User login
Advances in neuroimaging, cell biology, and post mortem analysis are starting to explain what happens in the brain of a person who develops schizophrenia. Schizophrenia appears to be a developmental disorder of disrupted neural connection within and between regions of the brain. These disruptions seem to result from genetic predispositions interacting with negative environmental events.
A matter of gray and white
Individuals with schizophrenia have deficits in gray matter and white matter, as illustrated by studies linking auditory hallucinations with brain regions associated with normal hearing (Box).
Gray matter. Magnetic resonance imaging (MRI) indicates that gray matter volume peaks in early adolescence and declines with age. The normal adolescent brain shrinks as inefficient neural connections are pruned away, a process that refines and matures gray matter. In individuals with schizophrenia, this reduction is more aggressive—perhaps because of excessive pruning—and occurs in the time frame when schizophrenia symptoms typically emerge.
Rapoport et al1 documented this process through sequential MRI scans in children with early-onset schizophrenia (mean age 14.5). Compared with age-matched healthy controls, youths with schizophrenia show greater and more rapid gray matter loss during late adolescence (Figure 1).2
Increased density. Reduced neuronal branching and spine formation also likely causes subtle reductions in gray matter volume (Figure 2). The resulting lack of dendritic connectivity may produce cognitive impairments and negative symptoms seen in schizophrenia.
Postmortem studies of gray matter cells show increased neuron density in patients with schizophrenia when compared with controls.3 Patients with schizophrenia have the same number of neurons as controls, but the neurons are more tightly packed because of reduced cell size, branching, and synapse formation.4
Research over the past decade has revealed schizophrenia to be a neurodegenerative disorder characterized by substantial brain tissue loss during first and subsequent psychotic episodes.5 Neuroimaging studies show that clinical and functional deterioration accompanies progressive loss of cortical gray matter volume and enlargement of cerebral ventricles. Thus, preventing relapses has come to be regarded as critical to long-term schizophrenia management.
Auditory hallucinations appear to emanate from the temporal lobe, the same brain region that processes external sound. Thus, it may be that patients experiencing hallucinations are misidentifying inner speech as coming from an outside source.
Using functional MRI to differentiate brain activity signals associated with hallucinating and nonhallucinating states, Dierks et al21 documented increased activity in auditory cortical gray matter during hallucinations in schizophrenia patients.
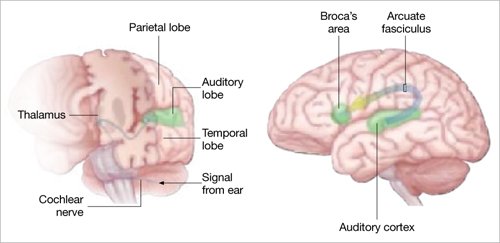
Auditory signals make synaptic connections in the thalamus (left) before reaching the auditory cortex. White matter fiber tracts called the arcuate fasciculus (right) connect the auditory cortex in the temporal lobe with Broca’s area in the frontal cortex.
Source: Adapted from reference 2
Using MR diffusion tensor imaging, Hubl et al22 identified white matter changes in the arcuate fasciculus of schizophrenia patients prone to hallucinations, compared with healthy controls and patients who had schizophrenia but not hallucinations.
These findings support the understanding that auditory hallucinations originate from altered connectivity of the same regions that process normal hearing and speech. The schizophrenia patient may perceive external voices from aberrant internal signals.
Figure 1 Rates of gray matter volume loss during adolescence
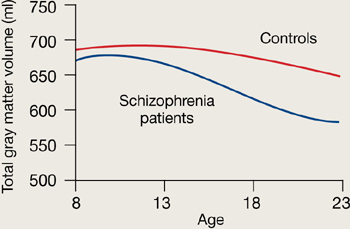
Youths with early-onset schizophrenia show greater gray matter volume loss during adolescence, compared with normal controls.
