User login
Gene therapy exceeds expectations in β-thalassemia

The gene therapy LentiGlobin can reduce or eliminate transfusion dependence in patients with β-thalassemia, according to a pair of phase 1/2 studies.
Fifteen of the 22 patients in these trials were able to discontinue red blood cell (RBC) transfusions after receiving LentiGlobin.
In the 9 patients with severe transfusion-dependent β-thalassemia (TDT), LentiGlobin reduced the transfusion volume by 73%.
There were 5 adverse events (AEs) considered possibly or probably related to LentiGlobin, all them grade 1.
“These study results exceeded our expectations, with clinical benefit for nearly all patients . . . ,” said Alexis Thompson, MD, of Ann & Robert H. Lurie Children’s Hospital of Chicago in Illinois.
“Since we saw such positive results, we are now enrolling patients as young as 5 years old on a phase 3 trial of gene therapy for transfusion-dependent thalassemia.”
Dr Thompson and her colleagues reported results from the phase 1/2 trials—known as HGB-204 and HGB-205—in NEJM. The studies were sponsored by Bluebird Bio, the company developing LentiGlobin.
Patients in HGB-204
HGB-204 (also known as Northstar) is a multicenter study that was recently completed. It included 18 patients with TDT. They had a median age of 20 (range, 12 to 35) at baseline, and 72% were female. Seventy-eight percent were Asian, and 22% were white.
Eight patients had a β0/β0 genotype, 6 had a βE/β0 genotype, and 4 had other genotypes.
The patients’ median monthly transfusion volume for 2 years before study enrollment was 13.6 ml/kg (range, 10.4 to 21.8). The median age at which patients started regular transfusions was 3.5 years (range, 0 to 26.0). Six patients had undergone splenectomy.
Patients in HGB-205
HGB-205 is an ongoing study being conducted at a single site in France. It was designed to evaluate LentiGlobin in patients with TDT or severe sickle cell disease.
The NEJM paper includes 4 patients with TDT from this study. They had a median age of 18 (range, 16 to 19) at baseline, and half were female. Half were Asian, and the other half were white.
Three patients had a βE/β0 genotype. The remaining patient was homozygous for the IVS1-110 mutation and had a severe clinical presentation similar to that seen in β0/β0 genotypes.
The patients’ median monthly transfusion volume for 2 years before study enrollment was 15.2 ml/kg (range, 11.6 to 15.7). The median age at which patients started regular transfusions was 1.8 years (range, 0 to 14.0). Three patients had undergone splenectomy.
Treatment
For both studies, the researchers harvested hematopoietic stem and progenitor cells (mobilized with filgrastim and plerixafor) from the patients.
CD34+ cells were transduced ex vivo with LentiGlobin BB305 vector, which encodes adult hemoglobin (HbA) with a T87Q amino acid substitution (HbAT87Q).
The patients underwent myeloablative conditioning with busulfan, and the final LentiGlobin product was infused into patients after a 72-hour washout period.
In HGB-205 only, patients received enhanced RBC transfusions for at least 3 months before stem cell mobilization and harvest to maintain a hemoglobin level of more than 11.0 g/dL.
Safety
In HGB-204, there were 5 grade 1 AEs considered possibly or probably related to LentiGlobin. These included abdominal pain (n=2), dyspnea (n=1), hot flush (n=1), and non-cardiac chest pain (n=1).
There were 9 serious AEs, including 2 episodes of grade 3 veno-occlusive liver disease that were attributed to busulfan.
The remaining serious AEs were Klebsiella infection, cardiac ventricular thrombosis, cellulitis, hyperglycemia, and gastroenteritis (all grade 3), as well as device-related thrombosis and infectious diarrhea (both grade 2).
In HGB-205, there were no AEs considered possibly or probably related to LentiGlobin.
The 3 serious AEs were tooth infection and major depression (both grade 3), as well as pneumonia (grade 2).
Efficacy
The median time to neutrophil engraftment was 18.5 days (range, 14.0 to 30.0) in HGB-204 and 16.5 days (range, 14.0 to 29.0) in HGB-205.
The median time to platelet engraftment was 39.5 days (range, 19.0 to 191.0) in HGB-204 and 23.0 days (range, 20.0 to 26.0) in HGB-205.
In both studies, the median follow-up was 26 months (range, 15 to 42) after LentiGlobin infusion.
At last follow-up, all but 1 of the 13 patients with a non-β0/β0 genotype had stopped receiving RBC transfusions.
At the last study visit (12 to 36 months post-treatment), the median HbAT87Q level in these patients was 6.0 g/dL (range, 3.4 to 10.0), and the median total hemoglobin was 11.2 g/dL (range, 8.2 to 13.7).
In the 8 patients with a β0/β0 genotype and the 1 patient with 2 copies of the IVS1-110 mutation, the median annualized transfusion volume decreased by 73% after LentiGlobin infusion.
Two patients with a β0/β0 genotype were able to stop receiving RBC transfusions, as was the patient with 2 copies of the IVS1-110 mutation.
At their most recent study visit (12 months to 30 months), these 3 patients had median HbAT87Q levels of 8.2 g/dL, 6.8 g/dL, and 6.6 g/dL, respectively. Their median total hemoglobin levels were 9.0 g/dL, 10.2 g/dL, and 8.3 g/dL, respectively.
For the 6 patients with a β0/β0 genotype who continued to receive RBC transfusions, the median HbAT87Q level was 4.2 g/dL (range, 0.3 to 8.7) at the last study visit.
“There is room for improvement, as we’d like to see the elimination of dependency on transfusion even for patients with the most severe form of the disease,” said study author Philippe Leboulch, MD, of Brigham and Women’s Hospital in Boston, Massachusetts.
“But there is also hope with protocol modifications we have introduced in our phase 3 trials.”
The gene therapy LentiGlobin can reduce or eliminate transfusion dependence in patients with β-thalassemia, according to a pair of phase 1/2 studies.
Fifteen of the 22 patients in these trials were able to discontinue red blood cell (RBC) transfusions after receiving LentiGlobin.
In the 9 patients with severe transfusion-dependent β-thalassemia (TDT), LentiGlobin reduced the transfusion volume by 73%.
There were 5 adverse events (AEs) considered possibly or probably related to LentiGlobin, all them grade 1.
“These study results exceeded our expectations, with clinical benefit for nearly all patients . . . ,” said Alexis Thompson, MD, of Ann & Robert H. Lurie Children’s Hospital of Chicago in Illinois.
“Since we saw such positive results, we are now enrolling patients as young as 5 years old on a phase 3 trial of gene therapy for transfusion-dependent thalassemia.”
Dr Thompson and her colleagues reported results from the phase 1/2 trials—known as HGB-204 and HGB-205—in NEJM. The studies were sponsored by Bluebird Bio, the company developing LentiGlobin.
Patients in HGB-204
HGB-204 (also known as Northstar) is a multicenter study that was recently completed. It included 18 patients with TDT. They had a median age of 20 (range, 12 to 35) at baseline, and 72% were female. Seventy-eight percent were Asian, and 22% were white.
Eight patients had a β0/β0 genotype, 6 had a βE/β0 genotype, and 4 had other genotypes.
The patients’ median monthly transfusion volume for 2 years before study enrollment was 13.6 ml/kg (range, 10.4 to 21.8). The median age at which patients started regular transfusions was 3.5 years (range, 0 to 26.0). Six patients had undergone splenectomy.
Patients in HGB-205
HGB-205 is an ongoing study being conducted at a single site in France. It was designed to evaluate LentiGlobin in patients with TDT or severe sickle cell disease.
The NEJM paper includes 4 patients with TDT from this study. They had a median age of 18 (range, 16 to 19) at baseline, and half were female. Half were Asian, and the other half were white.
Three patients had a βE/β0 genotype. The remaining patient was homozygous for the IVS1-110 mutation and had a severe clinical presentation similar to that seen in β0/β0 genotypes.
The patients’ median monthly transfusion volume for 2 years before study enrollment was 15.2 ml/kg (range, 11.6 to 15.7). The median age at which patients started regular transfusions was 1.8 years (range, 0 to 14.0). Three patients had undergone splenectomy.
Treatment
For both studies, the researchers harvested hematopoietic stem and progenitor cells (mobilized with filgrastim and plerixafor) from the patients.
CD34+ cells were transduced ex vivo with LentiGlobin BB305 vector, which encodes adult hemoglobin (HbA) with a T87Q amino acid substitution (HbAT87Q).
The patients underwent myeloablative conditioning with busulfan, and the final LentiGlobin product was infused into patients after a 72-hour washout period.
In HGB-205 only, patients received enhanced RBC transfusions for at least 3 months before stem cell mobilization and harvest to maintain a hemoglobin level of more than 11.0 g/dL.
Safety
In HGB-204, there were 5 grade 1 AEs considered possibly or probably related to LentiGlobin. These included abdominal pain (n=2), dyspnea (n=1), hot flush (n=1), and non-cardiac chest pain (n=1).
There were 9 serious AEs, including 2 episodes of grade 3 veno-occlusive liver disease that were attributed to busulfan.
The remaining serious AEs were Klebsiella infection, cardiac ventricular thrombosis, cellulitis, hyperglycemia, and gastroenteritis (all grade 3), as well as device-related thrombosis and infectious diarrhea (both grade 2).
In HGB-205, there were no AEs considered possibly or probably related to LentiGlobin.
The 3 serious AEs were tooth infection and major depression (both grade 3), as well as pneumonia (grade 2).
Efficacy
The median time to neutrophil engraftment was 18.5 days (range, 14.0 to 30.0) in HGB-204 and 16.5 days (range, 14.0 to 29.0) in HGB-205.
The median time to platelet engraftment was 39.5 days (range, 19.0 to 191.0) in HGB-204 and 23.0 days (range, 20.0 to 26.0) in HGB-205.
In both studies, the median follow-up was 26 months (range, 15 to 42) after LentiGlobin infusion.
At last follow-up, all but 1 of the 13 patients with a non-β0/β0 genotype had stopped receiving RBC transfusions.
At the last study visit (12 to 36 months post-treatment), the median HbAT87Q level in these patients was 6.0 g/dL (range, 3.4 to 10.0), and the median total hemoglobin was 11.2 g/dL (range, 8.2 to 13.7).
In the 8 patients with a β0/β0 genotype and the 1 patient with 2 copies of the IVS1-110 mutation, the median annualized transfusion volume decreased by 73% after LentiGlobin infusion.
