User login

The US Food and Drug Administration (FDA) has granted priority review and breakthrough therapy designation to vemurafenib (Zelboraf®) as a treatment for Erdheim-Chester disease (ECD) with BRAF V600 mutation.
Vemurafenib is a kinase inhibitor designed to inhibit some mutated forms of BRAF.
The drug is already FDA-approved to treat patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
The FDA is expected to make a decision on the approval of vemurafenib in ECD by December 7, 2017.
The supplemental new drug application for vemurafenib in this indication is supported by data from the phase 2 VE-BASKET study.
VE-BASKET was designed to investigate the use of vemurafenib in patients with BRAF V600 mutation-positive diseases, including ECD.
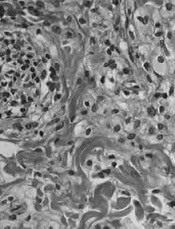
Final results for the 22 people with ECD showed a best overall response rate of 54.5%. The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up of 26.6 months.
The most common adverse events were joint pain, rash, hair loss, change in heart rhythm, fatigue, skin tags, diarrhea, and thickening of the skin. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain.
Initial results from this study were published in NEJM in August 2015.
About priority review
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted priority review and breakthrough therapy designation to vemurafenib (Zelboraf®) as a treatment for Erdheim-Chester disease (ECD) with BRAF V600 mutation.
Vemurafenib is a kinase inhibitor designed to inhibit some mutated forms of BRAF.
The drug is already FDA-approved to treat patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
The FDA is expected to make a decision on the approval of vemurafenib in ECD by December 7, 2017.
The supplemental new drug application for vemurafenib in this indication is supported by data from the phase 2 VE-BASKET study.
VE-BASKET was designed to investigate the use of vemurafenib in patients with BRAF V600 mutation-positive diseases, including ECD.
Final results for the 22 people with ECD showed a best overall response rate of 54.5%. The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up of 26.6 months.
The most common adverse events were joint pain, rash, hair loss, change in heart rhythm, fatigue, skin tags, diarrhea, and thickening of the skin. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain.
Initial results from this study were published in NEJM in August 2015.
About priority review
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()
The US Food and Drug Administration (FDA) has granted priority review and breakthrough therapy designation to vemurafenib (Zelboraf®) as a treatment for Erdheim-Chester disease (ECD) with BRAF V600 mutation.
Vemurafenib is a kinase inhibitor designed to inhibit some mutated forms of BRAF.
The drug is already FDA-approved to treat patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
The FDA is expected to make a decision on the approval of vemurafenib in ECD by December 7, 2017.
The supplemental new drug application for vemurafenib in this indication is supported by data from the phase 2 VE-BASKET study.
VE-BASKET was designed to investigate the use of vemurafenib in patients with BRAF V600 mutation-positive diseases, including ECD.
Final results for the 22 people with ECD showed a best overall response rate of 54.5%. The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up of 26.6 months.
The most common adverse events were joint pain, rash, hair loss, change in heart rhythm, fatigue, skin tags, diarrhea, and thickening of the skin. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain.
Initial results from this study were published in NEJM in August 2015.
About priority review
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need. ![]()