User login
Technique enables SCD detection with a smartphone

Photo by Daniel Sone
Researchers say they’ve developed a simple technique for diagnosing and monitoring sickle cell disease (SCD) that could be used in regions where advanced medical technology and training are scarce.
The team created a 3D-printed box that can be attached to an Android smartphone and used to test a small blood sample.
The testing method involves magnetic levitation, which allows the user to differentiate sickle cells from normal red blood cells with the naked eye.
Savas Tasoglu, PhD, of the University of Connecticut in Storrs, and his colleagues described this technique in Nature Scientific Reports.
First, a clinician takes a blood sample from a patient and mixes it with a common, salt-based solution that draws oxygen out of sickle cells, making them denser and easier to detect via magnetic levitation. The denser sickle cells will float at a lower height than healthy red blood cells, which are not affected by the solution.
The sample is then loaded into a disposable micro-capillary that is inserted into the tester attached to the smartphone. Inside the testing apparatus, the micro-capillary passes between 2 magnets that are aligned so that the same poles face each other, creating a magnetic field.
The capillary is then illuminated with an LED that is filtered through a ground glass diffuser and magnified by an internal lens.
The smartphone’s built-in camera captures the resulting image and presents it digitally on the phone’s external display. The blood cells floating inside the capillary—whether higher-floating healthy red blood cells or lower-floating sickle cells—can be easily observed.
The device also provides clinicians with a digital readout that assigns a numerical value to the sample density to assist with the diagnosis. The entire process takes less than 15 minutes.
“With this device, you’re getting much more specific information about your cells than some other tests,” said Stephanie Knowlton, a graduate student at the University of Connecticut.
“Rather than sending a sample to a lab and waiting 3 days to find out if you have this disease, with this device, you get on-site and portable results right away. We believe a device like this could be very helpful in third-world countries where laboratory resources may be limited.”
Dr Tasoglu’s lab has filed a provisional patent for the device and is working on expanding its capabilities so it can be applied to other diseases. ![]()
Photo by Daniel Sone
Researchers say they’ve developed a simple technique for diagnosing and monitoring sickle cell disease (SCD) that could be used in regions where advanced medical technology and training are scarce.
The team created a 3D-printed box that can be attached to an Android smartphone and used to test a small blood sample.
The testing method involves magnetic levitation, which allows the user to differentiate sickle cells from normal red blood cells with the naked eye.
Savas Tasoglu, PhD, of the University of Connecticut in Storrs, and his colleagues described this technique in Nature Scientific Reports.
First, a clinician takes a blood sample from a patient and mixes it with a common, salt-based solution that draws oxygen out of sickle cells, making them denser and easier to detect via magnetic levitation. The denser sickle cells will float at a lower height than healthy red blood cells, which are not affected by the solution.
The sample is then loaded into a disposable micro-capillary that is inserted into the tester attached to the smartphone. Inside the testing apparatus, the micro-capillary passes between 2 magnets that are aligned so that the same poles face each other, creating a magnetic field.
The capillary is then illuminated with an LED that is filtered through a ground glass diffuser and magnified by an internal lens.
The smartphone’s built-in camera captures the resulting image and presents it digitally on the phone’s external display. The blood cells floating inside the capillary—whether higher-floating healthy red blood cells or lower-floating sickle cells—can be easily observed.
The device also provides clinicians with a digital readout that assigns a numerical value to the sample density to assist with the diagnosis. The entire process takes less than 15 minutes.
“With this device, you’re getting much more specific information about your cells than some other tests,” said Stephanie Knowlton, a graduate student at the University of Connecticut.
“Rather than sending a sample to a lab and waiting 3 days to find out if you have this disease, with this device, you get on-site and portable results right away. We believe a device like this could be very helpful in third-world countries where laboratory resources may be limited.”
Dr Tasoglu’s lab has filed a provisional patent for the device and is working on expanding its capabilities so it can be applied to other diseases. ![]()
Photo by Daniel Sone
Researchers say they’ve developed a simple technique for diagnosing and monitoring sickle cell disease (SCD) that could be used in regions where advanced medical technology and training are scarce.
The team created a 3D-printed box that can be attached to an Android smartphone and used to test a small blood sample.
The testing method involves magnetic levitation, which allows the user to differentiate sickle cells from normal red blood cells with the naked eye.
Savas Tasoglu, PhD, of the University of Connecticut in Storrs, and his colleagues described this technique in Nature Scientific Reports.
First, a clinician takes a blood sample from a patient and mixes it with a common, salt-based solution that draws oxygen out of sickle cells, making them denser and easier to detect via magnetic levitation. The denser sickle cells will float at a lower height than healthy red blood cells, which are not affected by the solution.
The sample is then loaded into a disposable micro-capillary that is inserted into the tester attached to the smartphone. Inside the testing apparatus, the micro-capillary passes between 2 magnets that are aligned so that the same poles face each other, creating a magnetic field.
The capillary is then illuminated with an LED that is filtered through a ground glass diffuser and magnified by an internal lens.
The smartphone’s built-in camera captures the resulting image and presents it digitally on the phone’s external display. The blood cells floating inside the capillary—whether higher-floating healthy red blood cells or lower-floating sickle cells—can be easily observed.
The device also provides clinicians with a digital readout that assigns a numerical value to the sample density to assist with the diagnosis. The entire process takes less than 15 minutes.
“With this device, you’re getting much more specific information about your cells than some other tests,” said Stephanie Knowlton, a graduate student at the University of Connecticut.
“Rather than sending a sample to a lab and waiting 3 days to find out if you have this disease, with this device, you get on-site and portable results right away. We believe a device like this could be very helpful in third-world countries where laboratory resources may be limited.”
Dr Tasoglu’s lab has filed a provisional patent for the device and is working on expanding its capabilities so it can be applied to other diseases. ![]()
Immunosuppressant can treat autoimmune cytopenias

New research suggests the immunosuppressant sirolimus may be a promising treatment option for patients with refractory autoimmune cytopenias.
The drug proved particularly effective in children with autoimmune lymphoproliferative syndrome (ALPS), producing complete responses in all of the ALPS patients studied.
On the other hand, patients with single-lineage autoimmune cytopenias, such as immune thrombocytopenia (ITP), did not fare as well.
David T. Teachey, MD, of The Children’s Hospital of Philadelphia in Pennsylvania, and his colleagues reported these results in Blood.
The group studied sirolimus in 30 patients with refractory autoimmune cytopenias who were 5 to 19 years of age. All of the patients were refractory to or could not tolerate corticosteroids.
Twelve patients had ALPS, 6 had single-lineage autoimmune cytopenias (4 with ITP and 2 with autoimmune hemolytic anemia [AIHA]), and 12 patients had multi-lineage cytopenias secondary to common variable immune deficiency (n=2), Evans syndrome (n=8), or systemic lupus erythematosus (n=2).
The patients received 2 mg/m2 to 2.5 mg/m2 per day of sirolimus in liquid or tablet form for 6 months. After 6 months, those who benefited from the drug were allowed to continue treatment with follow-up appointments to monitor toxicities.
Of the 12 children with ALPS, 11 had complete responses—normalization of blood cell counts—from 1 to 3 months after receiving sirolimus. The remaining patient achieved a complete response after 18 months.
All ALPS patients were successfully weaned off steroids and discontinued all other medications within 1 week to 1 month after starting sirolimus.
The patients with multi-lineage cytopenias also responded well to sirolimus. Eight of the 12 patients had complete responses, although these occurred later than for most ALPS patients (after 3 months).
The 6 patients with single-lineage cytopenias had less robust results—1 complete response and 2 partial responses. One child with ITP achieved a partial response but had to discontinue therapy.
One of the patients with AIHA had a complete response by 6 months and was able to stop taking other medications within a month. The other child with AIHA achieved a partial response.
For all patients, the median time on sirolimus was 2 years (range, 1–4.5 years).
The most common adverse event observed in this study was grade 1-2 mucositis (n=10). Other toxicities included elevated triglycerides and elevated cholesterol (n=2), acne (n=1), sun sensitivity (n=1), and exacerbation of gastro-esophageal reflux disease (n=1).
One patient developed hypertension 2 years after starting sirolimus, but this was temporally related to starting a new psychiatric medication.
Another patient (with Evans syndrome) developed a headache with associated white matter changes (4 different lesions). The changes were attributed to disease-associated vasculitis, and the lesions resolved over a few months with the addition of steroids. The patient was eventually diagnosed with a primary T-cell immune deficiency and underwent hematopoietic stem cell transplant.