Two patients with a β0/β0 genotype were able to stop receiving RBC transfusions, as was the patient with 2 copies of the IVS1-110 mutation.
At their most recent study visit (12 months to 30 months), these 3 patients had median HbAT87Q levels of 8.2 g/dL, 6.8 g/dL, and 6.6 g/dL, respectively. Their median total hemoglobin levels were 9.0 g/dL, 10.2 g/dL, and 8.3 g/dL, respectively.
For the 6 patients with a β0/β0 genotype who continued to receive RBC transfusions, the median HbAT87Q level was 4.2 g/dL (range, 0.3 to 8.7) at the last study visit.
“There is room for improvement, as we’d like to see the elimination of dependency on transfusion even for patients with the most severe form of the disease,” said study author Philippe Leboulch, MD, of Brigham and Women’s Hospital in Boston, Massachusetts.
“But there is also hope with protocol modifications we have introduced in our phase 3 trials.”
The gene therapy LentiGlobin can reduce or eliminate transfusion dependence in patients with β-thalassemia, according to a pair of phase 1/2 studies.
Fifteen of the 22 patients in these trials were able to discontinue red blood cell (RBC) transfusions after receiving LentiGlobin.
In the 9 patients with severe transfusion-dependent β-thalassemia (TDT), LentiGlobin reduced the transfusion volume by 73%.
There were 5 adverse events (AEs) considered possibly or probably related to LentiGlobin, all them grade 1.
“These study results exceeded our expectations, with clinical benefit for nearly all patients . . . ,” said Alexis Thompson, MD, of Ann & Robert H. Lurie Children’s Hospital of Chicago in Illinois.
“Since we saw such positive results, we are now enrolling patients as young as 5 years old on a phase 3 trial of gene therapy for transfusion-dependent thalassemia.”
Dr Thompson and her colleagues reported results from the phase 1/2 trials—known as HGB-204 and HGB-205—in NEJM. The studies were sponsored by Bluebird Bio, the company developing LentiGlobin.
Patients in HGB-204
HGB-204 (also known as Northstar) is a multicenter study that was recently completed. It included 18 patients with TDT. They had a median age of 20 (range, 12 to 35) at baseline, and 72% were female. Seventy-eight percent were Asian, and 22% were white.
Eight patients had a β0/β0 genotype, 6 had a βE/β0 genotype, and 4 had other genotypes.
The patients’ median monthly transfusion volume for 2 years before study enrollment was 13.6 ml/kg (range, 10.4 to 21.8). The median age at which patients started regular transfusions was 3.5 years (range, 0 to 26.0). Six patients had undergone splenectomy.
Patients in HGB-205
HGB-205 is an ongoing study being conducted at a single site in France. It was designed to evaluate LentiGlobin in patients with TDT or severe sickle cell disease.
The NEJM paper includes 4 patients with TDT from this study. They had a median age of 18 (range, 16 to 19) at baseline, and half were female. Half were Asian, and the other half were white.
Three patients had a βE/β0 genotype. The remaining patient was homozygous for the IVS1-110 mutation and had a severe clinical presentation similar to that seen in β0/β0 genotypes.
The patients’ median monthly transfusion volume for 2 years before study enrollment was 15.2 ml/kg (range, 11.6 to 15.7). The median age at which patients started regular transfusions was 1.8 years (range, 0 to 14.0). Three patients had undergone splenectomy.
Treatment
For both studies, the researchers harvested hematopoietic stem and progenitor cells (mobilized with filgrastim and plerixafor) from the patients.
CD34+ cells were transduced ex vivo with LentiGlobin BB305 vector, which encodes adult hemoglobin (HbA) with a T87Q amino acid substitution (HbAT87Q).
The patients underwent myeloablative conditioning with busulfan, and the final LentiGlobin product was infused into patients after a 72-hour washout period.
In HGB-205 only, patients received enhanced RBC transfusions for at least 3 months before stem cell mobilization and harvest to maintain a hemoglobin level of more than 11.0 g/dL.
Safety
In HGB-204, there were 5 grade 1 AEs considered possibly or probably related to LentiGlobin. These included abdominal pain (n=2), dyspnea (n=1), hot flush (n=1), and non-cardiac chest pain (n=1).
There were 9 serious AEs, including 2 episodes of grade 3 veno-occlusive liver disease that were attributed to busulfan.
The remaining serious AEs were Klebsiella infection, cardiac ventricular thrombosis, cellulitis, hyperglycemia, and gastroenteritis (all grade 3), as well as device-related thrombosis and infectious diarrhea (both grade 2).
In HGB-205, there were no AEs considered possibly or probably related to LentiGlobin.
The 3 serious AEs were tooth infection and major depression (both grade 3), as well as pneumonia (grade 2).
Efficacy
The median time to neutrophil engraftment was 18.5 days (range, 14.0 to 30.0) in HGB-204 and 16.5 days (range, 14.0 to 29.0) in HGB-205.
The median time to platelet engraftment was 39.5 days (range, 19.0 to 191.0) in HGB-204 and 23.0 days (range, 20.0 to 26.0) in HGB-205.
In both studies, the median follow-up was 26 months (range, 15 to 42) after LentiGlobin infusion.
At last follow-up, all but 1 of the 13 patients with a non-β0/β0 genotype had stopped receiving RBC transfusions.
At the last study visit (12 to 36 months post-treatment), the median HbAT87Q level in these patients was 6.0 g/dL (range, 3.4 to 10.0), and the median total hemoglobin was 11.2 g/dL (range, 8.2 to 13.7).
In the 8 patients with a β0/β0 genotype and the 1 patient with 2 copies of the IVS1-110 mutation, the median annualized transfusion volume decreased by 73% after LentiGlobin infusion.
Two patients with a β0/β0 genotype were able to stop receiving RBC transfusions, as was the patient with 2 copies of the IVS1-110 mutation.
At their most recent study visit (12 months to 30 months), these 3 patients had median HbAT87Q levels of 8.2 g/dL, 6.8 g/dL, and 6.6 g/dL, respectively. Their median total hemoglobin levels were 9.0 g/dL, 10.2 g/dL, and 8.3 g/dL, respectively.
For the 6 patients with a β0/β0 genotype who continued to receive RBC transfusions, the median HbAT87Q level was 4.2 g/dL (range, 0.3 to 8.7) at the last study visit.
“There is room for improvement, as we’d like to see the elimination of dependency on transfusion even for patients with the most severe form of the disease,” said study author Philippe Leboulch, MD, of Brigham and Women’s Hospital in Boston, Massachusetts.
“But there is also hope with protocol modifications we have introduced in our phase 3 trials.”
Idarucizumab receives full FDA approval
The US Food and Drug Administration (FDA) has granted full approval to idarucizumab (Praxbind®), the specific reversal agent for dabigatran etexilate mesylate (Pradaxa®).
Idarucizumab received accelerated approval from the FDA in October 2015.
Now, the drug has full approval for use in reversing dabigatran’s anticoagulant effects in patients who require emergency surgery/urgent procedures and those who experience life-threatening or uncontrolled bleeding.
When the FDA granted idarucizumab accelerated approval, continued approval of the drug was contingent upon results from the phase 3 RE-VERSE AD™ trial.
Final results from RE-VERSE AD were published in NEJM in July 2017.
The trial enrolled 503 patients who required dabigatran reversal. They were divided into 2 groups:
- Group A included 301 patients with uncontrolled or life-threatening bleeding complications (eg, intracranial hemorrhage or severe trauma after a car accident)
- Group B included 202 patients requiring an invasive procedure or an emergency surgery or intervention (eg, surgery for an open fracture after a fall).
The study’s primary endpoint was the degree of reversal of the anticoagulant effect of dabigatran achieved by idarucizumab within 4 hours. The median maximum percentage reversal was 100%, as assessed on the basis of either the diluted thrombin time or the ecarin clotting time.
In group A, 68% of evaluable patients (134/203) had confirmed bleeding cessation within 24 hours of idarucizumab administration. Their median time to hemostasis after idarucizumab administration was 2.5 hours. (The time to the cessation of bleeding could not be assessed in 98 patients with intracranial bleeding.)
For patients in group B, their required procedures began a median of 1.6 hours from idarucizumab administration. Hemostasis during the procedure was described as normal for 93.4% of the patients, mildly abnormal in 5.1%, and moderately abnormal in 1.5%.
At 90 days, thrombotic events had occurred in 6.3% of patients in group A and 7.4% of those in group B. The 90-day mortality rates were 18.8% and 18.9%, respectively.
The US Food and Drug Administration (FDA) has granted full approval to idarucizumab (Praxbind®), the specific reversal agent for dabigatran etexilate mesylate (Pradaxa®).
Idarucizumab received accelerated approval from the FDA in October 2015.
Now, the drug has full approval for use in reversing dabigatran’s anticoagulant effects in patients who require emergency surgery/urgent procedures and those who experience life-threatening or uncontrolled bleeding.
When the FDA granted idarucizumab accelerated approval, continued approval of the drug was contingent upon results from the phase 3 RE-VERSE AD™ trial.
Final results from RE-VERSE AD were published in NEJM in July 2017.
The trial enrolled 503 patients who required dabigatran reversal. They were divided into 2 groups:
- Group A included 301 patients with uncontrolled or life-threatening bleeding complications (eg, intracranial hemorrhage or severe trauma after a car accident)
- Group B included 202 patients requiring an invasive procedure or an emergency surgery or intervention (eg, surgery for an open fracture after a fall).
The study’s primary endpoint was the degree of reversal of the anticoagulant effect of dabigatran achieved by idarucizumab within 4 hours. The median maximum percentage reversal was 100%, as assessed on the basis of either the diluted thrombin time or the ecarin clotting time.
In group A, 68% of evaluable patients (134/203) had confirmed bleeding cessation within 24 hours of idarucizumab administration. Their median time to hemostasis after idarucizumab administration was 2.5 hours. (The time to the cessation of bleeding could not be assessed in 98 patients with intracranial bleeding.)
For patients in group B, their required procedures began a median of 1.6 hours from idarucizumab administration. Hemostasis during the procedure was described as normal for 93.4% of the patients, mildly abnormal in 5.1%, and moderately abnormal in 1.5%.
At 90 days, thrombotic events had occurred in 6.3% of patients in group A and 7.4% of those in group B. The 90-day mortality rates were 18.8% and 18.9%, respectively.
