User login
Isatuximab added to RVd boosts response in new myeloma
ATLANTA -
The drug is isatuximab (Sarclisa, Sanofi), an anti-CD38 antibody that was approved last year for use in patients with advanced disease.
Now it has shown benefit in patients who have been newly diagnosed with the disease. When isatuximab was added onto a usual triplet therapy for myeloma, it increased the likelihood that patients would be negative for minimal residual disease (MRD) at the end of the induction phase of treatment, thereby increasing their chances for a successful autologous stem cell transplant (ASCT).
The new results come from the GMMG-HD7 trial, in which all patients were treated with the triplet combination of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd).
Some patients, after randomization, also received isatuximab, and in this group, the MRD-negativity rate was 50.1% at the end of induction therapy compared with 35.6% for patients treated with RVd alone.
Patients who are MRD-negative at the time of ASCT have significantly better outcomes than patients who remain MRD-positive.
“Isa-RVd is the first regimen to demonstrate significant MRD-negativity benefit at the end of induction versus RVd in a phase 3 trial,” reported Hartmut Goldschmidt, MD, from University Hospital Heidelberg, Germany.
“The benefits of the addition of Isa to RVd versus RVd regarding MRD negativity after induction therapy was consistent in all subgroups,” he added.
Dr. Goldschmidt spoke at a press briefing prior to his presentation of the data here at the annual meeting of the American Society of Hematology (ASH).
“I think that these data are encouraging, but they are preliminary, and we need mature data to be absolutely certain about whether this presents a major advance in treatment,” commented Ravi Vij, MD, from the Siteman Cancer Center and Washington University School of Medicine in St. Louis. Dr. Vij was not involved in the study.
“We know that for transplant-eligible patients, for whom this trial was conducted, the field is moving toward giving four drugs for induction,” he said in an interview with this news organization.
He noted that the combination of RVd with the other currently available anti-CD38 antibody, daratumumab (Darzalex), was approved for this indication in the United States in Jan. 2021.
Dr. Vij said that isatuximab has been slow to catch on in the United States both because it was approved after clinicians had already become familiar with daratumumab and because it is given intravenously, compared with subcutaneous administration of the latest formulation of daratumumab.
“Whereas isatuximab can take an hour-and-a-half with each infusion, daratumumab takes 5 minutes for an injection and the patient is out of there, so it is convenient both for the patient and the treating institution,” he said.
MRD vs. CR?
Dr. Goldschmidt was asked during the briefing about whether MRD-negativity or complete response rates are better predictors of progression-free survival (PFS). He replied that with current standardized sequencing techniques and sensitivity down to 10-6, “it’s a big benefit to analyze MRD negativity, and there is ongoing discussion between colleagues from the myeloma group with the Food and Drug Administration about how we can merge the data and predict PFS and overall survival.”
Laurie Sehn, MD, MPH, from the BC Cancer Centre for Lymphoid Cancer, Vancouver, who moderated the briefing, commented that “we’re desperately looking for surrogate markers to speed up answers to clinical trials, and I think MRD in myeloma is quickly becoming a very important surrogate marker.”
GMMG-7 results
For their trial, Dr. Goldschmidt and colleagues enrolled 662 patients with newly diagnosed multiple myeloma who were candidates for high-dose therapy and ASCT and after stratification by revised International Staging System (r-ISS) criteria, randomly assigned them six three-week cycles of induction therapy with Isa-RVd or RVd alone.
Following ASCT, patients were again randomized to maintenance with either isatuximab plus lenalidomide or lenalidomide alone.
As noted before, MRD rates at the end of induction were 50.1% with Isa-RVd versus 35.6% with RVd alone, translating to a hazard ratio favoring the four-drug combination of 1.83 (P < .001).
Treatment with Isa-RVd was the only significant predictor for the likelihood of MRD negativity in a multivariate analysis controlling for treatment group, r-ISS status, performance status, renal impairment, age, and sex.
Although the rate of complete responses at the end of induction was similar between the treatment groups, the rate of very good partial response or better was higher with the isatuximab-containing combination (77.3% vs. 60.5%; P < .001).
The respective rates of disease progression at the end of induction in the Isa-RVd and RVd groups were 1.5% versus 4.0%.
The rates of adverse events were generally similar between the groups, except a higher proportion of patients had leukocytopenia or neutropenia in the Isa-RVd than the RVdgroup (26.4% vs. 9.1%). There were four deaths in the Isa-RVd group and eight in the RVd group. Most of the deaths were attributable to disease progression or COVID-19, said Dr. Goldschmidt.
The study was funded by Sanofi. Dr. Goldschmidt has disclosed honoraria and research grants from Sanofi and others. Dr. Vij has disclosed honoraria or advisory board activities from various companies, including Sanofi. Dr. Sehn is a consultant for and has received honoraria from various companies, not including Sanofi.
A version of this article first appeared on Medscape.com.
ATLANTA -
The drug is isatuximab (Sarclisa, Sanofi), an anti-CD38 antibody that was approved last year for use in patients with advanced disease.
Now it has shown benefit in patients who have been newly diagnosed with the disease. When isatuximab was added onto a usual triplet therapy for myeloma, it increased the likelihood that patients would be negative for minimal residual disease (MRD) at the end of the induction phase of treatment, thereby increasing their chances for a successful autologous stem cell transplant (ASCT).
The new results come from the GMMG-HD7 trial, in which all patients were treated with the triplet combination of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd).
Some patients, after randomization, also received isatuximab, and in this group, the MRD-negativity rate was 50.1% at the end of induction therapy compared with 35.6% for patients treated with RVd alone.
Patients who are MRD-negative at the time of ASCT have significantly better outcomes than patients who remain MRD-positive.
“Isa-RVd is the first regimen to demonstrate significant MRD-negativity benefit at the end of induction versus RVd in a phase 3 trial,” reported Hartmut Goldschmidt, MD, from University Hospital Heidelberg, Germany.
“The benefits of the addition of Isa to RVd versus RVd regarding MRD negativity after induction therapy was consistent in all subgroups,” he added.
Dr. Goldschmidt spoke at a press briefing prior to his presentation of the data here at the annual meeting of the American Society of Hematology (ASH).
“I think that these data are encouraging, but they are preliminary, and we need mature data to be absolutely certain about whether this presents a major advance in treatment,” commented Ravi Vij, MD, from the Siteman Cancer Center and Washington University School of Medicine in St. Louis. Dr. Vij was not involved in the study.
“We know that for transplant-eligible patients, for whom this trial was conducted, the field is moving toward giving four drugs for induction,” he said in an interview with this news organization.
He noted that the combination of RVd with the other currently available anti-CD38 antibody, daratumumab (Darzalex), was approved for this indication in the United States in Jan. 2021.
Dr. Vij said that isatuximab has been slow to catch on in the United States both because it was approved after clinicians had already become familiar with daratumumab and because it is given intravenously, compared with subcutaneous administration of the latest formulation of daratumumab.
“Whereas isatuximab can take an hour-and-a-half with each infusion, daratumumab takes 5 minutes for an injection and the patient is out of there, so it is convenient both for the patient and the treating institution,” he said.
MRD vs. CR?
Dr. Goldschmidt was asked during the briefing about whether MRD-negativity or complete response rates are better predictors of progression-free survival (PFS). He replied that with current standardized sequencing techniques and sensitivity down to 10-6, “it’s a big benefit to analyze MRD negativity, and there is ongoing discussion between colleagues from the myeloma group with the Food and Drug Administration about how we can merge the data and predict PFS and overall survival.”
Laurie Sehn, MD, MPH, from the BC Cancer Centre for Lymphoid Cancer, Vancouver, who moderated the briefing, commented that “we’re desperately looking for surrogate markers to speed up answers to clinical trials, and I think MRD in myeloma is quickly becoming a very important surrogate marker.”
GMMG-7 results
For their trial, Dr. Goldschmidt and colleagues enrolled 662 patients with newly diagnosed multiple myeloma who were candidates for high-dose therapy and ASCT and after stratification by revised International Staging System (r-ISS) criteria, randomly assigned them six three-week cycles of induction therapy with Isa-RVd or RVd alone.
Following ASCT, patients were again randomized to maintenance with either isatuximab plus lenalidomide or lenalidomide alone.
As noted before, MRD rates at the end of induction were 50.1% with Isa-RVd versus 35.6% with RVd alone, translating to a hazard ratio favoring the four-drug combination of 1.83 (P < .001).
Treatment with Isa-RVd was the only significant predictor for the likelihood of MRD negativity in a multivariate analysis controlling for treatment group, r-ISS status, performance status, renal impairment, age, and sex.
Although the rate of complete responses at the end of induction was similar between the treatment groups, the rate of very good partial response or better was higher with the isatuximab-containing combination (77.3% vs. 60.5%; P < .001).
The respective rates of disease progression at the end of induction in the Isa-RVd and RVd groups were 1.5% versus 4.0%.
The rates of adverse events were generally similar between the groups, except a higher proportion of patients had leukocytopenia or neutropenia in the Isa-RVd than the RVdgroup (26.4% vs. 9.1%). There were four deaths in the Isa-RVd group and eight in the RVd group. Most of the deaths were attributable to disease progression or COVID-19, said Dr. Goldschmidt.
The study was funded by Sanofi. Dr. Goldschmidt has disclosed honoraria and research grants from Sanofi and others. Dr. Vij has disclosed honoraria or advisory board activities from various companies, including Sanofi. Dr. Sehn is a consultant for and has received honoraria from various companies, not including Sanofi.
A version of this article first appeared on Medscape.com.
ATLANTA -
The drug is isatuximab (Sarclisa, Sanofi), an anti-CD38 antibody that was approved last year for use in patients with advanced disease.
Now it has shown benefit in patients who have been newly diagnosed with the disease. When isatuximab was added onto a usual triplet therapy for myeloma, it increased the likelihood that patients would be negative for minimal residual disease (MRD) at the end of the induction phase of treatment, thereby increasing their chances for a successful autologous stem cell transplant (ASCT).
The new results come from the GMMG-HD7 trial, in which all patients were treated with the triplet combination of lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd).
Some patients, after randomization, also received isatuximab, and in this group, the MRD-negativity rate was 50.1% at the end of induction therapy compared with 35.6% for patients treated with RVd alone.
Patients who are MRD-negative at the time of ASCT have significantly better outcomes than patients who remain MRD-positive.
“Isa-RVd is the first regimen to demonstrate significant MRD-negativity benefit at the end of induction versus RVd in a phase 3 trial,” reported Hartmut Goldschmidt, MD, from University Hospital Heidelberg, Germany.
“The benefits of the addition of Isa to RVd versus RVd regarding MRD negativity after induction therapy was consistent in all subgroups,” he added.
Dr. Goldschmidt spoke at a press briefing prior to his presentation of the data here at the annual meeting of the American Society of Hematology (ASH).
“I think that these data are encouraging, but they are preliminary, and we need mature data to be absolutely certain about whether this presents a major advance in treatment,” commented Ravi Vij, MD, from the Siteman Cancer Center and Washington University School of Medicine in St. Louis. Dr. Vij was not involved in the study.
“We know that for transplant-eligible patients, for whom this trial was conducted, the field is moving toward giving four drugs for induction,” he said in an interview with this news organization.
He noted that the combination of RVd with the other currently available anti-CD38 antibody, daratumumab (Darzalex), was approved for this indication in the United States in Jan. 2021.
Dr. Vij said that isatuximab has been slow to catch on in the United States both because it was approved after clinicians had already become familiar with daratumumab and because it is given intravenously, compared with subcutaneous administration of the latest formulation of daratumumab.
“Whereas isatuximab can take an hour-and-a-half with each infusion, daratumumab takes 5 minutes for an injection and the patient is out of there, so it is convenient both for the patient and the treating institution,” he said.
MRD vs. CR?
Dr. Goldschmidt was asked during the briefing about whether MRD-negativity or complete response rates are better predictors of progression-free survival (PFS). He replied that with current standardized sequencing techniques and sensitivity down to 10-6, “it’s a big benefit to analyze MRD negativity, and there is ongoing discussion between colleagues from the myeloma group with the Food and Drug Administration about how we can merge the data and predict PFS and overall survival.”
