User login
SILVER SPRING, MD. – A permanently implanted device thought to reduce hunger pangs by delivering electrical pulses to the intra-abdominal vagus nerve could be the next treatment approved for treating obesity, considering a Food and Drug Administration advisory panel’s positive vote on the device.
At a meeting on June 17, the FDA’s Gastroenterology-Urology Devices Panel voted 6-2, with one abstention, that the benefits of the Maestro Rechargeable System outweighed its risks for the indication under FDA review: for weight reduction in obese adults, with a body mass index of at least 40 kg/m2; or in those with a BMI of at least 35 kg/m2 with one or more obesity-related comorbid conditions, who have failed at least one supervised weight-management program within the past 5 years. Although the two primary effectiveness endpoints in the pivotal 12-month trial were not met, the safety endpoint was met and panelists voting in favor agreed that the study provided evidence that it was effective in helping some patients lose weight.
This is among the devices currently being studied that, if approved, could start to fill the large gap in obesity treatment options that currently exists between lifestyle modifications and pharmacologic treatment and bariatric surgery.
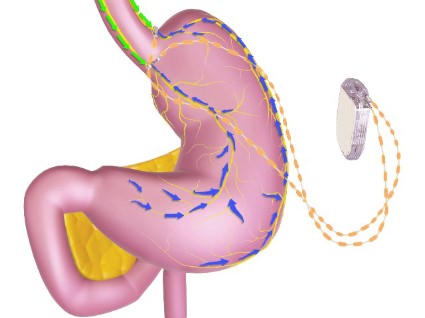
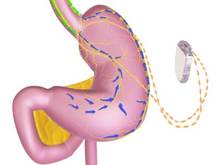
The main components of the device are a pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure under general anesthesia. External components include a mobile battery charger and a laptop computer used by clinicians to modify therapy and retrieve diagnostic information. By delivering intermittent electrical "blocking signals" to the anterior and posterior trunks of the intra-abdominal vagus nerve, the device "reduces sensations of hunger and produces satiety leading to weight loss," according to the manufacturer, EnteroMedics, which calls the treatment "VBLOC" therapy.
The ReCharge study, the main study submitted for approval, was a prospective, randomized, double-blind, sham-controlled study of 239 mostly female, white patients, whose mean age was about 47 years, and whose mean BMI at baseline was 41 (mean weight was about 250 lbs); about 5% had diabetes. A total of 162 had the device implanted and 77 had a nonfunctional neuroregulator implanted. At 12 months, almost 94% of the patients remained in the study.
One primary effectiveness endpoint was the mean percent excess weight loss (% EWL) among treated patients that was at least 10% greater than in the sham group (% EWL was calculated by dividing the weight lost by the excess weight). At 12 months, the mean % EWL was 24.4% in the treatment group, vs. 15.9% in the sham control group, an 8.5–percentage point difference. The second primary effectiveness endpoint, which applied only to those in the VBLOC group, had two components for the endpoint to be met: at least 55% of patients had least 20% EWL (which was met by 52.5% of patients); and at least 45% of patients had at least 25% EWL (which was met by 38.3% of patients).
Weight loss was maintained at 18 months, when the average % EWL in the VBLOC group was 25.2%, vs. 11.7% in the sham group, a 13.5% difference. However, 28% of the patients in the VBLOC group were lost to follow-up at that time, and the study was no longer blinded after 12 months.
With a 24% EWL after 1 year of treatment, an average patient – a 5’3"-tall woman weighing 225 lbs., with a BMI of 40 and 84 lbs. of excess weight – would lose 20 pounds, one of the FDA reviewers said at the meeting.
Over 12 months, among the 162 patients who received the device, there were 15 serious adverse events (9.3%): 9 related to the general surgery procedure (including 6 cases of nausea), 2 related to the implant or revision (one case of emesis and one case of atelectasis after the initial surgery), 3 device-related (pain at the neuroregulator site) and 1 treatment-related (gallbladder disease). Between 12 and 18 months, there was one gastric perforation during elective removal of the device.
Over 12 months, adverse events associated with the device included heartburn and dyspepsia, dysphagia, nausea, abdominal pain and pain at the neuroregulator site; nine surgical revisions were required in eight patients because of pain at the neuroregulator site (five cases), device malfunction (two cases), and readjustment of the neuroregulator because it was tilted (one case). In addition, five patients in the VBLOC group required surgical explantation of the device for jabbing pain during physical activity or heartburn symptoms.