The US Food and Drug Administration (FDA) has granted full approval to idarucizumab (Praxbind®), the specific reversal agent for dabigatran etexilate mesylate (Pradaxa®).
Idarucizumab received accelerated approval from the FDA in October 2015.
Now, the drug has full approval for use in reversing dabigatran’s anticoagulant effects in patients who require emergency surgery/urgent procedures and those who experience life-threatening or uncontrolled bleeding.
When the FDA granted idarucizumab accelerated approval, continued approval of the drug was contingent upon results from the phase 3 RE-VERSE AD™ trial.
Final results from RE-VERSE AD were published in NEJM in July 2017.
The trial enrolled 503 patients who required dabigatran reversal. They were divided into 2 groups:
- Group A included 301 patients with uncontrolled or life-threatening bleeding complications (eg, intracranial hemorrhage or severe trauma after a car accident)
- Group B included 202 patients requiring an invasive procedure or an emergency surgery or intervention (eg, surgery for an open fracture after a fall).
The study’s primary endpoint was the degree of reversal of the anticoagulant effect of dabigatran achieved by idarucizumab within 4 hours. The median maximum percentage reversal was 100%, as assessed on the basis of either the diluted thrombin time or the ecarin clotting time.
In group A, 68% of evaluable patients (134/203) had confirmed bleeding cessation within 24 hours of idarucizumab administration. Their median time to hemostasis after idarucizumab administration was 2.5 hours. (The time to the cessation of bleeding could not be assessed in 98 patients with intracranial bleeding.)
For patients in group B, their required procedures began a median of 1.6 hours from idarucizumab administration. Hemostasis during the procedure was described as normal for 93.4% of the patients, mildly abnormal in 5.1%, and moderately abnormal in 1.5%.
At 90 days, thrombotic events had occurred in 6.3% of patients in group A and 7.4% of those in group B. The 90-day mortality rates were 18.8% and 18.9%, respectively.
Antibody has ‘potent’ effects against AML

CHICAGO—The bispecific antibody APVO436 has demonstrated robust T-cell activation with limited cytokine release in acute myeloid leukemia (AML), according to researchers.
APVO436 binds CD123 and CD3 to redirect T-cell cytotoxicity against CD123-expressing tumor cells.
Researchers found that APVO436 induced T-cell cytotoxicity in AML cells in vitro and in mouse models.
In addition, levels of several cytokines were lower in experiments with APVO436 than in experiments with a comparator antibody.
These findings were presented in a poster at the AACR Annual Meeting 2018 (abstract 1786).
The research was conducted by employees of Aptevo Therapeutics Inc., the company developing APVO436.
“We are especially excited about these latest data for APVO436, which continue to show robust T-cell engagement and cytotoxic activity with reduced levels of cytokine release,” said Jane Gross, PhD, senior vice president and chief scientific officer for Aptevo.
Dr Gross and her colleagues found that APVO436 binds human CD123 and CD3-expressing cells and has “potent” target-specific activity against CD123-expressing AML cell lines (Molm-13 and KG-1a).
In addition, APVO436 induced endogenous T-cell activation and proliferation, accompanied by depletion of CD123-expressing cells, in samples from AML patients and healthy donors.
T cells from these cultures (both AML and non-AML) were expanded and co-cultured with Molm-13 cells and APVO436 or a control antibody. Again, the researchers observed “potent” cytotoxic activity in the presence of APVO436.
Dr Gross and her colleagues also tested APVO436, co-administered with human T cells, in mice with established disseminated Molm-13 tumors. The treatment resulted in a “rapid and significant” reduction in skeletal tumor burden.
Finally, the team compared APVO436 with an Aptevo-generated version of MGD006, a CD123 x CD3 dual-affinity re-targeting molecule being developed by Macrogenics, Inc.
The researchers took purified T cells from healthy donors and cultured them with Molm-13 cells, as well as APVO436, Aptevo’s version of MGD006, and a control antibody.
Both APVO436 and Aptevo’s version of MGD006 were effective at stimulating a tumor-directed immune response, inducing comparable levels of T-cell activation, proliferation, and cytotoxicity.
However, APVO436 induced lower levels of several cytokines—including IFNγ, IL-2, IL-6, and TNFα.
“Importantly, IFNγ, IL-6, and TNFα are considered to be the most relevant cytokines responsible for dosing toxicities observed in clinical studies with T-cell engaging molecules, which suggests that APVO436 could offer the potential for reduced toxicities compared to other CD123 x CD3 T-cell engagers at comparable or higher doses,” Dr Gross said.
She added that Aptevo is planning to launch a phase 1 trial of APVO436 in patients with AML and myelodysplastic syndromes later this year.
CHICAGO—The bispecific antibody APVO436 has demonstrated robust T-cell activation with limited cytokine release in acute myeloid leukemia (AML), according to researchers.
APVO436 binds CD123 and CD3 to redirect T-cell cytotoxicity against CD123-expressing tumor cells.
Researchers found that APVO436 induced T-cell cytotoxicity in AML cells in vitro and in mouse models.
In addition, levels of several cytokines were lower in experiments with APVO436 than in experiments with a comparator antibody.
These findings were presented in a poster at the AACR Annual Meeting 2018 (abstract 1786).
The research was conducted by employees of Aptevo Therapeutics Inc., the company developing APVO436.
“We are especially excited about these latest data for APVO436, which continue to show robust T-cell engagement and cytotoxic activity with reduced levels of cytokine release,” said Jane Gross, PhD, senior vice president and chief scientific officer for Aptevo.
Dr Gross and her colleagues found that APVO436 binds human CD123 and CD3-expressing cells and has “potent” target-specific activity against CD123-expressing AML cell lines (Molm-13 and KG-1a).
In addition, APVO436 induced endogenous T-cell activation and proliferation, accompanied by depletion of CD123-expressing cells, in samples from AML patients and healthy donors.
T cells from these cultures (both AML and non-AML) were expanded and co-cultured with Molm-13 cells and APVO436 or a control antibody. Again, the researchers observed “potent” cytotoxic activity in the presence of APVO436.
Dr Gross and her colleagues also tested APVO436, co-administered with human T cells, in mice with established disseminated Molm-13 tumors. The treatment resulted in a “rapid and significant” reduction in skeletal tumor burden.
Finally, the team compared APVO436 with an Aptevo-generated version of MGD006, a CD123 x CD3 dual-affinity re-targeting molecule being developed by Macrogenics, Inc.
The researchers took purified T cells from healthy donors and cultured them with Molm-13 cells, as well as APVO436, Aptevo’s version of MGD006, and a control antibody.
Both APVO436 and Aptevo’s version of MGD006 were effective at stimulating a tumor-directed immune response, inducing comparable levels of T-cell activation, proliferation, and cytotoxicity.
However, APVO436 induced lower levels of several cytokines—including IFNγ, IL-2, IL-6, and TNFα.
“Importantly, IFNγ, IL-6, and TNFα are considered to be the most relevant cytokines responsible for dosing toxicities observed in clinical studies with T-cell engaging molecules, which suggests that APVO436 could offer the potential for reduced toxicities compared to other CD123 x CD3 T-cell engagers at comparable or higher doses,” Dr Gross said.
She added that Aptevo is planning to launch a phase 1 trial of APVO436 in patients with AML and myelodysplastic syndromes later this year.
CHICAGO—The bispecific antibody APVO436 has demonstrated robust T-cell activation with limited cytokine release in acute myeloid leukemia (AML), according to researchers.
APVO436 binds CD123 and CD3 to redirect T-cell cytotoxicity against CD123-expressing tumor cells.
Researchers found that APVO436 induced T-cell cytotoxicity in AML cells in vitro and in mouse models.
In addition, levels of several cytokines were lower in experiments with APVO436 than in experiments with a comparator antibody.
These findings were presented in a poster at the AACR Annual Meeting 2018 (abstract 1786).
The research was conducted by employees of Aptevo Therapeutics Inc., the company developing APVO436.
“We are especially excited about these latest data for APVO436, which continue to show robust T-cell engagement and cytotoxic activity with reduced levels of cytokine release,” said Jane Gross, PhD, senior vice president and chief scientific officer for Aptevo.
Dr Gross and her colleagues found that APVO436 binds human CD123 and CD3-expressing cells and has “potent” target-specific activity against CD123-expressing AML cell lines (Molm-13 and KG-1a).
In addition, APVO436 induced endogenous T-cell activation and proliferation, accompanied by depletion of CD123-expressing cells, in samples from AML patients and healthy donors.
T cells from these cultures (both AML and non-AML) were expanded and co-cultured with Molm-13 cells and APVO436 or a control antibody. Again, the researchers observed “potent” cytotoxic activity in the presence of APVO436.
Dr Gross and her colleagues also tested APVO436, co-administered with human T cells, in mice with established disseminated Molm-13 tumors. The treatment resulted in a “rapid and significant” reduction in skeletal tumor burden.
Finally, the team compared APVO436 with an Aptevo-generated version of MGD006, a CD123 x CD3 dual-affinity re-targeting molecule being developed by Macrogenics, Inc.
The researchers took purified T cells from healthy donors and cultured them with Molm-13 cells, as well as APVO436, Aptevo’s version of MGD006, and a control antibody.
Both APVO436 and Aptevo’s version of MGD006 were effective at stimulating a tumor-directed immune response, inducing comparable levels of T-cell activation, proliferation, and cytotoxicity.
However, APVO436 induced lower levels of several cytokines—including IFNγ, IL-2, IL-6, and TNFα.
“Importantly, IFNγ, IL-6, and TNFα are considered to be the most relevant cytokines responsible for dosing toxicities observed in clinical studies with T-cell engaging molecules, which suggests that APVO436 could offer the potential for reduced toxicities compared to other CD123 x CD3 T-cell engagers at comparable or higher doses,” Dr Gross said.
She added that Aptevo is planning to launch a phase 1 trial of APVO436 in patients with AML and myelodysplastic syndromes later this year.
Fostamatinib approved to treat adults with chronic ITP
The US Food and Drug Administration (FDA) has granted approval for the oral SYK inhibitor fostamatinib disodium hexahydrate (Tavalisse™).
Fostamatinib is now approved for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment.
Rigel Pharmaceuticals, Inc., plans to launch fostamatinib in the US in late May 2018.
