User login
European Hematology Association (EHA): Annual Congress
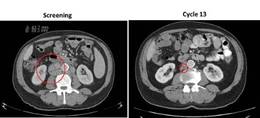
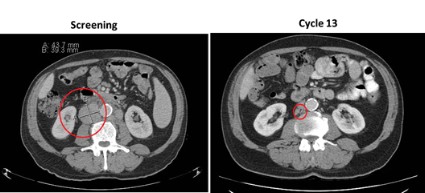
Surgical bleeding risk ‘remarkable’ with platelet disorders
VIENNA – Inherited platelet function disorders are associated with a significant bleeding risk during surgery, a retrospective, multicenter study shows.
“The frequency of excessive bleeding assessed by one of three criteria at surgery is pretty remarkable because we had rates ranging from 13% to about 19% depending on the assessment criteria,” Dr. Paolo Gresele said at the annual congress of the European Hematology Association.

Any excessive bleeding by any criteria took place in about 23% of patients.
Although conventionally considered rare, inherited platelet function disorders (IPFD) are more frequent than previously thought, and recent advances in their diagnosis have expanded the forms and number of reported patients.
Only a handful of case reports or small case series have evaluated the bleeding risk associated with surgery in patients with IPFD. As a result, the exact bleeding risk and most appropriate management options in IPFD are not known, Dr. Gresele of University of Perugia (Italy) said.
He reported on a retrospective study involving patients with IPFD enrolled at 42 centers from 16 countries. The median age of the 205 patients was 35 years, and 56% were women; 89 had Glanzmann thrombasthenia, 46 had primary secretion defect, 14 had combined alpha-delta granule deficiency, 11 had Hermansky-Pudlak syndrome, 9 had gray platelet syndrome, and 36 had other forms of IPFD.
Data were collected from 389 procedures, of which 54.5% were surgeries, 30% were dental procedures, and 15.5% were invasive procedures. The most frequent surgeries were otolaryngologic, abdominal, orthopedic, gynecologic, urologic, and dermatologic.
The frequency of excessive bleeding at surgery was about 19.7% by the Bleeding Academic Research Consortium (BARC) bleeding classification, 17% by subjective evaluation by the surgeon or patient, and 13% based on duration of bleeding at surgery of at least 6 or more hours, Dr. Gresele said.
The World Health Organization (WHO) bleeding scale before surgery was quite predictive of excessive bleeding at surgery, with patients at grade 3 and 4 having an odds ratio for excessive bleeding of 9.22 and 28, respectively, compared with patients at grade 0, he said.
Somewhat unexpectedly, the most serious bleeders based on a BARC of at least 2 or more at surgery were patients with Hermansky-Pudlak syndrome, Glanzmann thrombasthenia, and gray platelet syndrome. Those with primary secretion defect and combined alpha-delta granule deficiency had a relatively low risk of bleeding at surgery, he said.
The procedures associated with more excessive bleeding were urologic surgery, gynecologic surgery, and abdominal surgery.
Preparation for surgery was made in 82% of procedures and included prophylactic platelet transfusions in 38.5%, desmopressin (DDAVP) in about 19%, antifibrinolytic agents in about 12%, factor VIIa in less than 10%, and other interventions such as cryoprecipitate, fibrinogen, fresh frozen plasma, intravenous immunoglobulin, suture, and local hemostatic agent in less than 5%.
Generally all measures had a good ability to prevent excessive bleeding, bringing rates down to about 20% or less, Dr. Gresele said. The two exceptions were no prophylaxis and “other” interventions, where the bleeding rate reached a high of about 45% for the latter. In contrast, bleeding risk was significantly reduced when any prophylaxis (odds ratio, 0.38) or DDAVP (OR, 0.24) were used.
In 87 cases, however, emergency treatment for excessive bleeding was required, he said. Platelet transfusions were the most common treatment, given in roughly half of patients, followed by antifibrinolytic agents. The success rate was about 80% for all measures, except for “other” interventions, which had a success rate of about 65%.
“Treatment of excessive bleeding is made mostly with platelet transfusions or antifibrinolytic agents and is able to stop bleeding in many, but not all cases,” Dr. Gresele concluded.
On Twitter @pwendl
VIENNA – Inherited platelet function disorders are associated with a significant bleeding risk during surgery, a retrospective, multicenter study shows.
“The frequency of excessive bleeding assessed by one of three criteria at surgery is pretty remarkable because we had rates ranging from 13% to about 19% depending on the assessment criteria,” Dr. Paolo Gresele said at the annual congress of the European Hematology Association.
Any excessive bleeding by any criteria took place in about 23% of patients.
Although conventionally considered rare, inherited platelet function disorders (IPFD) are more frequent than previously thought, and recent advances in their diagnosis have expanded the forms and number of reported patients.
Only a handful of case reports or small case series have evaluated the bleeding risk associated with surgery in patients with IPFD. As a result, the exact bleeding risk and most appropriate management options in IPFD are not known, Dr. Gresele of University of Perugia (Italy) said.
He reported on a retrospective study involving patients with IPFD enrolled at 42 centers from 16 countries. The median age of the 205 patients was 35 years, and 56% were women; 89 had Glanzmann thrombasthenia, 46 had primary secretion defect, 14 had combined alpha-delta granule deficiency, 11 had Hermansky-Pudlak syndrome, 9 had gray platelet syndrome, and 36 had other forms of IPFD.
Data were collected from 389 procedures, of which 54.5% were surgeries, 30% were dental procedures, and 15.5% were invasive procedures. The most frequent surgeries were otolaryngologic, abdominal, orthopedic, gynecologic, urologic, and dermatologic.
The frequency of excessive bleeding at surgery was about 19.7% by the Bleeding Academic Research Consortium (BARC) bleeding classification, 17% by subjective evaluation by the surgeon or patient, and 13% based on duration of bleeding at surgery of at least 6 or more hours, Dr. Gresele said.
The World Health Organization (WHO) bleeding scale before surgery was quite predictive of excessive bleeding at surgery, with patients at grade 3 and 4 having an odds ratio for excessive bleeding of 9.22 and 28, respectively, compared with patients at grade 0, he said.
Somewhat unexpectedly, the most serious bleeders based on a BARC of at least 2 or more at surgery were patients with Hermansky-Pudlak syndrome, Glanzmann thrombasthenia, and gray platelet syndrome. Those with primary secretion defect and combined alpha-delta granule deficiency had a relatively low risk of bleeding at surgery, he said.
The procedures associated with more excessive bleeding were urologic surgery, gynecologic surgery, and abdominal surgery.
Preparation for surgery was made in 82% of procedures and included prophylactic platelet transfusions in 38.5%, desmopressin (DDAVP) in about 19%, antifibrinolytic agents in about 12%, factor VIIa in less than 10%, and other interventions such as cryoprecipitate, fibrinogen, fresh frozen plasma, intravenous immunoglobulin, suture, and local hemostatic agent in less than 5%.
Generally all measures had a good ability to prevent excessive bleeding, bringing rates down to about 20% or less, Dr. Gresele said. The two exceptions were no prophylaxis and “other” interventions, where the bleeding rate reached a high of about 45% for the latter. In contrast, bleeding risk was significantly reduced when any prophylaxis (odds ratio, 0.38) or DDAVP (OR, 0.24) were used.
In 87 cases, however, emergency treatment for excessive bleeding was required, he said. Platelet transfusions were the most common treatment, given in roughly half of patients, followed by antifibrinolytic agents. The success rate was about 80% for all measures, except for “other” interventions, which had a success rate of about 65%.
“Treatment of excessive bleeding is made mostly with platelet transfusions or antifibrinolytic agents and is able to stop bleeding in many, but not all cases,” Dr. Gresele concluded.
On Twitter @pwendl
VIENNA – Inherited platelet function disorders are associated with a significant bleeding risk during surgery, a retrospective, multicenter study shows.
“The frequency of excessive bleeding assessed by one of three criteria at surgery is pretty remarkable because we had rates ranging from 13% to about 19% depending on the assessment criteria,” Dr. Paolo Gresele said at the annual congress of the European Hematology Association.
Any excessive bleeding by any criteria took place in about 23% of patients.
Although conventionally considered rare, inherited platelet function disorders (IPFD) are more frequent than previously thought, and recent advances in their diagnosis have expanded the forms and number of reported patients.
Only a handful of case reports or small case series have evaluated the bleeding risk associated with surgery in patients with IPFD. As a result, the exact bleeding risk and most appropriate management options in IPFD are not known, Dr. Gresele of University of Perugia (Italy) said.
He reported on a retrospective study involving patients with IPFD enrolled at 42 centers from 16 countries. The median age of the 205 patients was 35 years, and 56% were women; 89 had Glanzmann thrombasthenia, 46 had primary secretion defect, 14 had combined alpha-delta granule deficiency, 11 had Hermansky-Pudlak syndrome, 9 had gray platelet syndrome, and 36 had other forms of IPFD.
Data were collected from 389 procedures, of which 54.5% were surgeries, 30% were dental procedures, and 15.5% were invasive procedures. The most frequent surgeries were otolaryngologic, abdominal, orthopedic, gynecologic, urologic, and dermatologic.
The frequency of excessive bleeding at surgery was about 19.7% by the Bleeding Academic Research Consortium (BARC) bleeding classification, 17% by subjective evaluation by the surgeon or patient, and 13% based on duration of bleeding at surgery of at least 6 or more hours, Dr. Gresele said.
The World Health Organization (WHO) bleeding scale before surgery was quite predictive of excessive bleeding at surgery, with patients at grade 3 and 4 having an odds ratio for excessive bleeding of 9.22 and 28, respectively, compared with patients at grade 0, he said.
Somewhat unexpectedly, the most serious bleeders based on a BARC of at least 2 or more at surgery were patients with Hermansky-Pudlak syndrome, Glanzmann thrombasthenia, and gray platelet syndrome. Those with primary secretion defect and combined alpha-delta granule deficiency had a relatively low risk of bleeding at surgery, he said.
The procedures associated with more excessive bleeding were urologic surgery, gynecologic surgery, and abdominal surgery.
Preparation for surgery was made in 82% of procedures and included prophylactic platelet transfusions in 38.5%, desmopressin (DDAVP) in about 19%, antifibrinolytic agents in about 12%, factor VIIa in less than 10%, and other interventions such as cryoprecipitate, fibrinogen, fresh frozen plasma, intravenous immunoglobulin, suture, and local hemostatic agent in less than 5%.
Generally all measures had a good ability to prevent excessive bleeding, bringing rates down to about 20% or less, Dr. Gresele said. The two exceptions were no prophylaxis and “other” interventions, where the bleeding rate reached a high of about 45% for the latter. In contrast, bleeding risk was significantly reduced when any prophylaxis (odds ratio, 0.38) or DDAVP (OR, 0.24) were used.
In 87 cases, however, emergency treatment for excessive bleeding was required, he said. Platelet transfusions were the most common treatment, given in roughly half of patients, followed by antifibrinolytic agents. The success rate was about 80% for all measures, except for “other” interventions, which had a success rate of about 65%.
“Treatment of excessive bleeding is made mostly with platelet transfusions or antifibrinolytic agents and is able to stop bleeding in many, but not all cases,” Dr. Gresele concluded.
