User login
Investigational treatments for cognitive impairment in schizophrenia
Available treatments for schizophrenia (eg, antipsychotics) are primarily effective on positive symptoms (hallucinations, delusions, etc.). It is, however, increasingly clear that schizophrenia also is a severe neuropsychiatric illness associated with deficits in cognitive function. These deficits represent a core feature of the disorder, and are a major determinant of long-term disability.1 Cognitive dysfunction is among the earliest signs of illness that, typically, presents in the prodromal phase.
Since the formulation of the dopaminergic model of schizophrenia, cognitive studies of the disease primarily have examined dysfunction in dopaminergic-rich regions of the brain, such as the prefrontal cortex, and, therefore, have focused largely on executive functioning. But neurocognitive deficits in schizophrenia are not limited to executive functioning; comparable deficits have been observed across multiple areas of cognition.2
More recent formulations of cognitive dysfunction in schizophrenia divide deficits into multiple domains. These include verbal, visual, and working memory; attention and vigilance; speed of processing, reasoning, and problem solving; and social cognition (Table). Neurocognitive impairments often are closely associated with deficits in early sensory processing and basic neurophysiology.3

The prevalence of cognitive dysfunction also can be estimated using baseline data from the large-scale Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) trial.4 Although cognitive dysfunction was not one of the inclusion criteria in CATIE, most patients who were enrolled had profound cognitive deficits.5 Furthermore, meta-analyses6 suggest that composite neurocognitive measures can explain as much as 60% of the variance of overall functioning in schizophrenia.
Antipsychotics aren’t the answer
The cognitive-enhancing benefits of antipsychotic medications are minimal.7 As evidence of a direct relationship between cognitive dysfunction and long-term functional outcome in schizophrenia becomes established, the need for safe and effective treatment for these symptoms becomes more urgent. Given the mechanistic complexity of the potential cause of poor cognitive performance, the search for an effective treatment is ongoing—but that search has not been successful.
Despite mixed results for recent novel mechanism trials (http://newsroom.lilly.com/releasedetail.cfm?releaseid=703018) and a number of companies ceasing drug development, the work to develop safe and effective treatments for cognitive dysfunction in schizophrenia continues, as exemplified by National Institute of Mental Health-initiated programs to spur development of drugs that work by a novel mechanism. Rather than simply assessing novel compounds with paper-and-pencil cognitive scales, such programs seek to assess the ability of the compound to engage with the intended receptor (target),9 using imaging or electrophysiological tools. Without utilization of a target engagement biomarker, there is no way to know whether 1) the drug simply does not get into the brain in sufficient concentration to be effective in humans or 2) the overall mechanism is wrong.
In this article, we review several promising targets and techniques that are the subject of active research on the treatment of cognitive disorders in schizophrenia. This list isn’t exhaustive; our aim is to highlight a few of the promising treatments now being studied in clinical trials.
Acetylcholine receptors
Acetylcholine receptors comprise two major families, nicotinic and muscarinic receptors; evidence implicates deficits of both families in schizophrenia.10 Following up on epidemiological studies11 of the high percentage of schizophrenia patients who smoke tobacco (60% to 90%), the role of alpha-7 nicotinic acetylcholine receptors (á7 nAchR) has been explored. Nicotine itself might normalize some disrupted auditory processes, as measured by electroencephalography.12
Several clinical trials of partial á7 nAchR agonists have been conducted, with EVP-6124 and TC-5619 furthest along in development.
EVP-6124. Information is unavailable publicly on EVP-6124, except for an abstract presented in 2011 at the 51st Annual Meeting of the American College of Neuropsychopharmacology.13 In that study, 319 patients with schizophrenia were randomized to EVP-6124 (0.3 mg/d or 1 mg/d [n = 213]) or placebo (n = 106) adjunctive to at least 4 weeks of non-clozapine antipsychotics. Efficacy was shown up to 1 mg, in a dose-responsive manner. Modest, but significant, improvements in cognition, clinical function, and negative symptoms were seen. The most commonly reported side effects were headache (3.8%), nausea (3.2%), and nasopharyngitis (2.5%). Phase III studies are underway.
TC-5619. This partial á7 nAchR also showed positive results recently in a Phase II trial. Significant (P < .05) improvement was demonstrated in executive function in the Groton Maze Learning Task of the CogState Schizophrenia Battery and the Scale for Assessment of Negative Symptoms.14
Strong anatomic links also exist between muscarinic acetylcholine receptors and the brain dopaminergic system, especially muscarinic type-1 and type-4 (M1 and M4) receptors. The potential utility of an M1, M4, or combined M/M4 agonist is also supported by studies of M1 and M4 knockout mice, with particular evidence of cognitive enhancement with the use of M1 agonists.15
GSK1034702. Administration of the M1 allosteric agonist GSK1034702 to healthy human smokers, using the nicotine abstinence model of cognitive dysfunction, resulted in improvements in immediate recall.16
Xanomeline. In a small pilot study of 20 schizophrenia patients, xanomeline, a mixed M1/M4 agonist, demonstrated significant improvements in verbal learning, short-term memory, and overall symptoms.17
Dopamine receptors
All marketed antipsychotics block the dopamine type-2 (D2) receptor18; they are primarily effective on positive symptoms.4 In contrast, a role for the dopamine type-1 (D1) receptor in cognition is suggested by studies that demonstrate reduced D1 and N-methyl-d-aspartate (NMDA) glutamate receptor function in the prefrontal cortex.19-22
In a model of cognitive impairment in non-human primates, low-dose intermittent dosing of D1-receptor agonists produced improvements in cognitive function.23 This strategy aims to sensitize, rather than induce tolerance, to the effects of the D1-receptor agonist. Benefits were primarily seen in working memory. Phase II trials of a potent D1-receptor agonist, DAR-100A, the active enantiomer of dihydrexidine24 are ongoing (www.clinicaltrials.gov/ct2/show/NCT01519557).
Glutamatergic receptors
Intoxication with NMDA antagonists (such as phencyclidine and ketamine) yields a phenotype with similarity to schizophrenia.25 More than 20 years of research has provided evidence for the role of glutamatergic NMDA receptors in the pathophysiology of schizophrenia.26,27
NMDA receptors are distributed widely in the brain, but specific glutamatergic processes are localized to areas that are associated with cognition. This relative distribution provides a convenient framework from which to view the pattern of cognitive dysfunction associated with schizophrenia:
• NMDA receptors in the hippocampus are involved in learning and memory acquisition
• NMDA receptors in the visual cortex and auditory cortex are fundamental for auditory and visual sensory memory.
Previous reviews of ketamine administration have described cognitive deficits in healthy control subjects, comparable to what is seen in schizophrenia.28 The deficits are noted primarily in measures of executive functioning, attention/vigilance, verbal fluency, and visual and verbal working memory.
Most treatment studies of glutamatergic-based drugs have focused on positive and negative symptoms. Two recent comprehensive meta-analyses29,30 of NMDA-based treatments support small-to-moderate effect size improvement in total symptoms and in negative symptoms, in patients with chronic schizophrenia, when the drugs are used in combination with non-clozapine antipsychotics.
Bitopertin. A novel glycine-transport inhibitor, bitopertin, showed significant improvement in negative symptoms as an adjunctive treatment in a large Phase II trial.31,32 In the “per protocol” population (ie, patients who completed 8 weeks of treatment without any major protocol violations [n = 231]), negative symptoms diminished to a significantly (P < .05) greater degree from baseline in the 10 mg/d and 30 mg/d dosage groups, compared with placebo. Phase III studies of bitopertin are ongoing (www.clinicaltrials.gov/ct2/show/NCT01192906).
Direct evidence of a cognitive benefit of glutamatergic-based drugs is limited. In a recent large, multicenter study, low dosage D-serine (~30 mg/kg/d) did not separate from placebo,33 but an open-label study suggests increased efficacy with dosages >30 mg/kg/d.34 In addition to symptomatic improvements, a highly significant, large effect-size improvement was seen for overall cognition for dosages ≥60 mg/kg/d, leading to a significant dose-by-time interaction (P < .01).
Combination approaches. The value of combining glutamatergic medication and a cognitive training program is supported by the role of NMDA receptors in learning. For example, D-cycloserine, a glycine-site partial agonist, has been shown in several studies to enhance learning and behavioral therapies in anxiety disorders.35 Although an initial study in schizophrenia was negative for the effectiveness of D-serine (a glycine-site full agonist) and combined cognitive training,36 further research is ongoing to evaluate a role for such combined therapy.37,38
Brain stimulation
Two nonpharmacotherapeutic brain stimulation techniques, repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS), have been applied in the study of schizophrenia symptoms, particularly for enhancing cognition.39 Both techniques use electric stimulation to influence activity of underlying brain regions: rTMS utilizes a magnetic coil and electromagnetic induction; tDCS, in contrast, utilizes constant low (<2 mA) direct current to specific regions of the scalp.
Cortical neuronal excitability is increased by anodal tDCS and high-frequency rTMS and reduced by cathodal tDCS and low-frequency rTMS. Both tDCS and rTMS appear to be NMDA receptor-dependent. tDCS is relatively inexpensive and requires less expertise to administer than rTMS does.
Both techniques might be efficacious for treating resistant auditory hallucinations.40,41 Applying rTMS over the left dorsolateral prefrontal cortex has led to improvement in verbal learning and visuomotor tracking in patients with schizophrenia.39 Stimulation of both sides of the prefrontal cortex with rTMS has brought improvement in visual memory, executive function, spatial working memory, and attention. Few papers have been published so far regarding enhancement of cognition with tDCS in schizophrenia,42 but beneficial effects of this technique have been seen across several disorders.43
Cognitive remediation techniques
A fundamental starting point for cognitive remediation is the idea that there is plasticity in the brain and that repetitive practice can lead to cognitive improvement. Cognitive remediation therapy often adopts computerized programs and exercises that attempt to improve psychosocial function by targeting structures of the brain that are involved in cognitive function, such as attention, working memory, executive functioning, planning, and cognitive flexibility.
In schizophrenia, cognitive remediation studies have traditionally targeted higher-order processes, such as attention and higher level processes, that might lead to improvement in overall cognition and function.44 Cognitive remediation typically is utilized complementary to pharmacotherapy, with some studies supporting the use of combined use of cognition-enhancing drugs and remediation programs.
A 2007 meta-analysis showed a medium-size but significant improvement in cognition through the use of cognitive remediation therapy45—especially when it is combined with psychiatric rehabilitation. More recent studies utilizing techniques that focus on bottom-up (auditory and visual processing) techniques has shown significant improvements.46-48 Several multicenter studies utilizing Posit Science programs combined with antipsychotic medication are ongoing (www.clinicaltrials.gov/ct2/show/NCT01173874 and www.clinicaltrials.gov/ct2/show/NCT01422902).
Bottom Line
Although cognitive dysfunction is a leading cause of disability in schizophrenia, no treatments are approved for this condition. Numerous novel-mechanism and nonpharmaceutical modalities are actively being studied for this difficult-to-treat problem, however—offering hope to patients.
Related Resources
Javitt DC, Zukin SR, Heresco-Levy U, et al. Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Has an angel shown the way? Schizophr Bull. 2012; 38(5):958-966.
