User login
WHY WE NEED TO UNDERSTAND MECHANISMS OF RECOVERY AFTER SEVERE BRAIN INJURY
The problem of recovery of consciousness after severe brain injury is one that easily captures the imagination of both the lay public and the professional. Puzzling reports continue to arise of late recovery of speech, language, memory, and other higher cognitive functions in rare patients, yet a scientific framework for the systematic assessment of these phenomena has been lacking.1–3 Some of these cases provide intriguing hints to the possible role of various medications (such as dopaminergic, serotoninergic, and noradrenergic agents) as well as spontaneous changes in brain function arising over time. As discussed below, the varying levels of recovery following coma seen after multifocal traumatic or nontraumatic brain injuries may share some common underlying mechanisms at the “circuit” level. Severe brain injuries producing coma have many causes (see Posner et al4 for a comprehensive review), but careful review reveals an overlap of structural pathologies and functional disturbances isolated to specific cerebral structures across several clinical syndromes grouped under the framework of “disorders of consciousness,” 5 with an emphasis on the role of particular substructures. Perhaps most important is a consideration of the pathologic, anatomic, and pathophysiologic role of the anterior forebrain, particularly the relationships of the brainstem and basal forebrain arousal systems, the central thalamus, and frontostriatal pathways, as reviewed below.
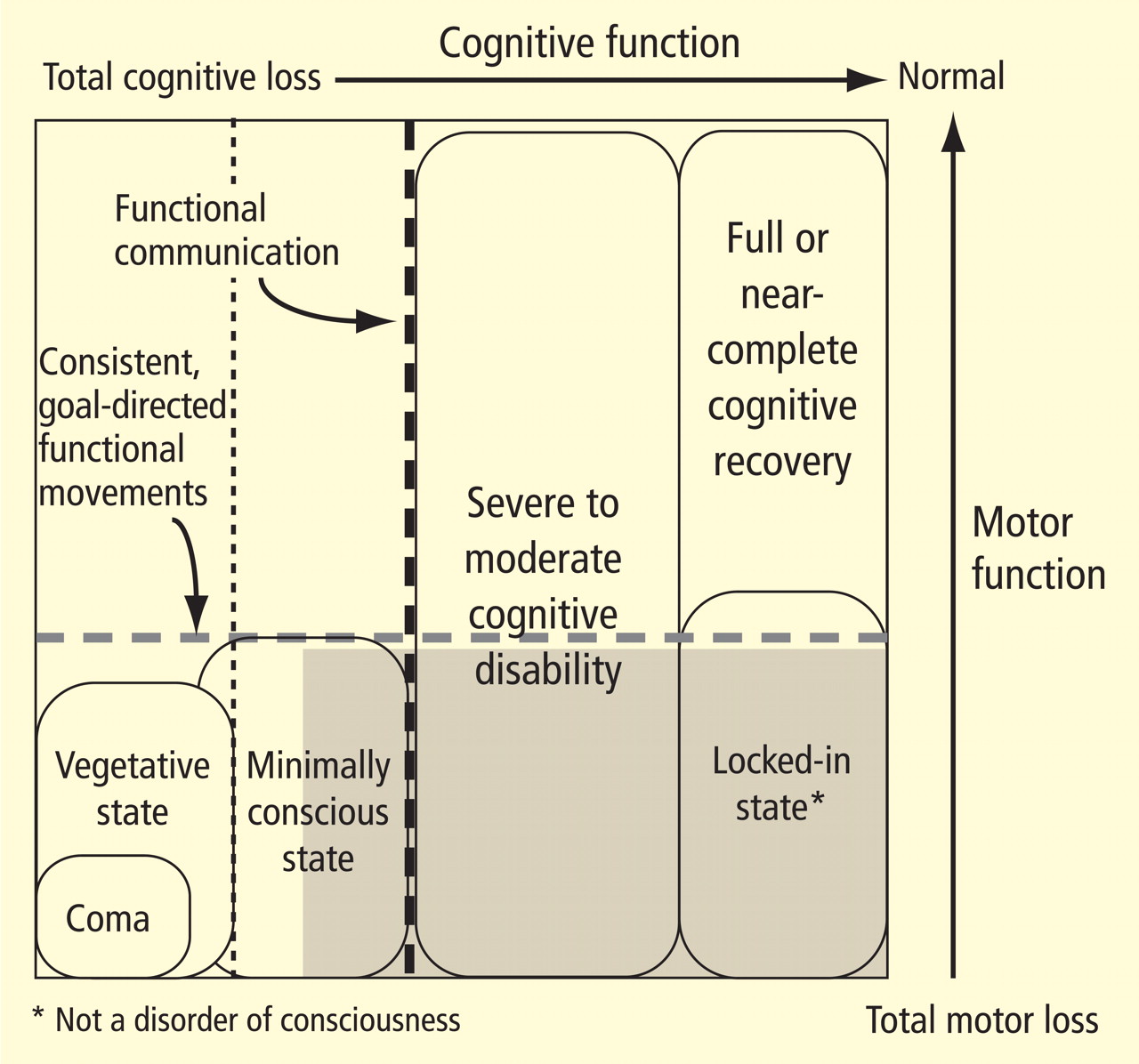
The large gray box in Figure 1 indicates the disquietingly high degree of uncertainty in assessing cognitive level in some patients who lack controllable motor output channels. The locked-in state (bottom right corner of figure) defines patients who retain total preservation of cognitive function but otherwise may appear no different from those in deep coma. Although locked-in state often arises in the context of neurologic injuries that selectively damage motor output pathways distal from their cortical origins or that slowly reduce primary motor neuron function, this syndrome and closely similar conditions arise in patients with complex brain injuries. Such patients likely retain full or nearly normal consciousness but unfortunately are unable to produce consistent goal-directed movements that allow for communication. In principle, such patients could retain significant cognitive capacity near the normal range of cognitive function yet be indistinguishable from patients in minimally conscious state.
ROLE OF THE CENTRAL THALAMUS IN SEVERE BRAIN INJURIES
Recent studies have yielded evidence for common anatomic pathologies following severe injuries associated with vegetative state10 and minimally conscious state11 as well as pathologies underlying severe to moderate cognitive disability.12 Autopsy studies of patients remaining in vegetative state at the time of death have identified widespread neuronal death throughout the thalamus as the common finding following either anoxia or diffuse axonal injury that produces widespread disruption of white matter connections.10 The severe bilateral thalamic damage after either trauma or anoxia seen in permanent vegetative state is not, however, invariably associated with diffuse neocortical neuronal cell death. This is particularly true of traumatic brain injury, in which only approximately 10% of brains at autopsy show widespread neocortical cell death.10 Specific subnuclei of the thalamus show the greatest neuronal cell loss following global and multifocal cerebral injuries produced by traumatic brain injuries.13 In particular, the central thalamic nuclei (intralaminar nuclei and related paralaminar nuclei) demonstrate progressive neuronal loss following severe traumatic brain injuries,13 and there is some evidence that a similar pattern might be identified in hypoxic-ischemic injuries.14
Progressively severe disability grades with neuronal loss along a rostrocaudal axis: the anterior intralaminar and surrounding regions initially show volume loss associated with moderate disability, while neuronal loss in the ventral and lateral nuclei of the central thalamus (posterior intralaminar group) appears with worsening disability associated with minimally conscious state and vegetative state.13 This progressive and relatively specific involvement of the nuclei of the central thalamus likely results from the unique geometry of these neurons, which have wide point-to-point connectivity across the cerebral hemisphere.15,16 The marked neuronal volume loss in these cells is likely due to their integration of the effects of neuronal cell death across large cerebral territories after diffuse trauma, hypoxia, and other nonselective severe brain injuries.
