User login
Ibrutinib sustains efficacy in CLL at 4-year follow-up

NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
BCMA emerging as a promising target in MM
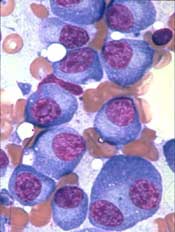
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
NEW YORK, NY—The B-cell maturation antigen (BCMA) is emerging as a promising target in multiple myeloma (MM), according to Adam D. Cohen, MD, of the University of Pennsylvania in Philadelphia.
BCMA is highly expressed on MM cells and, with its 2 ligands, is responsible for maintaining normal plasma cell homeostasis.
“And it’s really not expressed on any other normal tissues of the body,” Dr Cohen said.
“Importantly, BCMA is not just sitting on the cell surface as a target but actually promotes myeloma pathogenesis.”
Dr Cohen reviewed the progress being made using BCMA as a target in MM, paying particular attention to chimeric antigen receptor (CAR) T cells. He presented the update at Lymphoma & Myeloma 2017.
NCI BCMA-specific CARs
The first CAR to specifically target BCMA in MM was developed at the National Cancer Institute (NCI). It consisted of a murine single-chain variable fragment (scFv), CD3/CD28 signaling domains, and a gamma-retroviral vector.
Investigators conducted the first-in-human trial of this CAR T-cell therapy in 12 relapsed/refractory MM patients.
All patients received a lymphodepleting conditioning regimen of cyclophosphamide and fludarabine and a single infusion of 1 of 4 doses of the CAR T-cell therapy.
At higher dose levels, the BCMA-CAR produced objective responses “even in these highly refractory patients,” Dr Cohen said. “Some responses lasted 4 to 6 months.”
Patients who had the greatest degree of expansion of CAR T cells were the ones who had the best responses.
The BCMA-CAR is associated with the same toxicities as the CD19-directed CAR T-cell therapies now approved in acute lymphoblastic leukemia and non-Hodgkin lymphoma—cytokine release syndrome (CRS) and neurotoxicity.
The NCI study (NCT02215967) is ongoing.
Penn BCMA-specific CAR
A different BCMA CAR is being investigated at the University of Pennsylvania. It is a fully human CAR that consists of a human scFv, CD3/4-1BB costimulatory domains, and a lentiviral vector.
Investigators designed the first-in-human trial* (NCT02546167) with 3 different cohorts.
Patients in cohort 1 received 5 x 108 CAR T cells without any lymphodepleting chemotherapy.
The remaining patients received cyclophosphamide, followed by 5 x 107 CAR T cells in cohort 2 and 5 x 108 CAR T cells in cohort 3.
Dr Cohen reviewed current data from cohort 1, which included 9 patients. They were a median age of 57 (range, 44-70), and 67% were male. They were heavily pretreated with a median of 9 prior lines of therapy (range, 4-11).
All had high-risk cytogenetics, 67% had deletion 17p or TP53 mutation, and they had a median of 80% bone marrow plasma cells (range, 15%-95%).
“Despite this,” Dr Cohen said, “we were able to generate, successfully, CAR T cells from all patients, although 1 patient did require a second apheresis and manufacturing attempt.”
Four of the 9 patients achieved very good partial responses, and an additional 2 patients had minimal responses.
One patient had a stringent CR (sCR) for close to 2 years without having any intervening therapy.
“[The sCR] shows the potential for this [therapy] to create a durable remission in a patient without any other therapy,” Dr Cohen said. “And this patient still has circulating CAR cells detectable.”
Most of the other patients did not have as durable a response. Responses lasted a median of 3 to 5 months before the patients relapsed.
Dr Cohen noted that the Penn data confirm the NCI experience showing proof of principle.
“You can target BCMA with these cells and get objective responses that can lead to a durable one in a subset of patients,” he added.
Dr Cohen is scheduled to report the preliminary data on the cyclophosphamide cohorts (cohorts 2 and 3) at ASH 2017. Those cohorts, he said, are showing a bit more persistence and increased frequency of responses.
Other BCMA-specific CAR trials
Another BCMA CAR, bb2121, consists of a human anti-BCMA scFv, a lentiviral vector, and a CD3/4-1BB costimulatory domain.
In the first-in-human trial of bb2121* (NCT02658929), all 21 patients received cyclophosphamide and fludarabine conditioning first.
There were “very impressive response rates in this study,” Dr Cohen said.
Once patients were dosed at the higher levels of 150 million cells or greater, every patient responded.
“Many responses are still durable,” he pointed out, “some approaching a year.”
LCAR-B38M is structurally different from the other BCMA-specific CARs. It has 2 binding sites for BCMA.
Thirty-five patients enrolled on the trial of LCAR-B38M (NCT03090659), and 19 were evaluable for response.
Patients had a median of 3 or 4 lines of prior therapy. All received cyclophosphamide alone as conditioning.
All 19 evaluable patients responded, and 14 (74%) achieved an sCR.
Dr Cohen noted the differences among the 4 trials: costimulatory domains varied, the Penn study did not require a pre-existing level of BCMA on the MM cells, other studies excluded patients who didn’t meet a certain threshold level of BCMA expression, median lines of prior therapy differed, and conditioning regimens differed as well.
All trials, however, presented data on fewer than 20 patients.
“And so I think it’s a little too early to compare head-to-head between all of these, but they all are really demonstrating promising results so far,” he observed.
Toxicities
“Unfortunately, there’s no free lunch with CAR T cells,” Dr Cohen stated, “and these extraordinary responses do come at the cost of several somewhat unique toxicities that can be serious.”
Toxicities have included:
- Tumor lysis syndrome, which is expected and manageable
- B-cell aplasia, which is not as much of an issue with BCMA as with CD19-directed CAR Ts, since most B cells don’t express BCMA
- Hypogammaglobulinemia, which can be mitigated with IVIG
- CRS, which can be alleviated with tocilizumab
- Neurotoxicity and encephalopathy, which do not occur as frequently as CRS.
“These, I think, are things we still obviously need to learn more about and try to mitigate before this [BCMA-directed CAR therapy] is expanded and brought earlier to patients,” Dr Cohen said.
“The other issues with CAR T cells are logistical. Limited access (the procedure is available at only a few centers); manufacturing can take 2 to 4 weeks, during which time it is difficult to maintain disease control; and the cost is significant.”
“We are really in the early days of CAR cells for myeloma, but, certainly, this appears very bright in terms of its future.” ![]()
* Data from the presentation differ from the abstract.
Speaker advises caution in adding mAbs upfront in MM

NEW YORK, NY—Despite the attraction of incorporating monoclonal antibodies (mAbs) into upfront therapy for multiple myeloma (MM), a speaker at Lymphoma & Myeloma 2017 suggested mAbs are “not quite ready” for this use.
MAbs, particularly daratumumab, have shown single-agent activity in refractory MM and have been feasibly added to proteasome inhibitors and immunomodulatory drugs.
MAbs may even have the potential to enhance induction and shorten the time to minimal residual disease negativity.
“So the tendency is to simply add them to frontline therapy,” said the speaker, Joseph Mikhael, MD, from Mayo Clinic Arizona in Scottsdale.
However, he noted that there is little long-term experience with these agents.
“I’m going to suggest to you that they’re not quite ready [for upfront use] but will likely be ready in the future,” Dr Mikhael said. “We’ve had such a revolution in myeloma the last decade that it’s just easy for us to say, ‘Oh, throw it in there, just like we did, frankly, with rituximab in the lymphoma days. We added it to CVP, we added it to CHOP, we added it to bendamustine. It didn’t matter what we added it to, it just upgraded the response.”
“And, sometimes, I think we have the same approach with daratumumab or elotuzumab or some of the other mAbs that we have. I think we just have to do so cautiously.”
At present, the combination of a proteasome inhibitor and an immunomodulatory drug are the standard of care upfront in transplant-eligible and -ineligible MM patients.
Daratumumab plus KRd
Dr Mikhael described the experience of daratumumab added to carfilzomib, lenalidomide, and dexamethasone (KRd) in patients with newly diagnosed MM in the MMY1001 study.
Twenty-two patients were enrolled on the study, 91% achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR). The depth of response improved with the duration of treatment.
Eight patients (36%) discontinued treatment.
Dr Mikhael emphasized that the preliminary data included very small numbers.
“There is a little bit of a yellow flag that pops up here,” he added, “when I see that 36% discontinued treatment, even in small numbers.”
The safety profile was consistent with previous reports for daratumumab or KRd.
The most common hematologic grade 3-4 treatment-emergent adverse events (AEs) occurring in 30% or more of patients were lymphopenia (64%), thrombocytopenia (9%), anemia (9%), leukopenia (9%), and neutropenia (14%).
Diarrhea (14%), cough (5%), fatigue (5%), insomnia (5%), and increased ALT (9%) were the most common grade 3-4 nonhematologic treatment-emergent AEs occurring in 30% or more of patients.
