User login
Building evidence-based medicine skills in gynecology
Although evidence-based medicine, or EBM, is not a new concept, the phrase is tossed about frequently in today’s culture of quality improvement initiatives and metrics. What does EBM really mean, however, and how do we ensure we are practicing it?
At its heart, EBM integrates 3 components:
- the individual clinician’s expertise
- the patient’s values and preferences
- the best external evidence to guide treatment decisions.
Because each clinician’s skillset and each patient’s issues and preferences may be quite varied, in this article we target the third piece—determining the best external evidence.
Our focus on EBM is not meant to negate the importance of the clinician’s expertise, which has been gained through years of practice. Indeed, without expertise, “practice risks becoming tyrannized by evidence.”1 However, without current best evidence, “practice risks becoming rapidly out of date, to the detriment of patients.”1 With the integration of evidence, expertise, and patient choice, EBM is not “cookbook” medicine, and it is not conducted only from armchairs and ivory towers. Rather, EBM is, or should be, at the frontline of clinical care.
EBM begins with a specific clinical question, such as “What is the best treatment option for my patient?” The answer can be honed with the “PICO” approach, which considers Population, Intervention, Comparators, and Outcomes of interest. Specifically, in a particular patient population (similar to your own patient), how does an intervention impact key outcomes?
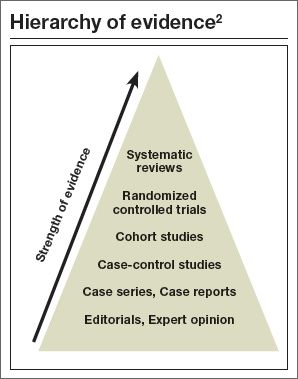
For directly comparing intervention options, such as surgery A versus surgery B, a randomized controlled trial (RCT) is one of the best methods to address clinical questions (FIGURE).2 Systematic reviews are more generalizable than single studies since they compare a range of relevant interventions across populations and settings. Evaluations of diagnostic test accuracy3,4or analyses of risk factors or natural history are best addressed by other study designs, which also can provide important evidence, but will not be discussed in depth here.
In this article, we focus on the benefits of RCTs and systematic reviews, as well as when to exhibit caution, for instance when RCTs report “surrogate outcomes” or make analyses drawn from subgroups of the original population. In addition, we discuss the inability to adequately assess treatment harms (versus benefits) from available evidence as well as the practicalities of how to apply EBM to patients.
RCTs: The good, the bad, and the ugly
RCTs are prospective experiments with a predefined protocol in which patients are randomly allocated to groups where the only difference is the intervention (vs comparators). This design helps to minimize the effects of known and unknown confounders and selection bias.
Ideally, the group into which a study participant is allocated is concealed from the patient and from the caregiver, minimizing the risk that the randomization is broken and the treatment allocation is biased. (Frequently this is not possible, however, particularly for surgical interventions.) Similarly, ideally, the outcome assessors are blinded to the treatment whenever possible. This minimizes the risk of a patient’s outcome being consciously or unconsciously altered due to the outcome assessor’s beliefs about the effectiveness of the intervention.
The reported clinical or surrogate outcomes (which will be discussed in more depth on the next page) for an RCT may be objective or subjective. Preferably, outcomes are patient-centered—important from the patient’s perspective of benefits and harms. Examples of these types of outcomes include survival, function, symptoms, and health-related quality of life, as well as impact on work and family, convenience, and cost. Patients likely are less interested in estimated blood loss, surgical time, biochemistry results, and other clinical or surrogate outcomes.
There are disadvantages to RCTs. For instance, each study provides only a snapshot of the evidence on a given topic. One study rarely, if ever, provides a definitive conclusion. The study’s findings are subject to random error and to biases introduced by study design or analytic methods, and they will not be generalizable to all patients and settings. In addition, the study likely has evaluated only 1 or 2 specific interventions among a plethora of available options, and is unlikely to have analyzed all outcomes of interest.
It becomes your burden to assess whether a trial’s findings are applicable to an actual patient (known as “external validity”). Because an RCT must artificially constrain the underlying clinical questions into a testable research question, translation to the specific patient is often flawed. Perhaps the patient does not precisely fit the inclusion criteria of the trial, for instance, or the exact intervention tested is not fully reproducible. From a practicality perspective, an RCT is often immensely costly to execute, which may be reflected in relatively small numbers of patients and short-term duration of follow-up. These disadvantages limit the ability of RCTs to assess harms, rare events, and long-term outcomes.
Surrogate outcomes
Outcomes measured in a trial should be relevant, easy to interpret and diagnose, sensitive to treatment differences, and measurable within a reasonable period of time. However, these characteristics are not always achievable for important clinical outcomes in an RCT. Therefore, a surrogate outcome may take the place of the true clinical efficacy measurement.
For example, in studies of interventions for infertility in patients with polycystic ovary syndrome (PCOS), common surrogates to the “true” desired outcome of a healthy live birth may include ovulation, implantation, or pregnancy rates. These surrogate outcomes may correlate with live birth but clearly ignore other factors extrinsic and intrinsic to PCOS that affect the chance for a healthy term delivery; the possible increased risk for miscarriage in PCOS; and increased risks of other pregnancy complications, such as preeclampsia and gestational diabetes.
Similarly, many trials of oral contraceptives that aim to study the clinical endpoint of pulmonary embolism or venous thromboembolism, which are rare events, instead use the surrogates of results of coagulation tests or levels of sex hormone-binding globulin. Clearly, caution must be exercised when interpreting studies that use surrogate outcomes. As the clinician, you must recognize that a change in a biologic or physical measurement may not be clinically relevant. Some judgment is required about causal pathways: The less that is known about the causal pathway of a disease, the less confident one should be in any surrogate outcome.
Finally, clinicians also must recognize that a valid surrogate for one treatment may not be valid for another treatment or another population.5 For example, ovulation inhibition would be an appropriate surrogate endpoint for contraceptive efficacy for a method that reliably prevents ovulation; however, this would not be a good surrogate outcome to evaluate the progestin-only pill, which fails to inhibit ovulation completely and yet is highly effective in contraceptive trials.
Avoiding pitfalls with subgroup analyses
It is common, particularly in large RCTs, to evaluate treatment effects for a specific endpoint in a subgroup of patients included in the trial. The goal is to determine whether the findings of the larger study apply more or less to a specific patient (who may differ from the total population by some important characteristic, such as age, weight, parity, or menopausal or smoking status). The variability in study results when stratified by these patient factors is known as heterogeneity of treatment effect, which may be quantitative or qualitative.6
In the former, one treatment is always better than the other, although by varying degrees depending on the subgroup. (For example, a stronger effect could be seen in those aged 65 and younger than in those older than 65.) In the latter, the treatment fares better than the comparator in one subgroup but worse or no different for another subgroup. In either case, the appropriate statistical tool to identify heterogeneity of treatment effect is a test for interaction between the characteristic and the treatment effect, rather than claiming heterogeneity on the basis of separate tests of treatment effects within the different subpopulations.
One problem with dividing the original population into smaller subpopulations is that the number of participants decreases—thus there is less power, or less statistical strength, to identify a treatment effect. More accurately, there is a greater likelihood of a type II error (a false negative) when these small subpopulations have too few patients to demonstrate a clinical treatment effect that actually may exist.
