User login
If a Chronic Wound Does Not Heal, Biopsy It: A Clinical Lesson on Underlying Malignancies
To the Editor:
Experience, subjective opinion, and relationships with patients are cornerstones of general practice but also can be pitfalls. It is common for a late-presenting patient to offer a seemingly rational explanation for a long-standing lesion. Unless an objective analysis of the clinical problem is undertaken, it can be easy to embark on an incorrect treatment pathway for the patient’s condition.
One of the luxuries of specialist hospital medicine or surgery is the ability to focus on a narrow range of clinical problems, which makes it easier to spot the anomaly, as long as it is within the purview of the practitioner. We report 2 cases of skin malignancies that were assumed to be chronic wounds of benign etiology.
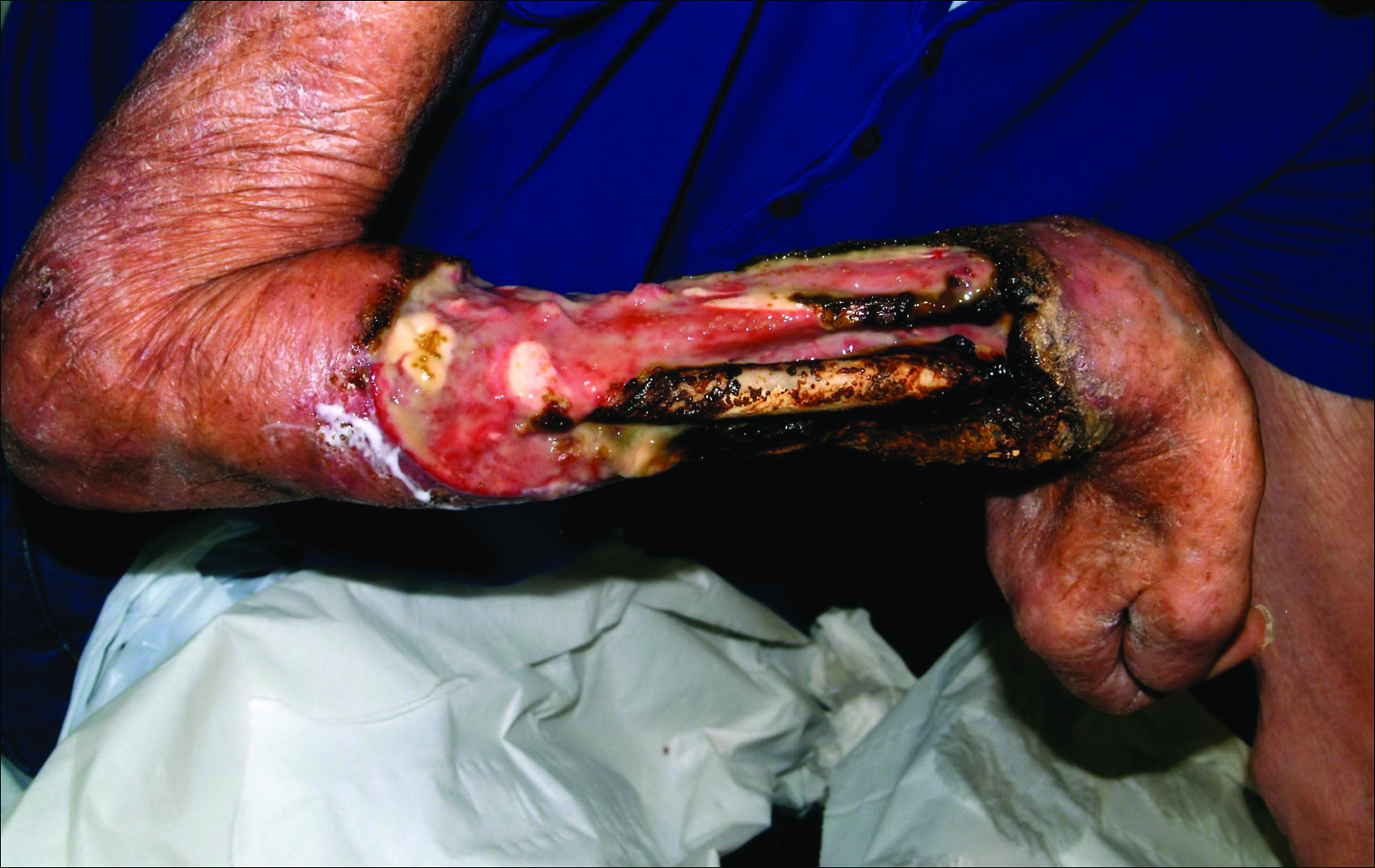
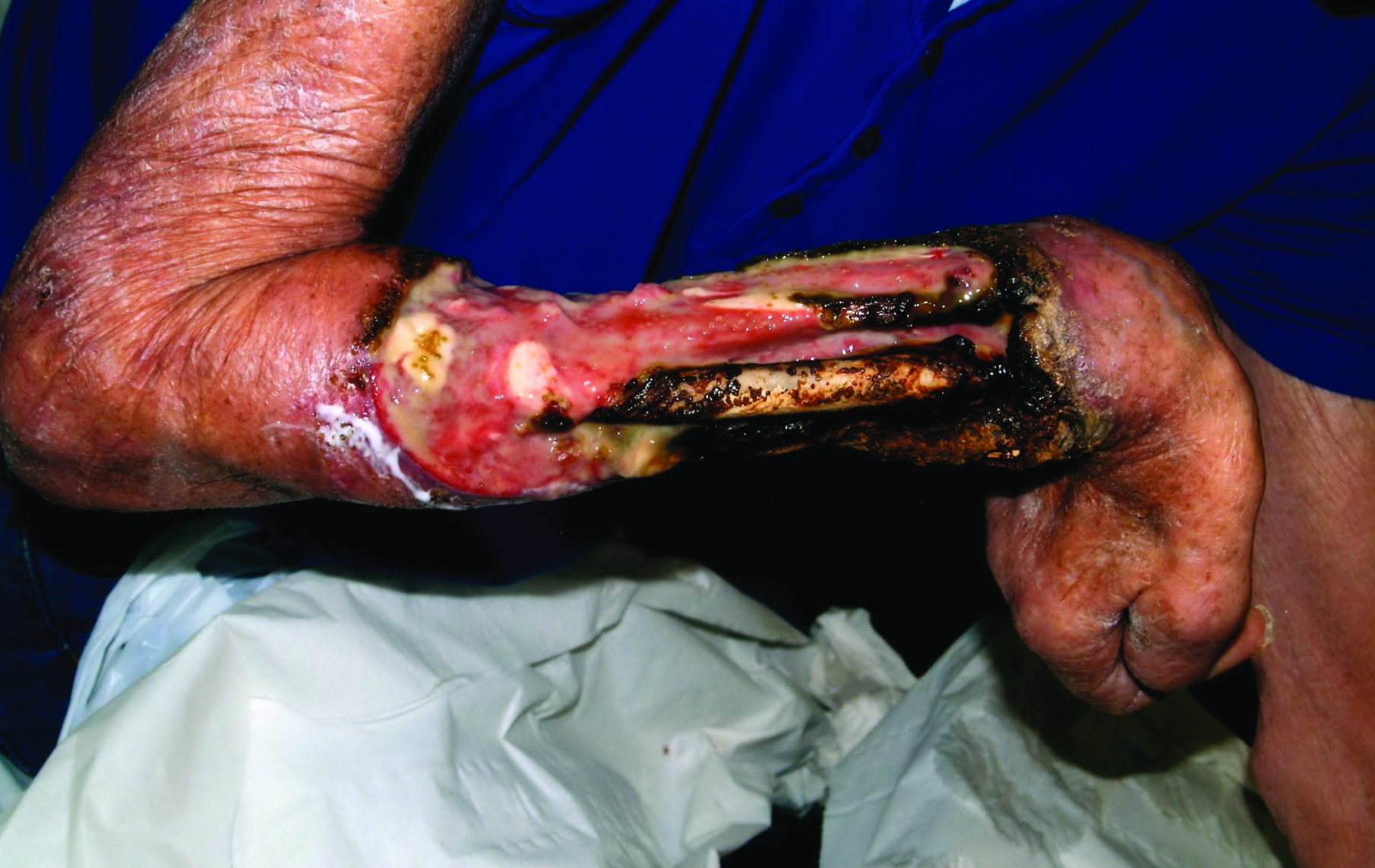
A 63-year-old builder was referred by his general practitioner with a chronic wound on the right forearm of 4 years’ duration. His medical history included psoriasis, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested possible incidental origin following a prior trauma or a psoriatic-related lesion. The patient reported that the lesion did not resemble prior psoriatic lesions and it had deteriorated substantially over the last 2 years. Furthermore, a small ulcer was starting to develop on the left forearm. Further advice was requested by the general practitioner regarding wound dressings. On examination a sloughy ulcer measuring 8.5×7.5 cm had eroded to expose necrotic tendons with surrounding induration and cellulitis (Figure 1A). In addition, a psoriatic lesion was found on the left forearm (Figure 1B). There were no palpable axillary lymph nodes. Clinical suspicion, incision biopsies, and subsequent histology confirmed cutaneous CD4+ T-cell lymphoma. This case was reviewed at a multidisciplinary team meeting and referred to the hematology-oncology department. The patient subsequently underwent chemotherapy with liposomal doxorubicin and radiotherapy over a period of 5 months. An elective right forearm amputation was planned due to erosion of the ulcer through tendons down to bone (Figure 2).
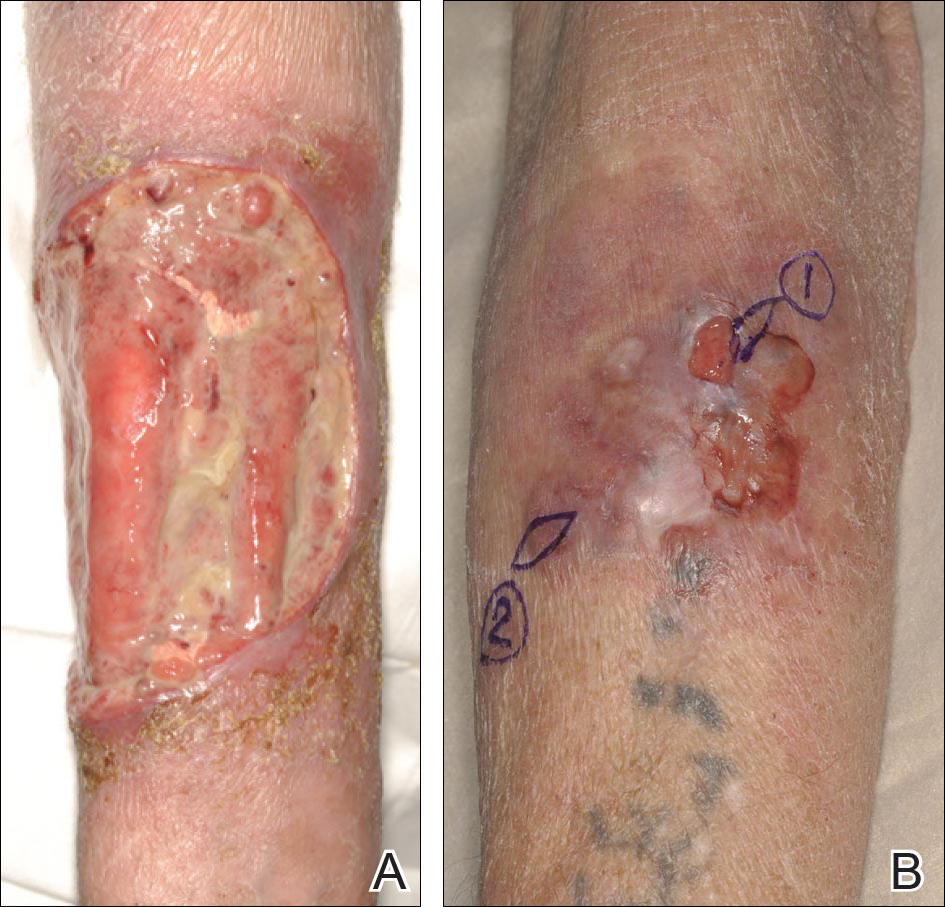
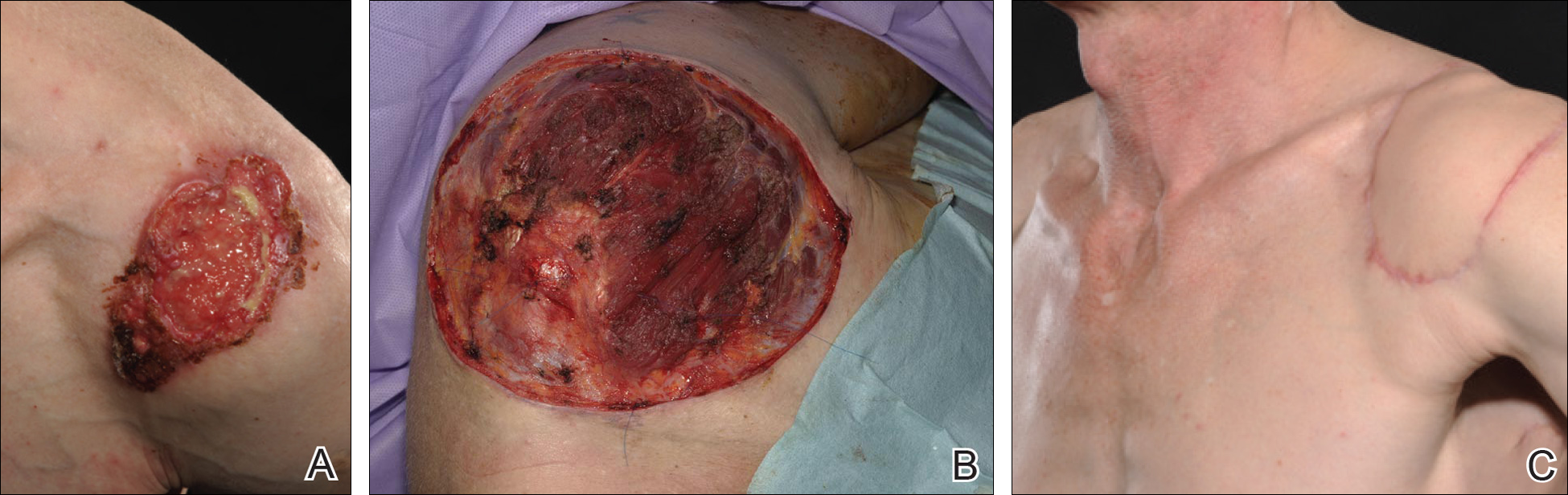
A 48-year-old Latvian lorry driver was referred by his general practitioner with a chronic wound on the left shoulder of 6 years’ duration. His medical history included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested the etiology was a burn from a hot metal rod 6 years prior. Advice was sought regarding dressings and suitability for a possible skin graft. Physical examination showed a 4.5×10-cm ulcer fixed to the underlying tissue on the anterior aspect of the left shoulder with no evidence of infection or presence of a foreign body (Figure 3A). Clinical suspicion, incision biopsies, and subsequent histology confirmed a highly infiltrative/morphoeic, partly nodular, and partly diffuse basal cell carcinoma (BCC) that measured 92 mm in diameter extending to the subcutis with no involvement of muscle or perineural or vascular invasion. The patient underwent wide local excision of the BCC with frozen section control. The BCC had eroded into the deltoid muscle and to the periosteum of the clavicle (Figure 3B). The defect was reconstructed with a pedicled muscle-sparing latissimus dorsi musculocutaneous flap. The patient presented for follow-up months following reconstruction with an uneventful recovery (Figure 3C).