On Twitter @pwendl
AT THE EHA CONGRESS
Key clinical point: Inherited platelet function disorders are associated with a significant surgical bleeding risk.
Major finding: The frequency of excessive bleeding ranged from about 13% to about 19%.
Data source: Retrospective, multicenter study in 205 patients with inherited platelet function disorders and 389 procedures.
Disclosures: Dr. Gesele reported no financial disclosures.

VIDEO: Are chemo-free regimens possible for CLL?
MILAN – The Food and Drug Administration has given full approval for ibrutinib for patients with chronic lymphocytic leukemia who have received at least one prior therapy and for those who have a deletion in chromosome 17 (17p deletion) and may or may not have received previous treatment.
At the annual congress of the European Hematology Association, Dr. Peter Hillmen discusses the results he presented from RESONATE, the study used to convert conditional, accelerated approval to full approval. The study evaluated daily ibrutinib monotherapy versus the anti-CD20 antibody ofatumumab, for patients with relapsed or refractory chronic lymphocytic leukemia (CLL).
Dr. Hillmen, professor of experimental hematology at the University of Leeds, England, also outlines some of the research in the works to evaluate ibrutinib as frontline therapy for patients with CLL and the possibility of chemotherapy-free regimens. He disclosed a consultancy role and honoraria from Pharmacyclics.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
MILAN – The Food and Drug Administration has given full approval for ibrutinib for patients with chronic lymphocytic leukemia who have received at least one prior therapy and for those who have a deletion in chromosome 17 (17p deletion) and may or may not have received previous treatment.
At the annual congress of the European Hematology Association, Dr. Peter Hillmen discusses the results he presented from RESONATE, the study used to convert conditional, accelerated approval to full approval. The study evaluated daily ibrutinib monotherapy versus the anti-CD20 antibody ofatumumab, for patients with relapsed or refractory chronic lymphocytic leukemia (CLL).
Dr. Hillmen, professor of experimental hematology at the University of Leeds, England, also outlines some of the research in the works to evaluate ibrutinib as frontline therapy for patients with CLL and the possibility of chemotherapy-free regimens. He disclosed a consultancy role and honoraria from Pharmacyclics.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
MILAN – The Food and Drug Administration has given full approval for ibrutinib for patients with chronic lymphocytic leukemia who have received at least one prior therapy and for those who have a deletion in chromosome 17 (17p deletion) and may or may not have received previous treatment.
At the annual congress of the European Hematology Association, Dr. Peter Hillmen discusses the results he presented from RESONATE, the study used to convert conditional, accelerated approval to full approval. The study evaluated daily ibrutinib monotherapy versus the anti-CD20 antibody ofatumumab, for patients with relapsed or refractory chronic lymphocytic leukemia (CLL).
Dr. Hillmen, professor of experimental hematology at the University of Leeds, England, also outlines some of the research in the works to evaluate ibrutinib as frontline therapy for patients with CLL and the possibility of chemotherapy-free regimens. He disclosed a consultancy role and honoraria from Pharmacyclics.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE EHA CONGRESS

VIDEO: ABT-199 alone and in combination shows promise against advanced CLL
MILAN – Dr. Andrew W. Roberts discusses the high response rates that have occurred with the Bcl-2 inhibitor ABT-199, alone and in combination, against refractory or relapsed chronic lymphocytic leukemia.
Drug-induced tumor lysis syndrome in some patients appears to have been avoided with a new modified dosing regimen, said Dr. Roberts, who presented phase Ib study results of ABT-199 in combination with rituximab at the annual congress of the European Hematology Association. In the interview, Dr. Roberts of the Royal Melbourne Hospital and Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia, describes the study results and future plans for ABT-199 research.
On Twitter @nikolaideslaura
MILAN – Dr. Andrew W. Roberts discusses the high response rates that have occurred with the Bcl-2 inhibitor ABT-199, alone and in combination, against refractory or relapsed chronic lymphocytic leukemia.
Drug-induced tumor lysis syndrome in some patients appears to have been avoided with a new modified dosing regimen, said Dr. Roberts, who presented phase Ib study results of ABT-199 in combination with rituximab at the annual congress of the European Hematology Association. In the interview, Dr. Roberts of the Royal Melbourne Hospital and Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia, describes the study results and future plans for ABT-199 research.
On Twitter @nikolaideslaura
MILAN – Dr. Andrew W. Roberts discusses the high response rates that have occurred with the Bcl-2 inhibitor ABT-199, alone and in combination, against refractory or relapsed chronic lymphocytic leukemia.
Drug-induced tumor lysis syndrome in some patients appears to have been avoided with a new modified dosing regimen, said Dr. Roberts, who presented phase Ib study results of ABT-199 in combination with rituximab at the annual congress of the European Hematology Association. In the interview, Dr. Roberts of the Royal Melbourne Hospital and Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia, describes the study results and future plans for ABT-199 research.
On Twitter @nikolaideslaura
AT THE EHA CONGRESS

Depth of molecular response factors into safe TKI withdrawal in CML
MILAN – A full 61.5% of patients with chronic myeloid leukemia in deep molecular remission for more than 1 year on long-term tyrosine kinase inhibitor therapy remained alive and free of relapse 6 months after stopping their TKI in the EURO-SKI trial.
Molecular relapse-free survival at 9 months was 58% and 55% at 12 months.
The preplanned interim analysis after 200 patients allowed the trialists to discard the study’s null hypothesis that molecular relapse-free survival at 6 months would be 40% or less (P less than .0001).
"With less strict inclusion and relapse criteria than the A-STIM study and other trials, stopping is safe and we can continue with the trial," Dr. Susanne Saussele said in a late-breaking abstract session at the annual meeting of the European Hematology Association.
Importantly, interim results from the EURO-SKI (European Stop TKI) study also suggest that the level of molecular response (MR) achieved prior to TKI withdrawal affects molecular relapse-free survival.
Among 197 patients with molecular laboratory results available for exact classification, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
"In the setting of standardized molecular testing within a CML [chronic myeloid leukemia] stopping trial, it seems that molecular remission has an impact on molecular free-survival," said Dr. Saussele of University Medical Centre Mannheim, Germany.
As no statistical test was performed and this was an interim analysis, MR4.5 or MR5 cannot yet be used as a criterion to select patients to withdraw from treatment, she said in an interview.
The findings do confirm results from the recent A-STIM (According to Stop Imatinib) study showing that loss of major molecular response can be used as a practical criterion for restarting imatinib (J. Clin. Oncol. 2014;32:424-30).
Several studies including the STIM (Stop Imatinib) trial and the STOP 2G-TKI (Stop Second Generation Tyrosine Kinase Inhibitors) study have shown that imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna) can be safely withdrawn in a substantial proportion of patients with CML in deep MR.
A number of questions remain open, however, such as which molecular level has to be reached before stopping TKI therapy, the minimal duration of TKI pretreatment or MR4 before stopping, and which prognostic factors influence molecular relapse-free survival, she said.
EURO-SKI was set up to define prognostic markers to increase the rate of patients in durable deep MR after stopping TKI treatment. Other aims are to evaluate methods of molecular monitoring, quality of life, and saved treatment costs per country.
Patients with chronic-phase CML from eight countries were eligible if they were on a TKI for at least 3 years and had a confirmed deep MR, defined as more than a 4 log reduction in BCR-ABL (breakpoint cluster region–Abelson) transcripts for more than 12 months confirmed by three consecutive polymerase chain reaction results.
MR4 status also had to be confirmed in an MR4-standardized laboratory according to criteria by Cross et al. (Leukemia 2012;26:2172-5)
Patients with previous or planned allogeneic stem cell transplantation or a prior TKI failure were excluded.
A total of 103 patients received pretreatment before TKI therapy, mostly with hydroxyurea alone (n = 71) or with interferon (n = 22). Their median age was 53 years.
First-line TKI was imatinib in 194 patients, nilotinib in 3, and dasatinib in 3.
The median duration of TKI therapy was 8 years and median MR4 duration before stopping TKI therapy was 5 years.
Dr. Saussele cautioned that adverse events and quality of life must be taken into account when considering TKI withdrawal in these patients.
A total of 222 adverse events were reported in 98 patients, with 57 events in 37 patients related to treatment stop. None were grade 4.
The most common adverse event was musculoskeletal or joint pain (39 events of all grades and 6 grade 3/4 events). This was first described in the Swedish EURO-SKI patients in 15-20% of patients after TKI withdrawal, prompting the investigators to send an advisory letter to all participating physicians, she remarked.
Other adverse events included sweating, skin disorders, folliculitis, depressive episodes, fatigue, urticaria, and weight loss.
The EURO-SKI trial was recently expanded to enroll 700 patients, with a quality of life analysis expected later this year. The next report on the primary endpoint is expected at the American Society of Hematology annual meeting, Dr. Saussele said.
Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
MILAN – A full 61.5% of patients with chronic myeloid leukemia in deep molecular remission for more than 1 year on long-term tyrosine kinase inhibitor therapy remained alive and free of relapse 6 months after stopping their TKI in the EURO-SKI trial.
Molecular relapse-free survival at 9 months was 58% and 55% at 12 months.
The preplanned interim analysis after 200 patients allowed the trialists to discard the study’s null hypothesis that molecular relapse-free survival at 6 months would be 40% or less (P less than .0001).
"With less strict inclusion and relapse criteria than the A-STIM study and other trials, stopping is safe and we can continue with the trial," Dr. Susanne Saussele said in a late-breaking abstract session at the annual meeting of the European Hematology Association.
Importantly, interim results from the EURO-SKI (European Stop TKI) study also suggest that the level of molecular response (MR) achieved prior to TKI withdrawal affects molecular relapse-free survival.
Among 197 patients with molecular laboratory results available for exact classification, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
"In the setting of standardized molecular testing within a CML [chronic myeloid leukemia] stopping trial, it seems that molecular remission has an impact on molecular free-survival," said Dr. Saussele of University Medical Centre Mannheim, Germany.
As no statistical test was performed and this was an interim analysis, MR4.5 or MR5 cannot yet be used as a criterion to select patients to withdraw from treatment, she said in an interview.
The findings do confirm results from the recent A-STIM (According to Stop Imatinib) study showing that loss of major molecular response can be used as a practical criterion for restarting imatinib (J. Clin. Oncol. 2014;32:424-30).
Several studies including the STIM (Stop Imatinib) trial and the STOP 2G-TKI (Stop Second Generation Tyrosine Kinase Inhibitors) study have shown that imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna) can be safely withdrawn in a substantial proportion of patients with CML in deep MR.
A number of questions remain open, however, such as which molecular level has to be reached before stopping TKI therapy, the minimal duration of TKI pretreatment or MR4 before stopping, and which prognostic factors influence molecular relapse-free survival, she said.
EURO-SKI was set up to define prognostic markers to increase the rate of patients in durable deep MR after stopping TKI treatment. Other aims are to evaluate methods of molecular monitoring, quality of life, and saved treatment costs per country.