False positives. Paradoxically, another problem with subgroup analyses is a greater chance for false positives due to the multiple statistical testing that is performed. The original study is rarely powered appropriately to do this (see “Error rates in subgroup analyses”). According to Wang and colleagues, “It is common practice to conduct a subgroup analysis for each of several (and often many) baseline characteristics, for each of several endpoints, or for both.”7 The more subgroup analyses performed, the more likely that differences found are due to chance only. Unfortunately, in unplanned post hoc analyses, the number of tests performed is often unreported; therefore, the error rates are unknown. There are statistical methods to try and correct for this “multiplicity” problem but, ideally, only a few key subgroup analyses are performed, and they are planned a priori in the original study design. In these cases, the study’s size can be adjusted accordingly. In most instances, findings from subgroup analyses, whether positive or negative, should be considered as “hypothesis generating” and interpreted with caution.
Error rates in subgroup analyses
With “k” independent subgroups and no difference in treatments, the probability of at least one “significant” subgroup (such as a false positive) is 1 – (1-α)k.
If α = 0.05 and there are k = 10 subgroups, then 1 – (0.95)10 = 0.40. That is, if 10 subgroup analyses are performed, there is a 40% likelihood that 1 will demonstrate a “significant” difference in treatment effect, even though no difference exists.
Systematic reviews: What, why, and how?
Systematic reviews aim to overcome the deficiencies of single studies in a comprehensive and unbiased manner. They critically evaluate, summarize, and, when possible, combine all available studies addressing a given topic. By comparing a range of relevant interventions across populations and settings, systematic reviews may be more generalizable than single studies. Meta-analysis, or quantitatively combining study results, increases sample size and usually provides more precise estimates of effect sizes than the single studies. Critical appraisal of the combined studies can highlight methodologic and other concerns about the body of evidence to assess the overall confidence in the included studies.
A systematic review, like a well-conducted RCT, has a protocol that lays out the scope of the review and defines a priori criteria and analytic plans—all with the goal of minimizing bias. It starts with a well-formulated research question, explicitly defining the PICO elements—population, interventions, comparators, outcomes—in addition to the setting and study designs of interest.8 Based on these eligibility criteria, several sources of evidence (such as electronic databases and reference lists) are searched to find all potentially eligible studies.
Typically, several thousand citations are found that must be matched against the eligibility criteria. Potentially eligible studies are then rescreened in full text to further scrutinize their eligibility. The goal is to be highly sensitive to avoid missing relevant studies—even at the time cost of screening many articles. The individual study designs (including the study eligibility criteria, interventions, outcomes, and analytic methods) and the results for all outcomes of interest are extracted from each study.
For most systematic reviews, researchers also will assess the quality, or risk of bias, of each study for each outcome.9,10 Study data are summarized across all included studies, with study results meta-analyzed and reasons for heterogeneity across studies explored. Several consensus statements detail the proper methodology to conduct and report a systematic review.11,12 Ultimately, the review’s conclusions are based on analyses of all available evidence. By contrast, narrative reviews typically start with a conclusion and then select evidence to support that conclusion, and are therefore more likely to be biased.13
As noted, systematic reviews often include meta-analysis, which may allow an exploration of some reasons for study heterogeneity. The meta-analysis is usually presented graphically in a forest plot, which displays point estimates for each study with their associated 95% confidence intervals and a description of each study.14 In a forest plot, one can see the estimate and precision of each study, assess the heterogeneity of results across studies, and compare individual studies to each other and to the overall summary estimate.
Systematic reviews should be read as critically as primary studies. Some important questions you should consider are:
- Did the review address the populations, interventions, comparators, outcomes, and settings relevant to your practice?
- Have studies been included in a nonbiased manner, and is the described body of evidence likely to be complete?
- Did the study authors evaluate and summarize the underlying risks of bias of the studies?
- Did the researchers avoid combining studies that are too different from each other to allow a coherent interpretation of the summary results?
- Did the researchers attempt to explain how and why studies differed from one another?
Of note, systematic reviews and meta-analyses are subject to the same biases as all retrospective studies. Also, the systematic reviewers’ own biases—due to factors such as funding source, researchers’ agendas, or specialties—may subtly affect systematic reviews just as biases may affect an individual study. Furthermore, the confidence you have in a systematic review’s conclusions may be limited by the quality and generalizability of the underlying studies.
Assessing harms
You make the ultimate management decisions for your patient (though, of course, with her participation). The likely benefit of a specific treatment—determined in an experimental trial and refined further in a systematic review and meta-analysis—must be balanced with the risk of harms. RCTs usually do not provide the highest quality evidence of harms due to their limited sample sizes and short follow-up duration. Rather, large observational studies, case series, and case reports commonly provide these important details. Increasingly, patient registries are being created to prospectively follow patients and gather uniform safety data. By providing a true denominator, more accurate estimates of adverse event incidence are possible. However, the disadvantages of all of these modalities are 1) there usually are no comparators (that is, “How does the adverse event incidence for surgery A compare to that for surgery B?”) and 2) data usually are gleaned from medical records and not directly from patients.
As a result, these studies typically lack information on subjective harms, such as impaired sexual function. The reporting of treatment harms suffers from inconsistent and imprecise terminology, making it hard to reliably gather all reports of similar adverse events. Adverse event reporting in clinical trials is often driven by regulatory definitions and requirements instead of patient-centered definitions. In fact, there has been little work to date that assesses which adverse events or complications may be most relevant or important from the patient perspective.
Taken together, it is clear that the medical literature tends to emphasize treatment benefits (with robust methodologies and data to detect these benefits) but does not reliably or adequately assess harms. For rare events, risk estimates always will be imprecise. Nonetheless, better systematic reviews and today’s larger comparative effectiveness reviews strive to gather harms data from the multiple available sources described above.
Applying the evidence and your expertise to your patient
Now that you have identified the best valid and important evidence to support or refute a clinical decision (TABLE15), and have coupled this with your own expert knowledge and judgment in shared decision making with your patient, you must communicate to her the personalized information about outcomes, probabilities, and scientific uncertainties of her available treatment options.15 Patients, in turn, should be allowed to communicate their values and the relative importance they place on benefits and harms.16 This conversation, of course, is built on the foundation of a sound physician–patient relationship and is a part of every informed consent process.
Is this evidence applicable to my patient? A decision guide.15
- Is my patient so different from those in the study that the trial results cannot be applied?
- Is the treatment feasible in my setting?
- What are my patient’s likely benefits and harms from the therapy?
- How will my patient’s values influence the final treatment decision?
Decision tools
Increasingly, decision-aid tools are being developed to support this process. These aids must express the helpful and harmful effects of a treatment, including alternative options, in statements that are valid and concise. Furthermore, they must be intelligible to both the clinician and patient and modifiable to the patient’s values and wishes.17 Two examples of counseling aids are the Gail model of breast cancer risk prediction18 and the Framingham Coronary Heart Disease Prediction Score.19 Web-based decision aids that can be accessed in real-time in busy clinical settings also are being developed for gynecology.20
Never stop re-evaluating
The final piece of EBM is to “close the loop”—meaning to evaluate the effectiveness of applying the evidence in clinical practice. To do this, watch for clinical practice guidelines that are based on systematic reviews and the EBM approach and stay abreast of ACOG’s and other professional societies’ guideline statements. Ultimately, guidelines beget performance measures. Organizations such as the National Quality Forum are working to define these standards of performance measurement and seek feedback from individual clinicians to ensure measures are meaningful and accurate. By 2017, 9% of all Medicare payments are scheduled to be performance based.21
Conclusion
During the course of reading medical literature, stay attuned to comparative effectiveness research and recognize studies with active comparators that examine clinical questions that could impact your day-to-day practice and that can be applied to your patient population. While there is no such thing as a perfect research study, and it is rare that one trial can address any one clinician’s specific patients precisely, increasingly we are seeing better systematic reviews and meta-analyses. It is these studies that provide the high quality data for you to couple with your clinical expertise and your patients’ values and preferences to truly deliver evidence-based medicine.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t. BMJ. 1996;312(7023):71–72.