“This study demonstrates that sirolimus is an effective and safe alternative to steroids, providing children with an improved quality of life as they continue treatment into adulthood,” Dr Teachey said. “While further studies are needed, sirolimus should be considered an early therapy option for patients with autoimmune blood disorders requiring ongoing therapy.” ![]()
New research suggests the immunosuppressant sirolimus may be a promising treatment option for patients with refractory autoimmune cytopenias.
The drug proved particularly effective in children with autoimmune lymphoproliferative syndrome (ALPS), producing complete responses in all of the ALPS patients studied.
On the other hand, patients with single-lineage autoimmune cytopenias, such as immune thrombocytopenia (ITP), did not fare as well.
David T. Teachey, MD, of The Children’s Hospital of Philadelphia in Pennsylvania, and his colleagues reported these results in Blood.
The group studied sirolimus in 30 patients with refractory autoimmune cytopenias who were 5 to 19 years of age. All of the patients were refractory to or could not tolerate corticosteroids.
Twelve patients had ALPS, 6 had single-lineage autoimmune cytopenias (4 with ITP and 2 with autoimmune hemolytic anemia [AIHA]), and 12 patients had multi-lineage cytopenias secondary to common variable immune deficiency (n=2), Evans syndrome (n=8), or systemic lupus erythematosus (n=2).
The patients received 2 mg/m2 to 2.5 mg/m2 per day of sirolimus in liquid or tablet form for 6 months. After 6 months, those who benefited from the drug were allowed to continue treatment with follow-up appointments to monitor toxicities.
Of the 12 children with ALPS, 11 had complete responses—normalization of blood cell counts—from 1 to 3 months after receiving sirolimus. The remaining patient achieved a complete response after 18 months.
All ALPS patients were successfully weaned off steroids and discontinued all other medications within 1 week to 1 month after starting sirolimus.
The patients with multi-lineage cytopenias also responded well to sirolimus. Eight of the 12 patients had complete responses, although these occurred later than for most ALPS patients (after 3 months).
The 6 patients with single-lineage cytopenias had less robust results—1 complete response and 2 partial responses. One child with ITP achieved a partial response but had to discontinue therapy.
One of the patients with AIHA had a complete response by 6 months and was able to stop taking other medications within a month. The other child with AIHA achieved a partial response.
For all patients, the median time on sirolimus was 2 years (range, 1–4.5 years).
The most common adverse event observed in this study was grade 1-2 mucositis (n=10). Other toxicities included elevated triglycerides and elevated cholesterol (n=2), acne (n=1), sun sensitivity (n=1), and exacerbation of gastro-esophageal reflux disease (n=1).
One patient developed hypertension 2 years after starting sirolimus, but this was temporally related to starting a new psychiatric medication.
Another patient (with Evans syndrome) developed a headache with associated white matter changes (4 different lesions). The changes were attributed to disease-associated vasculitis, and the lesions resolved over a few months with the addition of steroids. The patient was eventually diagnosed with a primary T-cell immune deficiency and underwent hematopoietic stem cell transplant.
“This study demonstrates that sirolimus is an effective and safe alternative to steroids, providing children with an improved quality of life as they continue treatment into adulthood,” Dr Teachey said. “While further studies are needed, sirolimus should be considered an early therapy option for patients with autoimmune blood disorders requiring ongoing therapy.” ![]()
New research suggests the immunosuppressant sirolimus may be a promising treatment option for patients with refractory autoimmune cytopenias.
The drug proved particularly effective in children with autoimmune lymphoproliferative syndrome (ALPS), producing complete responses in all of the ALPS patients studied.
On the other hand, patients with single-lineage autoimmune cytopenias, such as immune thrombocytopenia (ITP), did not fare as well.
David T. Teachey, MD, of The Children’s Hospital of Philadelphia in Pennsylvania, and his colleagues reported these results in Blood.
The group studied sirolimus in 30 patients with refractory autoimmune cytopenias who were 5 to 19 years of age. All of the patients were refractory to or could not tolerate corticosteroids.
Twelve patients had ALPS, 6 had single-lineage autoimmune cytopenias (4 with ITP and 2 with autoimmune hemolytic anemia [AIHA]), and 12 patients had multi-lineage cytopenias secondary to common variable immune deficiency (n=2), Evans syndrome (n=8), or systemic lupus erythematosus (n=2).
The patients received 2 mg/m2 to 2.5 mg/m2 per day of sirolimus in liquid or tablet form for 6 months. After 6 months, those who benefited from the drug were allowed to continue treatment with follow-up appointments to monitor toxicities.
Of the 12 children with ALPS, 11 had complete responses—normalization of blood cell counts—from 1 to 3 months after receiving sirolimus. The remaining patient achieved a complete response after 18 months.
All ALPS patients were successfully weaned off steroids and discontinued all other medications within 1 week to 1 month after starting sirolimus.
The patients with multi-lineage cytopenias also responded well to sirolimus. Eight of the 12 patients had complete responses, although these occurred later than for most ALPS patients (after 3 months).
The 6 patients with single-lineage cytopenias had less robust results—1 complete response and 2 partial responses. One child with ITP achieved a partial response but had to discontinue therapy.
One of the patients with AIHA had a complete response by 6 months and was able to stop taking other medications within a month. The other child with AIHA achieved a partial response.
For all patients, the median time on sirolimus was 2 years (range, 1–4.5 years).
The most common adverse event observed in this study was grade 1-2 mucositis (n=10). Other toxicities included elevated triglycerides and elevated cholesterol (n=2), acne (n=1), sun sensitivity (n=1), and exacerbation of gastro-esophageal reflux disease (n=1).
One patient developed hypertension 2 years after starting sirolimus, but this was temporally related to starting a new psychiatric medication.
Another patient (with Evans syndrome) developed a headache with associated white matter changes (4 different lesions). The changes were attributed to disease-associated vasculitis, and the lesions resolved over a few months with the addition of steroids. The patient was eventually diagnosed with a primary T-cell immune deficiency and underwent hematopoietic stem cell transplant.
“This study demonstrates that sirolimus is an effective and safe alternative to steroids, providing children with an improved quality of life as they continue treatment into adulthood,” Dr Teachey said. “While further studies are needed, sirolimus should be considered an early therapy option for patients with autoimmune blood disorders requiring ongoing therapy.” ![]()
How a cancer diagnosis affects income

receiving treatment
Photo by Rhoda Baer
A new study indicates that when American adults are diagnosed with
cancer, they experience significant decreases in the probability of
working, in the number of hours they work, and correspondingly, in their
incomes.
Additionally, these effects appear to be more pronounced among men than women.
Anna Zajacova, PhD, of the University of Wyoming in Laramie, and her colleagues reported these findings in Cancer.
The researchers analyzed data from the Panel Study of Income Dynamics, a nationally representative, prospective, population-based, observational study with individual and family level economic information. The data spanned the period from 1999 to 2009.
The team used models to estimate the impact of cancer on employment, hours worked, individual income, and total family income.
The results showed that, after a cancer diagnosis, the probability of a patient being employed dropped by almost 10 percentage points, and hours worked declined by up to 200 hours, or about 5 weeks of full-time work, in the first year.
Annual labor market earnings dropped almost 40% within 2 years of diagnosis, and they remained lower than before diagnosis. Total family income declined by 20%, although it recovered within 4 years of cancer diagnosis.
These effects were primarily driven by losses among male cancer survivors. For women diagnosed with cancer, the losses were largely not statistically significant.
“Fifteen million American adults are cancer survivors, and American families need economic support while they are dealing with the rigors of cancer treatment,” Dr Zajacova said.
“Our paper suggests that families where an adult—especially a working-age male—is diagnosed with cancer suffer short-term and long-term declines in their economic well-being. We need to improve workplace and insurance safety nets so families can focus on dealing with the cancer treatment rather than deal with the financial and employment fallout.” ![]()
receiving treatment
Photo by Rhoda Baer
A new study indicates that when American adults are diagnosed with
cancer, they experience significant decreases in the probability of
working, in the number of hours they work, and correspondingly, in their
incomes.
Additionally, these effects appear to be more pronounced among men than women.
Anna Zajacova, PhD, of the University of Wyoming in Laramie, and her colleagues reported these findings in Cancer.
The researchers analyzed data from the Panel Study of Income Dynamics, a nationally representative, prospective, population-based, observational study with individual and family level economic information. The data spanned the period from 1999 to 2009.
The team used models to estimate the impact of cancer on employment, hours worked, individual income, and total family income.
The results showed that, after a cancer diagnosis, the probability of a patient being employed dropped by almost 10 percentage points, and hours worked declined by up to 200 hours, or about 5 weeks of full-time work, in the first year.
Annual labor market earnings dropped almost 40% within 2 years of diagnosis, and they remained lower than before diagnosis. Total family income declined by 20%, although it recovered within 4 years of cancer diagnosis.
These effects were primarily driven by losses among male cancer survivors. For women diagnosed with cancer, the losses were largely not statistically significant.