Laurie Sehn, MD, MPH, from the BC Cancer Centre for Lymphoid Cancer, Vancouver, who moderated the briefing, commented that “we’re desperately looking for surrogate markers to speed up answers to clinical trials, and I think MRD in myeloma is quickly becoming a very important surrogate marker.”
GMMG-7 results
For their trial, Dr. Goldschmidt and colleagues enrolled 662 patients with newly diagnosed multiple myeloma who were candidates for high-dose therapy and ASCT and after stratification by revised International Staging System (r-ISS) criteria, randomly assigned them six three-week cycles of induction therapy with Isa-RVd or RVd alone.
Following ASCT, patients were again randomized to maintenance with either isatuximab plus lenalidomide or lenalidomide alone.
As noted before, MRD rates at the end of induction were 50.1% with Isa-RVd versus 35.6% with RVd alone, translating to a hazard ratio favoring the four-drug combination of 1.83 (P < .001).
Treatment with Isa-RVd was the only significant predictor for the likelihood of MRD negativity in a multivariate analysis controlling for treatment group, r-ISS status, performance status, renal impairment, age, and sex.
Although the rate of complete responses at the end of induction was similar between the treatment groups, the rate of very good partial response or better was higher with the isatuximab-containing combination (77.3% vs. 60.5%; P < .001).
The respective rates of disease progression at the end of induction in the Isa-RVd and RVd groups were 1.5% versus 4.0%.
The rates of adverse events were generally similar between the groups, except a higher proportion of patients had leukocytopenia or neutropenia in the Isa-RVd than the RVdgroup (26.4% vs. 9.1%). There were four deaths in the Isa-RVd group and eight in the RVd group. Most of the deaths were attributable to disease progression or COVID-19, said Dr. Goldschmidt.
The study was funded by Sanofi. Dr. Goldschmidt has disclosed honoraria and research grants from Sanofi and others. Dr. Vij has disclosed honoraria or advisory board activities from various companies, including Sanofi. Dr. Sehn is a consultant for and has received honoraria from various companies, not including Sanofi.
A version of this article first appeared on Medscape.com.
AT ASH 2021
Talk early to patients with high-risk AML about end-of-life decisions

The prognosis isn’t good for high-risk AML, defined in the study as either relapsing/recurrent disease or a diagnosis made past the age of 59 years. Almost 60% of the patients (114) died during the 7 years of the study, which started in 2014.
Therefore, it’s important to bring up end-of-life decisions when patients are still able to discuss them, so families aren’t left struggling to guess how aggressive their loved ones might have wanted their final care to be, said lead investigator Hannah Abrams, MD, an internal medicine resident at Massachusetts General. She presented these findings at the annual meeting of the American Society of Hematology.
Much of the time, however, end-of-life discussions come too late. The study team found that nearly 40% (45/114) of the patients who died during the study were not involved in their final code decisions, which most often were to administer comfort care only. Many patients were too ill to participate; the median time between the last code change and death was just 2 days.
Dr. Abrams said she’s seen how families agonize when patients haven’t addressed the issue beforehand. “Witnessing that made me think this is really important to look at. Having these conversations upfront is really important,” she said in an interview.
When asked for comment, hematologist-oncologist Toby Campbell, MD, chief of palliative care at the University of Wisconsin, Madison, agreed.
He called this issue a “missed opportunity for patient autonomy and self-determination. Patients with high-risk AML commonly experience rapid changes in their clinical condition, which catch everyone by surprise. Healthcare providers should do more to prepare patients and families, rather than allow them to be surprised,” Dr. Campbell said.
Part of the problem, Dr. Abrams said, is that end-of-life discussions can fall through the cracks amid urgent discussions about chemotherapy options and other matters.
“One of the biggest things to make this more feasible is to schedule and reimburse time in clinic for this to happen,” she said, noting a need to carve out and protect “15 minutes for patients and clinicians to talk about this.”
Another aspect is that patients are often overly optimistic about their prognoses, so end-of-life discussions don’t seem as pressing. Educational materials about the meaning of various code options and when they are appropriate could help, Dr. Abrams said.
As for the psychological impact of bringing up end-of-life decisions early on, Mikkael Sekeres, MD, chief of the division of hematology at the University of Miami, stressed the importance of telling patients, “We are having this conversation because you are doing well, not because you are doing poorly, and this is the time to have it.”
“Sometimes it does take a sentinel event like an ICU stay before some people want to engage in that conversation, and unfortunately, that is often too late,” said Dr. Sekeres, who moderated Dr. Abrams’ presentation at the meeting.
Among other findings, Dr. Abrams and her team reported that at diagnosis, 86.0% of patients were full-code, and 8.5% had restrictions on life-sustaining therapies. Overall, 57% (114/200) of patients experienced a code status transition, with a median of two transitions during their illness.
Among patients who died, older age and receipt of non-intensive chemotherapy were associated with earlier discussions about code status.
The next step in the project is to determine if palliative care consults yield earlier discussions and greater patient involvement.
There was no commercial funding for the study, and Dr. Abrams and Dr. Campbell didn’t have any relevant disclosures. Dr. Sekeres is an advisor to Novartis, Takeda, and BMS.
[email protected]
The prognosis isn’t good for high-risk AML, defined in the study as either relapsing/recurrent disease or a diagnosis made past the age of 59 years. Almost 60% of the patients (114) died during the 7 years of the study, which started in 2014.
Therefore, it’s important to bring up end-of-life decisions when patients are still able to discuss them, so families aren’t left struggling to guess how aggressive their loved ones might have wanted their final care to be, said lead investigator Hannah Abrams, MD, an internal medicine resident at Massachusetts General. She presented these findings at the annual meeting of the American Society of Hematology.
Much of the time, however, end-of-life discussions come too late. The study team found that nearly 40% (45/114) of the patients who died during the study were not involved in their final code decisions, which most often were to administer comfort care only. Many patients were too ill to participate; the median time between the last code change and death was just 2 days.
Dr. Abrams said she’s seen how families agonize when patients haven’t addressed the issue beforehand. “Witnessing that made me think this is really important to look at. Having these conversations upfront is really important,” she said in an interview.
When asked for comment, hematologist-oncologist Toby Campbell, MD, chief of palliative care at the University of Wisconsin, Madison, agreed.
He called this issue a “missed opportunity for patient autonomy and self-determination. Patients with high-risk AML commonly experience rapid changes in their clinical condition, which catch everyone by surprise. Healthcare providers should do more to prepare patients and families, rather than allow them to be surprised,” Dr. Campbell said.
Part of the problem, Dr. Abrams said, is that end-of-life discussions can fall through the cracks amid urgent discussions about chemotherapy options and other matters.
“One of the biggest things to make this more feasible is to schedule and reimburse time in clinic for this to happen,” she said, noting a need to carve out and protect “15 minutes for patients and clinicians to talk about this.”
Another aspect is that patients are often overly optimistic about their prognoses, so end-of-life discussions don’t seem as pressing. Educational materials about the meaning of various code options and when they are appropriate could help, Dr. Abrams said.
As for the psychological impact of bringing up end-of-life decisions early on, Mikkael Sekeres, MD, chief of the division of hematology at the University of Miami, stressed the importance of telling patients, “We are having this conversation because you are doing well, not because you are doing poorly, and this is the time to have it.”
“Sometimes it does take a sentinel event like an ICU stay before some people want to engage in that conversation, and unfortunately, that is often too late,” said Dr. Sekeres, who moderated Dr. Abrams’ presentation at the meeting.
Among other findings, Dr. Abrams and her team reported that at diagnosis, 86.0% of patients were full-code, and 8.5% had restrictions on life-sustaining therapies. Overall, 57% (114/200) of patients experienced a code status transition, with a median of two transitions during their illness.
Among patients who died, older age and receipt of non-intensive chemotherapy were associated with earlier discussions about code status.
The next step in the project is to determine if palliative care consults yield earlier discussions and greater patient involvement.
There was no commercial funding for the study, and Dr. Abrams and Dr. Campbell didn’t have any relevant disclosures. Dr. Sekeres is an advisor to Novartis, Takeda, and BMS.
[email protected]
The prognosis isn’t good for high-risk AML, defined in the study as either relapsing/recurrent disease or a diagnosis made past the age of 59 years. Almost 60% of the patients (114) died during the 7 years of the study, which started in 2014.
Therefore, it’s important to bring up end-of-life decisions when patients are still able to discuss them, so families aren’t left struggling to guess how aggressive their loved ones might have wanted their final care to be, said lead investigator Hannah Abrams, MD, an internal medicine resident at Massachusetts General. She presented these findings at the annual meeting of the American Society of Hematology.
Much of the time, however, end-of-life discussions come too late. The study team found that nearly 40% (45/114) of the patients who died during the study were not involved in their final code decisions, which most often were to administer comfort care only. Many patients were too ill to participate; the median time between the last code change and death was just 2 days.
Dr. Abrams said she’s seen how families agonize when patients haven’t addressed the issue beforehand. “Witnessing that made me think this is really important to look at. Having these conversations upfront is really important,” she said in an interview.
When asked for comment, hematologist-oncologist Toby Campbell, MD, chief of palliative care at the University of Wisconsin, Madison, agreed.
He called this issue a “missed opportunity for patient autonomy and self-determination. Patients with high-risk AML commonly experience rapid changes in their clinical condition, which catch everyone by surprise. Healthcare providers should do more to prepare patients and families, rather than allow them to be surprised,” Dr. Campbell said.
Part of the problem, Dr. Abrams said, is that end-of-life discussions can fall through the cracks amid urgent discussions about chemotherapy options and other matters.
“One of the biggest things to make this more feasible is to schedule and reimburse time in clinic for this to happen,” she said, noting a need to carve out and protect “15 minutes for patients and clinicians to talk about this.”
Another aspect is that patients are often overly optimistic about their prognoses, so end-of-life discussions don’t seem as pressing. Educational materials about the meaning of various code options and when they are appropriate could help, Dr. Abrams said.
As for the psychological impact of bringing up end-of-life decisions early on, Mikkael Sekeres, MD, chief of the division of hematology at the University of Miami, stressed the importance of telling patients, “We are having this conversation because you are doing well, not because you are doing poorly, and this is the time to have it.”
“Sometimes it does take a sentinel event like an ICU stay before some people want to engage in that conversation, and unfortunately, that is often too late,” said Dr. Sekeres, who moderated Dr. Abrams’ presentation at the meeting.
Among other findings, Dr. Abrams and her team reported that at diagnosis, 86.0% of patients were full-code, and 8.5% had restrictions on life-sustaining therapies. Overall, 57% (114/200) of patients experienced a code status transition, with a median of two transitions during their illness.
Among patients who died, older age and receipt of non-intensive chemotherapy were associated with earlier discussions about code status.
The next step in the project is to determine if palliative care consults yield earlier discussions and greater patient involvement.
There was no commercial funding for the study, and Dr. Abrams and Dr. Campbell didn’t have any relevant disclosures. Dr. Sekeres is an advisor to Novartis, Takeda, and BMS.
[email protected]
FROM ASH 2021
Beta-thalassemia gene therapy achieves lasting transfusion independence
, an investigator reported at the annual meeting of the American Society of Hematology.

Among patients who received betibeglogene autotemcel (beti-cel) in a phase 3 trial and enrolled in a long-term follow-up study, nearly 90% achieved durable transfusion independence, according to Alexis A. Thompson, MD, MPH, of the hematology section at the Ann & Robert H. Lurie Children’s Hospital of Chicago.
The median duration of ongoing transfusion independence was nearly 3 years as of this report, which Dr. Thompson described in a press conference at the meeting.
In a subanalysis of this international study, Dr. Thompson and co-investigators reported that in patients who achieve transfusion independence, chelation reduced iron, and iron markers stabilized even after chelation was stopped.
Beyond 2 years post-infusion, no adverse events related to the drug product were seen. This suggested that the therapy has a favorable long-term safety profile, according to Dr. Thompson.