The FDA’s approval of fostamatinib was supported by data from the FIT clinical program, which included 2 randomized, placebo-controlled, phase 3 trials—FIT-1 (NCT02076399) and FIT-2 (NCT02076412)—and an open-label extension study—FIT-3 (NCT02077192)—as well as an initial proof-of-concept study.
FIT-1 and FIT-2
FIT-1 and FIT-2 included 150 patients with persistent or chronic ITP who had an insufficient response to previous treatment. The patients had a median age of 54 (range, 20 to 88), 61% were female, and 93% were white.
Prior ITP treatments included corticosteroids (94%), immunoglobulins (53%), and thrombopoietin receptor agonists (48%). Thirty-five percent of patients had undergone splenectomy.
The patients’ median platelet count at baseline was 16 x 109/L, and 47% were on stable ITP therapy.
In each study, patients were randomized 2:1 to receive fostamatinib or placebo for 24 weeks. In FIT-1, 76 patients were randomized—51 to fostamatinib and 25 to placebo. In FIT-2, 74 patients were randomized—50 to fostamatinib and 24 to placebo.
All patients initially received fostamatinib at 100 mg twice daily. Most (88%) were escalated to 150 mg twice daily at week 4 or later. Patients could also receive stable concurrent ITP therapy—glucocorticoids (< 20 mg prednisone equivalent per day), azathioprine, or danazol—and rescue therapy if needed.
The efficacy of fostamatinib was based on stable platelet response, defined as a platelet count of at least 50 x 109/L on at least 4 of the 6 visits between weeks 14 to 24.
In FIT-1, 18% of patients in the fostamatinib arm and 0% of those in the placebo arm achieved a stable platelet response (P=0.03).
In FIT-2, 16% of patients in the fostamatinib arm and 4% in the placebo arm achieved a stable platelet response. In this trial, the between-arm difference was not statistically significant.
For both studies, the incidence of bleeding was 29% in the fostamatinib arm and 37% in the placebo arm. The rate of severe bleeding-related events was 1% and 6%, respectively, and the rate of serious bleeding-related events was 4% and 10%, respectively.
FIT-3
Patients from FIT-1 and FIT-2 could enroll in FIT-3 if they completed 24 weeks of treatment or did not respond to treatment any time after 12 weeks.
Patients who were in response at the time of roll over (those who had achieved a platelet count of at least 50 x 109/L) continued in FIT-3 at their current dose and regimen.
Non-responders received fostamatinib at 100 mg twice daily regardless of their dose and regimen in the prior study.
In all, 123 patients were enrolled—44 previously randomized to placebo and 79 previously randomized to fostamatinib. Half of patients (n=61) discontinued the study early.
Of the 44 patients with prior placebo, 10 (23%) had a stable platelet response to fostamatinib. This included a patient who was a responder to placebo in the prior study.
In FIT-3, stable platelet response was defined as having no 2 visits, at least 4 weeks apart, with a platelet count less than 50 x 109/L without an intervening visit with a platelet count of at least 50 x 109/L (unrelated to rescue therapy) within a period of 12 weeks after the patient’s initial achievement of the target platelet count.
Of all the responders in the FIT trials, there were 18 who maintained a platelet count of at least 50 x 109/L for 12 months or longer.
For more details on these trials and fostamatinib, see the full prescribing information, which is available at www.TAVALISSE.com.
The US Food and Drug Administration (FDA) has granted approval for the oral SYK inhibitor fostamatinib disodium hexahydrate (Tavalisse™).
Fostamatinib is now approved for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment.
Rigel Pharmaceuticals, Inc., plans to launch fostamatinib in the US in late May 2018.
The FDA’s approval of fostamatinib was supported by data from the FIT clinical program, which included 2 randomized, placebo-controlled, phase 3 trials—FIT-1 (NCT02076399) and FIT-2 (NCT02076412)—and an open-label extension study—FIT-3 (NCT02077192)—as well as an initial proof-of-concept study.
FIT-1 and FIT-2
FIT-1 and FIT-2 included 150 patients with persistent or chronic ITP who had an insufficient response to previous treatment. The patients had a median age of 54 (range, 20 to 88), 61% were female, and 93% were white.
Prior ITP treatments included corticosteroids (94%), immunoglobulins (53%), and thrombopoietin receptor agonists (48%). Thirty-five percent of patients had undergone splenectomy.
The patients’ median platelet count at baseline was 16 x 109/L, and 47% were on stable ITP therapy.
In each study, patients were randomized 2:1 to receive fostamatinib or placebo for 24 weeks. In FIT-1, 76 patients were randomized—51 to fostamatinib and 25 to placebo. In FIT-2, 74 patients were randomized—50 to fostamatinib and 24 to placebo.
All patients initially received fostamatinib at 100 mg twice daily. Most (88%) were escalated to 150 mg twice daily at week 4 or later. Patients could also receive stable concurrent ITP therapy—glucocorticoids (< 20 mg prednisone equivalent per day), azathioprine, or danazol—and rescue therapy if needed.
The efficacy of fostamatinib was based on stable platelet response, defined as a platelet count of at least 50 x 109/L on at least 4 of the 6 visits between weeks 14 to 24.
In FIT-1, 18% of patients in the fostamatinib arm and 0% of those in the placebo arm achieved a stable platelet response (P=0.03).
In FIT-2, 16% of patients in the fostamatinib arm and 4% in the placebo arm achieved a stable platelet response. In this trial, the between-arm difference was not statistically significant.
For both studies, the incidence of bleeding was 29% in the fostamatinib arm and 37% in the placebo arm. The rate of severe bleeding-related events was 1% and 6%, respectively, and the rate of serious bleeding-related events was 4% and 10%, respectively.
FIT-3
Patients from FIT-1 and FIT-2 could enroll in FIT-3 if they completed 24 weeks of treatment or did not respond to treatment any time after 12 weeks.
Patients who were in response at the time of roll over (those who had achieved a platelet count of at least 50 x 109/L) continued in FIT-3 at their current dose and regimen.
Non-responders received fostamatinib at 100 mg twice daily regardless of their dose and regimen in the prior study.
In all, 123 patients were enrolled—44 previously randomized to placebo and 79 previously randomized to fostamatinib. Half of patients (n=61) discontinued the study early.
Of the 44 patients with prior placebo, 10 (23%) had a stable platelet response to fostamatinib. This included a patient who was a responder to placebo in the prior study.
In FIT-3, stable platelet response was defined as having no 2 visits, at least 4 weeks apart, with a platelet count less than 50 x 109/L without an intervening visit with a platelet count of at least 50 x 109/L (unrelated to rescue therapy) within a period of 12 weeks after the patient’s initial achievement of the target platelet count.
Of all the responders in the FIT trials, there were 18 who maintained a platelet count of at least 50 x 109/L for 12 months or longer.
For more details on these trials and fostamatinib, see the full prescribing information, which is available at www.TAVALISSE.com.
The US Food and Drug Administration (FDA) has granted approval for the oral SYK inhibitor fostamatinib disodium hexahydrate (Tavalisse™).
Fostamatinib is now approved for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment.
Rigel Pharmaceuticals, Inc., plans to launch fostamatinib in the US in late May 2018.
The FDA’s approval of fostamatinib was supported by data from the FIT clinical program, which included 2 randomized, placebo-controlled, phase 3 trials—FIT-1 (NCT02076399) and FIT-2 (NCT02076412)—and an open-label extension study—FIT-3 (NCT02077192)—as well as an initial proof-of-concept study.
FIT-1 and FIT-2
FIT-1 and FIT-2 included 150 patients with persistent or chronic ITP who had an insufficient response to previous treatment. The patients had a median age of 54 (range, 20 to 88), 61% were female, and 93% were white.
Prior ITP treatments included corticosteroids (94%), immunoglobulins (53%), and thrombopoietin receptor agonists (48%). Thirty-five percent of patients had undergone splenectomy.
The patients’ median platelet count at baseline was 16 x 109/L, and 47% were on stable ITP therapy.
In each study, patients were randomized 2:1 to receive fostamatinib or placebo for 24 weeks. In FIT-1, 76 patients were randomized—51 to fostamatinib and 25 to placebo. In FIT-2, 74 patients were randomized—50 to fostamatinib and 24 to placebo.
All patients initially received fostamatinib at 100 mg twice daily. Most (88%) were escalated to 150 mg twice daily at week 4 or later. Patients could also receive stable concurrent ITP therapy—glucocorticoids (< 20 mg prednisone equivalent per day), azathioprine, or danazol—and rescue therapy if needed.
The efficacy of fostamatinib was based on stable platelet response, defined as a platelet count of at least 50 x 109/L on at least 4 of the 6 visits between weeks 14 to 24.
In FIT-1, 18% of patients in the fostamatinib arm and 0% of those in the placebo arm achieved a stable platelet response (P=0.03).
In FIT-2, 16% of patients in the fostamatinib arm and 4% in the placebo arm achieved a stable platelet response. In this trial, the between-arm difference was not statistically significant.
For both studies, the incidence of bleeding was 29% in the fostamatinib arm and 37% in the placebo arm. The rate of severe bleeding-related events was 1% and 6%, respectively, and the rate of serious bleeding-related events was 4% and 10%, respectively.
FIT-3
Patients from FIT-1 and FIT-2 could enroll in FIT-3 if they completed 24 weeks of treatment or did not respond to treatment any time after 12 weeks.
Patients who were in response at the time of roll over (those who had achieved a platelet count of at least 50 x 109/L) continued in FIT-3 at their current dose and regimen.
Non-responders received fostamatinib at 100 mg twice daily regardless of their dose and regimen in the prior study.
In all, 123 patients were enrolled—44 previously randomized to placebo and 79 previously randomized to fostamatinib. Half of patients (n=61) discontinued the study early.
Of the 44 patients with prior placebo, 10 (23%) had a stable platelet response to fostamatinib. This included a patient who was a responder to placebo in the prior study.
In FIT-3, stable platelet response was defined as having no 2 visits, at least 4 weeks apart, with a platelet count less than 50 x 109/L without an intervening visit with a platelet count of at least 50 x 109/L (unrelated to rescue therapy) within a period of 12 weeks after the patient’s initial achievement of the target platelet count.
Of all the responders in the FIT trials, there were 18 who maintained a platelet count of at least 50 x 109/L for 12 months or longer.
For more details on these trials and fostamatinib, see the full prescribing information, which is available at www.TAVALISSE.com.
