User login
Postoperative pulmonary complications of cardiac surgery
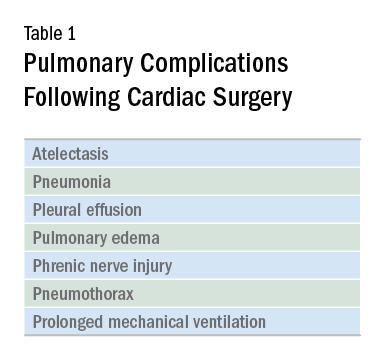
Cardiac surgery patients are sicker today than in previous decades due to an aging population and a rising complexity in medical care. There is an increasing reliance on noncardiac surgeons to care for these patients. The optimal postoperative providers and structure of the ICU where patients are cared for remain unclear, but what is irrefutable is patients’ increased postoperative morbidity. Pulmonary complications are a leading cause of morbidity in these patients, occurring in up to one-fifth of cases (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Common pulmonary complications of cardiac surgery are listed in Table 1. Those complications, captured by The Society of Thoracic Surgeons (STS) Cardiac Surgery Database, include receiving ventilation longer than 24 hours, pneumonia, pulmonary embolism, and pleural effusion requiring drainage (The Society of Thoracic Surgeons. STS National Database. https://www.sts.org/registries-research-center/sts-national-database. Accessed January 9, 2018).
It should come as no surprise that cardiac surgery can have pronounced effects on lung function. The anesthetic agents, chest wall alteration, and direct lung manipulation can all affect pulmonary parameters. Functional residual capacity (FRC) can decrease by up to 20% with anesthesia (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531), and the thoracic manipulation and alteration of rib cage mechanics with a classic median sternotomy approach can lead to decreases in forced vital capacity (FVC) and expiratory volume in the first second of forced expiration (FEV1) that can last for months after surgery. Use of the cardiopulmonary bypass circuit can also lead to bronchoconstriction. These changes in pulmonary function are less pronounced in alternative surgical approaches, such as partial sternotomies (Weissman C. Seminars in Cardiothoracic and Vascular Anesthesia: Pulmonary Complications After Cardiac Surgery. Glen Head, NY: Westminister Publications; 2004).
The most frequent pulmonary consequence of cardiac surgery is atelectasis, seen on postoperative chest radiographs in approximately 50% to 90% of patients (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Induction, apnea during cardiopulmonary bypass, manual compression of the lungs for surgical exposure, internal mammary harvesting, and pleurotomy can lead to atelectasis in the intraoperative setting while weak cough, poor inspiratory efforts, interstitial edema, and immobility further contribute postoperatively (Weissman 2004). While frequently seen, clinically significant pulmonary consequences from this radiographic finding alone are rare (Weissman 2004).
Pleural effusions are seen on immediate postoperative chest radiographs in the majority of patients. Additionally, 10% to 40% of patients develop pleural effusions 2 to 3 weeks after surgery secondary to postpericardiotomy syndrome. While some effusions require drainage and further intervention (eg, hemothorax), most effusions require no specific treatment and resolve over time (Weissman 2004).
The prevalence of pneumonia following cardiac surgery varies based on differences in study populations and diagnostic criteria, but it remains an important source of morbidity and mortality. In one series, postoperative pneumonia occurred in 3.1% of patients, with higher rates observed in patients who were older, had worse left ventricular ejection fraction, had COPD, experienced longer bypass times, and received more red blood cell transfusions in the operating room (Allou N, et al. Crit Care Med. 2014;42[5]:1150). A meta-analysis found that an average of 6.37% of patients developed ventilator-associated pneumonia (VAP), and this rose to 35.2% in those receiving ventilation for greater than 48 hours. Those who developed VAP had an odds ratio of dying of 15.18 (95% CI 5.81-39.68) compared with those who did not (He S, et al. J Thorac Cardiovasc Surg. 2014;148[6]:3148).
A small proportion of patients go on to develop ARDS. While relatively uncommon, ARDS carries a high mortality rate. Many possible etiologies for ARDS in cardiac surgery patients have been proposed, including an inflammatory response related to the cardiopulmonary bypass circuit, reperfusion injury secondary to reduced pulmonary blood flow during bypass, protamine administration, transfusion, hypothermia, and lack of ventilation during bypass (Weissman 2004); (Stephens RS, et al. Ann Thorac Surg. 2013;95[3]:1122). Type of surgery may also play a role, as patients who undergo aortic surgery are at an even greater risk (Stephens 2013). As with other cases of ARDS, treatment is supportive: low tidal volume ventilation and careful management of fluid balance, as well as paralysis, prone positioning, and consideration for extracorporeal membrane oxygenation (ECMO), as appropriate (Stephens 2013).

Therapies to prevent postoperative pulmonary complications have included early extubation, aggressive pain control, deep breathing, physical therapy, early mobilization, and noninvasive ventilation in the form of CPAP and intermittent positive pressure breathing. A meta-analysis of 18 trials looking at the use of various forms of prophylactic postoperative physiotherapy did not show a difference in any measured clinical outcome (Pasquina P, Walder B. Br Med J. 2003;327[7428]:1379).
