User login
Vascular signaling abnormalities in Alzheimer disease
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
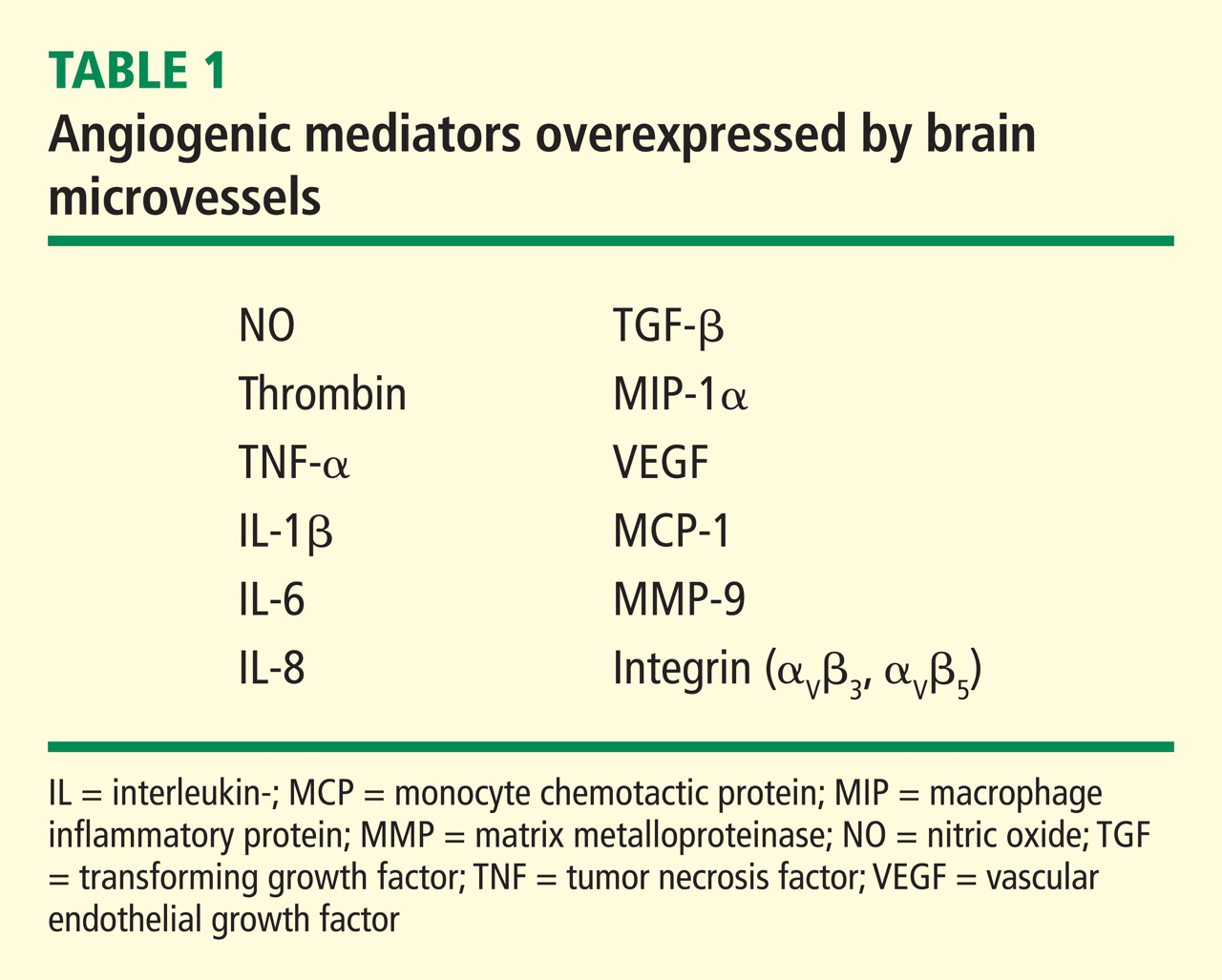
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
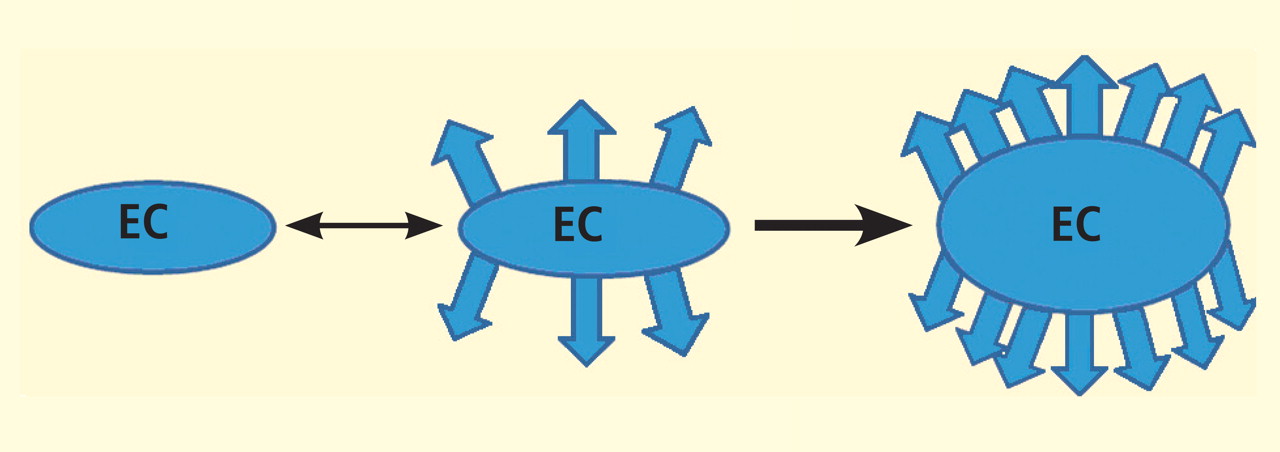
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
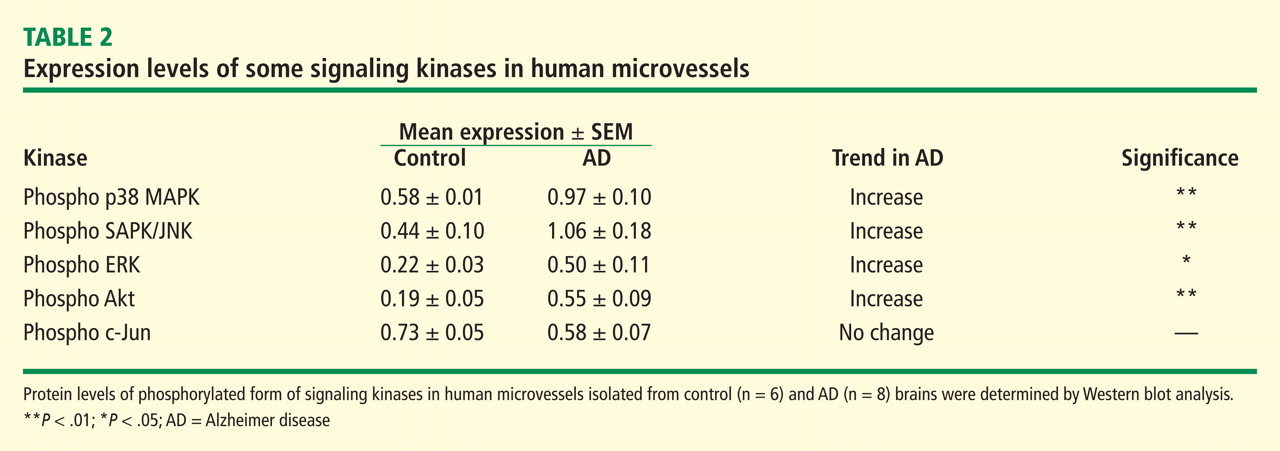
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
- 2010 Alzheimer’s facts and figures. Alzheimer’s Association Web site. http://www.alz.org/alzheimers_disease_facts_and_figures.asp. Updated January 5, 2011. Accessed February 10, 2011.
- Stewart R, Prince M, Mann A. Vascular risk factors and Alzheimer’s disease. Aust N Z J Psychiatry 1999; 33:809–813.
- Schmidt R, Schmidt H, Fasekas F. Vascular risk factors in dementia. J Neurol 2000; 247:81–87.
- Shi J, Perry G, Smith MA, Friedland RP. Vascular abnormalities: the insidious pathogenesis of Alzheimer’s disease. Neurobiol Aging 2000; 21:357–361.
- Pansari K, Gupta A, Thomas P. Alzheimer’s disease and vascular factors: facts and theories. Int J Clin Pract 2002; 56:197–203.
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33:1152–1162.