Patients with chronic-phase CML from eight countries were eligible if they were on a TKI for at least 3 years and had a confirmed deep MR, defined as more than a 4 log reduction in BCR-ABL (breakpoint cluster region–Abelson) transcripts for more than 12 months confirmed by three consecutive polymerase chain reaction results.
MR4 status also had to be confirmed in an MR4-standardized laboratory according to criteria by Cross et al. (Leukemia 2012;26:2172-5)
Patients with previous or planned allogeneic stem cell transplantation or a prior TKI failure were excluded.
A total of 103 patients received pretreatment before TKI therapy, mostly with hydroxyurea alone (n = 71) or with interferon (n = 22). Their median age was 53 years.
First-line TKI was imatinib in 194 patients, nilotinib in 3, and dasatinib in 3.
The median duration of TKI therapy was 8 years and median MR4 duration before stopping TKI therapy was 5 years.
Dr. Saussele cautioned that adverse events and quality of life must be taken into account when considering TKI withdrawal in these patients.
A total of 222 adverse events were reported in 98 patients, with 57 events in 37 patients related to treatment stop. None were grade 4.
The most common adverse event was musculoskeletal or joint pain (39 events of all grades and 6 grade 3/4 events). This was first described in the Swedish EURO-SKI patients in 15-20% of patients after TKI withdrawal, prompting the investigators to send an advisory letter to all participating physicians, she remarked.
Other adverse events included sweating, skin disorders, folliculitis, depressive episodes, fatigue, urticaria, and weight loss.
The EURO-SKI trial was recently expanded to enroll 700 patients, with a quality of life analysis expected later this year. The next report on the primary endpoint is expected at the American Society of Hematology annual meeting, Dr. Saussele said.
Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
MILAN – A full 61.5% of patients with chronic myeloid leukemia in deep molecular remission for more than 1 year on long-term tyrosine kinase inhibitor therapy remained alive and free of relapse 6 months after stopping their TKI in the EURO-SKI trial.
Molecular relapse-free survival at 9 months was 58% and 55% at 12 months.
The preplanned interim analysis after 200 patients allowed the trialists to discard the study’s null hypothesis that molecular relapse-free survival at 6 months would be 40% or less (P less than .0001).
"With less strict inclusion and relapse criteria than the A-STIM study and other trials, stopping is safe and we can continue with the trial," Dr. Susanne Saussele said in a late-breaking abstract session at the annual meeting of the European Hematology Association.
Importantly, interim results from the EURO-SKI (European Stop TKI) study also suggest that the level of molecular response (MR) achieved prior to TKI withdrawal affects molecular relapse-free survival.
Among 197 patients with molecular laboratory results available for exact classification, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
"In the setting of standardized molecular testing within a CML [chronic myeloid leukemia] stopping trial, it seems that molecular remission has an impact on molecular free-survival," said Dr. Saussele of University Medical Centre Mannheim, Germany.
As no statistical test was performed and this was an interim analysis, MR4.5 or MR5 cannot yet be used as a criterion to select patients to withdraw from treatment, she said in an interview.
The findings do confirm results from the recent A-STIM (According to Stop Imatinib) study showing that loss of major molecular response can be used as a practical criterion for restarting imatinib (J. Clin. Oncol. 2014;32:424-30).
Several studies including the STIM (Stop Imatinib) trial and the STOP 2G-TKI (Stop Second Generation Tyrosine Kinase Inhibitors) study have shown that imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna) can be safely withdrawn in a substantial proportion of patients with CML in deep MR.
A number of questions remain open, however, such as which molecular level has to be reached before stopping TKI therapy, the minimal duration of TKI pretreatment or MR4 before stopping, and which prognostic factors influence molecular relapse-free survival, she said.
EURO-SKI was set up to define prognostic markers to increase the rate of patients in durable deep MR after stopping TKI treatment. Other aims are to evaluate methods of molecular monitoring, quality of life, and saved treatment costs per country.
Patients with chronic-phase CML from eight countries were eligible if they were on a TKI for at least 3 years and had a confirmed deep MR, defined as more than a 4 log reduction in BCR-ABL (breakpoint cluster region–Abelson) transcripts for more than 12 months confirmed by three consecutive polymerase chain reaction results.
MR4 status also had to be confirmed in an MR4-standardized laboratory according to criteria by Cross et al. (Leukemia 2012;26:2172-5)
Patients with previous or planned allogeneic stem cell transplantation or a prior TKI failure were excluded.
A total of 103 patients received pretreatment before TKI therapy, mostly with hydroxyurea alone (n = 71) or with interferon (n = 22). Their median age was 53 years.
First-line TKI was imatinib in 194 patients, nilotinib in 3, and dasatinib in 3.
The median duration of TKI therapy was 8 years and median MR4 duration before stopping TKI therapy was 5 years.
Dr. Saussele cautioned that adverse events and quality of life must be taken into account when considering TKI withdrawal in these patients.
A total of 222 adverse events were reported in 98 patients, with 57 events in 37 patients related to treatment stop. None were grade 4.
The most common adverse event was musculoskeletal or joint pain (39 events of all grades and 6 grade 3/4 events). This was first described in the Swedish EURO-SKI patients in 15-20% of patients after TKI withdrawal, prompting the investigators to send an advisory letter to all participating physicians, she remarked.
Other adverse events included sweating, skin disorders, folliculitis, depressive episodes, fatigue, urticaria, and weight loss.
The EURO-SKI trial was recently expanded to enroll 700 patients, with a quality of life analysis expected later this year. The next report on the primary endpoint is expected at the American Society of Hematology annual meeting, Dr. Saussele said.
Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
AT THE EHA CONGRESS
Key clinical point: Long-time TKI therapy can be stopped in chronic myeloid leukemia in deep molecular remission. The level of molecular response prior to TKI withdrawal affects molecular relapse-free survival.
Major finding: After TKI withdrawal, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
Data source: First interim analysis from a prospective study in 200 CML patients in deep molecular remission.
Disclosures: Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
Depth of molecular response factors into safe TKI withdrawal in CML
MILAN – A full 61.5% of patients with chronic myeloid leukemia in deep molecular remission for more than 1 year on long-term tyrosine kinase inhibitor therapy remained alive and free of relapse 6 months after stopping their TKI in the EURO-SKI trial.
Molecular relapse-free survival at 9 months was 58% and 55% at 12 months.
The preplanned interim analysis after 200 patients allowed the trialists to discard the study’s null hypothesis that molecular relapse-free survival at 6 months would be 40% or less (P less than .0001).
"With less strict inclusion and relapse criteria than the A-STIM study and other trials, stopping is safe and we can continue with the trial," Dr. Susanne Saussele said in a late-breaking abstract session at the annual meeting of the European Hematology Association.
Importantly, interim results from the EURO-SKI (European Stop TKI) study also suggest that the level of molecular response (MR) achieved prior to TKI withdrawal affects molecular relapse-free survival.
Among 197 patients with molecular laboratory results available for exact classification, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
"In the setting of standardized molecular testing within a CML [chronic myeloid leukemia] stopping trial, it seems that molecular remission has an impact on molecular free-survival," said Dr. Saussele of University Medical Centre Mannheim, Germany.
As no statistical test was performed and this was an interim analysis, MR4.5 or MR5 cannot yet be used as a criterion to select patients to withdraw from treatment, she said in an interview.
The findings do confirm results from the recent A-STIM (According to Stop Imatinib) study showing that loss of major molecular response can be used as a practical criterion for restarting imatinib (J. Clin. Oncol. 2014;32:424-30).
Several studies including the STIM (Stop Imatinib) trial and the STOP 2G-TKI (Stop Second Generation Tyrosine Kinase Inhibitors) study have shown that imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna) can be safely withdrawn in a substantial proportion of patients with CML in deep MR.
A number of questions remain open, however, such as which molecular level has to be reached before stopping TKI therapy, the minimal duration of TKI pretreatment or MR4 before stopping, and which prognostic factors influence molecular relapse-free survival, she said.
EURO-SKI was set up to define prognostic markers to increase the rate of patients in durable deep MR after stopping TKI treatment. Other aims are to evaluate methods of molecular monitoring, quality of life, and saved treatment costs per country.
Patients with chronic-phase CML from eight countries were eligible if they were on a TKI for at least 3 years and had a confirmed deep MR, defined as more than a 4 log reduction in BCR-ABL (breakpoint cluster region–Abelson) transcripts for more than 12 months confirmed by three consecutive polymerase chain reaction results.
MR4 status also had to be confirmed in an MR4-standardized laboratory according to criteria by Cross et al. (Leukemia 2012;26:2172-5)
Patients with previous or planned allogeneic stem cell transplantation or a prior TKI failure were excluded.
A total of 103 patients received pretreatment before TKI therapy, mostly with hydroxyurea alone (n = 71) or with interferon (n = 22). Their median age was 53 years.
First-line TKI was imatinib in 194 patients, nilotinib in 3, and dasatinib in 3.
The median duration of TKI therapy was 8 years and median MR4 duration before stopping TKI therapy was 5 years.
Dr. Saussele cautioned that adverse events and quality of life must be taken into account when considering TKI withdrawal in these patients.
A total of 222 adverse events were reported in 98 patients, with 57 events in 37 patients related to treatment stop. None were grade 4.
The most common adverse event was musculoskeletal or joint pain (39 events of all grades and 6 grade 3/4 events). This was first described in the Swedish EURO-SKI patients in 15-20% of patients after TKI withdrawal, prompting the investigators to send an advisory letter to all participating physicians, she remarked.
Other adverse events included sweating, skin disorders, folliculitis, depressive episodes, fatigue, urticaria, and weight loss.
The EURO-SKI trial was recently expanded to enroll 700 patients, with a quality of life analysis expected later this year. The next report on the primary endpoint is expected at the American Society of Hematology annual meeting, Dr. Saussele said.
Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
MILAN – A full 61.5% of patients with chronic myeloid leukemia in deep molecular remission for more than 1 year on long-term tyrosine kinase inhibitor therapy remained alive and free of relapse 6 months after stopping their TKI in the EURO-SKI trial.
Molecular relapse-free survival at 9 months was 58% and 55% at 12 months.
The preplanned interim analysis after 200 patients allowed the trialists to discard the study’s null hypothesis that molecular relapse-free survival at 6 months would be 40% or less (P less than .0001).
"With less strict inclusion and relapse criteria than the A-STIM study and other trials, stopping is safe and we can continue with the trial," Dr. Susanne Saussele said in a late-breaking abstract session at the annual meeting of the European Hematology Association.
Importantly, interim results from the EURO-SKI (European Stop TKI) study also suggest that the level of molecular response (MR) achieved prior to TKI withdrawal affects molecular relapse-free survival.
Among 197 patients with molecular laboratory results available for exact classification, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
"In the setting of standardized molecular testing within a CML [chronic myeloid leukemia] stopping trial, it seems that molecular remission has an impact on molecular free-survival," said Dr. Saussele of University Medical Centre Mannheim, Germany.
As no statistical test was performed and this was an interim analysis, MR4.5 or MR5 cannot yet be used as a criterion to select patients to withdraw from treatment, she said in an interview.
The findings do confirm results from the recent A-STIM (According to Stop Imatinib) study showing that loss of major molecular response can be used as a practical criterion for restarting imatinib (J. Clin. Oncol. 2014;32:424-30).
