User login
Updated: 11/22/2010
COLLEGE PARK, MD. - A Food and Drug Administration advisory panel on Nov. 18 split on whether to support approval of a noninvasive device intended to help dermatologists and other physicians detect early melanomas.
At the meeting, the FDA's General and Plastic Surgery Devices Panel voted 8 to 7 with 1 abstention that the data available supported approval of the MelaFind device to evaluate clinically atypical cutaneous pigmented lesions that have one or more characteristics of melanoma, "when a physician chooses to obtain additional information before making a final decision to biopsy to rule out melanoma." That is the indication proposed for approval by the manufacturer, Mela Sciences Inc.
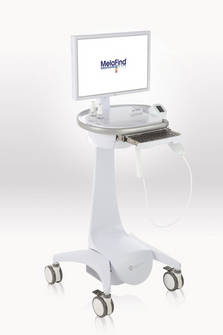
The first device of this kind, MelaFind is a multispectral computer vision system with a handheld component that captures the image of a lesion with a dermoscope through a thin layer of alcohol applied to the skin. The device has software that uses algorithms to analyze the image, indicating within 2 minutes whether a biopsy should be done, according to Mela Sciences. It is not intended to be used as a screening tool or in the evaluation of nonpigmented lesions; banal pigmented lesions; lesions considered definite melanomas; or mucosal, subungual and other lesions at different anatomic sites.
The device was used by dermatologists in a pivotal study of 1,257 patients (mean age 47 years) on 1,632 pigmented skin lesions that had at least one characteristic of melanoma, which were also biopsied. Of the 114 lesions that were positive for melanoma on biopsy, 112 were tagged as positive by MelaFind, for a sensitivity of 98.3%, which the company compared to the sensitivity of 70%-80% for dermatologists that has been reported in the literature. (Sensitivity for the dermatologists could not be determined because the decision to perform a biopsy had already been made by the clinician before referring to MelaFind.) Specificity was 9.5%.
Panelists voting for and against approval were concerned about the use of this device in the hands of nondermatologists, who, they said, might miss melanomas – a concern also raised by the FDA. But those voting in favor of approval said they believed with appropriate training, clinicians could learn to use the device effectively and that it would be a valuable tool. Concerns among those voting against approval included the need for more data, the risk of false negatives with the device, and issues with the data. It remained unclear how the results would guide decision-making.

Dermatologists on the panel voted on both sides of the approval question. "I would not mind having this in my practice," said Dr. Wilma Bergfeld, senior dermatologist and co-director of dermatopathology at the Cleveland Clinic, Cleveland, who voted in favor of approval. While clinical trial data and protocol were confusing, she said, "I believe that with the appropriate training and use, we might be able to use this instrument very effectively in the clinic to diagnose these equivocal clinical lesions."
Dr. Lynn Drake, lecturer in dermatology, Harvard Medical School, who voted against approval, said that while there is a need for such a device, "this falls short right now." Among her concerns were the need for more data, the high false-positive rate, and possible widespread use by clinicians not adequately trained once it becomes available. "We have to be very careful about approving something that might replace clinical judgement," she said.
FDA reviewers raised several concerns about the study, including possible selection bias, and concluded that the available data were inadequate to determine whether the use of MelaFind would add any true value to evaluating clinically atypical lesions – and that a prospective study was needed before approval.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts of interest prior to the meeting.
Updated: 11/22/2010
COLLEGE PARK, MD. - A Food and Drug Administration advisory panel on Nov. 18 split on whether to support approval of a noninvasive device intended to help dermatologists and other physicians detect early melanomas.
At the meeting, the FDA's General and Plastic Surgery Devices Panel voted 8 to 7 with 1 abstention that the data available supported approval of the MelaFind device to evaluate clinically atypical cutaneous pigmented lesions that have one or more characteristics of melanoma, "when a physician chooses to obtain additional information before making a final decision to biopsy to rule out melanoma." That is the indication proposed for approval by the manufacturer, Mela Sciences Inc.
The first device of this kind, MelaFind is a multispectral computer vision system with a handheld component that captures the image of a lesion with a dermoscope through a thin layer of alcohol applied to the skin. The device has software that uses algorithms to analyze the image, indicating within 2 minutes whether a biopsy should be done, according to Mela Sciences. It is not intended to be used as a screening tool or in the evaluation of nonpigmented lesions; banal pigmented lesions; lesions considered definite melanomas; or mucosal, subungual and other lesions at different anatomic sites.
The device was used by dermatologists in a pivotal study of 1,257 patients (mean age 47 years) on 1,632 pigmented skin lesions that had at least one characteristic of melanoma, which were also biopsied. Of the 114 lesions that were positive for melanoma on biopsy, 112 were tagged as positive by MelaFind, for a sensitivity of 98.3%, which the company compared to the sensitivity of 70%-80% for dermatologists that has been reported in the literature. (Sensitivity for the dermatologists could not be determined because the decision to perform a biopsy had already been made by the clinician before referring to MelaFind.) Specificity was 9.5%.
