User login
Simultaneous Cases of Carfilzomib-Induced Thrombotic Microangiopathy in 2 Patients With Multiple Myeloma
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
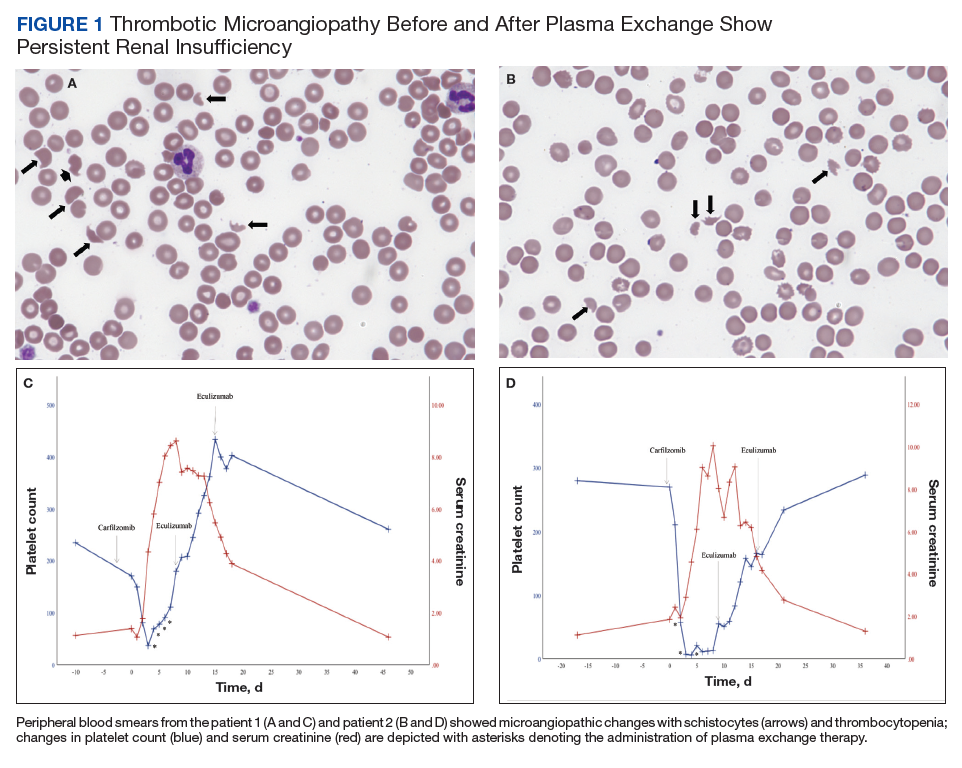
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
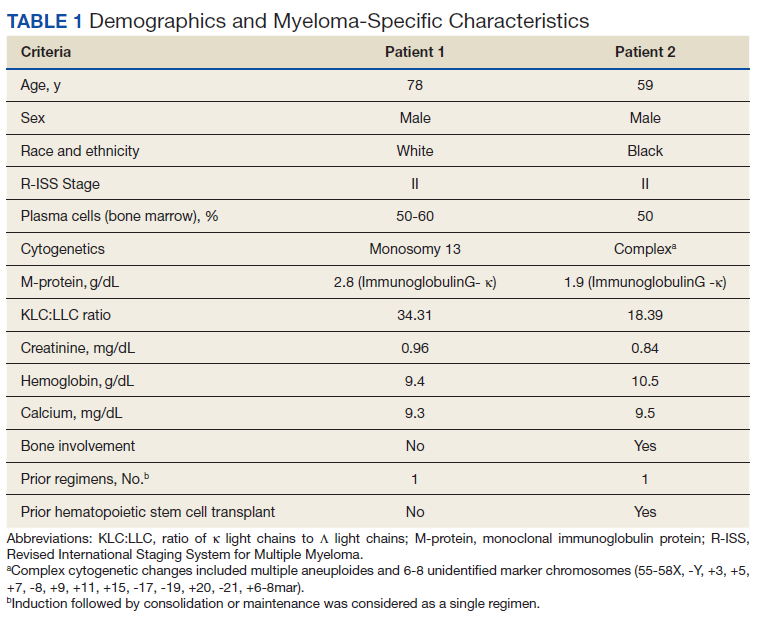
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
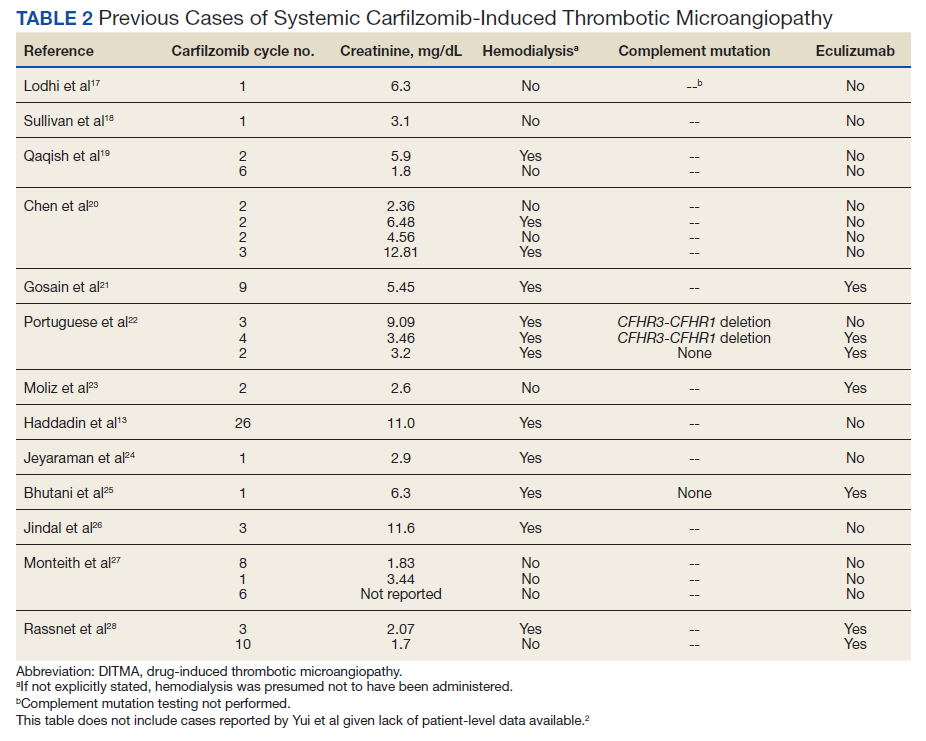
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
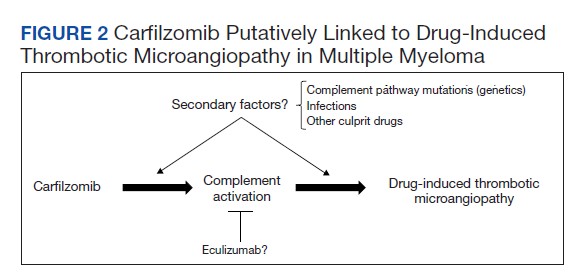
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
As a class of drugs, proteasome inhibitors are known to rarely cause drug-induced thrombotic microangiopathy (DITMA). In particular, carfilzomib is a second-generation, irreversible proteasome inhibitor approved for the treatment of relapsed, refractory multiple myeloma (MM) in combination with other therapeutic agents.1 Although generally well tolerated, carfilzomib has been associated with serious adverse events such as cardiovascular toxicity and DITMA.2-4 Thrombotic microangiopathy (TMA) is a life-threatening disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage.5 Its occurrence secondary to carfilzomib has been reported only rarely in clinical trials of MM, and the most effective management of the disorder as well as the concurrent risk factors that contribute to its development remain incompletely understood.6,7 As a result, given both the expanding use of carfilzomib in practice and the morbidity of TMA, descriptions of carfilzomib-induced TMA from the real-world setting continue to provide important contributions to our understanding of the disorder.
At our US Department of Veterans Affairs (VA) medical center, 2 patients developed severe carfilzomib-induced TMA within days of one another. The presentation of simultaneous cases was highly unexpected and offered the unique opportunity to compare clinical features in real time. Here, we describe our 2 cases in detail, review their presentations and management in the context of the prior literature, and discuss potential insights gained into the disease.
Case Presentation
Case 1
A 78-year-old male patient was diagnosed with monoclonal gammopathy of undetermined significance in 2012 that progressed to Revised International Staging System stage II IgG-κ MM in 2016 due to worsening anemia with a hemoglobin level < 10 g/dL (Table 1). He was treated initially with 8 cycles of first-line bortezomib, lenalidomide, and dexamethasone, to which he achieved a partial response with > 50% reduction in serum M-protein. He then received 3 cycles of maintenance bortezomib until relapse, at which time he was switched to second-line therapy consisting of carfilzomib 20 mg/m2 on days 1 and 2 and 56 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles plus dexamethasone 20 mg twice weekly every 28 days.
After the patient received cycle 3, day 1 of carfilzomib, he developed subjective fevers, chills, and diarrhea. He missed his day 2 infusion and instead presented to the VA emergency department, where his vital signs were stable and laboratory tests were notable for the following levels: leukocytosis of20.3 K/µL (91.7% neutrophils), hemoglobin 12.4 g/dL (prior 13.5 g/dL), platelet count 171 K/µL, and creatinine 1.39 mg/dL (prior 1.13 g/dL). A chest X-ray demonstrated diffuse bilateral opacities concerning for edema vs infection, and he was started empirically on vancomycin, piperacillin-tazobactam, and azithromycin. His outpatient medications, which included acyclovir, aspirin, finasteride, oxybutynin, ranitidine, omega-3 fatty acids, fish oil, vitamin D, and senna, were continued as indicated.
On hospital day 2, the patient’s platelet count dropped to 81 K/µL and creatinine level rose to 1.78 mg/dL. He developed dark urine (urinalysis [UA] 3+ blood, 6-11 red blood cells per high power field [RBC/HPF]) and had laboratory tests suggestive of hemolysis, including lactic dehydrogenase (LDH) > 1,200 IU/L (reference range, 60-250 IU/L), haptoglobin < 30 mg/dL (reference range, 44-215 mg/dL), total bilirubin 3.2 mg/dL (reference range, 0.2-1.3 mg/dL; indirect bilirubin, 2.6 mg/dL), and a peripheral blood smear demonstrating moderate microangiopathy (Figure 1).
Workup for alternative causes of thrombocytopenia included a negative heparin-induced thrombocytopenia panel and a disseminated intravascular coagulation (DIC) panel showing elevated fibrinogen (515 mg/dL; reference range, 200-400 mg/dL) and mildly elevated international normalized ratio (INR) (1.3). Blood cultures were negative, and a 22-pathogen gastrointestinal polymerase chain reaction (PCR) panel failed to identify viral or bacterial pathogens, including Escherichia coli O157:H7. C3 (81 mg/dL; reference range, 90-180 mg/dL) and C4 (16 mg/dL; reference range, 16-47 mg/dL) complement levels were borderline to mildly reduced.
Based on this constellation of findings, a diagnosis of TMA was made, and the patient was started empirically on plasma exchange and pulse-dosed steroids. After 4 cycles of plasma exchange, the platelet count had normalized from its nadir of 29 K/µL. ADAMTS13 activity (98% enzyme activity) ruled out thrombotic thrombocytopenic purpura (TTP), and the patient continued to have anuric renal failure (creatinine, 8.62 mg/dL) necessitating the initiation of hemodialysis. Given persistent renal insufficiency, a diagnosis of atypical hemolytic uremic syndrome (HUS) was considered, and eculizumab 900 mg was administered on days 8 and 15 with stabilization of renal function. By the time of discharge on day 18, the patient’s creatinine level had decreased to 3.89 mg/dL, and platelet count was 403 K/µL. Creatinine normalized to 1.07 mg/dL by day 46.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for mutations in the following genes associated with atypical HUS: CFH, CFI, MCP (CD46), THBD, CFB, C3, DGKE, ADAMTS13, C4BPA, C4BPB, LMNA, CFHR1, CFHR3, CFHR4, and CFHR5. The patient subsequently remained off all antimyeloma therapy for > 1 year until eventually starting third-line pomalidomide plus dexamethasone without reinitiation of proteasome inhibitor therapy.
Case 2
A 59-year-old male patient, diagnosed in 2013 with ISS stage I IgG-κ MM after presenting with compression fractures, completed 8 cycles of cyclophosphamide, bortezomib, and dexamethasone before undergoing autologous hematopoietic stem cell transplantation with complete response (Table 1). He subsequently received single-agent maintenance bortezomib until relapse nearly 2 years later, at which time he started second-line carfilzomib 20 mg/m2 on days 1 and 2 and 27 mg/m2 on days 8, 9, 15, and 16 for cycle 1, followed by 27 mg/m2 on days 8, 9, 15, and 16 for cycles 2 to 8, lenalidomide 25 mg on days 1 to 21, and dexamethasone 40 mg weekly every 28 days. Serum free light chain levels normalized after 9 cycles, and he subsequently began maintenance carfilzomib 70 mg/m2 on days 1 and 15 plus lenalidomide 10 mg on days 1 to 21 every 28 days.
On the morning before admission, the patient received C6D17 of maintenance carfilzomib, which had been delayed from day 15 because of the holiday. Later that evening, he developed nausea, vomiting, and fever of 101.3 °F. He presented to the VA emergency department and was tachycardic (108 beats per minute) and hypotensive (86/55 mm Hg). Laboratory tests were notable for hemoglobin level 9.9 g/dL (prior 11.6 g/dL), platelet count 270 K/µL, and creatinine level 1.86 mg/dL (prior 1.12 mg/dL). A respiratory viral panel was positive for influenza A, and antimicrobial agents were eventually broadened to piperacillin-tazobactam, azithromycin, and oseltamivir. His outpatient medications, which included acyclovir, zoledronic acid, sulfamethoxazole/trimethoprim, aspirin, amlodipine, atorvastatin, omeprazole, zolpidem, calcium, vitamin D, loratadine, ascorbic acid, and prochlorperazine, were continued as indicated.
On hospital day 2, the patient’s platelet count declined from 211 to 57 K/µL. He developed tea-colored urine (UA 2+ blood, 0-2 RBC/HPF) and had laboratory tests suggestive of hemolysis, including LDH 910 IU/L (reference range, 60-250 IU/L), total bilirubin 3.3 mg/dL (reference range, 0.2-1.3 mg/dL; no direct or indirect available), and a peripheral blood smear demonstrating moderate microangiopathy. Although haptoglobin level was normal at this time (206 mg/dL; reference range, 44-215 mg/dL), it decreased to 42 mg/dL by the following day. Additional workup included a negative direct Coombs and a DIC panel showing elevated fibrinogen (596 mg/dL; reference range, 200-400 mg/dL) and mildly elevated INR (1.16). Blood cultures remained negative, and a 22-pathogen GI PCR panel identified no viral or bacterial pathogens, including E coli O157:H7. C3 (114 mg/dL; reference range, 90-180 mg/dL) and C4 (40 mg/dL; reference range, 16-47 mg/dL) complement levels were both normal.
Based on these findings, empiric treatment was started with plasma exchange and pulse-dosed steroids. The patient received 3 cycles of plasma exchange until the results of the ADAMTS13 activity ruled out TTP (63% enzyme activity). Over the next 6 days, his platelet count reached a nadir of 6 K/µL and creatinine level peaked at 10.36 mg/dL, necessitating the initiation of hemodialysis. Given severe renal insufficiency, a diagnosis of atypical HUS was again considered, and eculizumab 900 mg was administered on days 9 and 16 with stabilization of renal function. By the time of discharge on day 17, the patient’s creatinine level had decreased to 4.17 mg/dL and platelet count was 164 K/µL. Creatinine level normalized to 1.02 mg/dL by day 72.
Outpatient genetic testing through the BloodCenter of Wisconsin Diagnostic Laboratories was negative for gene mutations associated with atypical HUS. Approximately 1 month after discharge, the patient resumed maintenance lenalidomide alone without reinitiation of proteasome inhibitor therapy.
Discussion
In this case series, we describe the uncommon drug-related adverse event of TMA occurring in 2 patients with MM after receiving carfilzomib. Although the incidence of TMA disorders is low, reaching up to 2.8% in patients receiving carfilzomib plus cyclophosphamide and dexamethasone in the phase 2 CARDAMON trial, our experience suggests that a high index of suspicion for carfilzomib-induced TMA is warranted in the real-world setting.8 TMA syndromes, including TTP, HUS, and DITMA, are characterized by microvascular endothelial injury and thrombosis leading to thrombocytopenia and microangiopathic hemolytic anemia.5,9 Several drug culprits of DITMA are recognized, including quinine, gemcitabine, tacrolimus, and proteasome inhibitors (bortezomib, carfilzomib, ixazomib).10-12 In a real-world series of patients receiving proteasome inhibitor therapy, either carfilzomib (n=8) or bortezomib (n=3), common clinical features of DITMA included thrombocytopenia, microangiopathic hemolytic anemia, gastrointestinal symptoms, and renal insufficiency with or without a need for hemodialysis.2 Although DITMA has been described primarily as an early event, its occurrence after 12 months of proteasome inhibitor therapy has also been reported, both in this series and elsewhere, thereby suggesting an ongoing risk for DITMA throughout the duration of carfilzomib treatment.2,13
The diagnosis of DITMA can be challenging given its nonspecific symptoms that overlap with other TMA syndromes. Previous studies have proposed that for a drug to be associated with DITMA, there should be: (1) evidence of clinical and/or pathologic findings of TMA; (2) exclusion of alternative causes of TMA; (3) no other new drug exposures other than the suspected culprit medication; and (4) a lack of recurrence of TMA in absence of the drug.10 In the case of patients with MM, other causes of TMA have also been described, including the underlying plasma cell disorder itself and stem cell transplantation.14 In the 2 cases we have described, these alternative causes were considered unlikely given that only 1 patient underwent transplantation remotely and neither had a previous history of TMA secondary to their disease. With respect to other TMA syndromes, ADAMTS13 levels > 10% and negative stool studies for E coli O157:H7 suggested against TTP or typical HUS, respectively. No other drug culprits were identified, and the close timing between the receipt of carfilzomib and symptom onset supported a causal relationship.
Because specific therapies are lacking, management of DITMA has traditionally included drug discontinuation and supportive care for end-organ injury.5 The terminal complement inhibitor, eculizumab, improves hematologic abnormalities and renal function in patients with atypical HUS but its use for treating patients with DITMA is not standard.15 Therefore, the decision to administer eculizumab to our 2 patients was driven by their severe renal insufficiency without improvement after plasma exchange, which suggested a phenotype similar to atypical HUS. After administration of eculizumab, renal function stabilized and then gradually improved over weeks to months, a time course similar to that described in cases of patients with DITMA secondary to other anticancer therapies treated with eculizumab.16 Although these results suggest a potential role for eculizumab in proteasome inhibitor–induced TMA, distinguishing the benefit of eculizumab over drug discontinuation alone remains challenging, and well-designed prospective investigations are needed.
The clustered occurrence of our 2 cases is unique from previous reports that describe carfilzomib-induced TMA as a sporadic event (Table 2).13,17-28 Both immune-mediated and direct toxic effects have been proposed as mechanisms of DITMA, and while our cases do not differentiate between these mechanisms, we considered whether a combined model of initiation, whereby patient or environmental risk factors modulate occurrence of the disease in conjunction with the inciting drug, could explain the clustered occurrence of cases. In this series, drug manufacturing was not a shared risk factor as each patient received carfilzomib from different lot numbers. Furthermore, other patients at our center received carfilzomib from the same batches without developing DITMA. We also considered the role of infection given that 1 patient was diagnosed with influenza A and both presented with nonspecific, viral-like symptoms during the winter season. Interestingly, concurrent viral infections have been reported in other cases of carfilzomib-induced DITMA as well and have also been discussed as a trigger of atypical HUS.20,29 Finally, genetic testing was negative for complement pathway mutations that might predispose to complement dysregulation.
The absence of complement mutations in our 2 patients differs from a recent series describing heterozygous CFHR3-CHFR1 deletions in association with carfilzomib-induced TMA.22 In that report, the authors hypothesized that carfilzomib decreases expression of complement factor H (CFH), a negative regulator of complement activation, thereby leading to complement dysregulation in patients who are genetically predisposed. In a second series, plasma from patients with DITMA secondary to carfilzomib induced the deposition of the complement complex, C5b-9, on endothelial cells in culture, suggesting activation of the complement pathway.30 The effective use of eculizumab would also point to a role for complement activation, and ongoing investigations should aim to identify the triggers and mechanisms of complement dysregulation in this setting, especially for patients like ours in whom genetic testing for complement pathway mutations is negative (Figure 2).
Conclusions
DITMA is a known risk of proteasome inhibitors and is listed as a safety warning in the prescribing information for bortezomib, carfilzomib, and ixazomib.12 Given the overall rarity of this adverse event, the simultaneous presentation of our 2 cases was unexpected and underscores the need for heightened awareness in clinical practice. In addition, while no underlying complement mutations were identified, eculizumab was used in both cases to successfully stabilize renal function. Further research investigating the efficacy of eculizumab and the role of complement activation in proteasome inhibitor–induced TMA will be valuable.
Acknowledgments
The authors would like to thank the patients whose histories are reported in this manuscript as well as the physicians and staff who provided care during the hospitalizations and beyond. We also thank Oscar Silva, MD, PhD, for his assistance in reviewing and formatting the peripheral blood smear images.
