User login
Idiopathic Follicular Mucinosis or Mycosis Fungoides? Classification and Diagnostic Challenges
When follicular mucinosis (FM) is defined as an epithelial reaction pattern characterized by intrafollicular and perifollicular mucin accumulation, it cannot be considered a distinct disease entity, as this pattern is ubiquitously present in various inflammatory and neoplastic skin conditions.1,2 The distinction between idiopathic FM and lymphoma-associated follicular mucinosis (LAFM) was made several years ago by authors who evaluated the differences in the clinical presentation of these entities, including patient age at onset, number of lesions, pattern of distribution, and most importantly clinical progression.1 In this article, we discuss the importance of close clinical follow-up in patients with FM or patch-stage mycosis fungoides (MF) in whom histopathologic evaluation is ambiguous or nondiagnostic. We also highlight the value of ancillary testing, including T-cell receptor gene rearrangement, flow cytometry, and immunohistochemistry, as a component in the diagnostic process rather than the sole diagnostic moiety. A review of the pertinent literature also is performed.
History of FM and MF
Pinkus3 first described an entity he termed alopecia mucinosa in 1957. Pinkus noted 3 distinct patterns: an idiopathic form of alopecia mucinosa, lymphoblastoma with associated FM, and alopecia mucinosa that later transformed into lymphoblastoma.4 In 1983, however, Pinkus4 described uncertainty if alopecia mucinosa represented the first stage of MF or if patients with alopecia mucinosa were simply at an increased risk for developing lymphoma. He believed there were too many cases of lymphoma following a diagnosis of alopecia mucinosa for the relation to be coincidental, yet he noted that many of the cases resolved either spontaneously or following treatment with x-rays or topical steroids. He concluded his report with a sentiment that is echoed in many current studies regarding this entity: “Many questions surrounding this entity are as unanswerable today as they were 25 years ago.”4
Jablonska et al5 were the first to coin the term mucinosis follicularis, now known as FM, to replace alopecia mucinosa because they felt the description was more accurate, as lesions also arise on non–hair-bearing skin. Although there is general agreement that there is a form of MF that has associated FM, this is where the agreement ends with regard to the diagnosis of MF versus FM. Böer et al6 discussed the historic evolution of these terms, mostly to highlight the origins of the confusion. The investigators proposed that FM should only be used as a descriptive term and that all cases of alopecia mucinosa represent MF. They also concluded that many benign dermatoses associated with a risk for evolution to MF (eg, small and large plaque psoriasis [LPP]) should simply be diagnosed as MF.6 Subsequently, the proposal that idiopathic FM and LAFM are not 2 distinct entities but rather a clinicopathologic continuum and that idiopathic FM is simply a variant of MF along this spectrum has gained some approval.6,7 However, this belief is not shared among all authorities in the field, and attempts to define diagnostic criteria that distinguish between a benign clinical course and a course that is more progressive and fatal continue. Currently, it is agreed upon that when distinguishing between these 2 clinical courses, primary (idiopathic) follicular mucinosis refers to a benign course with no overt sign of malignancy, and lymphoma-associated follicular mucinosis refers to a diagnostic malignant condition. Lymphoma-associated follicular mucinosis refers to FM associated with cutaneous T-cell lymphoma, the most common form of which is FM. Many authors8-15 have sought ancillary methodologies in addition to clinical parameters to assist in the evaluation between both disease courses. Methodologies have included assessment of T-cell receptor gene rearrangements, flow cytometry, and immunohistochemical staining, mostly as an effort to establish monoclonality as a defining characteristic of LAFM; however, monoclonality in cutaneous T-cell infiltrates should be interpreted with caution and should not be considered as a confirmation of malignancy due to recent findings of monoclonality in benign inflammatory dermatoses such as lichen planus. The Table outlines several of the most common benign inflammatory dermatoses that demonstrate monoclonality, but this list should not be considered exhaustive, as there are many others in which monoclonality is sometimes seen.8-15 The lack of definitive criteria to distinguish between the 2 groups has led to confusion and consternation regarding the diagnosis of idiopathic FM versus LAFM and has led many in the field to consider the 2 conditions to be one and the same.

Diagnosis of FM and MF: Clinicopathologic Features
The World Health Organization (WHO) defined MF as an epidermotropic primary cutaneous T-cell lymphoma (CTCL) characterized by infiltrates of small- to medium-sized T lymphocytes with cerebriform nuclei. Further, the WHO stated that the term mycosis fungoides should be exclusively reserved for classical cases typified by the evolution of cutaneous patches, plaques, and tumors, or for variants that show a similar clinical course.16 Mycosis fungoides is divided into 3 stages—patch, plaque, and tumor—which are solely clinical descriptors.17 The WHO also described a clinical staging system with pathologic emphasis placed only on lymph node involvement and identification of Sézary cells.16 It lists folliculotropic MF as a variant, with only some cases presenting with mucinous degeneration of hair follicles. A lack of consensus among pathologists regarding criteria for diagnosis in patch-stage MF remains, but diagnosis of plaque-stage disease is not regularly debated due to its more reliably present, well-developed histologic features (eg, haloed lymphocytes, epidermotropism of lymphocytes, lymphocytes with convoluted nuceli, Pautrier microabscesses).18 Although there have been specific histologic findings reported to be associated with patch-stage MF, they have only been present in a few cases and are therefore of limited usefulness in practice.1,19 The categorization of patients with subtle histologic features common to both MF and inflammatory conditions such as parapsoriasis en plaques (the term plaque in this case is a misnomer because the word plaque means patch in French) continues to be elusive. A lack of agreement regarding LPP persists in the current literature in the same manner as FM. Some researchers have contended for many years that LPP is a type of MF, while others remain unconvinced, mainly due to the lack of evidence that lumping a benign condition (LPP) with an increased risk for malignant transformation and a known malignancy (MF) together is of any benefit to the patient. Assessment of clinicopathologic correlation, immunohistochemistry, clonality, and T-cell gene rearrangement have failed to positively identify patients who are at risk for disease progression, whether the diagnosis is called LPP or early patch-stage MF.20
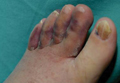
Mycosis fungoides is more common in males and its incidence increases with age; however, diagnosis should not be ruled out based on age or gender. Typical presentation of early-stage disease includes erythematous patches or plaques, often with light scaling.19 Lesions routinely are of long-standing duration (months to years), are located in areas that are infrequently exposed to sunlight, and often are 5 cm in diameter or larger with irregular borders.21 Associated poikiloderma is relatively specific to MF but rarely is seen in other CTCLs, connective-tissue diseases, and some genodermatoses. Poikiloderma commonly is identified in LPP, which shows the same telangiectasia, mottled pigmentation, and epidermal atrophy as MF-associated poikiloderma, leading some to believe that there is no separation between the 2 conditions. In all stages of MF, lesions frequently are numerous and occur on multiple sites. Plaques and tumors can show spontaneous ulceration. When lesions are folliculotropic, they can cause localized alopecia, follicular-based papules, and fungating pseudotumors in more advanced stages.1 The clinical presentation of FM substantially overlaps with folliculotropic MF, and although FM lesions often are solitary and are located on the face or scalp, they also can present as multiple lesions located elsewhere on the body. It also has been proposed that folliculotropic MF should not be separated from FM-associated MF (or LAFM).22
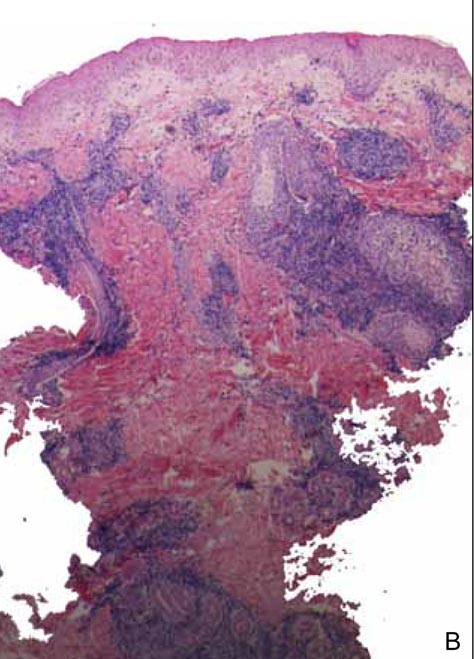
The characteristic histologic picture of LAFM in patch or plaque stage shows mucin deposition within hair follicles, similar to idiopathic FM. On histology, both conditions demonstrate dense lymphoid infiltrates around and within hair follicles as well as in the dermis (Figure). Most cases of LAFM show epidermotropism of lymphocytes between follicles, but this finding is not present in every case and often disappears when the disease advances to the tumor stage.1,19 Although Pautrier microabscesses (collections of lymphocytes within the superficial epidermis) are considered to be somewhat specific to MF, they are only present in a minority of cases.20 In a study by the International Society for Cutaneous Lymphomas,21 the only histopathologic criteria that showed any appreciable sensitivity or specificity in the diagnosis of MF were the presence of lymphoid cells with variable nuclear and cytoplasmic features and/or strikingly irregular nuclear contours with the presence of lymphocytes larger than those usually seen in inflammatory dermatoses. Despite these criteria, the study reported a high misclassification rate. A complicated scoring system for diagnosis of MF in patch- or early plaque-stage disease was proposed by the International Society for Cutaneous Lymphomas,21 which integrates clinical, histopathologic, molecular, and immunophenotypic criteria. However, these criteria have been continually debated in the literature and are only discussed in this article in relation to the association between MF and FM. Diagnosis of tumor-stage MF is not addressed in this article, as it is readily identified as lymphoma and is not easily confused with idiopathic FM.
|

Clinical assessment of a patient’s medical history to identify persistent and progressive disease is paramount to the diagnosis of MF. Although MF lesions tend to increase in size and number over time, this presentation is not without exception.21 In early patch-stage disease, eliminating some of the patient’s current medications may be sufficient in clearing cutaneous patches that cannot be conclusively identified as either MF or a benign inflammatory lymphoid infiltrate, which further emphasizes the importance of clinical assessment of the patient’s medical history in the diagnosis of MF. The shape of the lesions also is helpful in distinguishing between MF and other skin disorders, such as digitate dermatosis or LPP; unlike the latter, the waxing and waning nature of MF lesions often produces irregularly shaped patches with little coalescence. Again, there are some investigators who believe that these lesions represent varying presentations of MF.6
In a study by Cerroni et al,1 44 patients with FM were divided into 2 groups: (1) a cohort of 16 patients with no history or clinical evidence of MF or Sézary syndrome (ie, LAFM), and (2) a cohort of 28 patients with clinicopathologic evidence of CTCL. Patients in both groups were followed for a maximum of 20 years. Results indicated that that the presence of perifollicular or intrafollicular mucin, epidermotropism of lymphocytes, monoclonality, and epidemiologic characteristics (eg, age, sex, race) cannot reliably distinguish the 2 disease forms. Furthermore, it was suggested that these conditions are not mutually exclusive entities and are actually variants of CTCL. The observation that the 2 diseases share prognostic overlap adds further credence to the already puzzling conundrum. Nineteen of 28 patients with MF were alive and well at follow-up, and all patients in the idiopathic FM group were alive, with only 9 of 16 patients showing residual disease and none with CTCL.1
Other clinical factors that may be helpful in the diagnosis of MF are the presentation of lesions in non–sun-exposed areas of the skin and multiple lesions, as unilesional MF is exceedingly uncommon.21 No histologic features have been proven to predict which early patch- or plaque-stage MFs will progress to full-blown CTCL versus benign idiopathic FM; thus, great caution should be taken in patients with early-stage disease to ensure they are not prematurely and/or incorrectly classified as CTCL. Such a diagnosis has medical, social, and economical ramifications that should not be overlooked.
If idiopathic FM and LAFM were considered distinct disease processes, the ambiguity in making a definitive diagnosis should give the physician pause, and a diagnosis of LAFM may only be appropriate when there is unequivocal clinicopathologic evidence. Otherwise, a lymphoma diagnosis is somewhat superfluous and potentially harmful. Definitive diagnosis of LAFM also is complicated by reports of other hematologic malignancies presenting with FM-like histopathologic findings, such as chronic myelogenous leukemia, leukemia-associated eosinophilic folliculitis, and acute myeloblastic leukemia.23,24 Although MF is the most common malignancy associated with FM, it is important to consider other less common malignancies that also may be present.
Diagnosis: Patient Consequences
Accurate diagnosis of idiopathic FM versus LAFM is critical, as the ramifications of a cancer diagnosis can have broad implications. For example, patients who receive cancer diagnoses often experience emotional trauma and social stigma, even when adequate patient education has been provided. The incidence of depression and anxiety also can increase following a cancer diagnosis and can be complicated by medical treatments (eg, systemic steroids, interferon),25 which are known to increase the frequency of these psychological disturbances. Health insurance premiums likely will be higher if a patient is diagnosed with cancer versus a benign inflammatory condition. Hesitation of the pathologist to assign a cancer diagnosis when unequivocal evidence is not present should not be regarded as trickery, malpractice, or deceit of the health care bylaws, as benign language with suggestion of close clinical follow-up in the setting of diagnostic uncertainty will “first, do no harm” and secondly, serve as a vehicle for patient advocacy.
If there is a definitive distinction between idiopathic FM and LAFM, it requires further research before it can be fully understood. Currently, the WHO does not recognize a diagnosis of FM-associated MF (or LAFM) and acknowledges that folliculotropic MF is not always associated with FM.16,26 Given uncertainty and repercussions associated with a cancer diagnosis, however indolent, it may be morally responsible and medically favorable for physicians to consider FM in the differential diagnosis when applicable rather than making a diagnosis of MF outright. Given the importance of both clinical and histologic factors, it may be beneficial for definitive diagnosis of FM versus MF to lie with the clinician, while the pathologist serves as an adjunct in the diagnostic process. Because the prognosis of idiopathic FM often is marred by possible transformation into MF or other CTCLs, therapeutic decisions should be dictated by close clinical follow-up. Additionally, stage of disease, patient age, treatment compliance, comorbidities, and possible side effects of medications should all be considered when evaluating potential therapeutic regimens.27
Conclusion
Research is underway to more accurately identify patients with FM who are at risk for progression to LAFM versus those with benign remitting FM. Once the required diagnostic criteria are established to accurately classify these patients, with an emphasis on prognosis and suggested treatments, it might be necessary to establish new, less debated terminology so pathologists and clinicians alike can improve patient care. Continued histopathologic scrutiny, use of sophisticated molecular techniques, and knowledge of other currently undiscovered modalities will shed light on this important disease process and aid in proper disease management, which may be advantageous to both patients and physicians.
1. Cerroni L, Fink-Puches R, Bäck B, et al. Follicular mucinosis: a critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2002;138:182-189.
2. Parker SR, Murad E. Follicular mucinosis: clinical, histologic, and molecular remission with minocycline [published online ahead of print July 25, 2009]. J Am Acad Dermatol. 2010;62:139-141.
3. Pinkus H. Alopecia mucinosa; inflammatory plaques with alopecia characterized by root-sheath mucinosis. AMA Arch Dermatol. 1957;76:419-424, 424-426.
4. Pinkus H. Alopecia mucinosa. additional data in 1983. Arch Dermatol. 1983;119:698-699.
5. Jablonska S, Chorzelski T, Lancucki J. Mucinosis follicularis [in German]. Hautarzt. 1959;10:27-33.
6. Böer A, Guo Y, Ackerman AB. Alopecia mucinosa is mycosis fungoides. Am J Dermatopathol. 2004;26:33-52.
7. Brown HA, Gibson LE, Pujol RM, et al. Primary follicular mucinosis: long-term follow-up of patients younger than 40 years with and withoutclonal T-cell receptor gene rearrangement. J Am Acad Dermatol. 2002;47:856-862.
8. Schiller PI, Flaig MJ, Puchta U, et al. Detection of clonal T cells in lichen planus. Arch Dermatol Res. 2000;292:568-569.
9. Cerroni L, Kerl H. Primary follicular mucinosis and association with mycosis fungoides and other cutaneous T-cell lymphomas. J Am Acad Dermatol. 2004;51:146-147.
10. Dereure O, Levi E, Kadin ME. T-Cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
11. Haeffner AC, Smoller BR, Zepter K, et al. Differentiation and clonality of lesional lymphocytes in small plaque parapsoriasis. Arch Dermatol. 1995;131:321-324.
12. Schultz JC, Granados S, Vonderheid EC, et al. T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol. 2005;53:152-155.
13. Pfaltz K, Kerl K, Palmedo G, et al. Clonality in sarcoidosis, granuloma annulare, and granulomatous mycosis fungoides. Am J Dermatopathol. 2011;33:659-662.
14. Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
15. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
16. Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
17. Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides [published online ahead of print October 22, 2007]. Crit Rev Oncol Hematol. 2008;65:172-182.
18. Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
19. Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957.
20. Sarveswari KN, Yesudian P. The conundrum of parapsoriasis versus patch stage of mycosis fungoides. Indian J Dermatol Venereol Leprol. 2009;75:229-235.
21. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
22. Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides. a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
23. Rashid R, Hymes S. Folliculitis, follicular mucinosis, and papular mucinosis as a presentation of chronic myelomonocytic leukemia. Dermatol Online J. 2009;15:16.
24. Wada T, Yoshinaga E, Oiso N, et al. Adult T-cell leukemia-lymphoma associated with follicular mucinosis. J Dermatol. 2009;36:638-642.
25. Sampogna F, Frontani M, Baliva G, et al. Quality of life and psychological distress in patients with cutaneous lymphoma [published online ahead of print December 16, 2008]. Br J Dermatol. 2009;160:815-822.
26. Boone SL, Guitart J, Gerami P. Follicular mycosis fungoides: a histopathologic, immunohistochemical, and genotypic review. G Ital Dermatol Venereol. 2008;143:409-414.
27. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sézary syndrome [published online ahead of print August 20, 2009]. Blood. 2009;114:4337-4353.
When follicular mucinosis (FM) is defined as an epithelial reaction pattern characterized by intrafollicular and perifollicular mucin accumulation, it cannot be considered a distinct disease entity, as this pattern is ubiquitously present in various inflammatory and neoplastic skin conditions.1,2 The distinction between idiopathic FM and lymphoma-associated follicular mucinosis (LAFM) was made several years ago by authors who evaluated the differences in the clinical presentation of these entities, including patient age at onset, number of lesions, pattern of distribution, and most importantly clinical progression.1 In this article, we discuss the importance of close clinical follow-up in patients with FM or patch-stage mycosis fungoides (MF) in whom histopathologic evaluation is ambiguous or nondiagnostic. We also highlight the value of ancillary testing, including T-cell receptor gene rearrangement, flow cytometry, and immunohistochemistry, as a component in the diagnostic process rather than the sole diagnostic moiety. A review of the pertinent literature also is performed.
History of FM and MF
Pinkus3 first described an entity he termed alopecia mucinosa in 1957. Pinkus noted 3 distinct patterns: an idiopathic form of alopecia mucinosa, lymphoblastoma with associated FM, and alopecia mucinosa that later transformed into lymphoblastoma.4 In 1983, however, Pinkus4 described uncertainty if alopecia mucinosa represented the first stage of MF or if patients with alopecia mucinosa were simply at an increased risk for developing lymphoma. He believed there were too many cases of lymphoma following a diagnosis of alopecia mucinosa for the relation to be coincidental, yet he noted that many of the cases resolved either spontaneously or following treatment with x-rays or topical steroids. He concluded his report with a sentiment that is echoed in many current studies regarding this entity: “Many questions surrounding this entity are as unanswerable today as they were 25 years ago.”4
Jablonska et al5 were the first to coin the term mucinosis follicularis, now known as FM, to replace alopecia mucinosa because they felt the description was more accurate, as lesions also arise on non–hair-bearing skin. Although there is general agreement that there is a form of MF that has associated FM, this is where the agreement ends with regard to the diagnosis of MF versus FM. Böer et al6 discussed the historic evolution of these terms, mostly to highlight the origins of the confusion. The investigators proposed that FM should only be used as a descriptive term and that all cases of alopecia mucinosa represent MF. They also concluded that many benign dermatoses associated with a risk for evolution to MF (eg, small and large plaque psoriasis [LPP]) should simply be diagnosed as MF.6 Subsequently, the proposal that idiopathic FM and LAFM are not 2 distinct entities but rather a clinicopathologic continuum and that idiopathic FM is simply a variant of MF along this spectrum has gained some approval.6,7 However, this belief is not shared among all authorities in the field, and attempts to define diagnostic criteria that distinguish between a benign clinical course and a course that is more progressive and fatal continue. Currently, it is agreed upon that when distinguishing between these 2 clinical courses, primary (idiopathic) follicular mucinosis refers to a benign course with no overt sign of malignancy, and lymphoma-associated follicular mucinosis refers to a diagnostic malignant condition. Lymphoma-associated follicular mucinosis refers to FM associated with cutaneous T-cell lymphoma, the most common form of which is FM. Many authors8-15 have sought ancillary methodologies in addition to clinical parameters to assist in the evaluation between both disease courses. Methodologies have included assessment of T-cell receptor gene rearrangements, flow cytometry, and immunohistochemical staining, mostly as an effort to establish monoclonality as a defining characteristic of LAFM; however, monoclonality in cutaneous T-cell infiltrates should be interpreted with caution and should not be considered as a confirmation of malignancy due to recent findings of monoclonality in benign inflammatory dermatoses such as lichen planus. The Table outlines several of the most common benign inflammatory dermatoses that demonstrate monoclonality, but this list should not be considered exhaustive, as there are many others in which monoclonality is sometimes seen.8-15 The lack of definitive criteria to distinguish between the 2 groups has led to confusion and consternation regarding the diagnosis of idiopathic FM versus LAFM and has led many in the field to consider the 2 conditions to be one and the same.
Diagnosis of FM and MF: Clinicopathologic Features
The World Health Organization (WHO) defined MF as an epidermotropic primary cutaneous T-cell lymphoma (CTCL) characterized by infiltrates of small- to medium-sized T lymphocytes with cerebriform nuclei. Further, the WHO stated that the term mycosis fungoides should be exclusively reserved for classical cases typified by the evolution of cutaneous patches, plaques, and tumors, or for variants that show a similar clinical course.16 Mycosis fungoides is divided into 3 stages—patch, plaque, and tumor—which are solely clinical descriptors.17 The WHO also described a clinical staging system with pathologic emphasis placed only on lymph node involvement and identification of Sézary cells.16 It lists folliculotropic MF as a variant, with only some cases presenting with mucinous degeneration of hair follicles. A lack of consensus among pathologists regarding criteria for diagnosis in patch-stage MF remains, but diagnosis of plaque-stage disease is not regularly debated due to its more reliably present, well-developed histologic features (eg, haloed lymphocytes, epidermotropism of lymphocytes, lymphocytes with convoluted nuceli, Pautrier microabscesses).18 Although there have been specific histologic findings reported to be associated with patch-stage MF, they have only been present in a few cases and are therefore of limited usefulness in practice.1,19 The categorization of patients with subtle histologic features common to both MF and inflammatory conditions such as parapsoriasis en plaques (the term plaque in this case is a misnomer because the word plaque means patch in French) continues to be elusive. A lack of agreement regarding LPP persists in the current literature in the same manner as FM. Some researchers have contended for many years that LPP is a type of MF, while others remain unconvinced, mainly due to the lack of evidence that lumping a benign condition (LPP) with an increased risk for malignant transformation and a known malignancy (MF) together is of any benefit to the patient. Assessment of clinicopathologic correlation, immunohistochemistry, clonality, and T-cell gene rearrangement have failed to positively identify patients who are at risk for disease progression, whether the diagnosis is called LPP or early patch-stage MF.20
Mycosis fungoides is more common in males and its incidence increases with age; however, diagnosis should not be ruled out based on age or gender. Typical presentation of early-stage disease includes erythematous patches or plaques, often with light scaling.19 Lesions routinely are of long-standing duration (months to years), are located in areas that are infrequently exposed to sunlight, and often are 5 cm in diameter or larger with irregular borders.21 Associated poikiloderma is relatively specific to MF but rarely is seen in other CTCLs, connective-tissue diseases, and some genodermatoses. Poikiloderma commonly is identified in LPP, which shows the same telangiectasia, mottled pigmentation, and epidermal atrophy as MF-associated poikiloderma, leading some to believe that there is no separation between the 2 conditions. In all stages of MF, lesions frequently are numerous and occur on multiple sites. Plaques and tumors can show spontaneous ulceration. When lesions are folliculotropic, they can cause localized alopecia, follicular-based papules, and fungating pseudotumors in more advanced stages.1 The clinical presentation of FM substantially overlaps with folliculotropic MF, and although FM lesions often are solitary and are located on the face or scalp, they also can present as multiple lesions located elsewhere on the body. It also has been proposed that folliculotropic MF should not be separated from FM-associated MF (or LAFM).22
The characteristic histologic picture of LAFM in patch or plaque stage shows mucin deposition within hair follicles, similar to idiopathic FM. On histology, both conditions demonstrate dense lymphoid infiltrates around and within hair follicles as well as in the dermis (Figure). Most cases of LAFM show epidermotropism of lymphocytes between follicles, but this finding is not present in every case and often disappears when the disease advances to the tumor stage.1,19 Although Pautrier microabscesses (collections of lymphocytes within the superficial epidermis) are considered to be somewhat specific to MF, they are only present in a minority of cases.20 In a study by the International Society for Cutaneous Lymphomas,21 the only histopathologic criteria that showed any appreciable sensitivity or specificity in the diagnosis of MF were the presence of lymphoid cells with variable nuclear and cytoplasmic features and/or strikingly irregular nuclear contours with the presence of lymphocytes larger than those usually seen in inflammatory dermatoses. Despite these criteria, the study reported a high misclassification rate. A complicated scoring system for diagnosis of MF in patch- or early plaque-stage disease was proposed by the International Society for Cutaneous Lymphomas,21 which integrates clinical, histopathologic, molecular, and immunophenotypic criteria. However, these criteria have been continually debated in the literature and are only discussed in this article in relation to the association between MF and FM. Diagnosis of tumor-stage MF is not addressed in this article, as it is readily identified as lymphoma and is not easily confused with idiopathic FM.
|
Clinical assessment of a patient’s medical history to identify persistent and progressive disease is paramount to the diagnosis of MF. Although MF lesions tend to increase in size and number over time, this presentation is not without exception.21 In early patch-stage disease, eliminating some of the patient’s current medications may be sufficient in clearing cutaneous patches that cannot be conclusively identified as either MF or a benign inflammatory lymphoid infiltrate, which further emphasizes the importance of clinical assessment of the patient’s medical history in the diagnosis of MF. The shape of the lesions also is helpful in distinguishing between MF and other skin disorders, such as digitate dermatosis or LPP; unlike the latter, the waxing and waning nature of MF lesions often produces irregularly shaped patches with little coalescence. Again, there are some investigators who believe that these lesions represent varying presentations of MF.6
In a study by Cerroni et al,1 44 patients with FM were divided into 2 groups: (1) a cohort of 16 patients with no history or clinical evidence of MF or Sézary syndrome (ie, LAFM), and (2) a cohort of 28 patients with clinicopathologic evidence of CTCL. Patients in both groups were followed for a maximum of 20 years. Results indicated that that the presence of perifollicular or intrafollicular mucin, epidermotropism of lymphocytes, monoclonality, and epidemiologic characteristics (eg, age, sex, race) cannot reliably distinguish the 2 disease forms. Furthermore, it was suggested that these conditions are not mutually exclusive entities and are actually variants of CTCL. The observation that the 2 diseases share prognostic overlap adds further credence to the already puzzling conundrum. Nineteen of 28 patients with MF were alive and well at follow-up, and all patients in the idiopathic FM group were alive, with only 9 of 16 patients showing residual disease and none with CTCL.1
Other clinical factors that may be helpful in the diagnosis of MF are the presentation of lesions in non–sun-exposed areas of the skin and multiple lesions, as unilesional MF is exceedingly uncommon.21 No histologic features have been proven to predict which early patch- or plaque-stage MFs will progress to full-blown CTCL versus benign idiopathic FM; thus, great caution should be taken in patients with early-stage disease to ensure they are not prematurely and/or incorrectly classified as CTCL. Such a diagnosis has medical, social, and economical ramifications that should not be overlooked.
If idiopathic FM and LAFM were considered distinct disease processes, the ambiguity in making a definitive diagnosis should give the physician pause, and a diagnosis of LAFM may only be appropriate when there is unequivocal clinicopathologic evidence. Otherwise, a lymphoma diagnosis is somewhat superfluous and potentially harmful. Definitive diagnosis of LAFM also is complicated by reports of other hematologic malignancies presenting with FM-like histopathologic findings, such as chronic myelogenous leukemia, leukemia-associated eosinophilic folliculitis, and acute myeloblastic leukemia.23,24 Although MF is the most common malignancy associated with FM, it is important to consider other less common malignancies that also may be present.
Diagnosis: Patient Consequences
Accurate diagnosis of idiopathic FM versus LAFM is critical, as the ramifications of a cancer diagnosis can have broad implications. For example, patients who receive cancer diagnoses often experience emotional trauma and social stigma, even when adequate patient education has been provided. The incidence of depression and anxiety also can increase following a cancer diagnosis and can be complicated by medical treatments (eg, systemic steroids, interferon),25 which are known to increase the frequency of these psychological disturbances. Health insurance premiums likely will be higher if a patient is diagnosed with cancer versus a benign inflammatory condition. Hesitation of the pathologist to assign a cancer diagnosis when unequivocal evidence is not present should not be regarded as trickery, malpractice, or deceit of the health care bylaws, as benign language with suggestion of close clinical follow-up in the setting of diagnostic uncertainty will “first, do no harm” and secondly, serve as a vehicle for patient advocacy.
