User login
Drug gets orphan designation for BPDCN
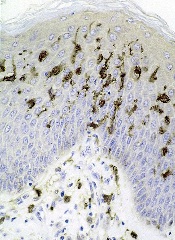
The European Medicines Agency (EMA) has granted orphan drug designation to SL-401 for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 is a targeted therapy directed to the interleukin-3 receptor (IL-3R), which is present on cancer stem cells and tumor bulk in a range of hematologic malignancies.
The drug is composed of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
SL-401 already has orphan designation from the EMA to treat acute myeloid leukemia (AML) and from the US Food and Drug Administration (FDA) for the treatment of AML and BPDCN. The drug is under development by Stemline Therapeutics, Inc.
SL-401 research
At ASH 2012 (abstract 3625), researchers reported results with SL-401 in a study of patients with AML, BPDCN, and myelodysplastic syndromes (MDS).
At that time, the study had enrolled 80 patients, including 59 with relapsed or refractory AML, 11 with de novo AML unfit for chemotherapy, 7 with high-risk MDS, and 3 with relapsed/refractory BPDCN.
Patients received a single cycle of SL-401 as a 15-minute intravenous infusion in 1 of 2 dosing regimens to determine the maximum tolerated dose (MTD) and assess antitumor activity.
With regimen A, 45 patients received doses ranging from 4 μg/kg to 12.5 μg/kg every other day for up to 6 doses. With regimen B, 35 patients received doses ranging from 7.1 μg/kg to 22.1 μg/kg daily for up to 5 doses.
Of the 59 patients with relapsed/refractory AML, 2 achieved complete responses (CRs), 5 had partial responses (PRs), and 8 had minor responses (MRs). One CR lasted more than 8 months, and the other lasted more than 25 months.
Of the 11 patients with AML who were not candidates for chemotherapy, 2 had PRs and 1 had an MR. Among the 7 patients with high-risk MDS, there was 1 PR and 1 MR.
And among the 3 patients with BPDCN, there were 2 CRs. One CR lasted more than 2 months, and the other lasted more than 4 months.
The MTD was not achieved with regimen A, but the MTD for regimen B was 16.6 μg/kg/day. The dose-limiting toxicities were a gastrointestinal bleed (n=1), transaminase and creatinine kinase elevations (n=1), and capillary leak syndrome (n=3). There was no evidence of treatment-related bone marrow suppression.
Last year, researchers reported additional results in BPDCN patients (Frankel et al, Blood 2014).
Eleven BPDCN patients received a single course of SL-401 (at 12.5 μg/kg intravenously over 15 minutes) daily for up to 5 doses. Three patients who had initial responses to SL-401 received a second course while in relapse.
Seven of 9 evaluable (78%) patients responded to a single course of SL-401. There were 5 CRs and 2 PRs. The median duration of responses was 5 months (range, 1-20+ months).
The most common adverse events were transient and included fever, chills, hypotension, edema, hypoalbuminemia, thrombocytopenia, and transaminasemia.
Three multicenter clinical trials of SL-401 are currently open in the following indications:
- BPDCN and relapsed/refractory AML
- AML patients in first complete remission with minimal residual disease
- Four types of advanced, high-risk myeloproliferative neoplasms, including systemic mastocytosis, advanced symptomatic hypereosinophilic disorder, myelofibrosis, and chronic myelomonocytic leukemia.
Additional SL-401 studies are planned for patients with myeloma, lymphomas, and other leukemias.
About orphan designation
In the European Union, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug in the European Union benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is on the market. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
In the US, orphan designation provides the sponsor of a drug with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
The European Medicines Agency (EMA) has granted orphan drug designation to SL-401 for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 is a targeted therapy directed to the interleukin-3 receptor (IL-3R), which is present on cancer stem cells and tumor bulk in a range of hematologic malignancies.
The drug is composed of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
SL-401 already has orphan designation from the EMA to treat acute myeloid leukemia (AML) and from the US Food and Drug Administration (FDA) for the treatment of AML and BPDCN. The drug is under development by Stemline Therapeutics, Inc.
SL-401 research
At ASH 2012 (abstract 3625), researchers reported results with SL-401 in a study of patients with AML, BPDCN, and myelodysplastic syndromes (MDS).
At that time, the study had enrolled 80 patients, including 59 with relapsed or refractory AML, 11 with de novo AML unfit for chemotherapy, 7 with high-risk MDS, and 3 with relapsed/refractory BPDCN.
Patients received a single cycle of SL-401 as a 15-minute intravenous infusion in 1 of 2 dosing regimens to determine the maximum tolerated dose (MTD) and assess antitumor activity.
With regimen A, 45 patients received doses ranging from 4 μg/kg to 12.5 μg/kg every other day for up to 6 doses. With regimen B, 35 patients received doses ranging from 7.1 μg/kg to 22.1 μg/kg daily for up to 5 doses.
Of the 59 patients with relapsed/refractory AML, 2 achieved complete responses (CRs), 5 had partial responses (PRs), and 8 had minor responses (MRs). One CR lasted more than 8 months, and the other lasted more than 25 months.
Of the 11 patients with AML who were not candidates for chemotherapy, 2 had PRs and 1 had an MR. Among the 7 patients with high-risk MDS, there was 1 PR and 1 MR.
And among the 3 patients with BPDCN, there were 2 CRs. One CR lasted more than 2 months, and the other lasted more than 4 months.
The MTD was not achieved with regimen A, but the MTD for regimen B was 16.6 μg/kg/day. The dose-limiting toxicities were a gastrointestinal bleed (n=1), transaminase and creatinine kinase elevations (n=1), and capillary leak syndrome (n=3). There was no evidence of treatment-related bone marrow suppression.
Last year, researchers reported additional results in BPDCN patients (Frankel et al, Blood 2014).
Eleven BPDCN patients received a single course of SL-401 (at 12.5 μg/kg intravenously over 15 minutes) daily for up to 5 doses. Three patients who had initial responses to SL-401 received a second course while in relapse.
Seven of 9 evaluable (78%) patients responded to a single course of SL-401. There were 5 CRs and 2 PRs. The median duration of responses was 5 months (range, 1-20+ months).
The most common adverse events were transient and included fever, chills, hypotension, edema, hypoalbuminemia, thrombocytopenia, and transaminasemia.
Three multicenter clinical trials of SL-401 are currently open in the following indications:
- BPDCN and relapsed/refractory AML
- AML patients in first complete remission with minimal residual disease
- Four types of advanced, high-risk myeloproliferative neoplasms, including systemic mastocytosis, advanced symptomatic hypereosinophilic disorder, myelofibrosis, and chronic myelomonocytic leukemia.
Additional SL-401 studies are planned for patients with myeloma, lymphomas, and other leukemias.
About orphan designation
In the European Union, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug in the European Union benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is on the market. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
In the US, orphan designation provides the sponsor of a drug with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
The European Medicines Agency (EMA) has granted orphan drug designation to SL-401 for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 is a targeted therapy directed to the interleukin-3 receptor (IL-3R), which is present on cancer stem cells and tumor bulk in a range of hematologic malignancies.
The drug is composed of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
SL-401 already has orphan designation from the EMA to treat acute myeloid leukemia (AML) and from the US Food and Drug Administration (FDA) for the treatment of AML and BPDCN. The drug is under development by Stemline Therapeutics, Inc.
SL-401 research
At ASH 2012 (abstract 3625), researchers reported results with SL-401 in a study of patients with AML, BPDCN, and myelodysplastic syndromes (MDS).
At that time, the study had enrolled 80 patients, including 59 with relapsed or refractory AML, 11 with de novo AML unfit for chemotherapy, 7 with high-risk MDS, and 3 with relapsed/refractory BPDCN.
Patients received a single cycle of SL-401 as a 15-minute intravenous infusion in 1 of 2 dosing regimens to determine the maximum tolerated dose (MTD) and assess antitumor activity.
With regimen A, 45 patients received doses ranging from 4 μg/kg to 12.5 μg/kg every other day for up to 6 doses. With regimen B, 35 patients received doses ranging from 7.1 μg/kg to 22.1 μg/kg daily for up to 5 doses.
Of the 59 patients with relapsed/refractory AML, 2 achieved complete responses (CRs), 5 had partial responses (PRs), and 8 had minor responses (MRs). One CR lasted more than 8 months, and the other lasted more than 25 months.
Of the 11 patients with AML who were not candidates for chemotherapy, 2 had PRs and 1 had an MR. Among the 7 patients with high-risk MDS, there was 1 PR and 1 MR.
