User login
A 69-year-old woman presented to the clinic with pain in the right great toe lasting several days. She was prescribed colchicine and indomethacin empirically for gout. She took one tablet of colchicine (0.6 mg) every hour until her stools became loose after the eighth tablet. Her toe pain resolved, but two days later she developed bilateral lower extremity pruritus and paresthesia and presented to the emergency department (ED). On physical examination, no rash, weakness, or sensory deficits were observed, and she was able to ambulate without assistance. Her patellar reflexes were normal. The complete blood count was notable for an absolute lymphocyte count of 6,120/µL (normal: 1,100-4,800), and the comprehensive metabolic panel was normal. Serum creatine kinase (CK) was 341 U/L (normal: 24-170) and uric acid 7.7 mg/dL (normal: 2.4-6.4). Her lower extremity symptoms were attributed to colchicine, which was discontinued. She was prescribed diphenhydramine and discharged home.
Monoarthritis of the hallux is the classic manifestation of gout, although other considerations include pseudogout, sesamoiditis, and trauma. The typical side effects of colchicine include diarrhea and myositis. Colchicine-induced muscle injury often results in a modest elevation of CK levels and is associated with myalgia.
Paresthesia is defined as abnormal sensory symptoms that most commonly localize to the peripheral nerves or spinal cord. Acute neuropathies or myelopathies might result from vasculitis, heavy metal toxicity, vitamin deficiencies, and paraneoplastic neurologic syndromes. The normal motor, sensory, and reflex examination, however, make these unlikely.
The neuro-anatomic localization of pruritus is poorly understood but is proposed to include peripheral nerves, spinothalamic tracts, and thalami. Acute pruritus (lasting <6 weeks) typically results from a primary dermatologic process such as a drug reaction, eczema, or xerosis. Less common causes include uremia, cholestasis, and thyroid disease. Pruritus can also be seen with malignancy, most commonly hematologic or paraneoplastic syndromes, or with connective tissue diseases. At this stage, it is unclear whether her pruritus and paresthesia are part of a unifying disease process.
Five days later she re-presented to the ED with nausea and emesis after eating at a restaurant. Her symptoms improved with intravenous fluids, and she was discharged. Four days later she returned with difficulty ambulating, bilateral leg cramping, and continued pruritus and paresthesia. The chemistry panel was normal except for a potassium level of 2.6 mmol/L and a bicarbonate level of 32 mmol/L. She was admitted to the hospital because of severe hypokalemia and impaired ability to ambulate. Her potassium was replenished. Her CK was elevated (3,551 U/L on hospital day 7). She was given cyclobenzaprine, gabapentin, oxycodone, acetaminophen, and prednisone (40 mg); her cramping only mildly improved, and she remained unable to walk. On hospital day five she had visual hallucinations and confusion, which did not resolve with administration of haloperidol; a head CT was unremarkable. On hospital day eight the patient, with her family’s support, left the hospital and presented to a different ED for a second opinion.
Difficulty ambulating often results from weakness, sensory impairment, cerebellar ataxia, extrapyramidal dysfunction (eg, parkinsonism), and pain. In this patient, leg cramping suggests pain or true weakness due to a myopathic process as a contributing factor. Symptoms of muscle disease include cramps, myalgia, and difficulty walking. Causes of elevated CK and myalgia include inflammatory myopathies, endocrinopathies, drugs, infections, and electrolyte abnormalities (eg, hypokalemia). Her age and acuity of presentation decrease the likelihood of a metabolic myopathy due to a disorder of glycogen storage, lipid metabolism, or mitochondrial function. Her hypokalemic metabolic alkalosis likely resulted from vomiting. Hypokalemic periodic paralysis is unlikely as exacerbations typically only last hours to days. As such, her difficulty ambulating, muscle cramps, and elevated CK strongly support a primary myopathic disorder, although additional information regarding the neurologic examination is still required.
Acute changes in mental status without corresponding changes in cranial nerve, motor, or sensory function are common in the hospital setting and frequently relate to delirium, which is the most likely explanation for her confusion. Her age and exposure to muscle relaxants, opiates, and corticosteroids increase her risk considerably. Other possible explanations for isolated changes in mental status include nonconvulsive seizures, central nervous system (CNS) infection, and strokes that involve the thalamus, nondominant parietal lobe, and reticular activating system. A shower of emboli resulting in small multifocal strokes can have the same effect.
She was re-evaluated by her new providers. Her only prior medical history was hypertension, which was treated at home with atenolol and amlodipine. She had emigrated from Nigeria to the US many years prior. She occasionally consumed alcohol and never smoked tobacco or used illicit drugs. She was unsure if she had received a tetanus booster in the past 10 years.
On physical examination, her temperature was 36°C, blood pressure 149/70 mm Hg, pulse 56 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 98% on ambient air. She was diaphoretic and appeared anxious, grabbing both bedrails out of fear of falling. Cardiovascular, pulmonary, abdominal, and skin examinations were normal. She was alert and oriented to her identity, her location, and the time. Cranial nerves II to XII were normal. Tone was normal in her upper extremities but markedly increased in her lower extremities and back. There were spontaneous and stimulus-induced painful spasms, predominantly involving her axial muscles and distal lower extremities. Muscle bulk was normal. Strength was normal in the upper extremities and could not be assessed in the lower extremities due to rigidity. Reflexes were 2+ and symmetric throughout with downgoing toes on Babinski testing. A sensory examination was normal. Gait could not be tested because of the severe muscle spasms. The patient was admitted to the hospital.
Localized muscle spasms may be caused by muscle overuse, but more generalized spasms are associated with systemic diseases such as electrolyte disturbances, toxidromes, tetanus, peripheral nerve hyperexcitability syndromes (including Isaacs syndrome and Morvan syndrome), or stiff person syndrome (SPS). Hypokalemia is unlikely the cause as its correction did not improve her symptoms. Although tetanus is rare in the United States, it remains endemic in the developing world and can cause focal as well as generalized stimulus-induced spasms. The patient should be asked about potential exposure to Clostridium tetani infection, such as incurring a puncture wound. It is also important to consider neuroleptic malignant syndrome and serotonin syndrome, which can cause confusion, elevated CK, and increased muscle tone. Her confusion, however, was transient and the elevated CK preceded the administration of haloperidol.
SPS and progressive encephalomyelitis with rigidity and myoclonus (PERM) provide better explanations for her presentation. Both diseases cause severe spasms, impaired ambulation, and stiffness. They differ in their acuity of onset, accompanying symptoms, antibody associations, and responses to treatment. The rapid onset, paresthesia, and confusion seen in this patient are atypical of SPS. SPS usually presents with subacute-to-chronic stiffness or soreness of muscles in the back and lower extremities, followed by the upper extremities. Rigidity, stimulation-provoked spasms, hyperlordosis, and difficulty ambulating are typically later-stage findings. Her rapid escalation of symptoms is more consistent with PERM, which is often more acute and progressive than typical SPS; however, unlike this patient, PERM commonly causes widespread CNS dysfunction, including persistent encephalopathy, cranial neuropathies, hyperreflexia, and autonomic instability. Both are rare diagnoses that can manifest as a paraneoplastic neurologic syndrome.