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
1. McBride A, Klaus JO, Stockeri-Goldstein K. Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am J Health Syst Pharm. 2015;72(5):353-360. doi:10.2146/ajhp130281
2. Yui JC, Van Keer J, Weiss BM, et al. Proteasome inhibitor associated thrombotic microangiopathy. Am J Hematol. 2016;91(9):E348-E352. doi:10.1002/ajh.24447
3. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017;1(7):449-454. doi:10.1182/bloodadvances.2016003269
4. Chari A, Stewart AK, Russell SD, et al. Analysis of carfilzomib cardiovascular safety profile across relapsed and/or refractory multiple myeloma clinical trials. Blood Adv. 2018;2(13):1633-1644. doi:10.1182/bloodadvances.2017015545
5. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666. doi:10.1056/NEJMra1312353
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38. doi:10.1016/S1470-2045(15)00464-7
7. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197. doi:10.1016/S0140-6736(20)30734-0
8. Camilleri M, Cuadrado M, Phillips E, et al. Thrombotic microangiopathy in untreated myeloma patients receiving carfilzomib, cyclophosphamide and dexamethasone on the CARDAMON study. Br J Haematol. 2021;193(4):750-760. doi:10.1111/bjh.17377
9. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129(21):2857-2863. doi:10.1182/blood-2016-11-743104
10. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systemic review of published reports. Blood. 2015;125(4):616-618. doi:10.1182/blood-2014-11-611335
11. Saleem R, Reese JA, George JN. Drug-induced thrombotic-microangiopathy: an updated systematic review, 2014-2018. Am J Hematol. 2018;93(9):E241-E243. doi:10.1002/ajh.25208
12 Nguyen MN, Nayernama A, Jones SC, Kanapuru B, Gormley N, Waldron PE. Proteasome inhibitor-associated thrombotic microangiopathy: a review of cases reported to the FDA adverse event reporting system and published in the literature. Am J Hematol. 2020;95(9):E218-E222. doi:10.1002/ajh.25832
13. Haddadin M, Al-Sadawi M, Madanat S, et al. Late presentation of carfilzomib associated thrombotic microangiopathy. Am J Med Case Rep. 2019;7(10):240-243. doi:10.12691/ajmcr-7-10-5
14 Portuguese AJ, Gleber C, Passero Jr FC, Lipe B. A review of thrombotic microangiopathies in multiple myeloma. Leuk Res. 2019;85:106195. doi:10.1016/j.leukres.2019.106195
15. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-2181. doi:10.1056/NEJMoa1208981
16. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96-107. doi:10.1159/000492033
17. Lodhi A, Kumar A, Saqlain MU, Suneja M. Thrombotic microangiopathy associated with proteasome inhibitors. Clin Kidney J. 2015;8(5):632-636. doi:10.1093/ckj/sfv059
18. Sullivan MR, Danilov AV, Lansigan F, Dunbar NM. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J Clin Apher., 2015;30(5):308-310. doi:10.1002/jca.21371
19. Qaqish I, Schlam IM, Chakkera HA, Fonseca R, Adamski J. Carfilzomib: a cause of drug associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54(3):401-404. doi:10.1016/j.transci.2016.03.002
20. Chen Y, Ooi M, Lim SF, et al. Thrombotic microangiopathy during carfilzomib use: case series in Singapore. Blood Cancer J. 2016;6(7):e450. doi:10.1038/bcj.2016.62
21. Gosain R, Gill A, Fuqua J, et al. Gemcitabine and carfilzomib induced thrombotic microangiopathy: eculizumab as a life-saving treatment. Clin Case Rep. 2017;5(12):1926-1930. doi:10.1002/ccr3.1214
22. Portuguese AJ, Lipe B. Carfilzomib-induced aHUS responds to early eculizumab and may be associated with heterozygrous CFHR3-CFHR1 deletion. Blood Adv. 2018;2(23):3443-3446. doi:10.1182/bloodadvances.2018027532
23. Moliz C, Gutiérrez E, Cavero T, Redondo B, Praga M. Eculizumab as a treatment for atypical hemolytic syndrome secondary to carfilzomib. Nefrologia (Engl Ed). 2019;39(1):86-88. doi:10.1016/j.nefro.2018.02.005
24. Jeyaraman P, Borah P, Singh A, et al., Thrombotic microangiopathy after carfilzomib in a very young myeloma patient. Blood Cells Mol Dis. 2020;81:102400. doi:10.1016/j.bcmd.2019.102400
25. Bhutani D, Assal A, Mapara MY, Prinzing S, Lentzsch S. Case report: carfilzomib-induced thrombotic microangiopathy with complement activation treated successfully with eculizumab. Clin Lymphoma Myeloma Leuk. 2020;20(4):e155-e157. doi:10.1016/j.clml.2020.01.016
26. Jindal N, Jandial A, Jain A, et al. Carfilzomib-induced thrombotic microangiopathy: a case based review. Hematol Oncol Stem Cell Ther. 2020;S1658-3876(20)30118-7. doi:10.1016/j.hemonc.2020.07.001
27. Monteith BE, Venner CP, Reece DE, et al. Drug-induced thrombotic microangiopathy with concurrent proteasome inhibitor use in the treatment of multiple myeloma: a case series and review of the literature. Clin Lymphoma Myeloma Leuk. 2020;20(11):e791-e780. doi:10.1016/j.clml.2020.04.014
28. Rassner M, Baur R, Wäsch R, et al. Two cases of carfilzomib-induced thrombotic microangiopathy successfully treated with eculizumab in multiple myeloma. BMC Nephrol. 2021;22(1):32. doi:10.1186/s12882-020-02226-5
29. Kavanagh D, Goodship THJ. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15-20. doi:10.1182/asheducation-2011.1.15
30. Blasco M, Martínez-Roca A, Rodríguez-Lobato LG, et al. Complement as the enabler of carfilzomib-induced thrombotic microangiopathy. Br J Haematol. 2021;193(1):181-187. doi:10.1111/bjh.16796
‘Extremely exciting’ study results guide MM treatment options
CHICAGO – New results from a trial in patients with newly diagnosed multiple myeloma (MM) offer some answers to questions about which treatment route to choose.
Patients who received the triplet of lenalidomide, bortezomib, and dexamethasone (RVD) plus ASCT had a median PFS of 67.5 months, compared with 46.2 months for those who received RVD but did not have a transplant soon after.
However, patients were just as likely to be alive more than 6 years after treatment regardless of whether or not they underwent an immediate stem cell transplant.
In addition, treatment-related adverse events of grade 3 or above were higher in the group that received the transplant immediately after the triplet therapy.
The results were presented during a plenary session at the American Society of Clinical Oncology annual meeting and simultaneously published in the New England Journal of Medicine.
“Our findings confirm the PFS benefit of transplantation as first-line treatment for patients with myeloma and confirms stem cell transplant as a standard of care with certain triplet therapy,” said lead author Paul G. Richardson, MD, professor of medicine, Harvard Medical School, and clinical program leader and director of clinical research at the Jerome Lipper Multiple Myeloma Center at Dana Farber Cancer Institute, Boston.
Another finding from the trial was that the use of maintenance lenalidomide in both groups continuously until progression conferred substantial clinical benefit.
“We can also say that the use of lenalidomide maintenance therapy is also a standard of care,” he added.
Study details
In this trial, Dr. Richardson and colleagues randomly assigned 873 patients newly diagnosed with multiple myeloma to the RVD-alone group (n = 357) or the transplantation group (n = 365). All patients had received one cycle of RVD prior to randomization and then received two additional RVD cycles plus stem-cell mobilization followed by either five additional RVD cycles (the RVD-alone group) or high-dose melphalan plus ASCT followed by two additional RVD cycles (the transplantation group). Lenalidomide was administered to all patients until disease progression, unacceptable side effects, or both.
At a median follow-up of 76.0 months, the risk of disease progression or death was 53% higher among patients who received RVD alone versus the transplantation group (hazard ratio [HR], 1.53; P < .001). The median duration of PFS among patients with a high-risk cytogenetic profile was 55.5 vs. 17.1 months, favoring the transplantation group.
The percentage of patients who were alive without progression at 5 years was 58.4% vs 41.6%, respectively (HR, 1.66) and median duration of response was 56.4 vs 38.9 months, also favoring transplantation (HR, 1.45).
The estimated 5-year overall survival was similar between groups: 80.7% for transplantation and 79.2% for RVD alone (HR for death, 1.10; P > .99). For patients with a high-risk cytogenetic profile, 5-year survival was 63.4% versus 54.3%, respectively.
“This tells us that for patients who had kept transplant in reserve, they had the same overall survival as those who had had a transplant right away, despite there being such impressive initial disease control for the patients in whom transplant was used early,” Dr. Richardson said in a press release from his institution.
Patients who did not undergo immediate transplant received treatment when their disease progressed with newer and active therapies, such as monoclonal antibodies and/or next-generation novel agents, he noted. Only 28% of patients used the reserve option of a transplant.
“It demonstrates the extent to which patients now have options and that we have new data to guide them in balancing the pluses and minuses of each approach,” he added.
When looking at safety, the authors noted that the most common treatment-related adverse events of grade 3 or higher occurred in 279 patients (78.2%) in the RVD-alone group and 344 patients (94.2%) in the transplantation group. Of those patients, 60.5% and 89.9%, respectively, reported hematologic events of grade 3 or higher (P < .001). The 5-year cumulative incidence of invasive second primary cancers was similar in both cohorts (RVD-alone group, 4.9%; transplantation group, 6.5%).
However, while the risk of secondary cancers was similar between groups, Dr. Richardson noted that there was a higher incidence of acute myeloid leukemia and myelodysplastic syndromes in the transplant cohort.
“There was also a significant drop in quality of life across transplant procedures, but the good news is that it was recoverable rapidly,” he said. “What is also really important is that we have prospective, multicenter, national comparative data on toxicity. That’s very important for providing patients with a choice as they move forward with their treatment plan.”
He noted that treatment continues to evolve. “This study was designed in 2009, begun in 2010, and now there is mature data in 2022,” Dr. Richardson said. “This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies and novel next-generation therapies. The results from these studies are extremely exciting.
“Now more than ever, treatment for multiple myeloma can be adapted for each patient,” Dr. Richardson said. “Our study provides important information about the benefits of transplant in the era of highly effective novel therapies and continuous maintenance, as well as the potential risks, to help patients and their physicians decide what approach may be best for them. This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies, such as RVD combined with daratumumab.”
Lack of difference in overall survival
These new results further support an already established role of autologous hematopoietic stem cell transplantation in the management of patients with multiple myeloma, said Samer Al-Homsi, MD, clinical professor of medicine and director of the blood and marrow transplant program at Perlmutter Cancer Center, NYU Langone, New York, who was approached for comment.
“The treatment regimen is applicable to patients who are determined by an expert in transplantation to be fit to receive autologous hematopoietic transplantation,” he added. “Although this study, like many others, establishes hematopoietic stem cell transplantation as part of the standard of care in multiple myeloma, only a fraction of patients are actually offered this important modality of treatment for a variety of reasons, including provider bias,” he noted. “In fact, although improvement in supportive care has enhanced the safety of the procedure, many patients are denied this therapy.”
Dr. Al-Homsi noted that the lack of difference in overall survival might be due to the fact that some patients (28%) in the RVD-alone group did end up undergoing transplantation at the time of progression. “Also, longer follow-up might reveal a difference in overall survival,” he said.
The toxicities are manageable, and the incidence of secondary malignancies was not significantly different between cohorts. “However,” he emphasized, “lenalidomide has been associated in other studies with increased incidence of secondary malignancies and it must be noted that this study used extended administration of lenalidomide until progression.”
Support for this study was provided by grants to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute, the National Cancer Institute, R. J. Corman Multiple Myeloma Foundation, Celgene/Bristol Myers Squibb, and Millennium/Takeda Pharmaceutical. Dr. Richardson has reported relationships with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, Secura Bio, Takeda, and Bristol Myers Squibb. Dr. Al-Homsi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CHICAGO – New results from a trial in patients with newly diagnosed multiple myeloma (MM) offer some answers to questions about which treatment route to choose.
Patients who received the triplet of lenalidomide, bortezomib, and dexamethasone (RVD) plus ASCT had a median PFS of 67.5 months, compared with 46.2 months for those who received RVD but did not have a transplant soon after.
However, patients were just as likely to be alive more than 6 years after treatment regardless of whether or not they underwent an immediate stem cell transplant.
In addition, treatment-related adverse events of grade 3 or above were higher in the group that received the transplant immediately after the triplet therapy.
The results were presented during a plenary session at the American Society of Clinical Oncology annual meeting and simultaneously published in the New England Journal of Medicine.
“Our findings confirm the PFS benefit of transplantation as first-line treatment for patients with myeloma and confirms stem cell transplant as a standard of care with certain triplet therapy,” said lead author Paul G. Richardson, MD, professor of medicine, Harvard Medical School, and clinical program leader and director of clinical research at the Jerome Lipper Multiple Myeloma Center at Dana Farber Cancer Institute, Boston.
Another finding from the trial was that the use of maintenance lenalidomide in both groups continuously until progression conferred substantial clinical benefit.
“We can also say that the use of lenalidomide maintenance therapy is also a standard of care,” he added.
Study details
In this trial, Dr. Richardson and colleagues randomly assigned 873 patients newly diagnosed with multiple myeloma to the RVD-alone group (n = 357) or the transplantation group (n = 365). All patients had received one cycle of RVD prior to randomization and then received two additional RVD cycles plus stem-cell mobilization followed by either five additional RVD cycles (the RVD-alone group) or high-dose melphalan plus ASCT followed by two additional RVD cycles (the transplantation group). Lenalidomide was administered to all patients until disease progression, unacceptable side effects, or both.
At a median follow-up of 76.0 months, the risk of disease progression or death was 53% higher among patients who received RVD alone versus the transplantation group (hazard ratio [HR], 1.53; P < .001). The median duration of PFS among patients with a high-risk cytogenetic profile was 55.5 vs. 17.1 months, favoring the transplantation group.
The percentage of patients who were alive without progression at 5 years was 58.4% vs 41.6%, respectively (HR, 1.66) and median duration of response was 56.4 vs 38.9 months, also favoring transplantation (HR, 1.45).
The estimated 5-year overall survival was similar between groups: 80.7% for transplantation and 79.2% for RVD alone (HR for death, 1.10; P > .99). For patients with a high-risk cytogenetic profile, 5-year survival was 63.4% versus 54.3%, respectively.
“This tells us that for patients who had kept transplant in reserve, they had the same overall survival as those who had had a transplant right away, despite there being such impressive initial disease control for the patients in whom transplant was used early,” Dr. Richardson said in a press release from his institution.
Patients who did not undergo immediate transplant received treatment when their disease progressed with newer and active therapies, such as monoclonal antibodies and/or next-generation novel agents, he noted. Only 28% of patients used the reserve option of a transplant.
“It demonstrates the extent to which patients now have options and that we have new data to guide them in balancing the pluses and minuses of each approach,” he added.
When looking at safety, the authors noted that the most common treatment-related adverse events of grade 3 or higher occurred in 279 patients (78.2%) in the RVD-alone group and 344 patients (94.2%) in the transplantation group. Of those patients, 60.5% and 89.9%, respectively, reported hematologic events of grade 3 or higher (P < .001). The 5-year cumulative incidence of invasive second primary cancers was similar in both cohorts (RVD-alone group, 4.9%; transplantation group, 6.5%).
However, while the risk of secondary cancers was similar between groups, Dr. Richardson noted that there was a higher incidence of acute myeloid leukemia and myelodysplastic syndromes in the transplant cohort.
“There was also a significant drop in quality of life across transplant procedures, but the good news is that it was recoverable rapidly,” he said. “What is also really important is that we have prospective, multicenter, national comparative data on toxicity. That’s very important for providing patients with a choice as they move forward with their treatment plan.”
He noted that treatment continues to evolve. “This study was designed in 2009, begun in 2010, and now there is mature data in 2022,” Dr. Richardson said. “This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies and novel next-generation therapies. The results from these studies are extremely exciting.
“Now more than ever, treatment for multiple myeloma can be adapted for each patient,” Dr. Richardson said. “Our study provides important information about the benefits of transplant in the era of highly effective novel therapies and continuous maintenance, as well as the potential risks, to help patients and their physicians decide what approach may be best for them. This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies, such as RVD combined with daratumumab.”
Lack of difference in overall survival
These new results further support an already established role of autologous hematopoietic stem cell transplantation in the management of patients with multiple myeloma, said Samer Al-Homsi, MD, clinical professor of medicine and director of the blood and marrow transplant program at Perlmutter Cancer Center, NYU Langone, New York, who was approached for comment.
“The treatment regimen is applicable to patients who are determined by an expert in transplantation to be fit to receive autologous hematopoietic transplantation,” he added. “Although this study, like many others, establishes hematopoietic stem cell transplantation as part of the standard of care in multiple myeloma, only a fraction of patients are actually offered this important modality of treatment for a variety of reasons, including provider bias,” he noted. “In fact, although improvement in supportive care has enhanced the safety of the procedure, many patients are denied this therapy.”
Dr. Al-Homsi noted that the lack of difference in overall survival might be due to the fact that some patients (28%) in the RVD-alone group did end up undergoing transplantation at the time of progression. “Also, longer follow-up might reveal a difference in overall survival,” he said.
The toxicities are manageable, and the incidence of secondary malignancies was not significantly different between cohorts. “However,” he emphasized, “lenalidomide has been associated in other studies with increased incidence of secondary malignancies and it must be noted that this study used extended administration of lenalidomide until progression.”
Support for this study was provided by grants to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute, the National Cancer Institute, R. J. Corman Multiple Myeloma Foundation, Celgene/Bristol Myers Squibb, and Millennium/Takeda Pharmaceutical. Dr. Richardson has reported relationships with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, Secura Bio, Takeda, and Bristol Myers Squibb. Dr. Al-Homsi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
CHICAGO – New results from a trial in patients with newly diagnosed multiple myeloma (MM) offer some answers to questions about which treatment route to choose.
Patients who received the triplet of lenalidomide, bortezomib, and dexamethasone (RVD) plus ASCT had a median PFS of 67.5 months, compared with 46.2 months for those who received RVD but did not have a transplant soon after.
However, patients were just as likely to be alive more than 6 years after treatment regardless of whether or not they underwent an immediate stem cell transplant.
In addition, treatment-related adverse events of grade 3 or above were higher in the group that received the transplant immediately after the triplet therapy.
The results were presented during a plenary session at the American Society of Clinical Oncology annual meeting and simultaneously published in the New England Journal of Medicine.
“Our findings confirm the PFS benefit of transplantation as first-line treatment for patients with myeloma and confirms stem cell transplant as a standard of care with certain triplet therapy,” said lead author Paul G. Richardson, MD, professor of medicine, Harvard Medical School, and clinical program leader and director of clinical research at the Jerome Lipper Multiple Myeloma Center at Dana Farber Cancer Institute, Boston.
Another finding from the trial was that the use of maintenance lenalidomide in both groups continuously until progression conferred substantial clinical benefit.
“We can also say that the use of lenalidomide maintenance therapy is also a standard of care,” he added.
Study details
In this trial, Dr. Richardson and colleagues randomly assigned 873 patients newly diagnosed with multiple myeloma to the RVD-alone group (n = 357) or the transplantation group (n = 365). All patients had received one cycle of RVD prior to randomization and then received two additional RVD cycles plus stem-cell mobilization followed by either five additional RVD cycles (the RVD-alone group) or high-dose melphalan plus ASCT followed by two additional RVD cycles (the transplantation group). Lenalidomide was administered to all patients until disease progression, unacceptable side effects, or both.
At a median follow-up of 76.0 months, the risk of disease progression or death was 53% higher among patients who received RVD alone versus the transplantation group (hazard ratio [HR], 1.53; P < .001). The median duration of PFS among patients with a high-risk cytogenetic profile was 55.5 vs. 17.1 months, favoring the transplantation group.
The percentage of patients who were alive without progression at 5 years was 58.4% vs 41.6%, respectively (HR, 1.66) and median duration of response was 56.4 vs 38.9 months, also favoring transplantation (HR, 1.45).
The estimated 5-year overall survival was similar between groups: 80.7% for transplantation and 79.2% for RVD alone (HR for death, 1.10; P > .99). For patients with a high-risk cytogenetic profile, 5-year survival was 63.4% versus 54.3%, respectively.
“This tells us that for patients who had kept transplant in reserve, they had the same overall survival as those who had had a transplant right away, despite there being such impressive initial disease control for the patients in whom transplant was used early,” Dr. Richardson said in a press release from his institution.
Patients who did not undergo immediate transplant received treatment when their disease progressed with newer and active therapies, such as monoclonal antibodies and/or next-generation novel agents, he noted. Only 28% of patients used the reserve option of a transplant.
“It demonstrates the extent to which patients now have options and that we have new data to guide them in balancing the pluses and minuses of each approach,” he added.
When looking at safety, the authors noted that the most common treatment-related adverse events of grade 3 or higher occurred in 279 patients (78.2%) in the RVD-alone group and 344 patients (94.2%) in the transplantation group. Of those patients, 60.5% and 89.9%, respectively, reported hematologic events of grade 3 or higher (P < .001). The 5-year cumulative incidence of invasive second primary cancers was similar in both cohorts (RVD-alone group, 4.9%; transplantation group, 6.5%).
However, while the risk of secondary cancers was similar between groups, Dr. Richardson noted that there was a higher incidence of acute myeloid leukemia and myelodysplastic syndromes in the transplant cohort.
“There was also a significant drop in quality of life across transplant procedures, but the good news is that it was recoverable rapidly,” he said. “What is also really important is that we have prospective, multicenter, national comparative data on toxicity. That’s very important for providing patients with a choice as they move forward with their treatment plan.”
He noted that treatment continues to evolve. “This study was designed in 2009, begun in 2010, and now there is mature data in 2022,” Dr. Richardson said. “This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies and novel next-generation therapies. The results from these studies are extremely exciting.
“Now more than ever, treatment for multiple myeloma can be adapted for each patient,” Dr. Richardson said. “Our study provides important information about the benefits of transplant in the era of highly effective novel therapies and continuous maintenance, as well as the potential risks, to help patients and their physicians decide what approach may be best for them. This is particularly relevant as we have now further improved the induction treatment for younger patients with newly diagnosed myeloma using quadruplet regimens incorporating monoclonal antibodies, such as RVD combined with daratumumab.”
Lack of difference in overall survival
These new results further support an already established role of autologous hematopoietic stem cell transplantation in the management of patients with multiple myeloma, said Samer Al-Homsi, MD, clinical professor of medicine and director of the blood and marrow transplant program at Perlmutter Cancer Center, NYU Langone, New York, who was approached for comment.
“The treatment regimen is applicable to patients who are determined by an expert in transplantation to be fit to receive autologous hematopoietic transplantation,” he added. “Although this study, like many others, establishes hematopoietic stem cell transplantation as part of the standard of care in multiple myeloma, only a fraction of patients are actually offered this important modality of treatment for a variety of reasons, including provider bias,” he noted. “In fact, although improvement in supportive care has enhanced the safety of the procedure, many patients are denied this therapy.”
Dr. Al-Homsi noted that the lack of difference in overall survival might be due to the fact that some patients (28%) in the RVD-alone group did end up undergoing transplantation at the time of progression. “Also, longer follow-up might reveal a difference in overall survival,” he said.
The toxicities are manageable, and the incidence of secondary malignancies was not significantly different between cohorts. “However,” he emphasized, “lenalidomide has been associated in other studies with increased incidence of secondary malignancies and it must be noted that this study used extended administration of lenalidomide until progression.”
Support for this study was provided by grants to the Blood and Marrow Transplant Clinical Trials Network from the National Heart, Lung, and Blood Institute, the National Cancer Institute, R. J. Corman Multiple Myeloma Foundation, Celgene/Bristol Myers Squibb, and Millennium/Takeda Pharmaceutical. Dr. Richardson has reported relationships with Celgene, Janssen, Jazz Pharmaceuticals, Karyopharm Therapeutics, Oncopeptides, Sanofi, Secura Bio, Takeda, and Bristol Myers Squibb. Dr. Al-Homsi has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ASCO 2022
Novel COVID-19 vaccine could fill the void for patients with blood cancers
according to study results presented at the annual meeting of the American Association for Cancer Research.
The phase 1/2 trial included 54 patients with a B-cell deficiency (mean age, 63 years; 28% female): 4 had congenital B-cell deficiency and 50 had a blood cancer (lymphocytic leukemia or lymphoma). T-cell immune responses were observed in 86% of patients 28 days after vaccination with a single CoVac-1 dose. The potency of CoVac-1–induced T-cell responses exceeded those seen typically with B cell–deficient patient responses after mRNA vaccine treatment and were comparable with those seen among nonimmunocompromised COVID-19 patients.
In the majority of individuals, currently approved SARS-CoV-2 vaccines induce a robust immune response, however, their efficacy, has been shown to be decreased among individuals who are immunocompromised. Patients treated for hematologic cancers, in particular, receive treatment regimens that damage healthy immune cells, particularly B cells, said Juliane Walz, MD, the study’s senior author and professor of medicine at University Hospital Tübingen (Germany).
“In the clinic, we see many cancer patients who do not mount sufficient humoral immune responses after vaccination with available SARS-CoV-2 vaccines,” Dr. Walz said. “These patients are at a high risk for a severe course of COVID-19.”
B-cell deficiency, she stated, can be compensated for by enhancing T-cell responses against SARS-CoV-2, which can then combat infections in the absence of neutralizing antibodies.
In a prior study of CoVac-1 among 36 adults without immune deficiency, the vaccine elicited T-cell responses that were still robust 3 months post vaccination, and that included responses against omicron and other key SARS-CoV-2 variants.
While mRNA-based or adenoviral vector-based vaccines are limited to the spike protein and are thus prone to loss of activity because of viral mutations, CoVac-1–induced T-cell immunity is far more intense and broader, Dr. Walz said.
CoVac-1 is a peptide vaccine that is injected directly rather than being encoded via mRNA and targets different viral components. It would not be given, however, to healthy, immunocompetent adults because it is important for them to have both B-cell antibody and T-cell response.
The patients with B-cell deficiency recruited for the study were given a single dose of CoVac-1 and assessed for safety and immunogenicity until day 56. Prior vaccinations with an approved SARS-CoV-2 vaccine had failed to elicit a humoral response in 87% of the subjects.
“Our vaccine does not induce antibody responses,” Dr. Walz said. “However, it could be used to induce broad T-cell responses as a complementary or additive vaccine for elderly adults. In the elderly, antibody responses decline very, very fast after vaccination.”