And among the 3 patients with BPDCN, there were 2 CRs. One CR lasted more than 2 months, and the other lasted more than 4 months.
The MTD was not achieved with regimen A, but the MTD for regimen B was 16.6 μg/kg/day. The dose-limiting toxicities were a gastrointestinal bleed (n=1), transaminase and creatinine kinase elevations (n=1), and capillary leak syndrome (n=3). There was no evidence of treatment-related bone marrow suppression.
Last year, researchers reported additional results in BPDCN patients (Frankel et al, Blood 2014).
Eleven BPDCN patients received a single course of SL-401 (at 12.5 μg/kg intravenously over 15 minutes) daily for up to 5 doses. Three patients who had initial responses to SL-401 received a second course while in relapse.
Seven of 9 evaluable (78%) patients responded to a single course of SL-401. There were 5 CRs and 2 PRs. The median duration of responses was 5 months (range, 1-20+ months).
The most common adverse events were transient and included fever, chills, hypotension, edema, hypoalbuminemia, thrombocytopenia, and transaminasemia.
Three multicenter clinical trials of SL-401 are currently open in the following indications:
- BPDCN and relapsed/refractory AML
- AML patients in first complete remission with minimal residual disease
- Four types of advanced, high-risk myeloproliferative neoplasms, including systemic mastocytosis, advanced symptomatic hypereosinophilic disorder, myelofibrosis, and chronic myelomonocytic leukemia.
Additional SL-401 studies are planned for patients with myeloma, lymphomas, and other leukemias.
About orphan designation
In the European Union, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug in the European Union benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is on the market. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
The FDA grants orphan designation to drugs that are intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
In the US, orphan designation provides the sponsor of a drug with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Iron chelator tablets may now be crushed

Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has approved a label change for Jadenu, an oral formulation of the iron chelator Exjade (deferasirox).
Jadenu comes in tablet form, and the previous label stated that Jadenu tablets must be swallowed whole.
Now, the medication can also be crushed to help simplify administration for patients who have difficulty swallowing whole tablets.
Jadenu tablets may be crushed and mixed with soft foods, such as yogurt or applesauce, immediately prior to use.
The label notes that commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be consumed immediately and not stored.
Jadenu was granted accelerated approval from the FDA earlier this year.
It is approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions, as well as to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
The full prescribing information for Jadenu can be found at http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has approved a label change for Jadenu, an oral formulation of the iron chelator Exjade (deferasirox).
Jadenu comes in tablet form, and the previous label stated that Jadenu tablets must be swallowed whole.
Now, the medication can also be crushed to help simplify administration for patients who have difficulty swallowing whole tablets.
Jadenu tablets may be crushed and mixed with soft foods, such as yogurt or applesauce, immediately prior to use.
The label notes that commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be consumed immediately and not stored.
Jadenu was granted accelerated approval from the FDA earlier this year.
It is approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions, as well as to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
The full prescribing information for Jadenu can be found at http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has approved a label change for Jadenu, an oral formulation of the iron chelator Exjade (deferasirox).
Jadenu comes in tablet form, and the previous label stated that Jadenu tablets must be swallowed whole.
Now, the medication can also be crushed to help simplify administration for patients who have difficulty swallowing whole tablets.
Jadenu tablets may be crushed and mixed with soft foods, such as yogurt or applesauce, immediately prior to use.
The label notes that commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be consumed immediately and not stored.
Jadenu was granted accelerated approval from the FDA earlier this year.
It is approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions, as well as to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
The full prescribing information for Jadenu can be found at http://www.pharma.us.novartis.com/product/pi/pdf/jadenu.pdf. ![]()
WHO recommends pilot projects for malaria vaccine

a malaria-endemic region
Photo by Sarah Mattison
More testing is needed before the malaria vaccine candidate RTS,S/AS01 (Mosquirix) can be put into widespread use, according to a pair of World Health Organization (WHO) advisory committees.
The WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) are recommending that RTS,S be introduced in 3 to 5 pilot projects to determine the best way to deliver the vaccine to young children.
The issue, according to the committees, is that the vaccine must be administered in 4 doses and therefore requires repeat contact with the healthcare system.
The first 3 doses are given 1 month apart, and the last dose is given 18 months later. Without the fourth dose, children in a phase 3 study of RTS,S had no overall reduction in severe malaria.
So SAGE and MPAC want to be sure this vaccination schedule is feasible.
“The question about how the malaria vaccine may best be delivered still needs to be answered,” said Jon S. Abramson, chair of SAGE. “After detailed assessment of all the evidence, we recommended that this question is best addressed by having 3 to 5 large pilot implementation projects.”
This could delay widespread implementation of RTS,S for 3 to 5 years. Alternatively, if it is not possible to deliver all 4 doses of RTS,S consistently, Abramson said the vaccine may not be used at all.
RTS,S is being assessed as a complementary malaria control tool that could potentially be added to—but not replace—the core package of proven malaria preventive, diagnostic, and treatment measures.
The vaccine acts against Plasmodium falciparum, the most deadly malaria parasite globally and the most prevalent in Africa.
In a phase 3 trial, young children who received 4 doses of RTS,S had a 36% reduction in the number of clinical episodes of malaria at 4 years. Infants who received 4 doses of RTS,S had a 26% reduction in the number of clinical malaria episodes over 3 years.
Children had a significantly lower incidence of severe malaria only if they received all 4 doses of RTS,S. The vaccine did not confer the same benefit in infants, regardless of the doses given.
Results of a subsequent study suggested that genetic variation influences the vaccine’s ability to ward off malaria in young children but not in infants.
RTS,S was recently granted a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) via Article 58 of Regulation No 726/2004.
This allows the CHMP, in cooperation with the WHO, to give a scientific opinion on a medicinal product intended for markets outside the European Union. This assessment requires products to meet the same standards as products intended for use in the European Union.
Once the CHMP issued a positive opinion of RTS,S, the WHO began formulating a policy recommendation on use of the vaccine in national immunization programs. RTS,S must pass the WHO pre-qualification process and be approved by national regulatory authorities before it can be used in such programs. ![]()
a malaria-endemic region
Photo by Sarah Mattison
More testing is needed before the malaria vaccine candidate RTS,S/AS01 (Mosquirix) can be put into widespread use, according to a pair of World Health Organization (WHO) advisory committees.
The WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) are recommending that RTS,S be introduced in 3 to 5 pilot projects to determine the best way to deliver the vaccine to young children.
The issue, according to the committees, is that the vaccine must be administered in 4 doses and therefore requires repeat contact with the healthcare system.
The first 3 doses are given 1 month apart, and the last dose is given 18 months later. Without the fourth dose, children in a phase 3 study of RTS,S had no overall reduction in severe malaria.
So SAGE and MPAC want to be sure this vaccination schedule is feasible.
“The question about how the malaria vaccine may best be delivered still needs to be answered,” said Jon S. Abramson, chair of SAGE. “After detailed assessment of all the evidence, we recommended that this question is best addressed by having 3 to 5 large pilot implementation projects.”
This could delay widespread implementation of RTS,S for 3 to 5 years. Alternatively, if it is not possible to deliver all 4 doses of RTS,S consistently, Abramson said the vaccine may not be used at all.
RTS,S is being assessed as a complementary malaria control tool that could potentially be added to—but not replace—the core package of proven malaria preventive, diagnostic, and treatment measures.
The vaccine acts against Plasmodium falciparum, the most deadly malaria parasite globally and the most prevalent in Africa.
In a phase 3 trial, young children who received 4 doses of RTS,S had a 36% reduction in the number of clinical episodes of malaria at 4 years. Infants who received 4 doses of RTS,S had a 26% reduction in the number of clinical malaria episodes over 3 years.
Children had a significantly lower incidence of severe malaria only if they received all 4 doses of RTS,S. The vaccine did not confer the same benefit in infants, regardless of the doses given.
Results of a subsequent study suggested that genetic variation influences the vaccine’s ability to ward off malaria in young children but not in infants.
RTS,S was recently granted a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) via Article 58 of Regulation No 726/2004.
This allows the CHMP, in cooperation with the WHO, to give a scientific opinion on a medicinal product intended for markets outside the European Union. This assessment requires products to meet the same standards as products intended for use in the European Union.
Once the CHMP issued a positive opinion of RTS,S, the WHO began formulating a policy recommendation on use of the vaccine in national immunization programs. RTS,S must pass the WHO pre-qualification process and be approved by national regulatory authorities before it can be used in such programs. ![]()
a malaria-endemic region
Photo by Sarah Mattison
More testing is needed before the malaria vaccine candidate RTS,S/AS01 (Mosquirix) can be put into widespread use, according to a pair of World Health Organization (WHO) advisory committees.