Importantly, however, focal bilateral injuries to these regions of the central thalamus are also associated with global disorders of consciousness (coma, vegetative state, and minimally conscious state).5,17 This observation indicates that these neurons also play a causal role in the production of disorders of consciousness. Abrupt injuries of the central thalamus on both sides of the brain are associated with acute coma, reflecting these cells’ key contribution to normal mechanisms of arousal regulation (reviewed by Schiff18). The central thalamus receives ascending projections from the brainstem/basal forebrain “arousal systems” that control the activity of many cortical and thalamic neurons during the sleep-wake cycle. Importantly, the central thalamus is strongly innervated by the cholinergic, serotoninergic, and noradrenergic afferents of the brainstem arousal systems (see Schiff18 for review). These same neurons also are innervated by descending projections from frontal cortical regions supporting “executive” functions that underlie goal-directed behaviors. Collectively, these ascending and descending influences on the central thalamus appear to modulate the level of arousal associated with generalized alertness and variations in cognitive effort, stress, sleep deprivation, and other variables affecting the wakeful state.15,18–22
Neuroimaging and electrophysiologic studies offer further evidence that the anatomic specializations of the central thalamus play an important role in regulating brain activation during attentive wakefulness. The central thalamus shows selective activation in normal subjects performing tasks requiring a short-term shift of attention,19,23 sustained cognitive demands of high vigilance,22 or memory holds over extended time periods.23,24 Central thalamic activation associated with varying levels of vigilance correlates with global cerebral blood flow19 and specifically covaries within the anterior cingulate cortex and pontomesencephalon. 22 Brain activity in the anterior cingulate cortex grades with increasing cognitive load and is recruited by a wide range of cognitive tasks, apparently reciprocally increasing activity along with the central thalamus in response to increasing demands of cognitive effort.20,22
CIRCUIT MECHANISMS UNDERLYING RECOVERY AFTER SEVERE BRAIN INJURY
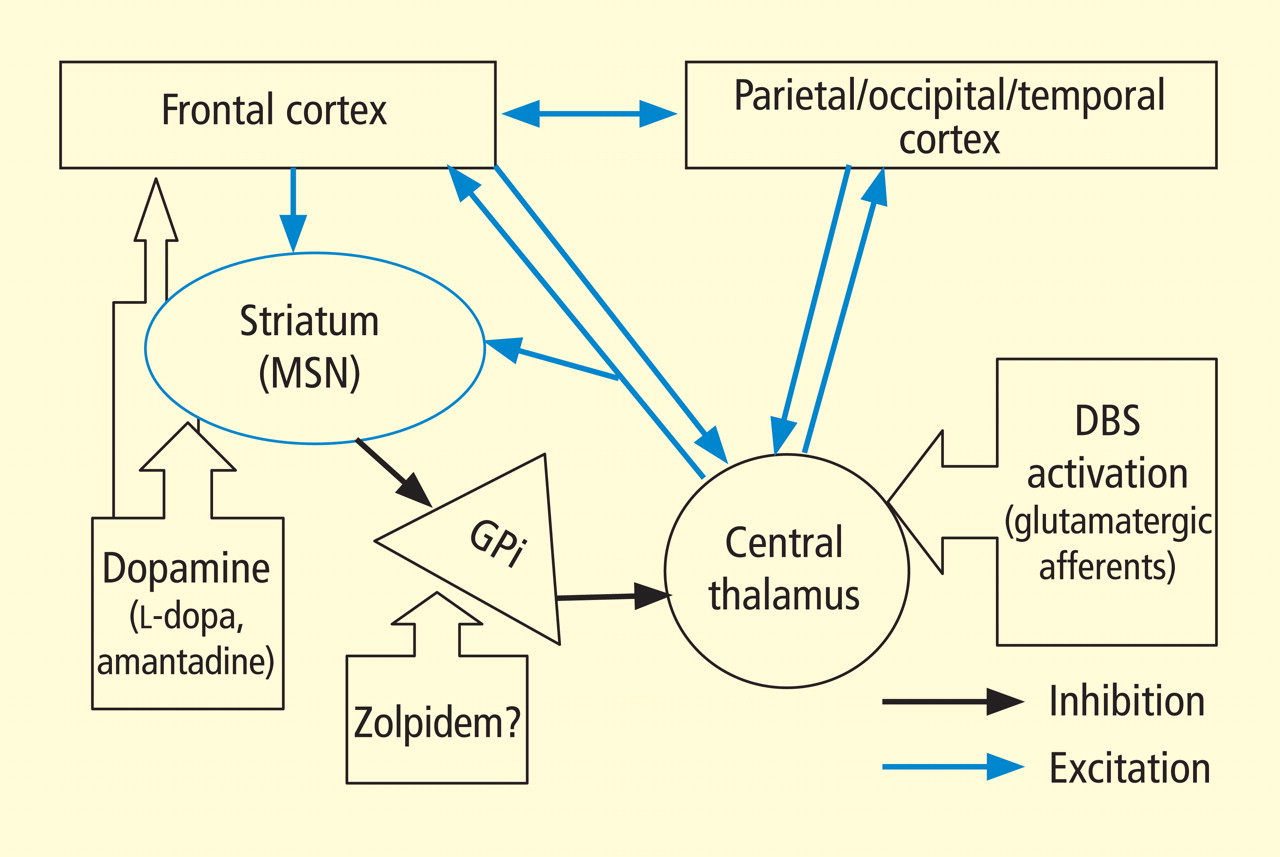
In addition to the wide point-to-point connections of the central thalamus with the cerebral cortex (predominantly connections to frontal and prefrontal cortices; see Van der Werf et al,15 Groenewegen and Berendse,28 Morel et al29), these neurons have important projections to the striatum that return via projections from the globus pallidus. 30 These projections from the central thalamus (both central lateral nucleus and parafascicularis nucleus) diffusely innervate the striatum and project onto the medium spiny neurons (MSNs), the output neuron of structure.31 Because the specific thalamostriatal projections from these central thalamic neurons use glutamate transmitters with a high probability of synaptic release,32 they likely also have a strong role in modulating background activity in the striatum.
The MSNs represent an important point of vulnerability in this anterior forebrain mesocircuit, as they have a key role in maintaining activity in the anterior forebrain through their inhibitory projections to the globus pallidus interna, which in turn inhibits the central thalamus. 33 MSNs have instrinsic cell membrane properties that keep them below their firing threshold unless a high level of spontaneous background synaptic activity arising from excitatory corticostriatal and thalamostriatal inputs is present in concert with sufficient concentrations of the neurotransmitter dopamine.33 In the setting of diffuse deafferentation or neuronal loss following severe brain injury of any type, it is expected that background excitatory synaptic activity is considerably reduced. Under these circumstances, a broad withdrawal of direct excitatory striatal projections from the central thalamus and corticostriatal inputs is likely to cause MSN output to shut down. Observations of regional changes in brain metabolism following severe brain injuries, specific responses to pharmacologic and electrophysiologic interventions in brain-injured subjects, and normal variations in brain state are all consistent with this mesocircuit model (see Schiff26 for comprehensive review). Similarly, a consistent pattern of selective metabolic downregulation within the anterior forebrain has been shown to specifically grade with severity of behavioral impairment following diffuse axonal injury,34 and application of dopaminergic agents in such patients often will produce behavioral facilitation.35,36 These medications may facilitate the output of the MSNs and directly modulate mesial frontal cortical neurons, possibly restoring anterior forebrain activity within the loop connections of the frontal cortex, striatum, pallidum, and central thalamus.
This model provides context for understanding another paradoxical observation—ie, the association of the sedative zolpidem (Ambien), a nonbenzodiazepine hypnotic that potentiates GABAA receptors, with behavioral improvement of alertness and interactive behavior in severely brain-injured patients.37–41 Zolpidem’s primary direct action in patient responders, as originally proposed by Schiff and Posner,27 may be upon on the globus pallidus interna, producing a release of tonic inhibition of the central thalamus in the setting of a broad reduction in background excitatory neurotransmission (as seen, for example, following diffuse hypoxic-ischemic injury) and leading to a shutdown of the inhibitory projection of the MSNs. The GABAA alpha-1 subunit is expressed in large quantity in the globus pallidus interna, and experimental studies support this mechanism of action.42
SINGLE-SUBJECT STUDY OF CENTRAL THALAMIC STIMULATION IN MINIMALLY CONSCIOUS STATE
A further implication of the mesocircuit model is that direct activation of the central thalamus is expected to be the causal step in reactivating a downregulated anterior forebrain system, suggesting that direct modulation of the central thalamus might facilitate behavioral responsiveness in some patients with severe brain injuries. A recent study offers evidence that direct electrical stimulation of the central thalamus can produce behavioral facilitation.
In this single-subject study of central thalamic deep brain stimulation (DBS), a 38-year-old man remained in minimally conscious state for 6 years following a severe closed head injury following blunt trauma to the right frontal lobe.43 After 3 months in a vegetative state, the patient exhibited the first evidence of clear behaviors in response to sensory stimulation consistent with minimally conscious state and advanced to eventually demonstrating a best behavioral response of inconsistent command-following and communication using eye movements. This behavioral level remained unchanged at the start of the DBS study 4 years later, as confirmed by evaluation with the Coma Recovery Scale–Revised (CRS-R), a formal behavioral assessment tool.