“At this point, we believe that beti-cel is potentially curative for patients with TDT [transfusion-dependent beta-thalassemia],” Dr. Thompson said in the press conference.
This study answers one of the major outstanding questions about beti-cel and iron metabolism, according to Arielle L. Langer, MD, MPH, an instructor in medicine at Harvard Medical School and attending physician for adult thalassemia patients at Brigham and Women’s and Dana Farber Cancer Institute, both in Boston.
“Seeing the restoration of iron metabolism, it really takes us a step closer to really thinking the term ‘cure’ might truly apply,” Dr. Langer said in an interview.
Dr. Langer said she looks forward to “very long-term outcomes” of beti-cel-treated patients to see whether endocrinopathies and other long-term sequelae of TDT are also abated.
“This [study] is a great intermediate point, but really, when we think about how thalassemia harms and kills our patients, we really sometimes measure that in decades,” she said.
Beta-thalassemia is caused by mutations in the beta-globin gene, resulting in reduced levels of hemoglobin. Patients with TDT, the most serious form of the disease, have severe anemia and are often dependent on red blood cell transfusions from infancy onward, Dr. Thompson said.
With chronic transfusions needed to maintain hemoglobin levels, TDT patients inevitably experience iron overload, which can lead to organ damage and can be fatal. Consequently, patients will require lifelong iron chelation therapy, she added.
Beti-cel, an investigational ex vivo gene addition therapy currently under review by the U.S. Food and Drug Administration, involves adding functional copies of a modified form of the beta-globin gene into a patient’s own hematopoietic stem cells. Once those cells are reinfused, patients may produce adult hemoglobin at levels that eliminate the need for transfusions, according to Dr. Thompson.
At the meeting, Dr. Thompson reported on patients from two phase 1/2 and two phase 3 beti-cel clinical trials who subsequently enrolled in LTF-303, a 13-year follow-up study of the gene therapy’s safety and efficacy.
A total of 57 patients were included in this report, making it the largest gene therapy program to date in any blood disorder, according to Dr. Thompson. Before receiving beti-cel, the patients, who had a broad range of thalassemia genotypes, were receiving between 10 and almost 40 red blood cell transfusions per year, she reported.
Patients ranged in age from 5 to 35 years. The median age in the phase 1/2 studies was 20 years, while in the phase 3 studies it was 15 years.
“The early experience in the phase 1/2 trials allowed us to be more comfortable with enrolling more children, and that has actually helped us to understand safety and efficacy and children in the phase 3 setting,” Dr. Thompson said.
Fertility preservation measures had been undertaken by about 59% of patients from the phase 1/2 studies and 71% of patients from the phase 3 studies, the data show.
Among patients from the phase 3 beti-cel studies who could be evaluated, 31 out of 35 (or 89%) achieved durable transfusion independence, according to the investigator.
The median duration of ongoing transfusion independence was 32 months, with a range of about 18 to 49 months, she added.
Dr. Thompson also reported a subanalysis intended to assess iron status in 16 patients who restarted and then stopped chelation. That subanalysis demonstrated iron reduction in response to chelation, and then stabilization of iron markers after chelation was stopped. Post-gene therapy chelation led to reductions in liver iron concentration and serum ferritin that remained relatively stable after chelation was stopped, she said.
Serious adverse events occurred in eight patients in the long-term follow-up study. However, adverse events related to beti-cel have been absent beyond 2 years post-infusion, according to Dr. Thompson, who added that there have been no reported cases of replication-competent lentivirus, no clonal expansion, no insertional oncogenesis, and no malignancies observed.
“Very reassuringly, there have been 2 male patients, one of whom underwent fertility preservation, who report having healthy children with their partners,” she added.
Dr. Thompson provided disclosures related to Baxalta, Biomarin, bluebird bio, Inc., Celgene/BMS, CRISPR Therapeutics, Vertex, Editas, Graphite Bio, Novartis, Agios, Beam, and Global Blood Therapeutics.
, an investigator reported at the annual meeting of the American Society of Hematology.
Among patients who received betibeglogene autotemcel (beti-cel) in a phase 3 trial and enrolled in a long-term follow-up study, nearly 90% achieved durable transfusion independence, according to Alexis A. Thompson, MD, MPH, of the hematology section at the Ann & Robert H. Lurie Children’s Hospital of Chicago.
The median duration of ongoing transfusion independence was nearly 3 years as of this report, which Dr. Thompson described in a press conference at the meeting.
In a subanalysis of this international study, Dr. Thompson and co-investigators reported that in patients who achieve transfusion independence, chelation reduced iron, and iron markers stabilized even after chelation was stopped.
Beyond 2 years post-infusion, no adverse events related to the drug product were seen. This suggested that the therapy has a favorable long-term safety profile, according to Dr. Thompson.
“At this point, we believe that beti-cel is potentially curative for patients with TDT [transfusion-dependent beta-thalassemia],” Dr. Thompson said in the press conference.
This study answers one of the major outstanding questions about beti-cel and iron metabolism, according to Arielle L. Langer, MD, MPH, an instructor in medicine at Harvard Medical School and attending physician for adult thalassemia patients at Brigham and Women’s and Dana Farber Cancer Institute, both in Boston.
“Seeing the restoration of iron metabolism, it really takes us a step closer to really thinking the term ‘cure’ might truly apply,” Dr. Langer said in an interview.
Dr. Langer said she looks forward to “very long-term outcomes” of beti-cel-treated patients to see whether endocrinopathies and other long-term sequelae of TDT are also abated.
“This [study] is a great intermediate point, but really, when we think about how thalassemia harms and kills our patients, we really sometimes measure that in decades,” she said.
Beta-thalassemia is caused by mutations in the beta-globin gene, resulting in reduced levels of hemoglobin. Patients with TDT, the most serious form of the disease, have severe anemia and are often dependent on red blood cell transfusions from infancy onward, Dr. Thompson said.
With chronic transfusions needed to maintain hemoglobin levels, TDT patients inevitably experience iron overload, which can lead to organ damage and can be fatal. Consequently, patients will require lifelong iron chelation therapy, she added.
Beti-cel, an investigational ex vivo gene addition therapy currently under review by the U.S. Food and Drug Administration, involves adding functional copies of a modified form of the beta-globin gene into a patient’s own hematopoietic stem cells. Once those cells are reinfused, patients may produce adult hemoglobin at levels that eliminate the need for transfusions, according to Dr. Thompson.
At the meeting, Dr. Thompson reported on patients from two phase 1/2 and two phase 3 beti-cel clinical trials who subsequently enrolled in LTF-303, a 13-year follow-up study of the gene therapy’s safety and efficacy.
A total of 57 patients were included in this report, making it the largest gene therapy program to date in any blood disorder, according to Dr. Thompson. Before receiving beti-cel, the patients, who had a broad range of thalassemia genotypes, were receiving between 10 and almost 40 red blood cell transfusions per year, she reported.
Patients ranged in age from 5 to 35 years. The median age in the phase 1/2 studies was 20 years, while in the phase 3 studies it was 15 years.
“The early experience in the phase 1/2 trials allowed us to be more comfortable with enrolling more children, and that has actually helped us to understand safety and efficacy and children in the phase 3 setting,” Dr. Thompson said.
Fertility preservation measures had been undertaken by about 59% of patients from the phase 1/2 studies and 71% of patients from the phase 3 studies, the data show.
Among patients from the phase 3 beti-cel studies who could be evaluated, 31 out of 35 (or 89%) achieved durable transfusion independence, according to the investigator.
The median duration of ongoing transfusion independence was 32 months, with a range of about 18 to 49 months, she added.
Dr. Thompson also reported a subanalysis intended to assess iron status in 16 patients who restarted and then stopped chelation. That subanalysis demonstrated iron reduction in response to chelation, and then stabilization of iron markers after chelation was stopped. Post-gene therapy chelation led to reductions in liver iron concentration and serum ferritin that remained relatively stable after chelation was stopped, she said.
Serious adverse events occurred in eight patients in the long-term follow-up study. However, adverse events related to beti-cel have been absent beyond 2 years post-infusion, according to Dr. Thompson, who added that there have been no reported cases of replication-competent lentivirus, no clonal expansion, no insertional oncogenesis, and no malignancies observed.
“Very reassuringly, there have been 2 male patients, one of whom underwent fertility preservation, who report having healthy children with their partners,” she added.
Dr. Thompson provided disclosures related to Baxalta, Biomarin, bluebird bio, Inc., Celgene/BMS, CRISPR Therapeutics, Vertex, Editas, Graphite Bio, Novartis, Agios, Beam, and Global Blood Therapeutics.
, an investigator reported at the annual meeting of the American Society of Hematology.
Among patients who received betibeglogene autotemcel (beti-cel) in a phase 3 trial and enrolled in a long-term follow-up study, nearly 90% achieved durable transfusion independence, according to Alexis A. Thompson, MD, MPH, of the hematology section at the Ann & Robert H. Lurie Children’s Hospital of Chicago.
The median duration of ongoing transfusion independence was nearly 3 years as of this report, which Dr. Thompson described in a press conference at the meeting.
In a subanalysis of this international study, Dr. Thompson and co-investigators reported that in patients who achieve transfusion independence, chelation reduced iron, and iron markers stabilized even after chelation was stopped.
Beyond 2 years post-infusion, no adverse events related to the drug product were seen. This suggested that the therapy has a favorable long-term safety profile, according to Dr. Thompson.
“At this point, we believe that beti-cel is potentially curative for patients with TDT [transfusion-dependent beta-thalassemia],” Dr. Thompson said in the press conference.
This study answers one of the major outstanding questions about beti-cel and iron metabolism, according to Arielle L. Langer, MD, MPH, an instructor in medicine at Harvard Medical School and attending physician for adult thalassemia patients at Brigham and Women’s and Dana Farber Cancer Institute, both in Boston.
“Seeing the restoration of iron metabolism, it really takes us a step closer to really thinking the term ‘cure’ might truly apply,” Dr. Langer said in an interview.
Dr. Langer said she looks forward to “very long-term outcomes” of beti-cel-treated patients to see whether endocrinopathies and other long-term sequelae of TDT are also abated.
“This [study] is a great intermediate point, but really, when we think about how thalassemia harms and kills our patients, we really sometimes measure that in decades,” she said.
Beta-thalassemia is caused by mutations in the beta-globin gene, resulting in reduced levels of hemoglobin. Patients with TDT, the most serious form of the disease, have severe anemia and are often dependent on red blood cell transfusions from infancy onward, Dr. Thompson said.
With chronic transfusions needed to maintain hemoglobin levels, TDT patients inevitably experience iron overload, which can lead to organ damage and can be fatal. Consequently, patients will require lifelong iron chelation therapy, she added.
Beti-cel, an investigational ex vivo gene addition therapy currently under review by the U.S. Food and Drug Administration, involves adding functional copies of a modified form of the beta-globin gene into a patient’s own hematopoietic stem cells. Once those cells are reinfused, patients may produce adult hemoglobin at levels that eliminate the need for transfusions, according to Dr. Thompson.
At the meeting, Dr. Thompson reported on patients from two phase 1/2 and two phase 3 beti-cel clinical trials who subsequently enrolled in LTF-303, a 13-year follow-up study of the gene therapy’s safety and efficacy.
A total of 57 patients were included in this report, making it the largest gene therapy program to date in any blood disorder, according to Dr. Thompson. Before receiving beti-cel, the patients, who had a broad range of thalassemia genotypes, were receiving between 10 and almost 40 red blood cell transfusions per year, she reported.
Patients ranged in age from 5 to 35 years. The median age in the phase 1/2 studies was 20 years, while in the phase 3 studies it was 15 years.
“The early experience in the phase 1/2 trials allowed us to be more comfortable with enrolling more children, and that has actually helped us to understand safety and efficacy and children in the phase 3 setting,” Dr. Thompson said.
Fertility preservation measures had been undertaken by about 59% of patients from the phase 1/2 studies and 71% of patients from the phase 3 studies, the data show.
