User login
GAITHERSBURG, MD – A Food and Drug Administration advisory panel has voted in favor of approving the Watchman Left Atrial Appendage Closure Device for patients with nonvalvular atrial fibrillation. One of the panelists described the device as "transformative technology," as it is the first device alternative to medical therapy for the prevention of stroke due to atrial fibrillation to be approved in the United States.
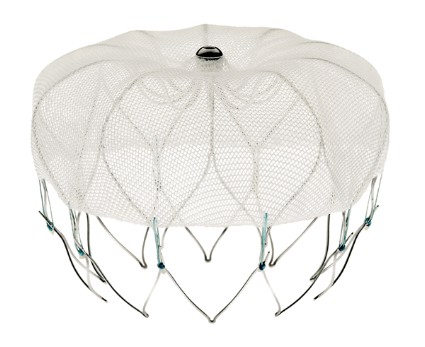
At a meeting on Dec. 11, the FDA Circulatory System Devices Panel of the Medical Devices Advisory Committee voted 13-1 to recommend approval of Boston Scientific’s Watchman Left Atrial Appendage Closure (LAAC) Therapy, a percutaneously delivered permanent cardiac implant placed in the left atrial appendage for the prevention of thromboembolism from the left atrial appendage. If approved, the indication will be for patients with nonvalvular atrial fibrillation who are eligible for warfarin therapy to reduce the risk of stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores. In addition, the panel recommended and the company echoed that the final indication language should make clear that the therapy should be only for warfarin-eligible patients who had a significant reason not to remain on chronic anticoagulation therapy.

Dr. Rachel Neubrander of the FDA detailed how the device previously was reviewed by an FDA panel in 2009, and ultimately was not approved because of the agency’s and an advisory panel’s significant concerns about the design of the pivotal PROTECT-AF trial, which included low-risk patients with a CHADS2 score of 1 and also included patients indicated for chronic clopidogrel therapy.
In concert with the FDA, Boston Scientific developed the PREVAIL trial to address those concerns. PREVAIL enrolled only high-risk subjects with CHADS2 scores of at least 2 and those with a CHADS2 score of 1 who had additional stroke risk factors present and were recommended for warfarin therapy by ACC/AHA/ESC 2006 guidelines.
In addition, PREVAIL excluded subjects indicated for chronic clopidogrel therapy. Because 56% of primary safety events in the device group occurred on the day of the procedure in the PROTECT-AF trial, and there was evidence of a significant learning curve, PREVAIL was designed to require at least 20% of the subjects to be implanted at new investigational sites and at least 25% of the subjects to be implanted by new operators.
At the current meeting, the device was recommended for approval based upon the totality of the data obtained from both the PROTECT-AF and PREVAIL trials, and information from the Continued Access to PREVAIL (CAP2) registry, according to the majority of the panelists, who indicated that the data from the PREVAIL trial alone was premature for such a determination based upon the current lack of long-term data, compared with the PROTECT-AF trial.
The panel stated that it was impressed by the greatly reduced incidence of perioperative primary safety events in PREVAIL compared with PROTECT-AF, and the success of Boston Scientific’s training protocol to ensure that new operators performed as well as or better than experienced operators. The panel thus suggested that some definition of the site requirements and training requirements instituted by the company for the PREVAIL trial be included in the final label.
Of concern, PREVAIL failed to meet its primary endpoint of noninferiority compared with warfarin with regard to the composite 18-month rate of stroke, systemic embolism, and cardiovascular or unexplained death. This was attributed to the fact that the PREVAIL trial had accumulated 18-month data on fewer than 30% of the patients at the point of analysis. The great majority of the panel concluded that the trend of PREVAIL, when combined in a Bayesien analysis with 50% discounted data from the patients with CHADS2 scores of at least 2 in the PROTECT-AF trial, and taking into account the CAP registry, was acceptable. The lone dissenting panel member said that this lack of complete PREVAIL data and the fact that it technically did not meet its primary endpoint were his reasons for voting against approval.
The panel concluded that the Watchman device data supported the hypothesis of left atrial appendage thromboembolism as the major source of stroke due to atrial fibrillation and that the device was noninferior to warfarin in preventing such strokes once the initial perioperative period of 7 days was passed. They said that because there was a periprocedural primary adverse event risk, the device should not be considered as a substitute for warfarin in all patients, but only in those for whom warfarin compliance over the long term was a significant problem whether from patient lifestyle or medical reasons.
Of additional concern, the panel noted that there was no evidence that the Watchman device was superior to warfarin therapy with regard to the elimination of major bleeding events. During the open portion of the panel meeting, patients and their doctors nearly unanimously indicated their preference for the Watchman technology over warfarin because of their fear of and experience of frequent and significant bleeds while on anticoagulation. The panel suggested that, because of this apparent common misperception, the lack of bleeding benefit should be pointed out in the final indication language.
