User login
Frustration has long plagued researchers who have sought to link the visual auras experienced by some migraineurs with the later onset of headache pain. But now, direct evidence from a new study in rats suggests that auras – presumed to be caused by waves of depression of spontaneous electrical activity that propagate slowly through the occipital lobe of the cortex – can trigger the activation of meningeal nociceptors.
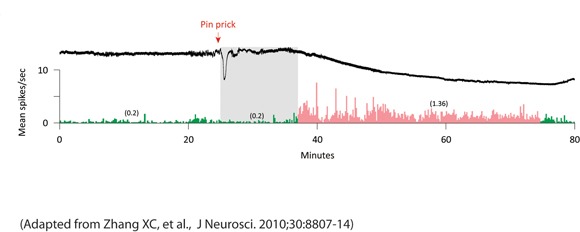
XiChun Zhang, Ph.D., and colleagues at Beth Israel Deaconess Medical Center, Boston, reported that those nociceptors in the trigeminal ganglion of rats became activated either immediately or after an average delay of 14 minutes following the administration of a pin prick, electrical pulses, or potassium chloride to the rats’ visual cortex to induce waves of cortical spreading depression (CSD).
The investigators conducted 83 trials of the three methods of cortical stimulation in 53 rats. Of 64 trials that induced waves of CSD, 31 resulted in increases in neuronal firing rates (25% or greater than baseline) that lasted at least 11 minutes. Sustained neuronal activation was not seen in all but 1 of the 19 trials that failed to produce CSD (J. Neurosci. 2010;30:8807-14).
Long-lasting neuronal activation coincided with the wave of CSD in 10 of the 31 trials in which both waves of CSD and increased neuronal firing were observed.
“The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache,” Dr. Zhang and associates wrote.
In the other 21 trials that recorded CSD and increased neuronal firing, long-lasting neuronal activation began a mean of 14 minutes after the CSD waves.
The delayed neuronal activation observed in those 21 trials “may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown,” wrote Dr. Zhang and coauthors. They noted that this observation may be the “most clinically promising” because “intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.”
There were no differences in the pattern of neuronal activation between C- and A-delta-nociceptors or between the methods of cortical stimulation. Responses to cortical stimulation in rats that had their ipsilateral sphenopalatine ganglion excised were no different from those in other rats, which indicated that the parasympathetic innervation of the dura does not contribute to the long-lasting activation of meningeal nociceptors.
The investigators proposed that the sustained activation of meningeal nociceptors could be the result of either a “short-lasting release of algesic molecules” during CSD that promotes an acute activation of the nociceptor and gives rise to an ongoing sensitization that typically outlasts the stimulus by 30-60 minutes, or an ongoing release of algesic molecules for up to 1 hour during CSD.
However, because this period of sustained activation of the meningeal nociceptors may not be sufficient “in and of itself” to explain the 4- to 72-hour duration of the headache phase of migraine, Dr. Zhang and associates proposed that the “duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours.”
Dr. Zhang’s research was funded by grants from the National Institutes of Health. Dr. Black has no relevant disclosures.
The relationship between CSD and migraine aura has a pathophysiological overlap that has led to the widely accepted theory that CSD is the electrophysiological substrate underlying the migraine aura. The question as to whether CSD is the brain mechanism causing the migraine headache is more controversial. Although circumstantial evidence and indirect data, including neuroimaging studies, provide strong evidence that CSD does occur in migraine, there is still no definitive demonstration in a migraine patient that CSD causes the proposed activation of nociceptors in the cranial pain-sensitive structures, specifically the meninges, large blood vessels, and large venous sinuses.
The fact that the majority of migraine attacks are not preceded by aura is explained by the theory that CSD events may have variations in expression and may occur in subcortical tissue such as the hippocampus, cerebellum, and striatum. In this situation, an intense and steady depolarization propagates centrifugally in grey matter and occurs within regions of the brain that remain clinically silent. The depolarization does not follow functional divisions or arterial territories, and quickly depresses brain function by transiently abolishing all spontaneous and evoked synaptic activity. Although CSD can be evoked experimentally in subcortical tissue, if this were to happen in a migraineur, one could postulate that the patient may not experience an aura but would still develop headache as a consequence of trigeminal activation. Thus, while CSD has been implicated in both aura and headache, it is less clear as to whether this pandepolarization actually initiates the migraine pain in humans.