Among patients from the phase 3 beti-cel studies who could be evaluated, 31 out of 35 (or 89%) achieved durable transfusion independence, according to the investigator.
The median duration of ongoing transfusion independence was 32 months, with a range of about 18 to 49 months, she added.
Dr. Thompson also reported a subanalysis intended to assess iron status in 16 patients who restarted and then stopped chelation. That subanalysis demonstrated iron reduction in response to chelation, and then stabilization of iron markers after chelation was stopped. Post-gene therapy chelation led to reductions in liver iron concentration and serum ferritin that remained relatively stable after chelation was stopped, she said.
Serious adverse events occurred in eight patients in the long-term follow-up study. However, adverse events related to beti-cel have been absent beyond 2 years post-infusion, according to Dr. Thompson, who added that there have been no reported cases of replication-competent lentivirus, no clonal expansion, no insertional oncogenesis, and no malignancies observed.
“Very reassuringly, there have been 2 male patients, one of whom underwent fertility preservation, who report having healthy children with their partners,” she added.
Dr. Thompson provided disclosures related to Baxalta, Biomarin, bluebird bio, Inc., Celgene/BMS, CRISPR Therapeutics, Vertex, Editas, Graphite Bio, Novartis, Agios, Beam, and Global Blood Therapeutics.
FROM ASH 2021
FDA approves time-saving combo for r/r multiple myeloma
The U.S. Food and Drug Administration who have had one to three prior lines of therapy.

Using the newly approved combination in this setting is a time-saver for patients and clinics, observed an investigator.
“The approval of subcutaneous daratumumab in combination with Kd will help clinicians address unmet patient needs by reducing the administration time from hours to just minutes and reducing the frequency of infusion-related reactions, as compared to the intravenous daratumumab formulation in combination with Kd,” said Ajai Chari, MD, of Mount Sinai Cancer Clinical Trials Office in New York City in a Janssen press statement.
Efficacy data for the new approval come from a single-arm cohort of PLEIADES, a multicohort, open-label trial. The cohort included 66 patients with relapsed or refractory multiple myeloma who had received one or more prior lines of therapy. Patients received daratumumab + hyaluronidase-fihj subcutaneously in combination with carfilzomib and dexamethasone.
The main efficacy outcome measure was overall response rate, which was 84.8%. At a median follow-up of 9.2 months, the median duration of response had not been reached.
The response rate with the new combination, which features a subcutaneous injection, was akin to those with the older combination, which features the more time-consuming IV administration and was FDA approved, according to the company press release.
The most common adverse reactions (≥20%) occurring in patients treated with Darzalex Faspro, Kyprolis, and dexamethasone were upper respiratory tract infections, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea, and edema peripheral.
A version of this article first appeared on Medscape.com .
The U.S. Food and Drug Administration who have had one to three prior lines of therapy.
Using the newly approved combination in this setting is a time-saver for patients and clinics, observed an investigator.
“The approval of subcutaneous daratumumab in combination with Kd will help clinicians address unmet patient needs by reducing the administration time from hours to just minutes and reducing the frequency of infusion-related reactions, as compared to the intravenous daratumumab formulation in combination with Kd,” said Ajai Chari, MD, of Mount Sinai Cancer Clinical Trials Office in New York City in a Janssen press statement.
Efficacy data for the new approval come from a single-arm cohort of PLEIADES, a multicohort, open-label trial. The cohort included 66 patients with relapsed or refractory multiple myeloma who had received one or more prior lines of therapy. Patients received daratumumab + hyaluronidase-fihj subcutaneously in combination with carfilzomib and dexamethasone.
The main efficacy outcome measure was overall response rate, which was 84.8%. At a median follow-up of 9.2 months, the median duration of response had not been reached.
The response rate with the new combination, which features a subcutaneous injection, was akin to those with the older combination, which features the more time-consuming IV administration and was FDA approved, according to the company press release.
The most common adverse reactions (≥20%) occurring in patients treated with Darzalex Faspro, Kyprolis, and dexamethasone were upper respiratory tract infections, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea, and edema peripheral.
A version of this article first appeared on Medscape.com .
The U.S. Food and Drug Administration who have had one to three prior lines of therapy.
Using the newly approved combination in this setting is a time-saver for patients and clinics, observed an investigator.
“The approval of subcutaneous daratumumab in combination with Kd will help clinicians address unmet patient needs by reducing the administration time from hours to just minutes and reducing the frequency of infusion-related reactions, as compared to the intravenous daratumumab formulation in combination with Kd,” said Ajai Chari, MD, of Mount Sinai Cancer Clinical Trials Office in New York City in a Janssen press statement.
Efficacy data for the new approval come from a single-arm cohort of PLEIADES, a multicohort, open-label trial. The cohort included 66 patients with relapsed or refractory multiple myeloma who had received one or more prior lines of therapy. Patients received daratumumab + hyaluronidase-fihj subcutaneously in combination with carfilzomib and dexamethasone.
The main efficacy outcome measure was overall response rate, which was 84.8%. At a median follow-up of 9.2 months, the median duration of response had not been reached.
The response rate with the new combination, which features a subcutaneous injection, was akin to those with the older combination, which features the more time-consuming IV administration and was FDA approved, according to the company press release.
The most common adverse reactions (≥20%) occurring in patients treated with Darzalex Faspro, Kyprolis, and dexamethasone were upper respiratory tract infections, fatigue, insomnia, hypertension, diarrhea, cough, dyspnea, headache, pyrexia, nausea, and edema peripheral.
A version of this article first appeared on Medscape.com .
Decades spent searching for genes linked to rare blood cancer
Mary Lou McMaster, MD, has spent her entire career at the National Cancer Institute (NCI) searching for the genetic underpinnings that give rise to Waldenstrom's macroglobulinemia (WM).
After searching for decades, she has yet to uncover a "smoking gun," though a few tantalizing clues have emerged along the way.
"Our questions are pretty basic: Why are some people more susceptible to developing WM, and why does WM sometimes cluster in families?" she explained. It turns out that the answers are not at all simple.
Dr. McMaster described some of the clues that her team at the Clinical Genetics Branch of the NCI has unearthed in a presentation at the recent International Waldenstrom's Macroglobulinemia Foundation (IWMF) 2021 Virtual Educational Forum.
Commenting after the presentation, Steven Treon, MD, PhD, professor of medicine, Harvard Medical School, Boston, who is collaborating with Dr. McMaster on this work, said: "From these familial studies, we can learn how familial genomics may give us insights into disease prevention and treatment."
Identifying affected families
Work began in 2001 to identify families in which two or more family members had been diagnosed with WM or in which there was one patient with WM and at least one other relative with a related B-cell cancer, such as chronic lymphocytic leukemia.
For a frame of reference, they enrolled some families with only one member with WM and in which there was no known family history of the disease.
"Overall, we have learned that familial WM is a rare disease but not nearly as rare as we first thought," Dr. McMaster said.
For example, in a referral hospital setting, 5% of WM patients will report having a family member with the same disorder, and up to 20% of WM patients report having a family member with a related but different B-cell cancer, she noted.
NCI researchers also discovered that environmental factors contribute to the development of WM. Notable chemical or occupational exposures include exposures to pesticides, herbicides, and fertilizers. Infections and autoimmune disease are additional factors.
"This was not a surprise," Dr. McMaster commented regarding the role of occupational exposures. The research community has known for decades that a "lymphoma belt" cuts through the Midwest farming states.
Focusing on genetic susceptibility, Dr. McMaster and colleagues first tried to identify a rare germline variant that can be passed down to offspring and that might confer high risk for the disease.
"We used our high-risk families to study these types of changes, although they may be modified by other genes and environmental factors," Dr. McMaster explained.
Much to their collective disappointment, the research team has been unable to identify any rare germline variant that could account for WM in many families. What they did find were many small changes in genes that are known to be important in B-cell development and function, but all of those would lead to only a small increase in WM risk.
"What is holding us back is that, so far, we are not seeing the same gene affected in more than one family, so this suggests to us either that this is not the mechanism behind the development of WM in families, or we have an unfortunate situation where each family is going to have a genetic change that is private to that family and which is not found in other families," Dr. McMaster acknowledged.
Sheer difficulty
Given the difficulty of determining whether these small genetic changes had any detrimental functional effect in each and every family with a member who had WM, Dr. McMaster and colleagues have now turned their attention to genes that exert only a small effect on disease risk.
"Here, we focused on specific genes that we knew were important in the function of the immune system," she explained. "We did find a few genes that may contribute to risk, but those have not yet been confirmed by us or others, and we cannot say they are causative without that confirmation," she said.
The team has gone on to scan the highway of our genetic material so as to isolate genetic "mile markers." They then examine the area around a particular marker that they suspect contains genes that may be involved in WM.
One study they conducted involved a cohort of 217 patients with WM in which numerous family members had WM and so was enriched with susceptibility genes. A second cohort comprised 312 WM patients in which there were few WM cases among family members. Both of these cohorts were compared with a group of healthy control persons.
From these genome studies, "we found there are at least two regions of the genome that can contribute to WM susceptibility, the largest effect being on the short arm of chromosome 6, and the other on the long arm of chromosome 14," Dr. McMaster reported. Dr. McMaster feels that there are probably more regions on the genome that also contribute to WM, although they do not yet understand how these regions contribute to susceptibility.
"It's more evidence that WM likely results from a combination of events rather than one single gene variant," she observed. Dr. McMaster and colleagues are now collaborating with a large consortium of WM researchers to confirm and extend their findings. Plans are underway to analyze data from approximately 1,350 WM patients and more than 20,000 control persons within the next year.
"Our hope is that we will confirm our original findings and, because we now have a much larger sample, we will be able to discover additional regions of the genome that are contributing to susceptibility," Dr. McMaster said.
"A single gene is not likely to account for all WM, as we've looked carefully and others have looked too," she commented.
"So the risk for WM depends on a combination of genes and environmental exposures and possibly lifestyle factors as well, although we still estimate that approximately 25% of the heritability of WM can be attributed to these kinds of genetic changes," Dr. McMaster predicted.
Dr. McMaster has disclosed no relevant financial relationships. Dr. Treon has served as a director, officer, partner, employee, advisor, consultant, or trustee for Janssen, Pfizer, PCYC, and BioGene.
A version of this article first appeared on Medscape.com
Mary Lou McMaster, MD, has spent her entire career at the National Cancer Institute (NCI) searching for the genetic underpinnings that give rise to Waldenstrom's macroglobulinemia (WM).
After searching for decades, she has yet to uncover a "smoking gun," though a few tantalizing clues have emerged along the way.
"Our questions are pretty basic: Why are some people more susceptible to developing WM, and why does WM sometimes cluster in families?" she explained. It turns out that the answers are not at all simple.
Dr. McMaster described some of the clues that her team at the Clinical Genetics Branch of the NCI has unearthed in a presentation at the recent International Waldenstrom's Macroglobulinemia Foundation (IWMF) 2021 Virtual Educational Forum.
Commenting after the presentation, Steven Treon, MD, PhD, professor of medicine, Harvard Medical School, Boston, who is collaborating with Dr. McMaster on this work, said: "From these familial studies, we can learn how familial genomics may give us insights into disease prevention and treatment."
Identifying affected families
Work began in 2001 to identify families in which two or more family members had been diagnosed with WM or in which there was one patient with WM and at least one other relative with a related B-cell cancer, such as chronic lymphocytic leukemia.
For a frame of reference, they enrolled some families with only one member with WM and in which there was no known family history of the disease.
"Overall, we have learned that familial WM is a rare disease but not nearly as rare as we first thought," Dr. McMaster said.
For example, in a referral hospital setting, 5% of WM patients will report having a family member with the same disorder, and up to 20% of WM patients report having a family member with a related but different B-cell cancer, she noted.
NCI researchers also discovered that environmental factors contribute to the development of WM. Notable chemical or occupational exposures include exposures to pesticides, herbicides, and fertilizers. Infections and autoimmune disease are additional factors.