However, the heterogeneity, short follow-up, and low quality of included studies made it difficult to draw meaningful conclusions on the benefit or lack thereof for these therapies. More recent studies have shown promise for chest physiotherapy started several weeks prior to elective coronary bypass graft surgery and extended CPAP via nasal CPAP mask immediately following extubation (Hulzebos EH. JAMA. 2006;296[15]:1851), (Stephens 2013).
Ongoing areas for improvement include further clarification and standardization of best practices for postcardiac surgery patients, including blood product transfusion, optimal tidal volumes for surgical and postsurgical ventilation, timing of extubation, and the use of preventive therapies in the pre- and postsurgical periods. As providers who care for these patients, understanding how we can improve their postoperative pulmonary recovery will allow us to enhance our patient’s experience.
Dr. Noel is a Critical Care Fellow, Cooper Medical School of Rowan University, Camden, New Jersey.
Cardiac surgery patients are sicker today than in previous decades due to an aging population and a rising complexity in medical care. There is an increasing reliance on noncardiac surgeons to care for these patients. The optimal postoperative providers and structure of the ICU where patients are cared for remain unclear, but what is irrefutable is patients’ increased postoperative morbidity. Pulmonary complications are a leading cause of morbidity in these patients, occurring in up to one-fifth of cases (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Common pulmonary complications of cardiac surgery are listed in Table 1. Those complications, captured by The Society of Thoracic Surgeons (STS) Cardiac Surgery Database, include receiving ventilation longer than 24 hours, pneumonia, pulmonary embolism, and pleural effusion requiring drainage (The Society of Thoracic Surgeons. STS National Database. https://www.sts.org/registries-research-center/sts-national-database. Accessed January 9, 2018).
It should come as no surprise that cardiac surgery can have pronounced effects on lung function. The anesthetic agents, chest wall alteration, and direct lung manipulation can all affect pulmonary parameters. Functional residual capacity (FRC) can decrease by up to 20% with anesthesia (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531), and the thoracic manipulation and alteration of rib cage mechanics with a classic median sternotomy approach can lead to decreases in forced vital capacity (FVC) and expiratory volume in the first second of forced expiration (FEV1) that can last for months after surgery. Use of the cardiopulmonary bypass circuit can also lead to bronchoconstriction. These changes in pulmonary function are less pronounced in alternative surgical approaches, such as partial sternotomies (Weissman C. Seminars in Cardiothoracic and Vascular Anesthesia: Pulmonary Complications After Cardiac Surgery. Glen Head, NY: Westminister Publications; 2004).
The most frequent pulmonary consequence of cardiac surgery is atelectasis, seen on postoperative chest radiographs in approximately 50% to 90% of patients (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Induction, apnea during cardiopulmonary bypass, manual compression of the lungs for surgical exposure, internal mammary harvesting, and pleurotomy can lead to atelectasis in the intraoperative setting while weak cough, poor inspiratory efforts, interstitial edema, and immobility further contribute postoperatively (Weissman 2004). While frequently seen, clinically significant pulmonary consequences from this radiographic finding alone are rare (Weissman 2004).
Pleural effusions are seen on immediate postoperative chest radiographs in the majority of patients. Additionally, 10% to 40% of patients develop pleural effusions 2 to 3 weeks after surgery secondary to postpericardiotomy syndrome. While some effusions require drainage and further intervention (eg, hemothorax), most effusions require no specific treatment and resolve over time (Weissman 2004).
The prevalence of pneumonia following cardiac surgery varies based on differences in study populations and diagnostic criteria, but it remains an important source of morbidity and mortality. In one series, postoperative pneumonia occurred in 3.1% of patients, with higher rates observed in patients who were older, had worse left ventricular ejection fraction, had COPD, experienced longer bypass times, and received more red blood cell transfusions in the operating room (Allou N, et al. Crit Care Med. 2014;42[5]:1150). A meta-analysis found that an average of 6.37% of patients developed ventilator-associated pneumonia (VAP), and this rose to 35.2% in those receiving ventilation for greater than 48 hours. Those who developed VAP had an odds ratio of dying of 15.18 (95% CI 5.81-39.68) compared with those who did not (He S, et al. J Thorac Cardiovasc Surg. 2014;148[6]:3148).
A small proportion of patients go on to develop ARDS. While relatively uncommon, ARDS carries a high mortality rate. Many possible etiologies for ARDS in cardiac surgery patients have been proposed, including an inflammatory response related to the cardiopulmonary bypass circuit, reperfusion injury secondary to reduced pulmonary blood flow during bypass, protamine administration, transfusion, hypothermia, and lack of ventilation during bypass (Weissman 2004); (Stephens RS, et al. Ann Thorac Surg. 2013;95[3]:1122). Type of surgery may also play a role, as patients who undergo aortic surgery are at an even greater risk (Stephens 2013). As with other cases of ARDS, treatment is supportive: low tidal volume ventilation and careful management of fluid balance, as well as paralysis, prone positioning, and consideration for extracorporeal membrane oxygenation (ECMO), as appropriate (Stephens 2013).