Source: Adapted from reference 2
Figure 2 Structural differences between neurons
in patients with schizophrenia and controls
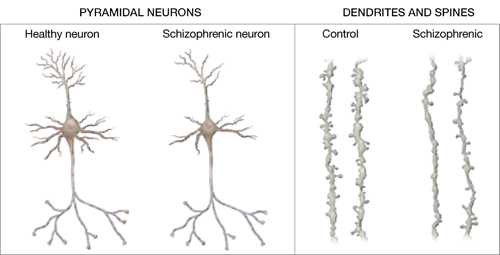
Schizophrenic neurons show reduced soma size, spine formation, and dendritic branching
Source: Adapted from reference 2White matter. Recent research suggests that white matter deficits also may be involved in schizophrenia’s pathophysiology. Studies using diffusion tensor imaging (DTI)—which measures the sum of vectors of water diffusion along axons—have documented white matter impairments in patients with schizophrenia.6
White matter tracks—myelinated axons that transport electrical signals among neurons—connect regions within the cortex and between the cortex and deeper brain structures. Disruption of white matter tracks may degrade signals and confuse neuronal communication.
Myelination. Genetic studies in patients with schizophrenia also have suggested that decreased neuron myelination may play a role in white matter deficits. Hakak et al8 examined more than 6,000 genes using microarray analysis and found only 17 genes were significantly down-regulated in patients with schizophrenia. Of those 17 genes, 6 were related to myelin and 11 showed no pattern.
Oligodendrocytes are glial cells that insulate axons with myelin and allow faster transmission of electrical impulses in the brain. In a postmortem study, Hof et al7 found 7 patients schizophrenia had 28% fewer oligodendrocytes per section of the superior frontal gyrus and 27% less white matter compared with 7 age-matched controls (Figure 3).
Figure 3 Reduced neuron myelination possible in schizophrenia
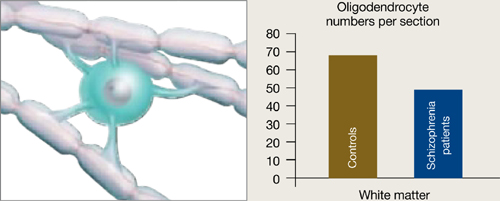
In a postmortem analysis, stained white matter sections taken from schizophrenia patients had fewer oligodendrocytes, cells that insulate axons with myelin and facilitate electrical transmission.
Source: Adapted from reference 2
Genes and the environment
Schizophrenia’s heritability is among the most repeated research findings in psychiatry.9 Other mechanisms besides genetics must be involved, however, as studies consistently show that monozygotic twins have a concordance rate of approximately 50% for the development of schizophrenia.
Environmental factors. Adverse environmental events may act in conjuction with genetic predisposition to trigger schizophrenia development. Ischemia or an impoverished diet, for example, have the potential to change DNA methylation.
Environmental factors associated with increased risk for schizophrenia include:
- maternal starvation during pregnancy10
- prenatal exposure to influenza11
- obstetrical complications with hypoxia12
- being born and raised in an urban environment13
- using marijuana during adolescence.14
Gene expression. Important genes may be silenced in individuals with increased DNA methylation and a susceptible genetic profile. Alterations in gene expression are the fundamental mechanism of behavioral change. Research shows that environmental events can alter gene expression without changing the genetic code, such as by adding methyl groups to DNA.15,16 The silencing of important developmental genes in this way can have devastating effects on development.
One explanation for the development of schizophrenia is that environmental events in susceptible individuals silence the production of proteins essential for maintaining neuronal connections through methylation of DNA. Postmortem analysis of brains of patients with schizophrenia show reduced mRNA of reelin,17 a protein produced in gamma-aminobutyric acid neurons involved in neuronal migration, axon branching, and synapse formation during brain development. Lowered production of proteins such as reelin may reduce connections between neurons and cause schizophrenia symptoms. Two research groups also have reported increased methylation of reelin DNA in postmortem studies of the brains of patients with schizophrenia.18,19 Increased methylation of DNA would silence production of this important protein.
Preventing neural disconnects? If schizophrenia is a developmental disorder resulting from failures in brain connectivity, then the ultimate treatment may be prevention. Recent research suggests that intervening with second-generation antipsychotics during the prodromal stage can prevent or delay the emergence of the disorder.20 Further research is needed to establish whether early intervention can prevent schizophrenia’s neuronal disruption.