"This was not a surprise," Dr. McMaster commented regarding the role of occupational exposures. The research community has known for decades that a "lymphoma belt" cuts through the Midwest farming states.
Focusing on genetic susceptibility, Dr. McMaster and colleagues first tried to identify a rare germline variant that can be passed down to offspring and that might confer high risk for the disease.
"We used our high-risk families to study these types of changes, although they may be modified by other genes and environmental factors," Dr. McMaster explained.
Much to their collective disappointment, the research team has been unable to identify any rare germline variant that could account for WM in many families. What they did find were many small changes in genes that are known to be important in B-cell development and function, but all of those would lead to only a small increase in WM risk.
"What is holding us back is that, so far, we are not seeing the same gene affected in more than one family, so this suggests to us either that this is not the mechanism behind the development of WM in families, or we have an unfortunate situation where each family is going to have a genetic change that is private to that family and which is not found in other families," Dr. McMaster acknowledged.
Sheer difficulty
Given the difficulty of determining whether these small genetic changes had any detrimental functional effect in each and every family with a member who had WM, Dr. McMaster and colleagues have now turned their attention to genes that exert only a small effect on disease risk.
"Here, we focused on specific genes that we knew were important in the function of the immune system," she explained. "We did find a few genes that may contribute to risk, but those have not yet been confirmed by us or others, and we cannot say they are causative without that confirmation," she said.
The team has gone on to scan the highway of our genetic material so as to isolate genetic "mile markers." They then examine the area around a particular marker that they suspect contains genes that may be involved in WM.
One study they conducted involved a cohort of 217 patients with WM in which numerous family members had WM and so was enriched with susceptibility genes. A second cohort comprised 312 WM patients in which there were few WM cases among family members. Both of these cohorts were compared with a group of healthy control persons.
From these genome studies, "we found there are at least two regions of the genome that can contribute to WM susceptibility, the largest effect being on the short arm of chromosome 6, and the other on the long arm of chromosome 14," Dr. McMaster reported. Dr. McMaster feels that there are probably more regions on the genome that also contribute to WM, although they do not yet understand how these regions contribute to susceptibility.
"It's more evidence that WM likely results from a combination of events rather than one single gene variant," she observed. Dr. McMaster and colleagues are now collaborating with a large consortium of WM researchers to confirm and extend their findings. Plans are underway to analyze data from approximately 1,350 WM patients and more than 20,000 control persons within the next year.
"Our hope is that we will confirm our original findings and, because we now have a much larger sample, we will be able to discover additional regions of the genome that are contributing to susceptibility," Dr. McMaster said.
"A single gene is not likely to account for all WM, as we've looked carefully and others have looked too," she commented.
"So the risk for WM depends on a combination of genes and environmental exposures and possibly lifestyle factors as well, although we still estimate that approximately 25% of the heritability of WM can be attributed to these kinds of genetic changes," Dr. McMaster predicted.
Dr. McMaster has disclosed no relevant financial relationships. Dr. Treon has served as a director, officer, partner, employee, advisor, consultant, or trustee for Janssen, Pfizer, PCYC, and BioGene.
A version of this article first appeared on Medscape.com
Mary Lou McMaster, MD, has spent her entire career at the National Cancer Institute (NCI) searching for the genetic underpinnings that give rise to Waldenstrom's macroglobulinemia (WM).
After searching for decades, she has yet to uncover a "smoking gun," though a few tantalizing clues have emerged along the way.
"Our questions are pretty basic: Why are some people more susceptible to developing WM, and why does WM sometimes cluster in families?" she explained. It turns out that the answers are not at all simple.
Dr. McMaster described some of the clues that her team at the Clinical Genetics Branch of the NCI has unearthed in a presentation at the recent International Waldenstrom's Macroglobulinemia Foundation (IWMF) 2021 Virtual Educational Forum.
Commenting after the presentation, Steven Treon, MD, PhD, professor of medicine, Harvard Medical School, Boston, who is collaborating with Dr. McMaster on this work, said: "From these familial studies, we can learn how familial genomics may give us insights into disease prevention and treatment."
Identifying affected families
Work began in 2001 to identify families in which two or more family members had been diagnosed with WM or in which there was one patient with WM and at least one other relative with a related B-cell cancer, such as chronic lymphocytic leukemia.
For a frame of reference, they enrolled some families with only one member with WM and in which there was no known family history of the disease.
"Overall, we have learned that familial WM is a rare disease but not nearly as rare as we first thought," Dr. McMaster said.
For example, in a referral hospital setting, 5% of WM patients will report having a family member with the same disorder, and up to 20% of WM patients report having a family member with a related but different B-cell cancer, she noted.
NCI researchers also discovered that environmental factors contribute to the development of WM. Notable chemical or occupational exposures include exposures to pesticides, herbicides, and fertilizers. Infections and autoimmune disease are additional factors.
"This was not a surprise," Dr. McMaster commented regarding the role of occupational exposures. The research community has known for decades that a "lymphoma belt" cuts through the Midwest farming states.
Focusing on genetic susceptibility, Dr. McMaster and colleagues first tried to identify a rare germline variant that can be passed down to offspring and that might confer high risk for the disease.
"We used our high-risk families to study these types of changes, although they may be modified by other genes and environmental factors," Dr. McMaster explained.
Much to their collective disappointment, the research team has been unable to identify any rare germline variant that could account for WM in many families. What they did find were many small changes in genes that are known to be important in B-cell development and function, but all of those would lead to only a small increase in WM risk.
"What is holding us back is that, so far, we are not seeing the same gene affected in more than one family, so this suggests to us either that this is not the mechanism behind the development of WM in families, or we have an unfortunate situation where each family is going to have a genetic change that is private to that family and which is not found in other families," Dr. McMaster acknowledged.
Sheer difficulty
Given the difficulty of determining whether these small genetic changes had any detrimental functional effect in each and every family with a member who had WM, Dr. McMaster and colleagues have now turned their attention to genes that exert only a small effect on disease risk.
"Here, we focused on specific genes that we knew were important in the function of the immune system," she explained. "We did find a few genes that may contribute to risk, but those have not yet been confirmed by us or others, and we cannot say they are causative without that confirmation," she said.
The team has gone on to scan the highway of our genetic material so as to isolate genetic "mile markers." They then examine the area around a particular marker that they suspect contains genes that may be involved in WM.
One study they conducted involved a cohort of 217 patients with WM in which numerous family members had WM and so was enriched with susceptibility genes. A second cohort comprised 312 WM patients in which there were few WM cases among family members. Both of these cohorts were compared with a group of healthy control persons.
From these genome studies, "we found there are at least two regions of the genome that can contribute to WM susceptibility, the largest effect being on the short arm of chromosome 6, and the other on the long arm of chromosome 14," Dr. McMaster reported. Dr. McMaster feels that there are probably more regions on the genome that also contribute to WM, although they do not yet understand how these regions contribute to susceptibility.
"It's more evidence that WM likely results from a combination of events rather than one single gene variant," she observed. Dr. McMaster and colleagues are now collaborating with a large consortium of WM researchers to confirm and extend their findings. Plans are underway to analyze data from approximately 1,350 WM patients and more than 20,000 control persons within the next year.
"Our hope is that we will confirm our original findings and, because we now have a much larger sample, we will be able to discover additional regions of the genome that are contributing to susceptibility," Dr. McMaster said.
"A single gene is not likely to account for all WM, as we've looked carefully and others have looked too," she commented.
"So the risk for WM depends on a combination of genes and environmental exposures and possibly lifestyle factors as well, although we still estimate that approximately 25% of the heritability of WM can be attributed to these kinds of genetic changes," Dr. McMaster predicted.
Dr. McMaster has disclosed no relevant financial relationships. Dr. Treon has served as a director, officer, partner, employee, advisor, consultant, or trustee for Janssen, Pfizer, PCYC, and BioGene.
A version of this article first appeared on Medscape.com
Orbital Varix Masquerading as an Intraorbital Lymphoma
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
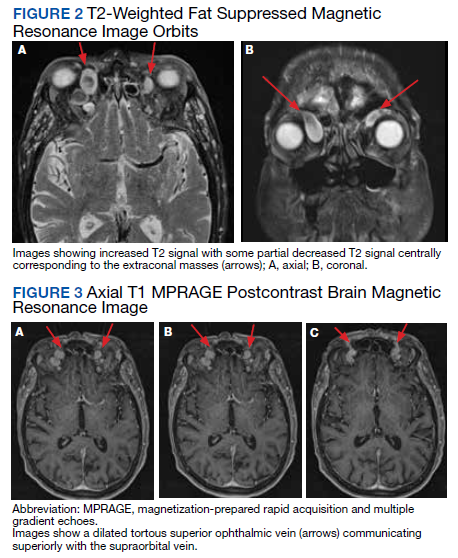
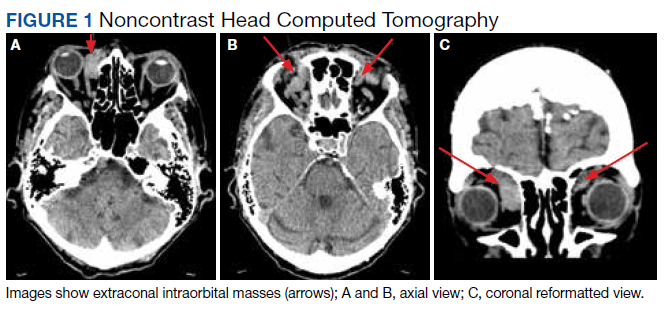
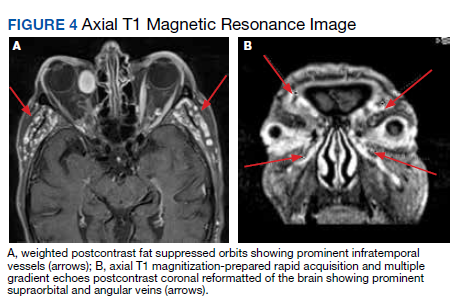
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
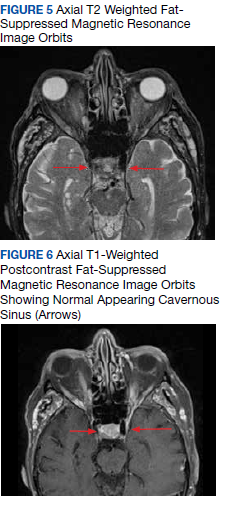
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
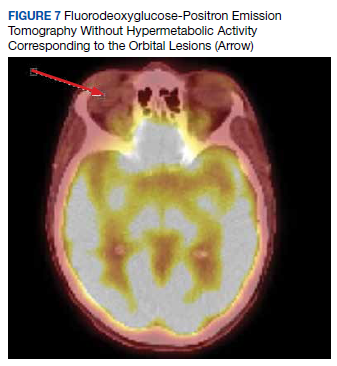
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
Evans’ Syndrome in Undiagnosed Small Lymphocytic Lymphoma: Case Report and Literature Review
Background
Evans’ syndrome is a rare entity characterized by concomitant or sequential multilineage cytopenia particularly autoimmune hemolytic anemia, ITP and very rarely autoimmune neutropenia. Although more common in young adults, it can occur in elderly usually associated with malignancies like CLL.
Case Report
A 74 years old Veteran presented with complaints of fatigue and worsening dyspnea on exertion. His physical exam was unremarkable except jaundice. His labs were significant for macrocytic anemia with Hemoglobin of 7.4g/dl compared to 11.7g/dl 6 months prior, MCV 106.9 fL, LDH 809U/L, indirect bilirubin 4.1mg/dl, absolute reticulocyte 0.16M/uL, Haptoglobin <15mg/dl and Positive DAT. Platelets were mildly decreased at 111K/ul. No lymphocytosis was noted. Initially, the hemolysis was thought to be cephalosporin- related given that the patient had taken cephalexin recently for cellulitis. As part of the workup for anemia, the patient underwent EGD and colonoscopy which was initially unrevealing. However, random biopsies from the descending colon and terminal ileum returned with a small lymphocytic infiltrate consistent with SLL/CLL. Cytogenetics showed trisomy-12 which is associated with intermediate prognosis for CLL. PET scan done subsequently revealed only a reactive marrow and an enlarged 15.8cm non-hypermetabolic spleen. This veteran having anemia, positive DAT, thrombocytopenia, and splenomegaly got diagnosed with Evans’s syndrome. This syndrome was the initial manifestation of his underlying CLL. We started the patient on a prednisone taper for 4 weeks to which anemia and thrombocytopenia barely responded, ultimately Rituximab 375mg/m2 x4 weekly doses was started which led to complete resolution of anemia and thrombocytopenia. We closely followed the patient and monitored CBC and hemolytic markers. The patient relapsed in two years which was subsequently managed with another course of Rituximab 375mg/m2 x4 weekly doses.