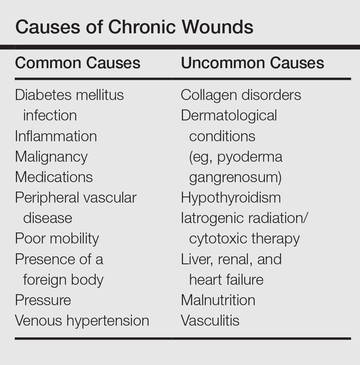
These 2 cases highlight easy pitfalls for an unsuspecting clinician. Although both cases had alternative plausible explanations, they proved to be cutaneous malignancies. The powerful message these cases send is that long-standing chronic wounds should be biopsied to exclude malignancy. Some of the other common underlying causes of wounds that may prevent healing are highlighted in the Table. Vascular insufficiency usually presents in characteristic patterns with a good clinical history and associated signs and findings on investigation. A foreign body, which can be anything from an orthopedic metal implant to a retained stitch from surgery or nonmedical material, may be the culprit and may be identified from a thorough medical history or appropriate imaging.
Infection is another possible explanation of a nonhealing wound. On the face, an underlying dental abscess with a sinus tracking from the root of the tooth to the skin of the cheek or jaw may be the source. Elsewhere on the body, chronic osteomyelitis may be the cause, which may be from any infective origin from Staphylococcus aureus to tuberculosis, and will most commonly present with a discharging sinus but also may present with a nonspecific ulcer.
Chronic wounds also may not heal because of a multitude of patient factors such as poor nutrition, diabetes mellitus, medication (eg, steroids, nonsteroidal anti-inflammatory drugs), other inflammatory causes, and poor mobility. Chronic wounds represent a substantial burden to patients, health care professionals, and the health care system. In the United States alone, they affect 5.7 million patients and cost an estimated $20 billion.1 Approximately 1% of the Western population will present with leg ulceration at some point in their lives.2
Physical examination of ulcers in any clinical setting can be difficult. We postulate that it can be made more difficult at times in primary care because the patient may add confounding elements for consideration or seemingly plausible explanations. However, whenever possible, a physician should ask, “Could there possibly be an underlying malignancy here?” If there is any chance of malignancy despite plausible explanations being offered, the lesion should be biopsied.
- Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing [published online July 7, 2008]. Burns. 2009;35:171-180.
- Callam MJ. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology. 1992;7(suppl 1):6-12.
To the Editor:
Experience, subjective opinion, and relationships with patients are cornerstones of general practice but also can be pitfalls. It is common for a late-presenting patient to offer a seemingly rational explanation for a long-standing lesion. Unless an objective analysis of the clinical problem is undertaken, it can be easy to embark on an incorrect treatment pathway for the patient’s condition.
One of the luxuries of specialist hospital medicine or surgery is the ability to focus on a narrow range of clinical problems, which makes it easier to spot the anomaly, as long as it is within the purview of the practitioner. We report 2 cases of skin malignancies that were assumed to be chronic wounds of benign etiology.
A 63-year-old builder was referred by his general practitioner with a chronic wound on the right forearm of 4 years’ duration. His medical history included psoriasis, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested possible incidental origin following a prior trauma or a psoriatic-related lesion. The patient reported that the lesion did not resemble prior psoriatic lesions and it had deteriorated substantially over the last 2 years. Furthermore, a small ulcer was starting to develop on the left forearm. Further advice was requested by the general practitioner regarding wound dressings. On examination a sloughy ulcer measuring 8.5×7.5 cm had eroded to expose necrotic tendons with surrounding induration and cellulitis (Figure 1A). In addition, a psoriatic lesion was found on the left forearm (Figure 1B). There were no palpable axillary lymph nodes. Clinical suspicion, incision biopsies, and subsequent histology confirmed cutaneous CD4+ T-cell lymphoma. This case was reviewed at a multidisciplinary team meeting and referred to the hematology-oncology department. The patient subsequently underwent chemotherapy with liposomal doxorubicin and radiotherapy over a period of 5 months. An elective right forearm amputation was planned due to erosion of the ulcer through tendons down to bone (Figure 2).
A 48-year-old Latvian lorry driver was referred by his general practitioner with a chronic wound on the left shoulder of 6 years’ duration. His medical history included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested the etiology was a burn from a hot metal rod 6 years prior. Advice was sought regarding dressings and suitability for a possible skin graft. Physical examination showed a 4.5×10-cm ulcer fixed to the underlying tissue on the anterior aspect of the left shoulder with no evidence of infection or presence of a foreign body (Figure 3A). Clinical suspicion, incision biopsies, and subsequent histology confirmed a highly infiltrative/morphoeic, partly nodular, and partly diffuse basal cell carcinoma (BCC) that measured 92 mm in diameter extending to the subcutis with no involvement of muscle or perineural or vascular invasion. The patient underwent wide local excision of the BCC with frozen section control. The BCC had eroded into the deltoid muscle and to the periosteum of the clavicle (Figure 3B). The defect was reconstructed with a pedicled muscle-sparing latissimus dorsi musculocutaneous flap. The patient presented for follow-up months following reconstruction with an uneventful recovery (Figure 3C).
These 2 cases highlight easy pitfalls for an unsuspecting clinician. Although both cases had alternative plausible explanations, they proved to be cutaneous malignancies. The powerful message these cases send is that long-standing chronic wounds should be biopsied to exclude malignancy. Some of the other common underlying causes of wounds that may prevent healing are highlighted in the Table. Vascular insufficiency usually presents in characteristic patterns with a good clinical history and associated signs and findings on investigation. A foreign body, which can be anything from an orthopedic metal implant to a retained stitch from surgery or nonmedical material, may be the culprit and may be identified from a thorough medical history or appropriate imaging.
Infection is another possible explanation of a nonhealing wound. On the face, an underlying dental abscess with a sinus tracking from the root of the tooth to the skin of the cheek or jaw may be the source. Elsewhere on the body, chronic osteomyelitis may be the cause, which may be from any infective origin from Staphylococcus aureus to tuberculosis, and will most commonly present with a discharging sinus but also may present with a nonspecific ulcer.
Chronic wounds also may not heal because of a multitude of patient factors such as poor nutrition, diabetes mellitus, medication (eg, steroids, nonsteroidal anti-inflammatory drugs), other inflammatory causes, and poor mobility. Chronic wounds represent a substantial burden to patients, health care professionals, and the health care system. In the United States alone, they affect 5.7 million patients and cost an estimated $20 billion.1 Approximately 1% of the Western population will present with leg ulceration at some point in their lives.2
Physical examination of ulcers in any clinical setting can be difficult. We postulate that it can be made more difficult at times in primary care because the patient may add confounding elements for consideration or seemingly plausible explanations. However, whenever possible, a physician should ask, “Could there possibly be an underlying malignancy here?” If there is any chance of malignancy despite plausible explanations being offered, the lesion should be biopsied.
To the Editor:
Experience, subjective opinion, and relationships with patients are cornerstones of general practice but also can be pitfalls. It is common for a late-presenting patient to offer a seemingly rational explanation for a long-standing lesion. Unless an objective analysis of the clinical problem is undertaken, it can be easy to embark on an incorrect treatment pathway for the patient’s condition.
One of the luxuries of specialist hospital medicine or surgery is the ability to focus on a narrow range of clinical problems, which makes it easier to spot the anomaly, as long as it is within the purview of the practitioner. We report 2 cases of skin malignancies that were assumed to be chronic wounds of benign etiology.
A 63-year-old builder was referred by his general practitioner with a chronic wound on the right forearm of 4 years’ duration. His medical history included psoriasis, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested possible incidental origin following a prior trauma or a psoriatic-related lesion. The patient reported that the lesion did not resemble prior psoriatic lesions and it had deteriorated substantially over the last 2 years. Furthermore, a small ulcer was starting to develop on the left forearm. Further advice was requested by the general practitioner regarding wound dressings. On examination a sloughy ulcer measuring 8.5×7.5 cm had eroded to expose necrotic tendons with surrounding induration and cellulitis (Figure 1A). In addition, a psoriatic lesion was found on the left forearm (Figure 1B). There were no palpable axillary lymph nodes. Clinical suspicion, incision biopsies, and subsequent histology confirmed cutaneous CD4+ T-cell lymphoma. This case was reviewed at a multidisciplinary team meeting and referred to the hematology-oncology department. The patient subsequently underwent chemotherapy with liposomal doxorubicin and radiotherapy over a period of 5 months. An elective right forearm amputation was planned due to erosion of the ulcer through tendons down to bone (Figure 2).
A 48-year-old Latvian lorry driver was referred by his general practitioner with a chronic wound on the left shoulder of 6 years’ duration. His medical history included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner included a partial gastrectomy for a peptic ulcer 18 years prior, and he did not have a history of diabetes mellitus or use of immunosuppressants. The general practitioner suggested the etiology was a burn from a hot metal rod 6 years prior. Advice was sought regarding dressings and suitability for a possible skin graft. Physical examination showed a 4.5×10-cm ulcer fixed to the underlying tissue on the anterior aspect of the left shoulder with no evidence of infection or presence of a foreign body (Figure 3A). Clinical suspicion, incision biopsies, and subsequent histology confirmed a highly infiltrative/morphoeic, partly nodular, and partly diffuse basal cell carcinoma (BCC) that measured 92 mm in diameter extending to the subcutis with no involvement of muscle or perineural or vascular invasion. The patient underwent wide local excision of the BCC with frozen section control. The BCC had eroded into the deltoid muscle and to the periosteum of the clavicle (Figure 3B). The defect was reconstructed with a pedicled muscle-sparing latissimus dorsi musculocutaneous flap. The patient presented for follow-up months following reconstruction with an uneventful recovery (Figure 3C).
These 2 cases highlight easy pitfalls for an unsuspecting clinician. Although both cases had alternative plausible explanations, they proved to be cutaneous malignancies. The powerful message these cases send is that long-standing chronic wounds should be biopsied to exclude malignancy. Some of the other common underlying causes of wounds that may prevent healing are highlighted in the Table. Vascular insufficiency usually presents in characteristic patterns with a good clinical history and associated signs and findings on investigation. A foreign body, which can be anything from an orthopedic metal implant to a retained stitch from surgery or nonmedical material, may be the culprit and may be identified from a thorough medical history or appropriate imaging.
Infection is another possible explanation of a nonhealing wound. On the face, an underlying dental abscess with a sinus tracking from the root of the tooth to the skin of the cheek or jaw may be the source. Elsewhere on the body, chronic osteomyelitis may be the cause, which may be from any infective origin from Staphylococcus aureus to tuberculosis, and will most commonly present with a discharging sinus but also may present with a nonspecific ulcer.
Chronic wounds also may not heal because of a multitude of patient factors such as poor nutrition, diabetes mellitus, medication (eg, steroids, nonsteroidal anti-inflammatory drugs), other inflammatory causes, and poor mobility. Chronic wounds represent a substantial burden to patients, health care professionals, and the health care system. In the United States alone, they affect 5.7 million patients and cost an estimated $20 billion.1 Approximately 1% of the Western population will present with leg ulceration at some point in their lives.2
Physical examination of ulcers in any clinical setting can be difficult. We postulate that it can be made more difficult at times in primary care because the patient may add confounding elements for consideration or seemingly plausible explanations. However, whenever possible, a physician should ask, “Could there possibly be an underlying malignancy here?” If there is any chance of malignancy despite plausible explanations being offered, the lesion should be biopsied.
- Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing [published online July 7, 2008]. Burns. 2009;35:171-180.
- Callam MJ. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology. 1992;7(suppl 1):6-12.
