User login
Omega-3 fatty acids for psychiatric illness
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.

Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.
Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.
Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
Treating ‘depression’ in patients with schizophrenia
Discuss this article at www.facebook.com/CurrentPsychiatry
Approximately 25% of schizophrenia patients experience course-related depression.1-4 Depression in patients with schizophrenia is linked to reduced social and vocational functioning, increased likelihood of psychotic relapse and rehospitalization, and other problems.2-4 Depression in patients with schizophrenia also has been linked to undesirable life events, especially “exit events” such as losing people in their lives, as well as suicidal ideation, suicide attempts, and completed suicides. Overall, it has been noted that approximately 10% of patients with schizophrenia commit suicide.5 Depressed schizophrenia patients are at particularly high risk for suicide the first few months after diagnosis and after hospital discharge.
Confirm the diagnosis
The best approach to treating depressive symptoms in schizophrenia patients is to formulate a thorough differential diagnosis (Table 1).
Table 1
Differential diagnosis of ‘depression’ in schizophrenia
| Organic factors |
| Antipsychotic-induced dysphoria |
| Akinesia |
| Akathisia |
| Negative symptoms |
| Acute disappointment reactions |
| Chronic disappointment reactions |
| Prodrome of psychotic relapse |
| Depression |
Antipsychotic-induced dysphoria. Blockade of dopamine receptors is an important feature of all antipsychotics; however, dopamine neurotransmission also is involved in the brain’s “pleasure” pathways. Individuals who take antipsychotics may experience reduced joy from once-pleasurable activities. Results of studies on the link between depression and antipsychotics have been mixed.2,4 Although some researchers have found depressed mood common among patients receiving antipsychotics, others have failed to show differences between patients treated with antipsychotics and those randomized to placebo.
Akinesia, a parkinsonian side effect of antipsychotics, can be blatant or subtle. The blatant form involves large muscle groups; these patients present with diminished arm swing, stooped posture, and parkinsonian gait. Easily spotted, such patients are unlikely to be considered depressed.
The more subtle form of akinesia is easier to confuse with depression. It can affect small muscle groups, such as in the face or vocal cords. Lack of responsiveness of facial expression is easily confused with blunted affect, low mood, lack of interest, or emotional unresponsiveness. Subtle akinesia also can impair a patient’s ability to initiate or sustain motor behavior. Many activities, from striking up a conversation to changing television channels, involve initiating and sustaining motor behavior, which these patients’ basal ganglia are underequipped to do. Life becomes boring and patients criticize themselves for “being lazy.” Patients with akinesia also are prone to dysphoria.6,7 When the lack of spontaneous motor behavior found in subtle akinesia is combined with diminished experience of pleasure due to antipsychotic blockade of dopamine, a patient may feel that “nothing is worth the effort.”
Akathisia is another movement disorder of the basal ganglia that can be triggered by antipsychotics. Whereas a patient with akinesia experiences having a “broken starter motor,” the akathisia patient experiences “a starter motor that won’t turn off.” Akathisia can be blatant or subtle. A patient with blatant akathisia has difficulty remaining seated and often paces. In subtle akathisia, increased motor activity is less dramatic, and patients may simply wander or talk excessively. Akathisia also has a dysphoric component that, when the movement is interpreted as restlessness or agitation, may look like depression.8
Negative symptoms. Primary negative symptoms in schizophrenia have several features in common with depression, which can create diagnostic challenges.9 These include anhedonia, social withdrawal, lack of initiative, lowered energy, diminished expectations and/or self confidence, and reduced speech or activity. The main feature that distinguishes the primary negative symptom syndrome from depression is prominent blue mood, which is present in depression but not in negative symptoms. Cognitive features—such as guilt, pessimism, and suicidal thoughts—are common in depression, but usually are absent in negative symptoms.
4 While the acute disappointment reaction is ongoing, the emotional burden may be substantial. With bereavement or grief reactions the loss is clear; however, be vigilant for situations where the patient’s loss may be idiosyncratic or symbolic.
Chronic disappointment reactions, also known as the demoralization syndrome, involve long-term convictions of defeat, despair, incompetence, and loss of control.10 These reactions can be devastating and prolonged. These reactions are important to identify because they may be ameliorated by rehabilitative interventions or other psychosocial supports.
Prodrome of psychotic relapse. Longitudinal observations of patients with schizophrenia have found depressive symptoms may occur during the early stages of psychotic decompensation.4,11,12 These symptoms include dysphoria, anxiety, agitation, sleep and/or appetite disturbances, impaired concentration, hopelessness, helplessness, feelings of loss of control or alienation, and social withdrawal. These features usually last a few days to a couple of weeks before they are overtaken by psychotic phenomena.
Treatment: A suggested approach
Based on my clinical experience in managing newly emergent “depression” episodes in patients correctly diagnosed with schizophrenia, I suggest the following approach:
First, assess the patient for medical disorders that could present with depressive features. Collaborate with the patient’s primary care physician to determine which medications the patient is taking and whether there have been any recent changes in these agents or their doses, including adherence issues, potential substance use or abuse, and changes between brand name and generic agents. Thoroughly evaluate the patient’s psychiatric status, including symptoms, suicidal risk, and changes in life circumstances. A patient who is at high risk of suicide may require hospitalization. Also assess for the presence of extrapyramidal side effects.
Do not change your patient’s medication regimen at this early stage, but provide him or her structure and support, and schedule an early appointment for the next visit (eg, 1 week later). A planned telephone call before the appointment may be helpful as well. If the “depression” is an acute disappointment reaction, it may run its course and resolve. However, if your patient’s depressive symptoms are a prodrome of psychotic relapse, the quick follow-up contact will improve the chances of preventing a psychotic episode by increasing the antipsychotic dosage or making other reasonable changes in pharmacotherapy.
If at the follow-up visit the patient’s psychotic symptoms have not progressed but depressive symptoms persist, evaluate for the possibility of parkinsonian symptoms, which may be subtle and difficult to rule out. If your patient is restless or tends to be physically active, a trial of a benzodiazepine can be added to treat akathisia. If the patient is underactive, consider a trial of an anticholinergic antiparkinsonian agent, such as benztropine, for akinesia. Dosages of benztropine can be raised in a stepwise manner up to 6 mg/d if there are no side effects, such as constipation, dry mouth, blurry vision, or memory impairment. Advantages of treating extrapyramidal side effects first include:
- response to antiparkinsonian medications occurs rapidly—if your patient shows no response within a week, future response at this dose is unlikely
- the presence of anticholinergic side effects is a biologic marker indicating that the treatment dose is adequate
- the clinician has more time to get to know the patient and his or her condition before committing to lowering, raising, or changing the antipsychotic dosage.
Table 2
Antidepressant effects of antipsychotics in schizophrenia patients
| Study | Design | Results |
|---|---|---|
| Marder et al, 199714 | In 2 double-blind trials, 513 patients with chronic schizophrenia received risperidone (2, 6, 10, or 16 mg/d), haloperidol (20 mg/d), or placebo for 8 weeks | Patients receiving risperidone showed greater reductions in anxiety and depression symptoms as measured by PANSS scores than patients receiving haloperidol or placebo |
| Tollefson et al, 199815 | In a prospective, blinded trial, 1,996 patients with schizophrenia received olanzapine (5 to 20 mg/d) or haloperidol (5 to 20 mg/d) | Among patients with depressive signs and symptoms, those who received olanzapine showed better improvement in MADRS scores than patients receiving haloperidol |
| Emsley et al, 200316 | Patients with schizophrenia (N = 269) who had not responded to 4 weeks of fluphenazine (20 mg/d) were randomized to receive quetiapine (600 mg/d) or haloperidol (20 mg/d) for 8 weeks | Quetiapine produced greater reduction on PANSS depression scores than haloperidol |
| Mauri et al, 200817 | In a retrospective study, 222 patients in the reexacerbation phase of schizophrenia received fluphenazine, haloperidol decanoate, haloperidol, clozapine, olanzapine, quetiapine, risperidone, or L-sulpiride monotherapy | All antipsychotics led to improvements in depressive symptoms as measured by the BPRS scale, but improvements were statistically significant only with fluphenazine, haloperidol, olanzapine, risperidone, and L-sulpiride |
| BPRS: Brief Psychiatric Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; PANSS: Positive and Negative Syndrome Scale | ||
Antidepressants. If depressive symptoms persist after lowering or changing the antipsychotic, consider a trial of an adjunctive antidepressant. Titrate antidepressants to the recommended dose over 1 month, and continue antiparkinsonian medications. See patients frequently, and ensure that they receive psychosocial support.
No randomized trials have compared the efficacy of antidepressants for treating patients with schizophrenia; therefore, it is unclear if there is a preferred agent. Newer antidepressants often are used in depressed patients with schizophrenia because they are less likely to cause anticholinergic side effects. However, anticholinergic activity may be desirable, eg, for patients with akinesia. Caution is required when combining a selective serotonin reuptake inhibitor with clozapine because metabolism interactions could lead to toxic clozapine levels in some patients.19
If your patient’s depressive symptoms improve after adding an antidepressant, continue that agent along with the antipsychotic and any antiparkinsonian medications. Only 1 study has evaluated maintenance adjunctive antidepressant therapy for depressed patients with schizophrenia who initially responded to antidepressants. It found that imipramine appeared to protect patients from depressive relapse, and patients who received maintenance adjunctive imipramine were less likely to experience worsening psychotic symptoms.20
Depressed schizophrenia patients are most likely to improve if they receive optimal psychosocial intervention,21 which consists of nonspecific support and, when indicated, psychosocial rehabilitation services. Change, even positive change, can be stressful, and patients with schizophrenia need every advantage they can get to be successful in moving their lives in a positive direction.
- Rybakowski JK, Vansteelandt K, Szafranski T, et al. Treatment of depression in first episode of schizophrenia: Results from EUFEST [published online ahead of print May 22, 2012]. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2012.04.001.
- Addington D, Addington J. Calgary Depression Scale for Schizophrenia. www.ucalgary.ca/cdss.
- Benztropine • Cogentin
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Siris reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Möller HJ. Drug treatment of depressive symptoms in schizophrenia. Clin Schizophr Relat Psychoses. 2008;1(4):328-340.
2. Buckley PF, Miller BJ, Lehrer DS, et al. Psychiatric comorbidities and schizophrenia. Schizophr Bull. 2009;35(2):383-402.
3. Hausmann A, Fleischhacker WW. Differential diagnosis of depressed mood in patients with schizophrenia: a diagnostic algorithm based on a review. Acta Psychiatr Scand. 2002;106(2):83-96.
4. Siris SG, Bench C. Depression and schizophrenia. In: Schizophrenia. Hirsch SR Weinberger DR, eds. Malden, MA: Blackwell Publishing Company; 2003:142-167.
5. Hawton K, Sutton L, Haw C, et al. Schizophrenia and suicide: systematic review of risk factors. Br J Psychiatry. 2005;187:9-20.
6. Rifkin A, Quitkin F, Klein DF. Akinesia: a poorly recognized drug-induced extrapyramidal behavioral disorder. Arch Gen Psychiatry. 1975;32(5):672-674.
7. Van Putten T, May RP. “Akinetic depression” in schizophrenia. Arch Gen Psychiatry. 1978;35(9):1101-1107.
8. Van Putten T. The many faces of akathisia. Compr Psychiatry. 1975;16(1):43-47.
9. Bermanzohn PC, Siris SG. Akinesia: a syndrome common to parkinsonism retarded depression, and negative symptoms of schizophrenia. Compr Psychiatry. 1992;33(4):221-232.
10. Clarke DM, Kissame DW. Demoralization: its phenomenology and importance. Aust N Z J Psychiatry. 2002;36(6):733-742.
11. Herz MI, Melville C. Relapse in schizophrenia. Am J Psychiatry. 1980;137(7):801-805.
12. Rosen JL, Miller TJ, D’Andrea JT, et al. Comorbid diagnoses in patients meeting criteria for the schizophrenia prodrome. Schizophr Res. 2006;85(1-3):124-131.
13. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
14. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
15. Tollefson GD, Sanger TM, Lu Y, et al. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250-258.
16. Emsley RA, Buckley P, Jones AM, et al. Differential effect of quetiapine on depressive symptoms in patients with partially responsive schizophrenia. J Psychopharmacol. 2003;17(2):210-215.
17. Mauri MC, Moliterno D, Rossattini M, et al. Depression in schizophrenia: comparison of first- and second-generation antipsychotic drugs. Schizophr Res. 2008;99(1-3):7-12.
18. Siris SG. Depression in schizophrenia: perspective in the era of “atypical” antipsychotic agents. Am J Psychiatry. 2000;157(9):1379-1389.
19. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
20. Siris SG, Bermanzohn PC, Mason SE, et al. Maintenance imipramine therapy for secondary depression in schizophrenia. A controlled trial. Arch Gen Psychiatry. 1994;51(2):109-115.
21. Bustillo J, Lauriello J, Horan W, et al. The psychosocial treatment of schizophrenia: an update. Am J Psychiatry. 2001;158(2):163-175.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approximately 25% of schizophrenia patients experience course-related depression.1-4 Depression in patients with schizophrenia is linked to reduced social and vocational functioning, increased likelihood of psychotic relapse and rehospitalization, and other problems.2-4 Depression in patients with schizophrenia also has been linked to undesirable life events, especially “exit events” such as losing people in their lives, as well as suicidal ideation, suicide attempts, and completed suicides. Overall, it has been noted that approximately 10% of patients with schizophrenia commit suicide.5 Depressed schizophrenia patients are at particularly high risk for suicide the first few months after diagnosis and after hospital discharge.
Confirm the diagnosis
The best approach to treating depressive symptoms in schizophrenia patients is to formulate a thorough differential diagnosis (Table 1).
Table 1
Differential diagnosis of ‘depression’ in schizophrenia
| Organic factors |
| Antipsychotic-induced dysphoria |
| Akinesia |
| Akathisia |
| Negative symptoms |
| Acute disappointment reactions |
| Chronic disappointment reactions |
| Prodrome of psychotic relapse |
| Depression |
Antipsychotic-induced dysphoria. Blockade of dopamine receptors is an important feature of all antipsychotics; however, dopamine neurotransmission also is involved in the brain’s “pleasure” pathways. Individuals who take antipsychotics may experience reduced joy from once-pleasurable activities. Results of studies on the link between depression and antipsychotics have been mixed.2,4 Although some researchers have found depressed mood common among patients receiving antipsychotics, others have failed to show differences between patients treated with antipsychotics and those randomized to placebo.
Akinesia, a parkinsonian side effect of antipsychotics, can be blatant or subtle. The blatant form involves large muscle groups; these patients present with diminished arm swing, stooped posture, and parkinsonian gait. Easily spotted, such patients are unlikely to be considered depressed.
The more subtle form of akinesia is easier to confuse with depression. It can affect small muscle groups, such as in the face or vocal cords. Lack of responsiveness of facial expression is easily confused with blunted affect, low mood, lack of interest, or emotional unresponsiveness. Subtle akinesia also can impair a patient’s ability to initiate or sustain motor behavior. Many activities, from striking up a conversation to changing television channels, involve initiating and sustaining motor behavior, which these patients’ basal ganglia are underequipped to do. Life becomes boring and patients criticize themselves for “being lazy.” Patients with akinesia also are prone to dysphoria.6,7 When the lack of spontaneous motor behavior found in subtle akinesia is combined with diminished experience of pleasure due to antipsychotic blockade of dopamine, a patient may feel that “nothing is worth the effort.”
Akathisia is another movement disorder of the basal ganglia that can be triggered by antipsychotics. Whereas a patient with akinesia experiences having a “broken starter motor,” the akathisia patient experiences “a starter motor that won’t turn off.” Akathisia can be blatant or subtle. A patient with blatant akathisia has difficulty remaining seated and often paces. In subtle akathisia, increased motor activity is less dramatic, and patients may simply wander or talk excessively. Akathisia also has a dysphoric component that, when the movement is interpreted as restlessness or agitation, may look like depression.8
Negative symptoms. Primary negative symptoms in schizophrenia have several features in common with depression, which can create diagnostic challenges.9 These include anhedonia, social withdrawal, lack of initiative, lowered energy, diminished expectations and/or self confidence, and reduced speech or activity. The main feature that distinguishes the primary negative symptom syndrome from depression is prominent blue mood, which is present in depression but not in negative symptoms. Cognitive features—such as guilt, pessimism, and suicidal thoughts—are common in depression, but usually are absent in negative symptoms.
4 While the acute disappointment reaction is ongoing, the emotional burden may be substantial. With bereavement or grief reactions the loss is clear; however, be vigilant for situations where the patient’s loss may be idiosyncratic or symbolic.
Chronic disappointment reactions, also known as the demoralization syndrome, involve long-term convictions of defeat, despair, incompetence, and loss of control.10 These reactions can be devastating and prolonged. These reactions are important to identify because they may be ameliorated by rehabilitative interventions or other psychosocial supports.
Prodrome of psychotic relapse. Longitudinal observations of patients with schizophrenia have found depressive symptoms may occur during the early stages of psychotic decompensation.4,11,12 These symptoms include dysphoria, anxiety, agitation, sleep and/or appetite disturbances, impaired concentration, hopelessness, helplessness, feelings of loss of control or alienation, and social withdrawal. These features usually last a few days to a couple of weeks before they are overtaken by psychotic phenomena.
Treatment: A suggested approach
Based on my clinical experience in managing newly emergent “depression” episodes in patients correctly diagnosed with schizophrenia, I suggest the following approach:
First, assess the patient for medical disorders that could present with depressive features. Collaborate with the patient’s primary care physician to determine which medications the patient is taking and whether there have been any recent changes in these agents or their doses, including adherence issues, potential substance use or abuse, and changes between brand name and generic agents. Thoroughly evaluate the patient’s psychiatric status, including symptoms, suicidal risk, and changes in life circumstances. A patient who is at high risk of suicide may require hospitalization. Also assess for the presence of extrapyramidal side effects.
Do not change your patient’s medication regimen at this early stage, but provide him or her structure and support, and schedule an early appointment for the next visit (eg, 1 week later). A planned telephone call before the appointment may be helpful as well. If the “depression” is an acute disappointment reaction, it may run its course and resolve. However, if your patient’s depressive symptoms are a prodrome of psychotic relapse, the quick follow-up contact will improve the chances of preventing a psychotic episode by increasing the antipsychotic dosage or making other reasonable changes in pharmacotherapy.
If at the follow-up visit the patient’s psychotic symptoms have not progressed but depressive symptoms persist, evaluate for the possibility of parkinsonian symptoms, which may be subtle and difficult to rule out. If your patient is restless or tends to be physically active, a trial of a benzodiazepine can be added to treat akathisia. If the patient is underactive, consider a trial of an anticholinergic antiparkinsonian agent, such as benztropine, for akinesia. Dosages of benztropine can be raised in a stepwise manner up to 6 mg/d if there are no side effects, such as constipation, dry mouth, blurry vision, or memory impairment. Advantages of treating extrapyramidal side effects first include:
- response to antiparkinsonian medications occurs rapidly—if your patient shows no response within a week, future response at this dose is unlikely
- the presence of anticholinergic side effects is a biologic marker indicating that the treatment dose is adequate
- the clinician has more time to get to know the patient and his or her condition before committing to lowering, raising, or changing the antipsychotic dosage.
Table 2
Antidepressant effects of antipsychotics in schizophrenia patients
| Study | Design | Results |
|---|---|---|
| Marder et al, 199714 | In 2 double-blind trials, 513 patients with chronic schizophrenia received risperidone (2, 6, 10, or 16 mg/d), haloperidol (20 mg/d), or placebo for 8 weeks | Patients receiving risperidone showed greater reductions in anxiety and depression symptoms as measured by PANSS scores than patients receiving haloperidol or placebo |
| Tollefson et al, 199815 | In a prospective, blinded trial, 1,996 patients with schizophrenia received olanzapine (5 to 20 mg/d) or haloperidol (5 to 20 mg/d) | Among patients with depressive signs and symptoms, those who received olanzapine showed better improvement in MADRS scores than patients receiving haloperidol |
| Emsley et al, 200316 | Patients with schizophrenia (N = 269) who had not responded to 4 weeks of fluphenazine (20 mg/d) were randomized to receive quetiapine (600 mg/d) or haloperidol (20 mg/d) for 8 weeks | Quetiapine produced greater reduction on PANSS depression scores than haloperidol |
| Mauri et al, 200817 | In a retrospective study, 222 patients in the reexacerbation phase of schizophrenia received fluphenazine, haloperidol decanoate, haloperidol, clozapine, olanzapine, quetiapine, risperidone, or L-sulpiride monotherapy | All antipsychotics led to improvements in depressive symptoms as measured by the BPRS scale, but improvements were statistically significant only with fluphenazine, haloperidol, olanzapine, risperidone, and L-sulpiride |
| BPRS: Brief Psychiatric Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; PANSS: Positive and Negative Syndrome Scale | ||
Antidepressants. If depressive symptoms persist after lowering or changing the antipsychotic, consider a trial of an adjunctive antidepressant. Titrate antidepressants to the recommended dose over 1 month, and continue antiparkinsonian medications. See patients frequently, and ensure that they receive psychosocial support.
No randomized trials have compared the efficacy of antidepressants for treating patients with schizophrenia; therefore, it is unclear if there is a preferred agent. Newer antidepressants often are used in depressed patients with schizophrenia because they are less likely to cause anticholinergic side effects. However, anticholinergic activity may be desirable, eg, for patients with akinesia. Caution is required when combining a selective serotonin reuptake inhibitor with clozapine because metabolism interactions could lead to toxic clozapine levels in some patients.19
If your patient’s depressive symptoms improve after adding an antidepressant, continue that agent along with the antipsychotic and any antiparkinsonian medications. Only 1 study has evaluated maintenance adjunctive antidepressant therapy for depressed patients with schizophrenia who initially responded to antidepressants. It found that imipramine appeared to protect patients from depressive relapse, and patients who received maintenance adjunctive imipramine were less likely to experience worsening psychotic symptoms.20
Depressed schizophrenia patients are most likely to improve if they receive optimal psychosocial intervention,21 which consists of nonspecific support and, when indicated, psychosocial rehabilitation services. Change, even positive change, can be stressful, and patients with schizophrenia need every advantage they can get to be successful in moving their lives in a positive direction.
- Rybakowski JK, Vansteelandt K, Szafranski T, et al. Treatment of depression in first episode of schizophrenia: Results from EUFEST [published online ahead of print May 22, 2012]. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2012.04.001.
- Addington D, Addington J. Calgary Depression Scale for Schizophrenia. www.ucalgary.ca/cdss.
- Benztropine • Cogentin
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Siris reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approximately 25% of schizophrenia patients experience course-related depression.1-4 Depression in patients with schizophrenia is linked to reduced social and vocational functioning, increased likelihood of psychotic relapse and rehospitalization, and other problems.2-4 Depression in patients with schizophrenia also has been linked to undesirable life events, especially “exit events” such as losing people in their lives, as well as suicidal ideation, suicide attempts, and completed suicides. Overall, it has been noted that approximately 10% of patients with schizophrenia commit suicide.5 Depressed schizophrenia patients are at particularly high risk for suicide the first few months after diagnosis and after hospital discharge.
Confirm the diagnosis
The best approach to treating depressive symptoms in schizophrenia patients is to formulate a thorough differential diagnosis (Table 1).
Table 1
Differential diagnosis of ‘depression’ in schizophrenia
| Organic factors |
| Antipsychotic-induced dysphoria |
| Akinesia |
| Akathisia |
| Negative symptoms |
| Acute disappointment reactions |
| Chronic disappointment reactions |
| Prodrome of psychotic relapse |
| Depression |
Antipsychotic-induced dysphoria. Blockade of dopamine receptors is an important feature of all antipsychotics; however, dopamine neurotransmission also is involved in the brain’s “pleasure” pathways. Individuals who take antipsychotics may experience reduced joy from once-pleasurable activities. Results of studies on the link between depression and antipsychotics have been mixed.2,4 Although some researchers have found depressed mood common among patients receiving antipsychotics, others have failed to show differences between patients treated with antipsychotics and those randomized to placebo.
Akinesia, a parkinsonian side effect of antipsychotics, can be blatant or subtle. The blatant form involves large muscle groups; these patients present with diminished arm swing, stooped posture, and parkinsonian gait. Easily spotted, such patients are unlikely to be considered depressed.
The more subtle form of akinesia is easier to confuse with depression. It can affect small muscle groups, such as in the face or vocal cords. Lack of responsiveness of facial expression is easily confused with blunted affect, low mood, lack of interest, or emotional unresponsiveness. Subtle akinesia also can impair a patient’s ability to initiate or sustain motor behavior. Many activities, from striking up a conversation to changing television channels, involve initiating and sustaining motor behavior, which these patients’ basal ganglia are underequipped to do. Life becomes boring and patients criticize themselves for “being lazy.” Patients with akinesia also are prone to dysphoria.6,7 When the lack of spontaneous motor behavior found in subtle akinesia is combined with diminished experience of pleasure due to antipsychotic blockade of dopamine, a patient may feel that “nothing is worth the effort.”
Akathisia is another movement disorder of the basal ganglia that can be triggered by antipsychotics. Whereas a patient with akinesia experiences having a “broken starter motor,” the akathisia patient experiences “a starter motor that won’t turn off.” Akathisia can be blatant or subtle. A patient with blatant akathisia has difficulty remaining seated and often paces. In subtle akathisia, increased motor activity is less dramatic, and patients may simply wander or talk excessively. Akathisia also has a dysphoric component that, when the movement is interpreted as restlessness or agitation, may look like depression.8
Negative symptoms. Primary negative symptoms in schizophrenia have several features in common with depression, which can create diagnostic challenges.9 These include anhedonia, social withdrawal, lack of initiative, lowered energy, diminished expectations and/or self confidence, and reduced speech or activity. The main feature that distinguishes the primary negative symptom syndrome from depression is prominent blue mood, which is present in depression but not in negative symptoms. Cognitive features—such as guilt, pessimism, and suicidal thoughts—are common in depression, but usually are absent in negative symptoms.
4 While the acute disappointment reaction is ongoing, the emotional burden may be substantial. With bereavement or grief reactions the loss is clear; however, be vigilant for situations where the patient’s loss may be idiosyncratic or symbolic.
Chronic disappointment reactions, also known as the demoralization syndrome, involve long-term convictions of defeat, despair, incompetence, and loss of control.10 These reactions can be devastating and prolonged. These reactions are important to identify because they may be ameliorated by rehabilitative interventions or other psychosocial supports.
Prodrome of psychotic relapse. Longitudinal observations of patients with schizophrenia have found depressive symptoms may occur during the early stages of psychotic decompensation.4,11,12 These symptoms include dysphoria, anxiety, agitation, sleep and/or appetite disturbances, impaired concentration, hopelessness, helplessness, feelings of loss of control or alienation, and social withdrawal. These features usually last a few days to a couple of weeks before they are overtaken by psychotic phenomena.
Treatment: A suggested approach
Based on my clinical experience in managing newly emergent “depression” episodes in patients correctly diagnosed with schizophrenia, I suggest the following approach:
First, assess the patient for medical disorders that could present with depressive features. Collaborate with the patient’s primary care physician to determine which medications the patient is taking and whether there have been any recent changes in these agents or their doses, including adherence issues, potential substance use or abuse, and changes between brand name and generic agents. Thoroughly evaluate the patient’s psychiatric status, including symptoms, suicidal risk, and changes in life circumstances. A patient who is at high risk of suicide may require hospitalization. Also assess for the presence of extrapyramidal side effects.
Do not change your patient’s medication regimen at this early stage, but provide him or her structure and support, and schedule an early appointment for the next visit (eg, 1 week later). A planned telephone call before the appointment may be helpful as well. If the “depression” is an acute disappointment reaction, it may run its course and resolve. However, if your patient’s depressive symptoms are a prodrome of psychotic relapse, the quick follow-up contact will improve the chances of preventing a psychotic episode by increasing the antipsychotic dosage or making other reasonable changes in pharmacotherapy.
If at the follow-up visit the patient’s psychotic symptoms have not progressed but depressive symptoms persist, evaluate for the possibility of parkinsonian symptoms, which may be subtle and difficult to rule out. If your patient is restless or tends to be physically active, a trial of a benzodiazepine can be added to treat akathisia. If the patient is underactive, consider a trial of an anticholinergic antiparkinsonian agent, such as benztropine, for akinesia. Dosages of benztropine can be raised in a stepwise manner up to 6 mg/d if there are no side effects, such as constipation, dry mouth, blurry vision, or memory impairment. Advantages of treating extrapyramidal side effects first include:
- response to antiparkinsonian medications occurs rapidly—if your patient shows no response within a week, future response at this dose is unlikely
- the presence of anticholinergic side effects is a biologic marker indicating that the treatment dose is adequate
- the clinician has more time to get to know the patient and his or her condition before committing to lowering, raising, or changing the antipsychotic dosage.
Table 2
Antidepressant effects of antipsychotics in schizophrenia patients
| Study | Design | Results |
|---|---|---|
| Marder et al, 199714 | In 2 double-blind trials, 513 patients with chronic schizophrenia received risperidone (2, 6, 10, or 16 mg/d), haloperidol (20 mg/d), or placebo for 8 weeks | Patients receiving risperidone showed greater reductions in anxiety and depression symptoms as measured by PANSS scores than patients receiving haloperidol or placebo |
| Tollefson et al, 199815 | In a prospective, blinded trial, 1,996 patients with schizophrenia received olanzapine (5 to 20 mg/d) or haloperidol (5 to 20 mg/d) | Among patients with depressive signs and symptoms, those who received olanzapine showed better improvement in MADRS scores than patients receiving haloperidol |
| Emsley et al, 200316 | Patients with schizophrenia (N = 269) who had not responded to 4 weeks of fluphenazine (20 mg/d) were randomized to receive quetiapine (600 mg/d) or haloperidol (20 mg/d) for 8 weeks | Quetiapine produced greater reduction on PANSS depression scores than haloperidol |
| Mauri et al, 200817 | In a retrospective study, 222 patients in the reexacerbation phase of schizophrenia received fluphenazine, haloperidol decanoate, haloperidol, clozapine, olanzapine, quetiapine, risperidone, or L-sulpiride monotherapy | All antipsychotics led to improvements in depressive symptoms as measured by the BPRS scale, but improvements were statistically significant only with fluphenazine, haloperidol, olanzapine, risperidone, and L-sulpiride |
| BPRS: Brief Psychiatric Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; PANSS: Positive and Negative Syndrome Scale | ||
Antidepressants. If depressive symptoms persist after lowering or changing the antipsychotic, consider a trial of an adjunctive antidepressant. Titrate antidepressants to the recommended dose over 1 month, and continue antiparkinsonian medications. See patients frequently, and ensure that they receive psychosocial support.
No randomized trials have compared the efficacy of antidepressants for treating patients with schizophrenia; therefore, it is unclear if there is a preferred agent. Newer antidepressants often are used in depressed patients with schizophrenia because they are less likely to cause anticholinergic side effects. However, anticholinergic activity may be desirable, eg, for patients with akinesia. Caution is required when combining a selective serotonin reuptake inhibitor with clozapine because metabolism interactions could lead to toxic clozapine levels in some patients.19
If your patient’s depressive symptoms improve after adding an antidepressant, continue that agent along with the antipsychotic and any antiparkinsonian medications. Only 1 study has evaluated maintenance adjunctive antidepressant therapy for depressed patients with schizophrenia who initially responded to antidepressants. It found that imipramine appeared to protect patients from depressive relapse, and patients who received maintenance adjunctive imipramine were less likely to experience worsening psychotic symptoms.20
Depressed schizophrenia patients are most likely to improve if they receive optimal psychosocial intervention,21 which consists of nonspecific support and, when indicated, psychosocial rehabilitation services. Change, even positive change, can be stressful, and patients with schizophrenia need every advantage they can get to be successful in moving their lives in a positive direction.
- Rybakowski JK, Vansteelandt K, Szafranski T, et al. Treatment of depression in first episode of schizophrenia: Results from EUFEST [published online ahead of print May 22, 2012]. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2012.04.001.
- Addington D, Addington J. Calgary Depression Scale for Schizophrenia. www.ucalgary.ca/cdss.
- Benztropine • Cogentin
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Siris reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Möller HJ. Drug treatment of depressive symptoms in schizophrenia. Clin Schizophr Relat Psychoses. 2008;1(4):328-340.
2. Buckley PF, Miller BJ, Lehrer DS, et al. Psychiatric comorbidities and schizophrenia. Schizophr Bull. 2009;35(2):383-402.
3. Hausmann A, Fleischhacker WW. Differential diagnosis of depressed mood in patients with schizophrenia: a diagnostic algorithm based on a review. Acta Psychiatr Scand. 2002;106(2):83-96.
4. Siris SG, Bench C. Depression and schizophrenia. In: Schizophrenia. Hirsch SR Weinberger DR, eds. Malden, MA: Blackwell Publishing Company; 2003:142-167.
5. Hawton K, Sutton L, Haw C, et al. Schizophrenia and suicide: systematic review of risk factors. Br J Psychiatry. 2005;187:9-20.
6. Rifkin A, Quitkin F, Klein DF. Akinesia: a poorly recognized drug-induced extrapyramidal behavioral disorder. Arch Gen Psychiatry. 1975;32(5):672-674.
7. Van Putten T, May RP. “Akinetic depression” in schizophrenia. Arch Gen Psychiatry. 1978;35(9):1101-1107.
8. Van Putten T. The many faces of akathisia. Compr Psychiatry. 1975;16(1):43-47.
9. Bermanzohn PC, Siris SG. Akinesia: a syndrome common to parkinsonism retarded depression, and negative symptoms of schizophrenia. Compr Psychiatry. 1992;33(4):221-232.
10. Clarke DM, Kissame DW. Demoralization: its phenomenology and importance. Aust N Z J Psychiatry. 2002;36(6):733-742.
11. Herz MI, Melville C. Relapse in schizophrenia. Am J Psychiatry. 1980;137(7):801-805.
12. Rosen JL, Miller TJ, D’Andrea JT, et al. Comorbid diagnoses in patients meeting criteria for the schizophrenia prodrome. Schizophr Res. 2006;85(1-3):124-131.
13. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
14. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
15. Tollefson GD, Sanger TM, Lu Y, et al. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250-258.
16. Emsley RA, Buckley P, Jones AM, et al. Differential effect of quetiapine on depressive symptoms in patients with partially responsive schizophrenia. J Psychopharmacol. 2003;17(2):210-215.
17. Mauri MC, Moliterno D, Rossattini M, et al. Depression in schizophrenia: comparison of first- and second-generation antipsychotic drugs. Schizophr Res. 2008;99(1-3):7-12.
18. Siris SG. Depression in schizophrenia: perspective in the era of “atypical” antipsychotic agents. Am J Psychiatry. 2000;157(9):1379-1389.
19. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
20. Siris SG, Bermanzohn PC, Mason SE, et al. Maintenance imipramine therapy for secondary depression in schizophrenia. A controlled trial. Arch Gen Psychiatry. 1994;51(2):109-115.
21. Bustillo J, Lauriello J, Horan W, et al. The psychosocial treatment of schizophrenia: an update. Am J Psychiatry. 2001;158(2):163-175.
1. Möller HJ. Drug treatment of depressive symptoms in schizophrenia. Clin Schizophr Relat Psychoses. 2008;1(4):328-340.
2. Buckley PF, Miller BJ, Lehrer DS, et al. Psychiatric comorbidities and schizophrenia. Schizophr Bull. 2009;35(2):383-402.
3. Hausmann A, Fleischhacker WW. Differential diagnosis of depressed mood in patients with schizophrenia: a diagnostic algorithm based on a review. Acta Psychiatr Scand. 2002;106(2):83-96.
4. Siris SG, Bench C. Depression and schizophrenia. In: Schizophrenia. Hirsch SR Weinberger DR, eds. Malden, MA: Blackwell Publishing Company; 2003:142-167.
5. Hawton K, Sutton L, Haw C, et al. Schizophrenia and suicide: systematic review of risk factors. Br J Psychiatry. 2005;187:9-20.
6. Rifkin A, Quitkin F, Klein DF. Akinesia: a poorly recognized drug-induced extrapyramidal behavioral disorder. Arch Gen Psychiatry. 1975;32(5):672-674.
7. Van Putten T, May RP. “Akinetic depression” in schizophrenia. Arch Gen Psychiatry. 1978;35(9):1101-1107.
8. Van Putten T. The many faces of akathisia. Compr Psychiatry. 1975;16(1):43-47.
9. Bermanzohn PC, Siris SG. Akinesia: a syndrome common to parkinsonism retarded depression, and negative symptoms of schizophrenia. Compr Psychiatry. 1992;33(4):221-232.
10. Clarke DM, Kissame DW. Demoralization: its phenomenology and importance. Aust N Z J Psychiatry. 2002;36(6):733-742.
11. Herz MI, Melville C. Relapse in schizophrenia. Am J Psychiatry. 1980;137(7):801-805.
12. Rosen JL, Miller TJ, D’Andrea JT, et al. Comorbid diagnoses in patients meeting criteria for the schizophrenia prodrome. Schizophr Res. 2006;85(1-3):124-131.
13. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
14. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
15. Tollefson GD, Sanger TM, Lu Y, et al. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250-258.
16. Emsley RA, Buckley P, Jones AM, et al. Differential effect of quetiapine on depressive symptoms in patients with partially responsive schizophrenia. J Psychopharmacol. 2003;17(2):210-215.
17. Mauri MC, Moliterno D, Rossattini M, et al. Depression in schizophrenia: comparison of first- and second-generation antipsychotic drugs. Schizophr Res. 2008;99(1-3):7-12.
18. Siris SG. Depression in schizophrenia: perspective in the era of “atypical” antipsychotic agents. Am J Psychiatry. 2000;157(9):1379-1389.
19. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
20. Siris SG, Bermanzohn PC, Mason SE, et al. Maintenance imipramine therapy for secondary depression in schizophrenia. A controlled trial. Arch Gen Psychiatry. 1994;51(2):109-115.
21. Bustillo J, Lauriello J, Horan W, et al. The psychosocial treatment of schizophrenia: an update. Am J Psychiatry. 2001;158(2):163-175.
Recognizing and treating complicated grief
Nearly 2.5 million persons die each year in the United States.1 For the bereaved, these deaths may be among the most painful and disruptive events they will experience. In this article, we evaluate the growing body of research on complicated grief (CG)—which also has been called prolonged grief, chronic grief, traumatic grief, and pathological grief—with an emphasis on how to identify CG and distinguish it from other adaptive and maladaptive reactions to the loss of a loved one. In addition, we review empirical evidence on treating CG, including psychotherapy, pharmacotherapy, and combined treatment approaches.
The bereavement-specific syndrome we refer to as CG currently is being reviewed for possible inclusion in DSM-5 as an official diagnosis. At press time, proposals for DSM-5 included a bereavement-related adjustment disorder within the new Trauma- and Stressor-Related Disorders category, as well as a provisional diagnosis of CG entitled Persistent Complex Bereavement-Related Disorder, which, upon acceptance, would be listed in Section III.2
What is ‘normal’ grief?
Grief is highly variable across individuals and time and may range from an absence of distress to severe and persistent pain and anguish. There’s no simple definition of “normal grief.” However, as clinicians, it’s necessary to understand the range of usual reactions. We recommend 2 considerations when evaluating grief reactions.
First, be aware that grief encompasses a range of cognitions, emotions, and behaviors. It may range from a relative lack of painful thoughts and emotions to intense and disruptive sadness, loneliness, anger, guilt, intrusive thoughts, difficulty concentrating, preoccupation with loss, social withdrawal, and a sense of being overwhelmed by the loss and its consequences. In the months after a loss, bereaved individuals may look for the deceased in a crowd, speak to them, or even experience auditory or visual hallucinations of the deceased. Nonetheless, positive feelings such as relief, peace, and happiness also are common following a loss.3 Moreover, laughter and smiling when discussing a lost loved one predicts reductions in grief symptoms over time.4 Overall, grief research suggests that, far from proceeding along standard and uniform stages,5 grief is complex and comprises a broad spectrum of thoughts, feelings, and behaviors that vary within and among individuals.
Second, note that in the absence of complications, grief progresses. For those who experience elevated levels of distress, the pain and disruption of loss initially may feel overwhelming but will subside in intensity over time for most individuals.5 This is not to say that an individual will never again feel sadness or longing for the deceased; elements of grief are likely to remain. Although the trajectory of grief symptoms varies among individuals and may progress in fits and starts, over time grief becomes more intermittent, less interfering, and is balanced with a sense of interest and purpose in life.
What is CG?
As research on grief experiences has grown, there’s increasing recognition that a minority of bereaved individuals experience more extreme grief symptoms that cause substantial, persistent distress and impairment despite the passage of many months or years. Shear et al6 proposed a set of CG diagnostic criteria (Table) in which a cluster of symptoms of intense and persistent separation distress are defined as core symptoms. Similar to other psychiatric diagnoses, the symptoms must be associated with significant distress or impairment.
Table
Proposed diagnostic criteria for complicated grief
| Symptom domain | Criteria |
|---|---|
| Separation distress | The patient has ≥1 of the following 4 symptoms: 1) Persistent, intense yearning or longing for the deceased 2) Frequent feelings of intense loneliness or emptiness 3) Recurrent negative thoughts about life without the deceased or recurrent urge to join the deceased 4) Preoccupying thoughts about the deceased that impair daily functioning |
| Thoughts | The patient has ≥2 of the following 8 symptoms: 1) Rumination about circumstances of the death 2) Frequent disbelief or inability to accept the death |
| Feelings | 3) Persistent feeling of being shocked, stunned, or emotionally numb since the death 4) Recurrent feelings of anger or bitterness regarding the death 5) Difficulty trusting or caring about others since the loss 6) Experiencing pain or other somatic symptoms the deceased person had, hearing the voice of the deceased, or seeing the deceased person 7) Intense emotional reactions to memories of the deceased |
| Behaviors | 8) Excessive avoidance or excessive preoccupation with places, people, and things related to the deceased or death |
| Source: Adapted from reference 6 | |
Assessing CG symptoms
Among those with persistent elevated distress, a CG diagnosis must be considered in the context of the individual’s social and cultural environment, time since the loss, and duration of symptoms. The hallmark symptom of CG is separation distress with a focus of cognitive, behavioral, and emotional symptoms on the loss and its consequences. CG is associated with substantial distress, functional impairment, and an increased risk for suicide. See the Box for a case study.
Many individuals with CG remain undiagnosed and untreated for years despite high levels of distress and impairment and high risk for negative consequences such as suicide.7 Accordingly, there’s a need for greater CG screening. Clinically useful tools for assessing CG include a brief, 5-item dimensional screening assessment6 and the patient-rated Inventory of Complicated Grief.8
Distinguishing complicated and uncomplicated grief. Exhibiting CG symptoms in the first several months after a loss does not mean an individual has or will develop CG. Most bereaved adults report painful thoughts and emotions in the weeks and months following the loss, including distressed yearning, waves of intense grief, persistent and intrusive thoughts, images related to the death, somatic distress, and a feeling of being disconnected from others. For most individuals, the intensity of this response diminishes within 6 to 18 months after the loved one’s death.5 Although the optimal length of time to wait before establishing a diagnosis remains debatable, the earliest CG should be diagnosed is 6 months after a loss.
It’s common for grief to occasionally rise in intensity for days or weeks. This surge may occur many months or years after the loss, even in people who exhibited relatively little distress or impairment. In particular, anniversaries, holidays, or periods of stress may trigger increased grief intensity. However, these surges typically subside naturally within a short time. Accordingly, CG should be diagnosed only when symptoms persist for >1 month.
CG vs other post-loss disorders. CG, major depressive disorder (MDD), and posttraumatic stress disorder (PTSD) often are comorbid in bereaved adults. Simon et al9 found 72% of CG patients in a treatment- seeking sample reported a lifetime history of MDD and 53% reported a lifetime history of PTSD. However, CG can be distinguished from these disorders. In the same study, 25% of CG patients had no other axis I diagnosis.9 After accounting for comorbid disorders, researchers associated CG severity with work and social impairment. These findings provide clear evidence for the incremental validity of CG—ie, a CG diagnosis gives clinicians additional information that predicts impairment above and beyond other disorders. However, future research needs to further examine CG and its overlap and differentiation from MDD and PTSD.
Distinguishing CG and MDD. Intense yearning or preoccupation with the deceased is a common symptom of CG but not MDD. In addition, CG symptoms possess intentionality. For example, emotional distress such as sadness and anger are prominent features of both CG and MDD. However, in CG, these symptoms are specific to the loss or circumstances of the loss, whereas in MDD they generally are more nebulous and generalized. Similarly, CG entails proximity seeking related to the deceased, and avoidance of reminders of the deceased, whereas MDD includes a more general social withdrawal and anhedonia.
Distinguishing CG and PTSD. CG and loss-related PTSD are distinguished by the predominant emotions and focus of concern associated with each disorder. The predominant emotion in PTSD is fear, whereas in CG it is sadness and longing. In PTSD, intrusive thoughts and memories associated with the trauma generally are associated with the event itself and produce an ongoing sense of threat.10 Avoidance in PTSD is intended to reduce this threat feeling. By contrast, in CG, intrusive memories focus on the deceased or the circumstances of the death, and avoidance is aimed at preventing painful reminders of the loss or its permanence. Importantly, both syndromes may be present.
Mr. C, age 67, presents to a local emergency department (ED) with his daughter. His daughter reports that he has not been himself since his wife died in a car accident 2 years ago. He continues to live in the house he shared with his wife, despite not needing the extra space and being unable to maintain it. Although Mr. C and his daughter used to talk about her mother a great deal, she says she now tries to avoid the subject because it upsets him. More recently she became concerned when Mr. C began to tell her that his life was meaningless without his wife. He said he frequently thinks about taking his own life to end his pain and loneliness.
Mr. C tells the ED psychiatrist he feels an intense wave of grief and loneliness every morning when he realizes his wife is not with him. He often stays in bed for hours, longing for her and thinking about their time together. At times, he thinks he hears her voice downstairs but when he searches for her, she is not there. Mr. C has been unable to go through his wife’s belongings, and feels nothing should be moved in their home. He will look at her photos, yet avoids other reminders of her (eg, partaking in their favorite hobbies, going to their favorite restaurants). He feels bitter and angry about his wife’s death, and becomes agitated when describing the car accident that took her life. Mr. C feels guilty for not being with his wife when she died. He assures the psychiatrist that he loves his children, but says he feels increasingly distant from them and doesn’t understand how they can move on after their mother’s death.
Mr. C reports symptoms consistent with a diagnosis of complicated grief. Further assessment is appropriate to determine if his symptoms are severe enough to warrant treatment.
Treating CG
When is treatment indicated? For years, bereavement theorists emphasized the need to work through emotions and memories related to the deceased with particular focus on negative material. However, evidence suggests that universal application of treatment to all bereaved individuals is unhelpful. In a recent meta-analysis, Neimeyer et al11 found that the outcomes of grief therapy applied indiscriminately to all bereaved adults or all members of high-risk populations—such as parents whose child experienced a violent death—were no better than would be expected by the passage of time. In contrast, grief therapy applied only to those who develop elevated and persistent distress (eg, CG) led to greater and more enduring improvement in post-loss distress than was observed in control conditions.
These results suggest that most grieving individuals who do not meet criteria for CG (or other psychiatric disorders) will not require intervention. Those who do seek treatment for grief-related distress in the acute grief period should be assessed for bereavement-related depression, anxiety, and suicidality, and treated or referred to professional or community-based resources for support or counseling as clinically indicated.
Evidence for psychotherapy. For those who meet CG criteria, psychotherapy targeting the specific symptoms of CG is helpful. The evidence is strongest for CG treatment (CGT), a 16-session, manualized psychotherapy developed by M. Katherine Shear, MD.12 CGT is based on an attachment model and cognitive-behavioral therapy (CBT) principles, and is informed by the dual-process theory proposed by Stroebe et al.13 According to this theory, natural healing following loss comprises 2 processes:
- a loss-oriented process in which the patient comes to terms with the loss, and
- a restoration-oriented process in which the patient reinvigorates a sense of purpose and meaning in life without the deceased.
CGT focuses on both processes. To address the former, it includes clinician-guided exercises in which the patient revisits the time of the death and planned activities in which the patient reengages with people, places, or thoughts that remind him or her of the deceased. CGT aims to allow the patient to gain an increased tolerance of the distressing thoughts and emotions associated with the loss so that these thoughts can be processed and the finality of the death and its circumstances can be accepted.
The restoration process is addressed by having patients generate and discuss personal goals and aspirations for the near and distant future, as well as scheduling pleasurable and rewarding events. This is accomplished by having patients imagine what they would want for themselves if their grief was less intense and planning concrete steps to take toward these goals. The restoration-oriented process is addressed concurrent with the loss-oriented process to encourage the oscillation between processes thought to be characteristic of a natural healing process following the loss of a loved one.
Other psychotherapy approaches (eg, support groups) may have a role for some individuals, and future research may suggest alternative approaches to CGT. To date, CGT is the most targeted evidence-based psychotherapy with randomized controlled data supporting its use in CG.
