User login
PRENATAL COUNSELING
Three important areas of research into stillbirth have evolved over the past year, furthering our understanding of the phenomenon and our ability to provide comprehensive, evidence-based care:
- Genetic studies. Karyotype analysis is useful in determining the cause of stillbirth, especially when analysis is based on a sample of amniotic fluid that was obtained before delivery. And array-based comparative genomic hybridization, which yields information on the chromosome count as well as micro-duplications and deletions, can be performed on nondividing cells.
- Risk factors. Further investigation implicates advanced maternal age, obesity, and African-American race.
- Classification. Paring down the more than three dozen systems that exist for classification of stillbirth was the main challenge addressed by an international consensus group in 2009 and the focus of a separate analysis.
The individual studies that contribute to our knowledge base in these areas are discussed in more detail in the articles that follow.
Stillbirth is broadly defined as fetal demise after 20 weeks’ gestation and with a fetal weight exceeding 350 g. In the United States, stillbirth occurs in 1 of every 160 live births (6 stillbirths for every 1,000 live births). Although the rate of neonatal demise has decreased over the past decade, the rate of stillbirth has declined less strikingly.
For an analysis of karyotype, amniotic fluid is best
Korteweg FJ, Bouman K, Erwich JJ, et al. Cytogenetic analysis after evaluation of 750 fetal deaths: proposal for diagnosis workup. Obstet Gynecol. 2008;111:865–874.
ACOG Practice Bulletin #102: Management of stillbirth. Obstet Gynecol. 2009;113:748–760.
When stillbirth occurs, determination of the cause of death fulfills several goals:
- It informs counseling of the parents, who must come to terms with the loss
- It aids in determining the risk of recurrence, which informs family planning
- It furthers research into stillbirth and facilitates the comparison of national and international data.
Chromosomal anomaly is one potential cause of stillbirth. Its frequency depends on the presence of structural malformation. For example, Korteweg and colleagues found a rate of chromosomal anomaly of 4.6% among stillbirths involving fetuses without structural abnormality, but the rate rose to 38% when anatomic malformation was present. The distribution of chromosomes among stillbirths mirrored the pattern seen in live births, including 45, X and trisomies of chromosome 21, 13, and 18.
The utility of karyotype assessment when ultrasonography (US) has not identified structural malformation has been debated. Given the 5% incidence of chromosomal anomaly in the absence of structural abnormality, and the limitations of US in detecting subtle dysmorphology, a karyotype seems advisable to assess all stillbirths.
Comparison of methods points to superiority of amniocentesis
Because fewer than 20% of skin biopsies result in a useful culture, postmortem skin biopsy for karyotype assessment is unreliable. Korteweg and colleagues evaluated other methods of obtaining cells for examination and found that a successful karyotype is most likely with predelivery amniocentesis (85%), followed by umbilical cord culture (32.1%). A karyotype of cells from fascia lata and skin biopsy yielded poor results, especially in the setting of maceration. Placental biopsy is likely to provide an adequate karyotype (71% probability) but findings may be confounded by confined placental mosaicism.1
ACOG also advocates predelivery amniocentesis
In its 2009 practice bulletin, ACOG supported inclusion of amniocentesis in the assessment of stillbirth and preparation for delivery. Once an epidural is placed, amniocentesis provides cells for karyotype assessment, polymerase chain reaction (PCR) for viral studies, and any other metabolic or specific genetic studies that may be indicated by fetopsy.
If amniocentesis is not performed, ACOG recommends umbilical cord culture as an alternative. Because nondividing cells can be utilized in fluorescence in situ hybridization (FISH) for chromosome 13, 18, 21, X, and Y, this method should be considered in any case involving culture failure (TABLE).2
TABLE
Genetic components of stillbirth assessment
| Type of assessment | Steps |
|---|---|
| Inspection of fetus and placenta | Measure head circumference and length of fetus |
| Weigh fetus and placenta | |
| Photograph fetus and placenta, including frontal and profile shots of whole body, face, extremities, palms, and any abnormality | |
| Document findings | |
| Cytologic analysis | Obtain consent from parents |
| Obtain acceptable specimens using one of the following sterile techniques: | |
| • Amniocentesis at the time of prenatal diagnosis of demise • Placental block (1 x 1 cm) taken from below the cord-insertion site on the unfixed placenta • Umbilical cord segment (1.5 cm) • Internal fetal tissue specimen, e.g., costochondral junction or patella (not skin) | |
| Preserve specimens in a sterile culture medium of lactated Ringer’s solution at room temperature during transfer to laboratory | |
| Fetopsy | Obtain parental consent; if no consent is given, send placenta for pathologic analysis |
| Perform autopsy and pathologic assessment of the placenta | |
| Consider whole-body fetal radiographs | |
| Source: ACOG Practice Bulletin #102 | |
Perform predelivery amniocentesis whenever possible at the time of diagnosis of demise to obtain a cell sample for karyotype analysis to determine the cause of death.
Array-based comparative genomic hybridization makes assessment of nondividing cells possible
Raca G, Artzer A, Thorson L, et al. Array-based comparative hybridization (aCGH) in the genetic evaluation of stillbirth. Am J Med Genet A. 2009;149A:2437–2443.
Array-based comparative genomic hybridization (aCGH) makes it possible to assess the chromosome count and perform a high-resolution search for microduplications and deletions. With known segments of the genome printed on slides, the clinical scientist can analyze DNA from nondividing cells from a stillbirth. The ability to use nondividing cells is important because no cell culture is required. (Cell culture is often difficult to obtain after stillbirth.) Depending on the array selected, the resolution can be as fine as a single nucleotide polymorphism.
aCGH can inform preconception counseling
Raca and colleagues used a range of arrays to assess 15 stillbirths that involved two or more malformations. Chromosomal abnormalities, including trisomy 21 and an unbalanced translocation, were detected by aCGH in two infants. Identification of these abnormalities helped inform counseling of the parents:
- In the case of trisomy 21, parental karyotypes revealed a nontranslocation event, making it possible to assure the parents that the risk of recurrence is low
- The unbalanced translocation resulted from a balanced chromosome translocation in the mother and was associated with a significant risk of recurrence (in this case, FISH would not have helped because chromosomes 13, 18, 21, X, and Y were not involved).
Limitations of aCGH
One limitation is an inability to detect polyploidy such as triploidy or tetraploidy. This problem can be circumvented through the use of a FISH preparation prior to aCGH.
In most centers, parental blood samples are drawn at the time of aCGH studies. Because aCGH offers greater resolution of chromosome regions, an increasing number of benign variations (i.e., present in one parent) are being identified. As aCGH technology advances, we are accumulating data on copy-number variations.
A large clinical trial is needed to assess the full potential of aCGH in this setting.
Use of array-basic comparative genomic hybridization to assess cells from a stillborn fetus can help determine the cause of death and inform counseling of the parents about the risk of recurrence.
Risk factors for stillbirth include
advanced maternal age, obesity, and black race
ACOG Practice Bulletin #102: Management of stillbirth. Obstet Gynecol. 2009;113:748–760.
Willinger M, Ko CW, Reddy UM. Racial disparities in stillbirth risk across gestation in the United States. Am J Obstet Gynecol. 2009;201:469.e1–469.e8.
Fretts RC. The study of stillbirth. Am J Obstet Gynecol. 2009;201:429–430.
Women who have diseases such as insulin-dependent diabetes and systemic lupus erythematosus have long been recognized as having a six- to 20-fold increase in the risk of stillbirth, compared with the general population. However, each of these disorders accounts for 2% and less than 1% of the pregnant population, respectively, so their overall contribution to stillbirth is small. Larger portions of the population have a lower—but still significant—risk of stillbirth:
- women older than 35 years
- women who have a body mass index (BMI) above 30
- non-Hispanic black women.
Each of these categories represents 15% or more of the typical obstetric population, and each group faces a risk of stillbirth approaching 1%. The ACOG practice bulletin and the study by Willinger and colleagues address these risks in detail.
Advanced maternal age is particularly risky among nulliparous women
Advanced maternal age (>35 years) is associated with increased rates of chromosomal abnormality and maternal morbidity, such as hypertension, that are known to raise the risk of stillbirth. Even when these and other variables associated with advanced maternal age, such as placenta previa, diabetes, and multiple gestation, are controlled, however, the increased risk of stillbirth remains.
Advanced maternal age in a first pregnancy carries a particularly elevated risk. For example, the risk of stillbirth in a 40-year-old nulliparous woman is more than twice the risk in a 40-year-old multiparous woman (1 in every 116 pregnancies vs 1 in every 304).3
The increased risk of stillbirth associated with advanced maternal age is present at all gestational ages, though it becomes most profound at 37 to 42 weeks’ gestation, notably for:
- women 35 to 39 years old (1 in every 382 pregnancies; relative risk [RR] of 1.32, compared with women <35 years old; 95% confidence interval [CI], 1.22, 1.43)
- women >40 years old (1 in every 267 pregnancies; RR, 1.88; 95% CI, 1.64, 2.16).
These numbers remain significant even after controlling for medical conditions.3
The utility of antepartum surveillance and induction of labor for delivery is unclear, given the risk of iatrogenic prematurity.
Risk of stillbirth is doubled among obese and markedly obese women
Although the number of adults who are overweight (BMI 25–30) has remained fairly constant over the past 20 years (30% to 35% of the population), the percentage of women of reproductive age who are obese (BMI >30) has risen markedly. Obesity is now present in 35% of the population, and marked obesity (BMI >40) affects an additional 6%. Both obese and markedly obese women face a twofold relative risk of stillbirth, compared with women of normal weight. The rate of stillbirth in this population is 12 to 18 for every 1,000 births—a 1.2% to 1.8% risk.
Although obesity-related stillbirth likely has multiple causes, the risk remains elevated even after exclusion of confounding factors such as smoking, gestational diabetes, and preeclampsia.
Race is an independent contributor
Racial differences in the rate of stillbirth remain despite a decrease in the overall stillbirth rate over the past 20 years ( FIGURE ). In 2003, the rate of stillbirth was 5 for every 1,000 births among non-Hispanic whites, 5.5 among Hispanics, and 12 among non-Hispanic blacks. In other words, the risk of stillbirth was 1 in 202, 1 in 183, and 1 in 87 births for white, Hispanic, and black women, respectively.
Willinger and colleagues utilized data from the National Center for Health Statistics and assessed 2001–2002 birth and infant death datasets for 36 states, examining the stillbirth hazard risk for more than 5 million singleton pregnancies. Stillbirth peaked at 20 to 23 weeks and 39 to 41 weeks’ gestation, as expected. However, at 20 to 23 weeks, the risk of stillbirth among non-Hispanic black women was more than twice the rate for non-Hispanic white women (RR, 2.8). Although it then declined as term approached, it remained greater than that of non-Hispanic white women (RR, 1.6).
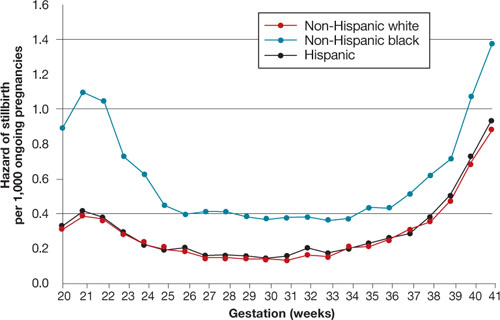
FIGURE Racial disparities in the risk of stillbirth
Hazard of stillbirth for singleton pregnancies by gestational age and race and ethnicity, 2001–2002. SOURCE: Willinger et al. Greater acceptance and use of induction of labor at term among whites merits attention
In an editorial accompanying the study by Willinger and colleagues, Fretts pointed out the higher rate of induction of labor at term among white women that has been observed in at least three studies of vital statistics. (Willinger and colleagues also pointed out this difference.) The acceptance and use of labor induction at term—and the lower stillbirth rate—among white women warrants further investigation.
Education appears to reduce the risk of stillbirth to a greater degree among whites than it does among blacks. Again, nulliparity and advanced maternal age were important contributors to the risk of stillbirth across all three races.
Counsel African-American gravidas and women older than 35 years that their risk of stillbirth is elevated.
Obese women should be advised to lose weight before conception if at all possible to reduce the risk of stillbirth.
Needed: Standardized analysis
and documentation of stillbirth
Reddy UM, Goldberg R, Silver R, et al. Stillbirth classification—developing an international consensus for research: executive summary of a National Institute of Child Health and Human Development workshop. Obstet Gynecol. 2009;114:901–914.
Flenady V, Frøen JF, Pinar H, et al. An evaluation of classification systems for stillbirth. BMC Pregnancy Childbirth. 2009;9:24.
Further guidance for the clinical management of stillbirth will come from investigations of the underlying pathologies and associated risk factors. Key to development of this guidance is the involvement of obstetricians in documenting the antenatal record and delivery information. Also needed is a standardized system for recording this information. More than three dozen systems have been developed to classify stillbirth, at the expense of uniformity of content.
An international consensus group published guidelines on how to describe the cause of death in research endeavors, recognizing the need to maintain the ability to attach a level of uncertainty. In addition, Flenady and colleagues compared the most widely used systems in clinical practice, assigning the highest score for components such as ease of use, inter observer variability, and proportion of unexplained stillbirths to CODAC [cause of death and two associated causes]. This system assigns a primary cause of death from a specified list of choices and allows inclusion of two possible contributing causes.
Both the international consensus classification and the CODAC scoring system are accessible through links embedded within the articles. Both systems require the establishment of standardized evaluation and review of stillbirth that should include obstetricians, pathologists, and geneticists.
Because assessment and classification of stillbirth are fundamental to its prevention, as well as a critical part of clinical practice, ObGyns should become familiar with the international consensus classification and CODAC scoring systems and adopt a standardized approach to assessment and documentation.
1. Rodgers CS, Creasy MR, Fitchett M, Maliszewska CT, Pratt NR, Waters JJ. Solid tissue culture for cytogenetic analysis: a collaborative survey for the Association of Clinical Cytogeneticists. J Clin Pathol. 1996;49:638-641.
2. Rivasi F, Schirosi L, Bettelli S, et al. FISH analysis in cell touch preparations and cytological specimens from formalin-fixed fetal autopsies. Diagn Cytopathol. 2008;36:633-636.
3. Reddy UM, Ko CW, Willinger M. Maternal age and the risk of stillbirth throughout pregnancy in the United States. Am J Obstet Gynecol. 2006;195:764-770.
4. MacDorman MF, Mathews TJ. NCHS Data Brief #9: Recent trends in infant mortality in the United States. Atlanta, Ga: National Center for Health Statistics; October 2008. Available at: http://www.cdc.gov/nchs/data/databriefs/db09.htm. Accessed Dec. 15, 2009.
Three important areas of research into stillbirth have evolved over the past year, furthering our understanding of the phenomenon and our ability to provide comprehensive, evidence-based care:
- Genetic studies. Karyotype analysis is useful in determining the cause of stillbirth, especially when analysis is based on a sample of amniotic fluid that was obtained before delivery. And array-based comparative genomic hybridization, which yields information on the chromosome count as well as micro-duplications and deletions, can be performed on nondividing cells.
- Risk factors. Further investigation implicates advanced maternal age, obesity, and African-American race.
- Classification. Paring down the more than three dozen systems that exist for classification of stillbirth was the main challenge addressed by an international consensus group in 2009 and the focus of a separate analysis.
The individual studies that contribute to our knowledge base in these areas are discussed in more detail in the articles that follow.
Stillbirth is broadly defined as fetal demise after 20 weeks’ gestation and with a fetal weight exceeding 350 g. In the United States, stillbirth occurs in 1 of every 160 live births (6 stillbirths for every 1,000 live births). Although the rate of neonatal demise has decreased over the past decade, the rate of stillbirth has declined less strikingly.
For an analysis of karyotype, amniotic fluid is best
Korteweg FJ, Bouman K, Erwich JJ, et al. Cytogenetic analysis after evaluation of 750 fetal deaths: proposal for diagnosis workup. Obstet Gynecol. 2008;111:865–874.
ACOG Practice Bulletin #102: Management of stillbirth. Obstet Gynecol. 2009;113:748–760.
When stillbirth occurs, determination of the cause of death fulfills several goals:
- It informs counseling of the parents, who must come to terms with the loss
- It aids in determining the risk of recurrence, which informs family planning
- It furthers research into stillbirth and facilitates the comparison of national and international data.
Chromosomal anomaly is one potential cause of stillbirth. Its frequency depends on the presence of structural malformation. For example, Korteweg and colleagues found a rate of chromosomal anomaly of 4.6% among stillbirths involving fetuses without structural abnormality, but the rate rose to 38% when anatomic malformation was present. The distribution of chromosomes among stillbirths mirrored the pattern seen in live births, including 45, X and trisomies of chromosome 21, 13, and 18.
The utility of karyotype assessment when ultrasonography (US) has not identified structural malformation has been debated. Given the 5% incidence of chromosomal anomaly in the absence of structural abnormality, and the limitations of US in detecting subtle dysmorphology, a karyotype seems advisable to assess all stillbirths.
Comparison of methods points to superiority of amniocentesis
Because fewer than 20% of skin biopsies result in a useful culture, postmortem skin biopsy for karyotype assessment is unreliable. Korteweg and colleagues evaluated other methods of obtaining cells for examination and found that a successful karyotype is most likely with predelivery amniocentesis (85%), followed by umbilical cord culture (32.1%). A karyotype of cells from fascia lata and skin biopsy yielded poor results, especially in the setting of maceration. Placental biopsy is likely to provide an adequate karyotype (71% probability) but findings may be confounded by confined placental mosaicism.1
ACOG also advocates predelivery amniocentesis
In its 2009 practice bulletin, ACOG supported inclusion of amniocentesis in the assessment of stillbirth and preparation for delivery. Once an epidural is placed, amniocentesis provides cells for karyotype assessment, polymerase chain reaction (PCR) for viral studies, and any other metabolic or specific genetic studies that may be indicated by fetopsy.
If amniocentesis is not performed, ACOG recommends umbilical cord culture as an alternative. Because nondividing cells can be utilized in fluorescence in situ hybridization (FISH) for chromosome 13, 18, 21, X, and Y, this method should be considered in any case involving culture failure (TABLE).2
TABLE
Genetic components of stillbirth assessment
| Type of assessment | Steps |
|---|---|
| Inspection of fetus and placenta | Measure head circumference and length of fetus |
| Weigh fetus and placenta | |
| Photograph fetus and placenta, including frontal and profile shots of whole body, face, extremities, palms, and any abnormality | |
| Document findings | |
| Cytologic analysis | Obtain consent from parents |
| Obtain acceptable specimens using one of the following sterile techniques: | |
| • Amniocentesis at the time of prenatal diagnosis of demise • Placental block (1 x 1 cm) taken from below the cord-insertion site on the unfixed placenta • Umbilical cord segment (1.5 cm) • Internal fetal tissue specimen, e.g., costochondral junction or patella (not skin) | |
| Preserve specimens in a sterile culture medium of lactated Ringer’s solution at room temperature during transfer to laboratory | |
| Fetopsy | Obtain parental consent; if no consent is given, send placenta for pathologic analysis |
| Perform autopsy and pathologic assessment of the placenta | |
| Consider whole-body fetal radiographs | |
| Source: ACOG Practice Bulletin #102 | |
Perform predelivery amniocentesis whenever possible at the time of diagnosis of demise to obtain a cell sample for karyotype analysis to determine the cause of death.
Array-based comparative genomic hybridization makes assessment of nondividing cells possible
Raca G, Artzer A, Thorson L, et al. Array-based comparative hybridization (aCGH) in the genetic evaluation of stillbirth. Am J Med Genet A. 2009;149A:2437–2443.
Array-based comparative genomic hybridization (aCGH) makes it possible to assess the chromosome count and perform a high-resolution search for microduplications and deletions. With known segments of the genome printed on slides, the clinical scientist can analyze DNA from nondividing cells from a stillbirth. The ability to use nondividing cells is important because no cell culture is required. (Cell culture is often difficult to obtain after stillbirth.) Depending on the array selected, the resolution can be as fine as a single nucleotide polymorphism.
aCGH can inform preconception counseling
Raca and colleagues used a range of arrays to assess 15 stillbirths that involved two or more malformations. Chromosomal abnormalities, including trisomy 21 and an unbalanced translocation, were detected by aCGH in two infants. Identification of these abnormalities helped inform counseling of the parents:
- In the case of trisomy 21, parental karyotypes revealed a nontranslocation event, making it possible to assure the parents that the risk of recurrence is low
- The unbalanced translocation resulted from a balanced chromosome translocation in the mother and was associated with a significant risk of recurrence (in this case, FISH would not have helped because chromosomes 13, 18, 21, X, and Y were not involved).
Limitations of aCGH
One limitation is an inability to detect polyploidy such as triploidy or tetraploidy. This problem can be circumvented through the use of a FISH preparation prior to aCGH.
In most centers, parental blood samples are drawn at the time of aCGH studies. Because aCGH offers greater resolution of chromosome regions, an increasing number of benign variations (i.e., present in one parent) are being identified. As aCGH technology advances, we are accumulating data on copy-number variations.
A large clinical trial is needed to assess the full potential of aCGH in this setting.
Use of array-basic comparative genomic hybridization to assess cells from a stillborn fetus can help determine the cause of death and inform counseling of the parents about the risk of recurrence.
Risk factors for stillbirth include
advanced maternal age, obesity, and black race
ACOG Practice Bulletin #102: Management of stillbirth. Obstet Gynecol. 2009;113:748–760.
Willinger M, Ko CW, Reddy UM. Racial disparities in stillbirth risk across gestation in the United States. Am J Obstet Gynecol. 2009;201:469.e1–469.e8.
Fretts RC. The study of stillbirth. Am J Obstet Gynecol. 2009;201:429–430.
Women who have diseases such as insulin-dependent diabetes and systemic lupus erythematosus have long been recognized as having a six- to 20-fold increase in the risk of stillbirth, compared with the general population. However, each of these disorders accounts for 2% and less than 1% of the pregnant population, respectively, so their overall contribution to stillbirth is small. Larger portions of the population have a lower—but still significant—risk of stillbirth:
- women older than 35 years
- women who have a body mass index (BMI) above 30
- non-Hispanic black women.
Each of these categories represents 15% or more of the typical obstetric population, and each group faces a risk of stillbirth approaching 1%. The ACOG practice bulletin and the study by Willinger and colleagues address these risks in detail.
Advanced maternal age is particularly risky among nulliparous women
Advanced maternal age (>35 years) is associated with increased rates of chromosomal abnormality and maternal morbidity, such as hypertension, that are known to raise the risk of stillbirth. Even when these and other variables associated with advanced maternal age, such as placenta previa, diabetes, and multiple gestation, are controlled, however, the increased risk of stillbirth remains.
Advanced maternal age in a first pregnancy carries a particularly elevated risk. For example, the risk of stillbirth in a 40-year-old nulliparous woman is more than twice the risk in a 40-year-old multiparous woman (1 in every 116 pregnancies vs 1 in every 304).3
The increased risk of stillbirth associated with advanced maternal age is present at all gestational ages, though it becomes most profound at 37 to 42 weeks’ gestation, notably for:
- women 35 to 39 years old (1 in every 382 pregnancies; relative risk [RR] of 1.32, compared with women <35 years old; 95% confidence interval [CI], 1.22, 1.43)
- women >40 years old (1 in every 267 pregnancies; RR, 1.88; 95% CI, 1.64, 2.16).
These numbers remain significant even after controlling for medical conditions.3
The utility of antepartum surveillance and induction of labor for delivery is unclear, given the risk of iatrogenic prematurity.
Risk of stillbirth is doubled among obese and markedly obese women
Although the number of adults who are overweight (BMI 25–30) has remained fairly constant over the past 20 years (30% to 35% of the population), the percentage of women of reproductive age who are obese (BMI >30) has risen markedly. Obesity is now present in 35% of the population, and marked obesity (BMI >40) affects an additional 6%. Both obese and markedly obese women face a twofold relative risk of stillbirth, compared with women of normal weight. The rate of stillbirth in this population is 12 to 18 for every 1,000 births—a 1.2% to 1.8% risk.
Although obesity-related stillbirth likely has multiple causes, the risk remains elevated even after exclusion of confounding factors such as smoking, gestational diabetes, and preeclampsia.
Race is an independent contributor
Racial differences in the rate of stillbirth remain despite a decrease in the overall stillbirth rate over the past 20 years ( FIGURE ). In 2003, the rate of stillbirth was 5 for every 1,000 births among non-Hispanic whites, 5.5 among Hispanics, and 12 among non-Hispanic blacks. In other words, the risk of stillbirth was 1 in 202, 1 in 183, and 1 in 87 births for white, Hispanic, and black women, respectively.
Willinger and colleagues utilized data from the National Center for Health Statistics and assessed 2001–2002 birth and infant death datasets for 36 states, examining the stillbirth hazard risk for more than 5 million singleton pregnancies. Stillbirth peaked at 20 to 23 weeks and 39 to 41 weeks’ gestation, as expected. However, at 20 to 23 weeks, the risk of stillbirth among non-Hispanic black women was more than twice the rate for non-Hispanic white women (RR, 2.8). Although it then declined as term approached, it remained greater than that of non-Hispanic white women (RR, 1.6).
FIGURE Racial disparities in the risk of stillbirth
Hazard of stillbirth for singleton pregnancies by gestational age and race and ethnicity, 2001–2002. SOURCE: Willinger et al. Greater acceptance and use of induction of labor at term among whites merits attention
In an editorial accompanying the study by Willinger and colleagues, Fretts pointed out the higher rate of induction of labor at term among white women that has been observed in at least three studies of vital statistics. (Willinger and colleagues also pointed out this difference.) The acceptance and use of labor induction at term—and the lower stillbirth rate—among white women warrants further investigation.
Education appears to reduce the risk of stillbirth to a greater degree among whites than it does among blacks. Again, nulliparity and advanced maternal age were important contributors to the risk of stillbirth across all three races.
Counsel African-American gravidas and women older than 35 years that their risk of stillbirth is elevated.
Obese women should be advised to lose weight before conception if at all possible to reduce the risk of stillbirth.
Needed: Standardized analysis
and documentation of stillbirth
Reddy UM, Goldberg R, Silver R, et al. Stillbirth classification—developing an international consensus for research: executive summary of a National Institute of Child Health and Human Development workshop. Obstet Gynecol. 2009;114:901–914.
Flenady V, Frøen JF, Pinar H, et al. An evaluation of classification systems for stillbirth. BMC Pregnancy Childbirth. 2009;9:24.
Further guidance for the clinical management of stillbirth will come from investigations of the underlying pathologies and associated risk factors. Key to development of this guidance is the involvement of obstetricians in documenting the antenatal record and delivery information. Also needed is a standardized system for recording this information. More than three dozen systems have been developed to classify stillbirth, at the expense of uniformity of content.
An international consensus group published guidelines on how to describe the cause of death in research endeavors, recognizing the need to maintain the ability to attach a level of uncertainty. In addition, Flenady and colleagues compared the most widely used systems in clinical practice, assigning the highest score for components such as ease of use, inter observer variability, and proportion of unexplained stillbirths to CODAC [cause of death and two associated causes]. This system assigns a primary cause of death from a specified list of choices and allows inclusion of two possible contributing causes.
Both the international consensus classification and the CODAC scoring system are accessible through links embedded within the articles. Both systems require the establishment of standardized evaluation and review of stillbirth that should include obstetricians, pathologists, and geneticists.
Because assessment and classification of stillbirth are fundamental to its prevention, as well as a critical part of clinical practice, ObGyns should become familiar with the international consensus classification and CODAC scoring systems and adopt a standardized approach to assessment and documentation.
Three important areas of research into stillbirth have evolved over the past year, furthering our understanding of the phenomenon and our ability to provide comprehensive, evidence-based care:
- Genetic studies. Karyotype analysis is useful in determining the cause of stillbirth, especially when analysis is based on a sample of amniotic fluid that was obtained before delivery. And array-based comparative genomic hybridization, which yields information on the chromosome count as well as micro-duplications and deletions, can be performed on nondividing cells.
- Risk factors. Further investigation implicates advanced maternal age, obesity, and African-American race.
- Classification. Paring down the more than three dozen systems that exist for classification of stillbirth was the main challenge addressed by an international consensus group in 2009 and the focus of a separate analysis.
The individual studies that contribute to our knowledge base in these areas are discussed in more detail in the articles that follow.
Stillbirth is broadly defined as fetal demise after 20 weeks’ gestation and with a fetal weight exceeding 350 g. In the United States, stillbirth occurs in 1 of every 160 live births (6 stillbirths for every 1,000 live births). Although the rate of neonatal demise has decreased over the past decade, the rate of stillbirth has declined less strikingly.
For an analysis of karyotype, amniotic fluid is best
Korteweg FJ, Bouman K, Erwich JJ, et al. Cytogenetic analysis after evaluation of 750 fetal deaths: proposal for diagnosis workup. Obstet Gynecol. 2008;111:865–874.