Dr. Walz said that CoVac-1 could find application in various syndromes associated with congenital B-cell deficiencies, in autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, or diseases treated with rituximab or other B cell–depleting therapies (for example, ofatumumab, blinatumomab, or chimeric antigen receptor T cells), and in transplant patients.
A phase 3 study of CoVac-1 versus placebo is under discussion and would require about 300-500 subjects, Dr. Walz said.
“CoVac-1 is designed to induce broad and long-lasting SARS-CoV-2 T-cell immunity, even in individuals who have impaired ability to mount sufficient immunity from a currently approved vaccine, and thus protect these high-risk patients from a severe course of COVID-19,” Dr. Walz said.
“Having an option for these patients is just critical – so this is significant work,” said Ana Maria Lopez, MD, MPH, of the Sidney Kimmel Cancer Center–Jefferson Health, Philadelphia.
Limitations of this study included the small sample size with low racial and ethnic diversity, Dr. Walz stated.
Funding was provided by the Ministry of Science, Research and the Arts of the state of Baden-Württemberg; the Federal Ministry of Research and Education in Germany; the German Research Foundation under Germany’s Excellence Strategy; and the Clinical Cooperation Unit Translational Immunology at University Hospital Tübingen. Dr. Walz holds the CoVac-1 patent.
according to study results presented at the annual meeting of the American Association for Cancer Research.
The phase 1/2 trial included 54 patients with a B-cell deficiency (mean age, 63 years; 28% female): 4 had congenital B-cell deficiency and 50 had a blood cancer (lymphocytic leukemia or lymphoma). T-cell immune responses were observed in 86% of patients 28 days after vaccination with a single CoVac-1 dose. The potency of CoVac-1–induced T-cell responses exceeded those seen typically with B cell–deficient patient responses after mRNA vaccine treatment and were comparable with those seen among nonimmunocompromised COVID-19 patients.
In the majority of individuals, currently approved SARS-CoV-2 vaccines induce a robust immune response, however, their efficacy, has been shown to be decreased among individuals who are immunocompromised. Patients treated for hematologic cancers, in particular, receive treatment regimens that damage healthy immune cells, particularly B cells, said Juliane Walz, MD, the study’s senior author and professor of medicine at University Hospital Tübingen (Germany).
“In the clinic, we see many cancer patients who do not mount sufficient humoral immune responses after vaccination with available SARS-CoV-2 vaccines,” Dr. Walz said. “These patients are at a high risk for a severe course of COVID-19.”
B-cell deficiency, she stated, can be compensated for by enhancing T-cell responses against SARS-CoV-2, which can then combat infections in the absence of neutralizing antibodies.
In a prior study of CoVac-1 among 36 adults without immune deficiency, the vaccine elicited T-cell responses that were still robust 3 months post vaccination, and that included responses against omicron and other key SARS-CoV-2 variants.
While mRNA-based or adenoviral vector-based vaccines are limited to the spike protein and are thus prone to loss of activity because of viral mutations, CoVac-1–induced T-cell immunity is far more intense and broader, Dr. Walz said.
CoVac-1 is a peptide vaccine that is injected directly rather than being encoded via mRNA and targets different viral components. It would not be given, however, to healthy, immunocompetent adults because it is important for them to have both B-cell antibody and T-cell response.
The patients with B-cell deficiency recruited for the study were given a single dose of CoVac-1 and assessed for safety and immunogenicity until day 56. Prior vaccinations with an approved SARS-CoV-2 vaccine had failed to elicit a humoral response in 87% of the subjects.
“Our vaccine does not induce antibody responses,” Dr. Walz said. “However, it could be used to induce broad T-cell responses as a complementary or additive vaccine for elderly adults. In the elderly, antibody responses decline very, very fast after vaccination.”
Dr. Walz said that CoVac-1 could find application in various syndromes associated with congenital B-cell deficiencies, in autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, or diseases treated with rituximab or other B cell–depleting therapies (for example, ofatumumab, blinatumomab, or chimeric antigen receptor T cells), and in transplant patients.
A phase 3 study of CoVac-1 versus placebo is under discussion and would require about 300-500 subjects, Dr. Walz said.
“CoVac-1 is designed to induce broad and long-lasting SARS-CoV-2 T-cell immunity, even in individuals who have impaired ability to mount sufficient immunity from a currently approved vaccine, and thus protect these high-risk patients from a severe course of COVID-19,” Dr. Walz said.
“Having an option for these patients is just critical – so this is significant work,” said Ana Maria Lopez, MD, MPH, of the Sidney Kimmel Cancer Center–Jefferson Health, Philadelphia.
Limitations of this study included the small sample size with low racial and ethnic diversity, Dr. Walz stated.
Funding was provided by the Ministry of Science, Research and the Arts of the state of Baden-Württemberg; the Federal Ministry of Research and Education in Germany; the German Research Foundation under Germany’s Excellence Strategy; and the Clinical Cooperation Unit Translational Immunology at University Hospital Tübingen. Dr. Walz holds the CoVac-1 patent.
according to study results presented at the annual meeting of the American Association for Cancer Research.
The phase 1/2 trial included 54 patients with a B-cell deficiency (mean age, 63 years; 28% female): 4 had congenital B-cell deficiency and 50 had a blood cancer (lymphocytic leukemia or lymphoma). T-cell immune responses were observed in 86% of patients 28 days after vaccination with a single CoVac-1 dose. The potency of CoVac-1–induced T-cell responses exceeded those seen typically with B cell–deficient patient responses after mRNA vaccine treatment and were comparable with those seen among nonimmunocompromised COVID-19 patients.
In the majority of individuals, currently approved SARS-CoV-2 vaccines induce a robust immune response, however, their efficacy, has been shown to be decreased among individuals who are immunocompromised. Patients treated for hematologic cancers, in particular, receive treatment regimens that damage healthy immune cells, particularly B cells, said Juliane Walz, MD, the study’s senior author and professor of medicine at University Hospital Tübingen (Germany).
“In the clinic, we see many cancer patients who do not mount sufficient humoral immune responses after vaccination with available SARS-CoV-2 vaccines,” Dr. Walz said. “These patients are at a high risk for a severe course of COVID-19.”
B-cell deficiency, she stated, can be compensated for by enhancing T-cell responses against SARS-CoV-2, which can then combat infections in the absence of neutralizing antibodies.
In a prior study of CoVac-1 among 36 adults without immune deficiency, the vaccine elicited T-cell responses that were still robust 3 months post vaccination, and that included responses against omicron and other key SARS-CoV-2 variants.
While mRNA-based or adenoviral vector-based vaccines are limited to the spike protein and are thus prone to loss of activity because of viral mutations, CoVac-1–induced T-cell immunity is far more intense and broader, Dr. Walz said.
CoVac-1 is a peptide vaccine that is injected directly rather than being encoded via mRNA and targets different viral components. It would not be given, however, to healthy, immunocompetent adults because it is important for them to have both B-cell antibody and T-cell response.
The patients with B-cell deficiency recruited for the study were given a single dose of CoVac-1 and assessed for safety and immunogenicity until day 56. Prior vaccinations with an approved SARS-CoV-2 vaccine had failed to elicit a humoral response in 87% of the subjects.
“Our vaccine does not induce antibody responses,” Dr. Walz said. “However, it could be used to induce broad T-cell responses as a complementary or additive vaccine for elderly adults. In the elderly, antibody responses decline very, very fast after vaccination.”
Dr. Walz said that CoVac-1 could find application in various syndromes associated with congenital B-cell deficiencies, in autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, or diseases treated with rituximab or other B cell–depleting therapies (for example, ofatumumab, blinatumomab, or chimeric antigen receptor T cells), and in transplant patients.
A phase 3 study of CoVac-1 versus placebo is under discussion and would require about 300-500 subjects, Dr. Walz said.
“CoVac-1 is designed to induce broad and long-lasting SARS-CoV-2 T-cell immunity, even in individuals who have impaired ability to mount sufficient immunity from a currently approved vaccine, and thus protect these high-risk patients from a severe course of COVID-19,” Dr. Walz said.
“Having an option for these patients is just critical – so this is significant work,” said Ana Maria Lopez, MD, MPH, of the Sidney Kimmel Cancer Center–Jefferson Health, Philadelphia.
Limitations of this study included the small sample size with low racial and ethnic diversity, Dr. Walz stated.
Funding was provided by the Ministry of Science, Research and the Arts of the state of Baden-Württemberg; the Federal Ministry of Research and Education in Germany; the German Research Foundation under Germany’s Excellence Strategy; and the Clinical Cooperation Unit Translational Immunology at University Hospital Tübingen. Dr. Walz holds the CoVac-1 patent.
FROM AACR 2022
Neutropenia and Leukopenia After Cross Taper From Quetiapine to Divalproex for the Treatment of Borderline Personality Disorder
Valproic acid (VPA) and its derivative, divalproex (DVP) are prescribed for a variety of indications, commonly for seizure control in patients with epilepsy, mood stabilization in patients with bipolar disorder, and migraine prophylaxis. Gastrointestinal distress and sedation are among the most reported adverse effects (AEs) with DVP therapy.1 Although serious hepatic and hematologic AEs are rare, monitoring is still recommended. DVP can cause various hematologic dyscrasias, the most common being thrombocytopenia.1,2 Neutropenia and leukopenia have been reported in isolated cases, most occurring in pediatric patients or patients with epilepsy.3-14
Several case reports of DVP-related neutropenia (absolute neutrophil count [ANC] < 1.50 103/mcL) and leukopenia (white blood cell count [WBC] < 4.0 103/mcL) were reviewed during our literature search, some caused by DVP monotherapy; others were thought to be related to concomitant use of DVP and another drug.15-25 Quetiapine was the antipsychotic most commonly implicated in causing hematologic abnormalities when combined with DVP. We report a case of neutropenia and leukopenia that presented after a cross taper from quetiapine to DVP for the treatment of borderline personality disorder (BPD).
Although no medications have been approved by the US Food and Drug Administration (FDA) for the treatment of BPD, mood stabilizers, including DVP, have literature to support their use for the treatment of affective dysregulation and impulsive behavioral dyscontrol.26-28 A therapeutic range for DVP in the treatment of BPD has not been defined; therefore, for this case report, the generally accepted range of 50 to 100 µg/mL will be considered therapeutic.1
Case Presentation
A 34-year-old male patient presented to the mental health clinic pharmacist reporting that his current psychotropic medication regimen was not effective. His medical history included posttraumatic stress disorder (PTSD), opioid use disorder, alcohol use disorder, stimulant use disorder, cannabis use, BPD, hypertension, hyperlipidemia, prediabetes, gastroesophageal reflex disease, and a pulmonary nodule. On initial presentation, the patient was prescribed buprenorphine 24 mg/naloxone 6 mg, quetiapine 400 mg, duloxetine 120 mg, and prazosin 15 mg per day. At the time of pharmacy consultation, last reported alcohol or nonprescribed opioid use was about 6 months prior, and methamphetamine use about 1 month prior, with ongoing cannabis use. The patient had a history of participating in cognitive processing therapy, dialectical behavior therapy (DBT), and residential treatment for both PTSD and substance use. Additionally, he was actively participating in contingency management for stimulant use disorder and self-management and recovery training group.
The patient reported ongoing mood lability, hypervigilance, and oversedation with current psychotropic regimen. The prescriber of his medication for opioid use disorder also reported the patient experienced labile mood, impulsive behavior, and anger outbursts. In the setting of intolerability due to oversedation with quetiapine, cardiometabolic risk, and lack of clear indication for use, the patient and health care practitioner (HCP) agreed to taper quetiapine and initiate a trial of DVP for affective dysregulation and impulsive-behavioral dyscontrol. To prevent cholinergic rebound and insomnia with abrupt discontinuation of quetiapine, DVP and quetiapine were cross tapered. The following cross taper was prescribed: quetiapine 300 mg and DVP 500 mg per day for week 1; quetiapine 200 mg and DVP 500 mg per day for week 2; quetiapine 100 mg and DVP 1000 mg per day for week 3; quetiapine 50 mg and DVP 1000 mg per day for week 4; followed by DVP 1000 mg per day and discontinuation of quetiapine.
During a 4-week follow-up appointment, the patient reported appropriate completion of cross taper but stopped taking the DVP 3 days prior to the appointment due to self-reported lack of efficacy. For this reason, serum VPA level was not obtained. After discussion with his HCP, the patient restarted DVP 1000 mg per day without retitration with plans to get laboratory tests in 1 week. The next week, laboratory tests were notable for VPA level 28.74 (reference range, 50-100) µg/mL, low WBC 3.51 (reference range, 4.00-10.00) 103/mcL, platelets 169 (reference range, 150-420) 103/mcL, and low ANC 1.00 (reference range, 1.50-7.40) 103/mcL (Table). This raised clinical concern as the patient had no history of documented neutropenia or leukopenia, with most recent complete blood count (CBC) prior to DVP initiation 3 months earlier while prescribed quetiapine.
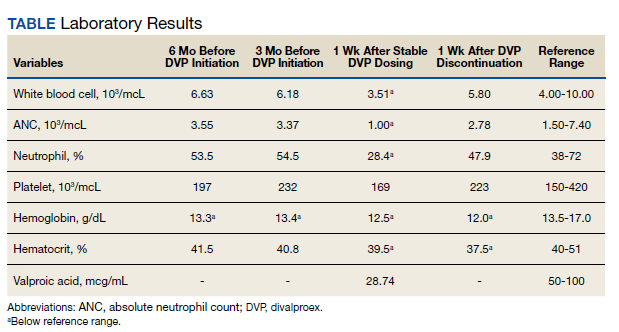
On further review, the HCP opted to cease administration of DVP and repeat CBC with differential in 1 week. Nine days later, laboratory tests were performed and compared with those collected the week before, revealing resolution of neutropenia and leukopenia. A score of 7 on the Naranjo Adverse Drug Reaction Probability Scale (NADRPS) was determined based on previous conclusive reports on the reaction (+1), appeared after suspected drug administration (+2), improved with drug discontinuation (+1), confirmed by objective evidence (+1), and no alternative causes could be found (+2).29 With a NADRPS score of 7, an AE of probable DVP-induced neutropenia was documented and medication was not resumed.
Discussion
Our case report describes isolated neutropenia and leukopenia that developed after a cross taper from quetiapine to DVP. Hematologic abnormalities resolved after discontinuation of DVP, suggesting a likely correlation. DVP has a well-established, dose-related prevalence of thrombocytopenia occurring in up to 27% of patients.1 Fewer case reports exist on neutropenia and leukopenia. DVP-induced neutropenia is thought to be a result of direct bone marrow suppression, whereas the more commonly occurring blood dyscrasia, thrombocytopenia, is thought to be caused by an antibody-mediated destruction of platelets.6
Management of DVP-induced thrombocytopenia is often dependent on the severity of the reaction. In mild-to-moderate cases, intervention may not be necessary as thrombocytopenia has been shown to resolve without adjustment to DVP therapy.1 In more severe or symptomatic cases, dose reduction or discontinuation of the offending agent is recommended, typically resulting in resolution shortly following pharmacologic intervention.
Guidance on the management of other drug-induced hematologic abnormalities, such as neutropenia and leukopenia are not as well established. A 2019 systematic review of idiosyncratic drug-induced neutropenia suggested that continuing the offending drug with strict monitoring could be considered in cases of mild neutropenia. In cases of moderate neutropenia, the author suggests temporary cessation of the drug and reinstatement once neutrophil count normalizes and definitive cessation of the drug in severe cases.30
In our case, continuing the offending agent with close monitoring was considered, similar to the well-established management of clozapine-induced neutropenia. However, due to the concern that the ANC was bordering moderate neutropenia in the absence of a therapeutic VPA level as well as a significant reduction in platelets, although not meeting criteria for thrombocytopenia, the decision was made to err on the side of caution and discontinue the most likely offending agent.
It is important to highlight that DVP was replacing quetiapine in the form of a cross taper. Quetiapine is structurally similar to clozapine. While clozapine has strict monitoring requirements related to neutropenia, blood dyscrasias with quetiapine therapy are rare. Quetiapine-induced hematologic abnormalities may be due to direct toxicity or to an immune-mediated mechanism, leading to bone marrow suppression.20 Case reports documenting blood dyscrasias with the combination of DVP and quetiapine were identified during literature review.15-19 Despite these case reports, we believe DVP was the primary offending agent in our case as the patient’s last dose of quetiapine was 2 weeks before obtaining the abnormal CBC. There was no history of blood dyscrasias with quetiapine monotherapy; however, the effect of the combination of DVP and quetiapine is unknown as no CBC was obtained during the cross-taper period.
Although there are no FDA-approved medications for the treatment of BPD, mood stabilizers, including DVP, have some research to support their use for the treatment of affective dysregulation and impulsive-behavioral dyscontrol.26-28 In our case, DVP was selected due to the evidence for use in BPD and ability to assess adherence with therapeutic monitoring. Although polypharmacy is a concern in patients with BPD, in our case we believed that the patient’s ongoing mood lability and impulsive behaviors warranted pharmacologic intervention. Additionally, DVP provided an advantage in its ability to quickly titrate to therapeutic dose when compared with lamotrigine and a lower risk of cognitive AEs when compared with topiramate.
Conclusions
To our knowledge, this case report demonstrates the first published case of neutropenia and leukopenia related to DVP therapy for the treatment of BPD. Routine CBC monitoring is recommended with DVP therapy, and our case highlights the importance of evaluating for not only thrombocytopenia, but also other blood dyscrasias during the titration phase even in the absence of a therapeutic VPA level. Further studies are warranted to determine incidence of DVP-related neutropenia and leukopenia and to evaluate the safety of continuing DVP in cases of mild-to-moderate neutropenia with close monitoring.
1. Depakote (valproic acid). Package insert. Abbott Laboratories; June 2000.
2. Conley EL, Coley KC, Pollock BG, Dapos SV, Maxwell R, Branch RA. Prevalence and risk of thrombocytopenia with valproic acid: experience at a psychiatric teaching hospital. Pharmacotherapy. 2001;21(11):1325-1330. doi:10.1592/phco.21.17.1325.34418
3. Jaeken J, van Goethem C, Casaer P, Devlieger H, Eggermont E, Pilet M. Neutropenia during sodium valproate therapy. Arch Dis Child. 1979;54(12):986-987. doi:10.1136/adc.54.12.986
4. Barr RD, Copeland SA, Stockwell MC, Morris N, Kelton JC. Valproic acid and immune thrombocytopenia. Arch Dis Child. 1982;57(9):681-684. doi:10.1136/adc.57.9.681
5. Symon DNK, Russell G. Sodium valproate and neutropenia (letter). Arch Dis Child. 1983;58:235. doi:10.1136/adc.58.3.235
6. Watts RG, Emanuel PD, Zuckerman KS, Howard TH. Valproic acid-induced cytopenias: evidence for a dose-related suppression of hematopoiesis. J Pediatr. 1990;117(3):495-499. doi:10.1016/s0022-3476(05)81105-9
7. Blackburn SC, Oliart AD, García-Rodríguez LA, Pérez Gutthann S. Antiepileptics and blood dyscrasias: a cohort study. Pharmacotherapy. 1998;18(6):1277-1283.
8. Acharya S, Bussel JB. Hematologic toxicity of sodium valproate. J Pediatr Hematol Oncol. 2000;22(1):62-65. doi:10.1097/00043426-200001000-00012
9. Vesta KS, Medina PJ. Valproic acid-induced neutropenia. Ann Pharmacother. 2003;37(6):819-821. doi:10.1345/aph.1C381
10. Kohli U, Gulati, S. Sodium valproate induced isolated neutropenia. Indian J Pediatr. 2006;73(9):844-844. doi:10.1007/BF02790401
11. Hsu HC, Tseng HK, Wang SC, Wang YY. Valproic acid-induced agranulocytosis. Int J Gerontol. 2009;3(2):137-139. doi:10.1016/S1873-9598(09)70036-5
12. Chakraborty S, Chakraborty J, Mandal S, Ghosal MK. A rare occurrence of isolated neutropenia with valproic acid: a case report. J Indian Med Assoc. 2011;109(5):345-346.
13. Stoner SC, Deal E, Lurk JT. Delayed-onset neutropenia with divalproex sodium. Ann Pharmacother. 2008;42(10):1507-1510. doi:10.1345/aph.1L239
14. Storch DD. Severe leukopenia with valproate. J Am Acad Child Adolesc Psychiatry. 2000;39(10):1208-1209. doi:10.1097/00004583-200010000-00003
15. Rahman A, Mican LM, Fischer C, Campbell AH. Evaluating the incidence of leukopenia and neutropenia with valproate, quetiapine, or the combination in children and adolescents. Ann Pharmacother. 2009;43:822-830. doi:10.1345/aph.1L617
16. Hung WC, Hsieh MH. Neutropenia associated with the comedication of quetiapine and valproate in 2 elderly patients. J Clin Psychopharmacol. 2012;32(3):416-417. doi:10.1097/JCP.0b013e3182549d2d
17. Park HJ, Kim JY. Incidence of neutropenia with valproate and quetiapine combination treatment in subjects with acquired brain injuries. Arch Phys Med Rehabil. 2016;97(2):183-188. doi:10.1016/j.apmr.2015.09.004
18. Estabrook KR, Pheister M. A case of quetiapine XR and divalproex-associated neutropenia followed by successful use of ziprasidone. J Clin Psychopharmacol. 2012;32(3):417-418. doi:10.1097/JCP.0b013e318253a071
19. Nair P, Lippmann S. Is leukopenia associated with divalproex and/or quetiapine? Psychosomatics. 2005;46(2):188-189. doi:10.1176/appi.psy.46.2.188
20. Cowan C, Oakley C. Leukopenia and neutropenia induced by quetiapine. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(1):292-294. doi:10.1016/j.pnpbp.2006.07.003
21. Fan KY, Chen WY, Huang MC. Quetiapine-associated leucopenia and thrombocytopenia: a case report. BMC Psychiatry. 2015;15:110. doi:10.1186/s12888-015-0495-9
22. Malik S, Lally J, Ajnakina O, et al. Sodium valproate and clozapine induced neutropenia: A case control study using register data. Schizophr Res. 2018;195:267-273. doi:10.1016/j.schres.2017.08.041
23. Pantelis C, Adesanya A. Increased risk of neutropaenia and agranulocytosis with sodium valproate used adjunctively with clozapine. Aust N Z J Psychiatry. 2001;35(4):544-545. doi:10.1046/j.1440-1614.2001.0911f.x
24. Madeb R, Hirschmann S, Kurs R, Turkie A, Modai I. Combined clozapine and valproic acid treatment-induced agranulocytosis. Eur Psychiatry. 2002;17(4):238-239. doi:10.1016/s0924-9338(02)00659-4
25. Dose M, Hellweg R, Yassouridis A, Theison M, Emrich HM. Combined treatment of schizophrenic psychoses with haloperidol and valproate. Pharmacopsychiatry. 1998;31(4):122-125. doi:10.1055/s-2007-979312
26. Ingenhoven T, Lafay P, Rinne T, Passchier J, Duivenvoorden H. Effectiveness of pharmacotherapy for severe personality disorders: meta-analyses of randomized controlled trials. J Clin Psychiatry. 2010;71:14. doi:10.4088/jcp.08r04526gre
27. Mercer D, Douglass AB, Links PS. Meta-analyses of mood stabilizers, antidepressants and antipsychotics in the treatment of borderline personality disorder: effectiveness for depression and anger symptoms. J Pers Disord. 2009;23(2):156-174. doi:10.1521/pedi.2009.23.2.156
28. Hollander E, Swann AC, Coccaro EF, Jiang P, Smith TB. Impact of trait impulsivity and state aggression on divalproex versus placebo response in borderline personality disorder. Am J Psychiatry. 2005;162(3):621-624. doi:10.1176/appi.ajp.162.3.621
29. Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245. doi:10.1038/clpt.1981.154
30. Andrès E, Villalba NL, Zulfiqar AA, Serraj K, Mourot-Cottet R, Gottenberg AJ. State of art of idiosyncratic drug-induced neutropenia or agranulocytosis, with a focus on biotherapies. J Clin Med. 2019;8(9):1351. doi:10.3390/jcm8091351
Valproic acid (VPA) and its derivative, divalproex (DVP) are prescribed for a variety of indications, commonly for seizure control in patients with epilepsy, mood stabilization in patients with bipolar disorder, and migraine prophylaxis. Gastrointestinal distress and sedation are among the most reported adverse effects (AEs) with DVP therapy.1 Although serious hepatic and hematologic AEs are rare, monitoring is still recommended. DVP can cause various hematologic dyscrasias, the most common being thrombocytopenia.1,2 Neutropenia and leukopenia have been reported in isolated cases, most occurring in pediatric patients or patients with epilepsy.3-14
Several case reports of DVP-related neutropenia (absolute neutrophil count [ANC] < 1.50 103/mcL) and leukopenia (white blood cell count [WBC] < 4.0 103/mcL) were reviewed during our literature search, some caused by DVP monotherapy; others were thought to be related to concomitant use of DVP and another drug.15-25 Quetiapine was the antipsychotic most commonly implicated in causing hematologic abnormalities when combined with DVP. We report a case of neutropenia and leukopenia that presented after a cross taper from quetiapine to DVP for the treatment of borderline personality disorder (BPD).