- Branski LK, Gauglitz GG, Herndon DN, et al. A review of gene and stem cell therapy in cutaneous wound healing [published online July 7, 2008]. Burns. 2009;35:171-180.
- Callam MJ. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology. 1992;7(suppl 1):6-12.
Practice Points
- Patients with chronic wounds should have a thorough history and examination, appropriate laboratory tests, and purposeful search to determine etiology.
- Long-standing chronic wounds should be biopsied to exclude malignancy.
What’s Less Noticeable: A Straight Scar or a Zigzag Scar?
One of the determinants of a successful surgical outcome is the perception, on the part of the patient, of the cosmesis of a scar. The use of Z-plasty is an accepted means by which to break a scar up into smaller geometric segments. In some instances, a Z-plasty is used for scar revision to elongate a scar that may be pulling. However, a study published online in JAMA Facial Plastic Surgery on April 7 mentions the lack of studies measuring the perception of these scars among the normal population after surgery.
Ratnarathorn et al designed a prospective Internet-based survey with a goal of 580 responses to give a power of 90%. The survey was distributed to a diverse sample of the US population. Using editing software, Ratnarathorn et al superimposed a mature linear scar and a mature zigzag scar onto the faces of standardized headshots from 4 individuals (2 males, 2 females). Each individual had 1 image of the linear scar and 1 image of the zigzag scars superimposed onto each of 3 anatomical areas—forehead (flat surface), cheek (convex surface), and temple (concave surface)—yielding 24 images for the respondents to assess.
A 24.5% (n=876) response rate was achieved with 3575 surveys distributed. Of the 876 respondents, 810 (92.5%) completed the survey (46.1% male, 53.9% female). Respondents were asked to rate the scars on a scale of 1 to 10 (1=normal skin; 10=worst scar imaginable).
Results were statistically significantly lower (better) for the linear scars compared to the zigzag scars in all 3 anatomic areas and across both male and female groups with a mean score of 2.9 versus 4.5 (P<.001). A multivariable regression model of respondent age, sex, educational level, and income showed no statistically significant effect on the rating of the scars.
What’s the issue?
This study highlights some interesting points. Coming from an academic practice, we oftentimes find ourselves teaching residents a variety of skin closure techniques to deal with defects from skin cancer excisions. It is both challenging and fun to design complex closures; however, we must keep in mind what is in the best interest of the patient. One of the points I try to emphasize is that we must understand that there are no true straight lines on the face. In fact, when scars from procedures appear as geometric shapes on the face, our eyes tend to be drawn to them. For this reason, it often is best to use curvilinear lines wherever possible. Ratnarathorn et al highlights that point exactly. More studies of this nature are needed to assess what is perceived as a successful outcome, by both physicians and patients.
As you follow your patients for the long-term, have you noticed that you perform more or fewer zigzag scars?
One of the determinants of a successful surgical outcome is the perception, on the part of the patient, of the cosmesis of a scar. The use of Z-plasty is an accepted means by which to break a scar up into smaller geometric segments. In some instances, a Z-plasty is used for scar revision to elongate a scar that may be pulling. However, a study published online in JAMA Facial Plastic Surgery on April 7 mentions the lack of studies measuring the perception of these scars among the normal population after surgery.
Ratnarathorn et al designed a prospective Internet-based survey with a goal of 580 responses to give a power of 90%. The survey was distributed to a diverse sample of the US population. Using editing software, Ratnarathorn et al superimposed a mature linear scar and a mature zigzag scar onto the faces of standardized headshots from 4 individuals (2 males, 2 females). Each individual had 1 image of the linear scar and 1 image of the zigzag scars superimposed onto each of 3 anatomical areas—forehead (flat surface), cheek (convex surface), and temple (concave surface)—yielding 24 images for the respondents to assess.
A 24.5% (n=876) response rate was achieved with 3575 surveys distributed. Of the 876 respondents, 810 (92.5%) completed the survey (46.1% male, 53.9% female). Respondents were asked to rate the scars on a scale of 1 to 10 (1=normal skin; 10=worst scar imaginable).
Results were statistically significantly lower (better) for the linear scars compared to the zigzag scars in all 3 anatomic areas and across both male and female groups with a mean score of 2.9 versus 4.5 (P<.001). A multivariable regression model of respondent age, sex, educational level, and income showed no statistically significant effect on the rating of the scars.
What’s the issue?
This study highlights some interesting points. Coming from an academic practice, we oftentimes find ourselves teaching residents a variety of skin closure techniques to deal with defects from skin cancer excisions. It is both challenging and fun to design complex closures; however, we must keep in mind what is in the best interest of the patient. One of the points I try to emphasize is that we must understand that there are no true straight lines on the face. In fact, when scars from procedures appear as geometric shapes on the face, our eyes tend to be drawn to them. For this reason, it often is best to use curvilinear lines wherever possible. Ratnarathorn et al highlights that point exactly. More studies of this nature are needed to assess what is perceived as a successful outcome, by both physicians and patients.
As you follow your patients for the long-term, have you noticed that you perform more or fewer zigzag scars?
One of the determinants of a successful surgical outcome is the perception, on the part of the patient, of the cosmesis of a scar. The use of Z-plasty is an accepted means by which to break a scar up into smaller geometric segments. In some instances, a Z-plasty is used for scar revision to elongate a scar that may be pulling. However, a study published online in JAMA Facial Plastic Surgery on April 7 mentions the lack of studies measuring the perception of these scars among the normal population after surgery.
Ratnarathorn et al designed a prospective Internet-based survey with a goal of 580 responses to give a power of 90%. The survey was distributed to a diverse sample of the US population. Using editing software, Ratnarathorn et al superimposed a mature linear scar and a mature zigzag scar onto the faces of standardized headshots from 4 individuals (2 males, 2 females). Each individual had 1 image of the linear scar and 1 image of the zigzag scars superimposed onto each of 3 anatomical areas—forehead (flat surface), cheek (convex surface), and temple (concave surface)—yielding 24 images for the respondents to assess.
A 24.5% (n=876) response rate was achieved with 3575 surveys distributed. Of the 876 respondents, 810 (92.5%) completed the survey (46.1% male, 53.9% female). Respondents were asked to rate the scars on a scale of 1 to 10 (1=normal skin; 10=worst scar imaginable).
Results were statistically significantly lower (better) for the linear scars compared to the zigzag scars in all 3 anatomic areas and across both male and female groups with a mean score of 2.9 versus 4.5 (P<.001). A multivariable regression model of respondent age, sex, educational level, and income showed no statistically significant effect on the rating of the scars.
What’s the issue?
This study highlights some interesting points. Coming from an academic practice, we oftentimes find ourselves teaching residents a variety of skin closure techniques to deal with defects from skin cancer excisions. It is both challenging and fun to design complex closures; however, we must keep in mind what is in the best interest of the patient. One of the points I try to emphasize is that we must understand that there are no true straight lines on the face. In fact, when scars from procedures appear as geometric shapes on the face, our eyes tend to be drawn to them. For this reason, it often is best to use curvilinear lines wherever possible. Ratnarathorn et al highlights that point exactly. More studies of this nature are needed to assess what is perceived as a successful outcome, by both physicians and patients.
As you follow your patients for the long-term, have you noticed that you perform more or fewer zigzag scars?

Regional Lymphomatoid Papulosis of the Breast Restricted to an Area of Prior Radiotherapy
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
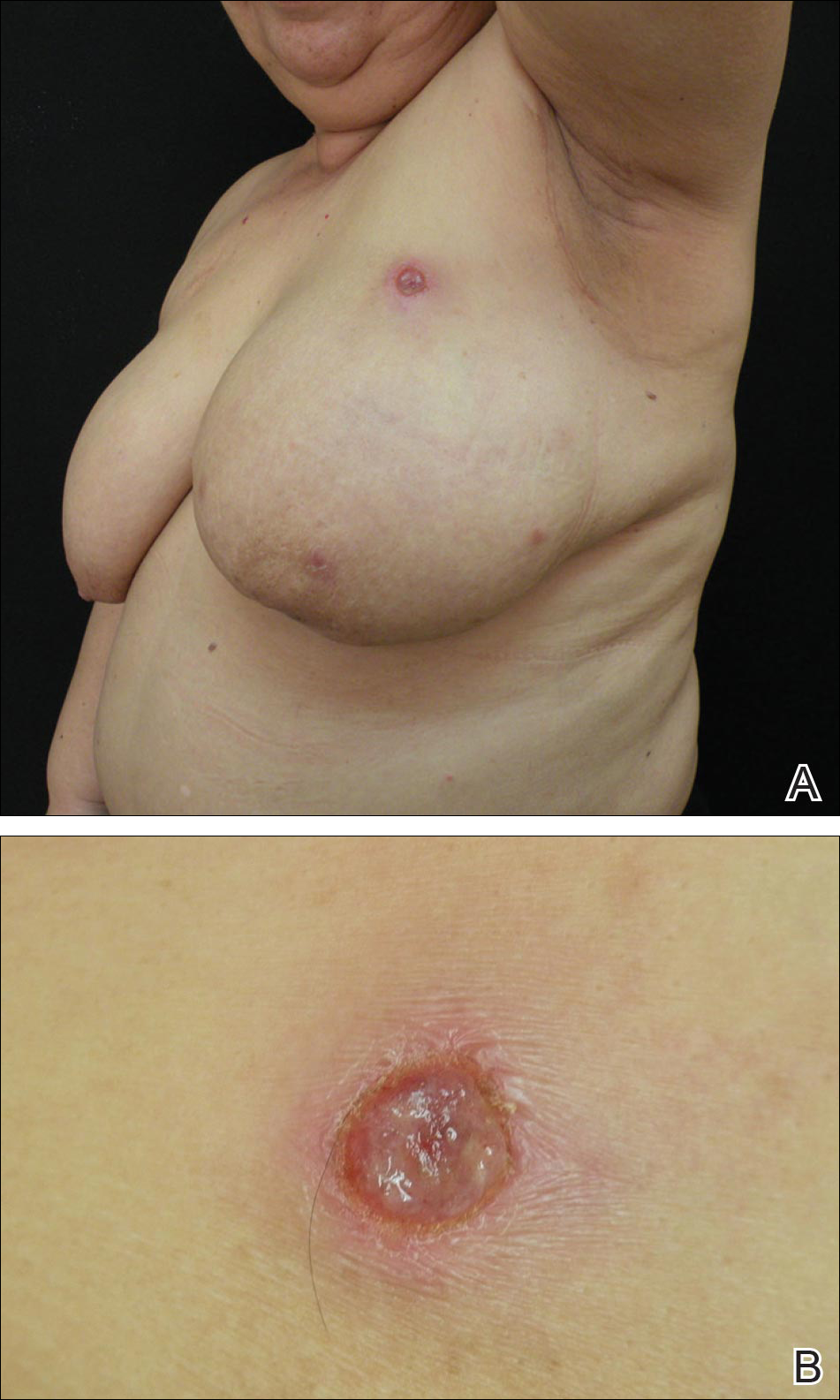
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.

The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.
The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
Lymphomatoid papulosis (LyP) is a clinicopathologic variant of CD30+ primary cutaneous T-cell lymphoproliferative disorder characterized by a chronic, recurrent, self-healing eruption of papules and small nodules. From a clinical point of view, LyP is not considered a malignant disorder despite demonstration of clonality in most cases.1 From a histopathologic point of view, there are 5 types of LyP: (1) type A, the most common type, which is characterized by a wedge-shaped infiltrate composed of clustered large atypical cells admixed with neutrophils, eosinophils, histiocytes, and small lymphocytes; (2) type B, a rare variant characterized by a bandlike infiltrate of small- to medium-sized pleomorphic and hyperchromatic lymphocytes involving the superficial dermis with epidermotropism; (3) type C, which consists of a nodular infiltrate of large atypical cells with a cohesive arrangement closely similar to anaplastic large-cell lymphoma; (4) type D, a variant with histopathologic features that resemble primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma, but neoplastic cells express CD30 and a T-cell cytotoxic phenotype (βF1+, CD3+, CD4‒, CD8+), and follow-up usually does not reveal development of systemic involvement or signs of other cutaneous lymphomas2; and (5) type E, which is characterized by oligolesional papules that rapidly ulcerate and evolve into large, necrotic, escharlike lesions with a diameter of 1 to 4 cm and an angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes expressing CD30 and frequently CD8.3
The clinical appearance of LyP usually is polymorphic, with lesions in different stages of evolution scattered all over the skin; however, the lesions are occasionally localized only to one area of the skin, the so-called regional or agminated LyP.4-14 We report a case of regional LyP that exclusively involved the skin of the left breast, which had previously received radiotherapy for treatment of breast carcinoma. Lymphomatoid papulosis with cutaneous lesions involving only an area of irradiated skin is rare.
Case Report
A 59-year-old woman presented with new-onset cutaneous lesions on the left breast. The patient had a history of invasive ductal carcinoma of the left breast, which had been treated 5 years prior with a partial mastectomy and radiotherapy (10 Gy per week for 5 consecutive weeks [50 Gy total]). Physical examination revealed a large nodular lesion with a necrotic surface on the upper half of the left breast as well as 3 small papular lesions with eroded surfaces on the lower half of the breast (Figure 1). A clinical diagnosis of cutaneous metastases from breast carcinoma was suspected.