ACOG Practice Bulletin #102: Management of stillbirth. Obstet Gynecol. 2009;113:748–760.
When stillbirth occurs, determination of the cause of death fulfills several goals:
- It informs counseling of the parents, who must come to terms with the loss
- It aids in determining the risk of recurrence, which informs family planning
- It furthers research into stillbirth and facilitates the comparison of national and international data.
Chromosomal anomaly is one potential cause of stillbirth. Its frequency depends on the presence of structural malformation. For example, Korteweg and colleagues found a rate of chromosomal anomaly of 4.6% among stillbirths involving fetuses without structural abnormality, but the rate rose to 38% when anatomic malformation was present. The distribution of chromosomes among stillbirths mirrored the pattern seen in live births, including 45, X and trisomies of chromosome 21, 13, and 18.
The utility of karyotype assessment when ultrasonography (US) has not identified structural malformation has been debated. Given the 5% incidence of chromosomal anomaly in the absence of structural abnormality, and the limitations of US in detecting subtle dysmorphology, a karyotype seems advisable to assess all stillbirths.
Comparison of methods points to superiority of amniocentesis
Because fewer than 20% of skin biopsies result in a useful culture, postmortem skin biopsy for karyotype assessment is unreliable. Korteweg and colleagues evaluated other methods of obtaining cells for examination and found that a successful karyotype is most likely with predelivery amniocentesis (85%), followed by umbilical cord culture (32.1%). A karyotype of cells from fascia lata and skin biopsy yielded poor results, especially in the setting of maceration. Placental biopsy is likely to provide an adequate karyotype (71% probability) but findings may be confounded by confined placental mosaicism.1
ACOG also advocates predelivery amniocentesis
In its 2009 practice bulletin, ACOG supported inclusion of amniocentesis in the assessment of stillbirth and preparation for delivery. Once an epidural is placed, amniocentesis provides cells for karyotype assessment, polymerase chain reaction (PCR) for viral studies, and any other metabolic or specific genetic studies that may be indicated by fetopsy.
If amniocentesis is not performed, ACOG recommends umbilical cord culture as an alternative. Because nondividing cells can be utilized in fluorescence in situ hybridization (FISH) for chromosome 13, 18, 21, X, and Y, this method should be considered in any case involving culture failure (TABLE).2
TABLE
Genetic components of stillbirth assessment
| Type of assessment | Steps |
|---|---|
| Inspection of fetus and placenta | Measure head circumference and length of fetus |
| Weigh fetus and placenta | |
| Photograph fetus and placenta, including frontal and profile shots of whole body, face, extremities, palms, and any abnormality | |
| Document findings | |
| Cytologic analysis | Obtain consent from parents |
| Obtain acceptable specimens using one of the following sterile techniques: | |
| • Amniocentesis at the time of prenatal diagnosis of demise • Placental block (1 x 1 cm) taken from below the cord-insertion site on the unfixed placenta • Umbilical cord segment (1.5 cm) • Internal fetal tissue specimen, e.g., costochondral junction or patella (not skin) | |
| Preserve specimens in a sterile culture medium of lactated Ringer’s solution at room temperature during transfer to laboratory | |
| Fetopsy | Obtain parental consent; if no consent is given, send placenta for pathologic analysis |
| Perform autopsy and pathologic assessment of the placenta | |
| Consider whole-body fetal radiographs | |
| Source: ACOG Practice Bulletin #102 | |
Perform predelivery amniocentesis whenever possible at the time of diagnosis of demise to obtain a cell sample for karyotype analysis to determine the cause of death.
Array-based comparative genomic hybridization makes assessment of nondividing cells possible
Raca G, Artzer A, Thorson L, et al. Array-based comparative hybridization (aCGH) in the genetic evaluation of stillbirth. Am J Med Genet A. 2009;149A:2437–2443.
Array-based comparative genomic hybridization (aCGH) makes it possible to assess the chromosome count and perform a high-resolution search for microduplications and deletions. With known segments of the genome printed on slides, the clinical scientist can analyze DNA from nondividing cells from a stillbirth. The ability to use nondividing cells is important because no cell culture is required. (Cell culture is often difficult to obtain after stillbirth.) Depending on the array selected, the resolution can be as fine as a single nucleotide polymorphism.
aCGH can inform preconception counseling
Raca and colleagues used a range of arrays to assess 15 stillbirths that involved two or more malformations. Chromosomal abnormalities, including trisomy 21 and an unbalanced translocation, were detected by aCGH in two infants. Identification of these abnormalities helped inform counseling of the parents:
- In the case of trisomy 21, parental karyotypes revealed a nontranslocation event, making it possible to assure the parents that the risk of recurrence is low
- The unbalanced translocation resulted from a balanced chromosome translocation in the mother and was associated with a significant risk of recurrence (in this case, FISH would not have helped because chromosomes 13, 18, 21, X, and Y were not involved).
Limitations of aCGH
One limitation is an inability to detect polyploidy such as triploidy or tetraploidy. This problem can be circumvented through the use of a FISH preparation prior to aCGH.
In most centers, parental blood samples are drawn at the time of aCGH studies. Because aCGH offers greater resolution of chromosome regions, an increasing number of benign variations (i.e., present in one parent) are being identified. As aCGH technology advances, we are accumulating data on copy-number variations.
A large clinical trial is needed to assess the full potential of aCGH in this setting.
Use of array-basic comparative genomic hybridization to assess cells from a stillborn fetus can help determine the cause of death and inform counseling of the parents about the risk of recurrence.
Risk factors for stillbirth include
advanced maternal age, obesity, and black race
ACOG Practice Bulletin #102: Management of stillbirth. Obstet Gynecol. 2009;113:748–760.
Willinger M, Ko CW, Reddy UM. Racial disparities in stillbirth risk across gestation in the United States. Am J Obstet Gynecol. 2009;201:469.e1–469.e8.
Fretts RC. The study of stillbirth. Am J Obstet Gynecol. 2009;201:429–430.
Women who have diseases such as insulin-dependent diabetes and systemic lupus erythematosus have long been recognized as having a six- to 20-fold increase in the risk of stillbirth, compared with the general population. However, each of these disorders accounts for 2% and less than 1% of the pregnant population, respectively, so their overall contribution to stillbirth is small. Larger portions of the population have a lower—but still significant—risk of stillbirth:
- women older than 35 years
- women who have a body mass index (BMI) above 30
- non-Hispanic black women.
Each of these categories represents 15% or more of the typical obstetric population, and each group faces a risk of stillbirth approaching 1%. The ACOG practice bulletin and the study by Willinger and colleagues address these risks in detail.
Advanced maternal age is particularly risky among nulliparous women
Advanced maternal age (>35 years) is associated with increased rates of chromosomal abnormality and maternal morbidity, such as hypertension, that are known to raise the risk of stillbirth. Even when these and other variables associated with advanced maternal age, such as placenta previa, diabetes, and multiple gestation, are controlled, however, the increased risk of stillbirth remains.
Advanced maternal age in a first pregnancy carries a particularly elevated risk. For example, the risk of stillbirth in a 40-year-old nulliparous woman is more than twice the risk in a 40-year-old multiparous woman (1 in every 116 pregnancies vs 1 in every 304).3
The increased risk of stillbirth associated with advanced maternal age is present at all gestational ages, though it becomes most profound at 37 to 42 weeks’ gestation, notably for:
- women 35 to 39 years old (1 in every 382 pregnancies; relative risk [RR] of 1.32, compared with women <35 years old; 95% confidence interval [CI], 1.22, 1.43)
- women >40 years old (1 in every 267 pregnancies; RR, 1.88; 95% CI, 1.64, 2.16).
These numbers remain significant even after controlling for medical conditions.3
The utility of antepartum surveillance and induction of labor for delivery is unclear, given the risk of iatrogenic prematurity.
Risk of stillbirth is doubled among obese and markedly obese women
Although the number of adults who are overweight (BMI 25–30) has remained fairly constant over the past 20 years (30% to 35% of the population), the percentage of women of reproductive age who are obese (BMI >30) has risen markedly. Obesity is now present in 35% of the population, and marked obesity (BMI >40) affects an additional 6%. Both obese and markedly obese women face a twofold relative risk of stillbirth, compared with women of normal weight. The rate of stillbirth in this population is 12 to 18 for every 1,000 births—a 1.2% to 1.8% risk.
Although obesity-related stillbirth likely has multiple causes, the risk remains elevated even after exclusion of confounding factors such as smoking, gestational diabetes, and preeclampsia.
Race is an independent contributor
Racial differences in the rate of stillbirth remain despite a decrease in the overall stillbirth rate over the past 20 years ( FIGURE ). In 2003, the rate of stillbirth was 5 for every 1,000 births among non-Hispanic whites, 5.5 among Hispanics, and 12 among non-Hispanic blacks. In other words, the risk of stillbirth was 1 in 202, 1 in 183, and 1 in 87 births for white, Hispanic, and black women, respectively.
Willinger and colleagues utilized data from the National Center for Health Statistics and assessed 2001–2002 birth and infant death datasets for 36 states, examining the stillbirth hazard risk for more than 5 million singleton pregnancies. Stillbirth peaked at 20 to 23 weeks and 39 to 41 weeks’ gestation, as expected. However, at 20 to 23 weeks, the risk of stillbirth among non-Hispanic black women was more than twice the rate for non-Hispanic white women (RR, 2.8). Although it then declined as term approached, it remained greater than that of non-Hispanic white women (RR, 1.6).
FIGURE Racial disparities in the risk of stillbirth
Hazard of stillbirth for singleton pregnancies by gestational age and race and ethnicity, 2001–2002. SOURCE: Willinger et al. Greater acceptance and use of induction of labor at term among whites merits attention
In an editorial accompanying the study by Willinger and colleagues, Fretts pointed out the higher rate of induction of labor at term among white women that has been observed in at least three studies of vital statistics. (Willinger and colleagues also pointed out this difference.) The acceptance and use of labor induction at term—and the lower stillbirth rate—among white women warrants further investigation.
Education appears to reduce the risk of stillbirth to a greater degree among whites than it does among blacks. Again, nulliparity and advanced maternal age were important contributors to the risk of stillbirth across all three races.
Counsel African-American gravidas and women older than 35 years that their risk of stillbirth is elevated.
Obese women should be advised to lose weight before conception if at all possible to reduce the risk of stillbirth.
Needed: Standardized analysis
and documentation of stillbirth
Reddy UM, Goldberg R, Silver R, et al. Stillbirth classification—developing an international consensus for research: executive summary of a National Institute of Child Health and Human Development workshop. Obstet Gynecol. 2009;114:901–914.
Flenady V, Frøen JF, Pinar H, et al. An evaluation of classification systems for stillbirth. BMC Pregnancy Childbirth. 2009;9:24.
Further guidance for the clinical management of stillbirth will come from investigations of the underlying pathologies and associated risk factors. Key to development of this guidance is the involvement of obstetricians in documenting the antenatal record and delivery information. Also needed is a standardized system for recording this information. More than three dozen systems have been developed to classify stillbirth, at the expense of uniformity of content.
An international consensus group published guidelines on how to describe the cause of death in research endeavors, recognizing the need to maintain the ability to attach a level of uncertainty. In addition, Flenady and colleagues compared the most widely used systems in clinical practice, assigning the highest score for components such as ease of use, inter observer variability, and proportion of unexplained stillbirths to CODAC [cause of death and two associated causes]. This system assigns a primary cause of death from a specified list of choices and allows inclusion of two possible contributing causes.
Both the international consensus classification and the CODAC scoring system are accessible through links embedded within the articles. Both systems require the establishment of standardized evaluation and review of stillbirth that should include obstetricians, pathologists, and geneticists.
Because assessment and classification of stillbirth are fundamental to its prevention, as well as a critical part of clinical practice, ObGyns should become familiar with the international consensus classification and CODAC scoring systems and adopt a standardized approach to assessment and documentation.
1. Rodgers CS, Creasy MR, Fitchett M, Maliszewska CT, Pratt NR, Waters JJ. Solid tissue culture for cytogenetic analysis: a collaborative survey for the Association of Clinical Cytogeneticists. J Clin Pathol. 1996;49:638-641.
2. Rivasi F, Schirosi L, Bettelli S, et al. FISH analysis in cell touch preparations and cytological specimens from formalin-fixed fetal autopsies. Diagn Cytopathol. 2008;36:633-636.
3. Reddy UM, Ko CW, Willinger M. Maternal age and the risk of stillbirth throughout pregnancy in the United States. Am J Obstet Gynecol. 2006;195:764-770.
4. MacDorman MF, Mathews TJ. NCHS Data Brief #9: Recent trends in infant mortality in the United States. Atlanta, Ga: National Center for Health Statistics; October 2008. Available at: http://www.cdc.gov/nchs/data/databriefs/db09.htm. Accessed Dec. 15, 2009.
1. Rodgers CS, Creasy MR, Fitchett M, Maliszewska CT, Pratt NR, Waters JJ. Solid tissue culture for cytogenetic analysis: a collaborative survey for the Association of Clinical Cytogeneticists. J Clin Pathol. 1996;49:638-641.
2. Rivasi F, Schirosi L, Bettelli S, et al. FISH analysis in cell touch preparations and cytological specimens from formalin-fixed fetal autopsies. Diagn Cytopathol. 2008;36:633-636.
3. Reddy UM, Ko CW, Willinger M. Maternal age and the risk of stillbirth throughout pregnancy in the United States. Am J Obstet Gynecol. 2006;195:764-770.
4. MacDorman MF, Mathews TJ. NCHS Data Brief #9: Recent trends in infant mortality in the United States. Atlanta, Ga: National Center for Health Statistics; October 2008. Available at: http://www.cdc.gov/nchs/data/databriefs/db09.htm. Accessed Dec. 15, 2009.
PRENATAL COUNSELING
Population-based screening for carriers of genetic diseases and advances in neonatal and pediatric genetic testing have resulted in more and more couples identified as at-risk for inherited disorders. Increasingly, women in these couples ask their ObGyn about their options for future pregnancies.
For some women, genetic testing of a pregnancy as early as possible—even before implantation—is desirable. In vitro fertilization affords such direct access to the genetic material of either gametes before fertilization (i.e., polar-body biopsy) or blastomeres once fertilization has occurred (blastomere biopsy). Complex genetic analysis of these single cells is now possible. Because polar-body biopsy is restricted to testing for maternal disease, blastomere biopsy has gained favor as the method of choice for genetic testing of preimplantation pregnancies.
The duality of genetic testing
Regardless of what genetic material is tested, preimplantation genetic testing encompasses two distinct categories: preimplantation genetic diagnosis, or PGD, and preimplantation genetic screening, or PGS.
What is PGD?
Here, testing is confined to women at risk of an offspring with an identified genetic abnormality. These women, or their partner, typically carry a gene mutation that, alone or in combination with another mutation in the same gene, would result in an identifiable outcome in their child (for example, autosomal-recessive, autosomal-dominant, and X-linked disorders).
PGD, by definition, also includes testing of women, or their partner, who possess a balanced chromosome rearrangement (translocation, inversion). Offspring of carriers of balanced chromosome rearrangements are at increased risk of particular genetic abnormalities, as a result of unbalanced segregation of chromosomes involved in their rearrangement.
How does PGS differ from PGD?
Screening, in contrast, focuses analysis on offspring of women who are theoretically at increased risk of a genetic abnormality based on their age or reproductive history, not on their genetic makeup. PGS looks specifically for chromosomal content, and is based on the premise that decreasing the rate of aneuploidy among the conceptions of women 1) of advanced maternal age, 2) who experience habitual miscarriage, or 3) who have failed multiple cycles of in vitro fertilization (IVF) would increase the rate of implantation and, ultimately, the live birth rate.
The articles below, beginning with a committee opinion from the American Society for Reproductive Medicine (ASRM), address the following:
- evidence in support of PGD for genetic disease
- caution about using PGS, in its current format, for aneuploidy screening.
Practice Committee of the Society for Assisted Reproductive Technology; Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril. 2007;88:1497–1504.
A gene mutation carried by one or both parents can increase the risk that their offspring will be affected with an inherited condition. Common examples include autosomal-recessive disorders such as cystic fibrosis; autosomal-dominant disorders such as neurofibromatosis; and X-linked disorders such as hemophilia A.
Recently, human leukocyte antigens (HLA) have been assessed in conjunction with testing for specific genetic diseases, such as Fanconi anemia. In these settings, the intent is to recognize not only the blastomeres that are free of Fanconi anemia, but also those that are potential HLA matches and, therefore, potential donors for an (older) affected sibling.
PGD has been extended to women, or their partner, who possess a gene mutation that places them at increased risk of cancer (such as BRCA-1) and who wish to avoid transmitting that risk-conferring gene to their offspring.
For these diseases, and for many others, knowledge of the specific genetic mutation enables similar molecular testing to be accomplished on a single cell, such as a blastomere.
Technical concerns of testing must be part
of the physician–patient discussion
Typically, PGD analysis is initiated by polymerase chain reaction (PCR) of DNA content extracted from the single cell. This is followed by application of mutation-appropriate molecular technology. Given 1) the short time in which these PGD results are needed (often, 24 to 48 hours) and 2) the limited amount of genetic material available for analysis, technical restraints on testing are recognized:
- Extraneous DNA contamination remains a problem with molecular technology, despite application of intracytoplasmic sperm injection
- Only partial amplification of the allele may occur, or allele “drop-out” may be present; both of these phenomena can cause false-negative results
- Error can occur dually: 1) Presumably unaffected embryos that are, indeed, affected are transferred and 2) actually normal embryos that have been interpreted incorrectly as abnormal are discarded
- The rate of misdiagnosis (false-negative results) ranges from 2% (with autosomal-recessive disorders) to 10% (with autosomal-dominant disorders), although this rate can be lessened with the use of linked markers.
PGD for investigating balanced chromosome rearrangements
These rearrangements represent another type of genetic abnormality in which PGD can reduce the likelihood of a conception that carries a specific genetic abnormality.
When one parent carries a balanced chromosome translocation, fluorescence in-situ hybridization (FISH) can be applied to assess the segregation of at-risk chromosomes in a single blastomere cell. In this technique, fluorescence-labeled DNA probes, selected for specificity to the translocation in question, are applied to the single cell fixed on a glass slide. Copies of the DNA segment and, by inference, the chromosomal segment in question are assessed by quantification of the sites of positive fluorescence.
Because translocation carriers are, theoretically, at high risk of transmission of an unbalanced segregant to the blastomere, as many as 10 blastomeres will often be screened until one or two are deemed normal for the FISH probes in question. When implantation does succeed after FISH analysis for a chromosome rearrangement, however, the pregnancy loss rate is lower and the likelihood of a live birth is higher.
Again, in-depth consultation is needed before PGD
Whether PGD is planned for investigating a single-gene disorder or a chromosome translocation, detailed consultation with the woman or the couple is important. This effort should include not only genetic counseling about inheritance, the natural history of the disorder in question, and other options for avoiding the transmission of the disorder—in addition, additional time should be spent describing:
- risks associated with IVF procedures and embryo biopsy (and with extended culture, if needed)
- technical limitations of the particular testing that is being considered
- options for prenatal testing during a pregnancy
- the possibility that embryos suitable for transfer will not be found (and that, potentially, erroneously tested normal embryos will not be transferred)
- disposition of embryos in which test results are inconclusive.
PGS for women at increased risk of aneuploidy isn’t supported by evidence; consider it investigational
Mastenbroek S, Twisk M, van Echten-Arends J, et al. In vitro fertilization with preimplantation genetic screening. N Engl J Med. 2007;357:9–17.
Mersereau JE, Pergament E, Zhang X, Milad MP. Preimplantation genetic screening to improve in vitro fertilization pregnancy rates: a prospective randomized controlled trial. Fertil Steril. 2008;90:1287–1289.
Aneuploidy contributes to pregnancy loss among women as they become older. Theoretically, avoiding aneuploid pregnancy among embryos transferred during IVF cycles—in older women and in women experiencing multiple pregnancy losses and failed IVF cycles—was expected to increase the implantation rate and decrease the rate of pregnancy loss.
This hypothesis was supported, at first, by observational trials. But at least one randomized study, by Staessen and colleagues,1 failed to demonstrate that PGS is beneficial in women of advanced maternal age.
Now, a large multicenter, randomized, double-blind, controlled trial conducted by Mastenbroek and co-workers provides further evidence that PGS does not increase the rate of pregnancy and, in fact, significantly reduces that rate among women of advanced maternal age.
The Mastenbroek study compared outcomes among 206 women who had PGS and 202 women who did not. Both groups were matched for maternal age older than 35 years. Blastomeres were analyzed for eight chromosomes, including those known to be highly associated with miscarriage (1, 16, 17, 13, 18, and 21; X and Y).
Among women who underwent PGS, 25% had an ongoing pregnancy of at least 12 weeks’ gestation, compared with 37% of unscreened women. A similar higher rate of live birth was seen among unscreened women (35%, versus 24% in the PGS group).
Mastenbroek’s results are comparable to what was reported from an earlier randomized trial of PGS,1 in which the implantation rate as the primary outcome among women who had PGS and among controls was not significantly different. Contributors to 1) the lack of success of PGS and 2) the apparent detriment of PGS to the ongoing pregnancy rate include:
- potential for damage to the embryo at biopsy
- limitations imposed by FISH technology on the number of probes that can be accurately assessed technically
- a growing knowledge that a significant percentage of embryos are chromosomal mosaics at this stage—a phenomenon that likely results in nontransfer of embryos that have the potential for developing karyotypically normally.
Does PGS improve outcomes?
More recently, Mersereau and colleagues reported pilot results from a prospective, randomized, controlled trial that assessed whether PGS could improve pregnancy outcomes. Here, selection of infertile women for the study was not restricted to poor prognosis categories, such as advanced maternal age and recurrent pregnancy loss.
Using the live birth rate as the outcome measure, PGS for seven chromosomes was determined not to be associated with a significantly increased live birth rate among screened pregnancies. Sample sizes had been calculated to establish, with significance, a 50% increase in live births—from 30% in the control (unscreened) population to 45% in the screened population. Secondary endpoints, such as the implantation rate and pregnancy loss, also did not differ significantly between the PGS cases and controls.
Again, technical difficulties of two-blastomere biopsy, with its potential for embryo damage, and the presence of underlying embryo mosaicism represent possible barriers to improving the live birth rate when utilizing PGS.
Technical limitations may be one of the largest obstacles
to applying PGS
Practice Committee of the Society for Assisted Reproductive Technology; Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril. 2007;88:1497–1504.
FISH probes can be chosen to reflect the nature of a given patient’s risk (advanced maternal age, recurrent pregnancy loss) when performing PGS, but the technique itself is limited by the number of probe sites that can be interpreted accurately at one time. Typically, analysis of more than five chromosomes requires two cycles of hybridization, with their associated time requirement and potential for degradation of the single cell.
Alternatively, advances in the analysis of all 23 chromosomes through comparative genomic hybridization may, ultimately, provide an avenue for applying PGS. At the moment, time limitations prohibit comparative genomic hybridization without embryo cryopreservation. Further investigation of other technical limitations, such as the high rate of mosaicism, has revealed that, when two cells are examined and found to be karyotypically discordant, further analysis of the entire embryo will reveal that more than 50% of embryos are, in fact, euploid—that is, chromosomally normal. Random biopsy of the abnormal cell solely would relegate the embryo to nontransfer, despite the predominance of an underlying euploid state.
Understanding of the potential that embryos have to self-correct early mosaicism is growing; we now know that almost one half of embryos identified as aneuploid at cleavage stage correct to euploid if they survive to blastocyst stage. A karyotypic abnormality in a single cell from a day-3 embryo does not always signal an abnormal embryo.
ASRM does not support PGS to improve the live birth rate
This determination by ASRM is based on available evidence about advanced maternal age, recurrent pregnancy loss, recurrent implantation failure, and recurrent aneuploidy loss:
- In women of advanced maternal age, many day-3 embryos display aneuploidy when studied by FISH. In theory, exclusion of these embryos for transfer should improve implantation and live birth rates, but evidence does not support that premise.
- Because almost 70% of spontaneous pregnancy loss is caused by a karyotypic abnormality, and women with karyotypically recurrent pregnancy loss are more likely to experience subsequent loss with karyotype abnormalities, the premise of preimplantation screening for aneuploidy also appeared to be well founded. Studies at this time are limited to retrospective series, without randomized controlled trials published.
- Among women who experience repeated implantation failure, a finding of more than 50% abnormal embryos isn’t uncommon, yet several studies have not supported an increased implantation rate or live birth rate after PGS.
A literature review of PGS calls its introduction “premature”
Gleicher N, Weghofer A, Barad D. Preimplantation genetic screening: “established” and ready for prime time? Fertil Steril. 2008;89:780–788.
After ASRM recognized PGD as an established technique in a 2001 committee opinion, extension of this status to PGS was inadvertently assumed. But PGS is a different testing modality—with different indications, risk/benefit profiles, and efficacy than PGD.
Today, FISH probes are utilized for PGS; the false-negative rate of FISH appears to be driven by the technical constraints of the technology. Potentially increasing the false-negative rate are inadequate hybridization and the use of increasing numbers of probes and hybridization cycles.
Conversely, the false-positive rate—the number of embryos not transferred that are, in fact, chromosomally normal—varies markedly from one study to another, and may be as high as 20% when discarded embryos are more completely assessed.
Similarly, laboratories utilize different methods of obtaining the genetic material. These methods range from biopsy of polar bodies to single-cell blastomere and routine two-cell blastomere biopsy—and, more recently, to blastocyst biopsy. The impact of these various embryo manipulations has yet to be fully considered. Whether biopsy affects the embryo has received little attention.
In fact, embryos that are of poor quality before biopsy—such as those found in women of advanced maternal age—may be more susceptible to the effects of biopsy. The outcome with such embryos may be of even greater detriment to the implantation rate (as discussed in regard to the Mastenbroek study earlier in this article).
The logic of performing PGS for aneuploidy in women of advanced maternal age was reasonable. But this group of women—in whom ovarian reserve is diminished, who respond poorly to ovulation induction, thereby limiting the total number of embryos for analysis and the poorer quality embryos possibly further impaired by the biopsy itself—represent the population that may be least amenable to PGS.
A further observation about PGS in women who have experienced recurrent pregnancy loss or IVF failure: Any impairment of embryos that is a consequence of the method of biopsy may further undermine the generally unsupportive results of PGS that have been documented in these patients.
Consensus on performing PGS
An assessment of European studies and practices reveals similar concerns voiced by the European Society for Human Reproduction and Embryology (ESHRE) PGD Consortium Steering Committee. The committee recently asserted a comparable opinion about “the insufficient data that demonstrate PGS is indeed a cost-effective alternative for standard IVF.”2 Gleicher and colleagues, in their review of the literature, conclude that the indications for PGS are currently undefined and, as such, screening should be considered experimental.
Gleicher’s sentiments echo the recommendations of ASRM that, when PGS is considered,
- patients undergo counseling about its limitations, risk of error, and lack of evidence that it improves the live-birth rate
- available evidence does not support improvement in the live birth rate in women of advanced maternal age, who have failed previous implantation, who have experienced recurrent pregnancy loss, or who have experienced recurrent pregnancy loss specifically related to aneuploidy
- decisions about management should not be based on aneuploidy results of prior PGS cycles for a woman who has experienced recurrent implantation failure.
1. Staessen C, Platteau P, Van Assche E, et al. Comparison of blastocyst transfer with and without preimplantation genetic diagnosis for aneuploidy screening in couples with advanced maternal age: a prospective randomized controlled trial. Hum Reprod. 2004;19:2849-2858.
2. Sermon KD, Michiels A, Harton G, et al. ESHRE PGD Consortium data collection VI: cycles from January to December 2003 with pregnancy follow-up to October 2004. Hum Reprod. 2007;22:323-336.
Population-based screening for carriers of genetic diseases and advances in neonatal and pediatric genetic testing have resulted in more and more couples identified as at-risk for inherited disorders. Increasingly, women in these couples ask their ObGyn about their options for future pregnancies.
For some women, genetic testing of a pregnancy as early as possible—even before implantation—is desirable. In vitro fertilization affords such direct access to the genetic material of either gametes before fertilization (i.e., polar-body biopsy) or blastomeres once fertilization has occurred (blastomere biopsy). Complex genetic analysis of these single cells is now possible. Because polar-body biopsy is restricted to testing for maternal disease, blastomere biopsy has gained favor as the method of choice for genetic testing of preimplantation pregnancies.
The duality of genetic testing
Regardless of what genetic material is tested, preimplantation genetic testing encompasses two distinct categories: preimplantation genetic diagnosis, or PGD, and preimplantation genetic screening, or PGS.
What is PGD?
Here, testing is confined to women at risk of an offspring with an identified genetic abnormality. These women, or their partner, typically carry a gene mutation that, alone or in combination with another mutation in the same gene, would result in an identifiable outcome in their child (for example, autosomal-recessive, autosomal-dominant, and X-linked disorders).