FDA expands approved use of product for VWD
The US Food and Drug Administration (FDA) has expanded the approved use of Vonvendi, a recombinant von Willebrand factor product.
Vonvendi is now approved for perioperative management of bleeding in adults (age 18 and older) with von Willebrand disease (VWD).
Vonvendi was previously FDA-approved for on-demand treatment and control of bleeding episodes in adults with VWD.
“Persons with von Willebrand disease face a heightened risk of bleeding during surgery and may require factor treatment before, during, or after surgery,” said Michael Tarantino, MD, of the Bleeding & Clotting Disorders Institute in Peoria, Illinois.
“For surgeries requiring repeated, frequent infusions with combined von Willebrand factor and factor VIII concentrates, an excessive rise in factor VIII levels may increase the risk of thromboembolic complications, such as blood clots. The expanded use for Vonvendi in surgical settings gives healthcare professionals flexibility in treating von Willebrand disease with an appropriate dose of von Willebrand factor, with or without recombinant factor VIII, based on each patient’s unique needs.”
The FDA’s approval of Vonvendi in surgical settings was based on results from a phase 3 trial. The study enrolled 15 adults with severe VWD who were undergoing elective surgical procedures (10 of them major procedures).
Patients received Vonvendi at 40 to 60 IU per kg of body weight 12 to 24 hours before surgery. Within 3 hours of surgery, each patient’s factor VIII level (FVIII:C) was assessed, with a target of 30 IU/dL for minor surgeries and 60 IU/dL for major surgeries.
Within an hour of surgery, patients received a dose of Vonvendi, with or without recombinant factor VIII, depending on the target FVIII:C levels at the 3-hour assessment.
The study’s primary endpoint was met. Vonvendi demonstrated overall hemostatic efficacy, as assessed 24 hours after the last perioperative infusion or the completion of the study visit, whichever occurred earlier.
Intra- and post-operative hemostasis was rated as “excellent” (as good as or better than expected) in 60% of patients and “good” (probably as good as expected) in 40% of patients.
One patient developed deep vein thrombosis 3 days after undergoing hip replacement surgery.
One patient tested positive for binding antibodies to von Willebrand factor. None of the patients developed binding antibodies against potential impurities such as rFurin, CHO-protein, or mouse IgG.
More details on this study are available in the full prescribing information for Vonvendi.
The US Food and Drug Administration (FDA) has expanded the approved use of Vonvendi, a recombinant von Willebrand factor product.
Vonvendi is now approved for perioperative management of bleeding in adults (age 18 and older) with von Willebrand disease (VWD).
Vonvendi was previously FDA-approved for on-demand treatment and control of bleeding episodes in adults with VWD.
“Persons with von Willebrand disease face a heightened risk of bleeding during surgery and may require factor treatment before, during, or after surgery,” said Michael Tarantino, MD, of the Bleeding & Clotting Disorders Institute in Peoria, Illinois.
“For surgeries requiring repeated, frequent infusions with combined von Willebrand factor and factor VIII concentrates, an excessive rise in factor VIII levels may increase the risk of thromboembolic complications, such as blood clots. The expanded use for Vonvendi in surgical settings gives healthcare professionals flexibility in treating von Willebrand disease with an appropriate dose of von Willebrand factor, with or without recombinant factor VIII, based on each patient’s unique needs.”
The FDA’s approval of Vonvendi in surgical settings was based on results from a phase 3 trial. The study enrolled 15 adults with severe VWD who were undergoing elective surgical procedures (10 of them major procedures).
Patients received Vonvendi at 40 to 60 IU per kg of body weight 12 to 24 hours before surgery. Within 3 hours of surgery, each patient’s factor VIII level (FVIII:C) was assessed, with a target of 30 IU/dL for minor surgeries and 60 IU/dL for major surgeries.
Within an hour of surgery, patients received a dose of Vonvendi, with or without recombinant factor VIII, depending on the target FVIII:C levels at the 3-hour assessment.
The study’s primary endpoint was met. Vonvendi demonstrated overall hemostatic efficacy, as assessed 24 hours after the last perioperative infusion or the completion of the study visit, whichever occurred earlier.
Intra- and post-operative hemostasis was rated as “excellent” (as good as or better than expected) in 60% of patients and “good” (probably as good as expected) in 40% of patients.
One patient developed deep vein thrombosis 3 days after undergoing hip replacement surgery.
One patient tested positive for binding antibodies to von Willebrand factor. None of the patients developed binding antibodies against potential impurities such as rFurin, CHO-protein, or mouse IgG.
More details on this study are available in the full prescribing information for Vonvendi.
The US Food and Drug Administration (FDA) has expanded the approved use of Vonvendi, a recombinant von Willebrand factor product.
Vonvendi is now approved for perioperative management of bleeding in adults (age 18 and older) with von Willebrand disease (VWD).
Vonvendi was previously FDA-approved for on-demand treatment and control of bleeding episodes in adults with VWD.
“Persons with von Willebrand disease face a heightened risk of bleeding during surgery and may require factor treatment before, during, or after surgery,” said Michael Tarantino, MD, of the Bleeding & Clotting Disorders Institute in Peoria, Illinois.
“For surgeries requiring repeated, frequent infusions with combined von Willebrand factor and factor VIII concentrates, an excessive rise in factor VIII levels may increase the risk of thromboembolic complications, such as blood clots. The expanded use for Vonvendi in surgical settings gives healthcare professionals flexibility in treating von Willebrand disease with an appropriate dose of von Willebrand factor, with or without recombinant factor VIII, based on each patient’s unique needs.”
The FDA’s approval of Vonvendi in surgical settings was based on results from a phase 3 trial. The study enrolled 15 adults with severe VWD who were undergoing elective surgical procedures (10 of them major procedures).
Patients received Vonvendi at 40 to 60 IU per kg of body weight 12 to 24 hours before surgery. Within 3 hours of surgery, each patient’s factor VIII level (FVIII:C) was assessed, with a target of 30 IU/dL for minor surgeries and 60 IU/dL for major surgeries.
Within an hour of surgery, patients received a dose of Vonvendi, with or without recombinant factor VIII, depending on the target FVIII:C levels at the 3-hour assessment.
The study’s primary endpoint was met. Vonvendi demonstrated overall hemostatic efficacy, as assessed 24 hours after the last perioperative infusion or the completion of the study visit, whichever occurred earlier.
Intra- and post-operative hemostasis was rated as “excellent” (as good as or better than expected) in 60% of patients and “good” (probably as good as expected) in 40% of patients.
One patient developed deep vein thrombosis 3 days after undergoing hip replacement surgery.
One patient tested positive for binding antibodies to von Willebrand factor. None of the patients developed binding antibodies against potential impurities such as rFurin, CHO-protein, or mouse IgG.
More details on this study are available in the full prescribing information for Vonvendi.
Emicizumab receives breakthrough designation

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to emicizumab (Hemlibra®) for patients who have hemophilia A without factor VIII inhibitors.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved by the FDA for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A with factor VIII inhibitors.
The FDA has granted emicizumab breakthrough designation for patients without inhibitors based on data from the phase 3 HAVEN 3 trial.
The trial enrolled 152 patients age 12 and older who had hemophilia A without inhibitors and were previously treated with factor VIII therapy either on-demand or for prophylaxis.
Patients received either no prophylaxis or emicizumab prophylaxis, dosed subcutaneously every week or every 2 weeks.
Detailed results from HAVEN 3 have not been released.
However, according to Genentech, prophylaxis with emicizumab produced a statistically significant and clinically meaningful reduction in treated bleeds, when compared to no prophylaxis.
In an intra-patient comparison, once-weekly emicizumab prophylaxis was superior to prior factor VIII prophylaxis, as demonstrated by a statistically significant and clinically meaningful reduction in treated bleeds.
The most common adverse events with emicizumab were injection site reactions, and no new safety signals were observed in this trial. No thrombotic microangiopathy or thrombotic events occurred.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to emicizumab (Hemlibra®) for patients who have hemophilia A without factor VIII inhibitors.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved by the FDA for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A with factor VIII inhibitors.
The FDA has granted emicizumab breakthrough designation for patients without inhibitors based on data from the phase 3 HAVEN 3 trial.
The trial enrolled 152 patients age 12 and older who had hemophilia A without inhibitors and were previously treated with factor VIII therapy either on-demand or for prophylaxis.
Patients received either no prophylaxis or emicizumab prophylaxis, dosed subcutaneously every week or every 2 weeks.
Detailed results from HAVEN 3 have not been released.
However, according to Genentech, prophylaxis with emicizumab produced a statistically significant and clinically meaningful reduction in treated bleeds, when compared to no prophylaxis.
In an intra-patient comparison, once-weekly emicizumab prophylaxis was superior to prior factor VIII prophylaxis, as demonstrated by a statistically significant and clinically meaningful reduction in treated bleeds.
The most common adverse events with emicizumab were injection site reactions, and no new safety signals were observed in this trial. No thrombotic microangiopathy or thrombotic events occurred.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to emicizumab (Hemlibra®) for patients who have hemophilia A without factor VIII inhibitors.
Emicizumab is a bispecific factor IXa- and factor X-directed antibody approved by the FDA for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A with factor VIII inhibitors.
The FDA has granted emicizumab breakthrough designation for patients without inhibitors based on data from the phase 3 HAVEN 3 trial.
The trial enrolled 152 patients age 12 and older who had hemophilia A without inhibitors and were previously treated with factor VIII therapy either on-demand or for prophylaxis.
Patients received either no prophylaxis or emicizumab prophylaxis, dosed subcutaneously every week or every 2 weeks.
Detailed results from HAVEN 3 have not been released.
However, according to Genentech, prophylaxis with emicizumab produced a statistically significant and clinically meaningful reduction in treated bleeds, when compared to no prophylaxis.
In an intra-patient comparison, once-weekly emicizumab prophylaxis was superior to prior factor VIII prophylaxis, as demonstrated by a statistically significant and clinically meaningful reduction in treated bleeds.
The most common adverse events with emicizumab were injection site reactions, and no new safety signals were observed in this trial. No thrombotic microangiopathy or thrombotic events occurred.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Generic melphalan available in US
Fresenius Kabi’s Melphalan Hydrochloride for Injection, a generic version of Alkeran®, is now available in the US.