In 1941, Karl Spencer Lashley, Ph.D., an eminent psychologist at Harvard University, published a classic paper, “Patterns of cerebral integration indicated by scatomas of migraine,” in which he described his own visual aura (Arch. Neurol. Psychiatry 1941;46:331-9). He meticulously mapped his aura and postulated that if his aura migrated, so did the underlying CNS process. Using those sketches, he was able to calculate that his migraine aura resulted from a wave of intense excitation of the visual cortex followed by complete inhibition of activity. He concluded that the cortical process progressed at a speed of 3 mm/min across the visual cortex. The migraine scotomata he drew have become known as “Lashley’s aura.”
Coincidentally, Aristides Le?o, a researcher from Brazil, was also at Harvard working toward his PhD in physiology. Early in his studies, he noted an interesting cortical response elicited by stimulation of the cortex in rabbits. The distinctive feature of the response was a spread of electrical activity, with a recovery of the initial pattern of spontaneous activity occurring after 5-10 minutes. The speed of the spread was in the range of 3 mm/min, exactly the rate reported by Lashley. Despite being totally unaware of Lashley’s publication 3 years earlier, Le?o noticed the similarity of this spreading depression to the migraine aura. In 1944, he published his research paper, “Spreading depression of activity in the cerebral cortex” (J. Neurophysiol. 1944;7:359-90). This original paper on CSD is still considered one of the classics in the field of neurophysiology. Dr. Lashley’s and Dr. Le?o’s observations changed the way migraine pathophysiology is scientifically conceived.
A review of the early contributors who set the foundation of our current understanding of headache would not be complete without reference to Dr. Harold G. Wolff. His enormous contribution to neurology included landmark studies that led to the modernization of the science of migraine and the anatomy of headache. One such contribution was the discovery that the brain itself is largely insensate. In a 1940 paper coauthored with neurosurgeon Dr. Bronson Ray, Dr. Wolff reported that mechanical stimulation of the brain parenchyma did not cause pain in patients during craniotomy, but that the crucial structures that produced pain were the dura mater, large intracranial blood vessels, and the venous sinuses (Arch. Surg. 1940;41:813-56). The innervation of the pain-sensitive intracranial structures is largely supplied by branches of the first division of the trigeminal nerve. Because the dura mater and large intracranial vessels are pain producing, they have been used in research to model trigeminovascular nociception.
It should also be noted that Dr. Wolff wrote the first textbook on headaches, entitled “Wolff’s Headache and Other Head Pain.” He wrote the first (1948) and second editions (1963) entirely by himself.
The recent study by Dr. Zhang and colleagues at Beth Israel Deaconess Medical Center, Boston, is an extension of earlier work by Dr. Lashley, Dr. Le?o, Dr. Wolff, and others. The purpose of this elegant study was to determine whether CSD can give rise to activation of nociceptors that innervate the meninges. The discovery that the induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of meningeal receptors is extremely important, especially if it is assumed that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway. It is important to recognize that we have no definitive demonstration to document that what occurs in the animal model directly translates into what takes place in the brain of a migraineur. However, it is this type of sophisticated research that allows us to work toward a better understanding of the mechanisms of migraine with and without aura. The recognition that CSD can trigger activation of nociceptive meningeal receptors in an animal model that has blood vessels and anatomy similar to humans provides important scientific data that could lead to beneficial clinical application.
Stuart B. Black, M.D., is chief of neurology and co-medical director of the neuroscience center at Baylor University Medical Center in Dallas. He has no relevant disclosures.
The relationship between CSD and migraine aura has a pathophysiological overlap that has led to the widely accepted theory that CSD is the electrophysiological substrate underlying the migraine aura. The question as to whether CSD is the brain mechanism causing the migraine headache is more controversial. Although circumstantial evidence and indirect data, including neuroimaging studies, provide strong evidence that CSD does occur in migraine, there is still no definitive demonstration in a migraine patient that CSD causes the proposed activation of nociceptors in the cranial pain-sensitive structures, specifically the meninges, large blood vessels, and large venous sinuses.
The fact that the majority of migraine attacks are not preceded by aura is explained by the theory that CSD events may have variations in expression and may occur in subcortical tissue such as the hippocampus, cerebellum, and striatum. In this situation, an intense and steady depolarization propagates centrifugally in grey matter and occurs within regions of the brain that remain clinically silent. The depolarization does not follow functional divisions or arterial territories, and quickly depresses brain function by transiently abolishing all spontaneous and evoked synaptic activity. Although CSD can be evoked experimentally in subcortical tissue, if this were to happen in a migraineur, one could postulate that the patient may not experience an aura but would still develop headache as a consequence of trigeminal activation. Thus, while CSD has been implicated in both aura and headache, it is less clear as to whether this pandepolarization actually initiates the migraine pain in humans.
