User login
Follicular lymphoma with histologic transformation may merit ASCT
Among patients with high tumor burden follicular lymphoma (FL) that responded to rituximab chemotherapy but then underwent histologic transformation, median overall survival was not reached when patients received autologous stem cell transplantation (ASCT), but was only 1.7 years otherwise, based on results of an ancillary study of a clinical trial.
In contrast, ASCT did not affect overall survival when patients progressed to untransformed FL, said Dr. Clémentine Sarkozy of Centre Hospitalier Lyon-Sud in Pierre Bénite, France, and her associates. Fully 58% of histologic transformations occurred in the first year of follow-up, highlighting “the necessity for biopsy at the first recurrence of FL,” they wrote online June 13 in the Journal of Clinical Oncology.
Histologic transformation in FL signifies progression to aggressive lymphoma. Studies of histologic transformation and subsequent overall survival in the rituximab era have been retrospective, with variable patient populations and initial management regimens, according to the investigators. Therefore, they followed 1,018 patients from the multicenter, randomized, phase III PRIMA (Primary Rituximab and Maintenance) trial, which evaluated maintenance rituximab therapy among patients with symptomatic FL who had responded to induction chemotherapy plus rituximab (J Clin Oncol. 2016 Jun. doi: 10.1200/JCO.2015.65.7163).
A total of 463 patients (45.5%) experienced disease recurrence or progression, and 194 (42%) were biopsied over a median follow-up time of 6 years. A total of 40 (20.6%) biopsies showed histologic transformation, while 154 (79.4%) had untransformed FL. Median time to recurrence was 9.6 months for patients with histologic transformation and 22.8 months for patients with untransformed FL (P = .02). Median overall survival with histologic transformation was worse than with untransformed FL (3.8 years vs. 6.4 years; hazard ratio, 3.9; 95% confidence interval, 2.2-6.9; P = .001). Furthermore, among patients who progressed within 12 months, median overall survival with histologic transformation was 2 years, compared with 6.4 years for patients with untransformed FL (P = .007).
After salvage therapy, 17 (42%) patients with histologic transformation underwent consolidation with high-dose chemotherapy and ASCT. Median overall survival for these patients was not reached, versus 1.7 years when they did not undergo ASCT. In contrast, ASCT did not improve overall survival among patients with untransformed FL. Results were similar after excluding patients with early progression and patients who were older than 65 years, the investigators reported.
Risk factors for histologic transformation in the univariate analysis included performance status, anemia, high lactate dehydrogenase level, “B” symptoms, histologic grade 3a, and high Follicular Lymphoma International Prognostic Index scores at diagnosis. However, only Eastern Cooperative Oncology Group performance status of 2 to 4 (HR, 5.6; 95% CI, 1.7-17.7), and anemia (HR, 3.7; 95% CI, 1.4-9.7) remained significant in the multivariate analysis. Neither the choice of induction regimen nor the quality of response seemed to affect the likelihood of histologic transformation, and rituximab maintenance therapy did not seem to alter response to salvage treatment or survival after histologic transformation. By necessity, the study excluded patients who did not respond to initial immunochemotherapy, which could have limited the generalizability of the findings, the investigators noted.
The study was funded by Sandoz and Takeda Pharmaceuticals. Dr. Sarkozy disclosed research funding from Sandoz and Takeda Pharmaceuticals and honoraria from Gilead Sciences. Twelve coinvestigators also disclosed ties to Takeda and a number of other pharmaceutical companies. The other seven coinvestigators had no disclosures.
In just 3 years, prospective observational studies and [this] clever ancillary analysis of a prospective clinical trial have better informed the lymphoma community about the expected incidence and timing of transformation in patients with follicular lymphoma after being treated with modern management strategies. But we are still limited by clumsy predictive tools for identifying patients at highest risk. Deeper understanding of biologic and genetic factors of FL subclonal populations as well as the tumor microenvironment will allow for more precise identification of patients truly at risk and potentially will provide actionable targets for abrogating that risk. Future [studies of] transformed lymphoma will hopefully replace variables such as anthracyclines, the Follicular Lymphoma International Prognostic Index, lactate dehydrogenase, and ASCT with promising new variables such as IRF-4, miR-31, bcl-2, pleuripotency, and nuclear factor kappa B pathway genes or new therapies that target these variables. Future analyses should not simply prognosticate who is at risk for transformation, but should predict a specific intervention to either prevent or treat such an event.
Dr. Brian K. Link is at the University of Iowa, Iowa City. He reported ties to AbbVie, Gilead Sciences, Genentech, Sandoz, Pharmacyclics, Millennium Pharmaceuticals, Genentech, Kite Pharma, Seattle Genetics, and Dynavax Technologies. These comments are from his editorial accompanying the report (J Clin Oncol. 2016 Jun. doi: 10.1200/JCO.2016.67.4234).
In just 3 years, prospective observational studies and [this] clever ancillary analysis of a prospective clinical trial have better informed the lymphoma community about the expected incidence and timing of transformation in patients with follicular lymphoma after being treated with modern management strategies. But we are still limited by clumsy predictive tools for identifying patients at highest risk. Deeper understanding of biologic and genetic factors of FL subclonal populations as well as the tumor microenvironment will allow for more precise identification of patients truly at risk and potentially will provide actionable targets for abrogating that risk. Future [studies of] transformed lymphoma will hopefully replace variables such as anthracyclines, the Follicular Lymphoma International Prognostic Index, lactate dehydrogenase, and ASCT with promising new variables such as IRF-4, miR-31, bcl-2, pleuripotency, and nuclear factor kappa B pathway genes or new therapies that target these variables. Future analyses should not simply prognosticate who is at risk for transformation, but should predict a specific intervention to either prevent or treat such an event.
Dr. Brian K. Link is at the University of Iowa, Iowa City. He reported ties to AbbVie, Gilead Sciences, Genentech, Sandoz, Pharmacyclics, Millennium Pharmaceuticals, Genentech, Kite Pharma, Seattle Genetics, and Dynavax Technologies. These comments are from his editorial accompanying the report (J Clin Oncol. 2016 Jun. doi: 10.1200/JCO.2016.67.4234).
In just 3 years, prospective observational studies and [this] clever ancillary analysis of a prospective clinical trial have better informed the lymphoma community about the expected incidence and timing of transformation in patients with follicular lymphoma after being treated with modern management strategies. But we are still limited by clumsy predictive tools for identifying patients at highest risk. Deeper understanding of biologic and genetic factors of FL subclonal populations as well as the tumor microenvironment will allow for more precise identification of patients truly at risk and potentially will provide actionable targets for abrogating that risk. Future [studies of] transformed lymphoma will hopefully replace variables such as anthracyclines, the Follicular Lymphoma International Prognostic Index, lactate dehydrogenase, and ASCT with promising new variables such as IRF-4, miR-31, bcl-2, pleuripotency, and nuclear factor kappa B pathway genes or new therapies that target these variables. Future analyses should not simply prognosticate who is at risk for transformation, but should predict a specific intervention to either prevent or treat such an event.
Dr. Brian K. Link is at the University of Iowa, Iowa City. He reported ties to AbbVie, Gilead Sciences, Genentech, Sandoz, Pharmacyclics, Millennium Pharmaceuticals, Genentech, Kite Pharma, Seattle Genetics, and Dynavax Technologies. These comments are from his editorial accompanying the report (J Clin Oncol. 2016 Jun. doi: 10.1200/JCO.2016.67.4234).
Among patients with high tumor burden follicular lymphoma (FL) that responded to rituximab chemotherapy but then underwent histologic transformation, median overall survival was not reached when patients received autologous stem cell transplantation (ASCT), but was only 1.7 years otherwise, based on results of an ancillary study of a clinical trial.
In contrast, ASCT did not affect overall survival when patients progressed to untransformed FL, said Dr. Clémentine Sarkozy of Centre Hospitalier Lyon-Sud in Pierre Bénite, France, and her associates. Fully 58% of histologic transformations occurred in the first year of follow-up, highlighting “the necessity for biopsy at the first recurrence of FL,” they wrote online June 13 in the Journal of Clinical Oncology.
Histologic transformation in FL signifies progression to aggressive lymphoma. Studies of histologic transformation and subsequent overall survival in the rituximab era have been retrospective, with variable patient populations and initial management regimens, according to the investigators. Therefore, they followed 1,018 patients from the multicenter, randomized, phase III PRIMA (Primary Rituximab and Maintenance) trial, which evaluated maintenance rituximab therapy among patients with symptomatic FL who had responded to induction chemotherapy plus rituximab (J Clin Oncol. 2016 Jun. doi: 10.1200/JCO.2015.65.7163).
A total of 463 patients (45.5%) experienced disease recurrence or progression, and 194 (42%) were biopsied over a median follow-up time of 6 years. A total of 40 (20.6%) biopsies showed histologic transformation, while 154 (79.4%) had untransformed FL. Median time to recurrence was 9.6 months for patients with histologic transformation and 22.8 months for patients with untransformed FL (P = .02). Median overall survival with histologic transformation was worse than with untransformed FL (3.8 years vs. 6.4 years; hazard ratio, 3.9; 95% confidence interval, 2.2-6.9; P = .001). Furthermore, among patients who progressed within 12 months, median overall survival with histologic transformation was 2 years, compared with 6.4 years for patients with untransformed FL (P = .007).
After salvage therapy, 17 (42%) patients with histologic transformation underwent consolidation with high-dose chemotherapy and ASCT. Median overall survival for these patients was not reached, versus 1.7 years when they did not undergo ASCT. In contrast, ASCT did not improve overall survival among patients with untransformed FL. Results were similar after excluding patients with early progression and patients who were older than 65 years, the investigators reported.
Risk factors for histologic transformation in the univariate analysis included performance status, anemia, high lactate dehydrogenase level, “B” symptoms, histologic grade 3a, and high Follicular Lymphoma International Prognostic Index scores at diagnosis. However, only Eastern Cooperative Oncology Group performance status of 2 to 4 (HR, 5.6; 95% CI, 1.7-17.7), and anemia (HR, 3.7; 95% CI, 1.4-9.7) remained significant in the multivariate analysis. Neither the choice of induction regimen nor the quality of response seemed to affect the likelihood of histologic transformation, and rituximab maintenance therapy did not seem to alter response to salvage treatment or survival after histologic transformation. By necessity, the study excluded patients who did not respond to initial immunochemotherapy, which could have limited the generalizability of the findings, the investigators noted.
The study was funded by Sandoz and Takeda Pharmaceuticals. Dr. Sarkozy disclosed research funding from Sandoz and Takeda Pharmaceuticals and honoraria from Gilead Sciences. Twelve coinvestigators also disclosed ties to Takeda and a number of other pharmaceutical companies. The other seven coinvestigators had no disclosures.
Among patients with high tumor burden follicular lymphoma (FL) that responded to rituximab chemotherapy but then underwent histologic transformation, median overall survival was not reached when patients received autologous stem cell transplantation (ASCT), but was only 1.7 years otherwise, based on results of an ancillary study of a clinical trial.