Melphalan Hydrochloride for Injection is available as a 2-vial kit containing 1 single-dose vial of melphalan hydrochloride equivalent to 50 mg of melphalan and 1 vial of sterile diluent.
Melphalan Hydrochloride for Injection is indicated for the palliative treatment of patients with multiple myeloma for whom oral therapy is not appropriate.
For more details on Melphalan Hydrochloride for Injection, see the full prescribing information.
Fresenius Kabi’s Melphalan Hydrochloride for Injection, a generic version of Alkeran®, is now available in the US.
Melphalan Hydrochloride for Injection is available as a 2-vial kit containing 1 single-dose vial of melphalan hydrochloride equivalent to 50 mg of melphalan and 1 vial of sterile diluent.
Melphalan Hydrochloride for Injection is indicated for the palliative treatment of patients with multiple myeloma for whom oral therapy is not appropriate.
For more details on Melphalan Hydrochloride for Injection, see the full prescribing information.
Fresenius Kabi’s Melphalan Hydrochloride for Injection, a generic version of Alkeran®, is now available in the US.
Melphalan Hydrochloride for Injection is available as a 2-vial kit containing 1 single-dose vial of melphalan hydrochloride equivalent to 50 mg of melphalan and 1 vial of sterile diluent.
Melphalan Hydrochloride for Injection is indicated for the palliative treatment of patients with multiple myeloma for whom oral therapy is not appropriate.
For more details on Melphalan Hydrochloride for Injection, see the full prescribing information.
Drug appears to aid chemo in AML

Adding an experimental compound to chemotherapy is a “promising” treatment approach for certain patients with acute myeloid leukemia (AML), according to researchers.
They tested the compound, CPI-613, in combination with high-dose cytarabine and mitoxantrone in a phase 1 trial of patients with relapsed or refractory AML.
The combination produced similar response rates in the overall patient population (50%), patients age 60 and older (47%), and those with poor-risk cytogenetics (46%).
The most common grade 3/4 adverse events (AEs) were hematologic toxicities, and there was 1 fatal AE—hypotension.
Mortality rates in this trial were similar to those observed in historical controls treated with high-dose cytarabine, mitoxantrone, and asparaginase.
“These data are very encouraging, especially for patients 60 years of age or older who have historically done very poorly with this disease,” said Timothy Pardee, MD, PhD, a professor at Wake Forest Baptist Health in Winston-Salem, North Carolina, and chief medical officer of Rafael Pharmaceuticals, Inc., the company developing CPI-613.
Dr Pardee and his colleagues reported these results in Clinical Cancer Research.
The researchers noted that CPI-613 is designed to target mitochondrial metabolism in cancer cells, and preclinical research showed that CPI-613 sensitized AML cells to chemotherapy.
To investigate this further, the team tested CPI-613 in combination with high-dose cytarabine and mitoxantrone in the phase 1 trial. The study included 66 patients with relapsed or refractory AML, as well as a patient with advanced-phase chronic myeloid leukemia (CML) who was mistakenly enrolled.
The patients’ median age was 60 (range, 21-79), and 54% were age 60 and older. Their median percentage of marrow blasts was 43%. Forty percent of patients had poor-risk cytogenetics, and 49% had intermediate-risk cytogenetics.
Most patients (72%) had no prior salvage therapy, 13% had 1 prior line of salvage, 10% had 2 prior lines, and 4% had more than 2. Thirty-one percent of patients had refractory disease.
Seven percent of patients had previously received high-dose cytarabine and mitoxantrone, and 25% had previous salvage including high-dose or intermediate-dose cytarabine.
Treatment
Patients received CPI-613, given over 2 hours, on days 1 to 5 of cycle 1. Doses ranged from 500 mg/m2 to 2750 mg/m2.
Starting on day 3, patients received 5 doses of cytarabine at 3 gm/m2 (for patients younger than 60) or 1.5 gm/m2 (for older patients) in 500 mL normal saline, over 3 hours, every 12 hours.
Patients also received 3 daily doses of mitoxantrone at 6 mg/m2 in 50 mL normal saline, given over 15 minutes, after the first, third, and fifth doses of cytarabine.
Patients were initially assigned to receive 1 cycle of treatment. Those with at least 5% blasts after the first cycle could receive a second course—either a full course or a 3-day course. And patients who responded to the first course could receive up to 2 cycles of the 3-day course.
Safety
There were 2 dose-limiting toxicities when CPI-613 was given at the 2750 mg/m2 dose. One of these toxicities was grade 3 diarrhea that didn’t respond to anti-diarrheals, and the other was grade 3 nausea that didn’t respond to antiemetics.
Because of these events, 2500 mg/m2 was deemed the maximum-tolerated dose. However, the recommended phase 2 dose is 2000 mg/m2.
The most common AEs—occurring in at least 50% of all patients who received CPI-613 (n=67)—included hemoglobin decrease (67%), hyperglycemia (67%), neutropenia (67%), thrombocytopenia (67%), hypomagnesemia (66%), leukopenia (66%), lymphopenia (66%), hypoalbuminemia (65%), hypokalemia (60%), hypocalcemia (57%), and diarrhea (55%).
All cases of neutropenia, thrombocytopenia, leukopenia, and lymphopenia were grade 3/4. Other common grade 3/4 AEs (occurring in at least 20% of patients) included hemoglobin decrease (62%), febrile neutropenia (28%), hypophosphatemia (24%), and hypokalemia (23%).
The only grade 5 AE was hypotension.
The mortality rate was 12% (n=8) at 30 days and 19% (n=13) at 60 days. The researchers said this was similar to the historical experience with high-dose cytarabine, mitoxantrone, and asparaginase. Mortality rates with this regimen were 13% at 30 days and 22% at 60 days.
Efficacy
Sixty-two patients were evaluable for response. Of the 5 patients who were not evaluable, 1 didn’t complete the first cycle of treatment, 1 was the CML patient, and 3 died before assessment.
The overall response rate was 50% (31/62). This included 26 patients with a complete response (CR) and 5 patients who had a CR with incomplete count recovery (CRi).
The rate of CR/CRi was 47% (15/32) in patients older than 60 years of age, 46% (11/24) in patients who had poor-risk cytogenetics, and 53% (8/15) when CPI-613 was given at the recommended phase 2 dose—2000 mg/m2.
The median overall survival (OS) was 6.7 months for all evaluable patients and 13.2 months for patients who achieved a CR/CRi.
The median OS was 6.9 months for patients age 60 and older, which was not significantly different from the median OS in younger patients (P=0.9642).
This study was sponsored by Wake Forest University Health Sciences and the National Cancer Institute.
Adding an experimental compound to chemotherapy is a “promising” treatment approach for certain patients with acute myeloid leukemia (AML), according to researchers.
They tested the compound, CPI-613, in combination with high-dose cytarabine and mitoxantrone in a phase 1 trial of patients with relapsed or refractory AML.
The combination produced similar response rates in the overall patient population (50%), patients age 60 and older (47%), and those with poor-risk cytogenetics (46%).
The most common grade 3/4 adverse events (AEs) were hematologic toxicities, and there was 1 fatal AE—hypotension.
Mortality rates in this trial were similar to those observed in historical controls treated with high-dose cytarabine, mitoxantrone, and asparaginase.
“These data are very encouraging, especially for patients 60 years of age or older who have historically done very poorly with this disease,” said Timothy Pardee, MD, PhD, a professor at Wake Forest Baptist Health in Winston-Salem, North Carolina, and chief medical officer of Rafael Pharmaceuticals, Inc., the company developing CPI-613.
Dr Pardee and his colleagues reported these results in Clinical Cancer Research.
The researchers noted that CPI-613 is designed to target mitochondrial metabolism in cancer cells, and preclinical research showed that CPI-613 sensitized AML cells to chemotherapy.
To investigate this further, the team tested CPI-613 in combination with high-dose cytarabine and mitoxantrone in the phase 1 trial. The study included 66 patients with relapsed or refractory AML, as well as a patient with advanced-phase chronic myeloid leukemia (CML) who was mistakenly enrolled.
The patients’ median age was 60 (range, 21-79), and 54% were age 60 and older. Their median percentage of marrow blasts was 43%. Forty percent of patients had poor-risk cytogenetics, and 49% had intermediate-risk cytogenetics.
Most patients (72%) had no prior salvage therapy, 13% had 1 prior line of salvage, 10% had 2 prior lines, and 4% had more than 2. Thirty-one percent of patients had refractory disease.
Seven percent of patients had previously received high-dose cytarabine and mitoxantrone, and 25% had previous salvage including high-dose or intermediate-dose cytarabine.
Treatment
Patients received CPI-613, given over 2 hours, on days 1 to 5 of cycle 1. Doses ranged from 500 mg/m2 to 2750 mg/m2.
Starting on day 3, patients received 5 doses of cytarabine at 3 gm/m2 (for patients younger than 60) or 1.5 gm/m2 (for older patients) in 500 mL normal saline, over 3 hours, every 12 hours.
Patients also received 3 daily doses of mitoxantrone at 6 mg/m2 in 50 mL normal saline, given over 15 minutes, after the first, third, and fifth doses of cytarabine.
Patients were initially assigned to receive 1 cycle of treatment. Those with at least 5% blasts after the first cycle could receive a second course—either a full course or a 3-day course. And patients who responded to the first course could receive up to 2 cycles of the 3-day course.
Safety
There were 2 dose-limiting toxicities when CPI-613 was given at the 2750 mg/m2 dose. One of these toxicities was grade 3 diarrhea that didn’t respond to anti-diarrheals, and the other was grade 3 nausea that didn’t respond to antiemetics.
Because of these events, 2500 mg/m2 was deemed the maximum-tolerated dose. However, the recommended phase 2 dose is 2000 mg/m2.
The most common AEs—occurring in at least 50% of all patients who received CPI-613 (n=67)—included hemoglobin decrease (67%), hyperglycemia (67%), neutropenia (67%), thrombocytopenia (67%), hypomagnesemia (66%), leukopenia (66%), lymphopenia (66%), hypoalbuminemia (65%), hypokalemia (60%), hypocalcemia (57%), and diarrhea (55%).
All cases of neutropenia, thrombocytopenia, leukopenia, and lymphopenia were grade 3/4. Other common grade 3/4 AEs (occurring in at least 20% of patients) included hemoglobin decrease (62%), febrile neutropenia (28%), hypophosphatemia (24%), and hypokalemia (23%).