Biopsies from one small papule and the large nodular lesion showed similar findings consisting of a necrotic epidermis covered by crusts and a wedge-shaped infiltrate involving the superficial dermis (Figure 2A). The infiltrate was mostly composed of large atypical mononuclear cells with oval to kidney-shaped nuclei, prominent nucleoli, and ample basophilic cytoplasm. Many mitotic figures were seen within the infiltrate (Figure 2B). The infiltrate of atypical cells was admixed with small lymphocytes, histiocytes, and some eosinophils. Immunohistochemically, the large atypical cells expressed CD2, CD3, CD4, CD45, CD30, and epithelial membrane antigen (Figures 2C and 2D). A few atypical cells also expressed CD8 and T-cell intracellular antigen 1. Approximately 60% of the nuclei of the atypical cells showed MIB-1 positivity, while CD20, CD56, AE1/AE3, S-100 protein, CD34, and CD31 were negative. The anaplastic lymphoma kinase was not expressed in atypical cells. Monoclonal rearrangement of the γ T-cell receptor was demonstrated on polymerase chain reaction. Physical examination showed no lymphadenopathy in any lymph node chains. Computed tomography of the chest and abdomen failed to demonstrate systemic involvement. On the basis of these clinical, histologic, immunohistochemical, and molecular results, a diagnosis of type A regional LyP was established.
The patient was treated with 2 daily applications of clobetasol propionate cream 0.5 mg/g and 10 mg of oral methotrexate per week for 4 weeks. After 4 weeks of treatment, the lesions on the left breast had resolved leaving slightly atrophic scars. Six months later, an episode of recurrent papular lesions occurred in the same area and responded to the same treatment, but no systemic involvement had been found.
Comment
Regional LyP is a rare variant, with only a few reported cases in the literature.4-18 Scarisbrick et al4 originally reported 4 patients with LyP limited to specific regions. Interestingly, one of the patients had mycosis fungoides and the LyP lesions were confined to the same region where the mycosis fungoides lesions were observed.4 In a review of LyP in patients from the Netherlands (n=118), lesions limited to a specific region of the body were observed in 13% of cases.5 Cases of LyP limited to acral skin also have been reported.6-8 Heald et al9 described 7 patients who had continuing eruptions of papulonodules with histopathologic features of LyP within well-circumscribed areas of the skin. The investigators interpreted this localized variant of LyP as an equivalent of the limited plaque stage of mycosis fungoides. Interestingly, one of the patients with LyP eventually developed plaques of mycosis fungoides in other areas of the skin not involved by LyP.9 Sharma et al10 described an additional example of regional LyP, and Nakahigashi et al11 described a patient with tumor-stage mycosis fungoides who subsequently developed regional LyP involving the right side of the chest. Kim et al12 described a patient with recurrent episodes of regional LyP exclusively involving the periorbital skin, and Torrelo et al13 reported a 12-year-old boy with persistent lesions of LyP involving the skin of the right side of the abdomen. Coelho et al14 reported a 13-year-old adolescent girl who presented with recurrent papules of LyP exclusively involving the left upper arm. Buder et al15 reported a case of LyP limited to Becker melanosis. Shang et al16 described an additional caseof regional LyP that was successfully controlled by interferon alfa-2b and nitrogen mustard solution. Haus et al17 reported type A LyP confined to the cutaneous area within a red tattoo. Finally, Wang et al18 reported a case of regional LyP in association with pseudoepitheliomatous hyperplasia
Several dermatoses may appear as specific isomorphic responses to various external stimuli, and it is possible that radiotherapy induces some damage that favors the location of the lesions because the irradiated skin behaves as a locus minoris resistentiae. Pemphigus vulgaris,19,20 Sweet syndrome,21 cutaneous angiosarcoma,22-32 and cutaneous metastases from malignant melanoma also have been reported to be confined to irradiated skin.33 However, in our PubMed search of articles indexed for MEDLINE using the terms lymphomatoid papules and regional, none of the previously reported cases of regional LyP had a history of radiotherapy, and in no instance did the lesions develop on a previously irradiated area of the skin.4-18 The localization of the lesions in our patient could have been the result of the so-called radiation recall phenomenon. Recall dermatitis is defined as a skin reaction in a previously irradiated field, usually subsequent to the administration of cytotoxic drugs or antibiotics.34 It may appear days to years after exposure to ionizing radiation and has mostly been associated with chemotherapy drugs, but recall dermatitis is neither exclusive of chemotherapy medications nor strictly radiotherapy induced. The concept of recall dermatitis has been expanded beyond radiation recall dermatitis to include dermatitis induced by other stimuli, including other drugs, contact irritants, and UV radiation, as well as residual herpes zoster. Nevertheless, in recall dermatitis the triggering drug or agent recalls a prior dermatitis in the involved area, such as sunburn or radiodermatitis. In our patient, there was no history of LyP prior to irradiation of the left breast; therefore, the most plausible interpretation of the peculiar localization of the lesions in our patient seems to be that the eruption resulted as expression of a locus minoris resistentiae.
Distinction between primary cutaneous anaplastic large-cell lymphoma and LyP may be difficult because the histopathologic and immunophenotypic features may overlap. In our case, the presence of several papular lesions and one large nodule are more consistent, from a clinical point of view, with a diagnosis of LyP rather than primary cutaneous anaplastic large-cell lymphoma, which usually presents with a solitary and often large, ulcerated, reddish brown tumor. In our patient, the absence of lymphadenopathy, negative results of the computed tomography of the chest and abdomen, and lack of expression for anaplastic lymphoma kinase in atypical cells of the infiltrate militate against a diagnosis of secondary cutaneous involvement from nodal disease.
The histopathologic differential diagnosis of the current case also included cutaneous CD30+ epithelioid angiosarcoma of the breast. Weed and Folpe35 reported the case of an 85-year-old woman who developed a CD30+ epithelioid angiosarcoma on the breast after undergoing breast-conserving surgery and adjuvant radiotherapy for treatment of an infiltrating ductal carcinoma of the breast. Histopathology showed a diffuse replacement of the dermis by a highly malignant-appearing epithelioid neoplasm growing in a solid sheet. Neoplastic cells expressed strong CD30 immunoreactivity with absence of immunoexpression for cytokeratins, S-100 protein, and CD45. Additional immunostaining demonstrated that neoplastic cells also expressed strong immunoreactivity for CD31 and the friend leukemia virus integration 1 gene, FLI-1, and focal positivity for von Willebrand factor, supporting a diagnosis of epithelioid angiosarcoma.35 In our patient, CD34 and CD31 were negative, which ruled out the endothelial nature of neoplastic cells.
Conclusion
In summary, we report an example of regional LyP limited to the left breast of a woman with a history of partial mastectomy and adjuvant radiotherapy for treatment of invasive ductal breast carcinoma. It is a rare case of regional LyP exclusively involving an irradiated area of the skin.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
- Ralfkiaer E, Willemze R, Paulli M, et al. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphomatoid Tissues. Lyon, France: IARC Press, 2008:300-301.
- Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma. description of 9 cases. Am J Surg Pathol. 2010;34:1168-1175.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Scarisbrick JJ, Evans AV, Woolford AJ, et al. Regional lymphomatoid papulosis: a report of four cases. Br J Dermatol. 1999;141:1125-1128.
- Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95:3653-3661.
- Thomas GJ, Conejo-Mir JS, Ruiz AP, et al. Lymphomatoid papulosis in childhood with exclusive acral involvement. Pediatr Dermatol. 1998;15:146-147.
- Deroo-Berger MC, Skowson F, Roner S, et al. Lymphomatoid papulosis: a localized form with acral pustular involvement. Dermatology. 2002;205:60-62.
- Kagaya M, Kondo S, Kamada A, et al. Localized lymphomatoid papulosis. Dermatology. 2002;204:72-74.
- Heald P, Subtil A, Breneman D, et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J Am Acad Dermatol. 2007;57:1005-1011.
- Sharma V, Xu G, Petronic-Rosic V, et al. Clinicopathologic challenge. regional lymphomatoid papulosis, type A. Int J Dermatol. 2007;46:905-909.
- Nakahigashi K, Ishida Y, Matsumura Y, et al. Large cell transformation mimicking regional lymphomatoid papulosis in a patient with mycosis fungoides. J Dermatol. 2008;35:283-288.
- Kim YJ, Rho YK, Yoo KH, et al. Case of regional lymphomatoid papulosis confined to the periorbital areas. J Dermatol. 2009;36:163-165.
- Torrelo A, Colmenero I, Hernández A, et al. Persistent agmination of lymphomatoid papulosis. Pediatr Dermatol. 2009;26:762-764.
- Coelho JD, Afonso A, Feio AB. Regional lymphomatoid papulosis in a child—treatment with a UVB phototherapy handpiece. J Cosmet Laser Ther. 2010;12:155-156.
- Buder K, Wendel AM, Cerroni L, et al. A case of lymphomatoid papulosis limited to Becker’s melanosis. Dermatology. 2013;226:124-127.
- Shang SX, Chen H, Sun JF, et al. Regional lymphomatoid papulosis successfully controlled by interferon α-2b and nitrogen mustard solution. Chin Med J (Engl). 2013;126:3194-3195.
- Haus G, Utikal J, Geraud C, et al. CD30-positive lymphoproliferative disorder in a red tattoo: regional lymphomatoid papulosis type C or pseudolymphoma? Br J Dermatol. 2014;171:668-670.
- Wang T, Guo CL, Xu CC, et al. Regional lymphomatoid papulosis in association with pseudoepitheliomatous hyperplasia: 13 years follow-up. J Eur Acad Dermatol Venereol. 2015;29:1853-1854.
- Davis M, Feverman EJ. Induction of pemphigus by X-ray irradiation. Clin Exp Dermatol. 1987;12:197-199.
- Crovato F, Descrello G, Nazzari G, et al. Liner pemphigus vulgaris after X-ray irradiation. Dermatologica. 1989;179:135-136.
- Vergara G, Vargas-Machuca I, Pastor MA, et al. Localized Sweet’s syndrome in radiation-induced locus minoris resistentae. J Am Acad Dermatol. 2003;49:907-909.
- Caldwell JB, Ryan MT, Benson PM, et al. Cutaneous angiosarcoma arising in the radiation site of a congenital hemangioma. J Am Acad Dermatol. 1995;33:865-870.
- Stone NM, Holden CA. Postirradiation angiosarcoma. Clin Exp Dermatol. 1997;22:46-47.
- Goette EK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12:922-926.
- Chen TK, Goffman KD, Hendricks EJ. Angiosarcoma following therapeutic irradiation. Cancer. 1979;44:2044-2048.
- Rubin E, Maddox WA, Mazur MT. Cutaneous angiosarcoma of the breast 7 years after lumpectomy and radiation therapy. Radiology. 1990;174:258-260.
- Stokkel MPM, Peterse HL. Angiosarcoma of the breast after lumpectomy and radiation therapy for adenocarcinoma. Cancer. 1992;69:2965-2968.
- Moskaluk CA, Merino MJ, Danforth DN, et al. Low-grade angiosarcoma of the skin of the breast: a complication of lumpectomy and radiation therapy for breast carcinoma. Hum Pathol. 1992;23:710-714.
- Parham DM, Fisher C. Angiosarcomas of the breast developing post radiotherapy. Histopathology. 1997;31:189-195.
- Rao J, DeKoven JG, Beatty JD, et al. Cutaneous angiosarcoma as a delayed complication of radiation therapy for carcinoma of the breast. J Am Acad Dermatol. 2003;49:532-538.
- Billings SD, McKenney JK, Folpe Al, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation. an analysis of 27 cases. Am J Surg Pathol. 2004;28:781-788.
- Fodor J, Orosz Z, Szabo E, et al. Angiosarcoma after conservation treatment for breast carcinoma: our experience and a review of the literature. J Am Acad Dermatol. 2006;54:499-504.
- Roses DP, Harris MN, Rigel D, et al. Local and in-transit metastases following definitive excision from primary cutaneous malignant melanoma. Ann Surg. 1983;198:65-69.
- Burris HA 3rd, Hurtig J. Radiation recall with anticancer agents. Oncologist. 2010;15:1227-1237.
- Weed BR, Folpe AL. Cutaneous CD30-positive epithelioid angiosarcoma following breast-conserving therapy and irradiation. a potential diagnostic pitfall. Am J Dermatopathol. 2008;30:370-372.
Practice Points
- Cutaneous lesions of lymphomatoid papulosis (LyP) sometimes are confined to only one area of the skin, which is known as regional LyP.
- Patients with regional LyP have the same prognosis as those with widespread LyP, and no specific association has been reported with this clinical variant.
- Lesions of regional LyP respond to the same treatments as widespread LyP.

Multiple Morphologically Distinct Cutaneous Granular Cell Tumors Occurring in a Single Patient
Case Report
A 27-year-old black man was admitted to the hospital with chills; night sweats; unintentional 25-lb weight loss; and multiple widespread, painful, progressively enlarging skin nodules of 3 months’ duration. The lesions had first developed on the back and later appeared on the face, trunk, arms, thighs, and genital region. He denied dysuria or urethral discharge. He had a remote history of adequately treated chlamydia infection but no other remarkable personal or family history.