Therapies to prevent postoperative pulmonary complications have included early extubation, aggressive pain control, deep breathing, physical therapy, early mobilization, and noninvasive ventilation in the form of CPAP and intermittent positive pressure breathing. A meta-analysis of 18 trials looking at the use of various forms of prophylactic postoperative physiotherapy did not show a difference in any measured clinical outcome (Pasquina P, Walder B. Br Med J. 2003;327[7428]:1379).
However, the heterogeneity, short follow-up, and low quality of included studies made it difficult to draw meaningful conclusions on the benefit or lack thereof for these therapies. More recent studies have shown promise for chest physiotherapy started several weeks prior to elective coronary bypass graft surgery and extended CPAP via nasal CPAP mask immediately following extubation (Hulzebos EH. JAMA. 2006;296[15]:1851), (Stephens 2013).
Ongoing areas for improvement include further clarification and standardization of best practices for postcardiac surgery patients, including blood product transfusion, optimal tidal volumes for surgical and postsurgical ventilation, timing of extubation, and the use of preventive therapies in the pre- and postsurgical periods. As providers who care for these patients, understanding how we can improve their postoperative pulmonary recovery will allow us to enhance our patient’s experience.
Dr. Noel is a Critical Care Fellow, Cooper Medical School of Rowan University, Camden, New Jersey.
Cardiac surgery patients are sicker today than in previous decades due to an aging population and a rising complexity in medical care. There is an increasing reliance on noncardiac surgeons to care for these patients. The optimal postoperative providers and structure of the ICU where patients are cared for remain unclear, but what is irrefutable is patients’ increased postoperative morbidity. Pulmonary complications are a leading cause of morbidity in these patients, occurring in up to one-fifth of cases (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Common pulmonary complications of cardiac surgery are listed in Table 1. Those complications, captured by The Society of Thoracic Surgeons (STS) Cardiac Surgery Database, include receiving ventilation longer than 24 hours, pneumonia, pulmonary embolism, and pleural effusion requiring drainage (The Society of Thoracic Surgeons. STS National Database. https://www.sts.org/registries-research-center/sts-national-database. Accessed January 9, 2018).
It should come as no surprise that cardiac surgery can have pronounced effects on lung function. The anesthetic agents, chest wall alteration, and direct lung manipulation can all affect pulmonary parameters. Functional residual capacity (FRC) can decrease by up to 20% with anesthesia (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531), and the thoracic manipulation and alteration of rib cage mechanics with a classic median sternotomy approach can lead to decreases in forced vital capacity (FVC) and expiratory volume in the first second of forced expiration (FEV1) that can last for months after surgery. Use of the cardiopulmonary bypass circuit can also lead to bronchoconstriction. These changes in pulmonary function are less pronounced in alternative surgical approaches, such as partial sternotomies (Weissman C. Seminars in Cardiothoracic and Vascular Anesthesia: Pulmonary Complications After Cardiac Surgery. Glen Head, NY: Westminister Publications; 2004).
The most frequent pulmonary consequence of cardiac surgery is atelectasis, seen on postoperative chest radiographs in approximately 50% to 90% of patients (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Induction, apnea during cardiopulmonary bypass, manual compression of the lungs for surgical exposure, internal mammary harvesting, and pleurotomy can lead to atelectasis in the intraoperative setting while weak cough, poor inspiratory efforts, interstitial edema, and immobility further contribute postoperatively (Weissman 2004). While frequently seen, clinically significant pulmonary consequences from this radiographic finding alone are rare (Weissman 2004).
Pleural effusions are seen on immediate postoperative chest radiographs in the majority of patients. Additionally, 10% to 40% of patients develop pleural effusions 2 to 3 weeks after surgery secondary to postpericardiotomy syndrome. While some effusions require drainage and further intervention (eg, hemothorax), most effusions require no specific treatment and resolve over time (Weissman 2004).
The prevalence of pneumonia following cardiac surgery varies based on differences in study populations and diagnostic criteria, but it remains an important source of morbidity and mortality. In one series, postoperative pneumonia occurred in 3.1% of patients, with higher rates observed in patients who were older, had worse left ventricular ejection fraction, had COPD, experienced longer bypass times, and received more red blood cell transfusions in the operating room (Allou N, et al. Crit Care Med. 2014;42[5]:1150). A meta-analysis found that an average of 6.37% of patients developed ventilator-associated pneumonia (VAP), and this rose to 35.2% in those receiving ventilation for greater than 48 hours. Those who developed VAP had an odds ratio of dying of 15.18 (95% CI 5.81-39.68) compared with those who did not (He S, et al. J Thorac Cardiovasc Surg. 2014;148[6]:3148).