Several studies including the STIM (Stop Imatinib) trial and the STOP 2G-TKI (Stop Second Generation Tyrosine Kinase Inhibitors) study have shown that imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna) can be safely withdrawn in a substantial proportion of patients with CML in deep MR.
A number of questions remain open, however, such as which molecular level has to be reached before stopping TKI therapy, the minimal duration of TKI pretreatment or MR4 before stopping, and which prognostic factors influence molecular relapse-free survival, she said.
EURO-SKI was set up to define prognostic markers to increase the rate of patients in durable deep MR after stopping TKI treatment. Other aims are to evaluate methods of molecular monitoring, quality of life, and saved treatment costs per country.
Patients with chronic-phase CML from eight countries were eligible if they were on a TKI for at least 3 years and had a confirmed deep MR, defined as more than a 4 log reduction in BCR-ABL (breakpoint cluster region–Abelson) transcripts for more than 12 months confirmed by three consecutive polymerase chain reaction results.
MR4 status also had to be confirmed in an MR4-standardized laboratory according to criteria by Cross et al. (Leukemia 2012;26:2172-5)
Patients with previous or planned allogeneic stem cell transplantation or a prior TKI failure were excluded.
A total of 103 patients received pretreatment before TKI therapy, mostly with hydroxyurea alone (n = 71) or with interferon (n = 22). Their median age was 53 years.
First-line TKI was imatinib in 194 patients, nilotinib in 3, and dasatinib in 3.
The median duration of TKI therapy was 8 years and median MR4 duration before stopping TKI therapy was 5 years.
Dr. Saussele cautioned that adverse events and quality of life must be taken into account when considering TKI withdrawal in these patients.
A total of 222 adverse events were reported in 98 patients, with 57 events in 37 patients related to treatment stop. None were grade 4.
The most common adverse event was musculoskeletal or joint pain (39 events of all grades and 6 grade 3/4 events). This was first described in the Swedish EURO-SKI patients in 15-20% of patients after TKI withdrawal, prompting the investigators to send an advisory letter to all participating physicians, she remarked.
Other adverse events included sweating, skin disorders, folliculitis, depressive episodes, fatigue, urticaria, and weight loss.
The EURO-SKI trial was recently expanded to enroll 700 patients, with a quality of life analysis expected later this year. The next report on the primary endpoint is expected at the American Society of Hematology annual meeting, Dr. Saussele said.
Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
MILAN – A full 61.5% of patients with chronic myeloid leukemia in deep molecular remission for more than 1 year on long-term tyrosine kinase inhibitor therapy remained alive and free of relapse 6 months after stopping their TKI in the EURO-SKI trial.
Molecular relapse-free survival at 9 months was 58% and 55% at 12 months.
The preplanned interim analysis after 200 patients allowed the trialists to discard the study’s null hypothesis that molecular relapse-free survival at 6 months would be 40% or less (P less than .0001).
"With less strict inclusion and relapse criteria than the A-STIM study and other trials, stopping is safe and we can continue with the trial," Dr. Susanne Saussele said in a late-breaking abstract session at the annual meeting of the European Hematology Association.
Importantly, interim results from the EURO-SKI (European Stop TKI) study also suggest that the level of molecular response (MR) achieved prior to TKI withdrawal affects molecular relapse-free survival.
Among 197 patients with molecular laboratory results available for exact classification, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
"In the setting of standardized molecular testing within a CML [chronic myeloid leukemia] stopping trial, it seems that molecular remission has an impact on molecular free-survival," said Dr. Saussele of University Medical Centre Mannheim, Germany.
As no statistical test was performed and this was an interim analysis, MR4.5 or MR5 cannot yet be used as a criterion to select patients to withdraw from treatment, she said in an interview.
The findings do confirm results from the recent A-STIM (According to Stop Imatinib) study showing that loss of major molecular response can be used as a practical criterion for restarting imatinib (J. Clin. Oncol. 2014;32:424-30).
Several studies including the STIM (Stop Imatinib) trial and the STOP 2G-TKI (Stop Second Generation Tyrosine Kinase Inhibitors) study have shown that imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna) can be safely withdrawn in a substantial proportion of patients with CML in deep MR.
A number of questions remain open, however, such as which molecular level has to be reached before stopping TKI therapy, the minimal duration of TKI pretreatment or MR4 before stopping, and which prognostic factors influence molecular relapse-free survival, she said.
EURO-SKI was set up to define prognostic markers to increase the rate of patients in durable deep MR after stopping TKI treatment. Other aims are to evaluate methods of molecular monitoring, quality of life, and saved treatment costs per country.
Patients with chronic-phase CML from eight countries were eligible if they were on a TKI for at least 3 years and had a confirmed deep MR, defined as more than a 4 log reduction in BCR-ABL (breakpoint cluster region–Abelson) transcripts for more than 12 months confirmed by three consecutive polymerase chain reaction results.
MR4 status also had to be confirmed in an MR4-standardized laboratory according to criteria by Cross et al. (Leukemia 2012;26:2172-5)
Patients with previous or planned allogeneic stem cell transplantation or a prior TKI failure were excluded.
A total of 103 patients received pretreatment before TKI therapy, mostly with hydroxyurea alone (n = 71) or with interferon (n = 22). Their median age was 53 years.
First-line TKI was imatinib in 194 patients, nilotinib in 3, and dasatinib in 3.
The median duration of TKI therapy was 8 years and median MR4 duration before stopping TKI therapy was 5 years.
Dr. Saussele cautioned that adverse events and quality of life must be taken into account when considering TKI withdrawal in these patients.
A total of 222 adverse events were reported in 98 patients, with 57 events in 37 patients related to treatment stop. None were grade 4.
The most common adverse event was musculoskeletal or joint pain (39 events of all grades and 6 grade 3/4 events). This was first described in the Swedish EURO-SKI patients in 15-20% of patients after TKI withdrawal, prompting the investigators to send an advisory letter to all participating physicians, she remarked.
Other adverse events included sweating, skin disorders, folliculitis, depressive episodes, fatigue, urticaria, and weight loss.
The EURO-SKI trial was recently expanded to enroll 700 patients, with a quality of life analysis expected later this year. The next report on the primary endpoint is expected at the American Society of Hematology annual meeting, Dr. Saussele said.
Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
AT THE EHA CONGRESS
Key clinical point: Long-time TKI therapy can be stopped in chronic myeloid leukemia in deep molecular remission. The level of molecular response prior to TKI withdrawal affects molecular relapse-free survival.
Major finding: After TKI withdrawal, 49% of patients in MR4 relapsed, compared with 39% in MR4.5 and 39% in MR5.
Data source: First interim analysis from a prospective study in 200 CML patients in deep molecular remission.
Disclosures: Dr. Saussele reported honoraria and research and travel support from BMS, Novartis, and Pfizer.
ABT-199 rebounds in advanced CLL after tackling tumor lysis syndrome
MILAN – The investigational Bcl-2 inhibitor ABT-199 continues to impress with substantial activity as single-agent or combination therapy in relapsed or refractory chronic lymphocytic leukemia, following dose-scheduling modifications to address the risk of tumor lysis syndrome.
ABT-199 monotherapy
Updated data from all 105 CLL patients in the phase I trial show the overall response rate remains high at 77%, with 23% of patients achieving complete remission.
Seven of 11 complete responders assessed had no detectable minimal residual disease, Dr. John F. Seymour said at the annual congress of the European Hematology Association.
Overall response rates were sustained in the 75%-80% range for high-risk patients with deletion 17p, fludarabine-refractory, or immunoglobulin heavy-chain variable (IGHV)-unmutated CLL; and complete response rates in these subgroups also did not differ from the overall group at 22% to 29%.
Prior results from the first-in-human trial dazzled the leukemia community, but tumor lysis syndrome (TLS) complications, including two fatal events, temporarily halted ABT-199 clinical trials and gave the advantage to its closest competitor, the recently approved and more tolerable CLL drug ibrutinib (Imbruvica).
Use of a modified, ramp-up dosing scheme and aggressive TLS prophylaxis appear to have ameliorated the risk of TLS, with no further clinically significant or grade 3 or 4 events reported in the 49 patients treated with this schema, said Dr. Seymour, director of hematology and cancer medicine at the Peter MacCallum Cancer Centre, East Melbourne, Australia.
Rather than using a 3-week schedule and 50-mg starting dose, the safety expansion cohort received once-daily oral ABT-199 beginning at 20 mg, with weekly adjustments to 50 mg, 100 mg, 200 mg, and 400 mg over 5 weeks.
The 400-mg dose has been identified as the phase II dose, with 59% of patients free of progression at 18 months and beyond on this dose, he said.
As of April 2014, 37 of the 105 patients discontinued treatment, 22 due to progressive disease and 12 for adverse events; in addition, two proceeded to allogeneic hematopoietic cell transplantation, and one needed Coumadin, which is not permitted on protocol.
The median duration of response has not yet been reached for patients treated at doses of 400 mg or above, he said.
The most common treatment-emergent adverse events of any grade were diarrhea (40% of patients), neutropenia (36%), and nausea (35%).
Neutropenia was the only grade 3/4 event occurring in more than 10% of patients (33%), followed by anemia in 10%.
Combination ABT-199
Of substantial interest to many was a second phase Ib study presented in the same session, evaluating the role of ABT-199 with the anti-CD20 antibody rituximab (Rituxan) in relapsed or refractory CLL.
After a median time on study of just 7.5 months, the overall response rate was 84% among 25 evaluable patients, including 9 complete responses (36%) and 12 partial responses (48%), said Dr. Andrew W. Roberts, with the Royal Melbourne (Australia) Hospital and Walter and Eliza Hall Institute of Medical Research.
Six of eight complete responders tested were negative for minimal residual disease by flow cytometry.
Preliminary pharmacokinetic results suggest no apparent effect of rituximab on ABT-199 exposure, he said.
Three patients discontinued ABT-199 after achieving a complete remission, including one with fludarabine-refractory disease, and all remain in complete remission at 8.6, 8.8, and 11.6 months after cessation.
Dose modifications were also made in this study following a fatal TLS event in December 2012 after a first dose of ABT-199 at 50 mg. Under the modified step-up dosing, ABT-199 was started at 20 mg, escalating up to 600 mg daily over 5 weeks, with rituximab 375 mg/m2 added on day 1 of week 5 and rituximab 500 mg/m2 added on day 1 of months 2-6.
The combination was well tolerated, and no new safety concerns were identified, Dr. Roberts said. The most common grade 3/4 adverse events among 45 patients evaluable for safety were neutropenia in 47%, anemia in 16%, thrombocytopenia in 13%, and febrile neutropenia in 7%. Grade 3/4 neutropenia was more common at 600 mg, with the 400-mg dose selected for the ongoing safety expansion cohort. Two serious TLS events occurred, but both were prior to schedule modifications, he said.
One patient in the combination study and 13 in the monotherapy study received treatment for small lymphocytic lymphoma. Response rates and tolerability were similar between CLL and SLL patients in the monotherapy study, Dr. Roberts said in an interview.