Conclusions
This case report aims to call attention to this relatively rare entity which is difficult to treat and often associated with frequent relapses. Though rare, physicians should maintain high suspicion for this syndrome in patients with multi-lineage cytopenia which are usually not even responding well to the common treatment for cytopenia. Furthermore, there is room for improvement in Evans’ syndrome management since mortality remains higher in these patients than in those with isolated autoimmuce cytopenias.
Background
Evans’ syndrome is a rare entity characterized by concomitant or sequential multilineage cytopenia particularly autoimmune hemolytic anemia, ITP and very rarely autoimmune neutropenia. Although more common in young adults, it can occur in elderly usually associated with malignancies like CLL.
Case Report
A 74 years old Veteran presented with complaints of fatigue and worsening dyspnea on exertion. His physical exam was unremarkable except jaundice. His labs were significant for macrocytic anemia with Hemoglobin of 7.4g/dl compared to 11.7g/dl 6 months prior, MCV 106.9 fL, LDH 809U/L, indirect bilirubin 4.1mg/dl, absolute reticulocyte 0.16M/uL, Haptoglobin <15mg/dl and Positive DAT. Platelets were mildly decreased at 111K/ul. No lymphocytosis was noted. Initially, the hemolysis was thought to be cephalosporin- related given that the patient had taken cephalexin recently for cellulitis. As part of the workup for anemia, the patient underwent EGD and colonoscopy which was initially unrevealing. However, random biopsies from the descending colon and terminal ileum returned with a small lymphocytic infiltrate consistent with SLL/CLL. Cytogenetics showed trisomy-12 which is associated with intermediate prognosis for CLL. PET scan done subsequently revealed only a reactive marrow and an enlarged 15.8cm non-hypermetabolic spleen. This veteran having anemia, positive DAT, thrombocytopenia, and splenomegaly got diagnosed with Evans’s syndrome. This syndrome was the initial manifestation of his underlying CLL. We started the patient on a prednisone taper for 4 weeks to which anemia and thrombocytopenia barely responded, ultimately Rituximab 375mg/m2 x4 weekly doses was started which led to complete resolution of anemia and thrombocytopenia. We closely followed the patient and monitored CBC and hemolytic markers. The patient relapsed in two years which was subsequently managed with another course of Rituximab 375mg/m2 x4 weekly doses.
Conclusions
This case report aims to call attention to this relatively rare entity which is difficult to treat and often associated with frequent relapses. Though rare, physicians should maintain high suspicion for this syndrome in patients with multi-lineage cytopenia which are usually not even responding well to the common treatment for cytopenia. Furthermore, there is room for improvement in Evans’ syndrome management since mortality remains higher in these patients than in those with isolated autoimmuce cytopenias.
Background
Evans’ syndrome is a rare entity characterized by concomitant or sequential multilineage cytopenia particularly autoimmune hemolytic anemia, ITP and very rarely autoimmune neutropenia. Although more common in young adults, it can occur in elderly usually associated with malignancies like CLL.
Case Report
A 74 years old Veteran presented with complaints of fatigue and worsening dyspnea on exertion. His physical exam was unremarkable except jaundice. His labs were significant for macrocytic anemia with Hemoglobin of 7.4g/dl compared to 11.7g/dl 6 months prior, MCV 106.9 fL, LDH 809U/L, indirect bilirubin 4.1mg/dl, absolute reticulocyte 0.16M/uL, Haptoglobin <15mg/dl and Positive DAT. Platelets were mildly decreased at 111K/ul. No lymphocytosis was noted. Initially, the hemolysis was thought to be cephalosporin- related given that the patient had taken cephalexin recently for cellulitis. As part of the workup for anemia, the patient underwent EGD and colonoscopy which was initially unrevealing. However, random biopsies from the descending colon and terminal ileum returned with a small lymphocytic infiltrate consistent with SLL/CLL. Cytogenetics showed trisomy-12 which is associated with intermediate prognosis for CLL. PET scan done subsequently revealed only a reactive marrow and an enlarged 15.8cm non-hypermetabolic spleen. This veteran having anemia, positive DAT, thrombocytopenia, and splenomegaly got diagnosed with Evans’s syndrome. This syndrome was the initial manifestation of his underlying CLL. We started the patient on a prednisone taper for 4 weeks to which anemia and thrombocytopenia barely responded, ultimately Rituximab 375mg/m2 x4 weekly doses was started which led to complete resolution of anemia and thrombocytopenia. We closely followed the patient and monitored CBC and hemolytic markers. The patient relapsed in two years which was subsequently managed with another course of Rituximab 375mg/m2 x4 weekly doses.
Conclusions
This case report aims to call attention to this relatively rare entity which is difficult to treat and often associated with frequent relapses. Though rare, physicians should maintain high suspicion for this syndrome in patients with multi-lineage cytopenia which are usually not even responding well to the common treatment for cytopenia. Furthermore, there is room for improvement in Evans’ syndrome management since mortality remains higher in these patients than in those with isolated autoimmuce cytopenias.
Hemophagocytic Lymphohistiocytosis: Early Treatment Leading to an Excellent Outcome
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
Targeted CLL treatments found effective in managing associated autoimmune cytopenias
Newer targeted drugs for chronic lymphocytic leukemia also appear to have a beneficial impact on underlying autoimmune cytopenias (AICs), results of a large retrospective study suggest.
Many autoimmune cytopenias improved or resolved during treatment with ibrutinib, idelalisib, or venetoclax, according to authors of the study, which appears in the journal Blood.
Treatment-emergent autoimmune cytopenias were seen in a “negligible portion” of patients overall, according to the report. The prevalence was about 1% each for patients treated with ibrutinib or idelalisib, though seen more frequently (at 7%) among patients who received venetoclax.
Nevertheless, treatment-emergent autoimmune cytopenias were “easily manageable” without interventions such as steroids and rituximab, and without need to interrupt the targeted treatment for chronic lymphocytic leukemia (CLL), according to the authors, led by Candida Vitale, MD, PhD, of the department of molecular biotechnology and health sciences at the University of Torino in Italy.
“Ibrutinib, idelalisib, and venetoclax have a beneficial impact on CLL-related preexisting AICs, achieving in most patients, in parallel with the consolidated antitumor efficacy, an effective control of the autoimmune phenomena,” Dr. Vitale and coauthors wrote in their report.
Study results
The retrospective study included 815 patients, of whom 572 were treated with ibrutinib, 143 with idelalisib plus rituximab, and 100 with venetoclax. Nine percent of ibrutinib-treated patients and 12% of venetoclax-treated patients also received an anti-CD20 monoclonal antibody, rituximab or obinutuzumab.
One hundred and four patients (13%) had preexisting autoimmune cytopenias, though the majority were resolved or controlled at the time targeted therapy was started.
Of patients with autoimmune cytopenias that were unresolved at the beginning of targeted therapy, 80% improved or resolved after starting targeted treatment, authors reported.
Most patients who developed autoimmune cytopenias on treatment had high-risk features such as unmutated IGHV, del(17)p, or TP53 mutation, according to the report.
Those treatment-emergent autoimmune cytopenias were seen in 1% of the ibrutinib group, 0.9% of the idelalisib group, and 7% of the venetoclax group. Out of 12 total treatment-emergent autoimmune cytopenias, all but 2 were resolved or controlled with dose reductions or temporary suspensions and use of steroids or rituximab, the report shows.
The higher incidence of autoimmune cytopenias in the venetoclax group held steady even when considering just the patients who had relapsed/refractory disease or had at least two prior lines of therapy, suggesting a “more meaningful” incidence, compared to what was observed for ibrutinib and idelalisib, the investigators said.
“However, the risk of autoimmune cytopenia episodes should not limit the use of venetoclax, considering the strong efficacy of this drug in treating patients with CLL, including those with high-risk features, and the possibility of effectively managing autoimmune complications, mostly without treatment interruption,” they concluded in their report.
Implications for patients with CLL
Findings of this study indicate that targeted therapies are effective for managing both CLL and autoimmune cytopenias, according to Carol Moreno, MD, PhD, of Hospital Sant Pau in Barcelona.

Moreover, the targeted therapies do not appear to change the overall prevalence of autoimmune cytopenias, compared to untreated patients, Dr. Moreno said in a commentary on the findings also published in Blood.
“These results are consistent with the concept that targeted therapies are not associated with a higher risk of autoimmune cytopenias, and that, if present, can be managed with immunosuppressive agents,” she wrote.
Autoimmune cytopenias and CLL
Autoimmune cytopenias are a relatively common complication of CLL, occurring in 5%-9% of CLL patients, Dr. Vitale and coauthors said in their report. The most common presentations include autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP).
Evidence from earlier studies suggests that treatment for CLL may trigger autoimmune cytopenias. Results of retrospective studies in the 1990s linked single-agent fludarabine to increased risk of AIHA, Dr. Moreno said in the commentary.
However, subsequent studies showed that fludarabine plus cyclophosphamide (FC) and fludarabine, cyclophosphamide, and rituximab (FCR) were associated with low proportions of AIHA.
“Taken together, these results convincingly suggest that rather than treatment, it is the lack of response to it that conveys a higher risk of AIC,” Dr. Moreno wrote.
Management considerations
There are currently no clinical practice guidelines that advise on how to manage patients who develop AICs during targeted treatment for CLL, Dr. Vitale and colleagues said in their report.
However, this new study data may help inform management of patients with CLL and an autoimmune cytopenia, Dr. Moreno said in the commentary.
If the patient doesn’t immediately require CLL treatment, patients can be managed according to existing guidelines for AIHA and ITP, she said. “Nonresponding patients should be given CLL therapy,” she added.
For CLL patients who do require therapy and have a preexisting or treatment-emergent AIC, a “CLL-oriented” treatment approach could be considered, according to Dr. Moreno.
“A reasonable approach consists of a short course (2 to 4 weeks) of corticosteroids followed by effective CLL therapy (i.e., FCR, bendamustine plus rituximab or ibrutinib), depending on the clinical situation,” she added.
Dr. Vitale reported receiving consultancy fees from Janssen outside the submitted work. Dr. Moreno declared no competing financial interests related to her commentary.
Newer targeted drugs for chronic lymphocytic leukemia also appear to have a beneficial impact on underlying autoimmune cytopenias (AICs), results of a large retrospective study suggest.
Many autoimmune cytopenias improved or resolved during treatment with ibrutinib, idelalisib, or venetoclax, according to authors of the study, which appears in the journal Blood.
Treatment-emergent autoimmune cytopenias were seen in a “negligible portion” of patients overall, according to the report. The prevalence was about 1% each for patients treated with ibrutinib or idelalisib, though seen more frequently (at 7%) among patients who received venetoclax.
Nevertheless, treatment-emergent autoimmune cytopenias were “easily manageable” without interventions such as steroids and rituximab, and without need to interrupt the targeted treatment for chronic lymphocytic leukemia (CLL), according to the authors, led by Candida Vitale, MD, PhD, of the department of molecular biotechnology and health sciences at the University of Torino in Italy.
“Ibrutinib, idelalisib, and venetoclax have a beneficial impact on CLL-related preexisting AICs, achieving in most patients, in parallel with the consolidated antitumor efficacy, an effective control of the autoimmune phenomena,” Dr. Vitale and coauthors wrote in their report.