Pharmacotherapy for CG. Early research suggested that antidepressants—in particular tricyclics—may effectively reduce depressive symptoms in bereavement-related depression; their effect on CG symptoms, however, may not be as strong.14 Research on pharmacologic treatment that targets CG symptoms is developing. Because of the overlap between CG, PTSD, and MDD, researchers have hypothesized that antidepressants may be effective. Two open-label studies reported that the selective serotonin reuptake inhibitor (SSRI) escitalopram may be effective for CG.15,16 Although a post-hoc comparison of paroxetine and nortriptyline17 showed significant reduction in CG and depressive symptoms with both agents, effects could not be separated from concomitant psychotherapy. Furthermore, an examination of naturalistic data on combining antidepressants with CGT suggested that antidepressants may improve outcomes for individuals receiving CGT.18 A multicenter, randomized controlled trial funded by the National Institute of Mental Health is examining the potential efficacy of citalopram, an SSRI, alone or in combination with CGT.19
The efficacy of benzodiazepines, which commonly are prescribed for bereaved individuals, has not been assessed in CG. However, recent research suggests they may not be useful for medically managing recent grief20 and that their use in the aftermath of a loss may lead to long-term dependence in geriatric patients.21
Related Resources
- Center for Anxiety and Traumatic Stress Disorders. Massachusetts General Hospital. www.bostongrief.com.
- Zisook S, Shear K. Grief and bereavement: what psychiatrists need to know. World Psychiatry. 2009;8(2):67-74.
- Bonanno G. The other side of sadness: what the new science of bereavement tells us about loss. New York, NY: Basic Books; 2009.
Drug Brand Names
- Citalopram • Celexa
- Nortriptyline • Aventyl, Pamelor
- Escitalopram • Lexapro
- Paroxetine • Paxil
Disclosures
Dr. Simon receives grant or research support from the American Cancer Society, the American Foundation for Suicide Prevention, the Department of Defense, Forest Laboratories, and the National Institute of Mental Health.
1. Kochanek KD, Xu J, Murphy SL, et al. U.S. Department of Health and Human Services. Deaths: preliminary data for 2009. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_04.pdf. Published March 16 2011. Accessed June 19, 2012.
2. American Psychiatric Association. Trauma- and stressor-related disorders. http://www.dsm5.org/ProposedRevision/Pages/TraumaandStressorRelatedDisorders.aspx. Accessed June 19 2012.
3. Bonanno GA, Kaltman S. Toward an integrative perspective on bereavement. Psychol Bull. 1999;125(6):760-776.
4. Bonanno GA, Keltner D. Facial expressions of emotion and the course of conjugal bereavement. J Abnorm Psychol. 1997;106(1):126-137.
5. Bonanno GA, Wortman CB, Lehman DR, et al. Resilience to loss and chronic grief: a prospective study from preloss to 18-months postloss. J Pers Soc Psychol. 2002;83(5):1150-1164.
6. Shear MK, Simon N, Wall M, et al. Complicated grief and related bereavement issues for DSM-5. Depress Anxiety. 2011;28(2):103-117.
7. Boelen PA, Prigerson HG. The influence of symptoms of prolonged grief disorder depression, and anxiety on quality of life among bereaved adults: a prospective study. Eur Arch Psychiatry Clin Neurosci. 2007;257(8):444-452.
8. Prigerson HG, Maciejewski PK, Reynolds CF, 3rd, et al. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res. 1995;59 (1-2):65-79.
9. Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry. 2007;48(5):395-399.
10. Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339-376.
11. Neimeyer RA, Currier JM. Grief therapy: evidence of efficacy and emerging directions. Curr Dir Psychol Sci. 2009;18(6):352-356.
12. Shear K, Frank E, Houck PR, et al. Treatment of complicated grief: a randomized controlled trial. JAMA. 2005;293(21):2601-2608.
13. Stroebe M, Schut H. The dual process model of coping with bereavement: rationale and description. Death Stud. 1999;23(3):197-224.
14. Reynolds CF, 3rd, Miller MD, Pasternak RE, et al. Treatment of bereavement-related major depressive episodes in later life: a controlled study of acute and continuation treatment with nortriptyline and interpersonal psychotherapy. Am J Psychiatry. 1999;156(2):202-208.
15. Simon NM, Thompson EH, Pollack MH, et al. Complicated grief: a case series using escitalopram. Am J Psychiatry. 2007;164(11):1760-1761.
16. Hensley PL, Slonimski CK, Uhlenhuth EH, et al. Escitalopram: an open-label study of bereavement-related depression and grief. J Affect Disord. 2009;113(1-2):142-149.
17. Zygmont M, Prigerson HG, Houck PR, et al. A post hoc comparison of paroxetine and nortriptyline for symptoms of traumatic grief. J Clin Psychiatry. 1998;59(5):241-245.
18. Simon NM, Shear MK, Fagiolini A, et al. Impact of concurrent naturalistic pharmacotherapy on psychotherapy of complicated grief. Psychiatry Res. 2008;159(1-2):31-36.
19. U.S. National Institutes of Health. A study of medication with or without psychotherapy for complicated grief (HEAL). http://clinicaltrials.gov/ct2/show/NCT01179568. Published June 24, 2012. Accessed June 25, 2012.
20. Warner J, Metcalfe C, King M. Evaluating the use of benzodiazepines following recent bereavement. Br J Psychiatry. 2001;178(1):36-41.
21. Cook JM, Biyanova T, Marshall R. Medicating grief with benzodiazepines: physician and patient perspectives. Arch Intern Med. 2007;167(18):2006-2007.
Nearly 2.5 million persons die each year in the United States.1 For the bereaved, these deaths may be among the most painful and disruptive events they will experience. In this article, we evaluate the growing body of research on complicated grief (CG)—which also has been called prolonged grief, chronic grief, traumatic grief, and pathological grief—with an emphasis on how to identify CG and distinguish it from other adaptive and maladaptive reactions to the loss of a loved one. In addition, we review empirical evidence on treating CG, including psychotherapy, pharmacotherapy, and combined treatment approaches.
The bereavement-specific syndrome we refer to as CG currently is being reviewed for possible inclusion in DSM-5 as an official diagnosis. At press time, proposals for DSM-5 included a bereavement-related adjustment disorder within the new Trauma- and Stressor-Related Disorders category, as well as a provisional diagnosis of CG entitled Persistent Complex Bereavement-Related Disorder, which, upon acceptance, would be listed in Section III.2
What is ‘normal’ grief?
Grief is highly variable across individuals and time and may range from an absence of distress to severe and persistent pain and anguish. There’s no simple definition of “normal grief.” However, as clinicians, it’s necessary to understand the range of usual reactions. We recommend 2 considerations when evaluating grief reactions.
First, be aware that grief encompasses a range of cognitions, emotions, and behaviors. It may range from a relative lack of painful thoughts and emotions to intense and disruptive sadness, loneliness, anger, guilt, intrusive thoughts, difficulty concentrating, preoccupation with loss, social withdrawal, and a sense of being overwhelmed by the loss and its consequences. In the months after a loss, bereaved individuals may look for the deceased in a crowd, speak to them, or even experience auditory or visual hallucinations of the deceased. Nonetheless, positive feelings such as relief, peace, and happiness also are common following a loss.3 Moreover, laughter and smiling when discussing a lost loved one predicts reductions in grief symptoms over time.4 Overall, grief research suggests that, far from proceeding along standard and uniform stages,5 grief is complex and comprises a broad spectrum of thoughts, feelings, and behaviors that vary within and among individuals.
Second, note that in the absence of complications, grief progresses. For those who experience elevated levels of distress, the pain and disruption of loss initially may feel overwhelming but will subside in intensity over time for most individuals.5 This is not to say that an individual will never again feel sadness or longing for the deceased; elements of grief are likely to remain. Although the trajectory of grief symptoms varies among individuals and may progress in fits and starts, over time grief becomes more intermittent, less interfering, and is balanced with a sense of interest and purpose in life.
What is CG?
As research on grief experiences has grown, there’s increasing recognition that a minority of bereaved individuals experience more extreme grief symptoms that cause substantial, persistent distress and impairment despite the passage of many months or years. Shear et al6 proposed a set of CG diagnostic criteria (Table) in which a cluster of symptoms of intense and persistent separation distress are defined as core symptoms. Similar to other psychiatric diagnoses, the symptoms must be associated with significant distress or impairment.
Table
Proposed diagnostic criteria for complicated grief
| Symptom domain | Criteria |
|---|---|
| Separation distress | The patient has ≥1 of the following 4 symptoms: 1) Persistent, intense yearning or longing for the deceased 2) Frequent feelings of intense loneliness or emptiness 3) Recurrent negative thoughts about life without the deceased or recurrent urge to join the deceased 4) Preoccupying thoughts about the deceased that impair daily functioning |
| Thoughts | The patient has ≥2 of the following 8 symptoms: 1) Rumination about circumstances of the death 2) Frequent disbelief or inability to accept the death |
| Feelings | 3) Persistent feeling of being shocked, stunned, or emotionally numb since the death 4) Recurrent feelings of anger or bitterness regarding the death 5) Difficulty trusting or caring about others since the loss 6) Experiencing pain or other somatic symptoms the deceased person had, hearing the voice of the deceased, or seeing the deceased person 7) Intense emotional reactions to memories of the deceased |
| Behaviors | 8) Excessive avoidance or excessive preoccupation with places, people, and things related to the deceased or death |
| Source: Adapted from reference 6 | |
Assessing CG symptoms
Among those with persistent elevated distress, a CG diagnosis must be considered in the context of the individual’s social and cultural environment, time since the loss, and duration of symptoms. The hallmark symptom of CG is separation distress with a focus of cognitive, behavioral, and emotional symptoms on the loss and its consequences. CG is associated with substantial distress, functional impairment, and an increased risk for suicide. See the Box for a case study.
Many individuals with CG remain undiagnosed and untreated for years despite high levels of distress and impairment and high risk for negative consequences such as suicide.7 Accordingly, there’s a need for greater CG screening. Clinically useful tools for assessing CG include a brief, 5-item dimensional screening assessment6 and the patient-rated Inventory of Complicated Grief.8
Distinguishing complicated and uncomplicated grief. Exhibiting CG symptoms in the first several months after a loss does not mean an individual has or will develop CG. Most bereaved adults report painful thoughts and emotions in the weeks and months following the loss, including distressed yearning, waves of intense grief, persistent and intrusive thoughts, images related to the death, somatic distress, and a feeling of being disconnected from others. For most individuals, the intensity of this response diminishes within 6 to 18 months after the loved one’s death.5 Although the optimal length of time to wait before establishing a diagnosis remains debatable, the earliest CG should be diagnosed is 6 months after a loss.
It’s common for grief to occasionally rise in intensity for days or weeks. This surge may occur many months or years after the loss, even in people who exhibited relatively little distress or impairment. In particular, anniversaries, holidays, or periods of stress may trigger increased grief intensity. However, these surges typically subside naturally within a short time. Accordingly, CG should be diagnosed only when symptoms persist for >1 month.
CG vs other post-loss disorders. CG, major depressive disorder (MDD), and posttraumatic stress disorder (PTSD) often are comorbid in bereaved adults. Simon et al9 found 72% of CG patients in a treatment- seeking sample reported a lifetime history of MDD and 53% reported a lifetime history of PTSD. However, CG can be distinguished from these disorders. In the same study, 25% of CG patients had no other axis I diagnosis.9 After accounting for comorbid disorders, researchers associated CG severity with work and social impairment. These findings provide clear evidence for the incremental validity of CG—ie, a CG diagnosis gives clinicians additional information that predicts impairment above and beyond other disorders. However, future research needs to further examine CG and its overlap and differentiation from MDD and PTSD.
Distinguishing CG and MDD. Intense yearning or preoccupation with the deceased is a common symptom of CG but not MDD. In addition, CG symptoms possess intentionality. For example, emotional distress such as sadness and anger are prominent features of both CG and MDD. However, in CG, these symptoms are specific to the loss or circumstances of the loss, whereas in MDD they generally are more nebulous and generalized. Similarly, CG entails proximity seeking related to the deceased, and avoidance of reminders of the deceased, whereas MDD includes a more general social withdrawal and anhedonia.
Distinguishing CG and PTSD. CG and loss-related PTSD are distinguished by the predominant emotions and focus of concern associated with each disorder. The predominant emotion in PTSD is fear, whereas in CG it is sadness and longing. In PTSD, intrusive thoughts and memories associated with the trauma generally are associated with the event itself and produce an ongoing sense of threat.10 Avoidance in PTSD is intended to reduce this threat feeling. By contrast, in CG, intrusive memories focus on the deceased or the circumstances of the death, and avoidance is aimed at preventing painful reminders of the loss or its permanence. Importantly, both syndromes may be present.
Mr. C, age 67, presents to a local emergency department (ED) with his daughter. His daughter reports that he has not been himself since his wife died in a car accident 2 years ago. He continues to live in the house he shared with his wife, despite not needing the extra space and being unable to maintain it. Although Mr. C and his daughter used to talk about her mother a great deal, she says she now tries to avoid the subject because it upsets him. More recently she became concerned when Mr. C began to tell her that his life was meaningless without his wife. He said he frequently thinks about taking his own life to end his pain and loneliness.
Mr. C tells the ED psychiatrist he feels an intense wave of grief and loneliness every morning when he realizes his wife is not with him. He often stays in bed for hours, longing for her and thinking about their time together. At times, he thinks he hears her voice downstairs but when he searches for her, she is not there. Mr. C has been unable to go through his wife’s belongings, and feels nothing should be moved in their home. He will look at her photos, yet avoids other reminders of her (eg, partaking in their favorite hobbies, going to their favorite restaurants). He feels bitter and angry about his wife’s death, and becomes agitated when describing the car accident that took her life. Mr. C feels guilty for not being with his wife when she died. He assures the psychiatrist that he loves his children, but says he feels increasingly distant from them and doesn’t understand how they can move on after their mother’s death.
Mr. C reports symptoms consistent with a diagnosis of complicated grief. Further assessment is appropriate to determine if his symptoms are severe enough to warrant treatment.
Treating CG
When is treatment indicated? For years, bereavement theorists emphasized the need to work through emotions and memories related to the deceased with particular focus on negative material. However, evidence suggests that universal application of treatment to all bereaved individuals is unhelpful. In a recent meta-analysis, Neimeyer et al11 found that the outcomes of grief therapy applied indiscriminately to all bereaved adults or all members of high-risk populations—such as parents whose child experienced a violent death—were no better than would be expected by the passage of time. In contrast, grief therapy applied only to those who develop elevated and persistent distress (eg, CG) led to greater and more enduring improvement in post-loss distress than was observed in control conditions.
These results suggest that most grieving individuals who do not meet criteria for CG (or other psychiatric disorders) will not require intervention. Those who do seek treatment for grief-related distress in the acute grief period should be assessed for bereavement-related depression, anxiety, and suicidality, and treated or referred to professional or community-based resources for support or counseling as clinically indicated.
Evidence for psychotherapy. For those who meet CG criteria, psychotherapy targeting the specific symptoms of CG is helpful. The evidence is strongest for CG treatment (CGT), a 16-session, manualized psychotherapy developed by M. Katherine Shear, MD.12 CGT is based on an attachment model and cognitive-behavioral therapy (CBT) principles, and is informed by the dual-process theory proposed by Stroebe et al.13 According to this theory, natural healing following loss comprises 2 processes:
- a loss-oriented process in which the patient comes to terms with the loss, and
- a restoration-oriented process in which the patient reinvigorates a sense of purpose and meaning in life without the deceased.
CGT focuses on both processes. To address the former, it includes clinician-guided exercises in which the patient revisits the time of the death and planned activities in which the patient reengages with people, places, or thoughts that remind him or her of the deceased. CGT aims to allow the patient to gain an increased tolerance of the distressing thoughts and emotions associated with the loss so that these thoughts can be processed and the finality of the death and its circumstances can be accepted.
The restoration process is addressed by having patients generate and discuss personal goals and aspirations for the near and distant future, as well as scheduling pleasurable and rewarding events. This is accomplished by having patients imagine what they would want for themselves if their grief was less intense and planning concrete steps to take toward these goals. The restoration-oriented process is addressed concurrent with the loss-oriented process to encourage the oscillation between processes thought to be characteristic of a natural healing process following the loss of a loved one.
Other psychotherapy approaches (eg, support groups) may have a role for some individuals, and future research may suggest alternative approaches to CGT. To date, CGT is the most targeted evidence-based psychotherapy with randomized controlled data supporting its use in CG.
Pharmacotherapy for CG. Early research suggested that antidepressants—in particular tricyclics—may effectively reduce depressive symptoms in bereavement-related depression; their effect on CG symptoms, however, may not be as strong.14 Research on pharmacologic treatment that targets CG symptoms is developing. Because of the overlap between CG, PTSD, and MDD, researchers have hypothesized that antidepressants may be effective. Two open-label studies reported that the selective serotonin reuptake inhibitor (SSRI) escitalopram may be effective for CG.15,16 Although a post-hoc comparison of paroxetine and nortriptyline17 showed significant reduction in CG and depressive symptoms with both agents, effects could not be separated from concomitant psychotherapy. Furthermore, an examination of naturalistic data on combining antidepressants with CGT suggested that antidepressants may improve outcomes for individuals receiving CGT.18 A multicenter, randomized controlled trial funded by the National Institute of Mental Health is examining the potential efficacy of citalopram, an SSRI, alone or in combination with CGT.19
The efficacy of benzodiazepines, which commonly are prescribed for bereaved individuals, has not been assessed in CG. However, recent research suggests they may not be useful for medically managing recent grief20 and that their use in the aftermath of a loss may lead to long-term dependence in geriatric patients.21
Related Resources
- Center for Anxiety and Traumatic Stress Disorders. Massachusetts General Hospital. www.bostongrief.com.
- Zisook S, Shear K. Grief and bereavement: what psychiatrists need to know. World Psychiatry. 2009;8(2):67-74.
- Bonanno G. The other side of sadness: what the new science of bereavement tells us about loss. New York, NY: Basic Books; 2009.
Drug Brand Names
- Citalopram • Celexa
- Nortriptyline • Aventyl, Pamelor
- Escitalopram • Lexapro
- Paroxetine • Paxil
Disclosures
Dr. Simon receives grant or research support from the American Cancer Society, the American Foundation for Suicide Prevention, the Department of Defense, Forest Laboratories, and the National Institute of Mental Health.
Nearly 2.5 million persons die each year in the United States.1 For the bereaved, these deaths may be among the most painful and disruptive events they will experience. In this article, we evaluate the growing body of research on complicated grief (CG)—which also has been called prolonged grief, chronic grief, traumatic grief, and pathological grief—with an emphasis on how to identify CG and distinguish it from other adaptive and maladaptive reactions to the loss of a loved one. In addition, we review empirical evidence on treating CG, including psychotherapy, pharmacotherapy, and combined treatment approaches.
The bereavement-specific syndrome we refer to as CG currently is being reviewed for possible inclusion in DSM-5 as an official diagnosis. At press time, proposals for DSM-5 included a bereavement-related adjustment disorder within the new Trauma- and Stressor-Related Disorders category, as well as a provisional diagnosis of CG entitled Persistent Complex Bereavement-Related Disorder, which, upon acceptance, would be listed in Section III.2
What is ‘normal’ grief?
Grief is highly variable across individuals and time and may range from an absence of distress to severe and persistent pain and anguish. There’s no simple definition of “normal grief.” However, as clinicians, it’s necessary to understand the range of usual reactions. We recommend 2 considerations when evaluating grief reactions.
First, be aware that grief encompasses a range of cognitions, emotions, and behaviors. It may range from a relative lack of painful thoughts and emotions to intense and disruptive sadness, loneliness, anger, guilt, intrusive thoughts, difficulty concentrating, preoccupation with loss, social withdrawal, and a sense of being overwhelmed by the loss and its consequences. In the months after a loss, bereaved individuals may look for the deceased in a crowd, speak to them, or even experience auditory or visual hallucinations of the deceased. Nonetheless, positive feelings such as relief, peace, and happiness also are common following a loss.3 Moreover, laughter and smiling when discussing a lost loved one predicts reductions in grief symptoms over time.4 Overall, grief research suggests that, far from proceeding along standard and uniform stages,5 grief is complex and comprises a broad spectrum of thoughts, feelings, and behaviors that vary within and among individuals.
Second, note that in the absence of complications, grief progresses. For those who experience elevated levels of distress, the pain and disruption of loss initially may feel overwhelming but will subside in intensity over time for most individuals.5 This is not to say that an individual will never again feel sadness or longing for the deceased; elements of grief are likely to remain. Although the trajectory of grief symptoms varies among individuals and may progress in fits and starts, over time grief becomes more intermittent, less interfering, and is balanced with a sense of interest and purpose in life.
What is CG?
As research on grief experiences has grown, there’s increasing recognition that a minority of bereaved individuals experience more extreme grief symptoms that cause substantial, persistent distress and impairment despite the passage of many months or years. Shear et al6 proposed a set of CG diagnostic criteria (Table) in which a cluster of symptoms of intense and persistent separation distress are defined as core symptoms. Similar to other psychiatric diagnoses, the symptoms must be associated with significant distress or impairment.
Table
Proposed diagnostic criteria for complicated grief
| Symptom domain | Criteria |
|---|---|
| Separation distress | The patient has ≥1 of the following 4 symptoms: 1) Persistent, intense yearning or longing for the deceased 2) Frequent feelings of intense loneliness or emptiness 3) Recurrent negative thoughts about life without the deceased or recurrent urge to join the deceased 4) Preoccupying thoughts about the deceased that impair daily functioning |
| Thoughts | The patient has ≥2 of the following 8 symptoms: 1) Rumination about circumstances of the death 2) Frequent disbelief or inability to accept the death |
| Feelings | 3) Persistent feeling of being shocked, stunned, or emotionally numb since the death 4) Recurrent feelings of anger or bitterness regarding the death 5) Difficulty trusting or caring about others since the loss 6) Experiencing pain or other somatic symptoms the deceased person had, hearing the voice of the deceased, or seeing the deceased person 7) Intense emotional reactions to memories of the deceased |
| Behaviors | 8) Excessive avoidance or excessive preoccupation with places, people, and things related to the deceased or death |
| Source: Adapted from reference 6 | |
Assessing CG symptoms
Among those with persistent elevated distress, a CG diagnosis must be considered in the context of the individual’s social and cultural environment, time since the loss, and duration of symptoms. The hallmark symptom of CG is separation distress with a focus of cognitive, behavioral, and emotional symptoms on the loss and its consequences. CG is associated with substantial distress, functional impairment, and an increased risk for suicide. See the Box for a case study.
Many individuals with CG remain undiagnosed and untreated for years despite high levels of distress and impairment and high risk for negative consequences such as suicide.7 Accordingly, there’s a need for greater CG screening. Clinically useful tools for assessing CG include a brief, 5-item dimensional screening assessment6 and the patient-rated Inventory of Complicated Grief.8
Distinguishing complicated and uncomplicated grief. Exhibiting CG symptoms in the first several months after a loss does not mean an individual has or will develop CG. Most bereaved adults report painful thoughts and emotions in the weeks and months following the loss, including distressed yearning, waves of intense grief, persistent and intrusive thoughts, images related to the death, somatic distress, and a feeling of being disconnected from others. For most individuals, the intensity of this response diminishes within 6 to 18 months after the loved one’s death.5 Although the optimal length of time to wait before establishing a diagnosis remains debatable, the earliest CG should be diagnosed is 6 months after a loss.
It’s common for grief to occasionally rise in intensity for days or weeks. This surge may occur many months or years after the loss, even in people who exhibited relatively little distress or impairment. In particular, anniversaries, holidays, or periods of stress may trigger increased grief intensity. However, these surges typically subside naturally within a short time. Accordingly, CG should be diagnosed only when symptoms persist for >1 month.
CG vs other post-loss disorders. CG, major depressive disorder (MDD), and posttraumatic stress disorder (PTSD) often are comorbid in bereaved adults. Simon et al9 found 72% of CG patients in a treatment- seeking sample reported a lifetime history of MDD and 53% reported a lifetime history of PTSD. However, CG can be distinguished from these disorders. In the same study, 25% of CG patients had no other axis I diagnosis.9 After accounting for comorbid disorders, researchers associated CG severity with work and social impairment. These findings provide clear evidence for the incremental validity of CG—ie, a CG diagnosis gives clinicians additional information that predicts impairment above and beyond other disorders. However, future research needs to further examine CG and its overlap and differentiation from MDD and PTSD.
Distinguishing CG and MDD. Intense yearning or preoccupation with the deceased is a common symptom of CG but not MDD. In addition, CG symptoms possess intentionality. For example, emotional distress such as sadness and anger are prominent features of both CG and MDD. However, in CG, these symptoms are specific to the loss or circumstances of the loss, whereas in MDD they generally are more nebulous and generalized. Similarly, CG entails proximity seeking related to the deceased, and avoidance of reminders of the deceased, whereas MDD includes a more general social withdrawal and anhedonia.
Distinguishing CG and PTSD. CG and loss-related PTSD are distinguished by the predominant emotions and focus of concern associated with each disorder. The predominant emotion in PTSD is fear, whereas in CG it is sadness and longing. In PTSD, intrusive thoughts and memories associated with the trauma generally are associated with the event itself and produce an ongoing sense of threat.10 Avoidance in PTSD is intended to reduce this threat feeling. By contrast, in CG, intrusive memories focus on the deceased or the circumstances of the death, and avoidance is aimed at preventing painful reminders of the loss or its permanence. Importantly, both syndromes may be present.
Mr. C, age 67, presents to a local emergency department (ED) with his daughter. His daughter reports that he has not been himself since his wife died in a car accident 2 years ago. He continues to live in the house he shared with his wife, despite not needing the extra space and being unable to maintain it. Although Mr. C and his daughter used to talk about her mother a great deal, she says she now tries to avoid the subject because it upsets him. More recently she became concerned when Mr. C began to tell her that his life was meaningless without his wife. He said he frequently thinks about taking his own life to end his pain and loneliness.
Mr. C tells the ED psychiatrist he feels an intense wave of grief and loneliness every morning when he realizes his wife is not with him. He often stays in bed for hours, longing for her and thinking about their time together. At times, he thinks he hears her voice downstairs but when he searches for her, she is not there. Mr. C has been unable to go through his wife’s belongings, and feels nothing should be moved in their home. He will look at her photos, yet avoids other reminders of her (eg, partaking in their favorite hobbies, going to their favorite restaurants). He feels bitter and angry about his wife’s death, and becomes agitated when describing the car accident that took her life. Mr. C feels guilty for not being with his wife when she died. He assures the psychiatrist that he loves his children, but says he feels increasingly distant from them and doesn’t understand how they can move on after their mother’s death.
Mr. C reports symptoms consistent with a diagnosis of complicated grief. Further assessment is appropriate to determine if his symptoms are severe enough to warrant treatment.
Treating CG
When is treatment indicated? For years, bereavement theorists emphasized the need to work through emotions and memories related to the deceased with particular focus on negative material. However, evidence suggests that universal application of treatment to all bereaved individuals is unhelpful. In a recent meta-analysis, Neimeyer et al11 found that the outcomes of grief therapy applied indiscriminately to all bereaved adults or all members of high-risk populations—such as parents whose child experienced a violent death—were no better than would be expected by the passage of time. In contrast, grief therapy applied only to those who develop elevated and persistent distress (eg, CG) led to greater and more enduring improvement in post-loss distress than was observed in control conditions.
These results suggest that most grieving individuals who do not meet criteria for CG (or other psychiatric disorders) will not require intervention. Those who do seek treatment for grief-related distress in the acute grief period should be assessed for bereavement-related depression, anxiety, and suicidality, and treated or referred to professional or community-based resources for support or counseling as clinically indicated.
Evidence for psychotherapy. For those who meet CG criteria, psychotherapy targeting the specific symptoms of CG is helpful. The evidence is strongest for CG treatment (CGT), a 16-session, manualized psychotherapy developed by M. Katherine Shear, MD.12 CGT is based on an attachment model and cognitive-behavioral therapy (CBT) principles, and is informed by the dual-process theory proposed by Stroebe et al.13 According to this theory, natural healing following loss comprises 2 processes:
- a loss-oriented process in which the patient comes to terms with the loss, and
- a restoration-oriented process in which the patient reinvigorates a sense of purpose and meaning in life without the deceased.
CGT focuses on both processes. To address the former, it includes clinician-guided exercises in which the patient revisits the time of the death and planned activities in which the patient reengages with people, places, or thoughts that remind him or her of the deceased. CGT aims to allow the patient to gain an increased tolerance of the distressing thoughts and emotions associated with the loss so that these thoughts can be processed and the finality of the death and its circumstances can be accepted.
The restoration process is addressed by having patients generate and discuss personal goals and aspirations for the near and distant future, as well as scheduling pleasurable and rewarding events. This is accomplished by having patients imagine what they would want for themselves if their grief was less intense and planning concrete steps to take toward these goals. The restoration-oriented process is addressed concurrent with the loss-oriented process to encourage the oscillation between processes thought to be characteristic of a natural healing process following the loss of a loved one.
Other psychotherapy approaches (eg, support groups) may have a role for some individuals, and future research may suggest alternative approaches to CGT. To date, CGT is the most targeted evidence-based psychotherapy with randomized controlled data supporting its use in CG.
Pharmacotherapy for CG. Early research suggested that antidepressants—in particular tricyclics—may effectively reduce depressive symptoms in bereavement-related depression; their effect on CG symptoms, however, may not be as strong.14 Research on pharmacologic treatment that targets CG symptoms is developing. Because of the overlap between CG, PTSD, and MDD, researchers have hypothesized that antidepressants may be effective. Two open-label studies reported that the selective serotonin reuptake inhibitor (SSRI) escitalopram may be effective for CG.15,16 Although a post-hoc comparison of paroxetine and nortriptyline17 showed significant reduction in CG and depressive symptoms with both agents, effects could not be separated from concomitant psychotherapy. Furthermore, an examination of naturalistic data on combining antidepressants with CGT suggested that antidepressants may improve outcomes for individuals receiving CGT.18 A multicenter, randomized controlled trial funded by the National Institute of Mental Health is examining the potential efficacy of citalopram, an SSRI, alone or in combination with CGT.19
The efficacy of benzodiazepines, which commonly are prescribed for bereaved individuals, has not been assessed in CG. However, recent research suggests they may not be useful for medically managing recent grief20 and that their use in the aftermath of a loss may lead to long-term dependence in geriatric patients.21
Related Resources
- Center for Anxiety and Traumatic Stress Disorders. Massachusetts General Hospital. www.bostongrief.com.
- Zisook S, Shear K. Grief and bereavement: what psychiatrists need to know. World Psychiatry. 2009;8(2):67-74.
- Bonanno G. The other side of sadness: what the new science of bereavement tells us about loss. New York, NY: Basic Books; 2009.
Drug Brand Names
- Citalopram • Celexa
- Nortriptyline • Aventyl, Pamelor
- Escitalopram • Lexapro
- Paroxetine • Paxil
Disclosures
Dr. Simon receives grant or research support from the American Cancer Society, the American Foundation for Suicide Prevention, the Department of Defense, Forest Laboratories, and the National Institute of Mental Health.
1. Kochanek KD, Xu J, Murphy SL, et al. U.S. Department of Health and Human Services. Deaths: preliminary data for 2009. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_04.pdf. Published March 16 2011. Accessed June 19, 2012.
2. American Psychiatric Association. Trauma- and stressor-related disorders. http://www.dsm5.org/ProposedRevision/Pages/TraumaandStressorRelatedDisorders.aspx. Accessed June 19 2012.
3. Bonanno GA, Kaltman S. Toward an integrative perspective on bereavement. Psychol Bull. 1999;125(6):760-776.
4. Bonanno GA, Keltner D. Facial expressions of emotion and the course of conjugal bereavement. J Abnorm Psychol. 1997;106(1):126-137.
5. Bonanno GA, Wortman CB, Lehman DR, et al. Resilience to loss and chronic grief: a prospective study from preloss to 18-months postloss. J Pers Soc Psychol. 2002;83(5):1150-1164.
6. Shear MK, Simon N, Wall M, et al. Complicated grief and related bereavement issues for DSM-5. Depress Anxiety. 2011;28(2):103-117.
7. Boelen PA, Prigerson HG. The influence of symptoms of prolonged grief disorder depression, and anxiety on quality of life among bereaved adults: a prospective study. Eur Arch Psychiatry Clin Neurosci. 2007;257(8):444-452.
8. Prigerson HG, Maciejewski PK, Reynolds CF, 3rd, et al. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res. 1995;59 (1-2):65-79.
9. Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry. 2007;48(5):395-399.
10. Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339-376.
11. Neimeyer RA, Currier JM. Grief therapy: evidence of efficacy and emerging directions. Curr Dir Psychol Sci. 2009;18(6):352-356.
12. Shear K, Frank E, Houck PR, et al. Treatment of complicated grief: a randomized controlled trial. JAMA. 2005;293(21):2601-2608.
13. Stroebe M, Schut H. The dual process model of coping with bereavement: rationale and description. Death Stud. 1999;23(3):197-224.
14. Reynolds CF, 3rd, Miller MD, Pasternak RE, et al. Treatment of bereavement-related major depressive episodes in later life: a controlled study of acute and continuation treatment with nortriptyline and interpersonal psychotherapy. Am J Psychiatry. 1999;156(2):202-208.
15. Simon NM, Thompson EH, Pollack MH, et al. Complicated grief: a case series using escitalopram. Am J Psychiatry. 2007;164(11):1760-1761.
16. Hensley PL, Slonimski CK, Uhlenhuth EH, et al. Escitalopram: an open-label study of bereavement-related depression and grief. J Affect Disord. 2009;113(1-2):142-149.
17. Zygmont M, Prigerson HG, Houck PR, et al. A post hoc comparison of paroxetine and nortriptyline for symptoms of traumatic grief. J Clin Psychiatry. 1998;59(5):241-245.
18. Simon NM, Shear MK, Fagiolini A, et al. Impact of concurrent naturalistic pharmacotherapy on psychotherapy of complicated grief. Psychiatry Res. 2008;159(1-2):31-36.
19. U.S. National Institutes of Health. A study of medication with or without psychotherapy for complicated grief (HEAL). http://clinicaltrials.gov/ct2/show/NCT01179568. Published June 24, 2012. Accessed June 25, 2012.
20. Warner J, Metcalfe C, King M. Evaluating the use of benzodiazepines following recent bereavement. Br J Psychiatry. 2001;178(1):36-41.
21. Cook JM, Biyanova T, Marshall R. Medicating grief with benzodiazepines: physician and patient perspectives. Arch Intern Med. 2007;167(18):2006-2007.
1. Kochanek KD, Xu J, Murphy SL, et al. U.S. Department of Health and Human Services. Deaths: preliminary data for 2009. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_04.pdf. Published March 16 2011. Accessed June 19, 2012.
2. American Psychiatric Association. Trauma- and stressor-related disorders. http://www.dsm5.org/ProposedRevision/Pages/TraumaandStressorRelatedDisorders.aspx. Accessed June 19 2012.
3. Bonanno GA, Kaltman S. Toward an integrative perspective on bereavement. Psychol Bull. 1999;125(6):760-776.
4. Bonanno GA, Keltner D. Facial expressions of emotion and the course of conjugal bereavement. J Abnorm Psychol. 1997;106(1):126-137.
5. Bonanno GA, Wortman CB, Lehman DR, et al. Resilience to loss and chronic grief: a prospective study from preloss to 18-months postloss. J Pers Soc Psychol. 2002;83(5):1150-1164.
6. Shear MK, Simon N, Wall M, et al. Complicated grief and related bereavement issues for DSM-5. Depress Anxiety. 2011;28(2):103-117.
7. Boelen PA, Prigerson HG. The influence of symptoms of prolonged grief disorder depression, and anxiety on quality of life among bereaved adults: a prospective study. Eur Arch Psychiatry Clin Neurosci. 2007;257(8):444-452.
8. Prigerson HG, Maciejewski PK, Reynolds CF, 3rd, et al. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res. 1995;59 (1-2):65-79.
9. Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry. 2007;48(5):395-399.
10. Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339-376.
11. Neimeyer RA, Currier JM. Grief therapy: evidence of efficacy and emerging directions. Curr Dir Psychol Sci. 2009;18(6):352-356.
12. Shear K, Frank E, Houck PR, et al. Treatment of complicated grief: a randomized controlled trial. JAMA. 2005;293(21):2601-2608.
13. Stroebe M, Schut H. The dual process model of coping with bereavement: rationale and description. Death Stud. 1999;23(3):197-224.
14. Reynolds CF, 3rd, Miller MD, Pasternak RE, et al. Treatment of bereavement-related major depressive episodes in later life: a controlled study of acute and continuation treatment with nortriptyline and interpersonal psychotherapy. Am J Psychiatry. 1999;156(2):202-208.
15. Simon NM, Thompson EH, Pollack MH, et al. Complicated grief: a case series using escitalopram. Am J Psychiatry. 2007;164(11):1760-1761.
16. Hensley PL, Slonimski CK, Uhlenhuth EH, et al. Escitalopram: an open-label study of bereavement-related depression and grief. J Affect Disord. 2009;113(1-2):142-149.
17. Zygmont M, Prigerson HG, Houck PR, et al. A post hoc comparison of paroxetine and nortriptyline for symptoms of traumatic grief. J Clin Psychiatry. 1998;59(5):241-245.
18. Simon NM, Shear MK, Fagiolini A, et al. Impact of concurrent naturalistic pharmacotherapy on psychotherapy of complicated grief. Psychiatry Res. 2008;159(1-2):31-36.
19. U.S. National Institutes of Health. A study of medication with or without psychotherapy for complicated grief (HEAL). http://clinicaltrials.gov/ct2/show/NCT01179568. Published June 24, 2012. Accessed June 25, 2012.
20. Warner J, Metcalfe C, King M. Evaluating the use of benzodiazepines following recent bereavement. Br J Psychiatry. 2001;178(1):36-41.
21. Cook JM, Biyanova T, Marshall R. Medicating grief with benzodiazepines: physician and patient perspectives. Arch Intern Med. 2007;167(18):2006-2007.
Prescription opioid use disorder: A complex clinical challenge
Discuss this article at www.facebook.com/CurrentPsychiatry
You’ve been treating Mr. H, a 54-year-old factory worker and tobacco user, for depression that developed after a work-related back injury and subsequent disability. His depression has had a fair response to an antidepressant. He also has been maintained on chronic opioids (morphine and oxycodone/acetaminophen) for 18 months by his primary care physician (PCP). At the end of your appointment, he asks you for a refill of the opioids because he “ran out” early because of increased night pain and resultant insomnia and “stress.” He clarifies he has asked for early refills before from his PCP, but lately he has been denied. Because you “seem to listen to me more,” he asks for your help. How should you manage Mr. H?
Opioids are among the most commonly misused prescription drugs in the United States.1 In 2008, poisoning was the leading cause of death from injury in the United States; roughly 90% of poisonings resulted from drug exposure, and >40% of these drug poisonings were from prescription opioids.2 The Centers for Disease Control and Prevention estimates that the number of emergency department (ED) visits for nonmedical use of opioids increased 111% between 2004 and 2008, from 144,600 to 305,900 visits.3 The highest number of visits were for use of oxycodone, hydrocodone, and methadone.3
Increased prescribing of opioids and overdose deaths attributable to prescribed opioids have raised concern among physicians about how to effectively treat pain as well as prevent, recognize, and manage aberrant medication-taking behaviors (AMTBs). Psychiatrists are well-positioned to screen and manage their own patients for prescription opioid use disorder (POUD) or collaborate with opioid prescribers to accomplish the same.
Clarifying terminology
Terminology used to describe POUD and related conditions often is poorly defined or loosely applied. Because emotions often enter discussions between patients and physicians about problems related to opioid therapy, nonstigmatizing and more objective terminology is needed, and clinicians are working toward standardizing this. Relevant terms are defined in Table 1.4
The DSM-5 Substance Use Disorders Work Group has proposed using the term opioid use disorder (OUD) to replace the term opioid dependence.5 The hope is that removing the word “dependence” from the diagnostic term will reduce confusion between “dependence” due to expected physical dependence (tolerance, withdrawal) on medically prescribed opioids vs true addiction (currently defined as “opioid dependence” in DSM-IV-TR). This Work Group also has proposed combining opioid abuse and opioid dependence criteria into a single diagnosis of OUD, and adding “craving” to the criteria. For the complete proposed criteria, see www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460.These changes are still under review. In this article, we use the term POUD.
Table 1
Terminology related to prescription opioid use disorder
| Term | Definition |
|---|---|
| Chronic paina | Pain that extends beyond the expected period for healing (6 months), initiated by tissue damage, but perpetrated by the interaction of physiologic, affective, and environmental factors |
| Chronic nonmalignant paina | Chronic pain associated with diverse diagnoses and syndromes that are not terminal but affect the patient’s function |
| Appropriate usea | Taking a prescription as prescribed, and only for the condition indicated |
| Misusea | Taking a prescription for a reason or at a dose or frequency other than for which it was prescribed; this may or may not reflect POUD |
| Drug-seeking behaviors | Patient behaviors directed toward obtaining controlled substances, driven not by amelioration of the condition for which the medication was indicated but rather by other maladaptive gains; this may or may not reflect POUD |
| Chemical coping | Taking a controlled substance medication to relieve psychological problems (eg, to relieve low mood, anxiety, insomnia) and for reasons other than the purpose for which it was prescribed; this may or may not reflect POUD |
| Aberrant medication-taking behaviorsa | Taking a controlled substance medication in a manner that is not prescribed; causes for this may include:
|
| Pseudoaddiction | An iatrogenic syndrome of “addiction-like” behaviors in which the patient seeks opioids to relieve pain—such as seeking different doctors, self-adjusting the opioid dose, early refills of opioids, etc.—rather than to achieve pleasure or other nonpain-related effect. At times mistaken for true addiction, these behaviors tend to resolve and function improves once analgesia is better addressed |
| a These terms and definitions are adapted from reference 4. The remaining terms and definitions were developed by the authors POUD: prescription opioid use disorder | |
POUD and chronic pain
The incidence of POUD during opioid therapy for pain is unknown.6 Some researchers have suggested it may be as low as 0.2%,7 while others estimate that rates of POUD in patients with chronic pain may be similar to those in the general population: 3% to 16%.8 When applying the proposed DSM-5 criteria to patients receiving long-term opioid therapy for noncancer pain, the lifetime prevalence of POUD may be as high as 35%.9
Prescribers may be contributing to POUD. Roughly 76% of opioids used for nonmedical purposes were prescribed to someone else, 20% were prescribed to the user, and 4% came from other sources.1 Strategies to reduce POUD risk may be underused. In a retrospective cohort study of 1,612 patient electronic medical records from 8 primary care clinics that managed patients with long-term opioids for chronic noncancer pain (average prescribing duration of 2 years duration, ≥3 monthly prescriptions in 6 months), researchers evaluated how often prescribers used 3 risk reduction practices:
- urine drug tests
- regular office visits (≥1 every 6 months and within 30 days of changing opioid treatment)
- restricted early refills (≤1 opioid refill more than a week early).10
Risk factors for opioid misuse included age 1 early refill. Researchers found that even for high-risk patients, these strategies were used infrequently. Less than one-quarter of patients with ≥3 risk factors ever had a drug test, and those at increased risk were more likely to receive >1 early refill but no more likely to have more frequent visits. Issues such as patient entitlement, lack of physician education, and time constraints may explain why these strategies are not used more often.11
No one procedure or set of variables is sufficient to identify chronic pain patients who may be at risk for POUD. However, a history of drug or alcohol use disorders may be a significant risk factor.12,13
Few tools have been developed to help identify those at risk of AMTBs or POUD, and all have limitations.4,14 Recommended self-report measures include the Current Opioid Misuse Measure and the Opioid Risk Tool.15 A review of studies in which these kinds of tools were developed revealed limited evidence for their use; most studies had methodological shortcomings, did not use standardized AMTB criteria, and provided little assessment of whether these tools changed clinician behaviors or improved patient outcomes.16
Evaluating AMTBs
Although diagnosing POUD in pain patients receiving chronic opioids can be challenging, assessing for AMTBs typically is helpful. Once AMTBs are identified, they can be examined to determine what drives their expression (Table 14 and Table 217). However, often it is easier to identify AMTBs than to interpret their origins; as much as 30% to 50% of patients who complain of chronic pain may have primary substance dependence to sedatives, opioids, or both.11
Table 2
Aberrant medication-taking behaviors and POUD risk
| Behaviors more suggestive of POUD |
|---|
| Deterioration in function (work, social) |
| Illegal activities (selling medication, forging prescriptions, buying from non-medical sources) |
| Altering the route of administration (snorting, injecting) |
| Multiple episodes of ‘lost’ or ‘stolen’ prescriptions |
| Resistance to change therapy despite negative outcomes |
| Refusal to comply with toxicology testing |
| Concurrent, active abuse of alcohol, illegal drugs |
| Use of multiple physicians or pharmacies to obtain the prescription |
| Behaviors less suggestive of POUD |
| Complaints for more medication |
| Medication hoarding |
| Requesting specific pain medications |
| Openly acquiring similar medications from other providers |
| Occasional unsanctioned dose escalation |
| Nonadherence to other recommendations for pain therapy |
| POUD: prescription opioid use disorder Source: Reference 17 |
Although AMTBs are common among chronic nonmalignant pain patients,18,19 how often AMTBs reflect underlying POUD is uncertain.7 It is critical to interpret AMTBs with a balance of caution and care: “react therapeutically, not punitively.”20 Categorizing a patient’s AMTB as more or less likely to support a POUD diagnosis can be helpful, but is not conclusive (Table 2).17 Clinical correlation often is required. No single AMTB alone is indicative of POUD. When evaluating AMTBs, the treating provider should use a nonjudgmental stance, and consider obtaining collateral data from people who can provide differing perspectives of the patient’s behaviors, such as other clinicians, significant others, family, etc. (a release of information from the patient may be required). Another source of collateral data is prescription monitoring databases. These databases typically are state-based and provide electronic access to prescription information, allowing you to search for patterns—ie, use of multiple prescribers or pharmacies, undisclosed prescriptions, etc. Interest in establishing a single, federal database has been increasing, but striking a balance between carefully monitoring for AMTBs and protecting privacy remains unresolved.
DSM-IV-TR diagnostic criteria for opioid dependence21 can be challenging to interpret in patients who are prescribed opioids for pain (Table 3
).6 To clarify interpretation, the Liaison Committee on Pain and Addiction of the American Society of Addiction Medicine (ASAM) has provided an outline of possible indicators of addiction in pain patients (Table 4).6 This was a consensus statement from the American Pain Society, the American Academy of Pain Medicine, and ASAM.
Assessment is primarily clinical and requires an awareness of appropriate terminology, an index of clinical suspicion, and expertise teasing apart pain, addiction, and pseudoaddiction. In our experience, it is helpful to ask a chronic pain patient whom you suspect might have POUD, “Have you ever used your prescribed opioids for reasons other than improving function or reducing pain, such as for getting a ‘high,’ managing stress, escaping from problems, etc.?” An affirmative response suggests an underlying problem with use of prescribed opioids, indicating a need for more careful questioning to determine if AMTBs or POUD coexist with chronic pain.
Drug testing can help determine if a patient is taking opioids that are not prescribed—as well as illicit drugs or alcohol—and confirm the presence of those that are prescribed. Toxicology screening should include opioids typically screened for (eg, morphine, codeine, heroin) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and hydrocodone and synthetics such as fentanyl).
Table 3
Identifying addiction in pain patients: Limitations of DSM-IV-TR
| DSM-IV-TR substance dependence criteria | Challenges in using criterion to diagnose prescription opioid use disorder |
|---|---|
| Tolerance | Expected with prolonged opioid compliance |
| Physical dependence, withdrawal | Expected with prolonged opioid compliance |
| Use of larger amounts or longer than initially intended | Emergence of pain may demand increased dose or prolonged use |
| Multiple failed attempts to cut down or control | Emergence of pain may deter dose reduction or cessation |
| Time spent finding, using, or recovering | Difficulty finding adequate pain treatment may increase time spent pursuing analgesics. However, time spent recovering from overuse may suggest addiction |
| Given up or reduced important activities | Valid criteria—engaging in activities is expected to increase, not decline, with effective pain treatment |
| Continued use despite knowledge of negative consequences | Valid criteria—no harm is anticipated from analgesic opioid use for pain (see Table 4) |
| Source: Adapted from reference 6 | |
Table 4
Possible indicators of addiction in pain patients
| ASAM-APS-AAPM behavioral criteria | Examples of specific behaviors in opioid therapy for pain |
|---|---|
| Impaired control over opioid use | Patient requests early refills, frequently reports loss or theft of medication. Withdrawal noted at follow-up appointments despite having an adequate quantity of medication prescribed |
| Continued use despite harm from opioids | Patient exhibits declining function, opioid intoxication, persistent oversedation from opioids |
| Preoccupation with opioids | Patient ignores non-opioid interventions for pain, makes recurrent requests for opioid dose escalation (or complains of increasing pain) despite absence of disease progression or despite opioid dose increase by provider |
| AAPM: American Academy of Pain Medicine; APS: American Pain Society; ASAM: American Society of Addiction Medicine Source: Adapted from reference 6 | |
Helping POUD patients
Goals of treatment include establishing a therapeutic alliance, educating patients about POUD, reducing relapse risk, and optimizing overall health (including pain and physical function). The ASAM Patient Placement Criteria22 provide guidance regarding level-of-care decisions. Treatment ideally includes a combination of education about POUD and its relationship to chronic pain, pharmacotherapy, psychotherapy—such as motivational enhancement therapy, 12-step facilitation therapy, cognitive-behavioral therapy, and relapse prevention—and referral to self-help groups such as Narcotics Anonymous or Pills Anonymous. Importantly, if pain is genuine, it requires treatment.
