User login
ACIP recommends Shingrix for younger immunocompromised adults; updates pneumococcal vaccine guidance
The U.S. Centers for Disease Control and Prevention Advisory Committee of Immunization Practices has voted to recommend Shingrix (zoster vaccine recombinant, adjuvanted) for the prevention of shingles in immunodeficient or immunosuppressed adults aged 19 or older. The recommendation was approved Oct. 20 by a unanimous vote.
Shingles is a reactivation of varicella zoster virus (VZV), the virus that causes chickenpox. There are about 1 million cases of shingles in the United States every year, according to CDC estimates, and one in three Americans will develop shingles over their lifetime. While adults older than 50 are one of the most vulnerable groups to reinfection – with about 99% having been infected with VZV – a weakened immune system is another common risk factor.
The Food and Drug Administration originally approved Shingrix in 2017 for the prevention of shingles in adults over 50; in July of this year, the vaccine was approved for immunodeficient adults aged 18 or older. The approval and subsequent recommendation by ACIP were based on clinical studies of Shingrix in adults being treated for hematologic malignancies or those who had undergone an autologous hematopoietic stem cell transplant.
According to a press statement from the FDA, “Further safety and immunogenicity data were generated in adults who were, or were anticipated to be, immunodeficient or immunosuppressed due to known disease or therapy, including patients with HIV, solid tumors, and renal transplants.”
For adults with functional immune systems, Shingrix is administered in two doses, 2-6 months apart. For immunocompromised individuals, the second dose can be given 1-2 months after the first dose.
During the same meeting, ACIP also voted to recommend pneumococcal vaccines for routine use in adults older than 65 and in adults aged 19-64 with chronic conditions such as diabetes, chronic heart disease, chronic liver disease, and HIV, and disease risk factors like smoking and alcoholism. The recommendation only applies to those who have not received a pneumococcal conjugate vaccine or whose vaccination history is unknown. The recommendation states that qualifying adults should be vaccinated with the 15-valent pneumococcal conjugate vaccine Vaxneuvance followed by Pneumovax23, or a single dose of the 20-valent pneumococcal conjugate vaccine Prevnar 20.
These ACIP recommendations will now be sent to the directors of the CDC and the U.S. Department of Health & Human Services for review and approval. If approved, the recommendations are considered finalized and will be published in a future Morbidity and Mortality Weekly Report.
A version of this article first appeared on Medscape.com.
The U.S. Centers for Disease Control and Prevention Advisory Committee of Immunization Practices has voted to recommend Shingrix (zoster vaccine recombinant, adjuvanted) for the prevention of shingles in immunodeficient or immunosuppressed adults aged 19 or older. The recommendation was approved Oct. 20 by a unanimous vote.
Shingles is a reactivation of varicella zoster virus (VZV), the virus that causes chickenpox. There are about 1 million cases of shingles in the United States every year, according to CDC estimates, and one in three Americans will develop shingles over their lifetime. While adults older than 50 are one of the most vulnerable groups to reinfection – with about 99% having been infected with VZV – a weakened immune system is another common risk factor.
The Food and Drug Administration originally approved Shingrix in 2017 for the prevention of shingles in adults over 50; in July of this year, the vaccine was approved for immunodeficient adults aged 18 or older. The approval and subsequent recommendation by ACIP were based on clinical studies of Shingrix in adults being treated for hematologic malignancies or those who had undergone an autologous hematopoietic stem cell transplant.
According to a press statement from the FDA, “Further safety and immunogenicity data were generated in adults who were, or were anticipated to be, immunodeficient or immunosuppressed due to known disease or therapy, including patients with HIV, solid tumors, and renal transplants.”
For adults with functional immune systems, Shingrix is administered in two doses, 2-6 months apart. For immunocompromised individuals, the second dose can be given 1-2 months after the first dose.
During the same meeting, ACIP also voted to recommend pneumococcal vaccines for routine use in adults older than 65 and in adults aged 19-64 with chronic conditions such as diabetes, chronic heart disease, chronic liver disease, and HIV, and disease risk factors like smoking and alcoholism. The recommendation only applies to those who have not received a pneumococcal conjugate vaccine or whose vaccination history is unknown. The recommendation states that qualifying adults should be vaccinated with the 15-valent pneumococcal conjugate vaccine Vaxneuvance followed by Pneumovax23, or a single dose of the 20-valent pneumococcal conjugate vaccine Prevnar 20.
These ACIP recommendations will now be sent to the directors of the CDC and the U.S. Department of Health & Human Services for review and approval. If approved, the recommendations are considered finalized and will be published in a future Morbidity and Mortality Weekly Report.
A version of this article first appeared on Medscape.com.
The U.S. Centers for Disease Control and Prevention Advisory Committee of Immunization Practices has voted to recommend Shingrix (zoster vaccine recombinant, adjuvanted) for the prevention of shingles in immunodeficient or immunosuppressed adults aged 19 or older. The recommendation was approved Oct. 20 by a unanimous vote.
Shingles is a reactivation of varicella zoster virus (VZV), the virus that causes chickenpox. There are about 1 million cases of shingles in the United States every year, according to CDC estimates, and one in three Americans will develop shingles over their lifetime. While adults older than 50 are one of the most vulnerable groups to reinfection – with about 99% having been infected with VZV – a weakened immune system is another common risk factor.
The Food and Drug Administration originally approved Shingrix in 2017 for the prevention of shingles in adults over 50; in July of this year, the vaccine was approved for immunodeficient adults aged 18 or older. The approval and subsequent recommendation by ACIP were based on clinical studies of Shingrix in adults being treated for hematologic malignancies or those who had undergone an autologous hematopoietic stem cell transplant.
According to a press statement from the FDA, “Further safety and immunogenicity data were generated in adults who were, or were anticipated to be, immunodeficient or immunosuppressed due to known disease or therapy, including patients with HIV, solid tumors, and renal transplants.”
For adults with functional immune systems, Shingrix is administered in two doses, 2-6 months apart. For immunocompromised individuals, the second dose can be given 1-2 months after the first dose.
During the same meeting, ACIP also voted to recommend pneumococcal vaccines for routine use in adults older than 65 and in adults aged 19-64 with chronic conditions such as diabetes, chronic heart disease, chronic liver disease, and HIV, and disease risk factors like smoking and alcoholism. The recommendation only applies to those who have not received a pneumococcal conjugate vaccine or whose vaccination history is unknown. The recommendation states that qualifying adults should be vaccinated with the 15-valent pneumococcal conjugate vaccine Vaxneuvance followed by Pneumovax23, or a single dose of the 20-valent pneumococcal conjugate vaccine Prevnar 20.
These ACIP recommendations will now be sent to the directors of the CDC and the U.S. Department of Health & Human Services for review and approval. If approved, the recommendations are considered finalized and will be published in a future Morbidity and Mortality Weekly Report.
A version of this article first appeared on Medscape.com.
CDC panel backs COVID-19 boosters for nearly all adults
Editor’s note: This story was updated with the CDC director’s endorsement.
Centers for Disease Control and Prevention (CDC) Director Rochelle Walensky, MD, has signed off on an advisory panel’s earlier unanimous vote to recommend boosters for the Moderna and Johnson and Johnson COVID vaccines.
The decision now means that millions of Americans are eligible to get a booster shot for either the Pfizer, Moderna, or J&J COVID vaccines.
“The evidence shows that all three COVID-19 vaccines authorized in the United States are safe – as demonstrated by the over 400 million vaccine doses already given. And, they are all highly effective in reducing the risk of severe disease, hospitalization, and death, even in the midst of the widely circulating Delta variant,” Dr. Walensky said in a CDC news release.
She also signed off on the panel’s suggestion that individuals can mix or match the booster from any one of the three available COVID-19 vaccines.
The Advisory Committee on Immunization Practices (ACIP) recommended in a late afternoon 15-0 vote that everyone over age 18 who are at least 2 months past their Johnson & Johnson vaccine should get a booster, an endorsement that affects an estimated 13 million Americans.
Those eligible for a booster at least 6 months after their last Moderna shot are the same groups who can get a Pfizer booster.
They are:
- Anyone over age 65.
- Those over age 18 with an underlying health condition that puts them at risk of severe COVID-19.
- Those over age 18 who may be at higher risk of a COVID-19 infection because they live or work in a risky setting.