- Sadowski M, Pankiewicz J, Scholtzova H, et al Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004; 29:1257–1266.
- Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol Aging 2000; 21:199–205.
- de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119–136.
- Roher AE, Esh C, Kokjohn TA, et al Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 2003; 23:2055–2062.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677–684.
- Yamakawa M, Liu LX, Date T, et al Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 2003; 93:664–673.
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental ischemia. Brain Res Mol Brain Res 1998; 62:101–105.
- Tarkowski E, Issa R, Sjogren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 2002; 23:237–243.
- Vagnucci AH, Li W. Alzheimer’s disease and angiogenesis. Lancet 2003; 361:605–608.
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport 2004; 15:1507–1510.
- Dorheim NA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun 1994; 30:659–665.
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 2001; 22:837–842.
- Grammas P, Ovase R. Cerebrovascular TGF-β contributes to inflammation in the Alzheimer’s brain. Am J Pathol 2002; 160:1583–1587.
- Grammas P, Ghatreh-Samany P, Thirmangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alz Dis 2006; 9:51–58.
- Thirumangakudi L, Ghatreh-Samany P, Owoso A, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alz Dis 2006; 10:111–118.
- Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 2010; 176:1600–1606.
- Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 2006; 38:333–357.
- Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J 2003; 24:586–603.
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000; 902:230–240.
- Magrane J, Christensen RA, Rosen KM, Veereshwarayya V, Querfurth HW. Dissociation of ERK and Akt signaling in endothelial cell angiogenic responses to beta-amyloid. Exp Cell Res 2006; 312:996–1010.
- Wu Z, Guo H, Chow N, et al Role of the MEOX2 gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005; 11:959–965.
- Gorski DH, Leal AJ. Inhibition of endothelial cell activation by the homeobox gene Gax. J Surg Res 2003; 111:91–99.
- Patel S, Leal AD, Gorski DH. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res 2005; 65:1414–1424.
- Edelber JM, Reed MJ. Aging and angiogenesis. Front Biosci 2003; 8:s1199–s1209.
- Buee L, Hof PR, Bouras C, et al Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480.
- Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann NY Acad Sci 1997; 826:7–24.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109:813–836.
- Paris D, Townsend K, Quadros A, et al Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004; 7:75–85.
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
Alzheimer disease (AD) is a progressive, irreversible, neurodegenerative disease that affects more than 5.3 million people in the United States.1 This number is significantly higher than the previous estimate of 4.5 million and is projected to increase sharply to nearly 8 million by 2030.1 At present, the few agents that are approved by the US Food and Drug Administration for treatment of AD have demonstrated only modest effects in modifying clinical symptoms for relatively short periods; none has shown a clear effect on disease progression. New therapeutic approaches are desperately needed.
VASCULAR DISEASE AND ALZHEIMER DISEASE
Although AD is classified as a neurodegenerative dementia, there is epidemiologic and pathologic evidence of an association with vascular risk factors and vascular disease.2–6 Vascular disease appears to lower the threshold for the clinical presentation of dementia at a given level of AD-related pathology.7 The possible association of AD with vascular disease suggests that there are important pathogenic mechanisms common to both AD and vascular disease. For example, there is increasing evidence that perturbations in cerebral vascular structure and function occur in AD.8
It has been suggested that cerebral hypoperfusion/hypoxia triggers hypometabolic, cognitive, and degenerative changes in the brain and contributes to the pathologic processes of AD.9 A study by Roher and colleagues reveals an association between severe circle of Willis atherosclerosis and sporadic AD.10 These observations suggest that atherosclerosis-induced brain hypoperfusion contributes to the clinical and pathologic manifestations of AD.