The treatment had no adverse impact on stem cell collection.
Elotuzumab plus VRd
Turning to elotuzumab in combination with bortezomib, lenalidomide, and dexamethasone (VRd), Dr Mikhael reviewed the phase 2a study (NCT02375555) presented at ASCO 2017 (abstract 8002).
Forty-one patients were enrolled on the study.
The overall response rate after 4 cycles was 100%, with 24% achieving a CR, 47% achieving a VGPR, and 29% a partial response.
Fatigue (60%), neuropathy (55%), musculoskeletal/joint pain (55%), infection (50%), back/neck pain (48%), diarrhea (45%), edema (38%), constipation (38%), cough (35%), mood alteration (35%), rash (35%), and insomnia (30%) occurred in 30% or more of patients.
“So again, not shocking,” Dr Mikhael said, “there was fatigue, there was neuropathy, and there were infections in 50% of patients.”
Grade 4 or greater AEs included thrombocytopenia, hyperglycemia, sepsis, cardiac arrest, and respiratory failure.
“However, here, [we have] maybe not even a yellow flag but a red flag of caution that there were 2 patients who died,” Dr Mikhael noted.
One patient died on study due to respiratory failure and sepsis that arose during cycle 2.
The other patient died more than 30 days after discontinuing study therapy due to febrile neutropenia and hypotension related to sepsis, followed by renal failure.
“Again, in a study that has such small numbers, I don’t want to overstate the case . . ., we don’t want to overreact, but whenever there is death involved, obviously, we have to be particularly cautious,” Dr Mikhael said.
Put into the context of 3 other VRd studies, he noted, the response rate with elotuzumab and VRd is relatively similar but not as good as the phase 3 study of VRd, which was a much larger study of 350 patients.
The situation with daratumumab and KRd is similar to elotuzumab, Dr Mikhael pointed out.
The initial response rates are impressive, but, when compared to other studies, “71% VGPR is good, only after 4 cycles, but we know that, in other studies, after a few more cycles, it was significantly higher.”
Cost
Dr Mikhael also considered cost in his assessment of daratumumab and elotuzumab integrated into frontline regimens.
Adding elotuzumab to VRd would almost double the cost of 12 weeks of therapy. And adding daratumumab to KRd would increase the cost even more.
“These costs are real,” Dr Mikhael said, “and, ultimately, if it’s the best thing for our patients, that’s what we are going to do. But until we have that convincing evidence, I think it’s critical to keep that in perspective. I would suggest that VRd, in many respects, is the standard of care for most patients.”
In terms of adding a mAb upfront, he said, “I don’t think we’re there yet. Do I think, in time, we will be? Quite likely, but I don’t think we are there yet.” ![]()
NEW YORK, NY—Despite the attraction of incorporating monoclonal antibodies (mAbs) into upfront therapy for multiple myeloma (MM), a speaker at Lymphoma & Myeloma 2017 suggested mAbs are “not quite ready” for this use.
MAbs, particularly daratumumab, have shown single-agent activity in refractory MM and have been feasibly added to proteasome inhibitors and immunomodulatory drugs.
MAbs may even have the potential to enhance induction and shorten the time to minimal residual disease negativity.
“So the tendency is to simply add them to frontline therapy,” said the speaker, Joseph Mikhael, MD, from Mayo Clinic Arizona in Scottsdale.
However, he noted that there is little long-term experience with these agents.
“I’m going to suggest to you that they’re not quite ready [for upfront use] but will likely be ready in the future,” Dr Mikhael said. “We’ve had such a revolution in myeloma the last decade that it’s just easy for us to say, ‘Oh, throw it in there, just like we did, frankly, with rituximab in the lymphoma days. We added it to CVP, we added it to CHOP, we added it to bendamustine. It didn’t matter what we added it to, it just upgraded the response.”
“And, sometimes, I think we have the same approach with daratumumab or elotuzumab or some of the other mAbs that we have. I think we just have to do so cautiously.”
At present, the combination of a proteasome inhibitor and an immunomodulatory drug are the standard of care upfront in transplant-eligible and -ineligible MM patients.
Daratumumab plus KRd
Dr Mikhael described the experience of daratumumab added to carfilzomib, lenalidomide, and dexamethasone (KRd) in patients with newly diagnosed MM in the MMY1001 study.
Twenty-two patients were enrolled on the study, 91% achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR). The depth of response improved with the duration of treatment.
Eight patients (36%) discontinued treatment.
Dr Mikhael emphasized that the preliminary data included very small numbers.
“There is a little bit of a yellow flag that pops up here,” he added, “when I see that 36% discontinued treatment, even in small numbers.”
The safety profile was consistent with previous reports for daratumumab or KRd.
The most common hematologic grade 3-4 treatment-emergent adverse events (AEs) occurring in 30% or more of patients were lymphopenia (64%), thrombocytopenia (9%), anemia (9%), leukopenia (9%), and neutropenia (14%).
Diarrhea (14%), cough (5%), fatigue (5%), insomnia (5%), and increased ALT (9%) were the most common grade 3-4 nonhematologic treatment-emergent AEs occurring in 30% or more of patients.
The treatment had no adverse impact on stem cell collection.
Elotuzumab plus VRd
Turning to elotuzumab in combination with bortezomib, lenalidomide, and dexamethasone (VRd), Dr Mikhael reviewed the phase 2a study (NCT02375555) presented at ASCO 2017 (abstract 8002).
Forty-one patients were enrolled on the study.
The overall response rate after 4 cycles was 100%, with 24% achieving a CR, 47% achieving a VGPR, and 29% a partial response.
Fatigue (60%), neuropathy (55%), musculoskeletal/joint pain (55%), infection (50%), back/neck pain (48%), diarrhea (45%), edema (38%), constipation (38%), cough (35%), mood alteration (35%), rash (35%), and insomnia (30%) occurred in 30% or more of patients.
“So again, not shocking,” Dr Mikhael said, “there was fatigue, there was neuropathy, and there were infections in 50% of patients.”
Grade 4 or greater AEs included thrombocytopenia, hyperglycemia, sepsis, cardiac arrest, and respiratory failure.
“However, here, [we have] maybe not even a yellow flag but a red flag of caution that there were 2 patients who died,” Dr Mikhael noted.
One patient died on study due to respiratory failure and sepsis that arose during cycle 2.
The other patient died more than 30 days after discontinuing study therapy due to febrile neutropenia and hypotension related to sepsis, followed by renal failure.
“Again, in a study that has such small numbers, I don’t want to overstate the case . . ., we don’t want to overreact, but whenever there is death involved, obviously, we have to be particularly cautious,” Dr Mikhael said.
Put into the context of 3 other VRd studies, he noted, the response rate with elotuzumab and VRd is relatively similar but not as good as the phase 3 study of VRd, which was a much larger study of 350 patients.
The situation with daratumumab and KRd is similar to elotuzumab, Dr Mikhael pointed out.
The initial response rates are impressive, but, when compared to other studies, “71% VGPR is good, only after 4 cycles, but we know that, in other studies, after a few more cycles, it was significantly higher.”
Cost
Dr Mikhael also considered cost in his assessment of daratumumab and elotuzumab integrated into frontline regimens.
Adding elotuzumab to VRd would almost double the cost of 12 weeks of therapy. And adding daratumumab to KRd would increase the cost even more.
“These costs are real,” Dr Mikhael said, “and, ultimately, if it’s the best thing for our patients, that’s what we are going to do. But until we have that convincing evidence, I think it’s critical to keep that in perspective. I would suggest that VRd, in many respects, is the standard of care for most patients.”
In terms of adding a mAb upfront, he said, “I don’t think we’re there yet. Do I think, in time, we will be? Quite likely, but I don’t think we are there yet.” ![]()
NEW YORK, NY—Despite the attraction of incorporating monoclonal antibodies (mAbs) into upfront therapy for multiple myeloma (MM), a speaker at Lymphoma & Myeloma 2017 suggested mAbs are “not quite ready” for this use.
MAbs, particularly daratumumab, have shown single-agent activity in refractory MM and have been feasibly added to proteasome inhibitors and immunomodulatory drugs.
MAbs may even have the potential to enhance induction and shorten the time to minimal residual disease negativity.
“So the tendency is to simply add them to frontline therapy,” said the speaker, Joseph Mikhael, MD, from Mayo Clinic Arizona in Scottsdale.
However, he noted that there is little long-term experience with these agents.
“I’m going to suggest to you that they’re not quite ready [for upfront use] but will likely be ready in the future,” Dr Mikhael said. “We’ve had such a revolution in myeloma the last decade that it’s just easy for us to say, ‘Oh, throw it in there, just like we did, frankly, with rituximab in the lymphoma days. We added it to CVP, we added it to CHOP, we added it to bendamustine. It didn’t matter what we added it to, it just upgraded the response.”
“And, sometimes, I think we have the same approach with daratumumab or elotuzumab or some of the other mAbs that we have. I think we just have to do so cautiously.”