In contrast, ASCT did not affect overall survival when patients progressed to untransformed FL, said Dr. Clémentine Sarkozy of Centre Hospitalier Lyon-Sud in Pierre Bénite, France, and her associates. Fully 58% of histologic transformations occurred in the first year of follow-up, highlighting “the necessity for biopsy at the first recurrence of FL,” they wrote online June 13 in the Journal of Clinical Oncology.
Histologic transformation in FL signifies progression to aggressive lymphoma. Studies of histologic transformation and subsequent overall survival in the rituximab era have been retrospective, with variable patient populations and initial management regimens, according to the investigators. Therefore, they followed 1,018 patients from the multicenter, randomized, phase III PRIMA (Primary Rituximab and Maintenance) trial, which evaluated maintenance rituximab therapy among patients with symptomatic FL who had responded to induction chemotherapy plus rituximab (J Clin Oncol. 2016 Jun. doi: 10.1200/JCO.2015.65.7163).
A total of 463 patients (45.5%) experienced disease recurrence or progression, and 194 (42%) were biopsied over a median follow-up time of 6 years. A total of 40 (20.6%) biopsies showed histologic transformation, while 154 (79.4%) had untransformed FL. Median time to recurrence was 9.6 months for patients with histologic transformation and 22.8 months for patients with untransformed FL (P = .02). Median overall survival with histologic transformation was worse than with untransformed FL (3.8 years vs. 6.4 years; hazard ratio, 3.9; 95% confidence interval, 2.2-6.9; P = .001). Furthermore, among patients who progressed within 12 months, median overall survival with histologic transformation was 2 years, compared with 6.4 years for patients with untransformed FL (P = .007).
After salvage therapy, 17 (42%) patients with histologic transformation underwent consolidation with high-dose chemotherapy and ASCT. Median overall survival for these patients was not reached, versus 1.7 years when they did not undergo ASCT. In contrast, ASCT did not improve overall survival among patients with untransformed FL. Results were similar after excluding patients with early progression and patients who were older than 65 years, the investigators reported.
Risk factors for histologic transformation in the univariate analysis included performance status, anemia, high lactate dehydrogenase level, “B” symptoms, histologic grade 3a, and high Follicular Lymphoma International Prognostic Index scores at diagnosis. However, only Eastern Cooperative Oncology Group performance status of 2 to 4 (HR, 5.6; 95% CI, 1.7-17.7), and anemia (HR, 3.7; 95% CI, 1.4-9.7) remained significant in the multivariate analysis. Neither the choice of induction regimen nor the quality of response seemed to affect the likelihood of histologic transformation, and rituximab maintenance therapy did not seem to alter response to salvage treatment or survival after histologic transformation. By necessity, the study excluded patients who did not respond to initial immunochemotherapy, which could have limited the generalizability of the findings, the investigators noted.
The study was funded by Sandoz and Takeda Pharmaceuticals. Dr. Sarkozy disclosed research funding from Sandoz and Takeda Pharmaceuticals and honoraria from Gilead Sciences. Twelve coinvestigators also disclosed ties to Takeda and a number of other pharmaceutical companies. The other seven coinvestigators had no disclosures.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Histologic transformation of follicular lymphoma tends to occur early and may merit intensive salvage with autologous stem cell transplantation.
Major finding: Median overall survival was not reached among patients who received ASCT and was 1.7 years in those who didn’t have ASCT.
Data source: A study of 1,018 patients from the multicenter, randomized, phase III PRIMA (Primary Rituximab and Maintenance) trial.
Disclosures: The study was funded by Sandoz and Takeda Pharmaceuticals. Dr. Sarkozy disclosed research funding from Sandoz and Takeda Pharmaceuticals and honoraria from Gilead Sciences. Twelve coinvestigators also disclosed ties to Takeda and a number of other pharmaceutical companies. The other seven coinvestigators had no disclosures.
Dr. Matt Kalaycio’s top 10 hematologic oncology abstracts for ASCO 2016
Hematology News’ Editor-in-Chief Matt Kalaycio selected the following as his “top 10” picks for hematologic oncology abstracts at ASCO 2016:
Abstract 7000: Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML
Comment: When any treatment appears to improve survival, compared with 7+3 for AML, all must take notice.
Abstract 7001: Treatment-free remission (TFR) in patients (pts) with chronic myeloid leukemia in chronic phase (CML-CP) treated with frontline nilotinib: Results from the ENESTFreedom study
Comment: About 50% of the CML patients treated with frontline nilotinib are eventually able to stop the drug and successfully stay off of it. That means more patients in treatment-free remission, compared with those initially treated with imatinib.
Link to abstract 7001
Abstract 7007: Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia
Abstract 7009: Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy
Comment: The response rates in these older AML patients are remarkable and challenge results typically seen with 7+3 in a younger population.
Link to abstract 7007 and 7009
Abstract 7501: A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL)
Comment: The response rate to mogamulizumab was outstanding in the largest randomized clinical trial thus far conducted for this cancer. Although rare in the USA, ATL is more common in Asia.
Link to abstract 7501
Abstract 7507: Effect of bortezomib on complete remission (CR) rate when added to bendamustine-rituximab (BR) in previously untreated high-risk (HR) follicular lymphoma (FL): A randomized phase II trial of the ECOG-ACRIN Cancer Research Group (E2408)
Comment: This interesting observation of improved complete remission needs longer follow-up.
Link to abstract 7507
Abstract 7519: Venetoclax activity in CLL patients who have relapsed after or are refractory to ibrutinib or idelalisib
Comment: This study has implications for practice. Venetoclax elicits a 50%-60% response rate after patients with CLL progress during treatment with B-cell receptor pathway inhibitors.
Link to abstract 7519
Abstract 7521: Acalabrutinib, a second-generation bruton tyrosine kinase (Btk) inhibitor, in previously untreated chronic lymphocytic leukemia (CLL)
Comment: This next-generation variation on ibrutinib was associated with a 96% overall response rate with fewer adverse effects such as atrial fibrillation.
Link to abstract 7521
Abstract 8000: Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): A randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial)
Comment: Other trials are underway to address the role of upfront ASCT for newly diagnosed multiple myeloma. While the last word on this issue has yet to be written, ASCT remains the standard of care for MM patients after induction.
Link to abstract 8000
LBA4: Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study
Comment: As predicted by most, the addition of daratumumab to bortezomib-based therapy increases response rates, compared with bortezomib-based alone. Efficacy is becoming less of a concern with myeloma treatment than is economics..
Look for the full, final text of this abstract to be posted online at 7:30 AM (EDT) on Sunday, June 5.
Hematology News’ Editor-in-Chief Matt Kalaycio selected the following as his “top 10” picks for hematologic oncology abstracts at ASCO 2016:
Abstract 7000: Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML
Comment: When any treatment appears to improve survival, compared with 7+3 for AML, all must take notice.
Abstract 7001: Treatment-free remission (TFR) in patients (pts) with chronic myeloid leukemia in chronic phase (CML-CP) treated with frontline nilotinib: Results from the ENESTFreedom study
Comment: About 50% of the CML patients treated with frontline nilotinib are eventually able to stop the drug and successfully stay off of it. That means more patients in treatment-free remission, compared with those initially treated with imatinib.
Link to abstract 7001
Abstract 7007: Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia
Abstract 7009: Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy
Comment: The response rates in these older AML patients are remarkable and challenge results typically seen with 7+3 in a younger population.
Link to abstract 7007 and 7009
Abstract 7501: A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL)
Comment: The response rate to mogamulizumab was outstanding in the largest randomized clinical trial thus far conducted for this cancer. Although rare in the USA, ATL is more common in Asia.
Link to abstract 7501
Abstract 7507: Effect of bortezomib on complete remission (CR) rate when added to bendamustine-rituximab (BR) in previously untreated high-risk (HR) follicular lymphoma (FL): A randomized phase II trial of the ECOG-ACRIN Cancer Research Group (E2408)
Comment: This interesting observation of improved complete remission needs longer follow-up.
Link to abstract 7507
Abstract 7519: Venetoclax activity in CLL patients who have relapsed after or are refractory to ibrutinib or idelalisib
Comment: This study has implications for practice. Venetoclax elicits a 50%-60% response rate after patients with CLL progress during treatment with B-cell receptor pathway inhibitors.
Link to abstract 7519
Abstract 7521: Acalabrutinib, a second-generation bruton tyrosine kinase (Btk) inhibitor, in previously untreated chronic lymphocytic leukemia (CLL)
Comment: This next-generation variation on ibrutinib was associated with a 96% overall response rate with fewer adverse effects such as atrial fibrillation.
Link to abstract 7521
Abstract 8000: Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): A randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial)
Comment: Other trials are underway to address the role of upfront ASCT for newly diagnosed multiple myeloma. While the last word on this issue has yet to be written, ASCT remains the standard of care for MM patients after induction.
Link to abstract 8000
LBA4: Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study
Comment: As predicted by most, the addition of daratumumab to bortezomib-based therapy increases response rates, compared with bortezomib-based alone. Efficacy is becoming less of a concern with myeloma treatment than is economics..
Look for the full, final text of this abstract to be posted online at 7:30 AM (EDT) on Sunday, June 5.
Hematology News’ Editor-in-Chief Matt Kalaycio selected the following as his “top 10” picks for hematologic oncology abstracts at ASCO 2016:
Abstract 7000: Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML
Comment: When any treatment appears to improve survival, compared with 7+3 for AML, all must take notice.
Abstract 7001: Treatment-free remission (TFR) in patients (pts) with chronic myeloid leukemia in chronic phase (CML-CP) treated with frontline nilotinib: Results from the ENESTFreedom study
Comment: About 50% of the CML patients treated with frontline nilotinib are eventually able to stop the drug and successfully stay off of it. That means more patients in treatment-free remission, compared with those initially treated with imatinib.
Link to abstract 7001
Abstract 7007: Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia
Abstract 7009: Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy
Comment: The response rates in these older AML patients are remarkable and challenge results typically seen with 7+3 in a younger population.
Link to abstract 7007 and 7009
Abstract 7501: A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL)
Comment: The response rate to mogamulizumab was outstanding in the largest randomized clinical trial thus far conducted for this cancer. Although rare in the USA, ATL is more common in Asia.
Link to abstract 7501
Abstract 7507: Effect of bortezomib on complete remission (CR) rate when added to bendamustine-rituximab (BR) in previously untreated high-risk (HR) follicular lymphoma (FL): A randomized phase II trial of the ECOG-ACRIN Cancer Research Group (E2408)
Comment: This interesting observation of improved complete remission needs longer follow-up.
Link to abstract 7507
Abstract 7519: Venetoclax activity in CLL patients who have relapsed after or are refractory to ibrutinib or idelalisib
Comment: This study has implications for practice. Venetoclax elicits a 50%-60% response rate after patients with CLL progress during treatment with B-cell receptor pathway inhibitors.
Link to abstract 7519
Abstract 7521: Acalabrutinib, a second-generation bruton tyrosine kinase (Btk) inhibitor, in previously untreated chronic lymphocytic leukemia (CLL)
Comment: This next-generation variation on ibrutinib was associated with a 96% overall response rate with fewer adverse effects such as atrial fibrillation.