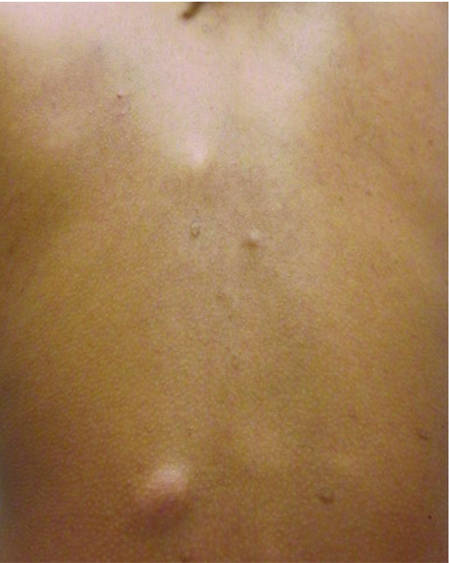
| ||
| Figure 1. Firm subcutaneous nodules on the back with no epidermal change. | ||
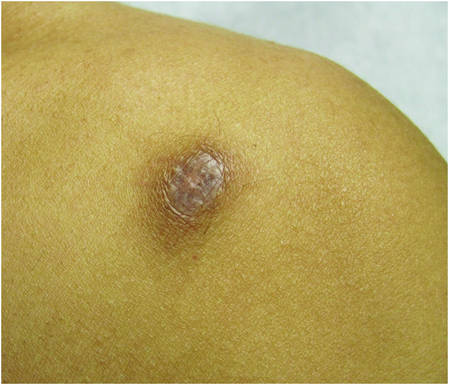
| ||
| Figure 2. Firm dermal papule on the anterior aspect of the left shoulder with violaceous hyperpigmentation (dermatofibromalike). |
Physical examination revealed a thin man with more than 20 lesions on the face, trunk, arms, thighs, and genital region ranging in size from 1 to 4 cm. Lesion morphologies varied greatly and included subcutaneous firm nodules with no epidermal change (Figure 1); dermatofibromalike nodules with overlying erythema and hyperpigmentation (Figure 2); condylomalike, verrucous, pink papulonodules (Figure 3); ulcerated angular plaques with rolled borders and palpable tumor extension deep (1–2 cm) to the subcutis (Figure 4); and a vegetative, eroded, exophytic tumor with palpable deep extension (Figure 4). A diffuse, erythematous, macular eruption also was noted on the trunk and bilateral arms and legs including the soles of both feet along with nontender cervical, axillary, and inguinal lymphadenopathy. The ocular, oral, and nasal mucosae were not affected.
The differential diagnosis for each lesion differed based on morphology. Infectious, inflammatory, and neoplastic processes were considered, including syphilis, dermatofibroma, dermatofibrosarcoma protuberans, metastatic disease, leukemia cutis, sarcoidosis, panniculitis, condyloma acuminatum, and vegetative herpes simplex virus infection (inguinal lesion).
Laboratory data revealed a reactive rapid plasma reagin with treponemal IgG titers of 1:64. Urine chlamydia RNA probe and lymphogranuloma venereum (LGV) serum antibodies also were positive. Human immunodeficiency virus screening was negative. Positron emission tomography–computerized tomography revealed enlarged and hypermetabolic lymphadenopathy above and below the diaphragm.
After therapy with intravenous penicillin G and oral doxycycline for concurrent secondary syphilis and LGV, the patient’s macular eruption and constitutional symptoms resolved within weeks of the initial presentation. His lymphadenopathy improved, his rapid plasma reagin titer decreased, and his chlamydia RNA became undetectable. However, the skin lesions remained unchanged.
Incisional biopsies of 4 clinically distinct skin lesions revealed well-delineated dermal proliferations of cells with eosinophilic granular cytoplasm and indistinct cell borders (Figure 5). Two specimens displayed marked epidermal hyperplasia (Figure 6).
No atypical mitotic figures were identified. Immunohistochemistry for S-100 protein was diffusely positive in the neoplastic cells. Immunohistochemistry for Treponema pallidum was negative.
No mycobacterial or fungal organisms were identified in acid-fast bacillus, periodic acid–Schiff, or Gomori methenamine-silver–stained sections. All 4 lesions had histopathologic findings characteristic of granular cell tumors (GCTs). A lesion in the left inguinal region (Figure 4 [medial lesion]), which initially was thought to be condyloma latum or a squamous cell carcinoma (SCC), also was later confirmed to be a GCT.
Repeat positron emission tomography–computerized tomography several weeks later confirmed resolution of the previously noted lymphadenopathy. Although 2 GCTs have not recurred after biopsy, the other 2, which the patient refused to have completely excised, continued to grow. Follow-up 2.5 years after hospitalization revealed persistence of the lesions with no remarkable morphological changes.
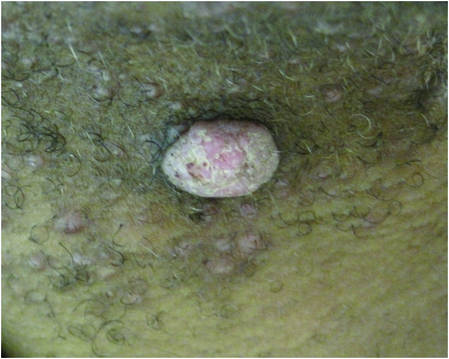
| 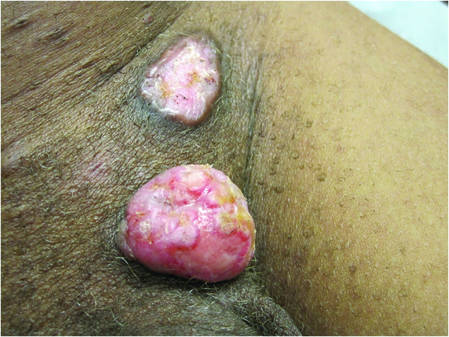
| |
| Figure 3. Verrucous pink papule on the right side of the neck. | Figure 4. Ulcerated angular plaque in the left inguinal/genital area with rolled borders and tumor extension deep to the subcutis adjacent to a vegetative, eroded, exophytic tumor with palpable deep extension. | |
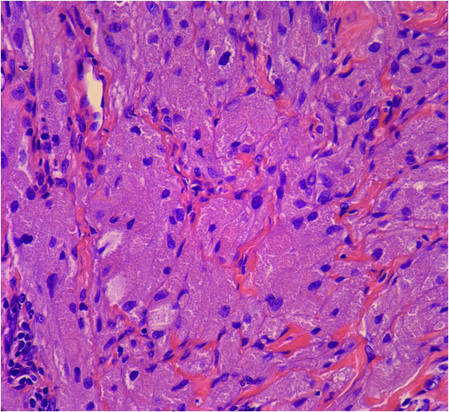
| 
| |
Figure 5. Large polygonal cells with eosinophilic granular cytoplasm, prominent bland nuclei, and indistinct cell borders (H&E, original magnification ×40). | Figure 6. Marked pseudoepitheliomatous hyperplasia (H&E, original magnification ×10). |
Comment
First described in 1854, GCTs are uncommon neoplasms of probable Schwann cell origin that can arise in almost any location of the body but most often appear on the skin and in the subcutaneous tissues and oral cavity.1,2 The commonly regarded rule of thirds describes its most favored locations: one-third on the tongue, one-third on the skin, and one-third in internal organs.3,4 Granular cell tumors occur with greater frequency in adults, females, and black individuals.1-5
Cutaneous GCTs usually present as solitary asymptomatic masses; however, multiple tumors have been noted in up to 25% of reported cases.4,6 In children, multiple cutaneous GCTs have been reported in the setting of neurofibromatosis type I as well as with other disorders.2,5,7-9
Cutaneous GCTs have been reported to range from sessile, pedunculated, or verrucous nodules to subcutaneous papules and nodules with no epidermal change. Our case not only illustrated the diverse clinical appearance of cutaneous GCTs but also demonstrated multiple morphologically distinct cutaneous GCTs occurring in a single patient. Of particular interest is our patient’s coexisting secondary syphilis and LGV infections, which can pose a diagnostic dilemma to the unsuspecting clinician. The manifold appearances of this patient’s GCTs resulted in a broad differential diagnosis. Syphilis (condyloma latum), condyloma acuminatum, LGV, metastatic disease, Kaposi sarcoma, lymphoma, dermatofibrosarcoma protuberans, leiomyoma, SCC, and deep fungal and atypical mycobacterial infection were all considerations. In 1981, Apisarnthanarax1 reviewed 88 cases of GCTs seen over a 15-year period and discovered that the preoperative clinical diagnoses were incorrect in all cases. Skin biopsy is necessary to diagnose GCT, and our patient’s case underscores the need for a thorough history, physical examination, and laboratory evaluation to rule out coexisting diseases.
Histopathology of cutaneous GCTs shows an unencapsulated dermal proliferation of large monotonous polygonal cells with blurred cell borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests. Nuclei are small, uniform, round, centrally located, and rarely contain mitoses.3 The presence of mitotic activity on histopathology does not necessarily portend malignant biological behavior.5 Overlying pseudoepitheliomatous hyperplasia has been reported in as many as 85% of GCTs and may mimic SCC.10 The neoplastic cells stain positively with S-100 protein, neuron-specific enolase, and peripheral nerve myelin proteins.3,4 The cytoplasmic granules are positive on periodic acid–Schiff staining and diastase resistant and will sometimes stain for CD68.1 Electron microscopy shows degraded myelinated axons intracellularly.4
Malignancy is rare and reportedly occurs in 1% to 3% of cases.4,5 Consideration of both clinical behavior and histopathology is important in distinguishing benign from malignant lesions. According to published reports, in GCTs that were regarded as malignant, size tended to be greater than 4 cm, growth was rapid, and metastases to regional lymph nodes were observed.4,5 Histologically, nuclear pleomorphism and atypia, cell spindling, vesicular nuclei with prominent nucleoli, necrosis, and high mitotic activity favor malignancy.1,3
Treatment is complete surgical excision. Observation is acceptable if tumors are asymptomatic and do not impede function. Regression of some GCTs has been induced with use of intralesional corticosteroids.5 Spontaneous regression is rare. Prior reports have emphasized the importance of long-term follow-up in patients with multiple GCTs to monitor for development of systemic lesions.4
1. Apisarnthanarax P. Granular cell tumor. an analysis of 16 cases and review of the literature. J Am Acad Dermatol. 1981;5:171-182.
2. Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol. 1991;24(2, pt 2):359-363.
3. Hazan C, Fangman W. Multiple cutaneous granular-cell tumors. Dermatol Online J. 2007;13:4.
4. Gross VL, Lynfield Y. Multiple cutaneous granular cell tumors: a case report and review of the literature. Cutis. 2002;69:343-346.
5. Martin RW 3rd, Neldner KH, Boyd AS, et al. Multiple cutaneous granular cell tumors and neurofibromatosis in childhood. a case report and review of the literature. Arch Dermatol. 1990;126:1051-1056.
6. Janousková G, Campr V, Konkol’ová R, et al. Multiple granular cell tumour. J Eur Acad Dermatol Venereol. 2004;18:347-349.
7. Gunson TH, Hashim N, Sharpe GR. Generalized lentiginosis, short stature, and multiple cutaneous nodules—quiz case. LEOPARD syndrome (LS) associated with multiple granular cell tumors (GCTs). Arch Dermatol. 2010;146:337-342.
8. De Raeve L, Roseeuw D, Otten J. Multiple cutaneous granular cell tumors in a child in remission for Hodgkin’s disease. J Am Acad Dermatol. 2002;47(2 suppl):S180-S182.
9. Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol. 2010;27:209-211.
10. Bangle R Jr. A morphological and histochemical study of the granular-cell myoblastoma. Cancer. 1952;5:950-965.
Case Report
A 27-year-old black man was admitted to the hospital with chills; night sweats; unintentional 25-lb weight loss; and multiple widespread, painful, progressively enlarging skin nodules of 3 months’ duration. The lesions had first developed on the back and later appeared on the face, trunk, arms, thighs, and genital region. He denied dysuria or urethral discharge. He had a remote history of adequately treated chlamydia infection but no other remarkable personal or family history.
|
| ||
| Figure 1. Firm subcutaneous nodules on the back with no epidermal change. | ||
|
| ||
| Figure 2. Firm dermal papule on the anterior aspect of the left shoulder with violaceous hyperpigmentation (dermatofibromalike). |
Physical examination revealed a thin man with more than 20 lesions on the face, trunk, arms, thighs, and genital region ranging in size from 1 to 4 cm. Lesion morphologies varied greatly and included subcutaneous firm nodules with no epidermal change (Figure 1); dermatofibromalike nodules with overlying erythema and hyperpigmentation (Figure 2); condylomalike, verrucous, pink papulonodules (Figure 3); ulcerated angular plaques with rolled borders and palpable tumor extension deep (1–2 cm) to the subcutis (Figure 4); and a vegetative, eroded, exophytic tumor with palpable deep extension (Figure 4). A diffuse, erythematous, macular eruption also was noted on the trunk and bilateral arms and legs including the soles of both feet along with nontender cervical, axillary, and inguinal lymphadenopathy. The ocular, oral, and nasal mucosae were not affected.
The differential diagnosis for each lesion differed based on morphology. Infectious, inflammatory, and neoplastic processes were considered, including syphilis, dermatofibroma, dermatofibrosarcoma protuberans, metastatic disease, leukemia cutis, sarcoidosis, panniculitis, condyloma acuminatum, and vegetative herpes simplex virus infection (inguinal lesion).
Laboratory data revealed a reactive rapid plasma reagin with treponemal IgG titers of 1:64. Urine chlamydia RNA probe and lymphogranuloma venereum (LGV) serum antibodies also were positive. Human immunodeficiency virus screening was negative. Positron emission tomography–computerized tomography revealed enlarged and hypermetabolic lymphadenopathy above and below the diaphragm.
After therapy with intravenous penicillin G and oral doxycycline for concurrent secondary syphilis and LGV, the patient’s macular eruption and constitutional symptoms resolved within weeks of the initial presentation. His lymphadenopathy improved, his rapid plasma reagin titer decreased, and his chlamydia RNA became undetectable. However, the skin lesions remained unchanged.
Incisional biopsies of 4 clinically distinct skin lesions revealed well-delineated dermal proliferations of cells with eosinophilic granular cytoplasm and indistinct cell borders (Figure 5). Two specimens displayed marked epidermal hyperplasia (Figure 6).