Keefe RS, Harvey PD. Cognitive impairment in schizophrenia. Handb Exp Pharmacol. 2012;(213):11-37.
Millan MJ, Agid Y, Brune M, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012; 11(2):141-168.
Drug Brand Names
D-cycloserine • Seromycin Ketamine • Ketalar
Xanomeline • Lumeron, Memcor
Disclosures
Dr. Kantrowitz receives grant or research support from EnVivo, the National Institute of Mental Health, Novartis, Pfizer, Roche-Genentech, the Stanley Foundation, and Sunovion; is a consultant to Health Advances, LLC, the Healthcare Advisory Board, Otsuka Pharmaceuticals, Strategic Edge Communications, and Vindico Medical Education; and owns a small number of shares of common stock in GlaxoSmithKline. Ms. Levy and Dr. Ballon report no financial relationships with manufacturers of any products mentioned in this article or with manufacturers of competing products.
1. Bowie CR, Reichenberg A, Patterson TL, et al. Determinants of real-world functional performance in schizophrenia subjects: correlations with cognition, functional capacity, and symptoms. Am J Psychiatry. 2006;163(3):418-425.
2. Kern RS, Gold JM, Dickinson D, et al. The MCCB impairment profile for schizophrenia outpatients: results from the MATRICS psychometric and standardization study. Schizophr Res. 2011;126(1-3):124-131.
3. Javitt DC, Spencer KM, Thaker GK, et al. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov. 2008;7(1):68-83.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1253.
5. Keefe RS, Bilder RM, Harvey PD, et al. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31(9):2033-2046.
6. Green MF, Kern RS, Braff DL, et al. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26(1):119-136.
7. Keefe RS, Bilder RM, Davis SM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64(6):633-647.
8. Yan J. NIMH tries to jumpstart drug innovations. Psychiatric News. 2013;48(1):8-10.
9. Javitt DC, Schoepp D, Kalivas PW, et al. Translating glutamate: from pathophysiology to treatment. Sci Transl Med. 2011;3(102):102mr2.
10. Foster DJ, Jones CK, Conn PJ. Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov Med. 2012;14(79):413-420.
11. D’Souza MS, Markou A. Schizophrenia and tobacco smoking comorbidity: nAChR agonists in the treatment of schizophrenia-associated cognitive deficits. Neuropharmacology. 2012;62(3):1564-1573.
12. Adler LE, Olincy A, Waldo M, et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998; 24(2):189-202.
13. Meltzer HY, Gawryl M, Ward S, et al. EVP-6124, an alpha-7 nicotinic partial agonist, reduces positive effects on cognition, clinical function, and negative symptoms in patients with chronic schizophrenia on stable antipsychotic therapy. Neuropsychopharmacology. 2011;36:S170-S171.
14. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
15. Digby GJ, Noetzel MJ, Bubser M, et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32(25):8532-8544.
16. Nathan PJ, Watson J, Lund J, et al. The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol. 2013;16(4):721-731.
17. Shekhar A, Potter WZ, Lightfoot J, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165(8):1033-1039.
18. Di Forti M, Lappin LM, Murray RM. Risk factors for schizophrenia—all roads lead to dopamine. Eur Neuropsychopharmacol. 2007;17(suppl 2):S101-S107.
19. Krystal JH, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl). 2003;169(3-4): 215-233.
20. Abi-Dargham A, Moore H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist. 2003;9(5):404-416.
21. Abi-Dargham A, Mawlawi O, Lombardo I, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22(9):3708-3719.
22. Martinez A, Ramanathan DS, Foxe JJ, et al. The role of spatial attention in the selection of real and illusory objects. J Neurosci. 2007;27(30):7963-7973.
23. Castner SA, Williams GV, Goldman-Rakic PS. Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science. 2000;287(5460):2020-2022.
24. Slifstein M, Suckow RF, Javitch JA, et al. Characterization of in vivo pharmacokinetic properties of the dopamine D1 receptor agonist DAR-0100A in nonhuman primates using PET with [11C] NNC112 and [11C] raclopride. J Cereb Blood Flow Metab. 2011;31(1):293-304.
25. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
26. Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4):108-121.
27. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4(3):189-200.
28. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Javitt DC, Kantrowitz JT, eds. Handbook of neurochemistry and molecular neurobiology. New York, NY: Springer; 2009:3-36.
29. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
30. Singh SP, Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs. 2011;25(10):859-868.
31. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropharmacology. 2010;35:S320-S321.
32. Pinard E, Alanine A, Alberati D, et al. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-(( S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J Med Chem. 2010;53(12):4603-4614.
33. Weiser M, Heresco-Levy U, Davidson M, et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry. 2012;73(6):e728-e734.
34. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
35. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
36. D’Souza DC, Radhakrishnan R, Perry E, et al. Feasibility, safety, and efficacy of the combination of D-serine and computerized cognitive retraining in schizophrenia: an international collaborative pilot study. Neuropsychopharmacology. 2013;38(3):492-503.
37. Gottlieb JD, Cather C, Shanahan M, et al. D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr Res. 2011;131(1-3):69-74.
38. Kantrowitz J, Sehatpour P, Oakman E, et al. D-Serine and NMDA based sensory modulation. Poster presented at: 3rd Biennial Schizophrenia International Research Conference; April 14-18, 2012; Florence, Italy.
39. Demirtas-Tatlidede, A, Vahabzadeh-Hagh AM, Pascual-Leone A. Can noninvasive brain stimulation enhance cognition in neuropsychiatric disorders? Neuropharmacology. 2013;64:566-578.
40. Brunelin J, Mondino M, Gassab L, et al. Examining transcranial direct-current stimulation (tDCS) as a treatment for hallucinations in schizophrenia. Am J Psychiatry. 2012;169(7):719-724.
41. Matheson SL, Green MJ, Loo C, et al. Quality assessment and comparison of evidence for electroconvulsive therapy and repetitive transcranial magnetic stimulation for schizophrenia: a systematic meta-review. Schizophr Res. 2012;118(1-3):201-210.
42. Vercammen A, Rushby JA, Loo C, et al. Transcranial direct current stimulation influences probabilistic association learning in schizophrenia. Schizophr Res. 2011;131(1-3):198-205.
43. Nitsche MA, Paulus W. Transcranial direct current stimulation--update 2011. Restor Neurol Neurosci. 2011; 29(6):463-492.
44. Keefe RS, Vinogradov S, Medalia A, et al. Report from the working group conference on multisite trial design for cognitive remediation in schizophrenia. Schizophr Bull. 2011;37(5):1057-1065.
45. McGurk SR, Twamley EW, Sitzer DI, et al. A meta-analysis of cognitive remediation in schizophrenia. Am J Psychiatry. 2007;164(12):1791-1802.
46. Fisher M, Holland C, Merzenich MM, et al. Using neuroplasticity-based auditory training to improve verbal memory in schizophrenia. Am J Psychiatry. 2009;166(7):805-811.
47. Norton DJ, McBain RK, Ongür D, et al. Perceptual training strongly improves visual motion perception in schizophrenia. Brain Cogn. 2011;77(2):248-256.
48. Kantrowitz JT, Revheim N, Pasternak R, et al. It’s all in the cards: effect of stimulus manipulation on Wisconsin Card Sorting Test performance in schizophrenia. Psychiatry Res. 2009;168(3):198-204.
Available treatments for schizophrenia (eg, antipsychotics) are primarily effective on positive symptoms (hallucinations, delusions, etc.). It is, however, increasingly clear that schizophrenia also is a severe neuropsychiatric illness associated with deficits in cognitive function. These deficits represent a core feature of the disorder, and are a major determinant of long-term disability.1 Cognitive dysfunction is among the earliest signs of illness that, typically, presents in the prodromal phase.
Since the formulation of the dopaminergic model of schizophrenia, cognitive studies of the disease primarily have examined dysfunction in dopaminergic-rich regions of the brain, such as the prefrontal cortex, and, therefore, have focused largely on executive functioning. But neurocognitive deficits in schizophrenia are not limited to executive functioning; comparable deficits have been observed across multiple areas of cognition.2
More recent formulations of cognitive dysfunction in schizophrenia divide deficits into multiple domains. These include verbal, visual, and working memory; attention and vigilance; speed of processing, reasoning, and problem solving; and social cognition (Table). Neurocognitive impairments often are closely associated with deficits in early sensory processing and basic neurophysiology.3
The prevalence of cognitive dysfunction also can be estimated using baseline data from the large-scale Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) trial.4 Although cognitive dysfunction was not one of the inclusion criteria in CATIE, most patients who were enrolled had profound cognitive deficits.5 Furthermore, meta-analyses6 suggest that composite neurocognitive measures can explain as much as 60% of the variance of overall functioning in schizophrenia.
Antipsychotics aren’t the answer
The cognitive-enhancing benefits of antipsychotic medications are minimal.7 As evidence of a direct relationship between cognitive dysfunction and long-term functional outcome in schizophrenia becomes established, the need for safe and effective treatment for these symptoms becomes more urgent. Given the mechanistic complexity of the potential cause of poor cognitive performance, the search for an effective treatment is ongoing—but that search has not been successful.
Despite mixed results for recent novel mechanism trials (http://newsroom.lilly.com/releasedetail.cfm?releaseid=703018) and a number of companies ceasing drug development, the work to develop safe and effective treatments for cognitive dysfunction in schizophrenia continues, as exemplified by National Institute of Mental Health-initiated programs to spur development of drugs that work by a novel mechanism. Rather than simply assessing novel compounds with paper-and-pencil cognitive scales, such programs seek to assess the ability of the compound to engage with the intended receptor (target),9 using imaging or electrophysiological tools. Without utilization of a target engagement biomarker, there is no way to know whether 1) the drug simply does not get into the brain in sufficient concentration to be effective in humans or 2) the overall mechanism is wrong.
In this article, we review several promising targets and techniques that are the subject of active research on the treatment of cognitive disorders in schizophrenia. This list isn’t exhaustive; our aim is to highlight a few of the promising treatments now being studied in clinical trials.
Acetylcholine receptors
Acetylcholine receptors comprise two major families, nicotinic and muscarinic receptors; evidence implicates deficits of both families in schizophrenia.10 Following up on epidemiological studies11 of the high percentage of schizophrenia patients who smoke tobacco (60% to 90%), the role of alpha-7 nicotinic acetylcholine receptors (á7 nAchR) has been explored. Nicotine itself might normalize some disrupted auditory processes, as measured by electroencephalography.12
Several clinical trials of partial á7 nAchR agonists have been conducted, with EVP-6124 and TC-5619 furthest along in development.
EVP-6124. Information is unavailable publicly on EVP-6124, except for an abstract presented in 2011 at the 51st Annual Meeting of the American College of Neuropsychopharmacology.13 In that study, 319 patients with schizophrenia were randomized to EVP-6124 (0.3 mg/d or 1 mg/d [n = 213]) or placebo (n = 106) adjunctive to at least 4 weeks of non-clozapine antipsychotics. Efficacy was shown up to 1 mg, in a dose-responsive manner. Modest, but significant, improvements in cognition, clinical function, and negative symptoms were seen. The most commonly reported side effects were headache (3.8%), nausea (3.2%), and nasopharyngitis (2.5%). Phase III studies are underway.