Hypoxia is also known to stimulate angiogenesis, especially via upregulation of hypoxia-inducible genes such as vascular endothelial growth factor (VEGF).11,12 VEGF, a critical mediator of angiogenesis, is present in the AD brain in the walls of intra-parenchymal vessels, in diffuse perivascular deposits, and in clusters of reactive astrocytes.13 In addition, intrathecal levels of VEGF in AD are related to clinical severity and intrathecal levels of amyloid-beta (Aβ).14 Emerging data support the idea that factors and processes characteristic of angiogenesis are found in the AD brain.15,16
ENDOTHELIAL ACTIVATION AND ANGIOGENESIS
The angiogenic process is complex and involves several discrete steps, such as endothelial activation, extracellular matrix degradation, proliferation and migration of endothelial cells, and morphologic differentiation of endothelial cells to form tubes. Stimuli known to initiate angiogenesis, including hypoxia, inflammation, and mechanical factors such as shear stress and stretch,23 either directly or indirectly activate endothelial cells. Activated endothelial cells elaborate adhesion molecules, cytokines and chemokines, growth factors, vasoactive molecules, major histocompatibility complex molecules, procoagulant and anticoagulant moieties, and a variety of other gene products with biologic activity.24 The activated endothelium exerts direct local effects by producing at least 20 paracrine factors that act on adjacent cells.25
ANGIOGENIC SIGNALING MECHANISMS IN BRAIN MICROVESSELS
Signaling mechanisms that have been identified as important to endothelial cell viability and angiogenesis include PI3K/Akt, p38 kinase, ERK, and JNK. In this regard, intracellular Aβ accumulation is toxic to endothelial cells and decreases PI3K/Akt.26 Extracellular Aβ peptides decrease phosphorylation and thus activation of ERK and p38 kinase.26 VEGF promotes endothelial survival, proliferation, and migration through numerous pathways, including activation of ERK, p38 kinase, JNK, and Rho GTPase family members.23
VASCULAR ACTIVATION IN ALZHEIMER DISEASE
Despite increases in several proangiogenic factors in the AD brain, evidence for increased vascularity in AD is lacking. On the contrary, it has been suggested that the angiogenic process is delayed or impaired in aged tissues, with several studies showing decreased microvascular density in the AD brain.30–33 Paris et al showed that wild-type Aβ peptides have antiangiogenic effects in vitro and in vivo.34
How can the data showing antiangiogenic effects of Aβ be reconciled with the presence or expression of a large number of proangiogenic proteins by brain microvessels in AD? These conflicting observations suggest an imbalance between proangiogenic and antiangiogeneic processes in the AD brain.
Preliminary experiments in our laboratory show that pharmacologic blockade of vascular activation improves cognitive function in an animal model of AD. Thus, “vascular activation” could be a novel, unexplored therapeutic target in AD.
Acknowledgment
The authors gratefully acknowledge the secretarial assistance of Terri Stahl.
- 2010 Alzheimer’s facts and figures. Alzheimer’s Association Web site. http://www.alz.org/alzheimers_disease_facts_and_figures.asp. Updated January 5, 2011. Accessed February 10, 2011.
- Stewart R, Prince M, Mann A. Vascular risk factors and Alzheimer’s disease. Aust N Z J Psychiatry 1999; 33:809–813.
- Schmidt R, Schmidt H, Fasekas F. Vascular risk factors in dementia. J Neurol 2000; 247:81–87.
- Shi J, Perry G, Smith MA, Friedland RP. Vascular abnormalities: the insidious pathogenesis of Alzheimer’s disease. Neurobiol Aging 2000; 21:357–361.
- Pansari K, Gupta A, Thomas P. Alzheimer’s disease and vascular factors: facts and theories. Int J Clin Pract 2002; 56:197–203.
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33:1152–1162.
- Sadowski M, Pankiewicz J, Scholtzova H, et al Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004; 29:1257–1266.
- Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol Aging 2000; 21:199–205.
- de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119–136.
- Roher AE, Esh C, Kokjohn TA, et al Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 2003; 23:2055–2062.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677–684.
- Yamakawa M, Liu LX, Date T, et al Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 2003; 93:664–673.
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental ischemia. Brain Res Mol Brain Res 1998; 62:101–105.
- Tarkowski E, Issa R, Sjogren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 2002; 23:237–243.
- Vagnucci AH, Li W. Alzheimer’s disease and angiogenesis. Lancet 2003; 361:605–608.
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport 2004; 15:1507–1510.
- Dorheim NA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun 1994; 30:659–665.
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 2001; 22:837–842.
- Grammas P, Ovase R. Cerebrovascular TGF-β contributes to inflammation in the Alzheimer’s brain. Am J Pathol 2002; 160:1583–1587.
- Grammas P, Ghatreh-Samany P, Thirmangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alz Dis 2006; 9:51–58.
- Thirumangakudi L, Ghatreh-Samany P, Owoso A, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alz Dis 2006; 10:111–118.
- Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 2010; 176:1600–1606.
- Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 2006; 38:333–357.
- Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J 2003; 24:586–603.
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000; 902:230–240.