- Sackett DL, et al. Evidence-Based Medicine: How to Practice and Teach EBM. 2nd ed. Edinburgh, UK: Churchill Livingstone; 2000.
- Knottnerus JA, van Weel C, Muris JW. Evaluation of diagnostic procedures. BMJ. 2002;324(7335):477–480.
- Bossuyt PM, Reitsma JB, Bruns DE, et al; Standards for Reporting of Diagnostic Accuracy Group. The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. Ann Intern Med. 2003;138(1):W1-W12.
- Grimes DA, Schulz KF, Raymond EG. Surrogate end points in women’s health research: science, protoscience, and pseudoscience. Fertil Steril. 2010;93(6):1731–1734.
- Lagakos SW. The challenge of subgroup analyses—reporting without distorting. N Engl J Med. 2006;354(16):1667–1669.
- Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine—reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357(21):2189–2194.
- Counsell C. Formulating questions and locating primary studies for inclusion in systematic reviews. Ann Intern Med. 1997;127(5):380–387.
- Higgins JP, Altman DG, Gøtzsche PC, et al; Cochrane Statistical Methods Group. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928.
- Berkman ND, Lohr KN, Ansari M, et al. Grading the Strength of a Body of Evidence When Assessing Health Care Interventions for the Effective Health Care Program of the Agency for Healthcare Research and Quality: An Update. Methods Guide for Effectiveness and Comparative Effectiveness Reviews. Rockville (MD)2008. AHRQ Methods for Effective Health Care. 2013 Nov 18.
- Liberati A1, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- Institute of Medicine of the National Academies. Finding What Works in Health Care: Standards for Systematic Reviews. iom.edu/Reports/2011/Finding-What-Works-in-Health-Care-Standards-for-Systematic-Reviews.aspx. Published March 23, 2011. Accessed March 20, 2015.
- Mulrow CD. The medical review article: state of the science. Ann Intern Med. 1987;106(3):485–488.
- Lewis S, Clarke M. Forest plots: trying to see the wood and the trees. BMJ. 2001;322(7300):1479–1480.
- Glasziou P, Guyatt GH, Dans AL, Dans LF, Straus S, Sackett DL. Applying the results of trials and systematic reviews to individual patients. ACP J Club. 1998;129(3):A15–A16.
- What is shared decision making? Informed Medical Decisions Foundation Web site. http://www.informed medicaldecisions.org/what-is-shared-decision-making. Published 2015. Accessed January 23, 2015.
- Straus SE, Sackett DL. Applying evidence to the individual patient. Ann Oncol. 1999;10(1):29–32.
- Rockhill B, Spiegelman D, Byrne C, Hunter DJ, Colditz GA. Validation of the Gail et al. model of breast cancer risk prediction and implications for chemoprevention. J Natl Cancer Inst. 2001;93(5):358–366.
- D’Agostino RB Sr, Grundy S, Sullivan LM, Wilson P; CHD Risk Prediction Group. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA. 2001;286(2):180–187.
- Jelovsek JE, Chagin K, Brubaker L, et al; Pelvic Floor Disorders Network. A model for predicting the risk of de novo stress urinary incontinence in women undergoing pelvic organ prolapse surgery. Obstet Gynecol. 2014;123(2 Pt 1):279–287.
- National Quality Forum. What we do. National Quality Forum Web sight. http://www.qualityforum.org/what_we_do.aspx. Published 2015. Accessed January 29, 2015.
David Rahn, MD; Vivian Sung, MD, MPH; and Ethan Balk, MD, MPH
Experts featured in this article
|
Dr. Rahn is Associate Professor, Department of Obstetrics & Gynecology, Division of Female Pelvic Medicine & Reconstructive Surgery, University of Texas Southwestern Medical Center, Dallas. |
| Dr. Sung is Associate Professor, Obstetrics and Gynecology, The Warren Alpert Medical School of Brown University, and Research Director, Division of Urogynecology and Reconstructive Pelvic Surgery, Women & Infants Hospital of Rhode Island, Providence. |
| Dr. Balk is Assistant Professor (Research), Center for Evidence-Based Medicine, and Associate Director, Brown Evidence-based Practice Center, Brown University School of Public Health, Providence, Rhode Island. |



Dr. Sung reports that she receives grant or research support from the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development. Drs. Balk and Rahn report no financial relationships relevant to this article.
Developed in partnership with the Society of Gynecologic Surgeons and based on workshop content supported by a grant from the American Board of Obstetrics and Gynecology
David Rahn, MD; Vivian Sung, MD, MPH; and Ethan Balk, MD, MPH
Experts featured in this article
|
Dr. Rahn is Associate Professor, Department of Obstetrics & Gynecology, Division of Female Pelvic Medicine & Reconstructive Surgery, University of Texas Southwestern Medical Center, Dallas. |
| Dr. Sung is Associate Professor, Obstetrics and Gynecology, The Warren Alpert Medical School of Brown University, and Research Director, Division of Urogynecology and Reconstructive Pelvic Surgery, Women & Infants Hospital of Rhode Island, Providence. |
| Dr. Balk is Assistant Professor (Research), Center for Evidence-Based Medicine, and Associate Director, Brown Evidence-based Practice Center, Brown University School of Public Health, Providence, Rhode Island. |
Dr. Sung reports that she receives grant or research support from the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development. Drs. Balk and Rahn report no financial relationships relevant to this article.
Developed in partnership with the Society of Gynecologic Surgeons and based on workshop content supported by a grant from the American Board of Obstetrics and Gynecology
David Rahn, MD; Vivian Sung, MD, MPH; and Ethan Balk, MD, MPH
Experts featured in this article
|
Dr. Rahn is Associate Professor, Department of Obstetrics & Gynecology, Division of Female Pelvic Medicine & Reconstructive Surgery, University of Texas Southwestern Medical Center, Dallas. |
| Dr. Sung is Associate Professor, Obstetrics and Gynecology, The Warren Alpert Medical School of Brown University, and Research Director, Division of Urogynecology and Reconstructive Pelvic Surgery, Women & Infants Hospital of Rhode Island, Providence. |
| Dr. Balk is Assistant Professor (Research), Center for Evidence-Based Medicine, and Associate Director, Brown Evidence-based Practice Center, Brown University School of Public Health, Providence, Rhode Island. |
Dr. Sung reports that she receives grant or research support from the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development. Drs. Balk and Rahn report no financial relationships relevant to this article.
Developed in partnership with the Society of Gynecologic Surgeons and based on workshop content supported by a grant from the American Board of Obstetrics and Gynecology
Although evidence-based medicine, or EBM, is not a new concept, the phrase is tossed about frequently in today’s culture of quality improvement initiatives and metrics. What does EBM really mean, however, and how do we ensure we are practicing it?
