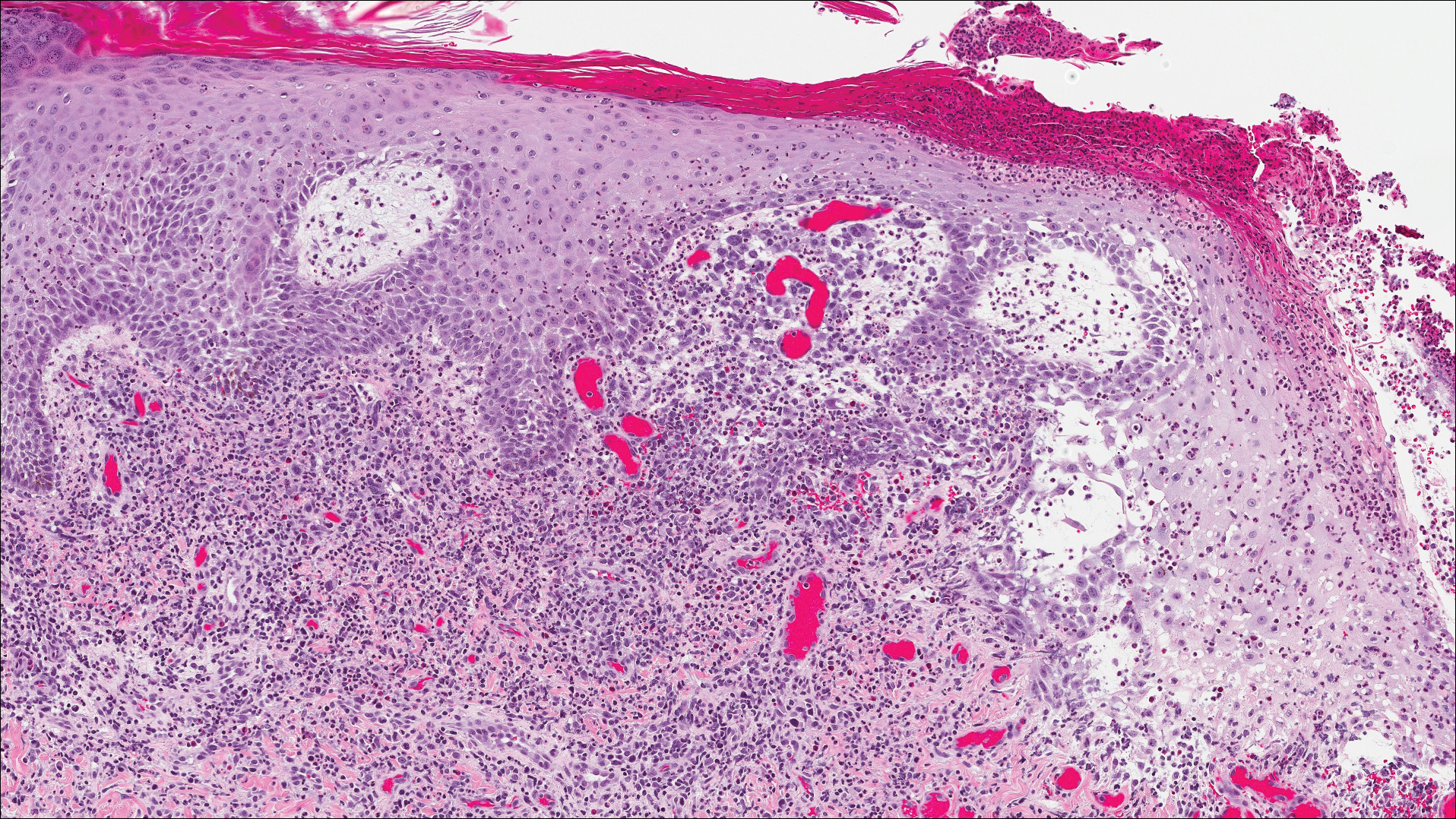
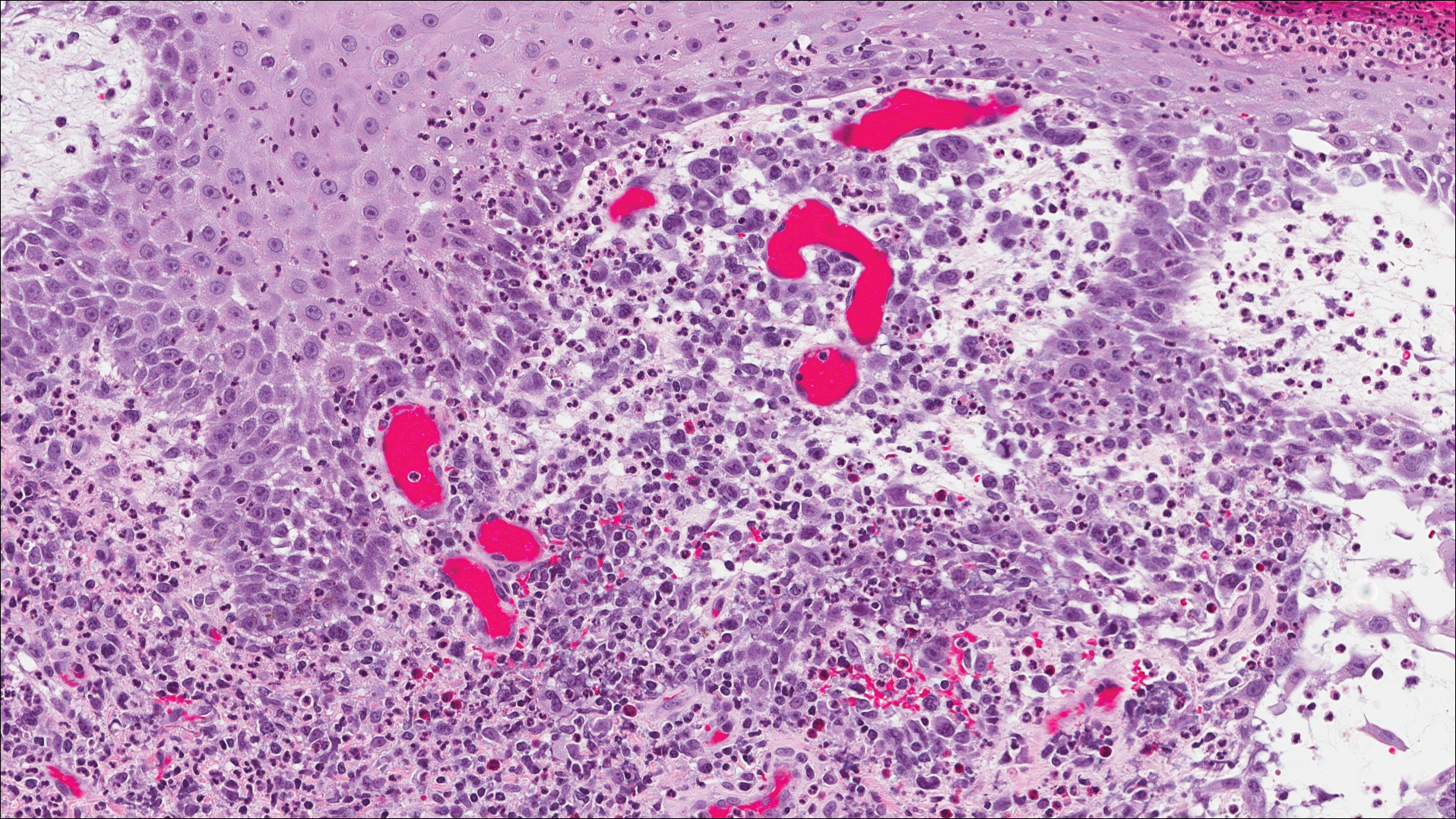
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
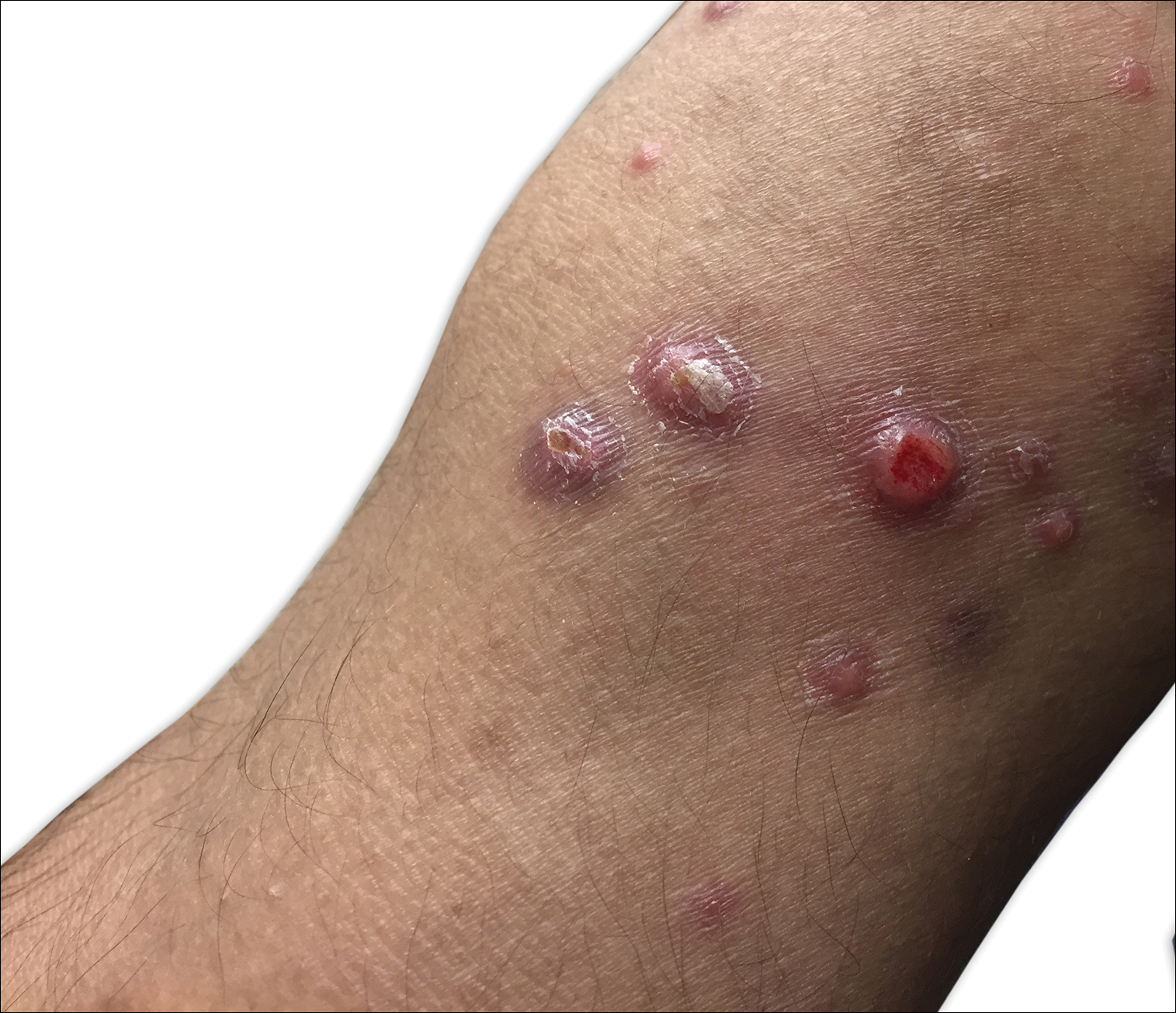
A 29-year-old man from Saudi Arabia presented with slightly tender skin lesions occurring in crops every few months over the last 7 years. The lesions typically would occur on the inguinal area, lower abdomen, buttocks, thighs, or arms, resolving within a few weeks despite no treatment. The patient denied having systemic symptoms such as fevers, chills, sweats, chest pain, shortness of breath, or unexpected weight loss. Physical examination revealed multiple erythematous papulonodules, some ulcerated with a superficial crust, grouped predominantly on the medial aspect of the right upper arm and left lower inguinal region. Isolated lesions also were present on the forearms, dorsal aspects of the hands, abdomen, and thighs. The grouped papulonodules were intermixed with faint hyperpigmented macules indicative of prior lesions. No oral lesions were noted, and there was no marked axillary or inguinal lymphadenopathy.