1. Gogtay N, Sporn A, Rapoport J. Structural brain MRI studies in childhood-onset schizophrenia and childhood atypical psychosis. In: Lawrie S, Johnstone E, Weinberger D, eds. Schizophrenia: from neuroimaging to neuroscience. New York, NY: Oxford University Press; 2004.
2. Higgins ES, George MS. The neuroscience of clinical psychiatry. Philadelphia: Lippincott, Williams, and Wilkins; 2007.
3. Selemon LD. Increased cortical neuronal density in schizophrenia. Am J Psychiatry 2004;161(9):1564.-
4. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57(1):65-73.
5. Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World Journal 2007;7:135-43.
6. Kubicki M, McCarley R, Westin CF, et al. A review of diffusion tensor imaging studies in schizophrenia. J Psychiatr Res 2007;41(1-2):15-30.
7. Hof PR, Haroutunian V, Friedrich VL, Jr, et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003;53(12):1075-85.
8. Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001;98(8):4746-51.
9. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatr 2004;16(4):260-83.
10. McClellan JM, Susser E, King MC. Maternal famine, de novo mutations, and schizophrenia. JAMA 2006;296(5):582-4.
11. Limosin F, Rouillon F, Payan C, et al. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003;107(5):331-5.
12. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002;159(7):1080-92.
13. Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry 2006;6:2.-
14. Arendt M, Rosenberg R, Foldager L, et al. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry 2005;187:510-5.
15. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics 2003;33(suppl):245-54.
16. Abdolmaleky HM, Smith CL, Faraone SV, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet 2004;127(1):51-9.
17. Fatemi SH, Stary JM, Earle JA, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005;72(2-3):109-22.
18. Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005;134(1):60-6.
19. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102(26):9341-6.
20. McGlashan TH, Zipursky RB, Perkins D, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 2006;163(5):790-799.
21. Dierks T, Linden DE, Jandl M, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron 1999;22(3):615-21.
22. Hubl D, Koenig T, Strik W, et al. Pathways that make voices: white matter changes in auditory hallucinations. Arch Gen Psychiatry 2004;61(7):658-68.
Adapted from The neuroscience of clinical psychiatry, by Edmund S. Higgins and Mark S. George. Philadelphia: Lippincott, Williams, and Wilkins; 2007:251-63.
Advances in neuroimaging, cell biology, and post mortem analysis are starting to explain what happens in the brain of a person who develops schizophrenia. Schizophrenia appears to be a developmental disorder of disrupted neural connection within and between regions of the brain. These disruptions seem to result from genetic predispositions interacting with negative environmental events.
A matter of gray and white
Individuals with schizophrenia have deficits in gray matter and white matter, as illustrated by studies linking auditory hallucinations with brain regions associated with normal hearing (Box).
Gray matter. Magnetic resonance imaging (MRI) indicates that gray matter volume peaks in early adolescence and declines with age. The normal adolescent brain shrinks as inefficient neural connections are pruned away, a process that refines and matures gray matter. In individuals with schizophrenia, this reduction is more aggressive—perhaps because of excessive pruning—and occurs in the time frame when schizophrenia symptoms typically emerge.
Rapoport et al1 documented this process through sequential MRI scans in children with early-onset schizophrenia (mean age 14.5). Compared with age-matched healthy controls, youths with schizophrenia show greater and more rapid gray matter loss during late adolescence (Figure 1).2
Increased density. Reduced neuronal branching and spine formation also likely causes subtle reductions in gray matter volume (Figure 2). The resulting lack of dendritic connectivity may produce cognitive impairments and negative symptoms seen in schizophrenia.
Postmortem studies of gray matter cells show increased neuron density in patients with schizophrenia when compared with controls.3 Patients with schizophrenia have the same number of neurons as controls, but the neurons are more tightly packed because of reduced cell size, branching, and synapse formation.4
Research over the past decade has revealed schizophrenia to be a neurodegenerative disorder characterized by substantial brain tissue loss during first and subsequent psychotic episodes.5 Neuroimaging studies show that clinical and functional deterioration accompanies progressive loss of cortical gray matter volume and enlargement of cerebral ventricles. Thus, preventing relapses has come to be regarded as critical to long-term schizophrenia management.