A small proportion of patients go on to develop ARDS. While relatively uncommon, ARDS carries a high mortality rate. Many possible etiologies for ARDS in cardiac surgery patients have been proposed, including an inflammatory response related to the cardiopulmonary bypass circuit, reperfusion injury secondary to reduced pulmonary blood flow during bypass, protamine administration, transfusion, hypothermia, and lack of ventilation during bypass (Weissman 2004); (Stephens RS, et al. Ann Thorac Surg. 2013;95[3]:1122). Type of surgery may also play a role, as patients who undergo aortic surgery are at an even greater risk (Stephens 2013). As with other cases of ARDS, treatment is supportive: low tidal volume ventilation and careful management of fluid balance, as well as paralysis, prone positioning, and consideration for extracorporeal membrane oxygenation (ECMO), as appropriate (Stephens 2013).
Therapies to prevent postoperative pulmonary complications have included early extubation, aggressive pain control, deep breathing, physical therapy, early mobilization, and noninvasive ventilation in the form of CPAP and intermittent positive pressure breathing. A meta-analysis of 18 trials looking at the use of various forms of prophylactic postoperative physiotherapy did not show a difference in any measured clinical outcome (Pasquina P, Walder B. Br Med J. 2003;327[7428]:1379).
However, the heterogeneity, short follow-up, and low quality of included studies made it difficult to draw meaningful conclusions on the benefit or lack thereof for these therapies. More recent studies have shown promise for chest physiotherapy started several weeks prior to elective coronary bypass graft surgery and extended CPAP via nasal CPAP mask immediately following extubation (Hulzebos EH. JAMA. 2006;296[15]:1851), (Stephens 2013).
Ongoing areas for improvement include further clarification and standardization of best practices for postcardiac surgery patients, including blood product transfusion, optimal tidal volumes for surgical and postsurgical ventilation, timing of extubation, and the use of preventive therapies in the pre- and postsurgical periods. As providers who care for these patients, understanding how we can improve their postoperative pulmonary recovery will allow us to enhance our patient’s experience.
Dr. Noel is a Critical Care Fellow, Cooper Medical School of Rowan University, Camden, New Jersey.
The rise and fall of treatment trials in group 3 pulmonary hypertension: Where do we go from here?
Treatment of fibrotic interstitial lung disease (ILD) is often dissatisfying to clinicians and patients. Despite significant advances in the field, particularly the validation of the efficacy of the antifibrotic drugs nintedanib (Richeldi L, et al. N Engl J Med. 2014;370[22]:2071) and pirfenidone (King TE Jr, et al. N Engl J Med. 2014;370[(22]:2083) in slowing the progression of idiopathic pulmonary fibrosis (IPF), we are still left with a paucity of therapeutic options to modulate the course of disease and improve functional outcomes. Given the difficulties in addressing the progression of parenchymal fibrosis, the pulmonary community has looked for alternative ways to approach treatment of ILD. One potential therapeutic inroad that has garnered substantial interest is the treatment of concurrent pulmonary hypertension (PH) or group 3 PH (Seeger W, et al. J Am Coll Cardiol. 2013;62 (25 Suppl):D109).
Group 3 PH – The rationale to treat
Group 3 PH has an indisputable association with adverse outcomes, including decreased functional status, increased need for supplemental oxygen, and decreased survival (King CS, Nathan SD. Pulmonary Hypertension and Interstitial Lung Disease. Ed 2. Ch 4.2017;67-84). In fact, PH is such a powerful predictor of survival in fibrotic ILD, the International Society of Heart and Lung Transplant (ISHLT) guidelines on candidate selection for lung transplantation cite development of PH as an indication for transplant listing (Weill D, et al. J Heart Lung Transplant. 2015;34:1). When one considers the strong association between group 3 PH and adverse outcomes, the numerous pulmonary vasodilator agents available to treat pulmonary arterial hypertension (PAH), and the success achieved in treating PAH, it is easy to see why group 3 PH is such a tempting therapeutic target.
Previous studies of pulmonary vasodilator therapy for group 3 PH

Over 20 studies assessing the effectiveness of pulmonary vasodilator therapy in ILD have been published (King CS, Nathan SD. Pulmonary Hypertension and Interstitial Lung Disease. Ed 2. Ch 4. 2017;67) The majority was small and unblinded with inherent limitations. To date, no randomized controlled trial (RCT) of therapy for group 3 PH has demonstrated efficacy. Several studies amongst the RCTs deserve highlighting. The most encouraging RCT of therapy for group 3 PH was STEP-IPF. This study compared sildenafil with placebo in 180 patients with advanced IPF. Though the study failed to demonstrate a difference in the primary endpoint of ≥ 20% increase in 6-minute walk test (6MWT) distance, it did show improvement in several secondary endpoints, including arterial oxygen saturation and quality of life measures (Zisman DA, et al. N Engl J Med. 2010;363[7]:620).