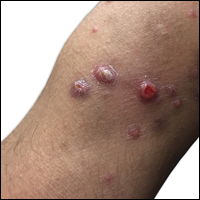
Ibrutinib and bleeding complications in Mohs surgery
Clinically significant bleeding events occurred in two elderly men who were taking ibrutinib and underwent Mohs micrographic surgery for squamous cell carcinomas, Cindy E. Parra and her colleagues reported in JAMA Dermatology.
On day 3 after his Mohs procedure, one 73-year-old man taking ibrutinib for Waldenstrom macroglobulinemia developed extensive bilateral periorbital ecchymosis that extended down to his upper chest. The other patient, an 88-year-old man taking ibrutinib for chronic lymphocytic leukemia, developed ecchymosis down to the chin. The first patient discontinued ibrutinib 3 days before his surgery; the second patient was taking ibrutinib at the time of his surgery.
“The increased incidence of nonmelanoma skin cancer and poorer outcomes in patients with non-Hodgkin lymphoma and CLL is well recognized, as is the importance of aggressive dermatologic management,” the researchers wrote (JAMA Dermatol. 2017 Jul 12. doi: 10.1001/jamadermatol.2017.1877). “It may be prudent to withhold ibrutinib treatment prior to dermatologic surgery to avoid potential bleeding complications.”
The findings argue for close collaboration between the dermatologic surgeon and the patient’s hematologist when scheduling extended-duration dermatologic procedures in patients taking ibrutinib.
Find the full summary here.
Clinically significant bleeding events occurred in two elderly men who were taking ibrutinib and underwent Mohs micrographic surgery for squamous cell carcinomas, Cindy E. Parra and her colleagues reported in JAMA Dermatology.
On day 3 after his Mohs procedure, one 73-year-old man taking ibrutinib for Waldenstrom macroglobulinemia developed extensive bilateral periorbital ecchymosis that extended down to his upper chest. The other patient, an 88-year-old man taking ibrutinib for chronic lymphocytic leukemia, developed ecchymosis down to the chin. The first patient discontinued ibrutinib 3 days before his surgery; the second patient was taking ibrutinib at the time of his surgery.
“The increased incidence of nonmelanoma skin cancer and poorer outcomes in patients with non-Hodgkin lymphoma and CLL is well recognized, as is the importance of aggressive dermatologic management,” the researchers wrote (JAMA Dermatol. 2017 Jul 12. doi: 10.1001/jamadermatol.2017.1877). “It may be prudent to withhold ibrutinib treatment prior to dermatologic surgery to avoid potential bleeding complications.”
The findings argue for close collaboration between the dermatologic surgeon and the patient’s hematologist when scheduling extended-duration dermatologic procedures in patients taking ibrutinib.
Find the full summary here.
Clinically significant bleeding events occurred in two elderly men who were taking ibrutinib and underwent Mohs micrographic surgery for squamous cell carcinomas, Cindy E. Parra and her colleagues reported in JAMA Dermatology.
On day 3 after his Mohs procedure, one 73-year-old man taking ibrutinib for Waldenstrom macroglobulinemia developed extensive bilateral periorbital ecchymosis that extended down to his upper chest. The other patient, an 88-year-old man taking ibrutinib for chronic lymphocytic leukemia, developed ecchymosis down to the chin. The first patient discontinued ibrutinib 3 days before his surgery; the second patient was taking ibrutinib at the time of his surgery.
“The increased incidence of nonmelanoma skin cancer and poorer outcomes in patients with non-Hodgkin lymphoma and CLL is well recognized, as is the importance of aggressive dermatologic management,” the researchers wrote (JAMA Dermatol. 2017 Jul 12. doi: 10.1001/jamadermatol.2017.1877). “It may be prudent to withhold ibrutinib treatment prior to dermatologic surgery to avoid potential bleeding complications.”
The findings argue for close collaboration between the dermatologic surgeon and the patient’s hematologist when scheduling extended-duration dermatologic procedures in patients taking ibrutinib.
Find the full summary here.
FROM JAMA DERMATOLOGY
Subsequent squamous cell carcinoma risk higher in HIV patients with low CD4 count
HIV-infected individuals who have experienced a nonmelanoma skin cancer may be at significantly greater risk of subsequent new squamous cell carcinoma if they have a lower CD4 cell count, a new study suggests.
In a study published online July 12 in JAMA Dermatology, researchers reported the results of a retrospective cohort study using medical record data from 455 HIV-infected and 1,952 HIV-uninfected patients who had previously been diagnosed with at least one nonmelanoma skin cancer.
Patients with CD4 cell counts below 200 cells/mcL had a 44% greater risk of a subsequent nonmelanoma skin cancer, compared with uninfected individuals (95% confidence interval, 1.10-1.88), while those with a viral load greater than 10,000 copies/mL had a 31% greater risk (95% CI, 1.00-1.72), after adjusting for age, sex, smoking status, and obesity.
The increase in nonmelanoma skin cancer risk was largely accounted for by an increased risk of squamous cell carcinoma (SCC). Among patients with lower CD4 cell counts and those with higher viral loads, the risk of SCC was more than twofold greater than among uninfected individuals. In contrast, while there was a trend toward a higher risk of basal cell carcinoma in those two groups, it did not reach significance (JAMA Dermatol. 2017 Jul 12. doi: 10.1001/jamadermatol.2017.1716).
Overall, HIV-infected individuals had a significant 15% increase in the risk of subsequent nonmelanoma skin cancer over an average follow-up period of 5 years, compared with uninfected individuals.
“This study addresses key questions regarding subsequent NMSC [nonmelanoma skin cancer] risk among a high-risk subgroup of HIV-infected population who, by virtue of having had a pathologically validated skin cancer, are at increased risk of subsequent NMSCs,” wrote Maryam M. Asgari, MD, of Massachusetts General Hospital, Boston, and her coauthors. “Specifically, it was previously not known precisely which NMSC subtype is increased in high-risk persons with HIV and whether biomarkers of HIV infections, such as degree of immune dysfunction, are associated with subsequent skin cancer risk.”
While the study wasn’t able to control for known skin cancer risk factors such as skin type, the patients were all non-Hispanic white, which the authors said selected for individuals with fair skin and some sun-exposure history.
The findings could have implications for skin cancer screening among individuals with HIV infection, with the authors suggesting more targeted monitoring for SCC among individuals with low CD4 counts or high viral loads.
“Our findings support immune dysfunction as a risk factor for SCCs and dovetail with SCC risk data from iatrogenic immunodeficiency states, such as organ transplantation and autoimmune diseases.”
The study was partly supported by Kaiser Permanente Northern California, and one author was supported by a grant from the National Cancer Institute. Two authors had previously served as investigators on studies funded by the pharmaceutical industry, one author declared research funding from the pharmaceutical industry, and one declared shares in two medical companies.
HIV-infected individuals who have experienced a nonmelanoma skin cancer may be at significantly greater risk of subsequent new squamous cell carcinoma if they have a lower CD4 cell count, a new study suggests.
In a study published online July 12 in JAMA Dermatology, researchers reported the results of a retrospective cohort study using medical record data from 455 HIV-infected and 1,952 HIV-uninfected patients who had previously been diagnosed with at least one nonmelanoma skin cancer.
Patients with CD4 cell counts below 200 cells/mcL had a 44% greater risk of a subsequent nonmelanoma skin cancer, compared with uninfected individuals (95% confidence interval, 1.10-1.88), while those with a viral load greater than 10,000 copies/mL had a 31% greater risk (95% CI, 1.00-1.72), after adjusting for age, sex, smoking status, and obesity.
The increase in nonmelanoma skin cancer risk was largely accounted for by an increased risk of squamous cell carcinoma (SCC). Among patients with lower CD4 cell counts and those with higher viral loads, the risk of SCC was more than twofold greater than among uninfected individuals. In contrast, while there was a trend toward a higher risk of basal cell carcinoma in those two groups, it did not reach significance (JAMA Dermatol. 2017 Jul 12. doi: 10.1001/jamadermatol.2017.1716).
Overall, HIV-infected individuals had a significant 15% increase in the risk of subsequent nonmelanoma skin cancer over an average follow-up period of 5 years, compared with uninfected individuals.
“This study addresses key questions regarding subsequent NMSC [nonmelanoma skin cancer] risk among a high-risk subgroup of HIV-infected population who, by virtue of having had a pathologically validated skin cancer, are at increased risk of subsequent NMSCs,” wrote Maryam M. Asgari, MD, of Massachusetts General Hospital, Boston, and her coauthors. “Specifically, it was previously not known precisely which NMSC subtype is increased in high-risk persons with HIV and whether biomarkers of HIV infections, such as degree of immune dysfunction, are associated with subsequent skin cancer risk.”
While the study wasn’t able to control for known skin cancer risk factors such as skin type, the patients were all non-Hispanic white, which the authors said selected for individuals with fair skin and some sun-exposure history.
The findings could have implications for skin cancer screening among individuals with HIV infection, with the authors suggesting more targeted monitoring for SCC among individuals with low CD4 counts or high viral loads.