PGD, by definition, also includes testing of women, or their partner, who possess a balanced chromosome rearrangement (translocation, inversion). Offspring of carriers of balanced chromosome rearrangements are at increased risk of particular genetic abnormalities, as a result of unbalanced segregation of chromosomes involved in their rearrangement.
How does PGS differ from PGD?
Screening, in contrast, focuses analysis on offspring of women who are theoretically at increased risk of a genetic abnormality based on their age or reproductive history, not on their genetic makeup. PGS looks specifically for chromosomal content, and is based on the premise that decreasing the rate of aneuploidy among the conceptions of women 1) of advanced maternal age, 2) who experience habitual miscarriage, or 3) who have failed multiple cycles of in vitro fertilization (IVF) would increase the rate of implantation and, ultimately, the live birth rate.
The articles below, beginning with a committee opinion from the American Society for Reproductive Medicine (ASRM), address the following:
- evidence in support of PGD for genetic disease
- caution about using PGS, in its current format, for aneuploidy screening.
Practice Committee of the Society for Assisted Reproductive Technology; Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril. 2007;88:1497–1504.
A gene mutation carried by one or both parents can increase the risk that their offspring will be affected with an inherited condition. Common examples include autosomal-recessive disorders such as cystic fibrosis; autosomal-dominant disorders such as neurofibromatosis; and X-linked disorders such as hemophilia A.
Recently, human leukocyte antigens (HLA) have been assessed in conjunction with testing for specific genetic diseases, such as Fanconi anemia. In these settings, the intent is to recognize not only the blastomeres that are free of Fanconi anemia, but also those that are potential HLA matches and, therefore, potential donors for an (older) affected sibling.
PGD has been extended to women, or their partner, who possess a gene mutation that places them at increased risk of cancer (such as BRCA-1) and who wish to avoid transmitting that risk-conferring gene to their offspring.
For these diseases, and for many others, knowledge of the specific genetic mutation enables similar molecular testing to be accomplished on a single cell, such as a blastomere.
Technical concerns of testing must be part
of the physician–patient discussion
Typically, PGD analysis is initiated by polymerase chain reaction (PCR) of DNA content extracted from the single cell. This is followed by application of mutation-appropriate molecular technology. Given 1) the short time in which these PGD results are needed (often, 24 to 48 hours) and 2) the limited amount of genetic material available for analysis, technical restraints on testing are recognized:
- Extraneous DNA contamination remains a problem with molecular technology, despite application of intracytoplasmic sperm injection
- Only partial amplification of the allele may occur, or allele “drop-out” may be present; both of these phenomena can cause false-negative results
- Error can occur dually: 1) Presumably unaffected embryos that are, indeed, affected are transferred and 2) actually normal embryos that have been interpreted incorrectly as abnormal are discarded
- The rate of misdiagnosis (false-negative results) ranges from 2% (with autosomal-recessive disorders) to 10% (with autosomal-dominant disorders), although this rate can be lessened with the use of linked markers.
PGD for investigating balanced chromosome rearrangements
These rearrangements represent another type of genetic abnormality in which PGD can reduce the likelihood of a conception that carries a specific genetic abnormality.
When one parent carries a balanced chromosome translocation, fluorescence in-situ hybridization (FISH) can be applied to assess the segregation of at-risk chromosomes in a single blastomere cell. In this technique, fluorescence-labeled DNA probes, selected for specificity to the translocation in question, are applied to the single cell fixed on a glass slide. Copies of the DNA segment and, by inference, the chromosomal segment in question are assessed by quantification of the sites of positive fluorescence.
Because translocation carriers are, theoretically, at high risk of transmission of an unbalanced segregant to the blastomere, as many as 10 blastomeres will often be screened until one or two are deemed normal for the FISH probes in question. When implantation does succeed after FISH analysis for a chromosome rearrangement, however, the pregnancy loss rate is lower and the likelihood of a live birth is higher.
Again, in-depth consultation is needed before PGD
Whether PGD is planned for investigating a single-gene disorder or a chromosome translocation, detailed consultation with the woman or the couple is important. This effort should include not only genetic counseling about inheritance, the natural history of the disorder in question, and other options for avoiding the transmission of the disorder—in addition, additional time should be spent describing:
- risks associated with IVF procedures and embryo biopsy (and with extended culture, if needed)
- technical limitations of the particular testing that is being considered
- options for prenatal testing during a pregnancy
- the possibility that embryos suitable for transfer will not be found (and that, potentially, erroneously tested normal embryos will not be transferred)
- disposition of embryos in which test results are inconclusive.
PGS for women at increased risk of aneuploidy isn’t supported by evidence; consider it investigational
Mastenbroek S, Twisk M, van Echten-Arends J, et al. In vitro fertilization with preimplantation genetic screening. N Engl J Med. 2007;357:9–17.
Mersereau JE, Pergament E, Zhang X, Milad MP. Preimplantation genetic screening to improve in vitro fertilization pregnancy rates: a prospective randomized controlled trial. Fertil Steril. 2008;90:1287–1289.
Aneuploidy contributes to pregnancy loss among women as they become older. Theoretically, avoiding aneuploid pregnancy among embryos transferred during IVF cycles—in older women and in women experiencing multiple pregnancy losses and failed IVF cycles—was expected to increase the implantation rate and decrease the rate of pregnancy loss.
This hypothesis was supported, at first, by observational trials. But at least one randomized study, by Staessen and colleagues,1 failed to demonstrate that PGS is beneficial in women of advanced maternal age.
Now, a large multicenter, randomized, double-blind, controlled trial conducted by Mastenbroek and co-workers provides further evidence that PGS does not increase the rate of pregnancy and, in fact, significantly reduces that rate among women of advanced maternal age.
The Mastenbroek study compared outcomes among 206 women who had PGS and 202 women who did not. Both groups were matched for maternal age older than 35 years. Blastomeres were analyzed for eight chromosomes, including those known to be highly associated with miscarriage (1, 16, 17, 13, 18, and 21; X and Y).
Among women who underwent PGS, 25% had an ongoing pregnancy of at least 12 weeks’ gestation, compared with 37% of unscreened women. A similar higher rate of live birth was seen among unscreened women (35%, versus 24% in the PGS group).
Mastenbroek’s results are comparable to what was reported from an earlier randomized trial of PGS,1 in which the implantation rate as the primary outcome among women who had PGS and among controls was not significantly different. Contributors to 1) the lack of success of PGS and 2) the apparent detriment of PGS to the ongoing pregnancy rate include:
- potential for damage to the embryo at biopsy
- limitations imposed by FISH technology on the number of probes that can be accurately assessed technically
- a growing knowledge that a significant percentage of embryos are chromosomal mosaics at this stage—a phenomenon that likely results in nontransfer of embryos that have the potential for developing karyotypically normally.
Does PGS improve outcomes?
More recently, Mersereau and colleagues reported pilot results from a prospective, randomized, controlled trial that assessed whether PGS could improve pregnancy outcomes. Here, selection of infertile women for the study was not restricted to poor prognosis categories, such as advanced maternal age and recurrent pregnancy loss.
Using the live birth rate as the outcome measure, PGS for seven chromosomes was determined not to be associated with a significantly increased live birth rate among screened pregnancies. Sample sizes had been calculated to establish, with significance, a 50% increase in live births—from 30% in the control (unscreened) population to 45% in the screened population. Secondary endpoints, such as the implantation rate and pregnancy loss, also did not differ significantly between the PGS cases and controls.
Again, technical difficulties of two-blastomere biopsy, with its potential for embryo damage, and the presence of underlying embryo mosaicism represent possible barriers to improving the live birth rate when utilizing PGS.
Technical limitations may be one of the largest obstacles
to applying PGS
Practice Committee of the Society for Assisted Reproductive Technology; Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril. 2007;88:1497–1504.
FISH probes can be chosen to reflect the nature of a given patient’s risk (advanced maternal age, recurrent pregnancy loss) when performing PGS, but the technique itself is limited by the number of probe sites that can be interpreted accurately at one time. Typically, analysis of more than five chromosomes requires two cycles of hybridization, with their associated time requirement and potential for degradation of the single cell.
Alternatively, advances in the analysis of all 23 chromosomes through comparative genomic hybridization may, ultimately, provide an avenue for applying PGS. At the moment, time limitations prohibit comparative genomic hybridization without embryo cryopreservation. Further investigation of other technical limitations, such as the high rate of mosaicism, has revealed that, when two cells are examined and found to be karyotypically discordant, further analysis of the entire embryo will reveal that more than 50% of embryos are, in fact, euploid—that is, chromosomally normal. Random biopsy of the abnormal cell solely would relegate the embryo to nontransfer, despite the predominance of an underlying euploid state.
Understanding of the potential that embryos have to self-correct early mosaicism is growing; we now know that almost one half of embryos identified as aneuploid at cleavage stage correct to euploid if they survive to blastocyst stage. A karyotypic abnormality in a single cell from a day-3 embryo does not always signal an abnormal embryo.
ASRM does not support PGS to improve the live birth rate
This determination by ASRM is based on available evidence about advanced maternal age, recurrent pregnancy loss, recurrent implantation failure, and recurrent aneuploidy loss:
- In women of advanced maternal age, many day-3 embryos display aneuploidy when studied by FISH. In theory, exclusion of these embryos for transfer should improve implantation and live birth rates, but evidence does not support that premise.
- Because almost 70% of spontaneous pregnancy loss is caused by a karyotypic abnormality, and women with karyotypically recurrent pregnancy loss are more likely to experience subsequent loss with karyotype abnormalities, the premise of preimplantation screening for aneuploidy also appeared to be well founded. Studies at this time are limited to retrospective series, without randomized controlled trials published.
- Among women who experience repeated implantation failure, a finding of more than 50% abnormal embryos isn’t uncommon, yet several studies have not supported an increased implantation rate or live birth rate after PGS.
A literature review of PGS calls its introduction “premature”
Gleicher N, Weghofer A, Barad D. Preimplantation genetic screening: “established” and ready for prime time? Fertil Steril. 2008;89:780–788.
After ASRM recognized PGD as an established technique in a 2001 committee opinion, extension of this status to PGS was inadvertently assumed. But PGS is a different testing modality—with different indications, risk/benefit profiles, and efficacy than PGD.
Today, FISH probes are utilized for PGS; the false-negative rate of FISH appears to be driven by the technical constraints of the technology. Potentially increasing the false-negative rate are inadequate hybridization and the use of increasing numbers of probes and hybridization cycles.
Conversely, the false-positive rate—the number of embryos not transferred that are, in fact, chromosomally normal—varies markedly from one study to another, and may be as high as 20% when discarded embryos are more completely assessed.
Similarly, laboratories utilize different methods of obtaining the genetic material. These methods range from biopsy of polar bodies to single-cell blastomere and routine two-cell blastomere biopsy—and, more recently, to blastocyst biopsy. The impact of these various embryo manipulations has yet to be fully considered. Whether biopsy affects the embryo has received little attention.
In fact, embryos that are of poor quality before biopsy—such as those found in women of advanced maternal age—may be more susceptible to the effects of biopsy. The outcome with such embryos may be of even greater detriment to the implantation rate (as discussed in regard to the Mastenbroek study earlier in this article).
The logic of performing PGS for aneuploidy in women of advanced maternal age was reasonable. But this group of women—in whom ovarian reserve is diminished, who respond poorly to ovulation induction, thereby limiting the total number of embryos for analysis and the poorer quality embryos possibly further impaired by the biopsy itself—represent the population that may be least amenable to PGS.
A further observation about PGS in women who have experienced recurrent pregnancy loss or IVF failure: Any impairment of embryos that is a consequence of the method of biopsy may further undermine the generally unsupportive results of PGS that have been documented in these patients.
Consensus on performing PGS
An assessment of European studies and practices reveals similar concerns voiced by the European Society for Human Reproduction and Embryology (ESHRE) PGD Consortium Steering Committee. The committee recently asserted a comparable opinion about “the insufficient data that demonstrate PGS is indeed a cost-effective alternative for standard IVF.”2 Gleicher and colleagues, in their review of the literature, conclude that the indications for PGS are currently undefined and, as such, screening should be considered experimental.
Gleicher’s sentiments echo the recommendations of ASRM that, when PGS is considered,
- patients undergo counseling about its limitations, risk of error, and lack of evidence that it improves the live-birth rate
- available evidence does not support improvement in the live birth rate in women of advanced maternal age, who have failed previous implantation, who have experienced recurrent pregnancy loss, or who have experienced recurrent pregnancy loss specifically related to aneuploidy
- decisions about management should not be based on aneuploidy results of prior PGS cycles for a woman who has experienced recurrent implantation failure.
Population-based screening for carriers of genetic diseases and advances in neonatal and pediatric genetic testing have resulted in more and more couples identified as at-risk for inherited disorders. Increasingly, women in these couples ask their ObGyn about their options for future pregnancies.
For some women, genetic testing of a pregnancy as early as possible—even before implantation—is desirable. In vitro fertilization affords such direct access to the genetic material of either gametes before fertilization (i.e., polar-body biopsy) or blastomeres once fertilization has occurred (blastomere biopsy). Complex genetic analysis of these single cells is now possible. Because polar-body biopsy is restricted to testing for maternal disease, blastomere biopsy has gained favor as the method of choice for genetic testing of preimplantation pregnancies.
The duality of genetic testing
Regardless of what genetic material is tested, preimplantation genetic testing encompasses two distinct categories: preimplantation genetic diagnosis, or PGD, and preimplantation genetic screening, or PGS.
What is PGD?
Here, testing is confined to women at risk of an offspring with an identified genetic abnormality. These women, or their partner, typically carry a gene mutation that, alone or in combination with another mutation in the same gene, would result in an identifiable outcome in their child (for example, autosomal-recessive, autosomal-dominant, and X-linked disorders).
PGD, by definition, also includes testing of women, or their partner, who possess a balanced chromosome rearrangement (translocation, inversion). Offspring of carriers of balanced chromosome rearrangements are at increased risk of particular genetic abnormalities, as a result of unbalanced segregation of chromosomes involved in their rearrangement.
How does PGS differ from PGD?
Screening, in contrast, focuses analysis on offspring of women who are theoretically at increased risk of a genetic abnormality based on their age or reproductive history, not on their genetic makeup. PGS looks specifically for chromosomal content, and is based on the premise that decreasing the rate of aneuploidy among the conceptions of women 1) of advanced maternal age, 2) who experience habitual miscarriage, or 3) who have failed multiple cycles of in vitro fertilization (IVF) would increase the rate of implantation and, ultimately, the live birth rate.
The articles below, beginning with a committee opinion from the American Society for Reproductive Medicine (ASRM), address the following:
- evidence in support of PGD for genetic disease
- caution about using PGS, in its current format, for aneuploidy screening.
Practice Committee of the Society for Assisted Reproductive Technology; Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril. 2007;88:1497–1504.
A gene mutation carried by one or both parents can increase the risk that their offspring will be affected with an inherited condition. Common examples include autosomal-recessive disorders such as cystic fibrosis; autosomal-dominant disorders such as neurofibromatosis; and X-linked disorders such as hemophilia A.
Recently, human leukocyte antigens (HLA) have been assessed in conjunction with testing for specific genetic diseases, such as Fanconi anemia. In these settings, the intent is to recognize not only the blastomeres that are free of Fanconi anemia, but also those that are potential HLA matches and, therefore, potential donors for an (older) affected sibling.
PGD has been extended to women, or their partner, who possess a gene mutation that places them at increased risk of cancer (such as BRCA-1) and who wish to avoid transmitting that risk-conferring gene to their offspring.
For these diseases, and for many others, knowledge of the specific genetic mutation enables similar molecular testing to be accomplished on a single cell, such as a blastomere.
Technical concerns of testing must be part
of the physician–patient discussion
Typically, PGD analysis is initiated by polymerase chain reaction (PCR) of DNA content extracted from the single cell. This is followed by application of mutation-appropriate molecular technology. Given 1) the short time in which these PGD results are needed (often, 24 to 48 hours) and 2) the limited amount of genetic material available for analysis, technical restraints on testing are recognized:
- Extraneous DNA contamination remains a problem with molecular technology, despite application of intracytoplasmic sperm injection
- Only partial amplification of the allele may occur, or allele “drop-out” may be present; both of these phenomena can cause false-negative results
- Error can occur dually: 1) Presumably unaffected embryos that are, indeed, affected are transferred and 2) actually normal embryos that have been interpreted incorrectly as abnormal are discarded
- The rate of misdiagnosis (false-negative results) ranges from 2% (with autosomal-recessive disorders) to 10% (with autosomal-dominant disorders), although this rate can be lessened with the use of linked markers.
PGD for investigating balanced chromosome rearrangements
These rearrangements represent another type of genetic abnormality in which PGD can reduce the likelihood of a conception that carries a specific genetic abnormality.
When one parent carries a balanced chromosome translocation, fluorescence in-situ hybridization (FISH) can be applied to assess the segregation of at-risk chromosomes in a single blastomere cell. In this technique, fluorescence-labeled DNA probes, selected for specificity to the translocation in question, are applied to the single cell fixed on a glass slide. Copies of the DNA segment and, by inference, the chromosomal segment in question are assessed by quantification of the sites of positive fluorescence.
Because translocation carriers are, theoretically, at high risk of transmission of an unbalanced segregant to the blastomere, as many as 10 blastomeres will often be screened until one or two are deemed normal for the FISH probes in question. When implantation does succeed after FISH analysis for a chromosome rearrangement, however, the pregnancy loss rate is lower and the likelihood of a live birth is higher.
Again, in-depth consultation is needed before PGD
Whether PGD is planned for investigating a single-gene disorder or a chromosome translocation, detailed consultation with the woman or the couple is important. This effort should include not only genetic counseling about inheritance, the natural history of the disorder in question, and other options for avoiding the transmission of the disorder—in addition, additional time should be spent describing:
- risks associated with IVF procedures and embryo biopsy (and with extended culture, if needed)
- technical limitations of the particular testing that is being considered
- options for prenatal testing during a pregnancy
- the possibility that embryos suitable for transfer will not be found (and that, potentially, erroneously tested normal embryos will not be transferred)
- disposition of embryos in which test results are inconclusive.
PGS for women at increased risk of aneuploidy isn’t supported by evidence; consider it investigational
Mastenbroek S, Twisk M, van Echten-Arends J, et al. In vitro fertilization with preimplantation genetic screening. N Engl J Med. 2007;357:9–17.
Mersereau JE, Pergament E, Zhang X, Milad MP. Preimplantation genetic screening to improve in vitro fertilization pregnancy rates: a prospective randomized controlled trial. Fertil Steril. 2008;90:1287–1289.
Aneuploidy contributes to pregnancy loss among women as they become older. Theoretically, avoiding aneuploid pregnancy among embryos transferred during IVF cycles—in older women and in women experiencing multiple pregnancy losses and failed IVF cycles—was expected to increase the implantation rate and decrease the rate of pregnancy loss.
This hypothesis was supported, at first, by observational trials. But at least one randomized study, by Staessen and colleagues,1 failed to demonstrate that PGS is beneficial in women of advanced maternal age.
Now, a large multicenter, randomized, double-blind, controlled trial conducted by Mastenbroek and co-workers provides further evidence that PGS does not increase the rate of pregnancy and, in fact, significantly reduces that rate among women of advanced maternal age.
The Mastenbroek study compared outcomes among 206 women who had PGS and 202 women who did not. Both groups were matched for maternal age older than 35 years. Blastomeres were analyzed for eight chromosomes, including those known to be highly associated with miscarriage (1, 16, 17, 13, 18, and 21; X and Y).
Among women who underwent PGS, 25% had an ongoing pregnancy of at least 12 weeks’ gestation, compared with 37% of unscreened women. A similar higher rate of live birth was seen among unscreened women (35%, versus 24% in the PGS group).
Mastenbroek’s results are comparable to what was reported from an earlier randomized trial of PGS,1 in which the implantation rate as the primary outcome among women who had PGS and among controls was not significantly different. Contributors to 1) the lack of success of PGS and 2) the apparent detriment of PGS to the ongoing pregnancy rate include:
- potential for damage to the embryo at biopsy
- limitations imposed by FISH technology on the number of probes that can be accurately assessed technically
- a growing knowledge that a significant percentage of embryos are chromosomal mosaics at this stage—a phenomenon that likely results in nontransfer of embryos that have the potential for developing karyotypically normally.
Does PGS improve outcomes?
More recently, Mersereau and colleagues reported pilot results from a prospective, randomized, controlled trial that assessed whether PGS could improve pregnancy outcomes. Here, selection of infertile women for the study was not restricted to poor prognosis categories, such as advanced maternal age and recurrent pregnancy loss.
Using the live birth rate as the outcome measure, PGS for seven chromosomes was determined not to be associated with a significantly increased live birth rate among screened pregnancies. Sample sizes had been calculated to establish, with significance, a 50% increase in live births—from 30% in the control (unscreened) population to 45% in the screened population. Secondary endpoints, such as the implantation rate and pregnancy loss, also did not differ significantly between the PGS cases and controls.
Again, technical difficulties of two-blastomere biopsy, with its potential for embryo damage, and the presence of underlying embryo mosaicism represent possible barriers to improving the live birth rate when utilizing PGS.
Technical limitations may be one of the largest obstacles
to applying PGS
Practice Committee of the Society for Assisted Reproductive Technology; Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril. 2007;88:1497–1504.
FISH probes can be chosen to reflect the nature of a given patient’s risk (advanced maternal age, recurrent pregnancy loss) when performing PGS, but the technique itself is limited by the number of probe sites that can be interpreted accurately at one time. Typically, analysis of more than five chromosomes requires two cycles of hybridization, with their associated time requirement and potential for degradation of the single cell.
Alternatively, advances in the analysis of all 23 chromosomes through comparative genomic hybridization may, ultimately, provide an avenue for applying PGS. At the moment, time limitations prohibit comparative genomic hybridization without embryo cryopreservation. Further investigation of other technical limitations, such as the high rate of mosaicism, has revealed that, when two cells are examined and found to be karyotypically discordant, further analysis of the entire embryo will reveal that more than 50% of embryos are, in fact, euploid—that is, chromosomally normal. Random biopsy of the abnormal cell solely would relegate the embryo to nontransfer, despite the predominance of an underlying euploid state.
Understanding of the potential that embryos have to self-correct early mosaicism is growing; we now know that almost one half of embryos identified as aneuploid at cleavage stage correct to euploid if they survive to blastocyst stage. A karyotypic abnormality in a single cell from a day-3 embryo does not always signal an abnormal embryo.
ASRM does not support PGS to improve the live birth rate
This determination by ASRM is based on available evidence about advanced maternal age, recurrent pregnancy loss, recurrent implantation failure, and recurrent aneuploidy loss:
- In women of advanced maternal age, many day-3 embryos display aneuploidy when studied by FISH. In theory, exclusion of these embryos for transfer should improve implantation and live birth rates, but evidence does not support that premise.
- Because almost 70% of spontaneous pregnancy loss is caused by a karyotypic abnormality, and women with karyotypically recurrent pregnancy loss are more likely to experience subsequent loss with karyotype abnormalities, the premise of preimplantation screening for aneuploidy also appeared to be well founded. Studies at this time are limited to retrospective series, without randomized controlled trials published.
- Among women who experience repeated implantation failure, a finding of more than 50% abnormal embryos isn’t uncommon, yet several studies have not supported an increased implantation rate or live birth rate after PGS.
A literature review of PGS calls its introduction “premature”
Gleicher N, Weghofer A, Barad D. Preimplantation genetic screening: “established” and ready for prime time? Fertil Steril. 2008;89:780–788.
After ASRM recognized PGD as an established technique in a 2001 committee opinion, extension of this status to PGS was inadvertently assumed. But PGS is a different testing modality—with different indications, risk/benefit profiles, and efficacy than PGD.
Today, FISH probes are utilized for PGS; the false-negative rate of FISH appears to be driven by the technical constraints of the technology. Potentially increasing the false-negative rate are inadequate hybridization and the use of increasing numbers of probes and hybridization cycles.
Conversely, the false-positive rate—the number of embryos not transferred that are, in fact, chromosomally normal—varies markedly from one study to another, and may be as high as 20% when discarded embryos are more completely assessed.
Similarly, laboratories utilize different methods of obtaining the genetic material. These methods range from biopsy of polar bodies to single-cell blastomere and routine two-cell blastomere biopsy—and, more recently, to blastocyst biopsy. The impact of these various embryo manipulations has yet to be fully considered. Whether biopsy affects the embryo has received little attention.
In fact, embryos that are of poor quality before biopsy—such as those found in women of advanced maternal age—may be more susceptible to the effects of biopsy. The outcome with such embryos may be of even greater detriment to the implantation rate (as discussed in regard to the Mastenbroek study earlier in this article).
The logic of performing PGS for aneuploidy in women of advanced maternal age was reasonable. But this group of women—in whom ovarian reserve is diminished, who respond poorly to ovulation induction, thereby limiting the total number of embryos for analysis and the poorer quality embryos possibly further impaired by the biopsy itself—represent the population that may be least amenable to PGS.
A further observation about PGS in women who have experienced recurrent pregnancy loss or IVF failure: Any impairment of embryos that is a consequence of the method of biopsy may further undermine the generally unsupportive results of PGS that have been documented in these patients.
Consensus on performing PGS
An assessment of European studies and practices reveals similar concerns voiced by the European Society for Human Reproduction and Embryology (ESHRE) PGD Consortium Steering Committee. The committee recently asserted a comparable opinion about “the insufficient data that demonstrate PGS is indeed a cost-effective alternative for standard IVF.”2 Gleicher and colleagues, in their review of the literature, conclude that the indications for PGS are currently undefined and, as such, screening should be considered experimental.
Gleicher’s sentiments echo the recommendations of ASRM that, when PGS is considered,
- patients undergo counseling about its limitations, risk of error, and lack of evidence that it improves the live-birth rate
- available evidence does not support improvement in the live birth rate in women of advanced maternal age, who have failed previous implantation, who have experienced recurrent pregnancy loss, or who have experienced recurrent pregnancy loss specifically related to aneuploidy
- decisions about management should not be based on aneuploidy results of prior PGS cycles for a woman who has experienced recurrent implantation failure.
1. Staessen C, Platteau P, Van Assche E, et al. Comparison of blastocyst transfer with and without preimplantation genetic diagnosis for aneuploidy screening in couples with advanced maternal age: a prospective randomized controlled trial. Hum Reprod. 2004;19:2849-2858.
2. Sermon KD, Michiels A, Harton G, et al. ESHRE PGD Consortium data collection VI: cycles from January to December 2003 with pregnancy follow-up to October 2004. Hum Reprod. 2007;22:323-336.
1. Staessen C, Platteau P, Van Assche E, et al. Comparison of blastocyst transfer with and without preimplantation genetic diagnosis for aneuploidy screening in couples with advanced maternal age: a prospective randomized controlled trial. Hum Reprod. 2004;19:2849-2858.
2. Sermon KD, Michiels A, Harton G, et al. ESHRE PGD Consortium data collection VI: cycles from January to December 2003 with pregnancy follow-up to October 2004. Hum Reprod. 2007;22:323-336.
PRENATAL COUNSELING
Investigations of maternal alcohol consumption have consistently produced the same finding: Even a low level of alcohol—especially in the first trimester—has a harmful effect on fetal development. The American College of Obstetricians and Gynecologists (ACOG), American Academy of Pediatricians, and the US Surgeon General now support the tenet that no lower limit of alcohol consumption is safe during pregnancy.
Although a specific fetal alcohol syndrome (FAS) was not identified until 1968, the adverse effects of alcohol during pregnancy have been observed for centuries. FAS is the most severe manifestation of maternal alcohol consumption and is estimated to affect 0.2 to 1.5 of every 1,000 births. The term refers to a “constellation of physical abnormalities” and “problems of behavior and cognition in children born to mothers who drank heavily during pregnancy.”1 The syndrome is also “completely preventable.”1
The US Surgeon General recommends that health professionals:
- routinely inquire about alcohol consumption in women of childbearing age
- inform them of the risks of alcohol consumption during pregnancy
- advise them not to drink during pregnancy.2
New drinking pattern emerges
Of special concern is binge drinking, initially defined as the consumption of five or more drinks during one session, even among women who do not chronically consume alcohol. Like lower levels of alcohol consumption during pregnancy, binge drinking increases the risk of developmental and growth delays in the child. The higher peak levels of alcohol associated with binge drinking appear particularly deleterious to fetal neurodevelopment. And because a woman may engage in binge drinking before she is aware that she is pregnant, the issue merits particular attention.
Hallmarks of FAS
FAS causes facial dysmorphia, including short palpebral fissures, flattened midfacies, epicanthal folds, and micrognathia. Defects of the central nervous system and cardiac, renal, and skeletal systems also can occur, along with prenatal and postnatal growth delay. In addition, developmental delay is present.