“Fifteen million American adults are cancer survivors, and American families need economic support while they are dealing with the rigors of cancer treatment,” Dr Zajacova said.
“Our paper suggests that families where an adult—especially a working-age male—is diagnosed with cancer suffer short-term and long-term declines in their economic well-being. We need to improve workplace and insurance safety nets so families can focus on dealing with the cancer treatment rather than deal with the financial and employment fallout.” ![]()
receiving treatment
Photo by Rhoda Baer
A new study indicates that when American adults are diagnosed with
cancer, they experience significant decreases in the probability of
working, in the number of hours they work, and correspondingly, in their
incomes.
Additionally, these effects appear to be more pronounced among men than women.
Anna Zajacova, PhD, of the University of Wyoming in Laramie, and her colleagues reported these findings in Cancer.
The researchers analyzed data from the Panel Study of Income Dynamics, a nationally representative, prospective, population-based, observational study with individual and family level economic information. The data spanned the period from 1999 to 2009.
The team used models to estimate the impact of cancer on employment, hours worked, individual income, and total family income.
The results showed that, after a cancer diagnosis, the probability of a patient being employed dropped by almost 10 percentage points, and hours worked declined by up to 200 hours, or about 5 weeks of full-time work, in the first year.
Annual labor market earnings dropped almost 40% within 2 years of diagnosis, and they remained lower than before diagnosis. Total family income declined by 20%, although it recovered within 4 years of cancer diagnosis.
These effects were primarily driven by losses among male cancer survivors. For women diagnosed with cancer, the losses were largely not statistically significant.
“Fifteen million American adults are cancer survivors, and American families need economic support while they are dealing with the rigors of cancer treatment,” Dr Zajacova said.
“Our paper suggests that families where an adult—especially a working-age male—is diagnosed with cancer suffer short-term and long-term declines in their economic well-being. We need to improve workplace and insurance safety nets so families can focus on dealing with the cancer treatment rather than deal with the financial and employment fallout.” ![]()
How a cancer diagnosis affects income
receiving treatment
Photo by Rhoda Baer
A new study indicates that when American adults are diagnosed with cancer, they experience significant decreases in the probability of working, in the number of hours they work, and correspondingly, in their incomes.
Additionally, these effects appear to be more pronounced among men than women.
Anna Zajacova, PhD, of the University of Wyoming in Laramie, and her colleagues reported these findings in Cancer.
The researchers analyzed data from the Panel Study of Income Dynamics, a nationally representative, prospective, population-based, observational study with individual and family level economic information. The data spanned the period from 1999 to 2009.
The team used models to estimate the impact of cancer on employment, hours worked, individual income, and total family income.
The results showed that, after a cancer diagnosis, the probability of a patient being employed dropped by almost 10 percentage points, and hours worked declined by up to 200 hours, or about 5 weeks of full-time work, in the first year.
Annual labor market earnings dropped almost 40% within 2 years of diagnosis, and they remained lower than before diagnosis. Total family income declined by 20%, although it recovered within 4 years of cancer diagnosis.
These effects were primarily driven by losses among male cancer survivors. For women diagnosed with cancer, the losses were largely not statistically significant.
“Fifteen million American adults are cancer survivors, and American families need economic support while they are dealing with the rigors of cancer treatment,” Dr Zajacova said.
“Our paper suggests that families where an adult—especially a working-age male—is diagnosed with cancer suffer short-term and long-term declines in their economic well-being. We need to improve workplace and insurance safety nets so families can focus on dealing with the cancer treatment rather than deal with the financial and employment fallout.” ![]()
receiving treatment
Photo by Rhoda Baer
A new study indicates that when American adults are diagnosed with cancer, they experience significant decreases in the probability of working, in the number of hours they work, and correspondingly, in their incomes.
Additionally, these effects appear to be more pronounced among men than women.
Anna Zajacova, PhD, of the University of Wyoming in Laramie, and her colleagues reported these findings in Cancer.
The researchers analyzed data from the Panel Study of Income Dynamics, a nationally representative, prospective, population-based, observational study with individual and family level economic information. The data spanned the period from 1999 to 2009.
The team used models to estimate the impact of cancer on employment, hours worked, individual income, and total family income.
The results showed that, after a cancer diagnosis, the probability of a patient being employed dropped by almost 10 percentage points, and hours worked declined by up to 200 hours, or about 5 weeks of full-time work, in the first year.
Annual labor market earnings dropped almost 40% within 2 years of diagnosis, and they remained lower than before diagnosis. Total family income declined by 20%, although it recovered within 4 years of cancer diagnosis.
These effects were primarily driven by losses among male cancer survivors. For women diagnosed with cancer, the losses were largely not statistically significant.
“Fifteen million American adults are cancer survivors, and American families need economic support while they are dealing with the rigors of cancer treatment,” Dr Zajacova said.
“Our paper suggests that families where an adult—especially a working-age male—is diagnosed with cancer suffer short-term and long-term declines in their economic well-being. We need to improve workplace and insurance safety nets so families can focus on dealing with the cancer treatment rather than deal with the financial and employment fallout.” ![]()
receiving treatment
Photo by Rhoda Baer
A new study indicates that when American adults are diagnosed with cancer, they experience significant decreases in the probability of working, in the number of hours they work, and correspondingly, in their incomes.
Additionally, these effects appear to be more pronounced among men than women.
Anna Zajacova, PhD, of the University of Wyoming in Laramie, and her colleagues reported these findings in Cancer.
The researchers analyzed data from the Panel Study of Income Dynamics, a nationally representative, prospective, population-based, observational study with individual and family level economic information. The data spanned the period from 1999 to 2009.
The team used models to estimate the impact of cancer on employment, hours worked, individual income, and total family income.
The results showed that, after a cancer diagnosis, the probability of a patient being employed dropped by almost 10 percentage points, and hours worked declined by up to 200 hours, or about 5 weeks of full-time work, in the first year.
Annual labor market earnings dropped almost 40% within 2 years of diagnosis, and they remained lower than before diagnosis. Total family income declined by 20%, although it recovered within 4 years of cancer diagnosis.
These effects were primarily driven by losses among male cancer survivors. For women diagnosed with cancer, the losses were largely not statistically significant.
“Fifteen million American adults are cancer survivors, and American families need economic support while they are dealing with the rigors of cancer treatment,” Dr Zajacova said.
“Our paper suggests that families where an adult—especially a working-age male—is diagnosed with cancer suffer short-term and long-term declines in their economic well-being. We need to improve workplace and insurance safety nets so families can focus on dealing with the cancer treatment rather than deal with the financial and employment fallout.” ![]()
Group identifies potential target for AML
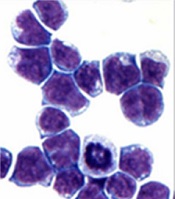
marrow cells generated by AE
Image courtesy of
The Rockefeller University
Preclinical research has revealed a potential therapeutic target for acute myeloid leukemia (AML)—the histone demethylase JMJD1C.
Investigators found that JMJD1C interacts with RUNX1–RUNX1T1 (formerly AML1-ETO and abbreviated here as AE), a transcription factor generated by the t(8;21) translocation in AML.
However, the team also found that JMJD1C is required for proliferation in many AML cell lines, not just those with AE.
Robert G. Roeder, PhD, of The Rockefeller University in New York, New York, and his colleagues detailed these findings in Genes and Development.
The investigators began this study by searching for proteins that interact with AE, and they identified JMJD1C. To investigate the relationship between JMJD1C and AE, the team explored the broader effects of removing JMJD1C.
“We found that numerous genes were downregulated upon loss of JMJD1C, and the set overlaps significantly with the genes that are normally activated by AE,” said study author Mo Chen, PhD, a researcher in Dr Roeder’s lab.
The investigators also found that AML cells are addicted to the presence of JMJD1C, and, without it, they cannot survive. The team observed an increase in apoptosis when JMJD1C was depleted from Kasumi-1 and SKNO-1 cell lines.
With subsequent experiments, the investigators confirmed that JMJD1C interacts with AE and demonstrated that JMJD1C is required for AE to exert its cancer-promoting effects. But they also found that JMJD1C plays an even broader role in AML.
“We were very surprised to find that JMJD1C is required for the proliferation of other acute myeloid leukemia cell lines, which do not have AE,” Dr Chen said.
The team found that depleting JMJD1C compromised the growth of the following cell lines: Kasumi-1 (AML1-ETO; AML M2), MOLM-13 (MLL-AF9, FLT3ITD; AML M5a), THP-1 (MLL-AF9, NRASmut; AML M5), HNT-34 (BCR-ABL1; AML M4), MV4-11 (MLL-AF4; AML M5), CMK (JAK3A527V; AML M7+ Down’s Syndrome), HEL (AML M6), NOMO-1 (MLL-AF9; AML M5a), NB4 (PML-RARα; AML M3), and HL-60 (MYC amplification; AML M2).