The WHO’s Strategic Advisory Group of Experts on Immunization (SAGE) and the Malaria Policy Advisory Committee (MPAC) are recommending that RTS,S be introduced in 3 to 5 pilot projects to determine the best way to deliver the vaccine to young children.
The issue, according to the committees, is that the vaccine must be administered in 4 doses and therefore requires repeat contact with the healthcare system.
The first 3 doses are given 1 month apart, and the last dose is given 18 months later. Without the fourth dose, children in a phase 3 study of RTS,S had no overall reduction in severe malaria.
So SAGE and MPAC want to be sure this vaccination schedule is feasible.
“The question about how the malaria vaccine may best be delivered still needs to be answered,” said Jon S. Abramson, chair of SAGE. “After detailed assessment of all the evidence, we recommended that this question is best addressed by having 3 to 5 large pilot implementation projects.”
This could delay widespread implementation of RTS,S for 3 to 5 years. Alternatively, if it is not possible to deliver all 4 doses of RTS,S consistently, Abramson said the vaccine may not be used at all.
RTS,S is being assessed as a complementary malaria control tool that could potentially be added to—but not replace—the core package of proven malaria preventive, diagnostic, and treatment measures.
The vaccine acts against Plasmodium falciparum, the most deadly malaria parasite globally and the most prevalent in Africa.
In a phase 3 trial, young children who received 4 doses of RTS,S had a 36% reduction in the number of clinical episodes of malaria at 4 years. Infants who received 4 doses of RTS,S had a 26% reduction in the number of clinical malaria episodes over 3 years.
Children had a significantly lower incidence of severe malaria only if they received all 4 doses of RTS,S. The vaccine did not confer the same benefit in infants, regardless of the doses given.
Results of a subsequent study suggested that genetic variation influences the vaccine’s ability to ward off malaria in young children but not in infants.
RTS,S was recently granted a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) via Article 58 of Regulation No 726/2004.
This allows the CHMP, in cooperation with the WHO, to give a scientific opinion on a medicinal product intended for markets outside the European Union. This assessment requires products to meet the same standards as products intended for use in the European Union.
Once the CHMP issued a positive opinion of RTS,S, the WHO began formulating a policy recommendation on use of the vaccine in national immunization programs. RTS,S must pass the WHO pre-qualification process and be approved by national regulatory authorities before it can be used in such programs. ![]()
New melphalan formulation denied approval
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has said that, at present, it cannot approve a propylene glycol-free melphalan formulation (Evomela) for use in patients with multiple myeloma (MM).
Spectrum Pharmaceuticals is seeking approval for Evomela as a high-dose conditioning treatment for MM patients undergoing hematopoietic stem cell transplant (HSCT) and for palliative treatment in MM patients for whom oral therapy is not appropriate.
The FDA issued a Complete Response Letter stating that the new drug application (NDA) for Evomela cannot be approved in its present form.
However, the FDA did not identify any clinical deficiency in the NDA package.
“We will work swiftly with the FDA to address the Complete Response Letter,” said Rajesh C. Shrotriya, MD, chairman and chief executive officer of Spectrum Pharmaceuticals. “We remain committed to bringing Evomela to the market for patients and plan to work closely with the FDA.”
About Evomela
Evomela is a Captisol-enabled, propylene glycol-free melphalan formulation. This formulation eliminates the need to use a propylene glycol-containing custom diluent, which is required with other intravenous melphalan formulations and has been reported to cause renal and cardiac side effects.
The use of Captisol technology to reformulate melphalan is reported to improve the drug’s stability, extending its use time to 5 hours. This is anticipated to simplify preparation and administration logistics and allow for slower infusion rates and longer administration durations for pre-transplant chemotherapy.
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs.
Spectrum Pharmaceuticals gained global development and commercialization rights to Evomela from Ligand Pharmaceuticals Incorporated in March 2013. Spectrum assumed responsibility for completing the pivotal phase 2 clinical trial and was responsible for filing the NDA. Spectrum filed the NDA in December 2014, and the FDA accepted the application the following March.
The FDA has granted Evomela orphan drug designation for use as a high-dose conditioning regimen for MM patients undergoing HSCT.
Phase 2 study
Researchers have evaluated Evomela in a phase 2, multicenter trial. Initial results from this trial (phase 2a) were published in Bone Marrow Transplantation in June 2014. Phase 2b results were published in Biology of Blood and Marrow Transplantation last month.
The latest publication includes data on 61 patients. Fifty-six had newly diagnosed MM, and 5 had relapsed MM following prior HSCT. The patients received Evomela at 200 mg/m2 given as 2 doses on Day -3 and Day -2 prior to HSCT (Day 0).
Efficacy was assessed by clinical response at Day +100. According to investigator assessment, the overall response rate was 95%, and the complete response (CR) rate was 31%.
According to independent pathology review, the overall response rate was 100%, and the CR rate was 21%. The lower rate of confirmed CRs in the independent review was due to missing data.
All 5 patients who had previously relapsed from a prior HSCT responded to Evomela.
All patients in the study achieved myeloablation with a median of 5 days post-HSCT. All patients had successful neutrophil and platelet engraftment at a median of 12 days and 13 days post-HSCT, respectively.
Treatment-related mortality was 0%, and non-hematologic adverse events were mostly grade 1 and 2 in severity. The incidence of grade 3 mucositis and grade 3 stomatitis were 10% and 5%, respectively, with no grade 4 mucositis or stomatitis reported.
Twenty percent of patients experienced treatment-emergent serious adverse events, most of which were grade 3 and consisted of events commonly reported in patients undergoing myeloablative chemotherapy. No new safety signals were identified. ![]()
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has said that, at present, it cannot approve a propylene glycol-free melphalan formulation (Evomela) for use in patients with multiple myeloma (MM).
Spectrum Pharmaceuticals is seeking approval for Evomela as a high-dose conditioning treatment for MM patients undergoing hematopoietic stem cell transplant (HSCT) and for palliative treatment in MM patients for whom oral therapy is not appropriate.
The FDA issued a Complete Response Letter stating that the new drug application (NDA) for Evomela cannot be approved in its present form.
However, the FDA did not identify any clinical deficiency in the NDA package.
“We will work swiftly with the FDA to address the Complete Response Letter,” said Rajesh C. Shrotriya, MD, chairman and chief executive officer of Spectrum Pharmaceuticals. “We remain committed to bringing Evomela to the market for patients and plan to work closely with the FDA.”
About Evomela
Evomela is a Captisol-enabled, propylene glycol-free melphalan formulation. This formulation eliminates the need to use a propylene glycol-containing custom diluent, which is required with other intravenous melphalan formulations and has been reported to cause renal and cardiac side effects.
The use of Captisol technology to reformulate melphalan is reported to improve the drug’s stability, extending its use time to 5 hours. This is anticipated to simplify preparation and administration logistics and allow for slower infusion rates and longer administration durations for pre-transplant chemotherapy.
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs.
Spectrum Pharmaceuticals gained global development and commercialization rights to Evomela from Ligand Pharmaceuticals Incorporated in March 2013. Spectrum assumed responsibility for completing the pivotal phase 2 clinical trial and was responsible for filing the NDA. Spectrum filed the NDA in December 2014, and the FDA accepted the application the following March.
The FDA has granted Evomela orphan drug designation for use as a high-dose conditioning regimen for MM patients undergoing HSCT.
Phase 2 study
Researchers have evaluated Evomela in a phase 2, multicenter trial. Initial results from this trial (phase 2a) were published in Bone Marrow Transplantation in June 2014. Phase 2b results were published in Biology of Blood and Marrow Transplantation last month.
The latest publication includes data on 61 patients. Fifty-six had newly diagnosed MM, and 5 had relapsed MM following prior HSCT. The patients received Evomela at 200 mg/m2 given as 2 doses on Day -3 and Day -2 prior to HSCT (Day 0).
Efficacy was assessed by clinical response at Day +100. According to investigator assessment, the overall response rate was 95%, and the complete response (CR) rate was 31%.
According to independent pathology review, the overall response rate was 100%, and the CR rate was 21%. The lower rate of confirmed CRs in the independent review was due to missing data.
All 5 patients who had previously relapsed from a prior HSCT responded to Evomela.
All patients in the study achieved myeloablation with a median of 5 days post-HSCT. All patients had successful neutrophil and platelet engraftment at a median of 12 days and 13 days post-HSCT, respectively.