The BUILD-3 study compared bosentan with placebo in 617 patients with IPF. Enrolled patients were not required to have PH. While bosentan was well tolerated, it failed to improve the primary endpoint of time to disease progression or death or secondary endpoints regarding quality of life or dyspnea (King TE Jr, et al. Am J Respir Crit Care Med. 2011; 184[1]:92). A smaller study comparing bosentan with placebo in 60 patients with fibrotic ILD with right-sided heart catheterization (RHC) confirmed PH failed to demonstrate any difference in pulmonary vascular hemodynamics, functional status, or symptoms (Corte TJ, et al. Am J Respir Crit Care Med. 2014;190[2]:208). Studies of the newer endothelin receptor antagonists, macitentan (Raghu, et al. Eur Respir J. 2013;42[6]:1622) and ambrisentan (Raghu, et al. Ann Int Med. 2013;158[9]:641), were conducted and failed to demonstrate improvements in outcomes, as well. Overall, the results of the available RCTs of pulmonary vasodilator therapy in group 3 PH have been disappointing, failing to conclusively improve the primary outcome in any of the studies performed.
Hot off the presses – RISE-IIP
The latest letdown in group 3 PH is “Riociguat for the Treatment of Pulmonary Hypertension in Idiopathic Interstitial Pneumonia (RISE-IIP). The results of the study were recently presented at the European Respiratory Society meeting in Milan, Italy, by my colleague from Inova Fairfax Hospital (Falls Church, VA), Dr. Steven Nathan. Riociguat is a soluble guanylate cyclase stimulator approved for use in PAH and chronic thromboembolic pulmonary hypertension. The rationale for the study was that riociguat would improve pulmonary hemodynamics leading to improved functional status. Additionally, several preclinical models have demonstrated antifibrotic effects of the drug (Geschka S, et al. PLoS One. 2011;6:e21853). Justification for the study was also bolstered by promising results from a pilot study conducted in 22 patients with RHC-confirmed PH with a mean pulmonary artery pressure (mPAP) > 30 and fibrotic lung disease. In this study, patients treated with riociguat had improved pulmonary vascular resistance, cardiac output, and 6MWT distance.
To be included in RISE-IIP, patients were required to have an idiopathic interstitial pneumonia, PH confirmed by RHC with a mPAP ≥ 25 mm Hg, World Health Organization Functional Class 2-4 symptoms, and a forced vital capacity (FVC) ≥ 45% predicted. Pertinent exclusion criteria included significant left-sided heart disease and extent of emphysema greater than fibrosis on HRCT. Patients with connective tissue disease, chronic hypersensitivity pneumonitis, occupational lung disease, and sarcoidosis were ineligible to participate. The placebo-controlled portion of the study lasted 26 weeks then crossed into an open label extension trial.
The study enrolled 147 total patients, with 73 receiving riociguat and 74 in the placebo arm. There was no significant improvement in the primary outcome of change in 6MWT distance or the secondary combined endpoint assessing clinical worsening. The study was terminated early for safety due to an increased number of deaths and adverse events in the treatment group. During the blinded phase of the study, eight deaths (11%) occurred in the riociguat arm as compared with three deaths (4%) in the placebo arm. Seventy patients entered the open label extension phase of the trial, and 9 of these patients died. Eight of these deaths occurred in the patients previously receiving placebo who were switched to riociguat. The authors of the study found no conclusive potential etiology to explain the increased mortality seen.
RISE’ing from the ashes – Where do we go from here?
So, what should we take away from the negative results of the RISE-IIP trial? Some may argue that treatment of group 3 PH is a flawed premise and should be abandoned. Perhaps development of group 3 PH is an adaptive response to worsening fibrotic lung disease, and treatment of the PH is unlikely to alter outcomes and introduces the possibility of harm through worsening hypoxemia due to increased ventilation/perfusion mismatch with nonselective pulmonary vasodilation. I suspect the truth is somewhat more nuanced. I believe there is a select population with severe or “out-of-proportion” PH that may still benefit from vasodilator therapy. Trials targeting patients with a higher mPAP or low cardiac index could test this hypothesis but will be difficult to enroll. Another possibility is that our mechanism of drug delivery in prior trials has been suboptimal. Inhaled pulmonary vasodilator therapy should minimize the risk of worsening ventilation/perfusion mismatch. An RCT assessing the response to inhaled treprostinil in group 3 PH (NCT02630316) is currently enrolling at 96 centers across the United States. Until data supporting positive effects from treating group 3 PH emerge, I would recommend against off-label treatment and encourage referral to clinical trials. Given the potential for harm, riociguat should be avoided in group 3 PH. If off-label therapy is being entertained in a patient with severe PH that is out of proportion to the extent of fibrotic lung disease, it should be initiated cautiously at a center experienced in treating PH. Finally, clinicians should refer appropriate candidates with ILD and group 3 PH for lung transplantation evaluation.
The great inventor Thomas Edison is credited with saying “I have not failed. I’ve just found 10,000 ways that won’t work.” While disappointing, negative studies are to be expected as we search for improved therapies for our patients. It’s essential that we reflect upon these studies, so we can improve future trial design.