If there is a definitive distinction between idiopathic FM and LAFM, it requires further research before it can be fully understood. Currently, the WHO does not recognize a diagnosis of FM-associated MF (or LAFM) and acknowledges that folliculotropic MF is not always associated with FM.16,26 Given uncertainty and repercussions associated with a cancer diagnosis, however indolent, it may be morally responsible and medically favorable for physicians to consider FM in the differential diagnosis when applicable rather than making a diagnosis of MF outright. Given the importance of both clinical and histologic factors, it may be beneficial for definitive diagnosis of FM versus MF to lie with the clinician, while the pathologist serves as an adjunct in the diagnostic process. Because the prognosis of idiopathic FM often is marred by possible transformation into MF or other CTCLs, therapeutic decisions should be dictated by close clinical follow-up. Additionally, stage of disease, patient age, treatment compliance, comorbidities, and possible side effects of medications should all be considered when evaluating potential therapeutic regimens.27
Conclusion
Research is underway to more accurately identify patients with FM who are at risk for progression to LAFM versus those with benign remitting FM. Once the required diagnostic criteria are established to accurately classify these patients, with an emphasis on prognosis and suggested treatments, it might be necessary to establish new, less debated terminology so pathologists and clinicians alike can improve patient care. Continued histopathologic scrutiny, use of sophisticated molecular techniques, and knowledge of other currently undiscovered modalities will shed light on this important disease process and aid in proper disease management, which may be advantageous to both patients and physicians.
When follicular mucinosis (FM) is defined as an epithelial reaction pattern characterized by intrafollicular and perifollicular mucin accumulation, it cannot be considered a distinct disease entity, as this pattern is ubiquitously present in various inflammatory and neoplastic skin conditions.1,2 The distinction between idiopathic FM and lymphoma-associated follicular mucinosis (LAFM) was made several years ago by authors who evaluated the differences in the clinical presentation of these entities, including patient age at onset, number of lesions, pattern of distribution, and most importantly clinical progression.1 In this article, we discuss the importance of close clinical follow-up in patients with FM or patch-stage mycosis fungoides (MF) in whom histopathologic evaluation is ambiguous or nondiagnostic. We also highlight the value of ancillary testing, including T-cell receptor gene rearrangement, flow cytometry, and immunohistochemistry, as a component in the diagnostic process rather than the sole diagnostic moiety. A review of the pertinent literature also is performed.
History of FM and MF
Pinkus3 first described an entity he termed alopecia mucinosa in 1957. Pinkus noted 3 distinct patterns: an idiopathic form of alopecia mucinosa, lymphoblastoma with associated FM, and alopecia mucinosa that later transformed into lymphoblastoma.4 In 1983, however, Pinkus4 described uncertainty if alopecia mucinosa represented the first stage of MF or if patients with alopecia mucinosa were simply at an increased risk for developing lymphoma. He believed there were too many cases of lymphoma following a diagnosis of alopecia mucinosa for the relation to be coincidental, yet he noted that many of the cases resolved either spontaneously or following treatment with x-rays or topical steroids. He concluded his report with a sentiment that is echoed in many current studies regarding this entity: “Many questions surrounding this entity are as unanswerable today as they were 25 years ago.”4
Jablonska et al5 were the first to coin the term mucinosis follicularis, now known as FM, to replace alopecia mucinosa because they felt the description was more accurate, as lesions also arise on non–hair-bearing skin. Although there is general agreement that there is a form of MF that has associated FM, this is where the agreement ends with regard to the diagnosis of MF versus FM. Böer et al6 discussed the historic evolution of these terms, mostly to highlight the origins of the confusion. The investigators proposed that FM should only be used as a descriptive term and that all cases of alopecia mucinosa represent MF. They also concluded that many benign dermatoses associated with a risk for evolution to MF (eg, small and large plaque psoriasis [LPP]) should simply be diagnosed as MF.6 Subsequently, the proposal that idiopathic FM and LAFM are not 2 distinct entities but rather a clinicopathologic continuum and that idiopathic FM is simply a variant of MF along this spectrum has gained some approval.6,7 However, this belief is not shared among all authorities in the field, and attempts to define diagnostic criteria that distinguish between a benign clinical course and a course that is more progressive and fatal continue. Currently, it is agreed upon that when distinguishing between these 2 clinical courses, primary (idiopathic) follicular mucinosis refers to a benign course with no overt sign of malignancy, and lymphoma-associated follicular mucinosis refers to a diagnostic malignant condition. Lymphoma-associated follicular mucinosis refers to FM associated with cutaneous T-cell lymphoma, the most common form of which is FM. Many authors8-15 have sought ancillary methodologies in addition to clinical parameters to assist in the evaluation between both disease courses. Methodologies have included assessment of T-cell receptor gene rearrangements, flow cytometry, and immunohistochemical staining, mostly as an effort to establish monoclonality as a defining characteristic of LAFM; however, monoclonality in cutaneous T-cell infiltrates should be interpreted with caution and should not be considered as a confirmation of malignancy due to recent findings of monoclonality in benign inflammatory dermatoses such as lichen planus. The Table outlines several of the most common benign inflammatory dermatoses that demonstrate monoclonality, but this list should not be considered exhaustive, as there are many others in which monoclonality is sometimes seen.8-15 The lack of definitive criteria to distinguish between the 2 groups has led to confusion and consternation regarding the diagnosis of idiopathic FM versus LAFM and has led many in the field to consider the 2 conditions to be one and the same.
Diagnosis of FM and MF: Clinicopathologic Features
The World Health Organization (WHO) defined MF as an epidermotropic primary cutaneous T-cell lymphoma (CTCL) characterized by infiltrates of small- to medium-sized T lymphocytes with cerebriform nuclei. Further, the WHO stated that the term mycosis fungoides should be exclusively reserved for classical cases typified by the evolution of cutaneous patches, plaques, and tumors, or for variants that show a similar clinical course.16 Mycosis fungoides is divided into 3 stages—patch, plaque, and tumor—which are solely clinical descriptors.17 The WHO also described a clinical staging system with pathologic emphasis placed only on lymph node involvement and identification of Sézary cells.16 It lists folliculotropic MF as a variant, with only some cases presenting with mucinous degeneration of hair follicles. A lack of consensus among pathologists regarding criteria for diagnosis in patch-stage MF remains, but diagnosis of plaque-stage disease is not regularly debated due to its more reliably present, well-developed histologic features (eg, haloed lymphocytes, epidermotropism of lymphocytes, lymphocytes with convoluted nuceli, Pautrier microabscesses).18 Although there have been specific histologic findings reported to be associated with patch-stage MF, they have only been present in a few cases and are therefore of limited usefulness in practice.1,19 The categorization of patients with subtle histologic features common to both MF and inflammatory conditions such as parapsoriasis en plaques (the term plaque in this case is a misnomer because the word plaque means patch in French) continues to be elusive. A lack of agreement regarding LPP persists in the current literature in the same manner as FM. Some researchers have contended for many years that LPP is a type of MF, while others remain unconvinced, mainly due to the lack of evidence that lumping a benign condition (LPP) with an increased risk for malignant transformation and a known malignancy (MF) together is of any benefit to the patient. Assessment of clinicopathologic correlation, immunohistochemistry, clonality, and T-cell gene rearrangement have failed to positively identify patients who are at risk for disease progression, whether the diagnosis is called LPP or early patch-stage MF.20
Mycosis fungoides is more common in males and its incidence increases with age; however, diagnosis should not be ruled out based on age or gender. Typical presentation of early-stage disease includes erythematous patches or plaques, often with light scaling.19 Lesions routinely are of long-standing duration (months to years), are located in areas that are infrequently exposed to sunlight, and often are 5 cm in diameter or larger with irregular borders.21 Associated poikiloderma is relatively specific to MF but rarely is seen in other CTCLs, connective-tissue diseases, and some genodermatoses. Poikiloderma commonly is identified in LPP, which shows the same telangiectasia, mottled pigmentation, and epidermal atrophy as MF-associated poikiloderma, leading some to believe that there is no separation between the 2 conditions. In all stages of MF, lesions frequently are numerous and occur on multiple sites. Plaques and tumors can show spontaneous ulceration. When lesions are folliculotropic, they can cause localized alopecia, follicular-based papules, and fungating pseudotumors in more advanced stages.1 The clinical presentation of FM substantially overlaps with folliculotropic MF, and although FM lesions often are solitary and are located on the face or scalp, they also can present as multiple lesions located elsewhere on the body. It also has been proposed that folliculotropic MF should not be separated from FM-associated MF (or LAFM).22
The characteristic histologic picture of LAFM in patch or plaque stage shows mucin deposition within hair follicles, similar to idiopathic FM. On histology, both conditions demonstrate dense lymphoid infiltrates around and within hair follicles as well as in the dermis (Figure). Most cases of LAFM show epidermotropism of lymphocytes between follicles, but this finding is not present in every case and often disappears when the disease advances to the tumor stage.1,19 Although Pautrier microabscesses (collections of lymphocytes within the superficial epidermis) are considered to be somewhat specific to MF, they are only present in a minority of cases.20 In a study by the International Society for Cutaneous Lymphomas,21 the only histopathologic criteria that showed any appreciable sensitivity or specificity in the diagnosis of MF were the presence of lymphoid cells with variable nuclear and cytoplasmic features and/or strikingly irregular nuclear contours with the presence of lymphocytes larger than those usually seen in inflammatory dermatoses. Despite these criteria, the study reported a high misclassification rate. A complicated scoring system for diagnosis of MF in patch- or early plaque-stage disease was proposed by the International Society for Cutaneous Lymphomas,21 which integrates clinical, histopathologic, molecular, and immunophenotypic criteria. However, these criteria have been continually debated in the literature and are only discussed in this article in relation to the association between MF and FM. Diagnosis of tumor-stage MF is not addressed in this article, as it is readily identified as lymphoma and is not easily confused with idiopathic FM.
|
Clinical assessment of a patient’s medical history to identify persistent and progressive disease is paramount to the diagnosis of MF. Although MF lesions tend to increase in size and number over time, this presentation is not without exception.21 In early patch-stage disease, eliminating some of the patient’s current medications may be sufficient in clearing cutaneous patches that cannot be conclusively identified as either MF or a benign inflammatory lymphoid infiltrate, which further emphasizes the importance of clinical assessment of the patient’s medical history in the diagnosis of MF. The shape of the lesions also is helpful in distinguishing between MF and other skin disorders, such as digitate dermatosis or LPP; unlike the latter, the waxing and waning nature of MF lesions often produces irregularly shaped patches with little coalescence. Again, there are some investigators who believe that these lesions represent varying presentations of MF.6
In a study by Cerroni et al,1 44 patients with FM were divided into 2 groups: (1) a cohort of 16 patients with no history or clinical evidence of MF or Sézary syndrome (ie, LAFM), and (2) a cohort of 28 patients with clinicopathologic evidence of CTCL. Patients in both groups were followed for a maximum of 20 years. Results indicated that that the presence of perifollicular or intrafollicular mucin, epidermotropism of lymphocytes, monoclonality, and epidemiologic characteristics (eg, age, sex, race) cannot reliably distinguish the 2 disease forms. Furthermore, it was suggested that these conditions are not mutually exclusive entities and are actually variants of CTCL. The observation that the 2 diseases share prognostic overlap adds further credence to the already puzzling conundrum. Nineteen of 28 patients with MF were alive and well at follow-up, and all patients in the idiopathic FM group were alive, with only 9 of 16 patients showing residual disease and none with CTCL.1
Other clinical factors that may be helpful in the diagnosis of MF are the presentation of lesions in non–sun-exposed areas of the skin and multiple lesions, as unilesional MF is exceedingly uncommon.21 No histologic features have been proven to predict which early patch- or plaque-stage MFs will progress to full-blown CTCL versus benign idiopathic FM; thus, great caution should be taken in patients with early-stage disease to ensure they are not prematurely and/or incorrectly classified as CTCL. Such a diagnosis has medical, social, and economical ramifications that should not be overlooked.
If idiopathic FM and LAFM were considered distinct disease processes, the ambiguity in making a definitive diagnosis should give the physician pause, and a diagnosis of LAFM may only be appropriate when there is unequivocal clinicopathologic evidence. Otherwise, a lymphoma diagnosis is somewhat superfluous and potentially harmful. Definitive diagnosis of LAFM also is complicated by reports of other hematologic malignancies presenting with FM-like histopathologic findings, such as chronic myelogenous leukemia, leukemia-associated eosinophilic folliculitis, and acute myeloblastic leukemia.23,24 Although MF is the most common malignancy associated with FM, it is important to consider other less common malignancies that also may be present.
Diagnosis: Patient Consequences
Accurate diagnosis of idiopathic FM versus LAFM is critical, as the ramifications of a cancer diagnosis can have broad implications. For example, patients who receive cancer diagnoses often experience emotional trauma and social stigma, even when adequate patient education has been provided. The incidence of depression and anxiety also can increase following a cancer diagnosis and can be complicated by medical treatments (eg, systemic steroids, interferon),25 which are known to increase the frequency of these psychological disturbances. Health insurance premiums likely will be higher if a patient is diagnosed with cancer versus a benign inflammatory condition. Hesitation of the pathologist to assign a cancer diagnosis when unequivocal evidence is not present should not be regarded as trickery, malpractice, or deceit of the health care bylaws, as benign language with suggestion of close clinical follow-up in the setting of diagnostic uncertainty will “first, do no harm” and secondly, serve as a vehicle for patient advocacy.
If there is a definitive distinction between idiopathic FM and LAFM, it requires further research before it can be fully understood. Currently, the WHO does not recognize a diagnosis of FM-associated MF (or LAFM) and acknowledges that folliculotropic MF is not always associated with FM.16,26 Given uncertainty and repercussions associated with a cancer diagnosis, however indolent, it may be morally responsible and medically favorable for physicians to consider FM in the differential diagnosis when applicable rather than making a diagnosis of MF outright. Given the importance of both clinical and histologic factors, it may be beneficial for definitive diagnosis of FM versus MF to lie with the clinician, while the pathologist serves as an adjunct in the diagnostic process. Because the prognosis of idiopathic FM often is marred by possible transformation into MF or other CTCLs, therapeutic decisions should be dictated by close clinical follow-up. Additionally, stage of disease, patient age, treatment compliance, comorbidities, and possible side effects of medications should all be considered when evaluating potential therapeutic regimens.27
Conclusion
Research is underway to more accurately identify patients with FM who are at risk for progression to LAFM versus those with benign remitting FM. Once the required diagnostic criteria are established to accurately classify these patients, with an emphasis on prognosis and suggested treatments, it might be necessary to establish new, less debated terminology so pathologists and clinicians alike can improve patient care. Continued histopathologic scrutiny, use of sophisticated molecular techniques, and knowledge of other currently undiscovered modalities will shed light on this important disease process and aid in proper disease management, which may be advantageous to both patients and physicians.
1. Cerroni L, Fink-Puches R, Bäck B, et al. Follicular mucinosis: a critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2002;138:182-189.
2. Parker SR, Murad E. Follicular mucinosis: clinical, histologic, and molecular remission with minocycline [published online ahead of print July 25, 2009]. J Am Acad Dermatol. 2010;62:139-141.
3. Pinkus H. Alopecia mucinosa; inflammatory plaques with alopecia characterized by root-sheath mucinosis. AMA Arch Dermatol. 1957;76:419-424, 424-426.
4. Pinkus H. Alopecia mucinosa. additional data in 1983. Arch Dermatol. 1983;119:698-699.
5. Jablonska S, Chorzelski T, Lancucki J. Mucinosis follicularis [in German]. Hautarzt. 1959;10:27-33.
6. Böer A, Guo Y, Ackerman AB. Alopecia mucinosa is mycosis fungoides. Am J Dermatopathol. 2004;26:33-52.
7. Brown HA, Gibson LE, Pujol RM, et al. Primary follicular mucinosis: long-term follow-up of patients younger than 40 years with and withoutclonal T-cell receptor gene rearrangement. J Am Acad Dermatol. 2002;47:856-862.
8. Schiller PI, Flaig MJ, Puchta U, et al. Detection of clonal T cells in lichen planus. Arch Dermatol Res. 2000;292:568-569.
9. Cerroni L, Kerl H. Primary follicular mucinosis and association with mycosis fungoides and other cutaneous T-cell lymphomas. J Am Acad Dermatol. 2004;51:146-147.
10. Dereure O, Levi E, Kadin ME. T-Cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
11. Haeffner AC, Smoller BR, Zepter K, et al. Differentiation and clonality of lesional lymphocytes in small plaque parapsoriasis. Arch Dermatol. 1995;131:321-324.
12. Schultz JC, Granados S, Vonderheid EC, et al. T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol. 2005;53:152-155.
13. Pfaltz K, Kerl K, Palmedo G, et al. Clonality in sarcoidosis, granuloma annulare, and granulomatous mycosis fungoides. Am J Dermatopathol. 2011;33:659-662.
14. Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
15. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
16. Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
17. Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides [published online ahead of print October 22, 2007]. Crit Rev Oncol Hematol. 2008;65:172-182.
18. Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
19. Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957.
20. Sarveswari KN, Yesudian P. The conundrum of parapsoriasis versus patch stage of mycosis fungoides. Indian J Dermatol Venereol Leprol. 2009;75:229-235.
21. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
22. Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides. a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
23. Rashid R, Hymes S. Folliculitis, follicular mucinosis, and papular mucinosis as a presentation of chronic myelomonocytic leukemia. Dermatol Online J. 2009;15:16.
24. Wada T, Yoshinaga E, Oiso N, et al. Adult T-cell leukemia-lymphoma associated with follicular mucinosis. J Dermatol. 2009;36:638-642.
25. Sampogna F, Frontani M, Baliva G, et al. Quality of life and psychological distress in patients with cutaneous lymphoma [published online ahead of print December 16, 2008]. Br J Dermatol. 2009;160:815-822.
26. Boone SL, Guitart J, Gerami P. Follicular mycosis fungoides: a histopathologic, immunohistochemical, and genotypic review. G Ital Dermatol Venereol. 2008;143:409-414.
27. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sézary syndrome [published online ahead of print August 20, 2009]. Blood. 2009;114:4337-4353.
1. Cerroni L, Fink-Puches R, Bäck B, et al. Follicular mucinosis: a critical reappraisal of clinicopathologic features and association with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2002;138:182-189.
2. Parker SR, Murad E. Follicular mucinosis: clinical, histologic, and molecular remission with minocycline [published online ahead of print July 25, 2009]. J Am Acad Dermatol. 2010;62:139-141.
3. Pinkus H. Alopecia mucinosa; inflammatory plaques with alopecia characterized by root-sheath mucinosis. AMA Arch Dermatol. 1957;76:419-424, 424-426.
4. Pinkus H. Alopecia mucinosa. additional data in 1983. Arch Dermatol. 1983;119:698-699.
5. Jablonska S, Chorzelski T, Lancucki J. Mucinosis follicularis [in German]. Hautarzt. 1959;10:27-33.
6. Böer A, Guo Y, Ackerman AB. Alopecia mucinosa is mycosis fungoides. Am J Dermatopathol. 2004;26:33-52.
7. Brown HA, Gibson LE, Pujol RM, et al. Primary follicular mucinosis: long-term follow-up of patients younger than 40 years with and withoutclonal T-cell receptor gene rearrangement. J Am Acad Dermatol. 2002;47:856-862.
8. Schiller PI, Flaig MJ, Puchta U, et al. Detection of clonal T cells in lichen planus. Arch Dermatol Res. 2000;292:568-569.
9. Cerroni L, Kerl H. Primary follicular mucinosis and association with mycosis fungoides and other cutaneous T-cell lymphomas. J Am Acad Dermatol. 2004;51:146-147.
10. Dereure O, Levi E, Kadin ME. T-Cell clonality in pityriasis lichenoides et varioliformis acuta: a heteroduplex analysis of 20 cases. Arch Dermatol. 2000;136:1483-1486.
11. Haeffner AC, Smoller BR, Zepter K, et al. Differentiation and clonality of lesional lymphocytes in small plaque parapsoriasis. Arch Dermatol. 1995;131:321-324.
12. Schultz JC, Granados S, Vonderheid EC, et al. T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol. 2005;53:152-155.
13. Pfaltz K, Kerl K, Palmedo G, et al. Clonality in sarcoidosis, granuloma annulare, and granulomatous mycosis fungoides. Am J Dermatopathol. 2011;33:659-662.
14. Weinberg JM, Kristal L, Chooback L, et al. The clonal nature of pityriasis lichenoides. Arch Dermatol. 2002;138:1063-1067.
15. Guitart J, Magro C. Cutaneous T-cell lymphoid dyscrasia: a unifying term for idiopathic chronic dermatoses with persistent T-cell clones. Arch Dermatol. 2007;143:921-932.
16. Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
17. Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides [published online ahead of print October 22, 2007]. Crit Rev Oncol Hematol. 2008;65:172-182.
18. Smoller BR, Bishop K, Glusac E, et al. Reassessment of histologic parameters in the diagnosis of mycosis fungoides. Am J Surg Pathol. 1995;19:1423-1430.
19. Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957.
20. Sarveswari KN, Yesudian P. The conundrum of parapsoriasis versus patch stage of mycosis fungoides. Indian J Dermatol Venereol Leprol. 2009;75:229-235.
21. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
22. Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides. a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525-530.
23. Rashid R, Hymes S. Folliculitis, follicular mucinosis, and papular mucinosis as a presentation of chronic myelomonocytic leukemia. Dermatol Online J. 2009;15:16.
24. Wada T, Yoshinaga E, Oiso N, et al. Adult T-cell leukemia-lymphoma associated with follicular mucinosis. J Dermatol. 2009;36:638-642.
25. Sampogna F, Frontani M, Baliva G, et al. Quality of life and psychological distress in patients with cutaneous lymphoma [published online ahead of print December 16, 2008]. Br J Dermatol. 2009;160:815-822.
26. Boone SL, Guitart J, Gerami P. Follicular mycosis fungoides: a histopathologic, immunohistochemical, and genotypic review. G Ital Dermatol Venereol. 2008;143:409-414.
27. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sézary syndrome [published online ahead of print August 20, 2009]. Blood. 2009;114:4337-4353.
Practice Points
- An isolated patch in the head or neck area is much more likely to be follicular mucinosis (FM) than mycosis fungoides (MF).
- Monoclonality does not reliably distinguish FM from MF.
- Younger patients are more likely to have FM with spontaneous remission, and older patients are more likely to develop MF.
- None of the clinicopathologic features of FM or MF are without overlap.
Primary Apocrine Adenocarcinoma of the Axilla
Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy, with most of the available information about this disease consolidated from anecdotal evidence of single case reports and small case series with fewer than 30 patients.1-11 Although certain histologic and immunohistochemical features have been suggested to be useful in the diagnosis of AA, there is no clear consensus on the required pathologic criteria.1,5,6,9,10,12,13 Additionally, the clinical presentation of AA is highly variable, which further adds to the challenge of making an accurate diagnosis.1-3,5,9,10,13
Apocrine adenocarcinoma usually arises in areas of high apocrine gland density such as the axillae or anogenital region.2,4,6 It also has been reported in areas such as the scalp, ear canal, eyelids, chest, nipples, arms, wrists, and fingers.4,8,10,14-16 Apocrine adenocarcinoma in unusual locations such as the eyelid and ear canal is thought to arise from modified apocrine glands such as the Moll glands of the eyelid and the ceruminous glands of the ear canal.9,10 The presence of ectopic apocrine glands may lead to AA in atypical sites such as the wrists and fingers.5,16 The areola is an apocrine-dense area; therefore, AA may present on the nipples or within supernumerary nipples anywhere along the milk lines.4
Apocrine adenocarcinoma clinically presents as an asymptomatic to slightly painful, slowly growing, and erythematous to violaceous nodule or tumor.4,6,9 However, in a minority of cases the initial presentation consists of a cystic or ulcerated mass with overlying granulation tissue and purulent discharge.6,9,11 A wide time frame from the onset of symptoms to diagnosis has been reported, ranging from weeks to decades.4,6-8 The conventional treatment of AA is wide local excision.2,4,6,9 Although AA often presents with local lymph node metastasis at the time of diagnosis, there is no consensus on the use of sentinel lymph node biopsy (SLNB), nodal dissection, or adjuvant chemoradiation therapy.1,3,8,9
We report the case of a 49-year-old man with primary AA of the left axilla; the clinical and histologic features of AA as well as the appropriate diagnostic and treatment modalities also are provided.
Case Report
A 49-year-old man with a slowly growing tender mass of the left axilla of 1 year’s duration was referred to our dermatology clinic for evaluation. A review of systems revealed loss of appetite, fatigue, and a 4-month history of unintentional weight loss (15–20 lb). The patient had a history of hepatitis C virus, intravenous drug use, alcohol abuse, and cigarette smoking (1 pack daily) for many years. Additionally, the patient reported a paternal family history of numerous visceral malignancies. Examination of the left axilla revealed a 1.5×5-cm ulcerated tumor that produced serosanguineous discharge and was tender to palpation (Figure 1). Two 1-cm, firm, freely mobile subcutaneous nodules with no overlying skin changes were palpable at the medial border of the ulcerated nodule. There was no additional cervical or axillary lymphadenopathy, and a breast examination was normal.
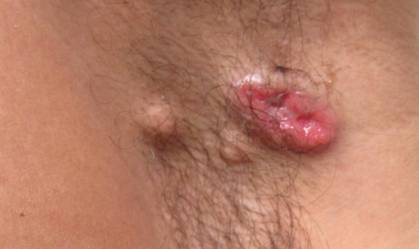
The differential diagnosis included primary squamous cell carcinoma or adnexal neoplasm, primary breast carcinoma, lymphoma, scrofuloderma, atypical mycobacterial infection, and cutaneous metastasis from an internal malignancy. Two 4-mm punch biopsies were performed and sent for routine histopathology and bacterial, fungal, and mycobacterial tissue cultures. To exclude a primary visceral malignancy or metastasis, computed tomography of the chest, abdomen, and pelvis; positron emission tomography (PET) from the base of the skull to the thighs; colonoscopy; magnetic resonance imaging of the brain; esophagogastroduodenoscopy; and mammography were conducted. Prominent left axillary lymphadenopathy was noted on computed tomography. Additionally, PET identified extranodal spread in the left axilla, left lateral chest wall, and the left sternocleidomastoid region. Furthermore, a 1-cm hypermetabolic nodule involving the right rectus abdominus muscle was noted on the PET scan. Based on their appearance, the nodules most likely represented metastasis from a primary skin malignancy. The rest of the studies were unremarkable. Serum tumor markers including prostate-specific antigen, cancer antigen 19-9, and carcinoembryonic antigen were within reference range. Immunostaining for estrogen receptor, progesterone receptor, and ERBB2 (formerly HER2/neu) was negative. The only abnormalities noted on serum chemistries were slight elevations in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and the a-fetoprotein tumor marker, which was attributed to chronic hepatitis C infection. Bacterial, fungal, and mycobacterial tissue cultures also were negative. These results ruled out infection and suggested against a primary visceral malignancy with cutaneous metastasis.
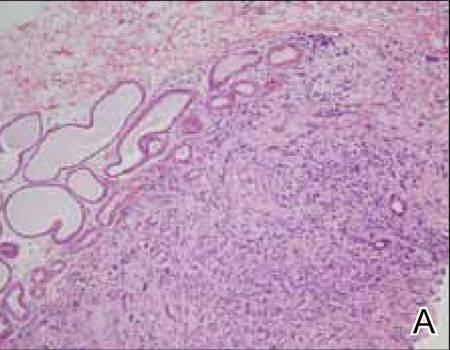
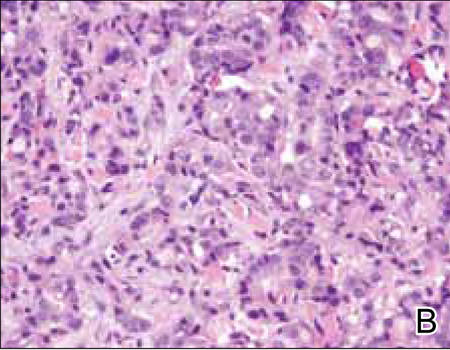
Histopathology revealed a moderately differentiated adenocarcinoma adjacent to healthy-appearing apocrine glands (Figure 2A). The normal glands were composed of cuboidal cells with abundant eosinophilic cytoplasm and prominent nuclei. The cells were arranged in a single layer in a glandular formation with prominent decapitation secretion. Adjacent to the normal apocrine glandular tissue was a focus of malignant epithelioid cells that extended to the lateral and inferior margins. The neoplastic cells were cuboidal to angulated in appearance with prominent nuclei and seemed to form ill-defined tubular or glandular structures that partially resembled apocrine glands (Figure 2B). Decapitation secretion is a feature of apocrine differentiation. Examination of additional tissue sections of the tumor did not reveal remarkable decapitation secretion in contrast to the adjacent healthy apocrine glands. Rather, a solid sheet arrangement was primarily noted in several sections (Figure 2B). Neither frequent mitoses nor prominent cellular atypia were seen, and there was no evidence of lymphatic, perineural, or vascular invasion.
|
Immunohistochemically, tumor cells reacted strongly to cytokeratin AE1/AE3 and CAM5.2, stains used to identify various cytokeratins present in epithelial tissue. Staining for epithelial membrane antigen and carcinoembryonic antigen revealed focal glandular differentiation, which further supported the epithelial origin of the neoplastic cells. Gross cystic disease fluid protein 15 (GCDFP-15) is a marker of apocrine differentiation and may indicate a carcinoma of apocrine or eccrine origin. In our case, staining for GCDFP-15 was negative in the cutaneous sections but highlighted tumor cells in 6 of 13 ipsilateral lymph nodes from locoregional metastasis. The cellular and structural morphology, immunohistochemistry, and absence of an alternative primary visceral malignancy supported the diagnosis of primary AA.