“Our findings support immune dysfunction as a risk factor for SCCs and dovetail with SCC risk data from iatrogenic immunodeficiency states, such as organ transplantation and autoimmune diseases.”
The study was partly supported by Kaiser Permanente Northern California, and one author was supported by a grant from the National Cancer Institute. Two authors had previously served as investigators on studies funded by the pharmaceutical industry, one author declared research funding from the pharmaceutical industry, and one declared shares in two medical companies.
HIV-infected individuals who have experienced a nonmelanoma skin cancer may be at significantly greater risk of subsequent new squamous cell carcinoma if they have a lower CD4 cell count, a new study suggests.
In a study published online July 12 in JAMA Dermatology, researchers reported the results of a retrospective cohort study using medical record data from 455 HIV-infected and 1,952 HIV-uninfected patients who had previously been diagnosed with at least one nonmelanoma skin cancer.
Patients with CD4 cell counts below 200 cells/mcL had a 44% greater risk of a subsequent nonmelanoma skin cancer, compared with uninfected individuals (95% confidence interval, 1.10-1.88), while those with a viral load greater than 10,000 copies/mL had a 31% greater risk (95% CI, 1.00-1.72), after adjusting for age, sex, smoking status, and obesity.
The increase in nonmelanoma skin cancer risk was largely accounted for by an increased risk of squamous cell carcinoma (SCC). Among patients with lower CD4 cell counts and those with higher viral loads, the risk of SCC was more than twofold greater than among uninfected individuals. In contrast, while there was a trend toward a higher risk of basal cell carcinoma in those two groups, it did not reach significance (JAMA Dermatol. 2017 Jul 12. doi: 10.1001/jamadermatol.2017.1716).
Overall, HIV-infected individuals had a significant 15% increase in the risk of subsequent nonmelanoma skin cancer over an average follow-up period of 5 years, compared with uninfected individuals.
“This study addresses key questions regarding subsequent NMSC [nonmelanoma skin cancer] risk among a high-risk subgroup of HIV-infected population who, by virtue of having had a pathologically validated skin cancer, are at increased risk of subsequent NMSCs,” wrote Maryam M. Asgari, MD, of Massachusetts General Hospital, Boston, and her coauthors. “Specifically, it was previously not known precisely which NMSC subtype is increased in high-risk persons with HIV and whether biomarkers of HIV infections, such as degree of immune dysfunction, are associated with subsequent skin cancer risk.”
While the study wasn’t able to control for known skin cancer risk factors such as skin type, the patients were all non-Hispanic white, which the authors said selected for individuals with fair skin and some sun-exposure history.
The findings could have implications for skin cancer screening among individuals with HIV infection, with the authors suggesting more targeted monitoring for SCC among individuals with low CD4 counts or high viral loads.
“Our findings support immune dysfunction as a risk factor for SCCs and dovetail with SCC risk data from iatrogenic immunodeficiency states, such as organ transplantation and autoimmune diseases.”
The study was partly supported by Kaiser Permanente Northern California, and one author was supported by a grant from the National Cancer Institute. Two authors had previously served as investigators on studies funded by the pharmaceutical industry, one author declared research funding from the pharmaceutical industry, and one declared shares in two medical companies.
FROM JAMA DERMATOLOGY
Key clinical point: HIV-infected people who have had a previous nonmelanoma skin cancer are at significantly higher risk of subsequent SCC if they have a lower CD4 count or higher viral load.
Major finding: HIV-infected people with a low CD4 cell count or high viral load have a greater than twofold increased risk of subsequent SCC after a primary nonmelanoma skin cancer than do uninfected people who have had a previous nonmelanoma skin cancer.
Data source: A retrospective cohort study in 455 HIV-infected and 1,945 HIV-uninfected patients.
Disclosures: The study was partly supported by Kaiser Permanente, Northern California, and one author was supported by a grant from the National Cancer Institute. Two authors had previously served as investigators on studies funded by the pharmaceutical industry, one author declared research funding from the pharmaceutical industry, and one declared shares in two medical companies.
Dermatofibrosarcoma Protuberans
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
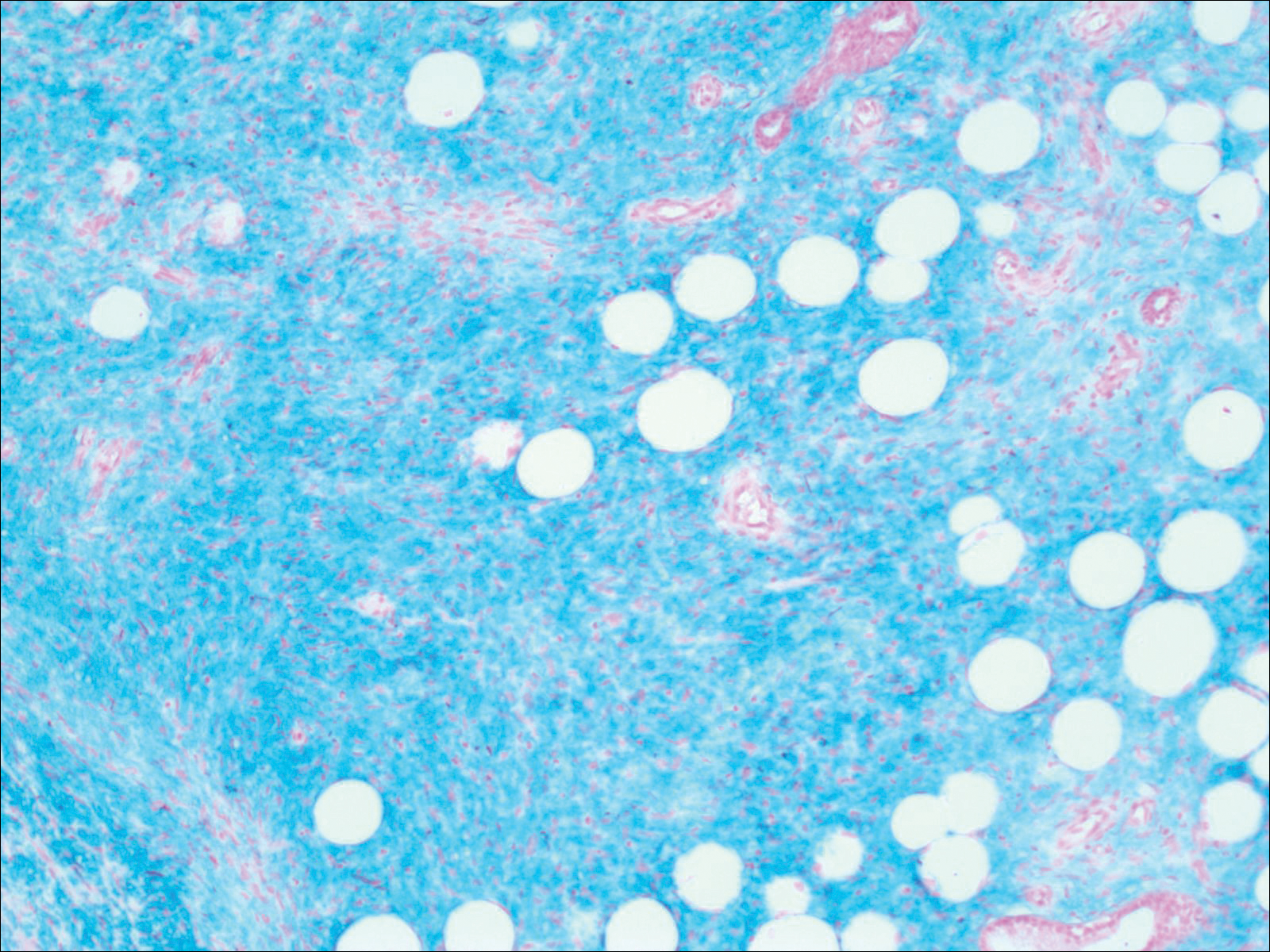
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
Practice Points
- Consider dermatofibrosarcoma protuberans for a keloidlike enlarging lesion when there is no history of trauma or prior keloid formation.
- Treatments such as Mohs micrographic surgery or oral imatinib mesylate can provide lower recurrence rates in appropriate patients as stand-alone or adjuvant therapy.
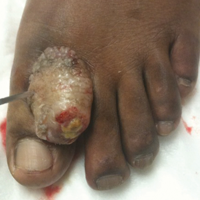
Eroded Plaque on the Lower Lip
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
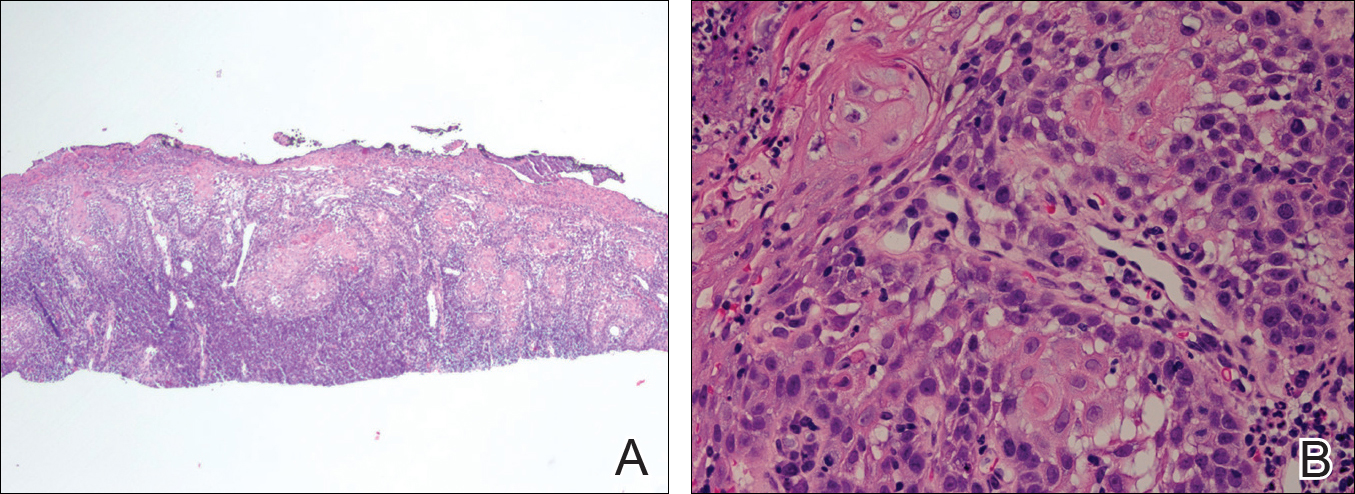
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.

The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
An 83-year-old man presented with a new-onset 1.2-cm eroded plaque on the vermilion border of the right lower lip that reportedly developed 2 weeks prior and was increasing in size. The plaque was moist and was composed of confluent glistening papules. Medical history was notable for the presence of both basal cell and squamous cell carcinomas.
