User login
Chorea is characterized by continuous, random, brief, involuntary muscle contractions that result from a variety of causes.1 These involuntary movements are nonstereotyped and irregular. Although choreic disorders are among the most common involuntary movement disorders, their diagnosis and treatment present several important challenges, including identifying and removing the cause if possible, controlling and preventing motor symptoms, and managing neuropsychologic complications.1 This article provides an overview of the diagnosis and treatment of choreic disorders, using Sydenham chorea to illustrate the management of autoimmune choreas and Huntington disease as the model for the management of heritable choreas.
Management of choreic disorders begins with a first-pass diagnosis and the use of symptomatic therapies. Even if this first pass yields no firm diagnosis, it at least rules out causes that have the most practical significance. A subsequent second-pass evaluation can be undertaken to look for rarer causes. Symptomatic therapies are continued throughout the diagnostic period. More specific therapies can be administered if an etiologic or pathogenic mechanism is determined (eg, postinfectious, autoimmune, metabolic).
CHOREIC DISORDERS: A PRACTICAL DIAGNOSTIC APPROACH
In general, choreic disorders may be subdivided into six categories:
1. Heredodegenerative disorders, such as Huntington disease and other genetically heterogeneous choreas, include Huntington disease–like 2 (HDL2) and benign hereditary chorea.1 Sporadic cases include those of unknown paternity; X-linked disorders (eg, McLeod syndrome); and autosomal-recessive disorders such as chorea-acanthocytosis, which is characterized by chorea, dystonia with prominent orofacial involvement, self-mutilation, myopathy, and neuropathy.2
2. Drug-induced choreas include neurolepticinduced tardive dyskinesia and nontardive hyperkinetic drug-related choreas, the most common of which is levodopa-induced dyskinesias.3 Tardive drug-induced choreas may occur while using the culprit drug, while tapering the drug, or after it has been discontinued. The culprit drugs are represented by dopamine-receptor blockers and include the first-generation neuroleptics (eg, phenothiazines, haloperidol), antidepressants (lox-apine), and gastrointestinal agents (metoclopramide, prochlorperazine). Drug-induced choreas are possible with a wide range of pharmacologic agents, including antiparkinsonian drugs (eg, levodopa, dopamine agonists, anticholinergics), sympathomimetics (eg, amphetamines, cocaine), anticonvulsants, calcium channel blockers, and oral contraceptives.1
3. Autoimmune choreas include Sydenham chorea, systemic lupus erythematosus, and antiphospholipid antibody syndromes. The latter encompass lupus anticoagulant and anticardiolipin antibodies.
4. Metabolic choreas are most often associated with hyperthyroidism, although case reports have described choreas in patients with vitamin B12 deficiency. A variety of hereditary metabolic diseases are also included in this category.
5. Vascular choreas include polycythemia vera and cerebrovascular accidents, the latter frequently presenting as hemiballismus. Polycythemia vera is associated with a high incidence of neurologic symptoms, including a reported incidence of chorea of 0.5% to 5%,4 and should be considered as a potential cause of chorea.
6. Other choreic disorders include a variety of entities such as rare paraneoplastic disorder/syndrome, and posttraumatic and postanoxic presentations.
The first-pass diagnostic approach includes a family history, drug history, and brain magnetic resonance imaging to identify potential structural causes of chorea. Genetic testing for Huntington disease or other choreic disorders may also be performed, although it is essential to consider the potential implications of a positive test result. Intensive pretest and posttest counseling is important both for the patient and for currently asymptomatic family members who may also be affected.1
Other testing includes:
- Complete blood count
- Creatine phosphokinase
- Peripheral smear for acanthocytes
- Comprehensive metabolic panel
- Ceruloplasmin level
- Measurement of thyroxine (T4) and triiodothyronine (T3)
- B12 tests
- Antinuclear antibody sedimentation rate
- Lupus anticoagulant-anticardiolipin antibodies
- Antistreptolysin O (ASO) titer
- Anti-DNase-B titer.
GENERAL CONSIDERATIONS FOR THE TREATMENT OF CHOREIC DISORDERS
In some cases, choreic disorders have a treatable underlying etiology, such as thyroid disease or vitamin B12 deficiency. Tardive syndromes may require treatment beyond drug discontinuation, including use of dopamine depleters for the classic tardive dyskinetic syndromes and anticholinergics for the tardive dystonic syndromes. Levodopa dyskinesia may be treated using amantadine, clozapine, or deep brain stimulation.5,6 The treatment of patients with autoimmune choreas is not well defined. It may include anticoagulation in patients with positive anticardiolipin antibodies to prevent venous or arterial thromboembolism,7 but the risk of arterial thromboembolism is uncertain, and it is unclear whether chorea is truly a harbinger of vascular events.
A negative ASO titer does not exclude Sydenham chorea, a result of childhood infection with group-A beta-hemolytic streptococcus, and antibiotics should be considered in the appropriate context. Some researchers have argued that immune responses associated with acute infections may result in autoimmune neuropsychiatric symptoms. In pediatric patients, this has been referred to as pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS).8
A related phenomenon has been proposed as a potential mechanism of some types of chorea, although the relationship between acute infection and chorea is controversial. Patients with elevated ASO titer or anti-DNase-B titers may be candidates for antibiotics. By 6 weeks after the onset of infection, these titers will fall and a diagnosis of Sydenham chorea can be postulated or based exclusively on clinical judgment.
SYDENHAM CHOREA
Unique to Sydenham chorea is the use of penicillin as prophylaxis. Other than that, the management of Sydenham chorea exemplifies the management approach for the larger category of autoimmune choreic disorders. Pathogenic-based treatment options include immune modulation with cortico steroids, intravenous immunoglobulin (IVIG), and plasma exchange; all treatments must be administered in the appropriate clinical context.
One double-blind clinical trial examined the effectiveness of corticosteroid treatment in children with Sydenham chorea randomly assigned to receive either prednisone (n = 22) or placebo (n = 15).9 Prednisone was administered at a dose of 2 mg/kg/day for 4 weeks, followed by gradual tapering and discontinuation. The median time to remission of chorea was significantly lower for patients in the prednisone group (54.3 days) compared with those in the placebo group (119.9 days; P < .001). Patients in the prednisone group also exhibited significantly better scores on a chorea intensity rating scale at 8 weeks and 12 weeks (P < .001). Potential limitations of this approach include relapse of chorea symptoms and corticosteroid-related adverse events (eg, Cushing syndrome, hypertension).