At its heart, EBM integrates 3 components:
- the individual clinician’s expertise
- the patient’s values and preferences
- the best external evidence to guide treatment decisions.
Because each clinician’s skillset and each patient’s issues and preferences may be quite varied, in this article we target the third piece—determining the best external evidence.
Our focus on EBM is not meant to negate the importance of the clinician’s expertise, which has been gained through years of practice. Indeed, without expertise, “practice risks becoming tyrannized by evidence.”1 However, without current best evidence, “practice risks becoming rapidly out of date, to the detriment of patients.”1 With the integration of evidence, expertise, and patient choice, EBM is not “cookbook” medicine, and it is not conducted only from armchairs and ivory towers. Rather, EBM is, or should be, at the frontline of clinical care.
EBM begins with a specific clinical question, such as “What is the best treatment option for my patient?” The answer can be honed with the “PICO” approach, which considers Population, Intervention, Comparators, and Outcomes of interest. Specifically, in a particular patient population (similar to your own patient), how does an intervention impact key outcomes?
For directly comparing intervention options, such as surgery A versus surgery B, a randomized controlled trial (RCT) is one of the best methods to address clinical questions (FIGURE).2 Systematic reviews are more generalizable than single studies since they compare a range of relevant interventions across populations and settings. Evaluations of diagnostic test accuracy3,4or analyses of risk factors or natural history are best addressed by other study designs, which also can provide important evidence, but will not be discussed in depth here.
In this article, we focus on the benefits of RCTs and systematic reviews, as well as when to exhibit caution, for instance when RCTs report “surrogate outcomes” or make analyses drawn from subgroups of the original population. In addition, we discuss the inability to adequately assess treatment harms (versus benefits) from available evidence as well as the practicalities of how to apply EBM to patients.
RCTs: The good, the bad, and the ugly
RCTs are prospective experiments with a predefined protocol in which patients are randomly allocated to groups where the only difference is the intervention (vs comparators). This design helps to minimize the effects of known and unknown confounders and selection bias.
Ideally, the group into which a study participant is allocated is concealed from the patient and from the caregiver, minimizing the risk that the randomization is broken and the treatment allocation is biased. (Frequently this is not possible, however, particularly for surgical interventions.) Similarly, ideally, the outcome assessors are blinded to the treatment whenever possible. This minimizes the risk of a patient’s outcome being consciously or unconsciously altered due to the outcome assessor’s beliefs about the effectiveness of the intervention.
The reported clinical or surrogate outcomes (which will be discussed in more depth on the next page) for an RCT may be objective or subjective. Preferably, outcomes are patient-centered—important from the patient’s perspective of benefits and harms. Examples of these types of outcomes include survival, function, symptoms, and health-related quality of life, as well as impact on work and family, convenience, and cost. Patients likely are less interested in estimated blood loss, surgical time, biochemistry results, and other clinical or surrogate outcomes.
There are disadvantages to RCTs. For instance, each study provides only a snapshot of the evidence on a given topic. One study rarely, if ever, provides a definitive conclusion. The study’s findings are subject to random error and to biases introduced by study design or analytic methods, and they will not be generalizable to all patients and settings. In addition, the study likely has evaluated only 1 or 2 specific interventions among a plethora of available options, and is unlikely to have analyzed all outcomes of interest.
It becomes your burden to assess whether a trial’s findings are applicable to an actual patient (known as “external validity”). Because an RCT must artificially constrain the underlying clinical questions into a testable research question, translation to the specific patient is often flawed. Perhaps the patient does not precisely fit the inclusion criteria of the trial, for instance, or the exact intervention tested is not fully reproducible. From a practicality perspective, an RCT is often immensely costly to execute, which may be reflected in relatively small numbers of patients and short-term duration of follow-up. These disadvantages limit the ability of RCTs to assess harms, rare events, and long-term outcomes.
Surrogate outcomes
Outcomes measured in a trial should be relevant, easy to interpret and diagnose, sensitive to treatment differences, and measurable within a reasonable period of time. However, these characteristics are not always achievable for important clinical outcomes in an RCT. Therefore, a surrogate outcome may take the place of the true clinical efficacy measurement.
For example, in studies of interventions for infertility in patients with polycystic ovary syndrome (PCOS), common surrogates to the “true” desired outcome of a healthy live birth may include ovulation, implantation, or pregnancy rates. These surrogate outcomes may correlate with live birth but clearly ignore other factors extrinsic and intrinsic to PCOS that affect the chance for a healthy term delivery; the possible increased risk for miscarriage in PCOS; and increased risks of other pregnancy complications, such as preeclampsia and gestational diabetes.
Similarly, many trials of oral contraceptives that aim to study the clinical endpoint of pulmonary embolism or venous thromboembolism, which are rare events, instead use the surrogates of results of coagulation tests or levels of sex hormone-binding globulin. Clearly, caution must be exercised when interpreting studies that use surrogate outcomes. As the clinician, you must recognize that a change in a biologic or physical measurement may not be clinically relevant. Some judgment is required about causal pathways: The less that is known about the causal pathway of a disease, the less confident one should be in any surrogate outcome.
Finally, clinicians also must recognize that a valid surrogate for one treatment may not be valid for another treatment or another population.5 For example, ovulation inhibition would be an appropriate surrogate endpoint for contraceptive efficacy for a method that reliably prevents ovulation; however, this would not be a good surrogate outcome to evaluate the progestin-only pill, which fails to inhibit ovulation completely and yet is highly effective in contraceptive trials.
Avoiding pitfalls with subgroup analyses
It is common, particularly in large RCTs, to evaluate treatment effects for a specific endpoint in a subgroup of patients included in the trial. The goal is to determine whether the findings of the larger study apply more or less to a specific patient (who may differ from the total population by some important characteristic, such as age, weight, parity, or menopausal or smoking status). The variability in study results when stratified by these patient factors is known as heterogeneity of treatment effect, which may be quantitative or qualitative.6
In the former, one treatment is always better than the other, although by varying degrees depending on the subgroup. (For example, a stronger effect could be seen in those aged 65 and younger than in those older than 65.) In the latter, the treatment fares better than the comparator in one subgroup but worse or no different for another subgroup. In either case, the appropriate statistical tool to identify heterogeneity of treatment effect is a test for interaction between the characteristic and the treatment effect, rather than claiming heterogeneity on the basis of separate tests of treatment effects within the different subpopulations.
One problem with dividing the original population into smaller subpopulations is that the number of participants decreases—thus there is less power, or less statistical strength, to identify a treatment effect. More accurately, there is a greater likelihood of a type II error (a false negative) when these small subpopulations have too few patients to demonstrate a clinical treatment effect that actually may exist.
False positives. Paradoxically, another problem with subgroup analyses is a greater chance for false positives due to the multiple statistical testing that is performed. The original study is rarely powered appropriately to do this (see “Error rates in subgroup analyses”). According to Wang and colleagues, “It is common practice to conduct a subgroup analysis for each of several (and often many) baseline characteristics, for each of several endpoints, or for both.”7 The more subgroup analyses performed, the more likely that differences found are due to chance only. Unfortunately, in unplanned post hoc analyses, the number of tests performed is often unreported; therefore, the error rates are unknown. There are statistical methods to try and correct for this “multiplicity” problem but, ideally, only a few key subgroup analyses are performed, and they are planned a priori in the original study design. In these cases, the study’s size can be adjusted accordingly. In most instances, findings from subgroup analyses, whether positive or negative, should be considered as “hypothesis generating” and interpreted with caution.