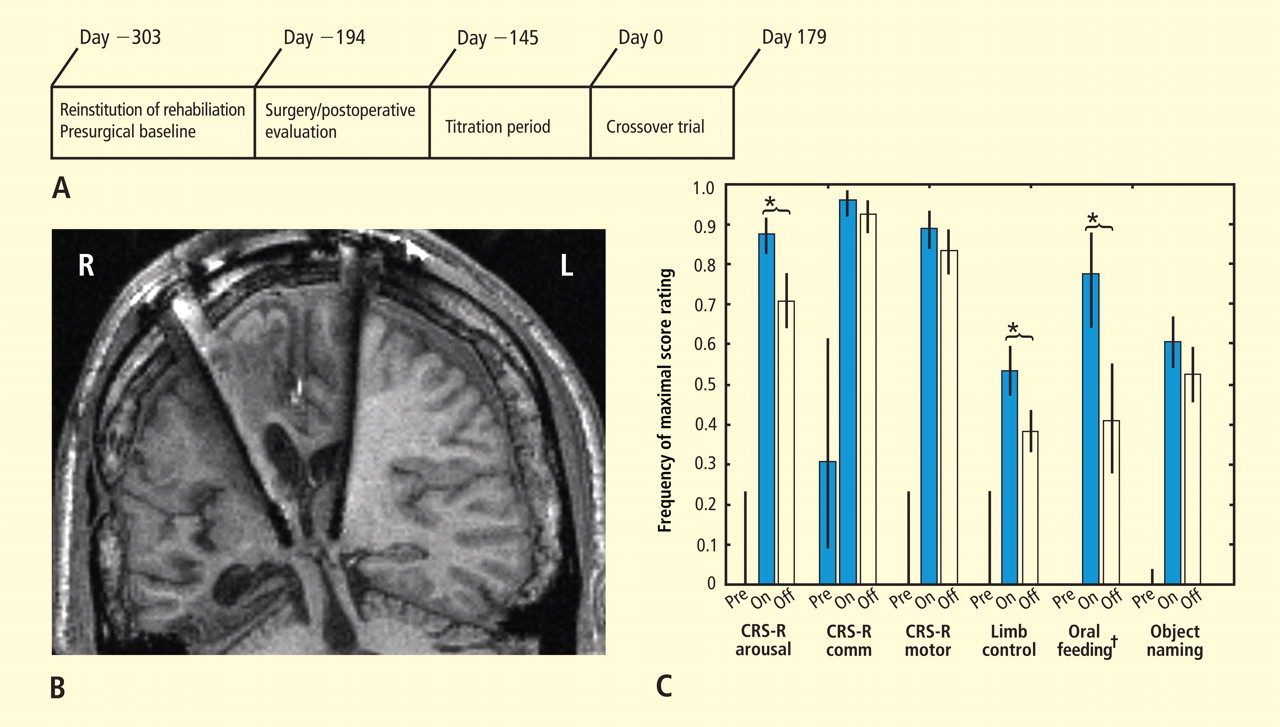
Figure 3C summarizes results of the alternating crossover study and compares the prestimulation baseline assessments of various behaviors with the “on” versus “off” testing of the DBS electrodes during the crossover phase. The results demonstrate the overall impact of DBS compared with approximately 6 months of ongoing rehabilitation efforts in the absence of DBS exposure. Overall the findings show marked improvement in behavioral responsiveness compared with prestimulation frequencies of the highest-level behavioral response across six categories. The primary outcome assessments were prospectively chosen from subscales of the CRS-R, which is a well-validated psychometric tool used in patients with disorders of consciousness. CRS-R subscale items that had shown variation during the presurgical baseline assessment were chosen prospectively as the primary outcome measures. Notably, the CRS-R oral motor subscale was not chosen because no variation in this measure had been identified during the baseline assessment period. In addition, an object-naming scale and two other tailored secondary measures were developed later, during the titration phase, as the patient’s behavior changed, and were calibrated to be tested using these secondary measurement scales. All six measures showed marked change from prestimulation baseline levels, with five of the six measures showing higher-level behaviors than those seen prior to stimulation, regardless of whether the electrodes were on or off.43
As shown in Figure 3C, the behaviors captured by secondary measures had never occurred before the titration phase of the study; ie, the patient initially lacked a capacity for object naming, oral feeding, and the complex controlled goal-directed movements captured in the secondary limb movement measure, thus setting a prestimulation baseline frequency of 0 for these measures (see supplementary material in Schiff et al43). Three outcome measures—one primary (CRS-R arousal subscale) and two secondary (oral feeding and limb control)—showed statistically significant dependence on DBS during the 6-month period, as indicated by a significantly higher frequency of maximal score rating during “on” versus “off” periods (Figure 3C). The continuation of improvements during the “off” periods of the crossover trial (relative to the prestimulation baseline assessments) showed that the DBS effects produced carryover changes that remained after the extensive exposure to DBS during the titration period (for further analysis of the dynamic of these data, see supplementary material in Schiff et al43).
Importantly, these observations are limited to a single human subject and do not provide a guide to their generalizability, 44,45 although they are consistent with the proposed mesocircuit model reviewed above. While the precise mechanism underlying this patient’s improved behavioral responsiveness with central thalamic DBS is unknown, it is likely that DBS served to partially reverse the markedly depressed cerebral global metabolism earlier measured in this patient using fluorodeoxyglucose position emission tomography (FDG-PET)46 and also seen in other patients in minimally conscious state.47 The depressed cerebral metabolism seen in minimally conscious state likely reflects volume loss of neurons, deafferentation of remaining neurons, and neuronal functional impairments. All of these mechanisms may result in low firing rates of neurons in the neocortex, thalamus, and striatum. The mesocircuit model in Figure 2 suggests that direct activation of the central thalamus in patients with such chronically downregulated background synaptic activity may produce excitatory output from central thalamic neurons that acts to partially normalize firing rates and possibly firing patterns within the corticostriatopallidal-thalamocortical system.
SPECULATIONS ON THE IMPORTANCE OF HEART-BRAIN RESEARCH IN FUTURE STUDIES OF RECOVERY OF CONSCIOUSNESS
As this conference is focused on the heart-brain interface, it is appropriate to consider the relevance of heart-brain research to the general set of problems reviewed above. In fact, the linkage is quite natural, and classical physiologic psychology research has shown that cerebral arousal regulation is associated with patterned modulation of cardiac rhythm and autonomic function linked to the behavioral state.48,49 Among the most relevant observations are demonstrations that sustained focused attention is associated with several stereotyped cardiac and autonomic changes, including anticipatory bradycardia,50,51 pupillary dilatation, 52 and others (eg, galvanic skin response). Neurologic cases have shown that such couplings of effort to reflex bradycardia, pupillary dilatation, and other autonomic markers are altered by focal cerebral lesions in the right frontal lobe53 and left anterior cingulate cortex.54
In the single-subject central thalamic DBS study reviewed above,43 there were several unpublished observations that are potentially relevant to these mechanisms. During initial bedside testing of the individual DBS electrode contacts in first 2 postoperative days, electrical stimulation above threshold voltages associated with visible arousal response (for details, see supplementary material in Schiff et al43) consistently produced marked changes in heart rate and audible modulations of heart rhythm during interactions with the patient. Notably, the patient’s basal heart rate rose from a stable level of approximately 50 to 55 beats per minute to approximately 70 to 75 beats per minute—a nearly 50% increase. While increases in blood pressure and heart rate typically accompany arousal, the heart rate change observed here may reflect a marked change in cerebral metabolic rates. Earlier quantitative FDG-PET imaging in this patient revealed a global metabolic rate across the brain of approximately half the normal level.46 Considering that the brain consumes approximately 23% of the cardiac output,55 the increased heart rate observed in this setting may reflect an increase in demand in cardiac output, possibly as much as 100%. At the same time that these changes occurred, there was an audible cardiac deceleration noted when the patient was attentionally engaged by the examiner (this occurred without scoreable variation in most of the quantitative neurobehavioral metrics; see supplementary material in Schiff et al43). Of note, although the patient had suffered a complex severe brain injury, the right ventral frontal lobe showed the largest structural lesion46; injuries to the right hemisphere are associated with loss of such anticipatory changes in heart rate during attentional task performance.53
These anecdotal observations suggest that future studies that include measures to track patterns of heart rate variation during recovery of consciousness might provide an indirect index of increasing brain demand for allocation of cardiac output or emergent neural control of mechanisms linking cardiovascular response to attentive behavior. Ongoing coupling of electroencephalographic measures to autonomic and basal cardiac rhythms may be particularly interesting to examine during social interactions,56 as it is likely that behavioral responsiveness is linked to social stimuli. Emotional reactivity has been proposed as an essential component of arousal per se,49 and although not formally studied in the DBS trial reviewed above, emotional reengagement seems to be a clear concomitant of the collection of gestural and verbal behavioral improvements operationally tracked using quantitative behavioral scales. Beyond tracking heart-brain interactions as an index of brain recovery, it is possible that the integrity of heart-brain interactions may also be a target for optimization in support of recovery of consciousness after nonprogressive brain injury. Moreover, studies of optimization of cardiac function in severely brain-injured patients may provide insight into the recovery process as well.
- Blake R. The Day Donny Herbert Woke Up: A True Story. New York, NY: Harmony Press; 2007.
- Schiff ND, Fins JJ. Hope for “comatose” patients. Cerebrum 2003; 5:7–24.
- Smothers R. Injured in ’88, officer awakes in ’96. New York Times. February 16, 1996. Available at: http://www.nytimes.com/1996/02/16/us/injured-in-88-officer-awakes-in-96.html. Accessed March 16, 2010.
- Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed. New York: Oxford University Press; 2007.
- Schiff ND, Plum F. The role of arousal and ‘gating’ systems in the neurology of impaired consciousness. J Clin Neurophysiol 2000; 17:438–452.
- Kobylarz EJ, Schiff ND. Neurophysiological correlates of persistent vegetative and minimally conscious states. Neuropsychol Rehabil 2005; 15:323–332.
- Giacino JT, Ashwal S, Childs N, et al The minimally conscious state: definition and diagnostic criteria. Neurology 2002; 58:349–353.
- Giacino JT, Whyte J. The vegetative and minimally conscious states: current knowledge and remaining questions. J Head Trauma Rehabil 2005; 20:30–50.
- Nakase-Richardson R, Yablon SA, Sherer M, Nick TG, Evans CC. Emergence from minimally conscious state: insights from evaluation of posttraumatic confusion. Neurology 2009; 73:1120–1126.
- Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after acute insult. Brain 2000; 123:1327–1338.
- Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury. Neurology 2001; 56:486–490.
- Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry 2001; 71:521–524.
- Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol 2006; 65:478–488.
- Kinney HC, Korein J, Panigrahy A, Dikkes P, Goode R. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med 1994; 330:1469–1475.
- Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev 2002; 39:107–140.
- Scannell JW, Burns GA, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex 1999; 9:277–299.
- Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol 1981; 10:127–148.
- Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci 2008; 1129:105–118.
- Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science 1996; 271:512–515.
- Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport 1998; 9:R37–47.
- Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage 2004; 21:1232–1241.
- Paus T, Zatorre RJ, Hofle N, et al Time-related changes in neural systems underlying attention and arousal during the performance of an auditory vigilance task. J Cogn Neurosci 1997; 9:392–408.
- Shah SA, Baker JL, Ryou JW, Purpura KP, Schiff ND. Modulation of arousal regulation with central thalamic deep brain stimulation. Conf Proc IEEE Eng Med Biol Soc 2009; 2009:3314–3317.
- Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol 2004; 91:2628–2648.
- Bohland JW, Wu C, Barbas H, et al A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput Biol 2009; 5:e1000334.
- Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci 2010; 33:1–9.
- Schiff ND, Posner JP. Another “Awakenings.” Ann Neurol 2007; 62:5–7.
- Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci 1994; 17:52–57.