The only cell line that was not affected by depletion of JMJD1C was KG-1 (NRASmut; AML).
To build upon this discovery, the investigators looked for transcription factors aside from AE that might be responsible for JMJD1C addiction. The team found at least 2—LYL1 and HEB—that can recruit JMJD1C to target genes in diseased cells that lack AE, fueling leukemia growth.
The investigators said these results suggest JMJD1C may play a general role in promoting the growth of myeloid leukemias.
“We are excited because this type of general phenomena is an ideal target for drug development,” Dr Roeder said. “Our work will facilitate the development of selective inhibitors against JMJD1C, which is a highly promising therapeutic target for multiple types of leukemia.” ![]()
marrow cells generated by AE
Image courtesy of
The Rockefeller University
Preclinical research has revealed a potential therapeutic target for acute myeloid leukemia (AML)—the histone demethylase JMJD1C.
Investigators found that JMJD1C interacts with RUNX1–RUNX1T1 (formerly AML1-ETO and abbreviated here as AE), a transcription factor generated by the t(8;21) translocation in AML.
However, the team also found that JMJD1C is required for proliferation in many AML cell lines, not just those with AE.
Robert G. Roeder, PhD, of The Rockefeller University in New York, New York, and his colleagues detailed these findings in Genes and Development.
The investigators began this study by searching for proteins that interact with AE, and they identified JMJD1C. To investigate the relationship between JMJD1C and AE, the team explored the broader effects of removing JMJD1C.
“We found that numerous genes were downregulated upon loss of JMJD1C, and the set overlaps significantly with the genes that are normally activated by AE,” said study author Mo Chen, PhD, a researcher in Dr Roeder’s lab.
The investigators also found that AML cells are addicted to the presence of JMJD1C, and, without it, they cannot survive. The team observed an increase in apoptosis when JMJD1C was depleted from Kasumi-1 and SKNO-1 cell lines.
With subsequent experiments, the investigators confirmed that JMJD1C interacts with AE and demonstrated that JMJD1C is required for AE to exert its cancer-promoting effects. But they also found that JMJD1C plays an even broader role in AML.
“We were very surprised to find that JMJD1C is required for the proliferation of other acute myeloid leukemia cell lines, which do not have AE,” Dr Chen said.
The team found that depleting JMJD1C compromised the growth of the following cell lines: Kasumi-1 (AML1-ETO; AML M2), MOLM-13 (MLL-AF9, FLT3ITD; AML M5a), THP-1 (MLL-AF9, NRASmut; AML M5), HNT-34 (BCR-ABL1; AML M4), MV4-11 (MLL-AF4; AML M5), CMK (JAK3A527V; AML M7+ Down’s Syndrome), HEL (AML M6), NOMO-1 (MLL-AF9; AML M5a), NB4 (PML-RARα; AML M3), and HL-60 (MYC amplification; AML M2).
The only cell line that was not affected by depletion of JMJD1C was KG-1 (NRASmut; AML).
To build upon this discovery, the investigators looked for transcription factors aside from AE that might be responsible for JMJD1C addiction. The team found at least 2—LYL1 and HEB—that can recruit JMJD1C to target genes in diseased cells that lack AE, fueling leukemia growth.
The investigators said these results suggest JMJD1C may play a general role in promoting the growth of myeloid leukemias.
“We are excited because this type of general phenomena is an ideal target for drug development,” Dr Roeder said. “Our work will facilitate the development of selective inhibitors against JMJD1C, which is a highly promising therapeutic target for multiple types of leukemia.” ![]()
marrow cells generated by AE
Image courtesy of
The Rockefeller University
Preclinical research has revealed a potential therapeutic target for acute myeloid leukemia (AML)—the histone demethylase JMJD1C.
Investigators found that JMJD1C interacts with RUNX1–RUNX1T1 (formerly AML1-ETO and abbreviated here as AE), a transcription factor generated by the t(8;21) translocation in AML.
However, the team also found that JMJD1C is required for proliferation in many AML cell lines, not just those with AE.
Robert G. Roeder, PhD, of The Rockefeller University in New York, New York, and his colleagues detailed these findings in Genes and Development.
The investigators began this study by searching for proteins that interact with AE, and they identified JMJD1C. To investigate the relationship between JMJD1C and AE, the team explored the broader effects of removing JMJD1C.
“We found that numerous genes were downregulated upon loss of JMJD1C, and the set overlaps significantly with the genes that are normally activated by AE,” said study author Mo Chen, PhD, a researcher in Dr Roeder’s lab.
The investigators also found that AML cells are addicted to the presence of JMJD1C, and, without it, they cannot survive. The team observed an increase in apoptosis when JMJD1C was depleted from Kasumi-1 and SKNO-1 cell lines.
With subsequent experiments, the investigators confirmed that JMJD1C interacts with AE and demonstrated that JMJD1C is required for AE to exert its cancer-promoting effects. But they also found that JMJD1C plays an even broader role in AML.
“We were very surprised to find that JMJD1C is required for the proliferation of other acute myeloid leukemia cell lines, which do not have AE,” Dr Chen said.
The team found that depleting JMJD1C compromised the growth of the following cell lines: Kasumi-1 (AML1-ETO; AML M2), MOLM-13 (MLL-AF9, FLT3ITD; AML M5a), THP-1 (MLL-AF9, NRASmut; AML M5), HNT-34 (BCR-ABL1; AML M4), MV4-11 (MLL-AF4; AML M5), CMK (JAK3A527V; AML M7+ Down’s Syndrome), HEL (AML M6), NOMO-1 (MLL-AF9; AML M5a), NB4 (PML-RARα; AML M3), and HL-60 (MYC amplification; AML M2).
The only cell line that was not affected by depletion of JMJD1C was KG-1 (NRASmut; AML).
To build upon this discovery, the investigators looked for transcription factors aside from AE that might be responsible for JMJD1C addiction. The team found at least 2—LYL1 and HEB—that can recruit JMJD1C to target genes in diseased cells that lack AE, fueling leukemia growth.
The investigators said these results suggest JMJD1C may play a general role in promoting the growth of myeloid leukemias.
“We are excited because this type of general phenomena is an ideal target for drug development,” Dr Roeder said. “Our work will facilitate the development of selective inhibitors against JMJD1C, which is a highly promising therapeutic target for multiple types of leukemia.” ![]()
WHO recommends pilot projects for malaria vaccine

a malaria-endemic region
Photo by Sarah Mattison
More testing is needed before the malaria vaccine candidate RTS,S/AS01 (Mosquirix) can be put into widespread use, according to a pair of World Health Organization (WHO) advisory committees.
The WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) are recommending that RTS,S be introduced in 3 to 5 pilot projects to determine the best way to deliver the vaccine to young children.
The issue, according to the committees, is that the vaccine must be administered in 4 doses and therefore requires repeat contact with the healthcare system.
The first 3 doses are given 1 month apart, and the last dose is given 18 months later. Without the fourth dose, children in a phase 3 study of RTS,S had no overall reduction in severe malaria.
So SAGE and MPAC want to be sure this vaccination schedule is feasible.
“The question about how the malaria vaccine may best be delivered still needs to be answered,” said Jon S. Abramson, chair of SAGE. “After detailed assessment of all the evidence, we recommended that this question is best addressed by having 3 to 5 large pilot implementation projects.”
This could delay widespread implementation of RTS,S for 3 to 5 years. Alternatively, if it is not possible to deliver all 4 doses of RTS,S consistently, Abramson said the vaccine may not be used at all.
RTS,S is being assessed as a complementary malaria control tool that could potentially be added to—but not replace—the core package of proven malaria preventive, diagnostic, and treatment measures.
The vaccine acts against Plasmodium falciparum, the most deadly malaria parasite globally and the most prevalent in Africa.
In a phase 3 trial, young children who received 4 doses of RTS,S had a 36% reduction in the number of clinical episodes of malaria at 4 years. Infants who received 4 doses of RTS,S had a 26% reduction in the number of clinical malaria episodes over 3 years.
Children had a significantly lower incidence of severe malaria only if they received all 4 doses of RTS,S. The vaccine did not confer the same benefit in infants, regardless of the doses given.
Results of a subsequent study suggested that genetic variation influences the vaccine’s ability to ward off malaria in young children but not in infants.
RTS,S was recently granted a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) via Article 58 of Regulation No 726/2004.
This allows the CHMP, in cooperation with the WHO, to give a scientific opinion on a medicinal product intended for markets outside the European Union. This assessment requires products to meet the same standards as products intended for use in the European Union.