Error rates in subgroup analyses
With “k” independent subgroups and no difference in treatments, the probability of at least one “significant” subgroup (such as a false positive) is 1 – (1-α)k.
If α = 0.05 and there are k = 10 subgroups, then 1 – (0.95)10 = 0.40. That is, if 10 subgroup analyses are performed, there is a 40% likelihood that 1 will demonstrate a “significant” difference in treatment effect, even though no difference exists.
Systematic reviews: What, why, and how?
Systematic reviews aim to overcome the deficiencies of single studies in a comprehensive and unbiased manner. They critically evaluate, summarize, and, when possible, combine all available studies addressing a given topic. By comparing a range of relevant interventions across populations and settings, systematic reviews may be more generalizable than single studies. Meta-analysis, or quantitatively combining study results, increases sample size and usually provides more precise estimates of effect sizes than the single studies. Critical appraisal of the combined studies can highlight methodologic and other concerns about the body of evidence to assess the overall confidence in the included studies.
A systematic review, like a well-conducted RCT, has a protocol that lays out the scope of the review and defines a priori criteria and analytic plans—all with the goal of minimizing bias. It starts with a well-formulated research question, explicitly defining the PICO elements—population, interventions, comparators, outcomes—in addition to the setting and study designs of interest.8 Based on these eligibility criteria, several sources of evidence (such as electronic databases and reference lists) are searched to find all potentially eligible studies.
Typically, several thousand citations are found that must be matched against the eligibility criteria. Potentially eligible studies are then rescreened in full text to further scrutinize their eligibility. The goal is to be highly sensitive to avoid missing relevant studies—even at the time cost of screening many articles. The individual study designs (including the study eligibility criteria, interventions, outcomes, and analytic methods) and the results for all outcomes of interest are extracted from each study.
For most systematic reviews, researchers also will assess the quality, or risk of bias, of each study for each outcome.9,10 Study data are summarized across all included studies, with study results meta-analyzed and reasons for heterogeneity across studies explored. Several consensus statements detail the proper methodology to conduct and report a systematic review.11,12 Ultimately, the review’s conclusions are based on analyses of all available evidence. By contrast, narrative reviews typically start with a conclusion and then select evidence to support that conclusion, and are therefore more likely to be biased.13
As noted, systematic reviews often include meta-analysis, which may allow an exploration of some reasons for study heterogeneity. The meta-analysis is usually presented graphically in a forest plot, which displays point estimates for each study with their associated 95% confidence intervals and a description of each study.14 In a forest plot, one can see the estimate and precision of each study, assess the heterogeneity of results across studies, and compare individual studies to each other and to the overall summary estimate.
Systematic reviews should be read as critically as primary studies. Some important questions you should consider are:
- Did the review address the populations, interventions, comparators, outcomes, and settings relevant to your practice?
- Have studies been included in a nonbiased manner, and is the described body of evidence likely to be complete?
- Did the study authors evaluate and summarize the underlying risks of bias of the studies?
- Did the researchers avoid combining studies that are too different from each other to allow a coherent interpretation of the summary results?
- Did the researchers attempt to explain how and why studies differed from one another?
Of note, systematic reviews and meta-analyses are subject to the same biases as all retrospective studies. Also, the systematic reviewers’ own biases—due to factors such as funding source, researchers’ agendas, or specialties—may subtly affect systematic reviews just as biases may affect an individual study. Furthermore, the confidence you have in a systematic review’s conclusions may be limited by the quality and generalizability of the underlying studies.
Assessing harms
You make the ultimate management decisions for your patient (though, of course, with her participation). The likely benefit of a specific treatment—determined in an experimental trial and refined further in a systematic review and meta-analysis—must be balanced with the risk of harms. RCTs usually do not provide the highest quality evidence of harms due to their limited sample sizes and short follow-up duration. Rather, large observational studies, case series, and case reports commonly provide these important details. Increasingly, patient registries are being created to prospectively follow patients and gather uniform safety data. By providing a true denominator, more accurate estimates of adverse event incidence are possible. However, the disadvantages of all of these modalities are 1) there usually are no comparators (that is, “How does the adverse event incidence for surgery A compare to that for surgery B?”) and 2) data usually are gleaned from medical records and not directly from patients.
As a result, these studies typically lack information on subjective harms, such as impaired sexual function. The reporting of treatment harms suffers from inconsistent and imprecise terminology, making it hard to reliably gather all reports of similar adverse events. Adverse event reporting in clinical trials is often driven by regulatory definitions and requirements instead of patient-centered definitions. In fact, there has been little work to date that assesses which adverse events or complications may be most relevant or important from the patient perspective.
Taken together, it is clear that the medical literature tends to emphasize treatment benefits (with robust methodologies and data to detect these benefits) but does not reliably or adequately assess harms. For rare events, risk estimates always will be imprecise. Nonetheless, better systematic reviews and today’s larger comparative effectiveness reviews strive to gather harms data from the multiple available sources described above.
Applying the evidence and your expertise to your patient
Now that you have identified the best valid and important evidence to support or refute a clinical decision (TABLE15), and have coupled this with your own expert knowledge and judgment in shared decision making with your patient, you must communicate to her the personalized information about outcomes, probabilities, and scientific uncertainties of her available treatment options.15 Patients, in turn, should be allowed to communicate their values and the relative importance they place on benefits and harms.16 This conversation, of course, is built on the foundation of a sound physician–patient relationship and is a part of every informed consent process.
Is this evidence applicable to my patient? A decision guide.15
- Is my patient so different from those in the study that the trial results cannot be applied?
- Is the treatment feasible in my setting?
- What are my patient’s likely benefits and harms from the therapy?
- How will my patient’s values influence the final treatment decision?
Decision tools
Increasingly, decision-aid tools are being developed to support this process. These aids must express the helpful and harmful effects of a treatment, including alternative options, in statements that are valid and concise. Furthermore, they must be intelligible to both the clinician and patient and modifiable to the patient’s values and wishes.17 Two examples of counseling aids are the Gail model of breast cancer risk prediction18 and the Framingham Coronary Heart Disease Prediction Score.19 Web-based decision aids that can be accessed in real-time in busy clinical settings also are being developed for gynecology.20
Never stop re-evaluating
The final piece of EBM is to “close the loop”—meaning to evaluate the effectiveness of applying the evidence in clinical practice. To do this, watch for clinical practice guidelines that are based on systematic reviews and the EBM approach and stay abreast of ACOG’s and other professional societies’ guideline statements. Ultimately, guidelines beget performance measures. Organizations such as the National Quality Forum are working to define these standards of performance measurement and seek feedback from individual clinicians to ensure measures are meaningful and accurate. By 2017, 9% of all Medicare payments are scheduled to be performance based.21
Conclusion
During the course of reading medical literature, stay attuned to comparative effectiveness research and recognize studies with active comparators that examine clinical questions that could impact your day-to-day practice and that can be applied to your patient population. While there is no such thing as a perfect research study, and it is rare that one trial can address any one clinician’s specific patients precisely, increasingly we are seeing better systematic reviews and meta-analyses. It is these studies that provide the high quality data for you to couple with your clinical expertise and your patients’ values and preferences to truly deliver evidence-based medicine.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Although evidence-based medicine, or EBM, is not a new concept, the phrase is tossed about frequently in today’s culture of quality improvement initiatives and metrics. What does EBM really mean, however, and how do we ensure we are practicing it?