- Morel A, Liu J, Wannier T, Jeanmonod D, Rouiller EM. Divergence and convergence of thalamocortical projections to premotor and supplementary motor cortex: a multiple tracing study in the macaque monkey. Eur J Neurosci 2005; 21:1007–1029.
- Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience 1996; 72:679–687.
- Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci 2007; 27:4374–4384.
- Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull 2009; 78:60–68.
- Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci 2005; 28:364–370.
- Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma 2007; 24:919–926.
- Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry 2003; 74:1571–1573.
- Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury–associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil 2002; 17:300–313.
- Brefel-Courbon C, Payoux P, Ory F, et al Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol 2007; 62:102–105.
- Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil 2009; 88:410–418.
- Shames JL, Ring H. Transient reversal of anoxic brain injury–related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil 2008; 89:386–388.
- Cohen SI, Duong TT. Increased arousal in a patient with anoxic brain injury after administration of zolpidem. Am J Phys Med Rehabil 2008; 87:229–231.
- Williams ST, Conte MM, Kobylarz EJ, Hersh JE, Victor JD, Schiff ND. Quantitative neurophysiologic characterization of a paradoxical response to zolpidem in a severely brain-injured human subject. Program No. 541.6/R9. 2009 Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2009. Online.
- Chen L, Savio Chan C, Yung WH. Electrophysiological and behavioral effects of zolpidem in rat globus pallidus. Exp Neurol 2004; 186:212–220.
- Schiff ND, Giacino JT, Kalmar K, et al Behavioral improvements with thalamic stimulation after severe traumatic brain injury. Nature 2007; 448:600–603.
- Victor JD, Schiff ND. Meeting rigorous statistical standards in case reports. Ann Neurol 2008; 64:592.
- Schiff ND, Giacino JT, Fins JJ. Deep brain stimulation, neuroethics, and the minimally conscious state: moving beyond proof of principle. Arch Neurol 2009; 66:697–702.
- Schiff ND, Rodriguez-Moreno D, Kamal A, et al fMRI reveals large-scale network activation in minimally conscious patients. Neurology 2005; 64:514–523.
- Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol 2004; 3:537–546.
- Obrist PA. Presidential address, 1975. The cardiovascular-behavioral interaction—as it appears today. Psychophysiology 1976; 13:95–107.
- Pfaff D. Brain Arousal and Information Theory: Neural and Genetic Mechanisms. Cambridge, MA: Harvard University Press; 2006.
- Lacey BC, Lacey JI. Presidential address, 1979. Cognitive modulation of time-dependent primary bradycardia. Psychophysiology 1980; 17:209–221.
- Obrist PA, Light KC, Langer AW, Gringnolo A, McCubbin JA. Behavioural-cardiac interactions: the psychosomatic hypothesis. J Psychosom Res 1978; 22:301–325.
- Kahneman D. Attention and Effort. Englewood Cliffs, NJ: Prentice-Hall; 1973.
- Yokoyama K, Jennings R, Ackles P, Hood P, Boller F. Lack of heart rate changes during an attention-demanding task after right hemisphere lesions. Neurology 1987; 37:624–630.
- Naccache L, Dehaene S, Cohen L, et al Effortless control: executive attention and conscious feeling of mental effort are dissociable. Neuropsychologia 2005; 43:1318–1328.
- Roland P. Brain Activation. New York, NY: Wiley Press; 1997.
- Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med 2009; 76( suppl 2):S86–S90.
WHY WE NEED TO UNDERSTAND MECHANISMS OF RECOVERY AFTER SEVERE BRAIN INJURY
The problem of recovery of consciousness after severe brain injury is one that easily captures the imagination of both the lay public and the professional. Puzzling reports continue to arise of late recovery of speech, language, memory, and other higher cognitive functions in rare patients, yet a scientific framework for the systematic assessment of these phenomena has been lacking.1–3 Some of these cases provide intriguing hints to the possible role of various medications (such as dopaminergic, serotoninergic, and noradrenergic agents) as well as spontaneous changes in brain function arising over time. As discussed below, the varying levels of recovery following coma seen after multifocal traumatic or nontraumatic brain injuries may share some common underlying mechanisms at the “circuit” level. Severe brain injuries producing coma have many causes (see Posner et al4 for a comprehensive review), but careful review reveals an overlap of structural pathologies and functional disturbances isolated to specific cerebral structures across several clinical syndromes grouped under the framework of “disorders of consciousness,” 5 with an emphasis on the role of particular substructures. Perhaps most important is a consideration of the pathologic, anatomic, and pathophysiologic role of the anterior forebrain, particularly the relationships of the brainstem and basal forebrain arousal systems, the central thalamus, and frontostriatal pathways, as reviewed below.
The large gray box in Figure 1 indicates the disquietingly high degree of uncertainty in assessing cognitive level in some patients who lack controllable motor output channels. The locked-in state (bottom right corner of figure) defines patients who retain total preservation of cognitive function but otherwise may appear no different from those in deep coma. Although locked-in state often arises in the context of neurologic injuries that selectively damage motor output pathways distal from their cortical origins or that slowly reduce primary motor neuron function, this syndrome and closely similar conditions arise in patients with complex brain injuries. Such patients likely retain full or nearly normal consciousness but unfortunately are unable to produce consistent goal-directed movements that allow for communication. In principle, such patients could retain significant cognitive capacity near the normal range of cognitive function yet be indistinguishable from patients in minimally conscious state.
ROLE OF THE CENTRAL THALAMUS IN SEVERE BRAIN INJURIES
Recent studies have yielded evidence for common anatomic pathologies following severe injuries associated with vegetative state10 and minimally conscious state11 as well as pathologies underlying severe to moderate cognitive disability.12 Autopsy studies of patients remaining in vegetative state at the time of death have identified widespread neuronal death throughout the thalamus as the common finding following either anoxia or diffuse axonal injury that produces widespread disruption of white matter connections.10 The severe bilateral thalamic damage after either trauma or anoxia seen in permanent vegetative state is not, however, invariably associated with diffuse neocortical neuronal cell death. This is particularly true of traumatic brain injury, in which only approximately 10% of brains at autopsy show widespread neocortical cell death.10 Specific subnuclei of the thalamus show the greatest neuronal cell loss following global and multifocal cerebral injuries produced by traumatic brain injuries.13 In particular, the central thalamic nuclei (intralaminar nuclei and related paralaminar nuclei) demonstrate progressive neuronal loss following severe traumatic brain injuries,13 and there is some evidence that a similar pattern might be identified in hypoxic-ischemic injuries.14
Progressively severe disability grades with neuronal loss along a rostrocaudal axis: the anterior intralaminar and surrounding regions initially show volume loss associated with moderate disability, while neuronal loss in the ventral and lateral nuclei of the central thalamus (posterior intralaminar group) appears with worsening disability associated with minimally conscious state and vegetative state.13 This progressive and relatively specific involvement of the nuclei of the central thalamus likely results from the unique geometry of these neurons, which have wide point-to-point connectivity across the cerebral hemisphere.15,16 The marked neuronal volume loss in these cells is likely due to their integration of the effects of neuronal cell death across large cerebral territories after diffuse trauma, hypoxia, and other nonselective severe brain injuries.
Importantly, however, focal bilateral injuries to these regions of the central thalamus are also associated with global disorders of consciousness (coma, vegetative state, and minimally conscious state).5,17 This observation indicates that these neurons also play a causal role in the production of disorders of consciousness. Abrupt injuries of the central thalamus on both sides of the brain are associated with acute coma, reflecting these cells’ key contribution to normal mechanisms of arousal regulation (reviewed by Schiff18). The central thalamus receives ascending projections from the brainstem/basal forebrain “arousal systems” that control the activity of many cortical and thalamic neurons during the sleep-wake cycle. Importantly, the central thalamus is strongly innervated by the cholinergic, serotoninergic, and noradrenergic afferents of the brainstem arousal systems (see Schiff18 for review). These same neurons also are innervated by descending projections from frontal cortical regions supporting “executive” functions that underlie goal-directed behaviors. Collectively, these ascending and descending influences on the central thalamus appear to modulate the level of arousal associated with generalized alertness and variations in cognitive effort, stress, sleep deprivation, and other variables affecting the wakeful state.15,18–22
Neuroimaging and electrophysiologic studies offer further evidence that the anatomic specializations of the central thalamus play an important role in regulating brain activation during attentive wakefulness. The central thalamus shows selective activation in normal subjects performing tasks requiring a short-term shift of attention,19,23 sustained cognitive demands of high vigilance,22 or memory holds over extended time periods.23,24 Central thalamic activation associated with varying levels of vigilance correlates with global cerebral blood flow19 and specifically covaries within the anterior cingulate cortex and pontomesencephalon. 22 Brain activity in the anterior cingulate cortex grades with increasing cognitive load and is recruited by a wide range of cognitive tasks, apparently reciprocally increasing activity along with the central thalamus in response to increasing demands of cognitive effort.20,22
CIRCUIT MECHANISMS UNDERLYING RECOVERY AFTER SEVERE BRAIN INJURY
In addition to the wide point-to-point connections of the central thalamus with the cerebral cortex (predominantly connections to frontal and prefrontal cortices; see Van der Werf et al,15 Groenewegen and Berendse,28 Morel et al29), these neurons have important projections to the striatum that return via projections from the globus pallidus. 30 These projections from the central thalamus (both central lateral nucleus and parafascicularis nucleus) diffusely innervate the striatum and project onto the medium spiny neurons (MSNs), the output neuron of structure.31 Because the specific thalamostriatal projections from these central thalamic neurons use glutamate transmitters with a high probability of synaptic release,32 they likely also have a strong role in modulating background activity in the striatum.
