User login
BY JEREMY UDKOFF AND CATALINA MATIZ, MD
Juvenile xanthogranuloma (JXG), a non–Langerhans cell histiocytosis, is a common pediatric tumor that most commonly presents either at birth, in infants, or in young children – with the majority of cases occurring before 2 years. There is a male predominance with a 50% increased prevalence for solitary lesion disease and a 12 times higher prevalence in multilesion disease.1 Few lesions are concerning, and spontaneous regression of cutaneous lesions over the subsequent 1-5 years is a hallmark feature of JXG.2
Clinically, the JXGs are initially smooth, pinkish papules that may enlarge to 1-cm nodules and become yellowish in appearance before resolving to become atrophic macules or patches.2,3 JXGs are firm but rubbery and may become scaly and/or ulcerate as the lesion progresses.2 The JXGs most frequently occur superficially on the scalp and flexural areas of the upper extremities but infrequently present in the subcutaneous soft tissue, central nervous system, liver/spleen, eye/orbit, oropharynx, and muscle tissue.3,4 A well-described and concerning extracutaneous manifestations of JXG is ocular involvement and may be associated with bleeding into the anterior chamber of the eye. Despite this potentially disabling complication, screening for ocular involvement is recommended only in patients under age 2 years and in those with multiple skin lesions.5
The etiology of JXG is largely unknown. However, an association between neurofibromatosis type 1 (NF1) and the development of JXG and other diseases such as juvenile myelomonocytic leukemia, previously called juvenile chronic myelomonocytic leukemia, exists. It was thought that patients with NF1 and JXGs had a higher risk to develop juvenile myelomonocytic leukemia.6 However, a recent study showed that JXG alone does not appear to confer an increased risk for developing malignancy in children with NF1.6,7
Differential diagnosis and work-up
The clinical differential diagnosis for JXG includes dermatofibromas, Langerhans cell histiocytosis, mastocytosis, Spitz nevus, hemangioendothelioma, and other xanthomas. Because of the concerning nature of these look-alikes, equivocal cases should be referred to a pediatric dermatologist.
Although, altered laboratory values may be seen with systemic JXG with solid organ involvement, there are no systemic tests that can be used to determine if a cutaneous lesion is JXG. Thus, biopsy is the gold standard diagnostic for confirmatory testing. As a histiocytic disorder, JXG will display various macrophages on histologic examination. Additionally, one may observe a dense dermal infiltrate of vacuolated cells, along with wreathlike giant cells (Touton cells) and eosinophils.3 Although these Touton cells are thought to be pathognomonic of JXG, early lesions may lack these cells.8 Thus, their absence does not exclude the diagnosis of JXG. These are more serious cases, and the work-up conducted depends upon the organ system(s) involved. Systemic disease occurs in approximately 5% of patients.
Treatment and prognosis
Clinical monitoring is the only therapy required if there are only a few cutaneous JXGs present. However, systemic JXG is a concerning disease and various chemotherapy regimens have been recommended.3 Additionally, the use of a vinca alkaloid in conjunction with a steroid is associated with better outcomes than either of these agents alone.9 As a word of caution, in one study of 12 patients who received therapeutic systemic chemotherapy or radiation therapy to the brain, eye, skin, or heart, the patients had long-term disabilities and 2 patients died of their disease.4 In another study, children with systemic JXG, again, had a poor prognosis: Despite courses of multiagent chemotherapy, 2 of 17 patients died.9
Despite the poor results associated with systemic JXG, the vast majority of JXG patients have localized disease, which is associated with an excellent prognosis. The majority of these lesions spontaneously regress.
In conclusion, JXG is typically a benign, cutaneous disease. It presents in infants and children and involutes over a 1-5 year period. Lesions that are not classic for JXG should be referred to a pediatric dermatologist, and biopsy is the gold standard diagnosis. Most manifestations of JXG do not require therapy. However, systemic JXG may be difficult to treat and is associated with poor outcomes.
Dr. Catalina Matiz is assistant professor of dermatology at Rady Children’s Hospital-San Diego, associated with the University of California, San Diego. Jeremy Udkoff is a medical student at the university. Neither Dr. Matiz nor Mr. Udkoff have relevant financial disclosures.
