User login
The history and findings in this case are suggestive of leukemic non-nodal mantle cell lymphoma (MCL).
MCL is a rare mature B-cell neoplasm characterized by t(11;14) (q13;q32) and cyclin D1 overexpression in more than 95% of cases. It accounts for approximately 5%-7% of all lymphomas, with an annual incidence of one case per 200,000 people. In North America and Europe, the incidence of MCL is like that of noncutaneous peripheral T-cell lymphomas. MCL occurs more frequently in men than in women (3:1), and the median age at diagnosis ranges from ages 60-70 years.
In recent years, MCL has been categorized into two major subgroups that have distinct clinical presentation and molecular features: nodal MCL and leukemic non-nodal MCL. Nodal MCL is a common variant with an aggressive disease course. Unmutated IGHV gene rearrangement, SOX11 overexpression, a higher degree of genomic instability (eg, ATM, CDKN2A, chromatin modifier mutations), and other oncogenic mutations and epigenetic modifications are seen in patients with this variant.
Leukemic non-nodal MCL is seen in 10%-20% of patients with MCL. Patients frequently present with lymphocytosis and splenomegaly. In most cases, it is associated with an indolent disease course and superior outcome. This subtype is largely IGHV mutated and mostly SOX11-negative, with positive expression of CD200, peripheral blood, bone marrow, and splenic involvement, low tumor burden, and a low Ki-67 index.
Recognition of the leukemic non-nodal MCL immunophenotype enables it to be differentiated from other CD5-positive B-cell cancers, particularly classical MCL and chronic lymphocytic leukemia (CLL). The overexpression of cyclin D1, the presence of the t(11;14) translocation, and the absence of chromosomal markers typically present in CLL differentiate leukemic non-nodal MCL from CLL. Moreover, CLL has high expression of CD23 and is negative for SOX11 and CD200.
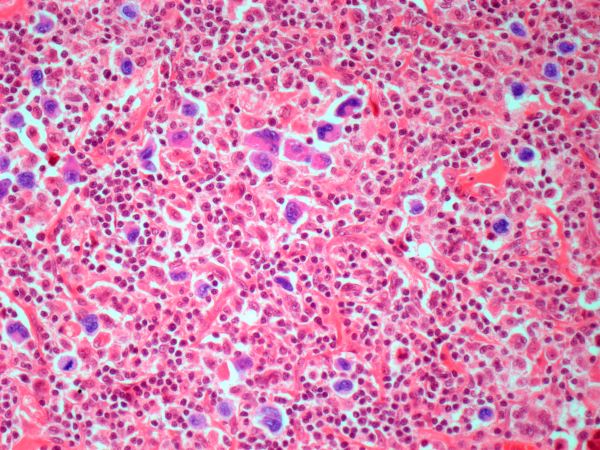
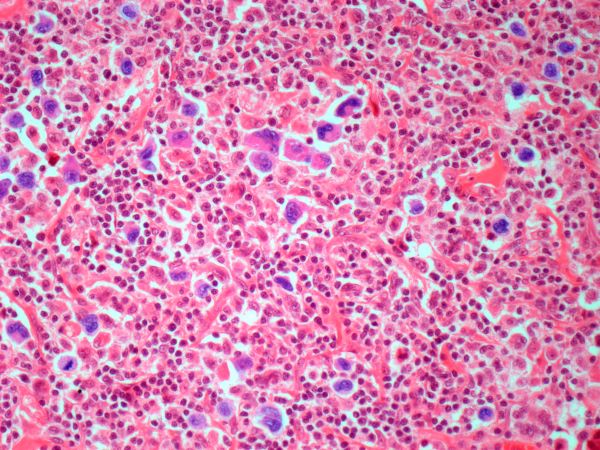
Pathologic features of MCL include small- to medium-size lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Observed growth patterns include diffuse, nodular (more vague and less discrete than that found in follicular lymphomas), mantle-zone lymphoma with expansion of mantle zones, and in situ mantle-cell neoplasia [typical cells with the characteristic t(11;14) translocation, scattered in the mantle zone of otherwise normal-appearing lymph nodes]. Cytologic subtypes include classic MCL, the blastoid subtype (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic subtype (cells of variable sizes, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli).
MCL is a challenging disease to treat. Despite treatment advances, it is largely incurable, with a median overall survival of 1.8-9.4 years, depending on whether it is aggressive or indolent MCL. The aggressiveness of the disease, the patient's performance status, age, and mantle cell international prognostic index score should all be considered when selecting treatment because there is no standard curative treatment.
According to the 2023 guidelines from the National Comprehensive Cancer Network (NCCN), for patients with indolent disease (eg, IGHV mutated and mostly SOX11-negative with leukemic and non-nodal presentation), observation is reasonable when patients are asymptomatic and have no indications for treatment. For patients with symptomatic disease or other indications for treatment, induction therapy with aggressive regimens is recommended when patients do not have a TP53 mutation. The optimum approach for patients with TP53 mutation is not yet known; induction therapy followed by high-dose therapy with autologous stem cell transplant or less aggressive regimens could be an option for these patients.