TC-5619. This partial á7 nAchR also showed positive results recently in a Phase II trial. Significant (P < .05) improvement was demonstrated in executive function in the Groton Maze Learning Task of the CogState Schizophrenia Battery and the Scale for Assessment of Negative Symptoms.14
Strong anatomic links also exist between muscarinic acetylcholine receptors and the brain dopaminergic system, especially muscarinic type-1 and type-4 (M1 and M4) receptors. The potential utility of an M1, M4, or combined M/M4 agonist is also supported by studies of M1 and M4 knockout mice, with particular evidence of cognitive enhancement with the use of M1 agonists.15
GSK1034702. Administration of the M1 allosteric agonist GSK1034702 to healthy human smokers, using the nicotine abstinence model of cognitive dysfunction, resulted in improvements in immediate recall.16
Xanomeline. In a small pilot study of 20 schizophrenia patients, xanomeline, a mixed M1/M4 agonist, demonstrated significant improvements in verbal learning, short-term memory, and overall symptoms.17
Dopamine receptors
All marketed antipsychotics block the dopamine type-2 (D2) receptor18; they are primarily effective on positive symptoms.4 In contrast, a role for the dopamine type-1 (D1) receptor in cognition is suggested by studies that demonstrate reduced D1 and N-methyl-d-aspartate (NMDA) glutamate receptor function in the prefrontal cortex.19-22
In a model of cognitive impairment in non-human primates, low-dose intermittent dosing of D1-receptor agonists produced improvements in cognitive function.23 This strategy aims to sensitize, rather than induce tolerance, to the effects of the D1-receptor agonist. Benefits were primarily seen in working memory. Phase II trials of a potent D1-receptor agonist, DAR-100A, the active enantiomer of dihydrexidine24 are ongoing (www.clinicaltrials.gov/ct2/show/NCT01519557).
Glutamatergic receptors
Intoxication with NMDA antagonists (such as phencyclidine and ketamine) yields a phenotype with similarity to schizophrenia.25 More than 20 years of research has provided evidence for the role of glutamatergic NMDA receptors in the pathophysiology of schizophrenia.26,27
NMDA receptors are distributed widely in the brain, but specific glutamatergic processes are localized to areas that are associated with cognition. This relative distribution provides a convenient framework from which to view the pattern of cognitive dysfunction associated with schizophrenia:
• NMDA receptors in the hippocampus are involved in learning and memory acquisition
• NMDA receptors in the visual cortex and auditory cortex are fundamental for auditory and visual sensory memory.
Previous reviews of ketamine administration have described cognitive deficits in healthy control subjects, comparable to what is seen in schizophrenia.28 The deficits are noted primarily in measures of executive functioning, attention/vigilance, verbal fluency, and visual and verbal working memory.
Most treatment studies of glutamatergic-based drugs have focused on positive and negative symptoms. Two recent comprehensive meta-analyses29,30 of NMDA-based treatments support small-to-moderate effect size improvement in total symptoms and in negative symptoms, in patients with chronic schizophrenia, when the drugs are used in combination with non-clozapine antipsychotics.
Bitopertin. A novel glycine-transport inhibitor, bitopertin, showed significant improvement in negative symptoms as an adjunctive treatment in a large Phase II trial.31,32 In the “per protocol” population (ie, patients who completed 8 weeks of treatment without any major protocol violations [n = 231]), negative symptoms diminished to a significantly (P < .05) greater degree from baseline in the 10 mg/d and 30 mg/d dosage groups, compared with placebo. Phase III studies of bitopertin are ongoing (www.clinicaltrials.gov/ct2/show/NCT01192906).
Direct evidence of a cognitive benefit of glutamatergic-based drugs is limited. In a recent large, multicenter study, low dosage D-serine (~30 mg/kg/d) did not separate from placebo,33 but an open-label study suggests increased efficacy with dosages >30 mg/kg/d.34 In addition to symptomatic improvements, a highly significant, large effect-size improvement was seen for overall cognition for dosages ≥60 mg/kg/d, leading to a significant dose-by-time interaction (P < .01).
Combination approaches. The value of combining glutamatergic medication and a cognitive training program is supported by the role of NMDA receptors in learning. For example, D-cycloserine, a glycine-site partial agonist, has been shown in several studies to enhance learning and behavioral therapies in anxiety disorders.35 Although an initial study in schizophrenia was negative for the effectiveness of D-serine (a glycine-site full agonist) and combined cognitive training,36 further research is ongoing to evaluate a role for such combined therapy.37,38
Brain stimulation
Two nonpharmacotherapeutic brain stimulation techniques, repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS), have been applied in the study of schizophrenia symptoms, particularly for enhancing cognition.39 Both techniques use electric stimulation to influence activity of underlying brain regions: rTMS utilizes a magnetic coil and electromagnetic induction; tDCS, in contrast, utilizes constant low (<2 mA) direct current to specific regions of the scalp.
Cortical neuronal excitability is increased by anodal tDCS and high-frequency rTMS and reduced by cathodal tDCS and low-frequency rTMS. Both tDCS and rTMS appear to be NMDA receptor-dependent. tDCS is relatively inexpensive and requires less expertise to administer than rTMS does.
Both techniques might be efficacious for treating resistant auditory hallucinations.40,41 Applying rTMS over the left dorsolateral prefrontal cortex has led to improvement in verbal learning and visuomotor tracking in patients with schizophrenia.39 Stimulation of both sides of the prefrontal cortex with rTMS has brought improvement in visual memory, executive function, spatial working memory, and attention. Few papers have been published so far regarding enhancement of cognition with tDCS in schizophrenia,42 but beneficial effects of this technique have been seen across several disorders.43
Cognitive remediation techniques
A fundamental starting point for cognitive remediation is the idea that there is plasticity in the brain and that repetitive practice can lead to cognitive improvement. Cognitive remediation therapy often adopts computerized programs and exercises that attempt to improve psychosocial function by targeting structures of the brain that are involved in cognitive function, such as attention, working memory, executive functioning, planning, and cognitive flexibility.
In schizophrenia, cognitive remediation studies have traditionally targeted higher-order processes, such as attention and higher level processes, that might lead to improvement in overall cognition and function.44 Cognitive remediation typically is utilized complementary to pharmacotherapy, with some studies supporting the use of combined use of cognition-enhancing drugs and remediation programs.
A 2007 meta-analysis showed a medium-size but significant improvement in cognition through the use of cognitive remediation therapy45—especially when it is combined with psychiatric rehabilitation. More recent studies utilizing techniques that focus on bottom-up (auditory and visual processing) techniques has shown significant improvements.46-48 Several multicenter studies utilizing Posit Science programs combined with antipsychotic medication are ongoing (www.clinicaltrials.gov/ct2/show/NCT01173874 and www.clinicaltrials.gov/ct2/show/NCT01422902).
Bottom Line
Although cognitive dysfunction is a leading cause of disability in schizophrenia, no treatments are approved for this condition. Numerous novel-mechanism and nonpharmaceutical modalities are actively being studied for this difficult-to-treat problem, however—offering hope to patients.
Related Resources
Javitt DC, Zukin SR, Heresco-Levy U, et al. Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Has an angel shown the way? Schizophr Bull. 2012; 38(5):958-966.
Keefe RS, Harvey PD. Cognitive impairment in schizophrenia. Handb Exp Pharmacol. 2012;(213):11-37.
Millan MJ, Agid Y, Brune M, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012; 11(2):141-168.
Drug Brand Names
D-cycloserine • Seromycin Ketamine • Ketalar
Xanomeline • Lumeron, Memcor
Disclosures
Dr. Kantrowitz receives grant or research support from EnVivo, the National Institute of Mental Health, Novartis, Pfizer, Roche-Genentech, the Stanley Foundation, and Sunovion; is a consultant to Health Advances, LLC, the Healthcare Advisory Board, Otsuka Pharmaceuticals, Strategic Edge Communications, and Vindico Medical Education; and owns a small number of shares of common stock in GlaxoSmithKline. Ms. Levy and Dr. Ballon report no financial relationships with manufacturers of any products mentioned in this article or with manufacturers of competing products.
Available treatments for schizophrenia (eg, antipsychotics) are primarily effective on positive symptoms (hallucinations, delusions, etc.). It is, however, increasingly clear that schizophrenia also is a severe neuropsychiatric illness associated with deficits in cognitive function. These deficits represent a core feature of the disorder, and are a major determinant of long-term disability.1 Cognitive dysfunction is among the earliest signs of illness that, typically, presents in the prodromal phase.
Since the formulation of the dopaminergic model of schizophrenia, cognitive studies of the disease primarily have examined dysfunction in dopaminergic-rich regions of the brain, such as the prefrontal cortex, and, therefore, have focused largely on executive functioning. But neurocognitive deficits in schizophrenia are not limited to executive functioning; comparable deficits have been observed across multiple areas of cognition.2
More recent formulations of cognitive dysfunction in schizophrenia divide deficits into multiple domains. These include verbal, visual, and working memory; attention and vigilance; speed of processing, reasoning, and problem solving; and social cognition (Table). Neurocognitive impairments often are closely associated with deficits in early sensory processing and basic neurophysiology.3
The prevalence of cognitive dysfunction also can be estimated using baseline data from the large-scale Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) trial.4 Although cognitive dysfunction was not one of the inclusion criteria in CATIE, most patients who were enrolled had profound cognitive deficits.5 Furthermore, meta-analyses6 suggest that composite neurocognitive measures can explain as much as 60% of the variance of overall functioning in schizophrenia.
Antipsychotics aren’t the answer
The cognitive-enhancing benefits of antipsychotic medications are minimal.7 As evidence of a direct relationship between cognitive dysfunction and long-term functional outcome in schizophrenia becomes established, the need for safe and effective treatment for these symptoms becomes more urgent. Given the mechanistic complexity of the potential cause of poor cognitive performance, the search for an effective treatment is ongoing—but that search has not been successful.
Despite mixed results for recent novel mechanism trials (http://newsroom.lilly.com/releasedetail.cfm?releaseid=703018) and a number of companies ceasing drug development, the work to develop safe and effective treatments for cognitive dysfunction in schizophrenia continues, as exemplified by National Institute of Mental Health-initiated programs to spur development of drugs that work by a novel mechanism. Rather than simply assessing novel compounds with paper-and-pencil cognitive scales, such programs seek to assess the ability of the compound to engage with the intended receptor (target),9 using imaging or electrophysiological tools. Without utilization of a target engagement biomarker, there is no way to know whether 1) the drug simply does not get into the brain in sufficient concentration to be effective in humans or 2) the overall mechanism is wrong.
In this article, we review several promising targets and techniques that are the subject of active research on the treatment of cognitive disorders in schizophrenia. This list isn’t exhaustive; our aim is to highlight a few of the promising treatments now being studied in clinical trials.
Acetylcholine receptors
Acetylcholine receptors comprise two major families, nicotinic and muscarinic receptors; evidence implicates deficits of both families in schizophrenia.10 Following up on epidemiological studies11 of the high percentage of schizophrenia patients who smoke tobacco (60% to 90%), the role of alpha-7 nicotinic acetylcholine receptors (á7 nAchR) has been explored. Nicotine itself might normalize some disrupted auditory processes, as measured by electroencephalography.12
Several clinical trials of partial á7 nAchR agonists have been conducted, with EVP-6124 and TC-5619 furthest along in development.