At present, the combination of a proteasome inhibitor and an immunomodulatory drug are the standard of care upfront in transplant-eligible and -ineligible MM patients.
Daratumumab plus KRd
Dr Mikhael described the experience of daratumumab added to carfilzomib, lenalidomide, and dexamethasone (KRd) in patients with newly diagnosed MM in the MMY1001 study.
Twenty-two patients were enrolled on the study, 91% achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR). The depth of response improved with the duration of treatment.
Eight patients (36%) discontinued treatment.
Dr Mikhael emphasized that the preliminary data included very small numbers.
“There is a little bit of a yellow flag that pops up here,” he added, “when I see that 36% discontinued treatment, even in small numbers.”
The safety profile was consistent with previous reports for daratumumab or KRd.
The most common hematologic grade 3-4 treatment-emergent adverse events (AEs) occurring in 30% or more of patients were lymphopenia (64%), thrombocytopenia (9%), anemia (9%), leukopenia (9%), and neutropenia (14%).
Diarrhea (14%), cough (5%), fatigue (5%), insomnia (5%), and increased ALT (9%) were the most common grade 3-4 nonhematologic treatment-emergent AEs occurring in 30% or more of patients.
The treatment had no adverse impact on stem cell collection.
Elotuzumab plus VRd
Turning to elotuzumab in combination with bortezomib, lenalidomide, and dexamethasone (VRd), Dr Mikhael reviewed the phase 2a study (NCT02375555) presented at ASCO 2017 (abstract 8002).
Forty-one patients were enrolled on the study.
The overall response rate after 4 cycles was 100%, with 24% achieving a CR, 47% achieving a VGPR, and 29% a partial response.
Fatigue (60%), neuropathy (55%), musculoskeletal/joint pain (55%), infection (50%), back/neck pain (48%), diarrhea (45%), edema (38%), constipation (38%), cough (35%), mood alteration (35%), rash (35%), and insomnia (30%) occurred in 30% or more of patients.
“So again, not shocking,” Dr Mikhael said, “there was fatigue, there was neuropathy, and there were infections in 50% of patients.”
Grade 4 or greater AEs included thrombocytopenia, hyperglycemia, sepsis, cardiac arrest, and respiratory failure.
“However, here, [we have] maybe not even a yellow flag but a red flag of caution that there were 2 patients who died,” Dr Mikhael noted.
One patient died on study due to respiratory failure and sepsis that arose during cycle 2.
The other patient died more than 30 days after discontinuing study therapy due to febrile neutropenia and hypotension related to sepsis, followed by renal failure.
“Again, in a study that has such small numbers, I don’t want to overstate the case . . ., we don’t want to overreact, but whenever there is death involved, obviously, we have to be particularly cautious,” Dr Mikhael said.
Put into the context of 3 other VRd studies, he noted, the response rate with elotuzumab and VRd is relatively similar but not as good as the phase 3 study of VRd, which was a much larger study of 350 patients.
The situation with daratumumab and KRd is similar to elotuzumab, Dr Mikhael pointed out.
The initial response rates are impressive, but, when compared to other studies, “71% VGPR is good, only after 4 cycles, but we know that, in other studies, after a few more cycles, it was significantly higher.”
Cost
Dr Mikhael also considered cost in his assessment of daratumumab and elotuzumab integrated into frontline regimens.
Adding elotuzumab to VRd would almost double the cost of 12 weeks of therapy. And adding daratumumab to KRd would increase the cost even more.
“These costs are real,” Dr Mikhael said, “and, ultimately, if it’s the best thing for our patients, that’s what we are going to do. But until we have that convincing evidence, I think it’s critical to keep that in perspective. I would suggest that VRd, in many respects, is the standard of care for most patients.”
In terms of adding a mAb upfront, he said, “I don’t think we’re there yet. Do I think, in time, we will be? Quite likely, but I don’t think we are there yet.” ![]()
X-GEM finds drug to have economic value in MM
NEW YORK, NY—Investigators have developed a model that suggests the clinical benefits of denosumab translate into economic value.
The investigators designed their model, X-GEM (Exgeva-Global Economic Model), using results from a large multiple myeloma (MM) trial that showed denosumab to be non-inferior to zoledronic acid (ZA) for skeletal-related events (SRE).
Noopur S. Raje, MD, of Massachusetts General Hospital Cancer Center in Boston, discussed this work at Lymphoma & Myeloma 2017.
The abstract was selected as the best clinical myeloma abstract of the meeting.
“I do think this is a very timely topic because people are not just concerned about the clinical outcomes in patients,” Dr Raje said. “[T]here’s a lot of buzz around the economic evaluation of what we do.”
Trial results
The phase 3 study (NCT01345019) on which X-GEM is based enrolled 1718 MM patients and randomized them to either denosumab or ZA.
The results, reported this year at ASCO, showed denosumab to be non-inferior to ZA in time to first on-study SRE, the primary endpoint.
Denosumab was also found to be non-inferior to ZA for the secondary endpoint of overall survival.
For the exploratory endpoint of progression-free survival (PFS), denosumab-treated patients experienced a significant benefit in terms of PFS. The median PFS was 46.09 months with denosumab and 35.38 months with ZA (difference, 10.71 months).
“Now, we’ve never really seen a survival difference or progression-free survival difference in patients, even in treatment studies, amounting to about 10.7 months,” Dr Raje said. “So this, we found, was quite remarkable in the study.”
Dr Raje also highlighted some safety features from the study. There was significantly less renal toxicity with denosumab than with ZA.
“And in patients who had a creatinine clearance of less than 60 [mL/min], there was almost a doubling of renal toxicity in patients getting zoledronic acid,” she said.
“[D]enosumab may, in fact, be the safer alternative, specifically, in our patients with multiple myeloma who we all know have this problem of renal toxicity throughout the course of their lifetime with myeloma.”
X-GEM
Investigators based X-GEM on the original model published by Stopeck et al. in 2012, which evaluated the cost-effectiveness of denosumab to prevent SREs compared with ZA in patients with solid tumors—prostate, breast, and lung cancer.
“[Stopeck’s] data did show that denosumab was, in fact, cost-effective when compared to zoledronic acid in respect to SREs,” Dr Raje noted.
The timeline for the economic analysis in MM patients spanned the time from diagnosis to death, and patients were evaluated for SREs every 4 weeks on study.
Investigators used 2 cost scenarios. The first was based on an average sales price for 28 days, which was $1928 for denosumab and $45 for ZA. The second was based on the wholesale acquisition cost, which was $2155 for denosumab and $922 for ZA.
“No surprise to anybody,” Dr Raje noted, “denosumab is a lot more expensive. But obviously, this does not tell the whole story. Built into the X-GEM model are a whole host of other factors.”
These include the costs of administration, adverse events, the number of SREs, treatment of an MM patient, and the quality-adjusted life year (QALY) gain with either denosumab or zoledronic acid.
“When you count up all these costs and calculate them based on the data set from the 1800 patients, we found that there was really a difference of zoledronic acid costing a little bit more than denosumab,” Dr Raje said.
From the payer perspective, when clinical outcomes were monetized, the net monetary benefit of denosumab compared with ZA was $5959.
And from a societal perspective, the model calculated the net monetary benefit for denosumab to be $10,259.
The societal perspective included SRE direct costs (hospital, outpatient, and emergency department visits, long-term care, hospice, physical therapy and devices, skilled nursing facility, and strong opioids), direct non-medical costs (driving for treatment, parking, and caregiver costs), and indirect costs (short-term disability and productivity loss).
The investigators concluded that denosumab is cost-effective below a willingness-to-pay threshold of $150,000/QALY regardless of ZA price, whether wholesale or average sales price.
“The bone-specific benefits and observed prolongation of progression-free survival in combination with the economic analysis provides denosumab as a valuable option for patients with multiple myeloma,” Dr Raje said. “In total, we still think it’s more cost-effective to use denosumab when compared to zoledronic acid in this patient population.”
At present, treatment options to prevent bone complications in MM patients are limited to bisphosphonates, including ZA. Denosumab is under review by the US Food and Drug Administration for an expanded indication to include MM.
Both the phase 3 and cost-effectiveness studies were funded by Amgen, Inc. Dr Raje and co-investigators of the study have either consulted for or are employed by Amgen, Inc. ![]()
NEW YORK, NY—Investigators have developed a model that suggests the clinical benefits of denosumab translate into economic value.
The investigators designed their model, X-GEM (Exgeva-Global Economic Model), using results from a large multiple myeloma (MM) trial that showed denosumab to be non-inferior to zoledronic acid (ZA) for skeletal-related events (SRE).
Noopur S. Raje, MD, of Massachusetts General Hospital Cancer Center in Boston, discussed this work at Lymphoma & Myeloma 2017.
The abstract was selected as the best clinical myeloma abstract of the meeting.
“I do think this is a very timely topic because people are not just concerned about the clinical outcomes in patients,” Dr Raje said. “[T]here’s a lot of buzz around the economic evaluation of what we do.”