Initially the patient was not considered to be a candidate for surgery due to the rapid growth of the tumor with metastases, fatigue, weight loss, and pain. Therefore, radiation therapy was started. The patient responded well to treatment with controlled pain and resolution of the palpable mass of the left axilla. Moreover, a follow-up PET scan revealed no residual tumor and persistent, albeit decreased, axillary lymphadenopathy. As the patient’s clinical status had improved, excision of the left axillary tumor with lymph node dissection was performed 10 months after initial presentation.
In this case, the differential diagnosis consisted of various cutaneous neoplasms, primary mammary carcinoma, cutaneous metastasis, and infection. Diagnostic imaging and laboratory testing failed to identify any primary internal malignancies. Similarly, the negative cultures ruled out an infectious process. Furthermore, the axillary mass was noted to be separate from the breast tissue on physical examination and mammography. Histologically, the tumor showed features that were suggestive of an anaplastic process as well as decapitation secretion and glandular formation that clearly resembled apocrine differentiation.
Comment
Apocrine adenocarcinoma arises from apocrine sweat glands and therefore is mostly reported in areas of high apocrine gland density such as the axillae and the anogenital region.2,4,6 However, AA also has been reported in unusual locations,1,5,10,14-16 and they may arise from a pre-existing nevus sebaceous or from supernumerary nipples, which can occur anywhere along the milk lines.4,15 Apocrine adenocarcinoma most commonly arises in individuals aged 40 to 50 years.3,17 A slight male predominance has been reported but no racial predilection.1,4-6 Although a few reports have described the development of AAs within pre-existing benign tumors such as apocrine adenomas, apocrine hyperplasias, cylindromas, and nevi sebaceous, they usually are thought to arise de novo.4-6
Clinical Presentation
Apocrine adenocarcinoma is highly variable in its clinical manifestation.1,6 Most cases arise as erythematous to violaceous, firm, solitary nodules. Nonetheless, AA also can present as erythematous patches of skin resembling erysipelas and ulcerated nodules with overlying granulation tissue and purulent exudate.4,6,9,11 Although AA typically is slow growing and indolent, the time frame reported from onset to diagnosis ranges from weeks to decades.1,6,7 Most cases present asymptomatically; when symptoms do occur, the most common ones are tenderness, purulent discharge, and restricted range of motion from extremely large tumors.3,9 Incidence of lymph node metastasis is reported at 40% to 50% at the time of presentation.4,6 Additionally, AA has a high rate of local recurrence, but extranodal metastasis rarely is seen.2,6 When metastasis does occur, it is via lymphatic and hematogenous spread.6,9 Metastatic dissemination of AA may occur in the liver, lungs, bone, brain, and parotid glands, as well as the skin via intraepidermal pagetoid spread.4,6,9,13
Histopathology
The histologic characteristics essential to the diagnosis of primary AA are anaplastic differentiation and apocrine origin.1,2,9,10,17 Apocrine units include coiled secretory glands that reside in the deep dermis connecting to a straight duct that empties into the isthmus of the hair follicle.9,13 These secretory glands have a single row of cuboidal secretory cells lining the tubular component and stratified squamous epithelium lining the straight intradermal component that opens onto the hair follicle.9 Contractile myoepithelial cells surround the secretory cell layer of the gland.9,13
The cuboidal secretory cells of the apocrine gland have abundant eosinophilic cytoplasm1,4,9 and are further characterized by glandular arrangement and decapitation secretion, 2 features that are strongly suggestive of apocrine differentiation.4-6 In contrast, the tumor cells of AA can be characterized by hyperchromatic nuclei, nuclear pleomorphism, mitotic figures, and a lack of decapitation secretion.1,2,6 In malignancy, erratic or poorly differentiated ductal structures may be seen,1,3-6 including papillary, cordlike, solid, or complex glandular patterns that can potentially invade the adjacent tissue without a clearly recognizable myoepithelial layer that contains them.1,3,4,6 Moreover, AA may progress with lymphatic, vascular, or neural invasion.1,13
Various stains may be used in immunohistochemical analysis to aid in the diagnosis of AA.1,5 Cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, smooth muscle antigen, periodic acid–Schiff positivity with diastase resistance, and GCDFP-15 are useful in supporting the diagnosis of AA.2,6,10,17 Cytokeratin AE1/AE3 and CAM5.2 stain various cytokeratins to confirm the epithelial origin of the tissue.2 Epithelial membrane antigen is an antigen present on the apical surface of glandular epithelial cells that also has been used to identify epithelial cells in AA.2 Additionally, smooth muscle actin may be used to detect the myoepithelial layer of cells surrounding the apocrine glands.17 The lack of a continuous layer surrounding the secretory cells suggests invasion into the adjacent tissue.1,9,17 Periodic acid–Schiff staining with diastase resistance can be used to identify the mucin stored in the intracytoplasmic granules of apocrine cells and the lumen.3 Some stains such as GCDFP-15 may highlight cells of multiple origins (eg, apocrine and eccrine).10 However, there is the possibility that poorly differentiated AAs would fail to be identified as such even with well-established apocrine markers, which may explain the differential GCDFP-15 staining patterns in our patient’s skin and lymph node sections.1,5 Therefore, there is not a single perfect set of immunohistological criteria to aid in the diagnosis of AA.6,10,12 Fundamentally, diagnosis requires detection of primary apocrine differentiation with features such as invasion or spread to adjacent tissue to suggest malignancy and rule out an alternate primary malignant process.1,2,9,10,17
Treatment and Prognosis
Primary treatment of AA consists of wide local excision with adjuvant options that include chemotherapy and radiation.2,6 Due to the high rate of lymph node metastases at presentation (40%–50%), SLNB is recommended. A positive SLNB should be followed with complete axillary lymphadenectomy4,6; however, there is a lack of consensus regarding the role of SLNB and lymph node dissection in detecting subclinical lymph node disease, which might improve local recurrence rate and prognosis.6 Similarly, research shows variable results with adjunctive treatment such as chemotherapy or radiation therapy.6,9,13 Adjuvant treatment with chemotherapy or radiation therapy should be considered in cases with large tumor size; perineural, lymphatic, or vascular invasion; or when complete removal of the tumor is not possible due to location or size.2,6 However, neither the role nor the efficacy of such treatments in AA is well established.6,9,13
There is little information in the literature regarding the prognosis of AA. Although no specific or well-documented prognostic criteria exist, it is generally believed that patients with well-differentiated AA will have higher cure rates or lower rates of local recurrence and lymph node metastasis than patients with poorly differentiated neoplasms.3,6,10 A few small case series with long-term follow-up of patients ranging from 2 to 10 years have shown that prognosis may be favorable for AA patients despite local recurrence and regional lymph node metastasis.1,5
Conclusion
Primary AA is a rare cutaneous neoplasm that most commonly occurs in the axillae and the anogenital region. Apocrine adenocarcinoma presents with highly variable clinical and histopathological findings that make diagnosis a challenge. Clinicians should keep this entity in their differential diagnosis for patients who present with nodules arising in apocrine gland–bearing skin. Ultimately, histopathology is critical to diagnosis, and special stains are often required. To make the diagnosis, a tissue biopsy demonstrating apocrine differentiation and anaplastic features to suggest a malignant process are required. Additionally, a careful workup to rule out other diagnoses should be performed. Testing modalities that detect the presence of useful markers such as apocrine or epithelial origin should be used, and the presence of positive findings should support the diagnosis of AA. However, immunohistochemical findings should be used in the context of the patient’s clinical presentation and other available data. Treatment includes wide local excision, and lymphadenectomy is recommended in the setting of nodal spread. For aggressive tumors or metastases, excision may be followed by radiation therapy and chemotherapy.
1. Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
2. Cham PM, Niehans GA, Foman N, et al. Primary cutaneous apocrine carcinoma presenting as carcinoma erysipeloides [published online ahead of print November 6, 2007]. Br J Dermatol. 2008;158:194-196.
3. Chamberlain RS, Huber K, White JC, et al. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22:131-135.
4. Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
5. Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
6. Katagiri Y, Ansai S. Two cases of cutaneous apocrine ductal carcinoma of the axilla. case report and review of the literature. Dermatology. 1999;199:332-337.
7. Maury G, Guillot B, Bessis D, et al. Unusual axillary apocrine carcinoma of the skin: histological diagnostic difficulties [article in French] [published online ahead of print July 7, 2010]. Ann Dermatol Venereol. 2010;137:555-559.
8. Alex G. Apocrine adenocarcinoma of the nipple: a case report. Cases J. 2008;1:88.
9. MacNeill KN, Riddell RH, Ghazarian D. Perianal apocrine adenocarcinoma arising in a benign apocrine adenoma; first case report and review of the literature. J Clin Pathol. 2005;58:217-219.
10. Shintaku M, Tsuta K, Yoshida H, et al. Apocrine adenocarcinoma of the eyelid with aggressive biological behavior: report of a case. Pathol Int. 2002;52:169-173.
11. Zehr KJ, Rubin M, Ratner L. Apocrine adenocarcinoma presenting as a large ulcerated axillary mass. Dermatol Surg. 1997;23:585-587.
12. Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Panonica Adriat. 2009;18:141-142.
13. Hernandez JM, Copeland EM 3rd. Infiltrating apocrine adenocarcinoma with extramammary pagetoid spread. Am Surg. 2007;73:307-309.
14. Dhawan SS, Nanda VS, Grekin S, et al. Apocrine adenocarcinoma: case report and review of the literature. J Dermatol Surg Oncol. 1990;16:468-470.
15. Hügel H, Requena L. Ductal carcinoma arising from a syringocystadenoma papilliferum in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 2003;25:490-493.
16. Stout AP, Cooley SG. Carcinoma of sweat glands. Cancer. 1951;4:521-536.
17. Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms—part 2: an approach to tumours of cutaneous sweat glands [published online ahead of print August 1, 2006]. J Clin Pathol. 2007;60:145-159.
Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy, with most of the available information about this disease consolidated from anecdotal evidence of single case reports and small case series with fewer than 30 patients.1-11 Although certain histologic and immunohistochemical features have been suggested to be useful in the diagnosis of AA, there is no clear consensus on the required pathologic criteria.1,5,6,9,10,12,13 Additionally, the clinical presentation of AA is highly variable, which further adds to the challenge of making an accurate diagnosis.1-3,5,9,10,13
Apocrine adenocarcinoma usually arises in areas of high apocrine gland density such as the axillae or anogenital region.2,4,6 It also has been reported in areas such as the scalp, ear canal, eyelids, chest, nipples, arms, wrists, and fingers.4,8,10,14-16 Apocrine adenocarcinoma in unusual locations such as the eyelid and ear canal is thought to arise from modified apocrine glands such as the Moll glands of the eyelid and the ceruminous glands of the ear canal.9,10 The presence of ectopic apocrine glands may lead to AA in atypical sites such as the wrists and fingers.5,16 The areola is an apocrine-dense area; therefore, AA may present on the nipples or within supernumerary nipples anywhere along the milk lines.4
Apocrine adenocarcinoma clinically presents as an asymptomatic to slightly painful, slowly growing, and erythematous to violaceous nodule or tumor.4,6,9 However, in a minority of cases the initial presentation consists of a cystic or ulcerated mass with overlying granulation tissue and purulent discharge.6,9,11 A wide time frame from the onset of symptoms to diagnosis has been reported, ranging from weeks to decades.4,6-8 The conventional treatment of AA is wide local excision.2,4,6,9 Although AA often presents with local lymph node metastasis at the time of diagnosis, there is no consensus on the use of sentinel lymph node biopsy (SLNB), nodal dissection, or adjuvant chemoradiation therapy.1,3,8,9
We report the case of a 49-year-old man with primary AA of the left axilla; the clinical and histologic features of AA as well as the appropriate diagnostic and treatment modalities also are provided.
Case Report
A 49-year-old man with a slowly growing tender mass of the left axilla of 1 year’s duration was referred to our dermatology clinic for evaluation. A review of systems revealed loss of appetite, fatigue, and a 4-month history of unintentional weight loss (15–20 lb). The patient had a history of hepatitis C virus, intravenous drug use, alcohol abuse, and cigarette smoking (1 pack daily) for many years. Additionally, the patient reported a paternal family history of numerous visceral malignancies. Examination of the left axilla revealed a 1.5×5-cm ulcerated tumor that produced serosanguineous discharge and was tender to palpation (Figure 1). Two 1-cm, firm, freely mobile subcutaneous nodules with no overlying skin changes were palpable at the medial border of the ulcerated nodule. There was no additional cervical or axillary lymphadenopathy, and a breast examination was normal.
The differential diagnosis included primary squamous cell carcinoma or adnexal neoplasm, primary breast carcinoma, lymphoma, scrofuloderma, atypical mycobacterial infection, and cutaneous metastasis from an internal malignancy. Two 4-mm punch biopsies were performed and sent for routine histopathology and bacterial, fungal, and mycobacterial tissue cultures. To exclude a primary visceral malignancy or metastasis, computed tomography of the chest, abdomen, and pelvis; positron emission tomography (PET) from the base of the skull to the thighs; colonoscopy; magnetic resonance imaging of the brain; esophagogastroduodenoscopy; and mammography were conducted. Prominent left axillary lymphadenopathy was noted on computed tomography. Additionally, PET identified extranodal spread in the left axilla, left lateral chest wall, and the left sternocleidomastoid region. Furthermore, a 1-cm hypermetabolic nodule involving the right rectus abdominus muscle was noted on the PET scan. Based on their appearance, the nodules most likely represented metastasis from a primary skin malignancy. The rest of the studies were unremarkable. Serum tumor markers including prostate-specific antigen, cancer antigen 19-9, and carcinoembryonic antigen were within reference range. Immunostaining for estrogen receptor, progesterone receptor, and ERBB2 (formerly HER2/neu) was negative. The only abnormalities noted on serum chemistries were slight elevations in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and the a-fetoprotein tumor marker, which was attributed to chronic hepatitis C infection. Bacterial, fungal, and mycobacterial tissue cultures also were negative. These results ruled out infection and suggested against a primary visceral malignancy with cutaneous metastasis.
Histopathology revealed a moderately differentiated adenocarcinoma adjacent to healthy-appearing apocrine glands (Figure 2A). The normal glands were composed of cuboidal cells with abundant eosinophilic cytoplasm and prominent nuclei. The cells were arranged in a single layer in a glandular formation with prominent decapitation secretion. Adjacent to the normal apocrine glandular tissue was a focus of malignant epithelioid cells that extended to the lateral and inferior margins. The neoplastic cells were cuboidal to angulated in appearance with prominent nuclei and seemed to form ill-defined tubular or glandular structures that partially resembled apocrine glands (Figure 2B). Decapitation secretion is a feature of apocrine differentiation. Examination of additional tissue sections of the tumor did not reveal remarkable decapitation secretion in contrast to the adjacent healthy apocrine glands. Rather, a solid sheet arrangement was primarily noted in several sections (Figure 2B). Neither frequent mitoses nor prominent cellular atypia were seen, and there was no evidence of lymphatic, perineural, or vascular invasion.
|
|
Immunohistochemically, tumor cells reacted strongly to cytokeratin AE1/AE3 and CAM5.2, stains used to identify various cytokeratins present in epithelial tissue. Staining for epithelial membrane antigen and carcinoembryonic antigen revealed focal glandular differentiation, which further supported the epithelial origin of the neoplastic cells. Gross cystic disease fluid protein 15 (GCDFP-15) is a marker of apocrine differentiation and may indicate a carcinoma of apocrine or eccrine origin. In our case, staining for GCDFP-15 was negative in the cutaneous sections but highlighted tumor cells in 6 of 13 ipsilateral lymph nodes from locoregional metastasis. The cellular and structural morphology, immunohistochemistry, and absence of an alternative primary visceral malignancy supported the diagnosis of primary AA.
Initially the patient was not considered to be a candidate for surgery due to the rapid growth of the tumor with metastases, fatigue, weight loss, and pain. Therefore, radiation therapy was started. The patient responded well to treatment with controlled pain and resolution of the palpable mass of the left axilla. Moreover, a follow-up PET scan revealed no residual tumor and persistent, albeit decreased, axillary lymphadenopathy. As the patient’s clinical status had improved, excision of the left axillary tumor with lymph node dissection was performed 10 months after initial presentation.
In this case, the differential diagnosis consisted of various cutaneous neoplasms, primary mammary carcinoma, cutaneous metastasis, and infection. Diagnostic imaging and laboratory testing failed to identify any primary internal malignancies. Similarly, the negative cultures ruled out an infectious process. Furthermore, the axillary mass was noted to be separate from the breast tissue on physical examination and mammography. Histologically, the tumor showed features that were suggestive of an anaplastic process as well as decapitation secretion and glandular formation that clearly resembled apocrine differentiation.
Comment
Apocrine adenocarcinoma arises from apocrine sweat glands and therefore is mostly reported in areas of high apocrine gland density such as the axillae and the anogenital region.2,4,6 However, AA also has been reported in unusual locations,1,5,10,14-16 and they may arise from a pre-existing nevus sebaceous or from supernumerary nipples, which can occur anywhere along the milk lines.4,15 Apocrine adenocarcinoma most commonly arises in individuals aged 40 to 50 years.3,17 A slight male predominance has been reported but no racial predilection.1,4-6 Although a few reports have described the development of AAs within pre-existing benign tumors such as apocrine adenomas, apocrine hyperplasias, cylindromas, and nevi sebaceous, they usually are thought to arise de novo.4-6
Clinical Presentation
Apocrine adenocarcinoma is highly variable in its clinical manifestation.1,6 Most cases arise as erythematous to violaceous, firm, solitary nodules. Nonetheless, AA also can present as erythematous patches of skin resembling erysipelas and ulcerated nodules with overlying granulation tissue and purulent exudate.4,6,9,11 Although AA typically is slow growing and indolent, the time frame reported from onset to diagnosis ranges from weeks to decades.1,6,7 Most cases present asymptomatically; when symptoms do occur, the most common ones are tenderness, purulent discharge, and restricted range of motion from extremely large tumors.3,9 Incidence of lymph node metastasis is reported at 40% to 50% at the time of presentation.4,6 Additionally, AA has a high rate of local recurrence, but extranodal metastasis rarely is seen.2,6 When metastasis does occur, it is via lymphatic and hematogenous spread.6,9 Metastatic dissemination of AA may occur in the liver, lungs, bone, brain, and parotid glands, as well as the skin via intraepidermal pagetoid spread.4,6,9,13
Histopathology
The histologic characteristics essential to the diagnosis of primary AA are anaplastic differentiation and apocrine origin.1,2,9,10,17 Apocrine units include coiled secretory glands that reside in the deep dermis connecting to a straight duct that empties into the isthmus of the hair follicle.9,13 These secretory glands have a single row of cuboidal secretory cells lining the tubular component and stratified squamous epithelium lining the straight intradermal component that opens onto the hair follicle.9 Contractile myoepithelial cells surround the secretory cell layer of the gland.9,13
The cuboidal secretory cells of the apocrine gland have abundant eosinophilic cytoplasm1,4,9 and are further characterized by glandular arrangement and decapitation secretion, 2 features that are strongly suggestive of apocrine differentiation.4-6 In contrast, the tumor cells of AA can be characterized by hyperchromatic nuclei, nuclear pleomorphism, mitotic figures, and a lack of decapitation secretion.1,2,6 In malignancy, erratic or poorly differentiated ductal structures may be seen,1,3-6 including papillary, cordlike, solid, or complex glandular patterns that can potentially invade the adjacent tissue without a clearly recognizable myoepithelial layer that contains them.1,3,4,6 Moreover, AA may progress with lymphatic, vascular, or neural invasion.1,13
Various stains may be used in immunohistochemical analysis to aid in the diagnosis of AA.1,5 Cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, smooth muscle antigen, periodic acid–Schiff positivity with diastase resistance, and GCDFP-15 are useful in supporting the diagnosis of AA.2,6,10,17 Cytokeratin AE1/AE3 and CAM5.2 stain various cytokeratins to confirm the epithelial origin of the tissue.2 Epithelial membrane antigen is an antigen present on the apical surface of glandular epithelial cells that also has been used to identify epithelial cells in AA.2 Additionally, smooth muscle actin may be used to detect the myoepithelial layer of cells surrounding the apocrine glands.17 The lack of a continuous layer surrounding the secretory cells suggests invasion into the adjacent tissue.1,9,17 Periodic acid–Schiff staining with diastase resistance can be used to identify the mucin stored in the intracytoplasmic granules of apocrine cells and the lumen.3 Some stains such as GCDFP-15 may highlight cells of multiple origins (eg, apocrine and eccrine).10 However, there is the possibility that poorly differentiated AAs would fail to be identified as such even with well-established apocrine markers, which may explain the differential GCDFP-15 staining patterns in our patient’s skin and lymph node sections.1,5 Therefore, there is not a single perfect set of immunohistological criteria to aid in the diagnosis of AA.6,10,12 Fundamentally, diagnosis requires detection of primary apocrine differentiation with features such as invasion or spread to adjacent tissue to suggest malignancy and rule out an alternate primary malignant process.1,2,9,10,17
Treatment and Prognosis
Primary treatment of AA consists of wide local excision with adjuvant options that include chemotherapy and radiation.2,6 Due to the high rate of lymph node metastases at presentation (40%–50%), SLNB is recommended. A positive SLNB should be followed with complete axillary lymphadenectomy4,6; however, there is a lack of consensus regarding the role of SLNB and lymph node dissection in detecting subclinical lymph node disease, which might improve local recurrence rate and prognosis.6 Similarly, research shows variable results with adjunctive treatment such as chemotherapy or radiation therapy.6,9,13 Adjuvant treatment with chemotherapy or radiation therapy should be considered in cases with large tumor size; perineural, lymphatic, or vascular invasion; or when complete removal of the tumor is not possible due to location or size.2,6 However, neither the role nor the efficacy of such treatments in AA is well established.6,9,13
There is little information in the literature regarding the prognosis of AA. Although no specific or well-documented prognostic criteria exist, it is generally believed that patients with well-differentiated AA will have higher cure rates or lower rates of local recurrence and lymph node metastasis than patients with poorly differentiated neoplasms.3,6,10 A few small case series with long-term follow-up of patients ranging from 2 to 10 years have shown that prognosis may be favorable for AA patients despite local recurrence and regional lymph node metastasis.1,5
Conclusion
Primary AA is a rare cutaneous neoplasm that most commonly occurs in the axillae and the anogenital region. Apocrine adenocarcinoma presents with highly variable clinical and histopathological findings that make diagnosis a challenge. Clinicians should keep this entity in their differential diagnosis for patients who present with nodules arising in apocrine gland–bearing skin. Ultimately, histopathology is critical to diagnosis, and special stains are often required. To make the diagnosis, a tissue biopsy demonstrating apocrine differentiation and anaplastic features to suggest a malignant process are required. Additionally, a careful workup to rule out other diagnoses should be performed. Testing modalities that detect the presence of useful markers such as apocrine or epithelial origin should be used, and the presence of positive findings should support the diagnosis of AA. However, immunohistochemical findings should be used in the context of the patient’s clinical presentation and other available data. Treatment includes wide local excision, and lymphadenectomy is recommended in the setting of nodal spread. For aggressive tumors or metastases, excision may be followed by radiation therapy and chemotherapy.
Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy, with most of the available information about this disease consolidated from anecdotal evidence of single case reports and small case series with fewer than 30 patients.1-11 Although certain histologic and immunohistochemical features have been suggested to be useful in the diagnosis of AA, there is no clear consensus on the required pathologic criteria.1,5,6,9,10,12,13 Additionally, the clinical presentation of AA is highly variable, which further adds to the challenge of making an accurate diagnosis.1-3,5,9,10,13
Apocrine adenocarcinoma usually arises in areas of high apocrine gland density such as the axillae or anogenital region.2,4,6 It also has been reported in areas such as the scalp, ear canal, eyelids, chest, nipples, arms, wrists, and fingers.4,8,10,14-16 Apocrine adenocarcinoma in unusual locations such as the eyelid and ear canal is thought to arise from modified apocrine glands such as the Moll glands of the eyelid and the ceruminous glands of the ear canal.9,10 The presence of ectopic apocrine glands may lead to AA in atypical sites such as the wrists and fingers.5,16 The areola is an apocrine-dense area; therefore, AA may present on the nipples or within supernumerary nipples anywhere along the milk lines.4
Apocrine adenocarcinoma clinically presents as an asymptomatic to slightly painful, slowly growing, and erythematous to violaceous nodule or tumor.4,6,9 However, in a minority of cases the initial presentation consists of a cystic or ulcerated mass with overlying granulation tissue and purulent discharge.6,9,11 A wide time frame from the onset of symptoms to diagnosis has been reported, ranging from weeks to decades.4,6-8 The conventional treatment of AA is wide local excision.2,4,6,9 Although AA often presents with local lymph node metastasis at the time of diagnosis, there is no consensus on the use of sentinel lymph node biopsy (SLNB), nodal dissection, or adjuvant chemoradiation therapy.1,3,8,9
We report the case of a 49-year-old man with primary AA of the left axilla; the clinical and histologic features of AA as well as the appropriate diagnostic and treatment modalities also are provided.
Case Report
A 49-year-old man with a slowly growing tender mass of the left axilla of 1 year’s duration was referred to our dermatology clinic for evaluation. A review of systems revealed loss of appetite, fatigue, and a 4-month history of unintentional weight loss (15–20 lb). The patient had a history of hepatitis C virus, intravenous drug use, alcohol abuse, and cigarette smoking (1 pack daily) for many years. Additionally, the patient reported a paternal family history of numerous visceral malignancies. Examination of the left axilla revealed a 1.5×5-cm ulcerated tumor that produced serosanguineous discharge and was tender to palpation (Figure 1). Two 1-cm, firm, freely mobile subcutaneous nodules with no overlying skin changes were palpable at the medial border of the ulcerated nodule. There was no additional cervical or axillary lymphadenopathy, and a breast examination was normal.
The differential diagnosis included primary squamous cell carcinoma or adnexal neoplasm, primary breast carcinoma, lymphoma, scrofuloderma, atypical mycobacterial infection, and cutaneous metastasis from an internal malignancy. Two 4-mm punch biopsies were performed and sent for routine histopathology and bacterial, fungal, and mycobacterial tissue cultures. To exclude a primary visceral malignancy or metastasis, computed tomography of the chest, abdomen, and pelvis; positron emission tomography (PET) from the base of the skull to the thighs; colonoscopy; magnetic resonance imaging of the brain; esophagogastroduodenoscopy; and mammography were conducted. Prominent left axillary lymphadenopathy was noted on computed tomography. Additionally, PET identified extranodal spread in the left axilla, left lateral chest wall, and the left sternocleidomastoid region. Furthermore, a 1-cm hypermetabolic nodule involving the right rectus abdominus muscle was noted on the PET scan. Based on their appearance, the nodules most likely represented metastasis from a primary skin malignancy. The rest of the studies were unremarkable. Serum tumor markers including prostate-specific antigen, cancer antigen 19-9, and carcinoembryonic antigen were within reference range. Immunostaining for estrogen receptor, progesterone receptor, and ERBB2 (formerly HER2/neu) was negative. The only abnormalities noted on serum chemistries were slight elevations in aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and the a-fetoprotein tumor marker, which was attributed to chronic hepatitis C infection. Bacterial, fungal, and mycobacterial tissue cultures also were negative. These results ruled out infection and suggested against a primary visceral malignancy with cutaneous metastasis.
Histopathology revealed a moderately differentiated adenocarcinoma adjacent to healthy-appearing apocrine glands (Figure 2A). The normal glands were composed of cuboidal cells with abundant eosinophilic cytoplasm and prominent nuclei. The cells were arranged in a single layer in a glandular formation with prominent decapitation secretion. Adjacent to the normal apocrine glandular tissue was a focus of malignant epithelioid cells that extended to the lateral and inferior margins. The neoplastic cells were cuboidal to angulated in appearance with prominent nuclei and seemed to form ill-defined tubular or glandular structures that partially resembled apocrine glands (Figure 2B). Decapitation secretion is a feature of apocrine differentiation. Examination of additional tissue sections of the tumor did not reveal remarkable decapitation secretion in contrast to the adjacent healthy apocrine glands. Rather, a solid sheet arrangement was primarily noted in several sections (Figure 2B). Neither frequent mitoses nor prominent cellular atypia were seen, and there was no evidence of lymphatic, perineural, or vascular invasion.
|
|
Immunohistochemically, tumor cells reacted strongly to cytokeratin AE1/AE3 and CAM5.2, stains used to identify various cytokeratins present in epithelial tissue. Staining for epithelial membrane antigen and carcinoembryonic antigen revealed focal glandular differentiation, which further supported the epithelial origin of the neoplastic cells. Gross cystic disease fluid protein 15 (GCDFP-15) is a marker of apocrine differentiation and may indicate a carcinoma of apocrine or eccrine origin. In our case, staining for GCDFP-15 was negative in the cutaneous sections but highlighted tumor cells in 6 of 13 ipsilateral lymph nodes from locoregional metastasis. The cellular and structural morphology, immunohistochemistry, and absence of an alternative primary visceral malignancy supported the diagnosis of primary AA.