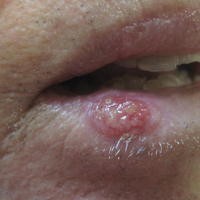
Cutaneous Myoepithelial Carcinoma With Disseminated Metastases
Cutaneous myoepithelial tumors are rare neoplasms but are being increasingly recognized and reported in the literature.1-7 Myoepithelial tumors are related to benign mixed tumors of the skin but lack the epithelial ductules that are present in mixed tumors. Cutaneous myoepithelial tumors may show a variety of architectural, cytological, and stromal features. Their immunophenotype usually is characterized by coexpression of an epithelial marker (eg, keratin, epithelial membrane antigen [EMA]) and S-100 protein; they also may express a variety of other myoepithelial markers, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin.7 EWS RNA binding protein 1 (EWSR1) and pleomorphic adenoma gene 1 (PLAG1) gene rearrangement has been detected in subsets of these tumors on in situ hybridization.8-10
Malignant myoepithelial tumors of the skin, also referred to as cutaneous myoepithelial carcinomas, are exceedingly rare. Including the current case, a search of PubMed articles indexed for MEDLINE and Google Scholar using the terms myoepithelial carcinoma and cutaneous revealed 12 cases that have been reported in the literature (Table).1-7,11-13 These tumors often occur in the head and neck areas and the lower extremities and display a bimodal age distribution, generally occurring in patients younger than 21 years and older than 50 years of age; they also show a slight female predominance. Available follow-up data from the literature have shown local recurrence or metastasis in 3 cases3,4,6; however, in one case the metastatic focus was not histologically identified.4 Cutaneous myoepithelial carcinoma presenting with metastatic disease further limits treatment options. Here, we describe a case of metastatic cutaneous myoepithelial carcinoma in a 47-year-old man, a rare example of cutaneous myoepithelial carcinoma with histologically documented metastatic disease at the initial presentation.
Case Report
A 47-year-old man who underwent a renal transplant 19 years prior presented with a weeping, ulcerated, mildly tender lesion on the scalp of 4 months’ duration with neck and back pain of 3 months’ duration. Physical examination demonstrated a 6-cm area of ulceration on the anterior crown of the scalp with adjacent enlarged keratoacanthomalike craters and satellite nodules (Figure 1). He was previously diagnosed with basal cell carcinoma (BCC) of the scalp at an outside institution 4 years prior and was treated with radiation therapy. The prior scalp biopsy for BCC diagnosis was unavailable for review. The patient had a history of chronic eczematous dermatitis in the waistband area that had been present for 19 years and another BCC with nodular and infiltrative patterns on the left helix. Of note, he also had been taking long-term immunosuppressant medications (ie, cyclosporine, azathioprine) for maintenance following the renal transplant.
Because of the extensive ulceration of the primary lesion, a shave biopsy of the scalp was performed on an adjacent satellite nodule. Histopathologic findings showed an intradermal neoplasm characterized by poorly cohesive cells exhibiting epithelioid to plasmacytoid morphologic features surrounded by abundant chondromyxoid stroma. Ductular differentiation was not identified (Figure 2A). The neoplastic cells displayed hyperchromatic nuclei with marked nuclear pleomorphism and atypical mitotic figures (Figure 2B). On immunohistochemistry the tumor cells stained positive for cytokeratin AE1/AE3 (Figure 3), S-100 protein (Figure 4), and p63, and were negative for calponin, desmin, melan-A, cytokeratin 7, and brachyury (Figure 5).
Radiographic imaging was performed due to the patient’s history of neck and back pain. Magnetic resonance imaging showed innumerable slightly expansile, T1-hypointense, T2-hyperintense, and robustly enhancing lesions involving the cervical, thoracic, lumbar, and sacral spine, as well as the thoracic ribs and bilateral iliac bones. There was no evidence of soft tissue tumor around the bone lesions. Ventral cervical spinal cord compression was detected at the C4 vertebra, causing a symptomatic radiculopathy; however, due to widely metastatic disease, the patient was not considered appropriate for neurosurgical intervention of the compression. Computerized tomography of the chest, abdomen, and pelvis did not identify any visceral source of malignancy, though multiple bilaterally enlarged cervical lymph nodes were identified on magnetic resonance imaging.
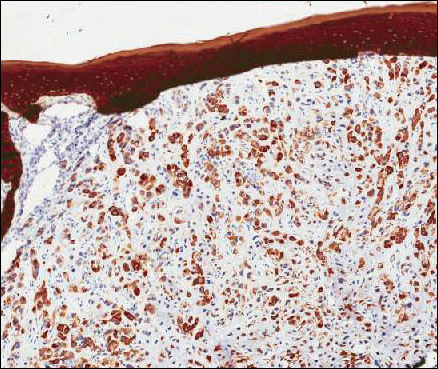
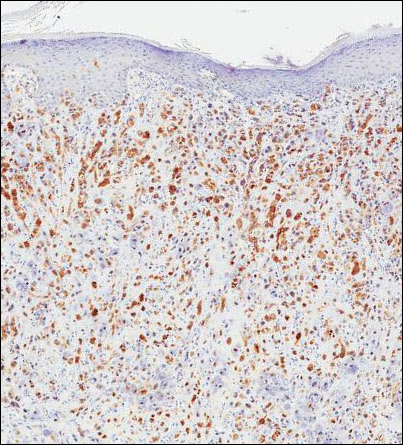

Fine needle aspiration of a left iliac bone lesion demonstrated neoplastic cells and chondromyxoid stroma essentially identical to the features shown in the skin biopsy (Figure 6). Given the morphologic features of the tumor and coexpression of cytokeratin and S-100 protein, the findings were interpreted as primary cutaneous myoepithelial carcinoma with disseminated metastatic lesions. The patient began treatment with carboplatin and paclitaxel chemotherapy. To combat the symptomatic bone pain and upper extremity radiculopathy, palliative radiation was administered to the cervical spine, lumbar spine, and right sacrum (30 Gy to each site in 10 fractions at 3 Gy per fraction). Despite the attempted chemotherapy and radiation, the patient continued to decline, and after 2 months, he elected to pursue palliative care. The patient died after 3 months in palliative care (5 months after the initial presentation).
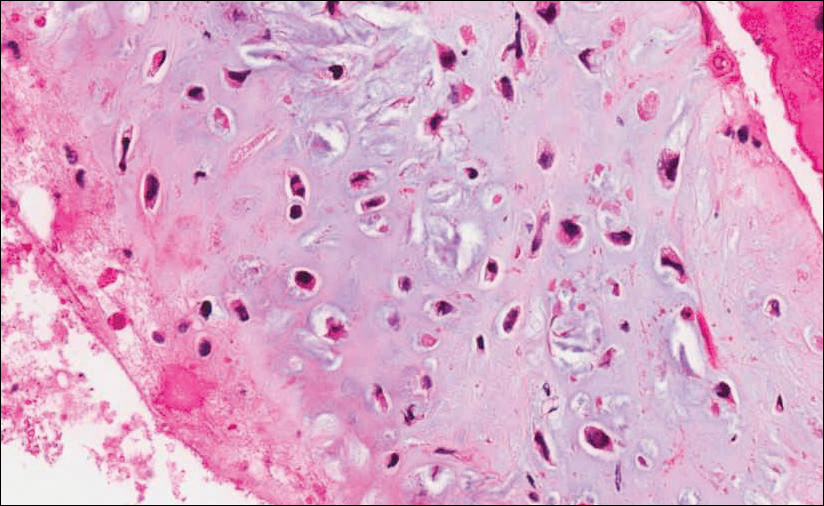
Comment
Myoepithelial cells normally surround ducts in secretory organs, such as the breasts, salivary glands, and cutaneous sweat glands. Myoepithelial neoplasms are well recognized in the salivary glands14,15; however, myoepithelial neoplasms also can arise in other sites, including the soft tissue4,5,16-18 and skin.1-3,7,11,19,20 Myoepithelioma of soft tissue was first described by Burke et al21 in 1995 and later described in the skin by Fernandez-Figueras et al22 in 1998. Since then, diagnostic criteria for cutaneous myoepithelial neoplasms have evolved, suggesting a spectrum of disease rather than a single distinct entity.11 Most often, cutaneous myoepithelial carcinomas arise as soft nodular lesions in the head and neck areas or extremities of adults. The nodules typically are nontender and range in size from 0.5 to 18.0 cm. Our review of the literature revealed 11 additional cases of cutaneous myoepithelial carcinomas have been reported, ranging in size from 0.7 to 7.0 cm (Table). In our case, the main lesion was 6 cm, mildly tender, ulcerated, and accompanied by satellite nodules.
Histologically, cutaneous myoepithelial tumors typically are well-defined, dermal-based nodules with no connection to the overlying epidermis. Similar to myoepithelial tumors of other sites, they can be diagnostically challenging due to the heterogeneity of both their architectural and cytological features. The presence of a chondromyxoid or hyalinized stroma is common but not always present. Neoplastic myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show growth patterns in clusters, cords, glands, or sheets. Focal epithelial cells can be present. Although benign myoepithelial neoplasms with overt ductal differentiation are consistent with cutaneous mixed tumors (chondroid syringomas), those without ducts are characterized as myoepitheliomas. It is uncertain if cases with only focal ductal differentiation should be classified as mixed tumors or as myoepitheliomas. Malignant myoepithelial tumors show infiltrative borders, nuclear pleomorphism, coarse nuclear chromatin, prominent nucleoli, and increased mitotic activity. A 2003 study by Hornick and Fletcher16 found that cytologic atypia was the primary predictor of malignant behavior for myoepithelial neoplasms of the soft tissue.