Blood tests showed a leukocyte count of 17,350/µL, neutrophils 8,720/µL (normal: 1,500–7,800), lymphocytes 6,130/µL, hemoglobin 11.3 g/dL, and platelets 231,000/µL. The basic metabolic panel was normal. Serum total protein was 6.7 g/dL with albumin 3.5 g/dL. Aspartate aminotransferase (AST) was 94 U/L (normal: 0-31), alanine aminotransferase (ALT) 56 U/L (normal: 0-31), alkaline phosphatase 45 U/L, and total bilirubin 1.1 mg/dL. Vitamin B12 was 868 pg/mL. Hemoglobin A1c and thyrotropin levels were normal. Creatine kinase was 3,757 U/L and lactate dehydrogenase (LDH) 435 U/L (normal: 122-220). The syphilis treponemal test and hepatitis B surface antigen were negative. HIV and hepatitis C antibodies were nonreactive. The anti-nuclear antibody screen was negative and complement C3 and C4 were normal.
Neutrophilia likely reflects glucocorticoid-induced demargination, as opposed to an infectious process, given the temporal association with steroid administration. Persistent mild lymphocytosis is nonspecific but more likely to reflect a reactive rather than a clonal process. Elevated LDH and CK, as well as a greater increase of AST relative to ALT, suggest muscle injury, although mild concomitant hepatic injury cannot be excluded. Normal or negative serum studies for TSH, HIV, ANA, peripheral blood smear, and creatinine eliminate many of the systemic causes of her pruritus, but malignancy and associated paraneoplastic etiologies remain considerations.
The initial work-up for SPS includes electromyography (EMG) which would show spontaneous muscle activity. Her poorly localized sensory abnormalities, transient vestibular symptoms, and confusion warrant an MRI of the brain and spine to evaluate for inflammation (eg, encephalomyelitis), which could be consistent with PERM.
An MRI of the brain and cervicothoracic spine without contrast was significantly limited by motion artifact but without obvious intracranial or cord signal abnormalities. Electromyography demonstrated spontaneous muscle activity in both lower extremities with co-contraction of agonist and antagonist muscles (hamstrings and quadriceps as well as medial gastrocnemius and tibialis anterior). Sensory and motor nerve conductions were normal. Cerebral spinal fluid (CSF) contained six leukocytes (96% lymphocytes) and three red blood cells per microliter; glucose was 67 mg/dL and protein 24 mg/dL. There were two oligoclonal bands unique to the CSF. Cytology was negative for malignant cells.
The EMG narrows the differential diagnosis considerably. Co-contraction of opposing flexor and extensor groups (with predominance of extensors) on EMG is a diagnostic criterion for SPS and explains the myalgia and elevated CK. Her normal MRI studies effectively ruled out any focal lesion and did not show signs of encephalitis. Oligoclonal bands in the CSF are a sensitive marker of intrathecal inflammation, although not specific to one diagnosis. The mildly elevated cell count also supports CNS inflammation. In the setting of a lymphocytic pleocytosis and unique oligoclonal bands, it is important to consider infectious, neoplastic, autoimmune, and paraneoplastic causes of neuroinflammatory disorders.
Serum analyses, including antiglutamic acid decarboxylase 65 (GAD65) antibody and anti-amphiphysin antibody, should be ordered. The anti-GAD65 antibody is most commonly elevated in the setting of autoimmune diabetes mellitus; the titer, however, is usually dramatically higher in SPS. The CSF titer of anti-GAD65 antibodies is more specific than the serum titer for SPS. Antibodies against amphiphysin are typically elevated in paraneoplastic SPS, and anti-glycine receptor antibodies are associated with PERM, which commonly does not have elevated anti-GAD65 antibodies.
The serum GAD65 antibody level was greater than 265,000 × 103 IU/µL (normal <5,000), and the CSF level was 11.2 nmol/L (normal: ≤0.02). Serum amphiphysin antibody testing was negative.
Significantly elevated serum and CSF anti-GAD65 antibody levels are highly suggestive of SPS. Stiff person syndrome with rapidly progressive clinical symptoms raises the concern of a paraneoplastic neurologic syndrome. Although anti-amphiphysin antibody – the antibody classically associated with breast cancer and SPS – was negative, anti-GAD65 antibody has been implicated in paraneoplastic SPS with thymoma, lymphoma, and thyroid carcinoma. Paraneoplastic neurologic syndrome can predate a detectable malignancy by several years. As SPS and lymphoma are associated with pruritus and lymphocytosis, imaging is indicated to search for malignancy. Antiglycine receptor antibody, associated with PERM, is not routinely available commercially.
Computed tomography of the chest, abdomen, and pelvis with intravenous contrast revealed a 3.9 × 8.0 × 7.0 cm anterior mediastinal mass (Figure 1, Panel A). Biopsy of the mass demonstrated a thymoma. Given that the patient exhibited no further signs of CNS involvement, her initial transiently altered mental status was attributed to opioids and steroids. As she did not meet the clinical criteria for PERM, testing of antiglycine antibodies was not pursued.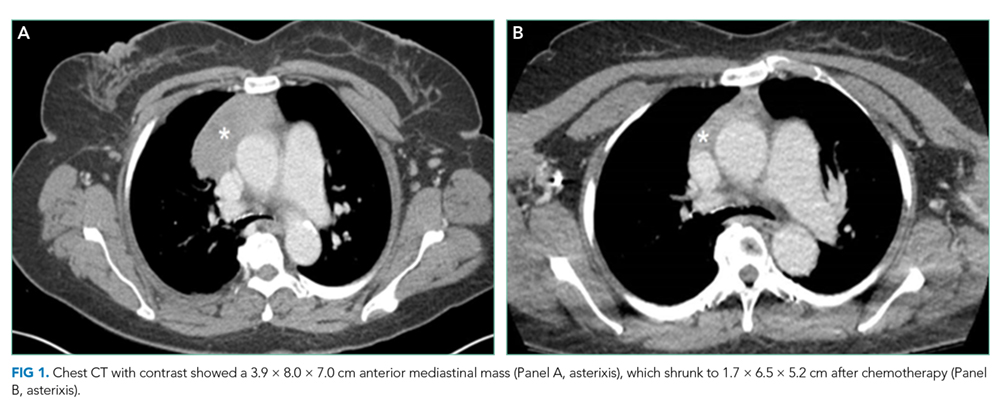
She received scheduled baclofen and diazepam with as needed cyclobenzaprine for continued muscle spasms. Over the next several days, her stiffness, spasms, and myoclonic jerks slowly improved, and she was able to attempt physical therapy (Appendix Video 1; https://youtu.be/d0gLpTgqaCs). She subsequently received intravenous immunoglobulin (IVIG) with further improvement. After five months of scheduled diazepam and baclofen, she was able to ambulate with minimal assistance (Appendix Video 2; https://youtu.be/I00i638u00o). Given the absence of safe tissue planes for resection, the patient received neoadjuvant chemotherapy with four cycles of cyclophosphamide, doxorubicin, and cisplatin. Tumor size decreased to 1.7 × 6.5 × 5.2 cm (Figure 1, Panel B), and she subsequently underwent resection (Figure 2). Pathological analysis demonstrated a type B1 thymoma.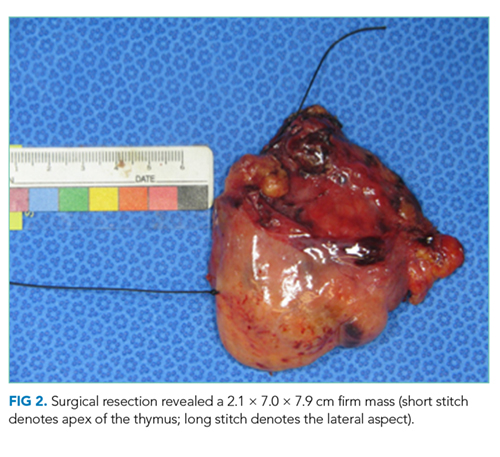
COMMENTARY
SPS is a condition of muscle stiffness and spasticity. Diagnosis is difficult and often delayed due to its rarity, with an approximate prevalence of one to two cases per million people.1 SPS typically occurs in middle age, and women are diagnosed twice as often as men. Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, high-titer antibody to GAD65 or amphiphysin, and the absence of an alternate diagnosis.2 Variant syndromes have been described, including a milder variant limited to the limbs, a severe variant with brainstem and spinal cord involvement, and a paraneoplastic variant.3 This patient’s clinical presentation, EMG findings, and extraordinarily high anti-GAD titers in the serum and CSF were diagnostic of SPS.