A second study compared the effectiveness of three modalities: IVIG at a dose of 1 g/kg/day for 2 days (n = 4), plasma exchange (n = 8), and prednisone (n = 6).10 Although differences between treatment groups were not statistically significant, the authors noted that the clinical improvement in chorea symptoms tended to be greater for patients who received IVIG or plasma exchange than for those who received prednisone. Mean chorea scores improved from baseline by 72% for the IVIG group, 50% for the plasma exchange group, and 29% for the prednisone group.
After etiology-dependent treatments have been considered, several other options may be effective regardless of the specific etiology. These include symptomatic treatments such as haloperidol, atypical neuroleptics, and amantadine.11 Antiepileptic medications or benzodiazepines may also help to control symptoms, although less information is available about the use of these agents for the treatment of Sydenham chorea. Tetrabenazine may be considered for patients who will require long-term treatment.
HUNTINGTON DISEASE
Pharmacotherapy of Huntington disease may be unnecessary if symptoms are mild or not bothersome. Symptomatic treatment options include tetrabenazine, amantadine, and either first-generation neuroleptics (eg, haloperidol) or second-generation atypical neuroleptics (eg, olanzapine, quetiapine, risperidone, ziprasidone).
Treating choreas with tetrabenazine or amantadine
Considerable recent attention has focused on the efficacy and safety of tetrabenazine for the treatment of Huntington disease and other choreic disorders. Tetrabenazine is a central monoamine depleter that reversibly binds to the type-2 vesicular monoamine transporter.12 The TETRA-HD study examined the efficacy and safety of tetrabenazine for the short- and long-term control of Huntington disease.12 An initial study compared tetrabenazine with placebo in 75 patients who were treated for up to 13 weeks. In an extension study, all patients received individualized tetrabenazine doses for up to 80 weeks.
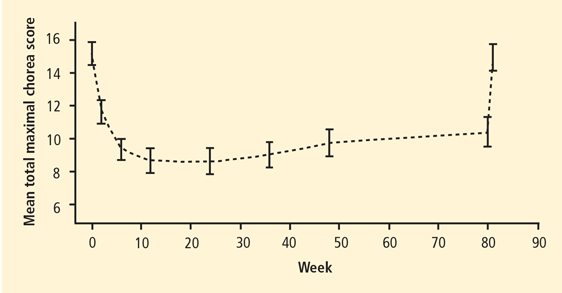
The mean total maximal chorea (TMC) scores from the Unified Huntington Disease Rating Scale (UHDRS) decreased markedly during the first 10 weeks of tetrabenazine treatment, remained lower than baseline throughout 80 weeks, and then returned to baseline levels after tetrabenazine discontinuation (Figure 1). At week 80, the mean TMC score was reduced by 4.6 UHDRS units compared with baseline (P < .001) The long-term extension phase was completed by 45 of 75 patients. Treatment-related adverse events that prompted discontinuation included depression, delusions, and vocal tics. The most commonly reported adverse events included sedation or somnolence (n = 18), depressed mood (n = 17), anxiety (n = 13), insomnia (n = 10), and akathisia (n = 9). Scores of parkinsonism and dysphagia increased significantly from baseline over the 80-week study.
Amantadine is an option for patients who cannot tolerate tetrabenazine. A double-blind, placebo-controlled study performed by researchers at the National Institutes of Health (NIH) examined the efficacy and safety of amantadine in 24 patients with Huntington disease.13 Patients were treated with oral amantadine 400 mg/day or placebo for 2 weeks, and were then crossed over to the other treatment. Amantadine was associated with a median reduction in extremity chorea score at rest of 36% from baseline (P = .04), versus 0% improvement with placebo. The mean improvement with amantadine was 56% for the 10 patients with the highest drug plasma levels.
Improvement in chorea scores from baseline for amantadine compared with placebo was rated with four different methods: (1) maximal chorea severity measured from video recordings; (2) maximal chorea severity measured by live raters; (3), chorea severity at rest measured from video recordings; and (4) extremity chorea at rest measured from video recordings. Amantadine was superior to placebo according to all four rating methods. Treatment was generally safe and well tolerated, and no consistent changes in cognitive function were noted with amantadine therapy.
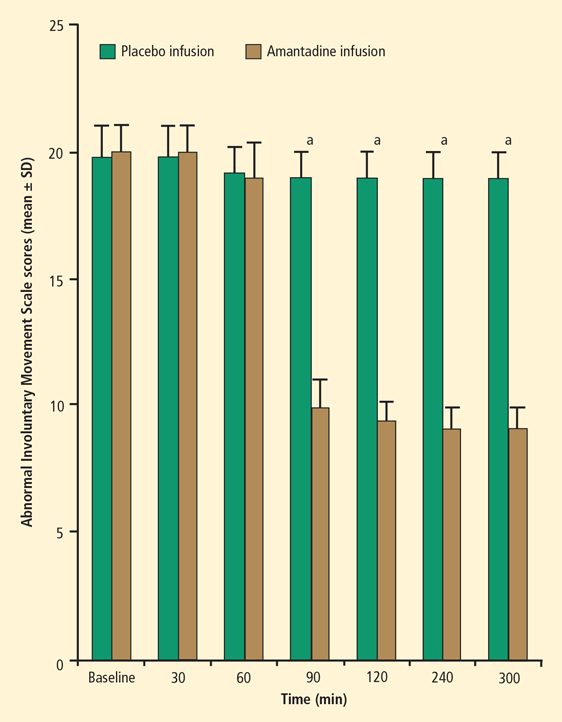
Managing nonmotor complications
In addition to addressing chorea, it is also important to manage nonmotor complications of Huntington disease, including cognition, mood, and thought disorders. Rivastigmine was assessed for the treatment of motor symptoms, functional disability, and cognitive impairment associated with Huntington disease in an open-label study of 18 patients; 11 received rivastigmine 6 mg/day and 7 control patients did not.15 Motor and cognitive function were assessed for up to 2 years by raters who were blinded to treatment assignment. Ratings on a global motor performance scale were significantly better for patients who received rivastigmine than for control subjects. Rivastigmine treatment was also associated with trends toward improvements in functional disability and cognitive impairment, although these differences were not statistically significant.
A small open-label study examined the effects of donepezil for movement and cognitive symptoms associated with Huntington disease.16 Donepezil did not significantly improve cognitive symptoms, although the study enrolled only eight patients. All patients tolerated oral donepezil at a dose of 5 mg/day, but four patients withdrew from the study when the dose was increased to 10 mg/day. In two patients, chorea worsened and falls increased, moderate to severe diarrhea developed in three patients, and one patient reported anxiety and irritability.