Treatment of fibrotic interstitial lung disease (ILD) is often dissatisfying to clinicians and patients. Despite significant advances in the field, particularly the validation of the efficacy of the antifibrotic drugs nintedanib (Richeldi L, et al. N Engl J Med. 2014;370[22]:2071) and pirfenidone (King TE Jr, et al. N Engl J Med. 2014;370[(22]:2083) in slowing the progression of idiopathic pulmonary fibrosis (IPF), we are still left with a paucity of therapeutic options to modulate the course of disease and improve functional outcomes. Given the difficulties in addressing the progression of parenchymal fibrosis, the pulmonary community has looked for alternative ways to approach treatment of ILD. One potential therapeutic inroad that has garnered substantial interest is the treatment of concurrent pulmonary hypertension (PH) or group 3 PH (Seeger W, et al. J Am Coll Cardiol. 2013;62 (25 Suppl):D109).
Group 3 PH – The rationale to treat
Group 3 PH has an indisputable association with adverse outcomes, including decreased functional status, increased need for supplemental oxygen, and decreased survival (King CS, Nathan SD. Pulmonary Hypertension and Interstitial Lung Disease. Ed 2. Ch 4.2017;67-84). In fact, PH is such a powerful predictor of survival in fibrotic ILD, the International Society of Heart and Lung Transplant (ISHLT) guidelines on candidate selection for lung transplantation cite development of PH as an indication for transplant listing (Weill D, et al. J Heart Lung Transplant. 2015;34:1). When one considers the strong association between group 3 PH and adverse outcomes, the numerous pulmonary vasodilator agents available to treat pulmonary arterial hypertension (PAH), and the success achieved in treating PAH, it is easy to see why group 3 PH is such a tempting therapeutic target.
Previous studies of pulmonary vasodilator therapy for group 3 PH
Over 20 studies assessing the effectiveness of pulmonary vasodilator therapy in ILD have been published (King CS, Nathan SD. Pulmonary Hypertension and Interstitial Lung Disease. Ed 2. Ch 4. 2017;67) The majority was small and unblinded with inherent limitations. To date, no randomized controlled trial (RCT) of therapy for group 3 PH has demonstrated efficacy. Several studies amongst the RCTs deserve highlighting. The most encouraging RCT of therapy for group 3 PH was STEP-IPF. This study compared sildenafil with placebo in 180 patients with advanced IPF. Though the study failed to demonstrate a difference in the primary endpoint of ≥ 20% increase in 6-minute walk test (6MWT) distance, it did show improvement in several secondary endpoints, including arterial oxygen saturation and quality of life measures (Zisman DA, et al. N Engl J Med. 2010;363[7]:620).
The BUILD-3 study compared bosentan with placebo in 617 patients with IPF. Enrolled patients were not required to have PH. While bosentan was well tolerated, it failed to improve the primary endpoint of time to disease progression or death or secondary endpoints regarding quality of life or dyspnea (King TE Jr, et al. Am J Respir Crit Care Med. 2011; 184[1]:92). A smaller study comparing bosentan with placebo in 60 patients with fibrotic ILD with right-sided heart catheterization (RHC) confirmed PH failed to demonstrate any difference in pulmonary vascular hemodynamics, functional status, or symptoms (Corte TJ, et al. Am J Respir Crit Care Med. 2014;190[2]:208). Studies of the newer endothelin receptor antagonists, macitentan (Raghu, et al. Eur Respir J. 2013;42[6]:1622) and ambrisentan (Raghu, et al. Ann Int Med. 2013;158[9]:641), were conducted and failed to demonstrate improvements in outcomes, as well. Overall, the results of the available RCTs of pulmonary vasodilator therapy in group 3 PH have been disappointing, failing to conclusively improve the primary outcome in any of the studies performed.
Hot off the presses – RISE-IIP
The latest letdown in group 3 PH is “Riociguat for the Treatment of Pulmonary Hypertension in Idiopathic Interstitial Pneumonia (RISE-IIP). The results of the study were recently presented at the European Respiratory Society meeting in Milan, Italy, by my colleague from Inova Fairfax Hospital (Falls Church, VA), Dr. Steven Nathan. Riociguat is a soluble guanylate cyclase stimulator approved for use in PAH and chronic thromboembolic pulmonary hypertension. The rationale for the study was that riociguat would improve pulmonary hemodynamics leading to improved functional status. Additionally, several preclinical models have demonstrated antifibrotic effects of the drug (Geschka S, et al. PLoS One. 2011;6:e21853). Justification for the study was also bolstered by promising results from a pilot study conducted in 22 patients with RHC-confirmed PH with a mean pulmonary artery pressure (mPAP) > 30 and fibrotic lung disease. In this study, patients treated with riociguat had improved pulmonary vascular resistance, cardiac output, and 6MWT distance.
To be included in RISE-IIP, patients were required to have an idiopathic interstitial pneumonia, PH confirmed by RHC with a mPAP ≥ 25 mm Hg, World Health Organization Functional Class 2-4 symptoms, and a forced vital capacity (FVC) ≥ 45% predicted. Pertinent exclusion criteria included significant left-sided heart disease and extent of emphysema greater than fibrosis on HRCT. Patients with connective tissue disease, chronic hypersensitivity pneumonitis, occupational lung disease, and sarcoidosis were ineligible to participate. The placebo-controlled portion of the study lasted 26 weeks then crossed into an open label extension trial.