EVP-6124. Information is unavailable publicly on EVP-6124, except for an abstract presented in 2011 at the 51st Annual Meeting of the American College of Neuropsychopharmacology.13 In that study, 319 patients with schizophrenia were randomized to EVP-6124 (0.3 mg/d or 1 mg/d [n = 213]) or placebo (n = 106) adjunctive to at least 4 weeks of non-clozapine antipsychotics. Efficacy was shown up to 1 mg, in a dose-responsive manner. Modest, but significant, improvements in cognition, clinical function, and negative symptoms were seen. The most commonly reported side effects were headache (3.8%), nausea (3.2%), and nasopharyngitis (2.5%). Phase III studies are underway.
TC-5619. This partial á7 nAchR also showed positive results recently in a Phase II trial. Significant (P < .05) improvement was demonstrated in executive function in the Groton Maze Learning Task of the CogState Schizophrenia Battery and the Scale for Assessment of Negative Symptoms.14
Strong anatomic links also exist between muscarinic acetylcholine receptors and the brain dopaminergic system, especially muscarinic type-1 and type-4 (M1 and M4) receptors. The potential utility of an M1, M4, or combined M/M4 agonist is also supported by studies of M1 and M4 knockout mice, with particular evidence of cognitive enhancement with the use of M1 agonists.15
GSK1034702. Administration of the M1 allosteric agonist GSK1034702 to healthy human smokers, using the nicotine abstinence model of cognitive dysfunction, resulted in improvements in immediate recall.16
Xanomeline. In a small pilot study of 20 schizophrenia patients, xanomeline, a mixed M1/M4 agonist, demonstrated significant improvements in verbal learning, short-term memory, and overall symptoms.17
Dopamine receptors
All marketed antipsychotics block the dopamine type-2 (D2) receptor18; they are primarily effective on positive symptoms.4 In contrast, a role for the dopamine type-1 (D1) receptor in cognition is suggested by studies that demonstrate reduced D1 and N-methyl-d-aspartate (NMDA) glutamate receptor function in the prefrontal cortex.19-22
In a model of cognitive impairment in non-human primates, low-dose intermittent dosing of D1-receptor agonists produced improvements in cognitive function.23 This strategy aims to sensitize, rather than induce tolerance, to the effects of the D1-receptor agonist. Benefits were primarily seen in working memory. Phase II trials of a potent D1-receptor agonist, DAR-100A, the active enantiomer of dihydrexidine24 are ongoing (www.clinicaltrials.gov/ct2/show/NCT01519557).
Glutamatergic receptors
Intoxication with NMDA antagonists (such as phencyclidine and ketamine) yields a phenotype with similarity to schizophrenia.25 More than 20 years of research has provided evidence for the role of glutamatergic NMDA receptors in the pathophysiology of schizophrenia.26,27
NMDA receptors are distributed widely in the brain, but specific glutamatergic processes are localized to areas that are associated with cognition. This relative distribution provides a convenient framework from which to view the pattern of cognitive dysfunction associated with schizophrenia:
• NMDA receptors in the hippocampus are involved in learning and memory acquisition
• NMDA receptors in the visual cortex and auditory cortex are fundamental for auditory and visual sensory memory.
Previous reviews of ketamine administration have described cognitive deficits in healthy control subjects, comparable to what is seen in schizophrenia.28 The deficits are noted primarily in measures of executive functioning, attention/vigilance, verbal fluency, and visual and verbal working memory.
Most treatment studies of glutamatergic-based drugs have focused on positive and negative symptoms. Two recent comprehensive meta-analyses29,30 of NMDA-based treatments support small-to-moderate effect size improvement in total symptoms and in negative symptoms, in patients with chronic schizophrenia, when the drugs are used in combination with non-clozapine antipsychotics.
Bitopertin. A novel glycine-transport inhibitor, bitopertin, showed significant improvement in negative symptoms as an adjunctive treatment in a large Phase II trial.31,32 In the “per protocol” population (ie, patients who completed 8 weeks of treatment without any major protocol violations [n = 231]), negative symptoms diminished to a significantly (P < .05) greater degree from baseline in the 10 mg/d and 30 mg/d dosage groups, compared with placebo. Phase III studies of bitopertin are ongoing (www.clinicaltrials.gov/ct2/show/NCT01192906).
Direct evidence of a cognitive benefit of glutamatergic-based drugs is limited. In a recent large, multicenter study, low dosage D-serine (~30 mg/kg/d) did not separate from placebo,33 but an open-label study suggests increased efficacy with dosages >30 mg/kg/d.34 In addition to symptomatic improvements, a highly significant, large effect-size improvement was seen for overall cognition for dosages ≥60 mg/kg/d, leading to a significant dose-by-time interaction (P < .01).
Combination approaches. The value of combining glutamatergic medication and a cognitive training program is supported by the role of NMDA receptors in learning. For example, D-cycloserine, a glycine-site partial agonist, has been shown in several studies to enhance learning and behavioral therapies in anxiety disorders.35 Although an initial study in schizophrenia was negative for the effectiveness of D-serine (a glycine-site full agonist) and combined cognitive training,36 further research is ongoing to evaluate a role for such combined therapy.37,38
Brain stimulation
Two nonpharmacotherapeutic brain stimulation techniques, repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS), have been applied in the study of schizophrenia symptoms, particularly for enhancing cognition.39 Both techniques use electric stimulation to influence activity of underlying brain regions: rTMS utilizes a magnetic coil and electromagnetic induction; tDCS, in contrast, utilizes constant low (<2 mA) direct current to specific regions of the scalp.
Cortical neuronal excitability is increased by anodal tDCS and high-frequency rTMS and reduced by cathodal tDCS and low-frequency rTMS. Both tDCS and rTMS appear to be NMDA receptor-dependent. tDCS is relatively inexpensive and requires less expertise to administer than rTMS does.
Both techniques might be efficacious for treating resistant auditory hallucinations.40,41 Applying rTMS over the left dorsolateral prefrontal cortex has led to improvement in verbal learning and visuomotor tracking in patients with schizophrenia.39 Stimulation of both sides of the prefrontal cortex with rTMS has brought improvement in visual memory, executive function, spatial working memory, and attention. Few papers have been published so far regarding enhancement of cognition with tDCS in schizophrenia,42 but beneficial effects of this technique have been seen across several disorders.43
Cognitive remediation techniques
A fundamental starting point for cognitive remediation is the idea that there is plasticity in the brain and that repetitive practice can lead to cognitive improvement. Cognitive remediation therapy often adopts computerized programs and exercises that attempt to improve psychosocial function by targeting structures of the brain that are involved in cognitive function, such as attention, working memory, executive functioning, planning, and cognitive flexibility.
In schizophrenia, cognitive remediation studies have traditionally targeted higher-order processes, such as attention and higher level processes, that might lead to improvement in overall cognition and function.44 Cognitive remediation typically is utilized complementary to pharmacotherapy, with some studies supporting the use of combined use of cognition-enhancing drugs and remediation programs.
A 2007 meta-analysis showed a medium-size but significant improvement in cognition through the use of cognitive remediation therapy45—especially when it is combined with psychiatric rehabilitation. More recent studies utilizing techniques that focus on bottom-up (auditory and visual processing) techniques has shown significant improvements.46-48 Several multicenter studies utilizing Posit Science programs combined with antipsychotic medication are ongoing (www.clinicaltrials.gov/ct2/show/NCT01173874 and www.clinicaltrials.gov/ct2/show/NCT01422902).
Bottom Line
Although cognitive dysfunction is a leading cause of disability in schizophrenia, no treatments are approved for this condition. Numerous novel-mechanism and nonpharmaceutical modalities are actively being studied for this difficult-to-treat problem, however—offering hope to patients.
Related Resources
Javitt DC, Zukin SR, Heresco-Levy U, et al. Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Has an angel shown the way? Schizophr Bull. 2012; 38(5):958-966.
Keefe RS, Harvey PD. Cognitive impairment in schizophrenia. Handb Exp Pharmacol. 2012;(213):11-37.
Millan MJ, Agid Y, Brune M, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012; 11(2):141-168.
Drug Brand Names
D-cycloserine • Seromycin Ketamine • Ketalar
Xanomeline • Lumeron, Memcor
Disclosures
Dr. Kantrowitz receives grant or research support from EnVivo, the National Institute of Mental Health, Novartis, Pfizer, Roche-Genentech, the Stanley Foundation, and Sunovion; is a consultant to Health Advances, LLC, the Healthcare Advisory Board, Otsuka Pharmaceuticals, Strategic Edge Communications, and Vindico Medical Education; and owns a small number of shares of common stock in GlaxoSmithKline. Ms. Levy and Dr. Ballon report no financial relationships with manufacturers of any products mentioned in this article or with manufacturers of competing products.
1. Bowie CR, Reichenberg A, Patterson TL, et al. Determinants of real-world functional performance in schizophrenia subjects: correlations with cognition, functional capacity, and symptoms. Am J Psychiatry. 2006;163(3):418-425.
2. Kern RS, Gold JM, Dickinson D, et al. The MCCB impairment profile for schizophrenia outpatients: results from the MATRICS psychometric and standardization study. Schizophr Res. 2011;126(1-3):124-131.
3. Javitt DC, Spencer KM, Thaker GK, et al. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov. 2008;7(1):68-83.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1253.
5. Keefe RS, Bilder RM, Harvey PD, et al. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31(9):2033-2046.
6. Green MF, Kern RS, Braff DL, et al. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26(1):119-136.
7. Keefe RS, Bilder RM, Davis SM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64(6):633-647.
8. Yan J. NIMH tries to jumpstart drug innovations. Psychiatric News. 2013;48(1):8-10.
9. Javitt DC, Schoepp D, Kalivas PW, et al. Translating glutamate: from pathophysiology to treatment. Sci Transl Med. 2011;3(102):102mr2.
10. Foster DJ, Jones CK, Conn PJ. Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov Med. 2012;14(79):413-420.
11. D’Souza MS, Markou A. Schizophrenia and tobacco smoking comorbidity: nAChR agonists in the treatment of schizophrenia-associated cognitive deficits. Neuropharmacology. 2012;62(3):1564-1573.
12. Adler LE, Olincy A, Waldo M, et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998; 24(2):189-202.
13. Meltzer HY, Gawryl M, Ward S, et al. EVP-6124, an alpha-7 nicotinic partial agonist, reduces positive effects on cognition, clinical function, and negative symptoms in patients with chronic schizophrenia on stable antipsychotic therapy. Neuropsychopharmacology. 2011;36:S170-S171.
14. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
15. Digby GJ, Noetzel MJ, Bubser M, et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32(25):8532-8544.
16. Nathan PJ, Watson J, Lund J, et al. The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol. 2013;16(4):721-731.
17. Shekhar A, Potter WZ, Lightfoot J, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165(8):1033-1039.
18. Di Forti M, Lappin LM, Murray RM. Risk factors for schizophrenia—all roads lead to dopamine. Eur Neuropsychopharmacol. 2007;17(suppl 2):S101-S107.