Despite the near unanimous vote on the risk-benefit question, the votes on safety and effectiveness were mixed. The panel voted 8-1 that there was reasonable assurance the device was safe for the indicated use, but voted 5-4 that there was not reasonable assurance that it was effective for that indication. Those voting in favor of risk-benefit were swayed by the evidence it was effective in some patients and was reasonably safe.
One of the panelists voting yes on the risk-benefit question, Dr. Karen Woods, a gastroenterologist at the Methodist Hospital, Houston, said that the data provided by the company indicated the device was beneficial and referred to the need for less-invasive devices to treat obesity. "I would say they have shown they have effectiveness, just not particularly meeting the exact criteria they created for the study," she noted. "I feel we’re filling in a little bit of an area of unmet need ... and it will either go on to be effective or it won’t be, but I don’t think we’re going to do a lot of harm."
The two panelists voting no on the risk-benefit question said they voted no because the effectiveness endpoints were not met, despite evidence of superior weight loss in the treated group. "It’s close, it’s approaching clinical and statistical significance, but it doesn’t reach it," said Dr. Gary Falk, a gastroenterologist and professor of medicine at the University of Pennsylvania, Philadelphia.
Concerns raised by the panelists included the lack of racial diversity and the low number of men enrolled in the study, which made it unclear whether the results could be applied to the general population of obese patients; possible detrimental effects on the vagus nerve after leads are explanted, and the lack of long-term follow-up data for a device that could remain in place for a lifetime. Another concern is that patients with the device cannot undergo magnetic resonance imaging.
The company expects an FDA decision on approval later this year, according to a statement released by EnteroMedics after the panel meeting. If the device is approved, the company plans postmarketing studies, a 5-year follow-up study of patients in the ReCharge trial and a registry study of 500 patients in the United States.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, but occasionally, a panelist is given a waiver. At this meeting, a waiver was granted Dr. Woods, who holds stock in a company with a competing product, according to the FDA briefing documents.
SILVER SPRING, MD. – A permanently implanted device thought to reduce hunger pangs by delivering electrical pulses to the intra-abdominal vagus nerve could be the next treatment approved for treating obesity, considering a Food and Drug Administration advisory panel’s positive vote on the device.
At a meeting on June 17, the FDA’s Gastroenterology-Urology Devices Panel voted 6-2, with one abstention, that the benefits of the Maestro Rechargeable System outweighed its risks for the indication under FDA review: for weight reduction in obese adults, with a body mass index of at least 40 kg/m2; or in those with a BMI of at least 35 kg/m2 with one or more obesity-related comorbid conditions, who have failed at least one supervised weight-management program within the past 5 years. Although the two primary effectiveness endpoints in the pivotal 12-month trial were not met, the safety endpoint was met and panelists voting in favor agreed that the study provided evidence that it was effective in helping some patients lose weight.
This is among the devices currently being studied that, if approved, could start to fill the large gap in obesity treatment options that currently exists between lifestyle modifications and pharmacologic treatment and bariatric surgery.
The main components of the device are a pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure under general anesthesia. External components include a mobile battery charger and a laptop computer used by clinicians to modify therapy and retrieve diagnostic information. By delivering intermittent electrical "blocking signals" to the anterior and posterior trunks of the intra-abdominal vagus nerve, the device "reduces sensations of hunger and produces satiety leading to weight loss," according to the manufacturer, EnteroMedics, which calls the treatment "VBLOC" therapy.
The ReCharge study, the main study submitted for approval, was a prospective, randomized, double-blind, sham-controlled study of 239 mostly female, white patients, whose mean age was about 47 years, and whose mean BMI at baseline was 41 (mean weight was about 250 lbs); about 5% had diabetes. A total of 162 had the device implanted and 77 had a nonfunctional neuroregulator implanted. At 12 months, almost 94% of the patients remained in the study.
One primary effectiveness endpoint was the mean percent excess weight loss (% EWL) among treated patients that was at least 10% greater than in the sham group (% EWL was calculated by dividing the weight lost by the excess weight). At 12 months, the mean % EWL was 24.4% in the treatment group, vs. 15.9% in the sham control group, an 8.5–percentage point difference. The second primary effectiveness endpoint, which applied only to those in the VBLOC group, had two components for the endpoint to be met: at least 55% of patients had least 20% EWL (which was met by 52.5% of patients); and at least 45% of patients had at least 25% EWL (which was met by 38.3% of patients).