The panel agreed with FDA’s concern that a postmarketing study should be conducted if the device is approved, and that the study should be large enough to answer some of the concerns brought up in the meeting as to long-term results, including safety and efficacy in a broader population than that studied in the two trials, which was overwhelmingly white, and predominately male.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
The Watchman device has been approved in Europe since 2005.
Dr. Rachel Neubrander,
GAITHERSBURG, MD – A Food and Drug Administration advisory panel has voted in favor of approving the Watchman Left Atrial Appendage Closure Device for patients with nonvalvular atrial fibrillation. One of the panelists described the device as "transformative technology," as it is the first device alternative to medical therapy for the prevention of stroke due to atrial fibrillation to be approved in the United States.
At a meeting on Dec. 11, the FDA Circulatory System Devices Panel of the Medical Devices Advisory Committee voted 13-1 to recommend approval of Boston Scientific’s Watchman Left Atrial Appendage Closure (LAAC) Therapy, a percutaneously delivered permanent cardiac implant placed in the left atrial appendage for the prevention of thromboembolism from the left atrial appendage. If approved, the indication will be for patients with nonvalvular atrial fibrillation who are eligible for warfarin therapy to reduce the risk of stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores. In addition, the panel recommended and the company echoed that the final indication language should make clear that the therapy should be only for warfarin-eligible patients who had a significant reason not to remain on chronic anticoagulation therapy.
Dr. Rachel Neubrander of the FDA detailed how the device previously was reviewed by an FDA panel in 2009, and ultimately was not approved because of the agency’s and an advisory panel’s significant concerns about the design of the pivotal PROTECT-AF trial, which included low-risk patients with a CHADS2 score of 1 and also included patients indicated for chronic clopidogrel therapy.
In concert with the FDA, Boston Scientific developed the PREVAIL trial to address those concerns. PREVAIL enrolled only high-risk subjects with CHADS2 scores of at least 2 and those with a CHADS2 score of 1 who had additional stroke risk factors present and were recommended for warfarin therapy by ACC/AHA/ESC 2006 guidelines.
In addition, PREVAIL excluded subjects indicated for chronic clopidogrel therapy. Because 56% of primary safety events in the device group occurred on the day of the procedure in the PROTECT-AF trial, and there was evidence of a significant learning curve, PREVAIL was designed to require at least 20% of the subjects to be implanted at new investigational sites and at least 25% of the subjects to be implanted by new operators.
At the current meeting, the device was recommended for approval based upon the totality of the data obtained from both the PROTECT-AF and PREVAIL trials, and information from the Continued Access to PREVAIL (CAP2) registry, according to the majority of the panelists, who indicated that the data from the PREVAIL trial alone was premature for such a determination based upon the current lack of long-term data, compared with the PROTECT-AF trial.
The panel stated that it was impressed by the greatly reduced incidence of perioperative primary safety events in PREVAIL compared with PROTECT-AF, and the success of Boston Scientific’s training protocol to ensure that new operators performed as well as or better than experienced operators. The panel thus suggested that some definition of the site requirements and training requirements instituted by the company for the PREVAIL trial be included in the final label.
Of concern, PREVAIL failed to meet its primary endpoint of noninferiority compared with warfarin with regard to the composite 18-month rate of stroke, systemic embolism, and cardiovascular or unexplained death. This was attributed to the fact that the PREVAIL trial had accumulated 18-month data on fewer than 30% of the patients at the point of analysis. The great majority of the panel concluded that the trend of PREVAIL, when combined in a Bayesien analysis with 50% discounted data from the patients with CHADS2 scores of at least 2 in the PROTECT-AF trial, and taking into account the CAP registry, was acceptable. The lone dissenting panel member said that this lack of complete PREVAIL data and the fact that it technically did not meet its primary endpoint were his reasons for voting against approval.
The panel concluded that the Watchman device data supported the hypothesis of left atrial appendage thromboembolism as the major source of stroke due to atrial fibrillation and that the device was noninferior to warfarin in preventing such strokes once the initial perioperative period of 7 days was passed. They said that because there was a periprocedural primary adverse event risk, the device should not be considered as a substitute for warfarin in all patients, but only in those for whom warfarin compliance over the long term was a significant problem whether from patient lifestyle or medical reasons.
Of additional concern, the panel noted that there was no evidence that the Watchman device was superior to warfarin therapy with regard to the elimination of major bleeding events. During the open portion of the panel meeting, patients and their doctors nearly unanimously indicated their preference for the Watchman technology over warfarin because of their fear of and experience of frequent and significant bleeds while on anticoagulation. The panel suggested that, because of this apparent common misperception, the lack of bleeding benefit should be pointed out in the final indication language.
The panel agreed with FDA’s concern that a postmarketing study should be conducted if the device is approved, and that the study should be large enough to answer some of the concerns brought up in the meeting as to long-term results, including safety and efficacy in a broader population than that studied in the two trials, which was overwhelmingly white, and predominately male.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
The Watchman device has been approved in Europe since 2005.