Although no medications have been approved by the US Food and Drug Administration (FDA) for the treatment of BPD, mood stabilizers, including DVP, have literature to support their use for the treatment of affective dysregulation and impulsive behavioral dyscontrol.26-28 A therapeutic range for DVP in the treatment of BPD has not been defined; therefore, for this case report, the generally accepted range of 50 to 100 µg/mL will be considered therapeutic.1
Case Presentation
A 34-year-old male patient presented to the mental health clinic pharmacist reporting that his current psychotropic medication regimen was not effective. His medical history included posttraumatic stress disorder (PTSD), opioid use disorder, alcohol use disorder, stimulant use disorder, cannabis use, BPD, hypertension, hyperlipidemia, prediabetes, gastroesophageal reflex disease, and a pulmonary nodule. On initial presentation, the patient was prescribed buprenorphine 24 mg/naloxone 6 mg, quetiapine 400 mg, duloxetine 120 mg, and prazosin 15 mg per day. At the time of pharmacy consultation, last reported alcohol or nonprescribed opioid use was about 6 months prior, and methamphetamine use about 1 month prior, with ongoing cannabis use. The patient had a history of participating in cognitive processing therapy, dialectical behavior therapy (DBT), and residential treatment for both PTSD and substance use. Additionally, he was actively participating in contingency management for stimulant use disorder and self-management and recovery training group.
The patient reported ongoing mood lability, hypervigilance, and oversedation with current psychotropic regimen. The prescriber of his medication for opioid use disorder also reported the patient experienced labile mood, impulsive behavior, and anger outbursts. In the setting of intolerability due to oversedation with quetiapine, cardiometabolic risk, and lack of clear indication for use, the patient and health care practitioner (HCP) agreed to taper quetiapine and initiate a trial of DVP for affective dysregulation and impulsive-behavioral dyscontrol. To prevent cholinergic rebound and insomnia with abrupt discontinuation of quetiapine, DVP and quetiapine were cross tapered. The following cross taper was prescribed: quetiapine 300 mg and DVP 500 mg per day for week 1; quetiapine 200 mg and DVP 500 mg per day for week 2; quetiapine 100 mg and DVP 1000 mg per day for week 3; quetiapine 50 mg and DVP 1000 mg per day for week 4; followed by DVP 1000 mg per day and discontinuation of quetiapine.
During a 4-week follow-up appointment, the patient reported appropriate completion of cross taper but stopped taking the DVP 3 days prior to the appointment due to self-reported lack of efficacy. For this reason, serum VPA level was not obtained. After discussion with his HCP, the patient restarted DVP 1000 mg per day without retitration with plans to get laboratory tests in 1 week. The next week, laboratory tests were notable for VPA level 28.74 (reference range, 50-100) µg/mL, low WBC 3.51 (reference range, 4.00-10.00) 103/mcL, platelets 169 (reference range, 150-420) 103/mcL, and low ANC 1.00 (reference range, 1.50-7.40) 103/mcL (Table). This raised clinical concern as the patient had no history of documented neutropenia or leukopenia, with most recent complete blood count (CBC) prior to DVP initiation 3 months earlier while prescribed quetiapine.
On further review, the HCP opted to cease administration of DVP and repeat CBC with differential in 1 week. Nine days later, laboratory tests were performed and compared with those collected the week before, revealing resolution of neutropenia and leukopenia. A score of 7 on the Naranjo Adverse Drug Reaction Probability Scale (NADRPS) was determined based on previous conclusive reports on the reaction (+1), appeared after suspected drug administration (+2), improved with drug discontinuation (+1), confirmed by objective evidence (+1), and no alternative causes could be found (+2).29 With a NADRPS score of 7, an AE of probable DVP-induced neutropenia was documented and medication was not resumed.
Discussion
Our case report describes isolated neutropenia and leukopenia that developed after a cross taper from quetiapine to DVP. Hematologic abnormalities resolved after discontinuation of DVP, suggesting a likely correlation. DVP has a well-established, dose-related prevalence of thrombocytopenia occurring in up to 27% of patients.1 Fewer case reports exist on neutropenia and leukopenia. DVP-induced neutropenia is thought to be a result of direct bone marrow suppression, whereas the more commonly occurring blood dyscrasia, thrombocytopenia, is thought to be caused by an antibody-mediated destruction of platelets.6
Management of DVP-induced thrombocytopenia is often dependent on the severity of the reaction. In mild-to-moderate cases, intervention may not be necessary as thrombocytopenia has been shown to resolve without adjustment to DVP therapy.1 In more severe or symptomatic cases, dose reduction or discontinuation of the offending agent is recommended, typically resulting in resolution shortly following pharmacologic intervention.
Guidance on the management of other drug-induced hematologic abnormalities, such as neutropenia and leukopenia are not as well established. A 2019 systematic review of idiosyncratic drug-induced neutropenia suggested that continuing the offending drug with strict monitoring could be considered in cases of mild neutropenia. In cases of moderate neutropenia, the author suggests temporary cessation of the drug and reinstatement once neutrophil count normalizes and definitive cessation of the drug in severe cases.30
In our case, continuing the offending agent with close monitoring was considered, similar to the well-established management of clozapine-induced neutropenia. However, due to the concern that the ANC was bordering moderate neutropenia in the absence of a therapeutic VPA level as well as a significant reduction in platelets, although not meeting criteria for thrombocytopenia, the decision was made to err on the side of caution and discontinue the most likely offending agent.
It is important to highlight that DVP was replacing quetiapine in the form of a cross taper. Quetiapine is structurally similar to clozapine. While clozapine has strict monitoring requirements related to neutropenia, blood dyscrasias with quetiapine therapy are rare. Quetiapine-induced hematologic abnormalities may be due to direct toxicity or to an immune-mediated mechanism, leading to bone marrow suppression.20 Case reports documenting blood dyscrasias with the combination of DVP and quetiapine were identified during literature review.15-19 Despite these case reports, we believe DVP was the primary offending agent in our case as the patient’s last dose of quetiapine was 2 weeks before obtaining the abnormal CBC. There was no history of blood dyscrasias with quetiapine monotherapy; however, the effect of the combination of DVP and quetiapine is unknown as no CBC was obtained during the cross-taper period.
Although there are no FDA-approved medications for the treatment of BPD, mood stabilizers, including DVP, have some research to support their use for the treatment of affective dysregulation and impulsive-behavioral dyscontrol.26-28 In our case, DVP was selected due to the evidence for use in BPD and ability to assess adherence with therapeutic monitoring. Although polypharmacy is a concern in patients with BPD, in our case we believed that the patient’s ongoing mood lability and impulsive behaviors warranted pharmacologic intervention. Additionally, DVP provided an advantage in its ability to quickly titrate to therapeutic dose when compared with lamotrigine and a lower risk of cognitive AEs when compared with topiramate.
Conclusions
To our knowledge, this case report demonstrates the first published case of neutropenia and leukopenia related to DVP therapy for the treatment of BPD. Routine CBC monitoring is recommended with DVP therapy, and our case highlights the importance of evaluating for not only thrombocytopenia, but also other blood dyscrasias during the titration phase even in the absence of a therapeutic VPA level. Further studies are warranted to determine incidence of DVP-related neutropenia and leukopenia and to evaluate the safety of continuing DVP in cases of mild-to-moderate neutropenia with close monitoring.
Valproic acid (VPA) and its derivative, divalproex (DVP) are prescribed for a variety of indications, commonly for seizure control in patients with epilepsy, mood stabilization in patients with bipolar disorder, and migraine prophylaxis. Gastrointestinal distress and sedation are among the most reported adverse effects (AEs) with DVP therapy.1 Although serious hepatic and hematologic AEs are rare, monitoring is still recommended. DVP can cause various hematologic dyscrasias, the most common being thrombocytopenia.1,2 Neutropenia and leukopenia have been reported in isolated cases, most occurring in pediatric patients or patients with epilepsy.3-14
Several case reports of DVP-related neutropenia (absolute neutrophil count [ANC] < 1.50 103/mcL) and leukopenia (white blood cell count [WBC] < 4.0 103/mcL) were reviewed during our literature search, some caused by DVP monotherapy; others were thought to be related to concomitant use of DVP and another drug.15-25 Quetiapine was the antipsychotic most commonly implicated in causing hematologic abnormalities when combined with DVP. We report a case of neutropenia and leukopenia that presented after a cross taper from quetiapine to DVP for the treatment of borderline personality disorder (BPD).
Although no medications have been approved by the US Food and Drug Administration (FDA) for the treatment of BPD, mood stabilizers, including DVP, have literature to support their use for the treatment of affective dysregulation and impulsive behavioral dyscontrol.26-28 A therapeutic range for DVP in the treatment of BPD has not been defined; therefore, for this case report, the generally accepted range of 50 to 100 µg/mL will be considered therapeutic.1
Case Presentation
A 34-year-old male patient presented to the mental health clinic pharmacist reporting that his current psychotropic medication regimen was not effective. His medical history included posttraumatic stress disorder (PTSD), opioid use disorder, alcohol use disorder, stimulant use disorder, cannabis use, BPD, hypertension, hyperlipidemia, prediabetes, gastroesophageal reflex disease, and a pulmonary nodule. On initial presentation, the patient was prescribed buprenorphine 24 mg/naloxone 6 mg, quetiapine 400 mg, duloxetine 120 mg, and prazosin 15 mg per day. At the time of pharmacy consultation, last reported alcohol or nonprescribed opioid use was about 6 months prior, and methamphetamine use about 1 month prior, with ongoing cannabis use. The patient had a history of participating in cognitive processing therapy, dialectical behavior therapy (DBT), and residential treatment for both PTSD and substance use. Additionally, he was actively participating in contingency management for stimulant use disorder and self-management and recovery training group.
The patient reported ongoing mood lability, hypervigilance, and oversedation with current psychotropic regimen. The prescriber of his medication for opioid use disorder also reported the patient experienced labile mood, impulsive behavior, and anger outbursts. In the setting of intolerability due to oversedation with quetiapine, cardiometabolic risk, and lack of clear indication for use, the patient and health care practitioner (HCP) agreed to taper quetiapine and initiate a trial of DVP for affective dysregulation and impulsive-behavioral dyscontrol. To prevent cholinergic rebound and insomnia with abrupt discontinuation of quetiapine, DVP and quetiapine were cross tapered. The following cross taper was prescribed: quetiapine 300 mg and DVP 500 mg per day for week 1; quetiapine 200 mg and DVP 500 mg per day for week 2; quetiapine 100 mg and DVP 1000 mg per day for week 3; quetiapine 50 mg and DVP 1000 mg per day for week 4; followed by DVP 1000 mg per day and discontinuation of quetiapine.
During a 4-week follow-up appointment, the patient reported appropriate completion of cross taper but stopped taking the DVP 3 days prior to the appointment due to self-reported lack of efficacy. For this reason, serum VPA level was not obtained. After discussion with his HCP, the patient restarted DVP 1000 mg per day without retitration with plans to get laboratory tests in 1 week. The next week, laboratory tests were notable for VPA level 28.74 (reference range, 50-100) µg/mL, low WBC 3.51 (reference range, 4.00-10.00) 103/mcL, platelets 169 (reference range, 150-420) 103/mcL, and low ANC 1.00 (reference range, 1.50-7.40) 103/mcL (Table). This raised clinical concern as the patient had no history of documented neutropenia or leukopenia, with most recent complete blood count (CBC) prior to DVP initiation 3 months earlier while prescribed quetiapine.
On further review, the HCP opted to cease administration of DVP and repeat CBC with differential in 1 week. Nine days later, laboratory tests were performed and compared with those collected the week before, revealing resolution of neutropenia and leukopenia. A score of 7 on the Naranjo Adverse Drug Reaction Probability Scale (NADRPS) was determined based on previous conclusive reports on the reaction (+1), appeared after suspected drug administration (+2), improved with drug discontinuation (+1), confirmed by objective evidence (+1), and no alternative causes could be found (+2).29 With a NADRPS score of 7, an AE of probable DVP-induced neutropenia was documented and medication was not resumed.
Discussion
Our case report describes isolated neutropenia and leukopenia that developed after a cross taper from quetiapine to DVP. Hematologic abnormalities resolved after discontinuation of DVP, suggesting a likely correlation. DVP has a well-established, dose-related prevalence of thrombocytopenia occurring in up to 27% of patients.1 Fewer case reports exist on neutropenia and leukopenia. DVP-induced neutropenia is thought to be a result of direct bone marrow suppression, whereas the more commonly occurring blood dyscrasia, thrombocytopenia, is thought to be caused by an antibody-mediated destruction of platelets.6
Management of DVP-induced thrombocytopenia is often dependent on the severity of the reaction. In mild-to-moderate cases, intervention may not be necessary as thrombocytopenia has been shown to resolve without adjustment to DVP therapy.1 In more severe or symptomatic cases, dose reduction or discontinuation of the offending agent is recommended, typically resulting in resolution shortly following pharmacologic intervention.
Guidance on the management of other drug-induced hematologic abnormalities, such as neutropenia and leukopenia are not as well established. A 2019 systematic review of idiosyncratic drug-induced neutropenia suggested that continuing the offending drug with strict monitoring could be considered in cases of mild neutropenia. In cases of moderate neutropenia, the author suggests temporary cessation of the drug and reinstatement once neutrophil count normalizes and definitive cessation of the drug in severe cases.30
In our case, continuing the offending agent with close monitoring was considered, similar to the well-established management of clozapine-induced neutropenia. However, due to the concern that the ANC was bordering moderate neutropenia in the absence of a therapeutic VPA level as well as a significant reduction in platelets, although not meeting criteria for thrombocytopenia, the decision was made to err on the side of caution and discontinue the most likely offending agent.
It is important to highlight that DVP was replacing quetiapine in the form of a cross taper. Quetiapine is structurally similar to clozapine. While clozapine has strict monitoring requirements related to neutropenia, blood dyscrasias with quetiapine therapy are rare. Quetiapine-induced hematologic abnormalities may be due to direct toxicity or to an immune-mediated mechanism, leading to bone marrow suppression.20 Case reports documenting blood dyscrasias with the combination of DVP and quetiapine were identified during literature review.15-19 Despite these case reports, we believe DVP was the primary offending agent in our case as the patient’s last dose of quetiapine was 2 weeks before obtaining the abnormal CBC. There was no history of blood dyscrasias with quetiapine monotherapy; however, the effect of the combination of DVP and quetiapine is unknown as no CBC was obtained during the cross-taper period.
Although there are no FDA-approved medications for the treatment of BPD, mood stabilizers, including DVP, have some research to support their use for the treatment of affective dysregulation and impulsive-behavioral dyscontrol.26-28 In our case, DVP was selected due to the evidence for use in BPD and ability to assess adherence with therapeutic monitoring. Although polypharmacy is a concern in patients with BPD, in our case we believed that the patient’s ongoing mood lability and impulsive behaviors warranted pharmacologic intervention. Additionally, DVP provided an advantage in its ability to quickly titrate to therapeutic dose when compared with lamotrigine and a lower risk of cognitive AEs when compared with topiramate.
Conclusions
To our knowledge, this case report demonstrates the first published case of neutropenia and leukopenia related to DVP therapy for the treatment of BPD. Routine CBC monitoring is recommended with DVP therapy, and our case highlights the importance of evaluating for not only thrombocytopenia, but also other blood dyscrasias during the titration phase even in the absence of a therapeutic VPA level. Further studies are warranted to determine incidence of DVP-related neutropenia and leukopenia and to evaluate the safety of continuing DVP in cases of mild-to-moderate neutropenia with close monitoring.
1. Depakote (valproic acid). Package insert. Abbott Laboratories; June 2000.
2. Conley EL, Coley KC, Pollock BG, Dapos SV, Maxwell R, Branch RA. Prevalence and risk of thrombocytopenia with valproic acid: experience at a psychiatric teaching hospital. Pharmacotherapy. 2001;21(11):1325-1330. doi:10.1592/phco.21.17.1325.34418
3. Jaeken J, van Goethem C, Casaer P, Devlieger H, Eggermont E, Pilet M. Neutropenia during sodium valproate therapy. Arch Dis Child. 1979;54(12):986-987. doi:10.1136/adc.54.12.986
4. Barr RD, Copeland SA, Stockwell MC, Morris N, Kelton JC. Valproic acid and immune thrombocytopenia. Arch Dis Child. 1982;57(9):681-684. doi:10.1136/adc.57.9.681
5. Symon DNK, Russell G. Sodium valproate and neutropenia (letter). Arch Dis Child. 1983;58:235. doi:10.1136/adc.58.3.235
6. Watts RG, Emanuel PD, Zuckerman KS, Howard TH. Valproic acid-induced cytopenias: evidence for a dose-related suppression of hematopoiesis. J Pediatr. 1990;117(3):495-499. doi:10.1016/s0022-3476(05)81105-9
7. Blackburn SC, Oliart AD, García-Rodríguez LA, Pérez Gutthann S. Antiepileptics and blood dyscrasias: a cohort study. Pharmacotherapy. 1998;18(6):1277-1283.
8. Acharya S, Bussel JB. Hematologic toxicity of sodium valproate. J Pediatr Hematol Oncol. 2000;22(1):62-65. doi:10.1097/00043426-200001000-00012
9. Vesta KS, Medina PJ. Valproic acid-induced neutropenia. Ann Pharmacother. 2003;37(6):819-821. doi:10.1345/aph.1C381
10. Kohli U, Gulati, S. Sodium valproate induced isolated neutropenia. Indian J Pediatr. 2006;73(9):844-844. doi:10.1007/BF02790401
11. Hsu HC, Tseng HK, Wang SC, Wang YY. Valproic acid-induced agranulocytosis. Int J Gerontol. 2009;3(2):137-139. doi:10.1016/S1873-9598(09)70036-5
12. Chakraborty S, Chakraborty J, Mandal S, Ghosal MK. A rare occurrence of isolated neutropenia with valproic acid: a case report. J Indian Med Assoc. 2011;109(5):345-346.
13. Stoner SC, Deal E, Lurk JT. Delayed-onset neutropenia with divalproex sodium. Ann Pharmacother. 2008;42(10):1507-1510. doi:10.1345/aph.1L239
14. Storch DD. Severe leukopenia with valproate. J Am Acad Child Adolesc Psychiatry. 2000;39(10):1208-1209. doi:10.1097/00004583-200010000-00003
15. Rahman A, Mican LM, Fischer C, Campbell AH. Evaluating the incidence of leukopenia and neutropenia with valproate, quetiapine, or the combination in children and adolescents. Ann Pharmacother. 2009;43:822-830. doi:10.1345/aph.1L617
16. Hung WC, Hsieh MH. Neutropenia associated with the comedication of quetiapine and valproate in 2 elderly patients. J Clin Psychopharmacol. 2012;32(3):416-417. doi:10.1097/JCP.0b013e3182549d2d
17. Park HJ, Kim JY. Incidence of neutropenia with valproate and quetiapine combination treatment in subjects with acquired brain injuries. Arch Phys Med Rehabil. 2016;97(2):183-188. doi:10.1016/j.apmr.2015.09.004
18. Estabrook KR, Pheister M. A case of quetiapine XR and divalproex-associated neutropenia followed by successful use of ziprasidone. J Clin Psychopharmacol. 2012;32(3):417-418. doi:10.1097/JCP.0b013e318253a071
19. Nair P, Lippmann S. Is leukopenia associated with divalproex and/or quetiapine? Psychosomatics. 2005;46(2):188-189. doi:10.1176/appi.psy.46.2.188
20. Cowan C, Oakley C. Leukopenia and neutropenia induced by quetiapine. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(1):292-294. doi:10.1016/j.pnpbp.2006.07.003
21. Fan KY, Chen WY, Huang MC. Quetiapine-associated leucopenia and thrombocytopenia: a case report. BMC Psychiatry. 2015;15:110. doi:10.1186/s12888-015-0495-9
22. Malik S, Lally J, Ajnakina O, et al. Sodium valproate and clozapine induced neutropenia: A case control study using register data. Schizophr Res. 2018;195:267-273. doi:10.1016/j.schres.2017.08.041
23. Pantelis C, Adesanya A. Increased risk of neutropaenia and agranulocytosis with sodium valproate used adjunctively with clozapine. Aust N Z J Psychiatry. 2001;35(4):544-545. doi:10.1046/j.1440-1614.2001.0911f.x
24. Madeb R, Hirschmann S, Kurs R, Turkie A, Modai I. Combined clozapine and valproic acid treatment-induced agranulocytosis. Eur Psychiatry. 2002;17(4):238-239. doi:10.1016/s0924-9338(02)00659-4
25. Dose M, Hellweg R, Yassouridis A, Theison M, Emrich HM. Combined treatment of schizophrenic psychoses with haloperidol and valproate. Pharmacopsychiatry. 1998;31(4):122-125. doi:10.1055/s-2007-979312
26. Ingenhoven T, Lafay P, Rinne T, Passchier J, Duivenvoorden H. Effectiveness of pharmacotherapy for severe personality disorders: meta-analyses of randomized controlled trials. J Clin Psychiatry. 2010;71:14. doi:10.4088/jcp.08r04526gre
27. Mercer D, Douglass AB, Links PS. Meta-analyses of mood stabilizers, antidepressants and antipsychotics in the treatment of borderline personality disorder: effectiveness for depression and anger symptoms. J Pers Disord. 2009;23(2):156-174. doi:10.1521/pedi.2009.23.2.156
28. Hollander E, Swann AC, Coccaro EF, Jiang P, Smith TB. Impact of trait impulsivity and state aggression on divalproex versus placebo response in borderline personality disorder. Am J Psychiatry. 2005;162(3):621-624. doi:10.1176/appi.ajp.162.3.621
29. Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245. doi:10.1038/clpt.1981.154
30. Andrès E, Villalba NL, Zulfiqar AA, Serraj K, Mourot-Cottet R, Gottenberg AJ. State of art of idiosyncratic drug-induced neutropenia or agranulocytosis, with a focus on biotherapies. J Clin Med. 2019;8(9):1351. doi:10.3390/jcm8091351
1. Depakote (valproic acid). Package insert. Abbott Laboratories; June 2000.
2. Conley EL, Coley KC, Pollock BG, Dapos SV, Maxwell R, Branch RA. Prevalence and risk of thrombocytopenia with valproic acid: experience at a psychiatric teaching hospital. Pharmacotherapy. 2001;21(11):1325-1330. doi:10.1592/phco.21.17.1325.34418
3. Jaeken J, van Goethem C, Casaer P, Devlieger H, Eggermont E, Pilet M. Neutropenia during sodium valproate therapy. Arch Dis Child. 1979;54(12):986-987. doi:10.1136/adc.54.12.986
4. Barr RD, Copeland SA, Stockwell MC, Morris N, Kelton JC. Valproic acid and immune thrombocytopenia. Arch Dis Child. 1982;57(9):681-684. doi:10.1136/adc.57.9.681
5. Symon DNK, Russell G. Sodium valproate and neutropenia (letter). Arch Dis Child. 1983;58:235. doi:10.1136/adc.58.3.235
6. Watts RG, Emanuel PD, Zuckerman KS, Howard TH. Valproic acid-induced cytopenias: evidence for a dose-related suppression of hematopoiesis. J Pediatr. 1990;117(3):495-499. doi:10.1016/s0022-3476(05)81105-9
7. Blackburn SC, Oliart AD, García-Rodríguez LA, Pérez Gutthann S. Antiepileptics and blood dyscrasias: a cohort study. Pharmacotherapy. 1998;18(6):1277-1283.