These recommendations are in line with the Food and Drug Administration’s Oct. 20 authorization of the boosters, along with the ability to mix-and-match vaccines.
There are an estimated 47 million Pfizer recipients and 39 million people vaccinated with Moderna who are now eligible for a booster dose, according to data presented by the CDC.
Questions, concerns
Before voting, some committee members expressed discomfort in broadly recommending boosters, stressing that there is very little evidence supporting the need for boosters in people younger than age 50.
“I can’t say that I am comfortable that anybody under 50 – an otherwise healthy individual – needs a booster vaccine at this time with either Moderna or Pfizer,” said ACIP member Sarah Long, MD, professor of pediatrics at Drexel University in Philadelphia.
She said she would try to mitigate any potential harm by having some kind of age restriction on the otherwise worried well.
“We don’t usually have the vaccines [for] the worried well. We give it because we have a need that’s worth the risk, and there’s a burden of severity of disease,” Dr. Long said.
The evidence to date shows that all the vaccines authorized for use in the U.S. continue to protect people well against severe COVID-19 outcomes, including hospitalization and death.
But breakthrough infections are on the rise, especially for people who initially received the Johnson and Johnson one-dose vaccine.
On Oct. 21, Pfizer released data from a study of more than 10,000 fully vaccinated people. Half were randomly assigned to get a booster of their Comirnaty vaccine, the other half were given a placebo.
Over the ensuing 2.5 months, there were 5 COVID-19 cases in the boosted group, and 109 in the group that got a placebo.
The data were posted in a press release and have not yet been peer reviewed, but are the first to show clinical effectiveness of boosters at preventing COVID-19 infections.
Data recently considered by the FDA and CDC for booster doses come from studies that were mostly shorter and smaller. These studies looked at biomarkers of immunity like the concentration of antibodies in a person’s blood and the percentage of study participants who saw a boost to those antibodies.
The studies demonstrated that boosters indeed restore high levels of antibodies, but unlike the newest Pfizer data they were not able to show that these antibodies prevented COVID-19.
These studies also weren’t powered to pick up on any less common safety problems that might arise after another dose of the shots.
“Real world” recommendations
In the end, however, the panel felt it was more important to be permissive in allowing boosters so that individuals and their doctors could be free to make their own decisions.
“The decision made by the FDA and the ACIP recommendations, I think, reflects the real world. The public is going to do what they feel driven to do. This at least adds a scientific review of the currently available data,” said Jay Varkey, MD, an infectious disease physician and associate professor at Emory University in Atlanta, who was not involved in the ACIP’s deliberations.
Dr. Varkey said he would recommend that anyone who is younger than 65, and who has no underlying medical conditions such as diabetes or obesity, speak with their doctor about their individual benefits and risks before getting a booster.
The CDC is planning to release a detailed suite of clinical considerations to help people weigh the risks and benefits of getting a booster.
Safety updates presented at the meeting show that serious adverse events after vaccination are extremely rare, but in some cases, they may rise above the risk for those problems generally seen in the population.
Those rare events include the disabling autoimmune condition Guillain-Barré syndrome and the platelet disorder thrombosis with thrombocytopenia (TTS), which causes blood clots along with the risk of excess bleeding because of a low platelet count.
Both can occur after the J&J vaccine. Out of 15.3 million doses of the vaccine given to date, there have been 47 cases of TTS and five deaths. These events are more common in younger women.
The mRNA vaccines, such as those from Pfizer and Moderna, can cause heart inflammation called myocarditis or pericarditis. This side effect is more common in men 18-24 years old. The reported rate of myocarditis after vaccination is 39 cases for every 1 million doses.
In voting to permit boosters, committee member Wilbur Chen, MD, professor at the University of Maryland’s Center for Vaccine Development, said he hoped boosters wouldn’t give Americans false confidence.
Dr. Chen stressed that ending the pandemic would depend on “a multilayered approach” that includes masking, social distancing, avoiding large crowds indoors, and convincing more Americans to take their first doses of the vaccines.
“We’re not just going to vaccinate ourselves out of this situation,” Dr. Chen said.
A version of this article first appeared on WebMD.com.
Editor’s note: This story was updated with the CDC director’s endorsement.
Centers for Disease Control and Prevention (CDC) Director Rochelle Walensky, MD, has signed off on an advisory panel’s earlier unanimous vote to recommend boosters for the Moderna and Johnson and Johnson COVID vaccines.
The decision now means that millions of Americans are eligible to get a booster shot for either the Pfizer, Moderna, or J&J COVID vaccines.
“The evidence shows that all three COVID-19 vaccines authorized in the United States are safe – as demonstrated by the over 400 million vaccine doses already given. And, they are all highly effective in reducing the risk of severe disease, hospitalization, and death, even in the midst of the widely circulating Delta variant,” Dr. Walensky said in a CDC news release.
She also signed off on the panel’s suggestion that individuals can mix or match the booster from any one of the three available COVID-19 vaccines.
The Advisory Committee on Immunization Practices (ACIP) recommended in a late afternoon 15-0 vote that everyone over age 18 who are at least 2 months past their Johnson & Johnson vaccine should get a booster, an endorsement that affects an estimated 13 million Americans.
Those eligible for a booster at least 6 months after their last Moderna shot are the same groups who can get a Pfizer booster.
They are:
- Anyone over age 65.
- Those over age 18 with an underlying health condition that puts them at risk of severe COVID-19.
- Those over age 18 who may be at higher risk of a COVID-19 infection because they live or work in a risky setting.
These recommendations are in line with the Food and Drug Administration’s Oct. 20 authorization of the boosters, along with the ability to mix-and-match vaccines.
There are an estimated 47 million Pfizer recipients and 39 million people vaccinated with Moderna who are now eligible for a booster dose, according to data presented by the CDC.
Questions, concerns
Before voting, some committee members expressed discomfort in broadly recommending boosters, stressing that there is very little evidence supporting the need for boosters in people younger than age 50.
“I can’t say that I am comfortable that anybody under 50 – an otherwise healthy individual – needs a booster vaccine at this time with either Moderna or Pfizer,” said ACIP member Sarah Long, MD, professor of pediatrics at Drexel University in Philadelphia.
She said she would try to mitigate any potential harm by having some kind of age restriction on the otherwise worried well.
“We don’t usually have the vaccines [for] the worried well. We give it because we have a need that’s worth the risk, and there’s a burden of severity of disease,” Dr. Long said.
The evidence to date shows that all the vaccines authorized for use in the U.S. continue to protect people well against severe COVID-19 outcomes, including hospitalization and death.
But breakthrough infections are on the rise, especially for people who initially received the Johnson and Johnson one-dose vaccine.
On Oct. 21, Pfizer released data from a study of more than 10,000 fully vaccinated people. Half were randomly assigned to get a booster of their Comirnaty vaccine, the other half were given a placebo.
Over the ensuing 2.5 months, there were 5 COVID-19 cases in the boosted group, and 109 in the group that got a placebo.
The data were posted in a press release and have not yet been peer reviewed, but are the first to show clinical effectiveness of boosters at preventing COVID-19 infections.
Data recently considered by the FDA and CDC for booster doses come from studies that were mostly shorter and smaller. These studies looked at biomarkers of immunity like the concentration of antibodies in a person’s blood and the percentage of study participants who saw a boost to those antibodies.
The studies demonstrated that boosters indeed restore high levels of antibodies, but unlike the newest Pfizer data they were not able to show that these antibodies prevented COVID-19.
These studies also weren’t powered to pick up on any less common safety problems that might arise after another dose of the shots.
“Real world” recommendations
In the end, however, the panel felt it was more important to be permissive in allowing boosters so that individuals and their doctors could be free to make their own decisions.
“The decision made by the FDA and the ACIP recommendations, I think, reflects the real world. The public is going to do what they feel driven to do. This at least adds a scientific review of the currently available data,” said Jay Varkey, MD, an infectious disease physician and associate professor at Emory University in Atlanta, who was not involved in the ACIP’s deliberations.
Dr. Varkey said he would recommend that anyone who is younger than 65, and who has no underlying medical conditions such as diabetes or obesity, speak with their doctor about their individual benefits and risks before getting a booster.
The CDC is planning to release a detailed suite of clinical considerations to help people weigh the risks and benefits of getting a booster.