During a press briefing at the meeting, Dr. Seymour said ABT-199 potentially could be combined with ibrutinib and that the combination was very potent in laboratory tests in both CLL and some forms of mantle cell lymphoma. Negotiations with the various companies involved are intricate, but there is agreement and commitment "to begin clinical trials of the combination later this year," he added.
ABT-199 is currently being evaluated in a phase II trial as monotherapy in deletion 17p relapsed CLL, as combination therapy with rituximab versus bendamustine plus rituximab in a phase III trial in relapsed/refractory CLL, and in combination trials with bendamustine/rituximab and obinutuzumab in relapsed/refractory CLL.
AbbVie and Genentech sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
MILAN – The investigational Bcl-2 inhibitor ABT-199 continues to impress with substantial activity as single-agent or combination therapy in relapsed or refractory chronic lymphocytic leukemia, following dose-scheduling modifications to address the risk of tumor lysis syndrome.
ABT-199 monotherapy
Updated data from all 105 CLL patients in the phase I trial show the overall response rate remains high at 77%, with 23% of patients achieving complete remission.
Seven of 11 complete responders assessed had no detectable minimal residual disease, Dr. John F. Seymour said at the annual congress of the European Hematology Association.
Overall response rates were sustained in the 75%-80% range for high-risk patients with deletion 17p, fludarabine-refractory, or immunoglobulin heavy-chain variable (IGHV)-unmutated CLL; and complete response rates in these subgroups also did not differ from the overall group at 22% to 29%.
Prior results from the first-in-human trial dazzled the leukemia community, but tumor lysis syndrome (TLS) complications, including two fatal events, temporarily halted ABT-199 clinical trials and gave the advantage to its closest competitor, the recently approved and more tolerable CLL drug ibrutinib (Imbruvica).
Use of a modified, ramp-up dosing scheme and aggressive TLS prophylaxis appear to have ameliorated the risk of TLS, with no further clinically significant or grade 3 or 4 events reported in the 49 patients treated with this schema, said Dr. Seymour, director of hematology and cancer medicine at the Peter MacCallum Cancer Centre, East Melbourne, Australia.
Rather than using a 3-week schedule and 50-mg starting dose, the safety expansion cohort received once-daily oral ABT-199 beginning at 20 mg, with weekly adjustments to 50 mg, 100 mg, 200 mg, and 400 mg over 5 weeks.
The 400-mg dose has been identified as the phase II dose, with 59% of patients free of progression at 18 months and beyond on this dose, he said.
As of April 2014, 37 of the 105 patients discontinued treatment, 22 due to progressive disease and 12 for adverse events; in addition, two proceeded to allogeneic hematopoietic cell transplantation, and one needed Coumadin, which is not permitted on protocol.
The median duration of response has not yet been reached for patients treated at doses of 400 mg or above, he said.
The most common treatment-emergent adverse events of any grade were diarrhea (40% of patients), neutropenia (36%), and nausea (35%).
Neutropenia was the only grade 3/4 event occurring in more than 10% of patients (33%), followed by anemia in 10%.
Combination ABT-199
Of substantial interest to many was a second phase Ib study presented in the same session, evaluating the role of ABT-199 with the anti-CD20 antibody rituximab (Rituxan) in relapsed or refractory CLL.
After a median time on study of just 7.5 months, the overall response rate was 84% among 25 evaluable patients, including 9 complete responses (36%) and 12 partial responses (48%), said Dr. Andrew W. Roberts, with the Royal Melbourne (Australia) Hospital and Walter and Eliza Hall Institute of Medical Research.
Six of eight complete responders tested were negative for minimal residual disease by flow cytometry.
Preliminary pharmacokinetic results suggest no apparent effect of rituximab on ABT-199 exposure, he said.
Three patients discontinued ABT-199 after achieving a complete remission, including one with fludarabine-refractory disease, and all remain in complete remission at 8.6, 8.8, and 11.6 months after cessation.
Dose modifications were also made in this study following a fatal TLS event in December 2012 after a first dose of ABT-199 at 50 mg. Under the modified step-up dosing, ABT-199 was started at 20 mg, escalating up to 600 mg daily over 5 weeks, with rituximab 375 mg/m2 added on day 1 of week 5 and rituximab 500 mg/m2 added on day 1 of months 2-6.
The combination was well tolerated, and no new safety concerns were identified, Dr. Roberts said. The most common grade 3/4 adverse events among 45 patients evaluable for safety were neutropenia in 47%, anemia in 16%, thrombocytopenia in 13%, and febrile neutropenia in 7%. Grade 3/4 neutropenia was more common at 600 mg, with the 400-mg dose selected for the ongoing safety expansion cohort. Two serious TLS events occurred, but both were prior to schedule modifications, he said.
One patient in the combination study and 13 in the monotherapy study received treatment for small lymphocytic lymphoma. Response rates and tolerability were similar between CLL and SLL patients in the monotherapy study, Dr. Roberts said in an interview.
During a press briefing at the meeting, Dr. Seymour said ABT-199 potentially could be combined with ibrutinib and that the combination was very potent in laboratory tests in both CLL and some forms of mantle cell lymphoma. Negotiations with the various companies involved are intricate, but there is agreement and commitment "to begin clinical trials of the combination later this year," he added.
ABT-199 is currently being evaluated in a phase II trial as monotherapy in deletion 17p relapsed CLL, as combination therapy with rituximab versus bendamustine plus rituximab in a phase III trial in relapsed/refractory CLL, and in combination trials with bendamustine/rituximab and obinutuzumab in relapsed/refractory CLL.
AbbVie and Genentech sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
MILAN – The investigational Bcl-2 inhibitor ABT-199 continues to impress with substantial activity as single-agent or combination therapy in relapsed or refractory chronic lymphocytic leukemia, following dose-scheduling modifications to address the risk of tumor lysis syndrome.
ABT-199 monotherapy
Updated data from all 105 CLL patients in the phase I trial show the overall response rate remains high at 77%, with 23% of patients achieving complete remission.
Seven of 11 complete responders assessed had no detectable minimal residual disease, Dr. John F. Seymour said at the annual congress of the European Hematology Association.
Overall response rates were sustained in the 75%-80% range for high-risk patients with deletion 17p, fludarabine-refractory, or immunoglobulin heavy-chain variable (IGHV)-unmutated CLL; and complete response rates in these subgroups also did not differ from the overall group at 22% to 29%.
Prior results from the first-in-human trial dazzled the leukemia community, but tumor lysis syndrome (TLS) complications, including two fatal events, temporarily halted ABT-199 clinical trials and gave the advantage to its closest competitor, the recently approved and more tolerable CLL drug ibrutinib (Imbruvica).
Use of a modified, ramp-up dosing scheme and aggressive TLS prophylaxis appear to have ameliorated the risk of TLS, with no further clinically significant or grade 3 or 4 events reported in the 49 patients treated with this schema, said Dr. Seymour, director of hematology and cancer medicine at the Peter MacCallum Cancer Centre, East Melbourne, Australia.
Rather than using a 3-week schedule and 50-mg starting dose, the safety expansion cohort received once-daily oral ABT-199 beginning at 20 mg, with weekly adjustments to 50 mg, 100 mg, 200 mg, and 400 mg over 5 weeks.
The 400-mg dose has been identified as the phase II dose, with 59% of patients free of progression at 18 months and beyond on this dose, he said.
As of April 2014, 37 of the 105 patients discontinued treatment, 22 due to progressive disease and 12 for adverse events; in addition, two proceeded to allogeneic hematopoietic cell transplantation, and one needed Coumadin, which is not permitted on protocol.
The median duration of response has not yet been reached for patients treated at doses of 400 mg or above, he said.
The most common treatment-emergent adverse events of any grade were diarrhea (40% of patients), neutropenia (36%), and nausea (35%).
Neutropenia was the only grade 3/4 event occurring in more than 10% of patients (33%), followed by anemia in 10%.
Combination ABT-199
Of substantial interest to many was a second phase Ib study presented in the same session, evaluating the role of ABT-199 with the anti-CD20 antibody rituximab (Rituxan) in relapsed or refractory CLL.
After a median time on study of just 7.5 months, the overall response rate was 84% among 25 evaluable patients, including 9 complete responses (36%) and 12 partial responses (48%), said Dr. Andrew W. Roberts, with the Royal Melbourne (Australia) Hospital and Walter and Eliza Hall Institute of Medical Research.
Six of eight complete responders tested were negative for minimal residual disease by flow cytometry.
Preliminary pharmacokinetic results suggest no apparent effect of rituximab on ABT-199 exposure, he said.
Three patients discontinued ABT-199 after achieving a complete remission, including one with fludarabine-refractory disease, and all remain in complete remission at 8.6, 8.8, and 11.6 months after cessation.
Dose modifications were also made in this study following a fatal TLS event in December 2012 after a first dose of ABT-199 at 50 mg. Under the modified step-up dosing, ABT-199 was started at 20 mg, escalating up to 600 mg daily over 5 weeks, with rituximab 375 mg/m2 added on day 1 of week 5 and rituximab 500 mg/m2 added on day 1 of months 2-6.
The combination was well tolerated, and no new safety concerns were identified, Dr. Roberts said. The most common grade 3/4 adverse events among 45 patients evaluable for safety were neutropenia in 47%, anemia in 16%, thrombocytopenia in 13%, and febrile neutropenia in 7%. Grade 3/4 neutropenia was more common at 600 mg, with the 400-mg dose selected for the ongoing safety expansion cohort. Two serious TLS events occurred, but both were prior to schedule modifications, he said.
One patient in the combination study and 13 in the monotherapy study received treatment for small lymphocytic lymphoma. Response rates and tolerability were similar between CLL and SLL patients in the monotherapy study, Dr. Roberts said in an interview.
During a press briefing at the meeting, Dr. Seymour said ABT-199 potentially could be combined with ibrutinib and that the combination was very potent in laboratory tests in both CLL and some forms of mantle cell lymphoma. Negotiations with the various companies involved are intricate, but there is agreement and commitment "to begin clinical trials of the combination later this year," he added.
ABT-199 is currently being evaluated in a phase II trial as monotherapy in deletion 17p relapsed CLL, as combination therapy with rituximab versus bendamustine plus rituximab in a phase III trial in relapsed/refractory CLL, and in combination trials with bendamustine/rituximab and obinutuzumab in relapsed/refractory CLL.
AbbVie and Genentech sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
AT THE EHA CONGRESS
Major finding: The overall response rate was 77% with ABT-199 monotherapy and 84% with the addition of rituximab in relapsed or refractory CLL.
Key clinical point: ABT-199 alone or as combination therapy with rituximab has substantial activity in relapsed/refractory CLL.
Data source: Two phase I trials in patients with CLL.
Disclosures: AbbVie and Genentech, codevelopers of ABT-199, sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
ONO-4059 delivered hit to relapsed/refractory CLL
MILAN – Monotherapy with the oral BTK inhibitor ONO-4059 showed good efficacy over a range of doses in relapsed or refractory and high-risk chronic lymphocytic leukemia, with a best overall response rate of 84% in a phase I trial.
Among 25 evaluable patients, 2 achieved complete responses with incomplete blood count recovery, 12 had partial responses, and 7 had partial responses with lymphocytosis.