Initially the patient was not considered to be a candidate for surgery due to the rapid growth of the tumor with metastases, fatigue, weight loss, and pain. Therefore, radiation therapy was started. The patient responded well to treatment with controlled pain and resolution of the palpable mass of the left axilla. Moreover, a follow-up PET scan revealed no residual tumor and persistent, albeit decreased, axillary lymphadenopathy. As the patient’s clinical status had improved, excision of the left axillary tumor with lymph node dissection was performed 10 months after initial presentation.
In this case, the differential diagnosis consisted of various cutaneous neoplasms, primary mammary carcinoma, cutaneous metastasis, and infection. Diagnostic imaging and laboratory testing failed to identify any primary internal malignancies. Similarly, the negative cultures ruled out an infectious process. Furthermore, the axillary mass was noted to be separate from the breast tissue on physical examination and mammography. Histologically, the tumor showed features that were suggestive of an anaplastic process as well as decapitation secretion and glandular formation that clearly resembled apocrine differentiation.
Comment
Apocrine adenocarcinoma arises from apocrine sweat glands and therefore is mostly reported in areas of high apocrine gland density such as the axillae and the anogenital region.2,4,6 However, AA also has been reported in unusual locations,1,5,10,14-16 and they may arise from a pre-existing nevus sebaceous or from supernumerary nipples, which can occur anywhere along the milk lines.4,15 Apocrine adenocarcinoma most commonly arises in individuals aged 40 to 50 years.3,17 A slight male predominance has been reported but no racial predilection.1,4-6 Although a few reports have described the development of AAs within pre-existing benign tumors such as apocrine adenomas, apocrine hyperplasias, cylindromas, and nevi sebaceous, they usually are thought to arise de novo.4-6
Clinical Presentation
Apocrine adenocarcinoma is highly variable in its clinical manifestation.1,6 Most cases arise as erythematous to violaceous, firm, solitary nodules. Nonetheless, AA also can present as erythematous patches of skin resembling erysipelas and ulcerated nodules with overlying granulation tissue and purulent exudate.4,6,9,11 Although AA typically is slow growing and indolent, the time frame reported from onset to diagnosis ranges from weeks to decades.1,6,7 Most cases present asymptomatically; when symptoms do occur, the most common ones are tenderness, purulent discharge, and restricted range of motion from extremely large tumors.3,9 Incidence of lymph node metastasis is reported at 40% to 50% at the time of presentation.4,6 Additionally, AA has a high rate of local recurrence, but extranodal metastasis rarely is seen.2,6 When metastasis does occur, it is via lymphatic and hematogenous spread.6,9 Metastatic dissemination of AA may occur in the liver, lungs, bone, brain, and parotid glands, as well as the skin via intraepidermal pagetoid spread.4,6,9,13
Histopathology
The histologic characteristics essential to the diagnosis of primary AA are anaplastic differentiation and apocrine origin.1,2,9,10,17 Apocrine units include coiled secretory glands that reside in the deep dermis connecting to a straight duct that empties into the isthmus of the hair follicle.9,13 These secretory glands have a single row of cuboidal secretory cells lining the tubular component and stratified squamous epithelium lining the straight intradermal component that opens onto the hair follicle.9 Contractile myoepithelial cells surround the secretory cell layer of the gland.9,13
The cuboidal secretory cells of the apocrine gland have abundant eosinophilic cytoplasm1,4,9 and are further characterized by glandular arrangement and decapitation secretion, 2 features that are strongly suggestive of apocrine differentiation.4-6 In contrast, the tumor cells of AA can be characterized by hyperchromatic nuclei, nuclear pleomorphism, mitotic figures, and a lack of decapitation secretion.1,2,6 In malignancy, erratic or poorly differentiated ductal structures may be seen,1,3-6 including papillary, cordlike, solid, or complex glandular patterns that can potentially invade the adjacent tissue without a clearly recognizable myoepithelial layer that contains them.1,3,4,6 Moreover, AA may progress with lymphatic, vascular, or neural invasion.1,13
Various stains may be used in immunohistochemical analysis to aid in the diagnosis of AA.1,5 Cytokeratin AE1/AE3, CAM5.2, epithelial membrane antigen, smooth muscle antigen, periodic acid–Schiff positivity with diastase resistance, and GCDFP-15 are useful in supporting the diagnosis of AA.2,6,10,17 Cytokeratin AE1/AE3 and CAM5.2 stain various cytokeratins to confirm the epithelial origin of the tissue.2 Epithelial membrane antigen is an antigen present on the apical surface of glandular epithelial cells that also has been used to identify epithelial cells in AA.2 Additionally, smooth muscle actin may be used to detect the myoepithelial layer of cells surrounding the apocrine glands.17 The lack of a continuous layer surrounding the secretory cells suggests invasion into the adjacent tissue.1,9,17 Periodic acid–Schiff staining with diastase resistance can be used to identify the mucin stored in the intracytoplasmic granules of apocrine cells and the lumen.3 Some stains such as GCDFP-15 may highlight cells of multiple origins (eg, apocrine and eccrine).10 However, there is the possibility that poorly differentiated AAs would fail to be identified as such even with well-established apocrine markers, which may explain the differential GCDFP-15 staining patterns in our patient’s skin and lymph node sections.1,5 Therefore, there is not a single perfect set of immunohistological criteria to aid in the diagnosis of AA.6,10,12 Fundamentally, diagnosis requires detection of primary apocrine differentiation with features such as invasion or spread to adjacent tissue to suggest malignancy and rule out an alternate primary malignant process.1,2,9,10,17
Treatment and Prognosis
Primary treatment of AA consists of wide local excision with adjuvant options that include chemotherapy and radiation.2,6 Due to the high rate of lymph node metastases at presentation (40%–50%), SLNB is recommended. A positive SLNB should be followed with complete axillary lymphadenectomy4,6; however, there is a lack of consensus regarding the role of SLNB and lymph node dissection in detecting subclinical lymph node disease, which might improve local recurrence rate and prognosis.6 Similarly, research shows variable results with adjunctive treatment such as chemotherapy or radiation therapy.6,9,13 Adjuvant treatment with chemotherapy or radiation therapy should be considered in cases with large tumor size; perineural, lymphatic, or vascular invasion; or when complete removal of the tumor is not possible due to location or size.2,6 However, neither the role nor the efficacy of such treatments in AA is well established.6,9,13
There is little information in the literature regarding the prognosis of AA. Although no specific or well-documented prognostic criteria exist, it is generally believed that patients with well-differentiated AA will have higher cure rates or lower rates of local recurrence and lymph node metastasis than patients with poorly differentiated neoplasms.3,6,10 A few small case series with long-term follow-up of patients ranging from 2 to 10 years have shown that prognosis may be favorable for AA patients despite local recurrence and regional lymph node metastasis.1,5
Conclusion
Primary AA is a rare cutaneous neoplasm that most commonly occurs in the axillae and the anogenital region. Apocrine adenocarcinoma presents with highly variable clinical and histopathological findings that make diagnosis a challenge. Clinicians should keep this entity in their differential diagnosis for patients who present with nodules arising in apocrine gland–bearing skin. Ultimately, histopathology is critical to diagnosis, and special stains are often required. To make the diagnosis, a tissue biopsy demonstrating apocrine differentiation and anaplastic features to suggest a malignant process are required. Additionally, a careful workup to rule out other diagnoses should be performed. Testing modalities that detect the presence of useful markers such as apocrine or epithelial origin should be used, and the presence of positive findings should support the diagnosis of AA. However, immunohistochemical findings should be used in the context of the patient’s clinical presentation and other available data. Treatment includes wide local excision, and lymphadenectomy is recommended in the setting of nodal spread. For aggressive tumors or metastases, excision may be followed by radiation therapy and chemotherapy.
1. Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
2. Cham PM, Niehans GA, Foman N, et al. Primary cutaneous apocrine carcinoma presenting as carcinoma erysipeloides [published online ahead of print November 6, 2007]. Br J Dermatol. 2008;158:194-196.
3. Chamberlain RS, Huber K, White JC, et al. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22:131-135.
4. Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
5. Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
6. Katagiri Y, Ansai S. Two cases of cutaneous apocrine ductal carcinoma of the axilla. case report and review of the literature. Dermatology. 1999;199:332-337.
7. Maury G, Guillot B, Bessis D, et al. Unusual axillary apocrine carcinoma of the skin: histological diagnostic difficulties [article in French] [published online ahead of print July 7, 2010]. Ann Dermatol Venereol. 2010;137:555-559.
8. Alex G. Apocrine adenocarcinoma of the nipple: a case report. Cases J. 2008;1:88.
9. MacNeill KN, Riddell RH, Ghazarian D. Perianal apocrine adenocarcinoma arising in a benign apocrine adenoma; first case report and review of the literature. J Clin Pathol. 2005;58:217-219.
10. Shintaku M, Tsuta K, Yoshida H, et al. Apocrine adenocarcinoma of the eyelid with aggressive biological behavior: report of a case. Pathol Int. 2002;52:169-173.
11. Zehr KJ, Rubin M, Ratner L. Apocrine adenocarcinoma presenting as a large ulcerated axillary mass. Dermatol Surg. 1997;23:585-587.
12. Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Panonica Adriat. 2009;18:141-142.
13. Hernandez JM, Copeland EM 3rd. Infiltrating apocrine adenocarcinoma with extramammary pagetoid spread. Am Surg. 2007;73:307-309.
14. Dhawan SS, Nanda VS, Grekin S, et al. Apocrine adenocarcinoma: case report and review of the literature. J Dermatol Surg Oncol. 1990;16:468-470.
15. Hügel H, Requena L. Ductal carcinoma arising from a syringocystadenoma papilliferum in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 2003;25:490-493.
16. Stout AP, Cooley SG. Carcinoma of sweat glands. Cancer. 1951;4:521-536.
17. Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms—part 2: an approach to tumours of cutaneous sweat glands [published online ahead of print August 1, 2006]. J Clin Pathol. 2007;60:145-159.
1. Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
2. Cham PM, Niehans GA, Foman N, et al. Primary cutaneous apocrine carcinoma presenting as carcinoma erysipeloides [published online ahead of print November 6, 2007]. Br J Dermatol. 2008;158:194-196.
3. Chamberlain RS, Huber K, White JC, et al. Apocrine gland carcinoma of the axilla: review of the literature and recommendations for treatment. Am J Clin Oncol. 1999;22:131-135.
4. Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
5. Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
6. Katagiri Y, Ansai S. Two cases of cutaneous apocrine ductal carcinoma of the axilla. case report and review of the literature. Dermatology. 1999;199:332-337.
7. Maury G, Guillot B, Bessis D, et al. Unusual axillary apocrine carcinoma of the skin: histological diagnostic difficulties [article in French] [published online ahead of print July 7, 2010]. Ann Dermatol Venereol. 2010;137:555-559.
8. Alex G. Apocrine adenocarcinoma of the nipple: a case report. Cases J. 2008;1:88.
9. MacNeill KN, Riddell RH, Ghazarian D. Perianal apocrine adenocarcinoma arising in a benign apocrine adenoma; first case report and review of the literature. J Clin Pathol. 2005;58:217-219.
10. Shintaku M, Tsuta K, Yoshida H, et al. Apocrine adenocarcinoma of the eyelid with aggressive biological behavior: report of a case. Pathol Int. 2002;52:169-173.
11. Zehr KJ, Rubin M, Ratner L. Apocrine adenocarcinoma presenting as a large ulcerated axillary mass. Dermatol Surg. 1997;23:585-587.
12. Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Panonica Adriat. 2009;18:141-142.
13. Hernandez JM, Copeland EM 3rd. Infiltrating apocrine adenocarcinoma with extramammary pagetoid spread. Am Surg. 2007;73:307-309.
14. Dhawan SS, Nanda VS, Grekin S, et al. Apocrine adenocarcinoma: case report and review of the literature. J Dermatol Surg Oncol. 1990;16:468-470.
15. Hügel H, Requena L. Ductal carcinoma arising from a syringocystadenoma papilliferum in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 2003;25:490-493.
16. Stout AP, Cooley SG. Carcinoma of sweat glands. Cancer. 1951;4:521-536.
17. Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms—part 2: an approach to tumours of cutaneous sweat glands [published online ahead of print August 1, 2006]. J Clin Pathol. 2007;60:145-159.
Practice Points
- Primary apocrine adenocarcinoma (AA) is a rare cutaneous malignancy with metastatic potential.
It arises in areas of high apocrine gland density including the axillae and anogenital region. - Apocrine adenocarcinoma must be differentiated from various infections and cutaneous metastases from internal malignancies.
- Primary apocrine differentiation with invasion to adjacent tissue is a key histopathologic feature of AA.
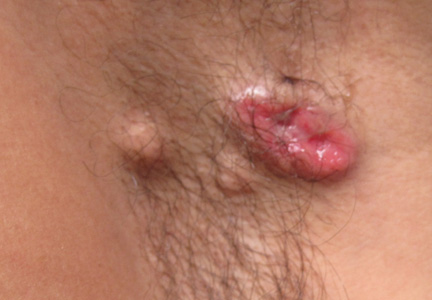
Nicotinamide cuts rate of nonmelanoma skin cancer in those at high risk
Nicotinamide, an inexpensive, over-the-counter form of vitamin B3, is safe and efficacious for the chemoprevention of nonmelanoma skin cancer in patients at high risk, according to data from the Australian Oral Nicotinamide to Reduce Actinic Cancer (ONTRAC) Study.
Results reported in a press briefing held before the annual meeting of the American Society of Clinical Oncology showed that patients taking nicotinamide were about one-fourth less likely than peers taking a placebo to develop new basal cell and squamous cell skin cancers. They also had a smaller reduction in new actinic keratoses.
“Nicotinamide, vitamin B3, significantly reduced nonmelanoma skin cancers and keratoses in just 12 months in a group of pretty high-risk patients. It’s safe, it’s almost obscenely inexpensive, and it’s already widely commercially available, so this one’s ready to go straight into the clinic,” commented senior investigator Dr. Diona Damian, professor of dermatology at the University of Sydney.
She cautioned that the results apply only to the population studied: adults who had experienced two or more nonmelanoma skin cancers in the past 5 years.
“These are the people we’d be recommending it for – people who have already got a skin cancer track record. It’s not something that we’d recommend at this stage for the general population,” she said. Likewise, the findings do not speak to patients at the other end of the spectrum who are in treatment for advanced or metastatic skin cancer, as they also were excluded.
That said, the researchers are planning additional studies in other populations, such patients who are at high risk because they have immunosuppression, according to Dr. Damian.
“We still need the overall skin cancer prevention strategies of sun-safe behavior, sunscreen, and regular skin surveillance,” she stressed, “but we now have an additional exciting opportunity for affordable skin cancer chemoprevention which we can instantly translate into clinical practice.”
Dr. Peter Paul Yu, ASCO President and a medical oncologist and hematologist who is director of cancer research at the Palo Alto Medical Foundation, Sunnyvale, Calif., commented, “This is a very exciting prevention trial. We all know that we clamor for preventing rather than treating diseases, and this is a major advance for us.”
Exposure to ultraviolet light packs a one-two punch to the skin, both damaging cellular DNA and suppressing the skin’s immune response, according to Dr. Damian. The investigators opted to test nicotinamide as it counters both of these events.
The 386 patients in ONTRAC had heavily sun damaged skin, with a mean of 8 nonmelanoma skin cancers in the past 5 years and 50 keratoses at baseline. They were randomized evenly to receive nicotinamide (500 mg twice daily) or placebo for 12 months.
Results showed that the average number of new nonmelanoma skin cancers per patient during the treatment period was 1.77 in the nicotinamide group and 2.42 in the placebo group. The difference translated to a 23% lower rate of new cancers with the vitamin.
“There were comparable reductions seen for both basal and squamous cell carcinomas,” Dr. Damian noted. “Interestingly, this reduction in skin cancers seemed to start as early as the first 3-month visit. And then when people stopped taking their tablets after 12 months, the benefit was no longer seen. In other words, you need to continue taking the tablets in order for them to be effective.”
The nicotinamide group also had a roughly 15% lower rate of new actinic keratoses, compared with the placebo group.
“Nicotinamide was very well tolerated. There was no difference in adverse events, blood parameters, or blood pressure in the two arms” of the study, reported Dr. Damian. She stressed that it is critically important to distinguish nicotinamide from niacin (nicotinic acid), another form of vitamin B3 that has a host of side effects such as headache and flushing.
“One of the great things about [nicotinamide] is that it really has hardly any drug interactions, which means that elderly patients who may be taking a whole cocktail of medications for their heart disease and their hypertension, and whatever else, the nicotinamide won’t interact with those,” she added.
Some evidence also has shown nonsteroidal anti-inflammatory drugs to reduce the risk of skin cancer. “The advantage of nicotinamide is that it doesn’t have the potential gastrointestinal bleeding or renal side effects of nonsteroidals, so it may be suitable for a group of people who aren’t suitable for taking nonsteroidals,” she said. “In our ONTRAC study, we didn’t find synergy or additional benefit in people who were coincidentally taking nonsteroidals for other indications.”
The trial’s results should be generalizable to similar high-risk patients in less sunny parts of the world, Dr. Damian said. “If their skin has shown that degree of damage to get skin cancer, then we suspect nicotinamide would offer benefits to them as well.”
Dr. Damian disclosed no relevant conflicts of interest. The study was funded by the National Health & Medical Research Council.
Nicotinamide, an inexpensive, over-the-counter form of vitamin B3, is safe and efficacious for the chemoprevention of nonmelanoma skin cancer in patients at high risk, according to data from the Australian Oral Nicotinamide to Reduce Actinic Cancer (ONTRAC) Study.
Results reported in a press briefing held before the annual meeting of the American Society of Clinical Oncology showed that patients taking nicotinamide were about one-fourth less likely than peers taking a placebo to develop new basal cell and squamous cell skin cancers. They also had a smaller reduction in new actinic keratoses.
“Nicotinamide, vitamin B3, significantly reduced nonmelanoma skin cancers and keratoses in just 12 months in a group of pretty high-risk patients. It’s safe, it’s almost obscenely inexpensive, and it’s already widely commercially available, so this one’s ready to go straight into the clinic,” commented senior investigator Dr. Diona Damian, professor of dermatology at the University of Sydney.
She cautioned that the results apply only to the population studied: adults who had experienced two or more nonmelanoma skin cancers in the past 5 years.
“These are the people we’d be recommending it for – people who have already got a skin cancer track record. It’s not something that we’d recommend at this stage for the general population,” she said. Likewise, the findings do not speak to patients at the other end of the spectrum who are in treatment for advanced or metastatic skin cancer, as they also were excluded.
That said, the researchers are planning additional studies in other populations, such patients who are at high risk because they have immunosuppression, according to Dr. Damian.
“We still need the overall skin cancer prevention strategies of sun-safe behavior, sunscreen, and regular skin surveillance,” she stressed, “but we now have an additional exciting opportunity for affordable skin cancer chemoprevention which we can instantly translate into clinical practice.”
Dr. Peter Paul Yu, ASCO President and a medical oncologist and hematologist who is director of cancer research at the Palo Alto Medical Foundation, Sunnyvale, Calif., commented, “This is a very exciting prevention trial. We all know that we clamor for preventing rather than treating diseases, and this is a major advance for us.”
Exposure to ultraviolet light packs a one-two punch to the skin, both damaging cellular DNA and suppressing the skin’s immune response, according to Dr. Damian. The investigators opted to test nicotinamide as it counters both of these events.
The 386 patients in ONTRAC had heavily sun damaged skin, with a mean of 8 nonmelanoma skin cancers in the past 5 years and 50 keratoses at baseline. They were randomized evenly to receive nicotinamide (500 mg twice daily) or placebo for 12 months.
Results showed that the average number of new nonmelanoma skin cancers per patient during the treatment period was 1.77 in the nicotinamide group and 2.42 in the placebo group. The difference translated to a 23% lower rate of new cancers with the vitamin.
“There were comparable reductions seen for both basal and squamous cell carcinomas,” Dr. Damian noted. “Interestingly, this reduction in skin cancers seemed to start as early as the first 3-month visit. And then when people stopped taking their tablets after 12 months, the benefit was no longer seen. In other words, you need to continue taking the tablets in order for them to be effective.”
The nicotinamide group also had a roughly 15% lower rate of new actinic keratoses, compared with the placebo group.
“Nicotinamide was very well tolerated. There was no difference in adverse events, blood parameters, or blood pressure in the two arms” of the study, reported Dr. Damian. She stressed that it is critically important to distinguish nicotinamide from niacin (nicotinic acid), another form of vitamin B3 that has a host of side effects such as headache and flushing.
“One of the great things about [nicotinamide] is that it really has hardly any drug interactions, which means that elderly patients who may be taking a whole cocktail of medications for their heart disease and their hypertension, and whatever else, the nicotinamide won’t interact with those,” she added.
Some evidence also has shown nonsteroidal anti-inflammatory drugs to reduce the risk of skin cancer. “The advantage of nicotinamide is that it doesn’t have the potential gastrointestinal bleeding or renal side effects of nonsteroidals, so it may be suitable for a group of people who aren’t suitable for taking nonsteroidals,” she said. “In our ONTRAC study, we didn’t find synergy or additional benefit in people who were coincidentally taking nonsteroidals for other indications.”
The trial’s results should be generalizable to similar high-risk patients in less sunny parts of the world, Dr. Damian said. “If their skin has shown that degree of damage to get skin cancer, then we suspect nicotinamide would offer benefits to them as well.”
Dr. Damian disclosed no relevant conflicts of interest. The study was funded by the National Health & Medical Research Council.
Nicotinamide, an inexpensive, over-the-counter form of vitamin B3, is safe and efficacious for the chemoprevention of nonmelanoma skin cancer in patients at high risk, according to data from the Australian Oral Nicotinamide to Reduce Actinic Cancer (ONTRAC) Study.
Results reported in a press briefing held before the annual meeting of the American Society of Clinical Oncology showed that patients taking nicotinamide were about one-fourth less likely than peers taking a placebo to develop new basal cell and squamous cell skin cancers. They also had a smaller reduction in new actinic keratoses.
“Nicotinamide, vitamin B3, significantly reduced nonmelanoma skin cancers and keratoses in just 12 months in a group of pretty high-risk patients. It’s safe, it’s almost obscenely inexpensive, and it’s already widely commercially available, so this one’s ready to go straight into the clinic,” commented senior investigator Dr. Diona Damian, professor of dermatology at the University of Sydney.
She cautioned that the results apply only to the population studied: adults who had experienced two or more nonmelanoma skin cancers in the past 5 years.
“These are the people we’d be recommending it for – people who have already got a skin cancer track record. It’s not something that we’d recommend at this stage for the general population,” she said. Likewise, the findings do not speak to patients at the other end of the spectrum who are in treatment for advanced or metastatic skin cancer, as they also were excluded.
That said, the researchers are planning additional studies in other populations, such patients who are at high risk because they have immunosuppression, according to Dr. Damian.
“We still need the overall skin cancer prevention strategies of sun-safe behavior, sunscreen, and regular skin surveillance,” she stressed, “but we now have an additional exciting opportunity for affordable skin cancer chemoprevention which we can instantly translate into clinical practice.”
Dr. Peter Paul Yu, ASCO President and a medical oncologist and hematologist who is director of cancer research at the Palo Alto Medical Foundation, Sunnyvale, Calif., commented, “This is a very exciting prevention trial. We all know that we clamor for preventing rather than treating diseases, and this is a major advance for us.”
Exposure to ultraviolet light packs a one-two punch to the skin, both damaging cellular DNA and suppressing the skin’s immune response, according to Dr. Damian. The investigators opted to test nicotinamide as it counters both of these events.
The 386 patients in ONTRAC had heavily sun damaged skin, with a mean of 8 nonmelanoma skin cancers in the past 5 years and 50 keratoses at baseline. They were randomized evenly to receive nicotinamide (500 mg twice daily) or placebo for 12 months.
Results showed that the average number of new nonmelanoma skin cancers per patient during the treatment period was 1.77 in the nicotinamide group and 2.42 in the placebo group. The difference translated to a 23% lower rate of new cancers with the vitamin.
“There were comparable reductions seen for both basal and squamous cell carcinomas,” Dr. Damian noted. “Interestingly, this reduction in skin cancers seemed to start as early as the first 3-month visit. And then when people stopped taking their tablets after 12 months, the benefit was no longer seen. In other words, you need to continue taking the tablets in order for them to be effective.”
The nicotinamide group also had a roughly 15% lower rate of new actinic keratoses, compared with the placebo group.
“Nicotinamide was very well tolerated. There was no difference in adverse events, blood parameters, or blood pressure in the two arms” of the study, reported Dr. Damian. She stressed that it is critically important to distinguish nicotinamide from niacin (nicotinic acid), another form of vitamin B3 that has a host of side effects such as headache and flushing.
“One of the great things about [nicotinamide] is that it really has hardly any drug interactions, which means that elderly patients who may be taking a whole cocktail of medications for their heart disease and their hypertension, and whatever else, the nicotinamide won’t interact with those,” she added.
Some evidence also has shown nonsteroidal anti-inflammatory drugs to reduce the risk of skin cancer. “The advantage of nicotinamide is that it doesn’t have the potential gastrointestinal bleeding or renal side effects of nonsteroidals, so it may be suitable for a group of people who aren’t suitable for taking nonsteroidals,” she said. “In our ONTRAC study, we didn’t find synergy or additional benefit in people who were coincidentally taking nonsteroidals for other indications.”
The trial’s results should be generalizable to similar high-risk patients in less sunny parts of the world, Dr. Damian said. “If their skin has shown that degree of damage to get skin cancer, then we suspect nicotinamide would offer benefits to them as well.”
Dr. Damian disclosed no relevant conflicts of interest. The study was funded by the National Health & Medical Research Council.
FROM THE ASCO 2015 PRESSCAST
Key clinical point: Nicotinamide, an inexpensive oral vitamin, protects against nonmelanoma skin cancer in patients at high risk.
Major finding: Patients taking nicotinamide had a 23% lower rate of new basal cell and squamous cell carcinomas.
Data source: A randomized, placebo-controlled phase III trial among 386 patients with past nonmelanoma skin cancers.
Disclosures: Dr. Damian disclosed no relevant conflicts of interest. The study was funded by the National Health & Medical Research Council.
Intralesional Vinblastine Injections for Treatment of Classic Kaposi Sarcoma in Diabetic Patients
Kaposi sarcoma (KS) is caused by infection with human herpesvirus 8, a DNA virus and member of the Gammaherpesvirinae subfamily.1,2 Cutaneous KS lesions generally appear as red to purple macules, plaques, and/or nodules that can become ulcerated, causing pain and bleeding. Lesions often are localized on the feet, which may impede walking and daily activities. Early diagnosis and management are important to avoid or minimize severe symptoms (eg, enlargement of lesions, ulceration with bleeding and pain). Injection therapy is recommended for the management of localized disease, while systemic therapy is appropriate for disseminated malignancy.3 There currently is no curative therapy available for KS; however, palliative therapy can avoid or reduce cosmetically unacceptable lesions and painful edema.
Vinblastine (VNB) is a natural vinca alkaloid whose primary cytotoxic effect is prevention of mitotic activity.4 However, antiangiogenetic activity has been demonstrated with low concentrations of VNB.5 The use of intralesional VNB injections for the management of oral KS in open-label studies4 using different doses and multiple courses has proven to be therapeutic.6-8 There are limited studies regarding the use of intralesional VNB injections in patients with cutaneous KS, particularly in those with associated type 2 diabetes mellitus.6-8 Thus, we evaluated the clinical efficacy of intralesional VNB injections in the treatment of KS lesions in diabetic patients with no visceral involvement.
Methods
This study was conducted in the department of dermatology from January 1, 2009, to December 31, 2011, and was approved by the institutional review board of the Fondazione IRCCS Policlinico San Matteo (Pavia, Italy). Six patients with type 2 diabetes mellitus and histologically diagnosed cutaneous KS were enrolled in the study. Inclusion criteria included histological diagnosis of KS, negative human immunodeficiency virus testing, history of type 2 diabetes mellitus, more than 5 cutaneous KS lesions, and no visceral involvement (confirmed by colonoscopy, gastroscopy, lymph node and abdominal echography, and chest radiograph). Informed written consent was obtained from all participants.
Treated plaques and nodules measured more than 1 to 2 cm in diameter. Intralesional vinblastine injections were administered, not perilesional, with a standard concentration of 0.5 mg/mL per 1-cm2 lesion (maximum dose, 2 mg daily). All participants received 1 injection per nodule and a maximum of 2 injections per plaque. Monitoring of the target lesions was done via photography and mapping. Clinical evaluation of the treated lesions and assessment of side effects was conducted at 1-week and 1-, 3-, and 6-month follow-up. Complete response was defined as 95% to 100% remission of the target lesions, partial response was defined as 50% to 95% remission, and minimal response was defined as less than 50% remission, all at 3-month follow-up. No response was defined as no change or increase in size of the target lesions. Side effects including pain, hyperpigmentation, and ulcer formation were evaluated.