Safety updates presented at the meeting show that serious adverse events after vaccination are extremely rare, but in some cases, they may rise above the risk for those problems generally seen in the population.
Those rare events include the disabling autoimmune condition Guillain-Barré syndrome and the platelet disorder thrombosis with thrombocytopenia (TTS), which causes blood clots along with the risk of excess bleeding because of a low platelet count.
Both can occur after the J&J vaccine. Out of 15.3 million doses of the vaccine given to date, there have been 47 cases of TTS and five deaths. These events are more common in younger women.
The mRNA vaccines, such as those from Pfizer and Moderna, can cause heart inflammation called myocarditis or pericarditis. This side effect is more common in men 18-24 years old. The reported rate of myocarditis after vaccination is 39 cases for every 1 million doses.
In voting to permit boosters, committee member Wilbur Chen, MD, professor at the University of Maryland’s Center for Vaccine Development, said he hoped boosters wouldn’t give Americans false confidence.
Dr. Chen stressed that ending the pandemic would depend on “a multilayered approach” that includes masking, social distancing, avoiding large crowds indoors, and convincing more Americans to take their first doses of the vaccines.
“We’re not just going to vaccinate ourselves out of this situation,” Dr. Chen said.
A version of this article first appeared on WebMD.com.
Editor’s note: This story was updated with the CDC director’s endorsement.
Centers for Disease Control and Prevention (CDC) Director Rochelle Walensky, MD, has signed off on an advisory panel’s earlier unanimous vote to recommend boosters for the Moderna and Johnson and Johnson COVID vaccines.
The decision now means that millions of Americans are eligible to get a booster shot for either the Pfizer, Moderna, or J&J COVID vaccines.
“The evidence shows that all three COVID-19 vaccines authorized in the United States are safe – as demonstrated by the over 400 million vaccine doses already given. And, they are all highly effective in reducing the risk of severe disease, hospitalization, and death, even in the midst of the widely circulating Delta variant,” Dr. Walensky said in a CDC news release.
She also signed off on the panel’s suggestion that individuals can mix or match the booster from any one of the three available COVID-19 vaccines.
The Advisory Committee on Immunization Practices (ACIP) recommended in a late afternoon 15-0 vote that everyone over age 18 who are at least 2 months past their Johnson & Johnson vaccine should get a booster, an endorsement that affects an estimated 13 million Americans.
Those eligible for a booster at least 6 months after their last Moderna shot are the same groups who can get a Pfizer booster.
They are:
- Anyone over age 65.
- Those over age 18 with an underlying health condition that puts them at risk of severe COVID-19.
- Those over age 18 who may be at higher risk of a COVID-19 infection because they live or work in a risky setting.
These recommendations are in line with the Food and Drug Administration’s Oct. 20 authorization of the boosters, along with the ability to mix-and-match vaccines.
There are an estimated 47 million Pfizer recipients and 39 million people vaccinated with Moderna who are now eligible for a booster dose, according to data presented by the CDC.
Questions, concerns
Before voting, some committee members expressed discomfort in broadly recommending boosters, stressing that there is very little evidence supporting the need for boosters in people younger than age 50.
“I can’t say that I am comfortable that anybody under 50 – an otherwise healthy individual – needs a booster vaccine at this time with either Moderna or Pfizer,” said ACIP member Sarah Long, MD, professor of pediatrics at Drexel University in Philadelphia.
She said she would try to mitigate any potential harm by having some kind of age restriction on the otherwise worried well.
“We don’t usually have the vaccines [for] the worried well. We give it because we have a need that’s worth the risk, and there’s a burden of severity of disease,” Dr. Long said.
The evidence to date shows that all the vaccines authorized for use in the U.S. continue to protect people well against severe COVID-19 outcomes, including hospitalization and death.
But breakthrough infections are on the rise, especially for people who initially received the Johnson and Johnson one-dose vaccine.
On Oct. 21, Pfizer released data from a study of more than 10,000 fully vaccinated people. Half were randomly assigned to get a booster of their Comirnaty vaccine, the other half were given a placebo.
Over the ensuing 2.5 months, there were 5 COVID-19 cases in the boosted group, and 109 in the group that got a placebo.
The data were posted in a press release and have not yet been peer reviewed, but are the first to show clinical effectiveness of boosters at preventing COVID-19 infections.
Data recently considered by the FDA and CDC for booster doses come from studies that were mostly shorter and smaller. These studies looked at biomarkers of immunity like the concentration of antibodies in a person’s blood and the percentage of study participants who saw a boost to those antibodies.
The studies demonstrated that boosters indeed restore high levels of antibodies, but unlike the newest Pfizer data they were not able to show that these antibodies prevented COVID-19.
These studies also weren’t powered to pick up on any less common safety problems that might arise after another dose of the shots.
“Real world” recommendations
In the end, however, the panel felt it was more important to be permissive in allowing boosters so that individuals and their doctors could be free to make their own decisions.
“The decision made by the FDA and the ACIP recommendations, I think, reflects the real world. The public is going to do what they feel driven to do. This at least adds a scientific review of the currently available data,” said Jay Varkey, MD, an infectious disease physician and associate professor at Emory University in Atlanta, who was not involved in the ACIP’s deliberations.
Dr. Varkey said he would recommend that anyone who is younger than 65, and who has no underlying medical conditions such as diabetes or obesity, speak with their doctor about their individual benefits and risks before getting a booster.
The CDC is planning to release a detailed suite of clinical considerations to help people weigh the risks and benefits of getting a booster.
Safety updates presented at the meeting show that serious adverse events after vaccination are extremely rare, but in some cases, they may rise above the risk for those problems generally seen in the population.
Those rare events include the disabling autoimmune condition Guillain-Barré syndrome and the platelet disorder thrombosis with thrombocytopenia (TTS), which causes blood clots along with the risk of excess bleeding because of a low platelet count.
Both can occur after the J&J vaccine. Out of 15.3 million doses of the vaccine given to date, there have been 47 cases of TTS and five deaths. These events are more common in younger women.
The mRNA vaccines, such as those from Pfizer and Moderna, can cause heart inflammation called myocarditis or pericarditis. This side effect is more common in men 18-24 years old. The reported rate of myocarditis after vaccination is 39 cases for every 1 million doses.
In voting to permit boosters, committee member Wilbur Chen, MD, professor at the University of Maryland’s Center for Vaccine Development, said he hoped boosters wouldn’t give Americans false confidence.
Dr. Chen stressed that ending the pandemic would depend on “a multilayered approach” that includes masking, social distancing, avoiding large crowds indoors, and convincing more Americans to take their first doses of the vaccines.
“We’re not just going to vaccinate ourselves out of this situation,” Dr. Chen said.
A version of this article first appeared on WebMD.com.
Children and COVID: Vaccinations lower than ever as cases continue to drop
As the COVID-19 vaccine heads toward approval for children under age 12 years, the number of older children receiving it dropped for the 10th consecutive week, based on data from the Centers for Disease Control and Prevention.
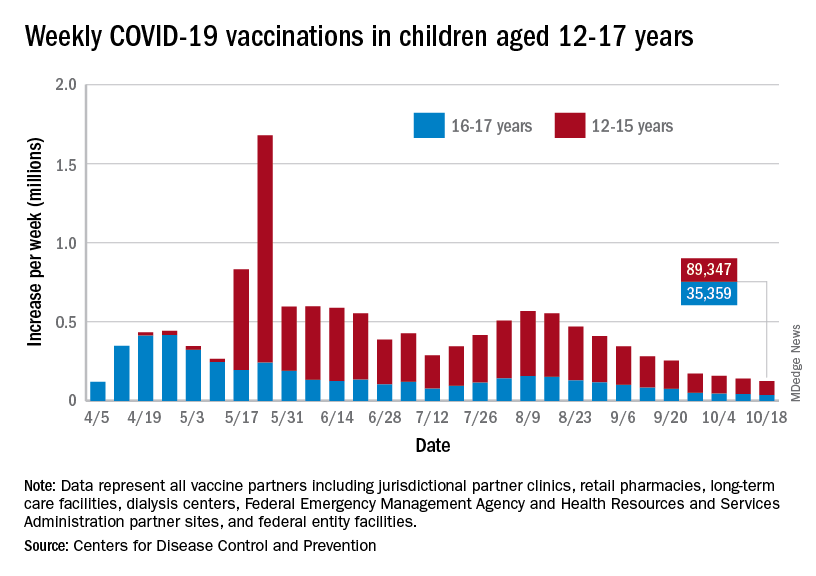
Over 47% of all children aged 12-17 years – that’s close to 12 million eligible individuals – have not received even one dose of COVID-19 vaccine, and less than 44% (about 11.1 million) were fully vaccinated as of Oct. 18, the CDC reported on its COVID Data Tracker.