Depression is another common complication of Huntington disease. The incidence of depression among patients with Huntington disease is approximately 40%, and the risk of suicide is at least eightfold greater than that among the general population.17 Treatment must be guided by clinical judgment. Selective serotonin reuptake inhibitor antidepressants have been recommended. Other options to manage depression include mirtazapine, monoamine oxidase inhibitors, or electroshock therapy. Mood-stabilizing agents (eg, carbamazepine, lamotrigine, valproate) may also be indicated in helping with impulse control. Haloperidol and second-generation antipsychotics are used for the treatment of a broad range of psychiatric conditions, many of which may overlap with Huntington disease, including schizophrenia and schizophreniform disorder, schizoaffective disorder, bipolar disorder, dementia, and disruptive behavior.18 The risk of tardive dyskinesia may be as much as fivefold lower with second-generation antipsychotics.18 Many patients with Huntington disease require treatment for aggression. A variety of approaches are available, including behavior modification, the antidepressant sertraline, buspirone, antipsychotic agents (eg, risperidone, olanzapine), propranolol, and lithium (combined with haloperidol).
Long-term care considerations
As a consequence of the diverse clinical manifestations of choreic disorders in movement, function, mood, and cognition, the treatment of Huntington disease requires a multidisciplinary approach that involves a number of different health care specialties across the long-term course of the disorder. Members of the Huntington disease treatment team may include neurologists, psychiatrists, nurses, social workers, geneticists, physical therapists, occupational therapists, speech therapists, dietitians, and other supporting groups or professional societies. The clinical manifestations of Huntington disease may evolve over time, as symptoms such as bradykinesia, dystonia, rigidity, cognitive decline, and gait instability become more significant.19 As a result, optimal management strategies for patients with Huntington disease may change significantly across the long-term course of the disease. During the early course of the disease, the typical clinical presentation is largely hyperkinesis, irritability, and distractibility. These patients will require initiation of drug therapy and linkage to sources of support. In the later stages of the disease, the presentation shifts to a more hypokinetic and apathetic profile, and patients are more likely to require drug regimen review and modification, nursing home placement, and palliative care services.19
Another important concern in Huntington disease treatment is care of the caregiver. Surveys show that the key concerns of caregivers include the expertise of the health care professionals who are treating the patient and the availability of sufficient services in the community.20 Several resources are available for Huntington disease caregivers, including local support groups, the Huntington’s Disease Society of America, Q Foundation, and the Huntington Study Group. The Lundbeck pharmaceutical company operates a patient assistance program (LundbeckShare. com) as well as an information center that can be accessed toll free at (888)457-4273. Approximately 90% of patients who request copayment assistance qualify for aid, regardless of the type of insurance they carry.
SUMMARY AND CONCLUSIONS
The approach to a patient with chorea starts with a search for specifically treatable etiologies. Autoimmune, metabolic, and vascular causes should be sought first and treated. The symptomatic treatment of all choreas is based on the model described here for Huntington disease, and includes attention to cognitive, psychiatric, and social support issues. The recommended approach is multidisciplinary, with a change in the mix of services as the disease progresses. It is also important to recognize the burden of Huntington disease on the caregiver and consider steps to make this burden more manageable.
- Cardoso F, Seppi K, Mair KJ, Wenning GK, Poewe W. Seminar on choreas. Lancet Neurol 2006; 5:589–602.
- Schneider SA, Walker RH, Bhatia KP. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 2007; 3:517–525.
- Zesiewicz TA, Sullivan KL. Drug-induced hyperkinetic movement disorders by nonneuroleptic agents. Handb Clin Neurol 2011; 100:347–363.
- Margolis RL, Ross CA. Diagnosis of Huntington disease. Clin Chem 2003; 49:1726–1732.
- Rascol O. The pharmacological therapeutic management of levodopa-induced dyskinesias in patients with Parkinson’s disease. J Neurol 2000; 247 (suppl 2):II51–II57.
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord 2007; 22:1379–1389.
- Lim W, Crowther MA, Eifelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Mink J, Kurlan R. Acute postinfectious movement and psychiatric disorders in children and adolescents. J Child Neurol 2011; 26:214–217.
- Paz JA, Silva CA, Marques-Dias MJ. Randomized double-blind study with prednisone in Sydenham’s chorea. Pediatr Neurol 2006; 34:264–269.
- Garvey MA, Snider LA, Leitman SF, Werden R, Swedo SE. Treatment of Sydenham’s chorea with intravenous immunoglobulin, plasma exchange, or prednisone. J Child Neurol 2005; 20:424–429.
- Walker KG, Wilmshurst JM. An update on the treatment of Sydenham’s chorea: the evidence for established and evolving interventions. Ther Adv Neurol Disord 2010; 3:301–309.
- Frank S. Tetrabenazine as anti-chorea therapy in Huntington disease: an open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol 2009; 9:62.
- Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002; 59:694–699.
- Lucetti C, Del Dotto P, Gamabaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology 2003; 60:1995–1997.
- de Tommaso M, Difruscolo O, Scirruicchio V, Specchio N, Livrea P. Two years’ follow-up of rivastigmine treatment in Huntington disease. Clin Neuropharmacol 2007; 30:43–46.
- Fernandez HH, Friedman JH, Grace J, Beasin-Hazen S. Donepezil for Huntington’s disease. Mov Disord 2000; 15:173–176.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry 2004; 161:414–425.
- Phillips W, Shannon KM, Barker RA. The current clinical management of Huntington’s disease. Mov Disord 2008; 23:1491–1504.
- Skirton H, Williams JK, Jackson Barnette J, Paulsen JS. Huntington disease: families’ experiences of healthcare services. J Adv Nursing 2010; 66:500–510.
Chorea is characterized by continuous, random, brief, involuntary muscle contractions that result from a variety of causes.1 These involuntary movements are nonstereotyped and irregular. Although choreic disorders are among the most common involuntary movement disorders, their diagnosis and treatment present several important challenges, including identifying and removing the cause if possible, controlling and preventing motor symptoms, and managing neuropsychologic complications.1 This article provides an overview of the diagnosis and treatment of choreic disorders, using Sydenham chorea to illustrate the management of autoimmune choreas and Huntington disease as the model for the management of heritable choreas.
Management of choreic disorders begins with a first-pass diagnosis and the use of symptomatic therapies. Even if this first pass yields no firm diagnosis, it at least rules out causes that have the most practical significance. A subsequent second-pass evaluation can be undertaken to look for rarer causes. Symptomatic therapies are continued throughout the diagnostic period. More specific therapies can be administered if an etiologic or pathogenic mechanism is determined (eg, postinfectious, autoimmune, metabolic).