Treatment-related mortality was 0%, and non-hematologic adverse events were mostly grade 1 and 2 in severity. The incidence of grade 3 mucositis and grade 3 stomatitis were 10% and 5%, respectively, with no grade 4 mucositis or stomatitis reported.
Twenty percent of patients experienced treatment-emergent serious adverse events, most of which were grade 3 and consisted of events commonly reported in patients undergoing myeloablative chemotherapy. No new safety signals were identified. ![]()
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has said that, at present, it cannot approve a propylene glycol-free melphalan formulation (Evomela) for use in patients with multiple myeloma (MM).
Spectrum Pharmaceuticals is seeking approval for Evomela as a high-dose conditioning treatment for MM patients undergoing hematopoietic stem cell transplant (HSCT) and for palliative treatment in MM patients for whom oral therapy is not appropriate.
The FDA issued a Complete Response Letter stating that the new drug application (NDA) for Evomela cannot be approved in its present form.
However, the FDA did not identify any clinical deficiency in the NDA package.
“We will work swiftly with the FDA to address the Complete Response Letter,” said Rajesh C. Shrotriya, MD, chairman and chief executive officer of Spectrum Pharmaceuticals. “We remain committed to bringing Evomela to the market for patients and plan to work closely with the FDA.”
About Evomela
Evomela is a Captisol-enabled, propylene glycol-free melphalan formulation. This formulation eliminates the need to use a propylene glycol-containing custom diluent, which is required with other intravenous melphalan formulations and has been reported to cause renal and cardiac side effects.
The use of Captisol technology to reformulate melphalan is reported to improve the drug’s stability, extending its use time to 5 hours. This is anticipated to simplify preparation and administration logistics and allow for slower infusion rates and longer administration durations for pre-transplant chemotherapy.
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs.
Spectrum Pharmaceuticals gained global development and commercialization rights to Evomela from Ligand Pharmaceuticals Incorporated in March 2013. Spectrum assumed responsibility for completing the pivotal phase 2 clinical trial and was responsible for filing the NDA. Spectrum filed the NDA in December 2014, and the FDA accepted the application the following March.
The FDA has granted Evomela orphan drug designation for use as a high-dose conditioning regimen for MM patients undergoing HSCT.
Phase 2 study
Researchers have evaluated Evomela in a phase 2, multicenter trial. Initial results from this trial (phase 2a) were published in Bone Marrow Transplantation in June 2014. Phase 2b results were published in Biology of Blood and Marrow Transplantation last month.
The latest publication includes data on 61 patients. Fifty-six had newly diagnosed MM, and 5 had relapsed MM following prior HSCT. The patients received Evomela at 200 mg/m2 given as 2 doses on Day -3 and Day -2 prior to HSCT (Day 0).
Efficacy was assessed by clinical response at Day +100. According to investigator assessment, the overall response rate was 95%, and the complete response (CR) rate was 31%.
According to independent pathology review, the overall response rate was 100%, and the CR rate was 21%. The lower rate of confirmed CRs in the independent review was due to missing data.
All 5 patients who had previously relapsed from a prior HSCT responded to Evomela.
All patients in the study achieved myeloablation with a median of 5 days post-HSCT. All patients had successful neutrophil and platelet engraftment at a median of 12 days and 13 days post-HSCT, respectively.
Treatment-related mortality was 0%, and non-hematologic adverse events were mostly grade 1 and 2 in severity. The incidence of grade 3 mucositis and grade 3 stomatitis were 10% and 5%, respectively, with no grade 4 mucositis or stomatitis reported.
Twenty percent of patients experienced treatment-emergent serious adverse events, most of which were grade 3 and consisted of events commonly reported in patients undergoing myeloablative chemotherapy. No new safety signals were identified. ![]()
FDA approves factor X concentrate

The US Food and Drug Administration (FDA) has approved a factor X product derived from human plasma (Coagadex) to treat patients with hereditary factor X deficiency who are 12 years of age and older.
Coagadex is approved for on-demand treatment and control of bleeding episodes in these patients as well as for perioperative management of bleeding in patients with mild hereditary factor X deficiency.
Prior to this approval, there was no specific coagulation factor replacement therapy available for patients with hereditary factor X deficiency in the US. The FDA previously granted Coagadex orphan product designation, fast track designation, and priority review.
The FDA based its approval of Coagadex on results from 2 phase 3 trials of patients age 12 and older.
The first trial included 16 patients who received Coagadex for pharmacokinetic evaluation, on-demand treatment and control of bleeding episodes, and/or perioperative management of minor surgical or dental procedures.
Coagadex was used to treat 208 bleeding episodes, and 187 of these episodes (in 15 patients) were evaluated for efficacy. Ninety-eight episodes were major bleeds, 88 were minor bleeds, and 1 was not assessed.
One hundred and fifty-five bleeds (83%) were treated with a single infusion of Coagadex, 28 (15%) were treated with 2 infusions, 3 bleeds (2%) required 3 infusions, and 1 bleed (0.5%) required 4 infusions. Four bleeding episodes in 2 patients were considered treatment failures.
The mean dose of Coagadex per infusion was 25.4 IU/kg, and the mean total dose was 30.4 IU/kg. The recommended dose of 25 IU/kg to treat a bleed was maintained for 14 of the 16 patients. The other 2 patients used doses of up to 30 IU/kg and 33 IU/kg.
There were 176 adverse events in this trial, but only 6 events in 2 patients were considered possibly related to Coagadex. This included 2 reports of infusion site erythema in 1 patient, 2 reports of fatigue in 1 patient, 1 report of back pain, and 1 report of infusion site pain.
The second trial included patients who received Coagadex for perioperative management. Five patients received Coagadex for 7 surgical procedures.
For major surgeries, a median of 13 infusions (range, 2-15) and a median cumulative dose of 181 IU/kg (range, 45-210 IU/kg) were required to maintain hemostasis. For minor surgeries, a median of 2.5 infusions (range, 1-4) and a median cumulative dose of 89 IU/kg (range, 51-127 IU/kg) were required to maintain hemostasis.
There were no adverse events related to Coagadex in this trial.
For more details on these trials, see the Coagadex package insert. Coagadex is manufactured by Bio Products Laboratory Limited in Elstree, Hertfordshire, UK. ![]()
The US Food and Drug Administration (FDA) has approved a factor X product derived from human plasma (Coagadex) to treat patients with hereditary factor X deficiency who are 12 years of age and older.
Coagadex is approved for on-demand treatment and control of bleeding episodes in these patients as well as for perioperative management of bleeding in patients with mild hereditary factor X deficiency.
Prior to this approval, there was no specific coagulation factor replacement therapy available for patients with hereditary factor X deficiency in the US. The FDA previously granted Coagadex orphan product designation, fast track designation, and priority review.
The FDA based its approval of Coagadex on results from 2 phase 3 trials of patients age 12 and older.
The first trial included 16 patients who received Coagadex for pharmacokinetic evaluation, on-demand treatment and control of bleeding episodes, and/or perioperative management of minor surgical or dental procedures.
Coagadex was used to treat 208 bleeding episodes, and 187 of these episodes (in 15 patients) were evaluated for efficacy. Ninety-eight episodes were major bleeds, 88 were minor bleeds, and 1 was not assessed.
One hundred and fifty-five bleeds (83%) were treated with a single infusion of Coagadex, 28 (15%) were treated with 2 infusions, 3 bleeds (2%) required 3 infusions, and 1 bleed (0.5%) required 4 infusions. Four bleeding episodes in 2 patients were considered treatment failures.
The mean dose of Coagadex per infusion was 25.4 IU/kg, and the mean total dose was 30.4 IU/kg. The recommended dose of 25 IU/kg to treat a bleed was maintained for 14 of the 16 patients. The other 2 patients used doses of up to 30 IU/kg and 33 IU/kg.
There were 176 adverse events in this trial, but only 6 events in 2 patients were considered possibly related to Coagadex. This included 2 reports of infusion site erythema in 1 patient, 2 reports of fatigue in 1 patient, 1 report of back pain, and 1 report of infusion site pain.
The second trial included patients who received Coagadex for perioperative management. Five patients received Coagadex for 7 surgical procedures.
For major surgeries, a median of 13 infusions (range, 2-15) and a median cumulative dose of 181 IU/kg (range, 45-210 IU/kg) were required to maintain hemostasis. For minor surgeries, a median of 2.5 infusions (range, 1-4) and a median cumulative dose of 89 IU/kg (range, 51-127 IU/kg) were required to maintain hemostasis.
There were no adverse events related to Coagadex in this trial.