Despite a wide variety of expression patterns, immunohistochemistry is critical in demonstrating myoepithelial differentiation and establishing a diagnosis of a myoepithelial neoplasm. Most cases display coexpression of epithelial markers, including keratins and/or EMA as well as S-100 protein. Myogenic markers also may be variably expressed; however, the absence of myogenic markers does not exclude the diagnosis of a myoepithelial tumor. Commonly expressed epithelial markers are cytokeratin AE1/AE3, cytokeratin 8/18, and EMA, while commonly expressed myogenic markers include muscle specific actin and smooth muscle actin.5,7,11,19 Myoepithelial tumors also may express calponin, p63, and glial fibrillary acidic protein.16
Molecular studies also can aid in the diagnosis of myoepithelial tumors. A study by Antonescu et al8 demonstrated EWSR1 gene rearrangement in 45% (30/66) of extrasalivary myoepithelial tumors and the absence of EWSR1 gene rearrangement in salivary gland myoepithelial tumors. The authors also showed that EWSR1-negative tumors were more likely to be superficially located, display ductal differentiation, and possess a benign clinical course.8 In another study, Bahrami et al23 suggested that a subset of mixed tumors, specifically those with tubuloductal differentiation, are genetically linked to salivary gland pleomorphic adenomas, which was achieved by the coexpression of the PLAG1 protein and PLAG1 gene rearrangement on immunohistochemistry and fluorescence in situ hybridization (FISH), respectively. Of the 19 cases evaluated, 11 (58%) expressed nuclear staining for PLAG1 immunohistochemistry; 8 of those 11 showed positive gene rearrangement for PLAG1 using FISH. These findings raise the possibility that cutaneous mixed tumors may be more closely related to those of the salivary glands, while deep myoepithelial tumors that lack ductal differentiation may represent a distinct group. Similar to the study by Antonescu et al,8 Flucke et al10 investigated EWSR1 gene rearrangement but limited their sample to cutaneous tumors, including myoepitheliomas, mixed tumors, and myoepithelial carcinoma. The authors found that 44% of cases (7/16) expressed EWSR1; this expression suggests that cutaneous myoepithelial tumors may have a genetic relationship to their soft tissue, bone, and visceral counterparts.10
Myoepithelial tumors display a broad spectrum of morphologic features; however, one of the most common growth patterns is that of oval to round cells forming cords and chains in a chondromyxoid stroma. As such, the histopathologic differential diagnosis for myoepithelial tumors includes other epithelioid or round-cell neoplasms with similar growth patterns including extraskeletal myxoid chondrosarcoma (EMC), ossifying fibromyxoid tumor of soft parts, and extra-axial soft tissue chordoma. Extraskeletal myxoid chondrosarcoma bears the closest similarity to myoepithelial tumors both histologically and by ancillary studies. It typically possesses cords or chains of small round tumor cells set in a chondromyxoid or myxoid background. In contrast to myoepithelial tumors, which typically have more abundant cytoplasm and can show at least focal areas of spindle cell growth, the cells of EMC are more uniform, small, round cells with relatively scant cytoplasm. Extraskeletal myxoid chondrosarcomas lack the typical myoepithelial coexpression of cytokeratin and S-100 protein, with a minority of EMCs expressing S-100 protein but rarely cytokeratin. Most cases of EMC possess a balanced t(9;22) translocation involving the EWSR1 gene,24 a finding that could lead to confusion with soft tissue myoepithelial tumors, which also may show EWSR1 rearrangement on FISH. Ossifying fibromyxoid tumor of soft parts is also composed of round cells arranged in cords in a myxoid or fibrous stroma; the majority of cases also display a peripheral rim of mature bone, a feature that is not typically seen in myoepithelial tumors. Similar to myoepithelial tumors, ossifying fibromyxoid tumor of soft parts often is positive for S-100 protein; however, it rarely is positive for cytokeratins. Ossifying fibromyxoid tumor of soft parts has been shown to have a rearrangement of the PHD finger protein 1 (PHF1) gene in approximately half of cases, a molecular finding that has not been reported for myoepithelial tumors.25 Finally, extra-axial soft tissue chordomas, though quite rare, may possess striking similarities to myoepithelial tumors both histopathologically and immunohistochemically. Chordomas are composed of epithelioid cells arranged in nests, nodules, and chains with a variably myxoid background. A variable amount of cells with bubbly cytoplasm (known as physaliphorous cells) can be seen. High mitotic activity is not a characteristic feature in chordomas. They classically coexpress cytokeratins and S-100 protein, similar to myoepithelial tumors.
Because cutaneous myoepithelial tumors are relatively rare, a well-defined standard of care for treatment is lacking. Surgical excision is the primary treatment method in most reported cases in the literature.17,19 Miller et al29 reported the successful treatment of recurrent cutaneous myoepitheliomas with Mohs micrographic surgery. Chemotherapy may be useful in the setting of metastatic myoepithelial carcinomas in adults, but reported results are inconsistent.30,31 Radiation treatment of recurrent or metastatic disease has not been shown to be effective. A study of children treated with surgical resection and chemotherapy using ifosfamide, cisplatin, and etoposide followed by radiation therapy showed positive results.32
Our case highlights several challenges that may arise in establishing a diagnosis of cutaneous myoepithelial carcinoma with disseminated metastases. The diagnostic difficulty in our case was compounded by the advanced nature of the lesion at the time of presentation. Given the rarity of metastatic cutaneous myoepithelial carcinomas and the lack of a prior primary diagnosis of a malignant myoepithelioma, the index of suspicion for this entity was not high. A report of myoepithelial carcinoma of the parotid gland metastatic to the skin has been reported,33 but in the absence of salivary gland involvement or other visceral lesions, metastasis from any source other than our patient’s cutaneous scalp lesion is unlikely. The histopathologic features in combination with the characteristic immunophenotype, unique clinical setting, and radiographic findings were essential to arriving at the correct diagnosis. Unlike previously reported metastatic lesions, our case is unique in that metastatic lesions were identified at the time of initial clinical presentation.
Conclusion
Cutaneous myoepithelial carcinomas are exceedingly rare tumors with a wide range of histopathologic and immunohistochemical findings. In challenging cases, studies for EWSR1 or PLAG1 gene rearrangement can be helpful. Furthermore, this case illustrates the potential for widespread dissemination of myoepithelial carcinomas requiring clinical evaluation and imaging studies to exclude metastatic lesions.
- Frost MW, Steiniche T, Damsgaard TE, et al. Primary cutaneous myoepithelial carcinoma: a case report and review of the literature. APMIS. 2014;122:369-379.
- Stojsic Z, Brasanac D, Boricic I, et al. Clear cell myoepithelial carcinoma of the skin. a case report. J Cutan Pathol. 2009;36:680-683.
- Tanahashi J, Kashima K, Daa T, et al. A case of cutaneous myoepithelial carcinoma. J Cutan Pathol. 2007;34:648-653.
- Michal M, Miettinen M. Myoepitheliomas of the skin and soft tissues. report of 12 cases. Virchows Arch. 1999;434:393-400.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Law RM, Viglione MP, Barrett TL. Metastatic myoepithelial carcinoma in a child. J Cutan Pathol. 2008;35:779-781.
- Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 2004;35:14-24.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. a molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114-1124.
- Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675-682.
- Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol. 2011;24:1444-1450.
- Mentzel T, Requena L, Kaddu S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30:294-302.
- Garcia-Sanchez S, Elices M, Nieto S. Cutaneous myoepithelial carcinoma (malignant myoepithelial tumor of skin). Virchows Archiv. 2009;455(suppl 1):1-482.
- Bajoghli A, Limpert J. Treatment of cutaneous malignant myoepithelioma on the nasal ala using Mohs micrographic surgery in a two and a half year old child. J Invest Dermatol. 2009;129:S44.
- Prasad AR, Savera AT, Gown AM, et al. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801-806.
- Savera AT, Sloman A, Huvos AG, et al. Myoepithelial carcinoma of the salivary glands. a clinicopathologic study of 25 patients. Am J Surg Pathol. 2000;24:761-774.
- Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183-1196.
- Kilpatrick SE, Hitchcock MG, Kraus MD, et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol. 1997;21:13-22.
- Neto AG, Pineda-Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: a case report and review of the literature. Head Neck. 2004;26:470-473.
- Kutzner H, Mentzel T, Kaddu S, et al. Cutaneous myoepithelioma: an under-recognized cutaneous neoplasm composed of myoepithelial cells. Am J Surg Pathol. 2001;25:348-355.
- Dix BT, Hentges MJ, Saltrick KR, et al. Cutaneous myoepithelioma in the foot: case report. Foot Ankle Spec. 2013;6:239-241.
- Burke T, Sahin A, Johnson DE, et al. Myoepithelioma of the retroperitoneum. Ultrastruct Pathol. 1995;19:269-274.
- Fernandez-Figueras MT, Puig L, Trias I, et al. Benign myoepithelioma of the skin. Am J Dermatopathol. 1998;20:208-212.
- Bahrami A, Dalton JD, Krane JF, et al. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140-148.
- Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340-352.
- Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: a fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am J Surg Pathol. 2013;37:1751-1755.
- Dabska M. Parachordoma: a new clinicopathologic entity. Cancer. 1977;40:1586-1592.
- Fletcher CDM, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Lauer SR, Edgar MA, Gardner JM, et al. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719-726.
- Miller TD, McCalmont T, Tope WD. Recurrent cutaneous myoepithelioma treated using Mohs micrographic surgery: case report and review of the literature. Dermatol Surg. 2009;35:139-143.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Noronha V, Cooper DL, Higgins SA, et al. Metastatic myoepithelial carcinoma of the vulva treated with carboplatin and paclitaxel. Lancet Oncol. 2006;7:270-271.
- Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer. 2014;61:643-646.
- He DQ, Hua CG, Tang XF, et al. Cutaneous metastasis from a parotid myoepithelial carcinoma: a case report and review of the literature. J Cutan Pathol. 2008;35:1138-1143.
Cutaneous myoepithelial tumors are rare neoplasms but are being increasingly recognized and reported in the literature.1-7 Myoepithelial tumors are related to benign mixed tumors of the skin but lack the epithelial ductules that are present in mixed tumors. Cutaneous myoepithelial tumors may show a variety of architectural, cytological, and stromal features. Their immunophenotype usually is characterized by coexpression of an epithelial marker (eg, keratin, epithelial membrane antigen [EMA]) and S-100 protein; they also may express a variety of other myoepithelial markers, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin.7 EWS RNA binding protein 1 (EWSR1) and pleomorphic adenoma gene 1 (PLAG1) gene rearrangement has been detected in subsets of these tumors on in situ hybridization.8-10
Malignant myoepithelial tumors of the skin, also referred to as cutaneous myoepithelial carcinomas, are exceedingly rare. Including the current case, a search of PubMed articles indexed for MEDLINE and Google Scholar using the terms myoepithelial carcinoma and cutaneous revealed 12 cases that have been reported in the literature (Table).1-7,11-13 These tumors often occur in the head and neck areas and the lower extremities and display a bimodal age distribution, generally occurring in patients younger than 21 years and older than 50 years of age; they also show a slight female predominance. Available follow-up data from the literature have shown local recurrence or metastasis in 3 cases3,4,6; however, in one case the metastatic focus was not histologically identified.4 Cutaneous myoepithelial carcinoma presenting with metastatic disease further limits treatment options. Here, we describe a case of metastatic cutaneous myoepithelial carcinoma in a 47-year-old man, a rare example of cutaneous myoepithelial carcinoma with histologically documented metastatic disease at the initial presentation.