The pathophysiology of SPS is associated with autoantibodies targeting proteins such as GAD65, amphiphysin, gephyrin, and GABAA receptor-associated protein (GABARAP). These proteins are critical to gamma-aminobutyric acid (GABA) signaling, the primary inhibitory neurotransmitter pathway in the CNS (Figure 3).4 The formation of GABA from glutamate is catalyzed by GAD65. Gamma-aminobutyric acid is loaded into secretory vesicles, and amphiphysin facilitates vesicle recycling from the synaptic space.5 In the postsynaptic neuron, GABA binds the GABAA receptor, leading to neuronal hyperpolarization and resistance to excitation. The GABAA receptor is clustered on the plasma membrane through a scaffold formed by gephyrin. GABARAP facilitates this clustering, in part by linking GABAA receptors and gephyrin.6 Autoantibodies to these proteins may be pathogenic; however, the direct effects on their targets are unclear. The end result is decreased GABAergic activity, leading to continuous activation of opposing muscle groups. The resulting stiffness is characteristic of this disorder. Colchicine is known to antagonize GABAA receptor signaling, and this may have brought the underlying diagnosis of SPS to clinical attention.7,8
Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines9 and baclofen10 are necessary. When symptoms are not controlled by GABAergic drugs, immunosuppression with corticosteroids and IVIG has been used, as have plasmapheresis and rituximab.11 The efficacy of the latter, however, was not supported by a randomized, placebo-controlled trial.12 This patient experienced significant improvement with benzodiazepines, baclofen, IVIG, and neoadjuvant chemotherapy prior to thymoma resection. The pruritus, paresthesia, and lymphocytosis also resolved with medical therapy. Interestingly, GABA signaling suppresses itch, suggesting that loss of GABAA signaling may have contributed to the development of pruritus.
SPS occasionally occurs as a paraneoplastic neurologic syndrome. Breast cancer is the most commonly associated malignancy, although associations between thymomas and SPS13 with anti-GAD65 antibodies14 have also been described. The presentation of thymomas is variable, with approximately one-third discovered incidentally on imaging, one-third producing symptoms of local compression, and one-third identified in the setting of another syndrome, most commonly myasthenia gravis. In addition to myasthenia gravis, thymomas have been associated with conditions such as hypogammaglobulinemia, pure red cell aplasia, and agranulocytosis. Stiff person syndrome is a known, albeit infrequently associated, condition.15
A critical step in arriving at the relevant differential diagnosis requires correctly framing the patient’s case.16 The treatment team’s initial frame was “a 69-year-old woman with weakness and elevated CK,” which prioritized causes of weakness and myositis. Stiff person syndrome does not cause weakness, but rather impaired movement from marked stiffness and spasms. The patient’s elevated CK was a result of continual muscle contractions. The physical exam and lack of motor deficit on EMG led the treatment team to reframe as “a 69-year-old woman with severe stiffness and spasms.” Egad! This correct frame was the key to diagnosis and confirmed by EMG and GAD65 antibody testing.
KEY LEARNING POINTS
- Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, and high-titer antibody to GAD65 or amphiphysin.
- SPS typically occurs in middle age, and women are diagnosed twice as often as men.
- Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines and baclofenare necessary.
- SPS occasionally occurs as a paraneoplastic neurologic syndrome, most commonly in association with breast cancer.
Acknowledgments
The authors wish to thank Jason Kern, MD for his preparation and interpretation of the pathologic image; and the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education, for supporting Reza Manesh, MD.
Disclosures
The authors have nothing to disclose.
Appendix Video 1: This video was taken during a physical therapy session after 1 week of scheduled benzodiazepine and 2 days of intravenous immunoglobuli
Appendix Video 2: This video was taken 5 months after scheduled diazepam and baclofen, and 1 week prior to thymectomy. (https://youtu.be/I00i638u00o)
1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9
3. Murinson BB. Stiff-person syndrome. Neurologist. 2004;10(3):131-137. doi: 10.1097/01.nrl.0000126587.37087.1a
4. Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45(5):623-634. doi: 10.1002/mus.23234
5. Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic. 2002;3(7):452-460. doi: 10.1034/j.1600-0854.2002.30702.x
6. Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15(3):141-156. doi: 10.1038/nrn3670
7. Bueno OF, Leidenheimer NJ. Colchicine inhibits GABA(A) receptors independently of microtubule depolymerization. Neuropharmacology. 1998;37(3):383-390. doi: 10.1016/S0028-3908(98)00020-3
8. Weiner JL, Buhler AV, Whatley VJ, Harris RA, Dunwiddie TV. Colchicine is a competitive antagonist at human recombinant γ-aminobutyric acidA receptors. J Pharmacol Exp Ther. 1998;284(1):95-102 . PubMed
9. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/S0025-6196(12)65339-7
10. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991
11. Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/WNL.57.5.780
12. Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002
13. Hagiwara H, Enomoto-Nakatani S, Sakai K, et al. Stiff-person syndrome associated with invasive thymoma: a case report. J Neurol Sci. 2001;193(1):59-62. doi: 10.1016/S0022-510X(01)00602-5
14. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270-7275. doi: 10.1158/1078-0432.CCR-04-0735 PubMed
15. Thomas CR, Wright CD, Loehrer PJ. Thymoma: state of the art. J Clin Oncol. 1999;17(7):2280-2289. doi: 10.1200/JCO.1999.17.7.2280 PubMed
16. Stuart S, Hartig JR, Willett L. The importance of framing. J Gen Intern Med. 2017;32(6):706-710. doi: 10.1007/s11606-016-3964-z PubMed
A 69-year-old woman presented to the clinic with pain in the right great toe lasting several days. She was prescribed colchicine and indomethacin empirically for gout. She took one tablet of colchicine (0.6 mg) every hour until her stools became loose after the eighth tablet. Her toe pain resolved, but two days later she developed bilateral lower extremity pruritus and paresthesia and presented to the emergency department (ED). On physical examination, no rash, weakness, or sensory deficits were observed, and she was able to ambulate without assistance. Her patellar reflexes were normal. The complete blood count was notable for an absolute lymphocyte count of 6,120/µL (normal: 1,100-4,800), and the comprehensive metabolic panel was normal. Serum creatine kinase (CK) was 341 U/L (normal: 24-170) and uric acid 7.7 mg/dL (normal: 2.4-6.4). Her lower extremity symptoms were attributed to colchicine, which was discontinued. She was prescribed diphenhydramine and discharged home.
Monoarthritis of the hallux is the classic manifestation of gout, although other considerations include pseudogout, sesamoiditis, and trauma. The typical side effects of colchicine include diarrhea and myositis. Colchicine-induced muscle injury often results in a modest elevation of CK levels and is associated with myalgia.