FAS can be present even if history of alcohol exposure is uncertain
In 1996, the Institute of Medicine broadened the classification of FAS to include:
- Category 1 – FAS with a confirmed history of maternal alcohol exposure
- Category 2 – FAS with no confirmed history of maternal alcohol exposure
- Category 3 – partial FAS with a history of maternal alcohol exposure
- Category 4 – alcohol-related birth defects (physical anomalies only)
- Category 5 – alcohol-related neurodevelopmental disorders.1
Alcohol exposure linked to a spectrum of effects
In 2005, the term “fetal alcohol spectrum disorder” (FASD) entered the lexicon. FASD is not intended to be used as a clinical diagnosis but to describe a spectrum of conditions that may result from prenatal alcohol exposure.
The prevalence of FASD is uncertain, although alcohol-related neurobehavioral abnormalities that affect learning and behavior may occur in three additional children for every one child who is given a diagnosis of classic FAS.
In this Update, I highlight recent studies or publications that:
- describe drinking patterns among women of reproductive age
- offer screening strategies or
- suggest a framework for counseling the patient to reduce or eliminate alcohol consumption.
Which women are most likely to drink during pregnancy?
Tsai J, Floyd RL, Green PP, Bouyle CA. Patterns and average volume of alcohol use among women of childbearing age. Matern Child Health J. 2007;11:437–445.
Tsai J, Floyd RL, Bertrand J. Tracking binge drinking among childbearing-age women. Prev Med. 2007; 44:298–302.
Caetano R, Ramisetty-Mikler S, Floyd L, McGrath C. The epidemiology of drinking among women of childbearing age. Alcohol Clin Exp Res. 2006;30:1023–1030.
Studies that led to the phenotypic description of FASD focused on women who had recognized alcohol dependency and who drank heavily. Additional research has identified another subset of women who are likely to continue alcohol consumption during pregnancy: binge drinkers. Many women who report binge drinking do not consider their alcohol consumption to be chronic or excessive.
Binge drinking is on the rise among women of childbearing age…
Binge drinking has increased steadily over the past 10 years despite public health initiatives and other programs developed to educate consumers. Tsai and colleagues used data from the Centers for Disease Control and Prevention (CDC) Behavior Risk Factor Surveillance System from 2001 to 2003 to calculate the magnitude of alcohol consumption among women of childbearing age. The rate of binge drinking increased from 10.3% to 13% between 1991 and 2003. In 2003, the highest prevalence of binge drinking was observed in the 18- to 24-year-old age group (20.5%), and among non-Hispanic white (15.5%), employed (14%), college-educated (13.3%), and unmarried women (18.7%). The highest number of binge sessions in the preceding month followed the same pattern.
In 2004, as it became clear that the adverse effects of binge alcohol consumption were more significant in women than men, at-risk binge drinking was redefined as more than three drinks in a single session.
…and also on the rise among pregnant women
In a separate study by Tsai and colleagues using the same data, one in 50 gravidas reported alcohol consumption in a binge fashion during the current pregnancy, with a background rate of 9% to 12% of pregnant women who reported any use of alcohol. More than 50% of the pregnant women who reported binge drinking said they had engaged in binge drinking at least twice during the preceding month.
Binge drinking and unplanned pregnancy—a risky combination
Binge drinking among women of reproductive age is especially risky because roughly half of all pregnancies in the United States are unplanned, so a woman may unwittingly engage in binge drinking during pregnancy. The rate of unintended pregnancy is highest among adolescents (82%) and 20- to 24-year-olds (61%), the groups with the highest rate of binge drinking (20%) and the most episodes in the preceding month (3.5). These figures suggest that efforts to prevent FAS should encompass the concept of binge drinking as an at-risk behavior and focus on all women of reproductive age, not just those known to be pregnant.
The typical binge drinker? She’s young, white, single, and employed
Utilizing the 2002 National Epidemiologic Survey on Alcohol and Related Conditions, Caetano and colleagues explored alcohol consumption among women of reproductive age before they recognized they were pregnant. Women of childbearing age who are social drinkers but develop a pattern of binge drinking represent a larger percentage of the female population than do women who consume alcohol daily, but both groups face an increased risk of bearing a child with alcohol-related neurodevelopmental difficulties.
Unplanned pregnancies were associated with a higher rate of preconception binge drinking than were planned gestations, and unmarried Caucasian women who smoked were most likely to engage in preconception binge drinking.
When the year preceding the study was assessed for both alcohol use and pregnancy, Caetano and associates found that 20% of women met the criteria for binge drinking or alcohol dependence. The high prevalence probably reflects the longer time span for acknowledgment of alcohol consumption (an entire year) and the lower drink limit for the redefined term “binge drinking” (in this study, it was defined as four drinks or more rather than five or more drinks on one occasion). The highest-risk women were young, single, and Caucasian, and had a higher income (>$40,000). White women had higher rates of binge drinking than black or Hispanic women at comparable ages, marital status, and income levels.
What’s the best way to screen for “at-risk” alcohol consumption?
Drinking and Reproductive Health: A Fetal Alcohol Spectrum Disorders Prevention Tool Kit. Washington, DC: American College of Obstetricians and Gynecologists; 2006. Available at: cdc.gov/ncbddd/fas/acog_toolkit.htm
In 2006, in collaboration with the CDC, ACOG developed a comprehensive educational tool kit for physicians. The kit, which can be downloaded from the CDC Web site, outlines office-based screening for at-risk drinking patterns in pregnant and nonpregnant women. It includes a screening tool—T-ACE—that has proved to be effective and can be incorporated into practice fairly efficiently. T-ACE and a similar tool—TWEAK—are presented in the TABLE.
ACOG recommends, and research supports, routine screening of all women of childbearing age. Studies assessing the prevalence of at-risk drinking and the efficacy of various interventions suggest that screening for alcohol use should be a routine part of prenatal care—as well as annual gynecologic care among women of childbearing age. One applicable approach is incorporation of a screening tool into the health-and-habits questionnaire administered to the patient.
Available as companion pieces to the tool kit are patient education sheets covering the risks of alcohol exposure and emphasizing basic concepts such as:
- alcohol equivalency (12 oz of beer=5 oz of wine=1 oz of liquor)
- risks of alcohol exposure before pregnancy is recognized
- goals for reducing or eliminating alcohol consumption.
Use these tools to screen for excessive alcohol consumption
| FOCUS | QUESTION | POINTS |
|---|---|---|
| T-ACE (a positive screen is ≥2 points) | ||
| (T) Tolerance | How many drinks does it take to make you feel high? | 1 point per drink |
| (A) Annoyed | Have people annoyed you by criticizing your drinking? | Yes = 1 point |
| (C) Cut down | Have you ever felt you ought to cut down on your drinking? | Yes = 1 point |
| (E) Eye-opener | Have you ever had a drink first thing in the morning to steady your nerves or get rid of a hangover? | Yes = 1 point |
| TWEAK (a positive screen is ≥2 points) | ||
| (T) Tolerance | Are more than two drinks necessary to make you feel high? | Yes = 2 points |
| (W) Worry | Are your friends or family worried about your level of alcohol consumption? | Yes = 1 point |
| (E) Eye-opener | Do you ever need to drink in the morning? | Yes = 1 point |
| (A) Amnesia | Do you ever black out when drinking? | Yes = 1 point |
| (K) Cut down | Do you believe you need to cut down on your drinking? | Yes = 1 point |
Are efforts to reduce alcohol use among gravidas successful?
Floyd RL, Sobell M, Velasquez M, et al; Project CHOICES Efficacy Study Group. Preventing alcohol-exposed pregnancies. A randomized controlled trial. Am J Prev Med. 2007;32:1–10.
Brief intervention has been a successful tool for changing the behavior of nonpregnant adults. It also appears to be effective and efficient in the pregnant population. A brief intervention typically consists of a time-limited motivational counseling session that aims to educate, recommend a change in habits, and help the patient set goals. Brief intervention has had special success among nondependent women and has been used effectively in obstetric clinics and among women of various racial, ethnic, and socioeconomic backgrounds.
This randomized, controlled trial by Floyd and colleagues focused on the pregnant population. Like three other brief intervention trials conducted between 2000 and 2006, it found that brief intervention reduced alcohol consumption, increased positive newborn outcomes, and decreased alcohol consumption in subsequent pregnancies.3-5
FRAMES model: 6 manageable steps
One successful brief intervention is the FRAMES model, which is included in the ACOG tool kit for physicians. It is based on concepts of:
- feedback (F) – compare the patient’s level of drinking with drinking patterns that are not risky
- responsibility (R) – emphasize that it is up to her to change her habits
- advice (A) – counsel her to change her behavior
- menu (M) – identify risky drinking situations and offer tactics for coping
- empathy (E) – be understanding
- self-efficacy (S) – encourage the patient to set goals and commit to change.
Use an individualized approach to change behavior
Despite widespread, population-based educational efforts throughout the 1990s, the prevalence of alcohol consumption among nonpregnant and pregnant women remains largely unchanged or even increased, particularly binge drinking. Other approaches are needed to avert the largest preventable contributor to birth defects and childhood neurodevelopmental disability.
With improved and validated office-based methods for identifying alcohol consumption, along with referrals when appropriate, it is possible to reduce maternal alcohol consumption during pregnancy. These simple methods are also easy to incorporate into an office routine. Equally important is incorporation of these methods into the office visit for the nonpregnant woman of reproductive age, with the aim of reducing alcohol consumption and increasing use of effective contraception.
1. Stratton K, Howe C, Battaglia F. eds. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. Washington, DC: National Academy Press; 1996. Available at: www.nap.edu/openbook.php?record_id=4991&page=R1. Accessed December 5, 2007.
2. US Department of Health and Human Services, Office of the Surgeon General. Surgeon General’s Advisory on Alcohol Use in Pregnancy. Available at: www.surgeongeneral.gov/pressreleases/sg02222005.html. Accessed December 5, 2007.
3. Manwell LB, Fleming MF, Mundt MP, Stauffacher EA, Barry KL. Treatment of problem alcohol use in women of childbearing age: results of a brief intervention trial. Alcohol Clin Exp Res. 2000;24:1517-1524.
4. Ingersoll KS, Ceperich SD, Nettleman MD, Karanda K, Brocksen S, Johnson BA. Reducing alcohol-exposed pregnancy risk in college women: initial outcomes of a clinical trial of a motivational intervention. J Subst Abuse Treat. 2005;29:173-189.
5. Chang G, Wilkins-Haug BS, Goetz MA. Brief interventions for alcohol use in pregnancy: a randomized trial. Addiction. 1999;94:1499-1508.
Investigations of maternal alcohol consumption have consistently produced the same finding: Even a low level of alcohol—especially in the first trimester—has a harmful effect on fetal development. The American College of Obstetricians and Gynecologists (ACOG), American Academy of Pediatricians, and the US Surgeon General now support the tenet that no lower limit of alcohol consumption is safe during pregnancy.
Although a specific fetal alcohol syndrome (FAS) was not identified until 1968, the adverse effects of alcohol during pregnancy have been observed for centuries. FAS is the most severe manifestation of maternal alcohol consumption and is estimated to affect 0.2 to 1.5 of every 1,000 births. The term refers to a “constellation of physical abnormalities” and “problems of behavior and cognition in children born to mothers who drank heavily during pregnancy.”1 The syndrome is also “completely preventable.”1
The US Surgeon General recommends that health professionals:
- routinely inquire about alcohol consumption in women of childbearing age
- inform them of the risks of alcohol consumption during pregnancy
- advise them not to drink during pregnancy.2
New drinking pattern emerges
Of special concern is binge drinking, initially defined as the consumption of five or more drinks during one session, even among women who do not chronically consume alcohol. Like lower levels of alcohol consumption during pregnancy, binge drinking increases the risk of developmental and growth delays in the child. The higher peak levels of alcohol associated with binge drinking appear particularly deleterious to fetal neurodevelopment. And because a woman may engage in binge drinking before she is aware that she is pregnant, the issue merits particular attention.
Hallmarks of FAS
FAS causes facial dysmorphia, including short palpebral fissures, flattened midfacies, epicanthal folds, and micrognathia. Defects of the central nervous system and cardiac, renal, and skeletal systems also can occur, along with prenatal and postnatal growth delay. In addition, developmental delay is present.
FAS can be present even if history of alcohol exposure is uncertain
In 1996, the Institute of Medicine broadened the classification of FAS to include:
- Category 1 – FAS with a confirmed history of maternal alcohol exposure
- Category 2 – FAS with no confirmed history of maternal alcohol exposure
- Category 3 – partial FAS with a history of maternal alcohol exposure
- Category 4 – alcohol-related birth defects (physical anomalies only)
- Category 5 – alcohol-related neurodevelopmental disorders.1
Alcohol exposure linked to a spectrum of effects
In 2005, the term “fetal alcohol spectrum disorder” (FASD) entered the lexicon. FASD is not intended to be used as a clinical diagnosis but to describe a spectrum of conditions that may result from prenatal alcohol exposure.
The prevalence of FASD is uncertain, although alcohol-related neurobehavioral abnormalities that affect learning and behavior may occur in three additional children for every one child who is given a diagnosis of classic FAS.
In this Update, I highlight recent studies or publications that:
- describe drinking patterns among women of reproductive age
- offer screening strategies or
- suggest a framework for counseling the patient to reduce or eliminate alcohol consumption.
Which women are most likely to drink during pregnancy?
Tsai J, Floyd RL, Green PP, Bouyle CA. Patterns and average volume of alcohol use among women of childbearing age. Matern Child Health J. 2007;11:437–445.
Tsai J, Floyd RL, Bertrand J. Tracking binge drinking among childbearing-age women. Prev Med. 2007; 44:298–302.
Caetano R, Ramisetty-Mikler S, Floyd L, McGrath C. The epidemiology of drinking among women of childbearing age. Alcohol Clin Exp Res. 2006;30:1023–1030.
Studies that led to the phenotypic description of FASD focused on women who had recognized alcohol dependency and who drank heavily. Additional research has identified another subset of women who are likely to continue alcohol consumption during pregnancy: binge drinkers. Many women who report binge drinking do not consider their alcohol consumption to be chronic or excessive.
Binge drinking is on the rise among women of childbearing age…
Binge drinking has increased steadily over the past 10 years despite public health initiatives and other programs developed to educate consumers. Tsai and colleagues used data from the Centers for Disease Control and Prevention (CDC) Behavior Risk Factor Surveillance System from 2001 to 2003 to calculate the magnitude of alcohol consumption among women of childbearing age. The rate of binge drinking increased from 10.3% to 13% between 1991 and 2003. In 2003, the highest prevalence of binge drinking was observed in the 18- to 24-year-old age group (20.5%), and among non-Hispanic white (15.5%), employed (14%), college-educated (13.3%), and unmarried women (18.7%). The highest number of binge sessions in the preceding month followed the same pattern.
In 2004, as it became clear that the adverse effects of binge alcohol consumption were more significant in women than men, at-risk binge drinking was redefined as more than three drinks in a single session.
…and also on the rise among pregnant women
In a separate study by Tsai and colleagues using the same data, one in 50 gravidas reported alcohol consumption in a binge fashion during the current pregnancy, with a background rate of 9% to 12% of pregnant women who reported any use of alcohol. More than 50% of the pregnant women who reported binge drinking said they had engaged in binge drinking at least twice during the preceding month.
Binge drinking and unplanned pregnancy—a risky combination
Binge drinking among women of reproductive age is especially risky because roughly half of all pregnancies in the United States are unplanned, so a woman may unwittingly engage in binge drinking during pregnancy. The rate of unintended pregnancy is highest among adolescents (82%) and 20- to 24-year-olds (61%), the groups with the highest rate of binge drinking (20%) and the most episodes in the preceding month (3.5). These figures suggest that efforts to prevent FAS should encompass the concept of binge drinking as an at-risk behavior and focus on all women of reproductive age, not just those known to be pregnant.
The typical binge drinker? She’s young, white, single, and employed
Utilizing the 2002 National Epidemiologic Survey on Alcohol and Related Conditions, Caetano and colleagues explored alcohol consumption among women of reproductive age before they recognized they were pregnant. Women of childbearing age who are social drinkers but develop a pattern of binge drinking represent a larger percentage of the female population than do women who consume alcohol daily, but both groups face an increased risk of bearing a child with alcohol-related neurodevelopmental difficulties.
Unplanned pregnancies were associated with a higher rate of preconception binge drinking than were planned gestations, and unmarried Caucasian women who smoked were most likely to engage in preconception binge drinking.
When the year preceding the study was assessed for both alcohol use and pregnancy, Caetano and associates found that 20% of women met the criteria for binge drinking or alcohol dependence. The high prevalence probably reflects the longer time span for acknowledgment of alcohol consumption (an entire year) and the lower drink limit for the redefined term “binge drinking” (in this study, it was defined as four drinks or more rather than five or more drinks on one occasion). The highest-risk women were young, single, and Caucasian, and had a higher income (>$40,000). White women had higher rates of binge drinking than black or Hispanic women at comparable ages, marital status, and income levels.
What’s the best way to screen for “at-risk” alcohol consumption?
Drinking and Reproductive Health: A Fetal Alcohol Spectrum Disorders Prevention Tool Kit. Washington, DC: American College of Obstetricians and Gynecologists; 2006. Available at: cdc.gov/ncbddd/fas/acog_toolkit.htm
In 2006, in collaboration with the CDC, ACOG developed a comprehensive educational tool kit for physicians. The kit, which can be downloaded from the CDC Web site, outlines office-based screening for at-risk drinking patterns in pregnant and nonpregnant women. It includes a screening tool—T-ACE—that has proved to be effective and can be incorporated into practice fairly efficiently. T-ACE and a similar tool—TWEAK—are presented in the TABLE.
ACOG recommends, and research supports, routine screening of all women of childbearing age. Studies assessing the prevalence of at-risk drinking and the efficacy of various interventions suggest that screening for alcohol use should be a routine part of prenatal care—as well as annual gynecologic care among women of childbearing age. One applicable approach is incorporation of a screening tool into the health-and-habits questionnaire administered to the patient.
Available as companion pieces to the tool kit are patient education sheets covering the risks of alcohol exposure and emphasizing basic concepts such as:
- alcohol equivalency (12 oz of beer=5 oz of wine=1 oz of liquor)
- risks of alcohol exposure before pregnancy is recognized
- goals for reducing or eliminating alcohol consumption.
Use these tools to screen for excessive alcohol consumption
| FOCUS | QUESTION | POINTS |
|---|---|---|
| T-ACE (a positive screen is ≥2 points) | ||
| (T) Tolerance | How many drinks does it take to make you feel high? | 1 point per drink |
| (A) Annoyed | Have people annoyed you by criticizing your drinking? | Yes = 1 point |
| (C) Cut down | Have you ever felt you ought to cut down on your drinking? | Yes = 1 point |
| (E) Eye-opener | Have you ever had a drink first thing in the morning to steady your nerves or get rid of a hangover? | Yes = 1 point |
| TWEAK (a positive screen is ≥2 points) | ||
| (T) Tolerance | Are more than two drinks necessary to make you feel high? | Yes = 2 points |
| (W) Worry | Are your friends or family worried about your level of alcohol consumption? | Yes = 1 point |
| (E) Eye-opener | Do you ever need to drink in the morning? | Yes = 1 point |
| (A) Amnesia | Do you ever black out when drinking? | Yes = 1 point |
| (K) Cut down | Do you believe you need to cut down on your drinking? | Yes = 1 point |
Are efforts to reduce alcohol use among gravidas successful?
Floyd RL, Sobell M, Velasquez M, et al; Project CHOICES Efficacy Study Group. Preventing alcohol-exposed pregnancies. A randomized controlled trial. Am J Prev Med. 2007;32:1–10.
Brief intervention has been a successful tool for changing the behavior of nonpregnant adults. It also appears to be effective and efficient in the pregnant population. A brief intervention typically consists of a time-limited motivational counseling session that aims to educate, recommend a change in habits, and help the patient set goals. Brief intervention has had special success among nondependent women and has been used effectively in obstetric clinics and among women of various racial, ethnic, and socioeconomic backgrounds.
This randomized, controlled trial by Floyd and colleagues focused on the pregnant population. Like three other brief intervention trials conducted between 2000 and 2006, it found that brief intervention reduced alcohol consumption, increased positive newborn outcomes, and decreased alcohol consumption in subsequent pregnancies.3-5
FRAMES model: 6 manageable steps
One successful brief intervention is the FRAMES model, which is included in the ACOG tool kit for physicians. It is based on concepts of:
- feedback (F) – compare the patient’s level of drinking with drinking patterns that are not risky
- responsibility (R) – emphasize that it is up to her to change her habits
- advice (A) – counsel her to change her behavior
- menu (M) – identify risky drinking situations and offer tactics for coping
- empathy (E) – be understanding
- self-efficacy (S) – encourage the patient to set goals and commit to change.
Use an individualized approach to change behavior
Despite widespread, population-based educational efforts throughout the 1990s, the prevalence of alcohol consumption among nonpregnant and pregnant women remains largely unchanged or even increased, particularly binge drinking. Other approaches are needed to avert the largest preventable contributor to birth defects and childhood neurodevelopmental disability.
With improved and validated office-based methods for identifying alcohol consumption, along with referrals when appropriate, it is possible to reduce maternal alcohol consumption during pregnancy. These simple methods are also easy to incorporate into an office routine. Equally important is incorporation of these methods into the office visit for the nonpregnant woman of reproductive age, with the aim of reducing alcohol consumption and increasing use of effective contraception.
Investigations of maternal alcohol consumption have consistently produced the same finding: Even a low level of alcohol—especially in the first trimester—has a harmful effect on fetal development. The American College of Obstetricians and Gynecologists (ACOG), American Academy of Pediatricians, and the US Surgeon General now support the tenet that no lower limit of alcohol consumption is safe during pregnancy.
Although a specific fetal alcohol syndrome (FAS) was not identified until 1968, the adverse effects of alcohol during pregnancy have been observed for centuries. FAS is the most severe manifestation of maternal alcohol consumption and is estimated to affect 0.2 to 1.5 of every 1,000 births. The term refers to a “constellation of physical abnormalities” and “problems of behavior and cognition in children born to mothers who drank heavily during pregnancy.”1 The syndrome is also “completely preventable.”1
The US Surgeon General recommends that health professionals:
- routinely inquire about alcohol consumption in women of childbearing age
- inform them of the risks of alcohol consumption during pregnancy
- advise them not to drink during pregnancy.2
New drinking pattern emerges
Of special concern is binge drinking, initially defined as the consumption of five or more drinks during one session, even among women who do not chronically consume alcohol. Like lower levels of alcohol consumption during pregnancy, binge drinking increases the risk of developmental and growth delays in the child. The higher peak levels of alcohol associated with binge drinking appear particularly deleterious to fetal neurodevelopment. And because a woman may engage in binge drinking before she is aware that she is pregnant, the issue merits particular attention.
Hallmarks of FAS
FAS causes facial dysmorphia, including short palpebral fissures, flattened midfacies, epicanthal folds, and micrognathia. Defects of the central nervous system and cardiac, renal, and skeletal systems also can occur, along with prenatal and postnatal growth delay. In addition, developmental delay is present.
FAS can be present even if history of alcohol exposure is uncertain
In 1996, the Institute of Medicine broadened the classification of FAS to include:
- Category 1 – FAS with a confirmed history of maternal alcohol exposure
- Category 2 – FAS with no confirmed history of maternal alcohol exposure
- Category 3 – partial FAS with a history of maternal alcohol exposure
- Category 4 – alcohol-related birth defects (physical anomalies only)
- Category 5 – alcohol-related neurodevelopmental disorders.1
Alcohol exposure linked to a spectrum of effects
In 2005, the term “fetal alcohol spectrum disorder” (FASD) entered the lexicon. FASD is not intended to be used as a clinical diagnosis but to describe a spectrum of conditions that may result from prenatal alcohol exposure.
The prevalence of FASD is uncertain, although alcohol-related neurobehavioral abnormalities that affect learning and behavior may occur in three additional children for every one child who is given a diagnosis of classic FAS.
In this Update, I highlight recent studies or publications that:
- describe drinking patterns among women of reproductive age
- offer screening strategies or
- suggest a framework for counseling the patient to reduce or eliminate alcohol consumption.
Which women are most likely to drink during pregnancy?
Tsai J, Floyd RL, Green PP, Bouyle CA. Patterns and average volume of alcohol use among women of childbearing age. Matern Child Health J. 2007;11:437–445.
Tsai J, Floyd RL, Bertrand J. Tracking binge drinking among childbearing-age women. Prev Med. 2007; 44:298–302.
Caetano R, Ramisetty-Mikler S, Floyd L, McGrath C. The epidemiology of drinking among women of childbearing age. Alcohol Clin Exp Res. 2006;30:1023–1030.
Studies that led to the phenotypic description of FASD focused on women who had recognized alcohol dependency and who drank heavily. Additional research has identified another subset of women who are likely to continue alcohol consumption during pregnancy: binge drinkers. Many women who report binge drinking do not consider their alcohol consumption to be chronic or excessive.
Binge drinking is on the rise among women of childbearing age…
Binge drinking has increased steadily over the past 10 years despite public health initiatives and other programs developed to educate consumers. Tsai and colleagues used data from the Centers for Disease Control and Prevention (CDC) Behavior Risk Factor Surveillance System from 2001 to 2003 to calculate the magnitude of alcohol consumption among women of childbearing age. The rate of binge drinking increased from 10.3% to 13% between 1991 and 2003. In 2003, the highest prevalence of binge drinking was observed in the 18- to 24-year-old age group (20.5%), and among non-Hispanic white (15.5%), employed (14%), college-educated (13.3%), and unmarried women (18.7%). The highest number of binge sessions in the preceding month followed the same pattern.
In 2004, as it became clear that the adverse effects of binge alcohol consumption were more significant in women than men, at-risk binge drinking was redefined as more than three drinks in a single session.
…and also on the rise among pregnant women
In a separate study by Tsai and colleagues using the same data, one in 50 gravidas reported alcohol consumption in a binge fashion during the current pregnancy, with a background rate of 9% to 12% of pregnant women who reported any use of alcohol. More than 50% of the pregnant women who reported binge drinking said they had engaged in binge drinking at least twice during the preceding month.
Binge drinking and unplanned pregnancy—a risky combination
Binge drinking among women of reproductive age is especially risky because roughly half of all pregnancies in the United States are unplanned, so a woman may unwittingly engage in binge drinking during pregnancy. The rate of unintended pregnancy is highest among adolescents (82%) and 20- to 24-year-olds (61%), the groups with the highest rate of binge drinking (20%) and the most episodes in the preceding month (3.5). These figures suggest that efforts to prevent FAS should encompass the concept of binge drinking as an at-risk behavior and focus on all women of reproductive age, not just those known to be pregnant.
The typical binge drinker? She’s young, white, single, and employed
Utilizing the 2002 National Epidemiologic Survey on Alcohol and Related Conditions, Caetano and colleagues explored alcohol consumption among women of reproductive age before they recognized they were pregnant. Women of childbearing age who are social drinkers but develop a pattern of binge drinking represent a larger percentage of the female population than do women who consume alcohol daily, but both groups face an increased risk of bearing a child with alcohol-related neurodevelopmental difficulties.
Unplanned pregnancies were associated with a higher rate of preconception binge drinking than were planned gestations, and unmarried Caucasian women who smoked were most likely to engage in preconception binge drinking.
When the year preceding the study was assessed for both alcohol use and pregnancy, Caetano and associates found that 20% of women met the criteria for binge drinking or alcohol dependence. The high prevalence probably reflects the longer time span for acknowledgment of alcohol consumption (an entire year) and the lower drink limit for the redefined term “binge drinking” (in this study, it was defined as four drinks or more rather than five or more drinks on one occasion). The highest-risk women were young, single, and Caucasian, and had a higher income (>$40,000). White women had higher rates of binge drinking than black or Hispanic women at comparable ages, marital status, and income levels.
What’s the best way to screen for “at-risk” alcohol consumption?
Drinking and Reproductive Health: A Fetal Alcohol Spectrum Disorders Prevention Tool Kit. Washington, DC: American College of Obstetricians and Gynecologists; 2006. Available at: cdc.gov/ncbddd/fas/acog_toolkit.htm
In 2006, in collaboration with the CDC, ACOG developed a comprehensive educational tool kit for physicians. The kit, which can be downloaded from the CDC Web site, outlines office-based screening for at-risk drinking patterns in pregnant and nonpregnant women. It includes a screening tool—T-ACE—that has proved to be effective and can be incorporated into practice fairly efficiently. T-ACE and a similar tool—TWEAK—are presented in the TABLE.
ACOG recommends, and research supports, routine screening of all women of childbearing age. Studies assessing the prevalence of at-risk drinking and the efficacy of various interventions suggest that screening for alcohol use should be a routine part of prenatal care—as well as annual gynecologic care among women of childbearing age. One applicable approach is incorporation of a screening tool into the health-and-habits questionnaire administered to the patient.