CHOREIC DISORDERS: A PRACTICAL DIAGNOSTIC APPROACH
In general, choreic disorders may be subdivided into six categories:
1. Heredodegenerative disorders, such as Huntington disease and other genetically heterogeneous choreas, include Huntington disease–like 2 (HDL2) and benign hereditary chorea.1 Sporadic cases include those of unknown paternity; X-linked disorders (eg, McLeod syndrome); and autosomal-recessive disorders such as chorea-acanthocytosis, which is characterized by chorea, dystonia with prominent orofacial involvement, self-mutilation, myopathy, and neuropathy.2
2. Drug-induced choreas include neurolepticinduced tardive dyskinesia and nontardive hyperkinetic drug-related choreas, the most common of which is levodopa-induced dyskinesias.3 Tardive drug-induced choreas may occur while using the culprit drug, while tapering the drug, or after it has been discontinued. The culprit drugs are represented by dopamine-receptor blockers and include the first-generation neuroleptics (eg, phenothiazines, haloperidol), antidepressants (lox-apine), and gastrointestinal agents (metoclopramide, prochlorperazine). Drug-induced choreas are possible with a wide range of pharmacologic agents, including antiparkinsonian drugs (eg, levodopa, dopamine agonists, anticholinergics), sympathomimetics (eg, amphetamines, cocaine), anticonvulsants, calcium channel blockers, and oral contraceptives.1
3. Autoimmune choreas include Sydenham chorea, systemic lupus erythematosus, and antiphospholipid antibody syndromes. The latter encompass lupus anticoagulant and anticardiolipin antibodies.
4. Metabolic choreas are most often associated with hyperthyroidism, although case reports have described choreas in patients with vitamin B12 deficiency. A variety of hereditary metabolic diseases are also included in this category.
5. Vascular choreas include polycythemia vera and cerebrovascular accidents, the latter frequently presenting as hemiballismus. Polycythemia vera is associated with a high incidence of neurologic symptoms, including a reported incidence of chorea of 0.5% to 5%,4 and should be considered as a potential cause of chorea.
6. Other choreic disorders include a variety of entities such as rare paraneoplastic disorder/syndrome, and posttraumatic and postanoxic presentations.
The first-pass diagnostic approach includes a family history, drug history, and brain magnetic resonance imaging to identify potential structural causes of chorea. Genetic testing for Huntington disease or other choreic disorders may also be performed, although it is essential to consider the potential implications of a positive test result. Intensive pretest and posttest counseling is important both for the patient and for currently asymptomatic family members who may also be affected.1
Other testing includes:
- Complete blood count
- Creatine phosphokinase
- Peripheral smear for acanthocytes
- Comprehensive metabolic panel
- Ceruloplasmin level
- Measurement of thyroxine (T4) and triiodothyronine (T3)
- B12 tests
- Antinuclear antibody sedimentation rate
- Lupus anticoagulant-anticardiolipin antibodies
- Antistreptolysin O (ASO) titer
- Anti-DNase-B titer.
GENERAL CONSIDERATIONS FOR THE TREATMENT OF CHOREIC DISORDERS
In some cases, choreic disorders have a treatable underlying etiology, such as thyroid disease or vitamin B12 deficiency. Tardive syndromes may require treatment beyond drug discontinuation, including use of dopamine depleters for the classic tardive dyskinetic syndromes and anticholinergics for the tardive dystonic syndromes. Levodopa dyskinesia may be treated using amantadine, clozapine, or deep brain stimulation.5,6 The treatment of patients with autoimmune choreas is not well defined. It may include anticoagulation in patients with positive anticardiolipin antibodies to prevent venous or arterial thromboembolism,7 but the risk of arterial thromboembolism is uncertain, and it is unclear whether chorea is truly a harbinger of vascular events.
A negative ASO titer does not exclude Sydenham chorea, a result of childhood infection with group-A beta-hemolytic streptococcus, and antibiotics should be considered in the appropriate context. Some researchers have argued that immune responses associated with acute infections may result in autoimmune neuropsychiatric symptoms. In pediatric patients, this has been referred to as pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS).8
A related phenomenon has been proposed as a potential mechanism of some types of chorea, although the relationship between acute infection and chorea is controversial. Patients with elevated ASO titer or anti-DNase-B titers may be candidates for antibiotics. By 6 weeks after the onset of infection, these titers will fall and a diagnosis of Sydenham chorea can be postulated or based exclusively on clinical judgment.
SYDENHAM CHOREA
Unique to Sydenham chorea is the use of penicillin as prophylaxis. Other than that, the management of Sydenham chorea exemplifies the management approach for the larger category of autoimmune choreic disorders. Pathogenic-based treatment options include immune modulation with cortico steroids, intravenous immunoglobulin (IVIG), and plasma exchange; all treatments must be administered in the appropriate clinical context.
One double-blind clinical trial examined the effectiveness of corticosteroid treatment in children with Sydenham chorea randomly assigned to receive either prednisone (n = 22) or placebo (n = 15).9 Prednisone was administered at a dose of 2 mg/kg/day for 4 weeks, followed by gradual tapering and discontinuation. The median time to remission of chorea was significantly lower for patients in the prednisone group (54.3 days) compared with those in the placebo group (119.9 days; P < .001). Patients in the prednisone group also exhibited significantly better scores on a chorea intensity rating scale at 8 weeks and 12 weeks (P < .001). Potential limitations of this approach include relapse of chorea symptoms and corticosteroid-related adverse events (eg, Cushing syndrome, hypertension).
A second study compared the effectiveness of three modalities: IVIG at a dose of 1 g/kg/day for 2 days (n = 4), plasma exchange (n = 8), and prednisone (n = 6).10 Although differences between treatment groups were not statistically significant, the authors noted that the clinical improvement in chorea symptoms tended to be greater for patients who received IVIG or plasma exchange than for those who received prednisone. Mean chorea scores improved from baseline by 72% for the IVIG group, 50% for the plasma exchange group, and 29% for the prednisone group.
After etiology-dependent treatments have been considered, several other options may be effective regardless of the specific etiology. These include symptomatic treatments such as haloperidol, atypical neuroleptics, and amantadine.11 Antiepileptic medications or benzodiazepines may also help to control symptoms, although less information is available about the use of these agents for the treatment of Sydenham chorea. Tetrabenazine may be considered for patients who will require long-term treatment.
HUNTINGTON DISEASE
Pharmacotherapy of Huntington disease may be unnecessary if symptoms are mild or not bothersome. Symptomatic treatment options include tetrabenazine, amantadine, and either first-generation neuroleptics (eg, haloperidol) or second-generation atypical neuroleptics (eg, olanzapine, quetiapine, risperidone, ziprasidone).