Once the CHMP issued a positive opinion of RTS,S, the WHO began formulating a policy recommendation on use of the vaccine in national immunization programs. RTS,S must pass the WHO pre-qualification process and be approved by national regulatory authorities before it can be used in such programs. ![]()
a malaria-endemic region
Photo by Sarah Mattison
More testing is needed before the malaria vaccine candidate RTS,S/AS01 (Mosquirix) can be put into widespread use, according to a pair of World Health Organization (WHO) advisory committees.
The WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) are recommending that RTS,S be introduced in 3 to 5 pilot projects to determine the best way to deliver the vaccine to young children.
The issue, according to the committees, is that the vaccine must be administered in 4 doses and therefore requires repeat contact with the healthcare system.
The first 3 doses are given 1 month apart, and the last dose is given 18 months later. Without the fourth dose, children in a phase 3 study of RTS,S had no overall reduction in severe malaria.
So SAGE and MPAC want to be sure this vaccination schedule is feasible.
“The question about how the malaria vaccine may best be delivered still needs to be answered,” said Jon S. Abramson, chair of SAGE. “After detailed assessment of all the evidence, we recommended that this question is best addressed by having 3 to 5 large pilot implementation projects.”
This could delay widespread implementation of RTS,S for 3 to 5 years. Alternatively, if it is not possible to deliver all 4 doses of RTS,S consistently, Abramson said the vaccine may not be used at all.
RTS,S is being assessed as a complementary malaria control tool that could potentially be added to—but not replace—the core package of proven malaria preventive, diagnostic, and treatment measures.
The vaccine acts against Plasmodium falciparum, the most deadly malaria parasite globally and the most prevalent in Africa.
In a phase 3 trial, young children who received 4 doses of RTS,S had a 36% reduction in the number of clinical episodes of malaria at 4 years. Infants who received 4 doses of RTS,S had a 26% reduction in the number of clinical malaria episodes over 3 years.
Children had a significantly lower incidence of severe malaria only if they received all 4 doses of RTS,S. The vaccine did not confer the same benefit in infants, regardless of the doses given.
Results of a subsequent study suggested that genetic variation influences the vaccine’s ability to ward off malaria in young children but not in infants.
RTS,S was recently granted a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) via Article 58 of Regulation No 726/2004.
This allows the CHMP, in cooperation with the WHO, to give a scientific opinion on a medicinal product intended for markets outside the European Union. This assessment requires products to meet the same standards as products intended for use in the European Union.
Once the CHMP issued a positive opinion of RTS,S, the WHO began formulating a policy recommendation on use of the vaccine in national immunization programs. RTS,S must pass the WHO pre-qualification process and be approved by national regulatory authorities before it can be used in such programs. ![]()
a malaria-endemic region
Photo by Sarah Mattison
More testing is needed before the malaria vaccine candidate RTS,S/AS01 (Mosquirix) can be put into widespread use, according to a pair of World Health Organization (WHO) advisory committees.
The WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) are recommending that RTS,S be introduced in 3 to 5 pilot projects to determine the best way to deliver the vaccine to young children.
The issue, according to the committees, is that the vaccine must be administered in 4 doses and therefore requires repeat contact with the healthcare system.
The first 3 doses are given 1 month apart, and the last dose is given 18 months later. Without the fourth dose, children in a phase 3 study of RTS,S had no overall reduction in severe malaria.
So SAGE and MPAC want to be sure this vaccination schedule is feasible.
“The question about how the malaria vaccine may best be delivered still needs to be answered,” said Jon S. Abramson, chair of SAGE. “After detailed assessment of all the evidence, we recommended that this question is best addressed by having 3 to 5 large pilot implementation projects.”
This could delay widespread implementation of RTS,S for 3 to 5 years. Alternatively, if it is not possible to deliver all 4 doses of RTS,S consistently, Abramson said the vaccine may not be used at all.
RTS,S is being assessed as a complementary malaria control tool that could potentially be added to—but not replace—the core package of proven malaria preventive, diagnostic, and treatment measures.
The vaccine acts against Plasmodium falciparum, the most deadly malaria parasite globally and the most prevalent in Africa.
In a phase 3 trial, young children who received 4 doses of RTS,S had a 36% reduction in the number of clinical episodes of malaria at 4 years. Infants who received 4 doses of RTS,S had a 26% reduction in the number of clinical malaria episodes over 3 years.
Children had a significantly lower incidence of severe malaria only if they received all 4 doses of RTS,S. The vaccine did not confer the same benefit in infants, regardless of the doses given.
Results of a subsequent study suggested that genetic variation influences the vaccine’s ability to ward off malaria in young children but not in infants.
RTS,S was recently granted a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) via Article 58 of Regulation No 726/2004.
This allows the CHMP, in cooperation with the WHO, to give a scientific opinion on a medicinal product intended for markets outside the European Union. This assessment requires products to meet the same standards as products intended for use in the European Union.
Once the CHMP issued a positive opinion of RTS,S, the WHO began formulating a policy recommendation on use of the vaccine in national immunization programs. RTS,S must pass the WHO pre-qualification process and be approved by national regulatory authorities before it can be used in such programs. ![]()
New melphalan formulation denied approval
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has said that, at present, it cannot approve a propylene glycol-free melphalan formulation (Evomela) for use in patients with multiple myeloma (MM).
Spectrum Pharmaceuticals is seeking approval for Evomela as a high-dose conditioning treatment for MM patients undergoing hematopoietic stem cell transplant (HSCT) and for palliative treatment in MM patients for whom oral therapy is not appropriate.
The FDA issued a Complete Response Letter stating that the new drug application (NDA) for Evomela cannot be approved in its present form.
However, the FDA did not identify any clinical deficiency in the NDA package.
“We will work swiftly with the FDA to address the Complete Response Letter,” said Rajesh C. Shrotriya, MD, chairman and chief executive officer of Spectrum Pharmaceuticals. “We remain committed to bringing Evomela to the market for patients and plan to work closely with the FDA.”
About Evomela
Evomela is a Captisol-enabled, propylene glycol-free melphalan formulation. This formulation eliminates the need to use a propylene glycol-containing custom diluent, which is required with other intravenous melphalan formulations and has been reported to cause renal and cardiac side effects.
The use of Captisol technology to reformulate melphalan is reported to improve the drug’s stability, extending its use time to 5 hours. This is anticipated to simplify preparation and administration logistics and allow for slower infusion rates and longer administration durations for pre-transplant chemotherapy.
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs.
Spectrum Pharmaceuticals gained global development and commercialization rights to Evomela from Ligand Pharmaceuticals Incorporated in March 2013. Spectrum assumed responsibility for completing the pivotal phase 2 clinical trial and was responsible for filing the NDA. Spectrum filed the NDA in December 2014, and the FDA accepted the application the following March.
The FDA has granted Evomela orphan drug designation for use as a high-dose conditioning regimen for MM patients undergoing HSCT.
Phase 2 study
Researchers have evaluated Evomela in a phase 2, multicenter trial. Initial results from this trial (phase 2a) were published in Bone Marrow Transplantation in June 2014. Phase 2b results were published in Biology of Blood and Marrow Transplantation last month.
The latest publication includes data on 61 patients. Fifty-six had newly diagnosed MM, and 5 had relapsed MM following prior HSCT. The patients received Evomela at 200 mg/m2 given as 2 doses on Day -3 and Day -2 prior to HSCT (Day 0).
Efficacy was assessed by clinical response at Day +100. According to investigator assessment, the overall response rate was 95%, and the complete response (CR) rate was 31%.
According to independent pathology review, the overall response rate was 100%, and the CR rate was 21%. The lower rate of confirmed CRs in the independent review was due to missing data.
All 5 patients who had previously relapsed from a prior HSCT responded to Evomela.
All patients in the study achieved myeloablation with a median of 5 days post-HSCT. All patients had successful neutrophil and platelet engraftment at a median of 12 days and 13 days post-HSCT, respectively.
Treatment-related mortality was 0%, and non-hematologic adverse events were mostly grade 1 and 2 in severity. The incidence of grade 3 mucositis and grade 3 stomatitis were 10% and 5%, respectively, with no grade 4 mucositis or stomatitis reported.
Twenty percent of patients experienced treatment-emergent serious adverse events, most of which were grade 3 and consisted of events commonly reported in patients undergoing myeloablative chemotherapy. No new safety signals were identified. ![]()
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has said that, at present, it cannot approve a propylene glycol-free melphalan formulation (Evomela) for use in patients with multiple myeloma (MM).
Spectrum Pharmaceuticals is seeking approval for Evomela as a high-dose conditioning treatment for MM patients undergoing hematopoietic stem cell transplant (HSCT) and for palliative treatment in MM patients for whom oral therapy is not appropriate.