The only grade 5 AE was hypotension.
The mortality rate was 12% (n=8) at 30 days and 19% (n=13) at 60 days. The researchers said this was similar to the historical experience with high-dose cytarabine, mitoxantrone, and asparaginase. Mortality rates with this regimen were 13% at 30 days and 22% at 60 days.
Efficacy
Sixty-two patients were evaluable for response. Of the 5 patients who were not evaluable, 1 didn’t complete the first cycle of treatment, 1 was the CML patient, and 3 died before assessment.
The overall response rate was 50% (31/62). This included 26 patients with a complete response (CR) and 5 patients who had a CR with incomplete count recovery (CRi).
The rate of CR/CRi was 47% (15/32) in patients older than 60 years of age, 46% (11/24) in patients who had poor-risk cytogenetics, and 53% (8/15) when CPI-613 was given at the recommended phase 2 dose—2000 mg/m2.
The median overall survival (OS) was 6.7 months for all evaluable patients and 13.2 months for patients who achieved a CR/CRi.
The median OS was 6.9 months for patients age 60 and older, which was not significantly different from the median OS in younger patients (P=0.9642).
This study was sponsored by Wake Forest University Health Sciences and the National Cancer Institute.
Adding an experimental compound to chemotherapy is a “promising” treatment approach for certain patients with acute myeloid leukemia (AML), according to researchers.
They tested the compound, CPI-613, in combination with high-dose cytarabine and mitoxantrone in a phase 1 trial of patients with relapsed or refractory AML.
The combination produced similar response rates in the overall patient population (50%), patients age 60 and older (47%), and those with poor-risk cytogenetics (46%).
The most common grade 3/4 adverse events (AEs) were hematologic toxicities, and there was 1 fatal AE—hypotension.
Mortality rates in this trial were similar to those observed in historical controls treated with high-dose cytarabine, mitoxantrone, and asparaginase.
“These data are very encouraging, especially for patients 60 years of age or older who have historically done very poorly with this disease,” said Timothy Pardee, MD, PhD, a professor at Wake Forest Baptist Health in Winston-Salem, North Carolina, and chief medical officer of Rafael Pharmaceuticals, Inc., the company developing CPI-613.
Dr Pardee and his colleagues reported these results in Clinical Cancer Research.
The researchers noted that CPI-613 is designed to target mitochondrial metabolism in cancer cells, and preclinical research showed that CPI-613 sensitized AML cells to chemotherapy.
To investigate this further, the team tested CPI-613 in combination with high-dose cytarabine and mitoxantrone in the phase 1 trial. The study included 66 patients with relapsed or refractory AML, as well as a patient with advanced-phase chronic myeloid leukemia (CML) who was mistakenly enrolled.
The patients’ median age was 60 (range, 21-79), and 54% were age 60 and older. Their median percentage of marrow blasts was 43%. Forty percent of patients had poor-risk cytogenetics, and 49% had intermediate-risk cytogenetics.
Most patients (72%) had no prior salvage therapy, 13% had 1 prior line of salvage, 10% had 2 prior lines, and 4% had more than 2. Thirty-one percent of patients had refractory disease.
Seven percent of patients had previously received high-dose cytarabine and mitoxantrone, and 25% had previous salvage including high-dose or intermediate-dose cytarabine.
Treatment
Patients received CPI-613, given over 2 hours, on days 1 to 5 of cycle 1. Doses ranged from 500 mg/m2 to 2750 mg/m2.
Starting on day 3, patients received 5 doses of cytarabine at 3 gm/m2 (for patients younger than 60) or 1.5 gm/m2 (for older patients) in 500 mL normal saline, over 3 hours, every 12 hours.
Patients also received 3 daily doses of mitoxantrone at 6 mg/m2 in 50 mL normal saline, given over 15 minutes, after the first, third, and fifth doses of cytarabine.
Patients were initially assigned to receive 1 cycle of treatment. Those with at least 5% blasts after the first cycle could receive a second course—either a full course or a 3-day course. And patients who responded to the first course could receive up to 2 cycles of the 3-day course.
Safety
There were 2 dose-limiting toxicities when CPI-613 was given at the 2750 mg/m2 dose. One of these toxicities was grade 3 diarrhea that didn’t respond to anti-diarrheals, and the other was grade 3 nausea that didn’t respond to antiemetics.
Because of these events, 2500 mg/m2 was deemed the maximum-tolerated dose. However, the recommended phase 2 dose is 2000 mg/m2.
The most common AEs—occurring in at least 50% of all patients who received CPI-613 (n=67)—included hemoglobin decrease (67%), hyperglycemia (67%), neutropenia (67%), thrombocytopenia (67%), hypomagnesemia (66%), leukopenia (66%), lymphopenia (66%), hypoalbuminemia (65%), hypokalemia (60%), hypocalcemia (57%), and diarrhea (55%).
All cases of neutropenia, thrombocytopenia, leukopenia, and lymphopenia were grade 3/4. Other common grade 3/4 AEs (occurring in at least 20% of patients) included hemoglobin decrease (62%), febrile neutropenia (28%), hypophosphatemia (24%), and hypokalemia (23%).
The only grade 5 AE was hypotension.
The mortality rate was 12% (n=8) at 30 days and 19% (n=13) at 60 days. The researchers said this was similar to the historical experience with high-dose cytarabine, mitoxantrone, and asparaginase. Mortality rates with this regimen were 13% at 30 days and 22% at 60 days.
Efficacy
Sixty-two patients were evaluable for response. Of the 5 patients who were not evaluable, 1 didn’t complete the first cycle of treatment, 1 was the CML patient, and 3 died before assessment.
The overall response rate was 50% (31/62). This included 26 patients with a complete response (CR) and 5 patients who had a CR with incomplete count recovery (CRi).
The rate of CR/CRi was 47% (15/32) in patients older than 60 years of age, 46% (11/24) in patients who had poor-risk cytogenetics, and 53% (8/15) when CPI-613 was given at the recommended phase 2 dose—2000 mg/m2.
The median overall survival (OS) was 6.7 months for all evaluable patients and 13.2 months for patients who achieved a CR/CRi.
The median OS was 6.9 months for patients age 60 and older, which was not significantly different from the median OS in younger patients (P=0.9642).
This study was sponsored by Wake Forest University Health Sciences and the National Cancer Institute.
Team modifies platelets to improve coagulation
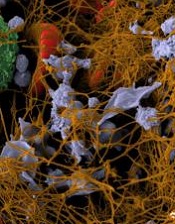
Researchers say they have found a way to improve the coagulability of platelets—by loading them with thrombin encapsulated in liposomes.
Platelets modified in this way decreased clotting time and enhanced clot strength in blood samples from healthy subjects.
The platelets also reduced clotting time in samples from patients with hemophilia A and decreased thrombin generation time in samples from patients with trauma-induced coagulopathy (TIC).
“Coagulation, which depends on a series of complex biochemical reactions, works great for scrapes and paper cuts,” said study author Christian Kastrup, PhD, of the University of British Columbia in Vancouver, Canada.
“But trauma often overwhelms this intricate, delicate process. We wanted to make it more resilient.”
Dr Kastrup and his colleagues described this endeavor in the Journal of Thrombosis and Haemostasis.
The researchers inserted thrombin into nanoliposomes and mixed them with platelets. After the platelets absorbed the nanoparticles, the team immersed the cells in blood samples for testing.
In platelet-rich plasma (PRP), the clot initiation time was about 26% faster with the modified platelets than with normal platelets—8.8 ± 1.7 min and 11.9 ± 2.5 min, respectively. And the clot strength was about 16% greater—13.4 ± 1.7 kdyne cm-1 and 11.5 ± 1.6 kdyne cm-1, respectively.
In whole blood, the clot initiation time was about 13% faster with modified platelets than with normal platelets—11.2 ± 1.2 min and 12.9 ± 1.2 min, respectively. And the clot strength was about 22% greater—5.0 ± 0.6 kdyne cm-1 and 4.1 ± 0.4 kdyne cm-1, respectively.
The researchers also found the modified platelets offset the effect of acidosis. Acidifying PRP increased thrombin generation time by 1.0 (± 0.4) min, but the modified platelets decreased thrombin generation time by 1.6 (± 0.2) min.
The modified platelets lessened the effects of antiplatelet agents as well. Aspirin or naproxen slowed thrombin generation time by 1.3 (± 0.5) min.
However, the modified platelets generated thrombin 0.4 (± 0.4) min faster in the presence of aspirin and 0.6 (± 0.4) min faster in the presence of naproxen, compared to normal platelets.
The researchers also tested the modified platelets in samples from patients with hemophilia A. Normal PRP had a clot initiation time of 5.3 (± 0.5) min, and clotting time for hemophilia A PRP was 33% slower.
In the presence of the modified platelets, clotting time was 17% slower in hemophilia A PRP than in normal PRP.
Finally, the researchers tested the modified platelets in plasma from 2 patients with TIC. With normal platelets, thrombin generation times were 5.0 (± 0.1) min and 6.7 (± 0.2) min.
With the modified platelets, thrombin generation times decreased to 3.9 (± 0.1) min and 5.9 (± 0.1) min, respectively.
If these modified platelets prove effective in subsequent preclinical and clinical testing, Dr Kastrup envisions that trauma centers could have bags of plasma containing modified platelets on hand for patients with severe bleeding. The modification could conceivably be done at the time donated blood is processed.
“Trauma is the leading killer of people under 45 years old, and blood loss is the second most common cause of such deaths,” Dr Kastrup noted. “By tweaking the body’s own clotting mechanism, we might be able to make trauma more survivable.”
Researchers say they have found a way to improve the coagulability of platelets—by loading them with thrombin encapsulated in liposomes.
Platelets modified in this way decreased clotting time and enhanced clot strength in blood samples from healthy subjects.
The platelets also reduced clotting time in samples from patients with hemophilia A and decreased thrombin generation time in samples from patients with trauma-induced coagulopathy (TIC).
“Coagulation, which depends on a series of complex biochemical reactions, works great for scrapes and paper cuts,” said study author Christian Kastrup, PhD, of the University of British Columbia in Vancouver, Canada.
“But trauma often overwhelms this intricate, delicate process. We wanted to make it more resilient.”
Dr Kastrup and his colleagues described this endeavor in the Journal of Thrombosis and Haemostasis.