The MSNs represent an important point of vulnerability in this anterior forebrain mesocircuit, as they have a key role in maintaining activity in the anterior forebrain through their inhibitory projections to the globus pallidus interna, which in turn inhibits the central thalamus. 33 MSNs have instrinsic cell membrane properties that keep them below their firing threshold unless a high level of spontaneous background synaptic activity arising from excitatory corticostriatal and thalamostriatal inputs is present in concert with sufficient concentrations of the neurotransmitter dopamine.33 In the setting of diffuse deafferentation or neuronal loss following severe brain injury of any type, it is expected that background excitatory synaptic activity is considerably reduced. Under these circumstances, a broad withdrawal of direct excitatory striatal projections from the central thalamus and corticostriatal inputs is likely to cause MSN output to shut down. Observations of regional changes in brain metabolism following severe brain injuries, specific responses to pharmacologic and electrophysiologic interventions in brain-injured subjects, and normal variations in brain state are all consistent with this mesocircuit model (see Schiff26 for comprehensive review). Similarly, a consistent pattern of selective metabolic downregulation within the anterior forebrain has been shown to specifically grade with severity of behavioral impairment following diffuse axonal injury,34 and application of dopaminergic agents in such patients often will produce behavioral facilitation.35,36 These medications may facilitate the output of the MSNs and directly modulate mesial frontal cortical neurons, possibly restoring anterior forebrain activity within the loop connections of the frontal cortex, striatum, pallidum, and central thalamus.
This model provides context for understanding another paradoxical observation—ie, the association of the sedative zolpidem (Ambien), a nonbenzodiazepine hypnotic that potentiates GABAA receptors, with behavioral improvement of alertness and interactive behavior in severely brain-injured patients.37–41 Zolpidem’s primary direct action in patient responders, as originally proposed by Schiff and Posner,27 may be upon on the globus pallidus interna, producing a release of tonic inhibition of the central thalamus in the setting of a broad reduction in background excitatory neurotransmission (as seen, for example, following diffuse hypoxic-ischemic injury) and leading to a shutdown of the inhibitory projection of the MSNs. The GABAA alpha-1 subunit is expressed in large quantity in the globus pallidus interna, and experimental studies support this mechanism of action.42
SINGLE-SUBJECT STUDY OF CENTRAL THALAMIC STIMULATION IN MINIMALLY CONSCIOUS STATE
A further implication of the mesocircuit model is that direct activation of the central thalamus is expected to be the causal step in reactivating a downregulated anterior forebrain system, suggesting that direct modulation of the central thalamus might facilitate behavioral responsiveness in some patients with severe brain injuries. A recent study offers evidence that direct electrical stimulation of the central thalamus can produce behavioral facilitation.
In this single-subject study of central thalamic deep brain stimulation (DBS), a 38-year-old man remained in minimally conscious state for 6 years following a severe closed head injury following blunt trauma to the right frontal lobe.43 After 3 months in a vegetative state, the patient exhibited the first evidence of clear behaviors in response to sensory stimulation consistent with minimally conscious state and advanced to eventually demonstrating a best behavioral response of inconsistent command-following and communication using eye movements. This behavioral level remained unchanged at the start of the DBS study 4 years later, as confirmed by evaluation with the Coma Recovery Scale–Revised (CRS-R), a formal behavioral assessment tool.
Figure 3C summarizes results of the alternating crossover study and compares the prestimulation baseline assessments of various behaviors with the “on” versus “off” testing of the DBS electrodes during the crossover phase. The results demonstrate the overall impact of DBS compared with approximately 6 months of ongoing rehabilitation efforts in the absence of DBS exposure. Overall the findings show marked improvement in behavioral responsiveness compared with prestimulation frequencies of the highest-level behavioral response across six categories. The primary outcome assessments were prospectively chosen from subscales of the CRS-R, which is a well-validated psychometric tool used in patients with disorders of consciousness. CRS-R subscale items that had shown variation during the presurgical baseline assessment were chosen prospectively as the primary outcome measures. Notably, the CRS-R oral motor subscale was not chosen because no variation in this measure had been identified during the baseline assessment period. In addition, an object-naming scale and two other tailored secondary measures were developed later, during the titration phase, as the patient’s behavior changed, and were calibrated to be tested using these secondary measurement scales. All six measures showed marked change from prestimulation baseline levels, with five of the six measures showing higher-level behaviors than those seen prior to stimulation, regardless of whether the electrodes were on or off.43
As shown in Figure 3C, the behaviors captured by secondary measures had never occurred before the titration phase of the study; ie, the patient initially lacked a capacity for object naming, oral feeding, and the complex controlled goal-directed movements captured in the secondary limb movement measure, thus setting a prestimulation baseline frequency of 0 for these measures (see supplementary material in Schiff et al43). Three outcome measures—one primary (CRS-R arousal subscale) and two secondary (oral feeding and limb control)—showed statistically significant dependence on DBS during the 6-month period, as indicated by a significantly higher frequency of maximal score rating during “on” versus “off” periods (Figure 3C). The continuation of improvements during the “off” periods of the crossover trial (relative to the prestimulation baseline assessments) showed that the DBS effects produced carryover changes that remained after the extensive exposure to DBS during the titration period (for further analysis of the dynamic of these data, see supplementary material in Schiff et al43).
Importantly, these observations are limited to a single human subject and do not provide a guide to their generalizability, 44,45 although they are consistent with the proposed mesocircuit model reviewed above. While the precise mechanism underlying this patient’s improved behavioral responsiveness with central thalamic DBS is unknown, it is likely that DBS served to partially reverse the markedly depressed cerebral global metabolism earlier measured in this patient using fluorodeoxyglucose position emission tomography (FDG-PET)46 and also seen in other patients in minimally conscious state.47 The depressed cerebral metabolism seen in minimally conscious state likely reflects volume loss of neurons, deafferentation of remaining neurons, and neuronal functional impairments. All of these mechanisms may result in low firing rates of neurons in the neocortex, thalamus, and striatum. The mesocircuit model in Figure 2 suggests that direct activation of the central thalamus in patients with such chronically downregulated background synaptic activity may produce excitatory output from central thalamic neurons that acts to partially normalize firing rates and possibly firing patterns within the corticostriatopallidal-thalamocortical system.
SPECULATIONS ON THE IMPORTANCE OF HEART-BRAIN RESEARCH IN FUTURE STUDIES OF RECOVERY OF CONSCIOUSNESS
As this conference is focused on the heart-brain interface, it is appropriate to consider the relevance of heart-brain research to the general set of problems reviewed above. In fact, the linkage is quite natural, and classical physiologic psychology research has shown that cerebral arousal regulation is associated with patterned modulation of cardiac rhythm and autonomic function linked to the behavioral state.48,49 Among the most relevant observations are demonstrations that sustained focused attention is associated with several stereotyped cardiac and autonomic changes, including anticipatory bradycardia,50,51 pupillary dilatation, 52 and others (eg, galvanic skin response). Neurologic cases have shown that such couplings of effort to reflex bradycardia, pupillary dilatation, and other autonomic markers are altered by focal cerebral lesions in the right frontal lobe53 and left anterior cingulate cortex.54
In the single-subject central thalamic DBS study reviewed above,43 there were several unpublished observations that are potentially relevant to these mechanisms. During initial bedside testing of the individual DBS electrode contacts in first 2 postoperative days, electrical stimulation above threshold voltages associated with visible arousal response (for details, see supplementary material in Schiff et al43) consistently produced marked changes in heart rate and audible modulations of heart rhythm during interactions with the patient. Notably, the patient’s basal heart rate rose from a stable level of approximately 50 to 55 beats per minute to approximately 70 to 75 beats per minute—a nearly 50% increase. While increases in blood pressure and heart rate typically accompany arousal, the heart rate change observed here may reflect a marked change in cerebral metabolic rates. Earlier quantitative FDG-PET imaging in this patient revealed a global metabolic rate across the brain of approximately half the normal level.46 Considering that the brain consumes approximately 23% of the cardiac output,55 the increased heart rate observed in this setting may reflect an increase in demand in cardiac output, possibly as much as 100%. At the same time that these changes occurred, there was an audible cardiac deceleration noted when the patient was attentionally engaged by the examiner (this occurred without scoreable variation in most of the quantitative neurobehavioral metrics; see supplementary material in Schiff et al43). Of note, although the patient had suffered a complex severe brain injury, the right ventral frontal lobe showed the largest structural lesion46; injuries to the right hemisphere are associated with loss of such anticipatory changes in heart rate during attentional task performance.53
These anecdotal observations suggest that future studies that include measures to track patterns of heart rate variation during recovery of consciousness might provide an indirect index of increasing brain demand for allocation of cardiac output or emergent neural control of mechanisms linking cardiovascular response to attentive behavior. Ongoing coupling of electroencephalographic measures to autonomic and basal cardiac rhythms may be particularly interesting to examine during social interactions,56 as it is likely that behavioral responsiveness is linked to social stimuli. Emotional reactivity has been proposed as an essential component of arousal per se,49 and although not formally studied in the DBS trial reviewed above, emotional reengagement seems to be a clear concomitant of the collection of gestural and verbal behavioral improvements operationally tracked using quantitative behavioral scales. Beyond tracking heart-brain interactions as an index of brain recovery, it is possible that the integrity of heart-brain interactions may also be a target for optimization in support of recovery of consciousness after nonprogressive brain injury. Moreover, studies of optimization of cardiac function in severely brain-injured patients may provide insight into the recovery process as well.