Study results
The retrospective study included 815 patients, of whom 572 were treated with ibrutinib, 143 with idelalisib plus rituximab, and 100 with venetoclax. Nine percent of ibrutinib-treated patients and 12% of venetoclax-treated patients also received an anti-CD20 monoclonal antibody, rituximab or obinutuzumab.
One hundred and four patients (13%) had preexisting autoimmune cytopenias, though the majority were resolved or controlled at the time targeted therapy was started.
Of patients with autoimmune cytopenias that were unresolved at the beginning of targeted therapy, 80% improved or resolved after starting targeted treatment, authors reported.
Most patients who developed autoimmune cytopenias on treatment had high-risk features such as unmutated IGHV, del(17)p, or TP53 mutation, according to the report.
Those treatment-emergent autoimmune cytopenias were seen in 1% of the ibrutinib group, 0.9% of the idelalisib group, and 7% of the venetoclax group. Out of 12 total treatment-emergent autoimmune cytopenias, all but 2 were resolved or controlled with dose reductions or temporary suspensions and use of steroids or rituximab, the report shows.
The higher incidence of autoimmune cytopenias in the venetoclax group held steady even when considering just the patients who had relapsed/refractory disease or had at least two prior lines of therapy, suggesting a “more meaningful” incidence, compared to what was observed for ibrutinib and idelalisib, the investigators said.
“However, the risk of autoimmune cytopenia episodes should not limit the use of venetoclax, considering the strong efficacy of this drug in treating patients with CLL, including those with high-risk features, and the possibility of effectively managing autoimmune complications, mostly without treatment interruption,” they concluded in their report.
Implications for patients with CLL
Findings of this study indicate that targeted therapies are effective for managing both CLL and autoimmune cytopenias, according to Carol Moreno, MD, PhD, of Hospital Sant Pau in Barcelona.
Moreover, the targeted therapies do not appear to change the overall prevalence of autoimmune cytopenias, compared to untreated patients, Dr. Moreno said in a commentary on the findings also published in Blood.
“These results are consistent with the concept that targeted therapies are not associated with a higher risk of autoimmune cytopenias, and that, if present, can be managed with immunosuppressive agents,” she wrote.
Autoimmune cytopenias and CLL
Autoimmune cytopenias are a relatively common complication of CLL, occurring in 5%-9% of CLL patients, Dr. Vitale and coauthors said in their report. The most common presentations include autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP).
Evidence from earlier studies suggests that treatment for CLL may trigger autoimmune cytopenias. Results of retrospective studies in the 1990s linked single-agent fludarabine to increased risk of AIHA, Dr. Moreno said in the commentary.
However, subsequent studies showed that fludarabine plus cyclophosphamide (FC) and fludarabine, cyclophosphamide, and rituximab (FCR) were associated with low proportions of AIHA.
“Taken together, these results convincingly suggest that rather than treatment, it is the lack of response to it that conveys a higher risk of AIC,” Dr. Moreno wrote.
Management considerations
There are currently no clinical practice guidelines that advise on how to manage patients who develop AICs during targeted treatment for CLL, Dr. Vitale and colleagues said in their report.
However, this new study data may help inform management of patients with CLL and an autoimmune cytopenia, Dr. Moreno said in the commentary.
If the patient doesn’t immediately require CLL treatment, patients can be managed according to existing guidelines for AIHA and ITP, she said. “Nonresponding patients should be given CLL therapy,” she added.
For CLL patients who do require therapy and have a preexisting or treatment-emergent AIC, a “CLL-oriented” treatment approach could be considered, according to Dr. Moreno.
“A reasonable approach consists of a short course (2 to 4 weeks) of corticosteroids followed by effective CLL therapy (i.e., FCR, bendamustine plus rituximab or ibrutinib), depending on the clinical situation,” she added.
Dr. Vitale reported receiving consultancy fees from Janssen outside the submitted work. Dr. Moreno declared no competing financial interests related to her commentary.
Newer targeted drugs for chronic lymphocytic leukemia also appear to have a beneficial impact on underlying autoimmune cytopenias (AICs), results of a large retrospective study suggest.
Many autoimmune cytopenias improved or resolved during treatment with ibrutinib, idelalisib, or venetoclax, according to authors of the study, which appears in the journal Blood.
Treatment-emergent autoimmune cytopenias were seen in a “negligible portion” of patients overall, according to the report. The prevalence was about 1% each for patients treated with ibrutinib or idelalisib, though seen more frequently (at 7%) among patients who received venetoclax.
Nevertheless, treatment-emergent autoimmune cytopenias were “easily manageable” without interventions such as steroids and rituximab, and without need to interrupt the targeted treatment for chronic lymphocytic leukemia (CLL), according to the authors, led by Candida Vitale, MD, PhD, of the department of molecular biotechnology and health sciences at the University of Torino in Italy.
“Ibrutinib, idelalisib, and venetoclax have a beneficial impact on CLL-related preexisting AICs, achieving in most patients, in parallel with the consolidated antitumor efficacy, an effective control of the autoimmune phenomena,” Dr. Vitale and coauthors wrote in their report.
Study results
The retrospective study included 815 patients, of whom 572 were treated with ibrutinib, 143 with idelalisib plus rituximab, and 100 with venetoclax. Nine percent of ibrutinib-treated patients and 12% of venetoclax-treated patients also received an anti-CD20 monoclonal antibody, rituximab or obinutuzumab.
One hundred and four patients (13%) had preexisting autoimmune cytopenias, though the majority were resolved or controlled at the time targeted therapy was started.
Of patients with autoimmune cytopenias that were unresolved at the beginning of targeted therapy, 80% improved or resolved after starting targeted treatment, authors reported.
Most patients who developed autoimmune cytopenias on treatment had high-risk features such as unmutated IGHV, del(17)p, or TP53 mutation, according to the report.
Those treatment-emergent autoimmune cytopenias were seen in 1% of the ibrutinib group, 0.9% of the idelalisib group, and 7% of the venetoclax group. Out of 12 total treatment-emergent autoimmune cytopenias, all but 2 were resolved or controlled with dose reductions or temporary suspensions and use of steroids or rituximab, the report shows.
The higher incidence of autoimmune cytopenias in the venetoclax group held steady even when considering just the patients who had relapsed/refractory disease or had at least two prior lines of therapy, suggesting a “more meaningful” incidence, compared to what was observed for ibrutinib and idelalisib, the investigators said.
“However, the risk of autoimmune cytopenia episodes should not limit the use of venetoclax, considering the strong efficacy of this drug in treating patients with CLL, including those with high-risk features, and the possibility of effectively managing autoimmune complications, mostly without treatment interruption,” they concluded in their report.
Implications for patients with CLL
Findings of this study indicate that targeted therapies are effective for managing both CLL and autoimmune cytopenias, according to Carol Moreno, MD, PhD, of Hospital Sant Pau in Barcelona.
Moreover, the targeted therapies do not appear to change the overall prevalence of autoimmune cytopenias, compared to untreated patients, Dr. Moreno said in a commentary on the findings also published in Blood.
“These results are consistent with the concept that targeted therapies are not associated with a higher risk of autoimmune cytopenias, and that, if present, can be managed with immunosuppressive agents,” she wrote.
Autoimmune cytopenias and CLL
Autoimmune cytopenias are a relatively common complication of CLL, occurring in 5%-9% of CLL patients, Dr. Vitale and coauthors said in their report. The most common presentations include autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP).
Evidence from earlier studies suggests that treatment for CLL may trigger autoimmune cytopenias. Results of retrospective studies in the 1990s linked single-agent fludarabine to increased risk of AIHA, Dr. Moreno said in the commentary.
However, subsequent studies showed that fludarabine plus cyclophosphamide (FC) and fludarabine, cyclophosphamide, and rituximab (FCR) were associated with low proportions of AIHA.
“Taken together, these results convincingly suggest that rather than treatment, it is the lack of response to it that conveys a higher risk of AIC,” Dr. Moreno wrote.
Management considerations
There are currently no clinical practice guidelines that advise on how to manage patients who develop AICs during targeted treatment for CLL, Dr. Vitale and colleagues said in their report.
However, this new study data may help inform management of patients with CLL and an autoimmune cytopenia, Dr. Moreno said in the commentary.
If the patient doesn’t immediately require CLL treatment, patients can be managed according to existing guidelines for AIHA and ITP, she said. “Nonresponding patients should be given CLL therapy,” she added.
For CLL patients who do require therapy and have a preexisting or treatment-emergent AIC, a “CLL-oriented” treatment approach could be considered, according to Dr. Moreno.
“A reasonable approach consists of a short course (2 to 4 weeks) of corticosteroids followed by effective CLL therapy (i.e., FCR, bendamustine plus rituximab or ibrutinib), depending on the clinical situation,” she added.
Dr. Vitale reported receiving consultancy fees from Janssen outside the submitted work. Dr. Moreno declared no competing financial interests related to her commentary.
FROM BLOOD
Experimental antibody-drug conjugate shown active against r/r DLBCL
Patients with relapsed or refractory B-cell non-Hodgkin lymphomas who are not candidates for hematopoietic stem cell transplant have a generally poor prognosis and few treatment options, but an experimental combination of the antibody-drug conjugate naratuximab with rituximab showed promising efficacy and acceptable safety in these patients in a phase 2 trial.
Among patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) the combination was associated with a 44.7% overall response rate, including 31.6% complete responses, and two-thirds of patients had responses lasting more than 12 months, reported Moshe Yair Levy, MD, from Texas Oncology–Baylor Charles A Sammons Cancer Center in Dallas.
“This is, in my viewpoint, very exciting therapy,” he said in a question-and-answer session following his presentation of the data in a late-breaking abstract session during the European Hematology Association annual congress. (Abstract LB1903).
Naratuximab emtansine is an investigational antibody-drug conjugate (ADC) consisting of a humanized monoclonal antibody against CD37, a surface marker on B lymphocytes that is highly expressed in non-Hodgkin lymphoma (NHL), conjugated to a cytotoxic derivative of maitansine.
CD37 is also an internalizable cell-surface antigen, making it an attractive candidate for an ADC approach.
In a phase 1 trial, naratuximab monotherapy showed a good safety profile and a 22% overall response rate, Dr. Levy noted.
“What they found is that, if you coadminister this ADC with rituximab, you’re actually going to get more internalization of the CD37 monoclonal, therefore more payload delivered to your target cells,” he said.
He reported results of a multicenter, adaptive phase 2 study of the combination in patients with DLBCL and other relapsed/refractory NHL.
DLBCL and others
The trial was divided into two parts, with the first consisting of a safety run-in phase with expansion in patients with confirmed diagnoses of relapsed/refractory NHL, including DLBCL, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma.
Patients with double- or triple-hit disease (with translocations in MYC plus either BCL2 and/or BCL6), bulky disease, or transformed lymphoma were eligible.
The second part consisted of two cohorts of patients with DLBCL treated with naratuximab and rituximab either weekly or every 3 weeks.
All patients in the study had received one to six prior lines of therapy, and had Eastern Cooperative Oncology Group performance status of 0-2. Patients with CNS lymphomas or prior anti-CD37 targeting therapy were excluded.
The safety population included 50 patients with DLBCL assigned to therapy every 3 weeks, 30 assigned to weekly therapy, and 20 patients with other NHL.
DLBCL efficacy
A total of 76 patients with DLBCL were evaluable for efficacy.
The ORR was 44% for patients in both the weekly and every 3 week cohorts, with 31.6% having complete responses.
Among 61 patients with nonbulky disease (longest diameter 7.5 cm or less), the ORR was 50.8%, and among 28 patients who had three or more prior lines of therapy the ORR was 46.4%, with 32.1% having a complete response.
Among responders followed for a median of 15 months, the median duration of response was not reached, and 66% had responses lasting beyond 12 months.
In the weekly dosing DLBCL cohort, 53.3% of patients discontinued treatment of both study drugs because of disease progression, as did 58% of those in the every 3 week cohort, and 30% of patients with other lymphomas. Only eight patients discontinued the combination because of treatment-emergent adverse events. Six patients had treatment-emergent adverse events leading to naratuximab dose reduction.