Pharmacotherapy. Methadone is recommended as the standard of care for OUD by the National Institutes of Health. Methadone is a full opioid agonist that decreases illicit opioid use, mortality, and related problems and requires highly structured treatment approaches under federal and state regulation. POUD patients may have higher rates of methadone maintenance treatment retention than heroin-dependent patients.23 Published trials of buprenorphine for OUD have shown good treatment retention and reduction in illicit drug use and adverse events.24 Buprenorphine also decreases mortality among OUD patients.
The first large-scale, randomized clinical trial of buprenorphine specifically for POUD included 653 treatment-seeking outpatients.25 This study was designed to approximate clinical practice and included buprenorphine/naloxone, recommended abstinence, and self-help; one-half of participants received intensive addiction counseling. POUD patients were most likely to reduce prescription opioid misuse during buprenorphine/naloxone treatment. If tapered off buprenorphine/naloxone, even after 12 weeks of treatment, the likelihood of an unsuccessful outcome was high. Moreover, opioid dependence counseling did not seem to afford any difference in outcomes. However, despite clinical effectiveness, over the last decade only 19% of patients admitted primarily for OUD treatment (other than heroin) were planned to be offered buprenorphine or methadone.26
A Cochrane review of oral naltrexone for OUD found that the drug was no better than placebo but concluded that available evidence does not allow an adequate evaluation.27 Opioid antagonists may be of value to patients who do not want to take agonists or partial agonists. Extended-release naltrexone also is available to treat OUD.
See the Box below that details steps the FDA and others have taken to prevent POUD and Table 5 for precautions to incorporate when prescribing opioids long-term.
The FDA has moved toward a risk evaluation and mitigation strategy (REMS) for opioids prescribed for pain that requires clinicians to receive training and certification in prescribing opioids for pain as well as identifying and reducing the risk for prescription opioid use disorder (POUD).a In 2011, the Obama administration developed an action plan to better address prescription drug abuse that required several federal agencies to develop programs and policies to address this growing problem; this plan was updated for 2012 (the complete National Drug Control Strategy 2012 is available at www.whitehouse.gov/sites/default/files/ondcp/2012_ndcs.pdf). The American Society of Addiction Medicine has issued a public policy statement that supports the federal approach and outlines other means to reduce POUD.b
Some pain specialists recommend requiring patients to sign an Opioid Pain Management Agreement that includes an “exit strategy” before the first opioid prescription is written. These agreements incorporate elements of “universal precautions” to take when prescribing opioids long term.c,d Although not well-studied, prescribing agreements may help educate patients and providers on how to interact in the management of pain with opioids in a way that is objective and empathic, and may reduce POUD risk.
References
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Opioid drugs and risk evaluation and mitigation strategies (REMS). http://www.fda.gov/drugs/drugsafety/informationbydrugclass/ucm163647.htm. Updated April 5, 2012. Accessed June 28, 2012.
- American Society of Addiction Medicine. Measures to counteract prescription drug diversion, misuse and addiction. http://www.asam.org/advocacy/find-a-policy-statement/view-policy-statement/public-policy-statements/2012/01/26/measures-to-counteract-prescription-drug-diversion-misuse-and-addiction. Published January 25, 2012. Accessed June 20, 2012.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112.
- Gourlay DL, Heit HA. Universal precautions revisited: managing the inherited pain patient. Pain Med. 2009; 10(suppl 2):S115-S123.
Table 5
Universal precautions with chronic opioid management
| Goals of therapy: partial pain relief and improvement in physical, emotional, and/or social functioning |
| Requirement for a single prescribing provider or treatment team |
| Limitation on dose and number of prescribed medications |
| Prohibition of changing dosage without discussion with the provider first |
| Monitoring patient adherence; discuss the use of ‘pill counts’ |
| Prohibition of use with alcohol, other sedating medications, or illegal drugs without discussion with the provider |
| Agreement not to drive or operate heavy machinery until abatement of medication-related drowsiness |
| Responsibility to keep medication safe and secure |
| Prohibition of selling, lending, sharing, or giving medication to others |
| Limitations on refills—only by appointment, in person, and no extra refills for running out early |
| Compliance with all components of overall treatment plan (including consultations and referrals) |
| Biological testing to screen for drugs of abuse or alcohol as well as to confirm the presence of prescribed opioids |
| Adverse effects and safety issues, such as the risk of physical dependence and addiction behaviors |
| The option of sharing information with family members and other providers, as necessary, with the patient’s consent |
| Need for periodic reevaluation of treatment |
| Reasons for stopping opioid therapy |
| Consequences of nonadherence with the treatment agreement |
| Source: Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112. |
CASE CONTINUED: A closer evaluation
After expressing your appreciation for Mr. H’s kind words and empathy for his chronic pain, you redirect him to his PCP. You ask him to sign a release of information so you and his other clinicians can coordinate his care. When discussing Mr. H with his PCP, you learn the patient has made limited requests for early refills and dose escalation primarily in relation to inadequate pain control and function, has genuine pain pathology, and is greatly distressed over his inability to work. No other AMTBs are present, and a check of the state prescribing database reveals that Mr. H did receive a small quantity of opioids from an ED on 1 occasion.
You and Mr. H’s PCP agree this is “pseudo-addiction” but want to watch Mr. H more closely and look for ways to coordinate his care. The PCP agrees to implement a prescribing agreement, start drug testing (including for the prescribed opioids), and reassess maximizing Mr. H’s function and pain management while you address his combined pain, depression, insomnia, and tobacco use.
Related Resources
- Ries RK, Fiellin D, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009.
- Department of Veterans Affairs. Department of Defense. VA/DoD clinical practice guideline for management of opioid therapy for chronic pain. Appendix C: sample opioid pain care agreement. http://www.healthquality.va.gov/COT_312_Full-er.pdf. Published May 2010. Accessed June 21, 2012.
- Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic non-malignant pain. J Addiction Med. 2007;1(1):2-10.
- Weaver M, Heit HA, Savage S, et al. Clinical case discussion: chronic pain management. J Addiction Med. 2007;1(1):11-14.
Drug Brand Names
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Codeine • Tylenol with codeine, others
- Fentanyl • Duragesic, Actiq
- Hydrocodone • Lortab, Vicodin, others
- Methadone • Dolophine, Methadose
- Morphine • Roxanol
- Naltrexone extended-release • Vivitrol
- Oxycodone • OxyContin, Roxicodone
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Frankowski’s time toward this project was provided by the American Board of Addiction Medicine-accredited Cincinnati VA Addiction Medicine Research Fellowship, affiliated with the CeTREAD, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs.
Acknowledgement
The authors thank Catherine Constance and Sandra Mason at the Cincinnati VA Medical Center for their administrative assistance.
1. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Office of Applied Studies. Results from the 2009 national survey on drug use and health: volume I. http://www.samhsa.gov/data/NSDUH/2k9NSDUH/2k9Results.htm. Accessed June 20, 2012.
2. Warner M, Chen LH, Makuc DM, et al. Drug poisoning deaths in the United States, 1980-2008. http://www.cdc.gov/nchs/data/databriefs/db81.htm. Published December 2011. Accessed June 20, 2012.
3. Centers for Disease Control and Prevention (CDC). Emergency department visits involving nonmedical use of selected prescription drugs - United States 2004-2008. MMWR Morb Mortal Wkly Rep. 2010;59(23):705-709.
4. Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic nonmalignant pain. J Addict Med. 2007;1(1):2-10.
5. American Psychiatric Association. R 19 opioid use disorder. http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460. Updated April 30 2012. Accessed June 20, 2012.
6. Savage SR, Horvath R. Opioid therapy of pain. In: Ries RK Fiellin DA, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009:1329-1351.
7. Fishbain DA, Cole B, Lewis J, et al. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008;9(4):444-459.
8. Gourlay DL, Heit HA. Pain and addiction: managing risk through comprehensive care. J Addict Dis. 2008;27(3):23-30.
9. Boscarino JA, Rukstalis MR, Hoffman SN, et al. Prevalence of prescription opioid-use disorder among chronic pain patients: comparison of the DSM-5 vs. DSM-4 diagnostic criteria. J Addict Dis. 2011;30(3):185-194.
10. Starrels JL, Becker WC, Weiner MG, et al. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med. 2011;26(9):958-964.
11. Miller NS. Failure of enforcement controlled substance laws in health policy for prescribing opiate medications: a painful assessment of morbidity and mortality. Am J Ther. 2006;13(6):527-533.
12. Turk DC, Swanson KS, Gatchel RJ. Predicting opioid misuse by chronic pain patients: a systematic review and literature synthesis. Clin J Pain. 2008;24(6):497-508.
13. Miller NS, Greenfeld A. Patient characteristics and risks factors for development of dependence on hydrocodone and oxycodone. Am J Ther. 2004;11(1):26-32.
14. Butler SF, Budman SH, Fernandez KC, et al. Cross-validation of a Screener to Predict Opioid Misuse in Chronic Pain Patients (SOAPP-R). J Addict Med. 2009;3(2):66-73.
15. Passik SD, Kirsh KL, Casper D. Addiction-related assessment tools and pain management: instruments for screening treatment planning, and monitoring compliance. Pain Med. 2008;9(suppl 2):S145-S166.
16. Chou R, Fanciullo GJ, Fine PG, et al. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain. 2009;10(2):131-146.
17. Alford DP, Liebschutz J, Jackson A, et al. Prescription drug abuse: an introduction. http://www.drugabuse.gov/sites/default/files/prescription-drug-abuse-alt.pdf. Published November 8, 2009. Accessed June 20, 2012.
18. Passik SD, Kirsh KL, Whitcomb L, et al. Monitoring outcomes during long-term opioid therapy for noncancer pain: results with the Pain Assessment and Documentation Tool. J Opioid Manag. 2005;1(5):257-266.
19. Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432-442.
20. Passik SD. Pain management misstatements: ceiling effects red and yellow flags. Pain Med. 2006;7(1):76-77.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington DC: American Psychiatric Association; 2000.
22. Mee-Lee D, Shulman GD, Fishman MJ, et al. eds. ASAM patient placement criteria for the treatment of substance-related disorders. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc.; 2001.
23. Banta-Green CJ, Maynard C, Koepsell TD, et al. Retention in methadone maintenance drug treatment for prescription-type opioid primary users compared to heroin users. Addiction. 2009;104(5):775-783.
24. Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med. 2007;22(4):527-530.
25. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68(12):1238-1246.
26. U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration (SAMHSA). Office of Applied Studies. Treatment Episode Data Set (TEDS). 1998 - 2008. National Admissions to Substance Abuse Treatment Services, DASIS Series: S-50, HHS Publication No. (SMA) 09-4471. Rockville, MD; 2010.
27. Minozzi S, Amato L, Vecchi S, et al. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011;(4):CD001333.-
Discuss this article at www.facebook.com/CurrentPsychiatry
You’ve been treating Mr. H, a 54-year-old factory worker and tobacco user, for depression that developed after a work-related back injury and subsequent disability. His depression has had a fair response to an antidepressant. He also has been maintained on chronic opioids (morphine and oxycodone/acetaminophen) for 18 months by his primary care physician (PCP). At the end of your appointment, he asks you for a refill of the opioids because he “ran out” early because of increased night pain and resultant insomnia and “stress.” He clarifies he has asked for early refills before from his PCP, but lately he has been denied. Because you “seem to listen to me more,” he asks for your help. How should you manage Mr. H?
Opioids are among the most commonly misused prescription drugs in the United States.1 In 2008, poisoning was the leading cause of death from injury in the United States; roughly 90% of poisonings resulted from drug exposure, and >40% of these drug poisonings were from prescription opioids.2 The Centers for Disease Control and Prevention estimates that the number of emergency department (ED) visits for nonmedical use of opioids increased 111% between 2004 and 2008, from 144,600 to 305,900 visits.3 The highest number of visits were for use of oxycodone, hydrocodone, and methadone.3
Increased prescribing of opioids and overdose deaths attributable to prescribed opioids have raised concern among physicians about how to effectively treat pain as well as prevent, recognize, and manage aberrant medication-taking behaviors (AMTBs). Psychiatrists are well-positioned to screen and manage their own patients for prescription opioid use disorder (POUD) or collaborate with opioid prescribers to accomplish the same.
Clarifying terminology
Terminology used to describe POUD and related conditions often is poorly defined or loosely applied. Because emotions often enter discussions between patients and physicians about problems related to opioid therapy, nonstigmatizing and more objective terminology is needed, and clinicians are working toward standardizing this. Relevant terms are defined in Table 1.4
The DSM-5 Substance Use Disorders Work Group has proposed using the term opioid use disorder (OUD) to replace the term opioid dependence.5 The hope is that removing the word “dependence” from the diagnostic term will reduce confusion between “dependence” due to expected physical dependence (tolerance, withdrawal) on medically prescribed opioids vs true addiction (currently defined as “opioid dependence” in DSM-IV-TR). This Work Group also has proposed combining opioid abuse and opioid dependence criteria into a single diagnosis of OUD, and adding “craving” to the criteria. For the complete proposed criteria, see www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460.These changes are still under review. In this article, we use the term POUD.
Table 1
Terminology related to prescription opioid use disorder
| Term | Definition |
|---|---|
| Chronic paina | Pain that extends beyond the expected period for healing (6 months), initiated by tissue damage, but perpetrated by the interaction of physiologic, affective, and environmental factors |
| Chronic nonmalignant paina | Chronic pain associated with diverse diagnoses and syndromes that are not terminal but affect the patient’s function |
| Appropriate usea | Taking a prescription as prescribed, and only for the condition indicated |
| Misusea | Taking a prescription for a reason or at a dose or frequency other than for which it was prescribed; this may or may not reflect POUD |
| Drug-seeking behaviors | Patient behaviors directed toward obtaining controlled substances, driven not by amelioration of the condition for which the medication was indicated but rather by other maladaptive gains; this may or may not reflect POUD |
| Chemical coping | Taking a controlled substance medication to relieve psychological problems (eg, to relieve low mood, anxiety, insomnia) and for reasons other than the purpose for which it was prescribed; this may or may not reflect POUD |
| Aberrant medication-taking behaviorsa | Taking a controlled substance medication in a manner that is not prescribed; causes for this may include:
|
| Pseudoaddiction | An iatrogenic syndrome of “addiction-like” behaviors in which the patient seeks opioids to relieve pain—such as seeking different doctors, self-adjusting the opioid dose, early refills of opioids, etc.—rather than to achieve pleasure or other nonpain-related effect. At times mistaken for true addiction, these behaviors tend to resolve and function improves once analgesia is better addressed |
| a These terms and definitions are adapted from reference 4. The remaining terms and definitions were developed by the authors POUD: prescription opioid use disorder | |
POUD and chronic pain
The incidence of POUD during opioid therapy for pain is unknown.6 Some researchers have suggested it may be as low as 0.2%,7 while others estimate that rates of POUD in patients with chronic pain may be similar to those in the general population: 3% to 16%.8 When applying the proposed DSM-5 criteria to patients receiving long-term opioid therapy for noncancer pain, the lifetime prevalence of POUD may be as high as 35%.9
Prescribers may be contributing to POUD. Roughly 76% of opioids used for nonmedical purposes were prescribed to someone else, 20% were prescribed to the user, and 4% came from other sources.1 Strategies to reduce POUD risk may be underused. In a retrospective cohort study of 1,612 patient electronic medical records from 8 primary care clinics that managed patients with long-term opioids for chronic noncancer pain (average prescribing duration of 2 years duration, ≥3 monthly prescriptions in 6 months), researchers evaluated how often prescribers used 3 risk reduction practices:
- urine drug tests
- regular office visits (≥1 every 6 months and within 30 days of changing opioid treatment)
- restricted early refills (≤1 opioid refill more than a week early).10
Risk factors for opioid misuse included age 1 early refill. Researchers found that even for high-risk patients, these strategies were used infrequently. Less than one-quarter of patients with ≥3 risk factors ever had a drug test, and those at increased risk were more likely to receive >1 early refill but no more likely to have more frequent visits. Issues such as patient entitlement, lack of physician education, and time constraints may explain why these strategies are not used more often.11
No one procedure or set of variables is sufficient to identify chronic pain patients who may be at risk for POUD. However, a history of drug or alcohol use disorders may be a significant risk factor.12,13
Few tools have been developed to help identify those at risk of AMTBs or POUD, and all have limitations.4,14 Recommended self-report measures include the Current Opioid Misuse Measure and the Opioid Risk Tool.15 A review of studies in which these kinds of tools were developed revealed limited evidence for their use; most studies had methodological shortcomings, did not use standardized AMTB criteria, and provided little assessment of whether these tools changed clinician behaviors or improved patient outcomes.16
Evaluating AMTBs
Although diagnosing POUD in pain patients receiving chronic opioids can be challenging, assessing for AMTBs typically is helpful. Once AMTBs are identified, they can be examined to determine what drives their expression (Table 14 and Table 217). However, often it is easier to identify AMTBs than to interpret their origins; as much as 30% to 50% of patients who complain of chronic pain may have primary substance dependence to sedatives, opioids, or both.11
Table 2
Aberrant medication-taking behaviors and POUD risk
| Behaviors more suggestive of POUD |
|---|
| Deterioration in function (work, social) |
| Illegal activities (selling medication, forging prescriptions, buying from non-medical sources) |
| Altering the route of administration (snorting, injecting) |
| Multiple episodes of ‘lost’ or ‘stolen’ prescriptions |
| Resistance to change therapy despite negative outcomes |
| Refusal to comply with toxicology testing |
| Concurrent, active abuse of alcohol, illegal drugs |
| Use of multiple physicians or pharmacies to obtain the prescription |
| Behaviors less suggestive of POUD |
| Complaints for more medication |
| Medication hoarding |
| Requesting specific pain medications |
| Openly acquiring similar medications from other providers |
| Occasional unsanctioned dose escalation |
| Nonadherence to other recommendations for pain therapy |
| POUD: prescription opioid use disorder Source: Reference 17 |
Although AMTBs are common among chronic nonmalignant pain patients,18,19 how often AMTBs reflect underlying POUD is uncertain.7 It is critical to interpret AMTBs with a balance of caution and care: “react therapeutically, not punitively.”20 Categorizing a patient’s AMTB as more or less likely to support a POUD diagnosis can be helpful, but is not conclusive (Table 2).17 Clinical correlation often is required. No single AMTB alone is indicative of POUD. When evaluating AMTBs, the treating provider should use a nonjudgmental stance, and consider obtaining collateral data from people who can provide differing perspectives of the patient’s behaviors, such as other clinicians, significant others, family, etc. (a release of information from the patient may be required). Another source of collateral data is prescription monitoring databases. These databases typically are state-based and provide electronic access to prescription information, allowing you to search for patterns—ie, use of multiple prescribers or pharmacies, undisclosed prescriptions, etc. Interest in establishing a single, federal database has been increasing, but striking a balance between carefully monitoring for AMTBs and protecting privacy remains unresolved.
DSM-IV-TR diagnostic criteria for opioid dependence21 can be challenging to interpret in patients who are prescribed opioids for pain (Table 3
).6 To clarify interpretation, the Liaison Committee on Pain and Addiction of the American Society of Addiction Medicine (ASAM) has provided an outline of possible indicators of addiction in pain patients (Table 4).6 This was a consensus statement from the American Pain Society, the American Academy of Pain Medicine, and ASAM.
Assessment is primarily clinical and requires an awareness of appropriate terminology, an index of clinical suspicion, and expertise teasing apart pain, addiction, and pseudoaddiction. In our experience, it is helpful to ask a chronic pain patient whom you suspect might have POUD, “Have you ever used your prescribed opioids for reasons other than improving function or reducing pain, such as for getting a ‘high,’ managing stress, escaping from problems, etc.?” An affirmative response suggests an underlying problem with use of prescribed opioids, indicating a need for more careful questioning to determine if AMTBs or POUD coexist with chronic pain.
Drug testing can help determine if a patient is taking opioids that are not prescribed—as well as illicit drugs or alcohol—and confirm the presence of those that are prescribed. Toxicology screening should include opioids typically screened for (eg, morphine, codeine, heroin) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and hydrocodone and synthetics such as fentanyl).
Table 3
Identifying addiction in pain patients: Limitations of DSM-IV-TR
| DSM-IV-TR substance dependence criteria | Challenges in using criterion to diagnose prescription opioid use disorder |
|---|---|
| Tolerance | Expected with prolonged opioid compliance |
| Physical dependence, withdrawal | Expected with prolonged opioid compliance |
| Use of larger amounts or longer than initially intended | Emergence of pain may demand increased dose or prolonged use |
| Multiple failed attempts to cut down or control | Emergence of pain may deter dose reduction or cessation |
| Time spent finding, using, or recovering | Difficulty finding adequate pain treatment may increase time spent pursuing analgesics. However, time spent recovering from overuse may suggest addiction |
| Given up or reduced important activities | Valid criteria—engaging in activities is expected to increase, not decline, with effective pain treatment |
| Continued use despite knowledge of negative consequences | Valid criteria—no harm is anticipated from analgesic opioid use for pain (see Table 4) |
| Source: Adapted from reference 6 | |
Table 4
Possible indicators of addiction in pain patients
| ASAM-APS-AAPM behavioral criteria | Examples of specific behaviors in opioid therapy for pain |
|---|---|
| Impaired control over opioid use | Patient requests early refills, frequently reports loss or theft of medication. Withdrawal noted at follow-up appointments despite having an adequate quantity of medication prescribed |
| Continued use despite harm from opioids | Patient exhibits declining function, opioid intoxication, persistent oversedation from opioids |
| Preoccupation with opioids | Patient ignores non-opioid interventions for pain, makes recurrent requests for opioid dose escalation (or complains of increasing pain) despite absence of disease progression or despite opioid dose increase by provider |
| AAPM: American Academy of Pain Medicine; APS: American Pain Society; ASAM: American Society of Addiction Medicine Source: Adapted from reference 6 | |
Helping POUD patients
Goals of treatment include establishing a therapeutic alliance, educating patients about POUD, reducing relapse risk, and optimizing overall health (including pain and physical function). The ASAM Patient Placement Criteria22 provide guidance regarding level-of-care decisions. Treatment ideally includes a combination of education about POUD and its relationship to chronic pain, pharmacotherapy, psychotherapy—such as motivational enhancement therapy, 12-step facilitation therapy, cognitive-behavioral therapy, and relapse prevention—and referral to self-help groups such as Narcotics Anonymous or Pills Anonymous. Importantly, if pain is genuine, it requires treatment.
Pharmacotherapy. Methadone is recommended as the standard of care for OUD by the National Institutes of Health. Methadone is a full opioid agonist that decreases illicit opioid use, mortality, and related problems and requires highly structured treatment approaches under federal and state regulation. POUD patients may have higher rates of methadone maintenance treatment retention than heroin-dependent patients.23 Published trials of buprenorphine for OUD have shown good treatment retention and reduction in illicit drug use and adverse events.24 Buprenorphine also decreases mortality among OUD patients.
The first large-scale, randomized clinical trial of buprenorphine specifically for POUD included 653 treatment-seeking outpatients.25 This study was designed to approximate clinical practice and included buprenorphine/naloxone, recommended abstinence, and self-help; one-half of participants received intensive addiction counseling. POUD patients were most likely to reduce prescription opioid misuse during buprenorphine/naloxone treatment. If tapered off buprenorphine/naloxone, even after 12 weeks of treatment, the likelihood of an unsuccessful outcome was high. Moreover, opioid dependence counseling did not seem to afford any difference in outcomes. However, despite clinical effectiveness, over the last decade only 19% of patients admitted primarily for OUD treatment (other than heroin) were planned to be offered buprenorphine or methadone.26
A Cochrane review of oral naltrexone for OUD found that the drug was no better than placebo but concluded that available evidence does not allow an adequate evaluation.27 Opioid antagonists may be of value to patients who do not want to take agonists or partial agonists. Extended-release naltrexone also is available to treat OUD.
See the Box below that details steps the FDA and others have taken to prevent POUD and Table 5 for precautions to incorporate when prescribing opioids long-term.
The FDA has moved toward a risk evaluation and mitigation strategy (REMS) for opioids prescribed for pain that requires clinicians to receive training and certification in prescribing opioids for pain as well as identifying and reducing the risk for prescription opioid use disorder (POUD).a In 2011, the Obama administration developed an action plan to better address prescription drug abuse that required several federal agencies to develop programs and policies to address this growing problem; this plan was updated for 2012 (the complete National Drug Control Strategy 2012 is available at www.whitehouse.gov/sites/default/files/ondcp/2012_ndcs.pdf). The American Society of Addiction Medicine has issued a public policy statement that supports the federal approach and outlines other means to reduce POUD.b
Some pain specialists recommend requiring patients to sign an Opioid Pain Management Agreement that includes an “exit strategy” before the first opioid prescription is written. These agreements incorporate elements of “universal precautions” to take when prescribing opioids long term.c,d Although not well-studied, prescribing agreements may help educate patients and providers on how to interact in the management of pain with opioids in a way that is objective and empathic, and may reduce POUD risk.
References
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Opioid drugs and risk evaluation and mitigation strategies (REMS). http://www.fda.gov/drugs/drugsafety/informationbydrugclass/ucm163647.htm. Updated April 5, 2012. Accessed June 28, 2012.
- American Society of Addiction Medicine. Measures to counteract prescription drug diversion, misuse and addiction. http://www.asam.org/advocacy/find-a-policy-statement/view-policy-statement/public-policy-statements/2012/01/26/measures-to-counteract-prescription-drug-diversion-misuse-and-addiction. Published January 25, 2012. Accessed June 20, 2012.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112.
- Gourlay DL, Heit HA. Universal precautions revisited: managing the inherited pain patient. Pain Med. 2009; 10(suppl 2):S115-S123.
Table 5
Universal precautions with chronic opioid management
| Goals of therapy: partial pain relief and improvement in physical, emotional, and/or social functioning |
| Requirement for a single prescribing provider or treatment team |
| Limitation on dose and number of prescribed medications |
| Prohibition of changing dosage without discussion with the provider first |
| Monitoring patient adherence; discuss the use of ‘pill counts’ |
| Prohibition of use with alcohol, other sedating medications, or illegal drugs without discussion with the provider |
| Agreement not to drive or operate heavy machinery until abatement of medication-related drowsiness |
| Responsibility to keep medication safe and secure |
| Prohibition of selling, lending, sharing, or giving medication to others |
| Limitations on refills—only by appointment, in person, and no extra refills for running out early |
| Compliance with all components of overall treatment plan (including consultations and referrals) |
| Biological testing to screen for drugs of abuse or alcohol as well as to confirm the presence of prescribed opioids |
| Adverse effects and safety issues, such as the risk of physical dependence and addiction behaviors |
| The option of sharing information with family members and other providers, as necessary, with the patient’s consent |
| Need for periodic reevaluation of treatment |
| Reasons for stopping opioid therapy |
| Consequences of nonadherence with the treatment agreement |
| Source: Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112. |
CASE CONTINUED: A closer evaluation
After expressing your appreciation for Mr. H’s kind words and empathy for his chronic pain, you redirect him to his PCP. You ask him to sign a release of information so you and his other clinicians can coordinate his care. When discussing Mr. H with his PCP, you learn the patient has made limited requests for early refills and dose escalation primarily in relation to inadequate pain control and function, has genuine pain pathology, and is greatly distressed over his inability to work. No other AMTBs are present, and a check of the state prescribing database reveals that Mr. H did receive a small quantity of opioids from an ED on 1 occasion.
You and Mr. H’s PCP agree this is “pseudo-addiction” but want to watch Mr. H more closely and look for ways to coordinate his care. The PCP agrees to implement a prescribing agreement, start drug testing (including for the prescribed opioids), and reassess maximizing Mr. H’s function and pain management while you address his combined pain, depression, insomnia, and tobacco use.
Related Resources
- Ries RK, Fiellin D, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009.
- Department of Veterans Affairs. Department of Defense. VA/DoD clinical practice guideline for management of opioid therapy for chronic pain. Appendix C: sample opioid pain care agreement. http://www.healthquality.va.gov/COT_312_Full-er.pdf. Published May 2010. Accessed June 21, 2012.
- Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic non-malignant pain. J Addiction Med. 2007;1(1):2-10.
- Weaver M, Heit HA, Savage S, et al. Clinical case discussion: chronic pain management. J Addiction Med. 2007;1(1):11-14.
Drug Brand Names
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Codeine • Tylenol with codeine, others
- Fentanyl • Duragesic, Actiq
- Hydrocodone • Lortab, Vicodin, others
- Methadone • Dolophine, Methadose
- Morphine • Roxanol
- Naltrexone extended-release • Vivitrol
- Oxycodone • OxyContin, Roxicodone
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Frankowski’s time toward this project was provided by the American Board of Addiction Medicine-accredited Cincinnati VA Addiction Medicine Research Fellowship, affiliated with the CeTREAD, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs.
Acknowledgement
The authors thank Catherine Constance and Sandra Mason at the Cincinnati VA Medical Center for their administrative assistance.
Discuss this article at www.facebook.com/CurrentPsychiatry
You’ve been treating Mr. H, a 54-year-old factory worker and tobacco user, for depression that developed after a work-related back injury and subsequent disability. His depression has had a fair response to an antidepressant. He also has been maintained on chronic opioids (morphine and oxycodone/acetaminophen) for 18 months by his primary care physician (PCP). At the end of your appointment, he asks you for a refill of the opioids because he “ran out” early because of increased night pain and resultant insomnia and “stress.” He clarifies he has asked for early refills before from his PCP, but lately he has been denied. Because you “seem to listen to me more,” he asks for your help. How should you manage Mr. H?
Opioids are among the most commonly misused prescription drugs in the United States.1 In 2008, poisoning was the leading cause of death from injury in the United States; roughly 90% of poisonings resulted from drug exposure, and >40% of these drug poisonings were from prescription opioids.2 The Centers for Disease Control and Prevention estimates that the number of emergency department (ED) visits for nonmedical use of opioids increased 111% between 2004 and 2008, from 144,600 to 305,900 visits.3 The highest number of visits were for use of oxycodone, hydrocodone, and methadone.3
Increased prescribing of opioids and overdose deaths attributable to prescribed opioids have raised concern among physicians about how to effectively treat pain as well as prevent, recognize, and manage aberrant medication-taking behaviors (AMTBs). Psychiatrists are well-positioned to screen and manage their own patients for prescription opioid use disorder (POUD) or collaborate with opioid prescribers to accomplish the same.
Clarifying terminology
Terminology used to describe POUD and related conditions often is poorly defined or loosely applied. Because emotions often enter discussions between patients and physicians about problems related to opioid therapy, nonstigmatizing and more objective terminology is needed, and clinicians are working toward standardizing this. Relevant terms are defined in Table 1.4
The DSM-5 Substance Use Disorders Work Group has proposed using the term opioid use disorder (OUD) to replace the term opioid dependence.5 The hope is that removing the word “dependence” from the diagnostic term will reduce confusion between “dependence” due to expected physical dependence (tolerance, withdrawal) on medically prescribed opioids vs true addiction (currently defined as “opioid dependence” in DSM-IV-TR). This Work Group also has proposed combining opioid abuse and opioid dependence criteria into a single diagnosis of OUD, and adding “craving” to the criteria. For the complete proposed criteria, see www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460.These changes are still under review. In this article, we use the term POUD.
Table 1
Terminology related to prescription opioid use disorder
| Term | Definition |
|---|---|
| Chronic paina | Pain that extends beyond the expected period for healing (6 months), initiated by tissue damage, but perpetrated by the interaction of physiologic, affective, and environmental factors |
| Chronic nonmalignant paina | Chronic pain associated with diverse diagnoses and syndromes that are not terminal but affect the patient’s function |
| Appropriate usea | Taking a prescription as prescribed, and only for the condition indicated |
| Misusea | Taking a prescription for a reason or at a dose or frequency other than for which it was prescribed; this may or may not reflect POUD |
| Drug-seeking behaviors | Patient behaviors directed toward obtaining controlled substances, driven not by amelioration of the condition for which the medication was indicated but rather by other maladaptive gains; this may or may not reflect POUD |
| Chemical coping | Taking a controlled substance medication to relieve psychological problems (eg, to relieve low mood, anxiety, insomnia) and for reasons other than the purpose for which it was prescribed; this may or may not reflect POUD |
| Aberrant medication-taking behaviorsa | Taking a controlled substance medication in a manner that is not prescribed; causes for this may include:
|
| Pseudoaddiction | An iatrogenic syndrome of “addiction-like” behaviors in which the patient seeks opioids to relieve pain—such as seeking different doctors, self-adjusting the opioid dose, early refills of opioids, etc.—rather than to achieve pleasure or other nonpain-related effect. At times mistaken for true addiction, these behaviors tend to resolve and function improves once analgesia is better addressed |
| a These terms and definitions are adapted from reference 4. The remaining terms and definitions were developed by the authors POUD: prescription opioid use disorder | |
POUD and chronic pain
The incidence of POUD during opioid therapy for pain is unknown.6 Some researchers have suggested it may be as low as 0.2%,7 while others estimate that rates of POUD in patients with chronic pain may be similar to those in the general population: 3% to 16%.8 When applying the proposed DSM-5 criteria to patients receiving long-term opioid therapy for noncancer pain, the lifetime prevalence of POUD may be as high as 35%.9
Prescribers may be contributing to POUD. Roughly 76% of opioids used for nonmedical purposes were prescribed to someone else, 20% were prescribed to the user, and 4% came from other sources.1 Strategies to reduce POUD risk may be underused. In a retrospective cohort study of 1,612 patient electronic medical records from 8 primary care clinics that managed patients with long-term opioids for chronic noncancer pain (average prescribing duration of 2 years duration, ≥3 monthly prescriptions in 6 months), researchers evaluated how often prescribers used 3 risk reduction practices:
- urine drug tests
- regular office visits (≥1 every 6 months and within 30 days of changing opioid treatment)
- restricted early refills (≤1 opioid refill more than a week early).10
Risk factors for opioid misuse included age 1 early refill. Researchers found that even for high-risk patients, these strategies were used infrequently. Less than one-quarter of patients with ≥3 risk factors ever had a drug test, and those at increased risk were more likely to receive >1 early refill but no more likely to have more frequent visits. Issues such as patient entitlement, lack of physician education, and time constraints may explain why these strategies are not used more often.11
No one procedure or set of variables is sufficient to identify chronic pain patients who may be at risk for POUD. However, a history of drug or alcohol use disorders may be a significant risk factor.12,13
Few tools have been developed to help identify those at risk of AMTBs or POUD, and all have limitations.4,14 Recommended self-report measures include the Current Opioid Misuse Measure and the Opioid Risk Tool.15 A review of studies in which these kinds of tools were developed revealed limited evidence for their use; most studies had methodological shortcomings, did not use standardized AMTB criteria, and provided little assessment of whether these tools changed clinician behaviors or improved patient outcomes.16
Evaluating AMTBs
Although diagnosing POUD in pain patients receiving chronic opioids can be challenging, assessing for AMTBs typically is helpful. Once AMTBs are identified, they can be examined to determine what drives their expression (Table 14 and Table 217). However, often it is easier to identify AMTBs than to interpret their origins; as much as 30% to 50% of patients who complain of chronic pain may have primary substance dependence to sedatives, opioids, or both.11
Table 2
Aberrant medication-taking behaviors and POUD risk
| Behaviors more suggestive of POUD |
|---|
| Deterioration in function (work, social) |
| Illegal activities (selling medication, forging prescriptions, buying from non-medical sources) |
| Altering the route of administration (snorting, injecting) |
| Multiple episodes of ‘lost’ or ‘stolen’ prescriptions |
| Resistance to change therapy despite negative outcomes |
| Refusal to comply with toxicology testing |
| Concurrent, active abuse of alcohol, illegal drugs |
| Use of multiple physicians or pharmacies to obtain the prescription |
| Behaviors less suggestive of POUD |
| Complaints for more medication |
| Medication hoarding |
| Requesting specific pain medications |
| Openly acquiring similar medications from other providers |
| Occasional unsanctioned dose escalation |
| Nonadherence to other recommendations for pain therapy |
| POUD: prescription opioid use disorder Source: Reference 17 |
Although AMTBs are common among chronic nonmalignant pain patients,18,19 how often AMTBs reflect underlying POUD is uncertain.7 It is critical to interpret AMTBs with a balance of caution and care: “react therapeutically, not punitively.”20 Categorizing a patient’s AMTB as more or less likely to support a POUD diagnosis can be helpful, but is not conclusive (Table 2).17 Clinical correlation often is required. No single AMTB alone is indicative of POUD. When evaluating AMTBs, the treating provider should use a nonjudgmental stance, and consider obtaining collateral data from people who can provide differing perspectives of the patient’s behaviors, such as other clinicians, significant others, family, etc. (a release of information from the patient may be required). Another source of collateral data is prescription monitoring databases. These databases typically are state-based and provide electronic access to prescription information, allowing you to search for patterns—ie, use of multiple prescribers or pharmacies, undisclosed prescriptions, etc. Interest in establishing a single, federal database has been increasing, but striking a balance between carefully monitoring for AMTBs and protecting privacy remains unresolved.
DSM-IV-TR diagnostic criteria for opioid dependence21 can be challenging to interpret in patients who are prescribed opioids for pain (Table 3
).6 To clarify interpretation, the Liaison Committee on Pain and Addiction of the American Society of Addiction Medicine (ASAM) has provided an outline of possible indicators of addiction in pain patients (Table 4).6 This was a consensus statement from the American Pain Society, the American Academy of Pain Medicine, and ASAM.
Assessment is primarily clinical and requires an awareness of appropriate terminology, an index of clinical suspicion, and expertise teasing apart pain, addiction, and pseudoaddiction. In our experience, it is helpful to ask a chronic pain patient whom you suspect might have POUD, “Have you ever used your prescribed opioids for reasons other than improving function or reducing pain, such as for getting a ‘high,’ managing stress, escaping from problems, etc.?” An affirmative response suggests an underlying problem with use of prescribed opioids, indicating a need for more careful questioning to determine if AMTBs or POUD coexist with chronic pain.
Drug testing can help determine if a patient is taking opioids that are not prescribed—as well as illicit drugs or alcohol—and confirm the presence of those that are prescribed. Toxicology screening should include opioids typically screened for (eg, morphine, codeine, heroin) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and hydrocodone and synthetics such as fentanyl).
Table 3
Identifying addiction in pain patients: Limitations of DSM-IV-TR
| DSM-IV-TR substance dependence criteria | Challenges in using criterion to diagnose prescription opioid use disorder |
|---|---|
| Tolerance | Expected with prolonged opioid compliance |
| Physical dependence, withdrawal | Expected with prolonged opioid compliance |
| Use of larger amounts or longer than initially intended | Emergence of pain may demand increased dose or prolonged use |
| Multiple failed attempts to cut down or control | Emergence of pain may deter dose reduction or cessation |
| Time spent finding, using, or recovering | Difficulty finding adequate pain treatment may increase time spent pursuing analgesics. However, time spent recovering from overuse may suggest addiction |
| Given up or reduced important activities | Valid criteria—engaging in activities is expected to increase, not decline, with effective pain treatment |
| Continued use despite knowledge of negative consequences | Valid criteria—no harm is anticipated from analgesic opioid use for pain (see Table 4) |
| Source: Adapted from reference 6 | |
Table 4
Possible indicators of addiction in pain patients
| ASAM-APS-AAPM behavioral criteria | Examples of specific behaviors in opioid therapy for pain |
|---|---|
| Impaired control over opioid use | Patient requests early refills, frequently reports loss or theft of medication. Withdrawal noted at follow-up appointments despite having an adequate quantity of medication prescribed |
| Continued use despite harm from opioids | Patient exhibits declining function, opioid intoxication, persistent oversedation from opioids |
| Preoccupation with opioids | Patient ignores non-opioid interventions for pain, makes recurrent requests for opioid dose escalation (or complains of increasing pain) despite absence of disease progression or despite opioid dose increase by provider |
| AAPM: American Academy of Pain Medicine; APS: American Pain Society; ASAM: American Society of Addiction Medicine Source: Adapted from reference 6 | |
Helping POUD patients
Goals of treatment include establishing a therapeutic alliance, educating patients about POUD, reducing relapse risk, and optimizing overall health (including pain and physical function). The ASAM Patient Placement Criteria22 provide guidance regarding level-of-care decisions. Treatment ideally includes a combination of education about POUD and its relationship to chronic pain, pharmacotherapy, psychotherapy—such as motivational enhancement therapy, 12-step facilitation therapy, cognitive-behavioral therapy, and relapse prevention—and referral to self-help groups such as Narcotics Anonymous or Pills Anonymous. Importantly, if pain is genuine, it requires treatment.
Pharmacotherapy. Methadone is recommended as the standard of care for OUD by the National Institutes of Health. Methadone is a full opioid agonist that decreases illicit opioid use, mortality, and related problems and requires highly structured treatment approaches under federal and state regulation. POUD patients may have higher rates of methadone maintenance treatment retention than heroin-dependent patients.23 Published trials of buprenorphine for OUD have shown good treatment retention and reduction in illicit drug use and adverse events.24 Buprenorphine also decreases mortality among OUD patients.
The first large-scale, randomized clinical trial of buprenorphine specifically for POUD included 653 treatment-seeking outpatients.25 This study was designed to approximate clinical practice and included buprenorphine/naloxone, recommended abstinence, and self-help; one-half of participants received intensive addiction counseling. POUD patients were most likely to reduce prescription opioid misuse during buprenorphine/naloxone treatment. If tapered off buprenorphine/naloxone, even after 12 weeks of treatment, the likelihood of an unsuccessful outcome was high. Moreover, opioid dependence counseling did not seem to afford any difference in outcomes. However, despite clinical effectiveness, over the last decade only 19% of patients admitted primarily for OUD treatment (other than heroin) were planned to be offered buprenorphine or methadone.26
A Cochrane review of oral naltrexone for OUD found that the drug was no better than placebo but concluded that available evidence does not allow an adequate evaluation.27 Opioid antagonists may be of value to patients who do not want to take agonists or partial agonists. Extended-release naltrexone also is available to treat OUD.
See the Box below that details steps the FDA and others have taken to prevent POUD and Table 5 for precautions to incorporate when prescribing opioids long-term.
The FDA has moved toward a risk evaluation and mitigation strategy (REMS) for opioids prescribed for pain that requires clinicians to receive training and certification in prescribing opioids for pain as well as identifying and reducing the risk for prescription opioid use disorder (POUD).a In 2011, the Obama administration developed an action plan to better address prescription drug abuse that required several federal agencies to develop programs and policies to address this growing problem; this plan was updated for 2012 (the complete National Drug Control Strategy 2012 is available at www.whitehouse.gov/sites/default/files/ondcp/2012_ndcs.pdf). The American Society of Addiction Medicine has issued a public policy statement that supports the federal approach and outlines other means to reduce POUD.b
Some pain specialists recommend requiring patients to sign an Opioid Pain Management Agreement that includes an “exit strategy” before the first opioid prescription is written. These agreements incorporate elements of “universal precautions” to take when prescribing opioids long term.c,d Although not well-studied, prescribing agreements may help educate patients and providers on how to interact in the management of pain with opioids in a way that is objective and empathic, and may reduce POUD risk.
References
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Opioid drugs and risk evaluation and mitigation strategies (REMS). http://www.fda.gov/drugs/drugsafety/informationbydrugclass/ucm163647.htm. Updated April 5, 2012. Accessed June 28, 2012.
- American Society of Addiction Medicine. Measures to counteract prescription drug diversion, misuse and addiction. http://www.asam.org/advocacy/find-a-policy-statement/view-policy-statement/public-policy-statements/2012/01/26/measures-to-counteract-prescription-drug-diversion-misuse-and-addiction. Published January 25, 2012. Accessed June 20, 2012.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112.
- Gourlay DL, Heit HA. Universal precautions revisited: managing the inherited pain patient. Pain Med. 2009; 10(suppl 2):S115-S123.
Table 5
Universal precautions with chronic opioid management
| Goals of therapy: partial pain relief and improvement in physical, emotional, and/or social functioning |
| Requirement for a single prescribing provider or treatment team |
| Limitation on dose and number of prescribed medications |
| Prohibition of changing dosage without discussion with the provider first |
| Monitoring patient adherence; discuss the use of ‘pill counts’ |
| Prohibition of use with alcohol, other sedating medications, or illegal drugs without discussion with the provider |
| Agreement not to drive or operate heavy machinery until abatement of medication-related drowsiness |
| Responsibility to keep medication safe and secure |
| Prohibition of selling, lending, sharing, or giving medication to others |
| Limitations on refills—only by appointment, in person, and no extra refills for running out early |
| Compliance with all components of overall treatment plan (including consultations and referrals) |
| Biological testing to screen for drugs of abuse or alcohol as well as to confirm the presence of prescribed opioids |
| Adverse effects and safety issues, such as the risk of physical dependence and addiction behaviors |
| The option of sharing information with family members and other providers, as necessary, with the patient’s consent |
| Need for periodic reevaluation of treatment |
| Reasons for stopping opioid therapy |
| Consequences of nonadherence with the treatment agreement |
| Source: Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112. |
CASE CONTINUED: A closer evaluation
After expressing your appreciation for Mr. H’s kind words and empathy for his chronic pain, you redirect him to his PCP. You ask him to sign a release of information so you and his other clinicians can coordinate his care. When discussing Mr. H with his PCP, you learn the patient has made limited requests for early refills and dose escalation primarily in relation to inadequate pain control and function, has genuine pain pathology, and is greatly distressed over his inability to work. No other AMTBs are present, and a check of the state prescribing database reveals that Mr. H did receive a small quantity of opioids from an ED on 1 occasion.
You and Mr. H’s PCP agree this is “pseudo-addiction” but want to watch Mr. H more closely and look for ways to coordinate his care. The PCP agrees to implement a prescribing agreement, start drug testing (including for the prescribed opioids), and reassess maximizing Mr. H’s function and pain management while you address his combined pain, depression, insomnia, and tobacco use.
Related Resources
- Ries RK, Fiellin D, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009.
- Department of Veterans Affairs. Department of Defense. VA/DoD clinical practice guideline for management of opioid therapy for chronic pain. Appendix C: sample opioid pain care agreement. http://www.healthquality.va.gov/COT_312_Full-er.pdf. Published May 2010. Accessed June 21, 2012.
- Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic non-malignant pain. J Addiction Med. 2007;1(1):2-10.
- Weaver M, Heit HA, Savage S, et al. Clinical case discussion: chronic pain management. J Addiction Med. 2007;1(1):11-14.
Drug Brand Names
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Codeine • Tylenol with codeine, others
- Fentanyl • Duragesic, Actiq
- Hydrocodone • Lortab, Vicodin, others
- Methadone • Dolophine, Methadose
- Morphine • Roxanol
- Naltrexone extended-release • Vivitrol
- Oxycodone • OxyContin, Roxicodone
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Frankowski’s time toward this project was provided by the American Board of Addiction Medicine-accredited Cincinnati VA Addiction Medicine Research Fellowship, affiliated with the CeTREAD, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs.
Acknowledgement
The authors thank Catherine Constance and Sandra Mason at the Cincinnati VA Medical Center for their administrative assistance.
1. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Office of Applied Studies. Results from the 2009 national survey on drug use and health: volume I. http://www.samhsa.gov/data/NSDUH/2k9NSDUH/2k9Results.htm. Accessed June 20, 2012.
2. Warner M, Chen LH, Makuc DM, et al. Drug poisoning deaths in the United States, 1980-2008. http://www.cdc.gov/nchs/data/databriefs/db81.htm. Published December 2011. Accessed June 20, 2012.
3. Centers for Disease Control and Prevention (CDC). Emergency department visits involving nonmedical use of selected prescription drugs - United States 2004-2008. MMWR Morb Mortal Wkly Rep. 2010;59(23):705-709.
4. Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic nonmalignant pain. J Addict Med. 2007;1(1):2-10.
5. American Psychiatric Association. R 19 opioid use disorder. http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460. Updated April 30 2012. Accessed June 20, 2012.
6. Savage SR, Horvath R. Opioid therapy of pain. In: Ries RK Fiellin DA, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009:1329-1351.
7. Fishbain DA, Cole B, Lewis J, et al. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008;9(4):444-459.
8. Gourlay DL, Heit HA. Pain and addiction: managing risk through comprehensive care. J Addict Dis. 2008;27(3):23-30.
9. Boscarino JA, Rukstalis MR, Hoffman SN, et al. Prevalence of prescription opioid-use disorder among chronic pain patients: comparison of the DSM-5 vs. DSM-4 diagnostic criteria. J Addict Dis. 2011;30(3):185-194.
10. Starrels JL, Becker WC, Weiner MG, et al. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med. 2011;26(9):958-964.
11. Miller NS. Failure of enforcement controlled substance laws in health policy for prescribing opiate medications: a painful assessment of morbidity and mortality. Am J Ther. 2006;13(6):527-533.
12. Turk DC, Swanson KS, Gatchel RJ. Predicting opioid misuse by chronic pain patients: a systematic review and literature synthesis. Clin J Pain. 2008;24(6):497-508.
13. Miller NS, Greenfeld A. Patient characteristics and risks factors for development of dependence on hydrocodone and oxycodone. Am J Ther. 2004;11(1):26-32.
14. Butler SF, Budman SH, Fernandez KC, et al. Cross-validation of a Screener to Predict Opioid Misuse in Chronic Pain Patients (SOAPP-R). J Addict Med. 2009;3(2):66-73.
15. Passik SD, Kirsh KL, Casper D. Addiction-related assessment tools and pain management: instruments for screening treatment planning, and monitoring compliance. Pain Med. 2008;9(suppl 2):S145-S166.