WHY WE NEED TO UNDERSTAND MECHANISMS OF RECOVERY AFTER SEVERE BRAIN INJURY
The problem of recovery of consciousness after severe brain injury is one that easily captures the imagination of both the lay public and the professional. Puzzling reports continue to arise of late recovery of speech, language, memory, and other higher cognitive functions in rare patients, yet a scientific framework for the systematic assessment of these phenomena has been lacking.1–3 Some of these cases provide intriguing hints to the possible role of various medications (such as dopaminergic, serotoninergic, and noradrenergic agents) as well as spontaneous changes in brain function arising over time. As discussed below, the varying levels of recovery following coma seen after multifocal traumatic or nontraumatic brain injuries may share some common underlying mechanisms at the “circuit” level. Severe brain injuries producing coma have many causes (see Posner et al4 for a comprehensive review), but careful review reveals an overlap of structural pathologies and functional disturbances isolated to specific cerebral structures across several clinical syndromes grouped under the framework of “disorders of consciousness,” 5 with an emphasis on the role of particular substructures. Perhaps most important is a consideration of the pathologic, anatomic, and pathophysiologic role of the anterior forebrain, particularly the relationships of the brainstem and basal forebrain arousal systems, the central thalamus, and frontostriatal pathways, as reviewed below.
The large gray box in Figure 1 indicates the disquietingly high degree of uncertainty in assessing cognitive level in some patients who lack controllable motor output channels. The locked-in state (bottom right corner of figure) defines patients who retain total preservation of cognitive function but otherwise may appear no different from those in deep coma. Although locked-in state often arises in the context of neurologic injuries that selectively damage motor output pathways distal from their cortical origins or that slowly reduce primary motor neuron function, this syndrome and closely similar conditions arise in patients with complex brain injuries. Such patients likely retain full or nearly normal consciousness but unfortunately are unable to produce consistent goal-directed movements that allow for communication. In principle, such patients could retain significant cognitive capacity near the normal range of cognitive function yet be indistinguishable from patients in minimally conscious state.
ROLE OF THE CENTRAL THALAMUS IN SEVERE BRAIN INJURIES
Recent studies have yielded evidence for common anatomic pathologies following severe injuries associated with vegetative state10 and minimally conscious state11 as well as pathologies underlying severe to moderate cognitive disability.12 Autopsy studies of patients remaining in vegetative state at the time of death have identified widespread neuronal death throughout the thalamus as the common finding following either anoxia or diffuse axonal injury that produces widespread disruption of white matter connections.10 The severe bilateral thalamic damage after either trauma or anoxia seen in permanent vegetative state is not, however, invariably associated with diffuse neocortical neuronal cell death. This is particularly true of traumatic brain injury, in which only approximately 10% of brains at autopsy show widespread neocortical cell death.10 Specific subnuclei of the thalamus show the greatest neuronal cell loss following global and multifocal cerebral injuries produced by traumatic brain injuries.13 In particular, the central thalamic nuclei (intralaminar nuclei and related paralaminar nuclei) demonstrate progressive neuronal loss following severe traumatic brain injuries,13 and there is some evidence that a similar pattern might be identified in hypoxic-ischemic injuries.14
Progressively severe disability grades with neuronal loss along a rostrocaudal axis: the anterior intralaminar and surrounding regions initially show volume loss associated with moderate disability, while neuronal loss in the ventral and lateral nuclei of the central thalamus (posterior intralaminar group) appears with worsening disability associated with minimally conscious state and vegetative state.13 This progressive and relatively specific involvement of the nuclei of the central thalamus likely results from the unique geometry of these neurons, which have wide point-to-point connectivity across the cerebral hemisphere.15,16 The marked neuronal volume loss in these cells is likely due to their integration of the effects of neuronal cell death across large cerebral territories after diffuse trauma, hypoxia, and other nonselective severe brain injuries.
Importantly, however, focal bilateral injuries to these regions of the central thalamus are also associated with global disorders of consciousness (coma, vegetative state, and minimally conscious state).5,17 This observation indicates that these neurons also play a causal role in the production of disorders of consciousness. Abrupt injuries of the central thalamus on both sides of the brain are associated with acute coma, reflecting these cells’ key contribution to normal mechanisms of arousal regulation (reviewed by Schiff18). The central thalamus receives ascending projections from the brainstem/basal forebrain “arousal systems” that control the activity of many cortical and thalamic neurons during the sleep-wake cycle. Importantly, the central thalamus is strongly innervated by the cholinergic, serotoninergic, and noradrenergic afferents of the brainstem arousal systems (see Schiff18 for review). These same neurons also are innervated by descending projections from frontal cortical regions supporting “executive” functions that underlie goal-directed behaviors. Collectively, these ascending and descending influences on the central thalamus appear to modulate the level of arousal associated with generalized alertness and variations in cognitive effort, stress, sleep deprivation, and other variables affecting the wakeful state.15,18–22
Neuroimaging and electrophysiologic studies offer further evidence that the anatomic specializations of the central thalamus play an important role in regulating brain activation during attentive wakefulness. The central thalamus shows selective activation in normal subjects performing tasks requiring a short-term shift of attention,19,23 sustained cognitive demands of high vigilance,22 or memory holds over extended time periods.23,24 Central thalamic activation associated with varying levels of vigilance correlates with global cerebral blood flow19 and specifically covaries within the anterior cingulate cortex and pontomesencephalon. 22 Brain activity in the anterior cingulate cortex grades with increasing cognitive load and is recruited by a wide range of cognitive tasks, apparently reciprocally increasing activity along with the central thalamus in response to increasing demands of cognitive effort.20,22
CIRCUIT MECHANISMS UNDERLYING RECOVERY AFTER SEVERE BRAIN INJURY
In addition to the wide point-to-point connections of the central thalamus with the cerebral cortex (predominantly connections to frontal and prefrontal cortices; see Van der Werf et al,15 Groenewegen and Berendse,28 Morel et al29), these neurons have important projections to the striatum that return via projections from the globus pallidus. 30 These projections from the central thalamus (both central lateral nucleus and parafascicularis nucleus) diffusely innervate the striatum and project onto the medium spiny neurons (MSNs), the output neuron of structure.31 Because the specific thalamostriatal projections from these central thalamic neurons use glutamate transmitters with a high probability of synaptic release,32 they likely also have a strong role in modulating background activity in the striatum.
The MSNs represent an important point of vulnerability in this anterior forebrain mesocircuit, as they have a key role in maintaining activity in the anterior forebrain through their inhibitory projections to the globus pallidus interna, which in turn inhibits the central thalamus. 33 MSNs have instrinsic cell membrane properties that keep them below their firing threshold unless a high level of spontaneous background synaptic activity arising from excitatory corticostriatal and thalamostriatal inputs is present in concert with sufficient concentrations of the neurotransmitter dopamine.33 In the setting of diffuse deafferentation or neuronal loss following severe brain injury of any type, it is expected that background excitatory synaptic activity is considerably reduced. Under these circumstances, a broad withdrawal of direct excitatory striatal projections from the central thalamus and corticostriatal inputs is likely to cause MSN output to shut down. Observations of regional changes in brain metabolism following severe brain injuries, specific responses to pharmacologic and electrophysiologic interventions in brain-injured subjects, and normal variations in brain state are all consistent with this mesocircuit model (see Schiff26 for comprehensive review). Similarly, a consistent pattern of selective metabolic downregulation within the anterior forebrain has been shown to specifically grade with severity of behavioral impairment following diffuse axonal injury,34 and application of dopaminergic agents in such patients often will produce behavioral facilitation.35,36 These medications may facilitate the output of the MSNs and directly modulate mesial frontal cortical neurons, possibly restoring anterior forebrain activity within the loop connections of the frontal cortex, striatum, pallidum, and central thalamus.
This model provides context for understanding another paradoxical observation—ie, the association of the sedative zolpidem (Ambien), a nonbenzodiazepine hypnotic that potentiates GABAA receptors, with behavioral improvement of alertness and interactive behavior in severely brain-injured patients.37–41 Zolpidem’s primary direct action in patient responders, as originally proposed by Schiff and Posner,27 may be upon on the globus pallidus interna, producing a release of tonic inhibition of the central thalamus in the setting of a broad reduction in background excitatory neurotransmission (as seen, for example, following diffuse hypoxic-ischemic injury) and leading to a shutdown of the inhibitory projection of the MSNs. The GABAA alpha-1 subunit is expressed in large quantity in the globus pallidus interna, and experimental studies support this mechanism of action.42
SINGLE-SUBJECT STUDY OF CENTRAL THALAMIC STIMULATION IN MINIMALLY CONSCIOUS STATE
A further implication of the mesocircuit model is that direct activation of the central thalamus is expected to be the causal step in reactivating a downregulated anterior forebrain system, suggesting that direct modulation of the central thalamus might facilitate behavioral responsiveness in some patients with severe brain injuries. A recent study offers evidence that direct electrical stimulation of the central thalamus can produce behavioral facilitation.
In this single-subject study of central thalamic deep brain stimulation (DBS), a 38-year-old man remained in minimally conscious state for 6 years following a severe closed head injury following blunt trauma to the right frontal lobe.43 After 3 months in a vegetative state, the patient exhibited the first evidence of clear behaviors in response to sensory stimulation consistent with minimally conscious state and advanced to eventually demonstrating a best behavioral response of inconsistent command-following and communication using eye movements. This behavioral level remained unchanged at the start of the DBS study 4 years later, as confirmed by evaluation with the Coma Recovery Scale–Revised (CRS-R), a formal behavioral assessment tool.