Case Report
A 47-year-old man who underwent a renal transplant 19 years prior presented with a weeping, ulcerated, mildly tender lesion on the scalp of 4 months’ duration with neck and back pain of 3 months’ duration. Physical examination demonstrated a 6-cm area of ulceration on the anterior crown of the scalp with adjacent enlarged keratoacanthomalike craters and satellite nodules (Figure 1). He was previously diagnosed with basal cell carcinoma (BCC) of the scalp at an outside institution 4 years prior and was treated with radiation therapy. The prior scalp biopsy for BCC diagnosis was unavailable for review. The patient had a history of chronic eczematous dermatitis in the waistband area that had been present for 19 years and another BCC with nodular and infiltrative patterns on the left helix. Of note, he also had been taking long-term immunosuppressant medications (ie, cyclosporine, azathioprine) for maintenance following the renal transplant.
Because of the extensive ulceration of the primary lesion, a shave biopsy of the scalp was performed on an adjacent satellite nodule. Histopathologic findings showed an intradermal neoplasm characterized by poorly cohesive cells exhibiting epithelioid to plasmacytoid morphologic features surrounded by abundant chondromyxoid stroma. Ductular differentiation was not identified (Figure 2A). The neoplastic cells displayed hyperchromatic nuclei with marked nuclear pleomorphism and atypical mitotic figures (Figure 2B). On immunohistochemistry the tumor cells stained positive for cytokeratin AE1/AE3 (Figure 3), S-100 protein (Figure 4), and p63, and were negative for calponin, desmin, melan-A, cytokeratin 7, and brachyury (Figure 5).
Radiographic imaging was performed due to the patient’s history of neck and back pain. Magnetic resonance imaging showed innumerable slightly expansile, T1-hypointense, T2-hyperintense, and robustly enhancing lesions involving the cervical, thoracic, lumbar, and sacral spine, as well as the thoracic ribs and bilateral iliac bones. There was no evidence of soft tissue tumor around the bone lesions. Ventral cervical spinal cord compression was detected at the C4 vertebra, causing a symptomatic radiculopathy; however, due to widely metastatic disease, the patient was not considered appropriate for neurosurgical intervention of the compression. Computerized tomography of the chest, abdomen, and pelvis did not identify any visceral source of malignancy, though multiple bilaterally enlarged cervical lymph nodes were identified on magnetic resonance imaging.
Fine needle aspiration of a left iliac bone lesion demonstrated neoplastic cells and chondromyxoid stroma essentially identical to the features shown in the skin biopsy (Figure 6). Given the morphologic features of the tumor and coexpression of cytokeratin and S-100 protein, the findings were interpreted as primary cutaneous myoepithelial carcinoma with disseminated metastatic lesions. The patient began treatment with carboplatin and paclitaxel chemotherapy. To combat the symptomatic bone pain and upper extremity radiculopathy, palliative radiation was administered to the cervical spine, lumbar spine, and right sacrum (30 Gy to each site in 10 fractions at 3 Gy per fraction). Despite the attempted chemotherapy and radiation, the patient continued to decline, and after 2 months, he elected to pursue palliative care. The patient died after 3 months in palliative care (5 months after the initial presentation).
Comment
Myoepithelial cells normally surround ducts in secretory organs, such as the breasts, salivary glands, and cutaneous sweat glands. Myoepithelial neoplasms are well recognized in the salivary glands14,15; however, myoepithelial neoplasms also can arise in other sites, including the soft tissue4,5,16-18 and skin.1-3,7,11,19,20 Myoepithelioma of soft tissue was first described by Burke et al21 in 1995 and later described in the skin by Fernandez-Figueras et al22 in 1998. Since then, diagnostic criteria for cutaneous myoepithelial neoplasms have evolved, suggesting a spectrum of disease rather than a single distinct entity.11 Most often, cutaneous myoepithelial carcinomas arise as soft nodular lesions in the head and neck areas or extremities of adults. The nodules typically are nontender and range in size from 0.5 to 18.0 cm. Our review of the literature revealed 11 additional cases of cutaneous myoepithelial carcinomas have been reported, ranging in size from 0.7 to 7.0 cm (Table). In our case, the main lesion was 6 cm, mildly tender, ulcerated, and accompanied by satellite nodules.
Histologically, cutaneous myoepithelial tumors typically are well-defined, dermal-based nodules with no connection to the overlying epidermis. Similar to myoepithelial tumors of other sites, they can be diagnostically challenging due to the heterogeneity of both their architectural and cytological features. The presence of a chondromyxoid or hyalinized stroma is common but not always present. Neoplastic myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show growth patterns in clusters, cords, glands, or sheets. Focal epithelial cells can be present. Although benign myoepithelial neoplasms with overt ductal differentiation are consistent with cutaneous mixed tumors (chondroid syringomas), those without ducts are characterized as myoepitheliomas. It is uncertain if cases with only focal ductal differentiation should be classified as mixed tumors or as myoepitheliomas. Malignant myoepithelial tumors show infiltrative borders, nuclear pleomorphism, coarse nuclear chromatin, prominent nucleoli, and increased mitotic activity. A 2003 study by Hornick and Fletcher16 found that cytologic atypia was the primary predictor of malignant behavior for myoepithelial neoplasms of the soft tissue.
Despite a wide variety of expression patterns, immunohistochemistry is critical in demonstrating myoepithelial differentiation and establishing a diagnosis of a myoepithelial neoplasm. Most cases display coexpression of epithelial markers, including keratins and/or EMA as well as S-100 protein. Myogenic markers also may be variably expressed; however, the absence of myogenic markers does not exclude the diagnosis of a myoepithelial tumor. Commonly expressed epithelial markers are cytokeratin AE1/AE3, cytokeratin 8/18, and EMA, while commonly expressed myogenic markers include muscle specific actin and smooth muscle actin.5,7,11,19 Myoepithelial tumors also may express calponin, p63, and glial fibrillary acidic protein.16
Molecular studies also can aid in the diagnosis of myoepithelial tumors. A study by Antonescu et al8 demonstrated EWSR1 gene rearrangement in 45% (30/66) of extrasalivary myoepithelial tumors and the absence of EWSR1 gene rearrangement in salivary gland myoepithelial tumors. The authors also showed that EWSR1-negative tumors were more likely to be superficially located, display ductal differentiation, and possess a benign clinical course.8 In another study, Bahrami et al23 suggested that a subset of mixed tumors, specifically those with tubuloductal differentiation, are genetically linked to salivary gland pleomorphic adenomas, which was achieved by the coexpression of the PLAG1 protein and PLAG1 gene rearrangement on immunohistochemistry and fluorescence in situ hybridization (FISH), respectively. Of the 19 cases evaluated, 11 (58%) expressed nuclear staining for PLAG1 immunohistochemistry; 8 of those 11 showed positive gene rearrangement for PLAG1 using FISH. These findings raise the possibility that cutaneous mixed tumors may be more closely related to those of the salivary glands, while deep myoepithelial tumors that lack ductal differentiation may represent a distinct group. Similar to the study by Antonescu et al,8 Flucke et al10 investigated EWSR1 gene rearrangement but limited their sample to cutaneous tumors, including myoepitheliomas, mixed tumors, and myoepithelial carcinoma. The authors found that 44% of cases (7/16) expressed EWSR1; this expression suggests that cutaneous myoepithelial tumors may have a genetic relationship to their soft tissue, bone, and visceral counterparts.10
Myoepithelial tumors display a broad spectrum of morphologic features; however, one of the most common growth patterns is that of oval to round cells forming cords and chains in a chondromyxoid stroma. As such, the histopathologic differential diagnosis for myoepithelial tumors includes other epithelioid or round-cell neoplasms with similar growth patterns including extraskeletal myxoid chondrosarcoma (EMC), ossifying fibromyxoid tumor of soft parts, and extra-axial soft tissue chordoma. Extraskeletal myxoid chondrosarcoma bears the closest similarity to myoepithelial tumors both histologically and by ancillary studies. It typically possesses cords or chains of small round tumor cells set in a chondromyxoid or myxoid background. In contrast to myoepithelial tumors, which typically have more abundant cytoplasm and can show at least focal areas of spindle cell growth, the cells of EMC are more uniform, small, round cells with relatively scant cytoplasm. Extraskeletal myxoid chondrosarcomas lack the typical myoepithelial coexpression of cytokeratin and S-100 protein, with a minority of EMCs expressing S-100 protein but rarely cytokeratin. Most cases of EMC possess a balanced t(9;22) translocation involving the EWSR1 gene,24 a finding that could lead to confusion with soft tissue myoepithelial tumors, which also may show EWSR1 rearrangement on FISH. Ossifying fibromyxoid tumor of soft parts is also composed of round cells arranged in cords in a myxoid or fibrous stroma; the majority of cases also display a peripheral rim of mature bone, a feature that is not typically seen in myoepithelial tumors. Similar to myoepithelial tumors, ossifying fibromyxoid tumor of soft parts often is positive for S-100 protein; however, it rarely is positive for cytokeratins. Ossifying fibromyxoid tumor of soft parts has been shown to have a rearrangement of the PHD finger protein 1 (PHF1) gene in approximately half of cases, a molecular finding that has not been reported for myoepithelial tumors.25 Finally, extra-axial soft tissue chordomas, though quite rare, may possess striking similarities to myoepithelial tumors both histopathologically and immunohistochemically. Chordomas are composed of epithelioid cells arranged in nests, nodules, and chains with a variably myxoid background. A variable amount of cells with bubbly cytoplasm (known as physaliphorous cells) can be seen. High mitotic activity is not a characteristic feature in chordomas. They classically coexpress cytokeratins and S-100 protein, similar to myoepithelial tumors.
Because cutaneous myoepithelial tumors are relatively rare, a well-defined standard of care for treatment is lacking. Surgical excision is the primary treatment method in most reported cases in the literature.17,19 Miller et al29 reported the successful treatment of recurrent cutaneous myoepitheliomas with Mohs micrographic surgery. Chemotherapy may be useful in the setting of metastatic myoepithelial carcinomas in adults, but reported results are inconsistent.30,31 Radiation treatment of recurrent or metastatic disease has not been shown to be effective. A study of children treated with surgical resection and chemotherapy using ifosfamide, cisplatin, and etoposide followed by radiation therapy showed positive results.32
Our case highlights several challenges that may arise in establishing a diagnosis of cutaneous myoepithelial carcinoma with disseminated metastases. The diagnostic difficulty in our case was compounded by the advanced nature of the lesion at the time of presentation. Given the rarity of metastatic cutaneous myoepithelial carcinomas and the lack of a prior primary diagnosis of a malignant myoepithelioma, the index of suspicion for this entity was not high. A report of myoepithelial carcinoma of the parotid gland metastatic to the skin has been reported,33 but in the absence of salivary gland involvement or other visceral lesions, metastasis from any source other than our patient’s cutaneous scalp lesion is unlikely. The histopathologic features in combination with the characteristic immunophenotype, unique clinical setting, and radiographic findings were essential to arriving at the correct diagnosis. Unlike previously reported metastatic lesions, our case is unique in that metastatic lesions were identified at the time of initial clinical presentation.