References
1. Am J Surg Pathol. 2005 Jan;29(1):21-8.
2. Int J Dermatol. 2015 Oct;54(10):1109-23.
4. J Pediatr. 1996 Aug;129(2):227-37.
5. J Am Acad Dermatol. 1996 Mar;34(3):445-9.
6. J Am Acad Dermatol. 2017 Feb 8. pii: S0190-9622(16)31196-3.
7. Pediatr Dermatol. 2004 Mar-Apr;21(2):97-101.
BY JEREMY UDKOFF AND CATALINA MATIZ, MD
Juvenile xanthogranuloma (JXG), a non–Langerhans cell histiocytosis, is a common pediatric tumor that most commonly presents either at birth, in infants, or in young children – with the majority of cases occurring before 2 years. There is a male predominance with a 50% increased prevalence for solitary lesion disease and a 12 times higher prevalence in multilesion disease.1 Few lesions are concerning, and spontaneous regression of cutaneous lesions over the subsequent 1-5 years is a hallmark feature of JXG.2
Clinically, the JXGs are initially smooth, pinkish papules that may enlarge to 1-cm nodules and become yellowish in appearance before resolving to become atrophic macules or patches.2,3 JXGs are firm but rubbery and may become scaly and/or ulcerate as the lesion progresses.2 The JXGs most frequently occur superficially on the scalp and flexural areas of the upper extremities but infrequently present in the subcutaneous soft tissue, central nervous system, liver/spleen, eye/orbit, oropharynx, and muscle tissue.3,4 A well-described and concerning extracutaneous manifestations of JXG is ocular involvement and may be associated with bleeding into the anterior chamber of the eye. Despite this potentially disabling complication, screening for ocular involvement is recommended only in patients under age 2 years and in those with multiple skin lesions.5
The etiology of JXG is largely unknown. However, an association between neurofibromatosis type 1 (NF1) and the development of JXG and other diseases such as juvenile myelomonocytic leukemia, previously called juvenile chronic myelomonocytic leukemia, exists. It was thought that patients with NF1 and JXGs had a higher risk to develop juvenile myelomonocytic leukemia.6 However, a recent study showed that JXG alone does not appear to confer an increased risk for developing malignancy in children with NF1.6,7
Differential diagnosis and work-up
The clinical differential diagnosis for JXG includes dermatofibromas, Langerhans cell histiocytosis, mastocytosis, Spitz nevus, hemangioendothelioma, and other xanthomas. Because of the concerning nature of these look-alikes, equivocal cases should be referred to a pediatric dermatologist.
Although, altered laboratory values may be seen with systemic JXG with solid organ involvement, there are no systemic tests that can be used to determine if a cutaneous lesion is JXG. Thus, biopsy is the gold standard diagnostic for confirmatory testing. As a histiocytic disorder, JXG will display various macrophages on histologic examination. Additionally, one may observe a dense dermal infiltrate of vacuolated cells, along with wreathlike giant cells (Touton cells) and eosinophils.3 Although these Touton cells are thought to be pathognomonic of JXG, early lesions may lack these cells.8 Thus, their absence does not exclude the diagnosis of JXG. These are more serious cases, and the work-up conducted depends upon the organ system(s) involved. Systemic disease occurs in approximately 5% of patients.
Treatment and prognosis
Clinical monitoring is the only therapy required if there are only a few cutaneous JXGs present. However, systemic JXG is a concerning disease and various chemotherapy regimens have been recommended.3 Additionally, the use of a vinca alkaloid in conjunction with a steroid is associated with better outcomes than either of these agents alone.9 As a word of caution, in one study of 12 patients who received therapeutic systemic chemotherapy or radiation therapy to the brain, eye, skin, or heart, the patients had long-term disabilities and 2 patients died of their disease.4 In another study, children with systemic JXG, again, had a poor prognosis: Despite courses of multiagent chemotherapy, 2 of 17 patients died.9
Despite the poor results associated with systemic JXG, the vast majority of JXG patients have localized disease, which is associated with an excellent prognosis. The majority of these lesions spontaneously regress.
In conclusion, JXG is typically a benign, cutaneous disease. It presents in infants and children and involutes over a 1-5 year period. Lesions that are not classic for JXG should be referred to a pediatric dermatologist, and biopsy is the gold standard diagnosis. Most manifestations of JXG do not require therapy. However, systemic JXG may be difficult to treat and is associated with poor outcomes.
Dr. Catalina Matiz is assistant professor of dermatology at Rady Children’s Hospital-San Diego, associated with the University of California, San Diego. Jeremy Udkoff is a medical student at the university. Neither Dr. Matiz nor Mr. Udkoff have relevant financial disclosures.