Treating choreas with tetrabenazine or amantadine
Considerable recent attention has focused on the efficacy and safety of tetrabenazine for the treatment of Huntington disease and other choreic disorders. Tetrabenazine is a central monoamine depleter that reversibly binds to the type-2 vesicular monoamine transporter.12 The TETRA-HD study examined the efficacy and safety of tetrabenazine for the short- and long-term control of Huntington disease.12 An initial study compared tetrabenazine with placebo in 75 patients who were treated for up to 13 weeks. In an extension study, all patients received individualized tetrabenazine doses for up to 80 weeks.
The mean total maximal chorea (TMC) scores from the Unified Huntington Disease Rating Scale (UHDRS) decreased markedly during the first 10 weeks of tetrabenazine treatment, remained lower than baseline throughout 80 weeks, and then returned to baseline levels after tetrabenazine discontinuation (Figure 1). At week 80, the mean TMC score was reduced by 4.6 UHDRS units compared with baseline (P < .001) The long-term extension phase was completed by 45 of 75 patients. Treatment-related adverse events that prompted discontinuation included depression, delusions, and vocal tics. The most commonly reported adverse events included sedation or somnolence (n = 18), depressed mood (n = 17), anxiety (n = 13), insomnia (n = 10), and akathisia (n = 9). Scores of parkinsonism and dysphagia increased significantly from baseline over the 80-week study.
Amantadine is an option for patients who cannot tolerate tetrabenazine. A double-blind, placebo-controlled study performed by researchers at the National Institutes of Health (NIH) examined the efficacy and safety of amantadine in 24 patients with Huntington disease.13 Patients were treated with oral amantadine 400 mg/day or placebo for 2 weeks, and were then crossed over to the other treatment. Amantadine was associated with a median reduction in extremity chorea score at rest of 36% from baseline (P = .04), versus 0% improvement with placebo. The mean improvement with amantadine was 56% for the 10 patients with the highest drug plasma levels.
Improvement in chorea scores from baseline for amantadine compared with placebo was rated with four different methods: (1) maximal chorea severity measured from video recordings; (2) maximal chorea severity measured by live raters; (3), chorea severity at rest measured from video recordings; and (4) extremity chorea at rest measured from video recordings. Amantadine was superior to placebo according to all four rating methods. Treatment was generally safe and well tolerated, and no consistent changes in cognitive function were noted with amantadine therapy.
Managing nonmotor complications
In addition to addressing chorea, it is also important to manage nonmotor complications of Huntington disease, including cognition, mood, and thought disorders. Rivastigmine was assessed for the treatment of motor symptoms, functional disability, and cognitive impairment associated with Huntington disease in an open-label study of 18 patients; 11 received rivastigmine 6 mg/day and 7 control patients did not.15 Motor and cognitive function were assessed for up to 2 years by raters who were blinded to treatment assignment. Ratings on a global motor performance scale were significantly better for patients who received rivastigmine than for control subjects. Rivastigmine treatment was also associated with trends toward improvements in functional disability and cognitive impairment, although these differences were not statistically significant.
A small open-label study examined the effects of donepezil for movement and cognitive symptoms associated with Huntington disease.16 Donepezil did not significantly improve cognitive symptoms, although the study enrolled only eight patients. All patients tolerated oral donepezil at a dose of 5 mg/day, but four patients withdrew from the study when the dose was increased to 10 mg/day. In two patients, chorea worsened and falls increased, moderate to severe diarrhea developed in three patients, and one patient reported anxiety and irritability.
Depression is another common complication of Huntington disease. The incidence of depression among patients with Huntington disease is approximately 40%, and the risk of suicide is at least eightfold greater than that among the general population.17 Treatment must be guided by clinical judgment. Selective serotonin reuptake inhibitor antidepressants have been recommended. Other options to manage depression include mirtazapine, monoamine oxidase inhibitors, or electroshock therapy. Mood-stabilizing agents (eg, carbamazepine, lamotrigine, valproate) may also be indicated in helping with impulse control. Haloperidol and second-generation antipsychotics are used for the treatment of a broad range of psychiatric conditions, many of which may overlap with Huntington disease, including schizophrenia and schizophreniform disorder, schizoaffective disorder, bipolar disorder, dementia, and disruptive behavior.18 The risk of tardive dyskinesia may be as much as fivefold lower with second-generation antipsychotics.18 Many patients with Huntington disease require treatment for aggression. A variety of approaches are available, including behavior modification, the antidepressant sertraline, buspirone, antipsychotic agents (eg, risperidone, olanzapine), propranolol, and lithium (combined with haloperidol).
Long-term care considerations
As a consequence of the diverse clinical manifestations of choreic disorders in movement, function, mood, and cognition, the treatment of Huntington disease requires a multidisciplinary approach that involves a number of different health care specialties across the long-term course of the disorder. Members of the Huntington disease treatment team may include neurologists, psychiatrists, nurses, social workers, geneticists, physical therapists, occupational therapists, speech therapists, dietitians, and other supporting groups or professional societies. The clinical manifestations of Huntington disease may evolve over time, as symptoms such as bradykinesia, dystonia, rigidity, cognitive decline, and gait instability become more significant.19 As a result, optimal management strategies for patients with Huntington disease may change significantly across the long-term course of the disease. During the early course of the disease, the typical clinical presentation is largely hyperkinesis, irritability, and distractibility. These patients will require initiation of drug therapy and linkage to sources of support. In the later stages of the disease, the presentation shifts to a more hypokinetic and apathetic profile, and patients are more likely to require drug regimen review and modification, nursing home placement, and palliative care services.19
Another important concern in Huntington disease treatment is care of the caregiver. Surveys show that the key concerns of caregivers include the expertise of the health care professionals who are treating the patient and the availability of sufficient services in the community.20 Several resources are available for Huntington disease caregivers, including local support groups, the Huntington’s Disease Society of America, Q Foundation, and the Huntington Study Group. The Lundbeck pharmaceutical company operates a patient assistance program (LundbeckShare. com) as well as an information center that can be accessed toll free at (888)457-4273. Approximately 90% of patients who request copayment assistance qualify for aid, regardless of the type of insurance they carry.
SUMMARY AND CONCLUSIONS
The approach to a patient with chorea starts with a search for specifically treatable etiologies. Autoimmune, metabolic, and vascular causes should be sought first and treated. The symptomatic treatment of all choreas is based on the model described here for Huntington disease, and includes attention to cognitive, psychiatric, and social support issues. The recommended approach is multidisciplinary, with a change in the mix of services as the disease progresses. It is also important to recognize the burden of Huntington disease on the caregiver and consider steps to make this burden more manageable.