Trial results
The phase 3 study (NCT01345019) on which X-GEM is based enrolled 1718 MM patients and randomized them to either denosumab or ZA.
The results, reported this year at ASCO, showed denosumab to be non-inferior to ZA in time to first on-study SRE, the primary endpoint.
Denosumab was also found to be non-inferior to ZA for the secondary endpoint of overall survival.
For the exploratory endpoint of progression-free survival (PFS), denosumab-treated patients experienced a significant benefit in terms of PFS. The median PFS was 46.09 months with denosumab and 35.38 months with ZA (difference, 10.71 months).
“Now, we’ve never really seen a survival difference or progression-free survival difference in patients, even in treatment studies, amounting to about 10.7 months,” Dr Raje said. “So this, we found, was quite remarkable in the study.”
Dr Raje also highlighted some safety features from the study. There was significantly less renal toxicity with denosumab than with ZA.
“And in patients who had a creatinine clearance of less than 60 [mL/min], there was almost a doubling of renal toxicity in patients getting zoledronic acid,” she said.
“[D]enosumab may, in fact, be the safer alternative, specifically, in our patients with multiple myeloma who we all know have this problem of renal toxicity throughout the course of their lifetime with myeloma.”
X-GEM
Investigators based X-GEM on the original model published by Stopeck et al. in 2012, which evaluated the cost-effectiveness of denosumab to prevent SREs compared with ZA in patients with solid tumors—prostate, breast, and lung cancer.
“[Stopeck’s] data did show that denosumab was, in fact, cost-effective when compared to zoledronic acid in respect to SREs,” Dr Raje noted.
The timeline for the economic analysis in MM patients spanned the time from diagnosis to death, and patients were evaluated for SREs every 4 weeks on study.
Investigators used 2 cost scenarios. The first was based on an average sales price for 28 days, which was $1928 for denosumab and $45 for ZA. The second was based on the wholesale acquisition cost, which was $2155 for denosumab and $922 for ZA.
“No surprise to anybody,” Dr Raje noted, “denosumab is a lot more expensive. But obviously, this does not tell the whole story. Built into the X-GEM model are a whole host of other factors.”
These include the costs of administration, adverse events, the number of SREs, treatment of an MM patient, and the quality-adjusted life year (QALY) gain with either denosumab or zoledronic acid.
“When you count up all these costs and calculate them based on the data set from the 1800 patients, we found that there was really a difference of zoledronic acid costing a little bit more than denosumab,” Dr Raje said.
From the payer perspective, when clinical outcomes were monetized, the net monetary benefit of denosumab compared with ZA was $5959.
And from a societal perspective, the model calculated the net monetary benefit for denosumab to be $10,259.
The societal perspective included SRE direct costs (hospital, outpatient, and emergency department visits, long-term care, hospice, physical therapy and devices, skilled nursing facility, and strong opioids), direct non-medical costs (driving for treatment, parking, and caregiver costs), and indirect costs (short-term disability and productivity loss).
The investigators concluded that denosumab is cost-effective below a willingness-to-pay threshold of $150,000/QALY regardless of ZA price, whether wholesale or average sales price.
“The bone-specific benefits and observed prolongation of progression-free survival in combination with the economic analysis provides denosumab as a valuable option for patients with multiple myeloma,” Dr Raje said. “In total, we still think it’s more cost-effective to use denosumab when compared to zoledronic acid in this patient population.”
At present, treatment options to prevent bone complications in MM patients are limited to bisphosphonates, including ZA. Denosumab is under review by the US Food and Drug Administration for an expanded indication to include MM.
Both the phase 3 and cost-effectiveness studies were funded by Amgen, Inc. Dr Raje and co-investigators of the study have either consulted for or are employed by Amgen, Inc. ![]()
NEW YORK, NY—Investigators have developed a model that suggests the clinical benefits of denosumab translate into economic value.
The investigators designed their model, X-GEM (Exgeva-Global Economic Model), using results from a large multiple myeloma (MM) trial that showed denosumab to be non-inferior to zoledronic acid (ZA) for skeletal-related events (SRE).
Noopur S. Raje, MD, of Massachusetts General Hospital Cancer Center in Boston, discussed this work at Lymphoma & Myeloma 2017.
The abstract was selected as the best clinical myeloma abstract of the meeting.
“I do think this is a very timely topic because people are not just concerned about the clinical outcomes in patients,” Dr Raje said. “[T]here’s a lot of buzz around the economic evaluation of what we do.”
Trial results
The phase 3 study (NCT01345019) on which X-GEM is based enrolled 1718 MM patients and randomized them to either denosumab or ZA.
The results, reported this year at ASCO, showed denosumab to be non-inferior to ZA in time to first on-study SRE, the primary endpoint.
Denosumab was also found to be non-inferior to ZA for the secondary endpoint of overall survival.
For the exploratory endpoint of progression-free survival (PFS), denosumab-treated patients experienced a significant benefit in terms of PFS. The median PFS was 46.09 months with denosumab and 35.38 months with ZA (difference, 10.71 months).
“Now, we’ve never really seen a survival difference or progression-free survival difference in patients, even in treatment studies, amounting to about 10.7 months,” Dr Raje said. “So this, we found, was quite remarkable in the study.”
Dr Raje also highlighted some safety features from the study. There was significantly less renal toxicity with denosumab than with ZA.
“And in patients who had a creatinine clearance of less than 60 [mL/min], there was almost a doubling of renal toxicity in patients getting zoledronic acid,” she said.
“[D]enosumab may, in fact, be the safer alternative, specifically, in our patients with multiple myeloma who we all know have this problem of renal toxicity throughout the course of their lifetime with myeloma.”
X-GEM
Investigators based X-GEM on the original model published by Stopeck et al. in 2012, which evaluated the cost-effectiveness of denosumab to prevent SREs compared with ZA in patients with solid tumors—prostate, breast, and lung cancer.
“[Stopeck’s] data did show that denosumab was, in fact, cost-effective when compared to zoledronic acid in respect to SREs,” Dr Raje noted.
The timeline for the economic analysis in MM patients spanned the time from diagnosis to death, and patients were evaluated for SREs every 4 weeks on study.
Investigators used 2 cost scenarios. The first was based on an average sales price for 28 days, which was $1928 for denosumab and $45 for ZA. The second was based on the wholesale acquisition cost, which was $2155 for denosumab and $922 for ZA.
“No surprise to anybody,” Dr Raje noted, “denosumab is a lot more expensive. But obviously, this does not tell the whole story. Built into the X-GEM model are a whole host of other factors.”
These include the costs of administration, adverse events, the number of SREs, treatment of an MM patient, and the quality-adjusted life year (QALY) gain with either denosumab or zoledronic acid.
“When you count up all these costs and calculate them based on the data set from the 1800 patients, we found that there was really a difference of zoledronic acid costing a little bit more than denosumab,” Dr Raje said.
From the payer perspective, when clinical outcomes were monetized, the net monetary benefit of denosumab compared with ZA was $5959.
And from a societal perspective, the model calculated the net monetary benefit for denosumab to be $10,259.
The societal perspective included SRE direct costs (hospital, outpatient, and emergency department visits, long-term care, hospice, physical therapy and devices, skilled nursing facility, and strong opioids), direct non-medical costs (driving for treatment, parking, and caregiver costs), and indirect costs (short-term disability and productivity loss).
The investigators concluded that denosumab is cost-effective below a willingness-to-pay threshold of $150,000/QALY regardless of ZA price, whether wholesale or average sales price.
“The bone-specific benefits and observed prolongation of progression-free survival in combination with the economic analysis provides denosumab as a valuable option for patients with multiple myeloma,” Dr Raje said. “In total, we still think it’s more cost-effective to use denosumab when compared to zoledronic acid in this patient population.”
At present, treatment options to prevent bone complications in MM patients are limited to bisphosphonates, including ZA. Denosumab is under review by the US Food and Drug Administration for an expanded indication to include MM.
Both the phase 3 and cost-effectiveness studies were funded by Amgen, Inc. Dr Raje and co-investigators of the study have either consulted for or are employed by Amgen, Inc. ![]()
Large MM trial finds denosumab non-inferior to ZA for SRE

CHICAGO—The largest international multiple myeloma (MM) trial ever conducted, according to the trial sponsor, met its primary endpoint, demonstrating that denosumab is non-inferior to zoledronic acid (ZA) in delaying the time to first on-study skeletal-related event (SRE) in patients with MM.
In addition to bone-specific benefits, denosumab-treated patients had significantly fewer renal adverse events and possible prolongation of progression-free survival.
Denosumab “may in fact be a new standard of care for multiple myeloma-related bone disease,” according to one of the investigators.
“The other important thing to note,” Noopur S. Raje, MD, said during her presentation at the ASCO 2017 Annual Meeting, “is denosumab can be administered despite renal function in patients with myeloma.” It does not need to be dose-adjusted, unlike bisphosphonates.