Link to abstract 7521
Abstract 8000: Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): A randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial)
Comment: Other trials are underway to address the role of upfront ASCT for newly diagnosed multiple myeloma. While the last word on this issue has yet to be written, ASCT remains the standard of care for MM patients after induction.
Link to abstract 8000
LBA4: Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study
Comment: As predicted by most, the addition of daratumumab to bortezomib-based therapy increases response rates, compared with bortezomib-based alone. Efficacy is becoming less of a concern with myeloma treatment than is economics..
Look for the full, final text of this abstract to be posted online at 7:30 AM (EDT) on Sunday, June 5.
Enzastaurin flops as maintenance in treated DLBCL
Maintenance therapy with enzastaurin, an experimental agent directed against B-cell malignancies, did not improve disease-free survival among patients with high-risk diffuse large B-cell lymphomas following complete responses to chemotherapy with rituximab.
In a randomized, double-blind trial, after a median follow-up of 48 months, the hazard ratio for disease-free survival with enzastaurin vs. placebo, the primary endpoint, was 0.92 (P = .541), reported Dr. Michael Crump of Princess Margaret Cancer Centre in Toronto, and colleagues.
“The risk of treatment failure, however defined, is likely to be different in the subpopulation of patients who achieve remission after that treatment. Furthermore, identifying the value of specific biomarkers in predicting therapeutic response to novel targeted agents may be necessary in guiding future trials within defined subgroups of patients with DLBCL,” they wrote in the study, published online May 23 in Journal of Clinical Oncology.
Enzastaurin is a selective inhibitor of the protein kinase C-beta isoform (PKC-beta) expressed in both normal and malignant B cells. It has been shown to decrease tumor proliferation and induced apoptosis in cancer cells, and has been shown to have activity against relapsed or refractory DLBCL, mantle cell lymphoma, and follicular lymphoma, the authors explained.
Dr. Crump and colleagues conducted a phase III study to determine whether enzastaurin could be effective as maintenance therapy in patients with DLBCL at high risk for relapse after having had complete responses to first-line therapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).
They enrolled 758 patients with stage II bulky DLBCL or stage III-IV disease who had three or more International Prognostic Index risk factors at diagnosis, and who had achieved either a confirmed or unconfirmed complete response after six to eight cycles of R-CHOP.
The patients were randomly assigned on a 2:1 basis to receive either oral enzastaurin 500 mg daily or placebo for 3 years, or until disease progression or unacceptable toxicity,
As noted, there was no significant difference in DFS with the active drug vs. placebo. In addition, in correlative analyses looking for biomarkers of response by cell of origin (i.e., germinal-center or non–germinal-center B cell) or by PKC-beta protein expression, the authors found no significant associations with either DFS or overall survival.
Enzastaurin was generally safe, with minor and manageable adverse events. More patients in the enzastaurin arm had episodes of QTc prolongation, but these did not require discontinuation of the drug.
Dr. Crump and coauthors disclosed consulting, advising, research funding and other relationships with various companies, including Eli Lilly. Five coauthors are Lilly employees.
Maintenance therapy with enzastaurin, an experimental agent directed against B-cell malignancies, did not improve disease-free survival among patients with high-risk diffuse large B-cell lymphomas following complete responses to chemotherapy with rituximab.
In a randomized, double-blind trial, after a median follow-up of 48 months, the hazard ratio for disease-free survival with enzastaurin vs. placebo, the primary endpoint, was 0.92 (P = .541), reported Dr. Michael Crump of Princess Margaret Cancer Centre in Toronto, and colleagues.
“The risk of treatment failure, however defined, is likely to be different in the subpopulation of patients who achieve remission after that treatment. Furthermore, identifying the value of specific biomarkers in predicting therapeutic response to novel targeted agents may be necessary in guiding future trials within defined subgroups of patients with DLBCL,” they wrote in the study, published online May 23 in Journal of Clinical Oncology.
Enzastaurin is a selective inhibitor of the protein kinase C-beta isoform (PKC-beta) expressed in both normal and malignant B cells. It has been shown to decrease tumor proliferation and induced apoptosis in cancer cells, and has been shown to have activity against relapsed or refractory DLBCL, mantle cell lymphoma, and follicular lymphoma, the authors explained.
Dr. Crump and colleagues conducted a phase III study to determine whether enzastaurin could be effective as maintenance therapy in patients with DLBCL at high risk for relapse after having had complete responses to first-line therapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).
They enrolled 758 patients with stage II bulky DLBCL or stage III-IV disease who had three or more International Prognostic Index risk factors at diagnosis, and who had achieved either a confirmed or unconfirmed complete response after six to eight cycles of R-CHOP.
The patients were randomly assigned on a 2:1 basis to receive either oral enzastaurin 500 mg daily or placebo for 3 years, or until disease progression or unacceptable toxicity,
As noted, there was no significant difference in DFS with the active drug vs. placebo. In addition, in correlative analyses looking for biomarkers of response by cell of origin (i.e., germinal-center or non–germinal-center B cell) or by PKC-beta protein expression, the authors found no significant associations with either DFS or overall survival.
Enzastaurin was generally safe, with minor and manageable adverse events. More patients in the enzastaurin arm had episodes of QTc prolongation, but these did not require discontinuation of the drug.
Dr. Crump and coauthors disclosed consulting, advising, research funding and other relationships with various companies, including Eli Lilly. Five coauthors are Lilly employees.
Maintenance therapy with enzastaurin, an experimental agent directed against B-cell malignancies, did not improve disease-free survival among patients with high-risk diffuse large B-cell lymphomas following complete responses to chemotherapy with rituximab.
In a randomized, double-blind trial, after a median follow-up of 48 months, the hazard ratio for disease-free survival with enzastaurin vs. placebo, the primary endpoint, was 0.92 (P = .541), reported Dr. Michael Crump of Princess Margaret Cancer Centre in Toronto, and colleagues.
“The risk of treatment failure, however defined, is likely to be different in the subpopulation of patients who achieve remission after that treatment. Furthermore, identifying the value of specific biomarkers in predicting therapeutic response to novel targeted agents may be necessary in guiding future trials within defined subgroups of patients with DLBCL,” they wrote in the study, published online May 23 in Journal of Clinical Oncology.
Enzastaurin is a selective inhibitor of the protein kinase C-beta isoform (PKC-beta) expressed in both normal and malignant B cells. It has been shown to decrease tumor proliferation and induced apoptosis in cancer cells, and has been shown to have activity against relapsed or refractory DLBCL, mantle cell lymphoma, and follicular lymphoma, the authors explained.
Dr. Crump and colleagues conducted a phase III study to determine whether enzastaurin could be effective as maintenance therapy in patients with DLBCL at high risk for relapse after having had complete responses to first-line therapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).
They enrolled 758 patients with stage II bulky DLBCL or stage III-IV disease who had three or more International Prognostic Index risk factors at diagnosis, and who had achieved either a confirmed or unconfirmed complete response after six to eight cycles of R-CHOP.
The patients were randomly assigned on a 2:1 basis to receive either oral enzastaurin 500 mg daily or placebo for 3 years, or until disease progression or unacceptable toxicity,
As noted, there was no significant difference in DFS with the active drug vs. placebo. In addition, in correlative analyses looking for biomarkers of response by cell of origin (i.e., germinal-center or non–germinal-center B cell) or by PKC-beta protein expression, the authors found no significant associations with either DFS or overall survival.
Enzastaurin was generally safe, with minor and manageable adverse events. More patients in the enzastaurin arm had episodes of QTc prolongation, but these did not require discontinuation of the drug.
Dr. Crump and coauthors disclosed consulting, advising, research funding and other relationships with various companies, including Eli Lilly. Five coauthors are Lilly employees.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The experimental agent enzastaurin was no better than placebo as maintenance therapy in patients who had achieved a complete response to R-CHOP.
Major finding: At 48 months median follow-up, the hazard ratio for enzastaurin was 0.92 (P = ns).
Data source: Randomized, double-blind, placebo controlled trial of 758 patients with diffuse large B-cell lymphoma in complete remission after first-line therapy with R-CHOP.
Disclosures: Dr. Crump and coauthors disclosed consulting, advising, research funding and other relationships with various companies, including Eli Lilly. Five coauthors are Lilly employees.
Study links radon and hematologic cancers in women

New research suggests there is a significant positive association between high levels of residential radon and the risk of hematologic malignancies in women.
The study is the first prospective, population-based study of residential radon exposure and hematologic malignancy risk.
Therefore, the researchers caution that it requires replication to better understand the association and whether it truly differs by sex.
Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this study and reported the results in Environmental Research.
Radon is a naturally occurring byproduct of the decay of radium and is a known human lung carcinogen. It is the second-leading cause of lung cancer in the US.
Modeling studies have shown that radon delivers a non-negligible dose of alpha radiation to the bone marrow and therefore could increase the risk of hematologic malignancies. However, studies investigating the link between radon and hematologic malignancies have produced inconsistent results.
For the current study, Dr Teras and her colleagues used data from the American Cancer Society Cancer Prevention Study-II Nutrition Cohort to examine the association between county-level residential radon exposure and the risk of hematologic cancer.
The analysis included 140,652 participants, including 3019 who had hematologic malignancies during 19 years of follow-up (1992 to 2011).
The researchers found that women living in counties with the highest mean radon concentration (> 148 Bq/m3) had a significantly higher risk of developing a hematologic malignancy than women living in counties with the lowest radon levels (< 74 Bq/m3).
The adjusted hazard ratio (adjusted for age, race, family history of hematologic malignancy, etc.) was 1.63 (P=0.0010).
The researchers also found evidence of a dose-response relationship, with an adjusted hazard ratio of 1.38 (P=0.001).
The team said there was evidence of a positive exposure-response relationship between radon concentration and the risk of all lymphoid malignancy subtypes in women. But the highest risk was observed for follicular lymphoma, with an adjusted hazard ratio of 2.74 (P=0.02).
On the other hand, there was a non-significant inverse association between radon and myeloid leukemias in women.
There was no association between hematologic malignancy and radon exposure among the men.
The researchers said a possible explanation for this finding is that men may have a higher baseline risk of hematologic malignancy, possibly because of more exposure to occupational or other risk factors, which would reduce the impact of any additional risk from residential radon.
In women, who have a smaller baseline risk, residential radon exposure might be a larger contributor to overall risk.
Another reason for the sex difference observed in this study may be that the women of this generation spent more time in their homes, so they had more residential exposure than men.
“The overall lifetime risk of hematological cancers in the United States is about 2%, so even a 60% relative increase would still mean a relatively small absolute risk,” Dr Teras noted.
“Nonetheless, radon is already associated with lung cancer, and if other studies confirm the link to blood cancers, we think it would warrant strengthened public health efforts to mitigate residential radon risks.” ![]()
New research suggests there is a significant positive association between high levels of residential radon and the risk of hematologic malignancies in women.
The study is the first prospective, population-based study of residential radon exposure and hematologic malignancy risk.
Therefore, the researchers caution that it requires replication to better understand the association and whether it truly differs by sex.
Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this study and reported the results in Environmental Research.
Radon is a naturally occurring byproduct of the decay of radium and is a known human lung carcinogen. It is the second-leading cause of lung cancer in the US.
Modeling studies have shown that radon delivers a non-negligible dose of alpha radiation to the bone marrow and therefore could increase the risk of hematologic malignancies. However, studies investigating the link between radon and hematologic malignancies have produced inconsistent results.
For the current study, Dr Teras and her colleagues used data from the American Cancer Society Cancer Prevention Study-II Nutrition Cohort to examine the association between county-level residential radon exposure and the risk of hematologic cancer.
The analysis included 140,652 participants, including 3019 who had hematologic malignancies during 19 years of follow-up (1992 to 2011).
The researchers found that women living in counties with the highest mean radon concentration (> 148 Bq/m3) had a significantly higher risk of developing a hematologic malignancy than women living in counties with the lowest radon levels (< 74 Bq/m3).
The adjusted hazard ratio (adjusted for age, race, family history of hematologic malignancy, etc.) was 1.63 (P=0.0010).
The researchers also found evidence of a dose-response relationship, with an adjusted hazard ratio of 1.38 (P=0.001).
The team said there was evidence of a positive exposure-response relationship between radon concentration and the risk of all lymphoid malignancy subtypes in women. But the highest risk was observed for follicular lymphoma, with an adjusted hazard ratio of 2.74 (P=0.02).
On the other hand, there was a non-significant inverse association between radon and myeloid leukemias in women.
There was no association between hematologic malignancy and radon exposure among the men.
The researchers said a possible explanation for this finding is that men may have a higher baseline risk of hematologic malignancy, possibly because of more exposure to occupational or other risk factors, which would reduce the impact of any additional risk from residential radon.
In women, who have a smaller baseline risk, residential radon exposure might be a larger contributor to overall risk.
Another reason for the sex difference observed in this study may be that the women of this generation spent more time in their homes, so they had more residential exposure than men.
“The overall lifetime risk of hematological cancers in the United States is about 2%, so even a 60% relative increase would still mean a relatively small absolute risk,” Dr Teras noted.
“Nonetheless, radon is already associated with lung cancer, and if other studies confirm the link to blood cancers, we think it would warrant strengthened public health efforts to mitigate residential radon risks.” ![]()
New research suggests there is a significant positive association between high levels of residential radon and the risk of hematologic malignancies in women.
The study is the first prospective, population-based study of residential radon exposure and hematologic malignancy risk.
Therefore, the researchers caution that it requires replication to better understand the association and whether it truly differs by sex.
Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this study and reported the results in Environmental Research.
Radon is a naturally occurring byproduct of the decay of radium and is a known human lung carcinogen. It is the second-leading cause of lung cancer in the US.
Modeling studies have shown that radon delivers a non-negligible dose of alpha radiation to the bone marrow and therefore could increase the risk of hematologic malignancies. However, studies investigating the link between radon and hematologic malignancies have produced inconsistent results.
For the current study, Dr Teras and her colleagues used data from the American Cancer Society Cancer Prevention Study-II Nutrition Cohort to examine the association between county-level residential radon exposure and the risk of hematologic cancer.
The analysis included 140,652 participants, including 3019 who had hematologic malignancies during 19 years of follow-up (1992 to 2011).
The researchers found that women living in counties with the highest mean radon concentration (> 148 Bq/m3) had a significantly higher risk of developing a hematologic malignancy than women living in counties with the lowest radon levels (< 74 Bq/m3).
The adjusted hazard ratio (adjusted for age, race, family history of hematologic malignancy, etc.) was 1.63 (P=0.0010).
The researchers also found evidence of a dose-response relationship, with an adjusted hazard ratio of 1.38 (P=0.001).
The team said there was evidence of a positive exposure-response relationship between radon concentration and the risk of all lymphoid malignancy subtypes in women. But the highest risk was observed for follicular lymphoma, with an adjusted hazard ratio of 2.74 (P=0.02).
On the other hand, there was a non-significant inverse association between radon and myeloid leukemias in women.
There was no association between hematologic malignancy and radon exposure among the men.
The researchers said a possible explanation for this finding is that men may have a higher baseline risk of hematologic malignancy, possibly because of more exposure to occupational or other risk factors, which would reduce the impact of any additional risk from residential radon.
In women, who have a smaller baseline risk, residential radon exposure might be a larger contributor to overall risk.
Another reason for the sex difference observed in this study may be that the women of this generation spent more time in their homes, so they had more residential exposure than men.
“The overall lifetime risk of hematological cancers in the United States is about 2%, so even a 60% relative increase would still mean a relatively small absolute risk,” Dr Teras noted.
“Nonetheless, radon is already associated with lung cancer, and if other studies confirm the link to blood cancers, we think it would warrant strengthened public health efforts to mitigate residential radon risks.” ![]()
CHMP recommends approving drug to treat FL

The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, be approved for use in patients with follicular lymphoma (FL).
The recommended indication is for obinutuzumab to be given first in combination with bendamustine and then as maintenance therapy in FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Based on the CHMP’s recommendation, a final decision regarding the approval of obinutuzumab in FL is expected from the European Commission in the coming months.
Obinutuzumab is already approved in the European Union for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche.
GADOLIN trial
The CHMP’s recommendation to approve obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, be approved for use in patients with follicular lymphoma (FL).
The recommended indication is for obinutuzumab to be given first in combination with bendamustine and then as maintenance therapy in FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Based on the CHMP’s recommendation, a final decision regarding the approval of obinutuzumab in FL is expected from the European Commission in the coming months.
Obinutuzumab is already approved in the European Union for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche.
GADOLIN trial
The CHMP’s recommendation to approve obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, be approved for use in patients with follicular lymphoma (FL).
The recommended indication is for obinutuzumab to be given first in combination with bendamustine and then as maintenance therapy in FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Based on the CHMP’s recommendation, a final decision regarding the approval of obinutuzumab in FL is expected from the European Commission in the coming months.
Obinutuzumab is already approved in the European Union for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche.
GADOLIN trial
The CHMP’s recommendation to approve obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
Low transformation rate in nodular lymphocyte–predominant Hodgkin lymphoma
Fewer than 8% of cases of nodular lymphocyte–predominant Hodgkin lymphoma (NLPHL) transformed to diffuse large B-cell lymphoma (DLBCL), based on a large prospective single-center study with long-term follow-up.
This rate was lower than the risk of transformation reported for transformed follicular lymphoma or chronic lymphocytic leukemia, according to Dr. Saad Kenderian and his associates at the Mayo Clinic, Rochester, Minn. Transformation was significantly associated with splenic involvement at presentation and with prior chemotherapy exposure, but did not worsen overall survival, they added.
“To our knowledge, this cohort represents the largest analysis to date of consecutive patients with NLPHL,” they said.
The study comprised 222 patients with newly diagnosed NLPHL who were treated at Mayo Clinic between 1970 and 2011. Median age at diagnosis was 40 years, and two-thirds of patients were men. The median follow-up period was 16 years (Blood 2016;12:1960-6. doi: 10.1182/blood-2015-08-665505).
During follow up, 17 cases (7.6%) transformed to DLBCL, for a transformation rate of 0.74 cases for every 100 patient-years, the investigators said. Median time to transformation was 35 months (range, 6-268 months). Predictors of transformation included any prior chemotherapy exposure (P = .04) and splenic involvement (P = .03). The rates of 40-year freedom from transformation were 87% when there was no splenic involvement and 21% when the spleen was involved, and were 87% if radiation therapy was used as a single modality compared with 77% in patients treated with prior chemotherapy or chemoradiation.
Five-year overall survival was 76% in patients with transformed disease, which was similar to overall survival among patients whose disease did not transform to DLBCL, the researchers noted.
Other studies of NLPHL have reported anywhere from a 2% to a 17% transformation rate, but those studies had smaller sample sizes, shorter follow-up periods, and less rigorous enrollment criteria and methods to confirm transformation, the investigators noted. “The finding of splenic involvement as a risk factor for transformation was reported by previous investigators. Interestingly, the association between exposure to prior chemotherapy and reduced freedom from transformation has not been reported in the past, but it has been observed in other low-grade lymphoma studies,” they added. “In contrast to follicular lymphoma, transformed NLPHL is not associated with an adverse impact on OS, suggesting a possibly different biology of transformation.”
The research was partially supported by Lymphoma SPORE and the Predolin Foundation. The investigators had no disclosures.
Kenderian et al. report a lower rate of transformation (7.6%) to diffuse large B-cell lymphoma for patients with nodular lymphocyte–predominant Hodgkin lymphoma compared with other series and found that transformation did not have a negative impact on overall survival. Reassuringly, even if transformation occurs, it is generally at a low rate. Also, these patients do well with additional treatment and do not have worse overall survival. At the MD Anderson Cancer Center, we have used a regimen based on R-CHOP and have not seen transformations. But only through large cooperative clinical trials can we determine whether R-CHOP or other more novel regimens are actually superior to ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) or rituximab (R)-ABVD for patients at high risk of transformation.
Dr. Michelle Fanale is at the University of Texas MD Anderson Cancer Center, Houston. She had no disclosures. These comments are from her editorial (Blood 2016;1927:1946-7 doi: 10.1182/blood-2016-03-699108).
Kenderian et al. report a lower rate of transformation (7.6%) to diffuse large B-cell lymphoma for patients with nodular lymphocyte–predominant Hodgkin lymphoma compared with other series and found that transformation did not have a negative impact on overall survival. Reassuringly, even if transformation occurs, it is generally at a low rate. Also, these patients do well with additional treatment and do not have worse overall survival. At the MD Anderson Cancer Center, we have used a regimen based on R-CHOP and have not seen transformations. But only through large cooperative clinical trials can we determine whether R-CHOP or other more novel regimens are actually superior to ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) or rituximab (R)-ABVD for patients at high risk of transformation.
Dr. Michelle Fanale is at the University of Texas MD Anderson Cancer Center, Houston. She had no disclosures. These comments are from her editorial (Blood 2016;1927:1946-7 doi: 10.1182/blood-2016-03-699108).
Kenderian et al. report a lower rate of transformation (7.6%) to diffuse large B-cell lymphoma for patients with nodular lymphocyte–predominant Hodgkin lymphoma compared with other series and found that transformation did not have a negative impact on overall survival. Reassuringly, even if transformation occurs, it is generally at a low rate. Also, these patients do well with additional treatment and do not have worse overall survival. At the MD Anderson Cancer Center, we have used a regimen based on R-CHOP and have not seen transformations. But only through large cooperative clinical trials can we determine whether R-CHOP or other more novel regimens are actually superior to ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) or rituximab (R)-ABVD for patients at high risk of transformation.
Dr. Michelle Fanale is at the University of Texas MD Anderson Cancer Center, Houston. She had no disclosures. These comments are from her editorial (Blood 2016;1927:1946-7 doi: 10.1182/blood-2016-03-699108).
Fewer than 8% of cases of nodular lymphocyte–predominant Hodgkin lymphoma (NLPHL) transformed to diffuse large B-cell lymphoma (DLBCL), based on a large prospective single-center study with long-term follow-up.