Chorea is characterized by continuous, random, brief, involuntary muscle contractions that result from a variety of causes.1 These involuntary movements are nonstereotyped and irregular. Although choreic disorders are among the most common involuntary movement disorders, their diagnosis and treatment present several important challenges, including identifying and removing the cause if possible, controlling and preventing motor symptoms, and managing neuropsychologic complications.1 This article provides an overview of the diagnosis and treatment of choreic disorders, using Sydenham chorea to illustrate the management of autoimmune choreas and Huntington disease as the model for the management of heritable choreas.
Management of choreic disorders begins with a first-pass diagnosis and the use of symptomatic therapies. Even if this first pass yields no firm diagnosis, it at least rules out causes that have the most practical significance. A subsequent second-pass evaluation can be undertaken to look for rarer causes. Symptomatic therapies are continued throughout the diagnostic period. More specific therapies can be administered if an etiologic or pathogenic mechanism is determined (eg, postinfectious, autoimmune, metabolic).
CHOREIC DISORDERS: A PRACTICAL DIAGNOSTIC APPROACH
In general, choreic disorders may be subdivided into six categories:
1. Heredodegenerative disorders, such as Huntington disease and other genetically heterogeneous choreas, include Huntington disease–like 2 (HDL2) and benign hereditary chorea.1 Sporadic cases include those of unknown paternity; X-linked disorders (eg, McLeod syndrome); and autosomal-recessive disorders such as chorea-acanthocytosis, which is characterized by chorea, dystonia with prominent orofacial involvement, self-mutilation, myopathy, and neuropathy.2
2. Drug-induced choreas include neurolepticinduced tardive dyskinesia and nontardive hyperkinetic drug-related choreas, the most common of which is levodopa-induced dyskinesias.3 Tardive drug-induced choreas may occur while using the culprit drug, while tapering the drug, or after it has been discontinued. The culprit drugs are represented by dopamine-receptor blockers and include the first-generation neuroleptics (eg, phenothiazines, haloperidol), antidepressants (lox-apine), and gastrointestinal agents (metoclopramide, prochlorperazine). Drug-induced choreas are possible with a wide range of pharmacologic agents, including antiparkinsonian drugs (eg, levodopa, dopamine agonists, anticholinergics), sympathomimetics (eg, amphetamines, cocaine), anticonvulsants, calcium channel blockers, and oral contraceptives.1
3. Autoimmune choreas include Sydenham chorea, systemic lupus erythematosus, and antiphospholipid antibody syndromes. The latter encompass lupus anticoagulant and anticardiolipin antibodies.
4. Metabolic choreas are most often associated with hyperthyroidism, although case reports have described choreas in patients with vitamin B12 deficiency. A variety of hereditary metabolic diseases are also included in this category.
5. Vascular choreas include polycythemia vera and cerebrovascular accidents, the latter frequently presenting as hemiballismus. Polycythemia vera is associated with a high incidence of neurologic symptoms, including a reported incidence of chorea of 0.5% to 5%,4 and should be considered as a potential cause of chorea.
6. Other choreic disorders include a variety of entities such as rare paraneoplastic disorder/syndrome, and posttraumatic and postanoxic presentations.
The first-pass diagnostic approach includes a family history, drug history, and brain magnetic resonance imaging to identify potential structural causes of chorea. Genetic testing for Huntington disease or other choreic disorders may also be performed, although it is essential to consider the potential implications of a positive test result. Intensive pretest and posttest counseling is important both for the patient and for currently asymptomatic family members who may also be affected.1
Other testing includes:
- Complete blood count
- Creatine phosphokinase
- Peripheral smear for acanthocytes
- Comprehensive metabolic panel
- Ceruloplasmin level
- Measurement of thyroxine (T4) and triiodothyronine (T3)
- B12 tests
- Antinuclear antibody sedimentation rate
- Lupus anticoagulant-anticardiolipin antibodies
- Antistreptolysin O (ASO) titer
- Anti-DNase-B titer.
GENERAL CONSIDERATIONS FOR THE TREATMENT OF CHOREIC DISORDERS
In some cases, choreic disorders have a treatable underlying etiology, such as thyroid disease or vitamin B12 deficiency. Tardive syndromes may require treatment beyond drug discontinuation, including use of dopamine depleters for the classic tardive dyskinetic syndromes and anticholinergics for the tardive dystonic syndromes. Levodopa dyskinesia may be treated using amantadine, clozapine, or deep brain stimulation.5,6 The treatment of patients with autoimmune choreas is not well defined. It may include anticoagulation in patients with positive anticardiolipin antibodies to prevent venous or arterial thromboembolism,7 but the risk of arterial thromboembolism is uncertain, and it is unclear whether chorea is truly a harbinger of vascular events.
A negative ASO titer does not exclude Sydenham chorea, a result of childhood infection with group-A beta-hemolytic streptococcus, and antibiotics should be considered in the appropriate context. Some researchers have argued that immune responses associated with acute infections may result in autoimmune neuropsychiatric symptoms. In pediatric patients, this has been referred to as pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS).8
A related phenomenon has been proposed as a potential mechanism of some types of chorea, although the relationship between acute infection and chorea is controversial. Patients with elevated ASO titer or anti-DNase-B titers may be candidates for antibiotics. By 6 weeks after the onset of infection, these titers will fall and a diagnosis of Sydenham chorea can be postulated or based exclusively on clinical judgment.
SYDENHAM CHOREA
Unique to Sydenham chorea is the use of penicillin as prophylaxis. Other than that, the management of Sydenham chorea exemplifies the management approach for the larger category of autoimmune choreic disorders. Pathogenic-based treatment options include immune modulation with cortico steroids, intravenous immunoglobulin (IVIG), and plasma exchange; all treatments must be administered in the appropriate clinical context.
One double-blind clinical trial examined the effectiveness of corticosteroid treatment in children with Sydenham chorea randomly assigned to receive either prednisone (n = 22) or placebo (n = 15).9 Prednisone was administered at a dose of 2 mg/kg/day for 4 weeks, followed by gradual tapering and discontinuation. The median time to remission of chorea was significantly lower for patients in the prednisone group (54.3 days) compared with those in the placebo group (119.9 days; P < .001). Patients in the prednisone group also exhibited significantly better scores on a chorea intensity rating scale at 8 weeks and 12 weeks (P < .001). Potential limitations of this approach include relapse of chorea symptoms and corticosteroid-related adverse events (eg, Cushing syndrome, hypertension).