Results
A total of 6 participants (5 men, 1 woman; age range, 66–82 years) completed the study. Baseline demographics and clinical characteristics of the participants are reported in Table 1. Three participants were currently undergoing diabetic therapy with metformin, 2 were receiving insulin injections, and 1 was following a diabetic diet. None of the participants had internal KS. All 5 participants with nodular KS lesions previously underwent surgical excision. One participant also underwent radiotherapy with a good initial outcome, but lesions recurred at the same site within 1 year.
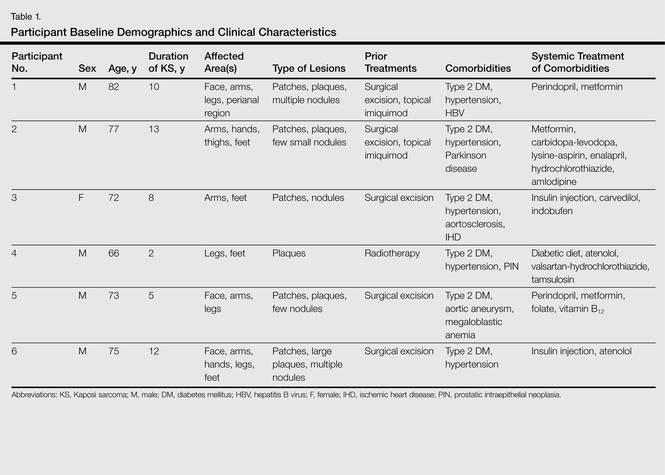
Complete response of nodular KS lesions and partial response of plaques to intralesional VNB injections was observed at 3-month follow-up in all treated lesions (Table 2). Five participants achieved a greater than 50% reduction (overall decrease in size and flattening of the plaque) of the KS plaque lesions. No participants showed minimal or no response.
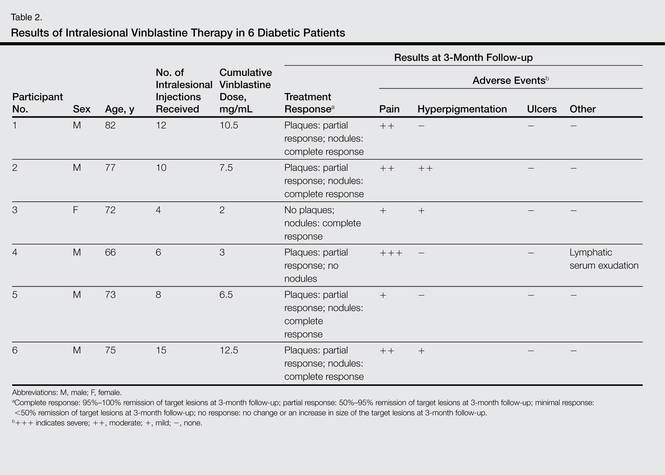
At 1-week follow-up, shrinkage of nodular lesions was evident with necrotic crusts. Participant 4 who presented with plaques under the feet developed lymphatic serum exudation in the first 3 days following treatment on the legs. All the treated nodular lesions resolved completely within 3 months after treatment (Figures 1 and 2), though some new lesions appeared in new locations. All the treated plaques showed partial response to treatment after 1- to 3-month follow-up (Figure 3).
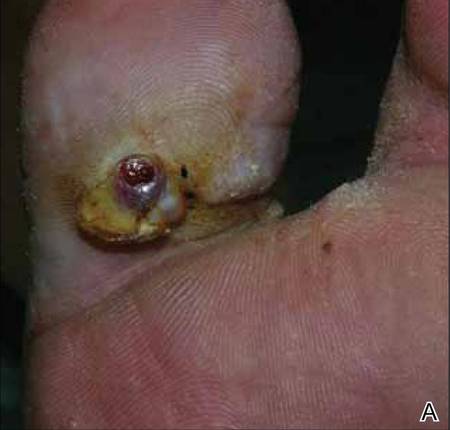
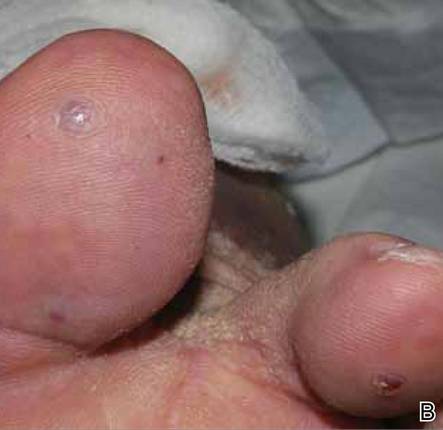
|
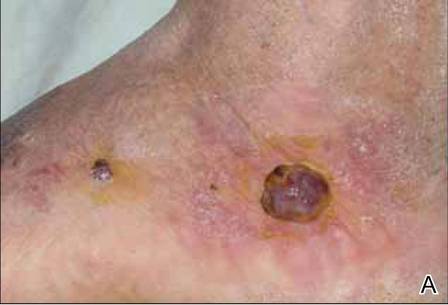
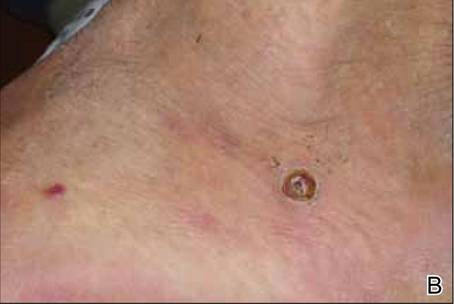
|
|
All participants reported pain on the second and third days posttreatment, but analgesics were only prescribed to participants 2 and 4. Hyperpigmentation developed after 3 to 6 months in 3 participants. Two participants developed small ulcers within 2 weeks following treatment that healed within 3 months.
Comment
Classic KS is characterized by a localized onset with slow progression of systemic involvement. Cutaneous lesions progress from mild to severe.9 Systemic therapeutic strategies currently employed for the management of KS include single-agent and multiagent cytotoxic chemotherapy.10 Because these drugs have been associated with remarkable systemic side effects and patients often are elderly individuals, systemic therapy may be unsuitable for long-term use. In an epidemiologic study of 37 patients with KS, 12 patients (32%) were found to have concurrent diabetes mellitus.11 Some authors suggest that KS in diabetic patients is a subtype of KS because of the immunosuppressive state caused by diabetes.12-15
Some of the reported therapies for lesions of classic KS include cryotherapy, surgical excision, radiation therapy, alitretinoin gel 0.1%, and infrared photocoagulation.3-16 Cryotherapy of a small papular lesion requires a 30- to 60-second freeze time that usually needs to be followed by further cryotherapy applications and is not known to be effective in lesions larger than 1 cm.17 Surgical procedures in the management of cutaneous KS may cause discomfort and bleeding and also are expensive.18 Radiotherapy is an alternative therapy, especially in plaque lesions.19 Alitretinoin gel 0.1% has been shown to be effective and well tolerated for the treatment of cutaneous lesions in patients with AIDS-related KS. Although Morganroth20 reported alitretinoin gel 0.1% as a promising therapy for control of classic KS cutaneous lesions, Rongioletti et al21 described failure of this topical treatment for classic KS. The efficacy of topical alitretinoin in classic KS needs to be confirmed by further studies. Short pulses of the infrared coagulator have been reported for the treatment of 10 cutaneous AIDS-related KS lesions in 7 patients. The infrared coagulator may be a useful addition in the palliative cosmetic treatment of KS, producing acceptable results in small (ie, <2 cm in diameter) lesions of the arms and trunk but not in those situated on the legs, as in our patients.16
The use of VNB in classic KS was pioneered in 1980 by Klein et al22 who described 14 patients with KS who were systemically treated with VNB sulfate at a low dose with excellent therapeutic results leading to regression of cutaneous lesions. In this study, intravenous VNB therapy was supplemented with intralesional VNB.22 In other reports, intralesional VNB injections have proved to be effective in reducing lesion size and symptoms in oral KS with mild adverse effects.19-29 For this reason, we decided to employ intralesional VNB injections in nodules and plaques of KS patients with diabetes. After treatment, lesion size was reduced in all 6 participants, with 4 participants achieving more than 50% reduction of the lesion size. In other studies reporting oral lesions,3 the highest proportion of complete responses was achieved with injections done periodically until evidence of clinical resolution was obtained, with a mean number of 2.4 injections per patient. In contrast, another study reported complete response of 48% in patients who received a single dose of VNB,7 as in our study.
Minimal complications following intralesional VNB injections have been reported. In our study, moderate pain following VNB injection was common during the first week posttreatment but typically resolved partially or completely during the following weeks. Additionally, pain described as needlelike during the procedure was minimal except for 1 participant who reported severe pain a day after the treatment. Unlike the study by Smith et al,30 lidocaine with epinephrine was not used in our study as a pretreatment.
At 1-month follow-up, clinical evaluation of the injection site revealed ulceration in 2 participants, but no ulcers were noted at 3-month follow-up. To avoid this potentially dangerous complication, which can involve the foot in diabetic patients, further studies of a larger sample of patients using a lower dose of VNB are needed.
In our small group of diabetic patients, intralesional injections of VNB proved to be effective in the management of cutaneous lesions of classic KS with minimal adverse side effects. Even in cases where only one intralesional injection was administered, improvement was observed in all treated lesions. Imiquimod also has been proposed for the treatment of KS. Célestin Schartz et al31 conducted a study of 17 patients who were treated with imiquimod cream applied under occlusion 3 times weekly for 24 weeks. Eight patients had an objective clinical response (2 complete and 6 partial responses), and tumor progression was noted in 6 patients.31 This treatment appears to be suitable for new small skin lesions in immunocompetent patients; however, no known comparative studies have been conducted, possibly because of the relatively high cost of treatment due to the chronic nature of the disease. Intralesional bleomycin also has been reported as useful in the treatment of cutaneous KS. The best results were obtained with macular lesions.32 However, the risk for necrosis is high. Therefore, bleomycin is not the first choice for diabetics, particularly those who have lesions localized on legs.
Conclusion
Standard treatments of KS lesions such as radiotherapy, surgery, and chemotherapy are associated with more severe adverse events, especially in diabetic patients, as well as higher costs due to the use of more sophisticated techniques and equipment. Because KS is a multicentric disease, local therapy should not be expected to prevent the development of new lesions on other sites of the skin or internal disease. The best candidates for localized therapy are patients affected by nodular classic KS without visceral involvement, with fewer than 10 lesions on acral sites.
1. Cathomas G. Human herpes virus 8: a new virus discloses its face. Virchows Arch. 2000;436:195-206.
2. Chang Y, Cesarman CE, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865-1869.
3. Tur E, Brenner S. Treatment of Kaposi’s sarcoma. Arch Dermatol. 1996;132:327-331.
4. Gorsky M, Epstein JB. A case series of acquired immunodeficiency syndrome patients with initial neoplastic diagnoses of intraoral Kaposi’s sarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:612-617.
5. Chaplin DJ. The effect of therapy on tumor vascular function. Int J Radiat Biol. 1991;60:311-325.
6. Donehower RC, Rowinsky EK. Anticancer drugs derived from plants. In: De Vita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 4th ed. Philadelphia, PA: Lippincott; 1993:409-417.
7. Epstein JB. Treatment of oral Kaposi’s sarcoma with intralesional vinblastine. Cancer. 1993;71:1722-1725.
8. McCormick SU. Intralesional vinblastine injections for the treatment of oral Kaposi’s sarcoma: report of 10 patients with 2-year follow-up. J Oral Maxillofac Surg. 1996;54:583-587.
9. Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
10. Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
11. Laor Y, Schwartz RA. Epidemiologic aspects of American Kaposi’s sarcoma. J Surg Oncol. 1979;12:299-303.
12. Gill K, Shah J. Kaposi sarcoma in patients with diabetes and wounds. Adv Skin Wound Care. 2006;19:196-198, 201.
13. Hurlbut W, Lincoln CS Jr. Multiple hemorrhagic sarcoma and diabetes mellitus; review of a series, with report of two cases. Arch Intern Med (Chic). 1949;84:738-750.
14. Fischer JW, Cohen DM. Simultaneous occurrence of Kaposi’s sarcoma, leukemia and diabetes mellitus; report of a case. Am J Clin Pathol. 1951;21:586-589.
15. Tankel R. Kaposi’s sarcoma and diabetes mellitus. Arch Dermatol. 1971;104:442-444.
16. Langtry JA, Bottomley DM, Phillips RH, et al. The infra-red coagulator in the treatment of AIDS-related Kaposi’s sarcoma and a comparison with radiotherapy. Clin Exp Dermatol. 1994;19:23-25.
17. Von Roenn JH, Cianfrocca M. Treatment of Kaposi’s sarcoma. Cancer Treat Res. 2001;104:127-148.
18. Dezube BJ. AIDS-related Kaposi sarcoma. the role of local therapy for a systemic disease. Arch Dermatol. 2000;136:1554-1556.
19. Oysul K, Beyzadeoglu M, Surenkok S, et al. A dose-response analysis for classical Kaposi’s sarcoma management by radiotherapy. Saudi Med J. 2008;29:837-840.
20. Morganroth GS. Topical 0.1% alitretinoin gel for classic Kaposi sarcoma. Arch Dermatol. 2002;138:542-543.
21. Rongioletti F, Zaccaria E, Viglizzo G. Failure of topical 0.1% alitretinoin gel for classic Kaposi sarcoma: first European experience. Br J Dermatol. 2006;155:856-857.
22. Klein E, Schwartz RA, Laor Y, et al. Treatment of Kaposi’s sarcoma with vinblastine. Cancer. 1980;45:427-431.
23. Flaitz CM, Nichols CM, Hicks MJ. Role of intralesional vinblastine administration in treatment of intraoral Kaposi’s sarcoma in AIDS. Eur J Cancer B Oral Oncol. 1995;31B:280-285.
24. Epstein JB, Lozada-Nur F, McLeod WA, et al. Oral Kaposi’s sarcoma in acquired immunodeficiency syndrome. review of management and report of the efficacy of intralesional vinblastine. Cancer. 1989;64:2424-2430.
25. Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342:1027-1038.
26. Ramírez-Amador V, Esquivel-Pedraza L, Lozada-Nur F, et al. Intralesional vinblastine vs. 3% sodium tetradecyl sulfate for the treatment of oral Kaposi’s sarcoma. a double blind, randomized clinical trial. Oral Oncol. 2002;38:460-467.
27. Boudreaux AA, Smith LL, Cosby CD, et al. Intralesional vinblastine for cutaneous Kaposi’s sarcoma associated with acquired immunodeficiency syndrome. a clinical trial to evaluate efficacy and discomfort associated with infection. J Am Acad Dermatol. 1993;28:61-65.
28. Odom RB, Goette DK. Treatment of cutaneous Kaposi’s sarcoma with intralesional vincristine. Arch Dermatol. 1978;114:1693-1694.
29. Tappero JW, Conant MA, Wolfe SF, et al. Kaposi’s sarcoma. epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
30. Smith KJ, Skelton HG, Turiansky G, et al. Hyaluronidase enhances the therapeutic effect of vinblastine in intralesional treatment of Kaposi’s sarcoma. Military Medical Consortium for the Advancement of Retroviral Research (MMCARR). J Am Acad Dermatol. 1997;36(2, pt 1):239-242.
31. Célestin Schartz NE, Chevret S, Paz C, et al. Imiquimod 5% cream for treatment of HIV-negative Kaposi’s sarcoma skin lesions: a phase I to II, open-label trial in 17 patients [published online ahead of print February 20, 2008]. J Am Acad Dermatol. 2008;58:585-591.
32. Poignonec S, Lachiver LD, Lamas G, et al. Intralesional bleomycin for acquired immunodeficiency syndrome-associated cutaneous Kaposi’s sarcoma. Arch Dermatol. 1995;131:228.
Kaposi sarcoma (KS) is caused by infection with human herpesvirus 8, a DNA virus and member of the Gammaherpesvirinae subfamily.1,2 Cutaneous KS lesions generally appear as red to purple macules, plaques, and/or nodules that can become ulcerated, causing pain and bleeding. Lesions often are localized on the feet, which may impede walking and daily activities. Early diagnosis and management are important to avoid or minimize severe symptoms (eg, enlargement of lesions, ulceration with bleeding and pain). Injection therapy is recommended for the management of localized disease, while systemic therapy is appropriate for disseminated malignancy.3 There currently is no curative therapy available for KS; however, palliative therapy can avoid or reduce cosmetically unacceptable lesions and painful edema.
Vinblastine (VNB) is a natural vinca alkaloid whose primary cytotoxic effect is prevention of mitotic activity.4 However, antiangiogenetic activity has been demonstrated with low concentrations of VNB.5 The use of intralesional VNB injections for the management of oral KS in open-label studies4 using different doses and multiple courses has proven to be therapeutic.6-8 There are limited studies regarding the use of intralesional VNB injections in patients with cutaneous KS, particularly in those with associated type 2 diabetes mellitus.6-8 Thus, we evaluated the clinical efficacy of intralesional VNB injections in the treatment of KS lesions in diabetic patients with no visceral involvement.
Methods
This study was conducted in the department of dermatology from January 1, 2009, to December 31, 2011, and was approved by the institutional review board of the Fondazione IRCCS Policlinico San Matteo (Pavia, Italy). Six patients with type 2 diabetes mellitus and histologically diagnosed cutaneous KS were enrolled in the study. Inclusion criteria included histological diagnosis of KS, negative human immunodeficiency virus testing, history of type 2 diabetes mellitus, more than 5 cutaneous KS lesions, and no visceral involvement (confirmed by colonoscopy, gastroscopy, lymph node and abdominal echography, and chest radiograph). Informed written consent was obtained from all participants.
Treated plaques and nodules measured more than 1 to 2 cm in diameter. Intralesional vinblastine injections were administered, not perilesional, with a standard concentration of 0.5 mg/mL per 1-cm2 lesion (maximum dose, 2 mg daily). All participants received 1 injection per nodule and a maximum of 2 injections per plaque. Monitoring of the target lesions was done via photography and mapping. Clinical evaluation of the treated lesions and assessment of side effects was conducted at 1-week and 1-, 3-, and 6-month follow-up. Complete response was defined as 95% to 100% remission of the target lesions, partial response was defined as 50% to 95% remission, and minimal response was defined as less than 50% remission, all at 3-month follow-up. No response was defined as no change or increase in size of the target lesions. Side effects including pain, hyperpigmentation, and ulcer formation were evaluated.
Results
A total of 6 participants (5 men, 1 woman; age range, 66–82 years) completed the study. Baseline demographics and clinical characteristics of the participants are reported in Table 1. Three participants were currently undergoing diabetic therapy with metformin, 2 were receiving insulin injections, and 1 was following a diabetic diet. None of the participants had internal KS. All 5 participants with nodular KS lesions previously underwent surgical excision. One participant also underwent radiotherapy with a good initial outcome, but lesions recurred at the same site within 1 year.
Complete response of nodular KS lesions and partial response of plaques to intralesional VNB injections was observed at 3-month follow-up in all treated lesions (Table 2). Five participants achieved a greater than 50% reduction (overall decrease in size and flattening of the plaque) of the KS plaque lesions. No participants showed minimal or no response.
At 1-week follow-up, shrinkage of nodular lesions was evident with necrotic crusts. Participant 4 who presented with plaques under the feet developed lymphatic serum exudation in the first 3 days following treatment on the legs. All the treated nodular lesions resolved completely within 3 months after treatment (Figures 1 and 2), though some new lesions appeared in new locations. All the treated plaques showed partial response to treatment after 1- to 3-month follow-up (Figure 3).
|
|
|
|
|
|
All participants reported pain on the second and third days posttreatment, but analgesics were only prescribed to participants 2 and 4. Hyperpigmentation developed after 3 to 6 months in 3 participants. Two participants developed small ulcers within 2 weeks following treatment that healed within 3 months.
Comment
Classic KS is characterized by a localized onset with slow progression of systemic involvement. Cutaneous lesions progress from mild to severe.9 Systemic therapeutic strategies currently employed for the management of KS include single-agent and multiagent cytotoxic chemotherapy.10 Because these drugs have been associated with remarkable systemic side effects and patients often are elderly individuals, systemic therapy may be unsuitable for long-term use. In an epidemiologic study of 37 patients with KS, 12 patients (32%) were found to have concurrent diabetes mellitus.11 Some authors suggest that KS in diabetic patients is a subtype of KS because of the immunosuppressive state caused by diabetes.12-15
Some of the reported therapies for lesions of classic KS include cryotherapy, surgical excision, radiation therapy, alitretinoin gel 0.1%, and infrared photocoagulation.3-16 Cryotherapy of a small papular lesion requires a 30- to 60-second freeze time that usually needs to be followed by further cryotherapy applications and is not known to be effective in lesions larger than 1 cm.17 Surgical procedures in the management of cutaneous KS may cause discomfort and bleeding and also are expensive.18 Radiotherapy is an alternative therapy, especially in plaque lesions.19 Alitretinoin gel 0.1% has been shown to be effective and well tolerated for the treatment of cutaneous lesions in patients with AIDS-related KS. Although Morganroth20 reported alitretinoin gel 0.1% as a promising therapy for control of classic KS cutaneous lesions, Rongioletti et al21 described failure of this topical treatment for classic KS. The efficacy of topical alitretinoin in classic KS needs to be confirmed by further studies. Short pulses of the infrared coagulator have been reported for the treatment of 10 cutaneous AIDS-related KS lesions in 7 patients. The infrared coagulator may be a useful addition in the palliative cosmetic treatment of KS, producing acceptable results in small (ie, <2 cm in diameter) lesions of the arms and trunk but not in those situated on the legs, as in our patients.16
The use of VNB in classic KS was pioneered in 1980 by Klein et al22 who described 14 patients with KS who were systemically treated with VNB sulfate at a low dose with excellent therapeutic results leading to regression of cutaneous lesions. In this study, intravenous VNB therapy was supplemented with intralesional VNB.22 In other reports, intralesional VNB injections have proved to be effective in reducing lesion size and symptoms in oral KS with mild adverse effects.19-29 For this reason, we decided to employ intralesional VNB injections in nodules and plaques of KS patients with diabetes. After treatment, lesion size was reduced in all 6 participants, with 4 participants achieving more than 50% reduction of the lesion size. In other studies reporting oral lesions,3 the highest proportion of complete responses was achieved with injections done periodically until evidence of clinical resolution was obtained, with a mean number of 2.4 injections per patient. In contrast, another study reported complete response of 48% in patients who received a single dose of VNB,7 as in our study.
Minimal complications following intralesional VNB injections have been reported. In our study, moderate pain following VNB injection was common during the first week posttreatment but typically resolved partially or completely during the following weeks. Additionally, pain described as needlelike during the procedure was minimal except for 1 participant who reported severe pain a day after the treatment. Unlike the study by Smith et al,30 lidocaine with epinephrine was not used in our study as a pretreatment.
At 1-month follow-up, clinical evaluation of the injection site revealed ulceration in 2 participants, but no ulcers were noted at 3-month follow-up. To avoid this potentially dangerous complication, which can involve the foot in diabetic patients, further studies of a larger sample of patients using a lower dose of VNB are needed.
In our small group of diabetic patients, intralesional injections of VNB proved to be effective in the management of cutaneous lesions of classic KS with minimal adverse side effects. Even in cases where only one intralesional injection was administered, improvement was observed in all treated lesions. Imiquimod also has been proposed for the treatment of KS. Célestin Schartz et al31 conducted a study of 17 patients who were treated with imiquimod cream applied under occlusion 3 times weekly for 24 weeks. Eight patients had an objective clinical response (2 complete and 6 partial responses), and tumor progression was noted in 6 patients.31 This treatment appears to be suitable for new small skin lesions in immunocompetent patients; however, no known comparative studies have been conducted, possibly because of the relatively high cost of treatment due to the chronic nature of the disease. Intralesional bleomycin also has been reported as useful in the treatment of cutaneous KS. The best results were obtained with macular lesions.32 However, the risk for necrosis is high. Therefore, bleomycin is not the first choice for diabetics, particularly those who have lesions localized on legs.
Conclusion
Standard treatments of KS lesions such as radiotherapy, surgery, and chemotherapy are associated with more severe adverse events, especially in diabetic patients, as well as higher costs due to the use of more sophisticated techniques and equipment. Because KS is a multicentric disease, local therapy should not be expected to prevent the development of new lesions on other sites of the skin or internal disease. The best candidates for localized therapy are patients affected by nodular classic KS without visceral involvement, with fewer than 10 lesions on acral sites.
Kaposi sarcoma (KS) is caused by infection with human herpesvirus 8, a DNA virus and member of the Gammaherpesvirinae subfamily.1,2 Cutaneous KS lesions generally appear as red to purple macules, plaques, and/or nodules that can become ulcerated, causing pain and bleeding. Lesions often are localized on the feet, which may impede walking and daily activities. Early diagnosis and management are important to avoid or minimize severe symptoms (eg, enlargement of lesions, ulceration with bleeding and pain). Injection therapy is recommended for the management of localized disease, while systemic therapy is appropriate for disseminated malignancy.3 There currently is no curative therapy available for KS; however, palliative therapy can avoid or reduce cosmetically unacceptable lesions and painful edema.
Vinblastine (VNB) is a natural vinca alkaloid whose primary cytotoxic effect is prevention of mitotic activity.4 However, antiangiogenetic activity has been demonstrated with low concentrations of VNB.5 The use of intralesional VNB injections for the management of oral KS in open-label studies4 using different doses and multiple courses has proven to be therapeutic.6-8 There are limited studies regarding the use of intralesional VNB injections in patients with cutaneous KS, particularly in those with associated type 2 diabetes mellitus.6-8 Thus, we evaluated the clinical efficacy of intralesional VNB injections in the treatment of KS lesions in diabetic patients with no visceral involvement.
Methods
This study was conducted in the department of dermatology from January 1, 2009, to December 31, 2011, and was approved by the institutional review board of the Fondazione IRCCS Policlinico San Matteo (Pavia, Italy). Six patients with type 2 diabetes mellitus and histologically diagnosed cutaneous KS were enrolled in the study. Inclusion criteria included histological diagnosis of KS, negative human immunodeficiency virus testing, history of type 2 diabetes mellitus, more than 5 cutaneous KS lesions, and no visceral involvement (confirmed by colonoscopy, gastroscopy, lymph node and abdominal echography, and chest radiograph). Informed written consent was obtained from all participants.
Treated plaques and nodules measured more than 1 to 2 cm in diameter. Intralesional vinblastine injections were administered, not perilesional, with a standard concentration of 0.5 mg/mL per 1-cm2 lesion (maximum dose, 2 mg daily). All participants received 1 injection per nodule and a maximum of 2 injections per plaque. Monitoring of the target lesions was done via photography and mapping. Clinical evaluation of the treated lesions and assessment of side effects was conducted at 1-week and 1-, 3-, and 6-month follow-up. Complete response was defined as 95% to 100% remission of the target lesions, partial response was defined as 50% to 95% remission, and minimal response was defined as less than 50% remission, all at 3-month follow-up. No response was defined as no change or increase in size of the target lesions. Side effects including pain, hyperpigmentation, and ulcer formation were evaluated.
Results
A total of 6 participants (5 men, 1 woman; age range, 66–82 years) completed the study. Baseline demographics and clinical characteristics of the participants are reported in Table 1. Three participants were currently undergoing diabetic therapy with metformin, 2 were receiving insulin injections, and 1 was following a diabetic diet. None of the participants had internal KS. All 5 participants with nodular KS lesions previously underwent surgical excision. One participant also underwent radiotherapy with a good initial outcome, but lesions recurred at the same site within 1 year.
Complete response of nodular KS lesions and partial response of plaques to intralesional VNB injections was observed at 3-month follow-up in all treated lesions (Table 2). Five participants achieved a greater than 50% reduction (overall decrease in size and flattening of the plaque) of the KS plaque lesions. No participants showed minimal or no response.
At 1-week follow-up, shrinkage of nodular lesions was evident with necrotic crusts. Participant 4 who presented with plaques under the feet developed lymphatic serum exudation in the first 3 days following treatment on the legs. All the treated nodular lesions resolved completely within 3 months after treatment (Figures 1 and 2), though some new lesions appeared in new locations. All the treated plaques showed partial response to treatment after 1- to 3-month follow-up (Figure 3).
|
|
|
|
|
|
All participants reported pain on the second and third days posttreatment, but analgesics were only prescribed to participants 2 and 4. Hyperpigmentation developed after 3 to 6 months in 3 participants. Two participants developed small ulcers within 2 weeks following treatment that healed within 3 months.