Figure 3C summarizes results of the alternating crossover study and compares the prestimulation baseline assessments of various behaviors with the “on” versus “off” testing of the DBS electrodes during the crossover phase. The results demonstrate the overall impact of DBS compared with approximately 6 months of ongoing rehabilitation efforts in the absence of DBS exposure. Overall the findings show marked improvement in behavioral responsiveness compared with prestimulation frequencies of the highest-level behavioral response across six categories. The primary outcome assessments were prospectively chosen from subscales of the CRS-R, which is a well-validated psychometric tool used in patients with disorders of consciousness. CRS-R subscale items that had shown variation during the presurgical baseline assessment were chosen prospectively as the primary outcome measures. Notably, the CRS-R oral motor subscale was not chosen because no variation in this measure had been identified during the baseline assessment period. In addition, an object-naming scale and two other tailored secondary measures were developed later, during the titration phase, as the patient’s behavior changed, and were calibrated to be tested using these secondary measurement scales. All six measures showed marked change from prestimulation baseline levels, with five of the six measures showing higher-level behaviors than those seen prior to stimulation, regardless of whether the electrodes were on or off.43
As shown in Figure 3C, the behaviors captured by secondary measures had never occurred before the titration phase of the study; ie, the patient initially lacked a capacity for object naming, oral feeding, and the complex controlled goal-directed movements captured in the secondary limb movement measure, thus setting a prestimulation baseline frequency of 0 for these measures (see supplementary material in Schiff et al43). Three outcome measures—one primary (CRS-R arousal subscale) and two secondary (oral feeding and limb control)—showed statistically significant dependence on DBS during the 6-month period, as indicated by a significantly higher frequency of maximal score rating during “on” versus “off” periods (Figure 3C). The continuation of improvements during the “off” periods of the crossover trial (relative to the prestimulation baseline assessments) showed that the DBS effects produced carryover changes that remained after the extensive exposure to DBS during the titration period (for further analysis of the dynamic of these data, see supplementary material in Schiff et al43).
Importantly, these observations are limited to a single human subject and do not provide a guide to their generalizability, 44,45 although they are consistent with the proposed mesocircuit model reviewed above. While the precise mechanism underlying this patient’s improved behavioral responsiveness with central thalamic DBS is unknown, it is likely that DBS served to partially reverse the markedly depressed cerebral global metabolism earlier measured in this patient using fluorodeoxyglucose position emission tomography (FDG-PET)46 and also seen in other patients in minimally conscious state.47 The depressed cerebral metabolism seen in minimally conscious state likely reflects volume loss of neurons, deafferentation of remaining neurons, and neuronal functional impairments. All of these mechanisms may result in low firing rates of neurons in the neocortex, thalamus, and striatum. The mesocircuit model in Figure 2 suggests that direct activation of the central thalamus in patients with such chronically downregulated background synaptic activity may produce excitatory output from central thalamic neurons that acts to partially normalize firing rates and possibly firing patterns within the corticostriatopallidal-thalamocortical system.
SPECULATIONS ON THE IMPORTANCE OF HEART-BRAIN RESEARCH IN FUTURE STUDIES OF RECOVERY OF CONSCIOUSNESS
As this conference is focused on the heart-brain interface, it is appropriate to consider the relevance of heart-brain research to the general set of problems reviewed above. In fact, the linkage is quite natural, and classical physiologic psychology research has shown that cerebral arousal regulation is associated with patterned modulation of cardiac rhythm and autonomic function linked to the behavioral state.48,49 Among the most relevant observations are demonstrations that sustained focused attention is associated with several stereotyped cardiac and autonomic changes, including anticipatory bradycardia,50,51 pupillary dilatation, 52 and others (eg, galvanic skin response). Neurologic cases have shown that such couplings of effort to reflex bradycardia, pupillary dilatation, and other autonomic markers are altered by focal cerebral lesions in the right frontal lobe53 and left anterior cingulate cortex.54
In the single-subject central thalamic DBS study reviewed above,43 there were several unpublished observations that are potentially relevant to these mechanisms. During initial bedside testing of the individual DBS electrode contacts in first 2 postoperative days, electrical stimulation above threshold voltages associated with visible arousal response (for details, see supplementary material in Schiff et al43) consistently produced marked changes in heart rate and audible modulations of heart rhythm during interactions with the patient. Notably, the patient’s basal heart rate rose from a stable level of approximately 50 to 55 beats per minute to approximately 70 to 75 beats per minute—a nearly 50% increase. While increases in blood pressure and heart rate typically accompany arousal, the heart rate change observed here may reflect a marked change in cerebral metabolic rates. Earlier quantitative FDG-PET imaging in this patient revealed a global metabolic rate across the brain of approximately half the normal level.46 Considering that the brain consumes approximately 23% of the cardiac output,55 the increased heart rate observed in this setting may reflect an increase in demand in cardiac output, possibly as much as 100%. At the same time that these changes occurred, there was an audible cardiac deceleration noted when the patient was attentionally engaged by the examiner (this occurred without scoreable variation in most of the quantitative neurobehavioral metrics; see supplementary material in Schiff et al43). Of note, although the patient had suffered a complex severe brain injury, the right ventral frontal lobe showed the largest structural lesion46; injuries to the right hemisphere are associated with loss of such anticipatory changes in heart rate during attentional task performance.53
These anecdotal observations suggest that future studies that include measures to track patterns of heart rate variation during recovery of consciousness might provide an indirect index of increasing brain demand for allocation of cardiac output or emergent neural control of mechanisms linking cardiovascular response to attentive behavior. Ongoing coupling of electroencephalographic measures to autonomic and basal cardiac rhythms may be particularly interesting to examine during social interactions,56 as it is likely that behavioral responsiveness is linked to social stimuli. Emotional reactivity has been proposed as an essential component of arousal per se,49 and although not formally studied in the DBS trial reviewed above, emotional reengagement seems to be a clear concomitant of the collection of gestural and verbal behavioral improvements operationally tracked using quantitative behavioral scales. Beyond tracking heart-brain interactions as an index of brain recovery, it is possible that the integrity of heart-brain interactions may also be a target for optimization in support of recovery of consciousness after nonprogressive brain injury. Moreover, studies of optimization of cardiac function in severely brain-injured patients may provide insight into the recovery process as well.
- Blake R. The Day Donny Herbert Woke Up: A True Story. New York, NY: Harmony Press; 2007.
- Schiff ND, Fins JJ. Hope for “comatose” patients. Cerebrum 2003; 5:7–24.
- Smothers R. Injured in ’88, officer awakes in ’96. New York Times. February 16, 1996. Available at: http://www.nytimes.com/1996/02/16/us/injured-in-88-officer-awakes-in-96.html. Accessed March 16, 2010.
- Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed. New York: Oxford University Press; 2007.
- Schiff ND, Plum F. The role of arousal and ‘gating’ systems in the neurology of impaired consciousness. J Clin Neurophysiol 2000; 17:438–452.
- Kobylarz EJ, Schiff ND. Neurophysiological correlates of persistent vegetative and minimally conscious states. Neuropsychol Rehabil 2005; 15:323–332.
- Giacino JT, Ashwal S, Childs N, et al The minimally conscious state: definition and diagnostic criteria. Neurology 2002; 58:349–353.
- Giacino JT, Whyte J. The vegetative and minimally conscious states: current knowledge and remaining questions. J Head Trauma Rehabil 2005; 20:30–50.
- Nakase-Richardson R, Yablon SA, Sherer M, Nick TG, Evans CC. Emergence from minimally conscious state: insights from evaluation of posttraumatic confusion. Neurology 2009; 73:1120–1126.
- Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after acute insult. Brain 2000; 123:1327–1338.
- Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury. Neurology 2001; 56:486–490.
- Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry 2001; 71:521–524.
- Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol 2006; 65:478–488.
- Kinney HC, Korein J, Panigrahy A, Dikkes P, Goode R. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med 1994; 330:1469–1475.
- Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev 2002; 39:107–140.
- Scannell JW, Burns GA, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex 1999; 9:277–299.
- Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol 1981; 10:127–148.
- Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci 2008; 1129:105–118.
- Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science 1996; 271:512–515.
- Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport 1998; 9:R37–47.
- Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage 2004; 21:1232–1241.
- Paus T, Zatorre RJ, Hofle N, et al Time-related changes in neural systems underlying attention and arousal during the performance of an auditory vigilance task. J Cogn Neurosci 1997; 9:392–408.
- Shah SA, Baker JL, Ryou JW, Purpura KP, Schiff ND. Modulation of arousal regulation with central thalamic deep brain stimulation. Conf Proc IEEE Eng Med Biol Soc 2009; 2009:3314–3317.
- Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol 2004; 91:2628–2648.
- Bohland JW, Wu C, Barbas H, et al A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput Biol 2009; 5:e1000334.
- Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci 2010; 33:1–9.
- Schiff ND, Posner JP. Another “Awakenings.” Ann Neurol 2007; 62:5–7.
- Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci 1994; 17:52–57.
- Morel A, Liu J, Wannier T, Jeanmonod D, Rouiller EM. Divergence and convergence of thalamocortical projections to premotor and supplementary motor cortex: a multiple tracing study in the macaque monkey. Eur J Neurosci 2005; 21:1007–1029.
- Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience 1996; 72:679–687.
- Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci 2007; 27:4374–4384.
- Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull 2009; 78:60–68.
- Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci 2005; 28:364–370.
- Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma 2007; 24:919–926.
- Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry 2003; 74:1571–1573.
- Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury–associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil 2002; 17:300–313.
- Brefel-Courbon C, Payoux P, Ory F, et al Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol 2007; 62:102–105.
- Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil 2009; 88:410–418.
- Shames JL, Ring H. Transient reversal of anoxic brain injury–related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil 2008; 89:386–388.
- Cohen SI, Duong TT. Increased arousal in a patient with anoxic brain injury after administration of zolpidem. Am J Phys Med Rehabil 2008; 87:229–231.
- Williams ST, Conte MM, Kobylarz EJ, Hersh JE, Victor JD, Schiff ND. Quantitative neurophysiologic characterization of a paradoxical response to zolpidem in a severely brain-injured human subject. Program No. 541.6/R9. 2009 Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2009. Online.