Auditory hallucinations appear to emanate from the temporal lobe, the same brain region that processes external sound. Thus, it may be that patients experiencing hallucinations are misidentifying inner speech as coming from an outside source.
Using functional MRI to differentiate brain activity signals associated with hallucinating and nonhallucinating states, Dierks et al21 documented increased activity in auditory cortical gray matter during hallucinations in schizophrenia patients.
Auditory signals make synaptic connections in the thalamus (left) before reaching the auditory cortex. White matter fiber tracts called the arcuate fasciculus (right) connect the auditory cortex in the temporal lobe with Broca’s area in the frontal cortex.
Source: Adapted from reference 2
Using MR diffusion tensor imaging, Hubl et al22 identified white matter changes in the arcuate fasciculus of schizophrenia patients prone to hallucinations, compared with healthy controls and patients who had schizophrenia but not hallucinations.
These findings support the understanding that auditory hallucinations originate from altered connectivity of the same regions that process normal hearing and speech. The schizophrenia patient may perceive external voices from aberrant internal signals.
Figure 1 Rates of gray matter volume loss during adolescence
Youths with early-onset schizophrenia show greater gray matter volume loss during adolescence, compared with normal controls.
Source: Adapted from reference 2
Figure 2 Structural differences between neurons
in patients with schizophrenia and controls
Schizophrenic neurons show reduced soma size, spine formation, and dendritic branching
Source: Adapted from reference 2White matter. Recent research suggests that white matter deficits also may be involved in schizophrenia’s pathophysiology. Studies using diffusion tensor imaging (DTI)—which measures the sum of vectors of water diffusion along axons—have documented white matter impairments in patients with schizophrenia.6
White matter tracks—myelinated axons that transport electrical signals among neurons—connect regions within the cortex and between the cortex and deeper brain structures. Disruption of white matter tracks may degrade signals and confuse neuronal communication.
Myelination. Genetic studies in patients with schizophrenia also have suggested that decreased neuron myelination may play a role in white matter deficits. Hakak et al8 examined more than 6,000 genes using microarray analysis and found only 17 genes were significantly down-regulated in patients with schizophrenia. Of those 17 genes, 6 were related to myelin and 11 showed no pattern.
Oligodendrocytes are glial cells that insulate axons with myelin and allow faster transmission of electrical impulses in the brain. In a postmortem study, Hof et al7 found 7 patients schizophrenia had 28% fewer oligodendrocytes per section of the superior frontal gyrus and 27% less white matter compared with 7 age-matched controls (Figure 3).
Figure 3 Reduced neuron myelination possible in schizophrenia
In a postmortem analysis, stained white matter sections taken from schizophrenia patients had fewer oligodendrocytes, cells that insulate axons with myelin and facilitate electrical transmission.
Source: Adapted from reference 2
Genes and the environment
Schizophrenia’s heritability is among the most repeated research findings in psychiatry.9 Other mechanisms besides genetics must be involved, however, as studies consistently show that monozygotic twins have a concordance rate of approximately 50% for the development of schizophrenia.
Environmental factors. Adverse environmental events may act in conjuction with genetic predisposition to trigger schizophrenia development. Ischemia or an impoverished diet, for example, have the potential to change DNA methylation.
Environmental factors associated with increased risk for schizophrenia include:
- maternal starvation during pregnancy10
- prenatal exposure to influenza11
- obstetrical complications with hypoxia12
- being born and raised in an urban environment13
- using marijuana during adolescence.14
Gene expression. Important genes may be silenced in individuals with increased DNA methylation and a susceptible genetic profile. Alterations in gene expression are the fundamental mechanism of behavioral change. Research shows that environmental events can alter gene expression without changing the genetic code, such as by adding methyl groups to DNA.15,16 The silencing of important developmental genes in this way can have devastating effects on development.