Comment
Classic KS is characterized by a localized onset with slow progression of systemic involvement. Cutaneous lesions progress from mild to severe.9 Systemic therapeutic strategies currently employed for the management of KS include single-agent and multiagent cytotoxic chemotherapy.10 Because these drugs have been associated with remarkable systemic side effects and patients often are elderly individuals, systemic therapy may be unsuitable for long-term use. In an epidemiologic study of 37 patients with KS, 12 patients (32%) were found to have concurrent diabetes mellitus.11 Some authors suggest that KS in diabetic patients is a subtype of KS because of the immunosuppressive state caused by diabetes.12-15
Some of the reported therapies for lesions of classic KS include cryotherapy, surgical excision, radiation therapy, alitretinoin gel 0.1%, and infrared photocoagulation.3-16 Cryotherapy of a small papular lesion requires a 30- to 60-second freeze time that usually needs to be followed by further cryotherapy applications and is not known to be effective in lesions larger than 1 cm.17 Surgical procedures in the management of cutaneous KS may cause discomfort and bleeding and also are expensive.18 Radiotherapy is an alternative therapy, especially in plaque lesions.19 Alitretinoin gel 0.1% has been shown to be effective and well tolerated for the treatment of cutaneous lesions in patients with AIDS-related KS. Although Morganroth20 reported alitretinoin gel 0.1% as a promising therapy for control of classic KS cutaneous lesions, Rongioletti et al21 described failure of this topical treatment for classic KS. The efficacy of topical alitretinoin in classic KS needs to be confirmed by further studies. Short pulses of the infrared coagulator have been reported for the treatment of 10 cutaneous AIDS-related KS lesions in 7 patients. The infrared coagulator may be a useful addition in the palliative cosmetic treatment of KS, producing acceptable results in small (ie, <2 cm in diameter) lesions of the arms and trunk but not in those situated on the legs, as in our patients.16
The use of VNB in classic KS was pioneered in 1980 by Klein et al22 who described 14 patients with KS who were systemically treated with VNB sulfate at a low dose with excellent therapeutic results leading to regression of cutaneous lesions. In this study, intravenous VNB therapy was supplemented with intralesional VNB.22 In other reports, intralesional VNB injections have proved to be effective in reducing lesion size and symptoms in oral KS with mild adverse effects.19-29 For this reason, we decided to employ intralesional VNB injections in nodules and plaques of KS patients with diabetes. After treatment, lesion size was reduced in all 6 participants, with 4 participants achieving more than 50% reduction of the lesion size. In other studies reporting oral lesions,3 the highest proportion of complete responses was achieved with injections done periodically until evidence of clinical resolution was obtained, with a mean number of 2.4 injections per patient. In contrast, another study reported complete response of 48% in patients who received a single dose of VNB,7 as in our study.
Minimal complications following intralesional VNB injections have been reported. In our study, moderate pain following VNB injection was common during the first week posttreatment but typically resolved partially or completely during the following weeks. Additionally, pain described as needlelike during the procedure was minimal except for 1 participant who reported severe pain a day after the treatment. Unlike the study by Smith et al,30 lidocaine with epinephrine was not used in our study as a pretreatment.
At 1-month follow-up, clinical evaluation of the injection site revealed ulceration in 2 participants, but no ulcers were noted at 3-month follow-up. To avoid this potentially dangerous complication, which can involve the foot in diabetic patients, further studies of a larger sample of patients using a lower dose of VNB are needed.
In our small group of diabetic patients, intralesional injections of VNB proved to be effective in the management of cutaneous lesions of classic KS with minimal adverse side effects. Even in cases where only one intralesional injection was administered, improvement was observed in all treated lesions. Imiquimod also has been proposed for the treatment of KS. Célestin Schartz et al31 conducted a study of 17 patients who were treated with imiquimod cream applied under occlusion 3 times weekly for 24 weeks. Eight patients had an objective clinical response (2 complete and 6 partial responses), and tumor progression was noted in 6 patients.31 This treatment appears to be suitable for new small skin lesions in immunocompetent patients; however, no known comparative studies have been conducted, possibly because of the relatively high cost of treatment due to the chronic nature of the disease. Intralesional bleomycin also has been reported as useful in the treatment of cutaneous KS. The best results were obtained with macular lesions.32 However, the risk for necrosis is high. Therefore, bleomycin is not the first choice for diabetics, particularly those who have lesions localized on legs.
Conclusion
Standard treatments of KS lesions such as radiotherapy, surgery, and chemotherapy are associated with more severe adverse events, especially in diabetic patients, as well as higher costs due to the use of more sophisticated techniques and equipment. Because KS is a multicentric disease, local therapy should not be expected to prevent the development of new lesions on other sites of the skin or internal disease. The best candidates for localized therapy are patients affected by nodular classic KS without visceral involvement, with fewer than 10 lesions on acral sites.
1. Cathomas G. Human herpes virus 8: a new virus discloses its face. Virchows Arch. 2000;436:195-206.
2. Chang Y, Cesarman CE, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865-1869.
3. Tur E, Brenner S. Treatment of Kaposi’s sarcoma. Arch Dermatol. 1996;132:327-331.
4. Gorsky M, Epstein JB. A case series of acquired immunodeficiency syndrome patients with initial neoplastic diagnoses of intraoral Kaposi’s sarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:612-617.
5. Chaplin DJ. The effect of therapy on tumor vascular function. Int J Radiat Biol. 1991;60:311-325.
6. Donehower RC, Rowinsky EK. Anticancer drugs derived from plants. In: De Vita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 4th ed. Philadelphia, PA: Lippincott; 1993:409-417.
7. Epstein JB. Treatment of oral Kaposi’s sarcoma with intralesional vinblastine. Cancer. 1993;71:1722-1725.
8. McCormick SU. Intralesional vinblastine injections for the treatment of oral Kaposi’s sarcoma: report of 10 patients with 2-year follow-up. J Oral Maxillofac Surg. 1996;54:583-587.
9. Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
10. Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
11. Laor Y, Schwartz RA. Epidemiologic aspects of American Kaposi’s sarcoma. J Surg Oncol. 1979;12:299-303.
12. Gill K, Shah J. Kaposi sarcoma in patients with diabetes and wounds. Adv Skin Wound Care. 2006;19:196-198, 201.
13. Hurlbut W, Lincoln CS Jr. Multiple hemorrhagic sarcoma and diabetes mellitus; review of a series, with report of two cases. Arch Intern Med (Chic). 1949;84:738-750.
14. Fischer JW, Cohen DM. Simultaneous occurrence of Kaposi’s sarcoma, leukemia and diabetes mellitus; report of a case. Am J Clin Pathol. 1951;21:586-589.
15. Tankel R. Kaposi’s sarcoma and diabetes mellitus. Arch Dermatol. 1971;104:442-444.
16. Langtry JA, Bottomley DM, Phillips RH, et al. The infra-red coagulator in the treatment of AIDS-related Kaposi’s sarcoma and a comparison with radiotherapy. Clin Exp Dermatol. 1994;19:23-25.
17. Von Roenn JH, Cianfrocca M. Treatment of Kaposi’s sarcoma. Cancer Treat Res. 2001;104:127-148.
18. Dezube BJ. AIDS-related Kaposi sarcoma. the role of local therapy for a systemic disease. Arch Dermatol. 2000;136:1554-1556.
19. Oysul K, Beyzadeoglu M, Surenkok S, et al. A dose-response analysis for classical Kaposi’s sarcoma management by radiotherapy. Saudi Med J. 2008;29:837-840.
20. Morganroth GS. Topical 0.1% alitretinoin gel for classic Kaposi sarcoma. Arch Dermatol. 2002;138:542-543.
21. Rongioletti F, Zaccaria E, Viglizzo G. Failure of topical 0.1% alitretinoin gel for classic Kaposi sarcoma: first European experience. Br J Dermatol. 2006;155:856-857.
22. Klein E, Schwartz RA, Laor Y, et al. Treatment of Kaposi’s sarcoma with vinblastine. Cancer. 1980;45:427-431.
23. Flaitz CM, Nichols CM, Hicks MJ. Role of intralesional vinblastine administration in treatment of intraoral Kaposi’s sarcoma in AIDS. Eur J Cancer B Oral Oncol. 1995;31B:280-285.
24. Epstein JB, Lozada-Nur F, McLeod WA, et al. Oral Kaposi’s sarcoma in acquired immunodeficiency syndrome. review of management and report of the efficacy of intralesional vinblastine. Cancer. 1989;64:2424-2430.
25. Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342:1027-1038.
26. Ramírez-Amador V, Esquivel-Pedraza L, Lozada-Nur F, et al. Intralesional vinblastine vs. 3% sodium tetradecyl sulfate for the treatment of oral Kaposi’s sarcoma. a double blind, randomized clinical trial. Oral Oncol. 2002;38:460-467.
27. Boudreaux AA, Smith LL, Cosby CD, et al. Intralesional vinblastine for cutaneous Kaposi’s sarcoma associated with acquired immunodeficiency syndrome. a clinical trial to evaluate efficacy and discomfort associated with infection. J Am Acad Dermatol. 1993;28:61-65.
28. Odom RB, Goette DK. Treatment of cutaneous Kaposi’s sarcoma with intralesional vincristine. Arch Dermatol. 1978;114:1693-1694.
29. Tappero JW, Conant MA, Wolfe SF, et al. Kaposi’s sarcoma. epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
30. Smith KJ, Skelton HG, Turiansky G, et al. Hyaluronidase enhances the therapeutic effect of vinblastine in intralesional treatment of Kaposi’s sarcoma. Military Medical Consortium for the Advancement of Retroviral Research (MMCARR). J Am Acad Dermatol. 1997;36(2, pt 1):239-242.
31. Célestin Schartz NE, Chevret S, Paz C, et al. Imiquimod 5% cream for treatment of HIV-negative Kaposi’s sarcoma skin lesions: a phase I to II, open-label trial in 17 patients [published online ahead of print February 20, 2008]. J Am Acad Dermatol. 2008;58:585-591.
32. Poignonec S, Lachiver LD, Lamas G, et al. Intralesional bleomycin for acquired immunodeficiency syndrome-associated cutaneous Kaposi’s sarcoma. Arch Dermatol. 1995;131:228.
1. Cathomas G. Human herpes virus 8: a new virus discloses its face. Virchows Arch. 2000;436:195-206.
2. Chang Y, Cesarman CE, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865-1869.
3. Tur E, Brenner S. Treatment of Kaposi’s sarcoma. Arch Dermatol. 1996;132:327-331.
4. Gorsky M, Epstein JB. A case series of acquired immunodeficiency syndrome patients with initial neoplastic diagnoses of intraoral Kaposi’s sarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:612-617.
5. Chaplin DJ. The effect of therapy on tumor vascular function. Int J Radiat Biol. 1991;60:311-325.
6. Donehower RC, Rowinsky EK. Anticancer drugs derived from plants. In: De Vita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 4th ed. Philadelphia, PA: Lippincott; 1993:409-417.
7. Epstein JB. Treatment of oral Kaposi’s sarcoma with intralesional vinblastine. Cancer. 1993;71:1722-1725.
8. McCormick SU. Intralesional vinblastine injections for the treatment of oral Kaposi’s sarcoma: report of 10 patients with 2-year follow-up. J Oral Maxillofac Surg. 1996;54:583-587.
9. Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
10. Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
11. Laor Y, Schwartz RA. Epidemiologic aspects of American Kaposi’s sarcoma. J Surg Oncol. 1979;12:299-303.
12. Gill K, Shah J. Kaposi sarcoma in patients with diabetes and wounds. Adv Skin Wound Care. 2006;19:196-198, 201.
13. Hurlbut W, Lincoln CS Jr. Multiple hemorrhagic sarcoma and diabetes mellitus; review of a series, with report of two cases. Arch Intern Med (Chic). 1949;84:738-750.
14. Fischer JW, Cohen DM. Simultaneous occurrence of Kaposi’s sarcoma, leukemia and diabetes mellitus; report of a case. Am J Clin Pathol. 1951;21:586-589.
15. Tankel R. Kaposi’s sarcoma and diabetes mellitus. Arch Dermatol. 1971;104:442-444.
16. Langtry JA, Bottomley DM, Phillips RH, et al. The infra-red coagulator in the treatment of AIDS-related Kaposi’s sarcoma and a comparison with radiotherapy. Clin Exp Dermatol. 1994;19:23-25.
17. Von Roenn JH, Cianfrocca M. Treatment of Kaposi’s sarcoma. Cancer Treat Res. 2001;104:127-148.
18. Dezube BJ. AIDS-related Kaposi sarcoma. the role of local therapy for a systemic disease. Arch Dermatol. 2000;136:1554-1556.
19. Oysul K, Beyzadeoglu M, Surenkok S, et al. A dose-response analysis for classical Kaposi’s sarcoma management by radiotherapy. Saudi Med J. 2008;29:837-840.
20. Morganroth GS. Topical 0.1% alitretinoin gel for classic Kaposi sarcoma. Arch Dermatol. 2002;138:542-543.
21. Rongioletti F, Zaccaria E, Viglizzo G. Failure of topical 0.1% alitretinoin gel for classic Kaposi sarcoma: first European experience. Br J Dermatol. 2006;155:856-857.
22. Klein E, Schwartz RA, Laor Y, et al. Treatment of Kaposi’s sarcoma with vinblastine. Cancer. 1980;45:427-431.
23. Flaitz CM, Nichols CM, Hicks MJ. Role of intralesional vinblastine administration in treatment of intraoral Kaposi’s sarcoma in AIDS. Eur J Cancer B Oral Oncol. 1995;31B:280-285.
24. Epstein JB, Lozada-Nur F, McLeod WA, et al. Oral Kaposi’s sarcoma in acquired immunodeficiency syndrome. review of management and report of the efficacy of intralesional vinblastine. Cancer. 1989;64:2424-2430.
25. Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342:1027-1038.
26. Ramírez-Amador V, Esquivel-Pedraza L, Lozada-Nur F, et al. Intralesional vinblastine vs. 3% sodium tetradecyl sulfate for the treatment of oral Kaposi’s sarcoma. a double blind, randomized clinical trial. Oral Oncol. 2002;38:460-467.
27. Boudreaux AA, Smith LL, Cosby CD, et al. Intralesional vinblastine for cutaneous Kaposi’s sarcoma associated with acquired immunodeficiency syndrome. a clinical trial to evaluate efficacy and discomfort associated with infection. J Am Acad Dermatol. 1993;28:61-65.
28. Odom RB, Goette DK. Treatment of cutaneous Kaposi’s sarcoma with intralesional vincristine. Arch Dermatol. 1978;114:1693-1694.
29. Tappero JW, Conant MA, Wolfe SF, et al. Kaposi’s sarcoma. epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
30. Smith KJ, Skelton HG, Turiansky G, et al. Hyaluronidase enhances the therapeutic effect of vinblastine in intralesional treatment of Kaposi’s sarcoma. Military Medical Consortium for the Advancement of Retroviral Research (MMCARR). J Am Acad Dermatol. 1997;36(2, pt 1):239-242.
31. Célestin Schartz NE, Chevret S, Paz C, et al. Imiquimod 5% cream for treatment of HIV-negative Kaposi’s sarcoma skin lesions: a phase I to II, open-label trial in 17 patients [published online ahead of print February 20, 2008]. J Am Acad Dermatol. 2008;58:585-591.
32. Poignonec S, Lachiver LD, Lamas G, et al. Intralesional bleomycin for acquired immunodeficiency syndrome-associated cutaneous Kaposi’s sarcoma. Arch Dermatol. 1995;131:228.
Practice Points
- Intralesional injection of low-dose vinblastine is a safe therapy for localized nodular Kaposi sarcoma (KS)(classic type).
- The best candidates for localized therapy are patients affected by nodular classic KS without visceral involvement, with fewer than 10 lesions on acral sites.
Marjolin Ulcer in a Surgical Scar
To the Editor:
Marjolin ulcers are malignancies arising in nonhealing cutaneous wounds. Although burn wounds are the most common type of cutaneous trauma associated with this entity, there are a multitude of possible lesions that may initiate this disease process including traumatic wounds, venous stasis ulcers, and vaccination sites.1,2 The most common type of malignancy reported in a Marjolin ulcer is an aggressive squamous cell carcinoma (SCC).1-3 Less commonly, basal cell carcinoma (BCC) also has been reported.1,3,4 However, cases of BCCs developing in surgical scars are exceedingly rare. We describe a case of a morphoeic BCC in a long-standing surgical scar in a 50-year-old woman with Crohn disease.
A 50-year-old woman presented with an intermittent ulceration within a horizontal surgical scar on the right side of the upper abdomen of 2 years’ duration that had not healed over the course of the last 6 months. The scar was present from surgeries conducted while she was a teenager for complications associated with Crohn disease. She underwent her first abdominal surgery for a partial gastric resection at 16 years of age, followed by multiple laparotomies from a perforated bile duct that occurred during the first surgery. The original incision created for the partial gastric resection was used for all subsequent surgeries.
The patient’s medical history was notable for central nervous system vasculitis with vision loss, chronic pancreatitis, Crohn disease, arthritis, multiple superficial BCCs on the back that were successfully treated with imiquimod cream, a nodular BCC on the neck that was surgically removed, and facial actinic keratoses treated with liquid nitrogen. She had Fitzpatrick skin type I. She grew up in a residential area in Southern Ontario and did not have a history of heavy sun exposure. She did not receive notable radiation from treatment of Crohn disease, and she usually wore a 1-piece bathing suit when swimming outdoors. According to the patient, she had never been exposed to arsenic. The patient’s family history was negative for skin cancer and she was a nonsmoker. She was taking methotrexate, prednisone, folic acid, pentazocine, and vitamin B12 injections at the time of presentation for the aforementioned conditions.
On physical examination a 1.5-cm honey-crusted ulcer with surrounding violaceous erythema in a long-standing surgical scar was observed (Figure 1). There was no palpable adenopathy in the inguinal or axillary regions. The suspected diagnosis prebiopsy was an SCC developing within the scar tissue. On histologic examination sections of small nests and strands of basal cell infiltrating thick collagen bundles were visualized. The appearance was consistent with a morphoeic BCC. The pathologist’s interpretation indicated that the lesion appeared to be a morphoeic BCC within the scar as opposed to a BCC that appeared morphoeic because of background scarring. Histologic images stained with hematoxylin and eosin showed small nests and strands of basal cells penetrating thick collagen bundles (Figure 2).
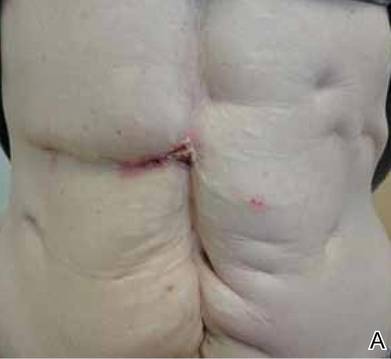

|
|
|

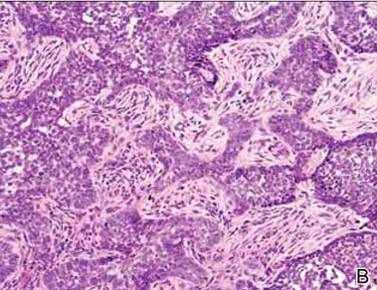
Marjolin ulcer was first described in 1828 by the French surgeon Jean-Nicolas Marjolin who published 4 cases of ulcers arising from scar tissue but did not appreciate their malignant capacity. However, the term Marjolin ulcer is now widely accepted as meaning any malignant tumor occurring within scar tissue or a chronic nonhealing wound.1,2
The exact incidence of malignant transformation in cutaneous wounds remains unknown, but this phenomenon can occur in individuals of all races and across all age groups.1,5-7 The most prevalent malignancy identified on biopsy is SCC, followed by BCC, melanoma, osteosarcoma, fibrosarcoma, and liposarcoma.1,2,6,7 With this entity infrequently occurring in the clinical setting, it often is overlooked or misdiagnosed.2 In addition, malignancies presenting as Marjolin ulcers have a greater tendency to metastasize and are reported to have a higher associated fatality rate.2,8 Thus, early recognition is essential, as a delay in diagnosis can potentially allow the tumor to progress to a life-threatening stage. In our patient, malignancy was clinically suspected based on the presence of an ulcer that was not healing despite adequate wound care and the location in a scar that was present for more than 30 years. The surgical scar had been a place of repeated trauma given the number of surgical procedures and the perforated bile duct, which can increase the potential for malignant transformation. Furthermore, the patient also was on immunosuppressive therapy for an extended period of time, possibly contributing to the development of this cancerous lesion and prior cutaneous malignancies.
The pathogenesis of a Marjolin ulcer is unclear, though many hypotheses have been suggested.1,2,6,9 Theories investigating decreased vascularity, lowered immune surveillance, decreased regenerative capacity, genetic mutations, and injury-related release of toxins have all been postulated as possible explanations for the increase in potential of malignant transformation.1-3,6,9 However, despite the pathogenesis, the mainstay of treatment remains wide local excision with at least 2-cm margins.1-3,10 Alternatively, Mohs micrographic surgery can be considered for Marjolin ulcers, but it is less frequently conducted in comparison to wide local excision. Radiation therapy often follows excision as adjuvant therapy, depending on the type of tumor.2,10 Prophylactic lymph node dissection is not indicated in most cases, but regional node dissection is suggested when palpable lymphadenopathy is present.1,2,10 Moreover, amputation is indicated with deep bone or joint involvement.1-3,10 Recurrence rates are high, ranging from 20% to 50%, and metastases to the brain, liver, lungs, kidneys, and lymph nodes have been reported.1,3 The prognosis of the cutaneous malignancy in this setting is not as favorable, and the 5-year survival rate is cited at approximately 60%.3 Overall prognosis depends on several factors including location, type of malignancy, immune status, progression of disease, and lymph node metastasis. Our patient’s presentation with a BCC instead of the more common SCC should carry a good overall prognosis, though she will need to be closely followed for recurrence after wide local excision.
This novel presentation of a morpheaform BCC in a surgical scar may serve as a reminder to consider this diagnosis and biopsy nonhealing ulcers within any type of chronic wound or scar.
1. Daya M, Balakrishan T. Advanced Marjolin’s ulcer of the scalp in a 13-year-old boy treated by excision and free tissue transfer: case report and review of literature. Indian J Plast Surg. 2009;42:106-111.
2. Pavlovic S, Wiley E, Guzman G, et al. Marjolin ulcer: an overlooked entity [published online ahead of print May 17, 2011]. Int Wound J. 2011;8:419-424.
3. Asuquo M, Ugare G, Ebughe G, et al. Marjolin’s ulcer: the importance of surgical management of chronic cutaneous ulcers. Int J Dermatol. 2007;46(suppl 2):S29-S32.
4. Ogawa B, Chen M, Margolis J, et al. Marjolin’s ulcer arising at the elbow: a case report and literature review. Hand (N Y). 2006;1:89-93.
5. Dupree MT, Boyer JD, Cobb MW. Marjolin’s ulcer arising in a burn scar. Cutis. 1998;62:49-51.
6. Er-Fan X, Li AO, Shi-ling W, et al. Burn scar carcinoma: case reports and review of the literature. Ann MBC. 1992;5:2.
7. Malheiro E, Pinto A, Choupina M, et al. Marjolin’s ulcer of the scalp: case report and literature review. Ann Burns and Fire Disasters. 2001;14:115-118.
8. Ozek C, Celik N, Bilkay U, et al. Marjolin’s ulcer of the scalp: report of 5 cases and review of the literature. J Burn Care Rehabil. 2001;22:65-69.
9. Thio D, Clarkson JH, Misra A, et al. Malignant change after 18 months in a lower limb ulcer: acute Marjolin’s revisited. Br J Plast Surg. 2003;56:825-828.
10. Aydogdu E, Yildirim S, Aköz T. Is surgery an effective and adequate treatmegnt in advanced Marjolin’s ulcer [published online ahead of print April 1, 2005]? Burns. 2005;31:421-431.
To the Editor:
Marjolin ulcers are malignancies arising in nonhealing cutaneous wounds. Although burn wounds are the most common type of cutaneous trauma associated with this entity, there are a multitude of possible lesions that may initiate this disease process including traumatic wounds, venous stasis ulcers, and vaccination sites.1,2 The most common type of malignancy reported in a Marjolin ulcer is an aggressive squamous cell carcinoma (SCC).1-3 Less commonly, basal cell carcinoma (BCC) also has been reported.1,3,4 However, cases of BCCs developing in surgical scars are exceedingly rare. We describe a case of a morphoeic BCC in a long-standing surgical scar in a 50-year-old woman with Crohn disease.
A 50-year-old woman presented with an intermittent ulceration within a horizontal surgical scar on the right side of the upper abdomen of 2 years’ duration that had not healed over the course of the last 6 months. The scar was present from surgeries conducted while she was a teenager for complications associated with Crohn disease. She underwent her first abdominal surgery for a partial gastric resection at 16 years of age, followed by multiple laparotomies from a perforated bile duct that occurred during the first surgery. The original incision created for the partial gastric resection was used for all subsequent surgeries.
The patient’s medical history was notable for central nervous system vasculitis with vision loss, chronic pancreatitis, Crohn disease, arthritis, multiple superficial BCCs on the back that were successfully treated with imiquimod cream, a nodular BCC on the neck that was surgically removed, and facial actinic keratoses treated with liquid nitrogen. She had Fitzpatrick skin type I. She grew up in a residential area in Southern Ontario and did not have a history of heavy sun exposure. She did not receive notable radiation from treatment of Crohn disease, and she usually wore a 1-piece bathing suit when swimming outdoors. According to the patient, she had never been exposed to arsenic. The patient’s family history was negative for skin cancer and she was a nonsmoker. She was taking methotrexate, prednisone, folic acid, pentazocine, and vitamin B12 injections at the time of presentation for the aforementioned conditions.
On physical examination a 1.5-cm honey-crusted ulcer with surrounding violaceous erythema in a long-standing surgical scar was observed (Figure 1). There was no palpable adenopathy in the inguinal or axillary regions. The suspected diagnosis prebiopsy was an SCC developing within the scar tissue. On histologic examination sections of small nests and strands of basal cell infiltrating thick collagen bundles were visualized. The appearance was consistent with a morphoeic BCC. The pathologist’s interpretation indicated that the lesion appeared to be a morphoeic BCC within the scar as opposed to a BCC that appeared morphoeic because of background scarring. Histologic images stained with hematoxylin and eosin showed small nests and strands of basal cells penetrating thick collagen bundles (Figure 2).
|
|
|
|
Marjolin ulcer was first described in 1828 by the French surgeon Jean-Nicolas Marjolin who published 4 cases of ulcers arising from scar tissue but did not appreciate their malignant capacity. However, the term Marjolin ulcer is now widely accepted as meaning any malignant tumor occurring within scar tissue or a chronic nonhealing wound.1,2
The exact incidence of malignant transformation in cutaneous wounds remains unknown, but this phenomenon can occur in individuals of all races and across all age groups.1,5-7 The most prevalent malignancy identified on biopsy is SCC, followed by BCC, melanoma, osteosarcoma, fibrosarcoma, and liposarcoma.1,2,6,7 With this entity infrequently occurring in the clinical setting, it often is overlooked or misdiagnosed.2 In addition, malignancies presenting as Marjolin ulcers have a greater tendency to metastasize and are reported to have a higher associated fatality rate.2,8 Thus, early recognition is essential, as a delay in diagnosis can potentially allow the tumor to progress to a life-threatening stage. In our patient, malignancy was clinically suspected based on the presence of an ulcer that was not healing despite adequate wound care and the location in a scar that was present for more than 30 years. The surgical scar had been a place of repeated trauma given the number of surgical procedures and the perforated bile duct, which can increase the potential for malignant transformation. Furthermore, the patient also was on immunosuppressive therapy for an extended period of time, possibly contributing to the development of this cancerous lesion and prior cutaneous malignancies.
The pathogenesis of a Marjolin ulcer is unclear, though many hypotheses have been suggested.1,2,6,9 Theories investigating decreased vascularity, lowered immune surveillance, decreased regenerative capacity, genetic mutations, and injury-related release of toxins have all been postulated as possible explanations for the increase in potential of malignant transformation.1-3,6,9 However, despite the pathogenesis, the mainstay of treatment remains wide local excision with at least 2-cm margins.1-3,10 Alternatively, Mohs micrographic surgery can be considered for Marjolin ulcers, but it is less frequently conducted in comparison to wide local excision. Radiation therapy often follows excision as adjuvant therapy, depending on the type of tumor.2,10 Prophylactic lymph node dissection is not indicated in most cases, but regional node dissection is suggested when palpable lymphadenopathy is present.1,2,10 Moreover, amputation is indicated with deep bone or joint involvement.1-3,10 Recurrence rates are high, ranging from 20% to 50%, and metastases to the brain, liver, lungs, kidneys, and lymph nodes have been reported.1,3 The prognosis of the cutaneous malignancy in this setting is not as favorable, and the 5-year survival rate is cited at approximately 60%.3 Overall prognosis depends on several factors including location, type of malignancy, immune status, progression of disease, and lymph node metastasis. Our patient’s presentation with a BCC instead of the more common SCC should carry a good overall prognosis, though she will need to be closely followed for recurrence after wide local excision.
This novel presentation of a morpheaform BCC in a surgical scar may serve as a reminder to consider this diagnosis and biopsy nonhealing ulcers within any type of chronic wound or scar.
To the Editor:
Marjolin ulcers are malignancies arising in nonhealing cutaneous wounds. Although burn wounds are the most common type of cutaneous trauma associated with this entity, there are a multitude of possible lesions that may initiate this disease process including traumatic wounds, venous stasis ulcers, and vaccination sites.1,2 The most common type of malignancy reported in a Marjolin ulcer is an aggressive squamous cell carcinoma (SCC).1-3 Less commonly, basal cell carcinoma (BCC) also has been reported.1,3,4 However, cases of BCCs developing in surgical scars are exceedingly rare. We describe a case of a morphoeic BCC in a long-standing surgical scar in a 50-year-old woman with Crohn disease.
A 50-year-old woman presented with an intermittent ulceration within a horizontal surgical scar on the right side of the upper abdomen of 2 years’ duration that had not healed over the course of the last 6 months. The scar was present from surgeries conducted while she was a teenager for complications associated with Crohn disease. She underwent her first abdominal surgery for a partial gastric resection at 16 years of age, followed by multiple laparotomies from a perforated bile duct that occurred during the first surgery. The original incision created for the partial gastric resection was used for all subsequent surgeries.