, when eligibility expanded to include 12- to 15-year-olds, according to the CDC data, which also show that weekly vaccinations have never been lower.
Fortunately, the decline in new cases also continued, as the national total fell for a 6th straight week. There were more than 130,000 child cases reported during the week of Oct. 8-14, compared with 148,000 the previous week and the high of almost 252,000 in late August/early September, the American Academy of Pediatrics and the Children’s Hospital Association said in their weekly COVID-19 report.
That brings the cumulative count to 6.18 million, with children accounting for 16.4% of all cases reported since the start of the pandemic. For the week of Oct. 8-14, children represented 25.5% of all COVID-19 cases in the 46 states with up-to-date online dashboards, the AAP and CHA said, noting that New York has never reported age ranges for cases and that Alabama, Nebraska, and Texas stopped reporting over the summer.
Current data indicate that child cases in California now exceed 671,000, more than any other state, followed by Florida with 439,000 (the state defines a child as someone aged 0-14 years) and Illinois with 301,000. Vermont has the highest proportion of COVID-19 cases occurring in children (24.3%), with Alaska (24.1%) and South Carolina (23.2%) just behind. The highest rate of cases – 15,569 per 100,000 children – can be found in South Carolina, while the lowest is in Hawaii (4,838 per 100,000), the AAP and CHA reported.
The total number of COVID-related deaths in children is 681 as of Oct. 18, according to the CDC, with the AAP/CHA reporting 558 as of Oct. 14, based on data from 45 states, New York City, Puerto Rico, and Guam. The CDC reports 65,655 admissions since Aug. 1, 2020, in children aged 0-17 years, and the AAP/CHA tally 23,582 since May 5, 2020, among children in 24 states and New York City.
As the COVID-19 vaccine heads toward approval for children under age 12 years, the number of older children receiving it dropped for the 10th consecutive week, based on data from the Centers for Disease Control and Prevention.
Over 47% of all children aged 12-17 years – that’s close to 12 million eligible individuals – have not received even one dose of COVID-19 vaccine, and less than 44% (about 11.1 million) were fully vaccinated as of Oct. 18, the CDC reported on its COVID Data Tracker.
, when eligibility expanded to include 12- to 15-year-olds, according to the CDC data, which also show that weekly vaccinations have never been lower.
Fortunately, the decline in new cases also continued, as the national total fell for a 6th straight week. There were more than 130,000 child cases reported during the week of Oct. 8-14, compared with 148,000 the previous week and the high of almost 252,000 in late August/early September, the American Academy of Pediatrics and the Children’s Hospital Association said in their weekly COVID-19 report.
That brings the cumulative count to 6.18 million, with children accounting for 16.4% of all cases reported since the start of the pandemic. For the week of Oct. 8-14, children represented 25.5% of all COVID-19 cases in the 46 states with up-to-date online dashboards, the AAP and CHA said, noting that New York has never reported age ranges for cases and that Alabama, Nebraska, and Texas stopped reporting over the summer.
Current data indicate that child cases in California now exceed 671,000, more than any other state, followed by Florida with 439,000 (the state defines a child as someone aged 0-14 years) and Illinois with 301,000. Vermont has the highest proportion of COVID-19 cases occurring in children (24.3%), with Alaska (24.1%) and South Carolina (23.2%) just behind. The highest rate of cases – 15,569 per 100,000 children – can be found in South Carolina, while the lowest is in Hawaii (4,838 per 100,000), the AAP and CHA reported.
The total number of COVID-related deaths in children is 681 as of Oct. 18, according to the CDC, with the AAP/CHA reporting 558 as of Oct. 14, based on data from 45 states, New York City, Puerto Rico, and Guam. The CDC reports 65,655 admissions since Aug. 1, 2020, in children aged 0-17 years, and the AAP/CHA tally 23,582 since May 5, 2020, among children in 24 states and New York City.
As the COVID-19 vaccine heads toward approval for children under age 12 years, the number of older children receiving it dropped for the 10th consecutive week, based on data from the Centers for Disease Control and Prevention.
Over 47% of all children aged 12-17 years – that’s close to 12 million eligible individuals – have not received even one dose of COVID-19 vaccine, and less than 44% (about 11.1 million) were fully vaccinated as of Oct. 18, the CDC reported on its COVID Data Tracker.
, when eligibility expanded to include 12- to 15-year-olds, according to the CDC data, which also show that weekly vaccinations have never been lower.
Fortunately, the decline in new cases also continued, as the national total fell for a 6th straight week. There were more than 130,000 child cases reported during the week of Oct. 8-14, compared with 148,000 the previous week and the high of almost 252,000 in late August/early September, the American Academy of Pediatrics and the Children’s Hospital Association said in their weekly COVID-19 report.
That brings the cumulative count to 6.18 million, with children accounting for 16.4% of all cases reported since the start of the pandemic. For the week of Oct. 8-14, children represented 25.5% of all COVID-19 cases in the 46 states with up-to-date online dashboards, the AAP and CHA said, noting that New York has never reported age ranges for cases and that Alabama, Nebraska, and Texas stopped reporting over the summer.
Current data indicate that child cases in California now exceed 671,000, more than any other state, followed by Florida with 439,000 (the state defines a child as someone aged 0-14 years) and Illinois with 301,000. Vermont has the highest proportion of COVID-19 cases occurring in children (24.3%), with Alaska (24.1%) and South Carolina (23.2%) just behind. The highest rate of cases – 15,569 per 100,000 children – can be found in South Carolina, while the lowest is in Hawaii (4,838 per 100,000), the AAP and CHA reported.
The total number of COVID-related deaths in children is 681 as of Oct. 18, according to the CDC, with the AAP/CHA reporting 558 as of Oct. 14, based on data from 45 states, New York City, Puerto Rico, and Guam. The CDC reports 65,655 admissions since Aug. 1, 2020, in children aged 0-17 years, and the AAP/CHA tally 23,582 since May 5, 2020, among children in 24 states and New York City.
FDA expands use of HIV drug to young children
The new lower dose is approved for children weighing from at least 14 kg (30 pounds) to 25 kg (55 pounds) who are virologically suppressed or new to antiretroviral therapy.
“Children living with HIV are in need of effective and accessible formulations of antiretroviral therapy,” said Merdad Parsey, MD, PhD, chief medical officer of Gilead Sciences, the company that produces Biktarvy, in a press release. “The New Drug Application approval is an important step in fulfilling Gilead’s commitment to a goal of bringing pediatric formulations of Biktarvy to children living with HIV around the world,” he said.
Although advances in treatment for pregnant women with HIV have lowered the likelihood of perinatal HIV transmission, pediatric HIV remains a global public health challenge. In 2020, about 1.7 million children younger than 15 years were living with HIV worldwide; 850 children become infected every day.
The approval, announced October 18, expands the use of Biktarvy to younger children. The medication was originally approved in February 2018 for treatment-naive or virologically suppressed adults. In June 2019, the FDA approved updating of the label to include pediatric patients weighing at least 25 kg. This new lower dose of Biktarvy is for a three-drug combo containing bictegravir 30 mg, emtricitabine 120 mg, and tenofovir alafenamide 15 mg. It is given once a day in tablet form.
The most recent expanded indication was based on data from an open-label, single-arm study that included 22 virologically suppressed children living with HIV. After switching to Biktarvy, 91% of participants (20 of 22) remained virologically suppressed at 24 weeks. HIV-1 RNA was not collected for two patients because of «pandemic-related study disruption,» the press release said.
“As children living with HIV will be on therapy for the foreseeable future and from such a young age, there are a number of factors I weigh as a clinician when prescribing the right HIV treatment option to my pediatric patients,” said Carina Rodriguez, MD, the division chief of pediatric infectious diseases at the University of South Florida, who was one of the study investigators. “Finding an efficacious treatment option is paramount, but tolerability and safety are keys to ensuring treatment success. With this expanded approval, clinicians can add Biktarvy to their arsenal of options to help ensure these children maintain virologic suppression with a treatment option that makes sense for them.”