The FDA issued a Complete Response Letter stating that the new drug application (NDA) for Evomela cannot be approved in its present form.
However, the FDA did not identify any clinical deficiency in the NDA package.
“We will work swiftly with the FDA to address the Complete Response Letter,” said Rajesh C. Shrotriya, MD, chairman and chief executive officer of Spectrum Pharmaceuticals. “We remain committed to bringing Evomela to the market for patients and plan to work closely with the FDA.”
About Evomela
Evomela is a Captisol-enabled, propylene glycol-free melphalan formulation. This formulation eliminates the need to use a propylene glycol-containing custom diluent, which is required with other intravenous melphalan formulations and has been reported to cause renal and cardiac side effects.
The use of Captisol technology to reformulate melphalan is reported to improve the drug’s stability, extending its use time to 5 hours. This is anticipated to simplify preparation and administration logistics and allow for slower infusion rates and longer administration durations for pre-transplant chemotherapy.
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs.
Spectrum Pharmaceuticals gained global development and commercialization rights to Evomela from Ligand Pharmaceuticals Incorporated in March 2013. Spectrum assumed responsibility for completing the pivotal phase 2 clinical trial and was responsible for filing the NDA. Spectrum filed the NDA in December 2014, and the FDA accepted the application the following March.
The FDA has granted Evomela orphan drug designation for use as a high-dose conditioning regimen for MM patients undergoing HSCT.
Phase 2 study
Researchers have evaluated Evomela in a phase 2, multicenter trial. Initial results from this trial (phase 2a) were published in Bone Marrow Transplantation in June 2014. Phase 2b results were published in Biology of Blood and Marrow Transplantation last month.
The latest publication includes data on 61 patients. Fifty-six had newly diagnosed MM, and 5 had relapsed MM following prior HSCT. The patients received Evomela at 200 mg/m2 given as 2 doses on Day -3 and Day -2 prior to HSCT (Day 0).
Efficacy was assessed by clinical response at Day +100. According to investigator assessment, the overall response rate was 95%, and the complete response (CR) rate was 31%.
According to independent pathology review, the overall response rate was 100%, and the CR rate was 21%. The lower rate of confirmed CRs in the independent review was due to missing data.
All 5 patients who had previously relapsed from a prior HSCT responded to Evomela.
All patients in the study achieved myeloablation with a median of 5 days post-HSCT. All patients had successful neutrophil and platelet engraftment at a median of 12 days and 13 days post-HSCT, respectively.
Treatment-related mortality was 0%, and non-hematologic adverse events were mostly grade 1 and 2 in severity. The incidence of grade 3 mucositis and grade 3 stomatitis were 10% and 5%, respectively, with no grade 4 mucositis or stomatitis reported.
Twenty percent of patients experienced treatment-emergent serious adverse events, most of which were grade 3 and consisted of events commonly reported in patients undergoing myeloablative chemotherapy. No new safety signals were identified. ![]()
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has said that, at present, it cannot approve a propylene glycol-free melphalan formulation (Evomela) for use in patients with multiple myeloma (MM).
Spectrum Pharmaceuticals is seeking approval for Evomela as a high-dose conditioning treatment for MM patients undergoing hematopoietic stem cell transplant (HSCT) and for palliative treatment in MM patients for whom oral therapy is not appropriate.
The FDA issued a Complete Response Letter stating that the new drug application (NDA) for Evomela cannot be approved in its present form.
However, the FDA did not identify any clinical deficiency in the NDA package.
“We will work swiftly with the FDA to address the Complete Response Letter,” said Rajesh C. Shrotriya, MD, chairman and chief executive officer of Spectrum Pharmaceuticals. “We remain committed to bringing Evomela to the market for patients and plan to work closely with the FDA.”
About Evomela
Evomela is a Captisol-enabled, propylene glycol-free melphalan formulation. This formulation eliminates the need to use a propylene glycol-containing custom diluent, which is required with other intravenous melphalan formulations and has been reported to cause renal and cardiac side effects.
The use of Captisol technology to reformulate melphalan is reported to improve the drug’s stability, extending its use time to 5 hours. This is anticipated to simplify preparation and administration logistics and allow for slower infusion rates and longer administration durations for pre-transplant chemotherapy.
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs.
Spectrum Pharmaceuticals gained global development and commercialization rights to Evomela from Ligand Pharmaceuticals Incorporated in March 2013. Spectrum assumed responsibility for completing the pivotal phase 2 clinical trial and was responsible for filing the NDA. Spectrum filed the NDA in December 2014, and the FDA accepted the application the following March.
The FDA has granted Evomela orphan drug designation for use as a high-dose conditioning regimen for MM patients undergoing HSCT.
Phase 2 study
Researchers have evaluated Evomela in a phase 2, multicenter trial. Initial results from this trial (phase 2a) were published in Bone Marrow Transplantation in June 2014. Phase 2b results were published in Biology of Blood and Marrow Transplantation last month.
The latest publication includes data on 61 patients. Fifty-six had newly diagnosed MM, and 5 had relapsed MM following prior HSCT. The patients received Evomela at 200 mg/m2 given as 2 doses on Day -3 and Day -2 prior to HSCT (Day 0).
Efficacy was assessed by clinical response at Day +100. According to investigator assessment, the overall response rate was 95%, and the complete response (CR) rate was 31%.
According to independent pathology review, the overall response rate was 100%, and the CR rate was 21%. The lower rate of confirmed CRs in the independent review was due to missing data.
All 5 patients who had previously relapsed from a prior HSCT responded to Evomela.
All patients in the study achieved myeloablation with a median of 5 days post-HSCT. All patients had successful neutrophil and platelet engraftment at a median of 12 days and 13 days post-HSCT, respectively.
Treatment-related mortality was 0%, and non-hematologic adverse events were mostly grade 1 and 2 in severity. The incidence of grade 3 mucositis and grade 3 stomatitis were 10% and 5%, respectively, with no grade 4 mucositis or stomatitis reported.
Twenty percent of patients experienced treatment-emergent serious adverse events, most of which were grade 3 and consisted of events commonly reported in patients undergoing myeloablative chemotherapy. No new safety signals were identified.
Radiation often underused in follicular lymphoma

woman for radiotherapy
Photo by Rhoda Baer
SAN ANTONIO—A new study indicates that patients with early stage follicular lymphoma (FL) are increasingly receiving no treatment or single-agent chemotherapy, despite evidence suggesting that radiation therapy can produce better outcomes.
Guidelines from the National Comprehensive Cancer Network and the European Society for Medical Oncology both list radiation therapy as the preferred treatment for low-grade FL.
However, investigators found that, in recent years, radiation has been replaced by alternative strategies.
“Our study highlights the increasing omission of radiation therapy in [FL] and its associated negative effect on overall survival at a national level,” said John Austin Vargo, MD, of the University of Pittsburg Cancer Institute in Pennsylvania.
“This increasing bias towards the omission of radiation therapy is despite proven efficacy and increasing adoption of lower radiation therapy doses and more modern radiation therapy techniques which decrease risk of side effects.”
Dr Vargo presented these findings at the 57th Annual Meeting of the American Society for Radiation Oncology (presentation #183).
He and his colleagues analyzed patterns of care and survival outcomes for 35,961 patients diagnosed with early stage FL as listed in the National Cancer Data Base. A majority of patients were older than 60 (61%), and most had stage I disease (63%).
The use of radiation therapy in this group of patients decreased from 37% in 1999 to 24% in 2012 (P<0.0001).
The use of observation increased from 34% in 1998 to 44% in 2012 (P<0.0001). And the use of single-agent chemotherapy increased from 5.4% in 1999 to 11.7% in 2006 (P=0.01).
The 5-year overall survival rate was 86% in patients who received radiation and 74% in those who did not (P<0.0001). Ten-year overall survival rates were 68% and 54%, respectively (P<0.0001).
In multivariate analysis, radiation therapy remained significantly associated with improved overall survival (P<0.0001).
woman for radiotherapy
Photo by Rhoda Baer
SAN ANTONIO—A new study indicates that patients with early stage follicular lymphoma (FL) are increasingly receiving no treatment or single-agent chemotherapy, despite evidence suggesting that radiation therapy can produce better outcomes.
Guidelines from the National Comprehensive Cancer Network and the European Society for Medical Oncology both list radiation therapy as the preferred treatment for low-grade FL.
However, investigators found that, in recent years, radiation has been replaced by alternative strategies.
“Our study highlights the increasing omission of radiation therapy in [FL] and its associated negative effect on overall survival at a national level,” said John Austin Vargo, MD, of the University of Pittsburg Cancer Institute in Pennsylvania.