8. Acharya S, Bussel JB. Hematologic toxicity of sodium valproate. J Pediatr Hematol Oncol. 2000;22(1):62-65. doi:10.1097/00043426-200001000-00012
9. Vesta KS, Medina PJ. Valproic acid-induced neutropenia. Ann Pharmacother. 2003;37(6):819-821. doi:10.1345/aph.1C381
10. Kohli U, Gulati, S. Sodium valproate induced isolated neutropenia. Indian J Pediatr. 2006;73(9):844-844. doi:10.1007/BF02790401
11. Hsu HC, Tseng HK, Wang SC, Wang YY. Valproic acid-induced agranulocytosis. Int J Gerontol. 2009;3(2):137-139. doi:10.1016/S1873-9598(09)70036-5
12. Chakraborty S, Chakraborty J, Mandal S, Ghosal MK. A rare occurrence of isolated neutropenia with valproic acid: a case report. J Indian Med Assoc. 2011;109(5):345-346.
13. Stoner SC, Deal E, Lurk JT. Delayed-onset neutropenia with divalproex sodium. Ann Pharmacother. 2008;42(10):1507-1510. doi:10.1345/aph.1L239
14. Storch DD. Severe leukopenia with valproate. J Am Acad Child Adolesc Psychiatry. 2000;39(10):1208-1209. doi:10.1097/00004583-200010000-00003
15. Rahman A, Mican LM, Fischer C, Campbell AH. Evaluating the incidence of leukopenia and neutropenia with valproate, quetiapine, or the combination in children and adolescents. Ann Pharmacother. 2009;43:822-830. doi:10.1345/aph.1L617
16. Hung WC, Hsieh MH. Neutropenia associated with the comedication of quetiapine and valproate in 2 elderly patients. J Clin Psychopharmacol. 2012;32(3):416-417. doi:10.1097/JCP.0b013e3182549d2d
17. Park HJ, Kim JY. Incidence of neutropenia with valproate and quetiapine combination treatment in subjects with acquired brain injuries. Arch Phys Med Rehabil. 2016;97(2):183-188. doi:10.1016/j.apmr.2015.09.004
18. Estabrook KR, Pheister M. A case of quetiapine XR and divalproex-associated neutropenia followed by successful use of ziprasidone. J Clin Psychopharmacol. 2012;32(3):417-418. doi:10.1097/JCP.0b013e318253a071
19. Nair P, Lippmann S. Is leukopenia associated with divalproex and/or quetiapine? Psychosomatics. 2005;46(2):188-189. doi:10.1176/appi.psy.46.2.188
20. Cowan C, Oakley C. Leukopenia and neutropenia induced by quetiapine. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(1):292-294. doi:10.1016/j.pnpbp.2006.07.003
21. Fan KY, Chen WY, Huang MC. Quetiapine-associated leucopenia and thrombocytopenia: a case report. BMC Psychiatry. 2015;15:110. doi:10.1186/s12888-015-0495-9
22. Malik S, Lally J, Ajnakina O, et al. Sodium valproate and clozapine induced neutropenia: A case control study using register data. Schizophr Res. 2018;195:267-273. doi:10.1016/j.schres.2017.08.041
23. Pantelis C, Adesanya A. Increased risk of neutropaenia and agranulocytosis with sodium valproate used adjunctively with clozapine. Aust N Z J Psychiatry. 2001;35(4):544-545. doi:10.1046/j.1440-1614.2001.0911f.x
24. Madeb R, Hirschmann S, Kurs R, Turkie A, Modai I. Combined clozapine and valproic acid treatment-induced agranulocytosis. Eur Psychiatry. 2002;17(4):238-239. doi:10.1016/s0924-9338(02)00659-4
25. Dose M, Hellweg R, Yassouridis A, Theison M, Emrich HM. Combined treatment of schizophrenic psychoses with haloperidol and valproate. Pharmacopsychiatry. 1998;31(4):122-125. doi:10.1055/s-2007-979312
26. Ingenhoven T, Lafay P, Rinne T, Passchier J, Duivenvoorden H. Effectiveness of pharmacotherapy for severe personality disorders: meta-analyses of randomized controlled trials. J Clin Psychiatry. 2010;71:14. doi:10.4088/jcp.08r04526gre
27. Mercer D, Douglass AB, Links PS. Meta-analyses of mood stabilizers, antidepressants and antipsychotics in the treatment of borderline personality disorder: effectiveness for depression and anger symptoms. J Pers Disord. 2009;23(2):156-174. doi:10.1521/pedi.2009.23.2.156
28. Hollander E, Swann AC, Coccaro EF, Jiang P, Smith TB. Impact of trait impulsivity and state aggression on divalproex versus placebo response in borderline personality disorder. Am J Psychiatry. 2005;162(3):621-624. doi:10.1176/appi.ajp.162.3.621
29. Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245. doi:10.1038/clpt.1981.154
30. Andrès E, Villalba NL, Zulfiqar AA, Serraj K, Mourot-Cottet R, Gottenberg AJ. State of art of idiosyncratic drug-induced neutropenia or agranulocytosis, with a focus on biotherapies. J Clin Med. 2019;8(9):1351. doi:10.3390/jcm8091351
Some leukemias detectable up to 16 years before diagnosis?
Previous analyses showed that monoclonal B-cell lymphocytosis (MBL), a CLL precursor state, has been detected up to 6 years before CLL diagnosis, the investigators explained, noting that “[a]nother prognostically relevant immunogenetic feature of CLL concerns the stereotype of the B-cell receptor immunoglobulins (BcR IG).”
“Indeed, distinct stereotyped subsets can be defined by the expression of shared sequence motifs and are associated with particular presentation and outcomes,” P. Martijn Kolijn, PhD, a researcher in the department of immunology at Erasmus Medical Center, Rotterdam, the Netherlands, and colleagues wrote in a brief report published online in Blood. In an effort to “gain insight into the composition of the BcR IG repertoire during the early stages of CLL,” the investigators utilized next-generation sequencing to analyze 124 blood samples taken from healthy individuals up to 22 years before they received a diagnosis of CLL or small lymphocytic leukemia (SLL). An additional 118 matched control samples were also analyzed.
Study subjects were participants in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort.
“First, unsurprisingly, we observed a significant difference in the frequency of the dominant clonotype in CLL patients versus controls with a median frequency of 54.9%, compared to only 0.38% in controls,” they wrote.
Among 28 patients whose lymphocyte counts were measured at baseline, 10 showed evidence of lymphocytosis up to 8 years before CLL diagnosis.
This suggests undiagnosed instances of high-count MBL (cases with a cell count above 0.5x 109 cells/L, which can progress to CLL) or asymptomatic CLL, they explained.
“In contrast, next-generation sequencing results showed detectable skewing of the IGH gene repertoire in 21/28 patients up to 15 years before CLL diagnosis, often in the absence of elevated lymphocyte counts,” they wrote. “Remarkably, some patients with CLL requiring treatment and clinical transformation to an aggressive B-cell lymphoma displayed considerable skewing in the IGH gene repertoire even 16 years before CLL diagnosis.”
Patients with a prediagnostic IGHV-unmutated dominant clonotype had significantly shorter overall survival after CLL diagnosis than did those with an IGHV-mutated clonotype, they noted.
“Furthermore, at early timepoints (>10 years before diagnosis), patients with a high dominant clonotype frequency were more likely to be IGHV mutated, whereas closer to diagnosis this tendency was lost, indicating that the prediagnostic phase may be even longer than 16 years for [mutated] CLL patients,” they added.
The investigators also found that:
- Twenty-five patients carried stereotyped BcR IG up to 17 years prior to CLL diagnosis, and of these, 10 clonotypes were assigned to minor subsets and 15 to major CLL subsets. Among the latter, 14 of the 15 belonged to high-risk subsets, and most of those showed a trend for faster disease evolution.
- High frequency of the dominant clonotype was evident in samples obtained less than 6 years before diagnosis, whereas high-risk stereotyped clonotypes found longer before diagnosis (as early as 16 years) tended to have a lower dominant clonotype frequency (<20% of IGH gene repertoire)
- The stereotyped BcR IG matched the clonotype at diagnosis for both patients with diagnostic material.
- No stereotyped subsets were identified among the dominant clonotypes of the healthy controls.
“To our knowledge, the dynamics of the emergence of biclonality in an MBL patient and subsequent progression to CLL have never been captured in such a convincing manner,” they noted.
The findings “extend current knowledge on the evolution of the IGH repertoire prior to CLL diagnosis, highlighting that even high-risk CLL subtypes may display a prolonged indolent preclinical stage,” they added, speculating that “somatic genetic aberrations, (auto)stimulation, epigenetic and/or microenvironmental influences are required for the transformation into overt CLL.”
The investigators also noted that since the observed skewing in the IGH gene repertoire often occurs prior to B-cell lymphocytosis, they consider the findings “a novel extension to the characterization of MBL.”
“Further studies may prove invaluable in the clinical distinction between ‘progressing’ MBL versus ‘stable’ MBL. Notwithstanding the above, we emphasize that early detection is only warranted if it provides clear benefits to patient care,” they concluded.
In a related commentary, Gerald Marti, MD, PhD, of the National Heart, Lung, and Blood Institute, emphasized that the findings “represent the earliest detection of a clonotypic precursor cell for CLL.” .
They also raise new questions and point to new directions for research, Dr. Marti noted.
“Where do we go from here? CLL has a long evolutionary history in which early branching may start as an oligoclonal process (antigen stimulation) and include driver mutations,” he wrote. “A long-term analysis of the B-cell repertoire in familial CLL might shed light on this process. Further clarification of the mechanisms of age-related immune senescence is also of interest.”
The study authors and Dr. Marti reported having no competing financial interests.
Previous analyses showed that monoclonal B-cell lymphocytosis (MBL), a CLL precursor state, has been detected up to 6 years before CLL diagnosis, the investigators explained, noting that “[a]nother prognostically relevant immunogenetic feature of CLL concerns the stereotype of the B-cell receptor immunoglobulins (BcR IG).”
“Indeed, distinct stereotyped subsets can be defined by the expression of shared sequence motifs and are associated with particular presentation and outcomes,” P. Martijn Kolijn, PhD, a researcher in the department of immunology at Erasmus Medical Center, Rotterdam, the Netherlands, and colleagues wrote in a brief report published online in Blood. In an effort to “gain insight into the composition of the BcR IG repertoire during the early stages of CLL,” the investigators utilized next-generation sequencing to analyze 124 blood samples taken from healthy individuals up to 22 years before they received a diagnosis of CLL or small lymphocytic leukemia (SLL). An additional 118 matched control samples were also analyzed.
Study subjects were participants in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort.
“First, unsurprisingly, we observed a significant difference in the frequency of the dominant clonotype in CLL patients versus controls with a median frequency of 54.9%, compared to only 0.38% in controls,” they wrote.
Among 28 patients whose lymphocyte counts were measured at baseline, 10 showed evidence of lymphocytosis up to 8 years before CLL diagnosis.
This suggests undiagnosed instances of high-count MBL (cases with a cell count above 0.5x 109 cells/L, which can progress to CLL) or asymptomatic CLL, they explained.
“In contrast, next-generation sequencing results showed detectable skewing of the IGH gene repertoire in 21/28 patients up to 15 years before CLL diagnosis, often in the absence of elevated lymphocyte counts,” they wrote. “Remarkably, some patients with CLL requiring treatment and clinical transformation to an aggressive B-cell lymphoma displayed considerable skewing in the IGH gene repertoire even 16 years before CLL diagnosis.”
Patients with a prediagnostic IGHV-unmutated dominant clonotype had significantly shorter overall survival after CLL diagnosis than did those with an IGHV-mutated clonotype, they noted.
“Furthermore, at early timepoints (>10 years before diagnosis), patients with a high dominant clonotype frequency were more likely to be IGHV mutated, whereas closer to diagnosis this tendency was lost, indicating that the prediagnostic phase may be even longer than 16 years for [mutated] CLL patients,” they added.
The investigators also found that:
- Twenty-five patients carried stereotyped BcR IG up to 17 years prior to CLL diagnosis, and of these, 10 clonotypes were assigned to minor subsets and 15 to major CLL subsets. Among the latter, 14 of the 15 belonged to high-risk subsets, and most of those showed a trend for faster disease evolution.
- High frequency of the dominant clonotype was evident in samples obtained less than 6 years before diagnosis, whereas high-risk stereotyped clonotypes found longer before diagnosis (as early as 16 years) tended to have a lower dominant clonotype frequency (<20% of IGH gene repertoire)
- The stereotyped BcR IG matched the clonotype at diagnosis for both patients with diagnostic material.
- No stereotyped subsets were identified among the dominant clonotypes of the healthy controls.
“To our knowledge, the dynamics of the emergence of biclonality in an MBL patient and subsequent progression to CLL have never been captured in such a convincing manner,” they noted.
The findings “extend current knowledge on the evolution of the IGH repertoire prior to CLL diagnosis, highlighting that even high-risk CLL subtypes may display a prolonged indolent preclinical stage,” they added, speculating that “somatic genetic aberrations, (auto)stimulation, epigenetic and/or microenvironmental influences are required for the transformation into overt CLL.”
The investigators also noted that since the observed skewing in the IGH gene repertoire often occurs prior to B-cell lymphocytosis, they consider the findings “a novel extension to the characterization of MBL.”
“Further studies may prove invaluable in the clinical distinction between ‘progressing’ MBL versus ‘stable’ MBL. Notwithstanding the above, we emphasize that early detection is only warranted if it provides clear benefits to patient care,” they concluded.
In a related commentary, Gerald Marti, MD, PhD, of the National Heart, Lung, and Blood Institute, emphasized that the findings “represent the earliest detection of a clonotypic precursor cell for CLL.” .
They also raise new questions and point to new directions for research, Dr. Marti noted.
“Where do we go from here? CLL has a long evolutionary history in which early branching may start as an oligoclonal process (antigen stimulation) and include driver mutations,” he wrote. “A long-term analysis of the B-cell repertoire in familial CLL might shed light on this process. Further clarification of the mechanisms of age-related immune senescence is also of interest.”
The study authors and Dr. Marti reported having no competing financial interests.
Previous analyses showed that monoclonal B-cell lymphocytosis (MBL), a CLL precursor state, has been detected up to 6 years before CLL diagnosis, the investigators explained, noting that “[a]nother prognostically relevant immunogenetic feature of CLL concerns the stereotype of the B-cell receptor immunoglobulins (BcR IG).”
“Indeed, distinct stereotyped subsets can be defined by the expression of shared sequence motifs and are associated with particular presentation and outcomes,” P. Martijn Kolijn, PhD, a researcher in the department of immunology at Erasmus Medical Center, Rotterdam, the Netherlands, and colleagues wrote in a brief report published online in Blood. In an effort to “gain insight into the composition of the BcR IG repertoire during the early stages of CLL,” the investigators utilized next-generation sequencing to analyze 124 blood samples taken from healthy individuals up to 22 years before they received a diagnosis of CLL or small lymphocytic leukemia (SLL). An additional 118 matched control samples were also analyzed.
Study subjects were participants in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort.
“First, unsurprisingly, we observed a significant difference in the frequency of the dominant clonotype in CLL patients versus controls with a median frequency of 54.9%, compared to only 0.38% in controls,” they wrote.
Among 28 patients whose lymphocyte counts were measured at baseline, 10 showed evidence of lymphocytosis up to 8 years before CLL diagnosis.
This suggests undiagnosed instances of high-count MBL (cases with a cell count above 0.5x 109 cells/L, which can progress to CLL) or asymptomatic CLL, they explained.
“In contrast, next-generation sequencing results showed detectable skewing of the IGH gene repertoire in 21/28 patients up to 15 years before CLL diagnosis, often in the absence of elevated lymphocyte counts,” they wrote. “Remarkably, some patients with CLL requiring treatment and clinical transformation to an aggressive B-cell lymphoma displayed considerable skewing in the IGH gene repertoire even 16 years before CLL diagnosis.”
Patients with a prediagnostic IGHV-unmutated dominant clonotype had significantly shorter overall survival after CLL diagnosis than did those with an IGHV-mutated clonotype, they noted.
“Furthermore, at early timepoints (>10 years before diagnosis), patients with a high dominant clonotype frequency were more likely to be IGHV mutated, whereas closer to diagnosis this tendency was lost, indicating that the prediagnostic phase may be even longer than 16 years for [mutated] CLL patients,” they added.
The investigators also found that:
- Twenty-five patients carried stereotyped BcR IG up to 17 years prior to CLL diagnosis, and of these, 10 clonotypes were assigned to minor subsets and 15 to major CLL subsets. Among the latter, 14 of the 15 belonged to high-risk subsets, and most of those showed a trend for faster disease evolution.
- High frequency of the dominant clonotype was evident in samples obtained less than 6 years before diagnosis, whereas high-risk stereotyped clonotypes found longer before diagnosis (as early as 16 years) tended to have a lower dominant clonotype frequency (<20% of IGH gene repertoire)
- The stereotyped BcR IG matched the clonotype at diagnosis for both patients with diagnostic material.
- No stereotyped subsets were identified among the dominant clonotypes of the healthy controls.
“To our knowledge, the dynamics of the emergence of biclonality in an MBL patient and subsequent progression to CLL have never been captured in such a convincing manner,” they noted.
The findings “extend current knowledge on the evolution of the IGH repertoire prior to CLL diagnosis, highlighting that even high-risk CLL subtypes may display a prolonged indolent preclinical stage,” they added, speculating that “somatic genetic aberrations, (auto)stimulation, epigenetic and/or microenvironmental influences are required for the transformation into overt CLL.”
The investigators also noted that since the observed skewing in the IGH gene repertoire often occurs prior to B-cell lymphocytosis, they consider the findings “a novel extension to the characterization of MBL.”
“Further studies may prove invaluable in the clinical distinction between ‘progressing’ MBL versus ‘stable’ MBL. Notwithstanding the above, we emphasize that early detection is only warranted if it provides clear benefits to patient care,” they concluded.
In a related commentary, Gerald Marti, MD, PhD, of the National Heart, Lung, and Blood Institute, emphasized that the findings “represent the earliest detection of a clonotypic precursor cell for CLL.” .
They also raise new questions and point to new directions for research, Dr. Marti noted.
“Where do we go from here? CLL has a long evolutionary history in which early branching may start as an oligoclonal process (antigen stimulation) and include driver mutations,” he wrote. “A long-term analysis of the B-cell repertoire in familial CLL might shed light on this process. Further clarification of the mechanisms of age-related immune senescence is also of interest.”
The study authors and Dr. Marti reported having no competing financial interests.
FROM BLOOD
Cancer Data Trends 2022
Federal Practitioner, in collaboration with the Association of VA Hematology/Oncology (AVAHO), present the 2022 edition of Cancer Data Trends (click to view the digital edition). This special issue provides updates on some of the top cancers and related concerns affecting veterans through original infographics and visual storytelling.

In this issue:
- Exposure-Related Cancers
- Cancer in Women
- Genitourinary Cancers
- Gastrointestinal Cancers
- Telehealth in Oncology
- Precision Oncology
- Palliative and Hospice Care
- Alcohol and Cancer
- Lung Cancer
- Oropharyngeal Cancer
- Hematologic Cancers
Federal Practitioner and AVAHO would like to thank the following experts for their contributions to this issue:
Anita Aggarwal, DO, PhD; Sara Ahmed, PhD; Katherine Faricy-Anderson, MD; Apar Kishor Ganti, MD, MS; Solomon A Graf, MD; Kate Hendricks Thomas, PhD; Michael Kelley, MD; Mark Klein, MD, Gina McWhirter, MSN, MBA, RN; Bruce Montgomery, MD; Vida Almario Passero, MD, MBA; Thomas D Rodgers, MD; Vlad C Sandulache, MD, PhD; David H Wang, MD, PhD.
Federal Practitioner, in collaboration with the Association of VA Hematology/Oncology (AVAHO), present the 2022 edition of Cancer Data Trends (click to view the digital edition). This special issue provides updates on some of the top cancers and related concerns affecting veterans through original infographics and visual storytelling.
In this issue:
- Exposure-Related Cancers
- Cancer in Women
- Genitourinary Cancers
- Gastrointestinal Cancers
- Telehealth in Oncology
- Precision Oncology
- Palliative and Hospice Care
- Alcohol and Cancer
- Lung Cancer
- Oropharyngeal Cancer
- Hematologic Cancers
Federal Practitioner and AVAHO would like to thank the following experts for their contributions to this issue:
Anita Aggarwal, DO, PhD; Sara Ahmed, PhD; Katherine Faricy-Anderson, MD; Apar Kishor Ganti, MD, MS; Solomon A Graf, MD; Kate Hendricks Thomas, PhD; Michael Kelley, MD; Mark Klein, MD, Gina McWhirter, MSN, MBA, RN; Bruce Montgomery, MD; Vida Almario Passero, MD, MBA; Thomas D Rodgers, MD; Vlad C Sandulache, MD, PhD; David H Wang, MD, PhD.
Federal Practitioner, in collaboration with the Association of VA Hematology/Oncology (AVAHO), present the 2022 edition of Cancer Data Trends (click to view the digital edition). This special issue provides updates on some of the top cancers and related concerns affecting veterans through original infographics and visual storytelling.
In this issue:
- Exposure-Related Cancers
- Cancer in Women
- Genitourinary Cancers
- Gastrointestinal Cancers
- Telehealth in Oncology
- Precision Oncology
- Palliative and Hospice Care
- Alcohol and Cancer
- Lung Cancer
- Oropharyngeal Cancer
- Hematologic Cancers
Federal Practitioner and AVAHO would like to thank the following experts for their contributions to this issue:
Anita Aggarwal, DO, PhD; Sara Ahmed, PhD; Katherine Faricy-Anderson, MD; Apar Kishor Ganti, MD, MS; Solomon A Graf, MD; Kate Hendricks Thomas, PhD; Michael Kelley, MD; Mark Klein, MD, Gina McWhirter, MSN, MBA, RN; Bruce Montgomery, MD; Vida Almario Passero, MD, MBA; Thomas D Rodgers, MD; Vlad C Sandulache, MD, PhD; David H Wang, MD, PhD.