16. Chou R, Fanciullo GJ, Fine PG, et al. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain. 2009;10(2):131-146.
17. Alford DP, Liebschutz J, Jackson A, et al. Prescription drug abuse: an introduction. http://www.drugabuse.gov/sites/default/files/prescription-drug-abuse-alt.pdf. Published November 8, 2009. Accessed June 20, 2012.
18. Passik SD, Kirsh KL, Whitcomb L, et al. Monitoring outcomes during long-term opioid therapy for noncancer pain: results with the Pain Assessment and Documentation Tool. J Opioid Manag. 2005;1(5):257-266.
19. Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432-442.
20. Passik SD. Pain management misstatements: ceiling effects red and yellow flags. Pain Med. 2006;7(1):76-77.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington DC: American Psychiatric Association; 2000.
22. Mee-Lee D, Shulman GD, Fishman MJ, et al. eds. ASAM patient placement criteria for the treatment of substance-related disorders. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc.; 2001.
23. Banta-Green CJ, Maynard C, Koepsell TD, et al. Retention in methadone maintenance drug treatment for prescription-type opioid primary users compared to heroin users. Addiction. 2009;104(5):775-783.
24. Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med. 2007;22(4):527-530.
25. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68(12):1238-1246.
26. U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration (SAMHSA). Office of Applied Studies. Treatment Episode Data Set (TEDS). 1998 - 2008. National Admissions to Substance Abuse Treatment Services, DASIS Series: S-50, HHS Publication No. (SMA) 09-4471. Rockville, MD; 2010.
27. Minozzi S, Amato L, Vecchi S, et al. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011;(4):CD001333.-
1. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Office of Applied Studies. Results from the 2009 national survey on drug use and health: volume I. http://www.samhsa.gov/data/NSDUH/2k9NSDUH/2k9Results.htm. Accessed June 20, 2012.
2. Warner M, Chen LH, Makuc DM, et al. Drug poisoning deaths in the United States, 1980-2008. http://www.cdc.gov/nchs/data/databriefs/db81.htm. Published December 2011. Accessed June 20, 2012.
3. Centers for Disease Control and Prevention (CDC). Emergency department visits involving nonmedical use of selected prescription drugs - United States 2004-2008. MMWR Morb Mortal Wkly Rep. 2010;59(23):705-709.
4. Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic nonmalignant pain. J Addict Med. 2007;1(1):2-10.
5. American Psychiatric Association. R 19 opioid use disorder. http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460. Updated April 30 2012. Accessed June 20, 2012.
6. Savage SR, Horvath R. Opioid therapy of pain. In: Ries RK Fiellin DA, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009:1329-1351.
7. Fishbain DA, Cole B, Lewis J, et al. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008;9(4):444-459.
8. Gourlay DL, Heit HA. Pain and addiction: managing risk through comprehensive care. J Addict Dis. 2008;27(3):23-30.
9. Boscarino JA, Rukstalis MR, Hoffman SN, et al. Prevalence of prescription opioid-use disorder among chronic pain patients: comparison of the DSM-5 vs. DSM-4 diagnostic criteria. J Addict Dis. 2011;30(3):185-194.
10. Starrels JL, Becker WC, Weiner MG, et al. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med. 2011;26(9):958-964.
11. Miller NS. Failure of enforcement controlled substance laws in health policy for prescribing opiate medications: a painful assessment of morbidity and mortality. Am J Ther. 2006;13(6):527-533.
12. Turk DC, Swanson KS, Gatchel RJ. Predicting opioid misuse by chronic pain patients: a systematic review and literature synthesis. Clin J Pain. 2008;24(6):497-508.
13. Miller NS, Greenfeld A. Patient characteristics and risks factors for development of dependence on hydrocodone and oxycodone. Am J Ther. 2004;11(1):26-32.
14. Butler SF, Budman SH, Fernandez KC, et al. Cross-validation of a Screener to Predict Opioid Misuse in Chronic Pain Patients (SOAPP-R). J Addict Med. 2009;3(2):66-73.
15. Passik SD, Kirsh KL, Casper D. Addiction-related assessment tools and pain management: instruments for screening treatment planning, and monitoring compliance. Pain Med. 2008;9(suppl 2):S145-S166.
16. Chou R, Fanciullo GJ, Fine PG, et al. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain. 2009;10(2):131-146.
17. Alford DP, Liebschutz J, Jackson A, et al. Prescription drug abuse: an introduction. http://www.drugabuse.gov/sites/default/files/prescription-drug-abuse-alt.pdf. Published November 8, 2009. Accessed June 20, 2012.
18. Passik SD, Kirsh KL, Whitcomb L, et al. Monitoring outcomes during long-term opioid therapy for noncancer pain: results with the Pain Assessment and Documentation Tool. J Opioid Manag. 2005;1(5):257-266.
19. Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432-442.
20. Passik SD. Pain management misstatements: ceiling effects red and yellow flags. Pain Med. 2006;7(1):76-77.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington DC: American Psychiatric Association; 2000.
22. Mee-Lee D, Shulman GD, Fishman MJ, et al. eds. ASAM patient placement criteria for the treatment of substance-related disorders. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc.; 2001.
23. Banta-Green CJ, Maynard C, Koepsell TD, et al. Retention in methadone maintenance drug treatment for prescription-type opioid primary users compared to heroin users. Addiction. 2009;104(5):775-783.
24. Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med. 2007;22(4):527-530.
25. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68(12):1238-1246.
26. U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration (SAMHSA). Office of Applied Studies. Treatment Episode Data Set (TEDS). 1998 - 2008. National Admissions to Substance Abuse Treatment Services, DASIS Series: S-50, HHS Publication No. (SMA) 09-4471. Rockville, MD; 2010.
27. Minozzi S, Amato L, Vecchi S, et al. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011;(4):CD001333.-
Is quetiapine effective for anxiety?
Discuss this article at www.facebook.com/CurrentPsychiatry
The rate of off-label prescribing of second-generation antipsychotics (SGAs) is estimated to have doubled in the past decade.1,2 In 2010, quetiapine was the most commonly used SGA in the United States with >10 million prescriptions dispensed.2 Clinical experience and reports from patients indicate quetiapine may be useful for treating anxiety. When making medication choices, it can be useful to combine anecdotal evidence with the facts (or lack thereof). Does evidence support or contradict the use of quetiapine for anxiety?
What the research shows
Quetiapine is FDA-approved for treating:
- adults and adolescents with schizophrenia
- adults, children, and adolescents with acute manic episodes associated with bipolar I disorder (BDI) as monotherapy or as an adjunct to lithium or divalproex
- adults with an acute depressive episode associated with bipolar disorder
- adjunctive treatment of major depressive disorder (MDD) in adults
- maintenance treatment of BDI as an adjunct to lithium or divalproex in adults.3
In addition, quetiapine extended-release (XR) is approved as an adjunctive treatment for MDD in adults.4
Neither the immediate-release or XR formulation is indicated for treating anxiety, but quetiapine has been studied as a treatment for several anxiety disorders, including posttraumatic stress disorder, social phobia, obsessive-compulsive disorder, and anxiety secondary to mood disorders. It has been most extensively studied as treatment for generalized anxiety disorder (GAD).
Three trials that involved >2,100 patients found quetiapine XR monotherapy is effective for GAD in doses of 50 to 300 mg/d.5-7 In 2 of the studies, quetiapine XR was as effective as paroxetine and escitalopram for GAD.5,6 Reviews of off-label SGA use have found that compared with placebo, quetiapine XR monotherapy is effective for GAD (number needed to treat=8).8,9 Side effects reported in clinical trials of quetiapine included headache, somnolence, sedation, fatigue, dizziness, dry mouth, weight gain, hyperlipidemia, and elevated glucose levels.
What did the FDA say?
In April 2009, the FDA’s Psychopharmacologic Drugs Advisory Committee reviewed whether evidence supported quetiapine XR for treating MDD and GAD.10 Although the committee found that quetiapine XR monotherapy effectively treated GAD, it concluded it was not acceptably safe.11 The committee expressed concerns over exposing a greatly expanded population to a drug with substantial metabolic side effects, including weight gain (incidence 3% to 23%), increased cholesterol (incidence 7% to 18%), and hyperglycemia.12-14 Weight gain and metabolic effects have been reported even when quetiapine is prescribed at low doses (≤100 mg/d).15,16 The FDA did not approve expanding the indication of quetiapine XR to include treatment of GAD.
Our opinion
Quetiapine XR is effective for treating GAD. However, even at low doses, it is associated with substantial side effects and should be reserved for patients with poor response or contraindications (eg, mania) to traditional GAD treatments such as selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors. Published studies assessed quetiapine XR only when used on a scheduled basis, and did not address use of quetiapine immediate release or XR on an as-needed basis for GAD.
Related Resources
- AstraZeneca. Seroquel prescribing information. www1.astrazeneca-us.com/pi/seroquel.pdf.
- AstraZeneca. Seroquel XR prescribing information. www1.astrazeneca-us.com/pi/seroquelxr.pdf.
Drug Brand Names
- Divalproex • Depakote
- Paroxetine • Paxil
- Escitalopram • Lexapro
- Quetiapine • Seroquel
- Lithium • Eskalith, Lithobid
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. Drug Topics. 2010 Top 200 branded drugs by total prescriptions. http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard/drugtopics/252011/727256/article.pdf. Published June 2011. Accessed June 26 2012.
3. Seroquel [package insert]. Wilmington DE: AstraZeneca; 2012.
4. Seroquel XR [package insert]. Wilmington DE: AstraZeneca; 2012.
5. Merideth C, Cutler A, Neijber A, et al. Efficacy and tolerability of extended release quetiapine fumarate monotherapy in the treatment of GAD. Eur Neuropsychopharmacol. 2008;18(suppl 4):S499-S500.
6. Bandelow B, Chouinard G, Bobes J, et al. Extended-release quetiapine fumarate (quetiapine XR): a once-daily monotherapy effective in generalized anxiety disorder. Data from a randomized, double-blind, placebo- and active-controlled study. Int J Neuropsychopharmacol. 2010;13(3):305-320.
7. Katzman MA, Brawman-Mintzer O, Reyes EB, et al. Extended release quetiapine fumarate (quetiapine XR) monotherapy as maintenance treatment for generalized anxiety disorder: a long-term, randomized, placebo-controlled trial. Int Clin Psychopharmacol. 2011;26(1):11-24.
8. Maglione M, Ruelaz Maher A, Hu J, et al. Agency for Healthcare Research and Quality. Off-label use of atypical antipsychotics: an update. http://www.effectivehealthcare.ahrq.gov/ehc/products/150/786/CER43_Off-LabelAntipsychotics_execsumm_20110928.pdf. Published September 2011. Accessed June 26 2012.
9. Maher AR, Maglione M, Bagley S, et al. Efficacy and comparative effectiveness of atypical antipsychotic medications for off-label uses in adults: a systematic review and meta-analysis. JAMA. 2011;306(12):1359-1369.
10. U.S. Food and Drug Administration. Psychopharmacologic Drugs Advisory Committee meeting announcement. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm136250.htm. Updated June 18, 2009. Accessed June 26, 2012.
11. FDA advisory committee recommendation on Seroquel XR supplemental new drug applications [news release]. Wilmington DE: AstraZeneca; April 9, 2009. http://www.astrazeneca.com/Media/Press-releases/Article/20090409—FDA-Advisory-Committee-Recommendation-on-Seroquel-XR-. Accessed June 26, 2012.
12. Meyer JM, Koro CE. The effects of antipsychotic therapy on serum lipids: a comprehensive review. Schizophr Res. 2004;70(1):1-17.
13. Newcomer JW. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. J Clin Psychiatry. 2007;68(suppl 1):20-27.
14. Chen WY, Chen CC, Hung GC. Hyperglycemic hyperosmolar state associated with low-dose quetiapine treatment in a patient with bipolar disorder. Curr Drug Saf. 2011;6(3):207-208.
15. Williams SG, Alinejad NA, Williams JA, et al. Statistically significant increase in weight caused by low-dose quetiapine. Pharmacotherapy. 2010;30(10):1011-1015.
16. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
Discuss this article at www.facebook.com/CurrentPsychiatry
The rate of off-label prescribing of second-generation antipsychotics (SGAs) is estimated to have doubled in the past decade.1,2 In 2010, quetiapine was the most commonly used SGA in the United States with >10 million prescriptions dispensed.2 Clinical experience and reports from patients indicate quetiapine may be useful for treating anxiety. When making medication choices, it can be useful to combine anecdotal evidence with the facts (or lack thereof). Does evidence support or contradict the use of quetiapine for anxiety?
What the research shows
Quetiapine is FDA-approved for treating:
- adults and adolescents with schizophrenia
- adults, children, and adolescents with acute manic episodes associated with bipolar I disorder (BDI) as monotherapy or as an adjunct to lithium or divalproex
- adults with an acute depressive episode associated with bipolar disorder
- adjunctive treatment of major depressive disorder (MDD) in adults
- maintenance treatment of BDI as an adjunct to lithium or divalproex in adults.3
In addition, quetiapine extended-release (XR) is approved as an adjunctive treatment for MDD in adults.4
Neither the immediate-release or XR formulation is indicated for treating anxiety, but quetiapine has been studied as a treatment for several anxiety disorders, including posttraumatic stress disorder, social phobia, obsessive-compulsive disorder, and anxiety secondary to mood disorders. It has been most extensively studied as treatment for generalized anxiety disorder (GAD).
Three trials that involved >2,100 patients found quetiapine XR monotherapy is effective for GAD in doses of 50 to 300 mg/d.5-7 In 2 of the studies, quetiapine XR was as effective as paroxetine and escitalopram for GAD.5,6 Reviews of off-label SGA use have found that compared with placebo, quetiapine XR monotherapy is effective for GAD (number needed to treat=8).8,9 Side effects reported in clinical trials of quetiapine included headache, somnolence, sedation, fatigue, dizziness, dry mouth, weight gain, hyperlipidemia, and elevated glucose levels.
What did the FDA say?
In April 2009, the FDA’s Psychopharmacologic Drugs Advisory Committee reviewed whether evidence supported quetiapine XR for treating MDD and GAD.10 Although the committee found that quetiapine XR monotherapy effectively treated GAD, it concluded it was not acceptably safe.11 The committee expressed concerns over exposing a greatly expanded population to a drug with substantial metabolic side effects, including weight gain (incidence 3% to 23%), increased cholesterol (incidence 7% to 18%), and hyperglycemia.12-14 Weight gain and metabolic effects have been reported even when quetiapine is prescribed at low doses (≤100 mg/d).15,16 The FDA did not approve expanding the indication of quetiapine XR to include treatment of GAD.
Our opinion
Quetiapine XR is effective for treating GAD. However, even at low doses, it is associated with substantial side effects and should be reserved for patients with poor response or contraindications (eg, mania) to traditional GAD treatments such as selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors. Published studies assessed quetiapine XR only when used on a scheduled basis, and did not address use of quetiapine immediate release or XR on an as-needed basis for GAD.
Related Resources
- AstraZeneca. Seroquel prescribing information. www1.astrazeneca-us.com/pi/seroquel.pdf.
- AstraZeneca. Seroquel XR prescribing information. www1.astrazeneca-us.com/pi/seroquelxr.pdf.
Drug Brand Names
- Divalproex • Depakote
- Paroxetine • Paxil
- Escitalopram • Lexapro
- Quetiapine • Seroquel
- Lithium • Eskalith, Lithobid
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
The rate of off-label prescribing of second-generation antipsychotics (SGAs) is estimated to have doubled in the past decade.1,2 In 2010, quetiapine was the most commonly used SGA in the United States with >10 million prescriptions dispensed.2 Clinical experience and reports from patients indicate quetiapine may be useful for treating anxiety. When making medication choices, it can be useful to combine anecdotal evidence with the facts (or lack thereof). Does evidence support or contradict the use of quetiapine for anxiety?
What the research shows
Quetiapine is FDA-approved for treating:
- adults and adolescents with schizophrenia
- adults, children, and adolescents with acute manic episodes associated with bipolar I disorder (BDI) as monotherapy or as an adjunct to lithium or divalproex
- adults with an acute depressive episode associated with bipolar disorder
- adjunctive treatment of major depressive disorder (MDD) in adults
- maintenance treatment of BDI as an adjunct to lithium or divalproex in adults.3
In addition, quetiapine extended-release (XR) is approved as an adjunctive treatment for MDD in adults.4
Neither the immediate-release or XR formulation is indicated for treating anxiety, but quetiapine has been studied as a treatment for several anxiety disorders, including posttraumatic stress disorder, social phobia, obsessive-compulsive disorder, and anxiety secondary to mood disorders. It has been most extensively studied as treatment for generalized anxiety disorder (GAD).
Three trials that involved >2,100 patients found quetiapine XR monotherapy is effective for GAD in doses of 50 to 300 mg/d.5-7 In 2 of the studies, quetiapine XR was as effective as paroxetine and escitalopram for GAD.5,6 Reviews of off-label SGA use have found that compared with placebo, quetiapine XR monotherapy is effective for GAD (number needed to treat=8).8,9 Side effects reported in clinical trials of quetiapine included headache, somnolence, sedation, fatigue, dizziness, dry mouth, weight gain, hyperlipidemia, and elevated glucose levels.
What did the FDA say?
In April 2009, the FDA’s Psychopharmacologic Drugs Advisory Committee reviewed whether evidence supported quetiapine XR for treating MDD and GAD.10 Although the committee found that quetiapine XR monotherapy effectively treated GAD, it concluded it was not acceptably safe.11 The committee expressed concerns over exposing a greatly expanded population to a drug with substantial metabolic side effects, including weight gain (incidence 3% to 23%), increased cholesterol (incidence 7% to 18%), and hyperglycemia.12-14 Weight gain and metabolic effects have been reported even when quetiapine is prescribed at low doses (≤100 mg/d).15,16 The FDA did not approve expanding the indication of quetiapine XR to include treatment of GAD.
Our opinion
Quetiapine XR is effective for treating GAD. However, even at low doses, it is associated with substantial side effects and should be reserved for patients with poor response or contraindications (eg, mania) to traditional GAD treatments such as selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors. Published studies assessed quetiapine XR only when used on a scheduled basis, and did not address use of quetiapine immediate release or XR on an as-needed basis for GAD.
Related Resources
- AstraZeneca. Seroquel prescribing information. www1.astrazeneca-us.com/pi/seroquel.pdf.
- AstraZeneca. Seroquel XR prescribing information. www1.astrazeneca-us.com/pi/seroquelxr.pdf.
Drug Brand Names
- Divalproex • Depakote
- Paroxetine • Paxil
- Escitalopram • Lexapro
- Quetiapine • Seroquel
- Lithium • Eskalith, Lithobid
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. Drug Topics. 2010 Top 200 branded drugs by total prescriptions. http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard/drugtopics/252011/727256/article.pdf. Published June 2011. Accessed June 26 2012.
3. Seroquel [package insert]. Wilmington DE: AstraZeneca; 2012.
4. Seroquel XR [package insert]. Wilmington DE: AstraZeneca; 2012.
5. Merideth C, Cutler A, Neijber A, et al. Efficacy and tolerability of extended release quetiapine fumarate monotherapy in the treatment of GAD. Eur Neuropsychopharmacol. 2008;18(suppl 4):S499-S500.
6. Bandelow B, Chouinard G, Bobes J, et al. Extended-release quetiapine fumarate (quetiapine XR): a once-daily monotherapy effective in generalized anxiety disorder. Data from a randomized, double-blind, placebo- and active-controlled study. Int J Neuropsychopharmacol. 2010;13(3):305-320.
7. Katzman MA, Brawman-Mintzer O, Reyes EB, et al. Extended release quetiapine fumarate (quetiapine XR) monotherapy as maintenance treatment for generalized anxiety disorder: a long-term, randomized, placebo-controlled trial. Int Clin Psychopharmacol. 2011;26(1):11-24.
8. Maglione M, Ruelaz Maher A, Hu J, et al. Agency for Healthcare Research and Quality. Off-label use of atypical antipsychotics: an update. http://www.effectivehealthcare.ahrq.gov/ehc/products/150/786/CER43_Off-LabelAntipsychotics_execsumm_20110928.pdf. Published September 2011. Accessed June 26 2012.
9. Maher AR, Maglione M, Bagley S, et al. Efficacy and comparative effectiveness of atypical antipsychotic medications for off-label uses in adults: a systematic review and meta-analysis. JAMA. 2011;306(12):1359-1369.
10. U.S. Food and Drug Administration. Psychopharmacologic Drugs Advisory Committee meeting announcement. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm136250.htm. Updated June 18, 2009. Accessed June 26, 2012.
11. FDA advisory committee recommendation on Seroquel XR supplemental new drug applications [news release]. Wilmington DE: AstraZeneca; April 9, 2009. http://www.astrazeneca.com/Media/Press-releases/Article/20090409—FDA-Advisory-Committee-Recommendation-on-Seroquel-XR-. Accessed June 26, 2012.
12. Meyer JM, Koro CE. The effects of antipsychotic therapy on serum lipids: a comprehensive review. Schizophr Res. 2004;70(1):1-17.
13. Newcomer JW. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. J Clin Psychiatry. 2007;68(suppl 1):20-27.
14. Chen WY, Chen CC, Hung GC. Hyperglycemic hyperosmolar state associated with low-dose quetiapine treatment in a patient with bipolar disorder. Curr Drug Saf. 2011;6(3):207-208.
15. Williams SG, Alinejad NA, Williams JA, et al. Statistically significant increase in weight caused by low-dose quetiapine. Pharmacotherapy. 2010;30(10):1011-1015.
16. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. Drug Topics. 2010 Top 200 branded drugs by total prescriptions. http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard/drugtopics/252011/727256/article.pdf. Published June 2011. Accessed June 26 2012.
3. Seroquel [package insert]. Wilmington DE: AstraZeneca; 2012.
4. Seroquel XR [package insert]. Wilmington DE: AstraZeneca; 2012.
5. Merideth C, Cutler A, Neijber A, et al. Efficacy and tolerability of extended release quetiapine fumarate monotherapy in the treatment of GAD. Eur Neuropsychopharmacol. 2008;18(suppl 4):S499-S500.
6. Bandelow B, Chouinard G, Bobes J, et al. Extended-release quetiapine fumarate (quetiapine XR): a once-daily monotherapy effective in generalized anxiety disorder. Data from a randomized, double-blind, placebo- and active-controlled study. Int J Neuropsychopharmacol. 2010;13(3):305-320.
7. Katzman MA, Brawman-Mintzer O, Reyes EB, et al. Extended release quetiapine fumarate (quetiapine XR) monotherapy as maintenance treatment for generalized anxiety disorder: a long-term, randomized, placebo-controlled trial. Int Clin Psychopharmacol. 2011;26(1):11-24.
8. Maglione M, Ruelaz Maher A, Hu J, et al. Agency for Healthcare Research and Quality. Off-label use of atypical antipsychotics: an update. http://www.effectivehealthcare.ahrq.gov/ehc/products/150/786/CER43_Off-LabelAntipsychotics_execsumm_20110928.pdf. Published September 2011. Accessed June 26 2012.
9. Maher AR, Maglione M, Bagley S, et al. Efficacy and comparative effectiveness of atypical antipsychotic medications for off-label uses in adults: a systematic review and meta-analysis. JAMA. 2011;306(12):1359-1369.
10. U.S. Food and Drug Administration. Psychopharmacologic Drugs Advisory Committee meeting announcement. http://www.fda.gov/AdvisoryCommittees/Calendar/ucm136250.htm. Updated June 18, 2009. Accessed June 26, 2012.
11. FDA advisory committee recommendation on Seroquel XR supplemental new drug applications [news release]. Wilmington DE: AstraZeneca; April 9, 2009. http://www.astrazeneca.com/Media/Press-releases/Article/20090409—FDA-Advisory-Committee-Recommendation-on-Seroquel-XR-. Accessed June 26, 2012.
12. Meyer JM, Koro CE. The effects of antipsychotic therapy on serum lipids: a comprehensive review. Schizophr Res. 2004;70(1):1-17.
13. Newcomer JW. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. J Clin Psychiatry. 2007;68(suppl 1):20-27.
14. Chen WY, Chen CC, Hung GC. Hyperglycemic hyperosmolar state associated with low-dose quetiapine treatment in a patient with bipolar disorder. Curr Drug Saf. 2011;6(3):207-208.
15. Williams SG, Alinejad NA, Williams JA, et al. Statistically significant increase in weight caused by low-dose quetiapine. Pharmacotherapy. 2010;30(10):1011-1015.
16. Simon V, van Winkel R, De Hert M. Are weight gain and metabolic side effects of atypical antipsychotics dose dependent? A literature review. J Clin Psychiatry. 2009;70(7):1041-1050.
How to manage depression in overweight or obese patients
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. G is a 52-year-old mother and teacher with a 20-year history of recurrent depressive episodes for which she has been treated with various antidepressants, including sertraline, fluoxetine, and citalopram. For some of her depressive recurrences, she also received adjunctive second-generation antipsychotics (SGAs), including quetiapine and olanzapine.
She describes feelings of “being defeated,” hopelessness, and boredom and frustration with her teaching. It takes her approximately 30 minutes to go to sleep each night, but she wakes up after 2 to 3 hours, and the remainder of her night’s sleep is markedly disrupted. Because of her hopeless feelings, she has given up on dieting and going to the gym. When feeling down she has donuts and coffee. She has gained 45 lbs over the past 10 years and now weighs 175 lbs. In addition to her disrupted mood, she complains of frequent headaches and sore muscles.
Mrs. G’s psychiatrist refers her to her primary care physician for evaluation of her physical complaints and recommendations regarding her weight gain. Her waistline measures 90 cm and her body mass index (BMI) is 29.1 kg/m2; a BMI of ≥30 is considered obese. Her blood pressure is 145/85 mmHg. Laboratory work reveals a total cholesterol level of 235 mg/dL, low-density lipoprotein of 146 mg/dL, and fasting blood sugar, 135 mg/dL.
Mrs. G’s case illustrates many of the issues psychiatrists face when caring for overweight or obese patients with depression (OW/OB-D). Both conditions can be challenging to manage, and may be especially difficult to treat when they co-occur. When depression and obesity co-occur, their capacity to inflict psychological and physical harm likely is greater than either condition alone. Data point to a “2-way street” of mutually destructive effects of being overweight/obese on depression and vice versa.1
This article summarizes ways that depression and obesity aggravate each other, and highlights research that suggests depression and obesity are manifestations of inflammatory processes. It also suggests a stepwise approach to treating OW/OB-D patients.
Mutually destructive processes
Self-esteem and body image. Lowered self-esteem is a hallmark of depression. In popular culture, “you can’t be too rich or too thin,” and the pressure to be slim is great. Therefore, OW/OB-D patients have 2 reasons to feel a depleted sense of self-worth: their psychiatric illness and their weight. Observant clinicians will recognize these dual sources of self-deprecation and tailor treatment to address both.
Increasing numbers of celebrities, performers, and prominent politicians are overweight or obese. Increased social acceptance of OW/OB individuals in our culture may be legitimizing weight gain and obesity. When OW/OB-D patients justify their weight by pointing to overweight celebrities, clinicians can counter this argument with data on the hazards of obesity on health and well-being, such as premature death, coronary artery disease, diabetes, arthritis, and some forms of cancer.
OW/OB patients tend to interact with other OW/OB individuals. Christakis et al2 reported that adults with obese friends were more likely to become obese than individuals without obese friends. Valente et al3 found that overweight teens were twice as likely to have overweight friends as non-overweight teens. This power of social connectedness can be harnessed when treating OW/OB-D patients, where therapeutic groups can help patients address both depression and weight gain.
Inactivity. OW/OB-D patients with psychomotor retardation or reduced activity may gain weight because they consume more calories than their body requires. Depressed patients may say they “have no energy” to participate in a clinician-recommended exercise program or that “it won’t do any good anyway.”
These tendencies are best dealt with by incorporating an exercise program into the comprehensive plan for OW/OB-D patients from the start of treatment. Several studies suggest that in addition to helping manage weight, exercise may have antidepressant effects. In a large, well-controlled trial of patients with major depressive disorder (MDD), Blumenthal et al4 found that an exercise program was as effective as fluoxetine, 20 mg/d, and the antidepressant effects persisted at 10-month follow-up for patients who continued to exercise.5 In a review of studies of exercise in depressed patients, Helmich et al6 concluded that in most studies exercise was beneficial. However, Mead et al7 found that nearly all trials of exercise and depression had substantial design flaws. Based on the 3 well-designed studies they reviewed, Mead et al concluded that the efficacy of exercise was comparable to that of cognitive therapy.
Although the evidence on exercise for treating depression is inconclusive, an exercise program is essential for OW/OB-D patients because it can help manage weight and improve cardiovascular fitness. Motivation is a key ingredient of successful programs.8 Encourage patients to make exercise enjoyable, perhaps by using video games or other interactive computer-based programs.9
Sleep disturbances. Disrupted sleep— another hallmark of depression—appears to be a risk factor for weight gain.10 Although the basis for this relationship is still under investigation, one possibility is that some patients with insomnia get up to eat more often than those without sleep disturbances. Research has shown that when sleep is curtailed in a sleep laboratory, patients consume approximately 20% more calories from snacks (1,086 calories) than non-sleep-deprived patients (866 calories).11 Although this 220-calorie increase may seem small, it would amount to approximately 2 lbs of additional weight per month.
Appetite. Although weight loss is a cardinal sign of MDD, increased appetite and weight gain can be seen in many depressed patients who do not meet diagnostic criteria for MDD as well as those with seasonal affective disorder and metabolic syndrome, a condition characterized by insulin resistance, glucose intolerance, atherogenic dyslipidemia, visceral adiposity, hypercoagulation, chronic inflammation, oxidative stress, and hypertension.12
Emerging information about the neuroendocrinology of appetite regulation may lead to a better understanding of weight management in OW/OB-D patients. Leptin, a hormone released by adipose tissue, increases when fat stores are high, leading to reduced appetite and fat stores. Conversely, when fat stores are low, plasma leptin levels decrease, producing increased appetite and reduced energy expenditure.13
Researchers have suggested that leptin insufficiency and/or leptin resistance may contribute to vulnerability to depression, and leptin may have antidepressant effects.14 Lawson et al15 found that leptin levels were inversely associated with Hamilton Depression Rating Scale scores in normal-weight (BMI ≤25) women.
Leptin levels also are significantly associated with comorbid depressed mood and sleep disturbance.16 In healthy volunteers, shortening sleep duration to 4 hours produced an approximately 20% reduction in leptin release compared with normal sleep duration.17 Because of the relationship between sleep disorders and depression, leptin may act on sleep regulatory mechanisms, depressogenic pathways, or both. But studies of leptin’s role in obesity, depression, and sleep have not yet found a single role for leptin that ties all 3 conditions to this hormone’s known physiological functions.
Nonadherence. Compared with non-depressed patients, depressed patients are 76% more likely to not adhere to treatment.18 Patients may report that they are not interested in the treatment program or lack hope that it will be successful. Furthermore, OW/OB-D patients may consider exercise programs to be too strenuous and diet programs too depriving.19
OW/OB-D patients may require special care in monitoring adherence. The presence of depression in patients enrolled in weight loss programs may prompt the treatment staff to modify the usual protocol by including the patient in an active depression treatment module.20
Effects of pharmacologic agents
Many antidepressant agents are associated with weight gain.21Tables 1 and 2 summarize the effects antidepressants and adjunctive medications used to treat depression have on weight.22,23 SGAs such as clozapine and olanzapine, which frequently are used as augmenting agents in patients with treatment-resistant depression (TRD), are associated with weight gain.22 Lamotrigine also is an effective adjunctive medication for TRD and is not associated with significant weight gain.24
Bupropion has antidepressant and weight-loss effects and may be a suitable primary medication for OW/OB-D patients.
Early weight gain with olanzapine/fluoxetine combination may be a strong indicator of substantial weight gain with longer-term treatment. A weight gain of >2 kg (4.4 lbs) during the first 2 weeks of treatment is a strong predictor of weight gain of ≥10 kg (22 lbs) at 26 weeks.25
Antidepressants may be associated with an increased risk of obesity, and strategies to offset this risk may be useful in clinical practice, particularly patient education on the risks of weight gain and early introduction of a diet and exercise program.
Evidence suggests that depression and obesity are associated with alterations in immune activity (Box). This suggests that anti-inflammatory agents might have a role in treating depression by reducing the release of cytokines that may lead to depressive symptoms.
Table 1
Pharmacotherapy and weight gain: Antidepressants
| Agent | Effect on weight |
|---|---|
| SSRIs | |
| Paroxetine | Moderate gain |
| Fluoxetine | Early: weight loss Long-term: moderate gain |
| SNRIs | |
| Duloxetine | Minimal gain |
| Escitalopram | Moderate gain |
| Other agents | |
| Imipramine (TCA) | Moderate gain |
| Selegiline (MAOI) | Moderate gain |
| Trazodone (tetracyclic) | Moderate gain |
| Bupropion (atypical) | Moderate loss |
| MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCA: tricyclic antidepressant Source: References 22,23 | |
Table 2
Pharmacotherapy and weight gain: Adjunctive agents
| Agent | Use in depression | Effect on weight |
|---|---|---|
| SGAs | ||
| Olanzapine | Psychotic depression | Large gain |
| Clozapine | Adjunct; psychotic depression | Large gain |
| Quetiapine | Primary; adjunct | Large gain |
| Aripiprazole | Adjunct | Small gain |
| Risperidone | Psychotic depression | Small gain |
| Ziprasidone | Psychotic depression | Small loss |
| Mood stabilizers | ||
| Divalproex | Treatment resistance, bipolar disorder | Moderate to large gain |
| Lamotrigine | Treatment resistance | Neutral |
| SGAs: second-generation antipsychotics Source: References 22,23 | ||
Research suggests that both depression and obesity are associated with immune dysregulation and inflammation.a-d Although the complexities of these interactions are beyond the scope of this article, having a model for understanding the role of inflammation in overweight or obese patients with depression (OW/OB-D) may be useful. Data supporting a role for immune dysregulation in OW/OB-D patients rests on the following findings:
Fat and muscle are endocrine organs: Fat is not just a storage organ for energy-rich lipids but also a rich source of cytokines, including monocyte chemotactic protein-1 (MCP-1), interleukin-2, and tumor necrosis factor-α (TNF-α). The increase in MCP-1 in fat tissue triggers a cascade of events that leads to chronic inflammation in adipose tissue. These substances can be released into circulation, stimulating inflammatory responses in other tissues. Data suggest that obesity’s effects on cardiovascular disease are mediated by these adipose-derived inflammatory hormones. There is a strong relationship between the volume of adipose tissue and the amount of pro-inflammatory hormones released; therefore, reducing weight reduces inflammatory burden on the body.
Pedersene pointed out that muscle also is an endocrine organ. Among the cytokines (or “myokines”) muscle produces are interleukin-6 (IL-6), interleukin-8, and brain-derived neurotrophic factor. During exercise, the amount of IL-6 released from muscles may increase by 100-fold. Although IL-6 usually is considered a pro-inflammatory regulator, it—or other muscle-derived myokines—may be responsible for some of exercise’s beneficial effects.e
If this hypothesis is correct, patients whose exercise includes resistance training—which increases muscle mass—are not just getting stronger or burning calories but may be facilitating release of hormones that could counteract obesity’s inflammatory effects.
Cytokine levels are elevated in depression and obesity: A substantial body of evidence shows that depressed patients have elevated circulating levels of inflammation markers. In particular, the proinflammatory cytokines IL-6 and interleukin-1β and the acute phase reactant C-reactive protein (CRP) are elevated in depressed patients.f Studies also show that blood levels of IL-6, TNF-α, and CRP are elevated in obese patients.g
Fat-derived cytokines alter metabolic pathways related to mood and inflammation: Among the many possible pathways linking cytokine actions and depression, the effects of TNF-α on serotonin metabolism have been studied extensively.h,i TNF-α activates brain indoleamine 2,3-dioxygenase, leading to rapid depletion of serotonin and exacerbation of depressive symptoms.j
Regarding physical problems, evidence suggests adipose-tissue-derived pro-inflammatory agents are involved in development of metabolic syndrome, a condition characterized by insulin resistance, glucose intolerance, atherogenic dyslipidemia, visceral adiposity, hypercoagulation, chronic inflammation, oxidative stress, and hypertension.k These conditions are strong risk factors for type II diabetes, coronary artery disease, hypertension, and stroke.
Anti-inflammatory agents for depression
Data suggest a model in which weight gain leads to an increase in pro-inflammatory cytokines. When released into the circulation, these cytokines produce a variety of deleterious effects, including blockade of serotonin synthesis in the brain that leads to depressive symptoms. Evidence suggests that anti-inflammatory agents might disrupt this process.
Celecoxib. The anti-inflammatory agent celecoxib acts by inhibiting cyclooxygenase-2, the rate-limiting enzyme in the synthesis of prostaglandin, a powerful inflammation mediator. Three double-blind, placebo-controlled trials have compared groups of:
- depressed patients receiving reboxetine with and without celecoxibl or fluoxetine, 40 mg/d, with and without celecoxibm
- bipolar disorder patients taking mood stabilizers or atypical antipsychotics with and without celecoxib, 400 mg/d.n
These studies suggest that celecoxib may accelerate improvement in depressive symptoms. Celecoxib’s potential for increased cardiovascular risk may limit its use.
Aspirin. Mendlewicz et alo conducted an open-label study in which 24 depressed patients who failed to respond to 4 weeks of antidepressant treatment received adjunctive acetylsalicylic acid, 160 mg/d, for another 4 weeks. They found that 52% of patients responded when aspirin was added to their regimen, and the improvement was seen during the first week of treatment.
References
- Shelton RC, Miller AH. Inflammation in depression: is adiposity a cause? Dialogues Clin Neurosci. 2011;13(1):41-53.
- Shelton RC, Miller AH. Eating ourselves to death (and despair): the contribution of adiposity and inflammation to depression. Prog Neurobiol. 2010;91(4):275-299.
- Soczynska JK, Kennedy SH, Woldeyohannes HO, et al. Mood disorders and obesity: understanding inflammation as a pathophysiological nexus. Neuromolecular Med. 2011;13(2):93-116.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111-2117.
- Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214(Pt 2):337-346.
- Maes M, Kubera M, Obuchowiczwa E, et al. Depression’s multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Neuro Endocrinol Lett. 2011;32(1):7-24.
- Khaodhiar L, Ling PR, Blackburn GL, et al. Serum levels of interleukin-6 and C-reactive protein correlate with body mass index across the broad range of obesity. JPEN J Parenter Enteral Nutr. 2004;28(6):410-415.
- Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130(2):226-238.
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
- O’Connor JC, André C, Wang Y, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. 2009;29(13):4200-4209.
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
- Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
- Akhondzadeh S, Jafari S, Raisi F, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety. 2009;26(7):607-611.
- Nery FG, Monkul ES, Hatch JP, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol. 2008;23(2):87-94.
- Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
Cognitive/behavioral approaches
Although large, well-designed studies of OW/OB-D patients are in the planning or pilot phases,26-28 a substantial database supports incorporating behavioral or cognitive-behavioral therapies when treating these patients.29 Patients in programs that combine behavioral approaches with diet and exercise achieve the greatest weight loss, and frequently show improved depression scores.30-32
In a randomized trial, 203 obese women with moderate to severe depression showed significant weight loss and decreased depression scores whether they were in a behavioral weight-loss program or one that combined behavioral weigh loss with cognitive-behavioral depression management.33 This study raises important questions: Did the behavioral weight-loss program effectively treat depression? Did patients’ depressive symptoms improve because of their improved sense of well-being as they lost weight? Did a putative reduction in cytokine production by fat cells improve their mood?
Treatment implications
Mrs. G has TRD, a BMI that borders on obesity, sleep problems, and lab values that suggest she may have metabolic syndrome. To best manage patients such as Mrs. G, consider the following steps:
- Select an antidepressant that is unlikely to cause further weight gain, such as bupropion, duloxetine, or fluoxetine.
- If necessary, add an augmenting agent that is not associated with weight gain, such as bupropion, aripiprazole, or lamotrigine.
- Verify that your patient is getting adequate sleep. Begin by reviewing the principles of sleep hygiene and, if necessary, prescribe a sedative or hypnotic medication.
- Although controlled clinical trials are lacking, consider including an anti-inflammatory agent such as aspirin to the pharmacologic armamentarium.
- Institute an exercise and diet program at the beginning of treatment. Exercise can begin with 20 to 30 minutes a day of walking. Tell patients that exercising in groups is a good way to address nonadherence and social isolation and reinforce positive lifestyle changes. Recommend that patients combine aerobic exercise to burn calories with resistance training to build muscle. Suggest that patients try to make exercising fun using video games or interactive computer-based programs.
- Encourage your patient to keep a journal to record his or her weight, amount and type of exercise, medication taken, and dietary intake. Review this information at every session to reinforce the importance of this integrated exercise and diet program.
Related Resources
- Markowitz S, Friedman MA, Arent SM. Understanding the relation between obesity and depression: causal mechanisms and implications for treatment. Clinical Psychology: Science and Practice. 2008:15(1):1-20.
- Burke LE, Wang J, Sevick MA. Self-monitoring in weight loss: a systematic review of the literature. J Am Diet Assoc. 2011;111(1):92-102.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Zyban
- Celecoxib • Celebrex
- Citalopram • Celexa
- Clozapine • Clozaril
- Divalproex • Depakote
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Selegiline • Emsam
- Sertraline • Zoloft
- Trazodone • Desyrel, Oleptro
- Ziprasidone • Geodon
Disclosure
Dr. Crayton reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Luppino FS, de Wit LM, Bouvy PF, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220-229.
2. Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007;357(4):370-379.
3. Valente TW, Fujimoto K, Chou CP, et al. Adolescent affiliations and adiposity: a social network analysis of friendships and obesity. J Adolesc Health. 2009;45(2):202-204.
4. Blumenthal JA, Babyak MA, Doraiswamy PM, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med. 2007;69(7):587-596.
5. Babyak M, Blumenthal JA, Herman S, et al. Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med. 2000;62(5):633-638.
6. Helmich I, Latini A, Sigwalt A, et al. Neurobiological alterations induced by exercise and their impact on depressive disorders [corrected]. Clin Pract Epidemiol Ment Health. 2010;6:115-125.
7. Mead GE, Morley W, Campbell P, et al. Exercise for depression. Cochrane Database Syst Rev. 2009;(3):CD004366.
8. Tse J, Chow E, Sultana-Cordero R, et al. Motivation-based interventions for obesity in serious mental illness. Psychiatric Ann. 2011;41(10):473-477.
9. Rosenberg D, Depp CA, Vahia IV, et al. Exergames for subsyndromal depression in older adults: a pilot study of a novel intervention. Am J Geriatr Psychiatry. 2010;18(3):221-226.
10. Knutson KL, Van Cauter E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci. 2008;1129:287-304.
11. Nedeltcheva AV, Kilkus JM, Imperial J, et al. Sleep curtailment is accompanied by increased intake of calories from snacks. Am J Clin Nutr. 2009;89(1):126-133.
12. Isomaa B. A major health hazard: the metabolic syndrome. Life Sci. 2003;73(19):2395-2411.
13. Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89(3):973S-979S.
14. Lu XY. The leptin hypothesis of depression: a potential link between mood disorders and obesity? Curr Opin Pharmacol. 2007;7(6):648-652.
15. Lawson EA, Miller KK, Blum JI, et al. Leptin levels are associated with decreased depressive symptoms in women across the weight spectrum, independent of body fat. Clin Endocrinol (Oxf). 2012;76(4):520-525.
16. Häfner S, Baumert J, Emeny RT, et al. Sleep disturbances and depressed mood: a harmful combination associated with increased leptin levels in women with normal weight. Biol Psychol. 2012;89(1):163-169.
17. Spiegel K, Leproult R, L’hermite-Balériaux M, et al. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89(11):5762-5771.
18. Grenard JL, Munjas BA, Adams JL, et al. Depression and medication adherence in the treatment of chronic diseases in the United States: a meta-analysis. J Gen Intern Med. 2011;26(10):1175-1182.
19. Gonzalez JS, Safren SA, Delahanty LM, et al. Symptoms of depression prospectively predict poorer self-care in patients with Type 2 diabetes. Diabet Med. 2008;25(9):1102-1107.
20. Somerset SM, Graham L, Markwell K. Depression scores predict adherence in a dietary weight loss intervention trial. Clin Nutr. 2011;30(5):593-598.
21. Patten SB, Williams JV, Lavorato DH, et al. Major depression, antidepressant medication and the risk of obesity. Psychother Psychosom. 2009;78(3):182-186.
22. Nihalani N, Schwartz TL, Siddiqui UA, et al. Weight gain, obesity, and psychotropic prescribing. J Obes. 2011;2011:893629.
23. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
24. Gabriel A. Lamotrigine adjunctive treatment in resistant unipolar depression: an open, descriptive study. Depress Anxiety. 2006;23(8):485-488.
25. Degenhardt EK, Jamal HH, Tormey S, et al. Early weight gain as a predictor of substantial weight gain with olanzapine/fluoxetine combination: an analysis of 2 adult studies in treatment-resistant depression. J Clin Psychopharmacol. 2011;31(3):337-340.
26. Faulconbridge LF, Wadden TA, Berkowitz RI, et al. Treatment of comorbid obesity and major depressive disorder: a prospective pilot study for their combined treatment. J Obes. 2011;2011:870385.
27. Schneider KL, Bodenlos JS, Ma Y, et al. Design and methods for a randomized clinical trial treating comorbid obesity and major depressive disorder. BMC Psychiatry. 2008;8:77.
28. Pagoto S, Bodenlos JS, Schneider KL, et al. Initial investigation of behavioral activation therapy for co-morbid major depressive disorder and obesity. Psychotherapy (Chic). 2008;45(3):410-415.
29. Shaw K, O’Rourke P, Del Mar C, et al. Psychological interventions for overweight or obesity. Cochrane Database Syst Rev. 2005;(2):CD003818.
30. Thieszen CL, Merrill RM, Aldana SG, et al. The Coronary Health Improvement Project (CHIP) for lowering weight and improving psychosocial health. Psychol Rep. 2011;109(1):338-352.
31. Fabricatore AN, Wadden TA, Higginbotham AJ, et al. Intentional weight loss and changes in symptoms of depression: a systematic review and meta-analysis. Int J Obes (Lond). 2011;35(11):1363-1376.
32. Simon GE, Rohde P, Ludman EJ, et al. Association between change in depression and change in weight among women enrolled in weight loss treatment. Gen Hosp Psychiatry. 2010;32(6):583-589.
33. Linde JA, Simon GE, Ludman EJ, et al. A randomized controlled trial of behavioral weight loss treatment versus combined weight loss/depression treatment among women with comorbid obesity and depression. Ann Behav Med. 2011;41(1):119-130.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. G is a 52-year-old mother and teacher with a 20-year history of recurrent depressive episodes for which she has been treated with various antidepressants, including sertraline, fluoxetine, and citalopram. For some of her depressive recurrences, she also received adjunctive second-generation antipsychotics (SGAs), including quetiapine and olanzapine.
She describes feelings of “being defeated,” hopelessness, and boredom and frustration with her teaching. It takes her approximately 30 minutes to go to sleep each night, but she wakes up after 2 to 3 hours, and the remainder of her night’s sleep is markedly disrupted. Because of her hopeless feelings, she has given up on dieting and going to the gym. When feeling down she has donuts and coffee. She has gained 45 lbs over the past 10 years and now weighs 175 lbs. In addition to her disrupted mood, she complains of frequent headaches and sore muscles.
Mrs. G’s psychiatrist refers her to her primary care physician for evaluation of her physical complaints and recommendations regarding her weight gain. Her waistline measures 90 cm and her body mass index (BMI) is 29.1 kg/m2; a BMI of ≥30 is considered obese. Her blood pressure is 145/85 mmHg. Laboratory work reveals a total cholesterol level of 235 mg/dL, low-density lipoprotein of 146 mg/dL, and fasting blood sugar, 135 mg/dL.
Mrs. G’s case illustrates many of the issues psychiatrists face when caring for overweight or obese patients with depression (OW/OB-D). Both conditions can be challenging to manage, and may be especially difficult to treat when they co-occur. When depression and obesity co-occur, their capacity to inflict psychological and physical harm likely is greater than either condition alone. Data point to a “2-way street” of mutually destructive effects of being overweight/obese on depression and vice versa.1
This article summarizes ways that depression and obesity aggravate each other, and highlights research that suggests depression and obesity are manifestations of inflammatory processes. It also suggests a stepwise approach to treating OW/OB-D patients.
Mutually destructive processes
Self-esteem and body image. Lowered self-esteem is a hallmark of depression. In popular culture, “you can’t be too rich or too thin,” and the pressure to be slim is great. Therefore, OW/OB-D patients have 2 reasons to feel a depleted sense of self-worth: their psychiatric illness and their weight. Observant clinicians will recognize these dual sources of self-deprecation and tailor treatment to address both.
Increasing numbers of celebrities, performers, and prominent politicians are overweight or obese. Increased social acceptance of OW/OB individuals in our culture may be legitimizing weight gain and obesity. When OW/OB-D patients justify their weight by pointing to overweight celebrities, clinicians can counter this argument with data on the hazards of obesity on health and well-being, such as premature death, coronary artery disease, diabetes, arthritis, and some forms of cancer.
OW/OB patients tend to interact with other OW/OB individuals. Christakis et al2 reported that adults with obese friends were more likely to become obese than individuals without obese friends. Valente et al3 found that overweight teens were twice as likely to have overweight friends as non-overweight teens. This power of social connectedness can be harnessed when treating OW/OB-D patients, where therapeutic groups can help patients address both depression and weight gain.