For more details on these trials, see the Coagadex package insert. Coagadex is manufactured by Bio Products Laboratory Limited in Elstree, Hertfordshire, UK. ![]()
The US Food and Drug Administration (FDA) has approved a factor X product derived from human plasma (Coagadex) to treat patients with hereditary factor X deficiency who are 12 years of age and older.
Coagadex is approved for on-demand treatment and control of bleeding episodes in these patients as well as for perioperative management of bleeding in patients with mild hereditary factor X deficiency.
Prior to this approval, there was no specific coagulation factor replacement therapy available for patients with hereditary factor X deficiency in the US. The FDA previously granted Coagadex orphan product designation, fast track designation, and priority review.
The FDA based its approval of Coagadex on results from 2 phase 3 trials of patients age 12 and older.
The first trial included 16 patients who received Coagadex for pharmacokinetic evaluation, on-demand treatment and control of bleeding episodes, and/or perioperative management of minor surgical or dental procedures.
Coagadex was used to treat 208 bleeding episodes, and 187 of these episodes (in 15 patients) were evaluated for efficacy. Ninety-eight episodes were major bleeds, 88 were minor bleeds, and 1 was not assessed.
One hundred and fifty-five bleeds (83%) were treated with a single infusion of Coagadex, 28 (15%) were treated with 2 infusions, 3 bleeds (2%) required 3 infusions, and 1 bleed (0.5%) required 4 infusions. Four bleeding episodes in 2 patients were considered treatment failures.
The mean dose of Coagadex per infusion was 25.4 IU/kg, and the mean total dose was 30.4 IU/kg. The recommended dose of 25 IU/kg to treat a bleed was maintained for 14 of the 16 patients. The other 2 patients used doses of up to 30 IU/kg and 33 IU/kg.
There were 176 adverse events in this trial, but only 6 events in 2 patients were considered possibly related to Coagadex. This included 2 reports of infusion site erythema in 1 patient, 2 reports of fatigue in 1 patient, 1 report of back pain, and 1 report of infusion site pain.
The second trial included patients who received Coagadex for perioperative management. Five patients received Coagadex for 7 surgical procedures.
For major surgeries, a median of 13 infusions (range, 2-15) and a median cumulative dose of 181 IU/kg (range, 45-210 IU/kg) were required to maintain hemostasis. For minor surgeries, a median of 2.5 infusions (range, 1-4) and a median cumulative dose of 89 IU/kg (range, 51-127 IU/kg) were required to maintain hemostasis.
There were no adverse events related to Coagadex in this trial.
For more details on these trials, see the Coagadex package insert. Coagadex is manufactured by Bio Products Laboratory Limited in Elstree, Hertfordshire, UK. ![]()
Drug granted breakthrough designation for ALL

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for inotuzumab ozogamicin to treat adults with acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin consists of a monoclonal antibody targeting CD22 and the cytotoxic agent calicheamicin.
When this antibody-drug conjugate binds to the CD22 antigen on malignant B cells, it is internalized, and calicheamicin is released to destroy the cell.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The FDA’s decision to grant inotuzumab ozogamicin breakthrough designation was based on results of the phase 3 INO-VATE ALL trial.
Results from this trial were presented at the 20th Congress of the European Hematology Association (EHA) last June (abstract LB2073*). The study is sponsored by Pfizer, the company developing inotuzumab ozogamicin.
This ongoing trial has enrolled 326 adult patients with relapsed or refractory, CD22-positive ALL. At EHA, Daniel DeAngelo, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, presented efficacy results in 205 patients and safety results in 259 patients.
Patients were assigned to receive inotuzumab ozogamicin (InO) or a defined set of chemotherapy choices (chemo). The InO schedule was once weekly for 3 weeks on a 3- to 4-week cycle for up to 6 cycles. Chemotherapy options included fludarabine, cytarabine, and G-CSF (FLAG); high-dose cytarabine (HIDAC); or cytarabine and mitoxantrone.
The primary endpoints of the study are hematologic remission, defined as a complete response with or without platelet and/or neutrophil recovery (CR/CRi), and overall survival. Survival data are not yet mature.
However, Dr DeAngelo reported that CR/CRi was significantly higher in the InO arm than the chemo arm—80.7% and 33.3%, respectively (P<0.0001). CR occurred in 35.8% and 19.8% of patients, respectively (P=0.0056), and CRi occurred in 45% and 13.5%, respectively (P<0.0001).
In both arms, most patients achieved CR/CRi during the first cycle of treatment—73% in the InO arm and 91% in the chemo arm.
The median duration of remission was 4.6 months in the InO arm and 3.1 months in the chemo arm (P=0.0169).
Overall, treatment-emergent adverse events (AEs) were similar between the arms. The incidence of any treatment-emergent AE was 98% in the InO arm and 99% in the chemo arm. The incidence of grade 3 or higher AEs was 91% and 95%, respectively. And the incidence of serious AEs was 48% and 46%, respectively.
Several AEs were more common in the chemo arm than the InO arm, including thrombocytopenia (61% vs 45%), anemia (53% vs 30%), febrile neutropenia (52% vs 27%), nausea (47% vs 32%), and pyrexia (42% vs 27%). The only AE that was more common in the InO arm than the chemo arm was AST increase (20% vs 10%).
There were 17 deaths in InO arm and 11 in the chemo arm. Four deaths in the InO arm and 2 in the chemo arm were considered treatment-related.
Causes of treatment-related deaths in the InO arm were acute respiratory distress syndrome as a terminal event of pneumonia (n=1), intestinal ischemia/septic shock (n=1), and veno-occlusive disease/ sinusoidal obstruction syndrome (n=2, both after post-study stem cell transplant). ![]()
*Information in the abstract differs from the presentation.
FDA approves reversal agent for dabigatran
treating a patient
Photo by Tom Watanabe
The US Food and Drug Administration (FDA) has granted accelerated approval for idarucizumab (Praxbind), the first reversal agent for the direct thrombin inhibitor dabigatran (Pradaxa).
Idarucizumab is now approved for use in emergency situations when there is a need to reverse the anticoagulant effect of dabigatran.
The FDA’s accelerated approval program allows the agency to approve drugs for serious conditions that fill an unmet medical need.
Accelerated approval is based on an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. So the company developing the drug is required to submit additional information after approval to confirm the drug’s clinical benefit.
About dabigatran and idarucizumab
Dabigatran is FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, as well as for the treatment and prevention of deep vein thrombosis and pulmonary embolism.
Idarucizumab is the first reversal agent approved specifically for dabigatran and works by binding to the drug compound to neutralize its effect. Idarucizumab is administered via intravenous injection.
Both idarucizumab and dabigatran are under development by Boehringer Ingelheim.
Idarucizumab has been studied in 3 randomized, double-blind, phase 1 trials of subjects who were not previously taking dabigatran and a phase 3 trial (RE-VERSE AD) of patients who were taking dabigatran and required reversal in an emergency setting.
Phase 1 trials
One phase 1 study (NCT01688830) enrolled 157 healthy male volunteers and consisted of 3 parts. Part 1 included 110 subjects who received placebo or idarucizumab at doses ranging from 20 mg to 8 g.
Idarucizumab (in the absence of dabigatran) was deemed safe and well tolerated. These results were published in Thrombosis and Haemostasis.
Parts 2 and 3 of the study included 47 subjects (part 2, n=35; part 3, n=12), and researchers investigated how well various doses of idarucizumab reversed the anticoagulant effect of dabigatran.
Results from parts 2 and 3 were published in The Lancet. The researchers said idarucizumab (given at 2 g or greater) provided immediate, complete, and sustained reversal of the anticoagulant effect of dabigatran, without producing serious adverse events.
In a second phase 1 study (NCT01955720), researchers evaluated idarucizumab in 46 subjects (males and females). This included healthy volunteers, elderly subjects, and participants with pre-existing mild or moderate kidney impairment.
Idarucizumab immediately and completely reversed dabigatran’s anticoagulant effect in these subjects, and they were able to restart dabigatran within 24 hours of receiving idarucizumab.
In addition, the researchers said there were no clinically relevant adverse events related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. These results were presented at the 2014 ASH Annual Meeting.
A third phase 1 study (NCT02028780) enrolled 80 healthy Japanese subjects. Researchers assessed the safety, tolerability, and pharmacokinetics of single, increasing doses of idarucizumab, administered both alone and after dabigatran.
Phase 3 trial
In the ongoing phase 3 trial, RE-VERSE AD, researchers are evaluating idarucizumab in emergency settings. The team reported interim results in 90 patients in NEJM and at the 2015 ISTH Congress.
Idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of patients who had uncontrolled or life-threatening bleeding complications while on dabigatran and in most patients who had to reverse dabigatran’s effects because they required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients experienced serious adverse events, 20% died, and several patients had thrombotic or bleeding events after receiving idarucizumab. ![]()
treating a patient
Photo by Tom Watanabe
The US Food and Drug Administration (FDA) has granted accelerated approval for idarucizumab (Praxbind), the first reversal agent for the direct thrombin inhibitor dabigatran (Pradaxa).
Idarucizumab is now approved for use in emergency situations when there is a need to reverse the anticoagulant effect of dabigatran.
The FDA’s accelerated approval program allows the agency to approve drugs for serious conditions that fill an unmet medical need.
Accelerated approval is based on an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. So the company developing the drug is required to submit additional information after approval to confirm the drug’s clinical benefit.
About dabigatran and idarucizumab
Dabigatran is FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, as well as for the treatment and prevention of deep vein thrombosis and pulmonary embolism.
Idarucizumab is the first reversal agent approved specifically for dabigatran and works by binding to the drug compound to neutralize its effect. Idarucizumab is administered via intravenous injection.
Both idarucizumab and dabigatran are under development by Boehringer Ingelheim.
Idarucizumab has been studied in 3 randomized, double-blind, phase 1 trials of subjects who were not previously taking dabigatran and a phase 3 trial (RE-VERSE AD) of patients who were taking dabigatran and required reversal in an emergency setting.
Phase 1 trials
One phase 1 study (NCT01688830) enrolled 157 healthy male volunteers and consisted of 3 parts. Part 1 included 110 subjects who received placebo or idarucizumab at doses ranging from 20 mg to 8 g.
Idarucizumab (in the absence of dabigatran) was deemed safe and well tolerated. These results were published in Thrombosis and Haemostasis.
Parts 2 and 3 of the study included 47 subjects (part 2, n=35; part 3, n=12), and researchers investigated how well various doses of idarucizumab reversed the anticoagulant effect of dabigatran.
Results from parts 2 and 3 were published in The Lancet. The researchers said idarucizumab (given at 2 g or greater) provided immediate, complete, and sustained reversal of the anticoagulant effect of dabigatran, without producing serious adverse events.
In a second phase 1 study (NCT01955720), researchers evaluated idarucizumab in 46 subjects (males and females). This included healthy volunteers, elderly subjects, and participants with pre-existing mild or moderate kidney impairment.
Idarucizumab immediately and completely reversed dabigatran’s anticoagulant effect in these subjects, and they were able to restart dabigatran within 24 hours of receiving idarucizumab.
In addition, the researchers said there were no clinically relevant adverse events related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. These results were presented at the 2014 ASH Annual Meeting.
A third phase 1 study (NCT02028780) enrolled 80 healthy Japanese subjects. Researchers assessed the safety, tolerability, and pharmacokinetics of single, increasing doses of idarucizumab, administered both alone and after dabigatran.
Phase 3 trial
In the ongoing phase 3 trial, RE-VERSE AD, researchers are evaluating idarucizumab in emergency settings. The team reported interim results in 90 patients in NEJM and at the 2015 ISTH Congress.
Idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of patients who had uncontrolled or life-threatening bleeding complications while on dabigatran and in most patients who had to reverse dabigatran’s effects because they required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients experienced serious adverse events, 20% died, and several patients had thrombotic or bleeding events after receiving idarucizumab. ![]()
treating a patient
Photo by Tom Watanabe
The US Food and Drug Administration (FDA) has granted accelerated approval for idarucizumab (Praxbind), the first reversal agent for the direct thrombin inhibitor dabigatran (Pradaxa).
Idarucizumab is now approved for use in emergency situations when there is a need to reverse the anticoagulant effect of dabigatran.
The FDA’s accelerated approval program allows the agency to approve drugs for serious conditions that fill an unmet medical need.
Accelerated approval is based on an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict a clinical benefit to patients. So the company developing the drug is required to submit additional information after approval to confirm the drug’s clinical benefit.
About dabigatran and idarucizumab
Dabigatran is FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, as well as for the treatment and prevention of deep vein thrombosis and pulmonary embolism.
Idarucizumab is the first reversal agent approved specifically for dabigatran and works by binding to the drug compound to neutralize its effect. Idarucizumab is administered via intravenous injection.
Both idarucizumab and dabigatran are under development by Boehringer Ingelheim.
Idarucizumab has been studied in 3 randomized, double-blind, phase 1 trials of subjects who were not previously taking dabigatran and a phase 3 trial (RE-VERSE AD) of patients who were taking dabigatran and required reversal in an emergency setting.
Phase 1 trials
One phase 1 study (NCT01688830) enrolled 157 healthy male volunteers and consisted of 3 parts. Part 1 included 110 subjects who received placebo or idarucizumab at doses ranging from 20 mg to 8 g.
Idarucizumab (in the absence of dabigatran) was deemed safe and well tolerated. These results were published in Thrombosis and Haemostasis.
Parts 2 and 3 of the study included 47 subjects (part 2, n=35; part 3, n=12), and researchers investigated how well various doses of idarucizumab reversed the anticoagulant effect of dabigatran.
Results from parts 2 and 3 were published in The Lancet. The researchers said idarucizumab (given at 2 g or greater) provided immediate, complete, and sustained reversal of the anticoagulant effect of dabigatran, without producing serious adverse events.
In a second phase 1 study (NCT01955720), researchers evaluated idarucizumab in 46 subjects (males and females). This included healthy volunteers, elderly subjects, and participants with pre-existing mild or moderate kidney impairment.
Idarucizumab immediately and completely reversed dabigatran’s anticoagulant effect in these subjects, and they were able to restart dabigatran within 24 hours of receiving idarucizumab.
In addition, the researchers said there were no clinically relevant adverse events related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. These results were presented at the 2014 ASH Annual Meeting.
A third phase 1 study (NCT02028780) enrolled 80 healthy Japanese subjects. Researchers assessed the safety, tolerability, and pharmacokinetics of single, increasing doses of idarucizumab, administered both alone and after dabigatran.
Phase 3 trial
In the ongoing phase 3 trial, RE-VERSE AD, researchers are evaluating idarucizumab in emergency settings. The team reported interim results in 90 patients in NEJM and at the 2015 ISTH Congress.
Idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of patients who had uncontrolled or life-threatening bleeding complications while on dabigatran and in most patients who had to reverse dabigatran’s effects because they required emergency surgery or an invasive procedure.
The researchers said there were no safety concerns related to idarucizumab. However, 23% of patients experienced serious adverse events, 20% died, and several patients had thrombotic or bleeding events after receiving idarucizumab.
Health Canada approves drug for acquired hemophilia A
Photo courtesy of
Baxter International Inc.
Health Canada has approved a recombinant porcine factor VIII (FVIII) product (Obizur) to treat bleeding episodes in patients with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Canada.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
Health Canada’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is currently approved for use in the US and is under regulatory review in the European Union, Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
Health Canada has approved a recombinant porcine factor VIII (FVIII) product (Obizur) to treat bleeding episodes in patients with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Canada.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
Health Canada’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is currently approved for use in the US and is under regulatory review in the European Union, Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
Health Canada has approved a recombinant porcine factor VIII (FVIII) product (Obizur) to treat bleeding episodes in patients with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Canada.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
Health Canada’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is currently approved for use in the US and is under regulatory review in the European Union, Switzerland, Australia, and Colombia.
COMP recommends orphan designations for KTE-C19

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has adopted positive opinions recommending orphan designation for KTE-C19 to treat acute lymphoblastic leukemia, chronic lymphocytic leukemia/small lymphocytic lymphoma, and follicular lymphoma.
KTE-C19 is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, a protein expressed on the surface of B cells.
The CAR T-cell therapy already has orphan designation for the treatment of diffuse large B-cell lymphoma in the US and the European Union (EU).
KTE-C19 also has COMP positive opinions for orphan designation in the EU for primary mediastinal B-cell lymphoma and mantle cell lymphoma.
About orphan designation
The COMP adopts an opinion on the granting of orphan designation, and that opinion is submitted to the European Commission for endorsement.
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
KTE-C19 research
Last year, researchers reported results with KTE-C19 in the Journal of Clinical Oncology. The study included 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of the CAR T-cell therapy. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR.
KTE-C19 was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or interleukin 6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
KTE-C19 is currently under investigation in a phase 1/2 trial (ZUMA-1) of patients with refractory, aggressive non-Hodgkin lymphomas. Kite Pharma, Inc., the company developing KTE-C19, plans to present top-line phase 1 data at the 2015 ASH Annual Meeting.