No atypical mitotic figures were identified. Immunohistochemistry for S-100 protein was diffusely positive in the neoplastic cells. Immunohistochemistry for Treponema pallidum was negative.
No mycobacterial or fungal organisms were identified in acid-fast bacillus, periodic acid–Schiff, or Gomori methenamine-silver–stained sections. All 4 lesions had histopathologic findings characteristic of granular cell tumors (GCTs). A lesion in the left inguinal region (Figure 4 [medial lesion]), which initially was thought to be condyloma latum or a squamous cell carcinoma (SCC), also was later confirmed to be a GCT.
Repeat positron emission tomography–computerized tomography several weeks later confirmed resolution of the previously noted lymphadenopathy. Although 2 GCTs have not recurred after biopsy, the other 2, which the patient refused to have completely excised, continued to grow. Follow-up 2.5 years after hospitalization revealed persistence of the lesions with no remarkable morphological changes.
|
|
| |
| Figure 3. Verrucous pink papule on the right side of the neck. | Figure 4. Ulcerated angular plaque in the left inguinal/genital area with rolled borders and tumor extension deep to the subcutis adjacent to a vegetative, eroded, exophytic tumor with palpable deep extension. | |
|
|
| |
Figure 5. Large polygonal cells with eosinophilic granular cytoplasm, prominent bland nuclei, and indistinct cell borders (H&E, original magnification ×40). | Figure 6. Marked pseudoepitheliomatous hyperplasia (H&E, original magnification ×10). |
Comment
First described in 1854, GCTs are uncommon neoplasms of probable Schwann cell origin that can arise in almost any location of the body but most often appear on the skin and in the subcutaneous tissues and oral cavity.1,2 The commonly regarded rule of thirds describes its most favored locations: one-third on the tongue, one-third on the skin, and one-third in internal organs.3,4 Granular cell tumors occur with greater frequency in adults, females, and black individuals.1-5
Cutaneous GCTs usually present as solitary asymptomatic masses; however, multiple tumors have been noted in up to 25% of reported cases.4,6 In children, multiple cutaneous GCTs have been reported in the setting of neurofibromatosis type I as well as with other disorders.2,5,7-9
Cutaneous GCTs have been reported to range from sessile, pedunculated, or verrucous nodules to subcutaneous papules and nodules with no epidermal change. Our case not only illustrated the diverse clinical appearance of cutaneous GCTs but also demonstrated multiple morphologically distinct cutaneous GCTs occurring in a single patient. Of particular interest is our patient’s coexisting secondary syphilis and LGV infections, which can pose a diagnostic dilemma to the unsuspecting clinician. The manifold appearances of this patient’s GCTs resulted in a broad differential diagnosis. Syphilis (condyloma latum), condyloma acuminatum, LGV, metastatic disease, Kaposi sarcoma, lymphoma, dermatofibrosarcoma protuberans, leiomyoma, SCC, and deep fungal and atypical mycobacterial infection were all considerations. In 1981, Apisarnthanarax1 reviewed 88 cases of GCTs seen over a 15-year period and discovered that the preoperative clinical diagnoses were incorrect in all cases. Skin biopsy is necessary to diagnose GCT, and our patient’s case underscores the need for a thorough history, physical examination, and laboratory evaluation to rule out coexisting diseases.
Histopathology of cutaneous GCTs shows an unencapsulated dermal proliferation of large monotonous polygonal cells with blurred cell borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests. Nuclei are small, uniform, round, centrally located, and rarely contain mitoses.3 The presence of mitotic activity on histopathology does not necessarily portend malignant biological behavior.5 Overlying pseudoepitheliomatous hyperplasia has been reported in as many as 85% of GCTs and may mimic SCC.10 The neoplastic cells stain positively with S-100 protein, neuron-specific enolase, and peripheral nerve myelin proteins.3,4 The cytoplasmic granules are positive on periodic acid–Schiff staining and diastase resistant and will sometimes stain for CD68.1 Electron microscopy shows degraded myelinated axons intracellularly.4
Malignancy is rare and reportedly occurs in 1% to 3% of cases.4,5 Consideration of both clinical behavior and histopathology is important in distinguishing benign from malignant lesions. According to published reports, in GCTs that were regarded as malignant, size tended to be greater than 4 cm, growth was rapid, and metastases to regional lymph nodes were observed.4,5 Histologically, nuclear pleomorphism and atypia, cell spindling, vesicular nuclei with prominent nucleoli, necrosis, and high mitotic activity favor malignancy.1,3
Treatment is complete surgical excision. Observation is acceptable if tumors are asymptomatic and do not impede function. Regression of some GCTs has been induced with use of intralesional corticosteroids.5 Spontaneous regression is rare. Prior reports have emphasized the importance of long-term follow-up in patients with multiple GCTs to monitor for development of systemic lesions.4
Case Report
A 27-year-old black man was admitted to the hospital with chills; night sweats; unintentional 25-lb weight loss; and multiple widespread, painful, progressively enlarging skin nodules of 3 months’ duration. The lesions had first developed on the back and later appeared on the face, trunk, arms, thighs, and genital region. He denied dysuria or urethral discharge. He had a remote history of adequately treated chlamydia infection but no other remarkable personal or family history.
|
| ||
| Figure 1. Firm subcutaneous nodules on the back with no epidermal change. | ||
|
| ||
| Figure 2. Firm dermal papule on the anterior aspect of the left shoulder with violaceous hyperpigmentation (dermatofibromalike). |
Physical examination revealed a thin man with more than 20 lesions on the face, trunk, arms, thighs, and genital region ranging in size from 1 to 4 cm. Lesion morphologies varied greatly and included subcutaneous firm nodules with no epidermal change (Figure 1); dermatofibromalike nodules with overlying erythema and hyperpigmentation (Figure 2); condylomalike, verrucous, pink papulonodules (Figure 3); ulcerated angular plaques with rolled borders and palpable tumor extension deep (1–2 cm) to the subcutis (Figure 4); and a vegetative, eroded, exophytic tumor with palpable deep extension (Figure 4). A diffuse, erythematous, macular eruption also was noted on the trunk and bilateral arms and legs including the soles of both feet along with nontender cervical, axillary, and inguinal lymphadenopathy. The ocular, oral, and nasal mucosae were not affected.
The differential diagnosis for each lesion differed based on morphology. Infectious, inflammatory, and neoplastic processes were considered, including syphilis, dermatofibroma, dermatofibrosarcoma protuberans, metastatic disease, leukemia cutis, sarcoidosis, panniculitis, condyloma acuminatum, and vegetative herpes simplex virus infection (inguinal lesion).
Laboratory data revealed a reactive rapid plasma reagin with treponemal IgG titers of 1:64. Urine chlamydia RNA probe and lymphogranuloma venereum (LGV) serum antibodies also were positive. Human immunodeficiency virus screening was negative. Positron emission tomography–computerized tomography revealed enlarged and hypermetabolic lymphadenopathy above and below the diaphragm.
After therapy with intravenous penicillin G and oral doxycycline for concurrent secondary syphilis and LGV, the patient’s macular eruption and constitutional symptoms resolved within weeks of the initial presentation. His lymphadenopathy improved, his rapid plasma reagin titer decreased, and his chlamydia RNA became undetectable. However, the skin lesions remained unchanged.
Incisional biopsies of 4 clinically distinct skin lesions revealed well-delineated dermal proliferations of cells with eosinophilic granular cytoplasm and indistinct cell borders (Figure 5). Two specimens displayed marked epidermal hyperplasia (Figure 6).
No atypical mitotic figures were identified. Immunohistochemistry for S-100 protein was diffusely positive in the neoplastic cells. Immunohistochemistry for Treponema pallidum was negative.
No mycobacterial or fungal organisms were identified in acid-fast bacillus, periodic acid–Schiff, or Gomori methenamine-silver–stained sections. All 4 lesions had histopathologic findings characteristic of granular cell tumors (GCTs). A lesion in the left inguinal region (Figure 4 [medial lesion]), which initially was thought to be condyloma latum or a squamous cell carcinoma (SCC), also was later confirmed to be a GCT.
Repeat positron emission tomography–computerized tomography several weeks later confirmed resolution of the previously noted lymphadenopathy. Although 2 GCTs have not recurred after biopsy, the other 2, which the patient refused to have completely excised, continued to grow. Follow-up 2.5 years after hospitalization revealed persistence of the lesions with no remarkable morphological changes.
|
|
| |
| Figure 3. Verrucous pink papule on the right side of the neck. | Figure 4. Ulcerated angular plaque in the left inguinal/genital area with rolled borders and tumor extension deep to the subcutis adjacent to a vegetative, eroded, exophytic tumor with palpable deep extension. | |
|
|
| |
Figure 5. Large polygonal cells with eosinophilic granular cytoplasm, prominent bland nuclei, and indistinct cell borders (H&E, original magnification ×40). | Figure 6. Marked pseudoepitheliomatous hyperplasia (H&E, original magnification ×10). |
Comment
First described in 1854, GCTs are uncommon neoplasms of probable Schwann cell origin that can arise in almost any location of the body but most often appear on the skin and in the subcutaneous tissues and oral cavity.1,2 The commonly regarded rule of thirds describes its most favored locations: one-third on the tongue, one-third on the skin, and one-third in internal organs.3,4 Granular cell tumors occur with greater frequency in adults, females, and black individuals.1-5
Cutaneous GCTs usually present as solitary asymptomatic masses; however, multiple tumors have been noted in up to 25% of reported cases.4,6 In children, multiple cutaneous GCTs have been reported in the setting of neurofibromatosis type I as well as with other disorders.2,5,7-9
Cutaneous GCTs have been reported to range from sessile, pedunculated, or verrucous nodules to subcutaneous papules and nodules with no epidermal change. Our case not only illustrated the diverse clinical appearance of cutaneous GCTs but also demonstrated multiple morphologically distinct cutaneous GCTs occurring in a single patient. Of particular interest is our patient’s coexisting secondary syphilis and LGV infections, which can pose a diagnostic dilemma to the unsuspecting clinician. The manifold appearances of this patient’s GCTs resulted in a broad differential diagnosis. Syphilis (condyloma latum), condyloma acuminatum, LGV, metastatic disease, Kaposi sarcoma, lymphoma, dermatofibrosarcoma protuberans, leiomyoma, SCC, and deep fungal and atypical mycobacterial infection were all considerations. In 1981, Apisarnthanarax1 reviewed 88 cases of GCTs seen over a 15-year period and discovered that the preoperative clinical diagnoses were incorrect in all cases. Skin biopsy is necessary to diagnose GCT, and our patient’s case underscores the need for a thorough history, physical examination, and laboratory evaluation to rule out coexisting diseases.
Histopathology of cutaneous GCTs shows an unencapsulated dermal proliferation of large monotonous polygonal cells with blurred cell borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests. Nuclei are small, uniform, round, centrally located, and rarely contain mitoses.3 The presence of mitotic activity on histopathology does not necessarily portend malignant biological behavior.5 Overlying pseudoepitheliomatous hyperplasia has been reported in as many as 85% of GCTs and may mimic SCC.10 The neoplastic cells stain positively with S-100 protein, neuron-specific enolase, and peripheral nerve myelin proteins.3,4 The cytoplasmic granules are positive on periodic acid–Schiff staining and diastase resistant and will sometimes stain for CD68.1 Electron microscopy shows degraded myelinated axons intracellularly.4
Malignancy is rare and reportedly occurs in 1% to 3% of cases.4,5 Consideration of both clinical behavior and histopathology is important in distinguishing benign from malignant lesions. According to published reports, in GCTs that were regarded as malignant, size tended to be greater than 4 cm, growth was rapid, and metastases to regional lymph nodes were observed.4,5 Histologically, nuclear pleomorphism and atypia, cell spindling, vesicular nuclei with prominent nucleoli, necrosis, and high mitotic activity favor malignancy.1,3
Treatment is complete surgical excision. Observation is acceptable if tumors are asymptomatic and do not impede function. Regression of some GCTs has been induced with use of intralesional corticosteroids.5 Spontaneous regression is rare. Prior reports have emphasized the importance of long-term follow-up in patients with multiple GCTs to monitor for development of systemic lesions.4
1. Apisarnthanarax P. Granular cell tumor. an analysis of 16 cases and review of the literature. J Am Acad Dermatol. 1981;5:171-182.
2. Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol. 1991;24(2, pt 2):359-363.
3. Hazan C, Fangman W. Multiple cutaneous granular-cell tumors. Dermatol Online J. 2007;13:4.
4. Gross VL, Lynfield Y. Multiple cutaneous granular cell tumors: a case report and review of the literature. Cutis. 2002;69:343-346.
5. Martin RW 3rd, Neldner KH, Boyd AS, et al. Multiple cutaneous granular cell tumors and neurofibromatosis in childhood. a case report and review of the literature. Arch Dermatol. 1990;126:1051-1056.
6. Janousková G, Campr V, Konkol’ová R, et al. Multiple granular cell tumour. J Eur Acad Dermatol Venereol. 2004;18:347-349.
7. Gunson TH, Hashim N, Sharpe GR. Generalized lentiginosis, short stature, and multiple cutaneous nodules—quiz case. LEOPARD syndrome (LS) associated with multiple granular cell tumors (GCTs). Arch Dermatol. 2010;146:337-342.
8. De Raeve L, Roseeuw D, Otten J. Multiple cutaneous granular cell tumors in a child in remission for Hodgkin’s disease. J Am Acad Dermatol. 2002;47(2 suppl):S180-S182.
9. Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol. 2010;27:209-211.