Inactivity. OW/OB-D patients with psychomotor retardation or reduced activity may gain weight because they consume more calories than their body requires. Depressed patients may say they “have no energy” to participate in a clinician-recommended exercise program or that “it won’t do any good anyway.”
These tendencies are best dealt with by incorporating an exercise program into the comprehensive plan for OW/OB-D patients from the start of treatment. Several studies suggest that in addition to helping manage weight, exercise may have antidepressant effects. In a large, well-controlled trial of patients with major depressive disorder (MDD), Blumenthal et al4 found that an exercise program was as effective as fluoxetine, 20 mg/d, and the antidepressant effects persisted at 10-month follow-up for patients who continued to exercise.5 In a review of studies of exercise in depressed patients, Helmich et al6 concluded that in most studies exercise was beneficial. However, Mead et al7 found that nearly all trials of exercise and depression had substantial design flaws. Based on the 3 well-designed studies they reviewed, Mead et al concluded that the efficacy of exercise was comparable to that of cognitive therapy.
Although the evidence on exercise for treating depression is inconclusive, an exercise program is essential for OW/OB-D patients because it can help manage weight and improve cardiovascular fitness. Motivation is a key ingredient of successful programs.8 Encourage patients to make exercise enjoyable, perhaps by using video games or other interactive computer-based programs.9
Sleep disturbances. Disrupted sleep— another hallmark of depression—appears to be a risk factor for weight gain.10 Although the basis for this relationship is still under investigation, one possibility is that some patients with insomnia get up to eat more often than those without sleep disturbances. Research has shown that when sleep is curtailed in a sleep laboratory, patients consume approximately 20% more calories from snacks (1,086 calories) than non-sleep-deprived patients (866 calories).11 Although this 220-calorie increase may seem small, it would amount to approximately 2 lbs of additional weight per month.
Appetite. Although weight loss is a cardinal sign of MDD, increased appetite and weight gain can be seen in many depressed patients who do not meet diagnostic criteria for MDD as well as those with seasonal affective disorder and metabolic syndrome, a condition characterized by insulin resistance, glucose intolerance, atherogenic dyslipidemia, visceral adiposity, hypercoagulation, chronic inflammation, oxidative stress, and hypertension.12
Emerging information about the neuroendocrinology of appetite regulation may lead to a better understanding of weight management in OW/OB-D patients. Leptin, a hormone released by adipose tissue, increases when fat stores are high, leading to reduced appetite and fat stores. Conversely, when fat stores are low, plasma leptin levels decrease, producing increased appetite and reduced energy expenditure.13
Researchers have suggested that leptin insufficiency and/or leptin resistance may contribute to vulnerability to depression, and leptin may have antidepressant effects.14 Lawson et al15 found that leptin levels were inversely associated with Hamilton Depression Rating Scale scores in normal-weight (BMI ≤25) women.
Leptin levels also are significantly associated with comorbid depressed mood and sleep disturbance.16 In healthy volunteers, shortening sleep duration to 4 hours produced an approximately 20% reduction in leptin release compared with normal sleep duration.17 Because of the relationship between sleep disorders and depression, leptin may act on sleep regulatory mechanisms, depressogenic pathways, or both. But studies of leptin’s role in obesity, depression, and sleep have not yet found a single role for leptin that ties all 3 conditions to this hormone’s known physiological functions.
Nonadherence. Compared with non-depressed patients, depressed patients are 76% more likely to not adhere to treatment.18 Patients may report that they are not interested in the treatment program or lack hope that it will be successful. Furthermore, OW/OB-D patients may consider exercise programs to be too strenuous and diet programs too depriving.19
OW/OB-D patients may require special care in monitoring adherence. The presence of depression in patients enrolled in weight loss programs may prompt the treatment staff to modify the usual protocol by including the patient in an active depression treatment module.20
Effects of pharmacologic agents
Many antidepressant agents are associated with weight gain.21Tables 1 and 2 summarize the effects antidepressants and adjunctive medications used to treat depression have on weight.22,23 SGAs such as clozapine and olanzapine, which frequently are used as augmenting agents in patients with treatment-resistant depression (TRD), are associated with weight gain.22 Lamotrigine also is an effective adjunctive medication for TRD and is not associated with significant weight gain.24
Bupropion has antidepressant and weight-loss effects and may be a suitable primary medication for OW/OB-D patients.
Early weight gain with olanzapine/fluoxetine combination may be a strong indicator of substantial weight gain with longer-term treatment. A weight gain of >2 kg (4.4 lbs) during the first 2 weeks of treatment is a strong predictor of weight gain of ≥10 kg (22 lbs) at 26 weeks.25
Antidepressants may be associated with an increased risk of obesity, and strategies to offset this risk may be useful in clinical practice, particularly patient education on the risks of weight gain and early introduction of a diet and exercise program.
Evidence suggests that depression and obesity are associated with alterations in immune activity (Box). This suggests that anti-inflammatory agents might have a role in treating depression by reducing the release of cytokines that may lead to depressive symptoms.
Table 1
Pharmacotherapy and weight gain: Antidepressants
| Agent | Effect on weight |
|---|---|
| SSRIs | |
| Paroxetine | Moderate gain |
| Fluoxetine | Early: weight loss Long-term: moderate gain |
| SNRIs | |
| Duloxetine | Minimal gain |
| Escitalopram | Moderate gain |
| Other agents | |
| Imipramine (TCA) | Moderate gain |
| Selegiline (MAOI) | Moderate gain |
| Trazodone (tetracyclic) | Moderate gain |
| Bupropion (atypical) | Moderate loss |
| MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCA: tricyclic antidepressant Source: References 22,23 | |
Table 2
Pharmacotherapy and weight gain: Adjunctive agents
| Agent | Use in depression | Effect on weight |
|---|---|---|
| SGAs | ||
| Olanzapine | Psychotic depression | Large gain |
| Clozapine | Adjunct; psychotic depression | Large gain |
| Quetiapine | Primary; adjunct | Large gain |
| Aripiprazole | Adjunct | Small gain |
| Risperidone | Psychotic depression | Small gain |
| Ziprasidone | Psychotic depression | Small loss |
| Mood stabilizers | ||
| Divalproex | Treatment resistance, bipolar disorder | Moderate to large gain |
| Lamotrigine | Treatment resistance | Neutral |
| SGAs: second-generation antipsychotics Source: References 22,23 | ||
Research suggests that both depression and obesity are associated with immune dysregulation and inflammation.a-d Although the complexities of these interactions are beyond the scope of this article, having a model for understanding the role of inflammation in overweight or obese patients with depression (OW/OB-D) may be useful. Data supporting a role for immune dysregulation in OW/OB-D patients rests on the following findings:
Fat and muscle are endocrine organs: Fat is not just a storage organ for energy-rich lipids but also a rich source of cytokines, including monocyte chemotactic protein-1 (MCP-1), interleukin-2, and tumor necrosis factor-α (TNF-α). The increase in MCP-1 in fat tissue triggers a cascade of events that leads to chronic inflammation in adipose tissue. These substances can be released into circulation, stimulating inflammatory responses in other tissues. Data suggest that obesity’s effects on cardiovascular disease are mediated by these adipose-derived inflammatory hormones. There is a strong relationship between the volume of adipose tissue and the amount of pro-inflammatory hormones released; therefore, reducing weight reduces inflammatory burden on the body.
Pedersene pointed out that muscle also is an endocrine organ. Among the cytokines (or “myokines”) muscle produces are interleukin-6 (IL-6), interleukin-8, and brain-derived neurotrophic factor. During exercise, the amount of IL-6 released from muscles may increase by 100-fold. Although IL-6 usually is considered a pro-inflammatory regulator, it—or other muscle-derived myokines—may be responsible for some of exercise’s beneficial effects.e
If this hypothesis is correct, patients whose exercise includes resistance training—which increases muscle mass—are not just getting stronger or burning calories but may be facilitating release of hormones that could counteract obesity’s inflammatory effects.
Cytokine levels are elevated in depression and obesity: A substantial body of evidence shows that depressed patients have elevated circulating levels of inflammation markers. In particular, the proinflammatory cytokines IL-6 and interleukin-1β and the acute phase reactant C-reactive protein (CRP) are elevated in depressed patients.f Studies also show that blood levels of IL-6, TNF-α, and CRP are elevated in obese patients.g
Fat-derived cytokines alter metabolic pathways related to mood and inflammation: Among the many possible pathways linking cytokine actions and depression, the effects of TNF-α on serotonin metabolism have been studied extensively.h,i TNF-α activates brain indoleamine 2,3-dioxygenase, leading to rapid depletion of serotonin and exacerbation of depressive symptoms.j
Regarding physical problems, evidence suggests adipose-tissue-derived pro-inflammatory agents are involved in development of metabolic syndrome, a condition characterized by insulin resistance, glucose intolerance, atherogenic dyslipidemia, visceral adiposity, hypercoagulation, chronic inflammation, oxidative stress, and hypertension.k These conditions are strong risk factors for type II diabetes, coronary artery disease, hypertension, and stroke.
Anti-inflammatory agents for depression
Data suggest a model in which weight gain leads to an increase in pro-inflammatory cytokines. When released into the circulation, these cytokines produce a variety of deleterious effects, including blockade of serotonin synthesis in the brain that leads to depressive symptoms. Evidence suggests that anti-inflammatory agents might disrupt this process.
Celecoxib. The anti-inflammatory agent celecoxib acts by inhibiting cyclooxygenase-2, the rate-limiting enzyme in the synthesis of prostaglandin, a powerful inflammation mediator. Three double-blind, placebo-controlled trials have compared groups of:
- depressed patients receiving reboxetine with and without celecoxibl or fluoxetine, 40 mg/d, with and without celecoxibm
- bipolar disorder patients taking mood stabilizers or atypical antipsychotics with and without celecoxib, 400 mg/d.n
These studies suggest that celecoxib may accelerate improvement in depressive symptoms. Celecoxib’s potential for increased cardiovascular risk may limit its use.
Aspirin. Mendlewicz et alo conducted an open-label study in which 24 depressed patients who failed to respond to 4 weeks of antidepressant treatment received adjunctive acetylsalicylic acid, 160 mg/d, for another 4 weeks. They found that 52% of patients responded when aspirin was added to their regimen, and the improvement was seen during the first week of treatment.
References
- Shelton RC, Miller AH. Inflammation in depression: is adiposity a cause? Dialogues Clin Neurosci. 2011;13(1):41-53.
- Shelton RC, Miller AH. Eating ourselves to death (and despair): the contribution of adiposity and inflammation to depression. Prog Neurobiol. 2010;91(4):275-299.
- Soczynska JK, Kennedy SH, Woldeyohannes HO, et al. Mood disorders and obesity: understanding inflammation as a pathophysiological nexus. Neuromolecular Med. 2011;13(2):93-116.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111-2117.
- Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214(Pt 2):337-346.
- Maes M, Kubera M, Obuchowiczwa E, et al. Depression’s multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Neuro Endocrinol Lett. 2011;32(1):7-24.
- Khaodhiar L, Ling PR, Blackburn GL, et al. Serum levels of interleukin-6 and C-reactive protein correlate with body mass index across the broad range of obesity. JPEN J Parenter Enteral Nutr. 2004;28(6):410-415.
- Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130(2):226-238.
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
- O’Connor JC, André C, Wang Y, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. 2009;29(13):4200-4209.
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
- Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
- Akhondzadeh S, Jafari S, Raisi F, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety. 2009;26(7):607-611.
- Nery FG, Monkul ES, Hatch JP, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol. 2008;23(2):87-94.
- Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
Cognitive/behavioral approaches
Although large, well-designed studies of OW/OB-D patients are in the planning or pilot phases,26-28 a substantial database supports incorporating behavioral or cognitive-behavioral therapies when treating these patients.29 Patients in programs that combine behavioral approaches with diet and exercise achieve the greatest weight loss, and frequently show improved depression scores.30-32
In a randomized trial, 203 obese women with moderate to severe depression showed significant weight loss and decreased depression scores whether they were in a behavioral weight-loss program or one that combined behavioral weigh loss with cognitive-behavioral depression management.33 This study raises important questions: Did the behavioral weight-loss program effectively treat depression? Did patients’ depressive symptoms improve because of their improved sense of well-being as they lost weight? Did a putative reduction in cytokine production by fat cells improve their mood?
Treatment implications
Mrs. G has TRD, a BMI that borders on obesity, sleep problems, and lab values that suggest she may have metabolic syndrome. To best manage patients such as Mrs. G, consider the following steps:
- Select an antidepressant that is unlikely to cause further weight gain, such as bupropion, duloxetine, or fluoxetine.
- If necessary, add an augmenting agent that is not associated with weight gain, such as bupropion, aripiprazole, or lamotrigine.
- Verify that your patient is getting adequate sleep. Begin by reviewing the principles of sleep hygiene and, if necessary, prescribe a sedative or hypnotic medication.
- Although controlled clinical trials are lacking, consider including an anti-inflammatory agent such as aspirin to the pharmacologic armamentarium.
- Institute an exercise and diet program at the beginning of treatment. Exercise can begin with 20 to 30 minutes a day of walking. Tell patients that exercising in groups is a good way to address nonadherence and social isolation and reinforce positive lifestyle changes. Recommend that patients combine aerobic exercise to burn calories with resistance training to build muscle. Suggest that patients try to make exercising fun using video games or interactive computer-based programs.
- Encourage your patient to keep a journal to record his or her weight, amount and type of exercise, medication taken, and dietary intake. Review this information at every session to reinforce the importance of this integrated exercise and diet program.
Related Resources
- Markowitz S, Friedman MA, Arent SM. Understanding the relation between obesity and depression: causal mechanisms and implications for treatment. Clinical Psychology: Science and Practice. 2008:15(1):1-20.
- Burke LE, Wang J, Sevick MA. Self-monitoring in weight loss: a systematic review of the literature. J Am Diet Assoc. 2011;111(1):92-102.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Zyban
- Celecoxib • Celebrex
- Citalopram • Celexa
- Clozapine • Clozaril
- Divalproex • Depakote
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Selegiline • Emsam
- Sertraline • Zoloft
- Trazodone • Desyrel, Oleptro
- Ziprasidone • Geodon
Disclosure
Dr. Crayton reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Mrs. G is a 52-year-old mother and teacher with a 20-year history of recurrent depressive episodes for which she has been treated with various antidepressants, including sertraline, fluoxetine, and citalopram. For some of her depressive recurrences, she also received adjunctive second-generation antipsychotics (SGAs), including quetiapine and olanzapine.
She describes feelings of “being defeated,” hopelessness, and boredom and frustration with her teaching. It takes her approximately 30 minutes to go to sleep each night, but she wakes up after 2 to 3 hours, and the remainder of her night’s sleep is markedly disrupted. Because of her hopeless feelings, she has given up on dieting and going to the gym. When feeling down she has donuts and coffee. She has gained 45 lbs over the past 10 years and now weighs 175 lbs. In addition to her disrupted mood, she complains of frequent headaches and sore muscles.
Mrs. G’s psychiatrist refers her to her primary care physician for evaluation of her physical complaints and recommendations regarding her weight gain. Her waistline measures 90 cm and her body mass index (BMI) is 29.1 kg/m2; a BMI of ≥30 is considered obese. Her blood pressure is 145/85 mmHg. Laboratory work reveals a total cholesterol level of 235 mg/dL, low-density lipoprotein of 146 mg/dL, and fasting blood sugar, 135 mg/dL.
Mrs. G’s case illustrates many of the issues psychiatrists face when caring for overweight or obese patients with depression (OW/OB-D). Both conditions can be challenging to manage, and may be especially difficult to treat when they co-occur. When depression and obesity co-occur, their capacity to inflict psychological and physical harm likely is greater than either condition alone. Data point to a “2-way street” of mutually destructive effects of being overweight/obese on depression and vice versa.1
This article summarizes ways that depression and obesity aggravate each other, and highlights research that suggests depression and obesity are manifestations of inflammatory processes. It also suggests a stepwise approach to treating OW/OB-D patients.
Mutually destructive processes
Self-esteem and body image. Lowered self-esteem is a hallmark of depression. In popular culture, “you can’t be too rich or too thin,” and the pressure to be slim is great. Therefore, OW/OB-D patients have 2 reasons to feel a depleted sense of self-worth: their psychiatric illness and their weight. Observant clinicians will recognize these dual sources of self-deprecation and tailor treatment to address both.
Increasing numbers of celebrities, performers, and prominent politicians are overweight or obese. Increased social acceptance of OW/OB individuals in our culture may be legitimizing weight gain and obesity. When OW/OB-D patients justify their weight by pointing to overweight celebrities, clinicians can counter this argument with data on the hazards of obesity on health and well-being, such as premature death, coronary artery disease, diabetes, arthritis, and some forms of cancer.
OW/OB patients tend to interact with other OW/OB individuals. Christakis et al2 reported that adults with obese friends were more likely to become obese than individuals without obese friends. Valente et al3 found that overweight teens were twice as likely to have overweight friends as non-overweight teens. This power of social connectedness can be harnessed when treating OW/OB-D patients, where therapeutic groups can help patients address both depression and weight gain.
Inactivity. OW/OB-D patients with psychomotor retardation or reduced activity may gain weight because they consume more calories than their body requires. Depressed patients may say they “have no energy” to participate in a clinician-recommended exercise program or that “it won’t do any good anyway.”
These tendencies are best dealt with by incorporating an exercise program into the comprehensive plan for OW/OB-D patients from the start of treatment. Several studies suggest that in addition to helping manage weight, exercise may have antidepressant effects. In a large, well-controlled trial of patients with major depressive disorder (MDD), Blumenthal et al4 found that an exercise program was as effective as fluoxetine, 20 mg/d, and the antidepressant effects persisted at 10-month follow-up for patients who continued to exercise.5 In a review of studies of exercise in depressed patients, Helmich et al6 concluded that in most studies exercise was beneficial. However, Mead et al7 found that nearly all trials of exercise and depression had substantial design flaws. Based on the 3 well-designed studies they reviewed, Mead et al concluded that the efficacy of exercise was comparable to that of cognitive therapy.
Although the evidence on exercise for treating depression is inconclusive, an exercise program is essential for OW/OB-D patients because it can help manage weight and improve cardiovascular fitness. Motivation is a key ingredient of successful programs.8 Encourage patients to make exercise enjoyable, perhaps by using video games or other interactive computer-based programs.9
Sleep disturbances. Disrupted sleep— another hallmark of depression—appears to be a risk factor for weight gain.10 Although the basis for this relationship is still under investigation, one possibility is that some patients with insomnia get up to eat more often than those without sleep disturbances. Research has shown that when sleep is curtailed in a sleep laboratory, patients consume approximately 20% more calories from snacks (1,086 calories) than non-sleep-deprived patients (866 calories).11 Although this 220-calorie increase may seem small, it would amount to approximately 2 lbs of additional weight per month.
Appetite. Although weight loss is a cardinal sign of MDD, increased appetite and weight gain can be seen in many depressed patients who do not meet diagnostic criteria for MDD as well as those with seasonal affective disorder and metabolic syndrome, a condition characterized by insulin resistance, glucose intolerance, atherogenic dyslipidemia, visceral adiposity, hypercoagulation, chronic inflammation, oxidative stress, and hypertension.12
Emerging information about the neuroendocrinology of appetite regulation may lead to a better understanding of weight management in OW/OB-D patients. Leptin, a hormone released by adipose tissue, increases when fat stores are high, leading to reduced appetite and fat stores. Conversely, when fat stores are low, plasma leptin levels decrease, producing increased appetite and reduced energy expenditure.13
Researchers have suggested that leptin insufficiency and/or leptin resistance may contribute to vulnerability to depression, and leptin may have antidepressant effects.14 Lawson et al15 found that leptin levels were inversely associated with Hamilton Depression Rating Scale scores in normal-weight (BMI ≤25) women.
Leptin levels also are significantly associated with comorbid depressed mood and sleep disturbance.16 In healthy volunteers, shortening sleep duration to 4 hours produced an approximately 20% reduction in leptin release compared with normal sleep duration.17 Because of the relationship between sleep disorders and depression, leptin may act on sleep regulatory mechanisms, depressogenic pathways, or both. But studies of leptin’s role in obesity, depression, and sleep have not yet found a single role for leptin that ties all 3 conditions to this hormone’s known physiological functions.
Nonadherence. Compared with non-depressed patients, depressed patients are 76% more likely to not adhere to treatment.18 Patients may report that they are not interested in the treatment program or lack hope that it will be successful. Furthermore, OW/OB-D patients may consider exercise programs to be too strenuous and diet programs too depriving.19
OW/OB-D patients may require special care in monitoring adherence. The presence of depression in patients enrolled in weight loss programs may prompt the treatment staff to modify the usual protocol by including the patient in an active depression treatment module.20
Effects of pharmacologic agents
Many antidepressant agents are associated with weight gain.21Tables 1 and 2 summarize the effects antidepressants and adjunctive medications used to treat depression have on weight.22,23 SGAs such as clozapine and olanzapine, which frequently are used as augmenting agents in patients with treatment-resistant depression (TRD), are associated with weight gain.22 Lamotrigine also is an effective adjunctive medication for TRD and is not associated with significant weight gain.24
Bupropion has antidepressant and weight-loss effects and may be a suitable primary medication for OW/OB-D patients.
Early weight gain with olanzapine/fluoxetine combination may be a strong indicator of substantial weight gain with longer-term treatment. A weight gain of >2 kg (4.4 lbs) during the first 2 weeks of treatment is a strong predictor of weight gain of ≥10 kg (22 lbs) at 26 weeks.25
Antidepressants may be associated with an increased risk of obesity, and strategies to offset this risk may be useful in clinical practice, particularly patient education on the risks of weight gain and early introduction of a diet and exercise program.
Evidence suggests that depression and obesity are associated with alterations in immune activity (Box). This suggests that anti-inflammatory agents might have a role in treating depression by reducing the release of cytokines that may lead to depressive symptoms.
Table 1
Pharmacotherapy and weight gain: Antidepressants
| Agent | Effect on weight |
|---|---|
| SSRIs | |
| Paroxetine | Moderate gain |
| Fluoxetine | Early: weight loss Long-term: moderate gain |
| SNRIs | |
| Duloxetine | Minimal gain |
| Escitalopram | Moderate gain |
| Other agents | |
| Imipramine (TCA) | Moderate gain |
| Selegiline (MAOI) | Moderate gain |
| Trazodone (tetracyclic) | Moderate gain |
| Bupropion (atypical) | Moderate loss |
| MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCA: tricyclic antidepressant Source: References 22,23 | |
Table 2
Pharmacotherapy and weight gain: Adjunctive agents
| Agent | Use in depression | Effect on weight |
|---|---|---|
| SGAs | ||
| Olanzapine | Psychotic depression | Large gain |
| Clozapine | Adjunct; psychotic depression | Large gain |
| Quetiapine | Primary; adjunct | Large gain |
| Aripiprazole | Adjunct | Small gain |
| Risperidone | Psychotic depression | Small gain |
| Ziprasidone | Psychotic depression | Small loss |
| Mood stabilizers | ||
| Divalproex | Treatment resistance, bipolar disorder | Moderate to large gain |
| Lamotrigine | Treatment resistance | Neutral |
| SGAs: second-generation antipsychotics Source: References 22,23 | ||
Research suggests that both depression and obesity are associated with immune dysregulation and inflammation.a-d Although the complexities of these interactions are beyond the scope of this article, having a model for understanding the role of inflammation in overweight or obese patients with depression (OW/OB-D) may be useful. Data supporting a role for immune dysregulation in OW/OB-D patients rests on the following findings:
Fat and muscle are endocrine organs: Fat is not just a storage organ for energy-rich lipids but also a rich source of cytokines, including monocyte chemotactic protein-1 (MCP-1), interleukin-2, and tumor necrosis factor-α (TNF-α). The increase in MCP-1 in fat tissue triggers a cascade of events that leads to chronic inflammation in adipose tissue. These substances can be released into circulation, stimulating inflammatory responses in other tissues. Data suggest that obesity’s effects on cardiovascular disease are mediated by these adipose-derived inflammatory hormones. There is a strong relationship between the volume of adipose tissue and the amount of pro-inflammatory hormones released; therefore, reducing weight reduces inflammatory burden on the body.
Pedersene pointed out that muscle also is an endocrine organ. Among the cytokines (or “myokines”) muscle produces are interleukin-6 (IL-6), interleukin-8, and brain-derived neurotrophic factor. During exercise, the amount of IL-6 released from muscles may increase by 100-fold. Although IL-6 usually is considered a pro-inflammatory regulator, it—or other muscle-derived myokines—may be responsible for some of exercise’s beneficial effects.e
If this hypothesis is correct, patients whose exercise includes resistance training—which increases muscle mass—are not just getting stronger or burning calories but may be facilitating release of hormones that could counteract obesity’s inflammatory effects.
Cytokine levels are elevated in depression and obesity: A substantial body of evidence shows that depressed patients have elevated circulating levels of inflammation markers. In particular, the proinflammatory cytokines IL-6 and interleukin-1β and the acute phase reactant C-reactive protein (CRP) are elevated in depressed patients.f Studies also show that blood levels of IL-6, TNF-α, and CRP are elevated in obese patients.g
Fat-derived cytokines alter metabolic pathways related to mood and inflammation: Among the many possible pathways linking cytokine actions and depression, the effects of TNF-α on serotonin metabolism have been studied extensively.h,i TNF-α activates brain indoleamine 2,3-dioxygenase, leading to rapid depletion of serotonin and exacerbation of depressive symptoms.j
Regarding physical problems, evidence suggests adipose-tissue-derived pro-inflammatory agents are involved in development of metabolic syndrome, a condition characterized by insulin resistance, glucose intolerance, atherogenic dyslipidemia, visceral adiposity, hypercoagulation, chronic inflammation, oxidative stress, and hypertension.k These conditions are strong risk factors for type II diabetes, coronary artery disease, hypertension, and stroke.
Anti-inflammatory agents for depression
Data suggest a model in which weight gain leads to an increase in pro-inflammatory cytokines. When released into the circulation, these cytokines produce a variety of deleterious effects, including blockade of serotonin synthesis in the brain that leads to depressive symptoms. Evidence suggests that anti-inflammatory agents might disrupt this process.
Celecoxib. The anti-inflammatory agent celecoxib acts by inhibiting cyclooxygenase-2, the rate-limiting enzyme in the synthesis of prostaglandin, a powerful inflammation mediator. Three double-blind, placebo-controlled trials have compared groups of:
- depressed patients receiving reboxetine with and without celecoxibl or fluoxetine, 40 mg/d, with and without celecoxibm
- bipolar disorder patients taking mood stabilizers or atypical antipsychotics with and without celecoxib, 400 mg/d.n
These studies suggest that celecoxib may accelerate improvement in depressive symptoms. Celecoxib’s potential for increased cardiovascular risk may limit its use.
Aspirin. Mendlewicz et alo conducted an open-label study in which 24 depressed patients who failed to respond to 4 weeks of antidepressant treatment received adjunctive acetylsalicylic acid, 160 mg/d, for another 4 weeks. They found that 52% of patients responded when aspirin was added to their regimen, and the improvement was seen during the first week of treatment.
References
- Shelton RC, Miller AH. Inflammation in depression: is adiposity a cause? Dialogues Clin Neurosci. 2011;13(1):41-53.
- Shelton RC, Miller AH. Eating ourselves to death (and despair): the contribution of adiposity and inflammation to depression. Prog Neurobiol. 2010;91(4):275-299.
- Soczynska JK, Kennedy SH, Woldeyohannes HO, et al. Mood disorders and obesity: understanding inflammation as a pathophysiological nexus. Neuromolecular Med. 2011;13(2):93-116.
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121(6):2111-2117.
- Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214(Pt 2):337-346.
- Maes M, Kubera M, Obuchowiczwa E, et al. Depression’s multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Neuro Endocrinol Lett. 2011;32(1):7-24.
- Khaodhiar L, Ling PR, Blackburn GL, et al. Serum levels of interleukin-6 and C-reactive protein correlate with body mass index across the broad range of obesity. JPEN J Parenter Enteral Nutr. 2004;28(6):410-415.
- Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130(2):226-238.
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
- O’Connor JC, André C, Wang Y, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. 2009;29(13):4200-4209.
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
- Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
- Akhondzadeh S, Jafari S, Raisi F, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety. 2009;26(7):607-611.
- Nery FG, Monkul ES, Hatch JP, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol. 2008;23(2):87-94.
- Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
Cognitive/behavioral approaches
Although large, well-designed studies of OW/OB-D patients are in the planning or pilot phases,26-28 a substantial database supports incorporating behavioral or cognitive-behavioral therapies when treating these patients.29 Patients in programs that combine behavioral approaches with diet and exercise achieve the greatest weight loss, and frequently show improved depression scores.30-32
In a randomized trial, 203 obese women with moderate to severe depression showed significant weight loss and decreased depression scores whether they were in a behavioral weight-loss program or one that combined behavioral weigh loss with cognitive-behavioral depression management.33 This study raises important questions: Did the behavioral weight-loss program effectively treat depression? Did patients’ depressive symptoms improve because of their improved sense of well-being as they lost weight? Did a putative reduction in cytokine production by fat cells improve their mood?
Treatment implications
Mrs. G has TRD, a BMI that borders on obesity, sleep problems, and lab values that suggest she may have metabolic syndrome. To best manage patients such as Mrs. G, consider the following steps:
- Select an antidepressant that is unlikely to cause further weight gain, such as bupropion, duloxetine, or fluoxetine.
- If necessary, add an augmenting agent that is not associated with weight gain, such as bupropion, aripiprazole, or lamotrigine.
- Verify that your patient is getting adequate sleep. Begin by reviewing the principles of sleep hygiene and, if necessary, prescribe a sedative or hypnotic medication.
- Although controlled clinical trials are lacking, consider including an anti-inflammatory agent such as aspirin to the pharmacologic armamentarium.
- Institute an exercise and diet program at the beginning of treatment. Exercise can begin with 20 to 30 minutes a day of walking. Tell patients that exercising in groups is a good way to address nonadherence and social isolation and reinforce positive lifestyle changes. Recommend that patients combine aerobic exercise to burn calories with resistance training to build muscle. Suggest that patients try to make exercising fun using video games or interactive computer-based programs.
- Encourage your patient to keep a journal to record his or her weight, amount and type of exercise, medication taken, and dietary intake. Review this information at every session to reinforce the importance of this integrated exercise and diet program.
Related Resources
- Markowitz S, Friedman MA, Arent SM. Understanding the relation between obesity and depression: causal mechanisms and implications for treatment. Clinical Psychology: Science and Practice. 2008:15(1):1-20.
- Burke LE, Wang J, Sevick MA. Self-monitoring in weight loss: a systematic review of the literature. J Am Diet Assoc. 2011;111(1):92-102.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Zyban
- Celecoxib • Celebrex
- Citalopram • Celexa
- Clozapine • Clozaril
- Divalproex • Depakote
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Selegiline • Emsam
- Sertraline • Zoloft
- Trazodone • Desyrel, Oleptro
- Ziprasidone • Geodon
Disclosure
Dr. Crayton reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Luppino FS, de Wit LM, Bouvy PF, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220-229.
2. Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007;357(4):370-379.
3. Valente TW, Fujimoto K, Chou CP, et al. Adolescent affiliations and adiposity: a social network analysis of friendships and obesity. J Adolesc Health. 2009;45(2):202-204.
4. Blumenthal JA, Babyak MA, Doraiswamy PM, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med. 2007;69(7):587-596.
5. Babyak M, Blumenthal JA, Herman S, et al. Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med. 2000;62(5):633-638.
6. Helmich I, Latini A, Sigwalt A, et al. Neurobiological alterations induced by exercise and their impact on depressive disorders [corrected]. Clin Pract Epidemiol Ment Health. 2010;6:115-125.
7. Mead GE, Morley W, Campbell P, et al. Exercise for depression. Cochrane Database Syst Rev. 2009;(3):CD004366.
8. Tse J, Chow E, Sultana-Cordero R, et al. Motivation-based interventions for obesity in serious mental illness. Psychiatric Ann. 2011;41(10):473-477.
9. Rosenberg D, Depp CA, Vahia IV, et al. Exergames for subsyndromal depression in older adults: a pilot study of a novel intervention. Am J Geriatr Psychiatry. 2010;18(3):221-226.
10. Knutson KL, Van Cauter E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci. 2008;1129:287-304.
11. Nedeltcheva AV, Kilkus JM, Imperial J, et al. Sleep curtailment is accompanied by increased intake of calories from snacks. Am J Clin Nutr. 2009;89(1):126-133.
12. Isomaa B. A major health hazard: the metabolic syndrome. Life Sci. 2003;73(19):2395-2411.
13. Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89(3):973S-979S.
14. Lu XY. The leptin hypothesis of depression: a potential link between mood disorders and obesity? Curr Opin Pharmacol. 2007;7(6):648-652.
15. Lawson EA, Miller KK, Blum JI, et al. Leptin levels are associated with decreased depressive symptoms in women across the weight spectrum, independent of body fat. Clin Endocrinol (Oxf). 2012;76(4):520-525.
16. Häfner S, Baumert J, Emeny RT, et al. Sleep disturbances and depressed mood: a harmful combination associated with increased leptin levels in women with normal weight. Biol Psychol. 2012;89(1):163-169.
17. Spiegel K, Leproult R, L’hermite-Balériaux M, et al. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89(11):5762-5771.
18. Grenard JL, Munjas BA, Adams JL, et al. Depression and medication adherence in the treatment of chronic diseases in the United States: a meta-analysis. J Gen Intern Med. 2011;26(10):1175-1182.
19. Gonzalez JS, Safren SA, Delahanty LM, et al. Symptoms of depression prospectively predict poorer self-care in patients with Type 2 diabetes. Diabet Med. 2008;25(9):1102-1107.
20. Somerset SM, Graham L, Markwell K. Depression scores predict adherence in a dietary weight loss intervention trial. Clin Nutr. 2011;30(5):593-598.
21. Patten SB, Williams JV, Lavorato DH, et al. Major depression, antidepressant medication and the risk of obesity. Psychother Psychosom. 2009;78(3):182-186.
22. Nihalani N, Schwartz TL, Siddiqui UA, et al. Weight gain, obesity, and psychotropic prescribing. J Obes. 2011;2011:893629.
23. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
24. Gabriel A. Lamotrigine adjunctive treatment in resistant unipolar depression: an open, descriptive study. Depress Anxiety. 2006;23(8):485-488.
25. Degenhardt EK, Jamal HH, Tormey S, et al. Early weight gain as a predictor of substantial weight gain with olanzapine/fluoxetine combination: an analysis of 2 adult studies in treatment-resistant depression. J Clin Psychopharmacol. 2011;31(3):337-340.
26. Faulconbridge LF, Wadden TA, Berkowitz RI, et al. Treatment of comorbid obesity and major depressive disorder: a prospective pilot study for their combined treatment. J Obes. 2011;2011:870385.
27. Schneider KL, Bodenlos JS, Ma Y, et al. Design and methods for a randomized clinical trial treating comorbid obesity and major depressive disorder. BMC Psychiatry. 2008;8:77.
28. Pagoto S, Bodenlos JS, Schneider KL, et al. Initial investigation of behavioral activation therapy for co-morbid major depressive disorder and obesity. Psychotherapy (Chic). 2008;45(3):410-415.
29. Shaw K, O’Rourke P, Del Mar C, et al. Psychological interventions for overweight or obesity. Cochrane Database Syst Rev. 2005;(2):CD003818.
30. Thieszen CL, Merrill RM, Aldana SG, et al. The Coronary Health Improvement Project (CHIP) for lowering weight and improving psychosocial health. Psychol Rep. 2011;109(1):338-352.
31. Fabricatore AN, Wadden TA, Higginbotham AJ, et al. Intentional weight loss and changes in symptoms of depression: a systematic review and meta-analysis. Int J Obes (Lond). 2011;35(11):1363-1376.
32. Simon GE, Rohde P, Ludman EJ, et al. Association between change in depression and change in weight among women enrolled in weight loss treatment. Gen Hosp Psychiatry. 2010;32(6):583-589.
33. Linde JA, Simon GE, Ludman EJ, et al. A randomized controlled trial of behavioral weight loss treatment versus combined weight loss/depression treatment among women with comorbid obesity and depression. Ann Behav Med. 2011;41(1):119-130.
1. Luppino FS, de Wit LM, Bouvy PF, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67(3):220-229.
2. Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007;357(4):370-379.
3. Valente TW, Fujimoto K, Chou CP, et al. Adolescent affiliations and adiposity: a social network analysis of friendships and obesity. J Adolesc Health. 2009;45(2):202-204.
4. Blumenthal JA, Babyak MA, Doraiswamy PM, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med. 2007;69(7):587-596.
5. Babyak M, Blumenthal JA, Herman S, et al. Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med. 2000;62(5):633-638.
6. Helmich I, Latini A, Sigwalt A, et al. Neurobiological alterations induced by exercise and their impact on depressive disorders [corrected]. Clin Pract Epidemiol Ment Health. 2010;6:115-125.
7. Mead GE, Morley W, Campbell P, et al. Exercise for depression. Cochrane Database Syst Rev. 2009;(3):CD004366.
8. Tse J, Chow E, Sultana-Cordero R, et al. Motivation-based interventions for obesity in serious mental illness. Psychiatric Ann. 2011;41(10):473-477.
9. Rosenberg D, Depp CA, Vahia IV, et al. Exergames for subsyndromal depression in older adults: a pilot study of a novel intervention. Am J Geriatr Psychiatry. 2010;18(3):221-226.
10. Knutson KL, Van Cauter E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci. 2008;1129:287-304.
11. Nedeltcheva AV, Kilkus JM, Imperial J, et al. Sleep curtailment is accompanied by increased intake of calories from snacks. Am J Clin Nutr. 2009;89(1):126-133.
12. Isomaa B. A major health hazard: the metabolic syndrome. Life Sci. 2003;73(19):2395-2411.
13. Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89(3):973S-979S.
14. Lu XY. The leptin hypothesis of depression: a potential link between mood disorders and obesity? Curr Opin Pharmacol. 2007;7(6):648-652.
15. Lawson EA, Miller KK, Blum JI, et al. Leptin levels are associated with decreased depressive symptoms in women across the weight spectrum, independent of body fat. Clin Endocrinol (Oxf). 2012;76(4):520-525.
16. Häfner S, Baumert J, Emeny RT, et al. Sleep disturbances and depressed mood: a harmful combination associated with increased leptin levels in women with normal weight. Biol Psychol. 2012;89(1):163-169.
17. Spiegel K, Leproult R, L’hermite-Balériaux M, et al. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89(11):5762-5771.
18. Grenard JL, Munjas BA, Adams JL, et al. Depression and medication adherence in the treatment of chronic diseases in the United States: a meta-analysis. J Gen Intern Med. 2011;26(10):1175-1182.
19. Gonzalez JS, Safren SA, Delahanty LM, et al. Symptoms of depression prospectively predict poorer self-care in patients with Type 2 diabetes. Diabet Med. 2008;25(9):1102-1107.
20. Somerset SM, Graham L, Markwell K. Depression scores predict adherence in a dietary weight loss intervention trial. Clin Nutr. 2011;30(5):593-598.
21. Patten SB, Williams JV, Lavorato DH, et al. Major depression, antidepressant medication and the risk of obesity. Psychother Psychosom. 2009;78(3):182-186.
22. Nihalani N, Schwartz TL, Siddiqui UA, et al. Weight gain, obesity, and psychotropic prescribing. J Obes. 2011;2011:893629.
23. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
24. Gabriel A. Lamotrigine adjunctive treatment in resistant unipolar depression: an open, descriptive study. Depress Anxiety. 2006;23(8):485-488.
25. Degenhardt EK, Jamal HH, Tormey S, et al. Early weight gain as a predictor of substantial weight gain with olanzapine/fluoxetine combination: an analysis of 2 adult studies in treatment-resistant depression. J Clin Psychopharmacol. 2011;31(3):337-340.
26. Faulconbridge LF, Wadden TA, Berkowitz RI, et al. Treatment of comorbid obesity and major depressive disorder: a prospective pilot study for their combined treatment. J Obes. 2011;2011:870385.
27. Schneider KL, Bodenlos JS, Ma Y, et al. Design and methods for a randomized clinical trial treating comorbid obesity and major depressive disorder. BMC Psychiatry. 2008;8:77.
28. Pagoto S, Bodenlos JS, Schneider KL, et al. Initial investigation of behavioral activation therapy for co-morbid major depressive disorder and obesity. Psychotherapy (Chic). 2008;45(3):410-415.
29. Shaw K, O’Rourke P, Del Mar C, et al. Psychological interventions for overweight or obesity. Cochrane Database Syst Rev. 2005;(2):CD003818.
30. Thieszen CL, Merrill RM, Aldana SG, et al. The Coronary Health Improvement Project (CHIP) for lowering weight and improving psychosocial health. Psychol Rep. 2011;109(1):338-352.
31. Fabricatore AN, Wadden TA, Higginbotham AJ, et al. Intentional weight loss and changes in symptoms of depression: a systematic review and meta-analysis. Int J Obes (Lond). 2011;35(11):1363-1376.
32. Simon GE, Rohde P, Ludman EJ, et al. Association between change in depression and change in weight among women enrolled in weight loss treatment. Gen Hosp Psychiatry. 2010;32(6):583-589.
33. Linde JA, Simon GE, Ludman EJ, et al. A randomized controlled trial of behavioral weight loss treatment versus combined weight loss/depression treatment among women with comorbid obesity and depression. Ann Behav Med. 2011;41(1):119-130.
Suicide rehearsals: A high-risk psychiatric emergency
A suicide rehearsal is a behavioral enactment of a suicide method, usually as part of a suicide plan. A mental suicide rehearsal is a process that evolves over time into a plan. Patients who are intent on attempting suicide usually do not reveal their plans. However, behavioral rehearsals display specific clinical characteristics that speak louder than the guarded patient’s denials, revealing the patient’s suicide plan (Table).
Suicide rehearsals may precede suicide attempts or suicide completions. The percentage of patients who stage suicide rehearsals before attempting or completing suicide is unknown; however, in my experience, suicide rehearsals are relatively common. This article describes suicide rehearsals, and offers 4 cases that illustrate what clinicians can learn from rehearsals to improve their patients’ safety.
Table
Clinical characteristics of suicide rehearsals
| Guarded patient |
| Behavioral enactment of a suicide method |
| Lethal means |
| Presumptive acute, high risk of suicide |
| Severe mental illness |
| Suicide attempt often within hours or days |
| Rehearsal usually covert |
| Rehearsal event or multiple events |
The psychology behind suicide rehearsals
Rehearsing suicidal behavior can lower the barrier to a suicide plan, thereby increasing a patient’s resolve and risk. Joiner1 notes that engaging in behavioral or mental suicide rehearsals increases the risk of suicide. Moreover, rehearsals diminish the prohibition against suicidal behavior and the fear of pain and dying. Examples of rehearsal psychology include:
- overcoming ambivalence about dying
- desensitizing anxiety about performing the suicide act
- testing or “perfecting” the method of a planned suicide
- firming one’s resolve to complete suicide.
Other non-lethal motivations include “a cry for help” and self-injurious behaviors motivated by external gains. Patients who do not intend to attempt suicide may openly rehearse low-risk methods, such as superficial cutting.
Rehearsal characteristics
Suicide rehearsals can be confused with aborted, interrupted, or failed suicide attempts. Suicide rehearsals usually are associated with severe psychiatric illness and high-risk lethal methods of attempting suicide. My experience is that suicide attempts or suicide completions often follow a rehearsal within a few hours or days. However, no short-term suicide risk factors—within hours, days, or weeks—can predict when or if a rehearsed suicide will proceed to a suicide attempt.2
A suicide rehearsal is presumptive evidence that the patient is at acute, high risk for suicide and immediate clinical intervention is necessary. A rehearsal allows the clinician to explore the various methods of suicide that the patient has considered, including prior rehearsals. Knowledge of prior rehearsals can inform the clinician’s management of the current suicide rehearsal.
Suicide rehearsals often are conducted covertly. On inpatient psychiatric units, the rehearsal usually is discovered by staff members or reported by other patients. In outpatient settings, the patient or a significant other may report a rehearsal.
The suicide method displayed in a rehearsal may change. A patient who is rehearsing a hanging may attempt suicide by overdose or a firearm. In a systematic review of prior suicide attempts (N = 1,397), Isometsä et al3 found that 82% of patients used 2 or more different methods in suicide attempts, including the completed suicide. However, in a cohort study of 48,649 individuals admitted to a hospital after an attempted suicide, Runeson et al4 found that patients who attempt suicide often used the same method in completed suicide (ie, >90% by hanging for both men and women). Therefore, when taking measures to restrict the patient’s access to lethal means, safety efforts should not be limited to the method used in the suicide rehearsal. Patients can always substitute methods.
Making overall preparations for suicide—for example, making a will, giving away valuable possessions, or putting financial affairs in order—could be confused with a suicide rehearsal, which displays the lethal method to be used in a suicide attempt, often after preparations are made. Suicide rehearsals tend to occur much closer in time to the suicide attempt than preparations for suicide. Similarly, a patient’s plan to hoard drugs for a suicide attempt is not the same as ingesting a sub-lethal dose of a drug to test his or her resolve to die.
By definition, impulsive suicide attempts are not rehearsed. However, an individual’s suicide rehearsal can impulsively segue into a suicide attempt. In a case control study (N = 153) Simon et al5 found that 24% of patients spent 6 found that 26% of individuals with lifetime suicide ideation transitioned from suicide ideation to an unplanned suicide attempt. In my experience, a suicide rehearsal before a suicide completion is presumptive evidence against an impulsive suicide.
Patients contemplating suicide may visit Web sites with instructions on “how to suicide,” providing “virtual” opportunities to rehearse suicide.7 Patients who are at risk for suicide should be asked if they have searched the Internet for suicide methodology.
What we can learn from rehearsals
Although the following case examples are fictional, they illustrate suicide rehearsals encountered in my clinical and forensic practice.
CASE 1: Looking for a location
Ms. B, a 28-year-old divorced mother of 2, is observed tarrying at the high point of a bridge on successive days. When police arrive and question her, she becomes agitated and distraught. Ms. B admits to “scoping out” the bridge and is taken to a hospital emergency room (ER). In the ER, Ms. B discloses, “I was looking for a good spot to jump.” She tells the triage nurse that she is very depressed but, “I couldn’t do it to my children.” Ms. B is placed in an unlocked room while she waits to be assessed by a psychiatrist. She leaves the ER, runs to a nearby parking garage, and jumps from the top level to her death.
Comment: A patient’s denial of suicide intent following a suicide rehearsal cannot be relied upon. Ms. B’s rehearsal revealed a plan with high-risk suicide intent and a lethal suicide method. Systematic suicide assessment that informs immediate clinical intervention is required.
CASE 2: Changing lethal means
Mr. N, a 43-year-old chief executive officer of a large company, is observed by an assistant loading and unloading a revolver at his desk. Alarmed, the assistant calls the company physician. Mr. N refuses psychiatric treatment, saying, “I’ll be all right; this is just a passing thing.” His wife tells the physician that her husband has a history of bipolar disorder but no prior suicide attempts. Guns and ammunition are removed from the home. One week later, Mr. N is found hanging in his garage. A loaded pistol is discovered in the glove compartment of his car.
Comment: There is no certainty that a subsequent suicide attempt will replicate the rehearsed method. A psychological autopsy was conducted, but no explanation was found for why Mr. N chose hanging after having rehearsed suicide with a loaded handgun. His wife thought that her husband, a very tidy person, did not want to leave a mess.
CASE 3: Grieving and depressed
Mr. O, age 67, is depressed after recently losing his wife. He considers a number of suicide methods. Mr. O decides to use a plastic bag to suffocate himself because he believes that this method will allow him to change his mind. Mr. O practices tying the bag tight around his neck. During this rehearsal, he realizes that he does not want to die. Instead, he pursues grief counseling.
Comment: For some patients, the act of rehearsing suicide can help them resolve ambivalent feelings about wanting to die in favor of wanting to live.
CASE 4: Suicide method and the Internet
Ms. S, a 22-year-old college student, is undergoing outpatient treatment for depression. She is accumulating prescription drugs to take as an overdose. Ms. S also searches the Internet for information about other suicide methods. Because she wants a “sure” method of suicide, she persuades an acquaintance to purchase a handgun. In private, Ms. S places the unloaded gun to her head and plays “Russian roulette,” pulling the trigger several times. Her mother discovers the gun and confronts her daughter. Ms. S is hospitalized on a closed psychiatric unit and tells a staff member, “I was practicing suicide with the gun.” Before Ms. S is discharged from the hospital, her parents are advised to watch for suicidal behaviors, especially the recurrence of rehearsals that indicate an acute, high suicide risk. Ms. S’s Internet use is restricted and monitored.
Comment: Suicide rehearsal with a gun reinforces the belief that a firearm death is quick and easy.8 Reaching for a loaded gun takes less time than most other methods of suicide. Patients who rehearse suicide with a gun should be prevented from having access to any firearms, weapons, or other highly lethal means of suicide.
Recognition and intervention
A guarded psychiatric inpatient who is intent on attempting suicide is unmasked when the discovery of a suicidal rehearsal reveals a suicide plan. This creates an opportunity for clinicians to intervene. The patient may attempt to cover up suicidal intent by stating, “I was just playing around” or “I just wanted to get attention.” Recognizing the emergency posed by a suicide rehearsal informs treatment. Safety measures—including 1-to-1 supervision—may be necessary during a period of acute, high suicide risk. The patient’s diagnosis, severity of illness, and treatment require reevaluation.