A version of this article first appeared on Medscape.com.
The new lower dose is approved for children weighing from at least 14 kg (30 pounds) to 25 kg (55 pounds) who are virologically suppressed or new to antiretroviral therapy.
“Children living with HIV are in need of effective and accessible formulations of antiretroviral therapy,” said Merdad Parsey, MD, PhD, chief medical officer of Gilead Sciences, the company that produces Biktarvy, in a press release. “The New Drug Application approval is an important step in fulfilling Gilead’s commitment to a goal of bringing pediatric formulations of Biktarvy to children living with HIV around the world,” he said.
Although advances in treatment for pregnant women with HIV have lowered the likelihood of perinatal HIV transmission, pediatric HIV remains a global public health challenge. In 2020, about 1.7 million children younger than 15 years were living with HIV worldwide; 850 children become infected every day.
The approval, announced October 18, expands the use of Biktarvy to younger children. The medication was originally approved in February 2018 for treatment-naive or virologically suppressed adults. In June 2019, the FDA approved updating of the label to include pediatric patients weighing at least 25 kg. This new lower dose of Biktarvy is for a three-drug combo containing bictegravir 30 mg, emtricitabine 120 mg, and tenofovir alafenamide 15 mg. It is given once a day in tablet form.
The most recent expanded indication was based on data from an open-label, single-arm study that included 22 virologically suppressed children living with HIV. After switching to Biktarvy, 91% of participants (20 of 22) remained virologically suppressed at 24 weeks. HIV-1 RNA was not collected for two patients because of «pandemic-related study disruption,» the press release said.
“As children living with HIV will be on therapy for the foreseeable future and from such a young age, there are a number of factors I weigh as a clinician when prescribing the right HIV treatment option to my pediatric patients,” said Carina Rodriguez, MD, the division chief of pediatric infectious diseases at the University of South Florida, who was one of the study investigators. “Finding an efficacious treatment option is paramount, but tolerability and safety are keys to ensuring treatment success. With this expanded approval, clinicians can add Biktarvy to their arsenal of options to help ensure these children maintain virologic suppression with a treatment option that makes sense for them.”
A version of this article first appeared on Medscape.com.
The new lower dose is approved for children weighing from at least 14 kg (30 pounds) to 25 kg (55 pounds) who are virologically suppressed or new to antiretroviral therapy.
“Children living with HIV are in need of effective and accessible formulations of antiretroviral therapy,” said Merdad Parsey, MD, PhD, chief medical officer of Gilead Sciences, the company that produces Biktarvy, in a press release. “The New Drug Application approval is an important step in fulfilling Gilead’s commitment to a goal of bringing pediatric formulations of Biktarvy to children living with HIV around the world,” he said.
Although advances in treatment for pregnant women with HIV have lowered the likelihood of perinatal HIV transmission, pediatric HIV remains a global public health challenge. In 2020, about 1.7 million children younger than 15 years were living with HIV worldwide; 850 children become infected every day.
The approval, announced October 18, expands the use of Biktarvy to younger children. The medication was originally approved in February 2018 for treatment-naive or virologically suppressed adults. In June 2019, the FDA approved updating of the label to include pediatric patients weighing at least 25 kg. This new lower dose of Biktarvy is for a three-drug combo containing bictegravir 30 mg, emtricitabine 120 mg, and tenofovir alafenamide 15 mg. It is given once a day in tablet form.
The most recent expanded indication was based on data from an open-label, single-arm study that included 22 virologically suppressed children living with HIV. After switching to Biktarvy, 91% of participants (20 of 22) remained virologically suppressed at 24 weeks. HIV-1 RNA was not collected for two patients because of «pandemic-related study disruption,» the press release said.
“As children living with HIV will be on therapy for the foreseeable future and from such a young age, there are a number of factors I weigh as a clinician when prescribing the right HIV treatment option to my pediatric patients,” said Carina Rodriguez, MD, the division chief of pediatric infectious diseases at the University of South Florida, who was one of the study investigators. “Finding an efficacious treatment option is paramount, but tolerability and safety are keys to ensuring treatment success. With this expanded approval, clinicians can add Biktarvy to their arsenal of options to help ensure these children maintain virologic suppression with a treatment option that makes sense for them.”
A version of this article first appeared on Medscape.com.
States can reserve COVID shots for kids 5-11 this week
States can preorder COVID-19 vaccine doses for younger children this week as they begin to set up vaccination campaigns for ages 5-11.
Vaccine advisory groups for the FDA and CDC are scheduled to discuss and approve the Pfizer shot for kids in the next three weeks. To help states and cities prepare for the rollout, the CDC issued guidance on how to set up expanded vaccination programs.
Immunization program managers can begin ordering doses on Wednesday, according to the guidance. The vials won’t be delivered until the FDA and CDC authorize the shot, but registering now will help federal officials ship doses quickly once they’re available.
Pharmacies in every state will be able to give COVID-19 shots to children, but they can only use doses that are prepared specifically for children. Ages 5-11 will need a 10-microgram dose, which is one-third of the dose administered to ages 12 and older. The guidance warns that doctors should not try to split up or fraction the adult doses.
The CDC guidance also recommends that pediatricians and family practice doctors should serve as primary places to give shots to kids. The document mentions other options, such as vaccination clinics at schools, but doesn’t endorse them as the first choice for vaccinating kids.
The CDC hasn’t yet addressed questions around whether kids should be required to get vaccinated to attend school. The decision will likely be left to state and city officials.
Federal health officials aren’t yet sure how many parents and guardians will seek shots for their younger kids right away, the AP reported. Demand may be high at first for some families, but it may not be as high as when shots first became available for adults, Marcus Plescia, MD, chief medical officer of the Association of State and Territorial Health Officials, told The Associated Press.
“We’re going to have potentially a very busy, and perhaps modestly chaotic time,” he said.
When vaccines were first authorized for adults, hospitals and pharmacies received priority for ordering shots. Some doctors felt left out. This time, however, the CDC has said that pediatricians will receive higher priority and be able to receive shipments quickly.
As the vaccine rollout begins, health officials should consider logistical concerns to address racial and economic disparities for younger kids, Richard Besser, MD, president and CEO of the Robert Wood Johnson Foundation and a former acting director of the CDC, told the AP.
If parents or guardians can’t leave work to take their kids to a pharmacy or doctor’s office, for instance, their kids may not receive a shot quickly – or at all.
“It’s really important that we recognize the barriers to vaccinations,” he said.
A version of this article first appeared on WebMD.com.
States can preorder COVID-19 vaccine doses for younger children this week as they begin to set up vaccination campaigns for ages 5-11.
Vaccine advisory groups for the FDA and CDC are scheduled to discuss and approve the Pfizer shot for kids in the next three weeks. To help states and cities prepare for the rollout, the CDC issued guidance on how to set up expanded vaccination programs.
Immunization program managers can begin ordering doses on Wednesday, according to the guidance. The vials won’t be delivered until the FDA and CDC authorize the shot, but registering now will help federal officials ship doses quickly once they’re available.
Pharmacies in every state will be able to give COVID-19 shots to children, but they can only use doses that are prepared specifically for children. Ages 5-11 will need a 10-microgram dose, which is one-third of the dose administered to ages 12 and older. The guidance warns that doctors should not try to split up or fraction the adult doses.
The CDC guidance also recommends that pediatricians and family practice doctors should serve as primary places to give shots to kids. The document mentions other options, such as vaccination clinics at schools, but doesn’t endorse them as the first choice for vaccinating kids.
The CDC hasn’t yet addressed questions around whether kids should be required to get vaccinated to attend school. The decision will likely be left to state and city officials.
Federal health officials aren’t yet sure how many parents and guardians will seek shots for their younger kids right away, the AP reported. Demand may be high at first for some families, but it may not be as high as when shots first became available for adults, Marcus Plescia, MD, chief medical officer of the Association of State and Territorial Health Officials, told The Associated Press.
“We’re going to have potentially a very busy, and perhaps modestly chaotic time,” he said.
When vaccines were first authorized for adults, hospitals and pharmacies received priority for ordering shots. Some doctors felt left out. This time, however, the CDC has said that pediatricians will receive higher priority and be able to receive shipments quickly.