Available as companion pieces to the tool kit are patient education sheets covering the risks of alcohol exposure and emphasizing basic concepts such as:
- alcohol equivalency (12 oz of beer=5 oz of wine=1 oz of liquor)
- risks of alcohol exposure before pregnancy is recognized
- goals for reducing or eliminating alcohol consumption.
Use these tools to screen for excessive alcohol consumption
| FOCUS | QUESTION | POINTS |
|---|---|---|
| T-ACE (a positive screen is ≥2 points) | ||
| (T) Tolerance | How many drinks does it take to make you feel high? | 1 point per drink |
| (A) Annoyed | Have people annoyed you by criticizing your drinking? | Yes = 1 point |
| (C) Cut down | Have you ever felt you ought to cut down on your drinking? | Yes = 1 point |
| (E) Eye-opener | Have you ever had a drink first thing in the morning to steady your nerves or get rid of a hangover? | Yes = 1 point |
| TWEAK (a positive screen is ≥2 points) | ||
| (T) Tolerance | Are more than two drinks necessary to make you feel high? | Yes = 2 points |
| (W) Worry | Are your friends or family worried about your level of alcohol consumption? | Yes = 1 point |
| (E) Eye-opener | Do you ever need to drink in the morning? | Yes = 1 point |
| (A) Amnesia | Do you ever black out when drinking? | Yes = 1 point |
| (K) Cut down | Do you believe you need to cut down on your drinking? | Yes = 1 point |
Are efforts to reduce alcohol use among gravidas successful?
Floyd RL, Sobell M, Velasquez M, et al; Project CHOICES Efficacy Study Group. Preventing alcohol-exposed pregnancies. A randomized controlled trial. Am J Prev Med. 2007;32:1–10.
Brief intervention has been a successful tool for changing the behavior of nonpregnant adults. It also appears to be effective and efficient in the pregnant population. A brief intervention typically consists of a time-limited motivational counseling session that aims to educate, recommend a change in habits, and help the patient set goals. Brief intervention has had special success among nondependent women and has been used effectively in obstetric clinics and among women of various racial, ethnic, and socioeconomic backgrounds.
This randomized, controlled trial by Floyd and colleagues focused on the pregnant population. Like three other brief intervention trials conducted between 2000 and 2006, it found that brief intervention reduced alcohol consumption, increased positive newborn outcomes, and decreased alcohol consumption in subsequent pregnancies.3-5
FRAMES model: 6 manageable steps
One successful brief intervention is the FRAMES model, which is included in the ACOG tool kit for physicians. It is based on concepts of:
- feedback (F) – compare the patient’s level of drinking with drinking patterns that are not risky
- responsibility (R) – emphasize that it is up to her to change her habits
- advice (A) – counsel her to change her behavior
- menu (M) – identify risky drinking situations and offer tactics for coping
- empathy (E) – be understanding
- self-efficacy (S) – encourage the patient to set goals and commit to change.
Use an individualized approach to change behavior
Despite widespread, population-based educational efforts throughout the 1990s, the prevalence of alcohol consumption among nonpregnant and pregnant women remains largely unchanged or even increased, particularly binge drinking. Other approaches are needed to avert the largest preventable contributor to birth defects and childhood neurodevelopmental disability.
With improved and validated office-based methods for identifying alcohol consumption, along with referrals when appropriate, it is possible to reduce maternal alcohol consumption during pregnancy. These simple methods are also easy to incorporate into an office routine. Equally important is incorporation of these methods into the office visit for the nonpregnant woman of reproductive age, with the aim of reducing alcohol consumption and increasing use of effective contraception.
1. Stratton K, Howe C, Battaglia F. eds. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. Washington, DC: National Academy Press; 1996. Available at: www.nap.edu/openbook.php?record_id=4991&page=R1. Accessed December 5, 2007.
2. US Department of Health and Human Services, Office of the Surgeon General. Surgeon General’s Advisory on Alcohol Use in Pregnancy. Available at: www.surgeongeneral.gov/pressreleases/sg02222005.html. Accessed December 5, 2007.
3. Manwell LB, Fleming MF, Mundt MP, Stauffacher EA, Barry KL. Treatment of problem alcohol use in women of childbearing age: results of a brief intervention trial. Alcohol Clin Exp Res. 2000;24:1517-1524.
4. Ingersoll KS, Ceperich SD, Nettleman MD, Karanda K, Brocksen S, Johnson BA. Reducing alcohol-exposed pregnancy risk in college women: initial outcomes of a clinical trial of a motivational intervention. J Subst Abuse Treat. 2005;29:173-189.
5. Chang G, Wilkins-Haug BS, Goetz MA. Brief interventions for alcohol use in pregnancy: a randomized trial. Addiction. 1999;94:1499-1508.
1. Stratton K, Howe C, Battaglia F. eds. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. Washington, DC: National Academy Press; 1996. Available at: www.nap.edu/openbook.php?record_id=4991&page=R1. Accessed December 5, 2007.
2. US Department of Health and Human Services, Office of the Surgeon General. Surgeon General’s Advisory on Alcohol Use in Pregnancy. Available at: www.surgeongeneral.gov/pressreleases/sg02222005.html. Accessed December 5, 2007.
3. Manwell LB, Fleming MF, Mundt MP, Stauffacher EA, Barry KL. Treatment of problem alcohol use in women of childbearing age: results of a brief intervention trial. Alcohol Clin Exp Res. 2000;24:1517-1524.
4. Ingersoll KS, Ceperich SD, Nettleman MD, Karanda K, Brocksen S, Johnson BA. Reducing alcohol-exposed pregnancy risk in college women: initial outcomes of a clinical trial of a motivational intervention. J Subst Abuse Treat. 2005;29:173-189.
5. Chang G, Wilkins-Haug BS, Goetz MA. Brief interventions for alcohol use in pregnancy: a randomized trial. Addiction. 1999;94:1499-1508.
Prenatal Counseling
Prenatal diagnosis became a reality almost 40 years ago, when advances in microscopy and cell culture made it possible to examine chromosomes in fetal cells drawn from amniotic fluid—the familiar karyotype analysis. Technical advances continue to sharpen the resolution of routine karyotype analysis on amniotic fluid or a specimen of chorionic villus, and to raise the level of detail obtained from such a study.
Yet examining chromosomes by light microscopy remains time- and labor-intensive. A cell culture typically requires 2 weeks before growth of cells is sufficient to undertake a karyotype analysis—after which the microscopic evaluation requires further time and significant skill to perform.
Change is coming to practice
Over the past 10 years, however, the human genome project has produced technologies that allow us to examine DNA at a level of resolution unattainable when chromosomes are evaluated under a light microscope. The exciting news is that these research technologies are being transferred to the clinical arena, where they will transform prenatal diagnosis and counseling in your practice.
One such technology that will have such a far-reaching effect, and that I focus on in this “Update,” is known as molecular karyotyping.
What is “molecular karyotyping”? How is it performed?
Refinements to hybridization technology yield new tools; a new term enters the lexicon of prenatal diagnosis
Vermeesch JR, Melotte C, Froyen G, et al. Molecular karyotyping: array CGH quality criteria for constitutional genetic diagnosis, J Histochem Cytochem. 2005;53:413–422.
Van den Veyver I, Beaudet A. Comparative genomic hybridization and prenatal diagnosis. Curr Opin Obstet Gynecol. 2006;18:185–191.
So-called molecular karyotyping utilizes the evolving technology of comparative genomic hybridization by microarray (or, simply, array CGH), which is a refinement of older CGH technology. Initial work with whole-genome hybridization involved applying fragmented and fluorescently labeled subject DNA to a normal metaphase chromosome spread. Deletions or duplications within the subject DNA were then made evident by reduced, or increased, fluorescence at complementary sites along the metaphase chromosomes. The resolution afforded by this approach was comparable to that of light microscopy—namely, alterations of at least 5 to 10 megabases (Mb) could be detected.
Array technology emerged in the late 1990s and increased the resolution of genome hybridization by at least 10-fold. How does it work?
FIGURE 1
Sample display of array CGH and corresponding FISH analysis
At left: Hybridization ratios of normal sex-matched control DNA (Cy5) to sample DNA (Cy3) are plotted as a function of Cy5/Cy3 signal intensity. (Note that ratios of deleted clones are greater than +3SD.)
At right: Fluorescence in situ hybridization (FISH) analysis demonstrates intact (arrows) and deleted (arrowheads) signals.
Bottom: Clones are summarized schematically.
Modified from Yamagata et al. Am J Med Genet. 2006;140A:205–211.
FIGURE 2
Array CGH reveals a duplicated chromosome 15q
At left: Analysis by array CGH demonstrates trisomy 16 and duplication of the Prader-Willi/Angelman syndrome region on chromosome 15q in this patient. Each clone is spotted in triplicate on the array; clones with a gain in the specimen are represented in green; those with a loss, in red; and those with a normal copy number, in gray. Green boxes mark chromosome 16 clones that demonstrate trisomy. White boxes highlight clones from the Prader-Willi/Angelman syndrome region that are duplicated; corresponding ratios are shown next to each target. Other red and green signals correspond to clones from, respectively, the X and Y chromosomes.
At right: Interphase FISH analysis confirms the interstitial duplication of chromosome 15q that was identified by array CGH. The small arrow in each cell points to the normal signal for the SNRPN (Prader-Willi) gene; the large arrow indicates duplicated chromosome 15q, which shows two hybridization signals for SNRPN.
Modified from Schaffer. Am J Human Genet. 2004;74:1168.
Array CGH is still new but already being improved
The 1st generation of array CGH slides covered the entire human genome with DNA fragments spaced approximately 1 Mb apart. Refinements have produced arrays of more than 30,000 overlapping DNA fragments. Such resolution allows detection of a gain or loss of segments as small as 100 to 200 kilobases (Kb). Compare this resolution with the best resolution of traditional microscopic cytogenetic analysis: approximately 5 Mb.
Into the clinical realm
Specialized “targeted” arrays can be applied to clinical work in several ways, including:
And consider what is anticipated: highly dense arrays that are capable of assessing single nucleotide alterations, making it possible to detect single-gene mutations.
Because array-CGH technology utilizes DNA and does not require cell culture, the time to results is significantly shorter. Furthermore, many aspects of the assessment are automated, providing both high resolution and rapid processing and reporting.
Array CGH uncovers genomic problems in the young
Causes of mental retardation, developmental deficits, congenital anomalies, and more are localized
Miyake N, Shimaokawa O, Harada N, et al. BAC array CGH genomic aberrations in idiopathic mental retardation. Am J Med Genet. 2006;140A:205–211.
Ming J, Geiger E, James A, et al. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum Mutat. 2006;27:467–473.
Early use of array CGH in the study of solid tumors was followed closely by its clinical application to children with mental retardation or developmental deficits, with or without birth defects. Historically, suspicion of a duplication or deletion syndrome despite a normal chromosome analysis in these children could prompt specific testing for that disorder. More often, however, it was impossible to delineate a specific syndrome, and disorder-by-disorder testing was not feasible. Today, estimates are that submicroscopic duplications and deletions on chromosomes, detected primarily by array CGH, occur in 1 of every 1, 000 births.
Initial work in the pediatric population by Vissers, in 2003, and Shaw-Smith, in 2004, showed that, with array CGH at a resolution of 1 Mb, 14% to 20% of children who were mentally retarded had duplications or deletions that could not be detected by routine karyotype analysis. Further detail on this approach, using an array with 1.4-Mb coverage, appears in the article by Miyake and co-workers. Among 30 children with idiopathic mental retardation and dysmorphic features, 17% (5 of 30) had submicroscopic deletions or duplications by array CGH. The imbalances ranged from 0.7 Mb to 1.0 Mb and spanned numerous and various chromosomes. The investigators emphasized the need to:
Numerous “copy number polymorphisms” have been uncovered—do they always matter?
Work with array CGH among the pediatric population was expanded by Ming and colleagues, who obtained greater resolution and coverage of the genome by utilizing a 2nd-generation array of oligonucleotides with >100,000 single-nucleotide polymorphisms. With this array, intermarker distance is estimated at 25 Kb—a resolution at which very small genomic imbalances can be identified. Of 10 children evaluated using this greater-density array, 2 (20%) had a previously unidentified genomic imbalance—both deletions.
Ming also put forward concerns that more non–disease-causing “copy number polymorphisms” (CNPs) will be uncovered as higher-density arrays increase the resolution of array CGH. These polymorphisms are encountered in healthy persons and are considered clinically insignificant. Consequently, when a copy number imbalance is detected by array, several actions are warranted: comparison with normal controls, evaluation of published CNP databases, and—most important—array CGH analysis of both parents’ DNA.
Such an approach adds to the labor-intensity of array CGH, but is necessary to ensure that imbalances that are clinically relevant and causative are distinguished from normal variants. With more than 250 discrete CNPs reported in normal controls, the use of denser arrays will uncover more CNPs than arrays targeted to significant fetal and pediatric disorders. Applying array CGH to clinical practice will entail (1) ongoing assessment of the technology and the results it provides and (2) perhaps, targeting of arrays to particular populations—the goal being to balance the yield of useful information against the increase in reported CNPs.
Where is the potential of array CGH in prenatal diagnosis?
Le Caignec C, Boceno M, Saugier-Veber P, et al. Detection of genomic imbalances by array based comparative genomic hybridization in fetuses with multiple malformations. J Med Genet. 2005;42:121–128.
Rickman L, Fiegler H, Shaw-Smith C, et al. Prenatal detection of unbalanced chromosomal rearrangements by array CGH. J Med Genet. 2006;43:353–361.
Sahoo T, Cheung S, Ward P, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med. 2006;8:719–727.
Prenatal diagnosis can be enhanced by array CGH
If ongoing research on array CGH can accomplish any of the following goals, it is likely that the technology will be propelled into clinical use as part of prenatal counseling within the next 5 years:
Le Caignec and colleagues’ work on applying array CGH to DNA specimens from fetuses that had multiple malformations—but in whom cytogenetic study was normal—have provided a foundation for subsequent prenatal studies. Using an array that targeted subtelomeres and specific DNA loci that are important in cytogenetic deletion–duplication syndromes, Le Caignec found that 5 of 49 (10.2%) fetuses studied had clinically significant genomic imbalances. These included:
The fetuses studied by Le Caignec had at least three malformations—variously in the cardiovascular, urogenital, skeletal, digestive, and central nervous systems. But when the list of identified anomalies was assessed, most of those fetuses, if examined by high-resolution ultrasonography, would have had anomalies identified in only 2 systems; the 3rd involved system would have been detectable only on fetopsy.
Rickman and colleagues used a custom array that focused on prenatal and pediatric abnormalities to examine the sensitivity and specificity of array CGH for detecting common aneuploidies in amniotic fluid specimens. All but 1 of the 30 subjects’ unbalanced chromosome rearrangements could be detected by array CGH—in some cases, from a specimen of amniotic fluid as small as 1 cc.
In Rickman’s hands, as well as in the hands of others, triploidy could not be detected, however—a problem that has been addressed in newer array platforms. In an additional 30 cases, no false positives were noted.
Similar results were obtained by Sahoo and co-workers: In 98 prenatal specimens (obtained by CVS or amniocentesis), there was complete concordance between the results of karyotype analysis and array CGH studies. In most cases, specimens were obtained because of advanced maternal age; only 19% represented concern over a sonographic abnormality. This study population included 4 cases of trisomy 21 and 1 case of an unbalanced translocation.
Notably, among the 98 specimens, 30 were thought to be characterized by gain or loss of copy number of 1 or more clones. Because these copy number repeats are recognized as normal variants (based on analyses of normal populations), they were considered copy number polymorphisms (CNPs) and without clinical significance to the fetus.
In addition, 12 cases contained a copy number imbalance that had not been recognized among normal controls. In 9 of those cases, the same loss or gain was demonstrated in 1 parent. In 1 other case, the parents elected not to be studied and, in the 2 others, the array finding was not confirmed on further testing (although low-level mosaicism could not be excluded). Sahoo’s team emphasizes both the targeted specificity of their custom array for well-characterized disorders, the reference to normal population databases being constructed for CNPs, the use of at least 3 clones for each disease locus, and the necessity for parental specimens to appropriately counsel the family about the presence of CNPs.
The work of Rickman and Sahoo reveals the potential for applying array CGH to a small volume of amniotic fluid or a specimen from direct CVS—a process that begins with whole-genome amplification. As this approach is refined to decrease the sample size and shorten the time to results even more, we can expect to see array CGH applied to areas where analysis has been constrained by the fact of small specimen size—such as preimplantation genetic screening.
Analysis of fetal loss will mean better counseling about recurrence
Fritz B, Hallerman C, Olert J, et al. Cytogenetic analyses of culture failures by comparative genomic hybridization (CGH)—re-evaluation of chromosome aberration rates in early spontaneous abortions. Eur J Hum Genet. 2001;9:539–547.
Schaeffer A, Chung J, Heretis K, et al. Comparative genomic hybridization-array analysis enhances the detection of aneuploidy and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet. 2004;74:1168–1174.
Approximately 50% of 1st-trimester pregnancy losses are considered to be the results of chromosomal abnormalities. Often, however, it isn’t productive to analyze the products of conception because fetal cells fail to grow in culture or are overgrown by maternal cells. And, although chromosomal abnormalities play less of a role in 2nd-trimester fetal loss or in stillbirth, the rate of nondiagnostic results from classical cytogenetic study in such cases is high.
Sampling of the placenta or amniocentesis at the time fetal loss/stillbirth is recognized can lower the no-growth rate, but these methods have not been incorporated into practice universally. With array CGH, however, results can be obtained from uncultured cells, and that capability offers the opportunity to assess a demised fetus for common aneuplodies.
Array CGH will also provide an assessment of genomic imbalances that aren’t otherwise detectable at the resolution of metaphase chromosomes. Identification of a genomic imbalance—during a 1st- or 2nd-trimester loss—would facilitate an appropriate workup and lead to more accurate counseling about the risk of recurrence.
Assessment of nondividing cells reveals an unexpectedly high rate of chromosomal abnormality
Fritz and co-workers used array CGH to assess 60 cases of 1st-trimester spontaneous loss in which culture did not yield a karyotype result. Utilizing the older methodology of genomic CGH (ie, resolution is comparable to that of karyotype analysis), 72% of fetuses were found to have an underlying chromosome abnormality.
The work of this team supports what is increasingly reported:
These data warrant expanding array CGH to the evaluation of loss in 2nd and 3rd trimesters.
Schaffer and colleagues assessed a population of 41 products of conception using conventional cytogenetic analysis and array CGH. The conventional karyotype study and the array CGH were concordant in 37 of 41 cases—with 100% concordance for normal karyotypes, 10 cases of trisomy, 2 cases of sex chromosome aneuploidy, and 2 cases of deletion. More important, 4 cases (9.8%) that had been interpreted as normal on a conventional karyotype study were found by array CGH to have submicroscopic genomic imbalances, including trisomic mosaicism, interstitial deletion, and subtelomeric deletion.
The author reports no financial relationships relevant to this article.
NEW DEVELOPMENTS THAT ARE CHANGING PATIENT CARE

Louise Wilkins-Haug, MD, PhD
Medical Director, Center for Fetal Medicine, Brigham and Women’s Hospital, and Associate Professor and Division Director, Maternal-Fetal Medicine and Reproductive Genetics, Department of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston
NEW DEVELOPMENTS THAT ARE CHANGING PATIENT CARE
Louise Wilkins-Haug, MD, PhD
Medical Director, Center for Fetal Medicine, Brigham and Women’s Hospital, and Associate Professor and Division Director, Maternal-Fetal Medicine and Reproductive Genetics, Department of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston
NEW DEVELOPMENTS THAT ARE CHANGING PATIENT CARE
Louise Wilkins-Haug, MD, PhD
Medical Director, Center for Fetal Medicine, Brigham and Women’s Hospital, and Associate Professor and Division Director, Maternal-Fetal Medicine and Reproductive Genetics, Department of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston
Prenatal diagnosis became a reality almost 40 years ago, when advances in microscopy and cell culture made it possible to examine chromosomes in fetal cells drawn from amniotic fluid—the familiar karyotype analysis. Technical advances continue to sharpen the resolution of routine karyotype analysis on amniotic fluid or a specimen of chorionic villus, and to raise the level of detail obtained from such a study.
Yet examining chromosomes by light microscopy remains time- and labor-intensive. A cell culture typically requires 2 weeks before growth of cells is sufficient to undertake a karyotype analysis—after which the microscopic evaluation requires further time and significant skill to perform.
Change is coming to practice
Over the past 10 years, however, the human genome project has produced technologies that allow us to examine DNA at a level of resolution unattainable when chromosomes are evaluated under a light microscope. The exciting news is that these research technologies are being transferred to the clinical arena, where they will transform prenatal diagnosis and counseling in your practice.
One such technology that will have such a far-reaching effect, and that I focus on in this “Update,” is known as molecular karyotyping.
What is “molecular karyotyping”? How is it performed?
Refinements to hybridization technology yield new tools; a new term enters the lexicon of prenatal diagnosis
Vermeesch JR, Melotte C, Froyen G, et al. Molecular karyotyping: array CGH quality criteria for constitutional genetic diagnosis, J Histochem Cytochem. 2005;53:413–422.
Van den Veyver I, Beaudet A. Comparative genomic hybridization and prenatal diagnosis. Curr Opin Obstet Gynecol. 2006;18:185–191.
So-called molecular karyotyping utilizes the evolving technology of comparative genomic hybridization by microarray (or, simply, array CGH), which is a refinement of older CGH technology. Initial work with whole-genome hybridization involved applying fragmented and fluorescently labeled subject DNA to a normal metaphase chromosome spread. Deletions or duplications within the subject DNA were then made evident by reduced, or increased, fluorescence at complementary sites along the metaphase chromosomes. The resolution afforded by this approach was comparable to that of light microscopy—namely, alterations of at least 5 to 10 megabases (Mb) could be detected.
Array technology emerged in the late 1990s and increased the resolution of genome hybridization by at least 10-fold. How does it work?
FIGURE 1
Sample display of array CGH and corresponding FISH analysis
At left: Hybridization ratios of normal sex-matched control DNA (Cy5) to sample DNA (Cy3) are plotted as a function of Cy5/Cy3 signal intensity. (Note that ratios of deleted clones are greater than +3SD.)
At right: Fluorescence in situ hybridization (FISH) analysis demonstrates intact (arrows) and deleted (arrowheads) signals.
Bottom: Clones are summarized schematically.
Modified from Yamagata et al. Am J Med Genet. 2006;140A:205–211.
FIGURE 2
Array CGH reveals a duplicated chromosome 15q
At left: Analysis by array CGH demonstrates trisomy 16 and duplication of the Prader-Willi/Angelman syndrome region on chromosome 15q in this patient. Each clone is spotted in triplicate on the array; clones with a gain in the specimen are represented in green; those with a loss, in red; and those with a normal copy number, in gray. Green boxes mark chromosome 16 clones that demonstrate trisomy. White boxes highlight clones from the Prader-Willi/Angelman syndrome region that are duplicated; corresponding ratios are shown next to each target. Other red and green signals correspond to clones from, respectively, the X and Y chromosomes.
At right: Interphase FISH analysis confirms the interstitial duplication of chromosome 15q that was identified by array CGH. The small arrow in each cell points to the normal signal for the SNRPN (Prader-Willi) gene; the large arrow indicates duplicated chromosome 15q, which shows two hybridization signals for SNRPN.
Modified from Schaffer. Am J Human Genet. 2004;74:1168.
Array CGH is still new but already being improved
The 1st generation of array CGH slides covered the entire human genome with DNA fragments spaced approximately 1 Mb apart. Refinements have produced arrays of more than 30,000 overlapping DNA fragments. Such resolution allows detection of a gain or loss of segments as small as 100 to 200 kilobases (Kb). Compare this resolution with the best resolution of traditional microscopic cytogenetic analysis: approximately 5 Mb.
Into the clinical realm
Specialized “targeted” arrays can be applied to clinical work in several ways, including:
And consider what is anticipated: highly dense arrays that are capable of assessing single nucleotide alterations, making it possible to detect single-gene mutations.
Because array-CGH technology utilizes DNA and does not require cell culture, the time to results is significantly shorter. Furthermore, many aspects of the assessment are automated, providing both high resolution and rapid processing and reporting.
Array CGH uncovers genomic problems in the young
Causes of mental retardation, developmental deficits, congenital anomalies, and more are localized
Miyake N, Shimaokawa O, Harada N, et al. BAC array CGH genomic aberrations in idiopathic mental retardation. Am J Med Genet. 2006;140A:205–211.
Ming J, Geiger E, James A, et al. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum Mutat. 2006;27:467–473.
Early use of array CGH in the study of solid tumors was followed closely by its clinical application to children with mental retardation or developmental deficits, with or without birth defects. Historically, suspicion of a duplication or deletion syndrome despite a normal chromosome analysis in these children could prompt specific testing for that disorder. More often, however, it was impossible to delineate a specific syndrome, and disorder-by-disorder testing was not feasible. Today, estimates are that submicroscopic duplications and deletions on chromosomes, detected primarily by array CGH, occur in 1 of every 1, 000 births.
Initial work in the pediatric population by Vissers, in 2003, and Shaw-Smith, in 2004, showed that, with array CGH at a resolution of 1 Mb, 14% to 20% of children who were mentally retarded had duplications or deletions that could not be detected by routine karyotype analysis. Further detail on this approach, using an array with 1.4-Mb coverage, appears in the article by Miyake and co-workers. Among 30 children with idiopathic mental retardation and dysmorphic features, 17% (5 of 30) had submicroscopic deletions or duplications by array CGH. The imbalances ranged from 0.7 Mb to 1.0 Mb and spanned numerous and various chromosomes. The investigators emphasized the need to:
Numerous “copy number polymorphisms” have been uncovered—do they always matter?
Work with array CGH among the pediatric population was expanded by Ming and colleagues, who obtained greater resolution and coverage of the genome by utilizing a 2nd-generation array of oligonucleotides with >100,000 single-nucleotide polymorphisms. With this array, intermarker distance is estimated at 25 Kb—a resolution at which very small genomic imbalances can be identified. Of 10 children evaluated using this greater-density array, 2 (20%) had a previously unidentified genomic imbalance—both deletions.
Ming also put forward concerns that more non–disease-causing “copy number polymorphisms” (CNPs) will be uncovered as higher-density arrays increase the resolution of array CGH. These polymorphisms are encountered in healthy persons and are considered clinically insignificant. Consequently, when a copy number imbalance is detected by array, several actions are warranted: comparison with normal controls, evaluation of published CNP databases, and—most important—array CGH analysis of both parents’ DNA.
Such an approach adds to the labor-intensity of array CGH, but is necessary to ensure that imbalances that are clinically relevant and causative are distinguished from normal variants. With more than 250 discrete CNPs reported in normal controls, the use of denser arrays will uncover more CNPs than arrays targeted to significant fetal and pediatric disorders. Applying array CGH to clinical practice will entail (1) ongoing assessment of the technology and the results it provides and (2) perhaps, targeting of arrays to particular populations—the goal being to balance the yield of useful information against the increase in reported CNPs.
Where is the potential of array CGH in prenatal diagnosis?
Le Caignec C, Boceno M, Saugier-Veber P, et al. Detection of genomic imbalances by array based comparative genomic hybridization in fetuses with multiple malformations. J Med Genet. 2005;42:121–128.
Rickman L, Fiegler H, Shaw-Smith C, et al. Prenatal detection of unbalanced chromosomal rearrangements by array CGH. J Med Genet. 2006;43:353–361.
Sahoo T, Cheung S, Ward P, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med. 2006;8:719–727.
Prenatal diagnosis can be enhanced by array CGH
If ongoing research on array CGH can accomplish any of the following goals, it is likely that the technology will be propelled into clinical use as part of prenatal counseling within the next 5 years:
Le Caignec and colleagues’ work on applying array CGH to DNA specimens from fetuses that had multiple malformations—but in whom cytogenetic study was normal—have provided a foundation for subsequent prenatal studies. Using an array that targeted subtelomeres and specific DNA loci that are important in cytogenetic deletion–duplication syndromes, Le Caignec found that 5 of 49 (10.2%) fetuses studied had clinically significant genomic imbalances. These included:
The fetuses studied by Le Caignec had at least three malformations—variously in the cardiovascular, urogenital, skeletal, digestive, and central nervous systems. But when the list of identified anomalies was assessed, most of those fetuses, if examined by high-resolution ultrasonography, would have had anomalies identified in only 2 systems; the 3rd involved system would have been detectable only on fetopsy.
Rickman and colleagues used a custom array that focused on prenatal and pediatric abnormalities to examine the sensitivity and specificity of array CGH for detecting common aneuploidies in amniotic fluid specimens. All but 1 of the 30 subjects’ unbalanced chromosome rearrangements could be detected by array CGH—in some cases, from a specimen of amniotic fluid as small as 1 cc.