An outpatient who performs a suicide rehearsal should be considered at acute, high risk for suicide, and immediate psychiatric hospitalization may be necessary. Whether as an inpatient or outpatient, the patient’s suicide intent and plan require careful exploration. The information gained will guide treatment and management decisions. Continuing systematic suicide risk assessment is essential.
Related Resources
- American Association of Suicidology. www.suicidology.org.
- Joiner T. Why people die by suicide. Cambridge, MA: Harvard University Press; 2007.
- American Psychiatric Association. Practice guideline for the assessment and treatment of patients with suicidal behaviors. Washington, DC: American Psychiatric Publishing, Inc. 2003.
- Simon RI. Preventing patient suicide: clinical assessment and management. Arlington, VA: American Psychiatric Publishing, Inc.; 2011.
Disclosure
Dr. Simon reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Joiner TE, Jr. The trajectory of suicidal behavior over time. Suicide Life Threat Behav. 2002;32(1):33-41.
2. Simon RI. Imminent suicide: the illusion of short-term prediction. Suicide Life Threat Behav. 2006;36(3):296-301.
3. Isometsä ET, Lönnqvist JK. Suicide attempts preceding completed suicide. Br J Psychiatry. 1998;173:531-535.
4. Runeson B, Tidemalm D, Dahlin M, et al. Method of attempted suicide as a predictor of subsequent successful suicide: national long-term cohort study. BMJ. 2010;341:c3222. doi:10.1136/bmj.63222.
5. Simon TR, Swann AC, Powell KE, et al. Characteristics of impulsive suicide attempts and attempters. Suicide Life Threat Behav. 2001;32(suppl):49-59.
6. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Study. Arch Gen Psychiatry. 1999;56(7):617-626.
7. Recupero PR. Suicide and the Internet. In: Simon RI Hales RE, eds. The American Psychiatric Publishing textbook of suicide assessment and management. 2nd ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2012:515–538.
8. Simon RI. Gun safety management with patients at risk for suicide. Suicide Life Threat Behav. 2007;37(5):518-526.
A suicide rehearsal is a behavioral enactment of a suicide method, usually as part of a suicide plan. A mental suicide rehearsal is a process that evolves over time into a plan. Patients who are intent on attempting suicide usually do not reveal their plans. However, behavioral rehearsals display specific clinical characteristics that speak louder than the guarded patient’s denials, revealing the patient’s suicide plan (Table).
Suicide rehearsals may precede suicide attempts or suicide completions. The percentage of patients who stage suicide rehearsals before attempting or completing suicide is unknown; however, in my experience, suicide rehearsals are relatively common. This article describes suicide rehearsals, and offers 4 cases that illustrate what clinicians can learn from rehearsals to improve their patients’ safety.
Table
Clinical characteristics of suicide rehearsals
| Guarded patient |
| Behavioral enactment of a suicide method |
| Lethal means |
| Presumptive acute, high risk of suicide |
| Severe mental illness |
| Suicide attempt often within hours or days |
| Rehearsal usually covert |
| Rehearsal event or multiple events |
The psychology behind suicide rehearsals
Rehearsing suicidal behavior can lower the barrier to a suicide plan, thereby increasing a patient’s resolve and risk. Joiner1 notes that engaging in behavioral or mental suicide rehearsals increases the risk of suicide. Moreover, rehearsals diminish the prohibition against suicidal behavior and the fear of pain and dying. Examples of rehearsal psychology include:
- overcoming ambivalence about dying
- desensitizing anxiety about performing the suicide act
- testing or “perfecting” the method of a planned suicide
- firming one’s resolve to complete suicide.
Other non-lethal motivations include “a cry for help” and self-injurious behaviors motivated by external gains. Patients who do not intend to attempt suicide may openly rehearse low-risk methods, such as superficial cutting.
Rehearsal characteristics
Suicide rehearsals can be confused with aborted, interrupted, or failed suicide attempts. Suicide rehearsals usually are associated with severe psychiatric illness and high-risk lethal methods of attempting suicide. My experience is that suicide attempts or suicide completions often follow a rehearsal within a few hours or days. However, no short-term suicide risk factors—within hours, days, or weeks—can predict when or if a rehearsed suicide will proceed to a suicide attempt.2
A suicide rehearsal is presumptive evidence that the patient is at acute, high risk for suicide and immediate clinical intervention is necessary. A rehearsal allows the clinician to explore the various methods of suicide that the patient has considered, including prior rehearsals. Knowledge of prior rehearsals can inform the clinician’s management of the current suicide rehearsal.
Suicide rehearsals often are conducted covertly. On inpatient psychiatric units, the rehearsal usually is discovered by staff members or reported by other patients. In outpatient settings, the patient or a significant other may report a rehearsal.
The suicide method displayed in a rehearsal may change. A patient who is rehearsing a hanging may attempt suicide by overdose or a firearm. In a systematic review of prior suicide attempts (N = 1,397), Isometsä et al3 found that 82% of patients used 2 or more different methods in suicide attempts, including the completed suicide. However, in a cohort study of 48,649 individuals admitted to a hospital after an attempted suicide, Runeson et al4 found that patients who attempt suicide often used the same method in completed suicide (ie, >90% by hanging for both men and women). Therefore, when taking measures to restrict the patient’s access to lethal means, safety efforts should not be limited to the method used in the suicide rehearsal. Patients can always substitute methods.
Making overall preparations for suicide—for example, making a will, giving away valuable possessions, or putting financial affairs in order—could be confused with a suicide rehearsal, which displays the lethal method to be used in a suicide attempt, often after preparations are made. Suicide rehearsals tend to occur much closer in time to the suicide attempt than preparations for suicide. Similarly, a patient’s plan to hoard drugs for a suicide attempt is not the same as ingesting a sub-lethal dose of a drug to test his or her resolve to die.
By definition, impulsive suicide attempts are not rehearsed. However, an individual’s suicide rehearsal can impulsively segue into a suicide attempt. In a case control study (N = 153) Simon et al5 found that 24% of patients spent 6 found that 26% of individuals with lifetime suicide ideation transitioned from suicide ideation to an unplanned suicide attempt. In my experience, a suicide rehearsal before a suicide completion is presumptive evidence against an impulsive suicide.
Patients contemplating suicide may visit Web sites with instructions on “how to suicide,” providing “virtual” opportunities to rehearse suicide.7 Patients who are at risk for suicide should be asked if they have searched the Internet for suicide methodology.
What we can learn from rehearsals
Although the following case examples are fictional, they illustrate suicide rehearsals encountered in my clinical and forensic practice.
CASE 1: Looking for a location
Ms. B, a 28-year-old divorced mother of 2, is observed tarrying at the high point of a bridge on successive days. When police arrive and question her, she becomes agitated and distraught. Ms. B admits to “scoping out” the bridge and is taken to a hospital emergency room (ER). In the ER, Ms. B discloses, “I was looking for a good spot to jump.” She tells the triage nurse that she is very depressed but, “I couldn’t do it to my children.” Ms. B is placed in an unlocked room while she waits to be assessed by a psychiatrist. She leaves the ER, runs to a nearby parking garage, and jumps from the top level to her death.
Comment: A patient’s denial of suicide intent following a suicide rehearsal cannot be relied upon. Ms. B’s rehearsal revealed a plan with high-risk suicide intent and a lethal suicide method. Systematic suicide assessment that informs immediate clinical intervention is required.
CASE 2: Changing lethal means
Mr. N, a 43-year-old chief executive officer of a large company, is observed by an assistant loading and unloading a revolver at his desk. Alarmed, the assistant calls the company physician. Mr. N refuses psychiatric treatment, saying, “I’ll be all right; this is just a passing thing.” His wife tells the physician that her husband has a history of bipolar disorder but no prior suicide attempts. Guns and ammunition are removed from the home. One week later, Mr. N is found hanging in his garage. A loaded pistol is discovered in the glove compartment of his car.
Comment: There is no certainty that a subsequent suicide attempt will replicate the rehearsed method. A psychological autopsy was conducted, but no explanation was found for why Mr. N chose hanging after having rehearsed suicide with a loaded handgun. His wife thought that her husband, a very tidy person, did not want to leave a mess.
CASE 3: Grieving and depressed
Mr. O, age 67, is depressed after recently losing his wife. He considers a number of suicide methods. Mr. O decides to use a plastic bag to suffocate himself because he believes that this method will allow him to change his mind. Mr. O practices tying the bag tight around his neck. During this rehearsal, he realizes that he does not want to die. Instead, he pursues grief counseling.
Comment: For some patients, the act of rehearsing suicide can help them resolve ambivalent feelings about wanting to die in favor of wanting to live.
CASE 4: Suicide method and the Internet
Ms. S, a 22-year-old college student, is undergoing outpatient treatment for depression. She is accumulating prescription drugs to take as an overdose. Ms. S also searches the Internet for information about other suicide methods. Because she wants a “sure” method of suicide, she persuades an acquaintance to purchase a handgun. In private, Ms. S places the unloaded gun to her head and plays “Russian roulette,” pulling the trigger several times. Her mother discovers the gun and confronts her daughter. Ms. S is hospitalized on a closed psychiatric unit and tells a staff member, “I was practicing suicide with the gun.” Before Ms. S is discharged from the hospital, her parents are advised to watch for suicidal behaviors, especially the recurrence of rehearsals that indicate an acute, high suicide risk. Ms. S’s Internet use is restricted and monitored.
Comment: Suicide rehearsal with a gun reinforces the belief that a firearm death is quick and easy.8 Reaching for a loaded gun takes less time than most other methods of suicide. Patients who rehearse suicide with a gun should be prevented from having access to any firearms, weapons, or other highly lethal means of suicide.
Recognition and intervention
A guarded psychiatric inpatient who is intent on attempting suicide is unmasked when the discovery of a suicidal rehearsal reveals a suicide plan. This creates an opportunity for clinicians to intervene. The patient may attempt to cover up suicidal intent by stating, “I was just playing around” or “I just wanted to get attention.” Recognizing the emergency posed by a suicide rehearsal informs treatment. Safety measures—including 1-to-1 supervision—may be necessary during a period of acute, high suicide risk. The patient’s diagnosis, severity of illness, and treatment require reevaluation.
An outpatient who performs a suicide rehearsal should be considered at acute, high risk for suicide, and immediate psychiatric hospitalization may be necessary. Whether as an inpatient or outpatient, the patient’s suicide intent and plan require careful exploration. The information gained will guide treatment and management decisions. Continuing systematic suicide risk assessment is essential.
Related Resources
- American Association of Suicidology. www.suicidology.org.
- Joiner T. Why people die by suicide. Cambridge, MA: Harvard University Press; 2007.
- American Psychiatric Association. Practice guideline for the assessment and treatment of patients with suicidal behaviors. Washington, DC: American Psychiatric Publishing, Inc. 2003.
- Simon RI. Preventing patient suicide: clinical assessment and management. Arlington, VA: American Psychiatric Publishing, Inc.; 2011.
Disclosure
Dr. Simon reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
A suicide rehearsal is a behavioral enactment of a suicide method, usually as part of a suicide plan. A mental suicide rehearsal is a process that evolves over time into a plan. Patients who are intent on attempting suicide usually do not reveal their plans. However, behavioral rehearsals display specific clinical characteristics that speak louder than the guarded patient’s denials, revealing the patient’s suicide plan (Table).
Suicide rehearsals may precede suicide attempts or suicide completions. The percentage of patients who stage suicide rehearsals before attempting or completing suicide is unknown; however, in my experience, suicide rehearsals are relatively common. This article describes suicide rehearsals, and offers 4 cases that illustrate what clinicians can learn from rehearsals to improve their patients’ safety.
Table
Clinical characteristics of suicide rehearsals
| Guarded patient |
| Behavioral enactment of a suicide method |
| Lethal means |
| Presumptive acute, high risk of suicide |
| Severe mental illness |
| Suicide attempt often within hours or days |
| Rehearsal usually covert |
| Rehearsal event or multiple events |
The psychology behind suicide rehearsals
Rehearsing suicidal behavior can lower the barrier to a suicide plan, thereby increasing a patient’s resolve and risk. Joiner1 notes that engaging in behavioral or mental suicide rehearsals increases the risk of suicide. Moreover, rehearsals diminish the prohibition against suicidal behavior and the fear of pain and dying. Examples of rehearsal psychology include:
- overcoming ambivalence about dying
- desensitizing anxiety about performing the suicide act
- testing or “perfecting” the method of a planned suicide
- firming one’s resolve to complete suicide.
Other non-lethal motivations include “a cry for help” and self-injurious behaviors motivated by external gains. Patients who do not intend to attempt suicide may openly rehearse low-risk methods, such as superficial cutting.
Rehearsal characteristics
Suicide rehearsals can be confused with aborted, interrupted, or failed suicide attempts. Suicide rehearsals usually are associated with severe psychiatric illness and high-risk lethal methods of attempting suicide. My experience is that suicide attempts or suicide completions often follow a rehearsal within a few hours or days. However, no short-term suicide risk factors—within hours, days, or weeks—can predict when or if a rehearsed suicide will proceed to a suicide attempt.2
A suicide rehearsal is presumptive evidence that the patient is at acute, high risk for suicide and immediate clinical intervention is necessary. A rehearsal allows the clinician to explore the various methods of suicide that the patient has considered, including prior rehearsals. Knowledge of prior rehearsals can inform the clinician’s management of the current suicide rehearsal.
Suicide rehearsals often are conducted covertly. On inpatient psychiatric units, the rehearsal usually is discovered by staff members or reported by other patients. In outpatient settings, the patient or a significant other may report a rehearsal.
The suicide method displayed in a rehearsal may change. A patient who is rehearsing a hanging may attempt suicide by overdose or a firearm. In a systematic review of prior suicide attempts (N = 1,397), Isometsä et al3 found that 82% of patients used 2 or more different methods in suicide attempts, including the completed suicide. However, in a cohort study of 48,649 individuals admitted to a hospital after an attempted suicide, Runeson et al4 found that patients who attempt suicide often used the same method in completed suicide (ie, >90% by hanging for both men and women). Therefore, when taking measures to restrict the patient’s access to lethal means, safety efforts should not be limited to the method used in the suicide rehearsal. Patients can always substitute methods.
Making overall preparations for suicide—for example, making a will, giving away valuable possessions, or putting financial affairs in order—could be confused with a suicide rehearsal, which displays the lethal method to be used in a suicide attempt, often after preparations are made. Suicide rehearsals tend to occur much closer in time to the suicide attempt than preparations for suicide. Similarly, a patient’s plan to hoard drugs for a suicide attempt is not the same as ingesting a sub-lethal dose of a drug to test his or her resolve to die.
By definition, impulsive suicide attempts are not rehearsed. However, an individual’s suicide rehearsal can impulsively segue into a suicide attempt. In a case control study (N = 153) Simon et al5 found that 24% of patients spent 6 found that 26% of individuals with lifetime suicide ideation transitioned from suicide ideation to an unplanned suicide attempt. In my experience, a suicide rehearsal before a suicide completion is presumptive evidence against an impulsive suicide.
Patients contemplating suicide may visit Web sites with instructions on “how to suicide,” providing “virtual” opportunities to rehearse suicide.7 Patients who are at risk for suicide should be asked if they have searched the Internet for suicide methodology.
What we can learn from rehearsals
Although the following case examples are fictional, they illustrate suicide rehearsals encountered in my clinical and forensic practice.
CASE 1: Looking for a location
Ms. B, a 28-year-old divorced mother of 2, is observed tarrying at the high point of a bridge on successive days. When police arrive and question her, she becomes agitated and distraught. Ms. B admits to “scoping out” the bridge and is taken to a hospital emergency room (ER). In the ER, Ms. B discloses, “I was looking for a good spot to jump.” She tells the triage nurse that she is very depressed but, “I couldn’t do it to my children.” Ms. B is placed in an unlocked room while she waits to be assessed by a psychiatrist. She leaves the ER, runs to a nearby parking garage, and jumps from the top level to her death.
Comment: A patient’s denial of suicide intent following a suicide rehearsal cannot be relied upon. Ms. B’s rehearsal revealed a plan with high-risk suicide intent and a lethal suicide method. Systematic suicide assessment that informs immediate clinical intervention is required.
CASE 2: Changing lethal means
Mr. N, a 43-year-old chief executive officer of a large company, is observed by an assistant loading and unloading a revolver at his desk. Alarmed, the assistant calls the company physician. Mr. N refuses psychiatric treatment, saying, “I’ll be all right; this is just a passing thing.” His wife tells the physician that her husband has a history of bipolar disorder but no prior suicide attempts. Guns and ammunition are removed from the home. One week later, Mr. N is found hanging in his garage. A loaded pistol is discovered in the glove compartment of his car.
Comment: There is no certainty that a subsequent suicide attempt will replicate the rehearsed method. A psychological autopsy was conducted, but no explanation was found for why Mr. N chose hanging after having rehearsed suicide with a loaded handgun. His wife thought that her husband, a very tidy person, did not want to leave a mess.
CASE 3: Grieving and depressed
Mr. O, age 67, is depressed after recently losing his wife. He considers a number of suicide methods. Mr. O decides to use a plastic bag to suffocate himself because he believes that this method will allow him to change his mind. Mr. O practices tying the bag tight around his neck. During this rehearsal, he realizes that he does not want to die. Instead, he pursues grief counseling.
Comment: For some patients, the act of rehearsing suicide can help them resolve ambivalent feelings about wanting to die in favor of wanting to live.
CASE 4: Suicide method and the Internet
Ms. S, a 22-year-old college student, is undergoing outpatient treatment for depression. She is accumulating prescription drugs to take as an overdose. Ms. S also searches the Internet for information about other suicide methods. Because she wants a “sure” method of suicide, she persuades an acquaintance to purchase a handgun. In private, Ms. S places the unloaded gun to her head and plays “Russian roulette,” pulling the trigger several times. Her mother discovers the gun and confronts her daughter. Ms. S is hospitalized on a closed psychiatric unit and tells a staff member, “I was practicing suicide with the gun.” Before Ms. S is discharged from the hospital, her parents are advised to watch for suicidal behaviors, especially the recurrence of rehearsals that indicate an acute, high suicide risk. Ms. S’s Internet use is restricted and monitored.
Comment: Suicide rehearsal with a gun reinforces the belief that a firearm death is quick and easy.8 Reaching for a loaded gun takes less time than most other methods of suicide. Patients who rehearse suicide with a gun should be prevented from having access to any firearms, weapons, or other highly lethal means of suicide.
Recognition and intervention
A guarded psychiatric inpatient who is intent on attempting suicide is unmasked when the discovery of a suicidal rehearsal reveals a suicide plan. This creates an opportunity for clinicians to intervene. The patient may attempt to cover up suicidal intent by stating, “I was just playing around” or “I just wanted to get attention.” Recognizing the emergency posed by a suicide rehearsal informs treatment. Safety measures—including 1-to-1 supervision—may be necessary during a period of acute, high suicide risk. The patient’s diagnosis, severity of illness, and treatment require reevaluation.
An outpatient who performs a suicide rehearsal should be considered at acute, high risk for suicide, and immediate psychiatric hospitalization may be necessary. Whether as an inpatient or outpatient, the patient’s suicide intent and plan require careful exploration. The information gained will guide treatment and management decisions. Continuing systematic suicide risk assessment is essential.
Related Resources
- American Association of Suicidology. www.suicidology.org.
- Joiner T. Why people die by suicide. Cambridge, MA: Harvard University Press; 2007.
- American Psychiatric Association. Practice guideline for the assessment and treatment of patients with suicidal behaviors. Washington, DC: American Psychiatric Publishing, Inc. 2003.
- Simon RI. Preventing patient suicide: clinical assessment and management. Arlington, VA: American Psychiatric Publishing, Inc.; 2011.
Disclosure
Dr. Simon reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Joiner TE, Jr. The trajectory of suicidal behavior over time. Suicide Life Threat Behav. 2002;32(1):33-41.
2. Simon RI. Imminent suicide: the illusion of short-term prediction. Suicide Life Threat Behav. 2006;36(3):296-301.
3. Isometsä ET, Lönnqvist JK. Suicide attempts preceding completed suicide. Br J Psychiatry. 1998;173:531-535.
4. Runeson B, Tidemalm D, Dahlin M, et al. Method of attempted suicide as a predictor of subsequent successful suicide: national long-term cohort study. BMJ. 2010;341:c3222. doi:10.1136/bmj.63222.
5. Simon TR, Swann AC, Powell KE, et al. Characteristics of impulsive suicide attempts and attempters. Suicide Life Threat Behav. 2001;32(suppl):49-59.
6. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Study. Arch Gen Psychiatry. 1999;56(7):617-626.
7. Recupero PR. Suicide and the Internet. In: Simon RI Hales RE, eds. The American Psychiatric Publishing textbook of suicide assessment and management. 2nd ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2012:515–538.
8. Simon RI. Gun safety management with patients at risk for suicide. Suicide Life Threat Behav. 2007;37(5):518-526.
1. Joiner TE, Jr. The trajectory of suicidal behavior over time. Suicide Life Threat Behav. 2002;32(1):33-41.
2. Simon RI. Imminent suicide: the illusion of short-term prediction. Suicide Life Threat Behav. 2006;36(3):296-301.
3. Isometsä ET, Lönnqvist JK. Suicide attempts preceding completed suicide. Br J Psychiatry. 1998;173:531-535.
4. Runeson B, Tidemalm D, Dahlin M, et al. Method of attempted suicide as a predictor of subsequent successful suicide: national long-term cohort study. BMJ. 2010;341:c3222. doi:10.1136/bmj.63222.
5. Simon TR, Swann AC, Powell KE, et al. Characteristics of impulsive suicide attempts and attempters. Suicide Life Threat Behav. 2001;32(suppl):49-59.
6. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Study. Arch Gen Psychiatry. 1999;56(7):617-626.
7. Recupero PR. Suicide and the Internet. In: Simon RI Hales RE, eds. The American Psychiatric Publishing textbook of suicide assessment and management. 2nd ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2012:515–538.
8. Simon RI. Gun safety management with patients at risk for suicide. Suicide Life Threat Behav. 2007;37(5):518-526.
Playing through the pain: Psychiatric risks among athletes
Discuss this article at www.facebook.com/CurrentPsychiatry

Suck it up. Tough it out. There is no “I” in team. These are a few of the messages athletes receive from coaches, teammates, and fans. There are norms, values, and expectations in every culture, including sports, that affect behavior and emotional expression. When taking a patient’s history, clinicians may ask about participation in sports because it provides health and lifestyle information. However, many clinicians fail to consider the extent to which sport participation can influence a person’s explanatory style, experience of injury, and attitude toward medications. Whether your patient is an elite athlete or someone who participates in sports solely for exercise, the extent to which he or she identifies as an athlete is worth exploring.
Research on athletes has focused on physical aspects of injury, but this may be just a small component of an athlete’s devastation after serious injury. In this article, we discuss athletes’:
- psychiatric risks after injury
- expression of pain
- risks of having an identity driven solely by sports
- distress tolerance.
We also provide tips for making a differential diagnosis and providing treatment. This information is based on our experience treating athletes, supplemented by relevant literature.
Psychiatric risks after injury
Research has explored eating disorders and substance use among athletes, but clinicians generally are less aware of the prevalence of mood and anxiety disorders in this population. Although participating in sports can protect against emotional distress, athletes who sustain an injury are at risk for major depressive disorder, posttraumatic stress disorder (PTSD), or an adjustment disorder.1 Only about 10% of injured athletes have severe, long-term psychological consequences,2 but the prevalence of anger and depression after an injury is well documented.3,4 Researchers have found that injured athletes experience clinically significant depression 6 times as often as non-injured athletes.5 Injured athletes also exhibit significantly greater anxiety and lower self-esteem than non-injured controls immediately after injury and at 2-month follow-up; those with more severe injuries are more likely to become depressed.6 Non-injured athletes seem to experience depression at the same rate as the general population.7
Injury and expression of pain
Psychiatric illnesses often are underreported and undertreated in athletes.8 This may be because athletes feel that admitting they have a psychiatric illness or symptoms could threaten their status with their team. One professional figure skater we treated failed to seek recommended treatment for a psychiatric disorder because she feared she would be asked to leave her skating company. Her symptoms dangerously escalated before she was hospitalized.
Based on our clinical experience, many athletes feel acute pressure to play through psychological and physical pain. Some athletes continue to play with an injury to hold on to a paycheck or scholarship. Some continue to play even though they no longer enjoy the sport to prevent letting down parents or coaches; others know no other way but to “tough it out.” Supporters such as coaches, parents, or teammates may encourage athletes to play with injury, and sometimes provide medication to do so.
Mostly, however, the pressure to continue to play despite injury comes from athletes themselves. The culture of sport may lead athletes to minimize pain, fear, and self doubt.9 Athletes who fuse the culture of sport with their own being may underreport physical and psychiatric symptoms. In a survey of National Collegiate Athletic Association Division I athletes, Nixon9 found that 70% of respondents reported having been injured at least once, and more than one-half felt pressure to play while injured. Feeling pressure to perform with injury was affected by “starter” status, and whites and men scored highest on pressure scales, although women showed a roughly equal probability of playing through injury. Students who received an athletic scholarship experienced more injuries that required surgery. There was no difference in pain expression between players of contact and non-contact sports. Finally, athletes may be less likely to seek pharmacologic treatments because of cultural messages that emphasize ideas such as “the body is a temple.”
Loss of identity
An athlete’s injury should be analyzed for meaning; what may seem insignificant to one person may be quite different for another. When injury makes athletic activity impossible, an athlete may suffer more distress than someone who does not exercise regularly. Understanding the significance of the experience for an athlete is crucial to achieving recovery.10 For example, to a non-athlete a fractured wrist may be an annoyance, but it may be disastrous to a collegiate pitcher who is forced to be inactive when scouts for Major League Baseball teams search for prospects.
To an athlete, injury can mean loss of identity. Whereas most people become competent in many aspects of life, and develop support systems across multiple contexts, an athlete—particularly an extraordinarily talented one—may have focused only on his or her sport. Although athletics can help young people develop confidence, participation also can be a trap. Individuals with strong athletic identities are less likely to explore other career, educational, and lifestyle options.11 In the context of team sports, an athlete may feel less emotionally supported if an injury results in the loss of his or her central role with the team. Helping athletes form an identity that is not based solely on sports is ideal because subsequent injuries could lead to recurrent struggles with loss of identity.
Athletes who achieve higher levels of success have higher levels of depression and higher suicidal ideation after injury.12 An athlete may attempt or complete suicide, particularly those who are injured (Box).13-16
Student athletes. When working with student athletes, it is crucial to understand the lifestyle that promotes forming a single-factor identity. Student athletes may be required to train 2 or 3 times a day, rarely spend their school breaks in tropical locations, often miss social events, and may forgo commencement ceremonies. When an injury suddenly makes these perpetual sacrifices seem to be in vain, the risk of psychiatric illness may increase.
Suicides by several high-profile athletes have called attention to the severity of psychiatric risks among athletes. In June 2002, 20-year-old Nathan Eisert died of a self-inflicted gunshot wound 5 weeks after being released from the Western Kentucky University basketball team for academic reasons; the year before, he had suffered a serious ankle injury.13 Former National Football League (NFL) player Kenny McKinley committed suicide in September 2010, after a knee injury sidelined him.14 In May 2012, former NFL star Junior Seau, who had retired in 2011, fatally shot himself.15
For some athletes, career-ending injuries lead to suicidal behaviors. A study of 5 athletes who attempted suicide after sustaining an injury found 5 common characteristics:
- all were successful in their sport before getting injured
- all sustained an injury severe enough to warrant surgery
- all endured a lengthy rehabilitation
- all were not as successful at their sport when they returned to play
- all were replaced by a teammate.16
Tolerating distress
Athletes often use their sport as an outlet for emotional expression. When an injury removes that outlet, an athlete may develop anxiety and disappointment. Left alone to manage these emotions, an athlete may become irritable, passive, socially isolated, depressed, or suicidal.17 Trying but failing to find socially acceptable ways to express these feelings may intensify depression or anger. Difficult life issues, such as avoided losses, relationship issues, or various insecurities, may come to the surface when an athlete’s primary coping skill is lost. In addition, without the support of the athletic “family” (eg, teammates, coaches, staff) many athletes turn to alcohol or drugs unless they have alternate coping strategies and social supports.18
Overtraining and stress
The differential diagnosis for athletes who present with psychiatric symptoms includes several mood and anxiety disorders and other conditions (Table). When evaluating athletes who have depressive symptoms, it is essential to rule out overtraining syndrome (OTS). A common phenomenon among athletes, OTS is characterized by athletic “staleness” and chronic fatigue.19 Although there are no official OTS diagnostic criteria, characteristic symptoms include decreased physical performance or stamina, fatigue, insomnia, change in appetite, irritability, restlessness, excitability, anxiety, weight loss, loss of motivation, and poor concentration.19 The primary distinction between OTS and depression is that OTS results from athletic endeavors and can be reversed by reducing activity.
Experiencing an injury—or even a near-miss—can be terrifying to a person who derives his or her identity from a fully functioning body and feels that a perfectly working body is essential to an acceptable life. Such athletes may develop acute stress disorder or PTSD.20,21 We treated a hockey player who just missed being involved in a serious incident on the ice. “I watched my whole athletic career up to that point flash before my eyes.… I keep getting flashes of that,” he said. After the incident, he experienced hypervigilance, avoidance, and anxiety—both on and off the ice—and was diagnosed with acute stress disorder. Similarly, we cared for a young running back whose physical symptoms had abated after experiencing a concussion. He developed an irrational fear that he would become injured again. Neither athlete had a history of psychiatric illness or serious injury, and both were paralyzed by the idea of returning to play. One of these athletes successfully engaged in exposure therapy, and the other experienced severe avoidance, hopelessness, depression, nightmares, and flashbacks before seeking treatment.
Table
Differential diagnosis of conditions associated with athletic injury
| Acute stress disorder |
| Adjustment disorder |
| Anxiety disorder NOS |
| Depressive disorder NOS |
| Major depressive disorder |
| Overtraining syndrome |
| Postconcussion syndrome |
| Posttraumatic stress disorder |
| NOS: Not otherwise specified |
Substance use: Common and risky
Anecdotal and clinical evidence suggests that athletes in different sports engage in different substance abuse patterns. Studies show that college athletes use alcohol at higher rates than non-athletes.22,23 In 2000, the American College of Sports Medicine reported that athletes’ abuse of recreational drugs far surpasses their abuse of performance-enhancing drugs.24 Some athletes may use prescription pain medications recreationally or to self-medicate emotional pain as a result of injury. Athletes may not understand the risks of recreational use of prescription medications or illicit substances—such as cocaine’s deleterious cardiovascular effects—and may hesitate to discuss their self-medicating with physicians.
Some athletes abuse performance-enhancing drugs, such as anabolic steroids, androstenedione, stimulants, diuretics, and creatine.25 Side effects of these substances include liver disease, brain hemorrhage, weight loss, and depression.25
Our recommendations
Working with athletes—particularly injured athletes who have internalized sports culture—requires informed clinical effort, whether your patient is a student athlete, elite athlete, leisure athlete, or former athlete. Successful diagnosis and treatment requires understanding the meaning of athletics in your patient’s life and the extent to which he or she has “back-up” stress relievers and support systems, and assessing for cognitive dysfunction that may contribute to mood or anxiety symptoms. During evaluation, take a careful history to distinguish major depression or adjustment disorders from OTS, and assess for PTSD symptoms. When treating an injured athlete, help the patient determine whether he or she can find another outlet—preferably more than one—to replace athletics.
For an athlete who has depressive symptoms, we recommend determining whether the patient’s symptoms remit after a brief period of rest before initiating pharmacotherapy. For patients who exhibit minimal neurovegetative features, we recommend psychotherapy as a first-line treatment. Many athletes are reluctant to take medication and would be more likely to follow through with cognitive-behavioral and biofeedback interventions.
If a patient requires pharmacotherapy, ask about his or her feelings toward medications that may impact adherence. For example, is a gymnast worried about weight gain? Is a sprinter concerned with lethargy? When prescribing, be aware of the prevalence of drug and alcohol problems among athletes, understand how habits and temptations differ among sports cultures, and provide patients with psychoeducation about substance abuse when appropriate.
Related Resources
- International Society for Sports Psychiatry. http://sportspsychiatry.org.
- Sabo D, Miller KE, Melnick MJ, et al. High school athletic participation and adolescent suicide: a nationwide U.S. study. Int Rev Sociol Sport. 2005;40(1):5-23. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2563797. Accessed June 7, 2012.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Brewer BW, Linder DE, Phelps CM. Situational correlates of emotional adjustment to athletic injury. Clin J Sport Med. 1995;5(4):241-245.
2. Brewer BW, Petrie TA. Psychopathology in sport and exercise. In: Van Raalte JL Brewer BW, eds. Exploring sport and exercise psychology. Washington, DC: American Psychological Association; 1996:257–274.
3. May J, Sieb G. Athletic injuries: psychosocial factors in the onset sequelae, rehabilitation and prevention. In: May JR, Asken MJ, eds. Sport psychology: the psychological health of the athlete. New York, NY: PMA Publishing; 1987: 157–185.
4. Quackenbush N, Crossman J. Injured athletes: a study of emotional responses. J Sport Behav. 1994;17:178-187.
5. Perna F, Roh J, Newcomer R, et al. Clinical depression among injured athletes: an empirical assessment. Presented at: Association for the Advancement of Applied Sport Psychology annual convention; September 1998; Hyannis, MA.
6. Leddy MH, Lambert MJ, Ogles BM. Psychological consequences of athletic injury among high-level competitors. Res Q Exerc Sport. 1994;65(4):347-354.
7. Schwenk TL. The stigmatisation and denial of mental illness in athletes. Br J Sports Med. 2000;34(1):4-5.
8. Rotella RJ, Heyman SR. Stress injury, and the psychological rehabilitation of athletes. In: Williams HR, ed. Applied sports psychology: personal growth to peak performance. 2nd ed. Mountain View, CA: Mayfield Publishing; 1993: 338–355.
9. Nixon HL, II. Explaining pain and injury attitudes and experiences in sport in terms of gender race, and sports status factors. Journal of Sport Social Issues. 1996;20(1):33-44.
10. Harris LL. Integrating and analyzing psychosocial and stage theories to challenge the development of the injured collegiate athlete. J Athl Train. 2003;38(1):75-82.
11. Brown C, Hartley DL. Athletic identity and career maturation of male college student athletes. International Journal of Sport Psychology. 1998;29(1):17-26.
12. Baum AL. Suicide in athletes: a review and commentary. Clin Sports Med. 2005;24(4):853-859, ix.
13. Ho J. Suicide on campus. CBS News. http://www.cbsnews.com/2100-500195_162-654130.html. Published February 11 2009. Accessed June 7, 2012.
14. Bunch J, Jones LH. Broncos WR Kenny McKinley found dead in apparent suicide. Denver Post. http://www.denverpost.com/sports/ci_16127852. Published September 20 2010. Accessed June 7, 2012.
15. Saraceno J. Junior Seau’s death came with ‘zero warning.’ USA Today. http://www.usatoday.com/sports/football/nfl/story/2012-05-02/junior-seau-dead-gunshot/54712488/1. Published May 3 2012. Accessed June 7, 2012.
16. Smith AM, Milliner EK. Injured athletes and the risk of suicide. J Athl Train. 1994;29(4):337-341.
17. Putukian M, Wilfert M. National Collegiate Athletic Association. Student-athletes also face dangers from depression. http://fs.ncaa.org/Docs/NCAANewsArchive/2004/Association-Wide/student-athletes+also+face+dangers+from+depression+-+4-12-04.html. Published April 12 2004. Accessed June 6, 2012.
18. Perna FM, Antoni MH, Baum A, et al. Cognitive behavioral stress management effects on injury and illness among competitive athletes: a randomized clinical trial. Ann Behav Med. 2003;25(1):66-73.
19. Armstrong LE, VanHeest JL. The unknown mechanism of the overtraining syndrome: clues from depression and psychoneuroimmunology. Sports Med. 2002;32(3):185-209.
20. Newcomer RR, Perna FM. Features of posttraumatic distress among adolescent athletes. J Athl Train. 2003;38(2):163-166.
21. Newcomer R, Roh J, Perna F, et al. Injury as a traumatic experience: Intrusive thoughts and avoidance behavior associated with injury among college student-athletes. J Appl Sport Psychol. 1998;10(suppl):S54.
22. Hildebrand KM, Johnson DJ, Bogle K. Comparison of patterns of alcohol use between high school and college athletes and non-athletes. College Student Journal. 2001;35:358-365.
23. Wilson GS, Pritchard ME, Schaffer J. Athletic status and drinking behavior in college students: the influence of gender and coping styles. J Am Coll Health. 2004;52(6):269-273.
24. Wadler GI. American College of Sports Medicine. Cocaine abuse in sports. http://www.acsm.org/docs/current-comments/cocainabuse.pdf. Accessed June 6, 2012.
25. Mayo Clinic. Performance-enhancing drugs: know the risks. http://www.mayoclinic.com/health/performance-enhancing-drugs/hq01105. Published December 23, 2010.
Discuss this article at www.facebook.com/CurrentPsychiatry
Suck it up. Tough it out. There is no “I” in team. These are a few of the messages athletes receive from coaches, teammates, and fans. There are norms, values, and expectations in every culture, including sports, that affect behavior and emotional expression. When taking a patient’s history, clinicians may ask about participation in sports because it provides health and lifestyle information. However, many clinicians fail to consider the extent to which sport participation can influence a person’s explanatory style, experience of injury, and attitude toward medications. Whether your patient is an elite athlete or someone who participates in sports solely for exercise, the extent to which he or she identifies as an athlete is worth exploring.
Research on athletes has focused on physical aspects of injury, but this may be just a small component of an athlete’s devastation after serious injury. In this article, we discuss athletes’:
- psychiatric risks after injury
- expression of pain
- risks of having an identity driven solely by sports
- distress tolerance.
We also provide tips for making a differential diagnosis and providing treatment. This information is based on our experience treating athletes, supplemented by relevant literature.
Psychiatric risks after injury
Research has explored eating disorders and substance use among athletes, but clinicians generally are less aware of the prevalence of mood and anxiety disorders in this population. Although participating in sports can protect against emotional distress, athletes who sustain an injury are at risk for major depressive disorder, posttraumatic stress disorder (PTSD), or an adjustment disorder.1 Only about 10% of injured athletes have severe, long-term psychological consequences,2 but the prevalence of anger and depression after an injury is well documented.3,4 Researchers have found that injured athletes experience clinically significant depression 6 times as often as non-injured athletes.5 Injured athletes also exhibit significantly greater anxiety and lower self-esteem than non-injured controls immediately after injury and at 2-month follow-up; those with more severe injuries are more likely to become depressed.6 Non-injured athletes seem to experience depression at the same rate as the general population.7
Injury and expression of pain
Psychiatric illnesses often are underreported and undertreated in athletes.8 This may be because athletes feel that admitting they have a psychiatric illness or symptoms could threaten their status with their team. One professional figure skater we treated failed to seek recommended treatment for a psychiatric disorder because she feared she would be asked to leave her skating company. Her symptoms dangerously escalated before she was hospitalized.
Based on our clinical experience, many athletes feel acute pressure to play through psychological and physical pain. Some athletes continue to play with an injury to hold on to a paycheck or scholarship. Some continue to play even though they no longer enjoy the sport to prevent letting down parents or coaches; others know no other way but to “tough it out.” Supporters such as coaches, parents, or teammates may encourage athletes to play with injury, and sometimes provide medication to do so.
Mostly, however, the pressure to continue to play despite injury comes from athletes themselves. The culture of sport may lead athletes to minimize pain, fear, and self doubt.9 Athletes who fuse the culture of sport with their own being may underreport physical and psychiatric symptoms. In a survey of National Collegiate Athletic Association Division I athletes, Nixon9 found that 70% of respondents reported having been injured at least once, and more than one-half felt pressure to play while injured. Feeling pressure to perform with injury was affected by “starter” status, and whites and men scored highest on pressure scales, although women showed a roughly equal probability of playing through injury. Students who received an athletic scholarship experienced more injuries that required surgery. There was no difference in pain expression between players of contact and non-contact sports. Finally, athletes may be less likely to seek pharmacologic treatments because of cultural messages that emphasize ideas such as “the body is a temple.”
Loss of identity
An athlete’s injury should be analyzed for meaning; what may seem insignificant to one person may be quite different for another. When injury makes athletic activity impossible, an athlete may suffer more distress than someone who does not exercise regularly. Understanding the significance of the experience for an athlete is crucial to achieving recovery.10 For example, to a non-athlete a fractured wrist may be an annoyance, but it may be disastrous to a collegiate pitcher who is forced to be inactive when scouts for Major League Baseball teams search for prospects.
To an athlete, injury can mean loss of identity. Whereas most people become competent in many aspects of life, and develop support systems across multiple contexts, an athlete—particularly an extraordinarily talented one—may have focused only on his or her sport. Although athletics can help young people develop confidence, participation also can be a trap. Individuals with strong athletic identities are less likely to explore other career, educational, and lifestyle options.11 In the context of team sports, an athlete may feel less emotionally supported if an injury results in the loss of his or her central role with the team. Helping athletes form an identity that is not based solely on sports is ideal because subsequent injuries could lead to recurrent struggles with loss of identity.
Athletes who achieve higher levels of success have higher levels of depression and higher suicidal ideation after injury.12 An athlete may attempt or complete suicide, particularly those who are injured (Box).13-16
Student athletes. When working with student athletes, it is crucial to understand the lifestyle that promotes forming a single-factor identity. Student athletes may be required to train 2 or 3 times a day, rarely spend their school breaks in tropical locations, often miss social events, and may forgo commencement ceremonies. When an injury suddenly makes these perpetual sacrifices seem to be in vain, the risk of psychiatric illness may increase.
Suicides by several high-profile athletes have called attention to the severity of psychiatric risks among athletes. In June 2002, 20-year-old Nathan Eisert died of a self-inflicted gunshot wound 5 weeks after being released from the Western Kentucky University basketball team for academic reasons; the year before, he had suffered a serious ankle injury.13 Former National Football League (NFL) player Kenny McKinley committed suicide in September 2010, after a knee injury sidelined him.14 In May 2012, former NFL star Junior Seau, who had retired in 2011, fatally shot himself.15
For some athletes, career-ending injuries lead to suicidal behaviors. A study of 5 athletes who attempted suicide after sustaining an injury found 5 common characteristics:
- all were successful in their sport before getting injured
- all sustained an injury severe enough to warrant surgery
- all endured a lengthy rehabilitation
- all were not as successful at their sport when they returned to play
- all were replaced by a teammate.16
Tolerating distress
Athletes often use their sport as an outlet for emotional expression. When an injury removes that outlet, an athlete may develop anxiety and disappointment. Left alone to manage these emotions, an athlete may become irritable, passive, socially isolated, depressed, or suicidal.17 Trying but failing to find socially acceptable ways to express these feelings may intensify depression or anger. Difficult life issues, such as avoided losses, relationship issues, or various insecurities, may come to the surface when an athlete’s primary coping skill is lost. In addition, without the support of the athletic “family” (eg, teammates, coaches, staff) many athletes turn to alcohol or drugs unless they have alternate coping strategies and social supports.18
Overtraining and stress
The differential diagnosis for athletes who present with psychiatric symptoms includes several mood and anxiety disorders and other conditions (Table). When evaluating athletes who have depressive symptoms, it is essential to rule out overtraining syndrome (OTS). A common phenomenon among athletes, OTS is characterized by athletic “staleness” and chronic fatigue.19 Although there are no official OTS diagnostic criteria, characteristic symptoms include decreased physical performance or stamina, fatigue, insomnia, change in appetite, irritability, restlessness, excitability, anxiety, weight loss, loss of motivation, and poor concentration.19 The primary distinction between OTS and depression is that OTS results from athletic endeavors and can be reversed by reducing activity.
Experiencing an injury—or even a near-miss—can be terrifying to a person who derives his or her identity from a fully functioning body and feels that a perfectly working body is essential to an acceptable life. Such athletes may develop acute stress disorder or PTSD.20,21 We treated a hockey player who just missed being involved in a serious incident on the ice. “I watched my whole athletic career up to that point flash before my eyes.… I keep getting flashes of that,” he said. After the incident, he experienced hypervigilance, avoidance, and anxiety—both on and off the ice—and was diagnosed with acute stress disorder. Similarly, we cared for a young running back whose physical symptoms had abated after experiencing a concussion. He developed an irrational fear that he would become injured again. Neither athlete had a history of psychiatric illness or serious injury, and both were paralyzed by the idea of returning to play. One of these athletes successfully engaged in exposure therapy, and the other experienced severe avoidance, hopelessness, depression, nightmares, and flashbacks before seeking treatment.
Table
Differential diagnosis of conditions associated with athletic injury
| Acute stress disorder |
| Adjustment disorder |
| Anxiety disorder NOS |
| Depressive disorder NOS |
| Major depressive disorder |
| Overtraining syndrome |
| Postconcussion syndrome |
| Posttraumatic stress disorder |
| NOS: Not otherwise specified |
Substance use: Common and risky
Anecdotal and clinical evidence suggests that athletes in different sports engage in different substance abuse patterns. Studies show that college athletes use alcohol at higher rates than non-athletes.22,23 In 2000, the American College of Sports Medicine reported that athletes’ abuse of recreational drugs far surpasses their abuse of performance-enhancing drugs.24 Some athletes may use prescription pain medications recreationally or to self-medicate emotional pain as a result of injury. Athletes may not understand the risks of recreational use of prescription medications or illicit substances—such as cocaine’s deleterious cardiovascular effects—and may hesitate to discuss their self-medicating with physicians.
Some athletes abuse performance-enhancing drugs, such as anabolic steroids, androstenedione, stimulants, diuretics, and creatine.25 Side effects of these substances include liver disease, brain hemorrhage, weight loss, and depression.25
Our recommendations
Working with athletes—particularly injured athletes who have internalized sports culture—requires informed clinical effort, whether your patient is a student athlete, elite athlete, leisure athlete, or former athlete. Successful diagnosis and treatment requires understanding the meaning of athletics in your patient’s life and the extent to which he or she has “back-up” stress relievers and support systems, and assessing for cognitive dysfunction that may contribute to mood or anxiety symptoms. During evaluation, take a careful history to distinguish major depression or adjustment disorders from OTS, and assess for PTSD symptoms. When treating an injured athlete, help the patient determine whether he or she can find another outlet—preferably more than one—to replace athletics.
For an athlete who has depressive symptoms, we recommend determining whether the patient’s symptoms remit after a brief period of rest before initiating pharmacotherapy. For patients who exhibit minimal neurovegetative features, we recommend psychotherapy as a first-line treatment. Many athletes are reluctant to take medication and would be more likely to follow through with cognitive-behavioral and biofeedback interventions.
If a patient requires pharmacotherapy, ask about his or her feelings toward medications that may impact adherence. For example, is a gymnast worried about weight gain? Is a sprinter concerned with lethargy? When prescribing, be aware of the prevalence of drug and alcohol problems among athletes, understand how habits and temptations differ among sports cultures, and provide patients with psychoeducation about substance abuse when appropriate.
Related Resources
- International Society for Sports Psychiatry. http://sportspsychiatry.org.
- Sabo D, Miller KE, Melnick MJ, et al. High school athletic participation and adolescent suicide: a nationwide U.S. study. Int Rev Sociol Sport. 2005;40(1):5-23. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2563797. Accessed June 7, 2012.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Suck it up. Tough it out. There is no “I” in team. These are a few of the messages athletes receive from coaches, teammates, and fans. There are norms, values, and expectations in every culture, including sports, that affect behavior and emotional expression. When taking a patient’s history, clinicians may ask about participation in sports because it provides health and lifestyle information. However, many clinicians fail to consider the extent to which sport participation can influence a person’s explanatory style, experience of injury, and attitude toward medications. Whether your patient is an elite athlete or someone who participates in sports solely for exercise, the extent to which he or she identifies as an athlete is worth exploring.
Research on athletes has focused on physical aspects of injury, but this may be just a small component of an athlete’s devastation after serious injury. In this article, we discuss athletes’:
- psychiatric risks after injury
- expression of pain
- risks of having an identity driven solely by sports
- distress tolerance.
We also provide tips for making a differential diagnosis and providing treatment. This information is based on our experience treating athletes, supplemented by relevant literature.
Psychiatric risks after injury
Research has explored eating disorders and substance use among athletes, but clinicians generally are less aware of the prevalence of mood and anxiety disorders in this population. Although participating in sports can protect against emotional distress, athletes who sustain an injury are at risk for major depressive disorder, posttraumatic stress disorder (PTSD), or an adjustment disorder.1 Only about 10% of injured athletes have severe, long-term psychological consequences,2 but the prevalence of anger and depression after an injury is well documented.3,4 Researchers have found that injured athletes experience clinically significant depression 6 times as often as non-injured athletes.5 Injured athletes also exhibit significantly greater anxiety and lower self-esteem than non-injured controls immediately after injury and at 2-month follow-up; those with more severe injuries are more likely to become depressed.6 Non-injured athletes seem to experience depression at the same rate as the general population.7
Injury and expression of pain
Psychiatric illnesses often are underreported and undertreated in athletes.8 This may be because athletes feel that admitting they have a psychiatric illness or symptoms could threaten their status with their team. One professional figure skater we treated failed to seek recommended treatment for a psychiatric disorder because she feared she would be asked to leave her skating company. Her symptoms dangerously escalated before she was hospitalized.