The most common grade 3 or 4 adverse events were neutropenia, leukopenia, lymphopenia and thrombocytopenias. Dr. Levy commented that the use of granulocyte colony-stimulating factor, which was not mandatory in the study, would likely have lowered the incidence of cytopenias.
There were 10 deaths during the study, 2 of which were considered to be treatment related, occurring in 1 patient each in the DLBCL dosing cohorts; 1 of the patients died from pneumonitis, and the other from left ventricular heart failure.
Other patients deaths were attributed to non–treatment-related cardiac arrest, acute renal failure, exacerbation of chronic heart failure, respiratory failure, multiorgan failure, lung infection, or colon adenocarcinoma.
Q 3 weeks suffices
In the question-and-answer session following the presentation, Kenny Lei, MD, from the Chinese University of Hong Kong asked Dr. Levy what the half-life of naratuximab is, and what was the investigator’s rationale for testing a weekly dosing schedule.
“I think the reason they checked the two different regimens, the Q week and the Q 3-week group, is that they noted that [naratuximab] was cleared relatively quickly, and they wanted to see whether or not, by giving Q weekly, when you get a continuous CD37 site occupancy if they would have a better outcome. But as you saw, in the groups there was really no clinically relevant difference in outcome,” Dr. Levy said.
Andrew Davies, MD, PhD, from the University of Southampton (England), asked whether the neutropenia seen in the study was related to myeloid expression of the target of from the off-target deconjugated payload.
“I don’t know that I necessarily have the answer to that,” Dr. Levy replied. “Remember there is the CD20 monoclonal rituximab which we know can cause neutropenia, as well as the CD37 and the target payload. I don’t know if we have enough information to attribute it to one specific component of the therapy,” he said.
The study was funded by Debiopharm International. Dr. Levy disclosed speaker activities for multiple companies, not including Debiopharm. Dr. Lei and Dr. Davies had no disclosures relevant to the study.
Patients with relapsed or refractory B-cell non-Hodgkin lymphomas who are not candidates for hematopoietic stem cell transplant have a generally poor prognosis and few treatment options, but an experimental combination of the antibody-drug conjugate naratuximab with rituximab showed promising efficacy and acceptable safety in these patients in a phase 2 trial.
Among patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) the combination was associated with a 44.7% overall response rate, including 31.6% complete responses, and two-thirds of patients had responses lasting more than 12 months, reported Moshe Yair Levy, MD, from Texas Oncology–Baylor Charles A Sammons Cancer Center in Dallas.
“This is, in my viewpoint, very exciting therapy,” he said in a question-and-answer session following his presentation of the data in a late-breaking abstract session during the European Hematology Association annual congress. (Abstract LB1903).
Naratuximab emtansine is an investigational antibody-drug conjugate (ADC) consisting of a humanized monoclonal antibody against CD37, a surface marker on B lymphocytes that is highly expressed in non-Hodgkin lymphoma (NHL), conjugated to a cytotoxic derivative of maitansine.
CD37 is also an internalizable cell-surface antigen, making it an attractive candidate for an ADC approach.
In a phase 1 trial, naratuximab monotherapy showed a good safety profile and a 22% overall response rate, Dr. Levy noted.
“What they found is that, if you coadminister this ADC with rituximab, you’re actually going to get more internalization of the CD37 monoclonal, therefore more payload delivered to your target cells,” he said.
He reported results of a multicenter, adaptive phase 2 study of the combination in patients with DLBCL and other relapsed/refractory NHL.
DLBCL and others
The trial was divided into two parts, with the first consisting of a safety run-in phase with expansion in patients with confirmed diagnoses of relapsed/refractory NHL, including DLBCL, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma.
Patients with double- or triple-hit disease (with translocations in MYC plus either BCL2 and/or BCL6), bulky disease, or transformed lymphoma were eligible.
The second part consisted of two cohorts of patients with DLBCL treated with naratuximab and rituximab either weekly or every 3 weeks.
All patients in the study had received one to six prior lines of therapy, and had Eastern Cooperative Oncology Group performance status of 0-2. Patients with CNS lymphomas or prior anti-CD37 targeting therapy were excluded.
The safety population included 50 patients with DLBCL assigned to therapy every 3 weeks, 30 assigned to weekly therapy, and 20 patients with other NHL.
DLBCL efficacy
A total of 76 patients with DLBCL were evaluable for efficacy.
The ORR was 44% for patients in both the weekly and every 3 week cohorts, with 31.6% having complete responses.
Among 61 patients with nonbulky disease (longest diameter 7.5 cm or less), the ORR was 50.8%, and among 28 patients who had three or more prior lines of therapy the ORR was 46.4%, with 32.1% having a complete response.
Among responders followed for a median of 15 months, the median duration of response was not reached, and 66% had responses lasting beyond 12 months.
In the weekly dosing DLBCL cohort, 53.3% of patients discontinued treatment of both study drugs because of disease progression, as did 58% of those in the every 3 week cohort, and 30% of patients with other lymphomas. Only eight patients discontinued the combination because of treatment-emergent adverse events. Six patients had treatment-emergent adverse events leading to naratuximab dose reduction.
The most common grade 3 or 4 adverse events were neutropenia, leukopenia, lymphopenia and thrombocytopenias. Dr. Levy commented that the use of granulocyte colony-stimulating factor, which was not mandatory in the study, would likely have lowered the incidence of cytopenias.
There were 10 deaths during the study, 2 of which were considered to be treatment related, occurring in 1 patient each in the DLBCL dosing cohorts; 1 of the patients died from pneumonitis, and the other from left ventricular heart failure.
Other patients deaths were attributed to non–treatment-related cardiac arrest, acute renal failure, exacerbation of chronic heart failure, respiratory failure, multiorgan failure, lung infection, or colon adenocarcinoma.
Q 3 weeks suffices
In the question-and-answer session following the presentation, Kenny Lei, MD, from the Chinese University of Hong Kong asked Dr. Levy what the half-life of naratuximab is, and what was the investigator’s rationale for testing a weekly dosing schedule.
“I think the reason they checked the two different regimens, the Q week and the Q 3-week group, is that they noted that [naratuximab] was cleared relatively quickly, and they wanted to see whether or not, by giving Q weekly, when you get a continuous CD37 site occupancy if they would have a better outcome. But as you saw, in the groups there was really no clinically relevant difference in outcome,” Dr. Levy said.
Andrew Davies, MD, PhD, from the University of Southampton (England), asked whether the neutropenia seen in the study was related to myeloid expression of the target of from the off-target deconjugated payload.
“I don’t know that I necessarily have the answer to that,” Dr. Levy replied. “Remember there is the CD20 monoclonal rituximab which we know can cause neutropenia, as well as the CD37 and the target payload. I don’t know if we have enough information to attribute it to one specific component of the therapy,” he said.
The study was funded by Debiopharm International. Dr. Levy disclosed speaker activities for multiple companies, not including Debiopharm. Dr. Lei and Dr. Davies had no disclosures relevant to the study.
Patients with relapsed or refractory B-cell non-Hodgkin lymphomas who are not candidates for hematopoietic stem cell transplant have a generally poor prognosis and few treatment options, but an experimental combination of the antibody-drug conjugate naratuximab with rituximab showed promising efficacy and acceptable safety in these patients in a phase 2 trial.
Among patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) the combination was associated with a 44.7% overall response rate, including 31.6% complete responses, and two-thirds of patients had responses lasting more than 12 months, reported Moshe Yair Levy, MD, from Texas Oncology–Baylor Charles A Sammons Cancer Center in Dallas.
“This is, in my viewpoint, very exciting therapy,” he said in a question-and-answer session following his presentation of the data in a late-breaking abstract session during the European Hematology Association annual congress. (Abstract LB1903).
Naratuximab emtansine is an investigational antibody-drug conjugate (ADC) consisting of a humanized monoclonal antibody against CD37, a surface marker on B lymphocytes that is highly expressed in non-Hodgkin lymphoma (NHL), conjugated to a cytotoxic derivative of maitansine.
CD37 is also an internalizable cell-surface antigen, making it an attractive candidate for an ADC approach.
In a phase 1 trial, naratuximab monotherapy showed a good safety profile and a 22% overall response rate, Dr. Levy noted.
“What they found is that, if you coadminister this ADC with rituximab, you’re actually going to get more internalization of the CD37 monoclonal, therefore more payload delivered to your target cells,” he said.
He reported results of a multicenter, adaptive phase 2 study of the combination in patients with DLBCL and other relapsed/refractory NHL.
DLBCL and others
The trial was divided into two parts, with the first consisting of a safety run-in phase with expansion in patients with confirmed diagnoses of relapsed/refractory NHL, including DLBCL, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma.
Patients with double- or triple-hit disease (with translocations in MYC plus either BCL2 and/or BCL6), bulky disease, or transformed lymphoma were eligible.
The second part consisted of two cohorts of patients with DLBCL treated with naratuximab and rituximab either weekly or every 3 weeks.
All patients in the study had received one to six prior lines of therapy, and had Eastern Cooperative Oncology Group performance status of 0-2. Patients with CNS lymphomas or prior anti-CD37 targeting therapy were excluded.
The safety population included 50 patients with DLBCL assigned to therapy every 3 weeks, 30 assigned to weekly therapy, and 20 patients with other NHL.
DLBCL efficacy
A total of 76 patients with DLBCL were evaluable for efficacy.
The ORR was 44% for patients in both the weekly and every 3 week cohorts, with 31.6% having complete responses.
Among 61 patients with nonbulky disease (longest diameter 7.5 cm or less), the ORR was 50.8%, and among 28 patients who had three or more prior lines of therapy the ORR was 46.4%, with 32.1% having a complete response.
Among responders followed for a median of 15 months, the median duration of response was not reached, and 66% had responses lasting beyond 12 months.
In the weekly dosing DLBCL cohort, 53.3% of patients discontinued treatment of both study drugs because of disease progression, as did 58% of those in the every 3 week cohort, and 30% of patients with other lymphomas. Only eight patients discontinued the combination because of treatment-emergent adverse events. Six patients had treatment-emergent adverse events leading to naratuximab dose reduction.
The most common grade 3 or 4 adverse events were neutropenia, leukopenia, lymphopenia and thrombocytopenias. Dr. Levy commented that the use of granulocyte colony-stimulating factor, which was not mandatory in the study, would likely have lowered the incidence of cytopenias.
There were 10 deaths during the study, 2 of which were considered to be treatment related, occurring in 1 patient each in the DLBCL dosing cohorts; 1 of the patients died from pneumonitis, and the other from left ventricular heart failure.
Other patients deaths were attributed to non–treatment-related cardiac arrest, acute renal failure, exacerbation of chronic heart failure, respiratory failure, multiorgan failure, lung infection, or colon adenocarcinoma.
Q 3 weeks suffices
In the question-and-answer session following the presentation, Kenny Lei, MD, from the Chinese University of Hong Kong asked Dr. Levy what the half-life of naratuximab is, and what was the investigator’s rationale for testing a weekly dosing schedule.
“I think the reason they checked the two different regimens, the Q week and the Q 3-week group, is that they noted that [naratuximab] was cleared relatively quickly, and they wanted to see whether or not, by giving Q weekly, when you get a continuous CD37 site occupancy if they would have a better outcome. But as you saw, in the groups there was really no clinically relevant difference in outcome,” Dr. Levy said.
Andrew Davies, MD, PhD, from the University of Southampton (England), asked whether the neutropenia seen in the study was related to myeloid expression of the target of from the off-target deconjugated payload.
“I don’t know that I necessarily have the answer to that,” Dr. Levy replied. “Remember there is the CD20 monoclonal rituximab which we know can cause neutropenia, as well as the CD37 and the target payload. I don’t know if we have enough information to attribute it to one specific component of the therapy,” he said.
The study was funded by Debiopharm International. Dr. Levy disclosed speaker activities for multiple companies, not including Debiopharm. Dr. Lei and Dr. Davies had no disclosures relevant to the study.
FROM EHA 2021