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has adopted positive opinions recommending orphan designation for KTE-C19 to treat acute lymphoblastic leukemia, chronic lymphocytic leukemia/small lymphocytic lymphoma, and follicular lymphoma.
KTE-C19 is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, a protein expressed on the surface of B cells.
The CAR T-cell therapy already has orphan designation for the treatment of diffuse large B-cell lymphoma in the US and the European Union (EU).
KTE-C19 also has COMP positive opinions for orphan designation in the EU for primary mediastinal B-cell lymphoma and mantle cell lymphoma.
About orphan designation
The COMP adopts an opinion on the granting of orphan designation, and that opinion is submitted to the European Commission for endorsement.
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
KTE-C19 research
Last year, researchers reported results with KTE-C19 in the Journal of Clinical Oncology. The study included 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of the CAR T-cell therapy. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR.
KTE-C19 was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or interleukin 6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
KTE-C19 is currently under investigation in a phase 1/2 trial (ZUMA-1) of patients with refractory, aggressive non-Hodgkin lymphomas. Kite Pharma, Inc., the company developing KTE-C19, plans to present top-line phase 1 data at the 2015 ASH Annual Meeting.
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has adopted positive opinions recommending orphan designation for KTE-C19 to treat acute lymphoblastic leukemia, chronic lymphocytic leukemia/small lymphocytic lymphoma, and follicular lymphoma.
KTE-C19 is an investigational chimeric antigen receptor (CAR) T-cell therapy designed to target CD19, a protein expressed on the surface of B cells.
The CAR T-cell therapy already has orphan designation for the treatment of diffuse large B-cell lymphoma in the US and the European Union (EU).
KTE-C19 also has COMP positive opinions for orphan designation in the EU for primary mediastinal B-cell lymphoma and mantle cell lymphoma.
About orphan designation
The COMP adopts an opinion on the granting of orphan designation, and that opinion is submitted to the European Commission for endorsement.
In the EU, orphan designation is granted to therapies intended to treat a life-threatening or chronically debilitating condition that affects no more than 5 in 10,000 persons and where no satisfactory treatment is available.
Companies that obtain orphan designation for a drug benefit from a number of incentives, including protocol assistance, a type of scientific advice specific for designated orphan medicines, and 10 years of market exclusivity once the medicine is approved. Fee reductions are also available, depending on the status of the sponsor and the type of service required.
KTE-C19 research
Last year, researchers reported results with KTE-C19 in the Journal of Clinical Oncology. The study included 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of the CAR T-cell therapy. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with chemotherapy-refractory diffuse large B-cell lymphoma, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Of the 4 patients with chronic lymphocytic leukemia, 3 had a CR, and 1 had a PR. Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR.
KTE-C19 was associated with fever, low blood pressure, focal neurological deficits, and delirium. Toxicities largely occurred in the first 2 weeks after infusion.
All but 2 patients experienced grade 3/4 adverse events. Four patients had grade 3/4 hypotension.
All patients had elevations in serum interferon gamma and/or interleukin 6 around the time of peak toxicity, but most did not develop elevations in serum tumor necrosis factor.
Neurologic toxicities included confusion and obtundation, which have been reported in previous studies. However, 3 patients developed unexpected neurologic abnormalities.
KTE-C19 is currently under investigation in a phase 1/2 trial (ZUMA-1) of patients with refractory, aggressive non-Hodgkin lymphomas. Kite Pharma, Inc., the company developing KTE-C19, plans to present top-line phase 1 data at the 2015 ASH Annual Meeting.
Watchdog condemns FDA oversight of dabigatran
The US Food and Drug Administration’s (FDA’s) oversight of the oral anticoagulant dabigatran (Pradaxa) raises questions about the agency’s reliability, according to a report by the Project On Government Oversight (POGO).
POGO’s report describes a series of “questionable” calls the FDA has made in its oversight of dabigatran.
The report calls attention to issues with the clinical trial used to support dabigatran’s approval.
It also details concerns regarding FDA advisory committee members, the distribution of potentially misleading information about dabigatran, the possibility that patients receiving dabigatran may actually need to be monitored, and other issues.
“The deeply disturbing part is that the public has to trust the FDA to keep it safe, and the way the agency handled Pradaxa doesn’t inspire confidence,” said Daniel Brian, POGO’s executive director.
This is not the first time the safety of dabigatran and results of dabigatran trials have been called into question.
Following post-marketing reports of adverse events and deaths related to dabigatran, the FDA conducted its own studies investigating the drug’s safety. The agency ultimately concluded that dabigatran’s benefits outweigh any detriments.
Still, a paper published in JAMA in 2012 suggested the FDA may have approved dabigatran too soon.
And several papers published in The BMJ last year indicated that Boehringer Ingelheim, the company developing dabigatran, underreported events associated with the drug and withheld data showing that monitoring and dose adjustment could improve the safety of dabigatran without compromising its efficacy.
The company has denied these allegations, but POGO’s report appears to support these claims, in addition to raising other concerns.
Trial issues
Dabigatran was initially approved by the FDA in 2010 on the basis of the RE-LY trial, in which researchers compared dabigatran and warfarin.
POGO’s report notes that this was an unblinded trial and suggests that investigators treated patients in the dabigatran arm differently than those in the warfarin arm. An FDA review showed that, when study subjects showed “signs of trouble,” those in the dabigatran arm were more likely to stop receiving treatment. This may have prevented adverse events such as hemorrhagic strokes.
FDA inspections also revealed that RE-LY investigators mismanaged parts of the trial. For example, they enrolled patients who were supposed to be excluded and failed to promptly inform Boehringer Ingelheim about possible adverse effects of dabigatran, among other infractions.
POGO’s report also cites FDA documents showing that the agency allowed Boehringer Ingelheim to finalize the scoring system for the RE-LY trial after it was over and the data had been gathered. A subsequent FDA review suggested that this allowance worked in dabigatran’s favor.
Advisory committee concerns
POGO’s report points out that members of the committee that advised the FDA on dabigatran’s approval have financial ties to the drug’s maker.
According to public databases, 2 of the advisory committee members who voted to approve dabigatran went on to receive tens of thousands of dollars from Boehringer Ingelheim.
POGO also found that when Boehringer Ingelheim held a practice session to prepare for questioning by the FDA advisory committee, a former member and a former chairman of that committee were paid to play roles in the rehearsal.
‘Misleading’ information
Another of POGO’s concerns is that the FDA redacted information from a public memo announcing and explaining its decision to approve dabigatran. The deleted section said that patients who were well-treated using warfarin had no reason to switch to dabigatran.
POGO’s report also points out that the FDA initially refused to allow Boehringer Ingelheim to claim that dabigatran was superior to warfarin but later changed its mind without providing much explanation.
In addition, the FDA allowed Boehringer Ingelheim to advertise that, in a clinical trial, dabigatran “reduced stroke risk 35% more than warfarin.” But after dabigatran had been on the market for more than 2.5 years, the FDA decided this claim was misleading.
The agency told Boehringer Ingelheim to add the following clarification: “That means that, in a large clinical study, 3.4% of patients taking warfarin had a stroke, compared to 2.2% of patients taking Pradaxa.” So, in absolute terms, the difference between the drugs was 1.2%.
Potential need for monitoring
According to POGO, Boehringer Ingelheim has been the target of thousands of lawsuits regarding adverse events and deaths thought to be related to dabigatran. In 2013, Boehringer Ingelheim was fined by a federal judge for withholding or failing to preserve records.
Some of these documents suggested that patients taking dabigatran may require monitoring to ensure they remain within a therapeutic range. A scientist at Boehringer Ingelheim, Thorsten Lehr, drafted a paper concluding that there is a therapeutic range for dabigatran.
But company emails indicated that other employees were against publishing this paper. An email from Boehringer Ingelheim’s Jutta Heinrich-Nols said publishing the paper would “make any defense of no monitoring . . . extremely difficult . . . and undermine our efforts to compete” with other new anticoagulants.
For more details, see POGO’s full report. POGO is a nonpartisan, independent watchdog that champions government reform.
The US Food and Drug Administration’s (FDA’s) oversight of the oral anticoagulant dabigatran (Pradaxa) raises questions about the agency’s reliability, according to a report by the Project On Government Oversight (POGO).
POGO’s report describes a series of “questionable” calls the FDA has made in its oversight of dabigatran.
The report calls attention to issues with the clinical trial used to support dabigatran’s approval.
It also details concerns regarding FDA advisory committee members, the distribution of potentially misleading information about dabigatran, the possibility that patients receiving dabigatran may actually need to be monitored, and other issues.