At its heart, EBM integrates 3 components:
- the individual clinician’s expertise
- the patient’s values and preferences
- the best external evidence to guide treatment decisions.
Because each clinician’s skillset and each patient’s issues and preferences may be quite varied, in this article we target the third piece—determining the best external evidence.
Our focus on EBM is not meant to negate the importance of the clinician’s expertise, which has been gained through years of practice. Indeed, without expertise, “practice risks becoming tyrannized by evidence.”1 However, without current best evidence, “practice risks becoming rapidly out of date, to the detriment of patients.”1 With the integration of evidence, expertise, and patient choice, EBM is not “cookbook” medicine, and it is not conducted only from armchairs and ivory towers. Rather, EBM is, or should be, at the frontline of clinical care.
EBM begins with a specific clinical question, such as “What is the best treatment option for my patient?” The answer can be honed with the “PICO” approach, which considers Population, Intervention, Comparators, and Outcomes of interest. Specifically, in a particular patient population (similar to your own patient), how does an intervention impact key outcomes?
For directly comparing intervention options, such as surgery A versus surgery B, a randomized controlled trial (RCT) is one of the best methods to address clinical questions (FIGURE).2 Systematic reviews are more generalizable than single studies since they compare a range of relevant interventions across populations and settings. Evaluations of diagnostic test accuracy3,4or analyses of risk factors or natural history are best addressed by other study designs, which also can provide important evidence, but will not be discussed in depth here.
In this article, we focus on the benefits of RCTs and systematic reviews, as well as when to exhibit caution, for instance when RCTs report “surrogate outcomes” or make analyses drawn from subgroups of the original population. In addition, we discuss the inability to adequately assess treatment harms (versus benefits) from available evidence as well as the practicalities of how to apply EBM to patients.
RCTs: The good, the bad, and the ugly
RCTs are prospective experiments with a predefined protocol in which patients are randomly allocated to groups where the only difference is the intervention (vs comparators). This design helps to minimize the effects of known and unknown confounders and selection bias.
Ideally, the group into which a study participant is allocated is concealed from the patient and from the caregiver, minimizing the risk that the randomization is broken and the treatment allocation is biased. (Frequently this is not possible, however, particularly for surgical interventions.) Similarly, ideally, the outcome assessors are blinded to the treatment whenever possible. This minimizes the risk of a patient’s outcome being consciously or unconsciously altered due to the outcome assessor’s beliefs about the effectiveness of the intervention.
The reported clinical or surrogate outcomes (which will be discussed in more depth on the next page) for an RCT may be objective or subjective. Preferably, outcomes are patient-centered—important from the patient’s perspective of benefits and harms. Examples of these types of outcomes include survival, function, symptoms, and health-related quality of life, as well as impact on work and family, convenience, and cost. Patients likely are less interested in estimated blood loss, surgical time, biochemistry results, and other clinical or surrogate outcomes.
There are disadvantages to RCTs. For instance, each study provides only a snapshot of the evidence on a given topic. One study rarely, if ever, provides a definitive conclusion. The study’s findings are subject to random error and to biases introduced by study design or analytic methods, and they will not be generalizable to all patients and settings. In addition, the study likely has evaluated only 1 or 2 specific interventions among a plethora of available options, and is unlikely to have analyzed all outcomes of interest.
It becomes your burden to assess whether a trial’s findings are applicable to an actual patient (known as “external validity”). Because an RCT must artificially constrain the underlying clinical questions into a testable research question, translation to the specific patient is often flawed. Perhaps the patient does not precisely fit the inclusion criteria of the trial, for instance, or the exact intervention tested is not fully reproducible. From a practicality perspective, an RCT is often immensely costly to execute, which may be reflected in relatively small numbers of patients and short-term duration of follow-up. These disadvantages limit the ability of RCTs to assess harms, rare events, and long-term outcomes.
Surrogate outcomes
Outcomes measured in a trial should be relevant, easy to interpret and diagnose, sensitive to treatment differences, and measurable within a reasonable period of time. However, these characteristics are not always achievable for important clinical outcomes in an RCT. Therefore, a surrogate outcome may take the place of the true clinical efficacy measurement.
For example, in studies of interventions for infertility in patients with polycystic ovary syndrome (PCOS), common surrogates to the “true” desired outcome of a healthy live birth may include ovulation, implantation, or pregnancy rates. These surrogate outcomes may correlate with live birth but clearly ignore other factors extrinsic and intrinsic to PCOS that affect the chance for a healthy term delivery; the possible increased risk for miscarriage in PCOS; and increased risks of other pregnancy complications, such as preeclampsia and gestational diabetes.
Similarly, many trials of oral contraceptives that aim to study the clinical endpoint of pulmonary embolism or venous thromboembolism, which are rare events, instead use the surrogates of results of coagulation tests or levels of sex hormone-binding globulin. Clearly, caution must be exercised when interpreting studies that use surrogate outcomes. As the clinician, you must recognize that a change in a biologic or physical measurement may not be clinically relevant. Some judgment is required about causal pathways: The less that is known about the causal pathway of a disease, the less confident one should be in any surrogate outcome.
Finally, clinicians also must recognize that a valid surrogate for one treatment may not be valid for another treatment or another population.5 For example, ovulation inhibition would be an appropriate surrogate endpoint for contraceptive efficacy for a method that reliably prevents ovulation; however, this would not be a good surrogate outcome to evaluate the progestin-only pill, which fails to inhibit ovulation completely and yet is highly effective in contraceptive trials.
Avoiding pitfalls with subgroup analyses
It is common, particularly in large RCTs, to evaluate treatment effects for a specific endpoint in a subgroup of patients included in the trial. The goal is to determine whether the findings of the larger study apply more or less to a specific patient (who may differ from the total population by some important characteristic, such as age, weight, parity, or menopausal or smoking status). The variability in study results when stratified by these patient factors is known as heterogeneity of treatment effect, which may be quantitative or qualitative.6
In the former, one treatment is always better than the other, although by varying degrees depending on the subgroup. (For example, a stronger effect could be seen in those aged 65 and younger than in those older than 65.) In the latter, the treatment fares better than the comparator in one subgroup but worse or no different for another subgroup. In either case, the appropriate statistical tool to identify heterogeneity of treatment effect is a test for interaction between the characteristic and the treatment effect, rather than claiming heterogeneity on the basis of separate tests of treatment effects within the different subpopulations.
One problem with dividing the original population into smaller subpopulations is that the number of participants decreases—thus there is less power, or less statistical strength, to identify a treatment effect. More accurately, there is a greater likelihood of a type II error (a false negative) when these small subpopulations have too few patients to demonstrate a clinical treatment effect that actually may exist.