- Magrane J, Christensen RA, Rosen KM, Veereshwarayya V, Querfurth HW. Dissociation of ERK and Akt signaling in endothelial cell angiogenic responses to beta-amyloid. Exp Cell Res 2006; 312:996–1010.
- Wu Z, Guo H, Chow N, et al Role of the MEOX2 gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005; 11:959–965.
- Gorski DH, Leal AJ. Inhibition of endothelial cell activation by the homeobox gene Gax. J Surg Res 2003; 111:91–99.
- Patel S, Leal AD, Gorski DH. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res 2005; 65:1414–1424.
- Edelber JM, Reed MJ. Aging and angiogenesis. Front Biosci 2003; 8:s1199–s1209.
- Buee L, Hof PR, Bouras C, et al Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480.
- Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann NY Acad Sci 1997; 826:7–24.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109:813–836.
- Paris D, Townsend K, Quadros A, et al Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004; 7:75–85.
- 2010 Alzheimer’s facts and figures. Alzheimer’s Association Web site. http://www.alz.org/alzheimers_disease_facts_and_figures.asp. Updated January 5, 2011. Accessed February 10, 2011.
- Stewart R, Prince M, Mann A. Vascular risk factors and Alzheimer’s disease. Aust N Z J Psychiatry 1999; 33:809–813.
- Schmidt R, Schmidt H, Fasekas F. Vascular risk factors in dementia. J Neurol 2000; 247:81–87.
- Shi J, Perry G, Smith MA, Friedland RP. Vascular abnormalities: the insidious pathogenesis of Alzheimer’s disease. Neurobiol Aging 2000; 21:357–361.
- Pansari K, Gupta A, Thomas P. Alzheimer’s disease and vascular factors: facts and theories. Int J Clin Pract 2002; 56:197–203.
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002; 33:1152–1162.
- Sadowski M, Pankiewicz J, Scholtzova H, et al Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 2004; 29:1257–1266.
- Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer’s disease. Neurobiol Aging 2000; 21:199–205.
- de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Rev 2000; 34:119–136.
- Roher AE, Esh C, Kokjohn TA, et al Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol 2003; 23:2055–2062.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9:677–684.
- Yamakawa M, Liu LX, Date T, et al Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 2003; 93:664–673.
- Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental ischemia. Brain Res Mol Brain Res 1998; 62:101–105.
- Tarkowski E, Issa R, Sjogren M, et al Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 2002; 23:237–243.
- Vagnucci AH, Li W. Alzheimer’s disease and angiogenesis. Lancet 2003; 361:605–608.
- Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport 2004; 15:1507–1510.
- Dorheim NA, Tracey WR, Pollock JS, Grammas P. Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer’s disease. Biochem Biophys Res Commun 1994; 30:659–665.
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 2001; 22:837–842.
- Grammas P, Ovase R. Cerebrovascular TGF-β contributes to inflammation in the Alzheimer’s brain. Am J Pathol 2002; 160:1583–1587.
- Grammas P, Ghatreh-Samany P, Thirmangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alz Dis 2006; 9:51–58.
- Thirumangakudi L, Ghatreh-Samany P, Owoso A, Grammas P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alz Dis 2006; 10:111–118.
- Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol 2010; 176:1600–1606.
- Milkiewicz M, Ispanovic E, Doyle JL, Haas TL. Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 2006; 38:333–357.
- Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J 2003; 24:586–603.
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000; 902:230–240.
- Magrane J, Christensen RA, Rosen KM, Veereshwarayya V, Querfurth HW. Dissociation of ERK and Akt signaling in endothelial cell angiogenic responses to beta-amyloid. Exp Cell Res 2006; 312:996–1010.
- Wu Z, Guo H, Chow N, et al Role of the MEOX2 gene in neurovascular dysfunction in Alzheimer disease. Nat Med 2005; 11:959–965.
- Gorski DH, Leal AJ. Inhibition of endothelial cell activation by the homeobox gene Gax. J Surg Res 2003; 111:91–99.
- Patel S, Leal AD, Gorski DH. The homeobox gene Gax inhibits angiogenesis through inhibition of nuclear factor-kappaB-dependent endothelial cell gene expression. Cancer Res 2005; 65:1414–1424.
- Edelber JM, Reed MJ. Aging and angiogenesis. Front Biosci 2003; 8:s1199–s1209.
- Buee L, Hof PR, Bouras C, et al Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480.
- Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann NY Acad Sci 1997; 826:7–24.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109:813–836.
- Paris D, Townsend K, Quadros A, et al Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004; 7:75–85.