Panelists voting for and against approval were concerned about the use of this device in the hands of nondermatologists, who, they said, might miss melanomas – a concern also raised by the FDA. But those voting in favor of approval said they believed with appropriate training, clinicians could learn to use the device effectively and that it would be a valuable tool. Concerns among those voting against approval included the need for more data, the risk of false negatives with the device, and issues with the data. It remained unclear how the results would guide decision-making.
Dermatologists on the panel voted on both sides of the approval question. "I would not mind having this in my practice," said Dr. Wilma Bergfeld, senior dermatologist and co-director of dermatopathology at the Cleveland Clinic, Cleveland, who voted in favor of approval. While clinical trial data and protocol were confusing, she said, "I believe that with the appropriate training and use, we might be able to use this instrument very effectively in the clinic to diagnose these equivocal clinical lesions."
Dr. Lynn Drake, lecturer in dermatology, Harvard Medical School, who voted against approval, said that while there is a need for such a device, "this falls short right now." Among her concerns were the need for more data, the high false-positive rate, and possible widespread use by clinicians not adequately trained once it becomes available. "We have to be very careful about approving something that might replace clinical judgement," she said.
FDA reviewers raised several concerns about the study, including possible selection bias, and concluded that the available data were inadequate to determine whether the use of MelaFind would add any true value to evaluating clinically atypical lesions – and that a prospective study was needed before approval.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts of interest prior to the meeting.
Updated: 11/22/2010
COLLEGE PARK, MD. - A Food and Drug Administration advisory panel on Nov. 18 split on whether to support approval of a noninvasive device intended to help dermatologists and other physicians detect early melanomas.
At the meeting, the FDA's General and Plastic Surgery Devices Panel voted 8 to 7 with 1 abstention that the data available supported approval of the MelaFind device to evaluate clinically atypical cutaneous pigmented lesions that have one or more characteristics of melanoma, "when a physician chooses to obtain additional information before making a final decision to biopsy to rule out melanoma." That is the indication proposed for approval by the manufacturer, Mela Sciences Inc.
The first device of this kind, MelaFind is a multispectral computer vision system with a handheld component that captures the image of a lesion with a dermoscope through a thin layer of alcohol applied to the skin. The device has software that uses algorithms to analyze the image, indicating within 2 minutes whether a biopsy should be done, according to Mela Sciences. It is not intended to be used as a screening tool or in the evaluation of nonpigmented lesions; banal pigmented lesions; lesions considered definite melanomas; or mucosal, subungual and other lesions at different anatomic sites.
The device was used by dermatologists in a pivotal study of 1,257 patients (mean age 47 years) on 1,632 pigmented skin lesions that had at least one characteristic of melanoma, which were also biopsied. Of the 114 lesions that were positive for melanoma on biopsy, 112 were tagged as positive by MelaFind, for a sensitivity of 98.3%, which the company compared to the sensitivity of 70%-80% for dermatologists that has been reported in the literature. (Sensitivity for the dermatologists could not be determined because the decision to perform a biopsy had already been made by the clinician before referring to MelaFind.) Specificity was 9.5%.
Panelists voting for and against approval were concerned about the use of this device in the hands of nondermatologists, who, they said, might miss melanomas – a concern also raised by the FDA. But those voting in favor of approval said they believed with appropriate training, clinicians could learn to use the device effectively and that it would be a valuable tool. Concerns among those voting against approval included the need for more data, the risk of false negatives with the device, and issues with the data. It remained unclear how the results would guide decision-making.
Dermatologists on the panel voted on both sides of the approval question. "I would not mind having this in my practice," said Dr. Wilma Bergfeld, senior dermatologist and co-director of dermatopathology at the Cleveland Clinic, Cleveland, who voted in favor of approval. While clinical trial data and protocol were confusing, she said, "I believe that with the appropriate training and use, we might be able to use this instrument very effectively in the clinic to diagnose these equivocal clinical lesions."
Dr. Lynn Drake, lecturer in dermatology, Harvard Medical School, who voted against approval, said that while there is a need for such a device, "this falls short right now." Among her concerns were the need for more data, the high false-positive rate, and possible widespread use by clinicians not adequately trained once it becomes available. "We have to be very careful about approving something that might replace clinical judgement," she said.
FDA reviewers raised several concerns about the study, including possible selection bias, and concluded that the available data were inadequate to determine whether the use of MelaFind would add any true value to evaluating clinically atypical lesions – and that a prospective study was needed before approval.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts of interest prior to the meeting.
FROM A MEETING OF THE FDA'S GENERAL AND PLASTIC SURGERY DEVICES PANEL