10. Bangle R Jr. A morphological and histochemical study of the granular-cell myoblastoma. Cancer. 1952;5:950-965.
1. Apisarnthanarax P. Granular cell tumor. an analysis of 16 cases and review of the literature. J Am Acad Dermatol. 1981;5:171-182.
2. Guiglia MC, Prendiville JS. Multiple granular cell tumors associated with giant speckled lentiginous nevus and nevus flammeus in a child. J Am Acad Dermatol. 1991;24(2, pt 2):359-363.
3. Hazan C, Fangman W. Multiple cutaneous granular-cell tumors. Dermatol Online J. 2007;13:4.
4. Gross VL, Lynfield Y. Multiple cutaneous granular cell tumors: a case report and review of the literature. Cutis. 2002;69:343-346.
5. Martin RW 3rd, Neldner KH, Boyd AS, et al. Multiple cutaneous granular cell tumors and neurofibromatosis in childhood. a case report and review of the literature. Arch Dermatol. 1990;126:1051-1056.
6. Janousková G, Campr V, Konkol’ová R, et al. Multiple granular cell tumour. J Eur Acad Dermatol Venereol. 2004;18:347-349.
7. Gunson TH, Hashim N, Sharpe GR. Generalized lentiginosis, short stature, and multiple cutaneous nodules—quiz case. LEOPARD syndrome (LS) associated with multiple granular cell tumors (GCTs). Arch Dermatol. 2010;146:337-342.
8. De Raeve L, Roseeuw D, Otten J. Multiple cutaneous granular cell tumors in a child in remission for Hodgkin’s disease. J Am Acad Dermatol. 2002;47(2 suppl):S180-S182.
9. Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol. 2010;27:209-211.
10. Bangle R Jr. A morphological and histochemical study of the granular-cell myoblastoma. Cancer. 1952;5:950-965.
Practice Points
- Granular cell tumors (GCTs) typically present as solitary lesions; however, multiple lesions occur in approximately 25% of cases.
- Granular cell tumors have a variable clinical appearance and may mimic malignant neoplasms (eg, squamous cell carcinoma) as well as infectious diseases (eg, condyloma, syphilis).
- The histological features of GCTs are distinctive, including an unencapsulated dermal proliferation of monotonous polygonal cells with indistinct borders and fine, granular, eosinophilic cytoplasm arranged in irregular sheets and nests.
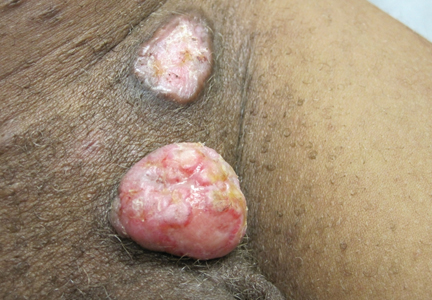
Exophytic Scalp Tumor
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
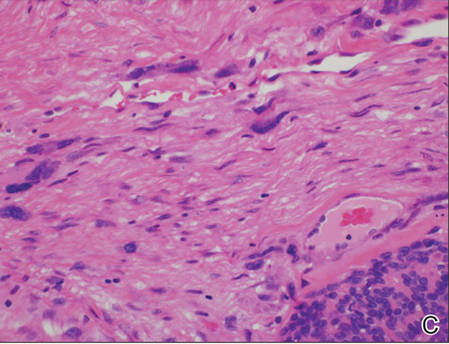
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
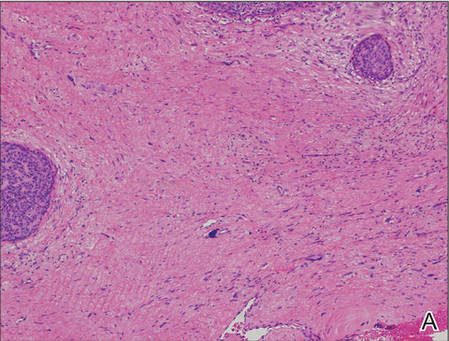
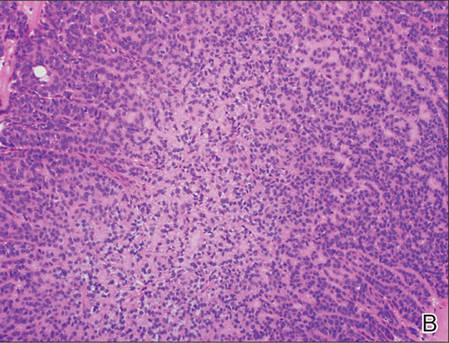
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
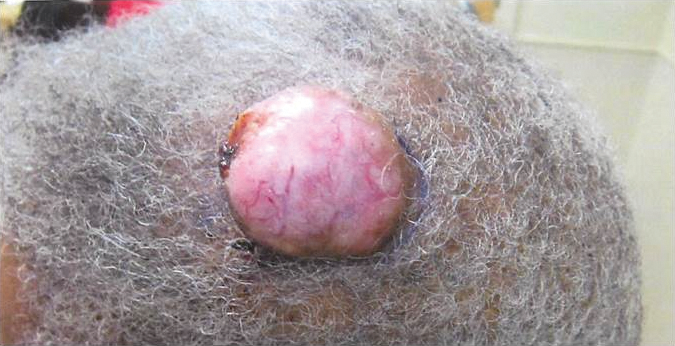
An 81-year-old man presented with a 3.5×3.0-cm pink exophytic tumor with an eroded surface and prominent vascularity on the left side of the parietal scalp. The patient reported that the tumor had been present for more than 30 years but recently had grown larger in size. He denied pain or pruritus in association with the lesion and did not report any systemic symptoms. He had received no prior treatments for the tumor.

Merkel Cell Carcinoma: A Review
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC

| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
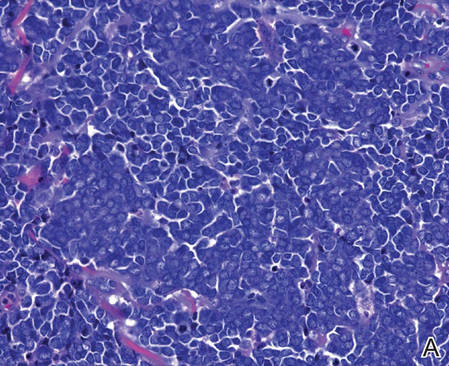
| ||
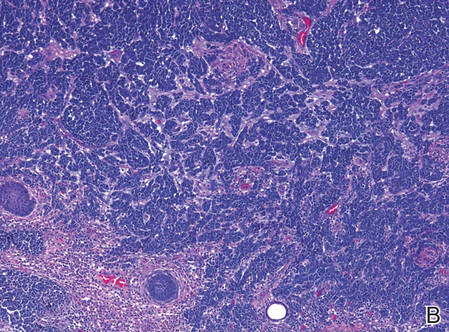
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
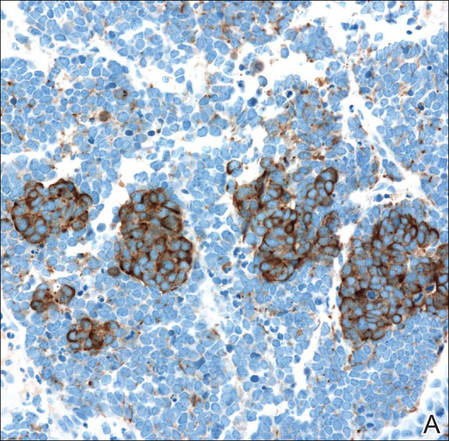
| ||
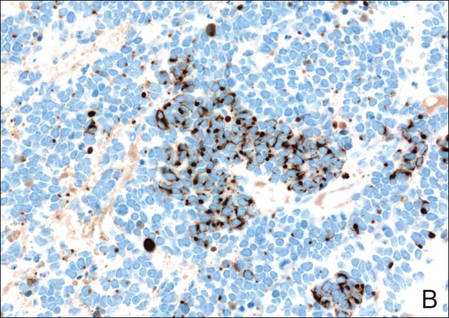
| ||
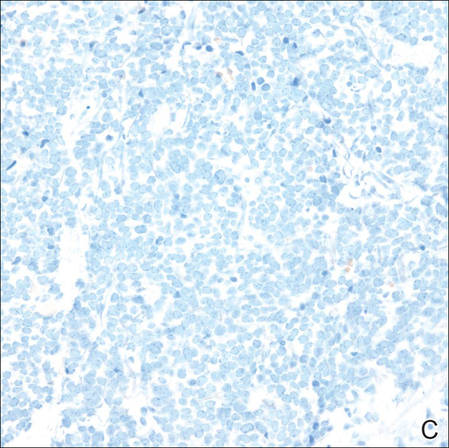
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
|
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
|
| ||
|
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
|
| ||
|
| ||
|
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
|
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
|
| ||
|
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
|
| ||
|
| ||
|
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Practice Points
- Merkel cell carcinoma has been associated with a novel polyomavirus.
- Merkel cell carcinoma follows a very aggressive course and is most likely metastatic at diagnosis.
FDA Proposes New Rule to Ban Use of Indoor Tanning Devices by Minors
The US Food and Drug Administration (FDA) has proposed 2 new rules to protect consumers from health risks associated with indoor tanning by banning use of indoor tanning devices by minors and imposing safety measures.
The first proposed rule restricts the use of indoor tanning devices to adults 18 years and older. It also requires indoor tanning facilities to inform adult users about the health risks of indoor tanning and obtain a signed risk acknowledgement from consumers before their first tanning session and every 6 months thereafter.
“Exposure to UV radiation from indoor tanning is a preventable cause of skin cancer,” explained Markham C. Luke, MD, PhD, deputy office director for the Office of Device Evaluation at the FDA’s Center for Devices and Radiological Health. “The FDA is committed to protecting public health by informing consumers of the risks of indoor tanning.”
The second proposed rule addresses performance standards, requiring manufacturers and indoor tanning facilities to take measures to improve the overall safety of tanning devices. Key changes would include:
- Make consumer warnings more prominent and easier to read on tanning devices.
- Require an easily accessible emergency shutoff switch (or panic button) on all tanning devices.
- Add requirements to limit the amount of light allowed through protective eyewear to protect the eyes.
- Improve labeling on replacement bulbs to ensure tanning facility operators use the proper bulbs to reduce risk for accidental burns.
- Prohibit tanning facilities from making dangerous device modifications (eg, installing stronger bulbs) without recertifying and reidentifying the device with the FDA.
The FDA reports that more than 1 million minors use indoor tanning facilities each year. According to the American Academy of Dermatology, consumers younger than 35 years who use indoor tanning facilities are 59% more likely to develop melanoma than those who have never tanned indoors. Because the effects of UV exposure are cumulative and add up over the course of one’s lifetime, minors who use indoor tanning devices are at an increased risk for developing melanoma and nonmelanoma skin cancers later in life.
In 2014 the FDA began requiring tanning devices to be labeled with a visible warning stating that individuals younger than 18 years should not use them. Additionally, several states have already passed laws prohibiting minors from indoor tanning; in Connecticut, New Jersey, New York, and Pennsylvania, tanning devices are banned in individuals younger than 17 years.
Dermatologists are in the position to discuss the health risks of indoor tanning with all patients regardless of age. Patients should be reminded that failure to wear appropriate protective eyewear can lead to short-term and long-term eye injury and that long exposures can lead to burning that may not be recognized until it is too late. It also is important to warn patients that tanning while using certain medications or cosmetics may cause increased sensitivity to UV radiation. Patients can be referred to the FDA website for more consumer updates about indoor tanning and the proposed rules.
The US Food and Drug Administration (FDA) has proposed 2 new rules to protect consumers from health risks associated with indoor tanning by banning use of indoor tanning devices by minors and imposing safety measures.
The first proposed rule restricts the use of indoor tanning devices to adults 18 years and older. It also requires indoor tanning facilities to inform adult users about the health risks of indoor tanning and obtain a signed risk acknowledgement from consumers before their first tanning session and every 6 months thereafter.
“Exposure to UV radiation from indoor tanning is a preventable cause of skin cancer,” explained Markham C. Luke, MD, PhD, deputy office director for the Office of Device Evaluation at the FDA’s Center for Devices and Radiological Health. “The FDA is committed to protecting public health by informing consumers of the risks of indoor tanning.”
The second proposed rule addresses performance standards, requiring manufacturers and indoor tanning facilities to take measures to improve the overall safety of tanning devices. Key changes would include:
- Make consumer warnings more prominent and easier to read on tanning devices.
- Require an easily accessible emergency shutoff switch (or panic button) on all tanning devices.
- Add requirements to limit the amount of light allowed through protective eyewear to protect the eyes.
- Improve labeling on replacement bulbs to ensure tanning facility operators use the proper bulbs to reduce risk for accidental burns.
- Prohibit tanning facilities from making dangerous device modifications (eg, installing stronger bulbs) without recertifying and reidentifying the device with the FDA.
The FDA reports that more than 1 million minors use indoor tanning facilities each year. According to the American Academy of Dermatology, consumers younger than 35 years who use indoor tanning facilities are 59% more likely to develop melanoma than those who have never tanned indoors. Because the effects of UV exposure are cumulative and add up over the course of one’s lifetime, minors who use indoor tanning devices are at an increased risk for developing melanoma and nonmelanoma skin cancers later in life.
In 2014 the FDA began requiring tanning devices to be labeled with a visible warning stating that individuals younger than 18 years should not use them. Additionally, several states have already passed laws prohibiting minors from indoor tanning; in Connecticut, New Jersey, New York, and Pennsylvania, tanning devices are banned in individuals younger than 17 years.