One explanation for the development of schizophrenia is that environmental events in susceptible individuals silence the production of proteins essential for maintaining neuronal connections through methylation of DNA. Postmortem analysis of brains of patients with schizophrenia show reduced mRNA of reelin,17 a protein produced in gamma-aminobutyric acid neurons involved in neuronal migration, axon branching, and synapse formation during brain development. Lowered production of proteins such as reelin may reduce connections between neurons and cause schizophrenia symptoms. Two research groups also have reported increased methylation of reelin DNA in postmortem studies of the brains of patients with schizophrenia.18,19 Increased methylation of DNA would silence production of this important protein.
Preventing neural disconnects? If schizophrenia is a developmental disorder resulting from failures in brain connectivity, then the ultimate treatment may be prevention. Recent research suggests that intervening with second-generation antipsychotics during the prodromal stage can prevent or delay the emergence of the disorder.20 Further research is needed to establish whether early intervention can prevent schizophrenia’s neuronal disruption.
Advances in neuroimaging, cell biology, and post mortem analysis are starting to explain what happens in the brain of a person who develops schizophrenia. Schizophrenia appears to be a developmental disorder of disrupted neural connection within and between regions of the brain. These disruptions seem to result from genetic predispositions interacting with negative environmental events.
A matter of gray and white
Individuals with schizophrenia have deficits in gray matter and white matter, as illustrated by studies linking auditory hallucinations with brain regions associated with normal hearing (Box).
Gray matter. Magnetic resonance imaging (MRI) indicates that gray matter volume peaks in early adolescence and declines with age. The normal adolescent brain shrinks as inefficient neural connections are pruned away, a process that refines and matures gray matter. In individuals with schizophrenia, this reduction is more aggressive—perhaps because of excessive pruning—and occurs in the time frame when schizophrenia symptoms typically emerge.
Rapoport et al1 documented this process through sequential MRI scans in children with early-onset schizophrenia (mean age 14.5). Compared with age-matched healthy controls, youths with schizophrenia show greater and more rapid gray matter loss during late adolescence (Figure 1).2
Increased density. Reduced neuronal branching and spine formation also likely causes subtle reductions in gray matter volume (Figure 2). The resulting lack of dendritic connectivity may produce cognitive impairments and negative symptoms seen in schizophrenia.
Postmortem studies of gray matter cells show increased neuron density in patients with schizophrenia when compared with controls.3 Patients with schizophrenia have the same number of neurons as controls, but the neurons are more tightly packed because of reduced cell size, branching, and synapse formation.4
Research over the past decade has revealed schizophrenia to be a neurodegenerative disorder characterized by substantial brain tissue loss during first and subsequent psychotic episodes.5 Neuroimaging studies show that clinical and functional deterioration accompanies progressive loss of cortical gray matter volume and enlargement of cerebral ventricles. Thus, preventing relapses has come to be regarded as critical to long-term schizophrenia management.
Auditory hallucinations appear to emanate from the temporal lobe, the same brain region that processes external sound. Thus, it may be that patients experiencing hallucinations are misidentifying inner speech as coming from an outside source.
Using functional MRI to differentiate brain activity signals associated with hallucinating and nonhallucinating states, Dierks et al21 documented increased activity in auditory cortical gray matter during hallucinations in schizophrenia patients.
Auditory signals make synaptic connections in the thalamus (left) before reaching the auditory cortex. White matter fiber tracts called the arcuate fasciculus (right) connect the auditory cortex in the temporal lobe with Broca’s area in the frontal cortex.
Source: Adapted from reference 2
Using MR diffusion tensor imaging, Hubl et al22 identified white matter changes in the arcuate fasciculus of schizophrenia patients prone to hallucinations, compared with healthy controls and patients who had schizophrenia but not hallucinations.
These findings support the understanding that auditory hallucinations originate from altered connectivity of the same regions that process normal hearing and speech. The schizophrenia patient may perceive external voices from aberrant internal signals.
Figure 1 Rates of gray matter volume loss during adolescence
Youths with early-onset schizophrenia show greater gray matter volume loss during adolescence, compared with normal controls.