One patient had stable disease and another progressive disease. Two patients withdrew due to adverse events.
ONO-4059 showed particularly good efficacy in patients with the deleterious 17p deletion (89%, or 8/9 patients) and in those refractory to their last therapy (91.6% or 11/12 patients), Dr. Franck Morschhauser, reported at the annual congress of the European Hematology Association.
ONO-4059 is a selective Bruton’s tyrosine kinase (BTK) inhibitor that has demonstrated greater than 90% inhibition of BTK at 12 hours in chronic lymphocytic leukemia (CLL) cells in vivo and antitumor activity in non-Hodgkin’s lymphoma.
In relapsed or refractory B-cell lymphoma, the same investigators reported a best overall response rate of 42% with ONO-4059 at doses of 40, 80, and 160 mg among 12 patients in a phase I study.
The current dose-escalation study evaluated the safety and tolerability of once-daily ONO-4059 at doses ranging from 20 to 600 mg for up to 2 years in patients with relapsed/refractory or high-risk CLL for whom no therapy of higher priority was available. Dose escalation was permitted upon completion of 6 months of treatment. The median duration of treatment was 11.5 cycles.
The 25 evaluable patients (median age, 67 years) had received a median of four prior therapies (range 2-9); 92% had prior exposure to rituximab (Rituxan) and 92% to fludarabine (Fludara); and 48% were refractory to their last therapy.
Lymph node reduction occurred early, between day 1 and 28 of the first cycle, and was accompanied by an improvement in hemoglobin and platelet counts within 3-4 months, said Dr. Morschhauser, head of the lymphoma unit, Centre Hospitalier Régional Universitaire–Claude Huriez Hospital, Lille, France.
"So far we do not have evidence that there is a dose-efficacy relationship, although there is quicker tumor shrinkage when you increase the dose," he said.
The majority of adverse events were grade 1 and 2, with a low incidence of diarrhea, fatigue, bruising, and rash and no dose-limiting toxicities, Dr. Morschhauser said. Five patients experienced grade 3/4 neutropenia: two grade 3 events at 20 and 40 mg and three grade 4 events at 20, 80, and 320 mg.
Eight ONO-4059–related serious adverse events were reported, including febrile neutropenia in the patient with grade 4 neutropenia at the 20-mg dose, pyrexia at 20 mg, rash at 80 mg, hepatitis E reactivation and neutropenia at 320 mg, lymphocytic infiltration of the right dorsal muscle and purpura at 400 mg, and spontaneous psoas hematoma at 500 mg. All events resolved, and four patients remain on study, he reported on behalf of lead author Dr. Christopher Fegan, clinical director of CLL, University Hospital of Wales, Cardiff.
The study was supported by Ono Pharmaceutical. The study authors reported no relevant financial disclosures.
MILAN – Monotherapy with the oral BTK inhibitor ONO-4059 showed good efficacy over a range of doses in relapsed or refractory and high-risk chronic lymphocytic leukemia, with a best overall response rate of 84% in a phase I trial.
Among 25 evaluable patients, 2 achieved complete responses with incomplete blood count recovery, 12 had partial responses, and 7 had partial responses with lymphocytosis.
One patient had stable disease and another progressive disease. Two patients withdrew due to adverse events.
ONO-4059 showed particularly good efficacy in patients with the deleterious 17p deletion (89%, or 8/9 patients) and in those refractory to their last therapy (91.6% or 11/12 patients), Dr. Franck Morschhauser, reported at the annual congress of the European Hematology Association.
ONO-4059 is a selective Bruton’s tyrosine kinase (BTK) inhibitor that has demonstrated greater than 90% inhibition of BTK at 12 hours in chronic lymphocytic leukemia (CLL) cells in vivo and antitumor activity in non-Hodgkin’s lymphoma.
In relapsed or refractory B-cell lymphoma, the same investigators reported a best overall response rate of 42% with ONO-4059 at doses of 40, 80, and 160 mg among 12 patients in a phase I study.
The current dose-escalation study evaluated the safety and tolerability of once-daily ONO-4059 at doses ranging from 20 to 600 mg for up to 2 years in patients with relapsed/refractory or high-risk CLL for whom no therapy of higher priority was available. Dose escalation was permitted upon completion of 6 months of treatment. The median duration of treatment was 11.5 cycles.
The 25 evaluable patients (median age, 67 years) had received a median of four prior therapies (range 2-9); 92% had prior exposure to rituximab (Rituxan) and 92% to fludarabine (Fludara); and 48% were refractory to their last therapy.
Lymph node reduction occurred early, between day 1 and 28 of the first cycle, and was accompanied by an improvement in hemoglobin and platelet counts within 3-4 months, said Dr. Morschhauser, head of the lymphoma unit, Centre Hospitalier Régional Universitaire–Claude Huriez Hospital, Lille, France.
"So far we do not have evidence that there is a dose-efficacy relationship, although there is quicker tumor shrinkage when you increase the dose," he said.
The majority of adverse events were grade 1 and 2, with a low incidence of diarrhea, fatigue, bruising, and rash and no dose-limiting toxicities, Dr. Morschhauser said. Five patients experienced grade 3/4 neutropenia: two grade 3 events at 20 and 40 mg and three grade 4 events at 20, 80, and 320 mg.
Eight ONO-4059–related serious adverse events were reported, including febrile neutropenia in the patient with grade 4 neutropenia at the 20-mg dose, pyrexia at 20 mg, rash at 80 mg, hepatitis E reactivation and neutropenia at 320 mg, lymphocytic infiltration of the right dorsal muscle and purpura at 400 mg, and spontaneous psoas hematoma at 500 mg. All events resolved, and four patients remain on study, he reported on behalf of lead author Dr. Christopher Fegan, clinical director of CLL, University Hospital of Wales, Cardiff.
The study was supported by Ono Pharmaceutical. The study authors reported no relevant financial disclosures.
MILAN – Monotherapy with the oral BTK inhibitor ONO-4059 showed good efficacy over a range of doses in relapsed or refractory and high-risk chronic lymphocytic leukemia, with a best overall response rate of 84% in a phase I trial.
Among 25 evaluable patients, 2 achieved complete responses with incomplete blood count recovery, 12 had partial responses, and 7 had partial responses with lymphocytosis.
One patient had stable disease and another progressive disease. Two patients withdrew due to adverse events.
ONO-4059 showed particularly good efficacy in patients with the deleterious 17p deletion (89%, or 8/9 patients) and in those refractory to their last therapy (91.6% or 11/12 patients), Dr. Franck Morschhauser, reported at the annual congress of the European Hematology Association.
ONO-4059 is a selective Bruton’s tyrosine kinase (BTK) inhibitor that has demonstrated greater than 90% inhibition of BTK at 12 hours in chronic lymphocytic leukemia (CLL) cells in vivo and antitumor activity in non-Hodgkin’s lymphoma.
In relapsed or refractory B-cell lymphoma, the same investigators reported a best overall response rate of 42% with ONO-4059 at doses of 40, 80, and 160 mg among 12 patients in a phase I study.
The current dose-escalation study evaluated the safety and tolerability of once-daily ONO-4059 at doses ranging from 20 to 600 mg for up to 2 years in patients with relapsed/refractory or high-risk CLL for whom no therapy of higher priority was available. Dose escalation was permitted upon completion of 6 months of treatment. The median duration of treatment was 11.5 cycles.
The 25 evaluable patients (median age, 67 years) had received a median of four prior therapies (range 2-9); 92% had prior exposure to rituximab (Rituxan) and 92% to fludarabine (Fludara); and 48% were refractory to their last therapy.
Lymph node reduction occurred early, between day 1 and 28 of the first cycle, and was accompanied by an improvement in hemoglobin and platelet counts within 3-4 months, said Dr. Morschhauser, head of the lymphoma unit, Centre Hospitalier Régional Universitaire–Claude Huriez Hospital, Lille, France.
"So far we do not have evidence that there is a dose-efficacy relationship, although there is quicker tumor shrinkage when you increase the dose," he said.
The majority of adverse events were grade 1 and 2, with a low incidence of diarrhea, fatigue, bruising, and rash and no dose-limiting toxicities, Dr. Morschhauser said. Five patients experienced grade 3/4 neutropenia: two grade 3 events at 20 and 40 mg and three grade 4 events at 20, 80, and 320 mg.
Eight ONO-4059–related serious adverse events were reported, including febrile neutropenia in the patient with grade 4 neutropenia at the 20-mg dose, pyrexia at 20 mg, rash at 80 mg, hepatitis E reactivation and neutropenia at 320 mg, lymphocytic infiltration of the right dorsal muscle and purpura at 400 mg, and spontaneous psoas hematoma at 500 mg. All events resolved, and four patients remain on study, he reported on behalf of lead author Dr. Christopher Fegan, clinical director of CLL, University Hospital of Wales, Cardiff.
The study was supported by Ono Pharmaceutical. The study authors reported no relevant financial disclosures.
AT THE EHA CONGRESS
Key clinical point: ONO-4059 induced responses in most patients with relapsed/refractory CLL, including those with a 17p deletion.
Major finding: The best response rate was 84% overall, 89% in 17p deletion patients, and 91.6% in refractory disease.
Data source: A phase I trial in 25 patients with relapsed/refractory or high-risk chronic lymphocytic leukemia.
Disclosures: The study was supported by Ono Pharmaceutical. The study authors reported no relevant financial disclosures.
De-escalation of ABVD chemotherapy fails in early-stage, favorable Hodgkin’s lymphoma
MILAN – Neither dacarbazine nor bleomycin can be safely omitted from ABVD chemotherapy without affecting efficacy in early-stage, favorable Hodgkin’s lymphoma, according to the final analysis of the German Hodgkin Study Group HD13 trial.
The primary endpoint of freedom from treatment failure (FFTF) at 5 years was 93.1% after ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, compared with 81.4% after ABV and 77.1% after AV.
All patients received two cycles of ABVD, ABV, AVD, or AV chemotherapy followed by 30 Gy involved-field radiation therapy.
"Dacarbazine cannot be omitted without considerable loss of efficacy," Dr. Karolin Behringer said at the annual congress of the European Hematology Association.
The findings confirm the inferiority of the ABV and AV arms, which were prematurely closed in 2006 and 2005 after a safety analysis detected a strong increase in events in the two arms compared with standard ABVD.
The four-armed HD13 trial, however, also sought to answer whether the AVD regimen is equivalent to ABVD chemotherapy.
The 5-year FFTF rate with AVD was 89.2%, or 3.9 percentage points lower than with ABVD (hazard ratio, 1.50; 95% confidence interval, 1.00-2.26).
This was largely due to more late relapses with AVD than with ABVD (37 vs. 20), said Dr. Behringer, of the University Hospital of Cologne, Germany.
As a result, AVD failed to meet the noninferiority test with a margin of 1.72 for hazard ratio, corresponding to a 6% difference in 5-year FFTF between ABVD and AVD.
"Bleomycin cannot be omitted with the predefined noninferiority margin of 6%," she said.
Importantly, the reduction in FFTF did not translate into poorer overall survival.