The study enrolled 147 total patients, with 73 receiving riociguat and 74 in the placebo arm. There was no significant improvement in the primary outcome of change in 6MWT distance or the secondary combined endpoint assessing clinical worsening. The study was terminated early for safety due to an increased number of deaths and adverse events in the treatment group. During the blinded phase of the study, eight deaths (11%) occurred in the riociguat arm as compared with three deaths (4%) in the placebo arm. Seventy patients entered the open label extension phase of the trial, and 9 of these patients died. Eight of these deaths occurred in the patients previously receiving placebo who were switched to riociguat. The authors of the study found no conclusive potential etiology to explain the increased mortality seen.
RISE’ing from the ashes – Where do we go from here?
So, what should we take away from the negative results of the RISE-IIP trial? Some may argue that treatment of group 3 PH is a flawed premise and should be abandoned. Perhaps development of group 3 PH is an adaptive response to worsening fibrotic lung disease, and treatment of the PH is unlikely to alter outcomes and introduces the possibility of harm through worsening hypoxemia due to increased ventilation/perfusion mismatch with nonselective pulmonary vasodilation. I suspect the truth is somewhat more nuanced. I believe there is a select population with severe or “out-of-proportion” PH that may still benefit from vasodilator therapy. Trials targeting patients with a higher mPAP or low cardiac index could test this hypothesis but will be difficult to enroll. Another possibility is that our mechanism of drug delivery in prior trials has been suboptimal. Inhaled pulmonary vasodilator therapy should minimize the risk of worsening ventilation/perfusion mismatch. An RCT assessing the response to inhaled treprostinil in group 3 PH (NCT02630316) is currently enrolling at 96 centers across the United States. Until data supporting positive effects from treating group 3 PH emerge, I would recommend against off-label treatment and encourage referral to clinical trials. Given the potential for harm, riociguat should be avoided in group 3 PH. If off-label therapy is being entertained in a patient with severe PH that is out of proportion to the extent of fibrotic lung disease, it should be initiated cautiously at a center experienced in treating PH. Finally, clinicians should refer appropriate candidates with ILD and group 3 PH for lung transplantation evaluation.
The great inventor Thomas Edison is credited with saying “I have not failed. I’ve just found 10,000 ways that won’t work.” While disappointing, negative studies are to be expected as we search for improved therapies for our patients. It’s essential that we reflect upon these studies, so we can improve future trial design.
Treatment of fibrotic interstitial lung disease (ILD) is often dissatisfying to clinicians and patients. Despite significant advances in the field, particularly the validation of the efficacy of the antifibrotic drugs nintedanib (Richeldi L, et al. N Engl J Med. 2014;370[22]:2071) and pirfenidone (King TE Jr, et al. N Engl J Med. 2014;370[(22]:2083) in slowing the progression of idiopathic pulmonary fibrosis (IPF), we are still left with a paucity of therapeutic options to modulate the course of disease and improve functional outcomes. Given the difficulties in addressing the progression of parenchymal fibrosis, the pulmonary community has looked for alternative ways to approach treatment of ILD. One potential therapeutic inroad that has garnered substantial interest is the treatment of concurrent pulmonary hypertension (PH) or group 3 PH (Seeger W, et al. J Am Coll Cardiol. 2013;62 (25 Suppl):D109).
Group 3 PH – The rationale to treat
Group 3 PH has an indisputable association with adverse outcomes, including decreased functional status, increased need for supplemental oxygen, and decreased survival (King CS, Nathan SD. Pulmonary Hypertension and Interstitial Lung Disease. Ed 2. Ch 4.2017;67-84). In fact, PH is such a powerful predictor of survival in fibrotic ILD, the International Society of Heart and Lung Transplant (ISHLT) guidelines on candidate selection for lung transplantation cite development of PH as an indication for transplant listing (Weill D, et al. J Heart Lung Transplant. 2015;34:1). When one considers the strong association between group 3 PH and adverse outcomes, the numerous pulmonary vasodilator agents available to treat pulmonary arterial hypertension (PAH), and the success achieved in treating PAH, it is easy to see why group 3 PH is such a tempting therapeutic target.
Previous studies of pulmonary vasodilator therapy for group 3 PH
Over 20 studies assessing the effectiveness of pulmonary vasodilator therapy in ILD have been published (King CS, Nathan SD. Pulmonary Hypertension and Interstitial Lung Disease. Ed 2. Ch 4. 2017;67) The majority was small and unblinded with inherent limitations. To date, no randomized controlled trial (RCT) of therapy for group 3 PH has demonstrated efficacy. Several studies amongst the RCTs deserve highlighting. The most encouraging RCT of therapy for group 3 PH was STEP-IPF. This study compared sildenafil with placebo in 180 patients with advanced IPF. Though the study failed to demonstrate a difference in the primary endpoint of ≥ 20% increase in 6-minute walk test (6MWT) distance, it did show improvement in several secondary endpoints, including arterial oxygen saturation and quality of life measures (Zisman DA, et al. N Engl J Med. 2010;363[7]:620).