19. Krystal JH, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl). 2003;169(3-4): 215-233.
20. Abi-Dargham A, Moore H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist. 2003;9(5):404-416.
21. Abi-Dargham A, Mawlawi O, Lombardo I, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22(9):3708-3719.
22. Martinez A, Ramanathan DS, Foxe JJ, et al. The role of spatial attention in the selection of real and illusory objects. J Neurosci. 2007;27(30):7963-7973.
23. Castner SA, Williams GV, Goldman-Rakic PS. Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science. 2000;287(5460):2020-2022.
24. Slifstein M, Suckow RF, Javitch JA, et al. Characterization of in vivo pharmacokinetic properties of the dopamine D1 receptor agonist DAR-0100A in nonhuman primates using PET with [11C] NNC112 and [11C] raclopride. J Cereb Blood Flow Metab. 2011;31(1):293-304.
25. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
26. Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4):108-121.
27. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4(3):189-200.
28. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Javitt DC, Kantrowitz JT, eds. Handbook of neurochemistry and molecular neurobiology. New York, NY: Springer; 2009:3-36.
29. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
30. Singh SP, Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs. 2011;25(10):859-868.
31. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropharmacology. 2010;35:S320-S321.
32. Pinard E, Alanine A, Alberati D, et al. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-(( S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J Med Chem. 2010;53(12):4603-4614.
33. Weiser M, Heresco-Levy U, Davidson M, et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry. 2012;73(6):e728-e734.
34. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
35. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
36. D’Souza DC, Radhakrishnan R, Perry E, et al. Feasibility, safety, and efficacy of the combination of D-serine and computerized cognitive retraining in schizophrenia: an international collaborative pilot study. Neuropsychopharmacology. 2013;38(3):492-503.
37. Gottlieb JD, Cather C, Shanahan M, et al. D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr Res. 2011;131(1-3):69-74.
38. Kantrowitz J, Sehatpour P, Oakman E, et al. D-Serine and NMDA based sensory modulation. Poster presented at: 3rd Biennial Schizophrenia International Research Conference; April 14-18, 2012; Florence, Italy.
39. Demirtas-Tatlidede, A, Vahabzadeh-Hagh AM, Pascual-Leone A. Can noninvasive brain stimulation enhance cognition in neuropsychiatric disorders? Neuropharmacology. 2013;64:566-578.
40. Brunelin J, Mondino M, Gassab L, et al. Examining transcranial direct-current stimulation (tDCS) as a treatment for hallucinations in schizophrenia. Am J Psychiatry. 2012;169(7):719-724.
41. Matheson SL, Green MJ, Loo C, et al. Quality assessment and comparison of evidence for electroconvulsive therapy and repetitive transcranial magnetic stimulation for schizophrenia: a systematic meta-review. Schizophr Res. 2012;118(1-3):201-210.
42. Vercammen A, Rushby JA, Loo C, et al. Transcranial direct current stimulation influences probabilistic association learning in schizophrenia. Schizophr Res. 2011;131(1-3):198-205.
43. Nitsche MA, Paulus W. Transcranial direct current stimulation--update 2011. Restor Neurol Neurosci. 2011; 29(6):463-492.
44. Keefe RS, Vinogradov S, Medalia A, et al. Report from the working group conference on multisite trial design for cognitive remediation in schizophrenia. Schizophr Bull. 2011;37(5):1057-1065.
45. McGurk SR, Twamley EW, Sitzer DI, et al. A meta-analysis of cognitive remediation in schizophrenia. Am J Psychiatry. 2007;164(12):1791-1802.
46. Fisher M, Holland C, Merzenich MM, et al. Using neuroplasticity-based auditory training to improve verbal memory in schizophrenia. Am J Psychiatry. 2009;166(7):805-811.
47. Norton DJ, McBain RK, Ongür D, et al. Perceptual training strongly improves visual motion perception in schizophrenia. Brain Cogn. 2011;77(2):248-256.
48. Kantrowitz JT, Revheim N, Pasternak R, et al. It’s all in the cards: effect of stimulus manipulation on Wisconsin Card Sorting Test performance in schizophrenia. Psychiatry Res. 2009;168(3):198-204.
1. Bowie CR, Reichenberg A, Patterson TL, et al. Determinants of real-world functional performance in schizophrenia subjects: correlations with cognition, functional capacity, and symptoms. Am J Psychiatry. 2006;163(3):418-425.
2. Kern RS, Gold JM, Dickinson D, et al. The MCCB impairment profile for schizophrenia outpatients: results from the MATRICS psychometric and standardization study. Schizophr Res. 2011;126(1-3):124-131.
3. Javitt DC, Spencer KM, Thaker GK, et al. Neurophysiological biomarkers for drug development in schizophrenia. Nat Rev Drug Discov. 2008;7(1):68-83.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1253.
5. Keefe RS, Bilder RM, Harvey PD, et al. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31(9):2033-2046.
6. Green MF, Kern RS, Braff DL, et al. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26(1):119-136.
7. Keefe RS, Bilder RM, Davis SM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64(6):633-647.
8. Yan J. NIMH tries to jumpstart drug innovations. Psychiatric News. 2013;48(1):8-10.
9. Javitt DC, Schoepp D, Kalivas PW, et al. Translating glutamate: from pathophysiology to treatment. Sci Transl Med. 2011;3(102):102mr2.
10. Foster DJ, Jones CK, Conn PJ. Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov Med. 2012;14(79):413-420.
11. D’Souza MS, Markou A. Schizophrenia and tobacco smoking comorbidity: nAChR agonists in the treatment of schizophrenia-associated cognitive deficits. Neuropharmacology. 2012;62(3):1564-1573.
12. Adler LE, Olincy A, Waldo M, et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998; 24(2):189-202.
13. Meltzer HY, Gawryl M, Ward S, et al. EVP-6124, an alpha-7 nicotinic partial agonist, reduces positive effects on cognition, clinical function, and negative symptoms in patients with chronic schizophrenia on stable antipsychotic therapy. Neuropsychopharmacology. 2011;36:S170-S171.
14. Lieberman JA, Dunbar G, Segreti AC, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38(6):968-975.
15. Digby GJ, Noetzel MJ, Bubser M, et al. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32(25):8532-8544.
16. Nathan PJ, Watson J, Lund J, et al. The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol. 2013;16(4):721-731.
17. Shekhar A, Potter WZ, Lightfoot J, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165(8):1033-1039.
18. Di Forti M, Lappin LM, Murray RM. Risk factors for schizophrenia—all roads lead to dopamine. Eur Neuropsychopharmacol. 2007;17(suppl 2):S101-S107.
19. Krystal JH, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl). 2003;169(3-4): 215-233.
20. Abi-Dargham A, Moore H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist. 2003;9(5):404-416.
21. Abi-Dargham A, Mawlawi O, Lombardo I, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22(9):3708-3719.
22. Martinez A, Ramanathan DS, Foxe JJ, et al. The role of spatial attention in the selection of real and illusory objects. J Neurosci. 2007;27(30):7963-7973.
23. Castner SA, Williams GV, Goldman-Rakic PS. Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science. 2000;287(5460):2020-2022.
24. Slifstein M, Suckow RF, Javitch JA, et al. Characterization of in vivo pharmacokinetic properties of the dopamine D1 receptor agonist DAR-0100A in nonhuman primates using PET with [11C] NNC112 and [11C] raclopride. J Cereb Blood Flow Metab. 2011;31(1):293-304.
25. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
26. Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4):108-121.
27. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4(3):189-200.
28. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Javitt DC, Kantrowitz JT, eds. Handbook of neurochemistry and molecular neurobiology. New York, NY: Springer; 2009:3-36.
29. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
30. Singh SP, Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs. 2011;25(10):859-868.
31. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropharmacology. 2010;35:S320-S321.
32. Pinard E, Alanine A, Alberati D, et al. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-(( S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J Med Chem. 2010;53(12):4603-4614.
33. Weiser M, Heresco-Levy U, Davidson M, et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry. 2012;73(6):e728-e734.
34. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
35. Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63(12):1118-1126.
36. D’Souza DC, Radhakrishnan R, Perry E, et al. Feasibility, safety, and efficacy of the combination of D-serine and computerized cognitive retraining in schizophrenia: an international collaborative pilot study. Neuropsychopharmacology. 2013;38(3):492-503.
37. Gottlieb JD, Cather C, Shanahan M, et al. D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr Res. 2011;131(1-3):69-74.
38. Kantrowitz J, Sehatpour P, Oakman E, et al. D-Serine and NMDA based sensory modulation. Poster presented at: 3rd Biennial Schizophrenia International Research Conference; April 14-18, 2012; Florence, Italy.
39. Demirtas-Tatlidede, A, Vahabzadeh-Hagh AM, Pascual-Leone A. Can noninvasive brain stimulation enhance cognition in neuropsychiatric disorders? Neuropharmacology. 2013;64:566-578.
40. Brunelin J, Mondino M, Gassab L, et al. Examining transcranial direct-current stimulation (tDCS) as a treatment for hallucinations in schizophrenia. Am J Psychiatry. 2012;169(7):719-724.
41. Matheson SL, Green MJ, Loo C, et al. Quality assessment and comparison of evidence for electroconvulsive therapy and repetitive transcranial magnetic stimulation for schizophrenia: a systematic meta-review. Schizophr Res. 2012;118(1-3):201-210.
42. Vercammen A, Rushby JA, Loo C, et al. Transcranial direct current stimulation influences probabilistic association learning in schizophrenia. Schizophr Res. 2011;131(1-3):198-205.
43. Nitsche MA, Paulus W. Transcranial direct current stimulation--update 2011. Restor Neurol Neurosci. 2011; 29(6):463-492.
44. Keefe RS, Vinogradov S, Medalia A, et al. Report from the working group conference on multisite trial design for cognitive remediation in schizophrenia. Schizophr Bull. 2011;37(5):1057-1065.
45. McGurk SR, Twamley EW, Sitzer DI, et al. A meta-analysis of cognitive remediation in schizophrenia. Am J Psychiatry. 2007;164(12):1791-1802.
46. Fisher M, Holland C, Merzenich MM, et al. Using neuroplasticity-based auditory training to improve verbal memory in schizophrenia. Am J Psychiatry. 2009;166(7):805-811.
47. Norton DJ, McBain RK, Ongür D, et al. Perceptual training strongly improves visual motion perception in schizophrenia. Brain Cogn. 2011;77(2):248-256.
48. Kantrowitz JT, Revheim N, Pasternak R, et al. It’s all in the cards: effect of stimulus manipulation on Wisconsin Card Sorting Test performance in schizophrenia. Psychiatry Res. 2009;168(3):198-204.

Glutamate: New hope for schizophrenia treatment
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
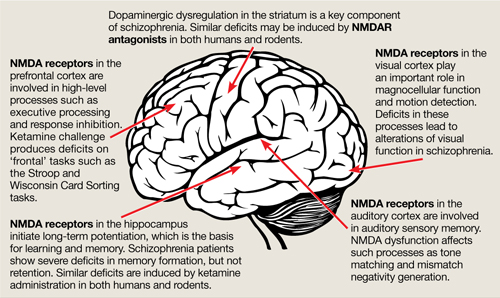
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6
NMDAR antagonists also reproduce the widespread neuropsychological abnormalities of schizophrenia (Figure).6 Ketamine infusion results in the severity and type of disorganized thinking seen in schizophrenia. Given the importance of neurocognitive dysfunction to the conceptualization of schizophrenia, these findings further support a glutamatergic model.