The patient’s medical history was notable for central nervous system vasculitis with vision loss, chronic pancreatitis, Crohn disease, arthritis, multiple superficial BCCs on the back that were successfully treated with imiquimod cream, a nodular BCC on the neck that was surgically removed, and facial actinic keratoses treated with liquid nitrogen. She had Fitzpatrick skin type I. She grew up in a residential area in Southern Ontario and did not have a history of heavy sun exposure. She did not receive notable radiation from treatment of Crohn disease, and she usually wore a 1-piece bathing suit when swimming outdoors. According to the patient, she had never been exposed to arsenic. The patient’s family history was negative for skin cancer and she was a nonsmoker. She was taking methotrexate, prednisone, folic acid, pentazocine, and vitamin B12 injections at the time of presentation for the aforementioned conditions.
On physical examination a 1.5-cm honey-crusted ulcer with surrounding violaceous erythema in a long-standing surgical scar was observed (Figure 1). There was no palpable adenopathy in the inguinal or axillary regions. The suspected diagnosis prebiopsy was an SCC developing within the scar tissue. On histologic examination sections of small nests and strands of basal cell infiltrating thick collagen bundles were visualized. The appearance was consistent with a morphoeic BCC. The pathologist’s interpretation indicated that the lesion appeared to be a morphoeic BCC within the scar as opposed to a BCC that appeared morphoeic because of background scarring. Histologic images stained with hematoxylin and eosin showed small nests and strands of basal cells penetrating thick collagen bundles (Figure 2).
|
|
|
|
Marjolin ulcer was first described in 1828 by the French surgeon Jean-Nicolas Marjolin who published 4 cases of ulcers arising from scar tissue but did not appreciate their malignant capacity. However, the term Marjolin ulcer is now widely accepted as meaning any malignant tumor occurring within scar tissue or a chronic nonhealing wound.1,2
The exact incidence of malignant transformation in cutaneous wounds remains unknown, but this phenomenon can occur in individuals of all races and across all age groups.1,5-7 The most prevalent malignancy identified on biopsy is SCC, followed by BCC, melanoma, osteosarcoma, fibrosarcoma, and liposarcoma.1,2,6,7 With this entity infrequently occurring in the clinical setting, it often is overlooked or misdiagnosed.2 In addition, malignancies presenting as Marjolin ulcers have a greater tendency to metastasize and are reported to have a higher associated fatality rate.2,8 Thus, early recognition is essential, as a delay in diagnosis can potentially allow the tumor to progress to a life-threatening stage. In our patient, malignancy was clinically suspected based on the presence of an ulcer that was not healing despite adequate wound care and the location in a scar that was present for more than 30 years. The surgical scar had been a place of repeated trauma given the number of surgical procedures and the perforated bile duct, which can increase the potential for malignant transformation. Furthermore, the patient also was on immunosuppressive therapy for an extended period of time, possibly contributing to the development of this cancerous lesion and prior cutaneous malignancies.
The pathogenesis of a Marjolin ulcer is unclear, though many hypotheses have been suggested.1,2,6,9 Theories investigating decreased vascularity, lowered immune surveillance, decreased regenerative capacity, genetic mutations, and injury-related release of toxins have all been postulated as possible explanations for the increase in potential of malignant transformation.1-3,6,9 However, despite the pathogenesis, the mainstay of treatment remains wide local excision with at least 2-cm margins.1-3,10 Alternatively, Mohs micrographic surgery can be considered for Marjolin ulcers, but it is less frequently conducted in comparison to wide local excision. Radiation therapy often follows excision as adjuvant therapy, depending on the type of tumor.2,10 Prophylactic lymph node dissection is not indicated in most cases, but regional node dissection is suggested when palpable lymphadenopathy is present.1,2,10 Moreover, amputation is indicated with deep bone or joint involvement.1-3,10 Recurrence rates are high, ranging from 20% to 50%, and metastases to the brain, liver, lungs, kidneys, and lymph nodes have been reported.1,3 The prognosis of the cutaneous malignancy in this setting is not as favorable, and the 5-year survival rate is cited at approximately 60%.3 Overall prognosis depends on several factors including location, type of malignancy, immune status, progression of disease, and lymph node metastasis. Our patient’s presentation with a BCC instead of the more common SCC should carry a good overall prognosis, though she will need to be closely followed for recurrence after wide local excision.
This novel presentation of a morpheaform BCC in a surgical scar may serve as a reminder to consider this diagnosis and biopsy nonhealing ulcers within any type of chronic wound or scar.
1. Daya M, Balakrishan T. Advanced Marjolin’s ulcer of the scalp in a 13-year-old boy treated by excision and free tissue transfer: case report and review of literature. Indian J Plast Surg. 2009;42:106-111.
2. Pavlovic S, Wiley E, Guzman G, et al. Marjolin ulcer: an overlooked entity [published online ahead of print May 17, 2011]. Int Wound J. 2011;8:419-424.
3. Asuquo M, Ugare G, Ebughe G, et al. Marjolin’s ulcer: the importance of surgical management of chronic cutaneous ulcers. Int J Dermatol. 2007;46(suppl 2):S29-S32.
4. Ogawa B, Chen M, Margolis J, et al. Marjolin’s ulcer arising at the elbow: a case report and literature review. Hand (N Y). 2006;1:89-93.
5. Dupree MT, Boyer JD, Cobb MW. Marjolin’s ulcer arising in a burn scar. Cutis. 1998;62:49-51.
6. Er-Fan X, Li AO, Shi-ling W, et al. Burn scar carcinoma: case reports and review of the literature. Ann MBC. 1992;5:2.
7. Malheiro E, Pinto A, Choupina M, et al. Marjolin’s ulcer of the scalp: case report and literature review. Ann Burns and Fire Disasters. 2001;14:115-118.
8. Ozek C, Celik N, Bilkay U, et al. Marjolin’s ulcer of the scalp: report of 5 cases and review of the literature. J Burn Care Rehabil. 2001;22:65-69.
9. Thio D, Clarkson JH, Misra A, et al. Malignant change after 18 months in a lower limb ulcer: acute Marjolin’s revisited. Br J Plast Surg. 2003;56:825-828.
10. Aydogdu E, Yildirim S, Aköz T. Is surgery an effective and adequate treatmegnt in advanced Marjolin’s ulcer [published online ahead of print April 1, 2005]? Burns. 2005;31:421-431.
1. Daya M, Balakrishan T. Advanced Marjolin’s ulcer of the scalp in a 13-year-old boy treated by excision and free tissue transfer: case report and review of literature. Indian J Plast Surg. 2009;42:106-111.
2. Pavlovic S, Wiley E, Guzman G, et al. Marjolin ulcer: an overlooked entity [published online ahead of print May 17, 2011]. Int Wound J. 2011;8:419-424.
3. Asuquo M, Ugare G, Ebughe G, et al. Marjolin’s ulcer: the importance of surgical management of chronic cutaneous ulcers. Int J Dermatol. 2007;46(suppl 2):S29-S32.
4. Ogawa B, Chen M, Margolis J, et al. Marjolin’s ulcer arising at the elbow: a case report and literature review. Hand (N Y). 2006;1:89-93.
5. Dupree MT, Boyer JD, Cobb MW. Marjolin’s ulcer arising in a burn scar. Cutis. 1998;62:49-51.
6. Er-Fan X, Li AO, Shi-ling W, et al. Burn scar carcinoma: case reports and review of the literature. Ann MBC. 1992;5:2.
7. Malheiro E, Pinto A, Choupina M, et al. Marjolin’s ulcer of the scalp: case report and literature review. Ann Burns and Fire Disasters. 2001;14:115-118.
8. Ozek C, Celik N, Bilkay U, et al. Marjolin’s ulcer of the scalp: report of 5 cases and review of the literature. J Burn Care Rehabil. 2001;22:65-69.
9. Thio D, Clarkson JH, Misra A, et al. Malignant change after 18 months in a lower limb ulcer: acute Marjolin’s revisited. Br J Plast Surg. 2003;56:825-828.
10. Aydogdu E, Yildirim S, Aköz T. Is surgery an effective and adequate treatmegnt in advanced Marjolin’s ulcer [published online ahead of print April 1, 2005]? Burns. 2005;31:421-431.
VIDEO: Updating the immune response to nonmelanoma skin cancer
ASHEVILLE, N.C. – Recent advances in basic science have shown how the local immune environment in tissue surrounding nonmelanoma skin cancer compares to adjacent normal tissue.
New Mexico Health Sciences Center’s Dr. Andrew Ondo reviewed the latest research in an interview at the annual meeting of the Noah Worcester Dermatological Society. “Each step along the way is a possible target for the treatment of squamous cell carcinoma,” said Dr. Ondo, who indicated that he had no financial conflicts to disclose.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ASHEVILLE, N.C. – Recent advances in basic science have shown how the local immune environment in tissue surrounding nonmelanoma skin cancer compares to adjacent normal tissue.
New Mexico Health Sciences Center’s Dr. Andrew Ondo reviewed the latest research in an interview at the annual meeting of the Noah Worcester Dermatological Society. “Each step along the way is a possible target for the treatment of squamous cell carcinoma,” said Dr. Ondo, who indicated that he had no financial conflicts to disclose.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ASHEVILLE, N.C. – Recent advances in basic science have shown how the local immune environment in tissue surrounding nonmelanoma skin cancer compares to adjacent normal tissue.
New Mexico Health Sciences Center’s Dr. Andrew Ondo reviewed the latest research in an interview at the annual meeting of the Noah Worcester Dermatological Society. “Each step along the way is a possible target for the treatment of squamous cell carcinoma,” said Dr. Ondo, who indicated that he had no financial conflicts to disclose.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM NOAH 57

Imaging guides BCC laser ablation
KISSIMMEE, FLA. – Reflective confocal microscopic imaging successfully guided carbon dioxide laser ablation of basal cell carcinomas, and imaging results fully matched those from Mohs histology, a small study found.
“Our results suggest that reflective confocal microscopy can accurately guide carbon dioxide laser ablation of superficial and early nodular basal cell carcinomas,” said Dr. Brian Hibler of Memorial Sloan Kettering Cancer Center in New York. The technique provides a real-time, noninvasive way to delineate the tumor area before ablation and to check for residual tumor between passes with the laser, he said at the annual meeting of the American Society for Laser Medicine and surgery.
While conventional and Mohs microscopic surgeries remain the gold standard for removing basal carcinomas (BCC), surgery is not an option for some patients because of tumor location, comorbidities, or personal preferences, Dr. Hibler noted. Past studies have reported good clinical and cosmetic outcomes with laser ablation of BCCs, but use of the modality has been limited because there was no way to assess response without excision or biopsies. Reflective confocal microscopy (RCM) uses a low-powered laser system that provides cellular-level imaging and can distinguish BCCs, he said.
Dr. Hibler and his colleagues performed baseline RCM of eight BCCs (three on the trunk, three on the extremities, and two on the head and neck) from seven patients aged 29-83 years. Two patients were men and five were women. The patients then underwent carbon dioxide laser ablation with a wavelength of 10,600 nm, pulse duration of 750 microseconds, and fluence of 7.5 J/cm2, using a square pattern and density of 30%. If RCM revealed residual BCC, the researchers repeated the process up to two more times, for a maximum of three passes. They then removed the entire lesion using Mohs micrographic surgery, performing vertical histologic sectioning of the tissue.
Reflective confocal microscopy generated reliable cellular-level images in real time on the tumor surface and up to 150 mcm deep, Dr. Hibler reported. Tissue from BCCs appears as dense nodular areas with adjacent spaces and red blood cell trafficking, he noted. Microscopy results were consistent with Mohs histology findings in all eight cases, including six in which the tumor was completely removed and two with residual tumor. One of these two cases was the only infiltrative BCC in the series, while the other might have been tissue artifact, Dr. Hibler said.
Patients experienced no adverse effects from the interventions, Dr. Hibler reported. “Future studies are planned are planned without Mohs, so we can use reflective confocal microscopy to longitudinally monitor for recurrence,” he added.
The study won an award at the meeting.
Dr. Hibler reported no funding sources and made no disclosures.
KISSIMMEE, FLA. – Reflective confocal microscopic imaging successfully guided carbon dioxide laser ablation of basal cell carcinomas, and imaging results fully matched those from Mohs histology, a small study found.
“Our results suggest that reflective confocal microscopy can accurately guide carbon dioxide laser ablation of superficial and early nodular basal cell carcinomas,” said Dr. Brian Hibler of Memorial Sloan Kettering Cancer Center in New York. The technique provides a real-time, noninvasive way to delineate the tumor area before ablation and to check for residual tumor between passes with the laser, he said at the annual meeting of the American Society for Laser Medicine and surgery.
While conventional and Mohs microscopic surgeries remain the gold standard for removing basal carcinomas (BCC), surgery is not an option for some patients because of tumor location, comorbidities, or personal preferences, Dr. Hibler noted. Past studies have reported good clinical and cosmetic outcomes with laser ablation of BCCs, but use of the modality has been limited because there was no way to assess response without excision or biopsies. Reflective confocal microscopy (RCM) uses a low-powered laser system that provides cellular-level imaging and can distinguish BCCs, he said.
Dr. Hibler and his colleagues performed baseline RCM of eight BCCs (three on the trunk, three on the extremities, and two on the head and neck) from seven patients aged 29-83 years. Two patients were men and five were women. The patients then underwent carbon dioxide laser ablation with a wavelength of 10,600 nm, pulse duration of 750 microseconds, and fluence of 7.5 J/cm2, using a square pattern and density of 30%. If RCM revealed residual BCC, the researchers repeated the process up to two more times, for a maximum of three passes. They then removed the entire lesion using Mohs micrographic surgery, performing vertical histologic sectioning of the tissue.
Reflective confocal microscopy generated reliable cellular-level images in real time on the tumor surface and up to 150 mcm deep, Dr. Hibler reported. Tissue from BCCs appears as dense nodular areas with adjacent spaces and red blood cell trafficking, he noted. Microscopy results were consistent with Mohs histology findings in all eight cases, including six in which the tumor was completely removed and two with residual tumor. One of these two cases was the only infiltrative BCC in the series, while the other might have been tissue artifact, Dr. Hibler said.
Patients experienced no adverse effects from the interventions, Dr. Hibler reported. “Future studies are planned are planned without Mohs, so we can use reflective confocal microscopy to longitudinally monitor for recurrence,” he added.
The study won an award at the meeting.
Dr. Hibler reported no funding sources and made no disclosures.
KISSIMMEE, FLA. – Reflective confocal microscopic imaging successfully guided carbon dioxide laser ablation of basal cell carcinomas, and imaging results fully matched those from Mohs histology, a small study found.
“Our results suggest that reflective confocal microscopy can accurately guide carbon dioxide laser ablation of superficial and early nodular basal cell carcinomas,” said Dr. Brian Hibler of Memorial Sloan Kettering Cancer Center in New York. The technique provides a real-time, noninvasive way to delineate the tumor area before ablation and to check for residual tumor between passes with the laser, he said at the annual meeting of the American Society for Laser Medicine and surgery.
While conventional and Mohs microscopic surgeries remain the gold standard for removing basal carcinomas (BCC), surgery is not an option for some patients because of tumor location, comorbidities, or personal preferences, Dr. Hibler noted. Past studies have reported good clinical and cosmetic outcomes with laser ablation of BCCs, but use of the modality has been limited because there was no way to assess response without excision or biopsies. Reflective confocal microscopy (RCM) uses a low-powered laser system that provides cellular-level imaging and can distinguish BCCs, he said.
Dr. Hibler and his colleagues performed baseline RCM of eight BCCs (three on the trunk, three on the extremities, and two on the head and neck) from seven patients aged 29-83 years. Two patients were men and five were women. The patients then underwent carbon dioxide laser ablation with a wavelength of 10,600 nm, pulse duration of 750 microseconds, and fluence of 7.5 J/cm2, using a square pattern and density of 30%. If RCM revealed residual BCC, the researchers repeated the process up to two more times, for a maximum of three passes. They then removed the entire lesion using Mohs micrographic surgery, performing vertical histologic sectioning of the tissue.
Reflective confocal microscopy generated reliable cellular-level images in real time on the tumor surface and up to 150 mcm deep, Dr. Hibler reported. Tissue from BCCs appears as dense nodular areas with adjacent spaces and red blood cell trafficking, he noted. Microscopy results were consistent with Mohs histology findings in all eight cases, including six in which the tumor was completely removed and two with residual tumor. One of these two cases was the only infiltrative BCC in the series, while the other might have been tissue artifact, Dr. Hibler said.
Patients experienced no adverse effects from the interventions, Dr. Hibler reported. “Future studies are planned are planned without Mohs, so we can use reflective confocal microscopy to longitudinally monitor for recurrence,” he added.
The study won an award at the meeting.
Dr. Hibler reported no funding sources and made no disclosures.
Key clinical point: Reflective confocal microscopy offers noninvasive, real-time imaging to guide laser ablation of basal cell carcinomas.
Major finding: Results from RCM matched those from Mohs histology in all patients.
Data source: Prospective study of eight BCCs in seven patients.
Disclosures: Dr. Hibler reported no funding sources and made no disclosures.
Intradermal ALA-PDT linked to long-term remission in BCC
KISSIMMEE, FLA. – Using a needle-free device to inject nodular basal cell carcinomas with intralesional 5-aminolevulinic acid before photodynamic therapy led to complete, years-long remissions and few side effects in a small case series.
“This approach represents an interesting alternative to Mohs, for sure,” Dr. Daniel Barolet said at the annual meeting of the American Society for Laser Medicine and Surgery. “The secret is in the injector nozzle, which lets you inject with multiple openings to get the best uniformity around the tumor.”
Mohs micrographic surgery remains the standard for basal cell carcinoma (BCC) in high-risk sites, and the number of Mohs surgeries has approximately doubled since 2001, said Dr. Barolet, adjunct professor of dermatology at McGill University in Montreal.
Mohs, however, can cause scarring, and BCCs recur in about 4% of patients. In contrast, photodynamic therapy (PDT) is associated with less scarring and pain, fewer complications, shorter recovery times, and lower costs, although the recurrence rate is about 14%, he noted.
Since PDT alone does not efficiently penetrate thick tumor volumes, it works best with pretreatment using agents such as aminolevulinic acid (ALA).
Using needles to inject the tumor, however, can cause pain, vascular damage, vasoconstriction, deep purpura, necrosis, and infection. “Because of this, no-needle injection is an interesting avenue for PDT,” he noted. Needle-free devices currently are used to inject insulin and to administer some vaccines. They are “virtually painless,” noninvasive, and tissue sparing, he said.
To explore the potential role for needle-free injection in ALA-PDT, Dr. Barolot used a prototype high-speed jet to deliver intralesional 5-ALA in the nodular facial BCCs of four patients. He then performed photoactivation with a red light–emitting diode, with continuous wave at 630 nm, irradiance at 50 mW/cm2, and total fluence 50-100 J/cm2.
Patients had no evidence of clinical or histopathologic recurrence for up to 7 years after treatment, Dr. Barolet reported. They experienced mild crusting at treated sites for up to a week after treatment, but no other adverse effects. Two patients needed a second treatment 2 months after the initial treatment to achieve complete remission. “Excellent cosmesis was obtained,” he added, pointing to before and after photos that showed no evidence of lesions several months after treatment.
Multicenter clinical trials are needed to further evaluate the modality, but the preliminary data suggest that intralesional PDT is a reasonable alternative to Mohs for BCCs in high-risk body sites, as long as lesions are few in number and do not affect large areas of the body, Dr. Barolet said.
The modality is especially well suited to “tricky” areas of the body that are difficult to treat with Mohs, he said.
“Developing a user-friendly, disposable no-needle injector will make it much easier for users,” he added. For low-risk BCCs in low-risk sites, conventional treatments such as surgical excision remain the best option, he said.
Dr. Barolet reported no funding sources for the study and said he had no relevant financial disclosures.
KISSIMMEE, FLA. – Using a needle-free device to inject nodular basal cell carcinomas with intralesional 5-aminolevulinic acid before photodynamic therapy led to complete, years-long remissions and few side effects in a small case series.
“This approach represents an interesting alternative to Mohs, for sure,” Dr. Daniel Barolet said at the annual meeting of the American Society for Laser Medicine and Surgery. “The secret is in the injector nozzle, which lets you inject with multiple openings to get the best uniformity around the tumor.”
Mohs micrographic surgery remains the standard for basal cell carcinoma (BCC) in high-risk sites, and the number of Mohs surgeries has approximately doubled since 2001, said Dr. Barolet, adjunct professor of dermatology at McGill University in Montreal.
Mohs, however, can cause scarring, and BCCs recur in about 4% of patients. In contrast, photodynamic therapy (PDT) is associated with less scarring and pain, fewer complications, shorter recovery times, and lower costs, although the recurrence rate is about 14%, he noted.
Since PDT alone does not efficiently penetrate thick tumor volumes, it works best with pretreatment using agents such as aminolevulinic acid (ALA).
Using needles to inject the tumor, however, can cause pain, vascular damage, vasoconstriction, deep purpura, necrosis, and infection. “Because of this, no-needle injection is an interesting avenue for PDT,” he noted. Needle-free devices currently are used to inject insulin and to administer some vaccines. They are “virtually painless,” noninvasive, and tissue sparing, he said.
To explore the potential role for needle-free injection in ALA-PDT, Dr. Barolot used a prototype high-speed jet to deliver intralesional 5-ALA in the nodular facial BCCs of four patients. He then performed photoactivation with a red light–emitting diode, with continuous wave at 630 nm, irradiance at 50 mW/cm2, and total fluence 50-100 J/cm2.
Patients had no evidence of clinical or histopathologic recurrence for up to 7 years after treatment, Dr. Barolet reported. They experienced mild crusting at treated sites for up to a week after treatment, but no other adverse effects. Two patients needed a second treatment 2 months after the initial treatment to achieve complete remission. “Excellent cosmesis was obtained,” he added, pointing to before and after photos that showed no evidence of lesions several months after treatment.
Multicenter clinical trials are needed to further evaluate the modality, but the preliminary data suggest that intralesional PDT is a reasonable alternative to Mohs for BCCs in high-risk body sites, as long as lesions are few in number and do not affect large areas of the body, Dr. Barolet said.
The modality is especially well suited to “tricky” areas of the body that are difficult to treat with Mohs, he said.
“Developing a user-friendly, disposable no-needle injector will make it much easier for users,” he added. For low-risk BCCs in low-risk sites, conventional treatments such as surgical excision remain the best option, he said.
Dr. Barolet reported no funding sources for the study and said he had no relevant financial disclosures.
KISSIMMEE, FLA. – Using a needle-free device to inject nodular basal cell carcinomas with intralesional 5-aminolevulinic acid before photodynamic therapy led to complete, years-long remissions and few side effects in a small case series.
“This approach represents an interesting alternative to Mohs, for sure,” Dr. Daniel Barolet said at the annual meeting of the American Society for Laser Medicine and Surgery. “The secret is in the injector nozzle, which lets you inject with multiple openings to get the best uniformity around the tumor.”
Mohs micrographic surgery remains the standard for basal cell carcinoma (BCC) in high-risk sites, and the number of Mohs surgeries has approximately doubled since 2001, said Dr. Barolet, adjunct professor of dermatology at McGill University in Montreal.
Mohs, however, can cause scarring, and BCCs recur in about 4% of patients. In contrast, photodynamic therapy (PDT) is associated with less scarring and pain, fewer complications, shorter recovery times, and lower costs, although the recurrence rate is about 14%, he noted.
Since PDT alone does not efficiently penetrate thick tumor volumes, it works best with pretreatment using agents such as aminolevulinic acid (ALA).
Using needles to inject the tumor, however, can cause pain, vascular damage, vasoconstriction, deep purpura, necrosis, and infection. “Because of this, no-needle injection is an interesting avenue for PDT,” he noted. Needle-free devices currently are used to inject insulin and to administer some vaccines. They are “virtually painless,” noninvasive, and tissue sparing, he said.
To explore the potential role for needle-free injection in ALA-PDT, Dr. Barolot used a prototype high-speed jet to deliver intralesional 5-ALA in the nodular facial BCCs of four patients. He then performed photoactivation with a red light–emitting diode, with continuous wave at 630 nm, irradiance at 50 mW/cm2, and total fluence 50-100 J/cm2.
Patients had no evidence of clinical or histopathologic recurrence for up to 7 years after treatment, Dr. Barolet reported. They experienced mild crusting at treated sites for up to a week after treatment, but no other adverse effects. Two patients needed a second treatment 2 months after the initial treatment to achieve complete remission. “Excellent cosmesis was obtained,” he added, pointing to before and after photos that showed no evidence of lesions several months after treatment.
Multicenter clinical trials are needed to further evaluate the modality, but the preliminary data suggest that intralesional PDT is a reasonable alternative to Mohs for BCCs in high-risk body sites, as long as lesions are few in number and do not affect large areas of the body, Dr. Barolet said.
The modality is especially well suited to “tricky” areas of the body that are difficult to treat with Mohs, he said.
“Developing a user-friendly, disposable no-needle injector will make it much easier for users,” he added. For low-risk BCCs in low-risk sites, conventional treatments such as surgical excision remain the best option, he said.
Dr. Barolet reported no funding sources for the study and said he had no relevant financial disclosures.
AT LASER 2015
Key clinical point: Intralesional 5-ALA-PDT is a potential alternative to Mohs micrographic surgery for treating basal cell carcinomas in high-risk sites.
Major finding: Four treated patients experienced resolution of recurrent basal cell carcinomas for up to 7 years.
Data source: Series of four cases of recurrent nodular facial basal cell carcinomas.
Disclosures: Dr. Barolet reported no funding sources and declared no relevant financial disclosures.
Trichoepithelioma and Spiradenoma Collision Tumor
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
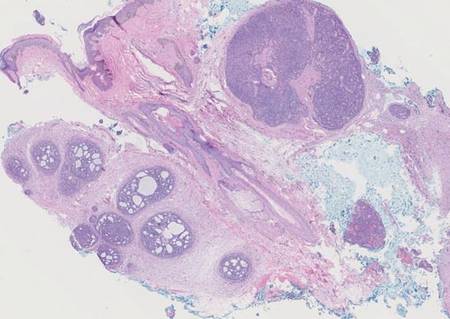
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
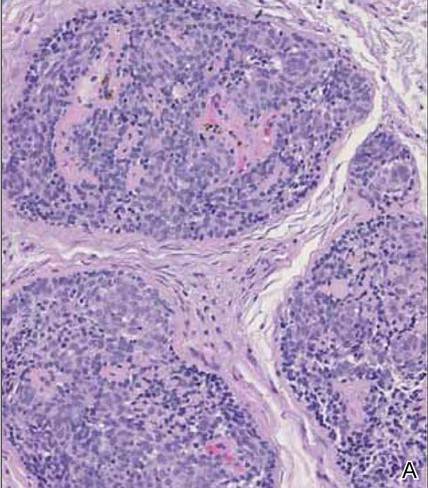
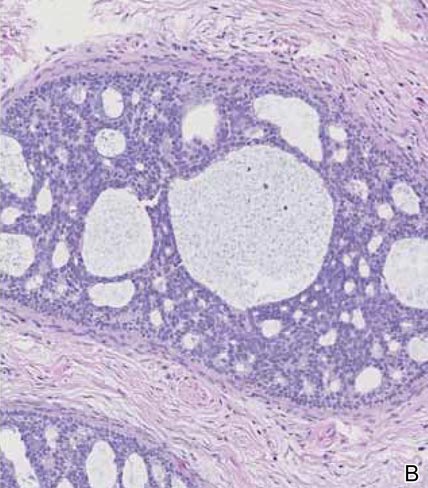
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.

| 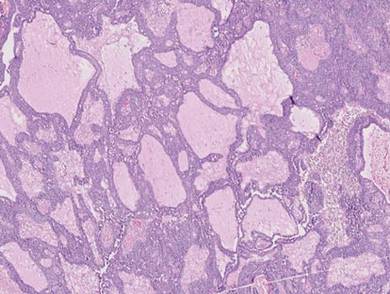
|
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
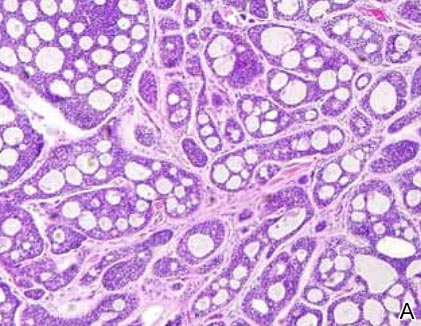
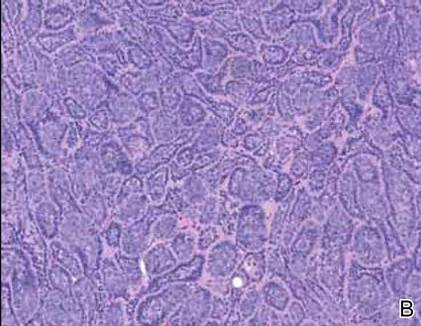
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
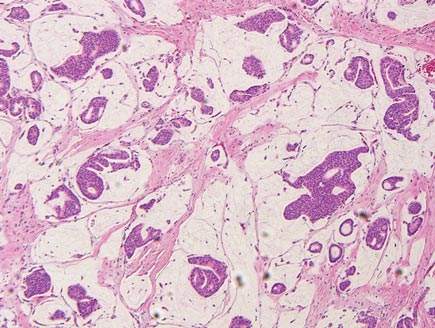
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
|
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
|
|
|
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
|
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
|
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
|
|
|
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
|
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
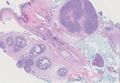
Mucocutaneous Presentation of Kaposi Sarcoma in an Asymptomatic Human Immunodeficiency Virus–Positive Man
Case Report
A 45-year-old man presented with persistent swelling and “black-and-blue” lesions on the legs, feet, and toes of 6 months’ duration. The painless lesions first appeared on the left lower leg and then began to appear on the right leg in recent months. Three weeks prior to presentation, he developed swelling of the left lower leg during hospitalization for a lumbar laminectomy. A venous ultrasound was negative for a deep vein thrombosis. He denied trauma or history of bleeding diathesis. He did not report symptoms of dyspnea, angina, or claudication, and a review of systems was unremarkable.