As the vaccine rollout begins, health officials should consider logistical concerns to address racial and economic disparities for younger kids, Richard Besser, MD, president and CEO of the Robert Wood Johnson Foundation and a former acting director of the CDC, told the AP.
If parents or guardians can’t leave work to take their kids to a pharmacy or doctor’s office, for instance, their kids may not receive a shot quickly – or at all.
“It’s really important that we recognize the barriers to vaccinations,” he said.
A version of this article first appeared on WebMD.com.
States can preorder COVID-19 vaccine doses for younger children this week as they begin to set up vaccination campaigns for ages 5-11.
Vaccine advisory groups for the FDA and CDC are scheduled to discuss and approve the Pfizer shot for kids in the next three weeks. To help states and cities prepare for the rollout, the CDC issued guidance on how to set up expanded vaccination programs.
Immunization program managers can begin ordering doses on Wednesday, according to the guidance. The vials won’t be delivered until the FDA and CDC authorize the shot, but registering now will help federal officials ship doses quickly once they’re available.
Pharmacies in every state will be able to give COVID-19 shots to children, but they can only use doses that are prepared specifically for children. Ages 5-11 will need a 10-microgram dose, which is one-third of the dose administered to ages 12 and older. The guidance warns that doctors should not try to split up or fraction the adult doses.
The CDC guidance also recommends that pediatricians and family practice doctors should serve as primary places to give shots to kids. The document mentions other options, such as vaccination clinics at schools, but doesn’t endorse them as the first choice for vaccinating kids.
The CDC hasn’t yet addressed questions around whether kids should be required to get vaccinated to attend school. The decision will likely be left to state and city officials.
Federal health officials aren’t yet sure how many parents and guardians will seek shots for their younger kids right away, the AP reported. Demand may be high at first for some families, but it may not be as high as when shots first became available for adults, Marcus Plescia, MD, chief medical officer of the Association of State and Territorial Health Officials, told The Associated Press.
“We’re going to have potentially a very busy, and perhaps modestly chaotic time,” he said.
When vaccines were first authorized for adults, hospitals and pharmacies received priority for ordering shots. Some doctors felt left out. This time, however, the CDC has said that pediatricians will receive higher priority and be able to receive shipments quickly.
As the vaccine rollout begins, health officials should consider logistical concerns to address racial and economic disparities for younger kids, Richard Besser, MD, president and CEO of the Robert Wood Johnson Foundation and a former acting director of the CDC, told the AP.
If parents or guardians can’t leave work to take their kids to a pharmacy or doctor’s office, for instance, their kids may not receive a shot quickly – or at all.
“It’s really important that we recognize the barriers to vaccinations,” he said.
A version of this article first appeared on WebMD.com.
Adalimumab biosimilar Cyltezo gets interchangeability designation
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
The Food and Drug Administration approved a supplement to the biologics license application of the adalimumab biosimilar drug Cyltezo (adalimumab-adbm) that makes it the first interchangeable biosimilar with Humira (adalimumab), the original branded version of the drug, its manufacturer Boehringer Ingelheim announced Oct. 15.
The FDA originally approved Cyltezo in 2017 for the treatment of multiple chronic inflammatory diseases, including seven of Humira’s nine indications for adults and pediatric patients: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis.
The interchangeability designation means that Cyltezo was tested in an additional clinical trial in which patients were successfully switched back and forth multiple times from Humira to Cyltezo and allows pharmacists to autosubstitute Humira with Cyltezo. In these cases, individual state laws control how and whether physicians will be notified of this switch.
Cyltezo is just the second biosimilar to be designated as interchangeable with its originator biologic product. The first approval, announced July 28, was for the interchangeability of Semglee (insulin glargine-yfgn) with the originator Lantus.
The agency based its decision on positive data from the VOLTAIRE-X study of 238 patients with moderate to severe chronic plaque psoriasis in which Cyltezo had no meaningful clinical differences from Humira in pharmacokinetics, efficacy, immunogenicity, and safety between the switching and continuous treatment groups.
Cyltezo will not be commercially available in the United States until July 1, 2023, according to Boehringer Ingelheim.
Anxiety, depression symptoms rose and fell with new COVID cases
Anxiety and depression symptoms increased in adults last winter as COVID-19 surged in the United States but declined in the spring as COVID activity approached its nadir, according to an analysis from the Centers for Disease Control and Prevention.
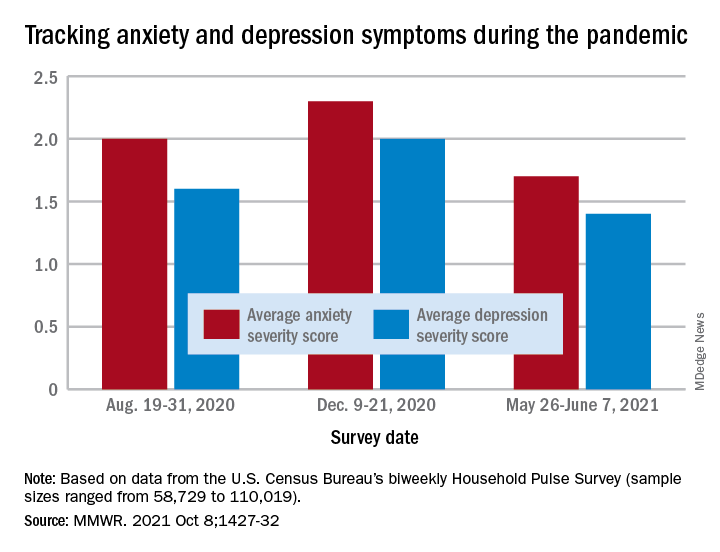
“The relative increases and decreases in frequency of reported symptoms of anxiety and depression at both the national and state levels mirrored the national weekly number of new COVID-19 cases during the same period,” Haomiao Jia, PhD, and associates wrote in the Morbidity and Mortality Weekly Report.
In a national survey conducted Aug. 19-31, 2020, the average anxiety severity score was 2.0 and the average depression score was 1.6 among adults in all 50 states. Those scores rose to 2.3 (+13.0%) and 2.0 (+14.8%), respectively, by Dec. 9-21, but then fell to 1.7 (–26.8%) and 1.4 (–24.8%) during the survey conducted from May 26 to June 7, 2021, the investigators reported.
Despite that decrease in the spring, however, “the frequency of symptoms ... in June 2021 remained elevated compared with estimates from” 2019, said Dr. Jia of Columbia University, New York, and associates. Data from the National Health Interview Survey put the prepandemic severity scores at 0.63 for anxiety and 0.51 for depression.
Weekly symptom frequency in the Household Pulse Survey, which began in April 2020, was assessed with the four-item Patient Health Questionnaire, which includes two questions on anxiety and two on depression. Each answer scored on a scale from 0 (no symptoms at all) to 3 (symptoms nearly every day), making a total of 6 possible for each severity score, they explained. Sample sizes for the biweekly surveys ranged from 58,729 to 110,019.
Among the states, there was something of a pattern involving the drop in scores during the fall and the rise over the winter and spring months. “States with larger increases in severity scores during August–December 2020 also tended to have larger decreases during January–June 2021,” the researchers noted.
That group includes Minnesota, Mississippi, South Dakota, and Utah for anxiety and Idaho, Michigan, Minnesota, and Wisconsin for depression, the survey data show.
Florida and New York had the smallest increases in depression and anxiety scores, respectively, from August to December, and New York had the smallest decrease in both anxiety and depression from January to June, Dr. Jia and associates said.
“ during national emergencies. The observed differences in severity score magnitude and peaks across states in this study indicate that these efforts are important at both the national and state levels,” they wrote.
Anxiety and depression symptoms increased in adults last winter as COVID-19 surged in the United States but declined in the spring as COVID activity approached its nadir, according to an analysis from the Centers for Disease Control and Prevention.
“The relative increases and decreases in frequency of reported symptoms of anxiety and depression at both the national and state levels mirrored the national weekly number of new COVID-19 cases during the same period,” Haomiao Jia, PhD, and associates wrote in the Morbidity and Mortality Weekly Report.