Dermatologists are in the position to discuss the health risks of indoor tanning with all patients regardless of age. Patients should be reminded that failure to wear appropriate protective eyewear can lead to short-term and long-term eye injury and that long exposures can lead to burning that may not be recognized until it is too late. It also is important to warn patients that tanning while using certain medications or cosmetics may cause increased sensitivity to UV radiation. Patients can be referred to the FDA website for more consumer updates about indoor tanning and the proposed rules.
The US Food and Drug Administration (FDA) has proposed 2 new rules to protect consumers from health risks associated with indoor tanning by banning use of indoor tanning devices by minors and imposing safety measures.
The first proposed rule restricts the use of indoor tanning devices to adults 18 years and older. It also requires indoor tanning facilities to inform adult users about the health risks of indoor tanning and obtain a signed risk acknowledgement from consumers before their first tanning session and every 6 months thereafter.
“Exposure to UV radiation from indoor tanning is a preventable cause of skin cancer,” explained Markham C. Luke, MD, PhD, deputy office director for the Office of Device Evaluation at the FDA’s Center for Devices and Radiological Health. “The FDA is committed to protecting public health by informing consumers of the risks of indoor tanning.”
The second proposed rule addresses performance standards, requiring manufacturers and indoor tanning facilities to take measures to improve the overall safety of tanning devices. Key changes would include:
- Make consumer warnings more prominent and easier to read on tanning devices.
- Require an easily accessible emergency shutoff switch (or panic button) on all tanning devices.
- Add requirements to limit the amount of light allowed through protective eyewear to protect the eyes.
- Improve labeling on replacement bulbs to ensure tanning facility operators use the proper bulbs to reduce risk for accidental burns.
- Prohibit tanning facilities from making dangerous device modifications (eg, installing stronger bulbs) without recertifying and reidentifying the device with the FDA.
The FDA reports that more than 1 million minors use indoor tanning facilities each year. According to the American Academy of Dermatology, consumers younger than 35 years who use indoor tanning facilities are 59% more likely to develop melanoma than those who have never tanned indoors. Because the effects of UV exposure are cumulative and add up over the course of one’s lifetime, minors who use indoor tanning devices are at an increased risk for developing melanoma and nonmelanoma skin cancers later in life.
In 2014 the FDA began requiring tanning devices to be labeled with a visible warning stating that individuals younger than 18 years should not use them. Additionally, several states have already passed laws prohibiting minors from indoor tanning; in Connecticut, New Jersey, New York, and Pennsylvania, tanning devices are banned in individuals younger than 17 years.
Dermatologists are in the position to discuss the health risks of indoor tanning with all patients regardless of age. Patients should be reminded that failure to wear appropriate protective eyewear can lead to short-term and long-term eye injury and that long exposures can lead to burning that may not be recognized until it is too late. It also is important to warn patients that tanning while using certain medications or cosmetics may cause increased sensitivity to UV radiation. Patients can be referred to the FDA website for more consumer updates about indoor tanning and the proposed rules.

Transition From Lichen Sclerosus to Squamous Cell Carcinoma in a Single Tissue Section
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
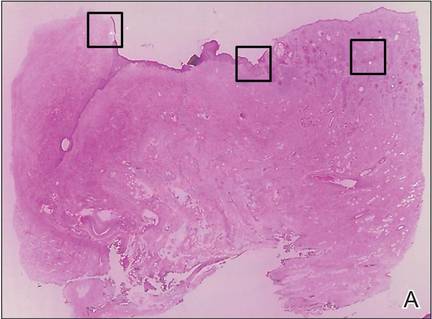
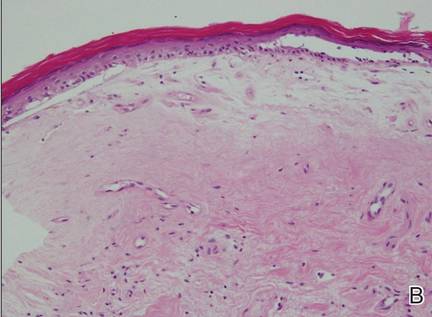
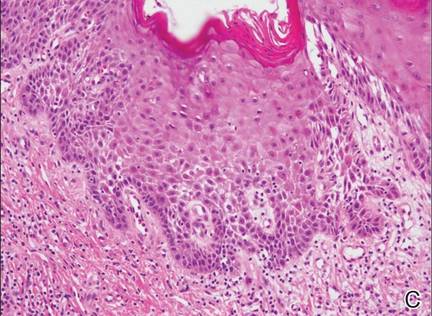

Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
Practice Points
- Lichen sclerosus has a considerable risk for malignant transformation and requires continuous follow-up in all patients.
- Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
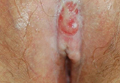
Intralesional interferon excels for challenging basal cell carcinomas
WAIKOLOA, HAWAII – Intralesional injection of interferon alfa-2b is an excellent option for the treatment of large problematic basal cell carcinomas in patients who aren’t interested in the higher-morbidity options, Dr. David L. Swanson said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“This is a really effective way to treat basal cell carcinomas. These things just melt away before your eyes. It’s really quite amazing,” observed Dr. Swanson of Mayo Clinic Scottsdale (Ariz.).

He finds this therapy particularly useful in frail elderly patients who have a large BCC on the head or neck. A good example would be an 89-year-old with multiple comorbid conditions who has a 2-cm BCC on the tip of the nose and doesn’t want anything done about it. The patient declines the options of Mohs micrographic surgery or radiotherapy.
“This is the patient who just wants to be left alone. That’s fine if they’re going to be dead within a year, but if they’re going to be around for several years, that basal cell carcinoma could become a major issue for them,” the dermatologist continued.
He and his colleagues at the Mayo Clinic follow a treatment regimen similar to one laid out by Turkish investigators more than a decade ago in one of the few long-term outcome studies of intralesional interferon for treatment of BCCs.
Although interferon alfa-2b is approved for the intralesional treatment of genital warts and subcutaneously for Kaposi’s sarcoma and malignant melanoma, among other conditions, it’s off-label therapy for BCCs. The treatment entails thrice-weekly intralesional injections for 3 weeks. The dosing is 1.5 million units per injection for BCCs smaller than 2 cm and 3 million units per injection for BCCs that are 2 cm or larger. The injections are given without anesthesia, but premedication with 500-1,000 mg of acetaminophen is advisable to minimize aches and fever.
Interferon alfa-2b (Intron A) comes in a vial containing 10 million units with 1 mL of diluent. It’s important to reconstitute it carefully, similar to onabotulinumtoxin. Don’t shake it, Dr. Swanson advised.
The Turkish report included 20 patients with histopathologically proven BCCs on the head or neck. At clinical and dermatopathologic follow-up 8 weeks after the last interferon injection, 11 BCCs showed complete clinical and histopathologic cure, six showed partial remission, two showed no response, and one actually increased in size during treatment.
The 11 patients with an initial complete cure were followed for 7 years. During that period, only one of the 11 skin cancers recurred, at the fifth year (Clin Drug Investig. 2005;25[10]:661-7).
Dr. Swanson reported having no financial conflicts regarding his presentation. SDEF and this news organization are owned by the same parent company.
WAIKOLOA, HAWAII – Intralesional injection of interferon alfa-2b is an excellent option for the treatment of large problematic basal cell carcinomas in patients who aren’t interested in the higher-morbidity options, Dr. David L. Swanson said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“This is a really effective way to treat basal cell carcinomas. These things just melt away before your eyes. It’s really quite amazing,” observed Dr. Swanson of Mayo Clinic Scottsdale (Ariz.).
He finds this therapy particularly useful in frail elderly patients who have a large BCC on the head or neck. A good example would be an 89-year-old with multiple comorbid conditions who has a 2-cm BCC on the tip of the nose and doesn’t want anything done about it. The patient declines the options of Mohs micrographic surgery or radiotherapy.
“This is the patient who just wants to be left alone. That’s fine if they’re going to be dead within a year, but if they’re going to be around for several years, that basal cell carcinoma could become a major issue for them,” the dermatologist continued.
He and his colleagues at the Mayo Clinic follow a treatment regimen similar to one laid out by Turkish investigators more than a decade ago in one of the few long-term outcome studies of intralesional interferon for treatment of BCCs.
Although interferon alfa-2b is approved for the intralesional treatment of genital warts and subcutaneously for Kaposi’s sarcoma and malignant melanoma, among other conditions, it’s off-label therapy for BCCs. The treatment entails thrice-weekly intralesional injections for 3 weeks. The dosing is 1.5 million units per injection for BCCs smaller than 2 cm and 3 million units per injection for BCCs that are 2 cm or larger. The injections are given without anesthesia, but premedication with 500-1,000 mg of acetaminophen is advisable to minimize aches and fever.
Interferon alfa-2b (Intron A) comes in a vial containing 10 million units with 1 mL of diluent. It’s important to reconstitute it carefully, similar to onabotulinumtoxin. Don’t shake it, Dr. Swanson advised.
The Turkish report included 20 patients with histopathologically proven BCCs on the head or neck. At clinical and dermatopathologic follow-up 8 weeks after the last interferon injection, 11 BCCs showed complete clinical and histopathologic cure, six showed partial remission, two showed no response, and one actually increased in size during treatment.
The 11 patients with an initial complete cure were followed for 7 years. During that period, only one of the 11 skin cancers recurred, at the fifth year (Clin Drug Investig. 2005;25[10]:661-7).
Dr. Swanson reported having no financial conflicts regarding his presentation. SDEF and this news organization are owned by the same parent company.
WAIKOLOA, HAWAII – Intralesional injection of interferon alfa-2b is an excellent option for the treatment of large problematic basal cell carcinomas in patients who aren’t interested in the higher-morbidity options, Dr. David L. Swanson said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“This is a really effective way to treat basal cell carcinomas. These things just melt away before your eyes. It’s really quite amazing,” observed Dr. Swanson of Mayo Clinic Scottsdale (Ariz.).
He finds this therapy particularly useful in frail elderly patients who have a large BCC on the head or neck. A good example would be an 89-year-old with multiple comorbid conditions who has a 2-cm BCC on the tip of the nose and doesn’t want anything done about it. The patient declines the options of Mohs micrographic surgery or radiotherapy.
“This is the patient who just wants to be left alone. That’s fine if they’re going to be dead within a year, but if they’re going to be around for several years, that basal cell carcinoma could become a major issue for them,” the dermatologist continued.
He and his colleagues at the Mayo Clinic follow a treatment regimen similar to one laid out by Turkish investigators more than a decade ago in one of the few long-term outcome studies of intralesional interferon for treatment of BCCs.
Although interferon alfa-2b is approved for the intralesional treatment of genital warts and subcutaneously for Kaposi’s sarcoma and malignant melanoma, among other conditions, it’s off-label therapy for BCCs. The treatment entails thrice-weekly intralesional injections for 3 weeks. The dosing is 1.5 million units per injection for BCCs smaller than 2 cm and 3 million units per injection for BCCs that are 2 cm or larger. The injections are given without anesthesia, but premedication with 500-1,000 mg of acetaminophen is advisable to minimize aches and fever.
Interferon alfa-2b (Intron A) comes in a vial containing 10 million units with 1 mL of diluent. It’s important to reconstitute it carefully, similar to onabotulinumtoxin. Don’t shake it, Dr. Swanson advised.
The Turkish report included 20 patients with histopathologically proven BCCs on the head or neck. At clinical and dermatopathologic follow-up 8 weeks after the last interferon injection, 11 BCCs showed complete clinical and histopathologic cure, six showed partial remission, two showed no response, and one actually increased in size during treatment.
The 11 patients with an initial complete cure were followed for 7 years. During that period, only one of the 11 skin cancers recurred, at the fifth year (Clin Drug Investig. 2005;25[10]:661-7).
Dr. Swanson reported having no financial conflicts regarding his presentation. SDEF and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR

Tools for Diagnosing Skin Cancer Earlier: Report From the AAD Meeting
At the 74th Annual Meeting of the American Academy of Dermatology, Dr. Orit Markowitz discussed noninvasive imaging tools that can help dermatologists diagnose skin cancers earlier. She provides highlights from this session, including the use of dermoscopy and optical coherence technology to detect features of early melanoma and nonmelanoma skin cancers as well as monitor skin cancer management. A lesion that is pink clinically but shows pigment dermoscopically should be biopsied, Dr. Markowtiz advises, as it may be an early amelanotic melanoma. She also notes that noninvasive imaging tools can be used to detect residual tumor cells in treated skin that otherwise looks clinically normal.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
At the 74th Annual Meeting of the American Academy of Dermatology, Dr. Orit Markowitz discussed noninvasive imaging tools that can help dermatologists diagnose skin cancers earlier. She provides highlights from this session, including the use of dermoscopy and optical coherence technology to detect features of early melanoma and nonmelanoma skin cancers as well as monitor skin cancer management. A lesion that is pink clinically but shows pigment dermoscopically should be biopsied, Dr. Markowtiz advises, as it may be an early amelanotic melanoma. She also notes that noninvasive imaging tools can be used to detect residual tumor cells in treated skin that otherwise looks clinically normal.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
At the 74th Annual Meeting of the American Academy of Dermatology, Dr. Orit Markowitz discussed noninvasive imaging tools that can help dermatologists diagnose skin cancers earlier. She provides highlights from this session, including the use of dermoscopy and optical coherence technology to detect features of early melanoma and nonmelanoma skin cancers as well as monitor skin cancer management. A lesion that is pink clinically but shows pigment dermoscopically should be biopsied, Dr. Markowtiz advises, as it may be an early amelanotic melanoma. She also notes that noninvasive imaging tools can be used to detect residual tumor cells in treated skin that otherwise looks clinically normal.