In 1941, Karl Spencer Lashley, Ph.D., an eminent psychologist at Harvard University, published a classic paper, “Patterns of cerebral integration indicated by scatomas of migraine,” in which he described his own visual aura (Arch. Neurol. Psychiatry 1941;46:331-9). He meticulously mapped his aura and postulated that if his aura migrated, so did the underlying CNS process. Using those sketches, he was able to calculate that his migraine aura resulted from a wave of intense excitation of the visual cortex followed by complete inhibition of activity. He concluded that the cortical process progressed at a speed of 3 mm/min across the visual cortex. The migraine scotomata he drew have become known as “Lashley’s aura.”
Coincidentally, Aristides Le?o, a researcher from Brazil, was also at Harvard working toward his PhD in physiology. Early in his studies, he noted an interesting cortical response elicited by stimulation of the cortex in rabbits. The distinctive feature of the response was a spread of electrical activity, with a recovery of the initial pattern of spontaneous activity occurring after 5-10 minutes. The speed of the spread was in the range of 3 mm/min, exactly the rate reported by Lashley. Despite being totally unaware of Lashley’s publication 3 years earlier, Le?o noticed the similarity of this spreading depression to the migraine aura. In 1944, he published his research paper, “Spreading depression of activity in the cerebral cortex” (J. Neurophysiol. 1944;7:359-90). This original paper on CSD is still considered one of the classics in the field of neurophysiology. Dr. Lashley’s and Dr. Le?o’s observations changed the way migraine pathophysiology is scientifically conceived.
A review of the early contributors who set the foundation of our current understanding of headache would not be complete without reference to Dr. Harold G. Wolff. His enormous contribution to neurology included landmark studies that led to the modernization of the science of migraine and the anatomy of headache. One such contribution was the discovery that the brain itself is largely insensate. In a 1940 paper coauthored with neurosurgeon Dr. Bronson Ray, Dr. Wolff reported that mechanical stimulation of the brain parenchyma did not cause pain in patients during craniotomy, but that the crucial structures that produced pain were the dura mater, large intracranial blood vessels, and the venous sinuses (Arch. Surg. 1940;41:813-56). The innervation of the pain-sensitive intracranial structures is largely supplied by branches of the first division of the trigeminal nerve. Because the dura mater and large intracranial vessels are pain producing, they have been used in research to model trigeminovascular nociception.
It should also be noted that Dr. Wolff wrote the first textbook on headaches, entitled “Wolff’s Headache and Other Head Pain.” He wrote the first (1948) and second editions (1963) entirely by himself.
The recent study by Dr. Zhang and colleagues at Beth Israel Deaconess Medical Center, Boston, is an extension of earlier work by Dr. Lashley, Dr. Le?o, Dr. Wolff, and others. The purpose of this elegant study was to determine whether CSD can give rise to activation of nociceptors that innervate the meninges. The discovery that the induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of meningeal receptors is extremely important, especially if it is assumed that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway. It is important to recognize that we have no definitive demonstration to document that what occurs in the animal model directly translates into what takes place in the brain of a migraineur. However, it is this type of sophisticated research that allows us to work toward a better understanding of the mechanisms of migraine with and without aura. The recognition that CSD can trigger activation of nociceptive meningeal receptors in an animal model that has blood vessels and anatomy similar to humans provides important scientific data that could lead to beneficial clinical application.
Stuart B. Black, M.D., is chief of neurology and co-medical director of the neuroscience center at Baylor University Medical Center in Dallas. He has no relevant disclosures.
The relationship between CSD and migraine aura has a pathophysiological overlap that has led to the widely accepted theory that CSD is the electrophysiological substrate underlying the migraine aura. The question as to whether CSD is the brain mechanism causing the migraine headache is more controversial. Although circumstantial evidence and indirect data, including neuroimaging studies, provide strong evidence that CSD does occur in migraine, there is still no definitive demonstration in a migraine patient that CSD causes the proposed activation of nociceptors in the cranial pain-sensitive structures, specifically the meninges, large blood vessels, and large venous sinuses.