This rate was lower than the risk of transformation reported for transformed follicular lymphoma or chronic lymphocytic leukemia, according to Dr. Saad Kenderian and his associates at the Mayo Clinic, Rochester, Minn. Transformation was significantly associated with splenic involvement at presentation and with prior chemotherapy exposure, but did not worsen overall survival, they added.
“To our knowledge, this cohort represents the largest analysis to date of consecutive patients with NLPHL,” they said.
The study comprised 222 patients with newly diagnosed NLPHL who were treated at Mayo Clinic between 1970 and 2011. Median age at diagnosis was 40 years, and two-thirds of patients were men. The median follow-up period was 16 years (Blood 2016;12:1960-6. doi: 10.1182/blood-2015-08-665505).
During follow up, 17 cases (7.6%) transformed to DLBCL, for a transformation rate of 0.74 cases for every 100 patient-years, the investigators said. Median time to transformation was 35 months (range, 6-268 months). Predictors of transformation included any prior chemotherapy exposure (P = .04) and splenic involvement (P = .03). The rates of 40-year freedom from transformation were 87% when there was no splenic involvement and 21% when the spleen was involved, and were 87% if radiation therapy was used as a single modality compared with 77% in patients treated with prior chemotherapy or chemoradiation.
Five-year overall survival was 76% in patients with transformed disease, which was similar to overall survival among patients whose disease did not transform to DLBCL, the researchers noted.
Other studies of NLPHL have reported anywhere from a 2% to a 17% transformation rate, but those studies had smaller sample sizes, shorter follow-up periods, and less rigorous enrollment criteria and methods to confirm transformation, the investigators noted. “The finding of splenic involvement as a risk factor for transformation was reported by previous investigators. Interestingly, the association between exposure to prior chemotherapy and reduced freedom from transformation has not been reported in the past, but it has been observed in other low-grade lymphoma studies,” they added. “In contrast to follicular lymphoma, transformed NLPHL is not associated with an adverse impact on OS, suggesting a possibly different biology of transformation.”
The research was partially supported by Lymphoma SPORE and the Predolin Foundation. The investigators had no disclosures.
Fewer than 8% of cases of nodular lymphocyte–predominant Hodgkin lymphoma (NLPHL) transformed to diffuse large B-cell lymphoma (DLBCL), based on a large prospective single-center study with long-term follow-up.
This rate was lower than the risk of transformation reported for transformed follicular lymphoma or chronic lymphocytic leukemia, according to Dr. Saad Kenderian and his associates at the Mayo Clinic, Rochester, Minn. Transformation was significantly associated with splenic involvement at presentation and with prior chemotherapy exposure, but did not worsen overall survival, they added.
“To our knowledge, this cohort represents the largest analysis to date of consecutive patients with NLPHL,” they said.
The study comprised 222 patients with newly diagnosed NLPHL who were treated at Mayo Clinic between 1970 and 2011. Median age at diagnosis was 40 years, and two-thirds of patients were men. The median follow-up period was 16 years (Blood 2016;12:1960-6. doi: 10.1182/blood-2015-08-665505).
During follow up, 17 cases (7.6%) transformed to DLBCL, for a transformation rate of 0.74 cases for every 100 patient-years, the investigators said. Median time to transformation was 35 months (range, 6-268 months). Predictors of transformation included any prior chemotherapy exposure (P = .04) and splenic involvement (P = .03). The rates of 40-year freedom from transformation were 87% when there was no splenic involvement and 21% when the spleen was involved, and were 87% if radiation therapy was used as a single modality compared with 77% in patients treated with prior chemotherapy or chemoradiation.
Five-year overall survival was 76% in patients with transformed disease, which was similar to overall survival among patients whose disease did not transform to DLBCL, the researchers noted.
Other studies of NLPHL have reported anywhere from a 2% to a 17% transformation rate, but those studies had smaller sample sizes, shorter follow-up periods, and less rigorous enrollment criteria and methods to confirm transformation, the investigators noted. “The finding of splenic involvement as a risk factor for transformation was reported by previous investigators. Interestingly, the association between exposure to prior chemotherapy and reduced freedom from transformation has not been reported in the past, but it has been observed in other low-grade lymphoma studies,” they added. “In contrast to follicular lymphoma, transformed NLPHL is not associated with an adverse impact on OS, suggesting a possibly different biology of transformation.”
The research was partially supported by Lymphoma SPORE and the Predolin Foundation. The investigators had no disclosures.
FROM BLOOD
Key clinical point: The risk of transformation to diffuse large B-cell lymphoma is low in patients with nodular lymphocyte–predominant Hodgkin lymphoma.
Major finding: Only 7.6% of cases transformed over a median of 16 years of follow-up, and transformation did not worsen overall survival.
Data source: A prospective single-center study of 222 consecutive adults with NLPHL.
Disclosures: The research was partially supported by Lymphoma SPORE and the Predolin Foundation. The investigators had no disclosures.
Treatment can produce durable responses in NHL

2016 AACR Annual Meeting
© AACR/Todd Buchanan
NEW ORLEANS—Administering an antibody-radionuclide conjugate after B-cell depletion with rituximab can produce lasting responses in patients with relapsed non-Hodgkin lymphoma (NHL), according to a phase 1/2 study.
The conjugate, 177Lu-DOTA-HH1 (Betalutin), consists of the tumor-specific antibody HH1, which targets the CD37 antigen on the surface of NHL cells, conjugated to the β-emitting isotope lutetium-177 (Lu-177) via the chemical linker DOTA.
In an ongoing phase 1/2 study, Betalutin given after rituximab produced an overall response rate of 63.2%.
The median duration of response has not yet been reached, and 1 patient has maintained a response for more than 36 months.
In addition, the researchers said Betalutin was well tolerated, with a predictable and manageable safety profile. Most adverse events were hematologic, and all have been transient and reversible.
These results were presented at the 2016 AACR Annual Meeting (abstract LB-252*). The study is sponsored by Nordic Nanovector ASA.
Patients and study design
The researchers presented data on 21 patients—19 with follicular lymphoma and 2 with mantle cell lymphoma. All tumors were positive for CD37.
The patients’ median age was 68 (range, 41-78). Sixty-seven percent were male, and they had received 1 to 8 prior treatment regimens.
In this dose-escalation study, patients received Betalutin at 3 different doses, but they were also divided into 2 arms according to predosing with cold HH1 antibody.
In Arm 1 (n=12), patients received rituximab (at 375 mg/m2) on day -28 and -21 to deplete circulating B cells. On day 0, predosing with 50 mg HH1 was given before Betalutin injection. Then, patients received Betalutin at 10 MBq/kg (n=3), 15 MBq/kg (n=6), or 20 MBq/kg (n=3).
In Arm 2 (n=4), patients received rituximab at the same dose and schedule as Arm 1, but Betalutin was administered without HH1 predosing on day 0 at either 10 MBq/kg (n=2) or 15 MBq/kg (n=2).
The first patient treated on this trial received 250 mg/m2 of rituximab on day -7 and day 0 prior to Betalutin administration and was included in the 10 MBq/kg group in Arm 2.
The 15 MBq/kg dose level of Arm 1 has been expanded into the phase 2 portion of the study, as dose-limiting toxicities occurred at the 20 MBq/kg dose. Five patients have been treated in the phase 2 portion.
Safety
Adverse events (AEs) from the phase 2 portion of the study were not reported, as the data are still being collected.
In the phase 1 portion, grade 3/4 AEs were hematologic in nature and included decreases in platelet counts (3 grade 3 and 6 grade 4) and neutrophil counts (5 grade 3 and 4 grade 4).
Serious AEs included decreases in platelet counts (n=2), atrial fibrillation (n=2), epistaxis (n=1), fractured sternum (n=1), decreased neutrophil count (n=1), pharyngitis (n=1), pneumonia (n=1), pulmonary embolism (n=1), and sepsis (n=1).
The pulmonary embolism was deemed unrelated to treatment, but the remaining events were considered possibly or probably related to Betalutin.
The researchers noted that 1 patient experienced pharyngitis, pneumonia, pulmonary embolism, epistaxis, sepsis, and a decrease in lymphocyte count.
All patients’ platelets and neutrophils recovered. Two patients required platelet transfusions—one patient in the 20 MBq/kg cohort of Arm 1 and one patient in the 15 MBq/kg cohort of Arm 2.
Efficacy
Nineteen patients were evaluable for response. The overall response rate was 63.2% (n=12) and included both complete (31.6%, n=6) and partial responses (31.6%, n=6). Progression occurred in 21.1% of patients (n=4), and 15.8% (n=3) had stable disease.
The researchers presented data on 9 patients treated at the recommended 15 MBq/kg dose level with 50 mg HH1 predosing. Five patients were treated in phase 1 and 4 in phase 2. One of these patients was excluded from the analysis due to transformed lymphoma.
Two patients in phase 1 responded—both complete responses—and 3 patients in phase 2 responded—2 complete and 1 partial response.
For the entire study cohort, the median duration of response has not yet been reached. Six responses are ongoing—2 for 3+ months, 1 for 6+ months, 1 for 18+ months, 1 for 24+ months, and 1 for 36+ months. ![]()
*Information in the abstract differs from that presented at the meeting.
2016 AACR Annual Meeting
© AACR/Todd Buchanan
NEW ORLEANS—Administering an antibody-radionuclide conjugate after B-cell depletion with rituximab can produce lasting responses in patients with relapsed non-Hodgkin lymphoma (NHL), according to a phase 1/2 study.
The conjugate, 177Lu-DOTA-HH1 (Betalutin), consists of the tumor-specific antibody HH1, which targets the CD37 antigen on the surface of NHL cells, conjugated to the β-emitting isotope lutetium-177 (Lu-177) via the chemical linker DOTA.
In an ongoing phase 1/2 study, Betalutin given after rituximab produced an overall response rate of 63.2%.
The median duration of response has not yet been reached, and 1 patient has maintained a response for more than 36 months.
In addition, the researchers said Betalutin was well tolerated, with a predictable and manageable safety profile. Most adverse events were hematologic, and all have been transient and reversible.
These results were presented at the 2016 AACR Annual Meeting (abstract LB-252*). The study is sponsored by Nordic Nanovector ASA.
Patients and study design
The researchers presented data on 21 patients—19 with follicular lymphoma and 2 with mantle cell lymphoma. All tumors were positive for CD37.
The patients’ median age was 68 (range, 41-78). Sixty-seven percent were male, and they had received 1 to 8 prior treatment regimens.
In this dose-escalation study, patients received Betalutin at 3 different doses, but they were also divided into 2 arms according to predosing with cold HH1 antibody.
In Arm 1 (n=12), patients received rituximab (at 375 mg/m2) on day -28 and -21 to deplete circulating B cells. On day 0, predosing with 50 mg HH1 was given before Betalutin injection. Then, patients received Betalutin at 10 MBq/kg (n=3), 15 MBq/kg (n=6), or 20 MBq/kg (n=3).