Source: Adapted from reference 2
Figure 2 Structural differences between neurons
in patients with schizophrenia and controls
Schizophrenic neurons show reduced soma size, spine formation, and dendritic branching
Source: Adapted from reference 2White matter. Recent research suggests that white matter deficits also may be involved in schizophrenia’s pathophysiology. Studies using diffusion tensor imaging (DTI)—which measures the sum of vectors of water diffusion along axons—have documented white matter impairments in patients with schizophrenia.6
White matter tracks—myelinated axons that transport electrical signals among neurons—connect regions within the cortex and between the cortex and deeper brain structures. Disruption of white matter tracks may degrade signals and confuse neuronal communication.
Myelination. Genetic studies in patients with schizophrenia also have suggested that decreased neuron myelination may play a role in white matter deficits. Hakak et al8 examined more than 6,000 genes using microarray analysis and found only 17 genes were significantly down-regulated in patients with schizophrenia. Of those 17 genes, 6 were related to myelin and 11 showed no pattern.
Oligodendrocytes are glial cells that insulate axons with myelin and allow faster transmission of electrical impulses in the brain. In a postmortem study, Hof et al7 found 7 patients schizophrenia had 28% fewer oligodendrocytes per section of the superior frontal gyrus and 27% less white matter compared with 7 age-matched controls (Figure 3).
Figure 3 Reduced neuron myelination possible in schizophrenia
In a postmortem analysis, stained white matter sections taken from schizophrenia patients had fewer oligodendrocytes, cells that insulate axons with myelin and facilitate electrical transmission.
Source: Adapted from reference 2
Genes and the environment
Schizophrenia’s heritability is among the most repeated research findings in psychiatry.9 Other mechanisms besides genetics must be involved, however, as studies consistently show that monozygotic twins have a concordance rate of approximately 50% for the development of schizophrenia.
Environmental factors. Adverse environmental events may act in conjuction with genetic predisposition to trigger schizophrenia development. Ischemia or an impoverished diet, for example, have the potential to change DNA methylation.
Environmental factors associated with increased risk for schizophrenia include:
- maternal starvation during pregnancy10
- prenatal exposure to influenza11
- obstetrical complications with hypoxia12
- being born and raised in an urban environment13
- using marijuana during adolescence.14
Gene expression. Important genes may be silenced in individuals with increased DNA methylation and a susceptible genetic profile. Alterations in gene expression are the fundamental mechanism of behavioral change. Research shows that environmental events can alter gene expression without changing the genetic code, such as by adding methyl groups to DNA.15,16 The silencing of important developmental genes in this way can have devastating effects on development.
One explanation for the development of schizophrenia is that environmental events in susceptible individuals silence the production of proteins essential for maintaining neuronal connections through methylation of DNA. Postmortem analysis of brains of patients with schizophrenia show reduced mRNA of reelin,17 a protein produced in gamma-aminobutyric acid neurons involved in neuronal migration, axon branching, and synapse formation during brain development. Lowered production of proteins such as reelin may reduce connections between neurons and cause schizophrenia symptoms. Two research groups also have reported increased methylation of reelin DNA in postmortem studies of the brains of patients with schizophrenia.18,19 Increased methylation of DNA would silence production of this important protein.
Preventing neural disconnects? If schizophrenia is a developmental disorder resulting from failures in brain connectivity, then the ultimate treatment may be prevention. Recent research suggests that intervening with second-generation antipsychotics during the prodromal stage can prevent or delay the emergence of the disorder.20 Further research is needed to establish whether early intervention can prevent schizophrenia’s neuronal disruption.
1. Gogtay N, Sporn A, Rapoport J. Structural brain MRI studies in childhood-onset schizophrenia and childhood atypical psychosis. In: Lawrie S, Johnstone E, Weinberger D, eds. Schizophrenia: from neuroimaging to neuroscience. New York, NY: Oxford University Press; 2004.
2. Higgins ES, George MS. The neuroscience of clinical psychiatry. Philadelphia: Lippincott, Williams, and Wilkins; 2007.