Five-year overall survival rates were 97.6% with ABVD, 94.1% with ABV, 97.6% with AVD, and 98.1% with AV, Dr. Behringer reported.
AVD patients had similar rates as those treated with ABVD chemotherapy as salvage therapy with stem cell transplantation (49% vs. 45%) or BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, and prednisone) chemotherapy (31% vs. 35%).
Patients in the AVD arm, however, experienced significantly less grade 3/4 toxicity than did those in the ABVD arm (26.3% vs. 32.7%; P = .03). Grade 3/4 events were similar in the ABV and AV arms (28.3% vs. 26.5%), she said.
The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
MILAN – Neither dacarbazine nor bleomycin can be safely omitted from ABVD chemotherapy without affecting efficacy in early-stage, favorable Hodgkin’s lymphoma, according to the final analysis of the German Hodgkin Study Group HD13 trial.
The primary endpoint of freedom from treatment failure (FFTF) at 5 years was 93.1% after ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, compared with 81.4% after ABV and 77.1% after AV.
All patients received two cycles of ABVD, ABV, AVD, or AV chemotherapy followed by 30 Gy involved-field radiation therapy.
"Dacarbazine cannot be omitted without considerable loss of efficacy," Dr. Karolin Behringer said at the annual congress of the European Hematology Association.
The findings confirm the inferiority of the ABV and AV arms, which were prematurely closed in 2006 and 2005 after a safety analysis detected a strong increase in events in the two arms compared with standard ABVD.
The four-armed HD13 trial, however, also sought to answer whether the AVD regimen is equivalent to ABVD chemotherapy.
The 5-year FFTF rate with AVD was 89.2%, or 3.9 percentage points lower than with ABVD (hazard ratio, 1.50; 95% confidence interval, 1.00-2.26).
This was largely due to more late relapses with AVD than with ABVD (37 vs. 20), said Dr. Behringer, of the University Hospital of Cologne, Germany.
As a result, AVD failed to meet the noninferiority test with a margin of 1.72 for hazard ratio, corresponding to a 6% difference in 5-year FFTF between ABVD and AVD.
"Bleomycin cannot be omitted with the predefined noninferiority margin of 6%," she said.
Importantly, the reduction in FFTF did not translate into poorer overall survival.
Five-year overall survival rates were 97.6% with ABVD, 94.1% with ABV, 97.6% with AVD, and 98.1% with AV, Dr. Behringer reported.
AVD patients had similar rates as those treated with ABVD chemotherapy as salvage therapy with stem cell transplantation (49% vs. 45%) or BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, and prednisone) chemotherapy (31% vs. 35%).
Patients in the AVD arm, however, experienced significantly less grade 3/4 toxicity than did those in the ABVD arm (26.3% vs. 32.7%; P = .03). Grade 3/4 events were similar in the ABV and AV arms (28.3% vs. 26.5%), she said.
The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
MILAN – Neither dacarbazine nor bleomycin can be safely omitted from ABVD chemotherapy without affecting efficacy in early-stage, favorable Hodgkin’s lymphoma, according to the final analysis of the German Hodgkin Study Group HD13 trial.
The primary endpoint of freedom from treatment failure (FFTF) at 5 years was 93.1% after ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, compared with 81.4% after ABV and 77.1% after AV.
All patients received two cycles of ABVD, ABV, AVD, or AV chemotherapy followed by 30 Gy involved-field radiation therapy.
"Dacarbazine cannot be omitted without considerable loss of efficacy," Dr. Karolin Behringer said at the annual congress of the European Hematology Association.
The findings confirm the inferiority of the ABV and AV arms, which were prematurely closed in 2006 and 2005 after a safety analysis detected a strong increase in events in the two arms compared with standard ABVD.
The four-armed HD13 trial, however, also sought to answer whether the AVD regimen is equivalent to ABVD chemotherapy.
The 5-year FFTF rate with AVD was 89.2%, or 3.9 percentage points lower than with ABVD (hazard ratio, 1.50; 95% confidence interval, 1.00-2.26).
This was largely due to more late relapses with AVD than with ABVD (37 vs. 20), said Dr. Behringer, of the University Hospital of Cologne, Germany.
As a result, AVD failed to meet the noninferiority test with a margin of 1.72 for hazard ratio, corresponding to a 6% difference in 5-year FFTF between ABVD and AVD.
"Bleomycin cannot be omitted with the predefined noninferiority margin of 6%," she said.
Importantly, the reduction in FFTF did not translate into poorer overall survival.
Five-year overall survival rates were 97.6% with ABVD, 94.1% with ABV, 97.6% with AVD, and 98.1% with AV, Dr. Behringer reported.
AVD patients had similar rates as those treated with ABVD chemotherapy as salvage therapy with stem cell transplantation (49% vs. 45%) or BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, and prednisone) chemotherapy (31% vs. 35%).
Patients in the AVD arm, however, experienced significantly less grade 3/4 toxicity than did those in the ABVD arm (26.3% vs. 32.7%; P = .03). Grade 3/4 events were similar in the ABV and AV arms (28.3% vs. 26.5%), she said.
The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
AT THE EHA CONGRESS
Major finding: The 5-year FFTF rate was 93.1% with ABVD and 89.2% with AVD (HR, 1.50; 95% confidence interval, 1.00-2.26).
Data source: A prospective, randomized study in 1,710 patients with early-stage, favorable Hodgkin’s lymphoma.
Key clinical point: Dacarbazine and bleomycin should not be omitted from ABVD chemotherapy in the treatment of early-stage, favorable Hodgkin’s lymphoma.
Disclosures: The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
De-escalation of ABVD chemotherapy fails in early-stage, favorable Hodgkin’s lymphoma
MILAN – Neither dacarbazine nor bleomycin can be safely omitted from ABVD chemotherapy without affecting efficacy in early-stage, favorable Hodgkin’s lymphoma, according to the final analysis of the German Hodgkin Study Group HD13 trial.
The primary endpoint of freedom from treatment failure (FFTF) at 5 years was 93.1% after ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, compared with 81.4% after ABV and 77.1% after AV.
All patients received two cycles of ABVD, ABV, AVD, or AV chemotherapy followed by 30 Gy involved-field radiation therapy.
"Dacarbazine cannot be omitted without considerable loss of efficacy," Dr. Karolin Behringer said at the annual congress of the European Hematology Association.
The findings confirm the inferiority of the ABV and AV arms, which were prematurely closed in 2006 and 2005 after a safety analysis detected a strong increase in events in the two arms compared with standard ABVD.
The four-armed HD13 trial, however, also sought to answer whether the AVD regimen is equivalent to ABVD chemotherapy.
The 5-year FFTF rate with AVD was 89.2%, or 3.9 percentage points lower than with ABVD (hazard ratio, 1.50; 95% confidence interval, 1.00-2.26).
This was largely due to more late relapses with AVD than with ABVD (37 vs. 20), said Dr. Behringer, of the University Hospital of Cologne, Germany.
As a result, AVD failed to meet the noninferiority test with a margin of 1.72 for hazard ratio, corresponding to a 6% difference in 5-year FFTF between ABVD and AVD.
"Bleomycin cannot be omitted with the predefined noninferiority margin of 6%," she said.
Importantly, the reduction in FFTF did not translate into poorer overall survival.
Five-year overall survival rates were 97.6% with ABVD, 94.1% with ABV, 97.6% with AVD, and 98.1% with AV, Dr. Behringer reported.
AVD patients had similar rates as those treated with ABVD chemotherapy as salvage therapy with stem cell transplantation (49% vs. 45%) or BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, and prednisone) chemotherapy (31% vs. 35%).
Patients in the AVD arm, however, experienced significantly less grade 3/4 toxicity than did those in the ABVD arm (26.3% vs. 32.7%; P = .03). Grade 3/4 events were similar in the ABV and AV arms (28.3% vs. 26.5%), she said.
The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
MILAN – Neither dacarbazine nor bleomycin can be safely omitted from ABVD chemotherapy without affecting efficacy in early-stage, favorable Hodgkin’s lymphoma, according to the final analysis of the German Hodgkin Study Group HD13 trial.
The primary endpoint of freedom from treatment failure (FFTF) at 5 years was 93.1% after ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, compared with 81.4% after ABV and 77.1% after AV.
All patients received two cycles of ABVD, ABV, AVD, or AV chemotherapy followed by 30 Gy involved-field radiation therapy.
"Dacarbazine cannot be omitted without considerable loss of efficacy," Dr. Karolin Behringer said at the annual congress of the European Hematology Association.
The findings confirm the inferiority of the ABV and AV arms, which were prematurely closed in 2006 and 2005 after a safety analysis detected a strong increase in events in the two arms compared with standard ABVD.
The four-armed HD13 trial, however, also sought to answer whether the AVD regimen is equivalent to ABVD chemotherapy.
The 5-year FFTF rate with AVD was 89.2%, or 3.9 percentage points lower than with ABVD (hazard ratio, 1.50; 95% confidence interval, 1.00-2.26).
This was largely due to more late relapses with AVD than with ABVD (37 vs. 20), said Dr. Behringer, of the University Hospital of Cologne, Germany.
As a result, AVD failed to meet the noninferiority test with a margin of 1.72 for hazard ratio, corresponding to a 6% difference in 5-year FFTF between ABVD and AVD.
"Bleomycin cannot be omitted with the predefined noninferiority margin of 6%," she said.
Importantly, the reduction in FFTF did not translate into poorer overall survival.
Five-year overall survival rates were 97.6% with ABVD, 94.1% with ABV, 97.6% with AVD, and 98.1% with AV, Dr. Behringer reported.
AVD patients had similar rates as those treated with ABVD chemotherapy as salvage therapy with stem cell transplantation (49% vs. 45%) or BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, and prednisone) chemotherapy (31% vs. 35%).
Patients in the AVD arm, however, experienced significantly less grade 3/4 toxicity than did those in the ABVD arm (26.3% vs. 32.7%; P = .03). Grade 3/4 events were similar in the ABV and AV arms (28.3% vs. 26.5%), she said.
The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
MILAN – Neither dacarbazine nor bleomycin can be safely omitted from ABVD chemotherapy without affecting efficacy in early-stage, favorable Hodgkin’s lymphoma, according to the final analysis of the German Hodgkin Study Group HD13 trial.
The primary endpoint of freedom from treatment failure (FFTF) at 5 years was 93.1% after ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, compared with 81.4% after ABV and 77.1% after AV.
All patients received two cycles of ABVD, ABV, AVD, or AV chemotherapy followed by 30 Gy involved-field radiation therapy.
"Dacarbazine cannot be omitted without considerable loss of efficacy," Dr. Karolin Behringer said at the annual congress of the European Hematology Association.
The findings confirm the inferiority of the ABV and AV arms, which were prematurely closed in 2006 and 2005 after a safety analysis detected a strong increase in events in the two arms compared with standard ABVD.
The four-armed HD13 trial, however, also sought to answer whether the AVD regimen is equivalent to ABVD chemotherapy.
The 5-year FFTF rate with AVD was 89.2%, or 3.9 percentage points lower than with ABVD (hazard ratio, 1.50; 95% confidence interval, 1.00-2.26).