“This increasing bias towards the omission of radiation therapy is despite proven efficacy and increasing adoption of lower radiation therapy doses and more modern radiation therapy techniques which decrease risk of side effects.”
Dr Vargo presented these findings at the 57th Annual Meeting of the American Society for Radiation Oncology (presentation #183).
He and his colleagues analyzed patterns of care and survival outcomes for 35,961 patients diagnosed with early stage FL as listed in the National Cancer Data Base. A majority of patients were older than 60 (61%), and most had stage I disease (63%).
The use of radiation therapy in this group of patients decreased from 37% in 1999 to 24% in 2012 (P<0.0001).
The use of observation increased from 34% in 1998 to 44% in 2012 (P<0.0001). And the use of single-agent chemotherapy increased from 5.4% in 1999 to 11.7% in 2006 (P=0.01).
The 5-year overall survival rate was 86% in patients who received radiation and 74% in those who did not (P<0.0001). Ten-year overall survival rates were 68% and 54%, respectively (P<0.0001).
In multivariate analysis, radiation therapy remained significantly associated with improved overall survival (P<0.0001).
woman for radiotherapy
Photo by Rhoda Baer
SAN ANTONIO—A new study indicates that patients with early stage follicular lymphoma (FL) are increasingly receiving no treatment or single-agent chemotherapy, despite evidence suggesting that radiation therapy can produce better outcomes.
Guidelines from the National Comprehensive Cancer Network and the European Society for Medical Oncology both list radiation therapy as the preferred treatment for low-grade FL.
However, investigators found that, in recent years, radiation has been replaced by alternative strategies.
“Our study highlights the increasing omission of radiation therapy in [FL] and its associated negative effect on overall survival at a national level,” said John Austin Vargo, MD, of the University of Pittsburg Cancer Institute in Pennsylvania.
“This increasing bias towards the omission of radiation therapy is despite proven efficacy and increasing adoption of lower radiation therapy doses and more modern radiation therapy techniques which decrease risk of side effects.”
Dr Vargo presented these findings at the 57th Annual Meeting of the American Society for Radiation Oncology (presentation #183).
He and his colleagues analyzed patterns of care and survival outcomes for 35,961 patients diagnosed with early stage FL as listed in the National Cancer Data Base. A majority of patients were older than 60 (61%), and most had stage I disease (63%).
The use of radiation therapy in this group of patients decreased from 37% in 1999 to 24% in 2012 (P<0.0001).
The use of observation increased from 34% in 1998 to 44% in 2012 (P<0.0001). And the use of single-agent chemotherapy increased from 5.4% in 1999 to 11.7% in 2006 (P=0.01).
The 5-year overall survival rate was 86% in patients who received radiation and 74% in those who did not (P<0.0001). Ten-year overall survival rates were 68% and 54%, respectively (P<0.0001).
In multivariate analysis, radiation therapy remained significantly associated with improved overall survival (P<0.0001).
Novel compound could treat leukemia
A small-molecule compound that has previously shown activity against Ewing sarcoma and prostate cancer may fight leukemia as well, according to preclinical research published in Oncotarget.
The compound, YK-4-279, inhibits the oncogenic activity of the fusion protein EWS-FLI1.
“EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma,” said study author Aykut Üren, MD, of Georgetown University Medical Center in Washington, DC.
“It also appears to drive cancer cell growth in some prostate cancers.”
ETS family fusion proteins are found in patients with acute myeloid leukemia and acute lymphoblastic leukemia as well.
So Dr Üren and his colleagues decided to create a mouse model of EWS-FLI1-induced leukemia and assess the activity of YK-4-279 in this model.
Mice with EWS-FLI1-induced leukemia presented with severe hepatomegaly, splenomegaly, and anemia, followed by rapid death.
The investigators treated these mice with injections of YK-4-279 five days a week for 2 weeks or vehicle intraperitoneal injections on the same schedule.
The team said treatment with YK-4-279 significantly reduced white blood cell counts, nucleated erythroblasts in the peripheral blood, splenomegaly, and hepatomegaly.
They noted that mice experienced reductions in the weight of their spleens and livers without experiencing reductions in total body weight.
In addition, mice that received YK-4-279 had significantly better overall survival than control mice. The median survival times were 60.5 days and 21 days, respectively.
The investigators also noted that treated mice did not exhibit overt toxicity in the liver, spleen, or bone marrow.
“The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can’t be answered until we conduct clinical trials,” Dr Üren said.
Nevertheless, he and his colleagues believe these results support the continued preclinical development of YK-4-279 for Ewing sarcoma, prostate cancers, and leukemias with highly homologous translocation products or with a clear ETS-driven gene signature.
A small-molecule compound that has previously shown activity against Ewing sarcoma and prostate cancer may fight leukemia as well, according to preclinical research published in Oncotarget.
The compound, YK-4-279, inhibits the oncogenic activity of the fusion protein EWS-FLI1.
“EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma,” said study author Aykut Üren, MD, of Georgetown University Medical Center in Washington, DC.
“It also appears to drive cancer cell growth in some prostate cancers.”
ETS family fusion proteins are found in patients with acute myeloid leukemia and acute lymphoblastic leukemia as well.
So Dr Üren and his colleagues decided to create a mouse model of EWS-FLI1-induced leukemia and assess the activity of YK-4-279 in this model.
Mice with EWS-FLI1-induced leukemia presented with severe hepatomegaly, splenomegaly, and anemia, followed by rapid death.
The investigators treated these mice with injections of YK-4-279 five days a week for 2 weeks or vehicle intraperitoneal injections on the same schedule.
The team said treatment with YK-4-279 significantly reduced white blood cell counts, nucleated erythroblasts in the peripheral blood, splenomegaly, and hepatomegaly.
They noted that mice experienced reductions in the weight of their spleens and livers without experiencing reductions in total body weight.
In addition, mice that received YK-4-279 had significantly better overall survival than control mice. The median survival times were 60.5 days and 21 days, respectively.
The investigators also noted that treated mice did not exhibit overt toxicity in the liver, spleen, or bone marrow.
“The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can’t be answered until we conduct clinical trials,” Dr Üren said.
Nevertheless, he and his colleagues believe these results support the continued preclinical development of YK-4-279 for Ewing sarcoma, prostate cancers, and leukemias with highly homologous translocation products or with a clear ETS-driven gene signature.
A small-molecule compound that has previously shown activity against Ewing sarcoma and prostate cancer may fight leukemia as well, according to preclinical research published in Oncotarget.
The compound, YK-4-279, inhibits the oncogenic activity of the fusion protein EWS-FLI1.
“EWS-FLI1 is already known to drive a rare but deadly bone cancer called Ewing sarcoma,” said study author Aykut Üren, MD, of Georgetown University Medical Center in Washington, DC.
“It also appears to drive cancer cell growth in some prostate cancers.”
ETS family fusion proteins are found in patients with acute myeloid leukemia and acute lymphoblastic leukemia as well.
So Dr Üren and his colleagues decided to create a mouse model of EWS-FLI1-induced leukemia and assess the activity of YK-4-279 in this model.
Mice with EWS-FLI1-induced leukemia presented with severe hepatomegaly, splenomegaly, and anemia, followed by rapid death.
The investigators treated these mice with injections of YK-4-279 five days a week for 2 weeks or vehicle intraperitoneal injections on the same schedule.
The team said treatment with YK-4-279 significantly reduced white blood cell counts, nucleated erythroblasts in the peripheral blood, splenomegaly, and hepatomegaly.
They noted that mice experienced reductions in the weight of their spleens and livers without experiencing reductions in total body weight.
In addition, mice that received YK-4-279 had significantly better overall survival than control mice. The median survival times were 60.5 days and 21 days, respectively.
The investigators also noted that treated mice did not exhibit overt toxicity in the liver, spleen, or bone marrow.
“The fact that treated mice did not get sick from the YK-4-279 gives us an early indication that it might be safe to use in humans, but that is a question that can’t be answered until we conduct clinical trials,” Dr Üren said.
Nevertheless, he and his colleagues believe these results support the continued preclinical development of YK-4-279 for Ewing sarcoma, prostate cancers, and leukemias with highly homologous translocation products or with a clear ETS-driven gene signature.
Team targets gene to increase RBC production

Researchers say they can increase the production of red blood cells (RBCs) in the lab by targeting a single gene—SH2B3.
The team used RNA interference (RNAi) to turn down SH2B3 in human hematopoietic stem and progenitor cells (HSPCs) and increased the yield of RBCs about 3- to 7-fold.
They also used CRISPR/Cas9 genome editing to shut off SH2B3 in human embryonic stem cell (hESC) lines, increasing the yield of RBCs about 3-fold.
The researchers noted that the method involving hESCs would be easier to use for large-scale production of RBCs.