Hematocrit, White Blood Cells, and Thrombotic Events in the Veteran Population With Polycythemia Vera
Polycythemia vera (PV) is a rare myeloproliferative neoplasm affecting 44 to 57 individuals per 100,000 in the United States.1,2 It is characterized by somatic mutations in the hematopoietic stem cell, resulting in hyperproliferation of mature myeloid lineage cells.2 Sustained erythrocytosis is a hallmark of PV, although many patients also have leukocytosis and thrombocytosis.2,3 These patients have increased inherent thrombotic risk with arterial events reported to occur at rates of 7 to 21/1000 person-years and venous thrombotic events at 5 to 20/1000 person-years.4-7 Thrombotic and cardiovascular events are leading causes of morbidity and mortality, resulting in a reduced overall survival of patients with PV compared with the general population.3,8-10
Blood Cell Counts and Thrombotic Events in PV
Treatment strategies for patients with PV mainly aim to prevent or manage thrombotic and bleeding complications through normalization of blood counts.11 Hematocrit (Hct) control has been reported to be associated with reduced thrombotic risk in patients with PV. This was shown and popularized by the prospective, randomized Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial in which participants were randomized 1:1 to maintaining either a low (< 45%) or high (45%-50%) Hct for 5 years to examine the long-term effects of more- or less-intensive cytoreductive therapy.12 Patients in the low-Hct group were found to have a lower rate of death from cardiovascular events or major thrombosis (1.1/100 person-years in the low-Hct group vs 4.4 in the high-Hct group; hazard ratio [HR], 3.91; 95% confidence interval [CI], 1.45-10.53; P = .007). Likewise, cardiovascular events occurred at a lower rate in patients in the low-Hct group compared with the high-Hct group (4.4% vs 10.9% of patients, respectively; HR, 2.69; 95% CI, 1.19-6.12; P = .02).12
Leukocytosis has also been linked to elevated risk for vascular events as shown in several studies, including the real-world European Collaboration on Low-Dose Aspirin in PV (ECLAP) observational study and a post hoc subanalysis of the CYTO-PV study.13,14 In a multivariate, time-dependent analysis in ECLAP, patients with white blood cell (WBC) counts > 15 × 109/L had a significant increase in the risk of thrombosis compared with those who had lower WBC counts, with higher WBC count more strongly associated with arterial than venous thromboembolism.13 In CYTO-PV, a significant correlation between elevated WBC count (≥ 11 × 109/L vs reference level of < 7 × 109/L) and time-dependent risk of major thrombosis was shown (HR, 3.9; 95% CI, 1.24-12.3; P = .02).14 Likewise, WBC count ≥ 11 × 109/L was found to be a predictor of subsequent venous events in a separate single-center multivariate analysis of patients with PV.8
Although CYTO-PV remains one of the largest prospective landmark studies in PV demonstrating the impact of Hct control on thrombosis, it is worthwhile to note that the patients in the high-Hct group who received less frequent myelosuppressive therapy with hydroxyurea than the low-Hct group also had higher WBC counts.12,15 Work is needed to determine the relative effects of high Hct and high WBC counts on PV independent of each other.
The Veteran Population with PV
Two recently published retrospective analyses from Parasuraman and colleagues used data from the Veterans Health Administration (VHA), the largest integrated health care system in the US, with an aim to replicate findings from CYTO-PV in a real-world population.16,17 The 2 analyses focused independently on the effects of Hct control and WBC count on the risk of a thrombotic event in patients with PV.
In the first retrospective analysis, 213 patients with PV and no prior thrombosis were placed into groups based on whether Hct levels were consistently either < 45% or ≥ 45% throughout the study period.17 The mean follow-up time was 2.3 years, during which 44.1% of patients experienced a thrombotic event (Figure 1). Patients with Hct levels < 45% had a lower rate of thrombotic events compared to those with levels ≥ 45% (40.3% vs 54.2%, respectively; HR, 1.61; 95% CI, 1.03-2.51; P = .04). In a sensitivity analysis that included patients with pre-index thrombotic events (N = 342), similar results were noted (55.6% vs 76.9% between the < 45% and ≥ 45% groups, respectively; HR, 1.95; 95% CI, 1.46-2.61; P < .001).
In the second analysis, the authors investigated the relationship between WBC counts and thrombotic events.16 Evaluable patients (N = 1565) were grouped into 1 of 4 cohorts based on the last WBC measurement taken during the study period before a thrombotic event or through the end of follow-up: (1) WBC < 7.0 × 109/L, (2) 7.0 to 8.4 × 109/L, (3) 8.5 to < 11.0 × 109/L, or (4) ≥ 11.0 × 109/L. Mean follow-up time ranged from 3.6 to 4.5 years among WBC count cohorts, during which 24.9% of patients experienced a thrombotic event. Compared with the reference cohort (WBC < 7.0 × 109/L), a significant positive association between WBC counts and thrombotic event occurrence was observed among patients with WBC counts of 8.5 to < 11.0 × 109/L (HR, 1.47; 95% CI, 1.10-1.96; P < .01) and ≥ 11 × 109/L (HR, 1.87; 95% CI, 1.44-2.43; P < .001) (Figure 2).16 When including all patients in a sensitivity analysis regardless of whether they experienced thrombotic events before the index date (N = 1876), similar results were obtained (7.0-8.4 × 109/L group: HR, 1.22; 95% CI, 0.97-1.55; P = .0959; 8.5 - 11.0 × 109/L group: HR, 1.41; 95% CI, 1.10-1.81; P = .0062; ≥ 11.0 × 109/L group: HR, 1.53; 95% CI, 1.23-1.91; P < .001; compared with < 7.0 × 109/L reference group). Rates of phlebotomy and cytoreductive treatments were similar across groups.16
Some limitations to these studies are attributable to their retrospective design, reliance on health records, and the VHA population characteristics, which differ from the general population. For example, in this analysis, patients with PV in the VHA population had significantly increased risk of thrombotic events, even at a lower WBC count threshold (≥ 8.5 × 109/L) compared with those reported in CYTO-PV (≥ 11 × 109/L). Furthermore, approximately one-third of patients had elevated WBC levels, compared with 25.5% in the CYTO-PV study.14,16 This is most likely due to the unique nature of the VHA patient population, who are predominantly older adult men and generally have a higher comorbidity burden. A notable pre-index comorbidity burden was reported in the VHA population in the Hct analysis, even when compared to patients with PV in the general US population (Charlson Comorbidity Index score, 1.3 vs 0.8).6,17 Comorbid conditions such as hypertension, diabetes, and tobacco use, which are most common among the VHA population, are independently associated with higher risk of cardiovascular and thrombotic events.18,19 However, whether these higher levels of comorbidities affected the type of treatments they received was not elucidated, and the effectiveness of treatments to maintain target Hct levels was not addressed in the study.
Current PV Management and Future Implications
The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology in myeloproliferative neoplasms recommend maintaining Hct levels < 45% in patients with PV.11 Patients with high-risk disease (age ≥ 60 years and/or history of thrombosis) are monitored for new thrombosis or bleeding and are managed for their cardiovascular risk factors. In addition, they receive low-dose aspirin (81-100 mg/day), undergo phlebotomy to maintain an Hct < 45%, and are managed with pharmacologic cytoreductive therapy. Cytoreductive therapy primarily consists of hydroxyurea or peginterferon alfa-2a for younger patients. Ruxolitinib, a Janus kinase (JAK1)/JAK2 inhibitor, is now approved by the US Food and Drug Administration as second-line treatment for those with PV that is intolerant or unresponsive to hydroxyurea or peginterferon alfa-2a treatments.11,20 However, the role of cytoreductive therapy is not clear for patients with low-risk disease (age < 60 years and no history of thrombosis). These patients are managed for their cardiovascular risk factors, undergo phlebotomy to maintain an Hct < 45%, are maintained on low-dose aspirin (81-100 mg/day), and are monitored for indications for cytoreductive therapy, which include any new thrombosis or disease-related major bleeding, frequent or persistent need for phlebotomy with poor tolerance for the procedure, splenomegaly, thrombocytosis, leukocytosis, and disease-related symptoms (eg, aquagenic pruritus, night sweats, fatigue).
Even though the current guidelines recommend maintaining a target Hct of < 45% in patients with high-risk PV, the role of Hct as the main determinant of thrombotic risk in patients with PV is still debated.21 In JAK2V617F-positive essential thrombocythemia, Hct levels are usually normal but risk of thrombosis is nevertheless still significant.22 The risk of thrombosis is significantly lower in primary familial and congenital polycythemia and much lower in secondary erythrocytosis such as cyanotic heart disease, long-term native dwellers of high altitude, and those with high-oxygen–affinity hemoglobins.21,23 In secondary erythrocytosis from hypoxia or upregulated hypoxic pathway such as hypoxia inducible factor-2α (HIF-2α) mutation and Chuvash erythrocytosis, the risk of thrombosis is more associated with the upregulated HIF pathway and its downstream consequences, rather than the elevated Hct level.24
However, most current literature supports the association of increased risk of thrombosis with higher Hct and high WBC count in patients with PV. In addition, the underlying mechanism of thrombogenesis still remains elusive; it is likely a complex process that involves interactions among multiple components, including elevated blood counts arising from clonal hematopoiesis, JAK2V617F allele burden, and platelet and WBC activation and their interaction with endothelial cells and inflammatory cytokines.25
Nevertheless, Hct control and aspirin use are current standard of care for patients with PV to mitigate thrombotic risk, and the results from the 2 analyses by Parasuraman and colleagues, using real-world data from the VHA, support the current practice guidelines to maintain Hct < 45% in these patients. They also provide additional support for considering WBC counts when determining patient risk and treatment plans. Although treatment response criteria from the European LeukemiaNet include achieving normal WBC levels to decrease the risk of thrombosis, current NCCN guidelines do not include WBC counts as a component for establishing patient risk or provide a target WBC count to guide patient management.11,26,27 Updates to these practice guidelines may be warranted. In addition, further study is needed to understand the mechanism of thrombogenesis in PV and other myeloproliferative disorders in order to develop novel therapeutic targets and improve patient outcomes.
Acknowledgments
Writing assistance was provided by Tania Iqbal, PhD, an employee of ICON (North Wales, PA), and was funded by Incyte Corporation (Wilmington, DE).
1. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595-600. doi:10.3109/10428194.2013.813500
2. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. doi:10.1182/blood-2016-03-643544
3. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-1881. doi:10.1038/leu.2013.163
4. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224-2232. doi:10.1200/JCO.2005.07.062
5. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110(3):840-846. doi:10.1182/blood-2006-12-064287
6. Goyal RK, Davis KL, Cote I, Mounedji N, Kaye JA. Increased incidence of thromboembolic event rates in patients diagnosed with polycythemia vera: results from an observational cohort study. Blood (ASH Annual Meeting Abstracts). 2014;124:4840. doi:10.1182/blood.V124.21.4840.4840
7. Barbui T, Carobbio A, Rumi E, et al. In contemporary patients with polycythemia vera, rates of thrombosis and risk factors delineate a new clinical epidemiology. Blood. 2014;124(19):3021-3023. doi:10.1182/blood-2014-07-591610 8. Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017;7(12):662. doi:10.1038/s41408-017-0035-6
9. Stein BL, Moliterno AR, Tiu RV. Polycythemia vera disease burden: contributing factors, impact on quality of life, and emerging treatment options. Ann Hematol. 2014;93(12):1965-1976. doi:10.1007/s00277-014-2205-y
10. Hultcrantz M, Kristinsson SY, Andersson TM-L, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol. 2012;30(24):2995-3001. doi:10.1200/JCO.2012.42.1925
11. National Comprehensive Cancer Network. NCCN clinical practice guidelines in myeloproliferative neoplasms (Version 1.2020). Accessed March 3, 2022. https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf
12. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. doi:10.1056/NEJMoa1208500
13. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109(6):2446-2452. doi:10.1182/blood-2006-08-042515
14. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126(4):560-561. doi:10.1182/blood-2015-04-638593
15. Prchal JT, Gordeuk VR. Treatment target in polycythemia vera. N Engl J Med. 2013;368(16):1555-1556. doi:10.1056/NEJMc1301262
16. Parasuraman S, Yu J, Paranagama D, et al. Elevated white blood cell levels and thrombotic events in patients with polycythemia vera: a real-world analysis of Veterans Health Administration data. Clin Lymphoma Myeloma Leuk. 2020;20(2):63-69. doi:10.1016/j.clml.2019.11.010
17. Parasuraman S, Yu J, Paranagama D, et al. Hematocrit levels and thrombotic events in patients with polycythemia vera: an analysis of Veterans Health Administration data. Ann Hematol. 2019;98(11):2533-2539. doi:10.1007/s00277-019-03793-w
18. WHO CVD Risk Chart Working Group. World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health. 2019;7(10):e1332-e1345. doi:10.1016/S2214-109X(19)30318-3.
19. D’Agostino RB Sr, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117(6):743-753. doi:10.1161/CIRCULATIONAHA.107.699579
20. Jakafi. Package insert. Incyte Corporation; 2020.
21. Gordeuk VR, Key NS, Prchal JT. Re-evaluation of hematocrit as a determinant of thrombotic risk in erythrocytosis. Haematologica. 2019;104(4):653-658. doi:10.3324/haematol.2018.210732
22. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859. doi:10.1182/blood-2011-02-339002
23. Perloff JK, Marelli AJ, Miner PD. Risk of stroke in adults with cyanotic congenital heart disease. Circulation. 1993;87(6):1954-1959. doi:10.1161/01.cir.87.6.1954
24. Gordeuk VR, Miasnikova GY, Sergueeva AI, et al. Thrombotic risk in congenital erythrocytosis due to up-regulated hypoxia sensing is not associated with elevated hematocrit. Haematologica. 2020;105(3):e87-e90. doi:10.3324/haematol.2019.216267
25. Kroll MH, Michaelis LC, Verstovsek S. Mechanisms of thrombogenesis in polycythemia vera. Blood Rev. 2015;29(4):215-221. doi:10.1016/j.blre.2014.12.002
26. Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057-1069. doi:10.1038/s41375-018-0077-1
27. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778-4781. doi:10.1182/blood-2013-01-478891
Polycythemia vera (PV) is a rare myeloproliferative neoplasm affecting 44 to 57 individuals per 100,000 in the United States.1,2 It is characterized by somatic mutations in the hematopoietic stem cell, resulting in hyperproliferation of mature myeloid lineage cells.2 Sustained erythrocytosis is a hallmark of PV, although many patients also have leukocytosis and thrombocytosis.2,3 These patients have increased inherent thrombotic risk with arterial events reported to occur at rates of 7 to 21/1000 person-years and venous thrombotic events at 5 to 20/1000 person-years.4-7 Thrombotic and cardiovascular events are leading causes of morbidity and mortality, resulting in a reduced overall survival of patients with PV compared with the general population.3,8-10
Blood Cell Counts and Thrombotic Events in PV
Treatment strategies for patients with PV mainly aim to prevent or manage thrombotic and bleeding complications through normalization of blood counts.11 Hematocrit (Hct) control has been reported to be associated with reduced thrombotic risk in patients with PV. This was shown and popularized by the prospective, randomized Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial in which participants were randomized 1:1 to maintaining either a low (< 45%) or high (45%-50%) Hct for 5 years to examine the long-term effects of more- or less-intensive cytoreductive therapy.12 Patients in the low-Hct group were found to have a lower rate of death from cardiovascular events or major thrombosis (1.1/100 person-years in the low-Hct group vs 4.4 in the high-Hct group; hazard ratio [HR], 3.91; 95% confidence interval [CI], 1.45-10.53; P = .007). Likewise, cardiovascular events occurred at a lower rate in patients in the low-Hct group compared with the high-Hct group (4.4% vs 10.9% of patients, respectively; HR, 2.69; 95% CI, 1.19-6.12; P = .02).12
Leukocytosis has also been linked to elevated risk for vascular events as shown in several studies, including the real-world European Collaboration on Low-Dose Aspirin in PV (ECLAP) observational study and a post hoc subanalysis of the CYTO-PV study.13,14 In a multivariate, time-dependent analysis in ECLAP, patients with white blood cell (WBC) counts > 15 × 109/L had a significant increase in the risk of thrombosis compared with those who had lower WBC counts, with higher WBC count more strongly associated with arterial than venous thromboembolism.13 In CYTO-PV, a significant correlation between elevated WBC count (≥ 11 × 109/L vs reference level of < 7 × 109/L) and time-dependent risk of major thrombosis was shown (HR, 3.9; 95% CI, 1.24-12.3; P = .02).14 Likewise, WBC count ≥ 11 × 109/L was found to be a predictor of subsequent venous events in a separate single-center multivariate analysis of patients with PV.8
Although CYTO-PV remains one of the largest prospective landmark studies in PV demonstrating the impact of Hct control on thrombosis, it is worthwhile to note that the patients in the high-Hct group who received less frequent myelosuppressive therapy with hydroxyurea than the low-Hct group also had higher WBC counts.12,15 Work is needed to determine the relative effects of high Hct and high WBC counts on PV independent of each other.
The Veteran Population with PV
Two recently published retrospective analyses from Parasuraman and colleagues used data from the Veterans Health Administration (VHA), the largest integrated health care system in the US, with an aim to replicate findings from CYTO-PV in a real-world population.16,17 The 2 analyses focused independently on the effects of Hct control and WBC count on the risk of a thrombotic event in patients with PV.
In the first retrospective analysis, 213 patients with PV and no prior thrombosis were placed into groups based on whether Hct levels were consistently either < 45% or ≥ 45% throughout the study period.17 The mean follow-up time was 2.3 years, during which 44.1% of patients experienced a thrombotic event (Figure 1). Patients with Hct levels < 45% had a lower rate of thrombotic events compared to those with levels ≥ 45% (40.3% vs 54.2%, respectively; HR, 1.61; 95% CI, 1.03-2.51; P = .04). In a sensitivity analysis that included patients with pre-index thrombotic events (N = 342), similar results were noted (55.6% vs 76.9% between the < 45% and ≥ 45% groups, respectively; HR, 1.95; 95% CI, 1.46-2.61; P < .001).
In the second analysis, the authors investigated the relationship between WBC counts and thrombotic events.16 Evaluable patients (N = 1565) were grouped into 1 of 4 cohorts based on the last WBC measurement taken during the study period before a thrombotic event or through the end of follow-up: (1) WBC < 7.0 × 109/L, (2) 7.0 to 8.4 × 109/L, (3) 8.5 to < 11.0 × 109/L, or (4) ≥ 11.0 × 109/L. Mean follow-up time ranged from 3.6 to 4.5 years among WBC count cohorts, during which 24.9% of patients experienced a thrombotic event. Compared with the reference cohort (WBC < 7.0 × 109/L), a significant positive association between WBC counts and thrombotic event occurrence was observed among patients with WBC counts of 8.5 to < 11.0 × 109/L (HR, 1.47; 95% CI, 1.10-1.96; P < .01) and ≥ 11 × 109/L (HR, 1.87; 95% CI, 1.44-2.43; P < .001) (Figure 2).16 When including all patients in a sensitivity analysis regardless of whether they experienced thrombotic events before the index date (N = 1876), similar results were obtained (7.0-8.4 × 109/L group: HR, 1.22; 95% CI, 0.97-1.55; P = .0959; 8.5 - 11.0 × 109/L group: HR, 1.41; 95% CI, 1.10-1.81; P = .0062; ≥ 11.0 × 109/L group: HR, 1.53; 95% CI, 1.23-1.91; P < .001; compared with < 7.0 × 109/L reference group). Rates of phlebotomy and cytoreductive treatments were similar across groups.16
Some limitations to these studies are attributable to their retrospective design, reliance on health records, and the VHA population characteristics, which differ from the general population. For example, in this analysis, patients with PV in the VHA population had significantly increased risk of thrombotic events, even at a lower WBC count threshold (≥ 8.5 × 109/L) compared with those reported in CYTO-PV (≥ 11 × 109/L). Furthermore, approximately one-third of patients had elevated WBC levels, compared with 25.5% in the CYTO-PV study.14,16 This is most likely due to the unique nature of the VHA patient population, who are predominantly older adult men and generally have a higher comorbidity burden. A notable pre-index comorbidity burden was reported in the VHA population in the Hct analysis, even when compared to patients with PV in the general US population (Charlson Comorbidity Index score, 1.3 vs 0.8).6,17 Comorbid conditions such as hypertension, diabetes, and tobacco use, which are most common among the VHA population, are independently associated with higher risk of cardiovascular and thrombotic events.18,19 However, whether these higher levels of comorbidities affected the type of treatments they received was not elucidated, and the effectiveness of treatments to maintain target Hct levels was not addressed in the study.
Current PV Management and Future Implications
The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology in myeloproliferative neoplasms recommend maintaining Hct levels < 45% in patients with PV.11 Patients with high-risk disease (age ≥ 60 years and/or history of thrombosis) are monitored for new thrombosis or bleeding and are managed for their cardiovascular risk factors. In addition, they receive low-dose aspirin (81-100 mg/day), undergo phlebotomy to maintain an Hct < 45%, and are managed with pharmacologic cytoreductive therapy. Cytoreductive therapy primarily consists of hydroxyurea or peginterferon alfa-2a for younger patients. Ruxolitinib, a Janus kinase (JAK1)/JAK2 inhibitor, is now approved by the US Food and Drug Administration as second-line treatment for those with PV that is intolerant or unresponsive to hydroxyurea or peginterferon alfa-2a treatments.11,20 However, the role of cytoreductive therapy is not clear for patients with low-risk disease (age < 60 years and no history of thrombosis). These patients are managed for their cardiovascular risk factors, undergo phlebotomy to maintain an Hct < 45%, are maintained on low-dose aspirin (81-100 mg/day), and are monitored for indications for cytoreductive therapy, which include any new thrombosis or disease-related major bleeding, frequent or persistent need for phlebotomy with poor tolerance for the procedure, splenomegaly, thrombocytosis, leukocytosis, and disease-related symptoms (eg, aquagenic pruritus, night sweats, fatigue).
Even though the current guidelines recommend maintaining a target Hct of < 45% in patients with high-risk PV, the role of Hct as the main determinant of thrombotic risk in patients with PV is still debated.21 In JAK2V617F-positive essential thrombocythemia, Hct levels are usually normal but risk of thrombosis is nevertheless still significant.22 The risk of thrombosis is significantly lower in primary familial and congenital polycythemia and much lower in secondary erythrocytosis such as cyanotic heart disease, long-term native dwellers of high altitude, and those with high-oxygen–affinity hemoglobins.21,23 In secondary erythrocytosis from hypoxia or upregulated hypoxic pathway such as hypoxia inducible factor-2α (HIF-2α) mutation and Chuvash erythrocytosis, the risk of thrombosis is more associated with the upregulated HIF pathway and its downstream consequences, rather than the elevated Hct level.24
However, most current literature supports the association of increased risk of thrombosis with higher Hct and high WBC count in patients with PV. In addition, the underlying mechanism of thrombogenesis still remains elusive; it is likely a complex process that involves interactions among multiple components, including elevated blood counts arising from clonal hematopoiesis, JAK2V617F allele burden, and platelet and WBC activation and their interaction with endothelial cells and inflammatory cytokines.25
Nevertheless, Hct control and aspirin use are current standard of care for patients with PV to mitigate thrombotic risk, and the results from the 2 analyses by Parasuraman and colleagues, using real-world data from the VHA, support the current practice guidelines to maintain Hct < 45% in these patients. They also provide additional support for considering WBC counts when determining patient risk and treatment plans. Although treatment response criteria from the European LeukemiaNet include achieving normal WBC levels to decrease the risk of thrombosis, current NCCN guidelines do not include WBC counts as a component for establishing patient risk or provide a target WBC count to guide patient management.11,26,27 Updates to these practice guidelines may be warranted. In addition, further study is needed to understand the mechanism of thrombogenesis in PV and other myeloproliferative disorders in order to develop novel therapeutic targets and improve patient outcomes.
Acknowledgments
Writing assistance was provided by Tania Iqbal, PhD, an employee of ICON (North Wales, PA), and was funded by Incyte Corporation (Wilmington, DE).