False positives. Paradoxically, another problem with subgroup analyses is a greater chance for false positives due to the multiple statistical testing that is performed. The original study is rarely powered appropriately to do this (see “Error rates in subgroup analyses”). According to Wang and colleagues, “It is common practice to conduct a subgroup analysis for each of several (and often many) baseline characteristics, for each of several endpoints, or for both.”7 The more subgroup analyses performed, the more likely that differences found are due to chance only. Unfortunately, in unplanned post hoc analyses, the number of tests performed is often unreported; therefore, the error rates are unknown. There are statistical methods to try and correct for this “multiplicity” problem but, ideally, only a few key subgroup analyses are performed, and they are planned a priori in the original study design. In these cases, the study’s size can be adjusted accordingly. In most instances, findings from subgroup analyses, whether positive or negative, should be considered as “hypothesis generating” and interpreted with caution.
Error rates in subgroup analyses
With “k” independent subgroups and no difference in treatments, the probability of at least one “significant” subgroup (such as a false positive) is 1 – (1-α)k.
If α = 0.05 and there are k = 10 subgroups, then 1 – (0.95)10 = 0.40. That is, if 10 subgroup analyses are performed, there is a 40% likelihood that 1 will demonstrate a “significant” difference in treatment effect, even though no difference exists.
Systematic reviews: What, why, and how?
Systematic reviews aim to overcome the deficiencies of single studies in a comprehensive and unbiased manner. They critically evaluate, summarize, and, when possible, combine all available studies addressing a given topic. By comparing a range of relevant interventions across populations and settings, systematic reviews may be more generalizable than single studies. Meta-analysis, or quantitatively combining study results, increases sample size and usually provides more precise estimates of effect sizes than the single studies. Critical appraisal of the combined studies can highlight methodologic and other concerns about the body of evidence to assess the overall confidence in the included studies.
A systematic review, like a well-conducted RCT, has a protocol that lays out the scope of the review and defines a priori criteria and analytic plans—all with the goal of minimizing bias. It starts with a well-formulated research question, explicitly defining the PICO elements—population, interventions, comparators, outcomes—in addition to the setting and study designs of interest.8 Based on these eligibility criteria, several sources of evidence (such as electronic databases and reference lists) are searched to find all potentially eligible studies.
Typically, several thousand citations are found that must be matched against the eligibility criteria. Potentially eligible studies are then rescreened in full text to further scrutinize their eligibility. The goal is to be highly sensitive to avoid missing relevant studies—even at the time cost of screening many articles. The individual study designs (including the study eligibility criteria, interventions, outcomes, and analytic methods) and the results for all outcomes of interest are extracted from each study.
For most systematic reviews, researchers also will assess the quality, or risk of bias, of each study for each outcome.9,10 Study data are summarized across all included studies, with study results meta-analyzed and reasons for heterogeneity across studies explored. Several consensus statements detail the proper methodology to conduct and report a systematic review.11,12 Ultimately, the review’s conclusions are based on analyses of all available evidence. By contrast, narrative reviews typically start with a conclusion and then select evidence to support that conclusion, and are therefore more likely to be biased.13
As noted, systematic reviews often include meta-analysis, which may allow an exploration of some reasons for study heterogeneity. The meta-analysis is usually presented graphically in a forest plot, which displays point estimates for each study with their associated 95% confidence intervals and a description of each study.14 In a forest plot, one can see the estimate and precision of each study, assess the heterogeneity of results across studies, and compare individual studies to each other and to the overall summary estimate.
Systematic reviews should be read as critically as primary studies. Some important questions you should consider are:
- Did the review address the populations, interventions, comparators, outcomes, and settings relevant to your practice?
- Have studies been included in a nonbiased manner, and is the described body of evidence likely to be complete?
- Did the study authors evaluate and summarize the underlying risks of bias of the studies?
- Did the researchers avoid combining studies that are too different from each other to allow a coherent interpretation of the summary results?
- Did the researchers attempt to explain how and why studies differed from one another?
Of note, systematic reviews and meta-analyses are subject to the same biases as all retrospective studies. Also, the systematic reviewers’ own biases—due to factors such as funding source, researchers’ agendas, or specialties—may subtly affect systematic reviews just as biases may affect an individual study. Furthermore, the confidence you have in a systematic review’s conclusions may be limited by the quality and generalizability of the underlying studies.
Assessing harms
You make the ultimate management decisions for your patient (though, of course, with her participation). The likely benefit of a specific treatment—determined in an experimental trial and refined further in a systematic review and meta-analysis—must be balanced with the risk of harms. RCTs usually do not provide the highest quality evidence of harms due to their limited sample sizes and short follow-up duration. Rather, large observational studies, case series, and case reports commonly provide these important details. Increasingly, patient registries are being created to prospectively follow patients and gather uniform safety data. By providing a true denominator, more accurate estimates of adverse event incidence are possible. However, the disadvantages of all of these modalities are 1) there usually are no comparators (that is, “How does the adverse event incidence for surgery A compare to that for surgery B?”) and 2) data usually are gleaned from medical records and not directly from patients.
As a result, these studies typically lack information on subjective harms, such as impaired sexual function. The reporting of treatment harms suffers from inconsistent and imprecise terminology, making it hard to reliably gather all reports of similar adverse events. Adverse event reporting in clinical trials is often driven by regulatory definitions and requirements instead of patient-centered definitions. In fact, there has been little work to date that assesses which adverse events or complications may be most relevant or important from the patient perspective.
Taken together, it is clear that the medical literature tends to emphasize treatment benefits (with robust methodologies and data to detect these benefits) but does not reliably or adequately assess harms. For rare events, risk estimates always will be imprecise. Nonetheless, better systematic reviews and today’s larger comparative effectiveness reviews strive to gather harms data from the multiple available sources described above.
Applying the evidence and your expertise to your patient
Now that you have identified the best valid and important evidence to support or refute a clinical decision (TABLE15), and have coupled this with your own expert knowledge and judgment in shared decision making with your patient, you must communicate to her the personalized information about outcomes, probabilities, and scientific uncertainties of her available treatment options.15 Patients, in turn, should be allowed to communicate their values and the relative importance they place on benefits and harms.16 This conversation, of course, is built on the foundation of a sound physician–patient relationship and is a part of every informed consent process.
Is this evidence applicable to my patient? A decision guide.15
- Is my patient so different from those in the study that the trial results cannot be applied?
- Is the treatment feasible in my setting?
- What are my patient’s likely benefits and harms from the therapy?
- How will my patient’s values influence the final treatment decision?
Decision tools
Increasingly, decision-aid tools are being developed to support this process. These aids must express the helpful and harmful effects of a treatment, including alternative options, in statements that are valid and concise. Furthermore, they must be intelligible to both the clinician and patient and modifiable to the patient’s values and wishes.17 Two examples of counseling aids are the Gail model of breast cancer risk prediction18 and the Framingham Coronary Heart Disease Prediction Score.19 Web-based decision aids that can be accessed in real-time in busy clinical settings also are being developed for gynecology.20
Never stop re-evaluating
The final piece of EBM is to “close the loop”—meaning to evaluate the effectiveness of applying the evidence in clinical practice. To do this, watch for clinical practice guidelines that are based on systematic reviews and the EBM approach and stay abreast of ACOG’s and other professional societies’ guideline statements. Ultimately, guidelines beget performance measures. Organizations such as the National Quality Forum are working to define these standards of performance measurement and seek feedback from individual clinicians to ensure measures are meaningful and accurate. By 2017, 9% of all Medicare payments are scheduled to be performance based.21
Conclusion
During the course of reading medical literature, stay attuned to comparative effectiveness research and recognize studies with active comparators that examine clinical questions that could impact your day-to-day practice and that can be applied to your patient population. While there is no such thing as a perfect research study, and it is rare that one trial can address any one clinician’s specific patients precisely, increasingly we are seeing better systematic reviews and meta-analyses. It is these studies that provide the high quality data for you to couple with your clinical expertise and your patients’ values and preferences to truly deliver evidence-based medicine.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t. BMJ. 1996;312(7023):71–72.