The fact that the majority of migraine attacks are not preceded by aura is explained by the theory that CSD events may have variations in expression and may occur in subcortical tissue such as the hippocampus, cerebellum, and striatum. In this situation, an intense and steady depolarization propagates centrifugally in grey matter and occurs within regions of the brain that remain clinically silent. The depolarization does not follow functional divisions or arterial territories, and quickly depresses brain function by transiently abolishing all spontaneous and evoked synaptic activity. Although CSD can be evoked experimentally in subcortical tissue, if this were to happen in a migraineur, one could postulate that the patient may not experience an aura but would still develop headache as a consequence of trigeminal activation. Thus, while CSD has been implicated in both aura and headache, it is less clear as to whether this pandepolarization actually initiates the migraine pain in humans.
In 1941, Karl Spencer Lashley, Ph.D., an eminent psychologist at Harvard University, published a classic paper, “Patterns of cerebral integration indicated by scatomas of migraine,” in which he described his own visual aura (Arch. Neurol. Psychiatry 1941;46:331-9). He meticulously mapped his aura and postulated that if his aura migrated, so did the underlying CNS process. Using those sketches, he was able to calculate that his migraine aura resulted from a wave of intense excitation of the visual cortex followed by complete inhibition of activity. He concluded that the cortical process progressed at a speed of 3 mm/min across the visual cortex. The migraine scotomata he drew have become known as “Lashley’s aura.”
Coincidentally, Aristides Le?o, a researcher from Brazil, was also at Harvard working toward his PhD in physiology. Early in his studies, he noted an interesting cortical response elicited by stimulation of the cortex in rabbits. The distinctive feature of the response was a spread of electrical activity, with a recovery of the initial pattern of spontaneous activity occurring after 5-10 minutes. The speed of the spread was in the range of 3 mm/min, exactly the rate reported by Lashley. Despite being totally unaware of Lashley’s publication 3 years earlier, Le?o noticed the similarity of this spreading depression to the migraine aura. In 1944, he published his research paper, “Spreading depression of activity in the cerebral cortex” (J. Neurophysiol. 1944;7:359-90). This original paper on CSD is still considered one of the classics in the field of neurophysiology. Dr. Lashley’s and Dr. Le?o’s observations changed the way migraine pathophysiology is scientifically conceived.
A review of the early contributors who set the foundation of our current understanding of headache would not be complete without reference to Dr. Harold G. Wolff. His enormous contribution to neurology included landmark studies that led to the modernization of the science of migraine and the anatomy of headache. One such contribution was the discovery that the brain itself is largely insensate. In a 1940 paper coauthored with neurosurgeon Dr. Bronson Ray, Dr. Wolff reported that mechanical stimulation of the brain parenchyma did not cause pain in patients during craniotomy, but that the crucial structures that produced pain were the dura mater, large intracranial blood vessels, and the venous sinuses (Arch. Surg. 1940;41:813-56). The innervation of the pain-sensitive intracranial structures is largely supplied by branches of the first division of the trigeminal nerve. Because the dura mater and large intracranial vessels are pain producing, they have been used in research to model trigeminovascular nociception.
It should also be noted that Dr. Wolff wrote the first textbook on headaches, entitled “Wolff’s Headache and Other Head Pain.” He wrote the first (1948) and second editions (1963) entirely by himself.
The recent study by Dr. Zhang and colleagues at Beth Israel Deaconess Medical Center, Boston, is an extension of earlier work by Dr. Lashley, Dr. Le?o, Dr. Wolff, and others. The purpose of this elegant study was to determine whether CSD can give rise to activation of nociceptors that innervate the meninges. The discovery that the induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of meningeal receptors is extremely important, especially if it is assumed that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway. It is important to recognize that we have no definitive demonstration to document that what occurs in the animal model directly translates into what takes place in the brain of a migraineur. However, it is this type of sophisticated research that allows us to work toward a better understanding of the mechanisms of migraine with and without aura. The recognition that CSD can trigger activation of nociceptive meningeal receptors in an animal model that has blood vessels and anatomy similar to humans provides important scientific data that could lead to beneficial clinical application.
Stuart B. Black, M.D., is chief of neurology and co-medical director of the neuroscience center at Baylor University Medical Center in Dallas. He has no relevant disclosures.
Frustration has long plagued researchers who have sought to link the visual auras experienced by some migraineurs with the later onset of headache pain. But now, direct evidence from a new study in rats suggests that auras – presumed to be caused by waves of depression of spontaneous electrical activity that propagate slowly through the occipital lobe of the cortex – can trigger the activation of meningeal nociceptors.
XiChun Zhang, Ph.D., and colleagues at Beth Israel Deaconess Medical Center, Boston, reported that those nociceptors in the trigeminal ganglion of rats became activated either immediately or after an average delay of 14 minutes following the administration of a pin prick, electrical pulses, or potassium chloride to the rats’ visual cortex to induce waves of cortical spreading depression (CSD).