Based on our clinical experience, many athletes feel acute pressure to play through psychological and physical pain. Some athletes continue to play with an injury to hold on to a paycheck or scholarship. Some continue to play even though they no longer enjoy the sport to prevent letting down parents or coaches; others know no other way but to “tough it out.” Supporters such as coaches, parents, or teammates may encourage athletes to play with injury, and sometimes provide medication to do so.
Mostly, however, the pressure to continue to play despite injury comes from athletes themselves. The culture of sport may lead athletes to minimize pain, fear, and self doubt.9 Athletes who fuse the culture of sport with their own being may underreport physical and psychiatric symptoms. In a survey of National Collegiate Athletic Association Division I athletes, Nixon9 found that 70% of respondents reported having been injured at least once, and more than one-half felt pressure to play while injured. Feeling pressure to perform with injury was affected by “starter” status, and whites and men scored highest on pressure scales, although women showed a roughly equal probability of playing through injury. Students who received an athletic scholarship experienced more injuries that required surgery. There was no difference in pain expression between players of contact and non-contact sports. Finally, athletes may be less likely to seek pharmacologic treatments because of cultural messages that emphasize ideas such as “the body is a temple.”
Loss of identity
An athlete’s injury should be analyzed for meaning; what may seem insignificant to one person may be quite different for another. When injury makes athletic activity impossible, an athlete may suffer more distress than someone who does not exercise regularly. Understanding the significance of the experience for an athlete is crucial to achieving recovery.10 For example, to a non-athlete a fractured wrist may be an annoyance, but it may be disastrous to a collegiate pitcher who is forced to be inactive when scouts for Major League Baseball teams search for prospects.
To an athlete, injury can mean loss of identity. Whereas most people become competent in many aspects of life, and develop support systems across multiple contexts, an athlete—particularly an extraordinarily talented one—may have focused only on his or her sport. Although athletics can help young people develop confidence, participation also can be a trap. Individuals with strong athletic identities are less likely to explore other career, educational, and lifestyle options.11 In the context of team sports, an athlete may feel less emotionally supported if an injury results in the loss of his or her central role with the team. Helping athletes form an identity that is not based solely on sports is ideal because subsequent injuries could lead to recurrent struggles with loss of identity.
Athletes who achieve higher levels of success have higher levels of depression and higher suicidal ideation after injury.12 An athlete may attempt or complete suicide, particularly those who are injured (Box).13-16
Student athletes. When working with student athletes, it is crucial to understand the lifestyle that promotes forming a single-factor identity. Student athletes may be required to train 2 or 3 times a day, rarely spend their school breaks in tropical locations, often miss social events, and may forgo commencement ceremonies. When an injury suddenly makes these perpetual sacrifices seem to be in vain, the risk of psychiatric illness may increase.
Suicides by several high-profile athletes have called attention to the severity of psychiatric risks among athletes. In June 2002, 20-year-old Nathan Eisert died of a self-inflicted gunshot wound 5 weeks after being released from the Western Kentucky University basketball team for academic reasons; the year before, he had suffered a serious ankle injury.13 Former National Football League (NFL) player Kenny McKinley committed suicide in September 2010, after a knee injury sidelined him.14 In May 2012, former NFL star Junior Seau, who had retired in 2011, fatally shot himself.15
For some athletes, career-ending injuries lead to suicidal behaviors. A study of 5 athletes who attempted suicide after sustaining an injury found 5 common characteristics:
- all were successful in their sport before getting injured
- all sustained an injury severe enough to warrant surgery
- all endured a lengthy rehabilitation
- all were not as successful at their sport when they returned to play
- all were replaced by a teammate.16
Tolerating distress
Athletes often use their sport as an outlet for emotional expression. When an injury removes that outlet, an athlete may develop anxiety and disappointment. Left alone to manage these emotions, an athlete may become irritable, passive, socially isolated, depressed, or suicidal.17 Trying but failing to find socially acceptable ways to express these feelings may intensify depression or anger. Difficult life issues, such as avoided losses, relationship issues, or various insecurities, may come to the surface when an athlete’s primary coping skill is lost. In addition, without the support of the athletic “family” (eg, teammates, coaches, staff) many athletes turn to alcohol or drugs unless they have alternate coping strategies and social supports.18
Overtraining and stress
The differential diagnosis for athletes who present with psychiatric symptoms includes several mood and anxiety disorders and other conditions (Table). When evaluating athletes who have depressive symptoms, it is essential to rule out overtraining syndrome (OTS). A common phenomenon among athletes, OTS is characterized by athletic “staleness” and chronic fatigue.19 Although there are no official OTS diagnostic criteria, characteristic symptoms include decreased physical performance or stamina, fatigue, insomnia, change in appetite, irritability, restlessness, excitability, anxiety, weight loss, loss of motivation, and poor concentration.19 The primary distinction between OTS and depression is that OTS results from athletic endeavors and can be reversed by reducing activity.
Experiencing an injury—or even a near-miss—can be terrifying to a person who derives his or her identity from a fully functioning body and feels that a perfectly working body is essential to an acceptable life. Such athletes may develop acute stress disorder or PTSD.20,21 We treated a hockey player who just missed being involved in a serious incident on the ice. “I watched my whole athletic career up to that point flash before my eyes.… I keep getting flashes of that,” he said. After the incident, he experienced hypervigilance, avoidance, and anxiety—both on and off the ice—and was diagnosed with acute stress disorder. Similarly, we cared for a young running back whose physical symptoms had abated after experiencing a concussion. He developed an irrational fear that he would become injured again. Neither athlete had a history of psychiatric illness or serious injury, and both were paralyzed by the idea of returning to play. One of these athletes successfully engaged in exposure therapy, and the other experienced severe avoidance, hopelessness, depression, nightmares, and flashbacks before seeking treatment.
Table
Differential diagnosis of conditions associated with athletic injury
| Acute stress disorder |
| Adjustment disorder |
| Anxiety disorder NOS |
| Depressive disorder NOS |
| Major depressive disorder |
| Overtraining syndrome |
| Postconcussion syndrome |
| Posttraumatic stress disorder |
| NOS: Not otherwise specified |
Substance use: Common and risky
Anecdotal and clinical evidence suggests that athletes in different sports engage in different substance abuse patterns. Studies show that college athletes use alcohol at higher rates than non-athletes.22,23 In 2000, the American College of Sports Medicine reported that athletes’ abuse of recreational drugs far surpasses their abuse of performance-enhancing drugs.24 Some athletes may use prescription pain medications recreationally or to self-medicate emotional pain as a result of injury. Athletes may not understand the risks of recreational use of prescription medications or illicit substances—such as cocaine’s deleterious cardiovascular effects—and may hesitate to discuss their self-medicating with physicians.
Some athletes abuse performance-enhancing drugs, such as anabolic steroids, androstenedione, stimulants, diuretics, and creatine.25 Side effects of these substances include liver disease, brain hemorrhage, weight loss, and depression.25
Our recommendations
Working with athletes—particularly injured athletes who have internalized sports culture—requires informed clinical effort, whether your patient is a student athlete, elite athlete, leisure athlete, or former athlete. Successful diagnosis and treatment requires understanding the meaning of athletics in your patient’s life and the extent to which he or she has “back-up” stress relievers and support systems, and assessing for cognitive dysfunction that may contribute to mood or anxiety symptoms. During evaluation, take a careful history to distinguish major depression or adjustment disorders from OTS, and assess for PTSD symptoms. When treating an injured athlete, help the patient determine whether he or she can find another outlet—preferably more than one—to replace athletics.
For an athlete who has depressive symptoms, we recommend determining whether the patient’s symptoms remit after a brief period of rest before initiating pharmacotherapy. For patients who exhibit minimal neurovegetative features, we recommend psychotherapy as a first-line treatment. Many athletes are reluctant to take medication and would be more likely to follow through with cognitive-behavioral and biofeedback interventions.
If a patient requires pharmacotherapy, ask about his or her feelings toward medications that may impact adherence. For example, is a gymnast worried about weight gain? Is a sprinter concerned with lethargy? When prescribing, be aware of the prevalence of drug and alcohol problems among athletes, understand how habits and temptations differ among sports cultures, and provide patients with psychoeducation about substance abuse when appropriate.
Related Resources
- International Society for Sports Psychiatry. http://sportspsychiatry.org.
- Sabo D, Miller KE, Melnick MJ, et al. High school athletic participation and adolescent suicide: a nationwide U.S. study. Int Rev Sociol Sport. 2005;40(1):5-23. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2563797. Accessed June 7, 2012.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Brewer BW, Linder DE, Phelps CM. Situational correlates of emotional adjustment to athletic injury. Clin J Sport Med. 1995;5(4):241-245.
2. Brewer BW, Petrie TA. Psychopathology in sport and exercise. In: Van Raalte JL Brewer BW, eds. Exploring sport and exercise psychology. Washington, DC: American Psychological Association; 1996:257–274.
3. May J, Sieb G. Athletic injuries: psychosocial factors in the onset sequelae, rehabilitation and prevention. In: May JR, Asken MJ, eds. Sport psychology: the psychological health of the athlete. New York, NY: PMA Publishing; 1987: 157–185.
4. Quackenbush N, Crossman J. Injured athletes: a study of emotional responses. J Sport Behav. 1994;17:178-187.
5. Perna F, Roh J, Newcomer R, et al. Clinical depression among injured athletes: an empirical assessment. Presented at: Association for the Advancement of Applied Sport Psychology annual convention; September 1998; Hyannis, MA.
6. Leddy MH, Lambert MJ, Ogles BM. Psychological consequences of athletic injury among high-level competitors. Res Q Exerc Sport. 1994;65(4):347-354.
7. Schwenk TL. The stigmatisation and denial of mental illness in athletes. Br J Sports Med. 2000;34(1):4-5.
8. Rotella RJ, Heyman SR. Stress injury, and the psychological rehabilitation of athletes. In: Williams HR, ed. Applied sports psychology: personal growth to peak performance. 2nd ed. Mountain View, CA: Mayfield Publishing; 1993: 338–355.
9. Nixon HL, II. Explaining pain and injury attitudes and experiences in sport in terms of gender race, and sports status factors. Journal of Sport Social Issues. 1996;20(1):33-44.
10. Harris LL. Integrating and analyzing psychosocial and stage theories to challenge the development of the injured collegiate athlete. J Athl Train. 2003;38(1):75-82.
11. Brown C, Hartley DL. Athletic identity and career maturation of male college student athletes. International Journal of Sport Psychology. 1998;29(1):17-26.
12. Baum AL. Suicide in athletes: a review and commentary. Clin Sports Med. 2005;24(4):853-859, ix.
13. Ho J. Suicide on campus. CBS News. http://www.cbsnews.com/2100-500195_162-654130.html. Published February 11 2009. Accessed June 7, 2012.
14. Bunch J, Jones LH. Broncos WR Kenny McKinley found dead in apparent suicide. Denver Post. http://www.denverpost.com/sports/ci_16127852. Published September 20 2010. Accessed June 7, 2012.
15. Saraceno J. Junior Seau’s death came with ‘zero warning.’ USA Today. http://www.usatoday.com/sports/football/nfl/story/2012-05-02/junior-seau-dead-gunshot/54712488/1. Published May 3 2012. Accessed June 7, 2012.
16. Smith AM, Milliner EK. Injured athletes and the risk of suicide. J Athl Train. 1994;29(4):337-341.
17. Putukian M, Wilfert M. National Collegiate Athletic Association. Student-athletes also face dangers from depression. http://fs.ncaa.org/Docs/NCAANewsArchive/2004/Association-Wide/student-athletes+also+face+dangers+from+depression+-+4-12-04.html. Published April 12 2004. Accessed June 6, 2012.
18. Perna FM, Antoni MH, Baum A, et al. Cognitive behavioral stress management effects on injury and illness among competitive athletes: a randomized clinical trial. Ann Behav Med. 2003;25(1):66-73.
19. Armstrong LE, VanHeest JL. The unknown mechanism of the overtraining syndrome: clues from depression and psychoneuroimmunology. Sports Med. 2002;32(3):185-209.
20. Newcomer RR, Perna FM. Features of posttraumatic distress among adolescent athletes. J Athl Train. 2003;38(2):163-166.
21. Newcomer R, Roh J, Perna F, et al. Injury as a traumatic experience: Intrusive thoughts and avoidance behavior associated with injury among college student-athletes. J Appl Sport Psychol. 1998;10(suppl):S54.
22. Hildebrand KM, Johnson DJ, Bogle K. Comparison of patterns of alcohol use between high school and college athletes and non-athletes. College Student Journal. 2001;35:358-365.
23. Wilson GS, Pritchard ME, Schaffer J. Athletic status and drinking behavior in college students: the influence of gender and coping styles. J Am Coll Health. 2004;52(6):269-273.
24. Wadler GI. American College of Sports Medicine. Cocaine abuse in sports. http://www.acsm.org/docs/current-comments/cocainabuse.pdf. Accessed June 6, 2012.
25. Mayo Clinic. Performance-enhancing drugs: know the risks. http://www.mayoclinic.com/health/performance-enhancing-drugs/hq01105. Published December 23, 2010.
1. Brewer BW, Linder DE, Phelps CM. Situational correlates of emotional adjustment to athletic injury. Clin J Sport Med. 1995;5(4):241-245.
2. Brewer BW, Petrie TA. Psychopathology in sport and exercise. In: Van Raalte JL Brewer BW, eds. Exploring sport and exercise psychology. Washington, DC: American Psychological Association; 1996:257–274.
3. May J, Sieb G. Athletic injuries: psychosocial factors in the onset sequelae, rehabilitation and prevention. In: May JR, Asken MJ, eds. Sport psychology: the psychological health of the athlete. New York, NY: PMA Publishing; 1987: 157–185.
4. Quackenbush N, Crossman J. Injured athletes: a study of emotional responses. J Sport Behav. 1994;17:178-187.
5. Perna F, Roh J, Newcomer R, et al. Clinical depression among injured athletes: an empirical assessment. Presented at: Association for the Advancement of Applied Sport Psychology annual convention; September 1998; Hyannis, MA.
6. Leddy MH, Lambert MJ, Ogles BM. Psychological consequences of athletic injury among high-level competitors. Res Q Exerc Sport. 1994;65(4):347-354.
7. Schwenk TL. The stigmatisation and denial of mental illness in athletes. Br J Sports Med. 2000;34(1):4-5.
8. Rotella RJ, Heyman SR. Stress injury, and the psychological rehabilitation of athletes. In: Williams HR, ed. Applied sports psychology: personal growth to peak performance. 2nd ed. Mountain View, CA: Mayfield Publishing; 1993: 338–355.
9. Nixon HL, II. Explaining pain and injury attitudes and experiences in sport in terms of gender race, and sports status factors. Journal of Sport Social Issues. 1996;20(1):33-44.
10. Harris LL. Integrating and analyzing psychosocial and stage theories to challenge the development of the injured collegiate athlete. J Athl Train. 2003;38(1):75-82.
11. Brown C, Hartley DL. Athletic identity and career maturation of male college student athletes. International Journal of Sport Psychology. 1998;29(1):17-26.
12. Baum AL. Suicide in athletes: a review and commentary. Clin Sports Med. 2005;24(4):853-859, ix.
13. Ho J. Suicide on campus. CBS News. http://www.cbsnews.com/2100-500195_162-654130.html. Published February 11 2009. Accessed June 7, 2012.
14. Bunch J, Jones LH. Broncos WR Kenny McKinley found dead in apparent suicide. Denver Post. http://www.denverpost.com/sports/ci_16127852. Published September 20 2010. Accessed June 7, 2012.
15. Saraceno J. Junior Seau’s death came with ‘zero warning.’ USA Today. http://www.usatoday.com/sports/football/nfl/story/2012-05-02/junior-seau-dead-gunshot/54712488/1. Published May 3 2012. Accessed June 7, 2012.
16. Smith AM, Milliner EK. Injured athletes and the risk of suicide. J Athl Train. 1994;29(4):337-341.
17. Putukian M, Wilfert M. National Collegiate Athletic Association. Student-athletes also face dangers from depression. http://fs.ncaa.org/Docs/NCAANewsArchive/2004/Association-Wide/student-athletes+also+face+dangers+from+depression+-+4-12-04.html. Published April 12 2004. Accessed June 6, 2012.
18. Perna FM, Antoni MH, Baum A, et al. Cognitive behavioral stress management effects on injury and illness among competitive athletes: a randomized clinical trial. Ann Behav Med. 2003;25(1):66-73.
19. Armstrong LE, VanHeest JL. The unknown mechanism of the overtraining syndrome: clues from depression and psychoneuroimmunology. Sports Med. 2002;32(3):185-209.
20. Newcomer RR, Perna FM. Features of posttraumatic distress among adolescent athletes. J Athl Train. 2003;38(2):163-166.
21. Newcomer R, Roh J, Perna F, et al. Injury as a traumatic experience: Intrusive thoughts and avoidance behavior associated with injury among college student-athletes. J Appl Sport Psychol. 1998;10(suppl):S54.
22. Hildebrand KM, Johnson DJ, Bogle K. Comparison of patterns of alcohol use between high school and college athletes and non-athletes. College Student Journal. 2001;35:358-365.
23. Wilson GS, Pritchard ME, Schaffer J. Athletic status and drinking behavior in college students: the influence of gender and coping styles. J Am Coll Health. 2004;52(6):269-273.
24. Wadler GI. American College of Sports Medicine. Cocaine abuse in sports. http://www.acsm.org/docs/current-comments/cocainabuse.pdf. Accessed June 6, 2012.
25. Mayo Clinic. Performance-enhancing drugs: know the risks. http://www.mayoclinic.com/health/performance-enhancing-drugs/hq01105. Published December 23, 2010.
When is off-label prescribing appropriate?
Discuss this article at www.facebook.com/CurrentPsychiatry
Off-label prescribing (OLP)—prescribing a medication in a manner different from that approved by the FDA—is common and can be clinically beneficial. A survey of 200 British psychiatrists found that 65% had prescribed a medication off-label within the previous month.1 However, OLP often is not supported by strong evidence and carries clinical risks, such as adverse effects and unproven efficacy. A 2006 study found that only 4% of off-label prescriptions of psychiatric medications were supported by strong scientific evidence.2
OLP may become unavoidable for several reasons, including:
- lack of data from quality trials on a specific indication or patient population
- patients seen in clinical practice vary from those evaluated in clinical trials (Figure 1)
- the need to treat patients who do not respond to first-line therapies or have treatment-resistant conditions.
At times, OLP may be a psychiatrist’s only option: >80% of DSM-IV-TR diagnoses have no FDA-approved medication.3
No practice guidelines are available to help clinicians decide when OLP is appropriate. Psychiatrists must rely on multiple, sometimes-conflicting sources to determine whether evidence is sufficient to support off-label use in a specific clinical scenario. This article looks at types of OLP and offers suggestions for clinicians who are considering prescribing a medication outside of its approved use.
Figure 1: Clinical trial patients vs ‘real world’ patients
The FDA’s role
Once the FDA approves a medication for a specific indication, physicians legally are permitted to prescribe that drug for indications, patient populations, or dosages not included on the FDA-approved label.2,4 The FDA oversees and regulates pharmaceutical companies, not physicians.4 Currently, pharmaceutical companies cannot market a drug for an indication not included on the label, but physicians are free to use the drug for any condition or disease.5
When is OLP used?
According to Baldwin et al,6 prescribing that is considered off-label generally falls into 1 of the following 4 categories:
Indication. This type of OLP is prescribing a medication for an indication other than those included on the FDA-approved label. The off-label indication may be a logical extension of an approved indication. For example, a medication approved for treating erectile dysfunction might be prescribed to a patient who is experiencing antidepressant-induced sexual dysfunction.
If a pharmaceutical company wishes to expand the indications of a medication they must seek supplemental approval from the FDA. This is a long, expensive process. In certain situations, it may be in the patient’s best interest to prescribe a medication off-label until that indication becomes approved. When clinicians identify new uses for existing medications while they care for patients, it is considered field discovery, and this innovative process may occur years before such indications receive FDA approval.
Dosage. The most common example of this type of OLP in psychiatry is prescribing higher-than-approved dosages of antidepressants or antipsychotics for patients who do not respond to the maximum approved dosages. The effectiveness of this strategy is unknown.6
Duration. This typically entails prescribing a medication for a period of time longer than specified on the label. For example, many antidepressants are approved only for treating depressive illness. Therefore, continuing an antidepressant as maintenance therapy for a patient who is in remission could be considered off-label. Benzodiazepines are approved primarily for short-term management of anxiety, but commonly are prescribed for patients with chronic, disabling anxiety disorders who do not respond to other medications.6
Patient age. The FDA approves medications for use in patients within a specified age range based on patients evaluated in clinical trials, and most trials of psychotropics include patients age 18 to 65. However, it is highly unlikely that a 17-year-old patient’s drug metabolism changes substantially when he or she reaches age 18, or when a 65-year-old turns 66. Research has demonstrated that medications can be effective outside of strict age ranges. For example, randomized controlled trials (RCTs) have shown that antidepressants are efficacious in treating depression in geriatric patients, and selective serotonin reuptake inhibitors are efficacious in treating obsessive-compulsive disorder (OCD) in children and adolescents.6
Few medications have been approved for treating geriatric patients with psychiatric illness. For example, no drugs are approved for treating psychotic, behavioral, and mood symptoms that may accompany dementia. For this reason, clinicians often prescribe psychotropics off-label to these patients.
The rate of psychotropic prescriptions to children and adolescents—particularly antidepressants and antipsychotics—has been increasing.7-9 The British Association for Psychopharmacology suggested that it may be reasonable to apply what we know regarding adults’ responses to drug treatment to children and adolescents with schizophrenia or OCD, but more caution is required in young patients with mood or anxiety disorders.10
Combination therapy is another type of OLP. Often a disease state consists of multiple underlying syndromes, and treating individual syndromes is a common strategy. For example, in addition to depressed mood, a patient with major depressive disorder also may have insomnia and poor concentration. A medication approved for treating depressed mood may not improve insomnia or poor concentration. Therefore, combination therapy may be necessary, but likely would be off-label. Combination therapy also may be tried when a patient does not respond to monotherapy. For example, although the evidence supporting the practice is inconclusive, clinicians commonly prescribe >1 antipsychotic to patients with schizophrenia or other psychotic disorders.
Help for making OLP decisions
Position statements/policies. The American Psychiatric Association (APA) and the American Medical Association support OLP when the practice is based on sound scientific evidence and medical opinion.11 The APA position statement encourages clinicians to use various compendia, including the American Hospital Formulary Service (AHFS) Drug Information, in conjunction with peer-reviewed literature to determine the medical acceptability of off-label uses.11
Evidence-based medicine includes a hierarchy of scientific and clinical evidence that can justify medical decisions. At the top of this hierarchy are large RCTs and smaller RCTs; cohort studies, case-control studies, poorly controlled or uncontrolled studies, case reports, and expert opinion are less valuable (Figure 2).4
Searching through all available resources for evidence supporting a specific off-label use is a cumbersome, time-consuming process. For this reason, clinicians may refer to compendia that evaluate and rate the available evidence supporting off-label use of medication, such as the AHFS Drug Information and DrugDex. Other resources include peer-reviewed medical journals. Physicians can contribute to knowledge of off-label uses by sharing their experiences, both good and bad, with their colleagues via presentations, publications, and/or initiating a study.
Other resources. Gazarian et al12 delineated 3 situations where OLP might be considered appropriate: use justified by high-quality evidence, use in research trials, and exceptional use justified by individual clinical circumstances. Exceptional use would require all of the following:
- the patient has a serious disease or condition
- evidence supports a potential beneficial effect of the off-label treatment
- potential benefits outweigh potential risks
- standard therapy has failed or is inappropriate
- an institutional drug committee approved the off-label use
- the patient provides written informed consent.12
Other authors13,14 have offered recommendations for psychiatrists considering OLP:
- Study available literature and assess whether sufficient evidence supports the proposed off-label use.
- If evidence is lacking, learn about the medication and its potential risks (interactions, adverse effects, and FDA “black-box” warnings). Also consult other resources for additional information and research, including peers and experts in the field.
- Consider and document risks and benefits of the proposed off-label use. Explain these, as well as uncertainties and potential costs, to patients and/or their families, and obtain and document informed consent.
- Cautiously initiate the off-label therapy, monitor patients closely, and meticulously document efficacy and tolerance.
Prescribing medications on-label does not guarantee safety or efficacy. Likewise, OLP does not imply a safety hazard or lack of efficacy. OLP may be in the best interest of the patient. Nonetheless, the practice must be carried out responsibly with utmost caution and consideration of acute and long-term burdens to patients, along with an assessment of the risk vs benefit of the proposed therapy.
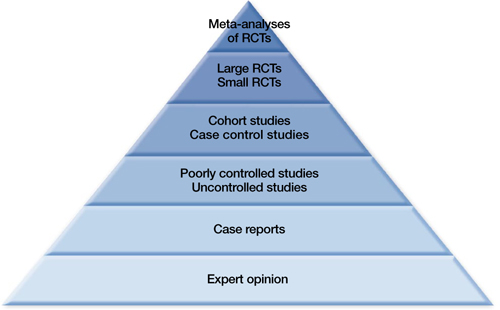
Figure 2: The hierarchy of sources for evidence-based medicine
RCTs: randomized controlled trials
Source: Reference 4Related Resource
- Mossman D. Why off-label isn’t off base. Current Psychiatry. 2009;8(2):19-22.
Disclosures
Dr. Ali receives research/grant support from Cyberonics and is a speaker for Merck.
Dr Ajmal reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Lowe-Ponsford F, Baldwin D. Off-label prescribing by psychiatrists. Psychiatric Bulletin. 2000;24:415-417.
2. Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026.
3. Devulapalli K, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders: the majority of psychiatric diagnoses have no approved drug. Asian Journal of Psychiatry. 2009;2(1):29-36.
4. Dresser R, Frader J. Off-label prescribing: a call for heightened professional and government oversight. J Law Med Ethics. 2009;37(3):476-486, 396.
5. Food, Drug, and Cosmetic Act, 21 USC §396 (2010).
6. Baldwin DS, Kosky N. Off-label prescribing in psychiatric practice. Advances in Psychiatric Treatment. 2007;13:414-422.
7. Zito JM, Safer DJ, dosReis S, et al. Trends in the prescribing of psychotropic medications to preschoolers. JAMA. 2000;283(8):1025-1030.
8. Tournier M, Greenfield B, Galbaud du Fort G, et al. Patterns of antidepressant use in Quebec children and adolescents: trends and predictors. Psychiatry Res. 2010;179(1):57-63.
9. Cooper WO, Arbogast PG, Ding H, et al. Trends in prescribing of antipsychotic medications for US children. Ambul Pediatr. 2006;6(2):79-83.
10. Child and learning disability psychopharmacology. J Psychopharmacol. 1997;11(4):291-294.
11. American Psychiatric Association. Position statement on patient access to treatments prescribed by their physicians. 2007. http://www.psychiatry.org/advocacy—newsroom/position-statements/apa-position-statements. Accessed May 24 2012.
12. Gazarian M, Kelly M, McPhee JR, et al. Off-label use of medicines: consensus recommendations for evaluating appropriateness. Med J Aust. 2006;185(10):544-548.
13. Kramer SI, McCall WV. Off-label prescribing: 7 steps for safer more effective prescribing. Current Psychiatry. 2006;5(4):14-28.
14. Royal College of Psychiatrists. Use of licensed medicines for unlicensed applications in psychiatric practice. College report CR142. http://www.rcpsych.ac.uk/files/pdfversion/cr142.pdf. Accessed May 24 2012.
Discuss this article at www.facebook.com/CurrentPsychiatry
Off-label prescribing (OLP)—prescribing a medication in a manner different from that approved by the FDA—is common and can be clinically beneficial. A survey of 200 British psychiatrists found that 65% had prescribed a medication off-label within the previous month.1 However, OLP often is not supported by strong evidence and carries clinical risks, such as adverse effects and unproven efficacy. A 2006 study found that only 4% of off-label prescriptions of psychiatric medications were supported by strong scientific evidence.2
OLP may become unavoidable for several reasons, including:
- lack of data from quality trials on a specific indication or patient population
- patients seen in clinical practice vary from those evaluated in clinical trials (Figure 1)
- the need to treat patients who do not respond to first-line therapies or have treatment-resistant conditions.
At times, OLP may be a psychiatrist’s only option: >80% of DSM-IV-TR diagnoses have no FDA-approved medication.3
No practice guidelines are available to help clinicians decide when OLP is appropriate. Psychiatrists must rely on multiple, sometimes-conflicting sources to determine whether evidence is sufficient to support off-label use in a specific clinical scenario. This article looks at types of OLP and offers suggestions for clinicians who are considering prescribing a medication outside of its approved use.
Figure 1: Clinical trial patients vs ‘real world’ patients
The FDA’s role
Once the FDA approves a medication for a specific indication, physicians legally are permitted to prescribe that drug for indications, patient populations, or dosages not included on the FDA-approved label.2,4 The FDA oversees and regulates pharmaceutical companies, not physicians.4 Currently, pharmaceutical companies cannot market a drug for an indication not included on the label, but physicians are free to use the drug for any condition or disease.5
When is OLP used?
According to Baldwin et al,6 prescribing that is considered off-label generally falls into 1 of the following 4 categories:
Indication. This type of OLP is prescribing a medication for an indication other than those included on the FDA-approved label. The off-label indication may be a logical extension of an approved indication. For example, a medication approved for treating erectile dysfunction might be prescribed to a patient who is experiencing antidepressant-induced sexual dysfunction.
If a pharmaceutical company wishes to expand the indications of a medication they must seek supplemental approval from the FDA. This is a long, expensive process. In certain situations, it may be in the patient’s best interest to prescribe a medication off-label until that indication becomes approved. When clinicians identify new uses for existing medications while they care for patients, it is considered field discovery, and this innovative process may occur years before such indications receive FDA approval.
Dosage. The most common example of this type of OLP in psychiatry is prescribing higher-than-approved dosages of antidepressants or antipsychotics for patients who do not respond to the maximum approved dosages. The effectiveness of this strategy is unknown.6
Duration. This typically entails prescribing a medication for a period of time longer than specified on the label. For example, many antidepressants are approved only for treating depressive illness. Therefore, continuing an antidepressant as maintenance therapy for a patient who is in remission could be considered off-label. Benzodiazepines are approved primarily for short-term management of anxiety, but commonly are prescribed for patients with chronic, disabling anxiety disorders who do not respond to other medications.6
Patient age. The FDA approves medications for use in patients within a specified age range based on patients evaluated in clinical trials, and most trials of psychotropics include patients age 18 to 65. However, it is highly unlikely that a 17-year-old patient’s drug metabolism changes substantially when he or she reaches age 18, or when a 65-year-old turns 66. Research has demonstrated that medications can be effective outside of strict age ranges. For example, randomized controlled trials (RCTs) have shown that antidepressants are efficacious in treating depression in geriatric patients, and selective serotonin reuptake inhibitors are efficacious in treating obsessive-compulsive disorder (OCD) in children and adolescents.6
Few medications have been approved for treating geriatric patients with psychiatric illness. For example, no drugs are approved for treating psychotic, behavioral, and mood symptoms that may accompany dementia. For this reason, clinicians often prescribe psychotropics off-label to these patients.
The rate of psychotropic prescriptions to children and adolescents—particularly antidepressants and antipsychotics—has been increasing.7-9 The British Association for Psychopharmacology suggested that it may be reasonable to apply what we know regarding adults’ responses to drug treatment to children and adolescents with schizophrenia or OCD, but more caution is required in young patients with mood or anxiety disorders.10
Combination therapy is another type of OLP. Often a disease state consists of multiple underlying syndromes, and treating individual syndromes is a common strategy. For example, in addition to depressed mood, a patient with major depressive disorder also may have insomnia and poor concentration. A medication approved for treating depressed mood may not improve insomnia or poor concentration. Therefore, combination therapy may be necessary, but likely would be off-label. Combination therapy also may be tried when a patient does not respond to monotherapy. For example, although the evidence supporting the practice is inconclusive, clinicians commonly prescribe >1 antipsychotic to patients with schizophrenia or other psychotic disorders.
Help for making OLP decisions
Position statements/policies. The American Psychiatric Association (APA) and the American Medical Association support OLP when the practice is based on sound scientific evidence and medical opinion.11 The APA position statement encourages clinicians to use various compendia, including the American Hospital Formulary Service (AHFS) Drug Information, in conjunction with peer-reviewed literature to determine the medical acceptability of off-label uses.11
Evidence-based medicine includes a hierarchy of scientific and clinical evidence that can justify medical decisions. At the top of this hierarchy are large RCTs and smaller RCTs; cohort studies, case-control studies, poorly controlled or uncontrolled studies, case reports, and expert opinion are less valuable (Figure 2).4
Searching through all available resources for evidence supporting a specific off-label use is a cumbersome, time-consuming process. For this reason, clinicians may refer to compendia that evaluate and rate the available evidence supporting off-label use of medication, such as the AHFS Drug Information and DrugDex. Other resources include peer-reviewed medical journals. Physicians can contribute to knowledge of off-label uses by sharing their experiences, both good and bad, with their colleagues via presentations, publications, and/or initiating a study.
Other resources. Gazarian et al12 delineated 3 situations where OLP might be considered appropriate: use justified by high-quality evidence, use in research trials, and exceptional use justified by individual clinical circumstances. Exceptional use would require all of the following:
- the patient has a serious disease or condition
- evidence supports a potential beneficial effect of the off-label treatment
- potential benefits outweigh potential risks
- standard therapy has failed or is inappropriate
- an institutional drug committee approved the off-label use
- the patient provides written informed consent.12
Other authors13,14 have offered recommendations for psychiatrists considering OLP:
- Study available literature and assess whether sufficient evidence supports the proposed off-label use.
- If evidence is lacking, learn about the medication and its potential risks (interactions, adverse effects, and FDA “black-box” warnings). Also consult other resources for additional information and research, including peers and experts in the field.
- Consider and document risks and benefits of the proposed off-label use. Explain these, as well as uncertainties and potential costs, to patients and/or their families, and obtain and document informed consent.
- Cautiously initiate the off-label therapy, monitor patients closely, and meticulously document efficacy and tolerance.
Prescribing medications on-label does not guarantee safety or efficacy. Likewise, OLP does not imply a safety hazard or lack of efficacy. OLP may be in the best interest of the patient. Nonetheless, the practice must be carried out responsibly with utmost caution and consideration of acute and long-term burdens to patients, along with an assessment of the risk vs benefit of the proposed therapy.
Figure 2: The hierarchy of sources for evidence-based medicine
RCTs: randomized controlled trials
Source: Reference 4Related Resource
- Mossman D. Why off-label isn’t off base. Current Psychiatry. 2009;8(2):19-22.
Disclosures
Dr. Ali receives research/grant support from Cyberonics and is a speaker for Merck.
Dr Ajmal reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Off-label prescribing (OLP)—prescribing a medication in a manner different from that approved by the FDA—is common and can be clinically beneficial. A survey of 200 British psychiatrists found that 65% had prescribed a medication off-label within the previous month.1 However, OLP often is not supported by strong evidence and carries clinical risks, such as adverse effects and unproven efficacy. A 2006 study found that only 4% of off-label prescriptions of psychiatric medications were supported by strong scientific evidence.2
OLP may become unavoidable for several reasons, including:
- lack of data from quality trials on a specific indication or patient population
- patients seen in clinical practice vary from those evaluated in clinical trials (Figure 1)
- the need to treat patients who do not respond to first-line therapies or have treatment-resistant conditions.
At times, OLP may be a psychiatrist’s only option: >80% of DSM-IV-TR diagnoses have no FDA-approved medication.3
No practice guidelines are available to help clinicians decide when OLP is appropriate. Psychiatrists must rely on multiple, sometimes-conflicting sources to determine whether evidence is sufficient to support off-label use in a specific clinical scenario. This article looks at types of OLP and offers suggestions for clinicians who are considering prescribing a medication outside of its approved use.
Figure 1: Clinical trial patients vs ‘real world’ patients
The FDA’s role
Once the FDA approves a medication for a specific indication, physicians legally are permitted to prescribe that drug for indications, patient populations, or dosages not included on the FDA-approved label.2,4 The FDA oversees and regulates pharmaceutical companies, not physicians.4 Currently, pharmaceutical companies cannot market a drug for an indication not included on the label, but physicians are free to use the drug for any condition or disease.5
When is OLP used?
According to Baldwin et al,6 prescribing that is considered off-label generally falls into 1 of the following 4 categories:
Indication. This type of OLP is prescribing a medication for an indication other than those included on the FDA-approved label. The off-label indication may be a logical extension of an approved indication. For example, a medication approved for treating erectile dysfunction might be prescribed to a patient who is experiencing antidepressant-induced sexual dysfunction.
If a pharmaceutical company wishes to expand the indications of a medication they must seek supplemental approval from the FDA. This is a long, expensive process. In certain situations, it may be in the patient’s best interest to prescribe a medication off-label until that indication becomes approved. When clinicians identify new uses for existing medications while they care for patients, it is considered field discovery, and this innovative process may occur years before such indications receive FDA approval.
Dosage. The most common example of this type of OLP in psychiatry is prescribing higher-than-approved dosages of antidepressants or antipsychotics for patients who do not respond to the maximum approved dosages. The effectiveness of this strategy is unknown.6
Duration. This typically entails prescribing a medication for a period of time longer than specified on the label. For example, many antidepressants are approved only for treating depressive illness. Therefore, continuing an antidepressant as maintenance therapy for a patient who is in remission could be considered off-label. Benzodiazepines are approved primarily for short-term management of anxiety, but commonly are prescribed for patients with chronic, disabling anxiety disorders who do not respond to other medications.6
Patient age. The FDA approves medications for use in patients within a specified age range based on patients evaluated in clinical trials, and most trials of psychotropics include patients age 18 to 65. However, it is highly unlikely that a 17-year-old patient’s drug metabolism changes substantially when he or she reaches age 18, or when a 65-year-old turns 66. Research has demonstrated that medications can be effective outside of strict age ranges. For example, randomized controlled trials (RCTs) have shown that antidepressants are efficacious in treating depression in geriatric patients, and selective serotonin reuptake inhibitors are efficacious in treating obsessive-compulsive disorder (OCD) in children and adolescents.6
Few medications have been approved for treating geriatric patients with psychiatric illness. For example, no drugs are approved for treating psychotic, behavioral, and mood symptoms that may accompany dementia. For this reason, clinicians often prescribe psychotropics off-label to these patients.
The rate of psychotropic prescriptions to children and adolescents—particularly antidepressants and antipsychotics—has been increasing.7-9 The British Association for Psychopharmacology suggested that it may be reasonable to apply what we know regarding adults’ responses to drug treatment to children and adolescents with schizophrenia or OCD, but more caution is required in young patients with mood or anxiety disorders.10
Combination therapy is another type of OLP. Often a disease state consists of multiple underlying syndromes, and treating individual syndromes is a common strategy. For example, in addition to depressed mood, a patient with major depressive disorder also may have insomnia and poor concentration. A medication approved for treating depressed mood may not improve insomnia or poor concentration. Therefore, combination therapy may be necessary, but likely would be off-label. Combination therapy also may be tried when a patient does not respond to monotherapy. For example, although the evidence supporting the practice is inconclusive, clinicians commonly prescribe >1 antipsychotic to patients with schizophrenia or other psychotic disorders.
Help for making OLP decisions
Position statements/policies. The American Psychiatric Association (APA) and the American Medical Association support OLP when the practice is based on sound scientific evidence and medical opinion.11 The APA position statement encourages clinicians to use various compendia, including the American Hospital Formulary Service (AHFS) Drug Information, in conjunction with peer-reviewed literature to determine the medical acceptability of off-label uses.11
Evidence-based medicine includes a hierarchy of scientific and clinical evidence that can justify medical decisions. At the top of this hierarchy are large RCTs and smaller RCTs; cohort studies, case-control studies, poorly controlled or uncontrolled studies, case reports, and expert opinion are less valuable (Figure 2).4
Searching through all available resources for evidence supporting a specific off-label use is a cumbersome, time-consuming process. For this reason, clinicians may refer to compendia that evaluate and rate the available evidence supporting off-label use of medication, such as the AHFS Drug Information and DrugDex. Other resources include peer-reviewed medical journals. Physicians can contribute to knowledge of off-label uses by sharing their experiences, both good and bad, with their colleagues via presentations, publications, and/or initiating a study.
Other resources. Gazarian et al12 delineated 3 situations where OLP might be considered appropriate: use justified by high-quality evidence, use in research trials, and exceptional use justified by individual clinical circumstances. Exceptional use would require all of the following:
- the patient has a serious disease or condition
- evidence supports a potential beneficial effect of the off-label treatment
- potential benefits outweigh potential risks
- standard therapy has failed or is inappropriate
- an institutional drug committee approved the off-label use
- the patient provides written informed consent.12
Other authors13,14 have offered recommendations for psychiatrists considering OLP:
- Study available literature and assess whether sufficient evidence supports the proposed off-label use.
- If evidence is lacking, learn about the medication and its potential risks (interactions, adverse effects, and FDA “black-box” warnings). Also consult other resources for additional information and research, including peers and experts in the field.
- Consider and document risks and benefits of the proposed off-label use. Explain these, as well as uncertainties and potential costs, to patients and/or their families, and obtain and document informed consent.
- Cautiously initiate the off-label therapy, monitor patients closely, and meticulously document efficacy and tolerance.
Prescribing medications on-label does not guarantee safety or efficacy. Likewise, OLP does not imply a safety hazard or lack of efficacy. OLP may be in the best interest of the patient. Nonetheless, the practice must be carried out responsibly with utmost caution and consideration of acute and long-term burdens to patients, along with an assessment of the risk vs benefit of the proposed therapy.
Figure 2: The hierarchy of sources for evidence-based medicine
RCTs: randomized controlled trials
Source: Reference 4Related Resource
- Mossman D. Why off-label isn’t off base. Current Psychiatry. 2009;8(2):19-22.
Disclosures
Dr. Ali receives research/grant support from Cyberonics and is a speaker for Merck.
Dr Ajmal reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Lowe-Ponsford F, Baldwin D. Off-label prescribing by psychiatrists. Psychiatric Bulletin. 2000;24:415-417.
2. Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026.
3. Devulapalli K, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders: the majority of psychiatric diagnoses have no approved drug. Asian Journal of Psychiatry. 2009;2(1):29-36.
4. Dresser R, Frader J. Off-label prescribing: a call for heightened professional and government oversight. J Law Med Ethics. 2009;37(3):476-486, 396.
5. Food, Drug, and Cosmetic Act, 21 USC §396 (2010).
6. Baldwin DS, Kosky N. Off-label prescribing in psychiatric practice. Advances in Psychiatric Treatment. 2007;13:414-422.
7. Zito JM, Safer DJ, dosReis S, et al. Trends in the prescribing of psychotropic medications to preschoolers. JAMA. 2000;283(8):1025-1030.
8. Tournier M, Greenfield B, Galbaud du Fort G, et al. Patterns of antidepressant use in Quebec children and adolescents: trends and predictors. Psychiatry Res. 2010;179(1):57-63.
9. Cooper WO, Arbogast PG, Ding H, et al. Trends in prescribing of antipsychotic medications for US children. Ambul Pediatr. 2006;6(2):79-83.
10. Child and learning disability psychopharmacology. J Psychopharmacol. 1997;11(4):291-294.
11. American Psychiatric Association. Position statement on patient access to treatments prescribed by their physicians. 2007. http://www.psychiatry.org/advocacy—newsroom/position-statements/apa-position-statements. Accessed May 24 2012.
12. Gazarian M, Kelly M, McPhee JR, et al. Off-label use of medicines: consensus recommendations for evaluating appropriateness. Med J Aust. 2006;185(10):544-548.
13. Kramer SI, McCall WV. Off-label prescribing: 7 steps for safer more effective prescribing. Current Psychiatry. 2006;5(4):14-28.
14. Royal College of Psychiatrists. Use of licensed medicines for unlicensed applications in psychiatric practice. College report CR142. http://www.rcpsych.ac.uk/files/pdfversion/cr142.pdf. Accessed May 24 2012.
1. Lowe-Ponsford F, Baldwin D. Off-label prescribing by psychiatrists. Psychiatric Bulletin. 2000;24:415-417.
2. Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026.
3. Devulapalli K, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders: the majority of psychiatric diagnoses have no approved drug. Asian Journal of Psychiatry. 2009;2(1):29-36.
4. Dresser R, Frader J. Off-label prescribing: a call for heightened professional and government oversight. J Law Med Ethics. 2009;37(3):476-486, 396.
5. Food, Drug, and Cosmetic Act, 21 USC §396 (2010).
6. Baldwin DS, Kosky N. Off-label prescribing in psychiatric practice. Advances in Psychiatric Treatment. 2007;13:414-422.
7. Zito JM, Safer DJ, dosReis S, et al. Trends in the prescribing of psychotropic medications to preschoolers. JAMA. 2000;283(8):1025-1030.
8. Tournier M, Greenfield B, Galbaud du Fort G, et al. Patterns of antidepressant use in Quebec children and adolescents: trends and predictors. Psychiatry Res. 2010;179(1):57-63.
9. Cooper WO, Arbogast PG, Ding H, et al. Trends in prescribing of antipsychotic medications for US children. Ambul Pediatr. 2006;6(2):79-83.
10. Child and learning disability psychopharmacology. J Psychopharmacol. 1997;11(4):291-294.
11. American Psychiatric Association. Position statement on patient access to treatments prescribed by their physicians. 2007. http://www.psychiatry.org/advocacy—newsroom/position-statements/apa-position-statements. Accessed May 24 2012.
12. Gazarian M, Kelly M, McPhee JR, et al. Off-label use of medicines: consensus recommendations for evaluating appropriateness. Med J Aust. 2006;185(10):544-548.
13. Kramer SI, McCall WV. Off-label prescribing: 7 steps for safer more effective prescribing. Current Psychiatry. 2006;5(4):14-28.
14. Royal College of Psychiatrists. Use of licensed medicines for unlicensed applications in psychiatric practice. College report CR142. http://www.rcpsych.ac.uk/files/pdfversion/cr142.pdf. Accessed May 24 2012.
High-dose donepezil or memantine: Next step for Alzheimer’s disease?
Although cholinesterase inhibitors (ChEIs) and memantine at standard doses may slow the progression of Alzheimer’s disease (AD) as assessed by cognitive, functional, and global measures, this effect is relatively modest. For the estimated 5.4 million Americans with AD1—more than one-half of whom have moderate to severe disease2—there is a great need for new approaches to slow AD progression.
High doses of donepezil or memantine may be the next step in achieving better results than standard pharmacologic treatments for AD. This article presents the possible benefits and indications for high doses of donepezil (23 mg/d) and memantine (28 mg/d) for managing moderate to severe AD and their safety and tolerability profiles.
Current treatments offer modest benefits
AD treatments comprise 2 categories: ChEIs (donepezil, rivastigmine, and galantamine) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine (Table 1).3,4 All ChEIs are FDA-approved for mild to moderate AD; donepezil also is approved for severe AD. Memantine is approved for moderate to severe AD, either alone or in combination with ChEIs. Until recently, the maximum FDA-approved doses were donepezil, 10 mg/d, and memantine, 20 mg/d. However, these dosages are associated with only modest beneficial effects in managing cognitive deterioration in patients with moderate to severe dementia.5,6 Studies have reported that combining a ChEI, such as donepezil, and memantine is well tolerated and may result in synergistic benefits by affecting different neurotransmitters in patients with moderate to severe AD.7,8
Recently, the FDA approved higher daily doses of donepezil (23 mg) and memantine (28 mg) for moderate to severe AD on the basis of positive phase III trial results.9-11 Donepezil, 23 mg/d, currently is marketed in the United States; the availability date for memantine, 28 mg/d, was undetermined at press time.