Polycythemia vera (PV) is a rare myeloproliferative neoplasm affecting 44 to 57 individuals per 100,000 in the United States.1,2 It is characterized by somatic mutations in the hematopoietic stem cell, resulting in hyperproliferation of mature myeloid lineage cells.2 Sustained erythrocytosis is a hallmark of PV, although many patients also have leukocytosis and thrombocytosis.2,3 These patients have increased inherent thrombotic risk with arterial events reported to occur at rates of 7 to 21/1000 person-years and venous thrombotic events at 5 to 20/1000 person-years.4-7 Thrombotic and cardiovascular events are leading causes of morbidity and mortality, resulting in a reduced overall survival of patients with PV compared with the general population.3,8-10
Blood Cell Counts and Thrombotic Events in PV
Treatment strategies for patients with PV mainly aim to prevent or manage thrombotic and bleeding complications through normalization of blood counts.11 Hematocrit (Hct) control has been reported to be associated with reduced thrombotic risk in patients with PV. This was shown and popularized by the prospective, randomized Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial in which participants were randomized 1:1 to maintaining either a low (< 45%) or high (45%-50%) Hct for 5 years to examine the long-term effects of more- or less-intensive cytoreductive therapy.12 Patients in the low-Hct group were found to have a lower rate of death from cardiovascular events or major thrombosis (1.1/100 person-years in the low-Hct group vs 4.4 in the high-Hct group; hazard ratio [HR], 3.91; 95% confidence interval [CI], 1.45-10.53; P = .007). Likewise, cardiovascular events occurred at a lower rate in patients in the low-Hct group compared with the high-Hct group (4.4% vs 10.9% of patients, respectively; HR, 2.69; 95% CI, 1.19-6.12; P = .02).12
Leukocytosis has also been linked to elevated risk for vascular events as shown in several studies, including the real-world European Collaboration on Low-Dose Aspirin in PV (ECLAP) observational study and a post hoc subanalysis of the CYTO-PV study.13,14 In a multivariate, time-dependent analysis in ECLAP, patients with white blood cell (WBC) counts > 15 × 109/L had a significant increase in the risk of thrombosis compared with those who had lower WBC counts, with higher WBC count more strongly associated with arterial than venous thromboembolism.13 In CYTO-PV, a significant correlation between elevated WBC count (≥ 11 × 109/L vs reference level of < 7 × 109/L) and time-dependent risk of major thrombosis was shown (HR, 3.9; 95% CI, 1.24-12.3; P = .02).14 Likewise, WBC count ≥ 11 × 109/L was found to be a predictor of subsequent venous events in a separate single-center multivariate analysis of patients with PV.8
Although CYTO-PV remains one of the largest prospective landmark studies in PV demonstrating the impact of Hct control on thrombosis, it is worthwhile to note that the patients in the high-Hct group who received less frequent myelosuppressive therapy with hydroxyurea than the low-Hct group also had higher WBC counts.12,15 Work is needed to determine the relative effects of high Hct and high WBC counts on PV independent of each other.
The Veteran Population with PV
Two recently published retrospective analyses from Parasuraman and colleagues used data from the Veterans Health Administration (VHA), the largest integrated health care system in the US, with an aim to replicate findings from CYTO-PV in a real-world population.16,17 The 2 analyses focused independently on the effects of Hct control and WBC count on the risk of a thrombotic event in patients with PV.
In the first retrospective analysis, 213 patients with PV and no prior thrombosis were placed into groups based on whether Hct levels were consistently either < 45% or ≥ 45% throughout the study period.17 The mean follow-up time was 2.3 years, during which 44.1% of patients experienced a thrombotic event (Figure 1). Patients with Hct levels < 45% had a lower rate of thrombotic events compared to those with levels ≥ 45% (40.3% vs 54.2%, respectively; HR, 1.61; 95% CI, 1.03-2.51; P = .04). In a sensitivity analysis that included patients with pre-index thrombotic events (N = 342), similar results were noted (55.6% vs 76.9% between the < 45% and ≥ 45% groups, respectively; HR, 1.95; 95% CI, 1.46-2.61; P < .001).
In the second analysis, the authors investigated the relationship between WBC counts and thrombotic events.16 Evaluable patients (N = 1565) were grouped into 1 of 4 cohorts based on the last WBC measurement taken during the study period before a thrombotic event or through the end of follow-up: (1) WBC < 7.0 × 109/L, (2) 7.0 to 8.4 × 109/L, (3) 8.5 to < 11.0 × 109/L, or (4) ≥ 11.0 × 109/L. Mean follow-up time ranged from 3.6 to 4.5 years among WBC count cohorts, during which 24.9% of patients experienced a thrombotic event. Compared with the reference cohort (WBC < 7.0 × 109/L), a significant positive association between WBC counts and thrombotic event occurrence was observed among patients with WBC counts of 8.5 to < 11.0 × 109/L (HR, 1.47; 95% CI, 1.10-1.96; P < .01) and ≥ 11 × 109/L (HR, 1.87; 95% CI, 1.44-2.43; P < .001) (Figure 2).16 When including all patients in a sensitivity analysis regardless of whether they experienced thrombotic events before the index date (N = 1876), similar results were obtained (7.0-8.4 × 109/L group: HR, 1.22; 95% CI, 0.97-1.55; P = .0959; 8.5 - 11.0 × 109/L group: HR, 1.41; 95% CI, 1.10-1.81; P = .0062; ≥ 11.0 × 109/L group: HR, 1.53; 95% CI, 1.23-1.91; P < .001; compared with < 7.0 × 109/L reference group). Rates of phlebotomy and cytoreductive treatments were similar across groups.16
Some limitations to these studies are attributable to their retrospective design, reliance on health records, and the VHA population characteristics, which differ from the general population. For example, in this analysis, patients with PV in the VHA population had significantly increased risk of thrombotic events, even at a lower WBC count threshold (≥ 8.5 × 109/L) compared with those reported in CYTO-PV (≥ 11 × 109/L). Furthermore, approximately one-third of patients had elevated WBC levels, compared with 25.5% in the CYTO-PV study.14,16 This is most likely due to the unique nature of the VHA patient population, who are predominantly older adult men and generally have a higher comorbidity burden. A notable pre-index comorbidity burden was reported in the VHA population in the Hct analysis, even when compared to patients with PV in the general US population (Charlson Comorbidity Index score, 1.3 vs 0.8).6,17 Comorbid conditions such as hypertension, diabetes, and tobacco use, which are most common among the VHA population, are independently associated with higher risk of cardiovascular and thrombotic events.18,19 However, whether these higher levels of comorbidities affected the type of treatments they received was not elucidated, and the effectiveness of treatments to maintain target Hct levels was not addressed in the study.
Current PV Management and Future Implications
The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology in myeloproliferative neoplasms recommend maintaining Hct levels < 45% in patients with PV.11 Patients with high-risk disease (age ≥ 60 years and/or history of thrombosis) are monitored for new thrombosis or bleeding and are managed for their cardiovascular risk factors. In addition, they receive low-dose aspirin (81-100 mg/day), undergo phlebotomy to maintain an Hct < 45%, and are managed with pharmacologic cytoreductive therapy. Cytoreductive therapy primarily consists of hydroxyurea or peginterferon alfa-2a for younger patients. Ruxolitinib, a Janus kinase (JAK1)/JAK2 inhibitor, is now approved by the US Food and Drug Administration as second-line treatment for those with PV that is intolerant or unresponsive to hydroxyurea or peginterferon alfa-2a treatments.11,20 However, the role of cytoreductive therapy is not clear for patients with low-risk disease (age < 60 years and no history of thrombosis). These patients are managed for their cardiovascular risk factors, undergo phlebotomy to maintain an Hct < 45%, are maintained on low-dose aspirin (81-100 mg/day), and are monitored for indications for cytoreductive therapy, which include any new thrombosis or disease-related major bleeding, frequent or persistent need for phlebotomy with poor tolerance for the procedure, splenomegaly, thrombocytosis, leukocytosis, and disease-related symptoms (eg, aquagenic pruritus, night sweats, fatigue).
Even though the current guidelines recommend maintaining a target Hct of < 45% in patients with high-risk PV, the role of Hct as the main determinant of thrombotic risk in patients with PV is still debated.21 In JAK2V617F-positive essential thrombocythemia, Hct levels are usually normal but risk of thrombosis is nevertheless still significant.22 The risk of thrombosis is significantly lower in primary familial and congenital polycythemia and much lower in secondary erythrocytosis such as cyanotic heart disease, long-term native dwellers of high altitude, and those with high-oxygen–affinity hemoglobins.21,23 In secondary erythrocytosis from hypoxia or upregulated hypoxic pathway such as hypoxia inducible factor-2α (HIF-2α) mutation and Chuvash erythrocytosis, the risk of thrombosis is more associated with the upregulated HIF pathway and its downstream consequences, rather than the elevated Hct level.24
However, most current literature supports the association of increased risk of thrombosis with higher Hct and high WBC count in patients with PV. In addition, the underlying mechanism of thrombogenesis still remains elusive; it is likely a complex process that involves interactions among multiple components, including elevated blood counts arising from clonal hematopoiesis, JAK2V617F allele burden, and platelet and WBC activation and their interaction with endothelial cells and inflammatory cytokines.25
Nevertheless, Hct control and aspirin use are current standard of care for patients with PV to mitigate thrombotic risk, and the results from the 2 analyses by Parasuraman and colleagues, using real-world data from the VHA, support the current practice guidelines to maintain Hct < 45% in these patients. They also provide additional support for considering WBC counts when determining patient risk and treatment plans. Although treatment response criteria from the European LeukemiaNet include achieving normal WBC levels to decrease the risk of thrombosis, current NCCN guidelines do not include WBC counts as a component for establishing patient risk or provide a target WBC count to guide patient management.11,26,27 Updates to these practice guidelines may be warranted. In addition, further study is needed to understand the mechanism of thrombogenesis in PV and other myeloproliferative disorders in order to develop novel therapeutic targets and improve patient outcomes.
Acknowledgments
Writing assistance was provided by Tania Iqbal, PhD, an employee of ICON (North Wales, PA), and was funded by Incyte Corporation (Wilmington, DE).
1. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595-600. doi:10.3109/10428194.2013.813500
2. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. doi:10.1182/blood-2016-03-643544
3. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-1881. doi:10.1038/leu.2013.163
4. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224-2232. doi:10.1200/JCO.2005.07.062
5. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110(3):840-846. doi:10.1182/blood-2006-12-064287
6. Goyal RK, Davis KL, Cote I, Mounedji N, Kaye JA. Increased incidence of thromboembolic event rates in patients diagnosed with polycythemia vera: results from an observational cohort study. Blood (ASH Annual Meeting Abstracts). 2014;124:4840. doi:10.1182/blood.V124.21.4840.4840
7. Barbui T, Carobbio A, Rumi E, et al. In contemporary patients with polycythemia vera, rates of thrombosis and risk factors delineate a new clinical epidemiology. Blood. 2014;124(19):3021-3023. doi:10.1182/blood-2014-07-591610 8. Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017;7(12):662. doi:10.1038/s41408-017-0035-6
9. Stein BL, Moliterno AR, Tiu RV. Polycythemia vera disease burden: contributing factors, impact on quality of life, and emerging treatment options. Ann Hematol. 2014;93(12):1965-1976. doi:10.1007/s00277-014-2205-y
10. Hultcrantz M, Kristinsson SY, Andersson TM-L, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol. 2012;30(24):2995-3001. doi:10.1200/JCO.2012.42.1925
11. National Comprehensive Cancer Network. NCCN clinical practice guidelines in myeloproliferative neoplasms (Version 1.2020). Accessed March 3, 2022. https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf
12. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. doi:10.1056/NEJMoa1208500
13. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109(6):2446-2452. doi:10.1182/blood-2006-08-042515
14. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126(4):560-561. doi:10.1182/blood-2015-04-638593
15. Prchal JT, Gordeuk VR. Treatment target in polycythemia vera. N Engl J Med. 2013;368(16):1555-1556. doi:10.1056/NEJMc1301262
16. Parasuraman S, Yu J, Paranagama D, et al. Elevated white blood cell levels and thrombotic events in patients with polycythemia vera: a real-world analysis of Veterans Health Administration data. Clin Lymphoma Myeloma Leuk. 2020;20(2):63-69. doi:10.1016/j.clml.2019.11.010
17. Parasuraman S, Yu J, Paranagama D, et al. Hematocrit levels and thrombotic events in patients with polycythemia vera: an analysis of Veterans Health Administration data. Ann Hematol. 2019;98(11):2533-2539. doi:10.1007/s00277-019-03793-w
18. WHO CVD Risk Chart Working Group. World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health. 2019;7(10):e1332-e1345. doi:10.1016/S2214-109X(19)30318-3.
19. D’Agostino RB Sr, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117(6):743-753. doi:10.1161/CIRCULATIONAHA.107.699579
20. Jakafi. Package insert. Incyte Corporation; 2020.
21. Gordeuk VR, Key NS, Prchal JT. Re-evaluation of hematocrit as a determinant of thrombotic risk in erythrocytosis. Haematologica. 2019;104(4):653-658. doi:10.3324/haematol.2018.210732
22. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859. doi:10.1182/blood-2011-02-339002
23. Perloff JK, Marelli AJ, Miner PD. Risk of stroke in adults with cyanotic congenital heart disease. Circulation. 1993;87(6):1954-1959. doi:10.1161/01.cir.87.6.1954
24. Gordeuk VR, Miasnikova GY, Sergueeva AI, et al. Thrombotic risk in congenital erythrocytosis due to up-regulated hypoxia sensing is not associated with elevated hematocrit. Haematologica. 2020;105(3):e87-e90. doi:10.3324/haematol.2019.216267
25. Kroll MH, Michaelis LC, Verstovsek S. Mechanisms of thrombogenesis in polycythemia vera. Blood Rev. 2015;29(4):215-221. doi:10.1016/j.blre.2014.12.002
26. Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057-1069. doi:10.1038/s41375-018-0077-1
27. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778-4781. doi:10.1182/blood-2013-01-478891
1. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595-600. doi:10.3109/10428194.2013.813500
2. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. doi:10.1182/blood-2016-03-643544
3. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-1881. doi:10.1038/leu.2013.163
4. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224-2232. doi:10.1200/JCO.2005.07.062
5. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110(3):840-846. doi:10.1182/blood-2006-12-064287
6. Goyal RK, Davis KL, Cote I, Mounedji N, Kaye JA. Increased incidence of thromboembolic event rates in patients diagnosed with polycythemia vera: results from an observational cohort study. Blood (ASH Annual Meeting Abstracts). 2014;124:4840. doi:10.1182/blood.V124.21.4840.4840
7. Barbui T, Carobbio A, Rumi E, et al. In contemporary patients with polycythemia vera, rates of thrombosis and risk factors delineate a new clinical epidemiology. Blood. 2014;124(19):3021-3023. doi:10.1182/blood-2014-07-591610 8. Cerquozzi S, Barraco D, Lasho T, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017;7(12):662. doi:10.1038/s41408-017-0035-6
9. Stein BL, Moliterno AR, Tiu RV. Polycythemia vera disease burden: contributing factors, impact on quality of life, and emerging treatment options. Ann Hematol. 2014;93(12):1965-1976. doi:10.1007/s00277-014-2205-y
10. Hultcrantz M, Kristinsson SY, Andersson TM-L, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol. 2012;30(24):2995-3001. doi:10.1200/JCO.2012.42.1925
11. National Comprehensive Cancer Network. NCCN clinical practice guidelines in myeloproliferative neoplasms (Version 1.2020). Accessed March 3, 2022. https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf
12. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33. doi:10.1056/NEJMoa1208500
13. Landolfi R, Di Gennaro L, Barbui T, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109(6):2446-2452. doi:10.1182/blood-2006-08-042515
14. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126(4):560-561. doi:10.1182/blood-2015-04-638593
15. Prchal JT, Gordeuk VR. Treatment target in polycythemia vera. N Engl J Med. 2013;368(16):1555-1556. doi:10.1056/NEJMc1301262
16. Parasuraman S, Yu J, Paranagama D, et al. Elevated white blood cell levels and thrombotic events in patients with polycythemia vera: a real-world analysis of Veterans Health Administration data. Clin Lymphoma Myeloma Leuk. 2020;20(2):63-69. doi:10.1016/j.clml.2019.11.010
17. Parasuraman S, Yu J, Paranagama D, et al. Hematocrit levels and thrombotic events in patients with polycythemia vera: an analysis of Veterans Health Administration data. Ann Hematol. 2019;98(11):2533-2539. doi:10.1007/s00277-019-03793-w
18. WHO CVD Risk Chart Working Group. World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health. 2019;7(10):e1332-e1345. doi:10.1016/S2214-109X(19)30318-3.
19. D’Agostino RB Sr, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117(6):743-753. doi:10.1161/CIRCULATIONAHA.107.699579
20. Jakafi. Package insert. Incyte Corporation; 2020.
21. Gordeuk VR, Key NS, Prchal JT. Re-evaluation of hematocrit as a determinant of thrombotic risk in erythrocytosis. Haematologica. 2019;104(4):653-658. doi:10.3324/haematol.2018.210732
22. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859. doi:10.1182/blood-2011-02-339002
23. Perloff JK, Marelli AJ, Miner PD. Risk of stroke in adults with cyanotic congenital heart disease. Circulation. 1993;87(6):1954-1959. doi:10.1161/01.cir.87.6.1954
24. Gordeuk VR, Miasnikova GY, Sergueeva AI, et al. Thrombotic risk in congenital erythrocytosis due to up-regulated hypoxia sensing is not associated with elevated hematocrit. Haematologica. 2020;105(3):e87-e90. doi:10.3324/haematol.2019.216267
25. Kroll MH, Michaelis LC, Verstovsek S. Mechanisms of thrombogenesis in polycythemia vera. Blood Rev. 2015;29(4):215-221. doi:10.1016/j.blre.2014.12.002
26. Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057-1069. doi:10.1038/s41375-018-0077-1
27. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778-4781. doi:10.1182/blood-2013-01-478891
Complex link between gut microbiome and immunotherapy response in advanced melanoma
A large-scale than previously thought.
Overall, researchers identified a panel of species, including Roseburia spp. and Akkermansia muciniphila, associated with responses to ICI therapy. However, no single species was a “fully consistent biomarker” across the studies, the authors explain.
This “machine learning analysis confirmed the link between the microbiome and overall response rates (ORRs) and progression-free survival (PFS) with ICIs but also revealed limited reproducibility of microbiome-based signatures across cohorts,” Karla A. Lee, PhD, a clinical research fellow at King’s College London, and colleagues report. The results suggest that “the microbiome is predictive of response in some, but not all, cohorts.”
The findings were published online Feb. 28 in Nature Medicine.
Despite recent advances in targeted therapies for melanoma, less than half of the those who receive a single-agent ICI respond, and those who receive combination ICI therapy often suffer from severe drug toxicity problems. That is why finding patients more likely to respond to a single-agent ICI has become a priority.
Previous studies have identified the gut microbiome as “a potential biomarker of response, as well as a therapeutic target” in melanoma and other malignancies, but “little consensus exists on which microbiome characteristics are associated with treatment responses in the human setting,” the authors explain.
To further clarify the microbiome–immunotherapy relationship, the researchers performed metagenomic sequencing of stool samples collected from 165 ICI-naive patients with unresectable stage III or IV cutaneous melanoma from 5 observational cohorts in the Netherlands, United Kingdom, and Spain. These data were integrated with 147 samples from publicly available datasets.
First, the authors highlighted the variability in findings across these observational studies. For instance, they analyzed stool samples from one UK-based observational study of patients with melanoma (PRIMM-UK) and found a small but statistically significant difference in the microbiome composition of immunotherapy responders versus nonresponders (P = .05) but did not find such an association in a parallel study in the Netherlands (PRIMM-NL, P = .61).
The investigators also explored biomarkers of response across different cohorts and found several standouts. In trials using ORR as an endpoint, two uncultivated Roseburia species (CAG:182 and CAG:471) were associated with responses to ICIs. For patients with available PFS data, Phascolarctobacterium succinatutens and Lactobacillus vaginalis were “enriched in responders” across 7 datasets and significant in 3 of the 8 meta-analysis approaches. A muciniphila and Dorea formicigenerans were also associated with ORR and PFS at 12 months in several meta-analyses.
However, “no single bacterium was a fully consistent biomarker of response across all datasets,” the authors wrote.
Still, the findings could have important implications for the more than 50% of patients with advanced melanoma who don’t respond to single-agent ICI therapy.
“Our study shows that studying the microbiome is important to improve and personalize immunotherapy treatments for melanoma,” study coauthor Nicola Segata, PhD, principal investigator in the Laboratory of Computational Metagenomics, University of Trento, Italy, said in a press release. “However, it also suggests that because of the person-to-person variability of the gut microbiome, even larger studies must be carried out to understand the specific gut microbial features that are more likely to lead to a positive response to immunotherapy.”
Coauthor Tim Spector, PhD, head of the Department of Twin Research & Genetic Epidemiology at King’s College London, added that “the ultimate goal is to identify which specific features of the microbiome are directly influencing the clinical benefits of immunotherapy to exploit these features in new personalized approaches to support cancer immunotherapy.”
In the meantime, he said, “this study highlights the potential impact of good diet and gut health on chances of survival in patients undergoing immunotherapy.”
This study was coordinated by King’s College London, CIBIO Department of the University of Trento and European Institute of Oncology in Italy, and the University of Groningen in the Netherlands, and was funded by the Seerave Foundation. Dr. Lee, Dr. Segata, and Dr. Spector have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A large-scale than previously thought.
Overall, researchers identified a panel of species, including Roseburia spp. and Akkermansia muciniphila, associated with responses to ICI therapy. However, no single species was a “fully consistent biomarker” across the studies, the authors explain.
This “machine learning analysis confirmed the link between the microbiome and overall response rates (ORRs) and progression-free survival (PFS) with ICIs but also revealed limited reproducibility of microbiome-based signatures across cohorts,” Karla A. Lee, PhD, a clinical research fellow at King’s College London, and colleagues report. The results suggest that “the microbiome is predictive of response in some, but not all, cohorts.”
The findings were published online Feb. 28 in Nature Medicine.
Despite recent advances in targeted therapies for melanoma, less than half of the those who receive a single-agent ICI respond, and those who receive combination ICI therapy often suffer from severe drug toxicity problems. That is why finding patients more likely to respond to a single-agent ICI has become a priority.
Previous studies have identified the gut microbiome as “a potential biomarker of response, as well as a therapeutic target” in melanoma and other malignancies, but “little consensus exists on which microbiome characteristics are associated with treatment responses in the human setting,” the authors explain.
To further clarify the microbiome–immunotherapy relationship, the researchers performed metagenomic sequencing of stool samples collected from 165 ICI-naive patients with unresectable stage III or IV cutaneous melanoma from 5 observational cohorts in the Netherlands, United Kingdom, and Spain. These data were integrated with 147 samples from publicly available datasets.
First, the authors highlighted the variability in findings across these observational studies. For instance, they analyzed stool samples from one UK-based observational study of patients with melanoma (PRIMM-UK) and found a small but statistically significant difference in the microbiome composition of immunotherapy responders versus nonresponders (P = .05) but did not find such an association in a parallel study in the Netherlands (PRIMM-NL, P = .61).
The investigators also explored biomarkers of response across different cohorts and found several standouts. In trials using ORR as an endpoint, two uncultivated Roseburia species (CAG:182 and CAG:471) were associated with responses to ICIs. For patients with available PFS data, Phascolarctobacterium succinatutens and Lactobacillus vaginalis were “enriched in responders” across 7 datasets and significant in 3 of the 8 meta-analysis approaches. A muciniphila and Dorea formicigenerans were also associated with ORR and PFS at 12 months in several meta-analyses.
However, “no single bacterium was a fully consistent biomarker of response across all datasets,” the authors wrote.
Still, the findings could have important implications for the more than 50% of patients with advanced melanoma who don’t respond to single-agent ICI therapy.
“Our study shows that studying the microbiome is important to improve and personalize immunotherapy treatments for melanoma,” study coauthor Nicola Segata, PhD, principal investigator in the Laboratory of Computational Metagenomics, University of Trento, Italy, said in a press release. “However, it also suggests that because of the person-to-person variability of the gut microbiome, even larger studies must be carried out to understand the specific gut microbial features that are more likely to lead to a positive response to immunotherapy.”
Coauthor Tim Spector, PhD, head of the Department of Twin Research & Genetic Epidemiology at King’s College London, added that “the ultimate goal is to identify which specific features of the microbiome are directly influencing the clinical benefits of immunotherapy to exploit these features in new personalized approaches to support cancer immunotherapy.”
In the meantime, he said, “this study highlights the potential impact of good diet and gut health on chances of survival in patients undergoing immunotherapy.”
This study was coordinated by King’s College London, CIBIO Department of the University of Trento and European Institute of Oncology in Italy, and the University of Groningen in the Netherlands, and was funded by the Seerave Foundation. Dr. Lee, Dr. Segata, and Dr. Spector have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A large-scale than previously thought.
Overall, researchers identified a panel of species, including Roseburia spp. and Akkermansia muciniphila, associated with responses to ICI therapy. However, no single species was a “fully consistent biomarker” across the studies, the authors explain.
This “machine learning analysis confirmed the link between the microbiome and overall response rates (ORRs) and progression-free survival (PFS) with ICIs but also revealed limited reproducibility of microbiome-based signatures across cohorts,” Karla A. Lee, PhD, a clinical research fellow at King’s College London, and colleagues report. The results suggest that “the microbiome is predictive of response in some, but not all, cohorts.”