Dr Raje, of the Massachusetts General Hospital Cancer Center in Boston, Massachusetts, presented the study results as abstract 8005.
Study design
The international, phase 3, randomized, double-blind study is evaluating the safety of denosumab compared with ZA in newly diagnosed MM patients.
Investigators enrolled 1718 patients from 259 sites and 29 countries.
They randomized 859 patients to receive denosumab 120 mg subcutaneously every 4 weeks plus intravenous placebo every 4 weeks, and 859 patients to the standard ZA dose of 4 mg intravenously plus subcutaneous placebo every 4 weeks.
Patients were stratified by whether they were on novel-based anti-myeloma therapy, whether they planned to have an autologous peripheral blood stem cell (PBSC) transplant, disease stage, and previous SRE.
“We were looking for 676 on-study SREs, and if we saw a benefit, patients would be offered open-label denosumab for up to 2 years after this,” Dr Raje said.
“Patients had to have radiographic evidence of bone disease, and this is different from some of the other bone disease studies that you’ve seen in the recent past,” she added.
In addition to documented evidence of MM, patients had to be 18 years or older, be ECOG status of 2 or better, have adequate organ function, and plan to receive or be receiving primary frontline anti-myeloma therapy.
Patients were excluded if they had nonsecretory MM, more than 30 days of previous treatment with anti-myeloma therapy prior to screening, prior use of denosumab, use of oral bisphosphonates with a cumulative dose of more than 1 year, more than 1 previous dose of intravenous bisphosphonate, or prior history or current evidence of osteonecrosis/osteomyelitis of the jaw.
The primary endpoint was time to first on-study SRE, “and the idea here was to look for non-inferiority,” Dr Raje explained.
Secondary endpoints included time to the first on-study SRE (superiority), time to the first-and-subsequent on-study SRE (superiority), and overall survival.
Investigators also included the exploratory objective of progression-free survival (PFS).
Patient demographics
Patients were well balanced across the 2 arms, Dr Raje noted, and the breakdown of myeloma disease stage at diagnosis was comparable between the ZA and denosumab arms.
About 32% of patients were stage I, 37% stage II, and 29% stage III. Stage was not available for 49 patients.
A little more than half (54%) were male, mean age was 63 years, and 82% were white.
Two thirds had prior SRE history, and 54% of patients intended to undergo autologous PBSC transplant.
Enrollment began May 2012 and continued through the end of March 2016. The primary analysis cutoff was July 19, 2016.
Results
The primary endpoint for non-inferiority for time to first on-study SRE was met by denosumab (HR=0.98, 95%CI: 0.85, 1.14; P=0.01).
“When we looked at the secondary endpoints for superiority, we were not able to confirm superiority in this analysis, either for time to first SRE or time to first-and subsequent SRE on this study,” Dr Raje said.
The investigators also did not observe a survival difference between denosumab and ZA, with a hazard ratio (HR) (95% CI) of 0.90 (0.70, 1.16), P=0.41.
“Importantly, we had an exploratory endpoint where we looked at progression-free survival in this newly diagnosed patient population,” she added, “and we saw an interestingly increased or prolonged progression-free survival in patients getting denosumab.”
“And that survival difference was more than 10 months between denosumab and zoledronic acid, favoring the denosumab arm,” she affirmed. The HR was 0.82, 95% CI: 0.68, 0.99, P=0.036 (descriptive).
Safety
“[I]f you look at all treatment-emergent adverse events between denosumab and zoledronic acid, we really could not find a big difference in either of these 2 groups of patients,” Dr Raje said.
“We saw that in general both denosumab and zoledronic acid were extremely well tolerated between the 2 groups of patients.”
The investigators “drilled down” on certain toxicity issues of interest and examined events such as atypical stress fractures, hypersensitivity reactions, musculoskeletal pain, infections and infestations, new primary malignancies, and acute phase reactions.
They observed no atypical femur fractures on the study, nor did they see any big differences with respect to hypersensitivity or acute phase reactions.
The investigators examined closely any renal issues because dosing of ZA specifically is impacted by renal function.
The data showed that treatment-emergent adverse event (TEAE) renal toxicity was significantly higher in the ZA group compared to the denosumab group, 17% and 10%, respectively (P<0.001).
“When you look at patients who had a creatinine clearance less than 60 mL per minute,” Dr Raje emphasized, “we saw an almost doubling of renal toxicity in the zoledronic acid arm (26.4%) compared to the denosumab arm (12.9%).”
Patients with a creatinine level greater than 2 mg/dL had a significant increase in creatinine in the ZA arm (P=0.010), which was also significantly increased if their creatinine clearance was less than 60 mL/minute (P=0.054).
“There was a doubling of creatinine from baseline, more so in the zoledronic acid arm compared to the patients with denosumab,” Dr Raje said. “And this was again more pronounced if you had a creatinine clearance of less than 60.”
Hypocalcemia was “not surprisingly” more common in the denosumab arm than the ZA arm (P=0.009) for all patients, and osteonecrosis of the jaw was equal in both arms (P=0.147), although numerically slightly higher with denosumab treatment.
Dr Raje summarized that there was no difference in overall survival at the time of this analysis, “but I will say that the follow-up for a newly diagnosed patient population is fairly short right now.”
“Progression-free survival, which we saw [cut] off 10.7 months, was actually quite striking when denosumab was compared to zoledronic acid, and this was statistically highly significant.”
“The bone-specific benefits in combination with significantly fewer renal adverse events and possible prolongation of PFS with denosumab therapy we do think is very promising,” she said, “and may in fact be a new standard of care for multiple myeloma-related bone disease.”
The study was funded by Amgen Inc.
Denosumab (XGEVA®) is indicated by the US Food and Drug Administration for the prevention of fractures and other SREs in patients with bone metastases from solid tumors. It is currently not indicated for the prevention of SREs in patients with MM. ![]()
CHICAGO—The largest international multiple myeloma (MM) trial ever conducted, according to the trial sponsor, met its primary endpoint, demonstrating that denosumab is non-inferior to zoledronic acid (ZA) in delaying the time to first on-study skeletal-related event (SRE) in patients with MM.
In addition to bone-specific benefits, denosumab-treated patients had significantly fewer renal adverse events and possible prolongation of progression-free survival.
Denosumab “may in fact be a new standard of care for multiple myeloma-related bone disease,” according to one of the investigators.
“The other important thing to note,” Noopur S. Raje, MD, said during her presentation at the ASCO 2017 Annual Meeting, “is denosumab can be administered despite renal function in patients with myeloma.” It does not need to be dose-adjusted, unlike bisphosphonates.
Dr Raje, of the Massachusetts General Hospital Cancer Center in Boston, Massachusetts, presented the study results as abstract 8005.
Study design
The international, phase 3, randomized, double-blind study is evaluating the safety of denosumab compared with ZA in newly diagnosed MM patients.
Investigators enrolled 1718 patients from 259 sites and 29 countries.
They randomized 859 patients to receive denosumab 120 mg subcutaneously every 4 weeks plus intravenous placebo every 4 weeks, and 859 patients to the standard ZA dose of 4 mg intravenously plus subcutaneous placebo every 4 weeks.
Patients were stratified by whether they were on novel-based anti-myeloma therapy, whether they planned to have an autologous peripheral blood stem cell (PBSC) transplant, disease stage, and previous SRE.
“We were looking for 676 on-study SREs, and if we saw a benefit, patients would be offered open-label denosumab for up to 2 years after this,” Dr Raje said.
“Patients had to have radiographic evidence of bone disease, and this is different from some of the other bone disease studies that you’ve seen in the recent past,” she added.
In addition to documented evidence of MM, patients had to be 18 years or older, be ECOG status of 2 or better, have adequate organ function, and plan to receive or be receiving primary frontline anti-myeloma therapy.
Patients were excluded if they had nonsecretory MM, more than 30 days of previous treatment with anti-myeloma therapy prior to screening, prior use of denosumab, use of oral bisphosphonates with a cumulative dose of more than 1 year, more than 1 previous dose of intravenous bisphosphonate, or prior history or current evidence of osteonecrosis/osteomyelitis of the jaw.
The primary endpoint was time to first on-study SRE, “and the idea here was to look for non-inferiority,” Dr Raje explained.
Secondary endpoints included time to the first on-study SRE (superiority), time to the first-and-subsequent on-study SRE (superiority), and overall survival.
Investigators also included the exploratory objective of progression-free survival (PFS).
Patient demographics
Patients were well balanced across the 2 arms, Dr Raje noted, and the breakdown of myeloma disease stage at diagnosis was comparable between the ZA and denosumab arms.
About 32% of patients were stage I, 37% stage II, and 29% stage III. Stage was not available for 49 patients.
A little more than half (54%) were male, mean age was 63 years, and 82% were white.
Two thirds had prior SRE history, and 54% of patients intended to undergo autologous PBSC transplant.