In a national survey conducted Aug. 19-31, 2020, the average anxiety severity score was 2.0 and the average depression score was 1.6 among adults in all 50 states. Those scores rose to 2.3 (+13.0%) and 2.0 (+14.8%), respectively, by Dec. 9-21, but then fell to 1.7 (–26.8%) and 1.4 (–24.8%) during the survey conducted from May 26 to June 7, 2021, the investigators reported.
Despite that decrease in the spring, however, “the frequency of symptoms ... in June 2021 remained elevated compared with estimates from” 2019, said Dr. Jia of Columbia University, New York, and associates. Data from the National Health Interview Survey put the prepandemic severity scores at 0.63 for anxiety and 0.51 for depression.
Weekly symptom frequency in the Household Pulse Survey, which began in April 2020, was assessed with the four-item Patient Health Questionnaire, which includes two questions on anxiety and two on depression. Each answer scored on a scale from 0 (no symptoms at all) to 3 (symptoms nearly every day), making a total of 6 possible for each severity score, they explained. Sample sizes for the biweekly surveys ranged from 58,729 to 110,019.
Among the states, there was something of a pattern involving the drop in scores during the fall and the rise over the winter and spring months. “States with larger increases in severity scores during August–December 2020 also tended to have larger decreases during January–June 2021,” the researchers noted.
That group includes Minnesota, Mississippi, South Dakota, and Utah for anxiety and Idaho, Michigan, Minnesota, and Wisconsin for depression, the survey data show.
Florida and New York had the smallest increases in depression and anxiety scores, respectively, from August to December, and New York had the smallest decrease in both anxiety and depression from January to June, Dr. Jia and associates said.
“ during national emergencies. The observed differences in severity score magnitude and peaks across states in this study indicate that these efforts are important at both the national and state levels,” they wrote.
Anxiety and depression symptoms increased in adults last winter as COVID-19 surged in the United States but declined in the spring as COVID activity approached its nadir, according to an analysis from the Centers for Disease Control and Prevention.
“The relative increases and decreases in frequency of reported symptoms of anxiety and depression at both the national and state levels mirrored the national weekly number of new COVID-19 cases during the same period,” Haomiao Jia, PhD, and associates wrote in the Morbidity and Mortality Weekly Report.
In a national survey conducted Aug. 19-31, 2020, the average anxiety severity score was 2.0 and the average depression score was 1.6 among adults in all 50 states. Those scores rose to 2.3 (+13.0%) and 2.0 (+14.8%), respectively, by Dec. 9-21, but then fell to 1.7 (–26.8%) and 1.4 (–24.8%) during the survey conducted from May 26 to June 7, 2021, the investigators reported.
Despite that decrease in the spring, however, “the frequency of symptoms ... in June 2021 remained elevated compared with estimates from” 2019, said Dr. Jia of Columbia University, New York, and associates. Data from the National Health Interview Survey put the prepandemic severity scores at 0.63 for anxiety and 0.51 for depression.
Weekly symptom frequency in the Household Pulse Survey, which began in April 2020, was assessed with the four-item Patient Health Questionnaire, which includes two questions on anxiety and two on depression. Each answer scored on a scale from 0 (no symptoms at all) to 3 (symptoms nearly every day), making a total of 6 possible for each severity score, they explained. Sample sizes for the biweekly surveys ranged from 58,729 to 110,019.
Among the states, there was something of a pattern involving the drop in scores during the fall and the rise over the winter and spring months. “States with larger increases in severity scores during August–December 2020 also tended to have larger decreases during January–June 2021,” the researchers noted.
That group includes Minnesota, Mississippi, South Dakota, and Utah for anxiety and Idaho, Michigan, Minnesota, and Wisconsin for depression, the survey data show.
Florida and New York had the smallest increases in depression and anxiety scores, respectively, from August to December, and New York had the smallest decrease in both anxiety and depression from January to June, Dr. Jia and associates said.
“ during national emergencies. The observed differences in severity score magnitude and peaks across states in this study indicate that these efforts are important at both the national and state levels,” they wrote.
FROM THE MMWR
FDA approves avacopan for rare ANCA autoimmune disease
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.

ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.
ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.
ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
FDA clears first mobile rapid test for concussion
, the company has announced.

Eye-Sync is a virtual reality eye-tracking platform that provides objective measurements to aid in the assessment of concussion. It’s the first mobile, rapid test for concussion that has been cleared by the FDA, the company said.
As reported by this news organization, Eye-Sync received breakthrough designation from the FDA for this indication in March 2019.
The FDA initially cleared the Eye-Sync platform for recording, viewing, and analyzing eye movements to help clinicians identify visual tracking impairment.
The Eye-Sync technology uses a series of 60-second eye tracking assessments, neurocognitive batteries, symptom inventories, and standardized patient inventories to identify the type and severity of impairment after concussion.
“The platform generates customizable and interpretive reports that support clinical decision making and offers visual and vestibular therapies to remedy deficits and monitor improvement over time,” the company said.
In support of the application for use in concussion, SyncThink enrolled 1,655 children and adults into a clinical study that collected comprehensive patient and concussion-related data for over 12 months.
The company used these data to develop proprietary algorithms and deep learning models to identify a positive or negative indication of concussion.
The study showed that Eye-Sinc had sensitivity greater than 82% and specificity greater than 93%, “thereby providing clinicians with significant and actionable data when evaluating individuals with concussion,” the company said in a news release.
“The outcome of this study very clearly shows the effectiveness of our technology at detecting concussion and definitively demonstrates the clinical utility of Eye-Sinc,” SyncThink Chief Clinical Officer Scott Anderson said in the release.
“It also shows that the future of concussion diagnosis is no longer purely symptom-based but that of a technology driven multi-modal approach,” Mr. Anderson said.
A version of this article first appeared on Medscape.com.
, the company has announced.
Eye-Sync is a virtual reality eye-tracking platform that provides objective measurements to aid in the assessment of concussion. It’s the first mobile, rapid test for concussion that has been cleared by the FDA, the company said.
As reported by this news organization, Eye-Sync received breakthrough designation from the FDA for this indication in March 2019.
The FDA initially cleared the Eye-Sync platform for recording, viewing, and analyzing eye movements to help clinicians identify visual tracking impairment.
The Eye-Sync technology uses a series of 60-second eye tracking assessments, neurocognitive batteries, symptom inventories, and standardized patient inventories to identify the type and severity of impairment after concussion.
“The platform generates customizable and interpretive reports that support clinical decision making and offers visual and vestibular therapies to remedy deficits and monitor improvement over time,” the company said.
In support of the application for use in concussion, SyncThink enrolled 1,655 children and adults into a clinical study that collected comprehensive patient and concussion-related data for over 12 months.
The company used these data to develop proprietary algorithms and deep learning models to identify a positive or negative indication of concussion.
The study showed that Eye-Sinc had sensitivity greater than 82% and specificity greater than 93%, “thereby providing clinicians with significant and actionable data when evaluating individuals with concussion,” the company said in a news release.
“The outcome of this study very clearly shows the effectiveness of our technology at detecting concussion and definitively demonstrates the clinical utility of Eye-Sinc,” SyncThink Chief Clinical Officer Scott Anderson said in the release.
“It also shows that the future of concussion diagnosis is no longer purely symptom-based but that of a technology driven multi-modal approach,” Mr. Anderson said.
A version of this article first appeared on Medscape.com.
, the company has announced.
Eye-Sync is a virtual reality eye-tracking platform that provides objective measurements to aid in the assessment of concussion. It’s the first mobile, rapid test for concussion that has been cleared by the FDA, the company said.
As reported by this news organization, Eye-Sync received breakthrough designation from the FDA for this indication in March 2019.
The FDA initially cleared the Eye-Sync platform for recording, viewing, and analyzing eye movements to help clinicians identify visual tracking impairment.
The Eye-Sync technology uses a series of 60-second eye tracking assessments, neurocognitive batteries, symptom inventories, and standardized patient inventories to identify the type and severity of impairment after concussion.
“The platform generates customizable and interpretive reports that support clinical decision making and offers visual and vestibular therapies to remedy deficits and monitor improvement over time,” the company said.
In support of the application for use in concussion, SyncThink enrolled 1,655 children and adults into a clinical study that collected comprehensive patient and concussion-related data for over 12 months.
The company used these data to develop proprietary algorithms and deep learning models to identify a positive or negative indication of concussion.