References
1. Am J Surg Pathol. 2005 Jan;29(1):21-8.
2. Int J Dermatol. 2015 Oct;54(10):1109-23.
4. J Pediatr. 1996 Aug;129(2):227-37.
5. J Am Acad Dermatol. 1996 Mar;34(3):445-9.
6. J Am Acad Dermatol. 2017 Feb 8. pii: S0190-9622(16)31196-3.
7. Pediatr Dermatol. 2004 Mar-Apr;21(2):97-101.
BY JEREMY UDKOFF AND CATALINA MATIZ, MD
Juvenile xanthogranuloma (JXG), a non–Langerhans cell histiocytosis, is a common pediatric tumor that most commonly presents either at birth, in infants, or in young children – with the majority of cases occurring before 2 years. There is a male predominance with a 50% increased prevalence for solitary lesion disease and a 12 times higher prevalence in multilesion disease.1 Few lesions are concerning, and spontaneous regression of cutaneous lesions over the subsequent 1-5 years is a hallmark feature of JXG.2
Clinically, the JXGs are initially smooth, pinkish papules that may enlarge to 1-cm nodules and become yellowish in appearance before resolving to become atrophic macules or patches.2,3 JXGs are firm but rubbery and may become scaly and/or ulcerate as the lesion progresses.2 The JXGs most frequently occur superficially on the scalp and flexural areas of the upper extremities but infrequently present in the subcutaneous soft tissue, central nervous system, liver/spleen, eye/orbit, oropharynx, and muscle tissue.3,4 A well-described and concerning extracutaneous manifestations of JXG is ocular involvement and may be associated with bleeding into the anterior chamber of the eye. Despite this potentially disabling complication, screening for ocular involvement is recommended only in patients under age 2 years and in those with multiple skin lesions.5
The etiology of JXG is largely unknown. However, an association between neurofibromatosis type 1 (NF1) and the development of JXG and other diseases such as juvenile myelomonocytic leukemia, previously called juvenile chronic myelomonocytic leukemia, exists. It was thought that patients with NF1 and JXGs had a higher risk to develop juvenile myelomonocytic leukemia.6 However, a recent study showed that JXG alone does not appear to confer an increased risk for developing malignancy in children with NF1.6,7
Differential diagnosis and work-up
The clinical differential diagnosis for JXG includes dermatofibromas, Langerhans cell histiocytosis, mastocytosis, Spitz nevus, hemangioendothelioma, and other xanthomas. Because of the concerning nature of these look-alikes, equivocal cases should be referred to a pediatric dermatologist.
Although, altered laboratory values may be seen with systemic JXG with solid organ involvement, there are no systemic tests that can be used to determine if a cutaneous lesion is JXG. Thus, biopsy is the gold standard diagnostic for confirmatory testing. As a histiocytic disorder, JXG will display various macrophages on histologic examination. Additionally, one may observe a dense dermal infiltrate of vacuolated cells, along with wreathlike giant cells (Touton cells) and eosinophils.3 Although these Touton cells are thought to be pathognomonic of JXG, early lesions may lack these cells.8 Thus, their absence does not exclude the diagnosis of JXG. These are more serious cases, and the work-up conducted depends upon the organ system(s) involved. Systemic disease occurs in approximately 5% of patients.
Treatment and prognosis
Clinical monitoring is the only therapy required if there are only a few cutaneous JXGs present. However, systemic JXG is a concerning disease and various chemotherapy regimens have been recommended.3 Additionally, the use of a vinca alkaloid in conjunction with a steroid is associated with better outcomes than either of these agents alone.9 As a word of caution, in one study of 12 patients who received therapeutic systemic chemotherapy or radiation therapy to the brain, eye, skin, or heart, the patients had long-term disabilities and 2 patients died of their disease.4 In another study, children with systemic JXG, again, had a poor prognosis: Despite courses of multiagent chemotherapy, 2 of 17 patients died.9
Despite the poor results associated with systemic JXG, the vast majority of JXG patients have localized disease, which is associated with an excellent prognosis. The majority of these lesions spontaneously regress.
In conclusion, JXG is typically a benign, cutaneous disease. It presents in infants and children and involutes over a 1-5 year period. Lesions that are not classic for JXG should be referred to a pediatric dermatologist, and biopsy is the gold standard diagnosis. Most manifestations of JXG do not require therapy. However, systemic JXG may be difficult to treat and is associated with poor outcomes.
Dr. Catalina Matiz is assistant professor of dermatology at Rady Children’s Hospital-San Diego, associated with the University of California, San Diego. Jeremy Udkoff is a medical student at the university. Neither Dr. Matiz nor Mr. Udkoff have relevant financial disclosures.
