User login
A 72-year-old man with a purpuric rash
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
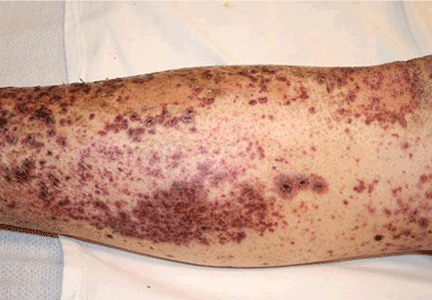
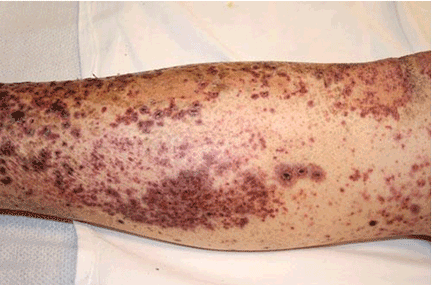
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
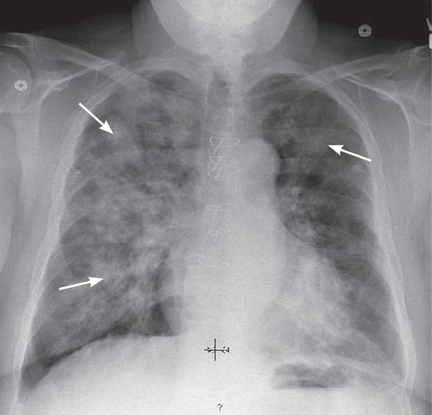
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
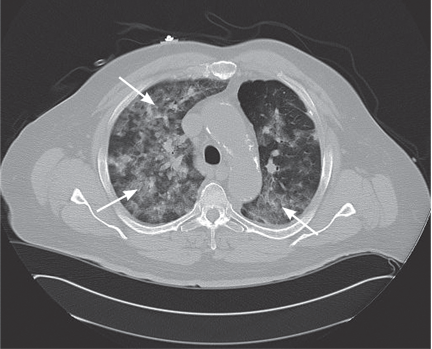
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.