Sensory processing deficits
A key difference between dopaminergic and glutamatergic models is prediction of sensory processing deficits. Traditionally, dopaminergic models have viewed cognitive deficits of schizophrenia as being driven “top down” from higher order brain regions such as the prefrontal cortex, or from local dysfunction within regions such as the striatum.11 In contrast, glutamatergic models predict that deficits also should be observed within sensory brain regions, such as the primary auditory and visual cortex.
Because of the focus on higher-level brain dysfunction, little research on sensory processing deficits was performed until recently. It has become increasingly clear that:
- patients with schizophrenia show severe deficits in early auditory and visual processing
- these deficits significantly contribute to patterns of cognitive dysfunction and psychosocial impairment.12,13
In the auditory system, patients show deficits in pitch perception and, specifically, the ability to match tones after a brief delay. Schizophrenia patients show dysfunction in a specific part of the visual system called the magnocellular visual system. Deficits in these regions lead to impaired ability to detect emotion based on vocal intonation or facial expression, among other deficits.
In addition, reading ability—which was once thought to be normal in patients with schizophrenia—has been found to be severely disturbed.14 As in developmental dyslexia, impairments relate to dysfunction of underlying auditory and visual brain regions. Administering NMDAR antagonists to humans or animals causes deficits in the auditory and visual system similar to those seen in schizophrenia, which confirms the importance of NMDA dysfunction.
Glutamate-based treatments
Because NMDAR antagonists can induce schizophrenia symptoms, the most straightforward approach for treatment is to develop compounds that stimulate glutamate or NMDAR function (Table). The NMDAR contains modulatory sites that may be appropriate targets for drug development, including one that binds the amino acids glycine and D-serine and a redox site that is sensitive to brain glutathione levels. Reductions in brain D-serine and glutathione levels have been reported in schizophrenia, which suggests that impaired NMDAR regulation may contribute directly to brain dysfunction.15 Other treatment approaches being developed include targeting glycine transporters, which indirectly regulate brain levels of glycine, or metabotropic glutamate receptors, which modulate both pre-synaptic glutamate release and post-synaptic NMDAR function.
Table
Glutamatergic drugs in development
| Target | Proposed mechanism | Proposed agents | Phase of development |
|---|---|---|---|
| Glycine/D-serine receptor | Allosteric modulator of the NMDA receptor | Glycine, D-serine, D-alanine, D-cycloserine | Phase II |
| Glycine-type I transport inhibitor | Blocks the reuptake of glycine, akin to SSRIs’ action on serotonin | Sarcosine, RG1678 | Phase II/III |
| Metabotropic glutamate type 2/3 (mGluR2/3) | Blocks presynaptic glutamate release | LY-2140023 | Phase II |
| Redox sensitive site | Allosteric modulator of the NMDA receptor | N-acetylcysteine | Phase II |
| D-amino acid oxidase (DAAO) inhibitors | Inhibits the enzyme that metabolizes D-serine | Remains in preclinical stage | |
| Tetrahydrobiopterin (BH4) | Indirectly modulates glutamatergic system | Remains in preclinical stage | |
| NMDA: N-methyl-D-aspartate; SSRIs: selective serotonin reuptake inhibitors | |||
Glycine/D-serine site agonists. To date, most studies have used glutamatergic drugs adjunctive to antipsychotics and targeted the glycine/D-serine modulatory site, in part because glycine and D-serine are natural compounds and therefore FDA approval for their use could be obtained without the extensive preclinical development usually required for new chemical entities.16 Unfortunately, these agents are less potent than traditional pharmaceuticals, and delivering optimal doses may be impossible. Nevertheless, positive studies with these compounds have provided proof-of-concept for development of agents with higher affinity and specificity.
Studies have used glycine administered at doses up to 60 g/d, D-serine up to 8 g/d, or D-alanine approximately 6 g/d. For glycine, 60 g/d is the highest dose that can be given because of concerns about tolerability and replacement of other essential amino acids. D-serine originally was tested at approximately 2 g/d with promising results, but a recent open-label trial suggested that higher doses may be more efficacious.17 D-serine doses are limited by potential renal toxicity, as demonstrated in rodents studies.
Although not all studies of glycine/D-serine site agonists have been positive, a recent meta-analysis suggests significant improvement in negative symptoms across studies.18 Variability in statistical results across studies is related primarily to degree of placebo effect within individual trials, with a mean improvement in negative symptoms of approximately 15%. Glycine/D-serine site agonists seem to be less effective when combined with clozapine, possibly because clozapine may already enhance the glutamatergic system and increase synaptic glycine levels.6
One study that evaluated effects of open-label glycine in individuals with schizophrenia symptoms observed a large effect-size improvement, including early remission in 3 of 10 patients.19 These data—if confirmed by double-blind trials—would indicate that glycine/d-serine site agonists might have utility in treating the schizophrenia prodrome.
Glycine transport inhibitors. A potential indirect approach to raising glycine levels in the brain is using GlyT1-type glycine transport inhibitors (GTIs). GlyT1 transporters are co-localized in brain with NMDARs and modulate local glycine levels. Rather than binding directly to the NMDAR glycine binding site, GTIs increase glycine levels in the synapse by preventing its removal by GlyT1 transporters. Their function is analogous to using selective serotonin reuptake inhibitors to increase serotonin levels in patients with depression.6
Sarcosine (N-methylglycine) is a naturally occurring GlyT1 inhibitor that has been used in early clinical trials in Taiwan. Initial studies with sarcosine showed efficacy similar to—and in some cases better than—that of direct glycine/D-serine site agonists when added to first-generation or non-clozapine second-generation antipsychotics.18 Sarcosine also has been found to be effective for acute treatment of schizophrenia.20 At present, however, sarcosine is not available for experimental use in the United States because of toxicity considerations.
Using high-affinity GTIs for schizophrenia was first proposed in the mid-1990s,6 but such compounds are only now entering clinical efficacy studies. Most recently, phase II results were presented for RG1678, a compound developed by Hoffman LaRoche.21 The study targeted persistent negative symptoms in patients receiving chronic antipsychotic treatment. Adding RG1678, 10 mg and 30 mg, to antipsychotics led to significant improvement in persistent negative symptoms vs placebo. These promising results are being followed up in phase III studies.
Other glutamatergic options. Few compounds are available to modulate NMDARs at sites other than the glycine/D-serine site. One study administered N-acetylcysteine, a glutathione precursor, as a potential treatment for persistent negative symptoms.22 Encouraging clinical results were observed in this double-blind study, along with improvement in electrophysiologic measures, negative symptoms, and overall functioning, but the study was limited by relatively high rates of noncompletion. Preclinical studies have combined D-serine with an inhibitor of D-amino acid oxidase to prevent D-serine breakdown.23 In rodents, this approach produces a 30-fold increase in D-serine potency.
Tetrahydrobiopterin (BH4) is a cofactor for enzymes responsible for the synthesis of dopamine and other monoamines, and presynaptic release of dopamine and glutamate. Reductions in BH4 levels have been reported in schizophrenia, which suggests that this compound may be etiologically important.24 Researchers have initiated a study of this compound in schizophrenia.
Other schizophrenia models propose that the crucial issue is not NMDA blockade but subsequent dysregulation of presynaptic glutamate release. Type 2/3 metabotropic glutamate receptors (mGluR2/3) are located on presynaptic glutamate terminals and inhibit presynaptic glutamate release. mGluR2/3 agonists have been shown to reverse ketamine’s effects in humans and in animal models,25,26 which suggests a potential role in schizophrenia treatment.
The first mGluR2/3 agonist entered into monotherapy clinical efficacy trials for schizophrenia was LY-2140023. In an initial trial, this compound showed significant efficacy in improving positive and negative symptoms, comparable to that of olanzapine.27 However, a follow-up study failed because of a large placebo effect,28 which leaves the efficacy question unresolved.
In contrast to mGluR2/3, type 5 metabotropic receptors (mGluR5) are co-localized with NMDA receptors and potentiate activation. Thus, mGluR5 agonists also may be effective for treating schizophrenia. These compounds remain in preclinical development. Other approaches, such as stimulating specific types of GABA receptors to overcome glutamatergic deficits, remain promising but have not been tested in definitive clinical trials.
Related Resources
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4): 108-121.
- Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT, Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Ketamine • Ketalar
- Olanzapine • Zyprexa
Disclosures
Dr. Javitt receives grant/research support from Jazz Pharmaceuticals, Pfizer Inc., and Roche and is a consultant to AstraZeneca, Cypress, Eli Lilly and Company, NPS Pharmaceuticals, Sepracor, Solvay, Sunovion, and Takeda. He holds intellectual property rights for use of glycine, D-serine, and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Dr. Kantrowitz receives grant/research support from Eli Lilly and Company, Jazz Pharmaceuticals, Pfizer Inc., Roche, and Sepracor.
Preparation of this manuscript was supported in part by National Institute of Health grants R01 DA03383, R37 MH49334, and P50 MH086385.
1. Fenton WS, McGlashan TH. Antecedents symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry. 1994;151(3):351-356.
2. Woodberry KA. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165(5):579-587.
3. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549-562.
4. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
5. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917-6922.
6. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
7. Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199-214.
8. Krystal J, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169(3-4):215-233.
9. Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141-150.
10. Lahti AC, Koffel B, LaPorte D, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9-19.
11. Lesh TA, Niendam TA, Minzenberg MJ, et al. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316-338.
12. Leitman DI, Laukka P, Juslin PN, et al. Getting the cue: sensory contributions to auditory emotion recognition impairments in schizophrenia. Schizophr Bull. 2010;36(3):545-556.
13. Butler PD, Abeles IY, Weiskopf NG, et al. Sensory contributions to impaired emotion processing in schizophrenia. Schizophr Bull. 2009;35(6):1095-1107.
14. Revheim N, Butler PD, Schechter I, et al. Reading impairment and visual processing deficits in schizophrenia. Schizophr Res. 2006;87(1-3):238-245.
15. Hashimoto K, Fukushima T, Shimizu E, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572-576.
16. Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29(2):300-307.
17. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
18. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
19. Woods SW, Thomas L, Tully E, et al. Effects of oral glycine in the schizophrenia prodrome. Schizophr Res. 2004;70(suppl 1):79.-
20. Lane HY, Liu YC, Huang CL, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63(1):9-12.
21. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacology. 2010;35:S320-S321.
22. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
23. Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3-9.
24. Richardson MA, Read LL, Reilly MA, et al. Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res. 2007;32(1):107-113.
25. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349-1352.
26. Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179(1):303-309.
27. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102-1107.
28. A multi-center, inpatient, phase 2, double-blind, placebo-controlled dose ranging study of LY2140023 in patients with DSM-IV schizophrenia. ClinicalTrials.gov identifier NCT00520923. Available at: http://clinicaltrials.gov/ct2/show/NCT00520923?intr=LY2140023&rank=1. Accessed February 23 2011.