The researchers inserted thrombin into nanoliposomes and mixed them with platelets. After the platelets absorbed the nanoparticles, the team immersed the cells in blood samples for testing.
In platelet-rich plasma (PRP), the clot initiation time was about 26% faster with the modified platelets than with normal platelets—8.8 ± 1.7 min and 11.9 ± 2.5 min, respectively. And the clot strength was about 16% greater—13.4 ± 1.7 kdyne cm-1 and 11.5 ± 1.6 kdyne cm-1, respectively.
In whole blood, the clot initiation time was about 13% faster with modified platelets than with normal platelets—11.2 ± 1.2 min and 12.9 ± 1.2 min, respectively. And the clot strength was about 22% greater—5.0 ± 0.6 kdyne cm-1 and 4.1 ± 0.4 kdyne cm-1, respectively.
The researchers also found the modified platelets offset the effect of acidosis. Acidifying PRP increased thrombin generation time by 1.0 (± 0.4) min, but the modified platelets decreased thrombin generation time by 1.6 (± 0.2) min.
The modified platelets lessened the effects of antiplatelet agents as well. Aspirin or naproxen slowed thrombin generation time by 1.3 (± 0.5) min.
However, the modified platelets generated thrombin 0.4 (± 0.4) min faster in the presence of aspirin and 0.6 (± 0.4) min faster in the presence of naproxen, compared to normal platelets.
The researchers also tested the modified platelets in samples from patients with hemophilia A. Normal PRP had a clot initiation time of 5.3 (± 0.5) min, and clotting time for hemophilia A PRP was 33% slower.
In the presence of the modified platelets, clotting time was 17% slower in hemophilia A PRP than in normal PRP.
Finally, the researchers tested the modified platelets in plasma from 2 patients with TIC. With normal platelets, thrombin generation times were 5.0 (± 0.1) min and 6.7 (± 0.2) min.
With the modified platelets, thrombin generation times decreased to 3.9 (± 0.1) min and 5.9 (± 0.1) min, respectively.
If these modified platelets prove effective in subsequent preclinical and clinical testing, Dr Kastrup envisions that trauma centers could have bags of plasma containing modified platelets on hand for patients with severe bleeding. The modification could conceivably be done at the time donated blood is processed.
“Trauma is the leading killer of people under 45 years old, and blood loss is the second most common cause of such deaths,” Dr Kastrup noted. “By tweaking the body’s own clotting mechanism, we might be able to make trauma more survivable.”
Researchers say they have found a way to improve the coagulability of platelets—by loading them with thrombin encapsulated in liposomes.
Platelets modified in this way decreased clotting time and enhanced clot strength in blood samples from healthy subjects.
The platelets also reduced clotting time in samples from patients with hemophilia A and decreased thrombin generation time in samples from patients with trauma-induced coagulopathy (TIC).
“Coagulation, which depends on a series of complex biochemical reactions, works great for scrapes and paper cuts,” said study author Christian Kastrup, PhD, of the University of British Columbia in Vancouver, Canada.
“But trauma often overwhelms this intricate, delicate process. We wanted to make it more resilient.”
Dr Kastrup and his colleagues described this endeavor in the Journal of Thrombosis and Haemostasis.
The researchers inserted thrombin into nanoliposomes and mixed them with platelets. After the platelets absorbed the nanoparticles, the team immersed the cells in blood samples for testing.
In platelet-rich plasma (PRP), the clot initiation time was about 26% faster with the modified platelets than with normal platelets—8.8 ± 1.7 min and 11.9 ± 2.5 min, respectively. And the clot strength was about 16% greater—13.4 ± 1.7 kdyne cm-1 and 11.5 ± 1.6 kdyne cm-1, respectively.
In whole blood, the clot initiation time was about 13% faster with modified platelets than with normal platelets—11.2 ± 1.2 min and 12.9 ± 1.2 min, respectively. And the clot strength was about 22% greater—5.0 ± 0.6 kdyne cm-1 and 4.1 ± 0.4 kdyne cm-1, respectively.
The researchers also found the modified platelets offset the effect of acidosis. Acidifying PRP increased thrombin generation time by 1.0 (± 0.4) min, but the modified platelets decreased thrombin generation time by 1.6 (± 0.2) min.
The modified platelets lessened the effects of antiplatelet agents as well. Aspirin or naproxen slowed thrombin generation time by 1.3 (± 0.5) min.
However, the modified platelets generated thrombin 0.4 (± 0.4) min faster in the presence of aspirin and 0.6 (± 0.4) min faster in the presence of naproxen, compared to normal platelets.
The researchers also tested the modified platelets in samples from patients with hemophilia A. Normal PRP had a clot initiation time of 5.3 (± 0.5) min, and clotting time for hemophilia A PRP was 33% slower.
In the presence of the modified platelets, clotting time was 17% slower in hemophilia A PRP than in normal PRP.
Finally, the researchers tested the modified platelets in plasma from 2 patients with TIC. With normal platelets, thrombin generation times were 5.0 (± 0.1) min and 6.7 (± 0.2) min.
With the modified platelets, thrombin generation times decreased to 3.9 (± 0.1) min and 5.9 (± 0.1) min, respectively.
If these modified platelets prove effective in subsequent preclinical and clinical testing, Dr Kastrup envisions that trauma centers could have bags of plasma containing modified platelets on hand for patients with severe bleeding. The modification could conceivably be done at the time donated blood is processed.
“Trauma is the leading killer of people under 45 years old, and blood loss is the second most common cause of such deaths,” Dr Kastrup noted. “By tweaking the body’s own clotting mechanism, we might be able to make trauma more survivable.”
FDA lifts hold on trials of BPX-501
The US Food and Drug Administration (FDA) has lifted the clinical hold on studies of BPX-501 in the US.
BPX-501 is an adjunct T-cell therapy intended to be given after allogeneic hematopoietic stem cell transplant (HSCT).
The product is being tested in phase 1/2 trials of children and adults with hematologic disorders.
The FDA placed all US trials of BPX-501 on hold earlier this year due to 3 cases of encephalopathy that were considered possibly related to BPX-501.
The hold did not affect a registrational trial of BPX-501—known as BP-004—that is underway in Europe.
Now, the FDA has lifted the hold on the US trials after discussions with Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
Bellicum agreed to amend study protocols to include guidance on monitoring and management of neurologic adverse events. The company said it will be working with US clinical sites to resume patient recruitment in BPX-501 trials based on the amended protocols.
About BPX-501
BPX-501 is intended to speed immune reconstitution, fight infection, and enhance the graft-versus-leukemia effect of HSCT while also minimizing graft-vs-host disease (GVHD) and other T-cell-mediated transplant complications.
BPX-501 contains a safety switch, CaspaCIDe®, that can be activated with the drug rimiducid. The drug can be used to eliminate alloreactive BPX-501 T cells if uncontrollable GVHD or other T-cell-mediated complications occur.
The FDA and the European Commission granted BPX-501 and rimiducid orphan drug designation for use in HSCT recipients.
BPX-501 and rimiducid have demonstrated efficacy in the BP-004 trial. Results from this trial have been presented at the EBMT Annual Meeting in 2016 (abstract WP16 and abstract O007), the 2017 EHA Congress, and the 2017 ASH Annual Meeting.
The US Food and Drug Administration (FDA) has lifted the clinical hold on studies of BPX-501 in the US.
BPX-501 is an adjunct T-cell therapy intended to be given after allogeneic hematopoietic stem cell transplant (HSCT).
The product is being tested in phase 1/2 trials of children and adults with hematologic disorders.
The FDA placed all US trials of BPX-501 on hold earlier this year due to 3 cases of encephalopathy that were considered possibly related to BPX-501.
The hold did not affect a registrational trial of BPX-501—known as BP-004—that is underway in Europe.
Now, the FDA has lifted the hold on the US trials after discussions with Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
Bellicum agreed to amend study protocols to include guidance on monitoring and management of neurologic adverse events. The company said it will be working with US clinical sites to resume patient recruitment in BPX-501 trials based on the amended protocols.
About BPX-501
BPX-501 is intended to speed immune reconstitution, fight infection, and enhance the graft-versus-leukemia effect of HSCT while also minimizing graft-vs-host disease (GVHD) and other T-cell-mediated transplant complications.
BPX-501 contains a safety switch, CaspaCIDe®, that can be activated with the drug rimiducid. The drug can be used to eliminate alloreactive BPX-501 T cells if uncontrollable GVHD or other T-cell-mediated complications occur.
The FDA and the European Commission granted BPX-501 and rimiducid orphan drug designation for use in HSCT recipients.
BPX-501 and rimiducid have demonstrated efficacy in the BP-004 trial. Results from this trial have been presented at the EBMT Annual Meeting in 2016 (abstract WP16 and abstract O007), the 2017 EHA Congress, and the 2017 ASH Annual Meeting.
The US Food and Drug Administration (FDA) has lifted the clinical hold on studies of BPX-501 in the US.
BPX-501 is an adjunct T-cell therapy intended to be given after allogeneic hematopoietic stem cell transplant (HSCT).
The product is being tested in phase 1/2 trials of children and adults with hematologic disorders.
The FDA placed all US trials of BPX-501 on hold earlier this year due to 3 cases of encephalopathy that were considered possibly related to BPX-501.
The hold did not affect a registrational trial of BPX-501—known as BP-004—that is underway in Europe.
Now, the FDA has lifted the hold on the US trials after discussions with Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
Bellicum agreed to amend study protocols to include guidance on monitoring and management of neurologic adverse events. The company said it will be working with US clinical sites to resume patient recruitment in BPX-501 trials based on the amended protocols.
About BPX-501
BPX-501 is intended to speed immune reconstitution, fight infection, and enhance the graft-versus-leukemia effect of HSCT while also minimizing graft-vs-host disease (GVHD) and other T-cell-mediated transplant complications.
BPX-501 contains a safety switch, CaspaCIDe®, that can be activated with the drug rimiducid. The drug can be used to eliminate alloreactive BPX-501 T cells if uncontrollable GVHD or other T-cell-mediated complications occur.
The FDA and the European Commission granted BPX-501 and rimiducid orphan drug designation for use in HSCT recipients.
BPX-501 and rimiducid have demonstrated efficacy in the BP-004 trial. Results from this trial have been presented at the EBMT Annual Meeting in 2016 (abstract WP16 and abstract O007), the 2017 EHA Congress, and the 2017 ASH Annual Meeting.