Conclusion
Cutaneous myoepithelial carcinomas are exceedingly rare tumors with a wide range of histopathologic and immunohistochemical findings. In challenging cases, studies for EWSR1 or PLAG1 gene rearrangement can be helpful. Furthermore, this case illustrates the potential for widespread dissemination of myoepithelial carcinomas requiring clinical evaluation and imaging studies to exclude metastatic lesions.
Cutaneous myoepithelial tumors are rare neoplasms but are being increasingly recognized and reported in the literature.1-7 Myoepithelial tumors are related to benign mixed tumors of the skin but lack the epithelial ductules that are present in mixed tumors. Cutaneous myoepithelial tumors may show a variety of architectural, cytological, and stromal features. Their immunophenotype usually is characterized by coexpression of an epithelial marker (eg, keratin, epithelial membrane antigen [EMA]) and S-100 protein; they also may express a variety of other myoepithelial markers, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin.7 EWS RNA binding protein 1 (EWSR1) and pleomorphic adenoma gene 1 (PLAG1) gene rearrangement has been detected in subsets of these tumors on in situ hybridization.8-10
Malignant myoepithelial tumors of the skin, also referred to as cutaneous myoepithelial carcinomas, are exceedingly rare. Including the current case, a search of PubMed articles indexed for MEDLINE and Google Scholar using the terms myoepithelial carcinoma and cutaneous revealed 12 cases that have been reported in the literature (Table).1-7,11-13 These tumors often occur in the head and neck areas and the lower extremities and display a bimodal age distribution, generally occurring in patients younger than 21 years and older than 50 years of age; they also show a slight female predominance. Available follow-up data from the literature have shown local recurrence or metastasis in 3 cases3,4,6; however, in one case the metastatic focus was not histologically identified.4 Cutaneous myoepithelial carcinoma presenting with metastatic disease further limits treatment options. Here, we describe a case of metastatic cutaneous myoepithelial carcinoma in a 47-year-old man, a rare example of cutaneous myoepithelial carcinoma with histologically documented metastatic disease at the initial presentation.
Case Report
A 47-year-old man who underwent a renal transplant 19 years prior presented with a weeping, ulcerated, mildly tender lesion on the scalp of 4 months’ duration with neck and back pain of 3 months’ duration. Physical examination demonstrated a 6-cm area of ulceration on the anterior crown of the scalp with adjacent enlarged keratoacanthomalike craters and satellite nodules (Figure 1). He was previously diagnosed with basal cell carcinoma (BCC) of the scalp at an outside institution 4 years prior and was treated with radiation therapy. The prior scalp biopsy for BCC diagnosis was unavailable for review. The patient had a history of chronic eczematous dermatitis in the waistband area that had been present for 19 years and another BCC with nodular and infiltrative patterns on the left helix. Of note, he also had been taking long-term immunosuppressant medications (ie, cyclosporine, azathioprine) for maintenance following the renal transplant.
Because of the extensive ulceration of the primary lesion, a shave biopsy of the scalp was performed on an adjacent satellite nodule. Histopathologic findings showed an intradermal neoplasm characterized by poorly cohesive cells exhibiting epithelioid to plasmacytoid morphologic features surrounded by abundant chondromyxoid stroma. Ductular differentiation was not identified (Figure 2A). The neoplastic cells displayed hyperchromatic nuclei with marked nuclear pleomorphism and atypical mitotic figures (Figure 2B). On immunohistochemistry the tumor cells stained positive for cytokeratin AE1/AE3 (Figure 3), S-100 protein (Figure 4), and p63, and were negative for calponin, desmin, melan-A, cytokeratin 7, and brachyury (Figure 5).
Radiographic imaging was performed due to the patient’s history of neck and back pain. Magnetic resonance imaging showed innumerable slightly expansile, T1-hypointense, T2-hyperintense, and robustly enhancing lesions involving the cervical, thoracic, lumbar, and sacral spine, as well as the thoracic ribs and bilateral iliac bones. There was no evidence of soft tissue tumor around the bone lesions. Ventral cervical spinal cord compression was detected at the C4 vertebra, causing a symptomatic radiculopathy; however, due to widely metastatic disease, the patient was not considered appropriate for neurosurgical intervention of the compression. Computerized tomography of the chest, abdomen, and pelvis did not identify any visceral source of malignancy, though multiple bilaterally enlarged cervical lymph nodes were identified on magnetic resonance imaging.
Fine needle aspiration of a left iliac bone lesion demonstrated neoplastic cells and chondromyxoid stroma essentially identical to the features shown in the skin biopsy (Figure 6). Given the morphologic features of the tumor and coexpression of cytokeratin and S-100 protein, the findings were interpreted as primary cutaneous myoepithelial carcinoma with disseminated metastatic lesions. The patient began treatment with carboplatin and paclitaxel chemotherapy. To combat the symptomatic bone pain and upper extremity radiculopathy, palliative radiation was administered to the cervical spine, lumbar spine, and right sacrum (30 Gy to each site in 10 fractions at 3 Gy per fraction). Despite the attempted chemotherapy and radiation, the patient continued to decline, and after 2 months, he elected to pursue palliative care. The patient died after 3 months in palliative care (5 months after the initial presentation).
Comment
Myoepithelial cells normally surround ducts in secretory organs, such as the breasts, salivary glands, and cutaneous sweat glands. Myoepithelial neoplasms are well recognized in the salivary glands14,15; however, myoepithelial neoplasms also can arise in other sites, including the soft tissue4,5,16-18 and skin.1-3,7,11,19,20 Myoepithelioma of soft tissue was first described by Burke et al21 in 1995 and later described in the skin by Fernandez-Figueras et al22 in 1998. Since then, diagnostic criteria for cutaneous myoepithelial neoplasms have evolved, suggesting a spectrum of disease rather than a single distinct entity.11 Most often, cutaneous myoepithelial carcinomas arise as soft nodular lesions in the head and neck areas or extremities of adults. The nodules typically are nontender and range in size from 0.5 to 18.0 cm. Our review of the literature revealed 11 additional cases of cutaneous myoepithelial carcinomas have been reported, ranging in size from 0.7 to 7.0 cm (Table). In our case, the main lesion was 6 cm, mildly tender, ulcerated, and accompanied by satellite nodules.
Histologically, cutaneous myoepithelial tumors typically are well-defined, dermal-based nodules with no connection to the overlying epidermis. Similar to myoepithelial tumors of other sites, they can be diagnostically challenging due to the heterogeneity of both their architectural and cytological features. The presence of a chondromyxoid or hyalinized stroma is common but not always present. Neoplastic myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show growth patterns in clusters, cords, glands, or sheets. Focal epithelial cells can be present. Although benign myoepithelial neoplasms with overt ductal differentiation are consistent with cutaneous mixed tumors (chondroid syringomas), those without ducts are characterized as myoepitheliomas. It is uncertain if cases with only focal ductal differentiation should be classified as mixed tumors or as myoepitheliomas. Malignant myoepithelial tumors show infiltrative borders, nuclear pleomorphism, coarse nuclear chromatin, prominent nucleoli, and increased mitotic activity. A 2003 study by Hornick and Fletcher16 found that cytologic atypia was the primary predictor of malignant behavior for myoepithelial neoplasms of the soft tissue.
Despite a wide variety of expression patterns, immunohistochemistry is critical in demonstrating myoepithelial differentiation and establishing a diagnosis of a myoepithelial neoplasm. Most cases display coexpression of epithelial markers, including keratins and/or EMA as well as S-100 protein. Myogenic markers also may be variably expressed; however, the absence of myogenic markers does not exclude the diagnosis of a myoepithelial tumor. Commonly expressed epithelial markers are cytokeratin AE1/AE3, cytokeratin 8/18, and EMA, while commonly expressed myogenic markers include muscle specific actin and smooth muscle actin.5,7,11,19 Myoepithelial tumors also may express calponin, p63, and glial fibrillary acidic protein.16
Molecular studies also can aid in the diagnosis of myoepithelial tumors. A study by Antonescu et al8 demonstrated EWSR1 gene rearrangement in 45% (30/66) of extrasalivary myoepithelial tumors and the absence of EWSR1 gene rearrangement in salivary gland myoepithelial tumors. The authors also showed that EWSR1-negative tumors were more likely to be superficially located, display ductal differentiation, and possess a benign clinical course.8 In another study, Bahrami et al23 suggested that a subset of mixed tumors, specifically those with tubuloductal differentiation, are genetically linked to salivary gland pleomorphic adenomas, which was achieved by the coexpression of the PLAG1 protein and PLAG1 gene rearrangement on immunohistochemistry and fluorescence in situ hybridization (FISH), respectively. Of the 19 cases evaluated, 11 (58%) expressed nuclear staining for PLAG1 immunohistochemistry; 8 of those 11 showed positive gene rearrangement for PLAG1 using FISH. These findings raise the possibility that cutaneous mixed tumors may be more closely related to those of the salivary glands, while deep myoepithelial tumors that lack ductal differentiation may represent a distinct group. Similar to the study by Antonescu et al,8 Flucke et al10 investigated EWSR1 gene rearrangement but limited their sample to cutaneous tumors, including myoepitheliomas, mixed tumors, and myoepithelial carcinoma. The authors found that 44% of cases (7/16) expressed EWSR1; this expression suggests that cutaneous myoepithelial tumors may have a genetic relationship to their soft tissue, bone, and visceral counterparts.10
Myoepithelial tumors display a broad spectrum of morphologic features; however, one of the most common growth patterns is that of oval to round cells forming cords and chains in a chondromyxoid stroma. As such, the histopathologic differential diagnosis for myoepithelial tumors includes other epithelioid or round-cell neoplasms with similar growth patterns including extraskeletal myxoid chondrosarcoma (EMC), ossifying fibromyxoid tumor of soft parts, and extra-axial soft tissue chordoma. Extraskeletal myxoid chondrosarcoma bears the closest similarity to myoepithelial tumors both histologically and by ancillary studies. It typically possesses cords or chains of small round tumor cells set in a chondromyxoid or myxoid background. In contrast to myoepithelial tumors, which typically have more abundant cytoplasm and can show at least focal areas of spindle cell growth, the cells of EMC are more uniform, small, round cells with relatively scant cytoplasm. Extraskeletal myxoid chondrosarcomas lack the typical myoepithelial coexpression of cytokeratin and S-100 protein, with a minority of EMCs expressing S-100 protein but rarely cytokeratin. Most cases of EMC possess a balanced t(9;22) translocation involving the EWSR1 gene,24 a finding that could lead to confusion with soft tissue myoepithelial tumors, which also may show EWSR1 rearrangement on FISH. Ossifying fibromyxoid tumor of soft parts is also composed of round cells arranged in cords in a myxoid or fibrous stroma; the majority of cases also display a peripheral rim of mature bone, a feature that is not typically seen in myoepithelial tumors. Similar to myoepithelial tumors, ossifying fibromyxoid tumor of soft parts often is positive for S-100 protein; however, it rarely is positive for cytokeratins. Ossifying fibromyxoid tumor of soft parts has been shown to have a rearrangement of the PHD finger protein 1 (PHF1) gene in approximately half of cases, a molecular finding that has not been reported for myoepithelial tumors.25 Finally, extra-axial soft tissue chordomas, though quite rare, may possess striking similarities to myoepithelial tumors both histopathologically and immunohistochemically. Chordomas are composed of epithelioid cells arranged in nests, nodules, and chains with a variably myxoid background. A variable amount of cells with bubbly cytoplasm (known as physaliphorous cells) can be seen. High mitotic activity is not a characteristic feature in chordomas. They classically coexpress cytokeratins and S-100 protein, similar to myoepithelial tumors.