Vijay Sankaran, MD, PhD, of the Broad Institute in Cambridge, Massachusetts, and his colleagues conducted this research and reported the results in Cell Stem Cell.
The researchers homed in on their target gene, SH2B3, after genome sequencing data revealed naturally occurring variations in SH2B3. These variations reduce the gene’s activity and increase RBC production.
“There’s a variation in SH2B3 found in about 40% of people that leads to modestly higher red blood cell counts,” Dr Sankaran said. “But if you look at people with really high red blood cell levels, they often have rare SH2B3 mutations. That said to us that here is a target where you can partially or completely eliminate its function as a way of increasing red blood cells robustly.”
So Dr Sankaran and his colleagues set out to see if they could use SH2B3 as a target to increase the yield of lab-based RBC production processes (as opposed to tweaking cells in culture by adding cytokines and other factors).
To do this, they first used RNAi to turn down SH2B3 in donated adult HSPCs and HSPCs from umbilical cord blood.
The team’s data confirmed that shutting off SH2B3 with RNAi skews an HSPC’s profile of cell production to favor RBCs. Adult HSPCs treated with RNAi produced 3- to 5-fold more RBCs than controls. And RNAi-treated HSPCs from cord blood produced 5- to 7-fold more RBCs than controls.
Using multiple tests, the researchers found the RBCs produced by RNAi were essentially indistinguishable from control cells.
Dr Sankaran and his colleagues recognized that this approach would be very difficult to scale up to a level that could impact the clinical need for RBCs. So, in a separate set of experiments, they used CRISPR to permanently shut off SH2B3 in hESC lines, which can be readily renewed in a lab.
The team then treated the edited cells with a cocktail of factors known to encourage blood cell production. Under these conditions, the edited hESCs produced 3 times more RBCs than controls. Again, the team could find no significant differences between RBCs from the edited stem cells and controls.
Dr Sankaran believes that SH2B3 enforces some kind of upper limit on how much RBC precursors respond to calls for more RBC production.
“This is a nice approach because it removes the brakes that normally keep cells restrained and limit how much red blood cell precursors respond to different laboratory conditions,” he said.
Dr Sankaran also believes that, with further development, the combination of CRISPR and hESCs could increase the yields and reduce the costs of producing RBCs in the lab to the level where commercial-scale manufacture could be feasible.
“This is allowing us to get close to the cost of normal donor-derived blood units,” he said. “If we can get the costs down to about $2000 per unit, that’s a reasonable cost.”
Previous research has shown it is possible to produce transfusion-grade RBCs, but the costs ranged from $8000 to $15,000 per unit of blood.
Researchers say they can increase the production of red blood cells (RBCs) in the lab by targeting a single gene—SH2B3.
The team used RNA interference (RNAi) to turn down SH2B3 in human hematopoietic stem and progenitor cells (HSPCs) and increased the yield of RBCs about 3- to 7-fold.
They also used CRISPR/Cas9 genome editing to shut off SH2B3 in human embryonic stem cell (hESC) lines, increasing the yield of RBCs about 3-fold.
The researchers noted that the method involving hESCs would be easier to use for large-scale production of RBCs.
Vijay Sankaran, MD, PhD, of the Broad Institute in Cambridge, Massachusetts, and his colleagues conducted this research and reported the results in Cell Stem Cell.
The researchers homed in on their target gene, SH2B3, after genome sequencing data revealed naturally occurring variations in SH2B3. These variations reduce the gene’s activity and increase RBC production.
“There’s a variation in SH2B3 found in about 40% of people that leads to modestly higher red blood cell counts,” Dr Sankaran said. “But if you look at people with really high red blood cell levels, they often have rare SH2B3 mutations. That said to us that here is a target where you can partially or completely eliminate its function as a way of increasing red blood cells robustly.”
So Dr Sankaran and his colleagues set out to see if they could use SH2B3 as a target to increase the yield of lab-based RBC production processes (as opposed to tweaking cells in culture by adding cytokines and other factors).
To do this, they first used RNAi to turn down SH2B3 in donated adult HSPCs and HSPCs from umbilical cord blood.
The team’s data confirmed that shutting off SH2B3 with RNAi skews an HSPC’s profile of cell production to favor RBCs. Adult HSPCs treated with RNAi produced 3- to 5-fold more RBCs than controls. And RNAi-treated HSPCs from cord blood produced 5- to 7-fold more RBCs than controls.
Using multiple tests, the researchers found the RBCs produced by RNAi were essentially indistinguishable from control cells.
Dr Sankaran and his colleagues recognized that this approach would be very difficult to scale up to a level that could impact the clinical need for RBCs. So, in a separate set of experiments, they used CRISPR to permanently shut off SH2B3 in hESC lines, which can be readily renewed in a lab.
The team then treated the edited cells with a cocktail of factors known to encourage blood cell production. Under these conditions, the edited hESCs produced 3 times more RBCs than controls. Again, the team could find no significant differences between RBCs from the edited stem cells and controls.
Dr Sankaran believes that SH2B3 enforces some kind of upper limit on how much RBC precursors respond to calls for more RBC production.
“This is a nice approach because it removes the brakes that normally keep cells restrained and limit how much red blood cell precursors respond to different laboratory conditions,” he said.
Dr Sankaran also believes that, with further development, the combination of CRISPR and hESCs could increase the yields and reduce the costs of producing RBCs in the lab to the level where commercial-scale manufacture could be feasible.
“This is allowing us to get close to the cost of normal donor-derived blood units,” he said. “If we can get the costs down to about $2000 per unit, that’s a reasonable cost.”
Previous research has shown it is possible to produce transfusion-grade RBCs, but the costs ranged from $8000 to $15,000 per unit of blood.
Researchers say they can increase the production of red blood cells (RBCs) in the lab by targeting a single gene—SH2B3.
The team used RNA interference (RNAi) to turn down SH2B3 in human hematopoietic stem and progenitor cells (HSPCs) and increased the yield of RBCs about 3- to 7-fold.
They also used CRISPR/Cas9 genome editing to shut off SH2B3 in human embryonic stem cell (hESC) lines, increasing the yield of RBCs about 3-fold.
The researchers noted that the method involving hESCs would be easier to use for large-scale production of RBCs.
Vijay Sankaran, MD, PhD, of the Broad Institute in Cambridge, Massachusetts, and his colleagues conducted this research and reported the results in Cell Stem Cell.
The researchers homed in on their target gene, SH2B3, after genome sequencing data revealed naturally occurring variations in SH2B3. These variations reduce the gene’s activity and increase RBC production.
“There’s a variation in SH2B3 found in about 40% of people that leads to modestly higher red blood cell counts,” Dr Sankaran said. “But if you look at people with really high red blood cell levels, they often have rare SH2B3 mutations. That said to us that here is a target where you can partially or completely eliminate its function as a way of increasing red blood cells robustly.”
So Dr Sankaran and his colleagues set out to see if they could use SH2B3 as a target to increase the yield of lab-based RBC production processes (as opposed to tweaking cells in culture by adding cytokines and other factors).
To do this, they first used RNAi to turn down SH2B3 in donated adult HSPCs and HSPCs from umbilical cord blood.
The team’s data confirmed that shutting off SH2B3 with RNAi skews an HSPC’s profile of cell production to favor RBCs. Adult HSPCs treated with RNAi produced 3- to 5-fold more RBCs than controls. And RNAi-treated HSPCs from cord blood produced 5- to 7-fold more RBCs than controls.
Using multiple tests, the researchers found the RBCs produced by RNAi were essentially indistinguishable from control cells.
Dr Sankaran and his colleagues recognized that this approach would be very difficult to scale up to a level that could impact the clinical need for RBCs. So, in a separate set of experiments, they used CRISPR to permanently shut off SH2B3 in hESC lines, which can be readily renewed in a lab.
The team then treated the edited cells with a cocktail of factors known to encourage blood cell production. Under these conditions, the edited hESCs produced 3 times more RBCs than controls. Again, the team could find no significant differences between RBCs from the edited stem cells and controls.
Dr Sankaran believes that SH2B3 enforces some kind of upper limit on how much RBC precursors respond to calls for more RBC production.
“This is a nice approach because it removes the brakes that normally keep cells restrained and limit how much red blood cell precursors respond to different laboratory conditions,” he said.
Dr Sankaran also believes that, with further development, the combination of CRISPR and hESCs could increase the yields and reduce the costs of producing RBCs in the lab to the level where commercial-scale manufacture could be feasible.
“This is allowing us to get close to the cost of normal donor-derived blood units,” he said. “If we can get the costs down to about $2000 per unit, that’s a reasonable cost.”
Previous research has shown it is possible to produce transfusion-grade RBCs, but the costs ranged from $8000 to $15,000 per unit of blood.