Treatment options for relapsed/refractory MCL include radiotherapy; traditional chemotherapy regimens, with or without rituximab; and newer targeted therapies. These include Bruton tyrosine kinase inhibitors (ibrutinib, zanubrutinib, acalabrutinib, pirtobrutinib), lenalidomide, bortezomib, the mammalian target of rapamycin inhibitors temsirolimus and everolimus, the phosphatidylinositol 3–kinase inhibitors idelalisib and umbralisib, and the B-cell lymphoma 2 inhibitor venetoclax. These agents are frequently administered in combination with rituximab or another anti-CD20 antibody.
For comprehensive guidance on the treatment of MCL, consult the complete NCCN guidelines.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of leukemic non-nodal mantle cell lymphoma (MCL).
MCL is a rare mature B-cell neoplasm characterized by t(11;14) (q13;q32) and cyclin D1 overexpression in more than 95% of cases. It accounts for approximately 5%-7% of all lymphomas, with an annual incidence of one case per 200,000 people. In North America and Europe, the incidence of MCL is like that of noncutaneous peripheral T-cell lymphomas. MCL occurs more frequently in men than in women (3:1), and the median age at diagnosis ranges from ages 60-70 years.
In recent years, MCL has been categorized into two major subgroups that have distinct clinical presentation and molecular features: nodal MCL and leukemic non-nodal MCL. Nodal MCL is a common variant with an aggressive disease course. Unmutated IGHV gene rearrangement, SOX11 overexpression, a higher degree of genomic instability (eg, ATM, CDKN2A, chromatin modifier mutations), and other oncogenic mutations and epigenetic modifications are seen in patients with this variant.
Leukemic non-nodal MCL is seen in 10%-20% of patients with MCL. Patients frequently present with lymphocytosis and splenomegaly. In most cases, it is associated with an indolent disease course and superior outcome. This subtype is largely IGHV mutated and mostly SOX11-negative, with positive expression of CD200, peripheral blood, bone marrow, and splenic involvement, low tumor burden, and a low Ki-67 index.
Recognition of the leukemic non-nodal MCL immunophenotype enables it to be differentiated from other CD5-positive B-cell cancers, particularly classical MCL and chronic lymphocytic leukemia (CLL). The overexpression of cyclin D1, the presence of the t(11;14) translocation, and the absence of chromosomal markers typically present in CLL differentiate leukemic non-nodal MCL from CLL. Moreover, CLL has high expression of CD23 and is negative for SOX11 and CD200.
Pathologic features of MCL include small- to medium-size lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Observed growth patterns include diffuse, nodular (more vague and less discrete than that found in follicular lymphomas), mantle-zone lymphoma with expansion of mantle zones, and in situ mantle-cell neoplasia [typical cells with the characteristic t(11;14) translocation, scattered in the mantle zone of otherwise normal-appearing lymph nodes]. Cytologic subtypes include classic MCL, the blastoid subtype (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic subtype (cells of variable sizes, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli).
MCL is a challenging disease to treat. Despite treatment advances, it is largely incurable, with a median overall survival of 1.8-9.4 years, depending on whether it is aggressive or indolent MCL. The aggressiveness of the disease, the patient's performance status, age, and mantle cell international prognostic index score should all be considered when selecting treatment because there is no standard curative treatment.
According to the 2023 guidelines from the National Comprehensive Cancer Network (NCCN), for patients with indolent disease (eg, IGHV mutated and mostly SOX11-negative with leukemic and non-nodal presentation), observation is reasonable when patients are asymptomatic and have no indications for treatment. For patients with symptomatic disease or other indications for treatment, induction therapy with aggressive regimens is recommended when patients do not have a TP53 mutation. The optimum approach for patients with TP53 mutation is not yet known; induction therapy followed by high-dose therapy with autologous stem cell transplant or less aggressive regimens could be an option for these patients.
Treatment options for relapsed/refractory MCL include radiotherapy; traditional chemotherapy regimens, with or without rituximab; and newer targeted therapies. These include Bruton tyrosine kinase inhibitors (ibrutinib, zanubrutinib, acalabrutinib, pirtobrutinib), lenalidomide, bortezomib, the mammalian target of rapamycin inhibitors temsirolimus and everolimus, the phosphatidylinositol 3–kinase inhibitors idelalisib and umbralisib, and the B-cell lymphoma 2 inhibitor venetoclax. These agents are frequently administered in combination with rituximab or another anti-CD20 antibody.
For comprehensive guidance on the treatment of MCL, consult the complete NCCN guidelines.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The history and findings in this case are suggestive of leukemic non-nodal mantle cell lymphoma (MCL).