The patient’s medical history included spinal stenosis, chronic back pain, osteoarthritis, and anxiety. His medications included oxycodone, zolpidem, and alprazolam. In addition to a recent lumbar laminectomy, he had undergone extensive dental work in the last 6 months. The patient denied the use of cigarettes, alcohol, or intravenous drugs.
Physical examination revealed scattered, purple, segmented patches on the dorsal and plantar aspects of the feet, both calves, both heels, and toes (Figure 1). Mild nonpitting edema was present below the left knee along with edema on the dorsum of the left foot. The distribution of the lesions was initially suggestive of cholesterol embolization syndrome; however, both the femoral and posterior tibial pulses were symmetric and palpable (+2). Well-demarcated violaceous plaques with central clearing and a rustlike discoloration were noted on the hard and soft palates. Cervical lymphadenopathy was not present.
|
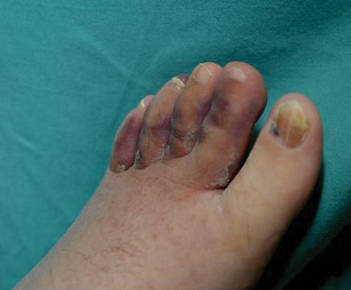
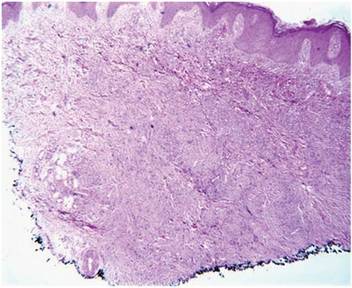
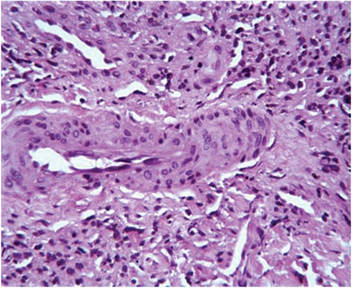
Laboratory tests including a repeat venous ultrasound of the left lower leg revealed no evidence of deep vein thrombosis. Ankle brachial index revealed no abnormalities and blood flow to the lower legs was adequate. Computed tomography scans of the chest, abdomen, and pelvis were unremarkable except for mild splenomegaly and moderate cardiomegaly. Lastly, human immunodeficiency virus (HIV) 1 and HIV-2 enzyme immunoassay was reactive.
Histopathologic examination of a punch biopsy from the right fourth toe was representative of the plaque stage of Kaposi sarcoma (KS) with a diffuse collection of extravasated erythrocytes and neoplastic vascular proliferation among a background of numerous plasma cells and hemosiderophages (Figure 2). Higher magnification illustrated the promontory sign, whereby native vessels encroach on neoplastic slitlike vascular spaces (Figure 3). A final diagnosis of AIDS-related KS was made. The patient was referred to an infectious disease specialist for evaluation of his CD4 levels and HIV management.
Comment
Kaposi sarcoma is a neoplastic proliferation of the blood vessels in the skin characterized by the formation of violaceous macules and papules that often appear on a single distal extremity, such as the foot. Over time the lesions can develop on the opposite extremity and coalesce into poorly demarcated plaques and nodules with accompanying stasis and lymphedema of the involved extremities.1 Evolution of the lesions depends on the KS subtype. The most common clinical variant of KS is the classic form, which primarily is seen in those of Mediterranean, Eastern European, or Ashkenazi Jewish descent, with a predilection for men and older adults.1,2 The endemic form of KS, or African KS, is more aggressive with rapid visceral involvement and rare skin lesions; it is common among prepubertal children in sub-Saharan Africa with no predilection for either sex.2 In the setting of severe immune suppression, organ transplantation, or chemotherapy, a third KS subtype known as iatrogenic KS can occur. The clinical course of iatrogenic KS may range from scattered cutaneous lesions to diffuse involvement secondary to increased dosages and long-term use of immunosuppressive agents.2
Our patient had AIDS-related or epidemic KS. AIDS-related KS is largely predominant among homosexual men, but due to the awareness of safe sexual practices and the introduction of highly active antiretroviral therapy (HAART), KS incidence in the United States has declined.1,2 However, despite recent advances in HIV therapy, AIDS-related KS is still the most common neoplasm seen in AIDS patients and is the presenting manifestation of AIDS in up to 30% of cases.3 Up to 22% of cases first appear on the gingiva, hard palate, and tongue, with concomitant dysphagia and airway obstruction in severe cases.4,5 More advanced cases of AIDS-related KS can present with initial symptoms such as abdominal pain, melena, dyspnea, lymphadenopathy, and weight loss, which suggests involvement of the gastrointestinal tract, lungs, and other organ systems.
Regardless of the subtype, the etiology of KS currently is thought to be secondary to infection with human herpesvirus 8 (HHV-8), also known as Kaposi sarcoma–associated herpesvirus (KSHV).1 Human immunodeficiency virus infection can enhance KSHV expression through the HIV transactivator protein, which activates KSHV oncogenes and angiogenic growth factors to promote the development of KS lesions.2,6Likewise, KSHV enhances HIV upregulation through latency-associated nuclear antigen, a protein that interacts with HIV Tat protein to further activate long terminal repeats of HIV-1.2
The differential diagnosis of KS is broad. The slightly elevated, pinkish reddish discolorations of KS may resemble verruca plana and/or squamous cell carcinoma on visual observation, whereas nodular KS may resemble giant cell granuloma, pyogenic granuloma, or hemangiopericytoma.4,7 Cases of KS with lymph node involvement may include a differential diagnosis of lymphoma, angiosarcoma, and bacillary angiomatosis.7 Other vascular pathologies that may be considered in the differential diagnosis include vascular tumors (eg, spindle cell hemangioma), fibrohistiocytic tumors (eg, dermatofibrosarcoma protuberans), and a collection of spindle cell mesenchymal tumors.8
Kaposi sarcoma progresses through several histologic stages beginning with the patch stage, then progressing to the plaque stage, and finally culminating in the nodular stage. The patch stage is the first stage in KS progression and a crowded dermis can be seen with the formation of slitlike vascular spaces lined by endothelial cells with red blood cell extravasation into the lumens of newly formed vascular channels, hence demonstrating the promontory sign.8 In the plaque stage, the promontory sign still is present and there is a greater presence of slitlike spaces, giving the micrograph an overall sievelike appearance. Erythrocytes can be found residing within the clear cytoplasm of spindled endothelial cells, leading to the development of autolumination. Finally, the nodular stage is characterized by a neoplastic proliferation of monomorphic spindle cells that form fasciclelike nests in the dermis.8 To distinguish KS from other angioproliferative tumors, one can stain for HHV-8 latent nuclear antigen-1, which is found in the nuclei of infected endothelial cells.1,8,9
Kaposi sarcoma is treated through a variety of mechanisms depending on the subtype. Classic KS lesions often can be observed as they a follow a benign and nonaggressive course.1 Highly active antiretroviral therapy is the mainstay of care in AIDS-related KS and has led to regression of lesions and a remarkable decline in the incidence of KS.3 The HAART regimen consists of a protease inhibitor or nonnucleoside reverse-transcriptase inhibitor with the addition of 2 nucleoside reverse-transcriptase inhibitors. More advanced or refractory cases of KS often require dual treatment with HAART and a chemotherapy agent such as pegylated liposomal doxorubicin. Combination therapy has been shown to result in stronger therapeutic responses and lower relapse rates in contrast to HAART alone.7 Patients also may consider other treatment modalities to manage KS lesions such as surgical removal of lesions, laser therapy, paclitaxel, interferon alfa, oral etoposide, thalidomide, and topical therapies such as imiquimod cream 5% and alitretinoin.1,7
Conclusion
Kaposi sarcoma is a rare but concerning dermatologic condition that signals the need for further diagnostic evaluation. Coexpression of viruses such as HIV and HHV-8 can result in a more virulent and rapid progression of KS to encompass both mucous membrane and systemic involvement. Our patient’s lesions were the first presenting sign of HIV infection despite being asymptomatic at the time of diagnosis, which is alarming in the sense that more than 21% of HIV-infected individuals in the United States have not been clinically diagnosed.10 Inquiry of HIV risk factors and routine screening for HIV should be performed in refractory cases of skin disease as an underlying clue to further investigate the immune system. We present our unique case of mucocutaneous development of KS in an asymptomatic HIV-positive man to stress the importance of KS recognition and management.
1. Jan MM, Laskas JW, Griffin TD. Eruptive Kaposi sarcoma: an unusual presentation in an HIV-negative patient. Cutis. 2011;87:34-38.
2. Geraminejad P, Memar O, Aronson I, et al. Kaposi’s sarcoma and other manifestations of human herpesvirus 8. J Am Acad Dermatol. 2002;47:641-655.
3. Kharkar V, Gutte RM, Khopkar U, et al. Kaposi’s sarcoma: a presenting manifestation of HIV infection in an Indian. Indian J Dermatol Venereol Leprol. 2009;75:391-393.
4. Naidu A, Havard DB, Ray JM, et al. Oral and maxillofacial pathology case of the month. Kaposi’s sarcoma. Tex Dent J. 2011;128:376-377, 382-383.
5. Lawson G, Matar N, Kesch S, et al. Laryngeal Kaposi sarcoma: case report and literature review. B-ENT. 2010;6:285-288.
6. Sullivan RJ, Pantanowitz L, Casper C, et al. HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47:1209-1215.
7. Uldrick TS, Whitby D. Update on KSHV epidemiology, Kaposi sarcoma pathogenesis, and treatment of Kaposi sarcoma [published online ahead of print March 4, 2011]. Cancer Lett. 2011;305:150-162.
8. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
9. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
10. Shiels MS, Pfeiffer RM, Hall HI, et al. Proportions of Kaposi sarcoma, selected non-Hodgkin lymphomas, and cervical cancer in the United States occurring in persons with AIDS, 1980-2007 [published correction appears in JAMA. 2011;306:1548]. JAMA. 2011;305:1450-1459.
Case Report
A 45-year-old man presented with persistent swelling and “black-and-blue” lesions on the legs, feet, and toes of 6 months’ duration. The painless lesions first appeared on the left lower leg and then began to appear on the right leg in recent months. Three weeks prior to presentation, he developed swelling of the left lower leg during hospitalization for a lumbar laminectomy. A venous ultrasound was negative for a deep vein thrombosis. He denied trauma or history of bleeding diathesis. He did not report symptoms of dyspnea, angina, or claudication, and a review of systems was unremarkable.
The patient’s medical history included spinal stenosis, chronic back pain, osteoarthritis, and anxiety. His medications included oxycodone, zolpidem, and alprazolam. In addition to a recent lumbar laminectomy, he had undergone extensive dental work in the last 6 months. The patient denied the use of cigarettes, alcohol, or intravenous drugs.
Physical examination revealed scattered, purple, segmented patches on the dorsal and plantar aspects of the feet, both calves, both heels, and toes (Figure 1). Mild nonpitting edema was present below the left knee along with edema on the dorsum of the left foot. The distribution of the lesions was initially suggestive of cholesterol embolization syndrome; however, both the femoral and posterior tibial pulses were symmetric and palpable (+2). Well-demarcated violaceous plaques with central clearing and a rustlike discoloration were noted on the hard and soft palates. Cervical lymphadenopathy was not present.
|
Laboratory tests including a repeat venous ultrasound of the left lower leg revealed no evidence of deep vein thrombosis. Ankle brachial index revealed no abnormalities and blood flow to the lower legs was adequate. Computed tomography scans of the chest, abdomen, and pelvis were unremarkable except for mild splenomegaly and moderate cardiomegaly. Lastly, human immunodeficiency virus (HIV) 1 and HIV-2 enzyme immunoassay was reactive.
Histopathologic examination of a punch biopsy from the right fourth toe was representative of the plaque stage of Kaposi sarcoma (KS) with a diffuse collection of extravasated erythrocytes and neoplastic vascular proliferation among a background of numerous plasma cells and hemosiderophages (Figure 2). Higher magnification illustrated the promontory sign, whereby native vessels encroach on neoplastic slitlike vascular spaces (Figure 3). A final diagnosis of AIDS-related KS was made. The patient was referred to an infectious disease specialist for evaluation of his CD4 levels and HIV management.
Comment
Kaposi sarcoma is a neoplastic proliferation of the blood vessels in the skin characterized by the formation of violaceous macules and papules that often appear on a single distal extremity, such as the foot. Over time the lesions can develop on the opposite extremity and coalesce into poorly demarcated plaques and nodules with accompanying stasis and lymphedema of the involved extremities.1 Evolution of the lesions depends on the KS subtype. The most common clinical variant of KS is the classic form, which primarily is seen in those of Mediterranean, Eastern European, or Ashkenazi Jewish descent, with a predilection for men and older adults.1,2 The endemic form of KS, or African KS, is more aggressive with rapid visceral involvement and rare skin lesions; it is common among prepubertal children in sub-Saharan Africa with no predilection for either sex.2 In the setting of severe immune suppression, organ transplantation, or chemotherapy, a third KS subtype known as iatrogenic KS can occur. The clinical course of iatrogenic KS may range from scattered cutaneous lesions to diffuse involvement secondary to increased dosages and long-term use of immunosuppressive agents.2
Our patient had AIDS-related or epidemic KS. AIDS-related KS is largely predominant among homosexual men, but due to the awareness of safe sexual practices and the introduction of highly active antiretroviral therapy (HAART), KS incidence in the United States has declined.1,2 However, despite recent advances in HIV therapy, AIDS-related KS is still the most common neoplasm seen in AIDS patients and is the presenting manifestation of AIDS in up to 30% of cases.3 Up to 22% of cases first appear on the gingiva, hard palate, and tongue, with concomitant dysphagia and airway obstruction in severe cases.4,5 More advanced cases of AIDS-related KS can present with initial symptoms such as abdominal pain, melena, dyspnea, lymphadenopathy, and weight loss, which suggests involvement of the gastrointestinal tract, lungs, and other organ systems.
Regardless of the subtype, the etiology of KS currently is thought to be secondary to infection with human herpesvirus 8 (HHV-8), also known as Kaposi sarcoma–associated herpesvirus (KSHV).1 Human immunodeficiency virus infection can enhance KSHV expression through the HIV transactivator protein, which activates KSHV oncogenes and angiogenic growth factors to promote the development of KS lesions.2,6Likewise, KSHV enhances HIV upregulation through latency-associated nuclear antigen, a protein that interacts with HIV Tat protein to further activate long terminal repeats of HIV-1.2
The differential diagnosis of KS is broad. The slightly elevated, pinkish reddish discolorations of KS may resemble verruca plana and/or squamous cell carcinoma on visual observation, whereas nodular KS may resemble giant cell granuloma, pyogenic granuloma, or hemangiopericytoma.4,7 Cases of KS with lymph node involvement may include a differential diagnosis of lymphoma, angiosarcoma, and bacillary angiomatosis.7 Other vascular pathologies that may be considered in the differential diagnosis include vascular tumors (eg, spindle cell hemangioma), fibrohistiocytic tumors (eg, dermatofibrosarcoma protuberans), and a collection of spindle cell mesenchymal tumors.8
Kaposi sarcoma progresses through several histologic stages beginning with the patch stage, then progressing to the plaque stage, and finally culminating in the nodular stage. The patch stage is the first stage in KS progression and a crowded dermis can be seen with the formation of slitlike vascular spaces lined by endothelial cells with red blood cell extravasation into the lumens of newly formed vascular channels, hence demonstrating the promontory sign.8 In the plaque stage, the promontory sign still is present and there is a greater presence of slitlike spaces, giving the micrograph an overall sievelike appearance. Erythrocytes can be found residing within the clear cytoplasm of spindled endothelial cells, leading to the development of autolumination. Finally, the nodular stage is characterized by a neoplastic proliferation of monomorphic spindle cells that form fasciclelike nests in the dermis.8 To distinguish KS from other angioproliferative tumors, one can stain for HHV-8 latent nuclear antigen-1, which is found in the nuclei of infected endothelial cells.1,8,9
Kaposi sarcoma is treated through a variety of mechanisms depending on the subtype. Classic KS lesions often can be observed as they a follow a benign and nonaggressive course.1 Highly active antiretroviral therapy is the mainstay of care in AIDS-related KS and has led to regression of lesions and a remarkable decline in the incidence of KS.3 The HAART regimen consists of a protease inhibitor or nonnucleoside reverse-transcriptase inhibitor with the addition of 2 nucleoside reverse-transcriptase inhibitors. More advanced or refractory cases of KS often require dual treatment with HAART and a chemotherapy agent such as pegylated liposomal doxorubicin. Combination therapy has been shown to result in stronger therapeutic responses and lower relapse rates in contrast to HAART alone.7 Patients also may consider other treatment modalities to manage KS lesions such as surgical removal of lesions, laser therapy, paclitaxel, interferon alfa, oral etoposide, thalidomide, and topical therapies such as imiquimod cream 5% and alitretinoin.1,7
Conclusion
Kaposi sarcoma is a rare but concerning dermatologic condition that signals the need for further diagnostic evaluation. Coexpression of viruses such as HIV and HHV-8 can result in a more virulent and rapid progression of KS to encompass both mucous membrane and systemic involvement. Our patient’s lesions were the first presenting sign of HIV infection despite being asymptomatic at the time of diagnosis, which is alarming in the sense that more than 21% of HIV-infected individuals in the United States have not been clinically diagnosed.10 Inquiry of HIV risk factors and routine screening for HIV should be performed in refractory cases of skin disease as an underlying clue to further investigate the immune system. We present our unique case of mucocutaneous development of KS in an asymptomatic HIV-positive man to stress the importance of KS recognition and management.
Case Report
A 45-year-old man presented with persistent swelling and “black-and-blue” lesions on the legs, feet, and toes of 6 months’ duration. The painless lesions first appeared on the left lower leg and then began to appear on the right leg in recent months. Three weeks prior to presentation, he developed swelling of the left lower leg during hospitalization for a lumbar laminectomy. A venous ultrasound was negative for a deep vein thrombosis. He denied trauma or history of bleeding diathesis. He did not report symptoms of dyspnea, angina, or claudication, and a review of systems was unremarkable.
The patient’s medical history included spinal stenosis, chronic back pain, osteoarthritis, and anxiety. His medications included oxycodone, zolpidem, and alprazolam. In addition to a recent lumbar laminectomy, he had undergone extensive dental work in the last 6 months. The patient denied the use of cigarettes, alcohol, or intravenous drugs.
Physical examination revealed scattered, purple, segmented patches on the dorsal and plantar aspects of the feet, both calves, both heels, and toes (Figure 1). Mild nonpitting edema was present below the left knee along with edema on the dorsum of the left foot. The distribution of the lesions was initially suggestive of cholesterol embolization syndrome; however, both the femoral and posterior tibial pulses were symmetric and palpable (+2). Well-demarcated violaceous plaques with central clearing and a rustlike discoloration were noted on the hard and soft palates. Cervical lymphadenopathy was not present.
|
Laboratory tests including a repeat venous ultrasound of the left lower leg revealed no evidence of deep vein thrombosis. Ankle brachial index revealed no abnormalities and blood flow to the lower legs was adequate. Computed tomography scans of the chest, abdomen, and pelvis were unremarkable except for mild splenomegaly and moderate cardiomegaly. Lastly, human immunodeficiency virus (HIV) 1 and HIV-2 enzyme immunoassay was reactive.
Histopathologic examination of a punch biopsy from the right fourth toe was representative of the plaque stage of Kaposi sarcoma (KS) with a diffuse collection of extravasated erythrocytes and neoplastic vascular proliferation among a background of numerous plasma cells and hemosiderophages (Figure 2). Higher magnification illustrated the promontory sign, whereby native vessels encroach on neoplastic slitlike vascular spaces (Figure 3). A final diagnosis of AIDS-related KS was made. The patient was referred to an infectious disease specialist for evaluation of his CD4 levels and HIV management.
Comment
Kaposi sarcoma is a neoplastic proliferation of the blood vessels in the skin characterized by the formation of violaceous macules and papules that often appear on a single distal extremity, such as the foot. Over time the lesions can develop on the opposite extremity and coalesce into poorly demarcated plaques and nodules with accompanying stasis and lymphedema of the involved extremities.1 Evolution of the lesions depends on the KS subtype. The most common clinical variant of KS is the classic form, which primarily is seen in those of Mediterranean, Eastern European, or Ashkenazi Jewish descent, with a predilection for men and older adults.1,2 The endemic form of KS, or African KS, is more aggressive with rapid visceral involvement and rare skin lesions; it is common among prepubertal children in sub-Saharan Africa with no predilection for either sex.2 In the setting of severe immune suppression, organ transplantation, or chemotherapy, a third KS subtype known as iatrogenic KS can occur. The clinical course of iatrogenic KS may range from scattered cutaneous lesions to diffuse involvement secondary to increased dosages and long-term use of immunosuppressive agents.2
Our patient had AIDS-related or epidemic KS. AIDS-related KS is largely predominant among homosexual men, but due to the awareness of safe sexual practices and the introduction of highly active antiretroviral therapy (HAART), KS incidence in the United States has declined.1,2 However, despite recent advances in HIV therapy, AIDS-related KS is still the most common neoplasm seen in AIDS patients and is the presenting manifestation of AIDS in up to 30% of cases.3 Up to 22% of cases first appear on the gingiva, hard palate, and tongue, with concomitant dysphagia and airway obstruction in severe cases.4,5 More advanced cases of AIDS-related KS can present with initial symptoms such as abdominal pain, melena, dyspnea, lymphadenopathy, and weight loss, which suggests involvement of the gastrointestinal tract, lungs, and other organ systems.
Regardless of the subtype, the etiology of KS currently is thought to be secondary to infection with human herpesvirus 8 (HHV-8), also known as Kaposi sarcoma–associated herpesvirus (KSHV).1 Human immunodeficiency virus infection can enhance KSHV expression through the HIV transactivator protein, which activates KSHV oncogenes and angiogenic growth factors to promote the development of KS lesions.2,6Likewise, KSHV enhances HIV upregulation through latency-associated nuclear antigen, a protein that interacts with HIV Tat protein to further activate long terminal repeats of HIV-1.2
The differential diagnosis of KS is broad. The slightly elevated, pinkish reddish discolorations of KS may resemble verruca plana and/or squamous cell carcinoma on visual observation, whereas nodular KS may resemble giant cell granuloma, pyogenic granuloma, or hemangiopericytoma.4,7 Cases of KS with lymph node involvement may include a differential diagnosis of lymphoma, angiosarcoma, and bacillary angiomatosis.7 Other vascular pathologies that may be considered in the differential diagnosis include vascular tumors (eg, spindle cell hemangioma), fibrohistiocytic tumors (eg, dermatofibrosarcoma protuberans), and a collection of spindle cell mesenchymal tumors.8
Kaposi sarcoma progresses through several histologic stages beginning with the patch stage, then progressing to the plaque stage, and finally culminating in the nodular stage. The patch stage is the first stage in KS progression and a crowded dermis can be seen with the formation of slitlike vascular spaces lined by endothelial cells with red blood cell extravasation into the lumens of newly formed vascular channels, hence demonstrating the promontory sign.8 In the plaque stage, the promontory sign still is present and there is a greater presence of slitlike spaces, giving the micrograph an overall sievelike appearance. Erythrocytes can be found residing within the clear cytoplasm of spindled endothelial cells, leading to the development of autolumination. Finally, the nodular stage is characterized by a neoplastic proliferation of monomorphic spindle cells that form fasciclelike nests in the dermis.8 To distinguish KS from other angioproliferative tumors, one can stain for HHV-8 latent nuclear antigen-1, which is found in the nuclei of infected endothelial cells.1,8,9
Kaposi sarcoma is treated through a variety of mechanisms depending on the subtype. Classic KS lesions often can be observed as they a follow a benign and nonaggressive course.1 Highly active antiretroviral therapy is the mainstay of care in AIDS-related KS and has led to regression of lesions and a remarkable decline in the incidence of KS.3 The HAART regimen consists of a protease inhibitor or nonnucleoside reverse-transcriptase inhibitor with the addition of 2 nucleoside reverse-transcriptase inhibitors. More advanced or refractory cases of KS often require dual treatment with HAART and a chemotherapy agent such as pegylated liposomal doxorubicin. Combination therapy has been shown to result in stronger therapeutic responses and lower relapse rates in contrast to HAART alone.7 Patients also may consider other treatment modalities to manage KS lesions such as surgical removal of lesions, laser therapy, paclitaxel, interferon alfa, oral etoposide, thalidomide, and topical therapies such as imiquimod cream 5% and alitretinoin.1,7
Conclusion
Kaposi sarcoma is a rare but concerning dermatologic condition that signals the need for further diagnostic evaluation. Coexpression of viruses such as HIV and HHV-8 can result in a more virulent and rapid progression of KS to encompass both mucous membrane and systemic involvement. Our patient’s lesions were the first presenting sign of HIV infection despite being asymptomatic at the time of diagnosis, which is alarming in the sense that more than 21% of HIV-infected individuals in the United States have not been clinically diagnosed.10 Inquiry of HIV risk factors and routine screening for HIV should be performed in refractory cases of skin disease as an underlying clue to further investigate the immune system. We present our unique case of mucocutaneous development of KS in an asymptomatic HIV-positive man to stress the importance of KS recognition and management.
1. Jan MM, Laskas JW, Griffin TD. Eruptive Kaposi sarcoma: an unusual presentation in an HIV-negative patient. Cutis. 2011;87:34-38.
2. Geraminejad P, Memar O, Aronson I, et al. Kaposi’s sarcoma and other manifestations of human herpesvirus 8. J Am Acad Dermatol. 2002;47:641-655.
3. Kharkar V, Gutte RM, Khopkar U, et al. Kaposi’s sarcoma: a presenting manifestation of HIV infection in an Indian. Indian J Dermatol Venereol Leprol. 2009;75:391-393.
4. Naidu A, Havard DB, Ray JM, et al. Oral and maxillofacial pathology case of the month. Kaposi’s sarcoma. Tex Dent J. 2011;128:376-377, 382-383.
5. Lawson G, Matar N, Kesch S, et al. Laryngeal Kaposi sarcoma: case report and literature review. B-ENT. 2010;6:285-288.
6. Sullivan RJ, Pantanowitz L, Casper C, et al. HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47:1209-1215.
7. Uldrick TS, Whitby D. Update on KSHV epidemiology, Kaposi sarcoma pathogenesis, and treatment of Kaposi sarcoma [published online ahead of print March 4, 2011]. Cancer Lett. 2011;305:150-162.
8. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
9. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
10. Shiels MS, Pfeiffer RM, Hall HI, et al. Proportions of Kaposi sarcoma, selected non-Hodgkin lymphomas, and cervical cancer in the United States occurring in persons with AIDS, 1980-2007 [published correction appears in JAMA. 2011;306:1548]. JAMA. 2011;305:1450-1459.
1. Jan MM, Laskas JW, Griffin TD. Eruptive Kaposi sarcoma: an unusual presentation in an HIV-negative patient. Cutis. 2011;87:34-38.
2. Geraminejad P, Memar O, Aronson I, et al. Kaposi’s sarcoma and other manifestations of human herpesvirus 8. J Am Acad Dermatol. 2002;47:641-655.
3. Kharkar V, Gutte RM, Khopkar U, et al. Kaposi’s sarcoma: a presenting manifestation of HIV infection in an Indian. Indian J Dermatol Venereol Leprol. 2009;75:391-393.
4. Naidu A, Havard DB, Ray JM, et al. Oral and maxillofacial pathology case of the month. Kaposi’s sarcoma. Tex Dent J. 2011;128:376-377, 382-383.
5. Lawson G, Matar N, Kesch S, et al. Laryngeal Kaposi sarcoma: case report and literature review. B-ENT. 2010;6:285-288.
6. Sullivan RJ, Pantanowitz L, Casper C, et al. HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47:1209-1215.
7. Uldrick TS, Whitby D. Update on KSHV epidemiology, Kaposi sarcoma pathogenesis, and treatment of Kaposi sarcoma [published online ahead of print March 4, 2011]. Cancer Lett. 2011;305:150-162.
8. Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
9. Cheuk W, Wong KO, Wong CS, et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
10. Shiels MS, Pfeiffer RM, Hall HI, et al. Proportions of Kaposi sarcoma, selected non-Hodgkin lymphomas, and cervical cancer in the United States occurring in persons with AIDS, 1980-2007 [published correction appears in JAMA. 2011;306:1548]. JAMA. 2011;305:1450-1459.
Practice Points
- Kaposi sarcoma is a rare malignant proliferation of endothelial cells with many subtypes.
- Kaposi sarcoma in patients with coexpression of human immunodeficiency virus and human herpesvirus 8 often have a more virulent and rapid progression of disease.