In Rickman’s hands, as well as in the hands of others, triploidy could not be detected, however—a problem that has been addressed in newer array platforms. In an additional 30 cases, no false positives were noted.
Similar results were obtained by Sahoo and co-workers: In 98 prenatal specimens (obtained by CVS or amniocentesis), there was complete concordance between the results of karyotype analysis and array CGH studies. In most cases, specimens were obtained because of advanced maternal age; only 19% represented concern over a sonographic abnormality. This study population included 4 cases of trisomy 21 and 1 case of an unbalanced translocation.
Notably, among the 98 specimens, 30 were thought to be characterized by gain or loss of copy number of 1 or more clones. Because these copy number repeats are recognized as normal variants (based on analyses of normal populations), they were considered copy number polymorphisms (CNPs) and without clinical significance to the fetus.
In addition, 12 cases contained a copy number imbalance that had not been recognized among normal controls. In 9 of those cases, the same loss or gain was demonstrated in 1 parent. In 1 other case, the parents elected not to be studied and, in the 2 others, the array finding was not confirmed on further testing (although low-level mosaicism could not be excluded). Sahoo’s team emphasizes both the targeted specificity of their custom array for well-characterized disorders, the reference to normal population databases being constructed for CNPs, the use of at least 3 clones for each disease locus, and the necessity for parental specimens to appropriately counsel the family about the presence of CNPs.
The work of Rickman and Sahoo reveals the potential for applying array CGH to a small volume of amniotic fluid or a specimen from direct CVS—a process that begins with whole-genome amplification. As this approach is refined to decrease the sample size and shorten the time to results even more, we can expect to see array CGH applied to areas where analysis has been constrained by the fact of small specimen size—such as preimplantation genetic screening.
Analysis of fetal loss will mean better counseling about recurrence
Fritz B, Hallerman C, Olert J, et al. Cytogenetic analyses of culture failures by comparative genomic hybridization (CGH)—re-evaluation of chromosome aberration rates in early spontaneous abortions. Eur J Hum Genet. 2001;9:539–547.
Schaeffer A, Chung J, Heretis K, et al. Comparative genomic hybridization-array analysis enhances the detection of aneuploidy and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet. 2004;74:1168–1174.
Approximately 50% of 1st-trimester pregnancy losses are considered to be the results of chromosomal abnormalities. Often, however, it isn’t productive to analyze the products of conception because fetal cells fail to grow in culture or are overgrown by maternal cells. And, although chromosomal abnormalities play less of a role in 2nd-trimester fetal loss or in stillbirth, the rate of nondiagnostic results from classical cytogenetic study in such cases is high.
Sampling of the placenta or amniocentesis at the time fetal loss/stillbirth is recognized can lower the no-growth rate, but these methods have not been incorporated into practice universally. With array CGH, however, results can be obtained from uncultured cells, and that capability offers the opportunity to assess a demised fetus for common aneuplodies.
Array CGH will also provide an assessment of genomic imbalances that aren’t otherwise detectable at the resolution of metaphase chromosomes. Identification of a genomic imbalance—during a 1st- or 2nd-trimester loss—would facilitate an appropriate workup and lead to more accurate counseling about the risk of recurrence.
Assessment of nondividing cells reveals an unexpectedly high rate of chromosomal abnormality
Fritz and co-workers used array CGH to assess 60 cases of 1st-trimester spontaneous loss in which culture did not yield a karyotype result. Utilizing the older methodology of genomic CGH (ie, resolution is comparable to that of karyotype analysis), 72% of fetuses were found to have an underlying chromosome abnormality.
The work of this team supports what is increasingly reported:
These data warrant expanding array CGH to the evaluation of loss in 2nd and 3rd trimesters.
Schaffer and colleagues assessed a population of 41 products of conception using conventional cytogenetic analysis and array CGH. The conventional karyotype study and the array CGH were concordant in 37 of 41 cases—with 100% concordance for normal karyotypes, 10 cases of trisomy, 2 cases of sex chromosome aneuploidy, and 2 cases of deletion. More important, 4 cases (9.8%) that had been interpreted as normal on a conventional karyotype study were found by array CGH to have submicroscopic genomic imbalances, including trisomic mosaicism, interstitial deletion, and subtelomeric deletion.
The author reports no financial relationships relevant to this article.
Prenatal diagnosis became a reality almost 40 years ago, when advances in microscopy and cell culture made it possible to examine chromosomes in fetal cells drawn from amniotic fluid—the familiar karyotype analysis. Technical advances continue to sharpen the resolution of routine karyotype analysis on amniotic fluid or a specimen of chorionic villus, and to raise the level of detail obtained from such a study.
Yet examining chromosomes by light microscopy remains time- and labor-intensive. A cell culture typically requires 2 weeks before growth of cells is sufficient to undertake a karyotype analysis—after which the microscopic evaluation requires further time and significant skill to perform.
Change is coming to practice
Over the past 10 years, however, the human genome project has produced technologies that allow us to examine DNA at a level of resolution unattainable when chromosomes are evaluated under a light microscope. The exciting news is that these research technologies are being transferred to the clinical arena, where they will transform prenatal diagnosis and counseling in your practice.
One such technology that will have such a far-reaching effect, and that I focus on in this “Update,” is known as molecular karyotyping.
What is “molecular karyotyping”? How is it performed?
Refinements to hybridization technology yield new tools; a new term enters the lexicon of prenatal diagnosis
Vermeesch JR, Melotte C, Froyen G, et al. Molecular karyotyping: array CGH quality criteria for constitutional genetic diagnosis, J Histochem Cytochem. 2005;53:413–422.
Van den Veyver I, Beaudet A. Comparative genomic hybridization and prenatal diagnosis. Curr Opin Obstet Gynecol. 2006;18:185–191.
So-called molecular karyotyping utilizes the evolving technology of comparative genomic hybridization by microarray (or, simply, array CGH), which is a refinement of older CGH technology. Initial work with whole-genome hybridization involved applying fragmented and fluorescently labeled subject DNA to a normal metaphase chromosome spread. Deletions or duplications within the subject DNA were then made evident by reduced, or increased, fluorescence at complementary sites along the metaphase chromosomes. The resolution afforded by this approach was comparable to that of light microscopy—namely, alterations of at least 5 to 10 megabases (Mb) could be detected.
Array technology emerged in the late 1990s and increased the resolution of genome hybridization by at least 10-fold. How does it work?
FIGURE 1
Sample display of array CGH and corresponding FISH analysis
At left: Hybridization ratios of normal sex-matched control DNA (Cy5) to sample DNA (Cy3) are plotted as a function of Cy5/Cy3 signal intensity. (Note that ratios of deleted clones are greater than +3SD.)
At right: Fluorescence in situ hybridization (FISH) analysis demonstrates intact (arrows) and deleted (arrowheads) signals.
Bottom: Clones are summarized schematically.
Modified from Yamagata et al. Am J Med Genet. 2006;140A:205–211.
FIGURE 2
Array CGH reveals a duplicated chromosome 15q
At left: Analysis by array CGH demonstrates trisomy 16 and duplication of the Prader-Willi/Angelman syndrome region on chromosome 15q in this patient. Each clone is spotted in triplicate on the array; clones with a gain in the specimen are represented in green; those with a loss, in red; and those with a normal copy number, in gray. Green boxes mark chromosome 16 clones that demonstrate trisomy. White boxes highlight clones from the Prader-Willi/Angelman syndrome region that are duplicated; corresponding ratios are shown next to each target. Other red and green signals correspond to clones from, respectively, the X and Y chromosomes.
At right: Interphase FISH analysis confirms the interstitial duplication of chromosome 15q that was identified by array CGH. The small arrow in each cell points to the normal signal for the SNRPN (Prader-Willi) gene; the large arrow indicates duplicated chromosome 15q, which shows two hybridization signals for SNRPN.
Modified from Schaffer. Am J Human Genet. 2004;74:1168.
Array CGH is still new but already being improved
The 1st generation of array CGH slides covered the entire human genome with DNA fragments spaced approximately 1 Mb apart. Refinements have produced arrays of more than 30,000 overlapping DNA fragments. Such resolution allows detection of a gain or loss of segments as small as 100 to 200 kilobases (Kb). Compare this resolution with the best resolution of traditional microscopic cytogenetic analysis: approximately 5 Mb.
Into the clinical realm
Specialized “targeted” arrays can be applied to clinical work in several ways, including:
And consider what is anticipated: highly dense arrays that are capable of assessing single nucleotide alterations, making it possible to detect single-gene mutations.
Because array-CGH technology utilizes DNA and does not require cell culture, the time to results is significantly shorter. Furthermore, many aspects of the assessment are automated, providing both high resolution and rapid processing and reporting.
Array CGH uncovers genomic problems in the young
Causes of mental retardation, developmental deficits, congenital anomalies, and more are localized
Miyake N, Shimaokawa O, Harada N, et al. BAC array CGH genomic aberrations in idiopathic mental retardation. Am J Med Genet. 2006;140A:205–211.
Ming J, Geiger E, James A, et al. Rapid detection of submicroscopic chromosomal rearrangements in children with multiple congenital anomalies using high density oligonucleotide arrays. Hum Mutat. 2006;27:467–473.
Early use of array CGH in the study of solid tumors was followed closely by its clinical application to children with mental retardation or developmental deficits, with or without birth defects. Historically, suspicion of a duplication or deletion syndrome despite a normal chromosome analysis in these children could prompt specific testing for that disorder. More often, however, it was impossible to delineate a specific syndrome, and disorder-by-disorder testing was not feasible. Today, estimates are that submicroscopic duplications and deletions on chromosomes, detected primarily by array CGH, occur in 1 of every 1, 000 births.
Initial work in the pediatric population by Vissers, in 2003, and Shaw-Smith, in 2004, showed that, with array CGH at a resolution of 1 Mb, 14% to 20% of children who were mentally retarded had duplications or deletions that could not be detected by routine karyotype analysis. Further detail on this approach, using an array with 1.4-Mb coverage, appears in the article by Miyake and co-workers. Among 30 children with idiopathic mental retardation and dysmorphic features, 17% (5 of 30) had submicroscopic deletions or duplications by array CGH. The imbalances ranged from 0.7 Mb to 1.0 Mb and spanned numerous and various chromosomes. The investigators emphasized the need to:
Numerous “copy number polymorphisms” have been uncovered—do they always matter?
Work with array CGH among the pediatric population was expanded by Ming and colleagues, who obtained greater resolution and coverage of the genome by utilizing a 2nd-generation array of oligonucleotides with >100,000 single-nucleotide polymorphisms. With this array, intermarker distance is estimated at 25 Kb—a resolution at which very small genomic imbalances can be identified. Of 10 children evaluated using this greater-density array, 2 (20%) had a previously unidentified genomic imbalance—both deletions.
Ming also put forward concerns that more non–disease-causing “copy number polymorphisms” (CNPs) will be uncovered as higher-density arrays increase the resolution of array CGH. These polymorphisms are encountered in healthy persons and are considered clinically insignificant. Consequently, when a copy number imbalance is detected by array, several actions are warranted: comparison with normal controls, evaluation of published CNP databases, and—most important—array CGH analysis of both parents’ DNA.
Such an approach adds to the labor-intensity of array CGH, but is necessary to ensure that imbalances that are clinically relevant and causative are distinguished from normal variants. With more than 250 discrete CNPs reported in normal controls, the use of denser arrays will uncover more CNPs than arrays targeted to significant fetal and pediatric disorders. Applying array CGH to clinical practice will entail (1) ongoing assessment of the technology and the results it provides and (2) perhaps, targeting of arrays to particular populations—the goal being to balance the yield of useful information against the increase in reported CNPs.
Where is the potential of array CGH in prenatal diagnosis?
Le Caignec C, Boceno M, Saugier-Veber P, et al. Detection of genomic imbalances by array based comparative genomic hybridization in fetuses with multiple malformations. J Med Genet. 2005;42:121–128.
Rickman L, Fiegler H, Shaw-Smith C, et al. Prenatal detection of unbalanced chromosomal rearrangements by array CGH. J Med Genet. 2006;43:353–361.
Sahoo T, Cheung S, Ward P, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med. 2006;8:719–727.
Prenatal diagnosis can be enhanced by array CGH
If ongoing research on array CGH can accomplish any of the following goals, it is likely that the technology will be propelled into clinical use as part of prenatal counseling within the next 5 years:
Le Caignec and colleagues’ work on applying array CGH to DNA specimens from fetuses that had multiple malformations—but in whom cytogenetic study was normal—have provided a foundation for subsequent prenatal studies. Using an array that targeted subtelomeres and specific DNA loci that are important in cytogenetic deletion–duplication syndromes, Le Caignec found that 5 of 49 (10.2%) fetuses studied had clinically significant genomic imbalances. These included:
The fetuses studied by Le Caignec had at least three malformations—variously in the cardiovascular, urogenital, skeletal, digestive, and central nervous systems. But when the list of identified anomalies was assessed, most of those fetuses, if examined by high-resolution ultrasonography, would have had anomalies identified in only 2 systems; the 3rd involved system would have been detectable only on fetopsy.
Rickman and colleagues used a custom array that focused on prenatal and pediatric abnormalities to examine the sensitivity and specificity of array CGH for detecting common aneuploidies in amniotic fluid specimens. All but 1 of the 30 subjects’ unbalanced chromosome rearrangements could be detected by array CGH—in some cases, from a specimen of amniotic fluid as small as 1 cc.
In Rickman’s hands, as well as in the hands of others, triploidy could not be detected, however—a problem that has been addressed in newer array platforms. In an additional 30 cases, no false positives were noted.
Similar results were obtained by Sahoo and co-workers: In 98 prenatal specimens (obtained by CVS or amniocentesis), there was complete concordance between the results of karyotype analysis and array CGH studies. In most cases, specimens were obtained because of advanced maternal age; only 19% represented concern over a sonographic abnormality. This study population included 4 cases of trisomy 21 and 1 case of an unbalanced translocation.
Notably, among the 98 specimens, 30 were thought to be characterized by gain or loss of copy number of 1 or more clones. Because these copy number repeats are recognized as normal variants (based on analyses of normal populations), they were considered copy number polymorphisms (CNPs) and without clinical significance to the fetus.
In addition, 12 cases contained a copy number imbalance that had not been recognized among normal controls. In 9 of those cases, the same loss or gain was demonstrated in 1 parent. In 1 other case, the parents elected not to be studied and, in the 2 others, the array finding was not confirmed on further testing (although low-level mosaicism could not be excluded). Sahoo’s team emphasizes both the targeted specificity of their custom array for well-characterized disorders, the reference to normal population databases being constructed for CNPs, the use of at least 3 clones for each disease locus, and the necessity for parental specimens to appropriately counsel the family about the presence of CNPs.
The work of Rickman and Sahoo reveals the potential for applying array CGH to a small volume of amniotic fluid or a specimen from direct CVS—a process that begins with whole-genome amplification. As this approach is refined to decrease the sample size and shorten the time to results even more, we can expect to see array CGH applied to areas where analysis has been constrained by the fact of small specimen size—such as preimplantation genetic screening.
Analysis of fetal loss will mean better counseling about recurrence
Fritz B, Hallerman C, Olert J, et al. Cytogenetic analyses of culture failures by comparative genomic hybridization (CGH)—re-evaluation of chromosome aberration rates in early spontaneous abortions. Eur J Hum Genet. 2001;9:539–547.
Schaeffer A, Chung J, Heretis K, et al. Comparative genomic hybridization-array analysis enhances the detection of aneuploidy and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet. 2004;74:1168–1174.
Approximately 50% of 1st-trimester pregnancy losses are considered to be the results of chromosomal abnormalities. Often, however, it isn’t productive to analyze the products of conception because fetal cells fail to grow in culture or are overgrown by maternal cells. And, although chromosomal abnormalities play less of a role in 2nd-trimester fetal loss or in stillbirth, the rate of nondiagnostic results from classical cytogenetic study in such cases is high.
Sampling of the placenta or amniocentesis at the time fetal loss/stillbirth is recognized can lower the no-growth rate, but these methods have not been incorporated into practice universally. With array CGH, however, results can be obtained from uncultured cells, and that capability offers the opportunity to assess a demised fetus for common aneuplodies.
Array CGH will also provide an assessment of genomic imbalances that aren’t otherwise detectable at the resolution of metaphase chromosomes. Identification of a genomic imbalance—during a 1st- or 2nd-trimester loss—would facilitate an appropriate workup and lead to more accurate counseling about the risk of recurrence.
Assessment of nondividing cells reveals an unexpectedly high rate of chromosomal abnormality
Fritz and co-workers used array CGH to assess 60 cases of 1st-trimester spontaneous loss in which culture did not yield a karyotype result. Utilizing the older methodology of genomic CGH (ie, resolution is comparable to that of karyotype analysis), 72% of fetuses were found to have an underlying chromosome abnormality.
The work of this team supports what is increasingly reported:
These data warrant expanding array CGH to the evaluation of loss in 2nd and 3rd trimesters.
Schaffer and colleagues assessed a population of 41 products of conception using conventional cytogenetic analysis and array CGH. The conventional karyotype study and the array CGH were concordant in 37 of 41 cases—with 100% concordance for normal karyotypes, 10 cases of trisomy, 2 cases of sex chromosome aneuploidy, and 2 cases of deletion. More important, 4 cases (9.8%) that had been interpreted as normal on a conventional karyotype study were found by array CGH to have submicroscopic genomic imbalances, including trisomic mosaicism, interstitial deletion, and subtelomeric deletion.
The author reports no financial relationships relevant to this article.
PRENATAL COUNSELING
- Screening for fragile X, the most common cause of mental retardation
- Fetal RhD genotyping using maternal plasma
Genetic medicine is fast gaining recognition as an essential component of clinical care. Since this Update last year, 2 areas have seen notable progress:
- New guidelines were issued for fragile X screening in women of reproductive age, and
- Research on free fetal DNA in the maternal circulation deepened our understanding of its diagnostic potential. Clinical applications are emerging in both avenues, and there will be further investigation and refinement. Be sure to check this column again next year!
New guidelines on who to test for mental retardation marker
Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7:584–587.
McConkie-Rosell A, Finucane B, Cronister A, Abrams L, Bennett RL, Petterson BJ. Genetic counseling for fragile X syndrome: updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2005;14:249–270.
Fragile X syndrome is the most common inherited cause of mental retardation. The condition can occur in both males and females and is characterized by a range of behavioral changes consistent with autism spectrum, mental retardation, and developmental delay, as well as a facial phenotype that tends to become more recognizable as the individual ages.
Test is not for everybody
New guidelines issued by the American College of Medical Genetics recommended general population screening only within the constructs of research protocols. In selected populations, however, screening should be considered (TABLE 1). Among preconception and prenatal patients, directed interrogation of the family history for findings suggestive of fragile X syndrome can be guided by these recommendations.
Prevalence. ObGyns should be aware of the increasing spectrum of full and premutation fragile X phenotypes and the relatively high prevalence of premutations among women.
Anatomy of fragile X
Changes in a specific region of the X chromosome known as the fragile X mental retardation-1 (FMR-1) gene are responsible for the syndrome. Elongation of an unstable CGG repeat sequence at the 5′ end of FMR-1 leads to hypermethylation, impaired translation, and altered production of the fragile X mental retardation protein. Investigations of knock-out mice reveal that this protein plays an important role in prenatal and postnatal brain development, especially in the area of dendrite maturation.
Among Caucasians, the characteristic features of fragile X syndrome occur in approximately 1 in 4,000 males and 1 in 8,000 females and are associated with elongation of the FMR-1 gene to more than 200 CGG repeats (a full mutation). Initial studies of other races suggest a similar range of full mutations in males and females.
TABLE 1
Fragile X syndrome: Diagnostic and carrier testing guidelines
| Both women and men with |
|
| Physical or behavioral characteristics of fragile X |
| or family history of fragile X |
| or a relative with undiagnosed mental retardation |
| Persons seeking reproductive counseling who have |
|
| Fetuses of carrier mothers |
| Affected individuals or relatives in whom the diagnosis was made by cytogenetic studies |
| Women with elevated follicle-stimulating hormone, especially with family history of |
|
| Men or women with late-onset intention tremor or ataxia…especially with family history of |
|
| Source: Sherman et al. |
Which offspring will inherit the gene?
In the general population, the FMR-1 region has variable lengths.1 In most individuals, 40 or fewer CGG repeats are present and the region remains stable when passed from either parent to the child. Occasionally, however, individuals inherit expansions of this repeat region—either slight (41–60 repeats, intermediate range) or larger (61–200, premutation range). Repeats in the premutation range are carried by 1 in 700 to 1,000 males and 1 in 113 to 350 females.
Expansion of the premutation to a full FMR-1 mutation depends on the sex of the transmitting parent, the length of repeats, and the frequency of AGG interspersion. Only the first 2 criteria are available for clinical interpretation. FMR-1 expansion occurs only in the X originating from the maternal cell line. The larger the premutation, the more likely it will expand to a full mutation (TABLE 2).
Timing of the maternal FMR-1 expansion can vary, with meiotic, postzygotic, and mitotic instability of CGG length all reported.
- Typically, all sons who inherit an expanded, full mutation exhibit features of fragile X syndrome.
- In daughters, however, a full mutation causes a range of features. In daughters with a full mutation, prognostication is limited. Studies indicate that at least 50%, and in some series 75%, have IQs in the borderline or mentally retarded range.
Longitudinal studies of asymptomatic females with full mutations have not been reported. Fathers with premutations pass the FMR-1 gene in a stable fashion to all offspring, occasionally with contraction to a smaller repeat size.
Note that the complex inheritance pattern, with premutations transmitted through both sexes but expansion limited to the maternal X chromosome, can confound interpretation of family histories of mental retardation or developmental delay.
TABLE 2
Number of CGG repeats influences mutation status
| PERCENT RISK OF EXPANSION TO FULL MUTATION (>200 REPEATS) | ||||
|---|---|---|---|---|
| MATERNAL REPEATS | NOLIN, 19962 | PESSO, 20003 | TOLEDANO-ALHADEF, 20014 | NOLIN, 20035 |
| 55–59 | 13 (3/22) | 0 (0/11) | 0 (0/22) | 4 (1/27) |
| 60–69 | 21 (7/34) | 12 (1/8) | 10 (2/20) | 5 (6/113) |
| 70–79 | 58 (59/102) | 50 (1/2) | 17 (1/6) | 31 (28/90) |
| 80–89 | 73 (78/107) | 50 (1/2) | — | 58 (81/140) |
| 90–99 | 94 (83/88) | 100 (1/1) | — | 80 (89/111) |
| 100–200 | 99 (177/179) | 75 (3/4) | — | 98 (194/197) |
| Values presented as percent (n/N). | ||||
| Data modified from Nolin et al. | ||||
Screening populations
Testing for the FMR-1 gene by determining the expansion size is possible via DNA analysis. Most labs utilize both Southern analyses, to measure the degree of methylation, and polymerase chain reaction, to discriminate at a more refined level the subtle differences in repeat sizes that distinguish intermediate and premutation sizes.
Screening the general population for premutations of the FMR-1 gene is not yet the standard of care. However, several authorities advocate fragile X screening among prenatal and preconception populations, given the relatively high rate of the carrier state (1 in 113 to 350), the sensitivity of testing, and the implications for mental retardation and disability in offspring.
In the United States, even assuming a relatively conservative premutation rate of 1 in 300 and an expansion rate of only 11.3%, such testing would be cost-effective, ranging from $99 to $300 per test.6
What are the risks?
Concerns include the implications of intermediate expansions and the substantial patient education needed to convey the risk of expansion to premutation (but not full mutation). No child with a full mutation has been born to a mother with 59 or fewer repeats. Also needing study is the variability of fragile X syndrome in women with a full mutation. These women are at substantial risk for learning impairment, but the degree of disability varies unpredictably.
Prenatal diagnosis requires DNA from amniocytes or chorionic villus sampling. If the latter, follow-up amniocentesis may be needed because methylation begins at variable times during placental development.7
Preimplantation genetic assessment for fragile X premutation carriers has been reported using a system of closely linked markers, circumventing the need to assess onset of methylation abnormalities.
Spectrum of symptoms
Previously, individuals with premutations were considered clinically asymptomatic. However, we now know that phenotypic expression of expansion sizes occurs along a spectrum.
Recent data indicate 2 phenotypes associated with premutations:
- In women, premature ovarian failure, defined as menopause before the age of 40 years, occurs in 13% to 24% of those with premutations of the FMR-1 gene, among families with fragile X syndrome. Conversely, among women with premature ovarian failure, premutations are found in 2% and 14% of sporadic and familial cases, respectively. Further, the size of the premutation may be directly correlated to the risk of premature ovarian failure.
- In men with premutations, a neurologic syndrome of tremors and ataxia is a newly described phenomenon. The fragile X-associated tremor/ataxia syndrome (FXTAS) is a progressive, neurodegenerative process with Parkinsonism and peripheral neuropathy, and penetrance appears to increase with age.8 The frequency of this diagnosis among older men with premutations is under study.
Fetal RhD genotyping now possible using maternal plasma
Gautier E, Benachi A, Giovangrandi Y, et al. Fetal RhD genotyping by maternal serum analyses: a two-year experience. Am J Obstet Gynecol. 2005;192:666–669.
Moise K. Fetal RhD typing with free DNA in maternal plasma. Am J Obstet Gynecol. 2005;192:663–665.
Fetal RhD typing using free fetal DNA (ffDNA) is routine in the United Kingdom but not yet in this country. Since 1997, when Lo identified ffDNA in maternal plasma,9 numerous studies have focused on the physiology, timing, and clinical application of fetal RhD typing using ffDNA. Previously, the focus was detection of fetal cells in maternal circulation.
Unlike intact fetal cells, ffDNA fragments are present in the maternal plasma in sufficient quantities to allow extensive investigation. While most maternal free DNA is composed of longer DNA fragments, shorter DNA fragments of fetal origin appear as the pregnancy advances and in some studies are first detectable as early as 32 days after conception.
Free fetal DNA increases throughout gestation, representing 3% of total DNA in maternal plasma during the second trimester, and increasing to 6% in the third trimester. Free fetal DNA fragments are cleared rapidly by the renal system, with a half-life of 16 minutes and no discernable levels as soon as 2 hours after delivery.
We now understand that ffDNA fragments are continuously deposited in the maternal circulation from early in pregnancy, perhaps even before fetal circulation develops. We also know that maternal levels of ffDNA depend on 2 forces: rate of deposition and rate of removal.10
Trophoblastic origin?
A placental source is suggested by evidence that ffDNA can be retrieved from maternal plasma prior to the development of fetal circulation. A trophoblastic origin is supported by identification in maternal plasma of fetal mRNA with specificity for genes expressed by the placenta. Moreover, ffDNA has been detected in maternal circulation as early as 14 days after conception, corroborating a trophoblastic origin, with programmed apoptosis of placental cells a likely mechanism.
Further increases in ffDNA throughout gestation may reflect direct contributions from the fetal circulation that are transferred to maternal circulation via the placenta. In later gestation, destruction of fetal cells within the maternal circulation may contribute to the pool of ffDNA in maternal plasma. The exact proportions of each contribution are unknown.11
ffDNA may help diagnose these disorders
Placental abnormalities
Recent work suggests sufficient quantities of ffDNA can be obtained for both quantitative and qualitative assessments.12 Used quantitatively, ffDNA reflects placental integrity, an active area of investigation.
Autosomal trisomies, preeclampsia, and fetal growth restriction are conditions thought to involve abnormal placental function. Increased levels of ffDNA have been found in these entities. Increases have been documented even before onset of preeclampsia.10
Gene defects
Use of ffDNA to identify specific gene defects is also under study. Sensitive microarray technology will likely be needed to assess fetal chromosome aneuploidy from maternal plasma.
The detection of single gene defects from ffDNA has been reported for paternally inherited myotonic dystrophy, Huntington disease, and achondroplasia.
For autosomal recessive disorders, genetic testing of ffDNA may be a first step to exclude inheritance of a paternal allele. For this application, discordant parental alleles will be needed so that exclusion of the paternal mutation in the ffDNA signifies an unaffected fetus or a heterozygotic carrier of the maternal allele. If the paternal allele is detected by ffDNA, further genetic testing by chorionic villus sampling or amniocentesis would be needed to differentiate heterozygotic carriers of the paternal mutation from homozygotic, affected fetuses.
RhD genotyping
Since 2001, ffDNA has been used clinically in the United Kingdom for fetal blood group genotyping in isoimmunized gravidas with heterozygous partners, through the International Blood Group Reference Laboratory (part of the National Blood Service), which brings us to the highlighted study. Gautier and colleagues added data affirming that the RhD genotype can be detected through ffDNA with high sensitivity and specificity. Among 285 RhD-negative women, the fetal RhD genotype was determined in 283. In 2 cases, the maternal RhD-negative phenotype did not result from a complete gene deletion; thus, the genotypes of fetus and mother could not be differentiated. Among the women with RhD-negative genotypes, all fetuses were accurately genotyped through ffDNA.