Enrollment began May 2012 and continued through the end of March 2016. The primary analysis cutoff was July 19, 2016.
Results
The primary endpoint for non-inferiority for time to first on-study SRE was met by denosumab (HR=0.98, 95%CI: 0.85, 1.14; P=0.01).
“When we looked at the secondary endpoints for superiority, we were not able to confirm superiority in this analysis, either for time to first SRE or time to first-and subsequent SRE on this study,” Dr Raje said.
The investigators also did not observe a survival difference between denosumab and ZA, with a hazard ratio (HR) (95% CI) of 0.90 (0.70, 1.16), P=0.41.
“Importantly, we had an exploratory endpoint where we looked at progression-free survival in this newly diagnosed patient population,” she added, “and we saw an interestingly increased or prolonged progression-free survival in patients getting denosumab.”
“And that survival difference was more than 10 months between denosumab and zoledronic acid, favoring the denosumab arm,” she affirmed. The HR was 0.82, 95% CI: 0.68, 0.99, P=0.036 (descriptive).
Safety
“[I]f you look at all treatment-emergent adverse events between denosumab and zoledronic acid, we really could not find a big difference in either of these 2 groups of patients,” Dr Raje said.
“We saw that in general both denosumab and zoledronic acid were extremely well tolerated between the 2 groups of patients.”
The investigators “drilled down” on certain toxicity issues of interest and examined events such as atypical stress fractures, hypersensitivity reactions, musculoskeletal pain, infections and infestations, new primary malignancies, and acute phase reactions.
They observed no atypical femur fractures on the study, nor did they see any big differences with respect to hypersensitivity or acute phase reactions.
The investigators examined closely any renal issues because dosing of ZA specifically is impacted by renal function.
The data showed that treatment-emergent adverse event (TEAE) renal toxicity was significantly higher in the ZA group compared to the denosumab group, 17% and 10%, respectively (P<0.001).
“When you look at patients who had a creatinine clearance less than 60 mL per minute,” Dr Raje emphasized, “we saw an almost doubling of renal toxicity in the zoledronic acid arm (26.4%) compared to the denosumab arm (12.9%).”
Patients with a creatinine level greater than 2 mg/dL had a significant increase in creatinine in the ZA arm (P=0.010), which was also significantly increased if their creatinine clearance was less than 60 mL/minute (P=0.054).
“There was a doubling of creatinine from baseline, more so in the zoledronic acid arm compared to the patients with denosumab,” Dr Raje said. “And this was again more pronounced if you had a creatinine clearance of less than 60.”
Hypocalcemia was “not surprisingly” more common in the denosumab arm than the ZA arm (P=0.009) for all patients, and osteonecrosis of the jaw was equal in both arms (P=0.147), although numerically slightly higher with denosumab treatment.
Dr Raje summarized that there was no difference in overall survival at the time of this analysis, “but I will say that the follow-up for a newly diagnosed patient population is fairly short right now.”
“Progression-free survival, which we saw [cut] off 10.7 months, was actually quite striking when denosumab was compared to zoledronic acid, and this was statistically highly significant.”
“The bone-specific benefits in combination with significantly fewer renal adverse events and possible prolongation of PFS with denosumab therapy we do think is very promising,” she said, “and may in fact be a new standard of care for multiple myeloma-related bone disease.”
The study was funded by Amgen Inc.
Denosumab (XGEVA®) is indicated by the US Food and Drug Administration for the prevention of fractures and other SREs in patients with bone metastases from solid tumors. It is currently not indicated for the prevention of SREs in patients with MM. ![]()
CHICAGO—The largest international multiple myeloma (MM) trial ever conducted, according to the trial sponsor, met its primary endpoint, demonstrating that denosumab is non-inferior to zoledronic acid (ZA) in delaying the time to first on-study skeletal-related event (SRE) in patients with MM.
In addition to bone-specific benefits, denosumab-treated patients had significantly fewer renal adverse events and possible prolongation of progression-free survival.
Denosumab “may in fact be a new standard of care for multiple myeloma-related bone disease,” according to one of the investigators.
“The other important thing to note,” Noopur S. Raje, MD, said during her presentation at the ASCO 2017 Annual Meeting, “is denosumab can be administered despite renal function in patients with myeloma.” It does not need to be dose-adjusted, unlike bisphosphonates.
Dr Raje, of the Massachusetts General Hospital Cancer Center in Boston, Massachusetts, presented the study results as abstract 8005.
Study design
The international, phase 3, randomized, double-blind study is evaluating the safety of denosumab compared with ZA in newly diagnosed MM patients.
Investigators enrolled 1718 patients from 259 sites and 29 countries.
They randomized 859 patients to receive denosumab 120 mg subcutaneously every 4 weeks plus intravenous placebo every 4 weeks, and 859 patients to the standard ZA dose of 4 mg intravenously plus subcutaneous placebo every 4 weeks.
Patients were stratified by whether they were on novel-based anti-myeloma therapy, whether they planned to have an autologous peripheral blood stem cell (PBSC) transplant, disease stage, and previous SRE.
“We were looking for 676 on-study SREs, and if we saw a benefit, patients would be offered open-label denosumab for up to 2 years after this,” Dr Raje said.
“Patients had to have radiographic evidence of bone disease, and this is different from some of the other bone disease studies that you’ve seen in the recent past,” she added.
In addition to documented evidence of MM, patients had to be 18 years or older, be ECOG status of 2 or better, have adequate organ function, and plan to receive or be receiving primary frontline anti-myeloma therapy.
Patients were excluded if they had nonsecretory MM, more than 30 days of previous treatment with anti-myeloma therapy prior to screening, prior use of denosumab, use of oral bisphosphonates with a cumulative dose of more than 1 year, more than 1 previous dose of intravenous bisphosphonate, or prior history or current evidence of osteonecrosis/osteomyelitis of the jaw.
The primary endpoint was time to first on-study SRE, “and the idea here was to look for non-inferiority,” Dr Raje explained.
Secondary endpoints included time to the first on-study SRE (superiority), time to the first-and-subsequent on-study SRE (superiority), and overall survival.
Investigators also included the exploratory objective of progression-free survival (PFS).
Patient demographics
Patients were well balanced across the 2 arms, Dr Raje noted, and the breakdown of myeloma disease stage at diagnosis was comparable between the ZA and denosumab arms.
About 32% of patients were stage I, 37% stage II, and 29% stage III. Stage was not available for 49 patients.
A little more than half (54%) were male, mean age was 63 years, and 82% were white.
Two thirds had prior SRE history, and 54% of patients intended to undergo autologous PBSC transplant.
Enrollment began May 2012 and continued through the end of March 2016. The primary analysis cutoff was July 19, 2016.
Results
The primary endpoint for non-inferiority for time to first on-study SRE was met by denosumab (HR=0.98, 95%CI: 0.85, 1.14; P=0.01).
“When we looked at the secondary endpoints for superiority, we were not able to confirm superiority in this analysis, either for time to first SRE or time to first-and subsequent SRE on this study,” Dr Raje said.
The investigators also did not observe a survival difference between denosumab and ZA, with a hazard ratio (HR) (95% CI) of 0.90 (0.70, 1.16), P=0.41.
“Importantly, we had an exploratory endpoint where we looked at progression-free survival in this newly diagnosed patient population,” she added, “and we saw an interestingly increased or prolonged progression-free survival in patients getting denosumab.”
“And that survival difference was more than 10 months between denosumab and zoledronic acid, favoring the denosumab arm,” she affirmed. The HR was 0.82, 95% CI: 0.68, 0.99, P=0.036 (descriptive).
Safety
“[I]f you look at all treatment-emergent adverse events between denosumab and zoledronic acid, we really could not find a big difference in either of these 2 groups of patients,” Dr Raje said.
“We saw that in general both denosumab and zoledronic acid were extremely well tolerated between the 2 groups of patients.”
The investigators “drilled down” on certain toxicity issues of interest and examined events such as atypical stress fractures, hypersensitivity reactions, musculoskeletal pain, infections and infestations, new primary malignancies, and acute phase reactions.
They observed no atypical femur fractures on the study, nor did they see any big differences with respect to hypersensitivity or acute phase reactions.
The investigators examined closely any renal issues because dosing of ZA specifically is impacted by renal function.
The data showed that treatment-emergent adverse event (TEAE) renal toxicity was significantly higher in the ZA group compared to the denosumab group, 17% and 10%, respectively (P<0.001).
“When you look at patients who had a creatinine clearance less than 60 mL per minute,” Dr Raje emphasized, “we saw an almost doubling of renal toxicity in the zoledronic acid arm (26.4%) compared to the denosumab arm (12.9%).”
Patients with a creatinine level greater than 2 mg/dL had a significant increase in creatinine in the ZA arm (P=0.010), which was also significantly increased if their creatinine clearance was less than 60 mL/minute (P=0.054).