GAITHERSBURG, MD – A Food and Drug Administration advisory panel has voted in favor of approving the Watchman Left Atrial Appendage Closure Device for patients with nonvalvular atrial fibrillation. One of the panelists described the device as "transformative technology," as it is the first device alternative to medical therapy for the prevention of stroke due to atrial fibrillation to be approved in the United States.
At a meeting on Dec. 11, the FDA Circulatory System Devices Panel of the Medical Devices Advisory Committee voted 13-1 to recommend approval of Boston Scientific’s Watchman Left Atrial Appendage Closure (LAAC) Therapy, a percutaneously delivered permanent cardiac implant placed in the left atrial appendage for the prevention of thromboembolism from the left atrial appendage. If approved, the indication will be for patients with nonvalvular atrial fibrillation who are eligible for warfarin therapy to reduce the risk of stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores. In addition, the panel recommended and the company echoed that the final indication language should make clear that the therapy should be only for warfarin-eligible patients who had a significant reason not to remain on chronic anticoagulation therapy.
Dr. Rachel Neubrander of the FDA detailed how the device previously was reviewed by an FDA panel in 2009, and ultimately was not approved because of the agency’s and an advisory panel’s significant concerns about the design of the pivotal PROTECT-AF trial, which included low-risk patients with a CHADS2 score of 1 and also included patients indicated for chronic clopidogrel therapy.
In concert with the FDA, Boston Scientific developed the PREVAIL trial to address those concerns. PREVAIL enrolled only high-risk subjects with CHADS2 scores of at least 2 and those with a CHADS2 score of 1 who had additional stroke risk factors present and were recommended for warfarin therapy by ACC/AHA/ESC 2006 guidelines.
In addition, PREVAIL excluded subjects indicated for chronic clopidogrel therapy. Because 56% of primary safety events in the device group occurred on the day of the procedure in the PROTECT-AF trial, and there was evidence of a significant learning curve, PREVAIL was designed to require at least 20% of the subjects to be implanted at new investigational sites and at least 25% of the subjects to be implanted by new operators.
At the current meeting, the device was recommended for approval based upon the totality of the data obtained from both the PROTECT-AF and PREVAIL trials, and information from the Continued Access to PREVAIL (CAP2) registry, according to the majority of the panelists, who indicated that the data from the PREVAIL trial alone was premature for such a determination based upon the current lack of long-term data, compared with the PROTECT-AF trial.
The panel stated that it was impressed by the greatly reduced incidence of perioperative primary safety events in PREVAIL compared with PROTECT-AF, and the success of Boston Scientific’s training protocol to ensure that new operators performed as well as or better than experienced operators. The panel thus suggested that some definition of the site requirements and training requirements instituted by the company for the PREVAIL trial be included in the final label.
Of concern, PREVAIL failed to meet its primary endpoint of noninferiority compared with warfarin with regard to the composite 18-month rate of stroke, systemic embolism, and cardiovascular or unexplained death. This was attributed to the fact that the PREVAIL trial had accumulated 18-month data on fewer than 30% of the patients at the point of analysis. The great majority of the panel concluded that the trend of PREVAIL, when combined in a Bayesien analysis with 50% discounted data from the patients with CHADS2 scores of at least 2 in the PROTECT-AF trial, and taking into account the CAP registry, was acceptable. The lone dissenting panel member said that this lack of complete PREVAIL data and the fact that it technically did not meet its primary endpoint were his reasons for voting against approval.
The panel concluded that the Watchman device data supported the hypothesis of left atrial appendage thromboembolism as the major source of stroke due to atrial fibrillation and that the device was noninferior to warfarin in preventing such strokes once the initial perioperative period of 7 days was passed. They said that because there was a periprocedural primary adverse event risk, the device should not be considered as a substitute for warfarin in all patients, but only in those for whom warfarin compliance over the long term was a significant problem whether from patient lifestyle or medical reasons.
Of additional concern, the panel noted that there was no evidence that the Watchman device was superior to warfarin therapy with regard to the elimination of major bleeding events. During the open portion of the panel meeting, patients and their doctors nearly unanimously indicated their preference for the Watchman technology over warfarin because of their fear of and experience of frequent and significant bleeds while on anticoagulation. The panel suggested that, because of this apparent common misperception, the lack of bleeding benefit should be pointed out in the final indication language.
The panel agreed with FDA’s concern that a postmarketing study should be conducted if the device is approved, and that the study should be large enough to answer some of the concerns brought up in the meeting as to long-term results, including safety and efficacy in a broader population than that studied in the two trials, which was overwhelmingly white, and predominately male.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
The Watchman device has been approved in Europe since 2005.
Dr. Rachel Neubrander,
Dr. Rachel Neubrander,
AT AN FDA ADVISORY COMMITTEE MEETING