- Sackett DL, et al. Evidence-Based Medicine: How to Practice and Teach EBM. 2nd ed. Edinburgh, UK: Churchill Livingstone; 2000.
- Knottnerus JA, van Weel C, Muris JW. Evaluation of diagnostic procedures. BMJ. 2002;324(7335):477–480.
- Bossuyt PM, Reitsma JB, Bruns DE, et al; Standards for Reporting of Diagnostic Accuracy Group. The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. Ann Intern Med. 2003;138(1):W1-W12.
- Grimes DA, Schulz KF, Raymond EG. Surrogate end points in women’s health research: science, protoscience, and pseudoscience. Fertil Steril. 2010;93(6):1731–1734.
- Lagakos SW. The challenge of subgroup analyses—reporting without distorting. N Engl J Med. 2006;354(16):1667–1669.
- Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine—reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357(21):2189–2194.
- Counsell C. Formulating questions and locating primary studies for inclusion in systematic reviews. Ann Intern Med. 1997;127(5):380–387.
- Higgins JP, Altman DG, Gøtzsche PC, et al; Cochrane Statistical Methods Group. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928.
- Berkman ND, Lohr KN, Ansari M, et al. Grading the Strength of a Body of Evidence When Assessing Health Care Interventions for the Effective Health Care Program of the Agency for Healthcare Research and Quality: An Update. Methods Guide for Effectiveness and Comparative Effectiveness Reviews. Rockville (MD)2008. AHRQ Methods for Effective Health Care. 2013 Nov 18.
- Liberati A1, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- Institute of Medicine of the National Academies. Finding What Works in Health Care: Standards for Systematic Reviews. iom.edu/Reports/2011/Finding-What-Works-in-Health-Care-Standards-for-Systematic-Reviews.aspx. Published March 23, 2011. Accessed March 20, 2015.
- Mulrow CD. The medical review article: state of the science. Ann Intern Med. 1987;106(3):485–488.
- Lewis S, Clarke M. Forest plots: trying to see the wood and the trees. BMJ. 2001;322(7300):1479–1480.
- Glasziou P, Guyatt GH, Dans AL, Dans LF, Straus S, Sackett DL. Applying the results of trials and systematic reviews to individual patients. ACP J Club. 1998;129(3):A15–A16.
- What is shared decision making? Informed Medical Decisions Foundation Web site. http://www.informed medicaldecisions.org/what-is-shared-decision-making. Published 2015. Accessed January 23, 2015.
- Straus SE, Sackett DL. Applying evidence to the individual patient. Ann Oncol. 1999;10(1):29–32.
- Rockhill B, Spiegelman D, Byrne C, Hunter DJ, Colditz GA. Validation of the Gail et al. model of breast cancer risk prediction and implications for chemoprevention. J Natl Cancer Inst. 2001;93(5):358–366.
- D’Agostino RB Sr, Grundy S, Sullivan LM, Wilson P; CHD Risk Prediction Group. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA. 2001;286(2):180–187.
- Jelovsek JE, Chagin K, Brubaker L, et al; Pelvic Floor Disorders Network. A model for predicting the risk of de novo stress urinary incontinence in women undergoing pelvic organ prolapse surgery. Obstet Gynecol. 2014;123(2 Pt 1):279–287.
- National Quality Forum. What we do. National Quality Forum Web sight. http://www.qualityforum.org/what_we_do.aspx. Published 2015. Accessed January 29, 2015.
- Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t. BMJ. 1996;312(7023):71–72.
- Sackett DL, et al. Evidence-Based Medicine: How to Practice and Teach EBM. 2nd ed. Edinburgh, UK: Churchill Livingstone; 2000.
- Knottnerus JA, van Weel C, Muris JW. Evaluation of diagnostic procedures. BMJ. 2002;324(7335):477–480.
- Bossuyt PM, Reitsma JB, Bruns DE, et al; Standards for Reporting of Diagnostic Accuracy Group. The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. Ann Intern Med. 2003;138(1):W1-W12.
- Grimes DA, Schulz KF, Raymond EG. Surrogate end points in women’s health research: science, protoscience, and pseudoscience. Fertil Steril. 2010;93(6):1731–1734.
- Lagakos SW. The challenge of subgroup analyses—reporting without distorting. N Engl J Med. 2006;354(16):1667–1669.
- Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine—reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357(21):2189–2194.
- Counsell C. Formulating questions and locating primary studies for inclusion in systematic reviews. Ann Intern Med. 1997;127(5):380–387.
- Higgins JP, Altman DG, Gøtzsche PC, et al; Cochrane Statistical Methods Group. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928.
- Berkman ND, Lohr KN, Ansari M, et al. Grading the Strength of a Body of Evidence When Assessing Health Care Interventions for the Effective Health Care Program of the Agency for Healthcare Research and Quality: An Update. Methods Guide for Effectiveness and Comparative Effectiveness Reviews. Rockville (MD)2008. AHRQ Methods for Effective Health Care. 2013 Nov 18.
- Liberati A1, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- Institute of Medicine of the National Academies. Finding What Works in Health Care: Standards for Systematic Reviews. iom.edu/Reports/2011/Finding-What-Works-in-Health-Care-Standards-for-Systematic-Reviews.aspx. Published March 23, 2011. Accessed March 20, 2015.
- Mulrow CD. The medical review article: state of the science. Ann Intern Med. 1987;106(3):485–488.
- Lewis S, Clarke M. Forest plots: trying to see the wood and the trees. BMJ. 2001;322(7300):1479–1480.
- Glasziou P, Guyatt GH, Dans AL, Dans LF, Straus S, Sackett DL. Applying the results of trials and systematic reviews to individual patients. ACP J Club. 1998;129(3):A15–A16.
- What is shared decision making? Informed Medical Decisions Foundation Web site. http://www.informed medicaldecisions.org/what-is-shared-decision-making. Published 2015. Accessed January 23, 2015.
- Straus SE, Sackett DL. Applying evidence to the individual patient. Ann Oncol. 1999;10(1):29–32.
- Rockhill B, Spiegelman D, Byrne C, Hunter DJ, Colditz GA. Validation of the Gail et al. model of breast cancer risk prediction and implications for chemoprevention. J Natl Cancer Inst. 2001;93(5):358–366.
- D’Agostino RB Sr, Grundy S, Sullivan LM, Wilson P; CHD Risk Prediction Group. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA. 2001;286(2):180–187.
- Jelovsek JE, Chagin K, Brubaker L, et al; Pelvic Floor Disorders Network. A model for predicting the risk of de novo stress urinary incontinence in women undergoing pelvic organ prolapse surgery. Obstet Gynecol. 2014;123(2 Pt 1):279–287.
- National Quality Forum. What we do. National Quality Forum Web sight. http://www.qualityforum.org/what_we_do.aspx. Published 2015. Accessed January 29, 2015.
In thIS article
- RCTs: The good, the bad, and the ugly
- Systematic reviews: What, why, and how?
- Assessing harms
- Applying the evidence and your expertise to your patient