This was largely due to more late relapses with AVD than with ABVD (37 vs. 20), said Dr. Behringer, of the University Hospital of Cologne, Germany.
As a result, AVD failed to meet the noninferiority test with a margin of 1.72 for hazard ratio, corresponding to a 6% difference in 5-year FFTF between ABVD and AVD.
"Bleomycin cannot be omitted with the predefined noninferiority margin of 6%," she said.
Importantly, the reduction in FFTF did not translate into poorer overall survival.
Five-year overall survival rates were 97.6% with ABVD, 94.1% with ABV, 97.6% with AVD, and 98.1% with AV, Dr. Behringer reported.
AVD patients had similar rates as those treated with ABVD chemotherapy as salvage therapy with stem cell transplantation (49% vs. 45%) or BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, and prednisone) chemotherapy (31% vs. 35%).
Patients in the AVD arm, however, experienced significantly less grade 3/4 toxicity than did those in the ABVD arm (26.3% vs. 32.7%; P = .03). Grade 3/4 events were similar in the ABV and AV arms (28.3% vs. 26.5%), she said.
The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
AT THE EHA CONGRESS
Major finding: The 5-year FFTF rate was 93.1% with ABVD and 89.2% with AVD (HR, 1.50; 95% confidence interval, 1.00-2.26).
Data source: A prospective, randomized study in 1,710 patients with early-stage, favorable Hodgkin’s lymphoma.
Key clinical point: Dacarbazine and bleomycin should not be omitted from ABVD chemotherapy in the treatment of early-stage, favorable Hodgkin’s lymphoma.
Disclosures: The study was conducted by the German Hodgkin Study Group. Dr. Behringer reported having no financial disclosures.
Study validates drug’s efficacy in CLL/SLL
MILAN—Results of the phase 3 RESONATE trial suggest ibrutinib can improve response and survival rates in patients with relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), when compared to ofatumumab.
Ibrutinib conferred these benefits irrespective of baseline clinical characteristics or molecular features, including 17p deletion.
Atrial fibrillation and bleeding-related events were more common with ibrutinib. But the rate of serious adverse events was similar between the treatment arms.
About 86% of patients remained on ibrutinib at last analysis, and roughly 29% of patients initially randomized to ofatumumab crossed over to the ibrutinib arm after disease progression.
“This study undoubtedly confirms that ibrutinib is a very effective agent—as a single-agent—in relapsed CLL patients,” said investigator Peter Hillmen, MD, PhD, of The Leeds Teaching Hospitals in the UK.
Dr Hillmen presented these results at the 19th Congress of the European Hematology Association (EHA) as abstract S693. The RESONATE trial was sponsored by Pharmacyclics and Janssen, the companies developing ibrutinib.
The trial included 391 patients with relapsed or refractory CLL/SLL. They were randomized to receive oral ibrutinib at 420 mg once daily until progression or unacceptable toxicity (n=195) or intravenous ofatumumab at an initial dose of 300 mg, followed by 11 doses of 2000 mg (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57).
The median age in both treatment arms was 67. Overall, roughly 50% of patients had received 3 or more prior therapies, including purine analogs, alkylating agents, and anti-CD20 antibodies. The proportion of patients with del17p was similar between the treatment arms—32% in the ibrutinib arm and 33% in the ofatumumab arm.
Response and survival
At the time of interim analysis, patients’ median time on study was 9.4 months. The best overall response among evaluable patients was 78% in the ibrutinib arm and 11% in the ofatumumab arm, according to an independent review committee.
In addition, ibrutinib significantly prolonged progression-free survival (PFS). The median PFS was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001). The improvement in PFS represents a 78% reduction in the risk of progression or death.
Dr Hillmen noted that PFS favored ibrutinib regardless of baseline characteristics such as refractoriness to purine analogs, del17p, age, gender, Rai stage, bulky disease, number of prior treatments, del11q, B2 microglobulin, and IgVH mutation status.
Ibrutinib significantly prolonged overall survival (OS) as well. The median OS was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049). The improvement in OS represents a 56% reduction in the risk of death in patients treated with ibrutinib.
Adverse events
Dr Hillmen pointed out that the median treatment duration was 8.6 months for ibrutinib and 5.3 months for ofatumumab, and this difference confounds the assessment of side effects.
Nevertheless, nearly all patients in both treatment arms experienced adverse events—99% in the ibrutinib arm and 98% in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation of any grade was more common in the ibrutinib arm (n=10) than in the ofatumumab arm (n=1), but 5 of the ibrutinib-treated patients had a prior history of atrial fibrillation. Bleeding-related events were also more common with ibrutinib (44% vs 12%), as were diarrhea (48% vs 18%) and arthralgia (17% vs 7%).
Events more common in the ofatumumab arm included infusion-related reactions (28% vs 0%), peripheral sensory neuropathy (13% vs 4%), urticaria (6% vs 1%), night sweats (13% vs 5%), and pruritus (9% vs 4%). ![]()
MILAN—Results of the phase 3 RESONATE trial suggest ibrutinib can improve response and survival rates in patients with relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), when compared to ofatumumab.
Ibrutinib conferred these benefits irrespective of baseline clinical characteristics or molecular features, including 17p deletion.
Atrial fibrillation and bleeding-related events were more common with ibrutinib. But the rate of serious adverse events was similar between the treatment arms.
About 86% of patients remained on ibrutinib at last analysis, and roughly 29% of patients initially randomized to ofatumumab crossed over to the ibrutinib arm after disease progression.
“This study undoubtedly confirms that ibrutinib is a very effective agent—as a single-agent—in relapsed CLL patients,” said investigator Peter Hillmen, MD, PhD, of The Leeds Teaching Hospitals in the UK.
Dr Hillmen presented these results at the 19th Congress of the European Hematology Association (EHA) as abstract S693. The RESONATE trial was sponsored by Pharmacyclics and Janssen, the companies developing ibrutinib.
The trial included 391 patients with relapsed or refractory CLL/SLL. They were randomized to receive oral ibrutinib at 420 mg once daily until progression or unacceptable toxicity (n=195) or intravenous ofatumumab at an initial dose of 300 mg, followed by 11 doses of 2000 mg (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57).
The median age in both treatment arms was 67. Overall, roughly 50% of patients had received 3 or more prior therapies, including purine analogs, alkylating agents, and anti-CD20 antibodies. The proportion of patients with del17p was similar between the treatment arms—32% in the ibrutinib arm and 33% in the ofatumumab arm.
Response and survival
At the time of interim analysis, patients’ median time on study was 9.4 months. The best overall response among evaluable patients was 78% in the ibrutinib arm and 11% in the ofatumumab arm, according to an independent review committee.
In addition, ibrutinib significantly prolonged progression-free survival (PFS). The median PFS was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001). The improvement in PFS represents a 78% reduction in the risk of progression or death.
Dr Hillmen noted that PFS favored ibrutinib regardless of baseline characteristics such as refractoriness to purine analogs, del17p, age, gender, Rai stage, bulky disease, number of prior treatments, del11q, B2 microglobulin, and IgVH mutation status.
Ibrutinib significantly prolonged overall survival (OS) as well. The median OS was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049). The improvement in OS represents a 56% reduction in the risk of death in patients treated with ibrutinib.
Adverse events
Dr Hillmen pointed out that the median treatment duration was 8.6 months for ibrutinib and 5.3 months for ofatumumab, and this difference confounds the assessment of side effects.
Nevertheless, nearly all patients in both treatment arms experienced adverse events—99% in the ibrutinib arm and 98% in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation of any grade was more common in the ibrutinib arm (n=10) than in the ofatumumab arm (n=1), but 5 of the ibrutinib-treated patients had a prior history of atrial fibrillation. Bleeding-related events were also more common with ibrutinib (44% vs 12%), as were diarrhea (48% vs 18%) and arthralgia (17% vs 7%).
Events more common in the ofatumumab arm included infusion-related reactions (28% vs 0%), peripheral sensory neuropathy (13% vs 4%), urticaria (6% vs 1%), night sweats (13% vs 5%), and pruritus (9% vs 4%). ![]()
MILAN—Results of the phase 3 RESONATE trial suggest ibrutinib can improve response and survival rates in patients with relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), when compared to ofatumumab.
Ibrutinib conferred these benefits irrespective of baseline clinical characteristics or molecular features, including 17p deletion.
Atrial fibrillation and bleeding-related events were more common with ibrutinib. But the rate of serious adverse events was similar between the treatment arms.
About 86% of patients remained on ibrutinib at last analysis, and roughly 29% of patients initially randomized to ofatumumab crossed over to the ibrutinib arm after disease progression.
“This study undoubtedly confirms that ibrutinib is a very effective agent—as a single-agent—in relapsed CLL patients,” said investigator Peter Hillmen, MD, PhD, of The Leeds Teaching Hospitals in the UK.
Dr Hillmen presented these results at the 19th Congress of the European Hematology Association (EHA) as abstract S693. The RESONATE trial was sponsored by Pharmacyclics and Janssen, the companies developing ibrutinib.
The trial included 391 patients with relapsed or refractory CLL/SLL. They were randomized to receive oral ibrutinib at 420 mg once daily until progression or unacceptable toxicity (n=195) or intravenous ofatumumab at an initial dose of 300 mg, followed by 11 doses of 2000 mg (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57).
The median age in both treatment arms was 67. Overall, roughly 50% of patients had received 3 or more prior therapies, including purine analogs, alkylating agents, and anti-CD20 antibodies. The proportion of patients with del17p was similar between the treatment arms—32% in the ibrutinib arm and 33% in the ofatumumab arm.
Response and survival
At the time of interim analysis, patients’ median time on study was 9.4 months. The best overall response among evaluable patients was 78% in the ibrutinib arm and 11% in the ofatumumab arm, according to an independent review committee.
In addition, ibrutinib significantly prolonged progression-free survival (PFS). The median PFS was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001). The improvement in PFS represents a 78% reduction in the risk of progression or death.
Dr Hillmen noted that PFS favored ibrutinib regardless of baseline characteristics such as refractoriness to purine analogs, del17p, age, gender, Rai stage, bulky disease, number of prior treatments, del11q, B2 microglobulin, and IgVH mutation status.
Ibrutinib significantly prolonged overall survival (OS) as well. The median OS was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049). The improvement in OS represents a 56% reduction in the risk of death in patients treated with ibrutinib.
Adverse events
Dr Hillmen pointed out that the median treatment duration was 8.6 months for ibrutinib and 5.3 months for ofatumumab, and this difference confounds the assessment of side effects.
Nevertheless, nearly all patients in both treatment arms experienced adverse events—99% in the ibrutinib arm and 98% in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation of any grade was more common in the ibrutinib arm (n=10) than in the ofatumumab arm (n=1), but 5 of the ibrutinib-treated patients had a prior history of atrial fibrillation. Bleeding-related events were also more common with ibrutinib (44% vs 12%), as were diarrhea (48% vs 18%) and arthralgia (17% vs 7%).
Events more common in the ofatumumab arm included infusion-related reactions (28% vs 0%), peripheral sensory neuropathy (13% vs 4%), urticaria (6% vs 1%), night sweats (13% vs 5%), and pruritus (9% vs 4%). ![]()