MCL is a rare mature B-cell neoplasm characterized by t(11;14) (q13;q32) and cyclin D1 overexpression in more than 95% of cases. It accounts for approximately 5%-7% of all lymphomas, with an annual incidence of one case per 200,000 people. In North America and Europe, the incidence of MCL is like that of noncutaneous peripheral T-cell lymphomas. MCL occurs more frequently in men than in women (3:1), and the median age at diagnosis ranges from ages 60-70 years.
In recent years, MCL has been categorized into two major subgroups that have distinct clinical presentation and molecular features: nodal MCL and leukemic non-nodal MCL. Nodal MCL is a common variant with an aggressive disease course. Unmutated IGHV gene rearrangement, SOX11 overexpression, a higher degree of genomic instability (eg, ATM, CDKN2A, chromatin modifier mutations), and other oncogenic mutations and epigenetic modifications are seen in patients with this variant.
Leukemic non-nodal MCL is seen in 10%-20% of patients with MCL. Patients frequently present with lymphocytosis and splenomegaly. In most cases, it is associated with an indolent disease course and superior outcome. This subtype is largely IGHV mutated and mostly SOX11-negative, with positive expression of CD200, peripheral blood, bone marrow, and splenic involvement, low tumor burden, and a low Ki-67 index.
Recognition of the leukemic non-nodal MCL immunophenotype enables it to be differentiated from other CD5-positive B-cell cancers, particularly classical MCL and chronic lymphocytic leukemia (CLL). The overexpression of cyclin D1, the presence of the t(11;14) translocation, and the absence of chromosomal markers typically present in CLL differentiate leukemic non-nodal MCL from CLL. Moreover, CLL has high expression of CD23 and is negative for SOX11 and CD200.
Pathologic features of MCL include small- to medium-size lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Observed growth patterns include diffuse, nodular (more vague and less discrete than that found in follicular lymphomas), mantle-zone lymphoma with expansion of mantle zones, and in situ mantle-cell neoplasia [typical cells with the characteristic t(11;14) translocation, scattered in the mantle zone of otherwise normal-appearing lymph nodes]. Cytologic subtypes include classic MCL, the blastoid subtype (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic subtype (cells of variable sizes, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli).
MCL is a challenging disease to treat. Despite treatment advances, it is largely incurable, with a median overall survival of 1.8-9.4 years, depending on whether it is aggressive or indolent MCL. The aggressiveness of the disease, the patient's performance status, age, and mantle cell international prognostic index score should all be considered when selecting treatment because there is no standard curative treatment.
According to the 2023 guidelines from the National Comprehensive Cancer Network (NCCN), for patients with indolent disease (eg, IGHV mutated and mostly SOX11-negative with leukemic and non-nodal presentation), observation is reasonable when patients are asymptomatic and have no indications for treatment. For patients with symptomatic disease or other indications for treatment, induction therapy with aggressive regimens is recommended when patients do not have a TP53 mutation. The optimum approach for patients with TP53 mutation is not yet known; induction therapy followed by high-dose therapy with autologous stem cell transplant or less aggressive regimens could be an option for these patients.
Treatment options for relapsed/refractory MCL include radiotherapy; traditional chemotherapy regimens, with or without rituximab; and newer targeted therapies. These include Bruton tyrosine kinase inhibitors (ibrutinib, zanubrutinib, acalabrutinib, pirtobrutinib), lenalidomide, bortezomib, the mammalian target of rapamycin inhibitors temsirolimus and everolimus, the phosphatidylinositol 3–kinase inhibitors idelalisib and umbralisib, and the B-cell lymphoma 2 inhibitor venetoclax. These agents are frequently administered in combination with rituximab or another anti-CD20 antibody.
For comprehensive guidance on the treatment of MCL, consult the complete NCCN guidelines.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 67-year-old White man presents for his annual physical examination. The patient is a physical therapist and reports regular exercise and adherence to a healthy diet. His previous medical history is unremarkable. There is a family history of non-Hodgkin lymphoma (paternal uncle). The patient has no complaints or concerns about his health.
Physical examination reveals non-tender abdominal distention and splenomegaly. Pertinent laboratory findings include hemoglobin = 10/g/dL; red blood cell = 3.28 M/mcL; mean corpuscular volume = 54.2 fL, hematocrit = 34%; and absolute lymphocyte count = 4820/µL.
Flow cytometry showed high positivity for CD5, no expression of SOX11, low expression of CD23 and CD200, and overexpression of cyclin D1. A bone marrow biopsy is performed and show an abnormal B-lymphoid infiltrate. Fluorescence in situ hybridization analysis revealed t(11;14)(q13;q32) and mutated IGHV. A blood smear showed abnormal mononuclear cells and atypical lymphocytes.