This study differs from prior investigations in its use of RhD-negative women who were not already sensitized, and suggests that ffDNA genotyping in RhD-negative women is sensitive enough to be incorporated into the distribution of Rh immune globulin.
2 problems
As Moise points out in an editorial accompanying the study, a robust, automated system for ffDNA assessment prior to administration of Rh immune globulin likely would be cost-effective. The Moise editorial also points out these 2 concerns:
False positives are a real possibility, as the 2 cases in the Gautier study illustrate. Free fetal DNA analysis for RhD genotyping assumes that the serologic finding that indicates RhD-negative status (lack of RhD on the fetal red blood cells) is due to deletion of the RhD locus. Thus, when RhD DNA fragments are detected in maternal plasma, they are presumed to be fetal in origin. However, we now know that pseudogene regions of the RhD locus occur with relatively high frequency—in particular, in more than half of African Americans, who serologically type as RhD-negative. Such pseudogenes cause a stop codon that effectively diminishes production of RhD antigen. Serologic typing of such individuals indicates an RhD-negative phenotype. Because the most common pseudogenes are within exon 4, inclusion of primers that assess multiple exons can reduce these false positives.
False negatives have graver clinical implications. Misidentification of an RhD-positive fetus as RhD-negative could prevent that fetus from receiving appropriate surveillance and intervention. False-negative assessments from ffDNA are probably caused by poor amplification of the test sample.
Safeguards have been used in most protocols, including tracer mouse DNA as an internal control to assure amplification. Simultaneous SRY gene testing assures amplification of male fetal DNA. For females, incorporation in the amplification assessment of highly polymorphic markers different from those of the maternal sample may verify fetal DNA amplification.
What is ahead?
Protocols to refine use of ffDNA for RhD genotyping are likely. Meanwhile, techniques are being modified to assure extraction of sufficient quantities of fetal DNA.
Future research will focus on quantitative changes in ffDNA as a marker for pregnancy complications, and development of noninvasive prenatal assessment of specific genes. Successful development of a noninvasive ffDNA diagnostic test will enhance prenatal evaluations without the risk of pregnancy loss currently associated with amniocentesis and chorionic villus sampling.
1. Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective [published correction appears in: Am J Hum Genet. 2004;75:352]. Am J Hum Genet. 2004;74:805-816.
2. Nolin SL, Lewis FA, 3rd, Ye LL, et al. Familial transmission of the FMR1 CGG repeat. Am J Hum Genet. 1996;59:1252-12561.
3. Pesso R, Berkenstadt M, Cuckle H, et al. Screening for fragile X syndrome in women of reproductive age. Prenat Diagn. 2000;20:611-614.
4. Toledano-Alhadef H, Basel-Vanagaite L, Magal N, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69:351-360.
5. Nolin SL, Brown WT, Glicksman A, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454-464.
6. Musci TJ, Caughey AB. Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol. 2005;192:1905-1915.
7. Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet. 2002;110:601-605.
8. Willemsen R, Mientjes E, Oostra BA. FXTAS: a progressive neurologic syndrome associated with fragile X premutation. Curr Neurol Neurosci Rep. 2005;5:405-410.
9. Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485-487.
10. Lo YM. Recent advances in fetal nucleic acids in maternal plasma. J Histochem Cytochem. 2005;53:293-296.
11. Illanes S, Avent N, Soothill PW. Cell-free fetal DNA in maternal plasma: an important advance to link fetal genetics to obstetric ultrasound. Ultrasound Obstet Gynecol. 2005;25:317-322.
12. Galbiati S, Smid M, Gambini D, et al. Fetal DNA detection in maternal plasma throughout gestation. Hum Genet. 2005;117:243-248.

Louise Wilkins-Haug, MD, PhD
Medical Director, Center for Fetal Medicine,
Brigham and Women’s Hospital,
and Associate Professor and Division Director,
Maternal-Fetal Medicine and Reproductive Genetics, Department of Obstetrics,
Gynecology and Reproductive Biology,
Harvard Medical School, Boston
Louise Wilkins-Haug, MD, PhD
Medical Director, Center for Fetal Medicine,
Brigham and Women’s Hospital,
and Associate Professor and Division Director,
Maternal-Fetal Medicine and Reproductive Genetics, Department of Obstetrics,
Gynecology and Reproductive Biology,
Harvard Medical School, Boston
Louise Wilkins-Haug, MD, PhD
Medical Director, Center for Fetal Medicine,
Brigham and Women’s Hospital,
and Associate Professor and Division Director,
Maternal-Fetal Medicine and Reproductive Genetics, Department of Obstetrics,
Gynecology and Reproductive Biology,
Harvard Medical School, Boston
- Screening for fragile X, the most common cause of mental retardation
- Fetal RhD genotyping using maternal plasma
Genetic medicine is fast gaining recognition as an essential component of clinical care. Since this Update last year, 2 areas have seen notable progress:
- New guidelines were issued for fragile X screening in women of reproductive age, and
- Research on free fetal DNA in the maternal circulation deepened our understanding of its diagnostic potential. Clinical applications are emerging in both avenues, and there will be further investigation and refinement. Be sure to check this column again next year!
New guidelines on who to test for mental retardation marker
Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7:584–587.
McConkie-Rosell A, Finucane B, Cronister A, Abrams L, Bennett RL, Petterson BJ. Genetic counseling for fragile X syndrome: updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2005;14:249–270.
Fragile X syndrome is the most common inherited cause of mental retardation. The condition can occur in both males and females and is characterized by a range of behavioral changes consistent with autism spectrum, mental retardation, and developmental delay, as well as a facial phenotype that tends to become more recognizable as the individual ages.
Test is not for everybody
New guidelines issued by the American College of Medical Genetics recommended general population screening only within the constructs of research protocols. In selected populations, however, screening should be considered (TABLE 1). Among preconception and prenatal patients, directed interrogation of the family history for findings suggestive of fragile X syndrome can be guided by these recommendations.
Prevalence. ObGyns should be aware of the increasing spectrum of full and premutation fragile X phenotypes and the relatively high prevalence of premutations among women.
Anatomy of fragile X
Changes in a specific region of the X chromosome known as the fragile X mental retardation-1 (FMR-1) gene are responsible for the syndrome. Elongation of an unstable CGG repeat sequence at the 5′ end of FMR-1 leads to hypermethylation, impaired translation, and altered production of the fragile X mental retardation protein. Investigations of knock-out mice reveal that this protein plays an important role in prenatal and postnatal brain development, especially in the area of dendrite maturation.
Among Caucasians, the characteristic features of fragile X syndrome occur in approximately 1 in 4,000 males and 1 in 8,000 females and are associated with elongation of the FMR-1 gene to more than 200 CGG repeats (a full mutation). Initial studies of other races suggest a similar range of full mutations in males and females.
TABLE 1
Fragile X syndrome: Diagnostic and carrier testing guidelines
| Both women and men with |
|
| Physical or behavioral characteristics of fragile X |
| or family history of fragile X |
| or a relative with undiagnosed mental retardation |
| Persons seeking reproductive counseling who have |
|
| Fetuses of carrier mothers |
| Affected individuals or relatives in whom the diagnosis was made by cytogenetic studies |
| Women with elevated follicle-stimulating hormone, especially with family history of |
|
| Men or women with late-onset intention tremor or ataxia…especially with family history of |
|
| Source: Sherman et al. |
Which offspring will inherit the gene?
In the general population, the FMR-1 region has variable lengths.1 In most individuals, 40 or fewer CGG repeats are present and the region remains stable when passed from either parent to the child. Occasionally, however, individuals inherit expansions of this repeat region—either slight (41–60 repeats, intermediate range) or larger (61–200, premutation range). Repeats in the premutation range are carried by 1 in 700 to 1,000 males and 1 in 113 to 350 females.
Expansion of the premutation to a full FMR-1 mutation depends on the sex of the transmitting parent, the length of repeats, and the frequency of AGG interspersion. Only the first 2 criteria are available for clinical interpretation. FMR-1 expansion occurs only in the X originating from the maternal cell line. The larger the premutation, the more likely it will expand to a full mutation (TABLE 2).
Timing of the maternal FMR-1 expansion can vary, with meiotic, postzygotic, and mitotic instability of CGG length all reported.
- Typically, all sons who inherit an expanded, full mutation exhibit features of fragile X syndrome.
- In daughters, however, a full mutation causes a range of features. In daughters with a full mutation, prognostication is limited. Studies indicate that at least 50%, and in some series 75%, have IQs in the borderline or mentally retarded range.
Longitudinal studies of asymptomatic females with full mutations have not been reported. Fathers with premutations pass the FMR-1 gene in a stable fashion to all offspring, occasionally with contraction to a smaller repeat size.
Note that the complex inheritance pattern, with premutations transmitted through both sexes but expansion limited to the maternal X chromosome, can confound interpretation of family histories of mental retardation or developmental delay.
TABLE 2
Number of CGG repeats influences mutation status
| PERCENT RISK OF EXPANSION TO FULL MUTATION (>200 REPEATS) | ||||
|---|---|---|---|---|
| MATERNAL REPEATS | NOLIN, 19962 | PESSO, 20003 | TOLEDANO-ALHADEF, 20014 | NOLIN, 20035 |
| 55–59 | 13 (3/22) | 0 (0/11) | 0 (0/22) | 4 (1/27) |
| 60–69 | 21 (7/34) | 12 (1/8) | 10 (2/20) | 5 (6/113) |
| 70–79 | 58 (59/102) | 50 (1/2) | 17 (1/6) | 31 (28/90) |
| 80–89 | 73 (78/107) | 50 (1/2) | — | 58 (81/140) |
| 90–99 | 94 (83/88) | 100 (1/1) | — | 80 (89/111) |
| 100–200 | 99 (177/179) | 75 (3/4) | — | 98 (194/197) |
| Values presented as percent (n/N). | ||||
| Data modified from Nolin et al. | ||||
Screening populations
Testing for the FMR-1 gene by determining the expansion size is possible via DNA analysis. Most labs utilize both Southern analyses, to measure the degree of methylation, and polymerase chain reaction, to discriminate at a more refined level the subtle differences in repeat sizes that distinguish intermediate and premutation sizes.
Screening the general population for premutations of the FMR-1 gene is not yet the standard of care. However, several authorities advocate fragile X screening among prenatal and preconception populations, given the relatively high rate of the carrier state (1 in 113 to 350), the sensitivity of testing, and the implications for mental retardation and disability in offspring.
In the United States, even assuming a relatively conservative premutation rate of 1 in 300 and an expansion rate of only 11.3%, such testing would be cost-effective, ranging from $99 to $300 per test.6
What are the risks?
Concerns include the implications of intermediate expansions and the substantial patient education needed to convey the risk of expansion to premutation (but not full mutation). No child with a full mutation has been born to a mother with 59 or fewer repeats. Also needing study is the variability of fragile X syndrome in women with a full mutation. These women are at substantial risk for learning impairment, but the degree of disability varies unpredictably.
Prenatal diagnosis requires DNA from amniocytes or chorionic villus sampling. If the latter, follow-up amniocentesis may be needed because methylation begins at variable times during placental development.7
Preimplantation genetic assessment for fragile X premutation carriers has been reported using a system of closely linked markers, circumventing the need to assess onset of methylation abnormalities.
Spectrum of symptoms
Previously, individuals with premutations were considered clinically asymptomatic. However, we now know that phenotypic expression of expansion sizes occurs along a spectrum.
Recent data indicate 2 phenotypes associated with premutations:
- In women, premature ovarian failure, defined as menopause before the age of 40 years, occurs in 13% to 24% of those with premutations of the FMR-1 gene, among families with fragile X syndrome. Conversely, among women with premature ovarian failure, premutations are found in 2% and 14% of sporadic and familial cases, respectively. Further, the size of the premutation may be directly correlated to the risk of premature ovarian failure.
- In men with premutations, a neurologic syndrome of tremors and ataxia is a newly described phenomenon. The fragile X-associated tremor/ataxia syndrome (FXTAS) is a progressive, neurodegenerative process with Parkinsonism and peripheral neuropathy, and penetrance appears to increase with age.8 The frequency of this diagnosis among older men with premutations is under study.
Fetal RhD genotyping now possible using maternal plasma
Gautier E, Benachi A, Giovangrandi Y, et al. Fetal RhD genotyping by maternal serum analyses: a two-year experience. Am J Obstet Gynecol. 2005;192:666–669.
Moise K. Fetal RhD typing with free DNA in maternal plasma. Am J Obstet Gynecol. 2005;192:663–665.
Fetal RhD typing using free fetal DNA (ffDNA) is routine in the United Kingdom but not yet in this country. Since 1997, when Lo identified ffDNA in maternal plasma,9 numerous studies have focused on the physiology, timing, and clinical application of fetal RhD typing using ffDNA. Previously, the focus was detection of fetal cells in maternal circulation.
Unlike intact fetal cells, ffDNA fragments are present in the maternal plasma in sufficient quantities to allow extensive investigation. While most maternal free DNA is composed of longer DNA fragments, shorter DNA fragments of fetal origin appear as the pregnancy advances and in some studies are first detectable as early as 32 days after conception.
Free fetal DNA increases throughout gestation, representing 3% of total DNA in maternal plasma during the second trimester, and increasing to 6% in the third trimester. Free fetal DNA fragments are cleared rapidly by the renal system, with a half-life of 16 minutes and no discernable levels as soon as 2 hours after delivery.
We now understand that ffDNA fragments are continuously deposited in the maternal circulation from early in pregnancy, perhaps even before fetal circulation develops. We also know that maternal levels of ffDNA depend on 2 forces: rate of deposition and rate of removal.10
Trophoblastic origin?
A placental source is suggested by evidence that ffDNA can be retrieved from maternal plasma prior to the development of fetal circulation. A trophoblastic origin is supported by identification in maternal plasma of fetal mRNA with specificity for genes expressed by the placenta. Moreover, ffDNA has been detected in maternal circulation as early as 14 days after conception, corroborating a trophoblastic origin, with programmed apoptosis of placental cells a likely mechanism.
Further increases in ffDNA throughout gestation may reflect direct contributions from the fetal circulation that are transferred to maternal circulation via the placenta. In later gestation, destruction of fetal cells within the maternal circulation may contribute to the pool of ffDNA in maternal plasma. The exact proportions of each contribution are unknown.11
ffDNA may help diagnose these disorders
Placental abnormalities
Recent work suggests sufficient quantities of ffDNA can be obtained for both quantitative and qualitative assessments.12 Used quantitatively, ffDNA reflects placental integrity, an active area of investigation.
Autosomal trisomies, preeclampsia, and fetal growth restriction are conditions thought to involve abnormal placental function. Increased levels of ffDNA have been found in these entities. Increases have been documented even before onset of preeclampsia.10
Gene defects
Use of ffDNA to identify specific gene defects is also under study. Sensitive microarray technology will likely be needed to assess fetal chromosome aneuploidy from maternal plasma.
The detection of single gene defects from ffDNA has been reported for paternally inherited myotonic dystrophy, Huntington disease, and achondroplasia.
For autosomal recessive disorders, genetic testing of ffDNA may be a first step to exclude inheritance of a paternal allele. For this application, discordant parental alleles will be needed so that exclusion of the paternal mutation in the ffDNA signifies an unaffected fetus or a heterozygotic carrier of the maternal allele. If the paternal allele is detected by ffDNA, further genetic testing by chorionic villus sampling or amniocentesis would be needed to differentiate heterozygotic carriers of the paternal mutation from homozygotic, affected fetuses.
RhD genotyping
Since 2001, ffDNA has been used clinically in the United Kingdom for fetal blood group genotyping in isoimmunized gravidas with heterozygous partners, through the International Blood Group Reference Laboratory (part of the National Blood Service), which brings us to the highlighted study. Gautier and colleagues added data affirming that the RhD genotype can be detected through ffDNA with high sensitivity and specificity. Among 285 RhD-negative women, the fetal RhD genotype was determined in 283. In 2 cases, the maternal RhD-negative phenotype did not result from a complete gene deletion; thus, the genotypes of fetus and mother could not be differentiated. Among the women with RhD-negative genotypes, all fetuses were accurately genotyped through ffDNA.
This study differs from prior investigations in its use of RhD-negative women who were not already sensitized, and suggests that ffDNA genotyping in RhD-negative women is sensitive enough to be incorporated into the distribution of Rh immune globulin.
2 problems
As Moise points out in an editorial accompanying the study, a robust, automated system for ffDNA assessment prior to administration of Rh immune globulin likely would be cost-effective. The Moise editorial also points out these 2 concerns:
False positives are a real possibility, as the 2 cases in the Gautier study illustrate. Free fetal DNA analysis for RhD genotyping assumes that the serologic finding that indicates RhD-negative status (lack of RhD on the fetal red blood cells) is due to deletion of the RhD locus. Thus, when RhD DNA fragments are detected in maternal plasma, they are presumed to be fetal in origin. However, we now know that pseudogene regions of the RhD locus occur with relatively high frequency—in particular, in more than half of African Americans, who serologically type as RhD-negative. Such pseudogenes cause a stop codon that effectively diminishes production of RhD antigen. Serologic typing of such individuals indicates an RhD-negative phenotype. Because the most common pseudogenes are within exon 4, inclusion of primers that assess multiple exons can reduce these false positives.
False negatives have graver clinical implications. Misidentification of an RhD-positive fetus as RhD-negative could prevent that fetus from receiving appropriate surveillance and intervention. False-negative assessments from ffDNA are probably caused by poor amplification of the test sample.
Safeguards have been used in most protocols, including tracer mouse DNA as an internal control to assure amplification. Simultaneous SRY gene testing assures amplification of male fetal DNA. For females, incorporation in the amplification assessment of highly polymorphic markers different from those of the maternal sample may verify fetal DNA amplification.
What is ahead?
Protocols to refine use of ffDNA for RhD genotyping are likely. Meanwhile, techniques are being modified to assure extraction of sufficient quantities of fetal DNA.
Future research will focus on quantitative changes in ffDNA as a marker for pregnancy complications, and development of noninvasive prenatal assessment of specific genes. Successful development of a noninvasive ffDNA diagnostic test will enhance prenatal evaluations without the risk of pregnancy loss currently associated with amniocentesis and chorionic villus sampling.
- Screening for fragile X, the most common cause of mental retardation
- Fetal RhD genotyping using maternal plasma
Genetic medicine is fast gaining recognition as an essential component of clinical care. Since this Update last year, 2 areas have seen notable progress:
- New guidelines were issued for fragile X screening in women of reproductive age, and
- Research on free fetal DNA in the maternal circulation deepened our understanding of its diagnostic potential. Clinical applications are emerging in both avenues, and there will be further investigation and refinement. Be sure to check this column again next year!
New guidelines on who to test for mental retardation marker
Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7:584–587.
McConkie-Rosell A, Finucane B, Cronister A, Abrams L, Bennett RL, Petterson BJ. Genetic counseling for fragile X syndrome: updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2005;14:249–270.
Fragile X syndrome is the most common inherited cause of mental retardation. The condition can occur in both males and females and is characterized by a range of behavioral changes consistent with autism spectrum, mental retardation, and developmental delay, as well as a facial phenotype that tends to become more recognizable as the individual ages.
Test is not for everybody
New guidelines issued by the American College of Medical Genetics recommended general population screening only within the constructs of research protocols. In selected populations, however, screening should be considered (TABLE 1). Among preconception and prenatal patients, directed interrogation of the family history for findings suggestive of fragile X syndrome can be guided by these recommendations.
Prevalence. ObGyns should be aware of the increasing spectrum of full and premutation fragile X phenotypes and the relatively high prevalence of premutations among women.
Anatomy of fragile X
Changes in a specific region of the X chromosome known as the fragile X mental retardation-1 (FMR-1) gene are responsible for the syndrome. Elongation of an unstable CGG repeat sequence at the 5′ end of FMR-1 leads to hypermethylation, impaired translation, and altered production of the fragile X mental retardation protein. Investigations of knock-out mice reveal that this protein plays an important role in prenatal and postnatal brain development, especially in the area of dendrite maturation.
Among Caucasians, the characteristic features of fragile X syndrome occur in approximately 1 in 4,000 males and 1 in 8,000 females and are associated with elongation of the FMR-1 gene to more than 200 CGG repeats (a full mutation). Initial studies of other races suggest a similar range of full mutations in males and females.
TABLE 1
Fragile X syndrome: Diagnostic and carrier testing guidelines
| Both women and men with |
|
| Physical or behavioral characteristics of fragile X |
| or family history of fragile X |
| or a relative with undiagnosed mental retardation |
| Persons seeking reproductive counseling who have |
|
| Fetuses of carrier mothers |
| Affected individuals or relatives in whom the diagnosis was made by cytogenetic studies |
| Women with elevated follicle-stimulating hormone, especially with family history of |
|
| Men or women with late-onset intention tremor or ataxia…especially with family history of |
|
| Source: Sherman et al. |
Which offspring will inherit the gene?
In the general population, the FMR-1 region has variable lengths.1 In most individuals, 40 or fewer CGG repeats are present and the region remains stable when passed from either parent to the child. Occasionally, however, individuals inherit expansions of this repeat region—either slight (41–60 repeats, intermediate range) or larger (61–200, premutation range). Repeats in the premutation range are carried by 1 in 700 to 1,000 males and 1 in 113 to 350 females.
Expansion of the premutation to a full FMR-1 mutation depends on the sex of the transmitting parent, the length of repeats, and the frequency of AGG interspersion. Only the first 2 criteria are available for clinical interpretation. FMR-1 expansion occurs only in the X originating from the maternal cell line. The larger the premutation, the more likely it will expand to a full mutation (TABLE 2).
Timing of the maternal FMR-1 expansion can vary, with meiotic, postzygotic, and mitotic instability of CGG length all reported.
- Typically, all sons who inherit an expanded, full mutation exhibit features of fragile X syndrome.
- In daughters, however, a full mutation causes a range of features. In daughters with a full mutation, prognostication is limited. Studies indicate that at least 50%, and in some series 75%, have IQs in the borderline or mentally retarded range.
Longitudinal studies of asymptomatic females with full mutations have not been reported. Fathers with premutations pass the FMR-1 gene in a stable fashion to all offspring, occasionally with contraction to a smaller repeat size.
Note that the complex inheritance pattern, with premutations transmitted through both sexes but expansion limited to the maternal X chromosome, can confound interpretation of family histories of mental retardation or developmental delay.
TABLE 2
Number of CGG repeats influences mutation status
| PERCENT RISK OF EXPANSION TO FULL MUTATION (>200 REPEATS) | ||||
|---|---|---|---|---|
| MATERNAL REPEATS | NOLIN, 19962 | PESSO, 20003 | TOLEDANO-ALHADEF, 20014 | NOLIN, 20035 |
| 55–59 | 13 (3/22) | 0 (0/11) | 0 (0/22) | 4 (1/27) |
| 60–69 | 21 (7/34) | 12 (1/8) | 10 (2/20) | 5 (6/113) |
| 70–79 | 58 (59/102) | 50 (1/2) | 17 (1/6) | 31 (28/90) |
| 80–89 | 73 (78/107) | 50 (1/2) | — | 58 (81/140) |
| 90–99 | 94 (83/88) | 100 (1/1) | — | 80 (89/111) |
| 100–200 | 99 (177/179) | 75 (3/4) | — | 98 (194/197) |
| Values presented as percent (n/N). | ||||
| Data modified from Nolin et al. | ||||
Screening populations
Testing for the FMR-1 gene by determining the expansion size is possible via DNA analysis. Most labs utilize both Southern analyses, to measure the degree of methylation, and polymerase chain reaction, to discriminate at a more refined level the subtle differences in repeat sizes that distinguish intermediate and premutation sizes.
Screening the general population for premutations of the FMR-1 gene is not yet the standard of care. However, several authorities advocate fragile X screening among prenatal and preconception populations, given the relatively high rate of the carrier state (1 in 113 to 350), the sensitivity of testing, and the implications for mental retardation and disability in offspring.
In the United States, even assuming a relatively conservative premutation rate of 1 in 300 and an expansion rate of only 11.3%, such testing would be cost-effective, ranging from $99 to $300 per test.6
What are the risks?
Concerns include the implications of intermediate expansions and the substantial patient education needed to convey the risk of expansion to premutation (but not full mutation). No child with a full mutation has been born to a mother with 59 or fewer repeats. Also needing study is the variability of fragile X syndrome in women with a full mutation. These women are at substantial risk for learning impairment, but the degree of disability varies unpredictably.
Prenatal diagnosis requires DNA from amniocytes or chorionic villus sampling. If the latter, follow-up amniocentesis may be needed because methylation begins at variable times during placental development.7
Preimplantation genetic assessment for fragile X premutation carriers has been reported using a system of closely linked markers, circumventing the need to assess onset of methylation abnormalities.
Spectrum of symptoms
Previously, individuals with premutations were considered clinically asymptomatic. However, we now know that phenotypic expression of expansion sizes occurs along a spectrum.
Recent data indicate 2 phenotypes associated with premutations:
- In women, premature ovarian failure, defined as menopause before the age of 40 years, occurs in 13% to 24% of those with premutations of the FMR-1 gene, among families with fragile X syndrome. Conversely, among women with premature ovarian failure, premutations are found in 2% and 14% of sporadic and familial cases, respectively. Further, the size of the premutation may be directly correlated to the risk of premature ovarian failure.
- In men with premutations, a neurologic syndrome of tremors and ataxia is a newly described phenomenon. The fragile X-associated tremor/ataxia syndrome (FXTAS) is a progressive, neurodegenerative process with Parkinsonism and peripheral neuropathy, and penetrance appears to increase with age.8 The frequency of this diagnosis among older men with premutations is under study.
Fetal RhD genotyping now possible using maternal plasma
Gautier E, Benachi A, Giovangrandi Y, et al. Fetal RhD genotyping by maternal serum analyses: a two-year experience. Am J Obstet Gynecol. 2005;192:666–669.
Moise K. Fetal RhD typing with free DNA in maternal plasma. Am J Obstet Gynecol. 2005;192:663–665.
Fetal RhD typing using free fetal DNA (ffDNA) is routine in the United Kingdom but not yet in this country. Since 1997, when Lo identified ffDNA in maternal plasma,9 numerous studies have focused on the physiology, timing, and clinical application of fetal RhD typing using ffDNA. Previously, the focus was detection of fetal cells in maternal circulation.
Unlike intact fetal cells, ffDNA fragments are present in the maternal plasma in sufficient quantities to allow extensive investigation. While most maternal free DNA is composed of longer DNA fragments, shorter DNA fragments of fetal origin appear as the pregnancy advances and in some studies are first detectable as early as 32 days after conception.
Free fetal DNA increases throughout gestation, representing 3% of total DNA in maternal plasma during the second trimester, and increasing to 6% in the third trimester. Free fetal DNA fragments are cleared rapidly by the renal system, with a half-life of 16 minutes and no discernable levels as soon as 2 hours after delivery.
We now understand that ffDNA fragments are continuously deposited in the maternal circulation from early in pregnancy, perhaps even before fetal circulation develops. We also know that maternal levels of ffDNA depend on 2 forces: rate of deposition and rate of removal.10
Trophoblastic origin?
A placental source is suggested by evidence that ffDNA can be retrieved from maternal plasma prior to the development of fetal circulation. A trophoblastic origin is supported by identification in maternal plasma of fetal mRNA with specificity for genes expressed by the placenta. Moreover, ffDNA has been detected in maternal circulation as early as 14 days after conception, corroborating a trophoblastic origin, with programmed apoptosis of placental cells a likely mechanism.
Further increases in ffDNA throughout gestation may reflect direct contributions from the fetal circulation that are transferred to maternal circulation via the placenta. In later gestation, destruction of fetal cells within the maternal circulation may contribute to the pool of ffDNA in maternal plasma. The exact proportions of each contribution are unknown.11
ffDNA may help diagnose these disorders
Placental abnormalities
Recent work suggests sufficient quantities of ffDNA can be obtained for both quantitative and qualitative assessments.12 Used quantitatively, ffDNA reflects placental integrity, an active area of investigation.
Autosomal trisomies, preeclampsia, and fetal growth restriction are conditions thought to involve abnormal placental function. Increased levels of ffDNA have been found in these entities. Increases have been documented even before onset of preeclampsia.10
Gene defects
Use of ffDNA to identify specific gene defects is also under study. Sensitive microarray technology will likely be needed to assess fetal chromosome aneuploidy from maternal plasma.
The detection of single gene defects from ffDNA has been reported for paternally inherited myotonic dystrophy, Huntington disease, and achondroplasia.
For autosomal recessive disorders, genetic testing of ffDNA may be a first step to exclude inheritance of a paternal allele. For this application, discordant parental alleles will be needed so that exclusion of the paternal mutation in the ffDNA signifies an unaffected fetus or a heterozygotic carrier of the maternal allele. If the paternal allele is detected by ffDNA, further genetic testing by chorionic villus sampling or amniocentesis would be needed to differentiate heterozygotic carriers of the paternal mutation from homozygotic, affected fetuses.