The investigators conducted 83 trials of the three methods of cortical stimulation in 53 rats. Of 64 trials that induced waves of CSD, 31 resulted in increases in neuronal firing rates (25% or greater than baseline) that lasted at least 11 minutes. Sustained neuronal activation was not seen in all but 1 of the 19 trials that failed to produce CSD (J. Neurosci. 2010;30:8807-14).
Long-lasting neuronal activation coincided with the wave of CSD in 10 of the 31 trials in which both waves of CSD and increased neuronal firing were observed.
“The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache,” Dr. Zhang and associates wrote.
In the other 21 trials that recorded CSD and increased neuronal firing, long-lasting neuronal activation began a mean of 14 minutes after the CSD waves.
The delayed neuronal activation observed in those 21 trials “may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown,” wrote Dr. Zhang and coauthors. They noted that this observation may be the “most clinically promising” because “intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.”
There were no differences in the pattern of neuronal activation between C- and A-delta-nociceptors or between the methods of cortical stimulation. Responses to cortical stimulation in rats that had their ipsilateral sphenopalatine ganglion excised were no different from those in other rats, which indicated that the parasympathetic innervation of the dura does not contribute to the long-lasting activation of meningeal nociceptors.
The investigators proposed that the sustained activation of meningeal nociceptors could be the result of either a “short-lasting release of algesic molecules” during CSD that promotes an acute activation of the nociceptor and gives rise to an ongoing sensitization that typically outlasts the stimulus by 30-60 minutes, or an ongoing release of algesic molecules for up to 1 hour during CSD.
However, because this period of sustained activation of the meningeal nociceptors may not be sufficient “in and of itself” to explain the 4- to 72-hour duration of the headache phase of migraine, Dr. Zhang and associates proposed that the “duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours.”
Dr. Zhang’s research was funded by grants from the National Institutes of Health. Dr. Black has no relevant disclosures.
Frustration has long plagued researchers who have sought to link the visual auras experienced by some migraineurs with the later onset of headache pain. But now, direct evidence from a new study in rats suggests that auras – presumed to be caused by waves of depression of spontaneous electrical activity that propagate slowly through the occipital lobe of the cortex – can trigger the activation of meningeal nociceptors.
XiChun Zhang, Ph.D., and colleagues at Beth Israel Deaconess Medical Center, Boston, reported that those nociceptors in the trigeminal ganglion of rats became activated either immediately or after an average delay of 14 minutes following the administration of a pin prick, electrical pulses, or potassium chloride to the rats’ visual cortex to induce waves of cortical spreading depression (CSD).
The investigators conducted 83 trials of the three methods of cortical stimulation in 53 rats. Of 64 trials that induced waves of CSD, 31 resulted in increases in neuronal firing rates (25% or greater than baseline) that lasted at least 11 minutes. Sustained neuronal activation was not seen in all but 1 of the 19 trials that failed to produce CSD (J. Neurosci. 2010;30:8807-14).
Long-lasting neuronal activation coincided with the wave of CSD in 10 of the 31 trials in which both waves of CSD and increased neuronal firing were observed.
“The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache,” Dr. Zhang and associates wrote.
In the other 21 trials that recorded CSD and increased neuronal firing, long-lasting neuronal activation began a mean of 14 minutes after the CSD waves.
The delayed neuronal activation observed in those 21 trials “may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown,” wrote Dr. Zhang and coauthors. They noted that this observation may be the “most clinically promising” because “intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.”
There were no differences in the pattern of neuronal activation between C- and A-delta-nociceptors or between the methods of cortical stimulation. Responses to cortical stimulation in rats that had their ipsilateral sphenopalatine ganglion excised were no different from those in other rats, which indicated that the parasympathetic innervation of the dura does not contribute to the long-lasting activation of meningeal nociceptors.
The investigators proposed that the sustained activation of meningeal nociceptors could be the result of either a “short-lasting release of algesic molecules” during CSD that promotes an acute activation of the nociceptor and gives rise to an ongoing sensitization that typically outlasts the stimulus by 30-60 minutes, or an ongoing release of algesic molecules for up to 1 hour during CSD.
However, because this period of sustained activation of the meningeal nociceptors may not be sufficient “in and of itself” to explain the 4- to 72-hour duration of the headache phase of migraine, Dr. Zhang and associates proposed that the “duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours.”
Dr. Zhang’s research was funded by grants from the National Institutes of Health. Dr. Black has no relevant disclosures.