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6
NMDAR antagonists also reproduce the widespread neuropsychological abnormalities of schizophrenia (Figure).6 Ketamine infusion results in the severity and type of disorganized thinking seen in schizophrenia. Given the importance of neurocognitive dysfunction to the conceptualization of schizophrenia, these findings further support a glutamatergic model.
Sensory processing deficits
A key difference between dopaminergic and glutamatergic models is prediction of sensory processing deficits. Traditionally, dopaminergic models have viewed cognitive deficits of schizophrenia as being driven “top down” from higher order brain regions such as the prefrontal cortex, or from local dysfunction within regions such as the striatum.11 In contrast, glutamatergic models predict that deficits also should be observed within sensory brain regions, such as the primary auditory and visual cortex.
Because of the focus on higher-level brain dysfunction, little research on sensory processing deficits was performed until recently. It has become increasingly clear that:
- patients with schizophrenia show severe deficits in early auditory and visual processing
- these deficits significantly contribute to patterns of cognitive dysfunction and psychosocial impairment.12,13
In the auditory system, patients show deficits in pitch perception and, specifically, the ability to match tones after a brief delay. Schizophrenia patients show dysfunction in a specific part of the visual system called the magnocellular visual system. Deficits in these regions lead to impaired ability to detect emotion based on vocal intonation or facial expression, among other deficits.
In addition, reading ability—which was once thought to be normal in patients with schizophrenia—has been found to be severely disturbed.14 As in developmental dyslexia, impairments relate to dysfunction of underlying auditory and visual brain regions. Administering NMDAR antagonists to humans or animals causes deficits in the auditory and visual system similar to those seen in schizophrenia, which confirms the importance of NMDA dysfunction.
Glutamate-based treatments
Because NMDAR antagonists can induce schizophrenia symptoms, the most straightforward approach for treatment is to develop compounds that stimulate glutamate or NMDAR function (Table). The NMDAR contains modulatory sites that may be appropriate targets for drug development, including one that binds the amino acids glycine and D-serine and a redox site that is sensitive to brain glutathione levels. Reductions in brain D-serine and glutathione levels have been reported in schizophrenia, which suggests that impaired NMDAR regulation may contribute directly to brain dysfunction.15 Other treatment approaches being developed include targeting glycine transporters, which indirectly regulate brain levels of glycine, or metabotropic glutamate receptors, which modulate both pre-synaptic glutamate release and post-synaptic NMDAR function.
Table
Glutamatergic drugs in development
| Target | Proposed mechanism | Proposed agents | Phase of development |
|---|---|---|---|
| Glycine/D-serine receptor | Allosteric modulator of the NMDA receptor | Glycine, D-serine, D-alanine, D-cycloserine | Phase II |
| Glycine-type I transport inhibitor | Blocks the reuptake of glycine, akin to SSRIs’ action on serotonin | Sarcosine, RG1678 | Phase II/III |
| Metabotropic glutamate type 2/3 (mGluR2/3) | Blocks presynaptic glutamate release | LY-2140023 | Phase II |
| Redox sensitive site | Allosteric modulator of the NMDA receptor | N-acetylcysteine | Phase II |
| D-amino acid oxidase (DAAO) inhibitors | Inhibits the enzyme that metabolizes D-serine | Remains in preclinical stage | |
| Tetrahydrobiopterin (BH4) | Indirectly modulates glutamatergic system | Remains in preclinical stage | |
| NMDA: N-methyl-D-aspartate; SSRIs: selective serotonin reuptake inhibitors | |||
Glycine/D-serine site agonists. To date, most studies have used glutamatergic drugs adjunctive to antipsychotics and targeted the glycine/D-serine modulatory site, in part because glycine and D-serine are natural compounds and therefore FDA approval for their use could be obtained without the extensive preclinical development usually required for new chemical entities.16 Unfortunately, these agents are less potent than traditional pharmaceuticals, and delivering optimal doses may be impossible. Nevertheless, positive studies with these compounds have provided proof-of-concept for development of agents with higher affinity and specificity.
Studies have used glycine administered at doses up to 60 g/d, D-serine up to 8 g/d, or D-alanine approximately 6 g/d. For glycine, 60 g/d is the highest dose that can be given because of concerns about tolerability and replacement of other essential amino acids. D-serine originally was tested at approximately 2 g/d with promising results, but a recent open-label trial suggested that higher doses may be more efficacious.17 D-serine doses are limited by potential renal toxicity, as demonstrated in rodents studies.
Although not all studies of glycine/D-serine site agonists have been positive, a recent meta-analysis suggests significant improvement in negative symptoms across studies.18 Variability in statistical results across studies is related primarily to degree of placebo effect within individual trials, with a mean improvement in negative symptoms of approximately 15%. Glycine/D-serine site agonists seem to be less effective when combined with clozapine, possibly because clozapine may already enhance the glutamatergic system and increase synaptic glycine levels.6
One study that evaluated effects of open-label glycine in individuals with schizophrenia symptoms observed a large effect-size improvement, including early remission in 3 of 10 patients.19 These data—if confirmed by double-blind trials—would indicate that glycine/d-serine site agonists might have utility in treating the schizophrenia prodrome.
Glycine transport inhibitors. A potential indirect approach to raising glycine levels in the brain is using GlyT1-type glycine transport inhibitors (GTIs). GlyT1 transporters are co-localized in brain with NMDARs and modulate local glycine levels. Rather than binding directly to the NMDAR glycine binding site, GTIs increase glycine levels in the synapse by preventing its removal by GlyT1 transporters. Their function is analogous to using selective serotonin reuptake inhibitors to increase serotonin levels in patients with depression.6
Sarcosine (N-methylglycine) is a naturally occurring GlyT1 inhibitor that has been used in early clinical trials in Taiwan. Initial studies with sarcosine showed efficacy similar to—and in some cases better than—that of direct glycine/D-serine site agonists when added to first-generation or non-clozapine second-generation antipsychotics.18 Sarcosine also has been found to be effective for acute treatment of schizophrenia.20 At present, however, sarcosine is not available for experimental use in the United States because of toxicity considerations.
Using high-affinity GTIs for schizophrenia was first proposed in the mid-1990s,6 but such compounds are only now entering clinical efficacy studies. Most recently, phase II results were presented for RG1678, a compound developed by Hoffman LaRoche.21 The study targeted persistent negative symptoms in patients receiving chronic antipsychotic treatment. Adding RG1678, 10 mg and 30 mg, to antipsychotics led to significant improvement in persistent negative symptoms vs placebo. These promising results are being followed up in phase III studies.
Other glutamatergic options. Few compounds are available to modulate NMDARs at sites other than the glycine/D-serine site. One study administered N-acetylcysteine, a glutathione precursor, as a potential treatment for persistent negative symptoms.22 Encouraging clinical results were observed in this double-blind study, along with improvement in electrophysiologic measures, negative symptoms, and overall functioning, but the study was limited by relatively high rates of noncompletion. Preclinical studies have combined D-serine with an inhibitor of D-amino acid oxidase to prevent D-serine breakdown.23 In rodents, this approach produces a 30-fold increase in D-serine potency.
Tetrahydrobiopterin (BH4) is a cofactor for enzymes responsible for the synthesis of dopamine and other monoamines, and presynaptic release of dopamine and glutamate. Reductions in BH4 levels have been reported in schizophrenia, which suggests that this compound may be etiologically important.24 Researchers have initiated a study of this compound in schizophrenia.
Other schizophrenia models propose that the crucial issue is not NMDA blockade but subsequent dysregulation of presynaptic glutamate release. Type 2/3 metabotropic glutamate receptors (mGluR2/3) are located on presynaptic glutamate terminals and inhibit presynaptic glutamate release. mGluR2/3 agonists have been shown to reverse ketamine’s effects in humans and in animal models,25,26 which suggests a potential role in schizophrenia treatment.
The first mGluR2/3 agonist entered into monotherapy clinical efficacy trials for schizophrenia was LY-2140023. In an initial trial, this compound showed significant efficacy in improving positive and negative symptoms, comparable to that of olanzapine.27 However, a follow-up study failed because of a large placebo effect,28 which leaves the efficacy question unresolved.
In contrast to mGluR2/3, type 5 metabotropic receptors (mGluR5) are co-localized with NMDA receptors and potentiate activation. Thus, mGluR5 agonists also may be effective for treating schizophrenia. These compounds remain in preclinical development. Other approaches, such as stimulating specific types of GABA receptors to overcome glutamatergic deficits, remain promising but have not been tested in definitive clinical trials.
Related Resources
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4): 108-121.
- Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT, Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Ketamine • Ketalar
- Olanzapine • Zyprexa
Disclosures
Dr. Javitt receives grant/research support from Jazz Pharmaceuticals, Pfizer Inc., and Roche and is a consultant to AstraZeneca, Cypress, Eli Lilly and Company, NPS Pharmaceuticals, Sepracor, Solvay, Sunovion, and Takeda. He holds intellectual property rights for use of glycine, D-serine, and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Dr. Kantrowitz receives grant/research support from Eli Lilly and Company, Jazz Pharmaceuticals, Pfizer Inc., Roche, and Sepracor.
Preparation of this manuscript was supported in part by National Institute of Health grants R01 DA03383, R37 MH49334, and P50 MH086385.
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6
NMDAR antagonists also reproduce the widespread neuropsychological abnormalities of schizophrenia (Figure).6 Ketamine infusion results in the severity and type of disorganized thinking seen in schizophrenia. Given the importance of neurocognitive dysfunction to the conceptualization of schizophrenia, these findings further support a glutamatergic model.
Sensory processing deficits
A key difference between dopaminergic and glutamatergic models is prediction of sensory processing deficits. Traditionally, dopaminergic models have viewed cognitive deficits of schizophrenia as being driven “top down” from higher order brain regions such as the prefrontal cortex, or from local dysfunction within regions such as the striatum.11 In contrast, glutamatergic models predict that deficits also should be observed within sensory brain regions, such as the primary auditory and visual cortex.
Because of the focus on higher-level brain dysfunction, little research on sensory processing deficits was performed until recently. It has become increasingly clear that:
- patients with schizophrenia show severe deficits in early auditory and visual processing
- these deficits significantly contribute to patterns of cognitive dysfunction and psychosocial impairment.12,13
In the auditory system, patients show deficits in pitch perception and, specifically, the ability to match tones after a brief delay. Schizophrenia patients show dysfunction in a specific part of the visual system called the magnocellular visual system. Deficits in these regions lead to impaired ability to detect emotion based on vocal intonation or facial expression, among other deficits.
In addition, reading ability—which was once thought to be normal in patients with schizophrenia—has been found to be severely disturbed.14 As in developmental dyslexia, impairments relate to dysfunction of underlying auditory and visual brain regions. Administering NMDAR antagonists to humans or animals causes deficits in the auditory and visual system similar to those seen in schizophrenia, which confirms the importance of NMDA dysfunction.