The study showed that Eye-Sinc had sensitivity greater than 82% and specificity greater than 93%, “thereby providing clinicians with significant and actionable data when evaluating individuals with concussion,” the company said in a news release.
“The outcome of this study very clearly shows the effectiveness of our technology at detecting concussion and definitively demonstrates the clinical utility of Eye-Sinc,” SyncThink Chief Clinical Officer Scott Anderson said in the release.
“It also shows that the future of concussion diagnosis is no longer purely symptom-based but that of a technology driven multi-modal approach,” Mr. Anderson said.
A version of this article first appeared on Medscape.com.
FDA approves first CAR T-cell for adult ALL: For patients with R/R B-cell disease
The therapy is the first chimeric antigen receptor (CAR) T-cell treatment approved for adults with ALL.
This is a “meaningful advance,” because “roughly half of all adults with B-ALL will relapse on currently available therapies,” said Bijal Shah, MD, of Moffitt Cancer Center, Tampa, Fla., in a press statement from the manufacturer, Kite.
“A single infusion of Tecartus has demonstrated durable responses, suggesting the potential for long-term remission and a new approach to care,” he added.
“Roughly half of all cases actually occur in adults, and unlike pediatric ALL, adult ALL has historically had a poor prognosis,” said Lee Greenberger, PhD, chief scientific officer at the Leukemia & Lymphoma Society, in the statement. The median overall survival (OS) is only about 8 months in this setting with current treatments, according to the company.
The new FDA approval, which is the fourth indication for brexucabtagene autoleucel, is based on results from ZUMA-3, a multicenter, single-arm study of 71 patients, with 54 efficacy-evaluable patients.
Efficacy was established on the basis of complete remission (CR) rate within 3 months after infusion and the duration of CR (DOCR). Twenty-eight (51.9%) of evaluable patients achieved CR, with a median follow-up for responders of 7.1 months. The median DOCR was not reached.
The median time to CR was 56 days. All 54 efficacy-evaluable patients had potential follow-up for 10 or more months with a median actual follow-up time of 12.3 months.
Among the 54 patients, the median time from leukapheresis to product delivery was 16 days and the median time from leukapheresis to infusion was 29 days.
Of the 17 study patients who did reach efficacy evaluation, 6 did not receive the agent because of manufacturing failure, 8 were not treated because of adverse events following leukapheresis, 2 underwent leukapheresis and received lymphodepleting chemotherapy but were not treated with the drug, and 1 treated patient was not evaluable for efficacy, per the prescribing information.
Among all patients treated with brexucabtagene autoleucel at its target dose, grade 3 or higher cytokine release syndrome (CRS) and neurologic events occurred in 26% and 35% of patients, respectively, and were generally well managed, according to the company.
The most common adverse reactions (≥20%) among ALL patients are fever, CRS, hypotension, encephalopathy, tachycardia, nausea, chills, headache, fatigue, febrile neutropenia, diarrhea, musculoskeletal pain, hypoxia, rash, edema, tremor, infection with pathogen unspecified, constipation, decreased appetite, and vomiting.
The prescribing information includes a boxed warning about the risks of CRS and neurologic toxicities; the drug is approved with a Risk Evaluation and Mitigation Strategy (REMS) because of these risks.
A version of this article first appeared on Medscape.com.
The therapy is the first chimeric antigen receptor (CAR) T-cell treatment approved for adults with ALL.
This is a “meaningful advance,” because “roughly half of all adults with B-ALL will relapse on currently available therapies,” said Bijal Shah, MD, of Moffitt Cancer Center, Tampa, Fla., in a press statement from the manufacturer, Kite.
“A single infusion of Tecartus has demonstrated durable responses, suggesting the potential for long-term remission and a new approach to care,” he added.
“Roughly half of all cases actually occur in adults, and unlike pediatric ALL, adult ALL has historically had a poor prognosis,” said Lee Greenberger, PhD, chief scientific officer at the Leukemia & Lymphoma Society, in the statement. The median overall survival (OS) is only about 8 months in this setting with current treatments, according to the company.
The new FDA approval, which is the fourth indication for brexucabtagene autoleucel, is based on results from ZUMA-3, a multicenter, single-arm study of 71 patients, with 54 efficacy-evaluable patients.
Efficacy was established on the basis of complete remission (CR) rate within 3 months after infusion and the duration of CR (DOCR). Twenty-eight (51.9%) of evaluable patients achieved CR, with a median follow-up for responders of 7.1 months. The median DOCR was not reached.
The median time to CR was 56 days. All 54 efficacy-evaluable patients had potential follow-up for 10 or more months with a median actual follow-up time of 12.3 months.
Among the 54 patients, the median time from leukapheresis to product delivery was 16 days and the median time from leukapheresis to infusion was 29 days.
Of the 17 study patients who did reach efficacy evaluation, 6 did not receive the agent because of manufacturing failure, 8 were not treated because of adverse events following leukapheresis, 2 underwent leukapheresis and received lymphodepleting chemotherapy but were not treated with the drug, and 1 treated patient was not evaluable for efficacy, per the prescribing information.
Among all patients treated with brexucabtagene autoleucel at its target dose, grade 3 or higher cytokine release syndrome (CRS) and neurologic events occurred in 26% and 35% of patients, respectively, and were generally well managed, according to the company.
The most common adverse reactions (≥20%) among ALL patients are fever, CRS, hypotension, encephalopathy, tachycardia, nausea, chills, headache, fatigue, febrile neutropenia, diarrhea, musculoskeletal pain, hypoxia, rash, edema, tremor, infection with pathogen unspecified, constipation, decreased appetite, and vomiting.
The prescribing information includes a boxed warning about the risks of CRS and neurologic toxicities; the drug is approved with a Risk Evaluation and Mitigation Strategy (REMS) because of these risks.
A version of this article first appeared on Medscape.com.
The therapy is the first chimeric antigen receptor (CAR) T-cell treatment approved for adults with ALL.
This is a “meaningful advance,” because “roughly half of all adults with B-ALL will relapse on currently available therapies,” said Bijal Shah, MD, of Moffitt Cancer Center, Tampa, Fla., in a press statement from the manufacturer, Kite.
“A single infusion of Tecartus has demonstrated durable responses, suggesting the potential for long-term remission and a new approach to care,” he added.
“Roughly half of all cases actually occur in adults, and unlike pediatric ALL, adult ALL has historically had a poor prognosis,” said Lee Greenberger, PhD, chief scientific officer at the Leukemia & Lymphoma Society, in the statement. The median overall survival (OS) is only about 8 months in this setting with current treatments, according to the company.
The new FDA approval, which is the fourth indication for brexucabtagene autoleucel, is based on results from ZUMA-3, a multicenter, single-arm study of 71 patients, with 54 efficacy-evaluable patients.
Efficacy was established on the basis of complete remission (CR) rate within 3 months after infusion and the duration of CR (DOCR). Twenty-eight (51.9%) of evaluable patients achieved CR, with a median follow-up for responders of 7.1 months. The median DOCR was not reached.
The median time to CR was 56 days. All 54 efficacy-evaluable patients had potential follow-up for 10 or more months with a median actual follow-up time of 12.3 months.
Among the 54 patients, the median time from leukapheresis to product delivery was 16 days and the median time from leukapheresis to infusion was 29 days.
Of the 17 study patients who did reach efficacy evaluation, 6 did not receive the agent because of manufacturing failure, 8 were not treated because of adverse events following leukapheresis, 2 underwent leukapheresis and received lymphodepleting chemotherapy but were not treated with the drug, and 1 treated patient was not evaluable for efficacy, per the prescribing information.
Among all patients treated with brexucabtagene autoleucel at its target dose, grade 3 or higher cytokine release syndrome (CRS) and neurologic events occurred in 26% and 35% of patients, respectively, and were generally well managed, according to the company.
The most common adverse reactions (≥20%) among ALL patients are fever, CRS, hypotension, encephalopathy, tachycardia, nausea, chills, headache, fatigue, febrile neutropenia, diarrhea, musculoskeletal pain, hypoxia, rash, edema, tremor, infection with pathogen unspecified, constipation, decreased appetite, and vomiting.
The prescribing information includes a boxed warning about the risks of CRS and neurologic toxicities; the drug is approved with a Risk Evaluation and Mitigation Strategy (REMS) because of these risks.
A version of this article first appeared on Medscape.com.