Weight loss was maintained at 18 months, when the average % EWL in the VBLOC group was 25.2%, vs. 11.7% in the sham group, a 13.5% difference. However, 28% of the patients in the VBLOC group were lost to follow-up at that time, and the study was no longer blinded after 12 months.
With a 24% EWL after 1 year of treatment, an average patient – a 5’3"-tall woman weighing 225 lbs., with a BMI of 40 and 84 lbs. of excess weight – would lose 20 pounds, one of the FDA reviewers said at the meeting.
Over 12 months, among the 162 patients who received the device, there were 15 serious adverse events (9.3%): 9 related to the general surgery procedure (including 6 cases of nausea), 2 related to the implant or revision (one case of emesis and one case of atelectasis after the initial surgery), 3 device-related (pain at the neuroregulator site) and 1 treatment-related (gallbladder disease). Between 12 and 18 months, there was one gastric perforation during elective removal of the device.
Over 12 months, adverse events associated with the device included heartburn and dyspepsia, dysphagia, nausea, abdominal pain and pain at the neuroregulator site; nine surgical revisions were required in eight patients because of pain at the neuroregulator site (five cases), device malfunction (two cases), and readjustment of the neuroregulator because it was tilted (one case). In addition, five patients in the VBLOC group required surgical explantation of the device for jabbing pain during physical activity or heartburn symptoms.
Despite the near unanimous vote on the risk-benefit question, the votes on safety and effectiveness were mixed. The panel voted 8-1 that there was reasonable assurance the device was safe for the indicated use, but voted 5-4 that there was not reasonable assurance that it was effective for that indication. Those voting in favor of risk-benefit were swayed by the evidence it was effective in some patients and was reasonably safe.
One of the panelists voting yes on the risk-benefit question, Dr. Karen Woods, a gastroenterologist at the Methodist Hospital, Houston, said that the data provided by the company indicated the device was beneficial and referred to the need for less-invasive devices to treat obesity. "I would say they have shown they have effectiveness, just not particularly meeting the exact criteria they created for the study," she noted. "I feel we’re filling in a little bit of an area of unmet need ... and it will either go on to be effective or it won’t be, but I don’t think we’re going to do a lot of harm."
The two panelists voting no on the risk-benefit question said they voted no because the effectiveness endpoints were not met, despite evidence of superior weight loss in the treated group. "It’s close, it’s approaching clinical and statistical significance, but it doesn’t reach it," said Dr. Gary Falk, a gastroenterologist and professor of medicine at the University of Pennsylvania, Philadelphia.
Concerns raised by the panelists included the lack of racial diversity and the low number of men enrolled in the study, which made it unclear whether the results could be applied to the general population of obese patients; possible detrimental effects on the vagus nerve after leads are explanted, and the lack of long-term follow-up data for a device that could remain in place for a lifetime. Another concern is that patients with the device cannot undergo magnetic resonance imaging.
The company expects an FDA decision on approval later this year, according to a statement released by EnteroMedics after the panel meeting. If the device is approved, the company plans postmarketing studies, a 5-year follow-up study of patients in the ReCharge trial and a registry study of 500 patients in the United States.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, but occasionally, a panelist is given a waiver. At this meeting, a waiver was granted Dr. Woods, who holds stock in a company with a competing product, according to the FDA briefing documents.
SILVER SPRING, MD. – A permanently implanted device thought to reduce hunger pangs by delivering electrical pulses to the intra-abdominal vagus nerve could be the next treatment approved for treating obesity, considering a Food and Drug Administration advisory panel’s positive vote on the device.
At a meeting on June 17, the FDA’s Gastroenterology-Urology Devices Panel voted 6-2, with one abstention, that the benefits of the Maestro Rechargeable System outweighed its risks for the indication under FDA review: for weight reduction in obese adults, with a body mass index of at least 40 kg/m2; or in those with a BMI of at least 35 kg/m2 with one or more obesity-related comorbid conditions, who have failed at least one supervised weight-management program within the past 5 years. Although the two primary effectiveness endpoints in the pivotal 12-month trial were not met, the safety endpoint was met and panelists voting in favor agreed that the study provided evidence that it was effective in helping some patients lose weight.
This is among the devices currently being studied that, if approved, could start to fill the large gap in obesity treatment options that currently exists between lifestyle modifications and pharmacologic treatment and bariatric surgery.
The main components of the device are a pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure under general anesthesia. External components include a mobile battery charger and a laptop computer used by clinicians to modify therapy and retrieve diagnostic information. By delivering intermittent electrical "blocking signals" to the anterior and posterior trunks of the intra-abdominal vagus nerve, the device "reduces sensations of hunger and produces satiety leading to weight loss," according to the manufacturer, EnteroMedics, which calls the treatment "VBLOC" therapy.