The BUILD-3 study compared bosentan with placebo in 617 patients with IPF. Enrolled patients were not required to have PH. While bosentan was well tolerated, it failed to improve the primary endpoint of time to disease progression or death or secondary endpoints regarding quality of life or dyspnea (King TE Jr, et al. Am J Respir Crit Care Med. 2011; 184[1]:92). A smaller study comparing bosentan with placebo in 60 patients with fibrotic ILD with right-sided heart catheterization (RHC) confirmed PH failed to demonstrate any difference in pulmonary vascular hemodynamics, functional status, or symptoms (Corte TJ, et al. Am J Respir Crit Care Med. 2014;190[2]:208). Studies of the newer endothelin receptor antagonists, macitentan (Raghu, et al. Eur Respir J. 2013;42[6]:1622) and ambrisentan (Raghu, et al. Ann Int Med. 2013;158[9]:641), were conducted and failed to demonstrate improvements in outcomes, as well. Overall, the results of the available RCTs of pulmonary vasodilator therapy in group 3 PH have been disappointing, failing to conclusively improve the primary outcome in any of the studies performed.
Hot off the presses – RISE-IIP
The latest letdown in group 3 PH is “Riociguat for the Treatment of Pulmonary Hypertension in Idiopathic Interstitial Pneumonia (RISE-IIP). The results of the study were recently presented at the European Respiratory Society meeting in Milan, Italy, by my colleague from Inova Fairfax Hospital (Falls Church, VA), Dr. Steven Nathan. Riociguat is a soluble guanylate cyclase stimulator approved for use in PAH and chronic thromboembolic pulmonary hypertension. The rationale for the study was that riociguat would improve pulmonary hemodynamics leading to improved functional status. Additionally, several preclinical models have demonstrated antifibrotic effects of the drug (Geschka S, et al. PLoS One. 2011;6:e21853). Justification for the study was also bolstered by promising results from a pilot study conducted in 22 patients with RHC-confirmed PH with a mean pulmonary artery pressure (mPAP) > 30 and fibrotic lung disease. In this study, patients treated with riociguat had improved pulmonary vascular resistance, cardiac output, and 6MWT distance.
To be included in RISE-IIP, patients were required to have an idiopathic interstitial pneumonia, PH confirmed by RHC with a mPAP ≥ 25 mm Hg, World Health Organization Functional Class 2-4 symptoms, and a forced vital capacity (FVC) ≥ 45% predicted. Pertinent exclusion criteria included significant left-sided heart disease and extent of emphysema greater than fibrosis on HRCT. Patients with connective tissue disease, chronic hypersensitivity pneumonitis, occupational lung disease, and sarcoidosis were ineligible to participate. The placebo-controlled portion of the study lasted 26 weeks then crossed into an open label extension trial.
The study enrolled 147 total patients, with 73 receiving riociguat and 74 in the placebo arm. There was no significant improvement in the primary outcome of change in 6MWT distance or the secondary combined endpoint assessing clinical worsening. The study was terminated early for safety due to an increased number of deaths and adverse events in the treatment group. During the blinded phase of the study, eight deaths (11%) occurred in the riociguat arm as compared with three deaths (4%) in the placebo arm. Seventy patients entered the open label extension phase of the trial, and 9 of these patients died. Eight of these deaths occurred in the patients previously receiving placebo who were switched to riociguat. The authors of the study found no conclusive potential etiology to explain the increased mortality seen.
RISE’ing from the ashes – Where do we go from here?
So, what should we take away from the negative results of the RISE-IIP trial? Some may argue that treatment of group 3 PH is a flawed premise and should be abandoned. Perhaps development of group 3 PH is an adaptive response to worsening fibrotic lung disease, and treatment of the PH is unlikely to alter outcomes and introduces the possibility of harm through worsening hypoxemia due to increased ventilation/perfusion mismatch with nonselective pulmonary vasodilation. I suspect the truth is somewhat more nuanced. I believe there is a select population with severe or “out-of-proportion” PH that may still benefit from vasodilator therapy. Trials targeting patients with a higher mPAP or low cardiac index could test this hypothesis but will be difficult to enroll. Another possibility is that our mechanism of drug delivery in prior trials has been suboptimal. Inhaled pulmonary vasodilator therapy should minimize the risk of worsening ventilation/perfusion mismatch. An RCT assessing the response to inhaled treprostinil in group 3 PH (NCT02630316) is currently enrolling at 96 centers across the United States. Until data supporting positive effects from treating group 3 PH emerge, I would recommend against off-label treatment and encourage referral to clinical trials. Given the potential for harm, riociguat should be avoided in group 3 PH. If off-label therapy is being entertained in a patient with severe PH that is out of proportion to the extent of fibrotic lung disease, it should be initiated cautiously at a center experienced in treating PH. Finally, clinicians should refer appropriate candidates with ILD and group 3 PH for lung transplantation evaluation.
The great inventor Thomas Edison is credited with saying “I have not failed. I’ve just found 10,000 ways that won’t work.” While disappointing, negative studies are to be expected as we search for improved therapies for our patients. It’s essential that we reflect upon these studies, so we can improve future trial design.