In Arm 2 (n=4), patients received rituximab at the same dose and schedule as Arm 1, but Betalutin was administered without HH1 predosing on day 0 at either 10 MBq/kg (n=2) or 15 MBq/kg (n=2).
The first patient treated on this trial received 250 mg/m2 of rituximab on day -7 and day 0 prior to Betalutin administration and was included in the 10 MBq/kg group in Arm 2.
The 15 MBq/kg dose level of Arm 1 has been expanded into the phase 2 portion of the study, as dose-limiting toxicities occurred at the 20 MBq/kg dose. Five patients have been treated in the phase 2 portion.
Safety
Adverse events (AEs) from the phase 2 portion of the study were not reported, as the data are still being collected.
In the phase 1 portion, grade 3/4 AEs were hematologic in nature and included decreases in platelet counts (3 grade 3 and 6 grade 4) and neutrophil counts (5 grade 3 and 4 grade 4).
Serious AEs included decreases in platelet counts (n=2), atrial fibrillation (n=2), epistaxis (n=1), fractured sternum (n=1), decreased neutrophil count (n=1), pharyngitis (n=1), pneumonia (n=1), pulmonary embolism (n=1), and sepsis (n=1).
The pulmonary embolism was deemed unrelated to treatment, but the remaining events were considered possibly or probably related to Betalutin.
The researchers noted that 1 patient experienced pharyngitis, pneumonia, pulmonary embolism, epistaxis, sepsis, and a decrease in lymphocyte count.
All patients’ platelets and neutrophils recovered. Two patients required platelet transfusions—one patient in the 20 MBq/kg cohort of Arm 1 and one patient in the 15 MBq/kg cohort of Arm 2.
Efficacy
Nineteen patients were evaluable for response. The overall response rate was 63.2% (n=12) and included both complete (31.6%, n=6) and partial responses (31.6%, n=6). Progression occurred in 21.1% of patients (n=4), and 15.8% (n=3) had stable disease.
The researchers presented data on 9 patients treated at the recommended 15 MBq/kg dose level with 50 mg HH1 predosing. Five patients were treated in phase 1 and 4 in phase 2. One of these patients was excluded from the analysis due to transformed lymphoma.
Two patients in phase 1 responded—both complete responses—and 3 patients in phase 2 responded—2 complete and 1 partial response.
For the entire study cohort, the median duration of response has not yet been reached. Six responses are ongoing—2 for 3+ months, 1 for 6+ months, 1 for 18+ months, 1 for 24+ months, and 1 for 36+ months. ![]()
*Information in the abstract differs from that presented at the meeting.
2016 AACR Annual Meeting
© AACR/Todd Buchanan
NEW ORLEANS—Administering an antibody-radionuclide conjugate after B-cell depletion with rituximab can produce lasting responses in patients with relapsed non-Hodgkin lymphoma (NHL), according to a phase 1/2 study.
The conjugate, 177Lu-DOTA-HH1 (Betalutin), consists of the tumor-specific antibody HH1, which targets the CD37 antigen on the surface of NHL cells, conjugated to the β-emitting isotope lutetium-177 (Lu-177) via the chemical linker DOTA.
In an ongoing phase 1/2 study, Betalutin given after rituximab produced an overall response rate of 63.2%.
The median duration of response has not yet been reached, and 1 patient has maintained a response for more than 36 months.
In addition, the researchers said Betalutin was well tolerated, with a predictable and manageable safety profile. Most adverse events were hematologic, and all have been transient and reversible.
These results were presented at the 2016 AACR Annual Meeting (abstract LB-252*). The study is sponsored by Nordic Nanovector ASA.
Patients and study design
The researchers presented data on 21 patients—19 with follicular lymphoma and 2 with mantle cell lymphoma. All tumors were positive for CD37.
The patients’ median age was 68 (range, 41-78). Sixty-seven percent were male, and they had received 1 to 8 prior treatment regimens.
In this dose-escalation study, patients received Betalutin at 3 different doses, but they were also divided into 2 arms according to predosing with cold HH1 antibody.
In Arm 1 (n=12), patients received rituximab (at 375 mg/m2) on day -28 and -21 to deplete circulating B cells. On day 0, predosing with 50 mg HH1 was given before Betalutin injection. Then, patients received Betalutin at 10 MBq/kg (n=3), 15 MBq/kg (n=6), or 20 MBq/kg (n=3).
In Arm 2 (n=4), patients received rituximab at the same dose and schedule as Arm 1, but Betalutin was administered without HH1 predosing on day 0 at either 10 MBq/kg (n=2) or 15 MBq/kg (n=2).
The first patient treated on this trial received 250 mg/m2 of rituximab on day -7 and day 0 prior to Betalutin administration and was included in the 10 MBq/kg group in Arm 2.
The 15 MBq/kg dose level of Arm 1 has been expanded into the phase 2 portion of the study, as dose-limiting toxicities occurred at the 20 MBq/kg dose. Five patients have been treated in the phase 2 portion.
Safety
Adverse events (AEs) from the phase 2 portion of the study were not reported, as the data are still being collected.
In the phase 1 portion, grade 3/4 AEs were hematologic in nature and included decreases in platelet counts (3 grade 3 and 6 grade 4) and neutrophil counts (5 grade 3 and 4 grade 4).
Serious AEs included decreases in platelet counts (n=2), atrial fibrillation (n=2), epistaxis (n=1), fractured sternum (n=1), decreased neutrophil count (n=1), pharyngitis (n=1), pneumonia (n=1), pulmonary embolism (n=1), and sepsis (n=1).
The pulmonary embolism was deemed unrelated to treatment, but the remaining events were considered possibly or probably related to Betalutin.
The researchers noted that 1 patient experienced pharyngitis, pneumonia, pulmonary embolism, epistaxis, sepsis, and a decrease in lymphocyte count.
All patients’ platelets and neutrophils recovered. Two patients required platelet transfusions—one patient in the 20 MBq/kg cohort of Arm 1 and one patient in the 15 MBq/kg cohort of Arm 2.
Efficacy
Nineteen patients were evaluable for response. The overall response rate was 63.2% (n=12) and included both complete (31.6%, n=6) and partial responses (31.6%, n=6). Progression occurred in 21.1% of patients (n=4), and 15.8% (n=3) had stable disease.
The researchers presented data on 9 patients treated at the recommended 15 MBq/kg dose level with 50 mg HH1 predosing. Five patients were treated in phase 1 and 4 in phase 2. One of these patients was excluded from the analysis due to transformed lymphoma.
Two patients in phase 1 responded—both complete responses—and 3 patients in phase 2 responded—2 complete and 1 partial response.
For the entire study cohort, the median duration of response has not yet been reached. Six responses are ongoing—2 for 3+ months, 1 for 6+ months, 1 for 18+ months, 1 for 24+ months, and 1 for 36+ months. ![]()
*Information in the abstract differs from that presented at the meeting.
Drug shows early promise for low-grade lymphoma

follicular lymphoma
NEW ORLEANS—The TLR9 agonist SD-101 has produced encouraging early results in patients with low-grade B-cell lymphoma, according to researchers.
In an ongoing phase 1/2 study, patients received low-dose radiation, followed by an injection of SD-101 into one of their tumors.
This prompted changes in the tumor microenvironment that potentially induced a systemic antitumor response and decreased the volume of both treated and untreated tumors.
In addition, SD-101 was considered well tolerated, with no dose-limiting toxicities or maximum-tolerated dose.
“We are pleased to have already demonstrated a safety profile, pharmacodynamics, and preliminary efficacy in this study,” said Ronald Levy, MD, of Stanford University in California.
Dr Levy and his colleagues presented these results at the 2016 AACR Annual Meeting (abstract CT047). The study is sponsored by Dynavax Technologies Corporation, the company developing SD-101.
The researchers reported data for 13 patients with untreated, low-grade B-cell lymphoma. They had a mean age of 63.2, and 53.8% were male.
Patients had follicular lymphoma (n=9), small lymphocytic lymphoma (n=2), chronic lymphocytic leukemia (n=1), and nodal marginal zone lymphoma (n=1).
At least 2 sites of measurable disease were required for participation in this study. One site was treated with low-dose radiation (2 Gy) and injected with SD-101 on days 1, 8, 15, 22, and 29. Other lesions received no treatment.
In Part 1—the dose-escalation portion of the study—patients received SD-101 at 1 mg (n=3), 2 mg (n=3), 4 mg (n=3), or 8 mg (n=4). The phase 2 expansion portion of the study is ongoing, with enrollment in 2 dose cohorts (1 mg and 8 mg).
“This clinical trial design is unique and takes advantage of the fact that lymphoma patients have easily injectable sites of disease,” Dr Levy said. “The local injections are conveniently added to low-dose radiotherapy, a standard treatment for low-grade lymphoma.”
Safety
All 13 patients experienced at least 1 adverse event (AEs), although nearly all were grade 1 or 2.
The most common treatment-related AEs were local injection-site reactions and flu-like symptoms, including fever, chills, and myalgia (n=11 for all 3). However, the researchers said these AEs were primarily short-lived and controlled by over-the-counter acetaminophen in most cases.
In the 8 mg dosing cohort, 1 patient had grade 3 neutropenia and 2 had grade 3 malaise, all of which were considered treatment-related. In addition, there was a case of grade 3 asymptomatic pulmonary embolism in the 4 mg dose cohort, which was deemed serious but unrelated to treatment.
Efficacy
The researchers observed induction of interferon-responsive genes at all dose levels 24 hours after the second dose of SD-101 was given (Day 9).
In addition, T-cell numbers increased at the treated site by Day 8. The total T cells increased in 7 of 10 evaluable patients by Day 8 (range, >300% to 18%).
CD4+ and CD8+ T cells increased simultaneously in 5 of 7 evaluable patients, regulatory T cells decreased in 8 of 10 evaluable patients by Day 8, and follicular T helper cells decreased in 9 of 9 evaluable patients by Day 8.
Furthermore, treated and untreated tumors decreased in volume across all dose groups.
At Day 90, 12 patients had a reduction of the product of diameters in treated tumors (median -45.3%; range, -87 to +100), and 11 patients had a reduction in untreated tumors (median -8.1%; range, -48 to +45).
In 9 patients, these abscopal effects were sustained for at least 180 to 360 days.
However, 6 patients discontinued from the study due to progression. Three had radiographic progression—1 at Day 92 (in the 4 mg cohort) and 2 at 1 year (1 in the 1 mg cohort and 1 in the 2 mg cohort).
Two patients had clinical progression—1 at Day 197 (4 mg) and 1 at Day 273 (2 mg). And 1 patient discontinued at Day 203 due to a combination of clinical and radiographic progression (8 mg).
The researchers pointed out that there was no evidence of a dose response with SD-101, but the study included a limited number of patients. ![]()
follicular lymphoma
NEW ORLEANS—The TLR9 agonist SD-101 has produced encouraging early results in patients with low-grade B-cell lymphoma, according to researchers.
In an ongoing phase 1/2 study, patients received low-dose radiation, followed by an injection of SD-101 into one of their tumors.
This prompted changes in the tumor microenvironment that potentially induced a systemic antitumor response and decreased the volume of both treated and untreated tumors.