3. Selemon LD. Increased cortical neuronal density in schizophrenia. Am J Psychiatry 2004;161(9):1564.-
4. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57(1):65-73.
5. Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World Journal 2007;7:135-43.
6. Kubicki M, McCarley R, Westin CF, et al. A review of diffusion tensor imaging studies in schizophrenia. J Psychiatr Res 2007;41(1-2):15-30.
7. Hof PR, Haroutunian V, Friedrich VL, Jr, et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003;53(12):1075-85.
8. Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001;98(8):4746-51.
9. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatr 2004;16(4):260-83.
10. McClellan JM, Susser E, King MC. Maternal famine, de novo mutations, and schizophrenia. JAMA 2006;296(5):582-4.
11. Limosin F, Rouillon F, Payan C, et al. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003;107(5):331-5.
12. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002;159(7):1080-92.
13. Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry 2006;6:2.-
14. Arendt M, Rosenberg R, Foldager L, et al. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry 2005;187:510-5.
15. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics 2003;33(suppl):245-54.
16. Abdolmaleky HM, Smith CL, Faraone SV, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet 2004;127(1):51-9.
17. Fatemi SH, Stary JM, Earle JA, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005;72(2-3):109-22.
18. Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005;134(1):60-6.
19. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102(26):9341-6.
20. McGlashan TH, Zipursky RB, Perkins D, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 2006;163(5):790-799.
21. Dierks T, Linden DE, Jandl M, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron 1999;22(3):615-21.
22. Hubl D, Koenig T, Strik W, et al. Pathways that make voices: white matter changes in auditory hallucinations. Arch Gen Psychiatry 2004;61(7):658-68.
Adapted from The neuroscience of clinical psychiatry, by Edmund S. Higgins and Mark S. George. Philadelphia: Lippincott, Williams, and Wilkins; 2007:251-63.
1. Gogtay N, Sporn A, Rapoport J. Structural brain MRI studies in childhood-onset schizophrenia and childhood atypical psychosis. In: Lawrie S, Johnstone E, Weinberger D, eds. Schizophrenia: from neuroimaging to neuroscience. New York, NY: Oxford University Press; 2004.
2. Higgins ES, George MS. The neuroscience of clinical psychiatry. Philadelphia: Lippincott, Williams, and Wilkins; 2007.
3. Selemon LD. Increased cortical neuronal density in schizophrenia. Am J Psychiatry 2004;161(9):1564.-
4. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57(1):65-73.
5. Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World Journal 2007;7:135-43.
6. Kubicki M, McCarley R, Westin CF, et al. A review of diffusion tensor imaging studies in schizophrenia. J Psychiatr Res 2007;41(1-2):15-30.
7. Hof PR, Haroutunian V, Friedrich VL, Jr, et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003;53(12):1075-85.
8. Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001;98(8):4746-51.
9. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatr 2004;16(4):260-83.
10. McClellan JM, Susser E, King MC. Maternal famine, de novo mutations, and schizophrenia. JAMA 2006;296(5):582-4.
11. Limosin F, Rouillon F, Payan C, et al. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003;107(5):331-5.
12. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002;159(7):1080-92.
13. Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry 2006;6:2.-
14. Arendt M, Rosenberg R, Foldager L, et al. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry 2005;187:510-5.
15. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics 2003;33(suppl):245-54.
16. Abdolmaleky HM, Smith CL, Faraone SV, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet 2004;127(1):51-9.
17. Fatemi SH, Stary JM, Earle JA, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005;72(2-3):109-22.
18. Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005;134(1):60-6.
19. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102(26):9341-6.
20. McGlashan TH, Zipursky RB, Perkins D, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 2006;163(5):790-799.
21. Dierks T, Linden DE, Jandl M, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron 1999;22(3):615-21.
22. Hubl D, Koenig T, Strik W, et al. Pathways that make voices: white matter changes in auditory hallucinations. Arch Gen Psychiatry 2004;61(7):658-68.
Adapted from The neuroscience of clinical psychiatry, by Edmund S. Higgins and Mark S. George. Philadelphia: Lippincott, Williams, and Wilkins; 2007:251-63.