The findings were published online Feb. 28 in Nature Medicine.
Despite recent advances in targeted therapies for melanoma, less than half of the those who receive a single-agent ICI respond, and those who receive combination ICI therapy often suffer from severe drug toxicity problems. That is why finding patients more likely to respond to a single-agent ICI has become a priority.
Previous studies have identified the gut microbiome as “a potential biomarker of response, as well as a therapeutic target” in melanoma and other malignancies, but “little consensus exists on which microbiome characteristics are associated with treatment responses in the human setting,” the authors explain.
To further clarify the microbiome–immunotherapy relationship, the researchers performed metagenomic sequencing of stool samples collected from 165 ICI-naive patients with unresectable stage III or IV cutaneous melanoma from 5 observational cohorts in the Netherlands, United Kingdom, and Spain. These data were integrated with 147 samples from publicly available datasets.
First, the authors highlighted the variability in findings across these observational studies. For instance, they analyzed stool samples from one UK-based observational study of patients with melanoma (PRIMM-UK) and found a small but statistically significant difference in the microbiome composition of immunotherapy responders versus nonresponders (P = .05) but did not find such an association in a parallel study in the Netherlands (PRIMM-NL, P = .61).
The investigators also explored biomarkers of response across different cohorts and found several standouts. In trials using ORR as an endpoint, two uncultivated Roseburia species (CAG:182 and CAG:471) were associated with responses to ICIs. For patients with available PFS data, Phascolarctobacterium succinatutens and Lactobacillus vaginalis were “enriched in responders” across 7 datasets and significant in 3 of the 8 meta-analysis approaches. A muciniphila and Dorea formicigenerans were also associated with ORR and PFS at 12 months in several meta-analyses.
However, “no single bacterium was a fully consistent biomarker of response across all datasets,” the authors wrote.
Still, the findings could have important implications for the more than 50% of patients with advanced melanoma who don’t respond to single-agent ICI therapy.
“Our study shows that studying the microbiome is important to improve and personalize immunotherapy treatments for melanoma,” study coauthor Nicola Segata, PhD, principal investigator in the Laboratory of Computational Metagenomics, University of Trento, Italy, said in a press release. “However, it also suggests that because of the person-to-person variability of the gut microbiome, even larger studies must be carried out to understand the specific gut microbial features that are more likely to lead to a positive response to immunotherapy.”
Coauthor Tim Spector, PhD, head of the Department of Twin Research & Genetic Epidemiology at King’s College London, added that “the ultimate goal is to identify which specific features of the microbiome are directly influencing the clinical benefits of immunotherapy to exploit these features in new personalized approaches to support cancer immunotherapy.”
In the meantime, he said, “this study highlights the potential impact of good diet and gut health on chances of survival in patients undergoing immunotherapy.”
This study was coordinated by King’s College London, CIBIO Department of the University of Trento and European Institute of Oncology in Italy, and the University of Groningen in the Netherlands, and was funded by the Seerave Foundation. Dr. Lee, Dr. Segata, and Dr. Spector have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FDA approves new CAR T-cell treatment for multiple myeloma
A new treatment option for patients with refractory/relapsed multiple myeloma who have already tried four or more therapies has been approved by the U.S. Food and Drug Administration.
There are already two other therapies on the market that target BCMA – another CAR T cell, idecabtagene vicleucel (Abecma), which was approved by the FDA in March 2021, and a drug conjugate, belantamab mafodotin (Blenrep), which was approved in August 2020.
The approval of cilta-cel was based on clinical data from the CARTITUDE-1 study, which were initially presented in December 2020 at the annual meeting of the American Society of Hematology, as reported at the time by this news organization.
The trial involved 97 patients with relapsed/refractory multiple myeloma who had already received a median of six previous treatments (range, three to 18), including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
“The treatment journey for the majority of patients living with multiple myeloma is a relentless cycle of remission and relapse, with fewer patients achieving a deep response as they progress through later lines of therapy,” commented Sundar Jagannath, MBBS, professor of medicine, hematology, and medical oncology at Mount Sinai, who was a principal investigator on the pivotal study.
“That is why I have been really excited about the results from the CARTITUDE-1 study, which has demonstrated that cilta-cel can provide deep and durable responses and long-term treatment-free intervals, even in this heavily pretreated multiple myeloma patient population,” he said.
“Today’s approval of Carvykti helps address a great unmet need for these patients,” he commented in a press release from the manufacturer.
Like other CAR T-cell therapies, ciltacabtagene autoleucel is a one-time treatment. It involves collecting blood from the patient, extracting T cells, genetically engineering them, then transfusing them back to the patient, who in the meantime has undergone conditioning.
The results from CARTITUDE-1 show that this one-time treatment resulted in deep and durable responses.
The overall response rate was 98%, and the majority of patients (78%) achieved a stringent complete response, in which physicians are unable to observe any signs or symptoms of disease via imaging or other tests after treatment.
At a median of 18 months’ follow-up, the median duration of response was 21.8 months.
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” commented Mr. Jagannath.
“The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time,” he added.
As with other CAR T-cell therapies, there were serious side effects, and these products are available only through restricted programs under a risk evaluation and mitigation strategy.
The product information for Cartykti includes a boxed warning that mentions cytokine release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome, parkinsonism, Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, and prolonged and/or recurrent cytopenias.
The most common adverse reactions (reported in greater than or equal to 20% of patients) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections–pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
A version of this article first appeared on Medscape.com.
A new treatment option for patients with refractory/relapsed multiple myeloma who have already tried four or more therapies has been approved by the U.S. Food and Drug Administration.
There are already two other therapies on the market that target BCMA – another CAR T cell, idecabtagene vicleucel (Abecma), which was approved by the FDA in March 2021, and a drug conjugate, belantamab mafodotin (Blenrep), which was approved in August 2020.
The approval of cilta-cel was based on clinical data from the CARTITUDE-1 study, which were initially presented in December 2020 at the annual meeting of the American Society of Hematology, as reported at the time by this news organization.
The trial involved 97 patients with relapsed/refractory multiple myeloma who had already received a median of six previous treatments (range, three to 18), including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
“The treatment journey for the majority of patients living with multiple myeloma is a relentless cycle of remission and relapse, with fewer patients achieving a deep response as they progress through later lines of therapy,” commented Sundar Jagannath, MBBS, professor of medicine, hematology, and medical oncology at Mount Sinai, who was a principal investigator on the pivotal study.
“That is why I have been really excited about the results from the CARTITUDE-1 study, which has demonstrated that cilta-cel can provide deep and durable responses and long-term treatment-free intervals, even in this heavily pretreated multiple myeloma patient population,” he said.
“Today’s approval of Carvykti helps address a great unmet need for these patients,” he commented in a press release from the manufacturer.
Like other CAR T-cell therapies, ciltacabtagene autoleucel is a one-time treatment. It involves collecting blood from the patient, extracting T cells, genetically engineering them, then transfusing them back to the patient, who in the meantime has undergone conditioning.
The results from CARTITUDE-1 show that this one-time treatment resulted in deep and durable responses.
The overall response rate was 98%, and the majority of patients (78%) achieved a stringent complete response, in which physicians are unable to observe any signs or symptoms of disease via imaging or other tests after treatment.
At a median of 18 months’ follow-up, the median duration of response was 21.8 months.
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” commented Mr. Jagannath.
“The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time,” he added.
As with other CAR T-cell therapies, there were serious side effects, and these products are available only through restricted programs under a risk evaluation and mitigation strategy.
The product information for Cartykti includes a boxed warning that mentions cytokine release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome, parkinsonism, Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, and prolonged and/or recurrent cytopenias.
The most common adverse reactions (reported in greater than or equal to 20% of patients) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections–pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
A version of this article first appeared on Medscape.com.
A new treatment option for patients with refractory/relapsed multiple myeloma who have already tried four or more therapies has been approved by the U.S. Food and Drug Administration.
There are already two other therapies on the market that target BCMA – another CAR T cell, idecabtagene vicleucel (Abecma), which was approved by the FDA in March 2021, and a drug conjugate, belantamab mafodotin (Blenrep), which was approved in August 2020.
The approval of cilta-cel was based on clinical data from the CARTITUDE-1 study, which were initially presented in December 2020 at the annual meeting of the American Society of Hematology, as reported at the time by this news organization.
The trial involved 97 patients with relapsed/refractory multiple myeloma who had already received a median of six previous treatments (range, three to 18), including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
“The treatment journey for the majority of patients living with multiple myeloma is a relentless cycle of remission and relapse, with fewer patients achieving a deep response as they progress through later lines of therapy,” commented Sundar Jagannath, MBBS, professor of medicine, hematology, and medical oncology at Mount Sinai, who was a principal investigator on the pivotal study.
“That is why I have been really excited about the results from the CARTITUDE-1 study, which has demonstrated that cilta-cel can provide deep and durable responses and long-term treatment-free intervals, even in this heavily pretreated multiple myeloma patient population,” he said.
“Today’s approval of Carvykti helps address a great unmet need for these patients,” he commented in a press release from the manufacturer.
Like other CAR T-cell therapies, ciltacabtagene autoleucel is a one-time treatment. It involves collecting blood from the patient, extracting T cells, genetically engineering them, then transfusing them back to the patient, who in the meantime has undergone conditioning.
The results from CARTITUDE-1 show that this one-time treatment resulted in deep and durable responses.
The overall response rate was 98%, and the majority of patients (78%) achieved a stringent complete response, in which physicians are unable to observe any signs or symptoms of disease via imaging or other tests after treatment.
At a median of 18 months’ follow-up, the median duration of response was 21.8 months.
“The responses in the CARTITUDE-1 study showed durability over time and resulted in the majority of heavily pretreated patients achieving deep responses after 18-month follow-up,” commented Mr. Jagannath.
“The approval of cilta-cel provides physicians an immunotherapy treatment option that offers patients an opportunity to be free from anti-myeloma therapies for a period of time,” he added.
As with other CAR T-cell therapies, there were serious side effects, and these products are available only through restricted programs under a risk evaluation and mitigation strategy.
The product information for Cartykti includes a boxed warning that mentions cytokine release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome, parkinsonism, Guillain-Barré syndrome, hemophagocytic lymphohistiocytosis/macrophage activation syndrome, and prolonged and/or recurrent cytopenias.
The most common adverse reactions (reported in greater than or equal to 20% of patients) are pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections–pathogens unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
A version of this article first appeared on Medscape.com.
AGILE: ‘Exciting’ survival results for IDH1-mutated AML
ATLANTA – Patients with acute myeloid leukemia (AML) bearing mutations in IDH1 who could not withstand the rigors of intensive therapy had improved event-free and overall survival when they were treated with the combination of ivosidenib (Tibsovo) and azacitidine (Onureg, Vidaza), compared with azacitidine alone.
The results come from the phase 3 AGILE trial., reported Hartmut Döhner, MD, from Ulm (Germany) University Hospital.
Median overall survival was 24 months with the combination, compared with 7.9 months for azacitidine-placebo, translating into a hazard ratio for death with the IVO-AZA of 0.44 (P = .0005).
“The IVO-AZA combination was safe and tolerable, with fewer infections reported relative to placebo plus AZA. Additionally, the clinical benefit of the combination was supported by favorable health-related quality of life,” he said.
Dr. Döhner spoke at a press briefing prior to the presentation of the trial results at the annual meeting of the American Society of Hematology.
“I’m really excited by the results from the AGILE trial,” commented Mikkael A. Sekeres, MD, chief of the division of hematology at the University of Miami’s Sylvester Comprehensive Cancer Center, who moderated the briefing.
The results show “survival that’s three times longer for a combination of ivosidenib plus azacitidine versus azacitidine alone in a very distinct population of patients who have an IDH1 mutation,” he said.
“The question that will arise is, if the standard of care is now to give azacitidine and venetoclax [Venclexta] to patients who don’t receive intensive chemotherapy in the inpatient setting, where does this trial end? And I would answer [by saying] that the combination of azacitidine is not truly nonintensive therapy, and it’s probably on a spectrum between nonintensive and intensive therapy, and probably closer to seven plus three than a lot of people recognize,” Dr. Sekeres said.
The combination of ivosidenib and azacitidine may therefore be a better choice for treatment of older patients with IDH1-mutated AML – particularly those with comorbidities – who may not be able to tolerate venetoclax plus azacitidine, he added.
AGILE results
For the analysis presented at the meeting, there was a data cutoff in March 2021, at which point 146 patients out of a planned 200 had been enrolled and randomly assigned. They received treatment with either oral ivosidenib 500 mg daily plus azacitidine 75 mg/m2, delivered subcutaneously or intravenously, or azacitidine plus placebo.
In May 2021, after an interim analysis, the independent data-monitoring committee recommended a halt to the trial because of significant improvements in outcomes among patients assigned to the combination, and those data are reported at the meeting.
The median patient age was about 76 years in each group. Approximately 75% of patients in each arm had de novo AML, and about 25% had AML secondary to treatment, myelodysplastic syndrome, or myeloproliferative neoplasms. The majority of patients in each group had intermediate cytogenetic risk disease.
The analysis was by intention to treat, with patients who did not have complete remission (CR) by week 24 considered to have had an event on day 1 of randomization.
At the time of the interim analysis, with the longest follow-up out to 29 months (the investigators did not report median follow-up time for the study), there were significantly fewer study events – defined as treatment failure by week 24, relapse from remission, or death from any cause – in the ivosidenib/azacitidine combination, with a hazard ratio of 0.33 (P = .0011).
The EFS benefit and the overall survival benefit were consistent across subgroups, the researchers noted, including in patients with de novo disease, demographics, baseline cytogenetic risk status, World Health Organization AML classification, baseline white blood cell count, and baseline percentage of bone marrow blasts.
Clinical and hematologic responses also favored the combination, with a complete response (CR) rate of 34%, compared with 11% for azacitidine alone (odds ratio, 4.8; P < .0001), and respective overall response rates of 45% versus 14% (odds ratio, 7.2; P < .0001).
Health-related quality of life measures also trended better with the combination across all subscales, and were significantly better at day 1 of cycle 5 in the diarrhea and appetite loss domains.
Treatment-emergent adverse events included grade 2 or higher differentiation syndrome, which occurred in 14.1% of patients treated with IVO-AZA versus 8.2% treated with AZA alone. Grade 3 or higher QT interval prolongation was also more frequent with the combination, at 9.9% versus 4.1%.
Any-grade infections were less common with IVO-AZA, however, at 28.2% versus 49.3% with AZA alone. At the briefing, this news organization asked coinvestigator Stephane de Botton, MD, Gustave Roussy Cancer Center, Villejuif, France, whether he could explain this seemingly paradoxical result.
He replied that the combination results in greater production of neutrophils and therefore better protection against infections, compared with azacitidine alone.
The study was funded by Agios Pharmaceuticals, now a part of Servier Pharmaceuticals. Dr. Döhner disclosed consultancy and other relationships with various companies. Dr. Sekeres disclosed consulting/advising for Novartis, Takea/Millennium, and Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
ATLANTA – Patients with acute myeloid leukemia (AML) bearing mutations in IDH1 who could not withstand the rigors of intensive therapy had improved event-free and overall survival when they were treated with the combination of ivosidenib (Tibsovo) and azacitidine (Onureg, Vidaza), compared with azacitidine alone.
The results come from the phase 3 AGILE trial., reported Hartmut Döhner, MD, from Ulm (Germany) University Hospital.
Median overall survival was 24 months with the combination, compared with 7.9 months for azacitidine-placebo, translating into a hazard ratio for death with the IVO-AZA of 0.44 (P = .0005).
“The IVO-AZA combination was safe and tolerable, with fewer infections reported relative to placebo plus AZA. Additionally, the clinical benefit of the combination was supported by favorable health-related quality of life,” he said.
Dr. Döhner spoke at a press briefing prior to the presentation of the trial results at the annual meeting of the American Society of Hematology.
“I’m really excited by the results from the AGILE trial,” commented Mikkael A. Sekeres, MD, chief of the division of hematology at the University of Miami’s Sylvester Comprehensive Cancer Center, who moderated the briefing.
The results show “survival that’s three times longer for a combination of ivosidenib plus azacitidine versus azacitidine alone in a very distinct population of patients who have an IDH1 mutation,” he said.
“The question that will arise is, if the standard of care is now to give azacitidine and venetoclax [Venclexta] to patients who don’t receive intensive chemotherapy in the inpatient setting, where does this trial end? And I would answer [by saying] that the combination of azacitidine is not truly nonintensive therapy, and it’s probably on a spectrum between nonintensive and intensive therapy, and probably closer to seven plus three than a lot of people recognize,” Dr. Sekeres said.
The combination of ivosidenib and azacitidine may therefore be a better choice for treatment of older patients with IDH1-mutated AML – particularly those with comorbidities – who may not be able to tolerate venetoclax plus azacitidine, he added.
AGILE results
For the analysis presented at the meeting, there was a data cutoff in March 2021, at which point 146 patients out of a planned 200 had been enrolled and randomly assigned. They received treatment with either oral ivosidenib 500 mg daily plus azacitidine 75 mg/m2, delivered subcutaneously or intravenously, or azacitidine plus placebo.
In May 2021, after an interim analysis, the independent data-monitoring committee recommended a halt to the trial because of significant improvements in outcomes among patients assigned to the combination, and those data are reported at the meeting.
The median patient age was about 76 years in each group. Approximately 75% of patients in each arm had de novo AML, and about 25% had AML secondary to treatment, myelodysplastic syndrome, or myeloproliferative neoplasms. The majority of patients in each group had intermediate cytogenetic risk disease.
The analysis was by intention to treat, with patients who did not have complete remission (CR) by week 24 considered to have had an event on day 1 of randomization.
At the time of the interim analysis, with the longest follow-up out to 29 months (the investigators did not report median follow-up time for the study), there were significantly fewer study events – defined as treatment failure by week 24, relapse from remission, or death from any cause – in the ivosidenib/azacitidine combination, with a hazard ratio of 0.33 (P = .0011).
The EFS benefit and the overall survival benefit were consistent across subgroups, the researchers noted, including in patients with de novo disease, demographics, baseline cytogenetic risk status, World Health Organization AML classification, baseline white blood cell count, and baseline percentage of bone marrow blasts.
Clinical and hematologic responses also favored the combination, with a complete response (CR) rate of 34%, compared with 11% for azacitidine alone (odds ratio, 4.8; P < .0001), and respective overall response rates of 45% versus 14% (odds ratio, 7.2; P < .0001).
Health-related quality of life measures also trended better with the combination across all subscales, and were significantly better at day 1 of cycle 5 in the diarrhea and appetite loss domains.
Treatment-emergent adverse events included grade 2 or higher differentiation syndrome, which occurred in 14.1% of patients treated with IVO-AZA versus 8.2% treated with AZA alone. Grade 3 or higher QT interval prolongation was also more frequent with the combination, at 9.9% versus 4.1%.
Any-grade infections were less common with IVO-AZA, however, at 28.2% versus 49.3% with AZA alone. At the briefing, this news organization asked coinvestigator Stephane de Botton, MD, Gustave Roussy Cancer Center, Villejuif, France, whether he could explain this seemingly paradoxical result.
He replied that the combination results in greater production of neutrophils and therefore better protection against infections, compared with azacitidine alone.
The study was funded by Agios Pharmaceuticals, now a part of Servier Pharmaceuticals. Dr. Döhner disclosed consultancy and other relationships with various companies. Dr. Sekeres disclosed consulting/advising for Novartis, Takea/Millennium, and Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
ATLANTA – Patients with acute myeloid leukemia (AML) bearing mutations in IDH1 who could not withstand the rigors of intensive therapy had improved event-free and overall survival when they were treated with the combination of ivosidenib (Tibsovo) and azacitidine (Onureg, Vidaza), compared with azacitidine alone.
The results come from the phase 3 AGILE trial., reported Hartmut Döhner, MD, from Ulm (Germany) University Hospital.
Median overall survival was 24 months with the combination, compared with 7.9 months for azacitidine-placebo, translating into a hazard ratio for death with the IVO-AZA of 0.44 (P = .0005).
“The IVO-AZA combination was safe and tolerable, with fewer infections reported relative to placebo plus AZA. Additionally, the clinical benefit of the combination was supported by favorable health-related quality of life,” he said.
Dr. Döhner spoke at a press briefing prior to the presentation of the trial results at the annual meeting of the American Society of Hematology.
“I’m really excited by the results from the AGILE trial,” commented Mikkael A. Sekeres, MD, chief of the division of hematology at the University of Miami’s Sylvester Comprehensive Cancer Center, who moderated the briefing.
The results show “survival that’s three times longer for a combination of ivosidenib plus azacitidine versus azacitidine alone in a very distinct population of patients who have an IDH1 mutation,” he said.
“The question that will arise is, if the standard of care is now to give azacitidine and venetoclax [Venclexta] to patients who don’t receive intensive chemotherapy in the inpatient setting, where does this trial end? And I would answer [by saying] that the combination of azacitidine is not truly nonintensive therapy, and it’s probably on a spectrum between nonintensive and intensive therapy, and probably closer to seven plus three than a lot of people recognize,” Dr. Sekeres said.
The combination of ivosidenib and azacitidine may therefore be a better choice for treatment of older patients with IDH1-mutated AML – particularly those with comorbidities – who may not be able to tolerate venetoclax plus azacitidine, he added.
AGILE results
For the analysis presented at the meeting, there was a data cutoff in March 2021, at which point 146 patients out of a planned 200 had been enrolled and randomly assigned. They received treatment with either oral ivosidenib 500 mg daily plus azacitidine 75 mg/m2, delivered subcutaneously or intravenously, or azacitidine plus placebo.
In May 2021, after an interim analysis, the independent data-monitoring committee recommended a halt to the trial because of significant improvements in outcomes among patients assigned to the combination, and those data are reported at the meeting.
The median patient age was about 76 years in each group. Approximately 75% of patients in each arm had de novo AML, and about 25% had AML secondary to treatment, myelodysplastic syndrome, or myeloproliferative neoplasms. The majority of patients in each group had intermediate cytogenetic risk disease.
The analysis was by intention to treat, with patients who did not have complete remission (CR) by week 24 considered to have had an event on day 1 of randomization.
At the time of the interim analysis, with the longest follow-up out to 29 months (the investigators did not report median follow-up time for the study), there were significantly fewer study events – defined as treatment failure by week 24, relapse from remission, or death from any cause – in the ivosidenib/azacitidine combination, with a hazard ratio of 0.33 (P = .0011).
The EFS benefit and the overall survival benefit were consistent across subgroups, the researchers noted, including in patients with de novo disease, demographics, baseline cytogenetic risk status, World Health Organization AML classification, baseline white blood cell count, and baseline percentage of bone marrow blasts.
Clinical and hematologic responses also favored the combination, with a complete response (CR) rate of 34%, compared with 11% for azacitidine alone (odds ratio, 4.8; P < .0001), and respective overall response rates of 45% versus 14% (odds ratio, 7.2; P < .0001).
Health-related quality of life measures also trended better with the combination across all subscales, and were significantly better at day 1 of cycle 5 in the diarrhea and appetite loss domains.
Treatment-emergent adverse events included grade 2 or higher differentiation syndrome, which occurred in 14.1% of patients treated with IVO-AZA versus 8.2% treated with AZA alone. Grade 3 or higher QT interval prolongation was also more frequent with the combination, at 9.9% versus 4.1%.
Any-grade infections were less common with IVO-AZA, however, at 28.2% versus 49.3% with AZA alone. At the briefing, this news organization asked coinvestigator Stephane de Botton, MD, Gustave Roussy Cancer Center, Villejuif, France, whether he could explain this seemingly paradoxical result.
He replied that the combination results in greater production of neutrophils and therefore better protection against infections, compared with azacitidine alone.
The study was funded by Agios Pharmaceuticals, now a part of Servier Pharmaceuticals. Dr. Döhner disclosed consultancy and other relationships with various companies. Dr. Sekeres disclosed consulting/advising for Novartis, Takea/Millennium, and Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
AT ASH 2021