In addition, SD-101 was considered well tolerated, with no dose-limiting toxicities or maximum-tolerated dose.
“We are pleased to have already demonstrated a safety profile, pharmacodynamics, and preliminary efficacy in this study,” said Ronald Levy, MD, of Stanford University in California.
Dr Levy and his colleagues presented these results at the 2016 AACR Annual Meeting (abstract CT047). The study is sponsored by Dynavax Technologies Corporation, the company developing SD-101.
The researchers reported data for 13 patients with untreated, low-grade B-cell lymphoma. They had a mean age of 63.2, and 53.8% were male.
Patients had follicular lymphoma (n=9), small lymphocytic lymphoma (n=2), chronic lymphocytic leukemia (n=1), and nodal marginal zone lymphoma (n=1).
At least 2 sites of measurable disease were required for participation in this study. One site was treated with low-dose radiation (2 Gy) and injected with SD-101 on days 1, 8, 15, 22, and 29. Other lesions received no treatment.
In Part 1—the dose-escalation portion of the study—patients received SD-101 at 1 mg (n=3), 2 mg (n=3), 4 mg (n=3), or 8 mg (n=4). The phase 2 expansion portion of the study is ongoing, with enrollment in 2 dose cohorts (1 mg and 8 mg).
“This clinical trial design is unique and takes advantage of the fact that lymphoma patients have easily injectable sites of disease,” Dr Levy said. “The local injections are conveniently added to low-dose radiotherapy, a standard treatment for low-grade lymphoma.”
Safety
All 13 patients experienced at least 1 adverse event (AEs), although nearly all were grade 1 or 2.
The most common treatment-related AEs were local injection-site reactions and flu-like symptoms, including fever, chills, and myalgia (n=11 for all 3). However, the researchers said these AEs were primarily short-lived and controlled by over-the-counter acetaminophen in most cases.
In the 8 mg dosing cohort, 1 patient had grade 3 neutropenia and 2 had grade 3 malaise, all of which were considered treatment-related. In addition, there was a case of grade 3 asymptomatic pulmonary embolism in the 4 mg dose cohort, which was deemed serious but unrelated to treatment.
Efficacy
The researchers observed induction of interferon-responsive genes at all dose levels 24 hours after the second dose of SD-101 was given (Day 9).
In addition, T-cell numbers increased at the treated site by Day 8. The total T cells increased in 7 of 10 evaluable patients by Day 8 (range, >300% to 18%).
CD4+ and CD8+ T cells increased simultaneously in 5 of 7 evaluable patients, regulatory T cells decreased in 8 of 10 evaluable patients by Day 8, and follicular T helper cells decreased in 9 of 9 evaluable patients by Day 8.
Furthermore, treated and untreated tumors decreased in volume across all dose groups.
At Day 90, 12 patients had a reduction of the product of diameters in treated tumors (median -45.3%; range, -87 to +100), and 11 patients had a reduction in untreated tumors (median -8.1%; range, -48 to +45).
In 9 patients, these abscopal effects were sustained for at least 180 to 360 days.
However, 6 patients discontinued from the study due to progression. Three had radiographic progression—1 at Day 92 (in the 4 mg cohort) and 2 at 1 year (1 in the 1 mg cohort and 1 in the 2 mg cohort).
Two patients had clinical progression—1 at Day 197 (4 mg) and 1 at Day 273 (2 mg). And 1 patient discontinued at Day 203 due to a combination of clinical and radiographic progression (8 mg).
The researchers pointed out that there was no evidence of a dose response with SD-101, but the study included a limited number of patients. ![]()
follicular lymphoma
NEW ORLEANS—The TLR9 agonist SD-101 has produced encouraging early results in patients with low-grade B-cell lymphoma, according to researchers.
In an ongoing phase 1/2 study, patients received low-dose radiation, followed by an injection of SD-101 into one of their tumors.
This prompted changes in the tumor microenvironment that potentially induced a systemic antitumor response and decreased the volume of both treated and untreated tumors.
In addition, SD-101 was considered well tolerated, with no dose-limiting toxicities or maximum-tolerated dose.
“We are pleased to have already demonstrated a safety profile, pharmacodynamics, and preliminary efficacy in this study,” said Ronald Levy, MD, of Stanford University in California.
Dr Levy and his colleagues presented these results at the 2016 AACR Annual Meeting (abstract CT047). The study is sponsored by Dynavax Technologies Corporation, the company developing SD-101.
The researchers reported data for 13 patients with untreated, low-grade B-cell lymphoma. They had a mean age of 63.2, and 53.8% were male.
Patients had follicular lymphoma (n=9), small lymphocytic lymphoma (n=2), chronic lymphocytic leukemia (n=1), and nodal marginal zone lymphoma (n=1).
At least 2 sites of measurable disease were required for participation in this study. One site was treated with low-dose radiation (2 Gy) and injected with SD-101 on days 1, 8, 15, 22, and 29. Other lesions received no treatment.
In Part 1—the dose-escalation portion of the study—patients received SD-101 at 1 mg (n=3), 2 mg (n=3), 4 mg (n=3), or 8 mg (n=4). The phase 2 expansion portion of the study is ongoing, with enrollment in 2 dose cohorts (1 mg and 8 mg).
“This clinical trial design is unique and takes advantage of the fact that lymphoma patients have easily injectable sites of disease,” Dr Levy said. “The local injections are conveniently added to low-dose radiotherapy, a standard treatment for low-grade lymphoma.”
Safety
All 13 patients experienced at least 1 adverse event (AEs), although nearly all were grade 1 or 2.
The most common treatment-related AEs were local injection-site reactions and flu-like symptoms, including fever, chills, and myalgia (n=11 for all 3). However, the researchers said these AEs were primarily short-lived and controlled by over-the-counter acetaminophen in most cases.
In the 8 mg dosing cohort, 1 patient had grade 3 neutropenia and 2 had grade 3 malaise, all of which were considered treatment-related. In addition, there was a case of grade 3 asymptomatic pulmonary embolism in the 4 mg dose cohort, which was deemed serious but unrelated to treatment.
Efficacy
The researchers observed induction of interferon-responsive genes at all dose levels 24 hours after the second dose of SD-101 was given (Day 9).
In addition, T-cell numbers increased at the treated site by Day 8. The total T cells increased in 7 of 10 evaluable patients by Day 8 (range, >300% to 18%).
CD4+ and CD8+ T cells increased simultaneously in 5 of 7 evaluable patients, regulatory T cells decreased in 8 of 10 evaluable patients by Day 8, and follicular T helper cells decreased in 9 of 9 evaluable patients by Day 8.
Furthermore, treated and untreated tumors decreased in volume across all dose groups.
At Day 90, 12 patients had a reduction of the product of diameters in treated tumors (median -45.3%; range, -87 to +100), and 11 patients had a reduction in untreated tumors (median -8.1%; range, -48 to +45).
In 9 patients, these abscopal effects were sustained for at least 180 to 360 days.
However, 6 patients discontinued from the study due to progression. Three had radiographic progression—1 at Day 92 (in the 4 mg cohort) and 2 at 1 year (1 in the 1 mg cohort and 1 in the 2 mg cohort).
Two patients had clinical progression—1 at Day 197 (4 mg) and 1 at Day 273 (2 mg). And 1 patient discontinued at Day 203 due to a combination of clinical and radiographic progression (8 mg).
The researchers pointed out that there was no evidence of a dose response with SD-101, but the study included a limited number of patients. ![]()
CUDC-907 enters phase II for relapsed or refractory lymphoma and multiple myeloma
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
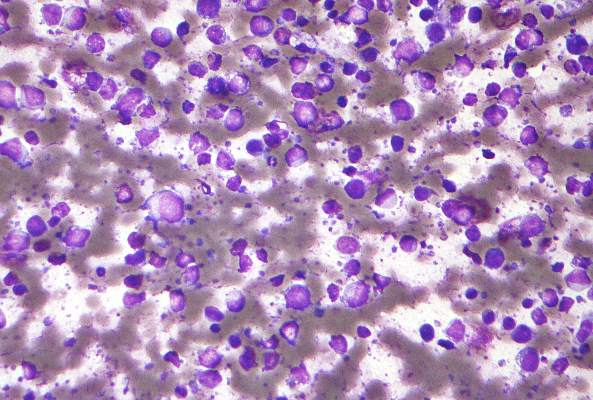
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
FROM THE LANCET ONCOLOGY
Key clinical point: The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule.
Major finding: Two complete responses and three partial responses were seen in the subgroup of nine patients with diffuse large B-cell lymphoma; three occurred in the five patients with transformed follicular disease.
Data source: A dose-escalation study involving 44 patients at four cancer centers.
Disclosures: The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
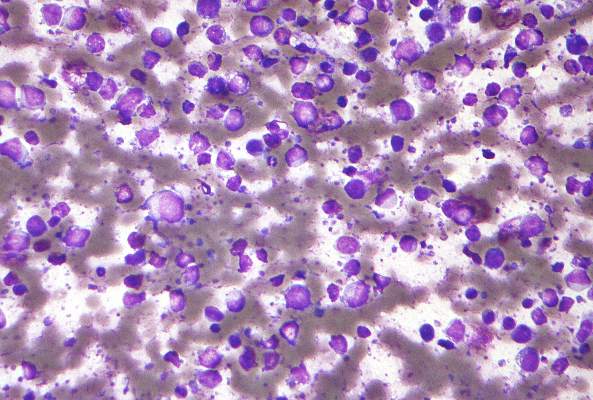
Dual inhibitor shows early promise for DLBCL

The small-molecule inhibitor CUDC-907 can provide disease control in patients with relapsed or refractory lymphoma and multiple myeloma (MM), according to researchers.
In a phase 1 trial, CUDC-907 produced responses in a small number of patients with diffuse large B-cell lymphoma (DLBCL).
And more than half of patients had stable disease while on CUDC-907, including those with MM, DLBCL, Hodgkin lymphoma (HL), and other lymphomas.
However, a majority of patients in this trial—84%—discontinued treatment due to confirmed progressive disease or signs of progression.
These results were published in The Lancet. The trial was sponsored by Curis, Inc., the company developing CUDC-907, and the Leukemia and Lymphoma Society.
“The data from the phase 1 monotherapy trial for CUDC-907, especially in heavily pretreated patients with relapsed/refractory DLBCL are very encouraging, and we look forward to data emerging from the current phase 2 trial in patients with MYC-altered DLBCL,” said study author Anas Younes, MD, of the Memorial Sloan Kettering Cancer Center in New York, New York.
CUDC-907 is an oral, dual inhibitor of class I and II histone deacetylases (HDACs), as well as class I PI3K enzymes. Specifically, CUDC-907 is designed to inhibit HDACs 1, 2, 3, 6, and 10 and PI3K-alpha, delta, and beta isoforms.
Between Jan 23, 2013, and July 27, 2015, the phase 1 trial of CUDC-907 enrolled 44 patients who were refractory to or had relapsed after 2 or more previous regimens. The patients’ median age was 63 (range, 22-83), and they had received a median of 5 prior treatments (range, 2-10).