Because cutaneous myoepithelial tumors are relatively rare, a well-defined standard of care for treatment is lacking. Surgical excision is the primary treatment method in most reported cases in the literature.17,19 Miller et al29 reported the successful treatment of recurrent cutaneous myoepitheliomas with Mohs micrographic surgery. Chemotherapy may be useful in the setting of metastatic myoepithelial carcinomas in adults, but reported results are inconsistent.30,31 Radiation treatment of recurrent or metastatic disease has not been shown to be effective. A study of children treated with surgical resection and chemotherapy using ifosfamide, cisplatin, and etoposide followed by radiation therapy showed positive results.32
Our case highlights several challenges that may arise in establishing a diagnosis of cutaneous myoepithelial carcinoma with disseminated metastases. The diagnostic difficulty in our case was compounded by the advanced nature of the lesion at the time of presentation. Given the rarity of metastatic cutaneous myoepithelial carcinomas and the lack of a prior primary diagnosis of a malignant myoepithelioma, the index of suspicion for this entity was not high. A report of myoepithelial carcinoma of the parotid gland metastatic to the skin has been reported,33 but in the absence of salivary gland involvement or other visceral lesions, metastasis from any source other than our patient’s cutaneous scalp lesion is unlikely. The histopathologic features in combination with the characteristic immunophenotype, unique clinical setting, and radiographic findings were essential to arriving at the correct diagnosis. Unlike previously reported metastatic lesions, our case is unique in that metastatic lesions were identified at the time of initial clinical presentation.
Conclusion
Cutaneous myoepithelial carcinomas are exceedingly rare tumors with a wide range of histopathologic and immunohistochemical findings. In challenging cases, studies for EWSR1 or PLAG1 gene rearrangement can be helpful. Furthermore, this case illustrates the potential for widespread dissemination of myoepithelial carcinomas requiring clinical evaluation and imaging studies to exclude metastatic lesions.
- Frost MW, Steiniche T, Damsgaard TE, et al. Primary cutaneous myoepithelial carcinoma: a case report and review of the literature. APMIS. 2014;122:369-379.
- Stojsic Z, Brasanac D, Boricic I, et al. Clear cell myoepithelial carcinoma of the skin. a case report. J Cutan Pathol. 2009;36:680-683.
- Tanahashi J, Kashima K, Daa T, et al. A case of cutaneous myoepithelial carcinoma. J Cutan Pathol. 2007;34:648-653.
- Michal M, Miettinen M. Myoepitheliomas of the skin and soft tissues. report of 12 cases. Virchows Arch. 1999;434:393-400.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Law RM, Viglione MP, Barrett TL. Metastatic myoepithelial carcinoma in a child. J Cutan Pathol. 2008;35:779-781.
- Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 2004;35:14-24.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. a molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114-1124.
- Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675-682.
- Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol. 2011;24:1444-1450.
- Mentzel T, Requena L, Kaddu S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30:294-302.
- Garcia-Sanchez S, Elices M, Nieto S. Cutaneous myoepithelial carcinoma (malignant myoepithelial tumor of skin). Virchows Archiv. 2009;455(suppl 1):1-482.
- Bajoghli A, Limpert J. Treatment of cutaneous malignant myoepithelioma on the nasal ala using Mohs micrographic surgery in a two and a half year old child. J Invest Dermatol. 2009;129:S44.
- Prasad AR, Savera AT, Gown AM, et al. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801-806.
- Savera AT, Sloman A, Huvos AG, et al. Myoepithelial carcinoma of the salivary glands. a clinicopathologic study of 25 patients. Am J Surg Pathol. 2000;24:761-774.
- Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183-1196.
- Kilpatrick SE, Hitchcock MG, Kraus MD, et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol. 1997;21:13-22.
- Neto AG, Pineda-Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: a case report and review of the literature. Head Neck. 2004;26:470-473.
- Kutzner H, Mentzel T, Kaddu S, et al. Cutaneous myoepithelioma: an under-recognized cutaneous neoplasm composed of myoepithelial cells. Am J Surg Pathol. 2001;25:348-355.
- Dix BT, Hentges MJ, Saltrick KR, et al. Cutaneous myoepithelioma in the foot: case report. Foot Ankle Spec. 2013;6:239-241.
- Burke T, Sahin A, Johnson DE, et al. Myoepithelioma of the retroperitoneum. Ultrastruct Pathol. 1995;19:269-274.
- Fernandez-Figueras MT, Puig L, Trias I, et al. Benign myoepithelioma of the skin. Am J Dermatopathol. 1998;20:208-212.
- Bahrami A, Dalton JD, Krane JF, et al. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140-148.
- Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340-352.
- Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: a fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am J Surg Pathol. 2013;37:1751-1755.
- Dabska M. Parachordoma: a new clinicopathologic entity. Cancer. 1977;40:1586-1592.
- Fletcher CDM, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Lauer SR, Edgar MA, Gardner JM, et al. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719-726.
- Miller TD, McCalmont T, Tope WD. Recurrent cutaneous myoepithelioma treated using Mohs micrographic surgery: case report and review of the literature. Dermatol Surg. 2009;35:139-143.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Noronha V, Cooper DL, Higgins SA, et al. Metastatic myoepithelial carcinoma of the vulva treated with carboplatin and paclitaxel. Lancet Oncol. 2006;7:270-271.
- Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer. 2014;61:643-646.
- He DQ, Hua CG, Tang XF, et al. Cutaneous metastasis from a parotid myoepithelial carcinoma: a case report and review of the literature. J Cutan Pathol. 2008;35:1138-1143.
- Frost MW, Steiniche T, Damsgaard TE, et al. Primary cutaneous myoepithelial carcinoma: a case report and review of the literature. APMIS. 2014;122:369-379.
- Stojsic Z, Brasanac D, Boricic I, et al. Clear cell myoepithelial carcinoma of the skin. a case report. J Cutan Pathol. 2009;36:680-683.
- Tanahashi J, Kashima K, Daa T, et al. A case of cutaneous myoepithelial carcinoma. J Cutan Pathol. 2007;34:648-653.
- Michal M, Miettinen M. Myoepitheliomas of the skin and soft tissues. report of 12 cases. Virchows Arch. 1999;434:393-400.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Law RM, Viglione MP, Barrett TL. Metastatic myoepithelial carcinoma in a child. J Cutan Pathol. 2008;35:779-781.
- Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 2004;35:14-24.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. a molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114-1124.
- Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675-682.
- Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol. 2011;24:1444-1450.
- Mentzel T, Requena L, Kaddu S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30:294-302.
- Garcia-Sanchez S, Elices M, Nieto S. Cutaneous myoepithelial carcinoma (malignant myoepithelial tumor of skin). Virchows Archiv. 2009;455(suppl 1):1-482.
- Bajoghli A, Limpert J. Treatment of cutaneous malignant myoepithelioma on the nasal ala using Mohs micrographic surgery in a two and a half year old child. J Invest Dermatol. 2009;129:S44.
- Prasad AR, Savera AT, Gown AM, et al. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801-806.
- Savera AT, Sloman A, Huvos AG, et al. Myoepithelial carcinoma of the salivary glands. a clinicopathologic study of 25 patients. Am J Surg Pathol. 2000;24:761-774.
- Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183-1196.
- Kilpatrick SE, Hitchcock MG, Kraus MD, et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol. 1997;21:13-22.
- Neto AG, Pineda-Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: a case report and review of the literature. Head Neck. 2004;26:470-473.
- Kutzner H, Mentzel T, Kaddu S, et al. Cutaneous myoepithelioma: an under-recognized cutaneous neoplasm composed of myoepithelial cells. Am J Surg Pathol. 2001;25:348-355.
- Dix BT, Hentges MJ, Saltrick KR, et al. Cutaneous myoepithelioma in the foot: case report. Foot Ankle Spec. 2013;6:239-241.
- Burke T, Sahin A, Johnson DE, et al. Myoepithelioma of the retroperitoneum. Ultrastruct Pathol. 1995;19:269-274.
- Fernandez-Figueras MT, Puig L, Trias I, et al. Benign myoepithelioma of the skin. Am J Dermatopathol. 1998;20:208-212.
- Bahrami A, Dalton JD, Krane JF, et al. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140-148.
- Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340-352.
- Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: a fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am J Surg Pathol. 2013;37:1751-1755.
- Dabska M. Parachordoma: a new clinicopathologic entity. Cancer. 1977;40:1586-1592.
- Fletcher CDM, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Lauer SR, Edgar MA, Gardner JM, et al. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719-726.
- Miller TD, McCalmont T, Tope WD. Recurrent cutaneous myoepithelioma treated using Mohs micrographic surgery: case report and review of the literature. Dermatol Surg. 2009;35:139-143.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Noronha V, Cooper DL, Higgins SA, et al. Metastatic myoepithelial carcinoma of the vulva treated with carboplatin and paclitaxel. Lancet Oncol. 2006;7:270-271.
- Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer. 2014;61:643-646.
- He DQ, Hua CG, Tang XF, et al. Cutaneous metastasis from a parotid myoepithelial carcinoma: a case report and review of the literature. J Cutan Pathol. 2008;35:1138-1143.
Practice Points
- Cutaneous myoepithelial carcinoma is a rare malignant adnexal neoplasm with metastatic potential that can present in the skin.
- Cutaneous myoepithelial carcinoma is a tumor that can occasionally show EWSR1 gene rearrangement.
- Excision with negative margins and close follow-up is recommended for cutaneous myoepithelial carcinoma.