“The deeply disturbing part is that the public has to trust the FDA to keep it safe, and the way the agency handled Pradaxa doesn’t inspire confidence,” said Daniel Brian, POGO’s executive director.
This is not the first time the safety of dabigatran and results of dabigatran trials have been called into question.
Following post-marketing reports of adverse events and deaths related to dabigatran, the FDA conducted its own studies investigating the drug’s safety. The agency ultimately concluded that dabigatran’s benefits outweigh any detriments.
Still, a paper published in JAMA in 2012 suggested the FDA may have approved dabigatran too soon.
And several papers published in The BMJ last year indicated that Boehringer Ingelheim, the company developing dabigatran, underreported events associated with the drug and withheld data showing that monitoring and dose adjustment could improve the safety of dabigatran without compromising its efficacy.
The company has denied these allegations, but POGO’s report appears to support these claims, in addition to raising other concerns.
Trial issues
Dabigatran was initially approved by the FDA in 2010 on the basis of the RE-LY trial, in which researchers compared dabigatran and warfarin.
POGO’s report notes that this was an unblinded trial and suggests that investigators treated patients in the dabigatran arm differently than those in the warfarin arm. An FDA review showed that, when study subjects showed “signs of trouble,” those in the dabigatran arm were more likely to stop receiving treatment. This may have prevented adverse events such as hemorrhagic strokes.
FDA inspections also revealed that RE-LY investigators mismanaged parts of the trial. For example, they enrolled patients who were supposed to be excluded and failed to promptly inform Boehringer Ingelheim about possible adverse effects of dabigatran, among other infractions.
POGO’s report also cites FDA documents showing that the agency allowed Boehringer Ingelheim to finalize the scoring system for the RE-LY trial after it was over and the data had been gathered. A subsequent FDA review suggested that this allowance worked in dabigatran’s favor.
Advisory committee concerns
POGO’s report points out that members of the committee that advised the FDA on dabigatran’s approval have financial ties to the drug’s maker.
According to public databases, 2 of the advisory committee members who voted to approve dabigatran went on to receive tens of thousands of dollars from Boehringer Ingelheim.
POGO also found that when Boehringer Ingelheim held a practice session to prepare for questioning by the FDA advisory committee, a former member and a former chairman of that committee were paid to play roles in the rehearsal.
‘Misleading’ information
Another of POGO’s concerns is that the FDA redacted information from a public memo announcing and explaining its decision to approve dabigatran. The deleted section said that patients who were well-treated using warfarin had no reason to switch to dabigatran.
POGO’s report also points out that the FDA initially refused to allow Boehringer Ingelheim to claim that dabigatran was superior to warfarin but later changed its mind without providing much explanation.
In addition, the FDA allowed Boehringer Ingelheim to advertise that, in a clinical trial, dabigatran “reduced stroke risk 35% more than warfarin.” But after dabigatran had been on the market for more than 2.5 years, the FDA decided this claim was misleading.
The agency told Boehringer Ingelheim to add the following clarification: “That means that, in a large clinical study, 3.4% of patients taking warfarin had a stroke, compared to 2.2% of patients taking Pradaxa.” So, in absolute terms, the difference between the drugs was 1.2%.
Potential need for monitoring
According to POGO, Boehringer Ingelheim has been the target of thousands of lawsuits regarding adverse events and deaths thought to be related to dabigatran. In 2013, Boehringer Ingelheim was fined by a federal judge for withholding or failing to preserve records.
Some of these documents suggested that patients taking dabigatran may require monitoring to ensure they remain within a therapeutic range. A scientist at Boehringer Ingelheim, Thorsten Lehr, drafted a paper concluding that there is a therapeutic range for dabigatran.
But company emails indicated that other employees were against publishing this paper. An email from Boehringer Ingelheim’s Jutta Heinrich-Nols said publishing the paper would “make any defense of no monitoring . . . extremely difficult . . . and undermine our efforts to compete” with other new anticoagulants.
For more details, see POGO’s full report. POGO is a nonpartisan, independent watchdog that champions government reform.
The US Food and Drug Administration’s (FDA’s) oversight of the oral anticoagulant dabigatran (Pradaxa) raises questions about the agency’s reliability, according to a report by the Project On Government Oversight (POGO).
POGO’s report describes a series of “questionable” calls the FDA has made in its oversight of dabigatran.
The report calls attention to issues with the clinical trial used to support dabigatran’s approval.
It also details concerns regarding FDA advisory committee members, the distribution of potentially misleading information about dabigatran, the possibility that patients receiving dabigatran may actually need to be monitored, and other issues.
“The deeply disturbing part is that the public has to trust the FDA to keep it safe, and the way the agency handled Pradaxa doesn’t inspire confidence,” said Daniel Brian, POGO’s executive director.
This is not the first time the safety of dabigatran and results of dabigatran trials have been called into question.
Following post-marketing reports of adverse events and deaths related to dabigatran, the FDA conducted its own studies investigating the drug’s safety. The agency ultimately concluded that dabigatran’s benefits outweigh any detriments.
Still, a paper published in JAMA in 2012 suggested the FDA may have approved dabigatran too soon.
And several papers published in The BMJ last year indicated that Boehringer Ingelheim, the company developing dabigatran, underreported events associated with the drug and withheld data showing that monitoring and dose adjustment could improve the safety of dabigatran without compromising its efficacy.
The company has denied these allegations, but POGO’s report appears to support these claims, in addition to raising other concerns.
Trial issues
Dabigatran was initially approved by the FDA in 2010 on the basis of the RE-LY trial, in which researchers compared dabigatran and warfarin.
POGO’s report notes that this was an unblinded trial and suggests that investigators treated patients in the dabigatran arm differently than those in the warfarin arm. An FDA review showed that, when study subjects showed “signs of trouble,” those in the dabigatran arm were more likely to stop receiving treatment. This may have prevented adverse events such as hemorrhagic strokes.
FDA inspections also revealed that RE-LY investigators mismanaged parts of the trial. For example, they enrolled patients who were supposed to be excluded and failed to promptly inform Boehringer Ingelheim about possible adverse effects of dabigatran, among other infractions.
POGO’s report also cites FDA documents showing that the agency allowed Boehringer Ingelheim to finalize the scoring system for the RE-LY trial after it was over and the data had been gathered. A subsequent FDA review suggested that this allowance worked in dabigatran’s favor.
Advisory committee concerns
POGO’s report points out that members of the committee that advised the FDA on dabigatran’s approval have financial ties to the drug’s maker.
According to public databases, 2 of the advisory committee members who voted to approve dabigatran went on to receive tens of thousands of dollars from Boehringer Ingelheim.
POGO also found that when Boehringer Ingelheim held a practice session to prepare for questioning by the FDA advisory committee, a former member and a former chairman of that committee were paid to play roles in the rehearsal.
‘Misleading’ information
Another of POGO’s concerns is that the FDA redacted information from a public memo announcing and explaining its decision to approve dabigatran. The deleted section said that patients who were well-treated using warfarin had no reason to switch to dabigatran.
POGO’s report also points out that the FDA initially refused to allow Boehringer Ingelheim to claim that dabigatran was superior to warfarin but later changed its mind without providing much explanation.
In addition, the FDA allowed Boehringer Ingelheim to advertise that, in a clinical trial, dabigatran “reduced stroke risk 35% more than warfarin.” But after dabigatran had been on the market for more than 2.5 years, the FDA decided this claim was misleading.
The agency told Boehringer Ingelheim to add the following clarification: “That means that, in a large clinical study, 3.4% of patients taking warfarin had a stroke, compared to 2.2% of patients taking Pradaxa.” So, in absolute terms, the difference between the drugs was 1.2%.
Potential need for monitoring
According to POGO, Boehringer Ingelheim has been the target of thousands of lawsuits regarding adverse events and deaths thought to be related to dabigatran. In 2013, Boehringer Ingelheim was fined by a federal judge for withholding or failing to preserve records.
Some of these documents suggested that patients taking dabigatran may require monitoring to ensure they remain within a therapeutic range. A scientist at Boehringer Ingelheim, Thorsten Lehr, drafted a paper concluding that there is a therapeutic range for dabigatran.
But company emails indicated that other employees were against publishing this paper. An email from Boehringer Ingelheim’s Jutta Heinrich-Nols said publishing the paper would “make any defense of no monitoring . . . extremely difficult . . . and undermine our efforts to compete” with other new anticoagulants.
For more details, see POGO’s full report. POGO is a nonpartisan, independent watchdog that champions government reform.