Paresthesia is defined as abnormal sensory symptoms that most commonly localize to the peripheral nerves or spinal cord. Acute neuropathies or myelopathies might result from vasculitis, heavy metal toxicity, vitamin deficiencies, and paraneoplastic neurologic syndromes. The normal motor, sensory, and reflex examination, however, make these unlikely.
The neuro-anatomic localization of pruritus is poorly understood but is proposed to include peripheral nerves, spinothalamic tracts, and thalami. Acute pruritus (lasting <6 weeks) typically results from a primary dermatologic process such as a drug reaction, eczema, or xerosis. Less common causes include uremia, cholestasis, and thyroid disease. Pruritus can also be seen with malignancy, most commonly hematologic or paraneoplastic syndromes, or with connective tissue diseases. At this stage, it is unclear whether her pruritus and paresthesia are part of a unifying disease process.
Five days later she re-presented to the ED with nausea and emesis after eating at a restaurant. Her symptoms improved with intravenous fluids, and she was discharged. Four days later she returned with difficulty ambulating, bilateral leg cramping, and continued pruritus and paresthesia. The chemistry panel was normal except for a potassium level of 2.6 mmol/L and a bicarbonate level of 32 mmol/L. She was admitted to the hospital because of severe hypokalemia and impaired ability to ambulate. Her potassium was replenished. Her CK was elevated (3,551 U/L on hospital day 7). She was given cyclobenzaprine, gabapentin, oxycodone, acetaminophen, and prednisone (40 mg); her cramping only mildly improved, and she remained unable to walk. On hospital day five she had visual hallucinations and confusion, which did not resolve with administration of haloperidol; a head CT was unremarkable. On hospital day eight the patient, with her family’s support, left the hospital and presented to a different ED for a second opinion.
Difficulty ambulating often results from weakness, sensory impairment, cerebellar ataxia, extrapyramidal dysfunction (eg, parkinsonism), and pain. In this patient, leg cramping suggests pain or true weakness due to a myopathic process as a contributing factor. Symptoms of muscle disease include cramps, myalgia, and difficulty walking. Causes of elevated CK and myalgia include inflammatory myopathies, endocrinopathies, drugs, infections, and electrolyte abnormalities (eg, hypokalemia). Her age and acuity of presentation decrease the likelihood of a metabolic myopathy due to a disorder of glycogen storage, lipid metabolism, or mitochondrial function. Her hypokalemic metabolic alkalosis likely resulted from vomiting. Hypokalemic periodic paralysis is unlikely as exacerbations typically only last hours to days. As such, her difficulty ambulating, muscle cramps, and elevated CK strongly support a primary myopathic disorder, although additional information regarding the neurologic examination is still required.
Acute changes in mental status without corresponding changes in cranial nerve, motor, or sensory function are common in the hospital setting and frequently relate to delirium, which is the most likely explanation for her confusion. Her age and exposure to muscle relaxants, opiates, and corticosteroids increase her risk considerably. Other possible explanations for isolated changes in mental status include nonconvulsive seizures, central nervous system (CNS) infection, and strokes that involve the thalamus, nondominant parietal lobe, and reticular activating system. A shower of emboli resulting in small multifocal strokes can have the same effect.
She was re-evaluated by her new providers. Her only prior medical history was hypertension, which was treated at home with atenolol and amlodipine. She had emigrated from Nigeria to the US many years prior. She occasionally consumed alcohol and never smoked tobacco or used illicit drugs. She was unsure if she had received a tetanus booster in the past 10 years.
On physical examination, her temperature was 36°C, blood pressure 149/70 mm Hg, pulse 56 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 98% on ambient air. She was diaphoretic and appeared anxious, grabbing both bedrails out of fear of falling. Cardiovascular, pulmonary, abdominal, and skin examinations were normal. She was alert and oriented to her identity, her location, and the time. Cranial nerves II to XII were normal. Tone was normal in her upper extremities but markedly increased in her lower extremities and back. There were spontaneous and stimulus-induced painful spasms, predominantly involving her axial muscles and distal lower extremities. Muscle bulk was normal. Strength was normal in the upper extremities and could not be assessed in the lower extremities due to rigidity. Reflexes were 2+ and symmetric throughout with downgoing toes on Babinski testing. A sensory examination was normal. Gait could not be tested because of the severe muscle spasms. The patient was admitted to the hospital.
Localized muscle spasms may be caused by muscle overuse, but more generalized spasms are associated with systemic diseases such as electrolyte disturbances, toxidromes, tetanus, peripheral nerve hyperexcitability syndromes (including Isaacs syndrome and Morvan syndrome), or stiff person syndrome (SPS). Hypokalemia is unlikely the cause as its correction did not improve her symptoms. Although tetanus is rare in the United States, it remains endemic in the developing world and can cause focal as well as generalized stimulus-induced spasms. The patient should be asked about potential exposure to Clostridium tetani infection, such as incurring a puncture wound. It is also important to consider neuroleptic malignant syndrome and serotonin syndrome, which can cause confusion, elevated CK, and increased muscle tone. Her confusion, however, was transient and the elevated CK preceded the administration of haloperidol.
SPS and progressive encephalomyelitis with rigidity and myoclonus (PERM) provide better explanations for her presentation. Both diseases cause severe spasms, impaired ambulation, and stiffness. They differ in their acuity of onset, accompanying symptoms, antibody associations, and responses to treatment. The rapid onset, paresthesia, and confusion seen in this patient are atypical of SPS. SPS usually presents with subacute-to-chronic stiffness or soreness of muscles in the back and lower extremities, followed by the upper extremities. Rigidity, stimulation-provoked spasms, hyperlordosis, and difficulty ambulating are typically later-stage findings. Her rapid escalation of symptoms is more consistent with PERM, which is often more acute and progressive than typical SPS; however, unlike this patient, PERM commonly causes widespread CNS dysfunction, including persistent encephalopathy, cranial neuropathies, hyperreflexia, and autonomic instability. Both are rare diagnoses that can manifest as a paraneoplastic neurologic syndrome.
Blood tests showed a leukocyte count of 17,350/µL, neutrophils 8,720/µL (normal: 1,500–7,800), lymphocytes 6,130/µL, hemoglobin 11.3 g/dL, and platelets 231,000/µL. The basic metabolic panel was normal. Serum total protein was 6.7 g/dL with albumin 3.5 g/dL. Aspartate aminotransferase (AST) was 94 U/L (normal: 0-31), alanine aminotransferase (ALT) 56 U/L (normal: 0-31), alkaline phosphatase 45 U/L, and total bilirubin 1.1 mg/dL. Vitamin B12 was 868 pg/mL. Hemoglobin A1c and thyrotropin levels were normal. Creatine kinase was 3,757 U/L and lactate dehydrogenase (LDH) 435 U/L (normal: 122-220). The syphilis treponemal test and hepatitis B surface antigen were negative. HIV and hepatitis C antibodies were nonreactive. The anti-nuclear antibody screen was negative and complement C3 and C4 were normal.
Neutrophilia likely reflects glucocorticoid-induced demargination, as opposed to an infectious process, given the temporal association with steroid administration. Persistent mild lymphocytosis is nonspecific but more likely to reflect a reactive rather than a clonal process. Elevated LDH and CK, as well as a greater increase of AST relative to ALT, suggest muscle injury, although mild concomitant hepatic injury cannot be excluded. Normal or negative serum studies for TSH, HIV, ANA, peripheral blood smear, and creatinine eliminate many of the systemic causes of her pruritus, but malignancy and associated paraneoplastic etiologies remain considerations.