References
1. Am J Surg Pathol. 2005 Jan;29(1):21-8.
2. Int J Dermatol. 2015 Oct;54(10):1109-23.
4. J Pediatr. 1996 Aug;129(2):227-37.
5. J Am Acad Dermatol. 1996 Mar;34(3):445-9.
6. J Am Acad Dermatol. 2017 Feb 8. pii: S0190-9622(16)31196-3.
7. Pediatr Dermatol. 2004 Mar-Apr;21(2):97-101.
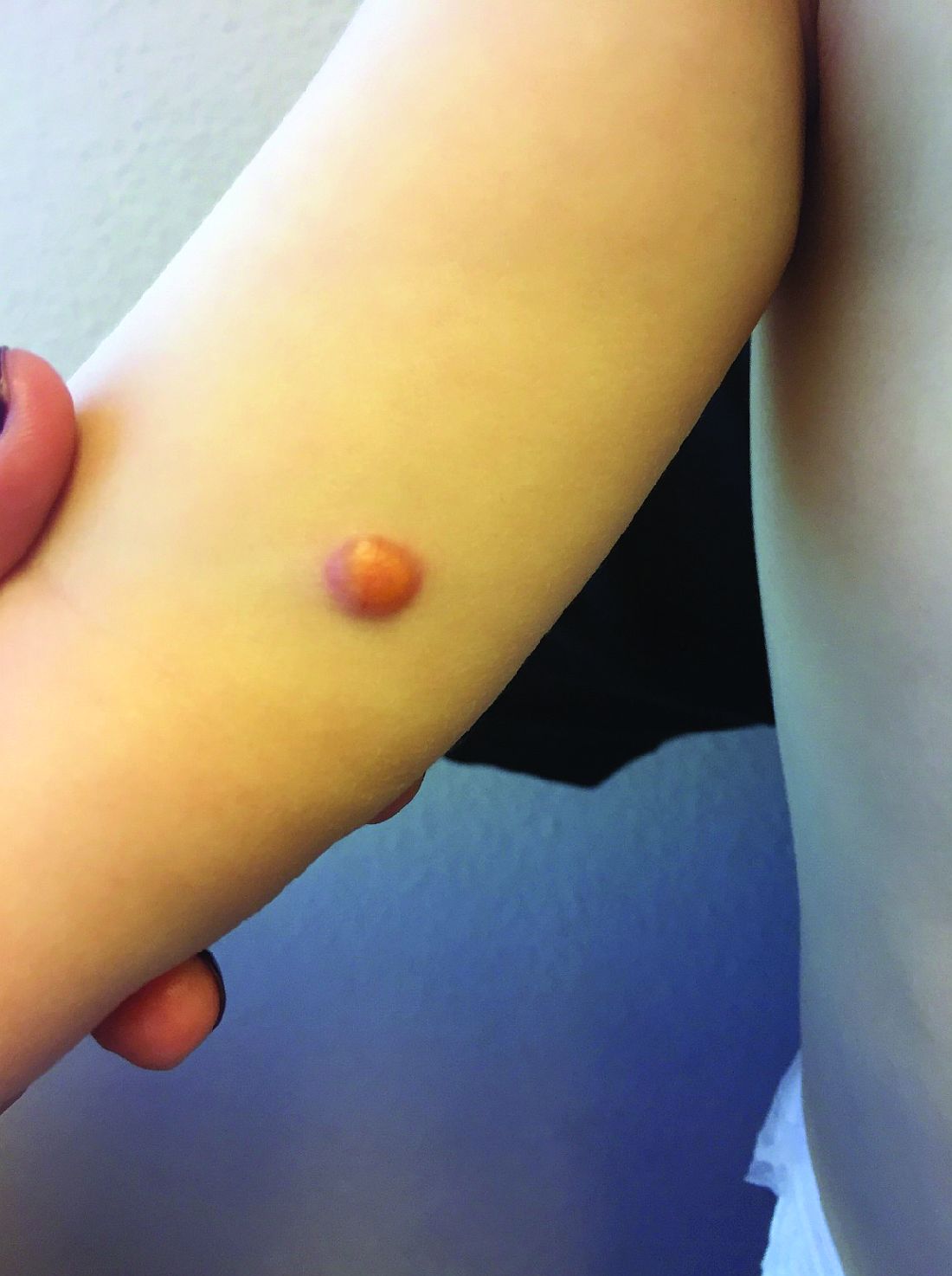
One month later, the patient returned to our clinic with an urgent concern. The patient accidentally scraped the lesion, and it bled heavily for 15 minutes before subsiding. The mother stated that the lesion grew rapidly since the prior visit and became painful. On repeat physical examination the lesion was a 1-cm fungiform, yellow to pink nodule, with central ulceration.