The ReCharge study, the main study submitted for approval, was a prospective, randomized, double-blind, sham-controlled study of 239 mostly female, white patients, whose mean age was about 47 years, and whose mean BMI at baseline was 41 (mean weight was about 250 lbs); about 5% had diabetes. A total of 162 had the device implanted and 77 had a nonfunctional neuroregulator implanted. At 12 months, almost 94% of the patients remained in the study.
One primary effectiveness endpoint was the mean percent excess weight loss (% EWL) among treated patients that was at least 10% greater than in the sham group (% EWL was calculated by dividing the weight lost by the excess weight). At 12 months, the mean % EWL was 24.4% in the treatment group, vs. 15.9% in the sham control group, an 8.5–percentage point difference. The second primary effectiveness endpoint, which applied only to those in the VBLOC group, had two components for the endpoint to be met: at least 55% of patients had least 20% EWL (which was met by 52.5% of patients); and at least 45% of patients had at least 25% EWL (which was met by 38.3% of patients).
Weight loss was maintained at 18 months, when the average % EWL in the VBLOC group was 25.2%, vs. 11.7% in the sham group, a 13.5% difference. However, 28% of the patients in the VBLOC group were lost to follow-up at that time, and the study was no longer blinded after 12 months.
With a 24% EWL after 1 year of treatment, an average patient – a 5’3"-tall woman weighing 225 lbs., with a BMI of 40 and 84 lbs. of excess weight – would lose 20 pounds, one of the FDA reviewers said at the meeting.
Over 12 months, among the 162 patients who received the device, there were 15 serious adverse events (9.3%): 9 related to the general surgery procedure (including 6 cases of nausea), 2 related to the implant or revision (one case of emesis and one case of atelectasis after the initial surgery), 3 device-related (pain at the neuroregulator site) and 1 treatment-related (gallbladder disease). Between 12 and 18 months, there was one gastric perforation during elective removal of the device.
Over 12 months, adverse events associated with the device included heartburn and dyspepsia, dysphagia, nausea, abdominal pain and pain at the neuroregulator site; nine surgical revisions were required in eight patients because of pain at the neuroregulator site (five cases), device malfunction (two cases), and readjustment of the neuroregulator because it was tilted (one case). In addition, five patients in the VBLOC group required surgical explantation of the device for jabbing pain during physical activity or heartburn symptoms.
Despite the near unanimous vote on the risk-benefit question, the votes on safety and effectiveness were mixed. The panel voted 8-1 that there was reasonable assurance the device was safe for the indicated use, but voted 5-4 that there was not reasonable assurance that it was effective for that indication. Those voting in favor of risk-benefit were swayed by the evidence it was effective in some patients and was reasonably safe.
One of the panelists voting yes on the risk-benefit question, Dr. Karen Woods, a gastroenterologist at the Methodist Hospital, Houston, said that the data provided by the company indicated the device was beneficial and referred to the need for less-invasive devices to treat obesity. "I would say they have shown they have effectiveness, just not particularly meeting the exact criteria they created for the study," she noted. "I feel we’re filling in a little bit of an area of unmet need ... and it will either go on to be effective or it won’t be, but I don’t think we’re going to do a lot of harm."
The two panelists voting no on the risk-benefit question said they voted no because the effectiveness endpoints were not met, despite evidence of superior weight loss in the treated group. "It’s close, it’s approaching clinical and statistical significance, but it doesn’t reach it," said Dr. Gary Falk, a gastroenterologist and professor of medicine at the University of Pennsylvania, Philadelphia.
Concerns raised by the panelists included the lack of racial diversity and the low number of men enrolled in the study, which made it unclear whether the results could be applied to the general population of obese patients; possible detrimental effects on the vagus nerve after leads are explanted, and the lack of long-term follow-up data for a device that could remain in place for a lifetime. Another concern is that patients with the device cannot undergo magnetic resonance imaging.
The company expects an FDA decision on approval later this year, according to a statement released by EnteroMedics after the panel meeting. If the device is approved, the company plans postmarketing studies, a 5-year follow-up study of patients in the ReCharge trial and a registry study of 500 patients in the United States.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, but occasionally, a panelist is given a waiver. At this meeting, a waiver was granted Dr. Woods, who holds stock in a company with a competing product, according to the FDA briefing documents.
AT AN FDA ADVISORY COMMITTEE MEETING