The initial work-up for SPS includes electromyography (EMG) which would show spontaneous muscle activity. Her poorly localized sensory abnormalities, transient vestibular symptoms, and confusion warrant an MRI of the brain and spine to evaluate for inflammation (eg, encephalomyelitis), which could be consistent with PERM.
An MRI of the brain and cervicothoracic spine without contrast was significantly limited by motion artifact but without obvious intracranial or cord signal abnormalities. Electromyography demonstrated spontaneous muscle activity in both lower extremities with co-contraction of agonist and antagonist muscles (hamstrings and quadriceps as well as medial gastrocnemius and tibialis anterior). Sensory and motor nerve conductions were normal. Cerebral spinal fluid (CSF) contained six leukocytes (96% lymphocytes) and three red blood cells per microliter; glucose was 67 mg/dL and protein 24 mg/dL. There were two oligoclonal bands unique to the CSF. Cytology was negative for malignant cells.
The EMG narrows the differential diagnosis considerably. Co-contraction of opposing flexor and extensor groups (with predominance of extensors) on EMG is a diagnostic criterion for SPS and explains the myalgia and elevated CK. Her normal MRI studies effectively ruled out any focal lesion and did not show signs of encephalitis. Oligoclonal bands in the CSF are a sensitive marker of intrathecal inflammation, although not specific to one diagnosis. The mildly elevated cell count also supports CNS inflammation. In the setting of a lymphocytic pleocytosis and unique oligoclonal bands, it is important to consider infectious, neoplastic, autoimmune, and paraneoplastic causes of neuroinflammatory disorders.
Serum analyses, including antiglutamic acid decarboxylase 65 (GAD65) antibody and anti-amphiphysin antibody, should be ordered. The anti-GAD65 antibody is most commonly elevated in the setting of autoimmune diabetes mellitus; the titer, however, is usually dramatically higher in SPS. The CSF titer of anti-GAD65 antibodies is more specific than the serum titer for SPS. Antibodies against amphiphysin are typically elevated in paraneoplastic SPS, and anti-glycine receptor antibodies are associated with PERM, which commonly does not have elevated anti-GAD65 antibodies.
The serum GAD65 antibody level was greater than 265,000 × 103 IU/µL (normal <5,000), and the CSF level was 11.2 nmol/L (normal: ≤0.02). Serum amphiphysin antibody testing was negative.
Significantly elevated serum and CSF anti-GAD65 antibody levels are highly suggestive of SPS. Stiff person syndrome with rapidly progressive clinical symptoms raises the concern of a paraneoplastic neurologic syndrome. Although anti-amphiphysin antibody – the antibody classically associated with breast cancer and SPS – was negative, anti-GAD65 antibody has been implicated in paraneoplastic SPS with thymoma, lymphoma, and thyroid carcinoma. Paraneoplastic neurologic syndrome can predate a detectable malignancy by several years. As SPS and lymphoma are associated with pruritus and lymphocytosis, imaging is indicated to search for malignancy. Antiglycine receptor antibody, associated with PERM, is not routinely available commercially.
Computed tomography of the chest, abdomen, and pelvis with intravenous contrast revealed a 3.9 × 8.0 × 7.0 cm anterior mediastinal mass (Figure 1, Panel A). Biopsy of the mass demonstrated a thymoma. Given that the patient exhibited no further signs of CNS involvement, her initial transiently altered mental status was attributed to opioids and steroids. As she did not meet the clinical criteria for PERM, testing of antiglycine antibodies was not pursued.
She received scheduled baclofen and diazepam with as needed cyclobenzaprine for continued muscle spasms. Over the next several days, her stiffness, spasms, and myoclonic jerks slowly improved, and she was able to attempt physical therapy (Appendix Video 1; https://youtu.be/d0gLpTgqaCs). She subsequently received intravenous immunoglobulin (IVIG) with further improvement. After five months of scheduled diazepam and baclofen, she was able to ambulate with minimal assistance (Appendix Video 2; https://youtu.be/I00i638u00o). Given the absence of safe tissue planes for resection, the patient received neoadjuvant chemotherapy with four cycles of cyclophosphamide, doxorubicin, and cisplatin. Tumor size decreased to 1.7 × 6.5 × 5.2 cm (Figure 1, Panel B), and she subsequently underwent resection (Figure 2). Pathological analysis demonstrated a type B1 thymoma.
COMMENTARY
SPS is a condition of muscle stiffness and spasticity. Diagnosis is difficult and often delayed due to its rarity, with an approximate prevalence of one to two cases per million people.1 SPS typically occurs in middle age, and women are diagnosed twice as often as men. Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, high-titer antibody to GAD65 or amphiphysin, and the absence of an alternate diagnosis.2 Variant syndromes have been described, including a milder variant limited to the limbs, a severe variant with brainstem and spinal cord involvement, and a paraneoplastic variant.3 This patient’s clinical presentation, EMG findings, and extraordinarily high anti-GAD titers in the serum and CSF were diagnostic of SPS.
The pathophysiology of SPS is associated with autoantibodies targeting proteins such as GAD65, amphiphysin, gephyrin, and GABAA receptor-associated protein (GABARAP). These proteins are critical to gamma-aminobutyric acid (GABA) signaling, the primary inhibitory neurotransmitter pathway in the CNS (Figure 3).4 The formation of GABA from glutamate is catalyzed by GAD65. Gamma-aminobutyric acid is loaded into secretory vesicles, and amphiphysin facilitates vesicle recycling from the synaptic space.5 In the postsynaptic neuron, GABA binds the GABAA receptor, leading to neuronal hyperpolarization and resistance to excitation. The GABAA receptor is clustered on the plasma membrane through a scaffold formed by gephyrin. GABARAP facilitates this clustering, in part by linking GABAA receptors and gephyrin.6 Autoantibodies to these proteins may be pathogenic; however, the direct effects on their targets are unclear. The end result is decreased GABAergic activity, leading to continuous activation of opposing muscle groups. The resulting stiffness is characteristic of this disorder. Colchicine is known to antagonize GABAA receptor signaling, and this may have brought the underlying diagnosis of SPS to clinical attention.7,8
Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines9 and baclofen10 are necessary. When symptoms are not controlled by GABAergic drugs, immunosuppression with corticosteroids and IVIG has been used, as have plasmapheresis and rituximab.11 The efficacy of the latter, however, was not supported by a randomized, placebo-controlled trial.12 This patient experienced significant improvement with benzodiazepines, baclofen, IVIG, and neoadjuvant chemotherapy prior to thymoma resection. The pruritus, paresthesia, and lymphocytosis also resolved with medical therapy. Interestingly, GABA signaling suppresses itch, suggesting that loss of GABAA signaling may have contributed to the development of pruritus.
SPS occasionally occurs as a paraneoplastic neurologic syndrome. Breast cancer is the most commonly associated malignancy, although associations between thymomas and SPS13 with anti-GAD65 antibodies14 have also been described. The presentation of thymomas is variable, with approximately one-third discovered incidentally on imaging, one-third producing symptoms of local compression, and one-third identified in the setting of another syndrome, most commonly myasthenia gravis. In addition to myasthenia gravis, thymomas have been associated with conditions such as hypogammaglobulinemia, pure red cell aplasia, and agranulocytosis. Stiff person syndrome is a known, albeit infrequently associated, condition.15
A critical step in arriving at the relevant differential diagnosis requires correctly framing the patient’s case.16 The treatment team’s initial frame was “a 69-year-old woman with weakness and elevated CK,” which prioritized causes of weakness and myositis. Stiff person syndrome does not cause weakness, but rather impaired movement from marked stiffness and spasms. The patient’s elevated CK was a result of continual muscle contractions. The physical exam and lack of motor deficit on EMG led the treatment team to reframe as “a 69-year-old woman with severe stiffness and spasms.” Egad! This correct frame was the key to diagnosis and confirmed by EMG and GAD65 antibody testing.