- Chen L, Savio Chan C, Yung WH. Electrophysiological and behavioral effects of zolpidem in rat globus pallidus. Exp Neurol 2004; 186:212–220.
- Schiff ND, Giacino JT, Kalmar K, et al Behavioral improvements with thalamic stimulation after severe traumatic brain injury. Nature 2007; 448:600–603.
- Victor JD, Schiff ND. Meeting rigorous statistical standards in case reports. Ann Neurol 2008; 64:592.
- Schiff ND, Giacino JT, Fins JJ. Deep brain stimulation, neuroethics, and the minimally conscious state: moving beyond proof of principle. Arch Neurol 2009; 66:697–702.
- Schiff ND, Rodriguez-Moreno D, Kamal A, et al fMRI reveals large-scale network activation in minimally conscious patients. Neurology 2005; 64:514–523.
- Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol 2004; 3:537–546.
- Obrist PA. Presidential address, 1975. The cardiovascular-behavioral interaction—as it appears today. Psychophysiology 1976; 13:95–107.
- Pfaff D. Brain Arousal and Information Theory: Neural and Genetic Mechanisms. Cambridge, MA: Harvard University Press; 2006.
- Lacey BC, Lacey JI. Presidential address, 1979. Cognitive modulation of time-dependent primary bradycardia. Psychophysiology 1980; 17:209–221.
- Obrist PA, Light KC, Langer AW, Gringnolo A, McCubbin JA. Behavioural-cardiac interactions: the psychosomatic hypothesis. J Psychosom Res 1978; 22:301–325.
- Kahneman D. Attention and Effort. Englewood Cliffs, NJ: Prentice-Hall; 1973.
- Yokoyama K, Jennings R, Ackles P, Hood P, Boller F. Lack of heart rate changes during an attention-demanding task after right hemisphere lesions. Neurology 1987; 37:624–630.
- Naccache L, Dehaene S, Cohen L, et al Effortless control: executive attention and conscious feeling of mental effort are dissociable. Neuropsychologia 2005; 43:1318–1328.
- Roland P. Brain Activation. New York, NY: Wiley Press; 1997.
- Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med 2009; 76( suppl 2):S86–S90.
- Blake R. The Day Donny Herbert Woke Up: A True Story. New York, NY: Harmony Press; 2007.
- Schiff ND, Fins JJ. Hope for “comatose” patients. Cerebrum 2003; 5:7–24.
- Smothers R. Injured in ’88, officer awakes in ’96. New York Times. February 16, 1996. Available at: http://www.nytimes.com/1996/02/16/us/injured-in-88-officer-awakes-in-96.html. Accessed March 16, 2010.
- Posner J, Saper C, Schiff N, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed. New York: Oxford University Press; 2007.
- Schiff ND, Plum F. The role of arousal and ‘gating’ systems in the neurology of impaired consciousness. J Clin Neurophysiol 2000; 17:438–452.
- Kobylarz EJ, Schiff ND. Neurophysiological correlates of persistent vegetative and minimally conscious states. Neuropsychol Rehabil 2005; 15:323–332.
- Giacino JT, Ashwal S, Childs N, et al The minimally conscious state: definition and diagnostic criteria. Neurology 2002; 58:349–353.
- Giacino JT, Whyte J. The vegetative and minimally conscious states: current knowledge and remaining questions. J Head Trauma Rehabil 2005; 20:30–50.
- Nakase-Richardson R, Yablon SA, Sherer M, Nick TG, Evans CC. Emergence from minimally conscious state: insights from evaluation of posttraumatic confusion. Neurology 2009; 73:1120–1126.
- Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after acute insult. Brain 2000; 123:1327–1338.
- Jennett B, Adams JH, Murray LS, Graham DI. Neuropathology in vegetative and severely disabled patients after head injury. Neurology 2001; 56:486–490.
- Adams JH, Graham DI, Jennett B. The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry 2001; 71:521–524.
- Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol 2006; 65:478–488.
- Kinney HC, Korein J, Panigrahy A, Dikkes P, Goode R. Neuropathological findings in the brain of Karen Ann Quinlan. The role of the thalamus in the persistent vegetative state. N Engl J Med 1994; 330:1469–1475.
- Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev 2002; 39:107–140.
- Scannell JW, Burns GA, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex 1999; 9:277–299.
- Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol 1981; 10:127–148.
- Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci 2008; 1129:105–118.
- Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science 1996; 271:512–515.
- Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport 1998; 9:R37–47.
- Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage 2004; 21:1232–1241.
- Paus T, Zatorre RJ, Hofle N, et al Time-related changes in neural systems underlying attention and arousal during the performance of an auditory vigilance task. J Cogn Neurosci 1997; 9:392–408.
- Shah SA, Baker JL, Ryou JW, Purpura KP, Schiff ND. Modulation of arousal regulation with central thalamic deep brain stimulation. Conf Proc IEEE Eng Med Biol Soc 2009; 2009:3314–3317.
- Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol 2004; 91:2628–2648.
- Bohland JW, Wu C, Barbas H, et al A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput Biol 2009; 5:e1000334.
- Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci 2010; 33:1–9.
- Schiff ND, Posner JP. Another “Awakenings.” Ann Neurol 2007; 62:5–7.
- Groenewegen HJ, Berendse HW. The specificity of the ‘nonspecific’ midline and intralaminar thalamic nuclei. Trends Neurosci 1994; 17:52–57.
- Morel A, Liu J, Wannier T, Jeanmonod D, Rouiller EM. Divergence and convergence of thalamocortical projections to premotor and supplementary motor cortex: a multiple tracing study in the macaque monkey. Eur J Neurosci 2005; 21:1007–1029.
- Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience 1996; 72:679–687.
- Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci 2007; 27:4374–4384.
- Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull 2009; 78:60–68.
- Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci 2005; 28:364–370.
- Kato T, Nakayama N, Yasokawa Y, Okumura A, Shinoda J, Iwama T. Statistical image analysis of cerebral glucose metabolism in patients with cognitive impairment following diffuse traumatic brain injury. J Neurotrauma 2007; 24:919–926.
- Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry 2003; 74:1571–1573.
- Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury–associated diffuse axonal injury: a pilot double-blind randomized trial. J Head Trauma Rehabil 2002; 17:300–313.
- Brefel-Courbon C, Payoux P, Ory F, et al Clinical and imaging evidence of zolpidem effect in hypoxic encephalopathy. Ann Neurol 2007; 62:102–105.
- Whyte J, Myers R. Incidence of clinically significant responses to zolpidem among patients with disorders of consciousness: a preliminary placebo controlled trial. Am J Phys Med Rehabil 2009; 88:410–418.
- Shames JL, Ring H. Transient reversal of anoxic brain injury–related minimally conscious state after zolpidem administration: a case report. Arch Phys Med Rehabil 2008; 89:386–388.
- Cohen SI, Duong TT. Increased arousal in a patient with anoxic brain injury after administration of zolpidem. Am J Phys Med Rehabil 2008; 87:229–231.
- Williams ST, Conte MM, Kobylarz EJ, Hersh JE, Victor JD, Schiff ND. Quantitative neurophysiologic characterization of a paradoxical response to zolpidem in a severely brain-injured human subject. Program No. 541.6/R9. 2009 Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2009. Online.
- Chen L, Savio Chan C, Yung WH. Electrophysiological and behavioral effects of zolpidem in rat globus pallidus. Exp Neurol 2004; 186:212–220.
- Schiff ND, Giacino JT, Kalmar K, et al Behavioral improvements with thalamic stimulation after severe traumatic brain injury. Nature 2007; 448:600–603.
- Victor JD, Schiff ND. Meeting rigorous statistical standards in case reports. Ann Neurol 2008; 64:592.
- Schiff ND, Giacino JT, Fins JJ. Deep brain stimulation, neuroethics, and the minimally conscious state: moving beyond proof of principle. Arch Neurol 2009; 66:697–702.
- Schiff ND, Rodriguez-Moreno D, Kamal A, et al fMRI reveals large-scale network activation in minimally conscious patients. Neurology 2005; 64:514–523.
- Laureys S, Owen AM, Schiff ND. Brain function in coma, vegetative state, and related disorders. Lancet Neurol 2004; 3:537–546.
- Obrist PA. Presidential address, 1975. The cardiovascular-behavioral interaction—as it appears today. Psychophysiology 1976; 13:95–107.
- Pfaff D. Brain Arousal and Information Theory: Neural and Genetic Mechanisms. Cambridge, MA: Harvard University Press; 2006.
- Lacey BC, Lacey JI. Presidential address, 1979. Cognitive modulation of time-dependent primary bradycardia. Psychophysiology 1980; 17:209–221.
- Obrist PA, Light KC, Langer AW, Gringnolo A, McCubbin JA. Behavioural-cardiac interactions: the psychosomatic hypothesis. J Psychosom Res 1978; 22:301–325.
- Kahneman D. Attention and Effort. Englewood Cliffs, NJ: Prentice-Hall; 1973.
- Yokoyama K, Jennings R, Ackles P, Hood P, Boller F. Lack of heart rate changes during an attention-demanding task after right hemisphere lesions. Neurology 1987; 37:624–630.
- Naccache L, Dehaene S, Cohen L, et al Effortless control: executive attention and conscious feeling of mental effort are dissociable. Neuropsychologia 2005; 43:1318–1328.
- Roland P. Brain Activation. New York, NY: Wiley Press; 1997.
- Porges SW. The polyvagal theory: new insights into adaptive reactions of the autonomic nervous system. Cleve Clin J Med 2009; 76( suppl 2):S86–S90.