A second study compared the effectiveness of three modalities: IVIG at a dose of 1 g/kg/day for 2 days (n = 4), plasma exchange (n = 8), and prednisone (n = 6).10 Although differences between treatment groups were not statistically significant, the authors noted that the clinical improvement in chorea symptoms tended to be greater for patients who received IVIG or plasma exchange than for those who received prednisone. Mean chorea scores improved from baseline by 72% for the IVIG group, 50% for the plasma exchange group, and 29% for the prednisone group.
After etiology-dependent treatments have been considered, several other options may be effective regardless of the specific etiology. These include symptomatic treatments such as haloperidol, atypical neuroleptics, and amantadine.11 Antiepileptic medications or benzodiazepines may also help to control symptoms, although less information is available about the use of these agents for the treatment of Sydenham chorea. Tetrabenazine may be considered for patients who will require long-term treatment.
HUNTINGTON DISEASE
Pharmacotherapy of Huntington disease may be unnecessary if symptoms are mild or not bothersome. Symptomatic treatment options include tetrabenazine, amantadine, and either first-generation neuroleptics (eg, haloperidol) or second-generation atypical neuroleptics (eg, olanzapine, quetiapine, risperidone, ziprasidone).
Treating choreas with tetrabenazine or amantadine
Considerable recent attention has focused on the efficacy and safety of tetrabenazine for the treatment of Huntington disease and other choreic disorders. Tetrabenazine is a central monoamine depleter that reversibly binds to the type-2 vesicular monoamine transporter.12 The TETRA-HD study examined the efficacy and safety of tetrabenazine for the short- and long-term control of Huntington disease.12 An initial study compared tetrabenazine with placebo in 75 patients who were treated for up to 13 weeks. In an extension study, all patients received individualized tetrabenazine doses for up to 80 weeks.
The mean total maximal chorea (TMC) scores from the Unified Huntington Disease Rating Scale (UHDRS) decreased markedly during the first 10 weeks of tetrabenazine treatment, remained lower than baseline throughout 80 weeks, and then returned to baseline levels after tetrabenazine discontinuation (Figure 1). At week 80, the mean TMC score was reduced by 4.6 UHDRS units compared with baseline (P < .001) The long-term extension phase was completed by 45 of 75 patients. Treatment-related adverse events that prompted discontinuation included depression, delusions, and vocal tics. The most commonly reported adverse events included sedation or somnolence (n = 18), depressed mood (n = 17), anxiety (n = 13), insomnia (n = 10), and akathisia (n = 9). Scores of parkinsonism and dysphagia increased significantly from baseline over the 80-week study.
Amantadine is an option for patients who cannot tolerate tetrabenazine. A double-blind, placebo-controlled study performed by researchers at the National Institutes of Health (NIH) examined the efficacy and safety of amantadine in 24 patients with Huntington disease.13 Patients were treated with oral amantadine 400 mg/day or placebo for 2 weeks, and were then crossed over to the other treatment. Amantadine was associated with a median reduction in extremity chorea score at rest of 36% from baseline (P = .04), versus 0% improvement with placebo. The mean improvement with amantadine was 56% for the 10 patients with the highest drug plasma levels.
Improvement in chorea scores from baseline for amantadine compared with placebo was rated with four different methods: (1) maximal chorea severity measured from video recordings; (2) maximal chorea severity measured by live raters; (3), chorea severity at rest measured from video recordings; and (4) extremity chorea at rest measured from video recordings. Amantadine was superior to placebo according to all four rating methods. Treatment was generally safe and well tolerated, and no consistent changes in cognitive function were noted with amantadine therapy.
Managing nonmotor complications
In addition to addressing chorea, it is also important to manage nonmotor complications of Huntington disease, including cognition, mood, and thought disorders. Rivastigmine was assessed for the treatment of motor symptoms, functional disability, and cognitive impairment associated with Huntington disease in an open-label study of 18 patients; 11 received rivastigmine 6 mg/day and 7 control patients did not.15 Motor and cognitive function were assessed for up to 2 years by raters who were blinded to treatment assignment. Ratings on a global motor performance scale were significantly better for patients who received rivastigmine than for control subjects. Rivastigmine treatment was also associated with trends toward improvements in functional disability and cognitive impairment, although these differences were not statistically significant.
A small open-label study examined the effects of donepezil for movement and cognitive symptoms associated with Huntington disease.16 Donepezil did not significantly improve cognitive symptoms, although the study enrolled only eight patients. All patients tolerated oral donepezil at a dose of 5 mg/day, but four patients withdrew from the study when the dose was increased to 10 mg/day. In two patients, chorea worsened and falls increased, moderate to severe diarrhea developed in three patients, and one patient reported anxiety and irritability.
Depression is another common complication of Huntington disease. The incidence of depression among patients with Huntington disease is approximately 40%, and the risk of suicide is at least eightfold greater than that among the general population.17 Treatment must be guided by clinical judgment. Selective serotonin reuptake inhibitor antidepressants have been recommended. Other options to manage depression include mirtazapine, monoamine oxidase inhibitors, or electroshock therapy. Mood-stabilizing agents (eg, carbamazepine, lamotrigine, valproate) may also be indicated in helping with impulse control. Haloperidol and second-generation antipsychotics are used for the treatment of a broad range of psychiatric conditions, many of which may overlap with Huntington disease, including schizophrenia and schizophreniform disorder, schizoaffective disorder, bipolar disorder, dementia, and disruptive behavior.18 The risk of tardive dyskinesia may be as much as fivefold lower with second-generation antipsychotics.18 Many patients with Huntington disease require treatment for aggression. A variety of approaches are available, including behavior modification, the antidepressant sertraline, buspirone, antipsychotic agents (eg, risperidone, olanzapine), propranolol, and lithium (combined with haloperidol).