Table 1
FDA-approved treatments for Alzheimer’s disease
| Drug | Maximum daily dose | Mechanism of action | Indication | Common side effects/comments |
|---|---|---|---|---|
| Tacrine | 160 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, loss of appetite, diarrhea. First ChEI to be approved, but rarely used because of associated possible hepatotoxicity |
| Donepezil | 10 mg/d | ChEI | All stages of AD | Nausea, vomiting, loss of appetite, diarrhea, sleep disturbance |
| Rivastigmine | 12 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Galantamine | 24 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Memantine | 20 mg/d | NMDA receptor antagonist | Moderate to severe AD | Dizziness, headache, constipation, confusion |
| Galantamine ER | 24 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Rivastigmine transdermal system | 9.5 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Donepezil 23 | 23 mg/d | ChEI | Moderate to severe AD | Nausea, vomiting, diarrhea |
| Memantine ER | 28 mg/d | NMDA receptor antagonist | Moderate to severe AD | Dizziness, headache, constipation, confusion |
| AD: Alzheimer’s disease; ChEI: cholinesterase inhibitor; ER: extended release; NMDA: N-methyl-D-aspartate Source: References 3,4 | ||||
High-dose donepezil (23 mg/d)
Cognitive decline with AD has been associated with increasing loss of cholinergic neurons and cholinergic activities, particularly in areas associated with memory/cognition and learning, including cortical areas involving the temporal lobe, hippocampus, and nucleus basalis of Meynert.12-14 In addition, evidence suggests that increasing levels of acetylcholine by using ChEIs can enhance cognitive function.13,15
Donepezil is a selective, reversible ChEI believed to enhance central cholinergic function.15 Randomized clinical trials assessing dose-response with donepezil, 5 mg/d and 10 mg/d, have demonstrated more benefit in cognition with either dose than placebo. The 10 mg/d dose was more effective than 5 mg/d in patients with mild to moderate and severe AD.16-18 In patients with advanced AD who are stable on 5 mg/d, increasing to 10 mg/d could slow the progression of cognitive decline.18
Rationale for higher doses. Positron emission tomography studies have shown that at stable doses of donepezil, 5 mg/d or 10 mg/d, average cortical acetylcholinesterase (AChE) inhibition was <30%.19,20 Based on these findings, researchers thought that cortical AChE inhibition may be suboptimal with donepezil, 10 mg/d, and that higher doses of ChEI may be required in patients with more advanced AD—and therefore more cholinergic loss—for adequate cholinesterase inhibition. In a pilot study of patients with mild to moderate AD, higher doses of donepezil (15 mg/d and 20 mg/d) were reported to be safe and well tolerated.21
The 23-mg/d donepezil formulation was developed to provide a higher dose administered once daily without a sharp rise in peak concentration. The FDA approved donepezil, 23 mg/d, for patients with moderate to severe AD on the basis of phase III trial results.9,22 In a randomized, double-blind, multicenter, head-to-head clinical trial, >1,400 patients with moderate to severe AD (Mini-Mental State Exam [MMSE]: 0 to 20) on a stable dose of donepezil, 10 mg/d, for ≥3 months were randomly assigned to receive high-dose donepezil (23 mg/d) or standard-dose donepezil (10 mg/d) for 24 weeks.9,22 Patients in the 23-mg/d group showed a statistically significant improvement in cognition compared with the 10-mg/d group. The difference between groups on a measure of global improvement was not significant.9,22 However, in a post-hoc analysis, it was demonstrated that a subgroup of patients with more severe cognitive impairment (baseline MMSE: 0 to 16), showed significant improvement in cognition as well as global functioning.9
Overall, treatment-emergent adverse events (TEAEs) during the study were higher in patients receiving 23 mg/d (74%) than those receiving 10 mg/d (64%). The most common TEAEs in the 23-mg/d and 10-mg/d groups were nausea (12% vs 3%, respectively), vomiting (9% vs 3%), and diarrhea (8% vs 5%) (Table 2).22 These gastrointestinal adverse effects were more frequent during the first month of treatment and were relatively infrequent beyond 1 month. Serious TEAEs, such as falls, urinary tract infection, pneumonia, syncope, aggression, and confusional state, were noted in a similar proportion of patients in the 23-mg/d and 10-mg/d groups; most of these were considered unrelated to treatment. No drug-related deaths occurred during the study. High-dose (23 mg/d) donepezil generally was well tolerated, with a typical ChEI safety profile but superior efficacy.
A recent commentary discussed the issue of effect size and whether a 2.2-point difference on a 100-point scale (the Severe Impairment Battery [SIB]) is clinically meaningful.23 As with all anti-dementia therapies, in any cohort some patients will gain considerably more than 2.2 points on the SIB, which is clinically significant. A 6-month trial is recommended to identify these optimal responders.
Table 2
High-dose vs standard-dose donepezil: Treatment-emergent adverse events
| Adverse event | Donepezil, 23 mg/d | Donepezil,10 mg/d |
|---|---|---|
| Nausea | 12% | 3% |
| Vomiting | 9% | 3% |
| Diarrhea | 8% | 5% |
| Anorexia | 5% | 2% |
| Dizziness | 5% | 3% |
| Weight decrease | 5% | 3% |
| Headache | 4% | 3% |
| Insomnia | 3% | 2% |
| Urinary incontinence | 3% | 1% |
| Fatigue | 2% | 1% |
| Weakness | 2% | 1% |
| Somnolence | 2% | 1% |
| Contusion | 2% | 0% |
| Source: Reference 22 | ||
High-dose memantine
Memantine is an NMDA receptor antagonist, which works on glutamate, an ubiquitous neurotransmitter in the brain that serves many functions. For reasons that are not fully understood, in AD glutamate becomes excitotoxic and causes neuronal death.
Some researchers have hypothesized that if safe and well tolerated, a memantine dose >20 mg/d may have better efficacy than a lower dose. Memantine’s manufacturer has developed an extended-release (ER), once-daily formulation of memantine, 28 mg/d, to improve adherence and possibly increase efficacy.10,11 Because of memantine ER’s relatively slow absorption rate and longer median Tmax, of 12 hours, there is minimal fluctuation in plasma levels during steady-state dosing intervals compared with the immediate-release (IR) formulation.10
In a phase I study of 24 healthy volunteers that investigated the safety, tolerability, and pharmacokinetics of memantine ER, 28 mg/d, TEAEs were mild; the most common were headache, somnolence, and dizziness.10 During memantine treatment, there were no serious adverse events, potential significant changes in patients’ vital signs, or deaths.
Memantine ER plus ChEI. A multicenter, multinational, randomized, double-blind study compared memantine ER, 28 mg/d, and placebo in patients with moderate to severe AD (MMSE: 3 to 14).11 All patients were receiving concurrent, stable ChEI treatment (donepezil, rivastigmine, or galantamine) for ≥3 months before the study. Patients treated with memantine ER, 28 mg/d, and ChEI (n = 342) showed a significant improvement compared with the placebo/ChEI group (n = 335) in cognition and global functioning. Patients receiving memantine/ChEI also showed statistically significant benefits on behavior and verbal fluency testing compared with patients receiving placebo/ChEI. Memantine was well tolerated; most adverse events were mild or moderate. The most common adverse events in the memantine/ChEI group that occurred at a higher rate relative to the placebo/ChEI group were headache (5.6% vs 5.1%, respectively), diarrhea (5.0% vs 3.9%), and dizziness (4.7% vs 1.5%). There were no deaths related to memantine (Table 3).11
Memantine ER, 28 mg/d, may be tolerated better than the IR formulation because of less plasma level fluctuation during the steady-state dosing interval. Also, memantine ER, 28 mg/d, may offer better efficacy over memantine IR, 20 mg/d, because of dose-dependent cognitive, global, and behavioral effects. In addition, once-daily dosing of memantine ER may improve adherence compared with the IR formulation.24
In patients with severe renal impairment, dosage of memantine IR should be reduced from 20 mg/d to 10 mg/d.25 However, there is no available information regarding the dosing, safety, and tolerability of memantine ER, 28 mg/d, in patients with renal disease.
Table3
High-dose memantine: Treatment-emergent adverse eventsa
| Adverse event | Placebo (n = 335) | Memantine ER (n = 341) |
|---|---|---|
| Any TEAE | 214 (63.9%) | 214 (62.8%) |
| Fall | 26 (7.8%) | 19 (5.6%) |
| Urinary tract infection | 24 (7.2%) | 19 (5.6%) |
| Headache | 17 (5.1%) | 19 (5.6%) |
| Diarrhea | 13 (3.9%) | 17 (5.0%) |
| Dizziness | 5 (1.5%) | 16 (4.7%) |
| Influenza | 9 (2.7%) | 15 (4.4%) |
| Insomnia | 16 (4.8%) | 14 (4.1%) |
| Agitation | 15 (4.5%) | 14 (4.1%) |
| Hypertension | 8 (2.4%) | 13 (3.8%) |
| Anxiety | 9 (2.7%) | 12 (3.5%) |
| Depression | 5 (1.5%) | 11 (3.2%) |
| Weight increased | 3 (0.9%) | 11 (3.2%) |
| Constipation | 4 (1.2%) | 10 (2.9%) |
| Somnolence | 4 (1.2%) | 10 (2.9%) |
| Back pain | 2 (0.6%) | 9 (2.6%) |
| Aggression | 5 (1.5%) | 8 (2.3%) |
| Hypotension | 5 (1.5%) | 7 (2.1%) |
| Vomiting | 4 (1.2%) | 7 (2.1%) |
| Abdominal pain | 2 (0.6%) | 7 (2.1%) |
| Nasopharyngitis | 10 (3.0%) | 6 (1.8%) |
| Confusional state | 7 (2.1%) | 6 (1.8%) |
| Weight decreased | 11 (3.3%) | 5 (1.5%) |
| Nausea | 7 (2.1%) | 5 (1.5%) |
| Irritability | 8 (2.4%) | 4 (1.2%) |
| Cough | 8 (2.4%) | 3 (0.9%) |
| aData [n (%)] include all adverse events experienced by ≥2% patients in either group (safety population). Adverse events that were experienced at twice the rate in 1 group compared with the other are indicated by bold type ER: extended-release (28 mg); TEAE: treatment-emergent adverse event Source: Reference 11 | ||
Recommendations
Because there are few FDA-approved treatments for AD, higher doses of donepezil or memantine may be an option for patients who have “maxed out” on their AD therapy or no longer respond to lower doses. Higher doses of donepezil (23 mg/d) and memantine (28 mg/d) could improve medication adherence because both are once-daily preparations. In clinical trials, donepezil, 23 mg/d, was more effective than donepezil, 10 mg/d.9 Whether memantine ER, 28 mg/d, is superior to memantine IR, 20 mg/d, needs to be investigated in head-to-head, double-blind, controlled studies.
For patients with moderate to severe AD, donepezil, 23 mg, is associated with greater benefits in cognition compared with donepezil, 10 mg/d.9 Similarly, because of potentially superior efficacy because of a higher dose, memantine ER, 28 mg, might best help patients with moderate to severe AD, specifically those who either don’t respond or lose response to memantine IR, 20 mg/d. Combining a ChEI, such as donepezil, with memantine is associated with slower cognitive decline and short and long-term benefits on measures of cognition, activities of daily living, global outcome, and behavior.7,26 However, additional clinical trials are needed to assess the safety, tolerability, and efficacy of combination therapy with higher doses of donepezil and memantine ER.
Related Resources
- Alzheimer’s Disease Education and Referral Center. www.nia.nih.gov/Alzheimers.
- Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med. 2006;57:513-533.
Drug Brand Names
- Donepezil • Aricept
- Galantamine • Razadyne
- Memantine • Namenda
- Rivastigmine • Exelon
- Tacrine • Cognex
Disclosures
Dr. Grossberg’s academic department has received research funding from Forest Pharmaceuticals and Pfizer Inc. Dr. Grossberg has received grant/research support from Baxter BioScience, Forest Pharmaceuticals, Janssen, the National Institutes of Health, Novartis, and Pfizer, Inc.; is a consultant to Baxter BioScience, Forest Pharmaceuticals, Merck, Novartis, and Otsuka; and is on the Safety Monitoring Committee for Merck.
Dr. Singh reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alzheimer’s Association, Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60(8):1119-1122.
3. Alzheimer’s Disease Education and Referral Center. Alzheimer’s disease medications. http://www.nia.nih.gov/alzheimers/publication/alzheimers-disease-medications-fact-sheet. Accessed May 10 2012.
4. Osborn GG, Saunders AV. Current treatments for patients with Alzheimer disease. J Am Osteopath Assoc. 2010;110(9 suppl 8):S16-S26.
5. Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379-397.
6. Cummings JL. Alzheimer’s disease. N Engl J Med. 2004;351(1):56-67.
7. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
8. Xiong G, Doraiswamy PM. Combination drug therapy for Alzheimer’s disease: what is evidence-based and what is not? Geriatrics. 2005;60(6):22-26.
9. Farlow MR, Salloway S, Tariot PN, et al. Effectiveness and tolerability of high (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week, randomized, double-blind study. Clin Ther. 2010;32(7):1234-1251.
10. Periclou A, Hu Y. Extended-release memantine capsule (28 mg once daily): a multiple dose, open-label study evaluating steady-state pharmacokinetics in healthy volunteers. Poster presented at 11th International Conference on Alzheimer’s Disease; July 26-31, 2008; Chicago, IL.
11. Grossberg GT, Manes F, Allegri R, et al. A multinational, randomized, double-blind, placebo-controlled, parallel-group trial of memantine extended-release capsule (28 mg, once daily) in patients with moderate to severe Alzheimer’s disease. Poster presented at 11th International Conference on Alzheimer’s Disease; July 26-31, 2008; Chicago, IL.
12. Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb Cortex. 1996;6(2):165-177.
13. Whitehouse PJ. The cholinergic deficit in Alzheimer’s disease. J Clin Psychiatry. 1998;59(suppl 13):19-22.
14. Teipel SJ, Flatz WH, Heinsen H, et al. Measurement of basal forebrain atrophy in Alzheimer’s disease using MRI. Brain. 2005;128(11):2626-2644.
15. Shintani EY, Uchida KM. Donepezil: an anticholinesterase inhibitor for Alzheimer’s disease. Am J Health Syst Pharm. 1997;54(24):2805-2810.
16. Homma A, Imai Y, Tago H, et al. Donepezil treatment of patients with severe Alzheimer’s disease in a Japanese population: results from a 24-week, double-blind, placebo-controlled, randomized trial. Dement Geriatr Cogn Disord. 2008;25(5):399-407.
17. Whitehead A, Perdomo C, Pratt RD, et al. Donepezil for the symptomatic treatment of patients with mild to moderate Alzheimer’s disease: a meta-analysis of individual patient data from randomised controlled trials. Int J Geriatr Psychiatry. 2004;19(7):624-633.
18. Nozawa M, Ichimiya Y, Nozawa E, et al. Clinical effects of high oral dose of donepezil for patients with Alzheimer’s disease in Japan. Psychogeriatrics. 2009;9(2):50-55.
19. Kuhl DE, Minoshima S, Frey KA, et al. Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann Neurol. 2000;48(3):391-395.
20. Bohnen NI, Kaufer DI, Hendrickson R, et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2005;76(3):315-319.
21. Doody RS, Corey-Bloom J, Zhang R, et al. Safety and tolerability of donepezil at doses up to 20 mg/day: results from a pilot study in patients with Alzheimer’s disease. Drugs Aging. 2008;25(2):163-174.
22. Aricept [package insert]. Woodcliff Lake NJ: Eisai Co.; 2012.
23. Schwartz LM, Woloshin S. How the FDA forgot the evidence: the case of donepezil 23 mg. BMJ. 2012;344:e1086.-doi: 10.1136/bmj.e1086.
24. Saini SD, Schoenfeld P, Kaulback K, et al. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manag Care. 2009;15(6):e22-e33.
25. Periclou A, Ventura D, Rao N, et al. Pharmacokinetic study of memantine in healthy and renally impaired subjects. Clin Pharmacol Ther. 2006;79(1):134-143.
26. Atri A, Shaughnessy LW, Locascio JJ, et al. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2008;22(3):209-221.
Although cholinesterase inhibitors (ChEIs) and memantine at standard doses may slow the progression of Alzheimer’s disease (AD) as assessed by cognitive, functional, and global measures, this effect is relatively modest. For the estimated 5.4 million Americans with AD1—more than one-half of whom have moderate to severe disease2—there is a great need for new approaches to slow AD progression.
High doses of donepezil or memantine may be the next step in achieving better results than standard pharmacologic treatments for AD. This article presents the possible benefits and indications for high doses of donepezil (23 mg/d) and memantine (28 mg/d) for managing moderate to severe AD and their safety and tolerability profiles.
Current treatments offer modest benefits
AD treatments comprise 2 categories: ChEIs (donepezil, rivastigmine, and galantamine) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine (Table 1).3,4 All ChEIs are FDA-approved for mild to moderate AD; donepezil also is approved for severe AD. Memantine is approved for moderate to severe AD, either alone or in combination with ChEIs. Until recently, the maximum FDA-approved doses were donepezil, 10 mg/d, and memantine, 20 mg/d. However, these dosages are associated with only modest beneficial effects in managing cognitive deterioration in patients with moderate to severe dementia.5,6 Studies have reported that combining a ChEI, such as donepezil, and memantine is well tolerated and may result in synergistic benefits by affecting different neurotransmitters in patients with moderate to severe AD.7,8
Recently, the FDA approved higher daily doses of donepezil (23 mg) and memantine (28 mg) for moderate to severe AD on the basis of positive phase III trial results.9-11 Donepezil, 23 mg/d, currently is marketed in the United States; the availability date for memantine, 28 mg/d, was undetermined at press time.
Table 1
FDA-approved treatments for Alzheimer’s disease
| Drug | Maximum daily dose | Mechanism of action | Indication | Common side effects/comments |
|---|---|---|---|---|
| Tacrine | 160 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, loss of appetite, diarrhea. First ChEI to be approved, but rarely used because of associated possible hepatotoxicity |
| Donepezil | 10 mg/d | ChEI | All stages of AD | Nausea, vomiting, loss of appetite, diarrhea, sleep disturbance |
| Rivastigmine | 12 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Galantamine | 24 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Memantine | 20 mg/d | NMDA receptor antagonist | Moderate to severe AD | Dizziness, headache, constipation, confusion |
| Galantamine ER | 24 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Rivastigmine transdermal system | 9.5 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Donepezil 23 | 23 mg/d | ChEI | Moderate to severe AD | Nausea, vomiting, diarrhea |
| Memantine ER | 28 mg/d | NMDA receptor antagonist | Moderate to severe AD | Dizziness, headache, constipation, confusion |
| AD: Alzheimer’s disease; ChEI: cholinesterase inhibitor; ER: extended release; NMDA: N-methyl-D-aspartate Source: References 3,4 | ||||
High-dose donepezil (23 mg/d)
Cognitive decline with AD has been associated with increasing loss of cholinergic neurons and cholinergic activities, particularly in areas associated with memory/cognition and learning, including cortical areas involving the temporal lobe, hippocampus, and nucleus basalis of Meynert.12-14 In addition, evidence suggests that increasing levels of acetylcholine by using ChEIs can enhance cognitive function.13,15
Donepezil is a selective, reversible ChEI believed to enhance central cholinergic function.15 Randomized clinical trials assessing dose-response with donepezil, 5 mg/d and 10 mg/d, have demonstrated more benefit in cognition with either dose than placebo. The 10 mg/d dose was more effective than 5 mg/d in patients with mild to moderate and severe AD.16-18 In patients with advanced AD who are stable on 5 mg/d, increasing to 10 mg/d could slow the progression of cognitive decline.18
Rationale for higher doses. Positron emission tomography studies have shown that at stable doses of donepezil, 5 mg/d or 10 mg/d, average cortical acetylcholinesterase (AChE) inhibition was <30%.19,20 Based on these findings, researchers thought that cortical AChE inhibition may be suboptimal with donepezil, 10 mg/d, and that higher doses of ChEI may be required in patients with more advanced AD—and therefore more cholinergic loss—for adequate cholinesterase inhibition. In a pilot study of patients with mild to moderate AD, higher doses of donepezil (15 mg/d and 20 mg/d) were reported to be safe and well tolerated.21
The 23-mg/d donepezil formulation was developed to provide a higher dose administered once daily without a sharp rise in peak concentration. The FDA approved donepezil, 23 mg/d, for patients with moderate to severe AD on the basis of phase III trial results.9,22 In a randomized, double-blind, multicenter, head-to-head clinical trial, >1,400 patients with moderate to severe AD (Mini-Mental State Exam [MMSE]: 0 to 20) on a stable dose of donepezil, 10 mg/d, for ≥3 months were randomly assigned to receive high-dose donepezil (23 mg/d) or standard-dose donepezil (10 mg/d) for 24 weeks.9,22 Patients in the 23-mg/d group showed a statistically significant improvement in cognition compared with the 10-mg/d group. The difference between groups on a measure of global improvement was not significant.9,22 However, in a post-hoc analysis, it was demonstrated that a subgroup of patients with more severe cognitive impairment (baseline MMSE: 0 to 16), showed significant improvement in cognition as well as global functioning.9
Overall, treatment-emergent adverse events (TEAEs) during the study were higher in patients receiving 23 mg/d (74%) than those receiving 10 mg/d (64%). The most common TEAEs in the 23-mg/d and 10-mg/d groups were nausea (12% vs 3%, respectively), vomiting (9% vs 3%), and diarrhea (8% vs 5%) (Table 2).22 These gastrointestinal adverse effects were more frequent during the first month of treatment and were relatively infrequent beyond 1 month. Serious TEAEs, such as falls, urinary tract infection, pneumonia, syncope, aggression, and confusional state, were noted in a similar proportion of patients in the 23-mg/d and 10-mg/d groups; most of these were considered unrelated to treatment. No drug-related deaths occurred during the study. High-dose (23 mg/d) donepezil generally was well tolerated, with a typical ChEI safety profile but superior efficacy.
A recent commentary discussed the issue of effect size and whether a 2.2-point difference on a 100-point scale (the Severe Impairment Battery [SIB]) is clinically meaningful.23 As with all anti-dementia therapies, in any cohort some patients will gain considerably more than 2.2 points on the SIB, which is clinically significant. A 6-month trial is recommended to identify these optimal responders.
Table 2
High-dose vs standard-dose donepezil: Treatment-emergent adverse events
| Adverse event | Donepezil, 23 mg/d | Donepezil,10 mg/d |
|---|---|---|
| Nausea | 12% | 3% |
| Vomiting | 9% | 3% |
| Diarrhea | 8% | 5% |
| Anorexia | 5% | 2% |
| Dizziness | 5% | 3% |
| Weight decrease | 5% | 3% |
| Headache | 4% | 3% |
| Insomnia | 3% | 2% |
| Urinary incontinence | 3% | 1% |
| Fatigue | 2% | 1% |
| Weakness | 2% | 1% |
| Somnolence | 2% | 1% |
| Contusion | 2% | 0% |
| Source: Reference 22 | ||
High-dose memantine
Memantine is an NMDA receptor antagonist, which works on glutamate, an ubiquitous neurotransmitter in the brain that serves many functions. For reasons that are not fully understood, in AD glutamate becomes excitotoxic and causes neuronal death.
Some researchers have hypothesized that if safe and well tolerated, a memantine dose >20 mg/d may have better efficacy than a lower dose. Memantine’s manufacturer has developed an extended-release (ER), once-daily formulation of memantine, 28 mg/d, to improve adherence and possibly increase efficacy.10,11 Because of memantine ER’s relatively slow absorption rate and longer median Tmax, of 12 hours, there is minimal fluctuation in plasma levels during steady-state dosing intervals compared with the immediate-release (IR) formulation.10
In a phase I study of 24 healthy volunteers that investigated the safety, tolerability, and pharmacokinetics of memantine ER, 28 mg/d, TEAEs were mild; the most common were headache, somnolence, and dizziness.10 During memantine treatment, there were no serious adverse events, potential significant changes in patients’ vital signs, or deaths.
Memantine ER plus ChEI. A multicenter, multinational, randomized, double-blind study compared memantine ER, 28 mg/d, and placebo in patients with moderate to severe AD (MMSE: 3 to 14).11 All patients were receiving concurrent, stable ChEI treatment (donepezil, rivastigmine, or galantamine) for ≥3 months before the study. Patients treated with memantine ER, 28 mg/d, and ChEI (n = 342) showed a significant improvement compared with the placebo/ChEI group (n = 335) in cognition and global functioning. Patients receiving memantine/ChEI also showed statistically significant benefits on behavior and verbal fluency testing compared with patients receiving placebo/ChEI. Memantine was well tolerated; most adverse events were mild or moderate. The most common adverse events in the memantine/ChEI group that occurred at a higher rate relative to the placebo/ChEI group were headache (5.6% vs 5.1%, respectively), diarrhea (5.0% vs 3.9%), and dizziness (4.7% vs 1.5%). There were no deaths related to memantine (Table 3).11
Memantine ER, 28 mg/d, may be tolerated better than the IR formulation because of less plasma level fluctuation during the steady-state dosing interval. Also, memantine ER, 28 mg/d, may offer better efficacy over memantine IR, 20 mg/d, because of dose-dependent cognitive, global, and behavioral effects. In addition, once-daily dosing of memantine ER may improve adherence compared with the IR formulation.24
In patients with severe renal impairment, dosage of memantine IR should be reduced from 20 mg/d to 10 mg/d.25 However, there is no available information regarding the dosing, safety, and tolerability of memantine ER, 28 mg/d, in patients with renal disease.
Table3
High-dose memantine: Treatment-emergent adverse eventsa
| Adverse event | Placebo (n = 335) | Memantine ER (n = 341) |
|---|---|---|
| Any TEAE | 214 (63.9%) | 214 (62.8%) |
| Fall | 26 (7.8%) | 19 (5.6%) |
| Urinary tract infection | 24 (7.2%) | 19 (5.6%) |
| Headache | 17 (5.1%) | 19 (5.6%) |
| Diarrhea | 13 (3.9%) | 17 (5.0%) |
| Dizziness | 5 (1.5%) | 16 (4.7%) |
| Influenza | 9 (2.7%) | 15 (4.4%) |
| Insomnia | 16 (4.8%) | 14 (4.1%) |
| Agitation | 15 (4.5%) | 14 (4.1%) |
| Hypertension | 8 (2.4%) | 13 (3.8%) |
| Anxiety | 9 (2.7%) | 12 (3.5%) |
| Depression | 5 (1.5%) | 11 (3.2%) |
| Weight increased | 3 (0.9%) | 11 (3.2%) |
| Constipation | 4 (1.2%) | 10 (2.9%) |
| Somnolence | 4 (1.2%) | 10 (2.9%) |
| Back pain | 2 (0.6%) | 9 (2.6%) |
| Aggression | 5 (1.5%) | 8 (2.3%) |
| Hypotension | 5 (1.5%) | 7 (2.1%) |
| Vomiting | 4 (1.2%) | 7 (2.1%) |
| Abdominal pain | 2 (0.6%) | 7 (2.1%) |
| Nasopharyngitis | 10 (3.0%) | 6 (1.8%) |
| Confusional state | 7 (2.1%) | 6 (1.8%) |
| Weight decreased | 11 (3.3%) | 5 (1.5%) |
| Nausea | 7 (2.1%) | 5 (1.5%) |
| Irritability | 8 (2.4%) | 4 (1.2%) |
| Cough | 8 (2.4%) | 3 (0.9%) |
| aData [n (%)] include all adverse events experienced by ≥2% patients in either group (safety population). Adverse events that were experienced at twice the rate in 1 group compared with the other are indicated by bold type ER: extended-release (28 mg); TEAE: treatment-emergent adverse event Source: Reference 11 | ||
Recommendations
Because there are few FDA-approved treatments for AD, higher doses of donepezil or memantine may be an option for patients who have “maxed out” on their AD therapy or no longer respond to lower doses. Higher doses of donepezil (23 mg/d) and memantine (28 mg/d) could improve medication adherence because both are once-daily preparations. In clinical trials, donepezil, 23 mg/d, was more effective than donepezil, 10 mg/d.9 Whether memantine ER, 28 mg/d, is superior to memantine IR, 20 mg/d, needs to be investigated in head-to-head, double-blind, controlled studies.
For patients with moderate to severe AD, donepezil, 23 mg, is associated with greater benefits in cognition compared with donepezil, 10 mg/d.9 Similarly, because of potentially superior efficacy because of a higher dose, memantine ER, 28 mg, might best help patients with moderate to severe AD, specifically those who either don’t respond or lose response to memantine IR, 20 mg/d. Combining a ChEI, such as donepezil, with memantine is associated with slower cognitive decline and short and long-term benefits on measures of cognition, activities of daily living, global outcome, and behavior.7,26 However, additional clinical trials are needed to assess the safety, tolerability, and efficacy of combination therapy with higher doses of donepezil and memantine ER.
Related Resources
- Alzheimer’s Disease Education and Referral Center. www.nia.nih.gov/Alzheimers.
- Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med. 2006;57:513-533.
Drug Brand Names
- Donepezil • Aricept
- Galantamine • Razadyne
- Memantine • Namenda
- Rivastigmine • Exelon
- Tacrine • Cognex
Disclosures
Dr. Grossberg’s academic department has received research funding from Forest Pharmaceuticals and Pfizer Inc. Dr. Grossberg has received grant/research support from Baxter BioScience, Forest Pharmaceuticals, Janssen, the National Institutes of Health, Novartis, and Pfizer, Inc.; is a consultant to Baxter BioScience, Forest Pharmaceuticals, Merck, Novartis, and Otsuka; and is on the Safety Monitoring Committee for Merck.
Dr. Singh reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Although cholinesterase inhibitors (ChEIs) and memantine at standard doses may slow the progression of Alzheimer’s disease (AD) as assessed by cognitive, functional, and global measures, this effect is relatively modest. For the estimated 5.4 million Americans with AD1—more than one-half of whom have moderate to severe disease2—there is a great need for new approaches to slow AD progression.
High doses of donepezil or memantine may be the next step in achieving better results than standard pharmacologic treatments for AD. This article presents the possible benefits and indications for high doses of donepezil (23 mg/d) and memantine (28 mg/d) for managing moderate to severe AD and their safety and tolerability profiles.
Current treatments offer modest benefits
AD treatments comprise 2 categories: ChEIs (donepezil, rivastigmine, and galantamine) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine (Table 1).3,4 All ChEIs are FDA-approved for mild to moderate AD; donepezil also is approved for severe AD. Memantine is approved for moderate to severe AD, either alone or in combination with ChEIs. Until recently, the maximum FDA-approved doses were donepezil, 10 mg/d, and memantine, 20 mg/d. However, these dosages are associated with only modest beneficial effects in managing cognitive deterioration in patients with moderate to severe dementia.5,6 Studies have reported that combining a ChEI, such as donepezil, and memantine is well tolerated and may result in synergistic benefits by affecting different neurotransmitters in patients with moderate to severe AD.7,8
Recently, the FDA approved higher daily doses of donepezil (23 mg) and memantine (28 mg) for moderate to severe AD on the basis of positive phase III trial results.9-11 Donepezil, 23 mg/d, currently is marketed in the United States; the availability date for memantine, 28 mg/d, was undetermined at press time.
Table 1
FDA-approved treatments for Alzheimer’s disease
| Drug | Maximum daily dose | Mechanism of action | Indication | Common side effects/comments |
|---|---|---|---|---|
| Tacrine | 160 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, loss of appetite, diarrhea. First ChEI to be approved, but rarely used because of associated possible hepatotoxicity |
| Donepezil | 10 mg/d | ChEI | All stages of AD | Nausea, vomiting, loss of appetite, diarrhea, sleep disturbance |
| Rivastigmine | 12 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Galantamine | 24 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Memantine | 20 mg/d | NMDA receptor antagonist | Moderate to severe AD | Dizziness, headache, constipation, confusion |
| Galantamine ER | 24 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Rivastigmine transdermal system | 9.5 mg/d | ChEI | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite |
| Donepezil 23 | 23 mg/d | ChEI | Moderate to severe AD | Nausea, vomiting, diarrhea |
| Memantine ER | 28 mg/d | NMDA receptor antagonist | Moderate to severe AD | Dizziness, headache, constipation, confusion |
| AD: Alzheimer’s disease; ChEI: cholinesterase inhibitor; ER: extended release; NMDA: N-methyl-D-aspartate Source: References 3,4 | ||||
High-dose donepezil (23 mg/d)
Cognitive decline with AD has been associated with increasing loss of cholinergic neurons and cholinergic activities, particularly in areas associated with memory/cognition and learning, including cortical areas involving the temporal lobe, hippocampus, and nucleus basalis of Meynert.12-14 In addition, evidence suggests that increasing levels of acetylcholine by using ChEIs can enhance cognitive function.13,15
Donepezil is a selective, reversible ChEI believed to enhance central cholinergic function.15 Randomized clinical trials assessing dose-response with donepezil, 5 mg/d and 10 mg/d, have demonstrated more benefit in cognition with either dose than placebo. The 10 mg/d dose was more effective than 5 mg/d in patients with mild to moderate and severe AD.16-18 In patients with advanced AD who are stable on 5 mg/d, increasing to 10 mg/d could slow the progression of cognitive decline.18
Rationale for higher doses. Positron emission tomography studies have shown that at stable doses of donepezil, 5 mg/d or 10 mg/d, average cortical acetylcholinesterase (AChE) inhibition was <30%.19,20 Based on these findings, researchers thought that cortical AChE inhibition may be suboptimal with donepezil, 10 mg/d, and that higher doses of ChEI may be required in patients with more advanced AD—and therefore more cholinergic loss—for adequate cholinesterase inhibition. In a pilot study of patients with mild to moderate AD, higher doses of donepezil (15 mg/d and 20 mg/d) were reported to be safe and well tolerated.21
The 23-mg/d donepezil formulation was developed to provide a higher dose administered once daily without a sharp rise in peak concentration. The FDA approved donepezil, 23 mg/d, for patients with moderate to severe AD on the basis of phase III trial results.9,22 In a randomized, double-blind, multicenter, head-to-head clinical trial, >1,400 patients with moderate to severe AD (Mini-Mental State Exam [MMSE]: 0 to 20) on a stable dose of donepezil, 10 mg/d, for ≥3 months were randomly assigned to receive high-dose donepezil (23 mg/d) or standard-dose donepezil (10 mg/d) for 24 weeks.9,22 Patients in the 23-mg/d group showed a statistically significant improvement in cognition compared with the 10-mg/d group. The difference between groups on a measure of global improvement was not significant.9,22 However, in a post-hoc analysis, it was demonstrated that a subgroup of patients with more severe cognitive impairment (baseline MMSE: 0 to 16), showed significant improvement in cognition as well as global functioning.9
Overall, treatment-emergent adverse events (TEAEs) during the study were higher in patients receiving 23 mg/d (74%) than those receiving 10 mg/d (64%). The most common TEAEs in the 23-mg/d and 10-mg/d groups were nausea (12% vs 3%, respectively), vomiting (9% vs 3%), and diarrhea (8% vs 5%) (Table 2).22 These gastrointestinal adverse effects were more frequent during the first month of treatment and were relatively infrequent beyond 1 month. Serious TEAEs, such as falls, urinary tract infection, pneumonia, syncope, aggression, and confusional state, were noted in a similar proportion of patients in the 23-mg/d and 10-mg/d groups; most of these were considered unrelated to treatment. No drug-related deaths occurred during the study. High-dose (23 mg/d) donepezil generally was well tolerated, with a typical ChEI safety profile but superior efficacy.
A recent commentary discussed the issue of effect size and whether a 2.2-point difference on a 100-point scale (the Severe Impairment Battery [SIB]) is clinically meaningful.23 As with all anti-dementia therapies, in any cohort some patients will gain considerably more than 2.2 points on the SIB, which is clinically significant. A 6-month trial is recommended to identify these optimal responders.
Table 2
High-dose vs standard-dose donepezil: Treatment-emergent adverse events
| Adverse event | Donepezil, 23 mg/d | Donepezil,10 mg/d |
|---|---|---|
| Nausea | 12% | 3% |
| Vomiting | 9% | 3% |
| Diarrhea | 8% | 5% |
| Anorexia | 5% | 2% |
| Dizziness | 5% | 3% |
| Weight decrease | 5% | 3% |
| Headache | 4% | 3% |
| Insomnia | 3% | 2% |
| Urinary incontinence | 3% | 1% |
| Fatigue | 2% | 1% |
| Weakness | 2% | 1% |
| Somnolence | 2% | 1% |
| Contusion | 2% | 0% |
| Source: Reference 22 | ||
High-dose memantine
Memantine is an NMDA receptor antagonist, which works on glutamate, an ubiquitous neurotransmitter in the brain that serves many functions. For reasons that are not fully understood, in AD glutamate becomes excitotoxic and causes neuronal death.
Some researchers have hypothesized that if safe and well tolerated, a memantine dose >20 mg/d may have better efficacy than a lower dose. Memantine’s manufacturer has developed an extended-release (ER), once-daily formulation of memantine, 28 mg/d, to improve adherence and possibly increase efficacy.10,11 Because of memantine ER’s relatively slow absorption rate and longer median Tmax, of 12 hours, there is minimal fluctuation in plasma levels during steady-state dosing intervals compared with the immediate-release (IR) formulation.10
In a phase I study of 24 healthy volunteers that investigated the safety, tolerability, and pharmacokinetics of memantine ER, 28 mg/d, TEAEs were mild; the most common were headache, somnolence, and dizziness.10 During memantine treatment, there were no serious adverse events, potential significant changes in patients’ vital signs, or deaths.
Memantine ER plus ChEI. A multicenter, multinational, randomized, double-blind study compared memantine ER, 28 mg/d, and placebo in patients with moderate to severe AD (MMSE: 3 to 14).11 All patients were receiving concurrent, stable ChEI treatment (donepezil, rivastigmine, or galantamine) for ≥3 months before the study. Patients treated with memantine ER, 28 mg/d, and ChEI (n = 342) showed a significant improvement compared with the placebo/ChEI group (n = 335) in cognition and global functioning. Patients receiving memantine/ChEI also showed statistically significant benefits on behavior and verbal fluency testing compared with patients receiving placebo/ChEI. Memantine was well tolerated; most adverse events were mild or moderate. The most common adverse events in the memantine/ChEI group that occurred at a higher rate relative to the placebo/ChEI group were headache (5.6% vs 5.1%, respectively), diarrhea (5.0% vs 3.9%), and dizziness (4.7% vs 1.5%). There were no deaths related to memantine (Table 3).11
Memantine ER, 28 mg/d, may be tolerated better than the IR formulation because of less plasma level fluctuation during the steady-state dosing interval. Also, memantine ER, 28 mg/d, may offer better efficacy over memantine IR, 20 mg/d, because of dose-dependent cognitive, global, and behavioral effects. In addition, once-daily dosing of memantine ER may improve adherence compared with the IR formulation.24
In patients with severe renal impairment, dosage of memantine IR should be reduced from 20 mg/d to 10 mg/d.25 However, there is no available information regarding the dosing, safety, and tolerability of memantine ER, 28 mg/d, in patients with renal disease.
Table3
High-dose memantine: Treatment-emergent adverse eventsa
| Adverse event | Placebo (n = 335) | Memantine ER (n = 341) |
|---|---|---|
| Any TEAE | 214 (63.9%) | 214 (62.8%) |
| Fall | 26 (7.8%) | 19 (5.6%) |
| Urinary tract infection | 24 (7.2%) | 19 (5.6%) |
| Headache | 17 (5.1%) | 19 (5.6%) |
| Diarrhea | 13 (3.9%) | 17 (5.0%) |
| Dizziness | 5 (1.5%) | 16 (4.7%) |
| Influenza | 9 (2.7%) | 15 (4.4%) |
| Insomnia | 16 (4.8%) | 14 (4.1%) |
| Agitation | 15 (4.5%) | 14 (4.1%) |
| Hypertension | 8 (2.4%) | 13 (3.8%) |
| Anxiety | 9 (2.7%) | 12 (3.5%) |
| Depression | 5 (1.5%) | 11 (3.2%) |
| Weight increased | 3 (0.9%) | 11 (3.2%) |
| Constipation | 4 (1.2%) | 10 (2.9%) |
| Somnolence | 4 (1.2%) | 10 (2.9%) |
| Back pain | 2 (0.6%) | 9 (2.6%) |
| Aggression | 5 (1.5%) | 8 (2.3%) |
| Hypotension | 5 (1.5%) | 7 (2.1%) |
| Vomiting | 4 (1.2%) | 7 (2.1%) |
| Abdominal pain | 2 (0.6%) | 7 (2.1%) |
| Nasopharyngitis | 10 (3.0%) | 6 (1.8%) |
| Confusional state | 7 (2.1%) | 6 (1.8%) |
| Weight decreased | 11 (3.3%) | 5 (1.5%) |
| Nausea | 7 (2.1%) | 5 (1.5%) |
| Irritability | 8 (2.4%) | 4 (1.2%) |
| Cough | 8 (2.4%) | 3 (0.9%) |
| aData [n (%)] include all adverse events experienced by ≥2% patients in either group (safety population). Adverse events that were experienced at twice the rate in 1 group compared with the other are indicated by bold type ER: extended-release (28 mg); TEAE: treatment-emergent adverse event Source: Reference 11 | ||
Recommendations
Because there are few FDA-approved treatments for AD, higher doses of donepezil or memantine may be an option for patients who have “maxed out” on their AD therapy or no longer respond to lower doses. Higher doses of donepezil (23 mg/d) and memantine (28 mg/d) could improve medication adherence because both are once-daily preparations. In clinical trials, donepezil, 23 mg/d, was more effective than donepezil, 10 mg/d.9 Whether memantine ER, 28 mg/d, is superior to memantine IR, 20 mg/d, needs to be investigated in head-to-head, double-blind, controlled studies.
For patients with moderate to severe AD, donepezil, 23 mg, is associated with greater benefits in cognition compared with donepezil, 10 mg/d.9 Similarly, because of potentially superior efficacy because of a higher dose, memantine ER, 28 mg, might best help patients with moderate to severe AD, specifically those who either don’t respond or lose response to memantine IR, 20 mg/d. Combining a ChEI, such as donepezil, with memantine is associated with slower cognitive decline and short and long-term benefits on measures of cognition, activities of daily living, global outcome, and behavior.7,26 However, additional clinical trials are needed to assess the safety, tolerability, and efficacy of combination therapy with higher doses of donepezil and memantine ER.
Related Resources
- Alzheimer’s Disease Education and Referral Center. www.nia.nih.gov/Alzheimers.
- Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med. 2006;57:513-533.
Drug Brand Names
- Donepezil • Aricept
- Galantamine • Razadyne
- Memantine • Namenda
- Rivastigmine • Exelon
- Tacrine • Cognex
Disclosures
Dr. Grossberg’s academic department has received research funding from Forest Pharmaceuticals and Pfizer Inc. Dr. Grossberg has received grant/research support from Baxter BioScience, Forest Pharmaceuticals, Janssen, the National Institutes of Health, Novartis, and Pfizer, Inc.; is a consultant to Baxter BioScience, Forest Pharmaceuticals, Merck, Novartis, and Otsuka; and is on the Safety Monitoring Committee for Merck.
Dr. Singh reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alzheimer’s Association, Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60(8):1119-1122.
3. Alzheimer’s Disease Education and Referral Center. Alzheimer’s disease medications. http://www.nia.nih.gov/alzheimers/publication/alzheimers-disease-medications-fact-sheet. Accessed May 10 2012.
4. Osborn GG, Saunders AV. Current treatments for patients with Alzheimer disease. J Am Osteopath Assoc. 2010;110(9 suppl 8):S16-S26.
5. Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379-397.
6. Cummings JL. Alzheimer’s disease. N Engl J Med. 2004;351(1):56-67.
7. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
8. Xiong G, Doraiswamy PM. Combination drug therapy for Alzheimer’s disease: what is evidence-based and what is not? Geriatrics. 2005;60(6):22-26.
9. Farlow MR, Salloway S, Tariot PN, et al. Effectiveness and tolerability of high (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week, randomized, double-blind study. Clin Ther. 2010;32(7):1234-1251.
10. Periclou A, Hu Y. Extended-release memantine capsule (28 mg once daily): a multiple dose, open-label study evaluating steady-state pharmacokinetics in healthy volunteers. Poster presented at 11th International Conference on Alzheimer’s Disease; July 26-31, 2008; Chicago, IL.
11. Grossberg GT, Manes F, Allegri R, et al. A multinational, randomized, double-blind, placebo-controlled, parallel-group trial of memantine extended-release capsule (28 mg, once daily) in patients with moderate to severe Alzheimer’s disease. Poster presented at 11th International Conference on Alzheimer’s Disease; July 26-31, 2008; Chicago, IL.
12. Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb Cortex. 1996;6(2):165-177.
13. Whitehouse PJ. The cholinergic deficit in Alzheimer’s disease. J Clin Psychiatry. 1998;59(suppl 13):19-22.
14. Teipel SJ, Flatz WH, Heinsen H, et al. Measurement of basal forebrain atrophy in Alzheimer’s disease using MRI. Brain. 2005;128(11):2626-2644.
15. Shintani EY, Uchida KM. Donepezil: an anticholinesterase inhibitor for Alzheimer’s disease. Am J Health Syst Pharm. 1997;54(24):2805-2810.
16. Homma A, Imai Y, Tago H, et al. Donepezil treatment of patients with severe Alzheimer’s disease in a Japanese population: results from a 24-week, double-blind, placebo-controlled, randomized trial. Dement Geriatr Cogn Disord. 2008;25(5):399-407.
17. Whitehead A, Perdomo C, Pratt RD, et al. Donepezil for the symptomatic treatment of patients with mild to moderate Alzheimer’s disease: a meta-analysis of individual patient data from randomised controlled trials. Int J Geriatr Psychiatry. 2004;19(7):624-633.
18. Nozawa M, Ichimiya Y, Nozawa E, et al. Clinical effects of high oral dose of donepezil for patients with Alzheimer’s disease in Japan. Psychogeriatrics. 2009;9(2):50-55.
19. Kuhl DE, Minoshima S, Frey KA, et al. Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann Neurol. 2000;48(3):391-395.
20. Bohnen NI, Kaufer DI, Hendrickson R, et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2005;76(3):315-319.
21. Doody RS, Corey-Bloom J, Zhang R, et al. Safety and tolerability of donepezil at doses up to 20 mg/day: results from a pilot study in patients with Alzheimer’s disease. Drugs Aging. 2008;25(2):163-174.
22. Aricept [package insert]. Woodcliff Lake NJ: Eisai Co.; 2012.
23. Schwartz LM, Woloshin S. How the FDA forgot the evidence: the case of donepezil 23 mg. BMJ. 2012;344:e1086.-doi: 10.1136/bmj.e1086.
24. Saini SD, Schoenfeld P, Kaulback K, et al. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manag Care. 2009;15(6):e22-e33.
25. Periclou A, Ventura D, Rao N, et al. Pharmacokinetic study of memantine in healthy and renally impaired subjects. Clin Pharmacol Ther. 2006;79(1):134-143.
26. Atri A, Shaughnessy LW, Locascio JJ, et al. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2008;22(3):209-221.
1. Alzheimer’s Association, Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60(8):1119-1122.
3. Alzheimer’s Disease Education and Referral Center. Alzheimer’s disease medications. http://www.nia.nih.gov/alzheimers/publication/alzheimers-disease-medications-fact-sheet. Accessed May 10 2012.
4. Osborn GG, Saunders AV. Current treatments for patients with Alzheimer disease. J Am Osteopath Assoc. 2010;110(9 suppl 8):S16-S26.
5. Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379-397.
6. Cummings JL. Alzheimer’s disease. N Engl J Med. 2004;351(1):56-67.
7. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine Study Group. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
8. Xiong G, Doraiswamy PM. Combination drug therapy for Alzheimer’s disease: what is evidence-based and what is not? Geriatrics. 2005;60(6):22-26.
9. Farlow MR, Salloway S, Tariot PN, et al. Effectiveness and tolerability of high (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: a 24-week, randomized, double-blind study. Clin Ther. 2010;32(7):1234-1251.
10. Periclou A, Hu Y. Extended-release memantine capsule (28 mg once daily): a multiple dose, open-label study evaluating steady-state pharmacokinetics in healthy volunteers. Poster presented at 11th International Conference on Alzheimer’s Disease; July 26-31, 2008; Chicago, IL.
11. Grossberg GT, Manes F, Allegri R, et al. A multinational, randomized, double-blind, placebo-controlled, parallel-group trial of memantine extended-release capsule (28 mg, once daily) in patients with moderate to severe Alzheimer’s disease. Poster presented at 11th International Conference on Alzheimer’s Disease; July 26-31, 2008; Chicago, IL.
12. Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb Cortex. 1996;6(2):165-177.
13. Whitehouse PJ. The cholinergic deficit in Alzheimer’s disease. J Clin Psychiatry. 1998;59(suppl 13):19-22.
14. Teipel SJ, Flatz WH, Heinsen H, et al. Measurement of basal forebrain atrophy in Alzheimer’s disease using MRI. Brain. 2005;128(11):2626-2644.
15. Shintani EY, Uchida KM. Donepezil: an anticholinesterase inhibitor for Alzheimer’s disease. Am J Health Syst Pharm. 1997;54(24):2805-2810.
16. Homma A, Imai Y, Tago H, et al. Donepezil treatment of patients with severe Alzheimer’s disease in a Japanese population: results from a 24-week, double-blind, placebo-controlled, randomized trial. Dement Geriatr Cogn Disord. 2008;25(5):399-407.
17. Whitehead A, Perdomo C, Pratt RD, et al. Donepezil for the symptomatic treatment of patients with mild to moderate Alzheimer’s disease: a meta-analysis of individual patient data from randomised controlled trials. Int J Geriatr Psychiatry. 2004;19(7):624-633.
18. Nozawa M, Ichimiya Y, Nozawa E, et al. Clinical effects of high oral dose of donepezil for patients with Alzheimer’s disease in Japan. Psychogeriatrics. 2009;9(2):50-55.
19. Kuhl DE, Minoshima S, Frey KA, et al. Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann Neurol. 2000;48(3):391-395.
20. Bohnen NI, Kaufer DI, Hendrickson R, et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2005;76(3):315-319.
21. Doody RS, Corey-Bloom J, Zhang R, et al. Safety and tolerability of donepezil at doses up to 20 mg/day: results from a pilot study in patients with Alzheimer’s disease. Drugs Aging. 2008;25(2):163-174.
22. Aricept [package insert]. Woodcliff Lake NJ: Eisai Co.; 2012.
23. Schwartz LM, Woloshin S. How the FDA forgot the evidence: the case of donepezil 23 mg. BMJ. 2012;344:e1086.-doi: 10.1136/bmj.e1086.
24. Saini SD, Schoenfeld P, Kaulback K, et al. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manag Care. 2009;15(6):e22-e33.
25. Periclou A, Ventura D, Rao N, et al. Pharmacokinetic study of memantine in healthy and renally impaired subjects. Clin Pharmacol Ther. 2006;79(1):134-143.
26. Atri A, Shaughnessy LW, Locascio JJ, et al. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2008;22(3):209-221.