Four patients had MM, 12 had HL, and 12 had DLBCL. The remaining 16 patients had other types of lymphoma, including lymphoplasmacytic lymphoma (n=3), small lymphocytic lymphoma (n=3), mantle cell lymphoma (n=3), follicular lymphoma (n=2), T-cell lymphoma (n=2), marginal zone lymphoma (n=1), Burkitt lymphoma (n=1), and gray zone lymphoma (n=1).
Treatment
CUDC-907 was given in a standard 3 + 3 dose-escalation design at 4 different dosing schedules—once daily, twice weekly, 3 times weekly, and daily for 5 days followed by a 2-day break (5/2)—in 21-day cycles.
Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase 2 dose.
Ten patients were sequentially assigned to CUDC-907 once-daily (MTD 60 mg), 12 to twice-weekly (MTD 150 mg), 15 to 3-times-weekly (MTD 150 mg), and 7 to the 5/2 dosing schedule (MTD 60 mg).
Safety
Four dose-limiting toxicities (DLTs) occurred in 3 of 40 DLT-evaluable patients. The DLTs were diarrhea and hyperglycemia in 1 patient on 60 mg once daily, hyperglycemia in 1 patient on 150 mg twice weekly, and diarrhea in 1 patient on 150 mg 3 times weekly. There were no DLTs in patients on the 5/2 schedule.
The incidence of grade 3 or higher adverse events (AEs) was 43% (19/44). The most common of these AEs were thrombocytopenia (20%, n=9), neutropenia (7%, n=3), and hyperglycemia (7%, n=3).
Twenty-five percent of patients (11/44) had serious AEs. Three of these events were considered treatment-related. They were epistaxis and the DLTs of diarrhea and hyperglycemia.
AEs led to dose reductions in 6 patients (14%) and treatment discontinuation in 7 patients (16%).
Efficacy
Thirty-seven patients were evaluable for response, and 5 of these patients responded (14%). All responses—2 complete and 3 partial responses—occurred in patients with DLBCL.
Twenty-one of the response-evaluable patients (57%) had stable disease. This included 1 patient with DLBCL, 2 with MM, 8 with HL, and 10 with the “other” types of lymphoma.
The remaining 11 patients progressed (30%)—3 with DLBCL, 2 with MM, 2 with HL, and 4 with other lymphomas.
Thirty-seven patients (84%) discontinued CUDC-907 because of progressive disease or clinical signs of progressive disease at the data cutoff.
Based on the clinical activity of CUDC-907 in patients with relapsed/refractory DLBCL, particularly those with MYC alterations, Curis has initiated a phase 2 trial of the drug in these patients. The recommended phase 2 dose is 60 mg on the 5/2 dosing schedule. ![]()
The small-molecule inhibitor CUDC-907 can provide disease control in patients with relapsed or refractory lymphoma and multiple myeloma (MM), according to researchers.
In a phase 1 trial, CUDC-907 produced responses in a small number of patients with diffuse large B-cell lymphoma (DLBCL).
And more than half of patients had stable disease while on CUDC-907, including those with MM, DLBCL, Hodgkin lymphoma (HL), and other lymphomas.
However, a majority of patients in this trial—84%—discontinued treatment due to confirmed progressive disease or signs of progression.
These results were published in The Lancet. The trial was sponsored by Curis, Inc., the company developing CUDC-907, and the Leukemia and Lymphoma Society.
“The data from the phase 1 monotherapy trial for CUDC-907, especially in heavily pretreated patients with relapsed/refractory DLBCL are very encouraging, and we look forward to data emerging from the current phase 2 trial in patients with MYC-altered DLBCL,” said study author Anas Younes, MD, of the Memorial Sloan Kettering Cancer Center in New York, New York.
CUDC-907 is an oral, dual inhibitor of class I and II histone deacetylases (HDACs), as well as class I PI3K enzymes. Specifically, CUDC-907 is designed to inhibit HDACs 1, 2, 3, 6, and 10 and PI3K-alpha, delta, and beta isoforms.
Between Jan 23, 2013, and July 27, 2015, the phase 1 trial of CUDC-907 enrolled 44 patients who were refractory to or had relapsed after 2 or more previous regimens. The patients’ median age was 63 (range, 22-83), and they had received a median of 5 prior treatments (range, 2-10).
Four patients had MM, 12 had HL, and 12 had DLBCL. The remaining 16 patients had other types of lymphoma, including lymphoplasmacytic lymphoma (n=3), small lymphocytic lymphoma (n=3), mantle cell lymphoma (n=3), follicular lymphoma (n=2), T-cell lymphoma (n=2), marginal zone lymphoma (n=1), Burkitt lymphoma (n=1), and gray zone lymphoma (n=1).
Treatment
CUDC-907 was given in a standard 3 + 3 dose-escalation design at 4 different dosing schedules—once daily, twice weekly, 3 times weekly, and daily for 5 days followed by a 2-day break (5/2)—in 21-day cycles.
Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase 2 dose.
Ten patients were sequentially assigned to CUDC-907 once-daily (MTD 60 mg), 12 to twice-weekly (MTD 150 mg), 15 to 3-times-weekly (MTD 150 mg), and 7 to the 5/2 dosing schedule (MTD 60 mg).
Safety
Four dose-limiting toxicities (DLTs) occurred in 3 of 40 DLT-evaluable patients. The DLTs were diarrhea and hyperglycemia in 1 patient on 60 mg once daily, hyperglycemia in 1 patient on 150 mg twice weekly, and diarrhea in 1 patient on 150 mg 3 times weekly. There were no DLTs in patients on the 5/2 schedule.
The incidence of grade 3 or higher adverse events (AEs) was 43% (19/44). The most common of these AEs were thrombocytopenia (20%, n=9), neutropenia (7%, n=3), and hyperglycemia (7%, n=3).
Twenty-five percent of patients (11/44) had serious AEs. Three of these events were considered treatment-related. They were epistaxis and the DLTs of diarrhea and hyperglycemia.
AEs led to dose reductions in 6 patients (14%) and treatment discontinuation in 7 patients (16%).
Efficacy
Thirty-seven patients were evaluable for response, and 5 of these patients responded (14%). All responses—2 complete and 3 partial responses—occurred in patients with DLBCL.
Twenty-one of the response-evaluable patients (57%) had stable disease. This included 1 patient with DLBCL, 2 with MM, 8 with HL, and 10 with the “other” types of lymphoma.
The remaining 11 patients progressed (30%)—3 with DLBCL, 2 with MM, 2 with HL, and 4 with other lymphomas.
Thirty-seven patients (84%) discontinued CUDC-907 because of progressive disease or clinical signs of progressive disease at the data cutoff.
Based on the clinical activity of CUDC-907 in patients with relapsed/refractory DLBCL, particularly those with MYC alterations, Curis has initiated a phase 2 trial of the drug in these patients. The recommended phase 2 dose is 60 mg on the 5/2 dosing schedule. ![]()
The small-molecule inhibitor CUDC-907 can provide disease control in patients with relapsed or refractory lymphoma and multiple myeloma (MM), according to researchers.
In a phase 1 trial, CUDC-907 produced responses in a small number of patients with diffuse large B-cell lymphoma (DLBCL).
And more than half of patients had stable disease while on CUDC-907, including those with MM, DLBCL, Hodgkin lymphoma (HL), and other lymphomas.
However, a majority of patients in this trial—84%—discontinued treatment due to confirmed progressive disease or signs of progression.
These results were published in The Lancet. The trial was sponsored by Curis, Inc., the company developing CUDC-907, and the Leukemia and Lymphoma Society.
“The data from the phase 1 monotherapy trial for CUDC-907, especially in heavily pretreated patients with relapsed/refractory DLBCL are very encouraging, and we look forward to data emerging from the current phase 2 trial in patients with MYC-altered DLBCL,” said study author Anas Younes, MD, of the Memorial Sloan Kettering Cancer Center in New York, New York.
CUDC-907 is an oral, dual inhibitor of class I and II histone deacetylases (HDACs), as well as class I PI3K enzymes. Specifically, CUDC-907 is designed to inhibit HDACs 1, 2, 3, 6, and 10 and PI3K-alpha, delta, and beta isoforms.
Between Jan 23, 2013, and July 27, 2015, the phase 1 trial of CUDC-907 enrolled 44 patients who were refractory to or had relapsed after 2 or more previous regimens. The patients’ median age was 63 (range, 22-83), and they had received a median of 5 prior treatments (range, 2-10).
Four patients had MM, 12 had HL, and 12 had DLBCL. The remaining 16 patients had other types of lymphoma, including lymphoplasmacytic lymphoma (n=3), small lymphocytic lymphoma (n=3), mantle cell lymphoma (n=3), follicular lymphoma (n=2), T-cell lymphoma (n=2), marginal zone lymphoma (n=1), Burkitt lymphoma (n=1), and gray zone lymphoma (n=1).
Treatment
CUDC-907 was given in a standard 3 + 3 dose-escalation design at 4 different dosing schedules—once daily, twice weekly, 3 times weekly, and daily for 5 days followed by a 2-day break (5/2)—in 21-day cycles.
Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase 2 dose.
Ten patients were sequentially assigned to CUDC-907 once-daily (MTD 60 mg), 12 to twice-weekly (MTD 150 mg), 15 to 3-times-weekly (MTD 150 mg), and 7 to the 5/2 dosing schedule (MTD 60 mg).
Safety
Four dose-limiting toxicities (DLTs) occurred in 3 of 40 DLT-evaluable patients. The DLTs were diarrhea and hyperglycemia in 1 patient on 60 mg once daily, hyperglycemia in 1 patient on 150 mg twice weekly, and diarrhea in 1 patient on 150 mg 3 times weekly. There were no DLTs in patients on the 5/2 schedule.
The incidence of grade 3 or higher adverse events (AEs) was 43% (19/44). The most common of these AEs were thrombocytopenia (20%, n=9), neutropenia (7%, n=3), and hyperglycemia (7%, n=3).
Twenty-five percent of patients (11/44) had serious AEs. Three of these events were considered treatment-related. They were epistaxis and the DLTs of diarrhea and hyperglycemia.
AEs led to dose reductions in 6 patients (14%) and treatment discontinuation in 7 patients (16%).
Efficacy
Thirty-seven patients were evaluable for response, and 5 of these patients responded (14%). All responses—2 complete and 3 partial responses—occurred in patients with DLBCL.
Twenty-one of the response-evaluable patients (57%) had stable disease. This included 1 patient with DLBCL, 2 with MM, 8 with HL, and 10 with the “other” types of lymphoma.
The remaining 11 patients progressed (30%)—3 with DLBCL, 2 with MM, 2 with HL, and 4 with other lymphomas.
Thirty-seven patients (84%) discontinued CUDC-907 because of progressive disease or clinical signs of progressive disease at the data cutoff.
Based on the clinical activity of CUDC-907 in patients with relapsed/refractory DLBCL, particularly those with MYC alterations, Curis has initiated a phase 2 trial of the drug in these patients. The recommended phase 2 dose is 60 mg on the 5/2 dosing schedule. ![]()