Long-term care considerations
As a consequence of the diverse clinical manifestations of choreic disorders in movement, function, mood, and cognition, the treatment of Huntington disease requires a multidisciplinary approach that involves a number of different health care specialties across the long-term course of the disorder. Members of the Huntington disease treatment team may include neurologists, psychiatrists, nurses, social workers, geneticists, physical therapists, occupational therapists, speech therapists, dietitians, and other supporting groups or professional societies. The clinical manifestations of Huntington disease may evolve over time, as symptoms such as bradykinesia, dystonia, rigidity, cognitive decline, and gait instability become more significant.19 As a result, optimal management strategies for patients with Huntington disease may change significantly across the long-term course of the disease. During the early course of the disease, the typical clinical presentation is largely hyperkinesis, irritability, and distractibility. These patients will require initiation of drug therapy and linkage to sources of support. In the later stages of the disease, the presentation shifts to a more hypokinetic and apathetic profile, and patients are more likely to require drug regimen review and modification, nursing home placement, and palliative care services.19
Another important concern in Huntington disease treatment is care of the caregiver. Surveys show that the key concerns of caregivers include the expertise of the health care professionals who are treating the patient and the availability of sufficient services in the community.20 Several resources are available for Huntington disease caregivers, including local support groups, the Huntington’s Disease Society of America, Q Foundation, and the Huntington Study Group. The Lundbeck pharmaceutical company operates a patient assistance program (LundbeckShare. com) as well as an information center that can be accessed toll free at (888)457-4273. Approximately 90% of patients who request copayment assistance qualify for aid, regardless of the type of insurance they carry.
SUMMARY AND CONCLUSIONS
The approach to a patient with chorea starts with a search for specifically treatable etiologies. Autoimmune, metabolic, and vascular causes should be sought first and treated. The symptomatic treatment of all choreas is based on the model described here for Huntington disease, and includes attention to cognitive, psychiatric, and social support issues. The recommended approach is multidisciplinary, with a change in the mix of services as the disease progresses. It is also important to recognize the burden of Huntington disease on the caregiver and consider steps to make this burden more manageable.
- Cardoso F, Seppi K, Mair KJ, Wenning GK, Poewe W. Seminar on choreas. Lancet Neurol 2006; 5:589–602.
- Schneider SA, Walker RH, Bhatia KP. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 2007; 3:517–525.
- Zesiewicz TA, Sullivan KL. Drug-induced hyperkinetic movement disorders by nonneuroleptic agents. Handb Clin Neurol 2011; 100:347–363.
- Margolis RL, Ross CA. Diagnosis of Huntington disease. Clin Chem 2003; 49:1726–1732.
- Rascol O. The pharmacological therapeutic management of levodopa-induced dyskinesias in patients with Parkinson’s disease. J Neurol 2000; 247 (suppl 2):II51–II57.
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord 2007; 22:1379–1389.
- Lim W, Crowther MA, Eifelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Mink J, Kurlan R. Acute postinfectious movement and psychiatric disorders in children and adolescents. J Child Neurol 2011; 26:214–217.
- Paz JA, Silva CA, Marques-Dias MJ. Randomized double-blind study with prednisone in Sydenham’s chorea. Pediatr Neurol 2006; 34:264–269.
- Garvey MA, Snider LA, Leitman SF, Werden R, Swedo SE. Treatment of Sydenham’s chorea with intravenous immunoglobulin, plasma exchange, or prednisone. J Child Neurol 2005; 20:424–429.
- Walker KG, Wilmshurst JM. An update on the treatment of Sydenham’s chorea: the evidence for established and evolving interventions. Ther Adv Neurol Disord 2010; 3:301–309.
- Frank S. Tetrabenazine as anti-chorea therapy in Huntington disease: an open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol 2009; 9:62.
- Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002; 59:694–699.
- Lucetti C, Del Dotto P, Gamabaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology 2003; 60:1995–1997.
- de Tommaso M, Difruscolo O, Scirruicchio V, Specchio N, Livrea P. Two years’ follow-up of rivastigmine treatment in Huntington disease. Clin Neuropharmacol 2007; 30:43–46.
- Fernandez HH, Friedman JH, Grace J, Beasin-Hazen S. Donepezil for Huntington’s disease. Mov Disord 2000; 15:173–176.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry 2004; 161:414–425.
- Phillips W, Shannon KM, Barker RA. The current clinical management of Huntington’s disease. Mov Disord 2008; 23:1491–1504.
- Skirton H, Williams JK, Jackson Barnette J, Paulsen JS. Huntington disease: families’ experiences of healthcare services. J Adv Nursing 2010; 66:500–510.
- Cardoso F, Seppi K, Mair KJ, Wenning GK, Poewe W. Seminar on choreas. Lancet Neurol 2006; 5:589–602.
- Schneider SA, Walker RH, Bhatia KP. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 2007; 3:517–525.
- Zesiewicz TA, Sullivan KL. Drug-induced hyperkinetic movement disorders by nonneuroleptic agents. Handb Clin Neurol 2011; 100:347–363.
- Margolis RL, Ross CA. Diagnosis of Huntington disease. Clin Chem 2003; 49:1726–1732.
- Rascol O. The pharmacological therapeutic management of levodopa-induced dyskinesias in patients with Parkinson’s disease. J Neurol 2000; 247 (suppl 2):II51–II57.
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord 2007; 22:1379–1389.
- Lim W, Crowther MA, Eifelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Mink J, Kurlan R. Acute postinfectious movement and psychiatric disorders in children and adolescents. J Child Neurol 2011; 26:214–217.
- Paz JA, Silva CA, Marques-Dias MJ. Randomized double-blind study with prednisone in Sydenham’s chorea. Pediatr Neurol 2006; 34:264–269.
- Garvey MA, Snider LA, Leitman SF, Werden R, Swedo SE. Treatment of Sydenham’s chorea with intravenous immunoglobulin, plasma exchange, or prednisone. J Child Neurol 2005; 20:424–429.
- Walker KG, Wilmshurst JM. An update on the treatment of Sydenham’s chorea: the evidence for established and evolving interventions. Ther Adv Neurol Disord 2010; 3:301–309.
- Frank S. Tetrabenazine as anti-chorea therapy in Huntington disease: an open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol 2009; 9:62.
- Verhagen Metman L, Morris MJ, Farmer C, et al. Huntington’s disease: a randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002; 59:694–699.
- Lucetti C, Del Dotto P, Gamabaccini G, et al. IV amantadine improves chorea in Huntington’s disease: an acute randomized, controlled study. Neurology 2003; 60:1995–1997.
- de Tommaso M, Difruscolo O, Scirruicchio V, Specchio N, Livrea P. Two years’ follow-up of rivastigmine treatment in Huntington disease. Clin Neuropharmacol 2007; 30:43–46.
- Fernandez HH, Friedman JH, Grace J, Beasin-Hazen S. Donepezil for Huntington’s disease. Mov Disord 2000; 15:173–176.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry 2004; 161:414–425.
- Phillips W, Shannon KM, Barker RA. The current clinical management of Huntington’s disease. Mov Disord 2008; 23:1491–1504.
- Skirton H, Williams JK, Jackson Barnette J, Paulsen JS. Huntington disease: families’ experiences of healthcare services. J Adv Nursing 2010; 66:500–510.