Glutamate-based treatments
Because NMDAR antagonists can induce schizophrenia symptoms, the most straightforward approach for treatment is to develop compounds that stimulate glutamate or NMDAR function (Table). The NMDAR contains modulatory sites that may be appropriate targets for drug development, including one that binds the amino acids glycine and D-serine and a redox site that is sensitive to brain glutathione levels. Reductions in brain D-serine and glutathione levels have been reported in schizophrenia, which suggests that impaired NMDAR regulation may contribute directly to brain dysfunction.15 Other treatment approaches being developed include targeting glycine transporters, which indirectly regulate brain levels of glycine, or metabotropic glutamate receptors, which modulate both pre-synaptic glutamate release and post-synaptic NMDAR function.
Table
Glutamatergic drugs in development
| Target | Proposed mechanism | Proposed agents | Phase of development |
|---|---|---|---|
| Glycine/D-serine receptor | Allosteric modulator of the NMDA receptor | Glycine, D-serine, D-alanine, D-cycloserine | Phase II |
| Glycine-type I transport inhibitor | Blocks the reuptake of glycine, akin to SSRIs’ action on serotonin | Sarcosine, RG1678 | Phase II/III |
| Metabotropic glutamate type 2/3 (mGluR2/3) | Blocks presynaptic glutamate release | LY-2140023 | Phase II |
| Redox sensitive site | Allosteric modulator of the NMDA receptor | N-acetylcysteine | Phase II |
| D-amino acid oxidase (DAAO) inhibitors | Inhibits the enzyme that metabolizes D-serine | Remains in preclinical stage | |
| Tetrahydrobiopterin (BH4) | Indirectly modulates glutamatergic system | Remains in preclinical stage | |
| NMDA: N-methyl-D-aspartate; SSRIs: selective serotonin reuptake inhibitors | |||
Glycine/D-serine site agonists. To date, most studies have used glutamatergic drugs adjunctive to antipsychotics and targeted the glycine/D-serine modulatory site, in part because glycine and D-serine are natural compounds and therefore FDA approval for their use could be obtained without the extensive preclinical development usually required for new chemical entities.16 Unfortunately, these agents are less potent than traditional pharmaceuticals, and delivering optimal doses may be impossible. Nevertheless, positive studies with these compounds have provided proof-of-concept for development of agents with higher affinity and specificity.
Studies have used glycine administered at doses up to 60 g/d, D-serine up to 8 g/d, or D-alanine approximately 6 g/d. For glycine, 60 g/d is the highest dose that can be given because of concerns about tolerability and replacement of other essential amino acids. D-serine originally was tested at approximately 2 g/d with promising results, but a recent open-label trial suggested that higher doses may be more efficacious.17 D-serine doses are limited by potential renal toxicity, as demonstrated in rodents studies.
Although not all studies of glycine/D-serine site agonists have been positive, a recent meta-analysis suggests significant improvement in negative symptoms across studies.18 Variability in statistical results across studies is related primarily to degree of placebo effect within individual trials, with a mean improvement in negative symptoms of approximately 15%. Glycine/D-serine site agonists seem to be less effective when combined with clozapine, possibly because clozapine may already enhance the glutamatergic system and increase synaptic glycine levels.6
One study that evaluated effects of open-label glycine in individuals with schizophrenia symptoms observed a large effect-size improvement, including early remission in 3 of 10 patients.19 These data—if confirmed by double-blind trials—would indicate that glycine/d-serine site agonists might have utility in treating the schizophrenia prodrome.
Glycine transport inhibitors. A potential indirect approach to raising glycine levels in the brain is using GlyT1-type glycine transport inhibitors (GTIs). GlyT1 transporters are co-localized in brain with NMDARs and modulate local glycine levels. Rather than binding directly to the NMDAR glycine binding site, GTIs increase glycine levels in the synapse by preventing its removal by GlyT1 transporters. Their function is analogous to using selective serotonin reuptake inhibitors to increase serotonin levels in patients with depression.6
Sarcosine (N-methylglycine) is a naturally occurring GlyT1 inhibitor that has been used in early clinical trials in Taiwan. Initial studies with sarcosine showed efficacy similar to—and in some cases better than—that of direct glycine/D-serine site agonists when added to first-generation or non-clozapine second-generation antipsychotics.18 Sarcosine also has been found to be effective for acute treatment of schizophrenia.20 At present, however, sarcosine is not available for experimental use in the United States because of toxicity considerations.
Using high-affinity GTIs for schizophrenia was first proposed in the mid-1990s,6 but such compounds are only now entering clinical efficacy studies. Most recently, phase II results were presented for RG1678, a compound developed by Hoffman LaRoche.21 The study targeted persistent negative symptoms in patients receiving chronic antipsychotic treatment. Adding RG1678, 10 mg and 30 mg, to antipsychotics led to significant improvement in persistent negative symptoms vs placebo. These promising results are being followed up in phase III studies.
Other glutamatergic options. Few compounds are available to modulate NMDARs at sites other than the glycine/D-serine site. One study administered N-acetylcysteine, a glutathione precursor, as a potential treatment for persistent negative symptoms.22 Encouraging clinical results were observed in this double-blind study, along with improvement in electrophysiologic measures, negative symptoms, and overall functioning, but the study was limited by relatively high rates of noncompletion. Preclinical studies have combined D-serine with an inhibitor of D-amino acid oxidase to prevent D-serine breakdown.23 In rodents, this approach produces a 30-fold increase in D-serine potency.
Tetrahydrobiopterin (BH4) is a cofactor for enzymes responsible for the synthesis of dopamine and other monoamines, and presynaptic release of dopamine and glutamate. Reductions in BH4 levels have been reported in schizophrenia, which suggests that this compound may be etiologically important.24 Researchers have initiated a study of this compound in schizophrenia.
Other schizophrenia models propose that the crucial issue is not NMDA blockade but subsequent dysregulation of presynaptic glutamate release. Type 2/3 metabotropic glutamate receptors (mGluR2/3) are located on presynaptic glutamate terminals and inhibit presynaptic glutamate release. mGluR2/3 agonists have been shown to reverse ketamine’s effects in humans and in animal models,25,26 which suggests a potential role in schizophrenia treatment.
The first mGluR2/3 agonist entered into monotherapy clinical efficacy trials for schizophrenia was LY-2140023. In an initial trial, this compound showed significant efficacy in improving positive and negative symptoms, comparable to that of olanzapine.27 However, a follow-up study failed because of a large placebo effect,28 which leaves the efficacy question unresolved.
In contrast to mGluR2/3, type 5 metabotropic receptors (mGluR5) are co-localized with NMDA receptors and potentiate activation. Thus, mGluR5 agonists also may be effective for treating schizophrenia. These compounds remain in preclinical development. Other approaches, such as stimulating specific types of GABA receptors to overcome glutamatergic deficits, remain promising but have not been tested in definitive clinical trials.
Related Resources
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4): 108-121.
- Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT, Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Ketamine • Ketalar
- Olanzapine • Zyprexa
Disclosures
Dr. Javitt receives grant/research support from Jazz Pharmaceuticals, Pfizer Inc., and Roche and is a consultant to AstraZeneca, Cypress, Eli Lilly and Company, NPS Pharmaceuticals, Sepracor, Solvay, Sunovion, and Takeda. He holds intellectual property rights for use of glycine, D-serine, and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Dr. Kantrowitz receives grant/research support from Eli Lilly and Company, Jazz Pharmaceuticals, Pfizer Inc., Roche, and Sepracor.
Preparation of this manuscript was supported in part by National Institute of Health grants R01 DA03383, R37 MH49334, and P50 MH086385.
1. Fenton WS, McGlashan TH. Antecedents symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry. 1994;151(3):351-356.
2. Woodberry KA. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165(5):579-587.
3. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549-562.
4. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
5. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917-6922.
6. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
7. Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199-214.
8. Krystal J, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169(3-4):215-233.
9. Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141-150.
10. Lahti AC, Koffel B, LaPorte D, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9-19.
11. Lesh TA, Niendam TA, Minzenberg MJ, et al. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316-338.
12. Leitman DI, Laukka P, Juslin PN, et al. Getting the cue: sensory contributions to auditory emotion recognition impairments in schizophrenia. Schizophr Bull. 2010;36(3):545-556.
13. Butler PD, Abeles IY, Weiskopf NG, et al. Sensory contributions to impaired emotion processing in schizophrenia. Schizophr Bull. 2009;35(6):1095-1107.
14. Revheim N, Butler PD, Schechter I, et al. Reading impairment and visual processing deficits in schizophrenia. Schizophr Res. 2006;87(1-3):238-245.
15. Hashimoto K, Fukushima T, Shimizu E, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572-576.
16. Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29(2):300-307.
17. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
18. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
19. Woods SW, Thomas L, Tully E, et al. Effects of oral glycine in the schizophrenia prodrome. Schizophr Res. 2004;70(suppl 1):79.-
20. Lane HY, Liu YC, Huang CL, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63(1):9-12.
21. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacology. 2010;35:S320-S321.
22. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
23. Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3-9.
24. Richardson MA, Read LL, Reilly MA, et al. Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res. 2007;32(1):107-113.
25. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349-1352.
26. Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179(1):303-309.
27. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102-1107.
28. A multi-center, inpatient, phase 2, double-blind, placebo-controlled dose ranging study of LY2140023 in patients with DSM-IV schizophrenia. ClinicalTrials.gov identifier NCT00520923. Available at: http://clinicaltrials.gov/ct2/show/NCT00520923?intr=LY2140023&rank=1. Accessed February 23 2011.
1. Fenton WS, McGlashan TH. Antecedents symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry. 1994;151(3):351-356.
2. Woodberry KA. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165(5):579-587.
3. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549-562.
4. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
5. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917-6922.
6. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
7. Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199-214.
8. Krystal J, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169(3-4):215-233.
9. Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141-150.
10. Lahti AC, Koffel B, LaPorte D, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9-19.
11. Lesh TA, Niendam TA, Minzenberg MJ, et al. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316-338.
12. Leitman DI, Laukka P, Juslin PN, et al. Getting the cue: sensory contributions to auditory emotion recognition impairments in schizophrenia. Schizophr Bull. 2010;36(3):545-556.
13. Butler PD, Abeles IY, Weiskopf NG, et al. Sensory contributions to impaired emotion processing in schizophrenia. Schizophr Bull. 2009;35(6):1095-1107.
14. Revheim N, Butler PD, Schechter I, et al. Reading impairment and visual processing deficits in schizophrenia. Schizophr Res. 2006;87(1-3):238-245.
15. Hashimoto K, Fukushima T, Shimizu E, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572-576.
16. Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29(2):300-307.
17. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
18. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
19. Woods SW, Thomas L, Tully E, et al. Effects of oral glycine in the schizophrenia prodrome. Schizophr Res. 2004;70(suppl 1):79.-
20. Lane HY, Liu YC, Huang CL, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63(1):9-12.
21. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacology. 2010;35:S320-S321.
22. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
23. Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3-9.
24. Richardson MA, Read LL, Reilly MA, et al. Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res. 2007;32(1):107-113.
25. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349-1352.
26. Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179(1):303-309.
27. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102-1107.
28. A multi-center, inpatient, phase 2, double-blind, placebo-controlled dose ranging study of LY2140023 in patients with DSM-IV schizophrenia. ClinicalTrials.gov identifier NCT00520923. Available at: http://clinicaltrials.gov/ct2/show/NCT00520923?intr=LY2140023&rank=1. Accessed February 23 2011.