KEY LEARNING POINTS
- Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, and high-titer antibody to GAD65 or amphiphysin.
- SPS typically occurs in middle age, and women are diagnosed twice as often as men.
- Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines and baclofenare necessary.
- SPS occasionally occurs as a paraneoplastic neurologic syndrome, most commonly in association with breast cancer.
Acknowledgments
The authors wish to thank Jason Kern, MD for his preparation and interpretation of the pathologic image; and the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education, for supporting Reza Manesh, MD.
Disclosures
The authors have nothing to disclose.
Appendix Video 1: This video was taken during a physical therapy session after 1 week of scheduled benzodiazepine and 2 days of intravenous immunoglobuli
Appendix Video 2: This video was taken 5 months after scheduled diazepam and baclofen, and 1 week prior to thymectomy. (https://youtu.be/I00i638u00o)
A 69-year-old woman presented to the clinic with pain in the right great toe lasting several days. She was prescribed colchicine and indomethacin empirically for gout. She took one tablet of colchicine (0.6 mg) every hour until her stools became loose after the eighth tablet. Her toe pain resolved, but two days later she developed bilateral lower extremity pruritus and paresthesia and presented to the emergency department (ED). On physical examination, no rash, weakness, or sensory deficits were observed, and she was able to ambulate without assistance. Her patellar reflexes were normal. The complete blood count was notable for an absolute lymphocyte count of 6,120/µL (normal: 1,100-4,800), and the comprehensive metabolic panel was normal. Serum creatine kinase (CK) was 341 U/L (normal: 24-170) and uric acid 7.7 mg/dL (normal: 2.4-6.4). Her lower extremity symptoms were attributed to colchicine, which was discontinued. She was prescribed diphenhydramine and discharged home.
Monoarthritis of the hallux is the classic manifestation of gout, although other considerations include pseudogout, sesamoiditis, and trauma. The typical side effects of colchicine include diarrhea and myositis. Colchicine-induced muscle injury often results in a modest elevation of CK levels and is associated with myalgia.
Paresthesia is defined as abnormal sensory symptoms that most commonly localize to the peripheral nerves or spinal cord. Acute neuropathies or myelopathies might result from vasculitis, heavy metal toxicity, vitamin deficiencies, and paraneoplastic neurologic syndromes. The normal motor, sensory, and reflex examination, however, make these unlikely.
The neuro-anatomic localization of pruritus is poorly understood but is proposed to include peripheral nerves, spinothalamic tracts, and thalami. Acute pruritus (lasting <6 weeks) typically results from a primary dermatologic process such as a drug reaction, eczema, or xerosis. Less common causes include uremia, cholestasis, and thyroid disease. Pruritus can also be seen with malignancy, most commonly hematologic or paraneoplastic syndromes, or with connective tissue diseases. At this stage, it is unclear whether her pruritus and paresthesia are part of a unifying disease process.
Five days later she re-presented to the ED with nausea and emesis after eating at a restaurant. Her symptoms improved with intravenous fluids, and she was discharged. Four days later she returned with difficulty ambulating, bilateral leg cramping, and continued pruritus and paresthesia. The chemistry panel was normal except for a potassium level of 2.6 mmol/L and a bicarbonate level of 32 mmol/L. She was admitted to the hospital because of severe hypokalemia and impaired ability to ambulate. Her potassium was replenished. Her CK was elevated (3,551 U/L on hospital day 7). She was given cyclobenzaprine, gabapentin, oxycodone, acetaminophen, and prednisone (40 mg); her cramping only mildly improved, and she remained unable to walk. On hospital day five she had visual hallucinations and confusion, which did not resolve with administration of haloperidol; a head CT was unremarkable. On hospital day eight the patient, with her family’s support, left the hospital and presented to a different ED for a second opinion.
Difficulty ambulating often results from weakness, sensory impairment, cerebellar ataxia, extrapyramidal dysfunction (eg, parkinsonism), and pain. In this patient, leg cramping suggests pain or true weakness due to a myopathic process as a contributing factor. Symptoms of muscle disease include cramps, myalgia, and difficulty walking. Causes of elevated CK and myalgia include inflammatory myopathies, endocrinopathies, drugs, infections, and electrolyte abnormalities (eg, hypokalemia). Her age and acuity of presentation decrease the likelihood of a metabolic myopathy due to a disorder of glycogen storage, lipid metabolism, or mitochondrial function. Her hypokalemic metabolic alkalosis likely resulted from vomiting. Hypokalemic periodic paralysis is unlikely as exacerbations typically only last hours to days. As such, her difficulty ambulating, muscle cramps, and elevated CK strongly support a primary myopathic disorder, although additional information regarding the neurologic examination is still required.
Acute changes in mental status without corresponding changes in cranial nerve, motor, or sensory function are common in the hospital setting and frequently relate to delirium, which is the most likely explanation for her confusion. Her age and exposure to muscle relaxants, opiates, and corticosteroids increase her risk considerably. Other possible explanations for isolated changes in mental status include nonconvulsive seizures, central nervous system (CNS) infection, and strokes that involve the thalamus, nondominant parietal lobe, and reticular activating system. A shower of emboli resulting in small multifocal strokes can have the same effect.
She was re-evaluated by her new providers. Her only prior medical history was hypertension, which was treated at home with atenolol and amlodipine. She had emigrated from Nigeria to the US many years prior. She occasionally consumed alcohol and never smoked tobacco or used illicit drugs. She was unsure if she had received a tetanus booster in the past 10 years.
On physical examination, her temperature was 36°C, blood pressure 149/70 mm Hg, pulse 56 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 98% on ambient air. She was diaphoretic and appeared anxious, grabbing both bedrails out of fear of falling. Cardiovascular, pulmonary, abdominal, and skin examinations were normal. She was alert and oriented to her identity, her location, and the time. Cranial nerves II to XII were normal. Tone was normal in her upper extremities but markedly increased in her lower extremities and back. There were spontaneous and stimulus-induced painful spasms, predominantly involving her axial muscles and distal lower extremities. Muscle bulk was normal. Strength was normal in the upper extremities and could not be assessed in the lower extremities due to rigidity. Reflexes were 2+ and symmetric throughout with downgoing toes on Babinski testing. A sensory examination was normal. Gait could not be tested because of the severe muscle spasms. The patient was admitted to the hospital.
Localized muscle spasms may be caused by muscle overuse, but more generalized spasms are associated with systemic diseases such as electrolyte disturbances, toxidromes, tetanus, peripheral nerve hyperexcitability syndromes (including Isaacs syndrome and Morvan syndrome), or stiff person syndrome (SPS). Hypokalemia is unlikely the cause as its correction did not improve her symptoms. Although tetanus is rare in the United States, it remains endemic in the developing world and can cause focal as well as generalized stimulus-induced spasms. The patient should be asked about potential exposure to Clostridium tetani infection, such as incurring a puncture wound. It is also important to consider neuroleptic malignant syndrome and serotonin syndrome, which can cause confusion, elevated CK, and increased muscle tone. Her confusion, however, was transient and the elevated CK preceded the administration of haloperidol.