RhD genotyping
Since 2001, ffDNA has been used clinically in the United Kingdom for fetal blood group genotyping in isoimmunized gravidas with heterozygous partners, through the International Blood Group Reference Laboratory (part of the National Blood Service), which brings us to the highlighted study. Gautier and colleagues added data affirming that the RhD genotype can be detected through ffDNA with high sensitivity and specificity. Among 285 RhD-negative women, the fetal RhD genotype was determined in 283. In 2 cases, the maternal RhD-negative phenotype did not result from a complete gene deletion; thus, the genotypes of fetus and mother could not be differentiated. Among the women with RhD-negative genotypes, all fetuses were accurately genotyped through ffDNA.
This study differs from prior investigations in its use of RhD-negative women who were not already sensitized, and suggests that ffDNA genotyping in RhD-negative women is sensitive enough to be incorporated into the distribution of Rh immune globulin.
2 problems
As Moise points out in an editorial accompanying the study, a robust, automated system for ffDNA assessment prior to administration of Rh immune globulin likely would be cost-effective. The Moise editorial also points out these 2 concerns:
False positives are a real possibility, as the 2 cases in the Gautier study illustrate. Free fetal DNA analysis for RhD genotyping assumes that the serologic finding that indicates RhD-negative status (lack of RhD on the fetal red blood cells) is due to deletion of the RhD locus. Thus, when RhD DNA fragments are detected in maternal plasma, they are presumed to be fetal in origin. However, we now know that pseudogene regions of the RhD locus occur with relatively high frequency—in particular, in more than half of African Americans, who serologically type as RhD-negative. Such pseudogenes cause a stop codon that effectively diminishes production of RhD antigen. Serologic typing of such individuals indicates an RhD-negative phenotype. Because the most common pseudogenes are within exon 4, inclusion of primers that assess multiple exons can reduce these false positives.
False negatives have graver clinical implications. Misidentification of an RhD-positive fetus as RhD-negative could prevent that fetus from receiving appropriate surveillance and intervention. False-negative assessments from ffDNA are probably caused by poor amplification of the test sample.
Safeguards have been used in most protocols, including tracer mouse DNA as an internal control to assure amplification. Simultaneous SRY gene testing assures amplification of male fetal DNA. For females, incorporation in the amplification assessment of highly polymorphic markers different from those of the maternal sample may verify fetal DNA amplification.
What is ahead?
Protocols to refine use of ffDNA for RhD genotyping are likely. Meanwhile, techniques are being modified to assure extraction of sufficient quantities of fetal DNA.
Future research will focus on quantitative changes in ffDNA as a marker for pregnancy complications, and development of noninvasive prenatal assessment of specific genes. Successful development of a noninvasive ffDNA diagnostic test will enhance prenatal evaluations without the risk of pregnancy loss currently associated with amniocentesis and chorionic villus sampling.
1. Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective [published correction appears in: Am J Hum Genet. 2004;75:352]. Am J Hum Genet. 2004;74:805-816.
2. Nolin SL, Lewis FA, 3rd, Ye LL, et al. Familial transmission of the FMR1 CGG repeat. Am J Hum Genet. 1996;59:1252-12561.
3. Pesso R, Berkenstadt M, Cuckle H, et al. Screening for fragile X syndrome in women of reproductive age. Prenat Diagn. 2000;20:611-614.
4. Toledano-Alhadef H, Basel-Vanagaite L, Magal N, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69:351-360.
5. Nolin SL, Brown WT, Glicksman A, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454-464.
6. Musci TJ, Caughey AB. Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol. 2005;192:1905-1915.
7. Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet. 2002;110:601-605.
8. Willemsen R, Mientjes E, Oostra BA. FXTAS: a progressive neurologic syndrome associated with fragile X premutation. Curr Neurol Neurosci Rep. 2005;5:405-410.
9. Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485-487.
10. Lo YM. Recent advances in fetal nucleic acids in maternal plasma. J Histochem Cytochem. 2005;53:293-296.
11. Illanes S, Avent N, Soothill PW. Cell-free fetal DNA in maternal plasma: an important advance to link fetal genetics to obstetric ultrasound. Ultrasound Obstet Gynecol. 2005;25:317-322.
12. Galbiati S, Smid M, Gambini D, et al. Fetal DNA detection in maternal plasma throughout gestation. Hum Genet. 2005;117:243-248.
1. Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective [published correction appears in: Am J Hum Genet. 2004;75:352]. Am J Hum Genet. 2004;74:805-816.
2. Nolin SL, Lewis FA, 3rd, Ye LL, et al. Familial transmission of the FMR1 CGG repeat. Am J Hum Genet. 1996;59:1252-12561.
3. Pesso R, Berkenstadt M, Cuckle H, et al. Screening for fragile X syndrome in women of reproductive age. Prenat Diagn. 2000;20:611-614.
4. Toledano-Alhadef H, Basel-Vanagaite L, Magal N, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69:351-360.
5. Nolin SL, Brown WT, Glicksman A, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454-464.
6. Musci TJ, Caughey AB. Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol. 2005;192:1905-1915.
7. Willemsen R, Bontekoe CJ, Severijnen LA, Oostra BA. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum Genet. 2002;110:601-605.
8. Willemsen R, Mientjes E, Oostra BA. FXTAS: a progressive neurologic syndrome associated with fragile X premutation. Curr Neurol Neurosci Rep. 2005;5:405-410.
9. Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485-487.
10. Lo YM. Recent advances in fetal nucleic acids in maternal plasma. J Histochem Cytochem. 2005;53:293-296.
11. Illanes S, Avent N, Soothill PW. Cell-free fetal DNA in maternal plasma: an important advance to link fetal genetics to obstetric ultrasound. Ultrasound Obstet Gynecol. 2005;25:317-322.
12. Galbiati S, Smid M, Gambini D, et al. Fetal DNA detection in maternal plasma throughout gestation. Hum Genet. 2005;117:243-248.
New screening basics for the generalist
Overall, you must determine the extent to which you will provide and interpret genetic testing and when to refer patients to a specialist. This article aims to simplify that decision by reviewing guidelines and key studies in 3 areas:
- For genetic carrier screening for people of Ashkenazi Jewish heritage, add familial dysautonomia to the list of screened diseases.
- Screening for Down syndrome is now possible in the first trimester.
- Greater genetic risks may be present among children born as a result of assisted reproductive technology (ART), although it’s unclear whether the cause is their parents’ infertility or ART itself.
On the plus side, molecular DNA diagnostics are increasingly sophisticated, readily available, and cost-efficient. The downside: As the list of recommended studies grows, successful testing programs are harder to achieve because of the need to educate patients—and yourself—about each test.
Preconception testing may be especially advisable in women with infertility because it can identify carriers and detect conditions related to infertility or its treatment. With 1% of US births attributable to ART, the possibility of genetic effects continues to raise concern.
Add another disease to genetic carrier screening
ACOG Committee Opinion #298: Prenatal and preconception carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2004;104:425–428.
Add familial dysautonomia to carrier screening when patients—or their partners—are of Ashkenazi Jewish heritage. That’s the advice from an American College of Obstetricians and Gynecologists (ACOG) committee opinion. Also conduct previously recommended screening for Tay-Sachs disease, Canavan disease, and cystic fibrosis, and advise patients that testing is available for several other diseases as well (TABLE 1). For Tay-Sachs disease, screening also is urged for patients of French Canadian and Cajun descent.
ACOG emphasizes the importance of assessing these risks prior to pregnancy to allow time for the partner to be tested, if necessary.
Among the Ashkenazi Jewish population, DNA testing detects more than 95% of carriers of autosomal disorders by analyzing the small number of mutations responsible. Tay-Sachs was the first disease for which mutations were identified.
Familial dysautonomia is caused by a single mutation in the gene IKBKAP in more than 99% of affected patients. It has a carrier rate (1/32) similar to that of Tay-Sachs disease (1/30) and involves substantial morbidity of the autonomic and sensory nervous system, with symptoms such as abnormal sweating, pain/temperature insensitivity, and labile blood pressure. Treatment may relieve symptoms, but does not cure the disease.
Refer non-Ashkenazi partners of identified carriers. Although non-Ashkenazi partners are less likely to be carriers, the exact carrier frequency and detection rates for these people are unknown (except for Tay-Sachs disease and cystic fibrosis). In these situations, it may be wise to refer the patient and her partner for genetic counseling to clarify the sensitivity of DNA analysis and the advisability of possible alternative testing by enzyme analysis.
What a generalist should offer. Because the availability of genetic testing will continue to increase, ACOG recommends that generalists provide:
- patient education on the disorders,
- referral sources for additional counseling and prenatal diagnostic testing,
- informed consent when obtaining samples for genetic testing, and
- assurance of confidentiality.
Screen for Down syndrome in the first trimester
Screening for Down syndrome is now available in the first trimester; ACOG recommends using ultrasound and maternal serum screening, with 3 criteria:
- standardized, continuous quality assurance,
- ability to counsel patients about the testing options, and
- access to appropriate diagnostic testing.
TABLE 2).
Integrated versus contingency screening. “Integrated” screening combines information from the first trimester (nuchal lucency and serum screen) with serum screening in the second trimester. This approach yields the lowest screen-positive rate (2.6%) and a high detection rate (90%), but has an important shortcoming: The results are not disclosed until the second trimester.
“Contingency” screening is emerging as an alternative: High first-trimester risks are relayed to the patient, while women with low screen values are excused from further testing. Patients with intermediate risk proceed to second-trimester serum screening.
Disadvantages of this approach include the need to coordinate the various steps and adequately inform the patient of them.
Added value of first-trimester nuchal lucency screening. Increased nuchal translucency alone is an important screen for structural abnormalities and adverse pregnancy outcomes. If a karyotypically normal fetus has an increased first-trimester nuchal lucency, the possibility of a structural anomaly on second-trimester ultrasound increases 2-to 10-fold. Absolute risk rises with increasing nuchal lucency.
Since an average of 10% to 15% of the identified anomalies are cardiac defects, fetal echocardiogram and a comprehensive fetal survey are appropriate in the second trimester.
TABLE 2
Detecting Down syndrome: Which test is best?
| MODALITY | SCREEN-POSITIVE RATE | DETECTION RATE |
|---|---|---|
| Maternal age >35 years | 18% | 30% |
| Triple screen (MSAFP, beta-hCG, estriol) | 5% | 65% |
| Quad screen (triple plus inhibin) | 5% | 75% |
| First-trimester (nuchal lucency, PAPP-A, free beta-hCG) | 5% | 80% |
| Integrated (first-trimester nuchal lucency and serum screen combined with second-trimester serum screen) | 2.5% | 90% |
| MSAFP = maternal serum alpha-fetoprotein, PAPP-A = pregnancy-associated plasma protein-A | ||
REFERENCE
1. Wapner R, Thom E, Simpsoon JL, et al. First-trimester screening for trisomies 21 and 18. N Engl J Med. 2003;349:1405-1413.
Are children conceived with ART at increased risk?
Children born as a result of ART may face a higher risk of inherited disorders and congenital malformations, but it is unclear whether the risks are due to their parents’ infertility or to ART.
For this reason, it may be wise to refer ART patients for additional genetic counseling and fetal structural surveillance by ultrasound.
Schieve and colleagues attempted to clarify the risks by reviewing the theoretical and empiric literature. Two studies provide the bulk of evidence. In Western Australia, the background risk of birth defects doubled in infants conceived with ART: 9% risk in both intracytoplasmic sperm injection and IVF patients, compared with 4% with spontaneous conception.1 This study is notable because ART programs are more highly regulated in Australia and similar methods were used to ascertain congenital anomalies in both groups.
A comparable study2 in Sweden also noted an increased risk, but attributed it to the underlying cause of the parents’ infertility rather than to ART itself. The reason: The increased risk disappeared when the authors adjusted for the period of “involuntary childlessness.” However, they provided very little detail on how involuntary childlessness was defined and “whether and how strongly this measure is correlated with infertility severity in Sweden.”2
Imprinting disorders among ART offspring. Schieve et al also explored imprinting disorders, since diseases such as Beckwith-Wiedemann syndrome are attributed to them. Imprinting is an epigenetic phenomenon in which the allele of only 1 parent is active at a particular gene locus. The inactive—or imprinted—allele is rendered nonfunctional, often through methylation. Gametogenesis and preimplantation are times of increased imprinting. Identified imprinted genes include those that control embryonic growth and differentiation.
Analyses of Beckwith-Wiedemann syndrome registries in the United States, France, and the UK3-5 revealed a 3- to 6-fold increase in ART conception among infants with the syndrome. Case reports of other rare imprinted disorders such as Angelman syndrome and retinoblastoma are also beginning to appear.
Direct treatment effect not established. According to Schieve et al and others, evidence of an increased risk of defects following ART does not indicate whether a direct treatment effect is present. Future studies that address methodological flaws are sure to be time-consuming; they also will require large sample sizes and consistent ascertainment and ART treatment.
Dr. Wilkins-Haug reports no relevant financial relationships.
1. Hansen M, Kurinczuk JJ, Bower C, Webb S. The risk of major birth defects after intracytoplasmic sperm injection and in vitro fertilization. N Engl J Med. 2002;346:725-730.
2. Ericson A, Kallen B. Congenital malformations in infants born after IVF: a population-based study. Hum Reprod. 2001;16:504-509.
3. DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72:156-160.
4. Maher ER, Brueton LA, Bowdin SC, et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART) [published erratum appears in J Med Genet. 2003;40:304]. J Med Genet. 2003;40:62-64.
5. Gicquel C, Gaston V, Mandelbaum J, et al. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003;72:1338-1341.
Overall, you must determine the extent to which you will provide and interpret genetic testing and when to refer patients to a specialist. This article aims to simplify that decision by reviewing guidelines and key studies in 3 areas:
- For genetic carrier screening for people of Ashkenazi Jewish heritage, add familial dysautonomia to the list of screened diseases.
- Screening for Down syndrome is now possible in the first trimester.
- Greater genetic risks may be present among children born as a result of assisted reproductive technology (ART), although it’s unclear whether the cause is their parents’ infertility or ART itself.
On the plus side, molecular DNA diagnostics are increasingly sophisticated, readily available, and cost-efficient. The downside: As the list of recommended studies grows, successful testing programs are harder to achieve because of the need to educate patients—and yourself—about each test.
Preconception testing may be especially advisable in women with infertility because it can identify carriers and detect conditions related to infertility or its treatment. With 1% of US births attributable to ART, the possibility of genetic effects continues to raise concern.
Add another disease to genetic carrier screening
ACOG Committee Opinion #298: Prenatal and preconception carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2004;104:425–428.
Add familial dysautonomia to carrier screening when patients—or their partners—are of Ashkenazi Jewish heritage. That’s the advice from an American College of Obstetricians and Gynecologists (ACOG) committee opinion. Also conduct previously recommended screening for Tay-Sachs disease, Canavan disease, and cystic fibrosis, and advise patients that testing is available for several other diseases as well (TABLE 1). For Tay-Sachs disease, screening also is urged for patients of French Canadian and Cajun descent.
ACOG emphasizes the importance of assessing these risks prior to pregnancy to allow time for the partner to be tested, if necessary.
Among the Ashkenazi Jewish population, DNA testing detects more than 95% of carriers of autosomal disorders by analyzing the small number of mutations responsible. Tay-Sachs was the first disease for which mutations were identified.
Familial dysautonomia is caused by a single mutation in the gene IKBKAP in more than 99% of affected patients. It has a carrier rate (1/32) similar to that of Tay-Sachs disease (1/30) and involves substantial morbidity of the autonomic and sensory nervous system, with symptoms such as abnormal sweating, pain/temperature insensitivity, and labile blood pressure. Treatment may relieve symptoms, but does not cure the disease.
Refer non-Ashkenazi partners of identified carriers. Although non-Ashkenazi partners are less likely to be carriers, the exact carrier frequency and detection rates for these people are unknown (except for Tay-Sachs disease and cystic fibrosis). In these situations, it may be wise to refer the patient and her partner for genetic counseling to clarify the sensitivity of DNA analysis and the advisability of possible alternative testing by enzyme analysis.
What a generalist should offer. Because the availability of genetic testing will continue to increase, ACOG recommends that generalists provide:
- patient education on the disorders,
- referral sources for additional counseling and prenatal diagnostic testing,
- informed consent when obtaining samples for genetic testing, and
- assurance of confidentiality.
Screen for Down syndrome in the first trimester
Screening for Down syndrome is now available in the first trimester; ACOG recommends using ultrasound and maternal serum screening, with 3 criteria:
- standardized, continuous quality assurance,
- ability to counsel patients about the testing options, and
- access to appropriate diagnostic testing.
TABLE 2).
Integrated versus contingency screening. “Integrated” screening combines information from the first trimester (nuchal lucency and serum screen) with serum screening in the second trimester. This approach yields the lowest screen-positive rate (2.6%) and a high detection rate (90%), but has an important shortcoming: The results are not disclosed until the second trimester.
“Contingency” screening is emerging as an alternative: High first-trimester risks are relayed to the patient, while women with low screen values are excused from further testing. Patients with intermediate risk proceed to second-trimester serum screening.
Disadvantages of this approach include the need to coordinate the various steps and adequately inform the patient of them.
Added value of first-trimester nuchal lucency screening. Increased nuchal translucency alone is an important screen for structural abnormalities and adverse pregnancy outcomes. If a karyotypically normal fetus has an increased first-trimester nuchal lucency, the possibility of a structural anomaly on second-trimester ultrasound increases 2-to 10-fold. Absolute risk rises with increasing nuchal lucency.
Since an average of 10% to 15% of the identified anomalies are cardiac defects, fetal echocardiogram and a comprehensive fetal survey are appropriate in the second trimester.
TABLE 2
Detecting Down syndrome: Which test is best?
| MODALITY | SCREEN-POSITIVE RATE | DETECTION RATE |
|---|---|---|
| Maternal age >35 years | 18% | 30% |
| Triple screen (MSAFP, beta-hCG, estriol) | 5% | 65% |
| Quad screen (triple plus inhibin) | 5% | 75% |
| First-trimester (nuchal lucency, PAPP-A, free beta-hCG) | 5% | 80% |
| Integrated (first-trimester nuchal lucency and serum screen combined with second-trimester serum screen) | 2.5% | 90% |
| MSAFP = maternal serum alpha-fetoprotein, PAPP-A = pregnancy-associated plasma protein-A | ||
REFERENCE
1. Wapner R, Thom E, Simpsoon JL, et al. First-trimester screening for trisomies 21 and 18. N Engl J Med. 2003;349:1405-1413.
Are children conceived with ART at increased risk?
Children born as a result of ART may face a higher risk of inherited disorders and congenital malformations, but it is unclear whether the risks are due to their parents’ infertility or to ART.
For this reason, it may be wise to refer ART patients for additional genetic counseling and fetal structural surveillance by ultrasound.
Schieve and colleagues attempted to clarify the risks by reviewing the theoretical and empiric literature. Two studies provide the bulk of evidence. In Western Australia, the background risk of birth defects doubled in infants conceived with ART: 9% risk in both intracytoplasmic sperm injection and IVF patients, compared with 4% with spontaneous conception.1 This study is notable because ART programs are more highly regulated in Australia and similar methods were used to ascertain congenital anomalies in both groups.
A comparable study2 in Sweden also noted an increased risk, but attributed it to the underlying cause of the parents’ infertility rather than to ART itself. The reason: The increased risk disappeared when the authors adjusted for the period of “involuntary childlessness.” However, they provided very little detail on how involuntary childlessness was defined and “whether and how strongly this measure is correlated with infertility severity in Sweden.”2
Imprinting disorders among ART offspring. Schieve et al also explored imprinting disorders, since diseases such as Beckwith-Wiedemann syndrome are attributed to them. Imprinting is an epigenetic phenomenon in which the allele of only 1 parent is active at a particular gene locus. The inactive—or imprinted—allele is rendered nonfunctional, often through methylation. Gametogenesis and preimplantation are times of increased imprinting. Identified imprinted genes include those that control embryonic growth and differentiation.
Analyses of Beckwith-Wiedemann syndrome registries in the United States, France, and the UK3-5 revealed a 3- to 6-fold increase in ART conception among infants with the syndrome. Case reports of other rare imprinted disorders such as Angelman syndrome and retinoblastoma are also beginning to appear.
Direct treatment effect not established. According to Schieve et al and others, evidence of an increased risk of defects following ART does not indicate whether a direct treatment effect is present. Future studies that address methodological flaws are sure to be time-consuming; they also will require large sample sizes and consistent ascertainment and ART treatment.
Dr. Wilkins-Haug reports no relevant financial relationships.
Overall, you must determine the extent to which you will provide and interpret genetic testing and when to refer patients to a specialist. This article aims to simplify that decision by reviewing guidelines and key studies in 3 areas:
- For genetic carrier screening for people of Ashkenazi Jewish heritage, add familial dysautonomia to the list of screened diseases.
- Screening for Down syndrome is now possible in the first trimester.
- Greater genetic risks may be present among children born as a result of assisted reproductive technology (ART), although it’s unclear whether the cause is their parents’ infertility or ART itself.
On the plus side, molecular DNA diagnostics are increasingly sophisticated, readily available, and cost-efficient. The downside: As the list of recommended studies grows, successful testing programs are harder to achieve because of the need to educate patients—and yourself—about each test.
Preconception testing may be especially advisable in women with infertility because it can identify carriers and detect conditions related to infertility or its treatment. With 1% of US births attributable to ART, the possibility of genetic effects continues to raise concern.
Add another disease to genetic carrier screening
ACOG Committee Opinion #298: Prenatal and preconception carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2004;104:425–428.
Add familial dysautonomia to carrier screening when patients—or their partners—are of Ashkenazi Jewish heritage. That’s the advice from an American College of Obstetricians and Gynecologists (ACOG) committee opinion. Also conduct previously recommended screening for Tay-Sachs disease, Canavan disease, and cystic fibrosis, and advise patients that testing is available for several other diseases as well (TABLE 1). For Tay-Sachs disease, screening also is urged for patients of French Canadian and Cajun descent.
ACOG emphasizes the importance of assessing these risks prior to pregnancy to allow time for the partner to be tested, if necessary.
Among the Ashkenazi Jewish population, DNA testing detects more than 95% of carriers of autosomal disorders by analyzing the small number of mutations responsible. Tay-Sachs was the first disease for which mutations were identified.
Familial dysautonomia is caused by a single mutation in the gene IKBKAP in more than 99% of affected patients. It has a carrier rate (1/32) similar to that of Tay-Sachs disease (1/30) and involves substantial morbidity of the autonomic and sensory nervous system, with symptoms such as abnormal sweating, pain/temperature insensitivity, and labile blood pressure. Treatment may relieve symptoms, but does not cure the disease.
Refer non-Ashkenazi partners of identified carriers. Although non-Ashkenazi partners are less likely to be carriers, the exact carrier frequency and detection rates for these people are unknown (except for Tay-Sachs disease and cystic fibrosis). In these situations, it may be wise to refer the patient and her partner for genetic counseling to clarify the sensitivity of DNA analysis and the advisability of possible alternative testing by enzyme analysis.
What a generalist should offer. Because the availability of genetic testing will continue to increase, ACOG recommends that generalists provide:
- patient education on the disorders,
- referral sources for additional counseling and prenatal diagnostic testing,
- informed consent when obtaining samples for genetic testing, and
- assurance of confidentiality.
Screen for Down syndrome in the first trimester
Screening for Down syndrome is now available in the first trimester; ACOG recommends using ultrasound and maternal serum screening, with 3 criteria:
- standardized, continuous quality assurance,
- ability to counsel patients about the testing options, and
- access to appropriate diagnostic testing.
TABLE 2).
Integrated versus contingency screening. “Integrated” screening combines information from the first trimester (nuchal lucency and serum screen) with serum screening in the second trimester. This approach yields the lowest screen-positive rate (2.6%) and a high detection rate (90%), but has an important shortcoming: The results are not disclosed until the second trimester.
“Contingency” screening is emerging as an alternative: High first-trimester risks are relayed to the patient, while women with low screen values are excused from further testing. Patients with intermediate risk proceed to second-trimester serum screening.
Disadvantages of this approach include the need to coordinate the various steps and adequately inform the patient of them.
Added value of first-trimester nuchal lucency screening. Increased nuchal translucency alone is an important screen for structural abnormalities and adverse pregnancy outcomes. If a karyotypically normal fetus has an increased first-trimester nuchal lucency, the possibility of a structural anomaly on second-trimester ultrasound increases 2-to 10-fold. Absolute risk rises with increasing nuchal lucency.
Since an average of 10% to 15% of the identified anomalies are cardiac defects, fetal echocardiogram and a comprehensive fetal survey are appropriate in the second trimester.
TABLE 2
Detecting Down syndrome: Which test is best?
| MODALITY | SCREEN-POSITIVE RATE | DETECTION RATE |
|---|---|---|
| Maternal age >35 years | 18% | 30% |
| Triple screen (MSAFP, beta-hCG, estriol) | 5% | 65% |
| Quad screen (triple plus inhibin) | 5% | 75% |
| First-trimester (nuchal lucency, PAPP-A, free beta-hCG) | 5% | 80% |
| Integrated (first-trimester nuchal lucency and serum screen combined with second-trimester serum screen) | 2.5% | 90% |
| MSAFP = maternal serum alpha-fetoprotein, PAPP-A = pregnancy-associated plasma protein-A | ||
REFERENCE
1. Wapner R, Thom E, Simpsoon JL, et al. First-trimester screening for trisomies 21 and 18. N Engl J Med. 2003;349:1405-1413.
Are children conceived with ART at increased risk?
Children born as a result of ART may face a higher risk of inherited disorders and congenital malformations, but it is unclear whether the risks are due to their parents’ infertility or to ART.
For this reason, it may be wise to refer ART patients for additional genetic counseling and fetal structural surveillance by ultrasound.
Schieve and colleagues attempted to clarify the risks by reviewing the theoretical and empiric literature. Two studies provide the bulk of evidence. In Western Australia, the background risk of birth defects doubled in infants conceived with ART: 9% risk in both intracytoplasmic sperm injection and IVF patients, compared with 4% with spontaneous conception.1 This study is notable because ART programs are more highly regulated in Australia and similar methods were used to ascertain congenital anomalies in both groups.
A comparable study2 in Sweden also noted an increased risk, but attributed it to the underlying cause of the parents’ infertility rather than to ART itself. The reason: The increased risk disappeared when the authors adjusted for the period of “involuntary childlessness.” However, they provided very little detail on how involuntary childlessness was defined and “whether and how strongly this measure is correlated with infertility severity in Sweden.”2
Imprinting disorders among ART offspring. Schieve et al also explored imprinting disorders, since diseases such as Beckwith-Wiedemann syndrome are attributed to them. Imprinting is an epigenetic phenomenon in which the allele of only 1 parent is active at a particular gene locus. The inactive—or imprinted—allele is rendered nonfunctional, often through methylation. Gametogenesis and preimplantation are times of increased imprinting. Identified imprinted genes include those that control embryonic growth and differentiation.
Analyses of Beckwith-Wiedemann syndrome registries in the United States, France, and the UK3-5 revealed a 3- to 6-fold increase in ART conception among infants with the syndrome. Case reports of other rare imprinted disorders such as Angelman syndrome and retinoblastoma are also beginning to appear.
Direct treatment effect not established. According to Schieve et al and others, evidence of an increased risk of defects following ART does not indicate whether a direct treatment effect is present. Future studies that address methodological flaws are sure to be time-consuming; they also will require large sample sizes and consistent ascertainment and ART treatment.
Dr. Wilkins-Haug reports no relevant financial relationships.
1. Hansen M, Kurinczuk JJ, Bower C, Webb S. The risk of major birth defects after intracytoplasmic sperm injection and in vitro fertilization. N Engl J Med. 2002;346:725-730.
2. Ericson A, Kallen B. Congenital malformations in infants born after IVF: a population-based study. Hum Reprod. 2001;16:504-509.
3. DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72:156-160.
4. Maher ER, Brueton LA, Bowdin SC, et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART) [published erratum appears in J Med Genet. 2003;40:304]. J Med Genet. 2003;40:62-64.
5. Gicquel C, Gaston V, Mandelbaum J, et al. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003;72:1338-1341.
1. Hansen M, Kurinczuk JJ, Bower C, Webb S. The risk of major birth defects after intracytoplasmic sperm injection and in vitro fertilization. N Engl J Med. 2002;346:725-730.
2. Ericson A, Kallen B. Congenital malformations in infants born after IVF: a population-based study. Hum Reprod. 2001;16:504-509.
3. DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72:156-160.
4. Maher ER, Brueton LA, Bowdin SC, et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART) [published erratum appears in J Med Genet. 2003;40:304]. J Med Genet. 2003;40:62-64.
5. Gicquel C, Gaston V, Mandelbaum J, et al. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003;72:1338-1341.