“There was a doubling of creatinine from baseline, more so in the zoledronic acid arm compared to the patients with denosumab,” Dr Raje said. “And this was again more pronounced if you had a creatinine clearance of less than 60.”
Hypocalcemia was “not surprisingly” more common in the denosumab arm than the ZA arm (P=0.009) for all patients, and osteonecrosis of the jaw was equal in both arms (P=0.147), although numerically slightly higher with denosumab treatment.
Dr Raje summarized that there was no difference in overall survival at the time of this analysis, “but I will say that the follow-up for a newly diagnosed patient population is fairly short right now.”
“Progression-free survival, which we saw [cut] off 10.7 months, was actually quite striking when denosumab was compared to zoledronic acid, and this was statistically highly significant.”
“The bone-specific benefits in combination with significantly fewer renal adverse events and possible prolongation of PFS with denosumab therapy we do think is very promising,” she said, “and may in fact be a new standard of care for multiple myeloma-related bone disease.”
The study was funded by Amgen Inc.
Denosumab (XGEVA®) is indicated by the US Food and Drug Administration for the prevention of fractures and other SREs in patients with bone metastases from solid tumors. It is currently not indicated for the prevention of SREs in patients with MM. ![]()
‘Admirable’ overall survival attainable in AML with enasidenib

CHICAGO—The experimental mutant IDH2 (mIDH2) inhibitor enasidenib has produced “admirable” overall survival in patients with mIDH2 relapsed or refractory acute myeloid leukemia (AML), according to Eytan M. Stein, MD, an investigator on the phase 1 dose escalation and expansion study.
Patients who achieved a complete remission (CR) had a median overall survival (OS) of 19.7 months and non-CR responders, 13.8 months.
“I really want to make the point,” Dr Stein said, “this is a group of patients that are highly refractory, either refractory to induction chemotherapy, refractory to standard of care approaches for patients who are unable to get induction chemotherapy, so refractory to hypomethylating agents or low-dose cytarabine.”
Mutations in IDH2 occur in approximately 12% of AML patients.
Dr Stein explained that the mutant protein converts alpha ketoglutarate to beta hydroxyglutarate (2-HG). And increased levels of intracellular 2-HG lead to methylation changes in the cell that cause a block in myeloid differentiation.
Enasidenib, also known as AG-221, is a selective, oral, potent inhibitor of the mIDH2 enzyyme.
Dr Stein, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the results during the ASCO 2017 Annual Meeting (abstract 7004).
The clinical and translational papers were published simultaneously in Blood.
Study design
The phase 1/2 study had a large dose-escalation component, with 113 patients enrolled. Patients had to have an advanced hematologic malignancy with an IDH2 mutation.
Patients received cumulative daily doses of 50 mg – 650 mg of enasidenib in continuous 28-day cycles.
Four expansion arms were added, with 126 patients.
Two expansion arms were in relapsed/refractory AML patients: one in patients 60 years or older or any age if they had relapsed after bone marrow transplant (BMT), and the other in patients younger than 60 excluding those relapsed after BMT.
The other 2 expansion arms were in untreated AML patients and in patients with any hematologic malignancy ineligible for the other arms.
Dr Stein presented results for the relapsed/refractory AML patients in the dose escalation and expansion phases of the study.
The key endpoints were safety, tolerability, maximum tolerated dose (MTD), and dose-limiting toxicities; response rates as assessed by the local investigator according to IWG criteria; and assessment of clinical activity.
Dr Stein noted the phase 2 study is now completely accrued (n=91) and the recommended enasidenib dose is 100 mg/day in relapsed/refractory AML.
The MTD was not reached at doses up to 650 mg/day.
Baseline characteristics
Median age of all 239 phase 1 patients was 70 years (range, 19-100), 57% were male, and almost all patients had intermediate- or poor-risk disease.
The investigators were also interested in the co-occurring mutations in patients on screening and whether there were differences between patients with mIDH2 at R172 and R140.
Seventy-five percent of the patients (n=179) had R140 and 24% had R172 (n=57).
There was a statistically significant difference in the number of co-occurring mutations in the R140 and R172 patients, with the R140 patients having a higher co-mutation burden compared with the R172 patients, (P=0.020).
The most frequent mutations co-occurring in R140 patients were SRSF2, followed by, in descending order of frequency, DNMT3A, RUNX1, ASXL1, and 24 others.
SFSR2 does not occur in R172 patients. DNMT3A was the most frequently co-occurring mutation in R172, followed by ASXL1, BCOR, NRAS, RUNX1, KMT2A, KRAS, and STAG2.
Safety
The most common treatment-emergent adverse events (TEAE) that occurred in 20% or more of all patients of any grade included nausea (46%), hyperbilirubinemia (45%), diarrhea and fatigue (40% each), decreased appetite (38%), vomiting (32%), dyspnea (31%), cough (29%), pyrexia and febrile neutropenia (28% each), thrombocytopenia, anemia, constipation, hypokalemia, and peripheral edema (27% each), pneumonia (21%), and hyperuricemia (20%).
The only 2 grade 3/4 TEAEs that rose above the level of 5% were hyperbilirubinemia (12%) and thrombocytopenia (6%).
“The hyperbilirubinemia, as I’ve mentioned in a number of meetings before this,” Dr Stein clarified, “is one that occurs because the enzyme is an off-target effect of inhibiting the UGT1A1 enzyme, which conjugates bilirubin.”
“So a patient who goes on this study who has a defect in bilirubin conjugation because they have Gilbert’s disease, they will have a higher level of bilirubin compared to a patient who doesn’t have Gilbert’s disease. This does not appear to have any clinical sequelae. You’ll also notice AST, ALT, alkaline phosphatase or any liver failure is not on this [TEAE] list.”
Response
The overall response rate for the patients who received enasidenib 100 mg/day was 38.5% (42/109) and for all doses 40.3% (71/176).
The true CR rate was 20.2% (100 mg/day) and 19.3% for all doses.
An additional 20% achieved a CR with incomplete hematologic recovery, CR with incomplete platelet recovery, partial response (PR), and morphologic leukemia-free state with either 100 mg enasidenib daily or all doses.
“Time to first response is not immediate,” Dr Stein pointed out. “It takes a median of 1.9 months to get there, and the time to complete remission takes even longer, a median of 3.7 months in the 100-mg experience, 3.8 months in all doses, to get to that best response.”
“I think the clinical importance of this is,” he added, “for a patient that one might have who is on this drug, it is important to keep them on the drug for a prolonged period of time so that they have the opportunity to have that response.”
Hematologic parameters also improved gradually.
Increases in platelet count, absolute neutrophil count, and hemoglobin level did not rise exponentially upon administration of study drug, but rather they slowly rose, “again getting to this point, that the drug takes time to work,” Dr Stein emphasized.
Patients in CR had very high transfusion independence rates, “which is what I would expect,” Dr Stein said. “If you are in complete remission, you should be transfusion independent.”
“What’s a little bit more interesting, though,” Dr Stein added, “is those patients who are non-CR responders. [I]n those patients who have responded but have less than a complete remission, 50% of them are independent of red cell transfusions and 50% of them are independent of platelet transfusions.”
Survival
The CR data and transfusion independence data translated into a median OS in these relapsed and refractory AML patients of 9.3 months.
And about 10% - 15% of the patients had prolonged survival up to 2 years and longer on the single agent.
Analysis of OS by best response revealed that for patients with a CR, “they really have an admirable overall survival of 19.7 months, almost 20 months,” Dr Stein said.
Patients who had a non-CR response had a median OS of 13.8 months, and non-responders had a median OS of 7.0 months.
And there was a qualitative improvement in response over time: the number of patients with CRs and PRs increased, while the number with stable disease decreased.
“Again, I think getting at the point it takes time for these responses to occur,” Dr Stein iterated.
Over the course of therapy, some responders had a differentiation of myeloblasts, so that by cycle 3, the marrow looked largely normal.
The investigators did not observe any morphological evidence of cytotoxicity or cellular aplasia.
But they did observe myeloid differentiation using FISH.
Trisomy 8 that was evident at the time of screening in responders’ myeloblasts, persisted in the promyelocytes and mature granulocyte population, and was no longer evident in the lymphoid compartment.
Baseline 2-HG levels and mIDH2 variant allele frequency were similar for responding and non-responding patients.
The investigators believe that differentiation of myeloblsts, not cytotoxicity, may drive the clinical efficacy of enasidenib.
A phase 3 trial of enasidenib monotherapy versus conventional care regimens is underway in older patients with late-stage AML, and phase 1/2 studies of enasidenib combinations are ongoing in newly diagnosed AML patients.
Enasidenib, which also has efficacy in myelodysplastic syndromes, has been granted priority review for relapsed/refractory AML by the US Food and Drug Administration. ![]()
CHICAGO—The experimental mutant IDH2 (mIDH2) inhibitor enasidenib has produced “admirable” overall survival in patients with mIDH2 relapsed or refractory acute myeloid leukemia (AML), according to Eytan M. Stein, MD, an investigator on the phase 1 dose escalation and expansion study.