SPS and progressive encephalomyelitis with rigidity and myoclonus (PERM) provide better explanations for her presentation. Both diseases cause severe spasms, impaired ambulation, and stiffness. They differ in their acuity of onset, accompanying symptoms, antibody associations, and responses to treatment. The rapid onset, paresthesia, and confusion seen in this patient are atypical of SPS. SPS usually presents with subacute-to-chronic stiffness or soreness of muscles in the back and lower extremities, followed by the upper extremities. Rigidity, stimulation-provoked spasms, hyperlordosis, and difficulty ambulating are typically later-stage findings. Her rapid escalation of symptoms is more consistent with PERM, which is often more acute and progressive than typical SPS; however, unlike this patient, PERM commonly causes widespread CNS dysfunction, including persistent encephalopathy, cranial neuropathies, hyperreflexia, and autonomic instability. Both are rare diagnoses that can manifest as a paraneoplastic neurologic syndrome.
Blood tests showed a leukocyte count of 17,350/µL, neutrophils 8,720/µL (normal: 1,500–7,800), lymphocytes 6,130/µL, hemoglobin 11.3 g/dL, and platelets 231,000/µL. The basic metabolic panel was normal. Serum total protein was 6.7 g/dL with albumin 3.5 g/dL. Aspartate aminotransferase (AST) was 94 U/L (normal: 0-31), alanine aminotransferase (ALT) 56 U/L (normal: 0-31), alkaline phosphatase 45 U/L, and total bilirubin 1.1 mg/dL. Vitamin B12 was 868 pg/mL. Hemoglobin A1c and thyrotropin levels were normal. Creatine kinase was 3,757 U/L and lactate dehydrogenase (LDH) 435 U/L (normal: 122-220). The syphilis treponemal test and hepatitis B surface antigen were negative. HIV and hepatitis C antibodies were nonreactive. The anti-nuclear antibody screen was negative and complement C3 and C4 were normal.
Neutrophilia likely reflects glucocorticoid-induced demargination, as opposed to an infectious process, given the temporal association with steroid administration. Persistent mild lymphocytosis is nonspecific but more likely to reflect a reactive rather than a clonal process. Elevated LDH and CK, as well as a greater increase of AST relative to ALT, suggest muscle injury, although mild concomitant hepatic injury cannot be excluded. Normal or negative serum studies for TSH, HIV, ANA, peripheral blood smear, and creatinine eliminate many of the systemic causes of her pruritus, but malignancy and associated paraneoplastic etiologies remain considerations.
The initial work-up for SPS includes electromyography (EMG) which would show spontaneous muscle activity. Her poorly localized sensory abnormalities, transient vestibular symptoms, and confusion warrant an MRI of the brain and spine to evaluate for inflammation (eg, encephalomyelitis), which could be consistent with PERM.
An MRI of the brain and cervicothoracic spine without contrast was significantly limited by motion artifact but without obvious intracranial or cord signal abnormalities. Electromyography demonstrated spontaneous muscle activity in both lower extremities with co-contraction of agonist and antagonist muscles (hamstrings and quadriceps as well as medial gastrocnemius and tibialis anterior). Sensory and motor nerve conductions were normal. Cerebral spinal fluid (CSF) contained six leukocytes (96% lymphocytes) and three red blood cells per microliter; glucose was 67 mg/dL and protein 24 mg/dL. There were two oligoclonal bands unique to the CSF. Cytology was negative for malignant cells.
The EMG narrows the differential diagnosis considerably. Co-contraction of opposing flexor and extensor groups (with predominance of extensors) on EMG is a diagnostic criterion for SPS and explains the myalgia and elevated CK. Her normal MRI studies effectively ruled out any focal lesion and did not show signs of encephalitis. Oligoclonal bands in the CSF are a sensitive marker of intrathecal inflammation, although not specific to one diagnosis. The mildly elevated cell count also supports CNS inflammation. In the setting of a lymphocytic pleocytosis and unique oligoclonal bands, it is important to consider infectious, neoplastic, autoimmune, and paraneoplastic causes of neuroinflammatory disorders.
Serum analyses, including antiglutamic acid decarboxylase 65 (GAD65) antibody and anti-amphiphysin antibody, should be ordered. The anti-GAD65 antibody is most commonly elevated in the setting of autoimmune diabetes mellitus; the titer, however, is usually dramatically higher in SPS. The CSF titer of anti-GAD65 antibodies is more specific than the serum titer for SPS. Antibodies against amphiphysin are typically elevated in paraneoplastic SPS, and anti-glycine receptor antibodies are associated with PERM, which commonly does not have elevated anti-GAD65 antibodies.
The serum GAD65 antibody level was greater than 265,000 × 103 IU/µL (normal <5,000), and the CSF level was 11.2 nmol/L (normal: ≤0.02). Serum amphiphysin antibody testing was negative.
Significantly elevated serum and CSF anti-GAD65 antibody levels are highly suggestive of SPS. Stiff person syndrome with rapidly progressive clinical symptoms raises the concern of a paraneoplastic neurologic syndrome. Although anti-amphiphysin antibody – the antibody classically associated with breast cancer and SPS – was negative, anti-GAD65 antibody has been implicated in paraneoplastic SPS with thymoma, lymphoma, and thyroid carcinoma. Paraneoplastic neurologic syndrome can predate a detectable malignancy by several years. As SPS and lymphoma are associated with pruritus and lymphocytosis, imaging is indicated to search for malignancy. Antiglycine receptor antibody, associated with PERM, is not routinely available commercially.
Computed tomography of the chest, abdomen, and pelvis with intravenous contrast revealed a 3.9 × 8.0 × 7.0 cm anterior mediastinal mass (Figure 1, Panel A). Biopsy of the mass demonstrated a thymoma. Given that the patient exhibited no further signs of CNS involvement, her initial transiently altered mental status was attributed to opioids and steroids. As she did not meet the clinical criteria for PERM, testing of antiglycine antibodies was not pursued.
She received scheduled baclofen and diazepam with as needed cyclobenzaprine for continued muscle spasms. Over the next several days, her stiffness, spasms, and myoclonic jerks slowly improved, and she was able to attempt physical therapy (Appendix Video 1; https://youtu.be/d0gLpTgqaCs). She subsequently received intravenous immunoglobulin (IVIG) with further improvement. After five months of scheduled diazepam and baclofen, she was able to ambulate with minimal assistance (Appendix Video 2; https://youtu.be/I00i638u00o). Given the absence of safe tissue planes for resection, the patient received neoadjuvant chemotherapy with four cycles of cyclophosphamide, doxorubicin, and cisplatin. Tumor size decreased to 1.7 × 6.5 × 5.2 cm (Figure 1, Panel B), and she subsequently underwent resection (Figure 2). Pathological analysis demonstrated a type B1 thymoma.
COMMENTARY
SPS is a condition of muscle stiffness and spasticity. Diagnosis is difficult and often delayed due to its rarity, with an approximate prevalence of one to two cases per million people.1 SPS typically occurs in middle age, and women are diagnosed twice as often as men. Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, high-titer antibody to GAD65 or amphiphysin, and the absence of an alternate diagnosis.2 Variant syndromes have been described, including a milder variant limited to the limbs, a severe variant with brainstem and spinal cord involvement, and a paraneoplastic variant.3 This patient’s clinical presentation, EMG findings, and extraordinarily high anti-GAD titers in the serum and CSF were diagnostic of SPS.
The pathophysiology of SPS is associated with autoantibodies targeting proteins such as GAD65, amphiphysin, gephyrin, and GABAA receptor-associated protein (GABARAP). These proteins are critical to gamma-aminobutyric acid (GABA) signaling, the primary inhibitory neurotransmitter pathway in the CNS (Figure 3).4 The formation of GABA from glutamate is catalyzed by GAD65. Gamma-aminobutyric acid is loaded into secretory vesicles, and amphiphysin facilitates vesicle recycling from the synaptic space.5 In the postsynaptic neuron, GABA binds the GABAA receptor, leading to neuronal hyperpolarization and resistance to excitation. The GABAA receptor is clustered on the plasma membrane through a scaffold formed by gephyrin. GABARAP facilitates this clustering, in part by linking GABAA receptors and gephyrin.6 Autoantibodies to these proteins may be pathogenic; however, the direct effects on their targets are unclear. The end result is decreased GABAergic activity, leading to continuous activation of opposing muscle groups. The resulting stiffness is characteristic of this disorder. Colchicine is known to antagonize GABAA receptor signaling, and this may have brought the underlying diagnosis of SPS to clinical attention.7,8
Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines9 and baclofen10 are necessary. When symptoms are not controlled by GABAergic drugs, immunosuppression with corticosteroids and IVIG has been used, as have plasmapheresis and rituximab.11 The efficacy of the latter, however, was not supported by a randomized, placebo-controlled trial.12 This patient experienced significant improvement with benzodiazepines, baclofen, IVIG, and neoadjuvant chemotherapy prior to thymoma resection. The pruritus, paresthesia, and lymphocytosis also resolved with medical therapy. Interestingly, GABA signaling suppresses itch, suggesting that loss of GABAA signaling may have contributed to the development of pruritus.
SPS occasionally occurs as a paraneoplastic neurologic syndrome. Breast cancer is the most commonly associated malignancy, although associations between thymomas and SPS13 with anti-GAD65 antibodies14 have also been described. The presentation of thymomas is variable, with approximately one-third discovered incidentally on imaging, one-third producing symptoms of local compression, and one-third identified in the setting of another syndrome, most commonly myasthenia gravis. In addition to myasthenia gravis, thymomas have been associated with conditions such as hypogammaglobulinemia, pure red cell aplasia, and agranulocytosis. Stiff person syndrome is a known, albeit infrequently associated, condition.15
A critical step in arriving at the relevant differential diagnosis requires correctly framing the patient’s case.16 The treatment team’s initial frame was “a 69-year-old woman with weakness and elevated CK,” which prioritized causes of weakness and myositis. Stiff person syndrome does not cause weakness, but rather impaired movement from marked stiffness and spasms. The patient’s elevated CK was a result of continual muscle contractions. The physical exam and lack of motor deficit on EMG led the treatment team to reframe as “a 69-year-old woman with severe stiffness and spasms.” Egad! This correct frame was the key to diagnosis and confirmed by EMG and GAD65 antibody testing.
KEY LEARNING POINTS
- Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, and high-titer antibody to GAD65 or amphiphysin.
- SPS typically occurs in middle age, and women are diagnosed twice as often as men.
- Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines and baclofenare necessary.
- SPS occasionally occurs as a paraneoplastic neurologic syndrome, most commonly in association with breast cancer.
Acknowledgments
The authors wish to thank Jason Kern, MD for his preparation and interpretation of the pathologic image; and the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education, for supporting Reza Manesh, MD.
Disclosures
The authors have nothing to disclose.
Appendix Video 1: This video was taken during a physical therapy session after 1 week of scheduled benzodiazepine and 2 days of intravenous immunoglobuli
Appendix Video 2: This video was taken 5 months after scheduled diazepam and baclofen, and 1 week prior to thymectomy. (https://youtu.be/I00i638u00o)
1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9
3. Murinson BB. Stiff-person syndrome. Neurologist. 2004;10(3):131-137. doi: 10.1097/01.nrl.0000126587.37087.1a
4. Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45(5):623-634. doi: 10.1002/mus.23234
5. Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic. 2002;3(7):452-460. doi: 10.1034/j.1600-0854.2002.30702.x
6. Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15(3):141-156. doi: 10.1038/nrn3670
7. Bueno OF, Leidenheimer NJ. Colchicine inhibits GABA(A) receptors independently of microtubule depolymerization. Neuropharmacology. 1998;37(3):383-390. doi: 10.1016/S0028-3908(98)00020-3
8. Weiner JL, Buhler AV, Whatley VJ, Harris RA, Dunwiddie TV. Colchicine is a competitive antagonist at human recombinant γ-aminobutyric acidA receptors. J Pharmacol Exp Ther. 1998;284(1):95-102 . PubMed
9. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/S0025-6196(12)65339-7
10. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991
11. Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/WNL.57.5.780
12. Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002
13. Hagiwara H, Enomoto-Nakatani S, Sakai K, et al. Stiff-person syndrome associated with invasive thymoma: a case report. J Neurol Sci. 2001;193(1):59-62. doi: 10.1016/S0022-510X(01)00602-5
14. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270-7275. doi: 10.1158/1078-0432.CCR-04-0735 PubMed
15. Thomas CR, Wright CD, Loehrer PJ. Thymoma: state of the art. J Clin Oncol. 1999;17(7):2280-2289. doi: 10.1200/JCO.1999.17.7.2280 PubMed
16. Stuart S, Hartig JR, Willett L. The importance of framing. J Gen Intern Med. 2017;32(6):706-710. doi: 10.1007/s11606-016-3964-z PubMed
1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9
3. Murinson BB. Stiff-person syndrome. Neurologist. 2004;10(3):131-137. doi: 10.1097/01.nrl.0000126587.37087.1a
4. Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45(5):623-634. doi: 10.1002/mus.23234
5. Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic. 2002;3(7):452-460. doi: 10.1034/j.1600-0854.2002.30702.x
6. Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15(3):141-156. doi: 10.1038/nrn3670
7. Bueno OF, Leidenheimer NJ. Colchicine inhibits GABA(A) receptors independently of microtubule depolymerization. Neuropharmacology. 1998;37(3):383-390. doi: 10.1016/S0028-3908(98)00020-3
8. Weiner JL, Buhler AV, Whatley VJ, Harris RA, Dunwiddie TV. Colchicine is a competitive antagonist at human recombinant γ-aminobutyric acidA receptors. J Pharmacol Exp Ther. 1998;284(1):95-102 . PubMed
9. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/S0025-6196(12)65339-7
10. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991
11. Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/WNL.57.5.780
12. Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002
13. Hagiwara H, Enomoto-Nakatani S, Sakai K, et al. Stiff-person syndrome associated with invasive thymoma: a case report. J Neurol Sci. 2001;193(1):59-62. doi: 10.1016/S0022-510X(01)00602-5
14. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270-7275. doi: 10.1158/1078-0432.CCR-04-0735 PubMed
15. Thomas CR, Wright CD, Loehrer PJ. Thymoma: state of the art. J Clin Oncol. 1999;17(7):2280-2289. doi: 10.1200/JCO.1999.17.7.2280 PubMed
16. Stuart S, Hartig JR, Willett L. The importance of framing. J Gen Intern Med. 2017;32(6):706-710. doi: 10.1007/s11606-016-3964-z PubMed
© 2019 Society of Hospital Medicine