Patients who achieved a complete remission (CR) had a median overall survival (OS) of 19.7 months and non-CR responders, 13.8 months.
“I really want to make the point,” Dr Stein said, “this is a group of patients that are highly refractory, either refractory to induction chemotherapy, refractory to standard of care approaches for patients who are unable to get induction chemotherapy, so refractory to hypomethylating agents or low-dose cytarabine.”
Mutations in IDH2 occur in approximately 12% of AML patients.
Dr Stein explained that the mutant protein converts alpha ketoglutarate to beta hydroxyglutarate (2-HG). And increased levels of intracellular 2-HG lead to methylation changes in the cell that cause a block in myeloid differentiation.
Enasidenib, also known as AG-221, is a selective, oral, potent inhibitor of the mIDH2 enzyyme.
Dr Stein, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the results during the ASCO 2017 Annual Meeting (abstract 7004).
The clinical and translational papers were published simultaneously in Blood.
Study design
The phase 1/2 study had a large dose-escalation component, with 113 patients enrolled. Patients had to have an advanced hematologic malignancy with an IDH2 mutation.
Patients received cumulative daily doses of 50 mg – 650 mg of enasidenib in continuous 28-day cycles.
Four expansion arms were added, with 126 patients.
Two expansion arms were in relapsed/refractory AML patients: one in patients 60 years or older or any age if they had relapsed after bone marrow transplant (BMT), and the other in patients younger than 60 excluding those relapsed after BMT.
The other 2 expansion arms were in untreated AML patients and in patients with any hematologic malignancy ineligible for the other arms.
Dr Stein presented results for the relapsed/refractory AML patients in the dose escalation and expansion phases of the study.
The key endpoints were safety, tolerability, maximum tolerated dose (MTD), and dose-limiting toxicities; response rates as assessed by the local investigator according to IWG criteria; and assessment of clinical activity.
Dr Stein noted the phase 2 study is now completely accrued (n=91) and the recommended enasidenib dose is 100 mg/day in relapsed/refractory AML.
The MTD was not reached at doses up to 650 mg/day.
Baseline characteristics
Median age of all 239 phase 1 patients was 70 years (range, 19-100), 57% were male, and almost all patients had intermediate- or poor-risk disease.
The investigators were also interested in the co-occurring mutations in patients on screening and whether there were differences between patients with mIDH2 at R172 and R140.
Seventy-five percent of the patients (n=179) had R140 and 24% had R172 (n=57).
There was a statistically significant difference in the number of co-occurring mutations in the R140 and R172 patients, with the R140 patients having a higher co-mutation burden compared with the R172 patients, (P=0.020).
The most frequent mutations co-occurring in R140 patients were SRSF2, followed by, in descending order of frequency, DNMT3A, RUNX1, ASXL1, and 24 others.
SFSR2 does not occur in R172 patients. DNMT3A was the most frequently co-occurring mutation in R172, followed by ASXL1, BCOR, NRAS, RUNX1, KMT2A, KRAS, and STAG2.
Safety
The most common treatment-emergent adverse events (TEAE) that occurred in 20% or more of all patients of any grade included nausea (46%), hyperbilirubinemia (45%), diarrhea and fatigue (40% each), decreased appetite (38%), vomiting (32%), dyspnea (31%), cough (29%), pyrexia and febrile neutropenia (28% each), thrombocytopenia, anemia, constipation, hypokalemia, and peripheral edema (27% each), pneumonia (21%), and hyperuricemia (20%).
The only 2 grade 3/4 TEAEs that rose above the level of 5% were hyperbilirubinemia (12%) and thrombocytopenia (6%).
“The hyperbilirubinemia, as I’ve mentioned in a number of meetings before this,” Dr Stein clarified, “is one that occurs because the enzyme is an off-target effect of inhibiting the UGT1A1 enzyme, which conjugates bilirubin.”
“So a patient who goes on this study who has a defect in bilirubin conjugation because they have Gilbert’s disease, they will have a higher level of bilirubin compared to a patient who doesn’t have Gilbert’s disease. This does not appear to have any clinical sequelae. You’ll also notice AST, ALT, alkaline phosphatase or any liver failure is not on this [TEAE] list.”
Response
The overall response rate for the patients who received enasidenib 100 mg/day was 38.5% (42/109) and for all doses 40.3% (71/176).
The true CR rate was 20.2% (100 mg/day) and 19.3% for all doses.
An additional 20% achieved a CR with incomplete hematologic recovery, CR with incomplete platelet recovery, partial response (PR), and morphologic leukemia-free state with either 100 mg enasidenib daily or all doses.
“Time to first response is not immediate,” Dr Stein pointed out. “It takes a median of 1.9 months to get there, and the time to complete remission takes even longer, a median of 3.7 months in the 100-mg experience, 3.8 months in all doses, to get to that best response.”
“I think the clinical importance of this is,” he added, “for a patient that one might have who is on this drug, it is important to keep them on the drug for a prolonged period of time so that they have the opportunity to have that response.”
Hematologic parameters also improved gradually.
Increases in platelet count, absolute neutrophil count, and hemoglobin level did not rise exponentially upon administration of study drug, but rather they slowly rose, “again getting to this point, that the drug takes time to work,” Dr Stein emphasized.
Patients in CR had very high transfusion independence rates, “which is what I would expect,” Dr Stein said. “If you are in complete remission, you should be transfusion independent.”
“What’s a little bit more interesting, though,” Dr Stein added, “is those patients who are non-CR responders. [I]n those patients who have responded but have less than a complete remission, 50% of them are independent of red cell transfusions and 50% of them are independent of platelet transfusions.”
Survival
The CR data and transfusion independence data translated into a median OS in these relapsed and refractory AML patients of 9.3 months.
And about 10% - 15% of the patients had prolonged survival up to 2 years and longer on the single agent.
Analysis of OS by best response revealed that for patients with a CR, “they really have an admirable overall survival of 19.7 months, almost 20 months,” Dr Stein said.
Patients who had a non-CR response had a median OS of 13.8 months, and non-responders had a median OS of 7.0 months.
And there was a qualitative improvement in response over time: the number of patients with CRs and PRs increased, while the number with stable disease decreased.
“Again, I think getting at the point it takes time for these responses to occur,” Dr Stein iterated.
Over the course of therapy, some responders had a differentiation of myeloblasts, so that by cycle 3, the marrow looked largely normal.
The investigators did not observe any morphological evidence of cytotoxicity or cellular aplasia.
But they did observe myeloid differentiation using FISH.
Trisomy 8 that was evident at the time of screening in responders’ myeloblasts, persisted in the promyelocytes and mature granulocyte population, and was no longer evident in the lymphoid compartment.
Baseline 2-HG levels and mIDH2 variant allele frequency were similar for responding and non-responding patients.
The investigators believe that differentiation of myeloblsts, not cytotoxicity, may drive the clinical efficacy of enasidenib.
A phase 3 trial of enasidenib monotherapy versus conventional care regimens is underway in older patients with late-stage AML, and phase 1/2 studies of enasidenib combinations are ongoing in newly diagnosed AML patients.
Enasidenib, which also has efficacy in myelodysplastic syndromes, has been granted priority review for relapsed/refractory AML by the US Food and Drug Administration. ![]()
CHICAGO—The experimental mutant IDH2 (mIDH2) inhibitor enasidenib has produced “admirable” overall survival in patients with mIDH2 relapsed or refractory acute myeloid leukemia (AML), according to Eytan M. Stein, MD, an investigator on the phase 1 dose escalation and expansion study.
Patients who achieved a complete remission (CR) had a median overall survival (OS) of 19.7 months and non-CR responders, 13.8 months.
“I really want to make the point,” Dr Stein said, “this is a group of patients that are highly refractory, either refractory to induction chemotherapy, refractory to standard of care approaches for patients who are unable to get induction chemotherapy, so refractory to hypomethylating agents or low-dose cytarabine.”
Mutations in IDH2 occur in approximately 12% of AML patients.
Dr Stein explained that the mutant protein converts alpha ketoglutarate to beta hydroxyglutarate (2-HG). And increased levels of intracellular 2-HG lead to methylation changes in the cell that cause a block in myeloid differentiation.
Enasidenib, also known as AG-221, is a selective, oral, potent inhibitor of the mIDH2 enzyyme.
Dr Stein, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the results during the ASCO 2017 Annual Meeting (abstract 7004).
The clinical and translational papers were published simultaneously in Blood.
Study design
The phase 1/2 study had a large dose-escalation component, with 113 patients enrolled. Patients had to have an advanced hematologic malignancy with an IDH2 mutation.
Patients received cumulative daily doses of 50 mg – 650 mg of enasidenib in continuous 28-day cycles.
Four expansion arms were added, with 126 patients.
Two expansion arms were in relapsed/refractory AML patients: one in patients 60 years or older or any age if they had relapsed after bone marrow transplant (BMT), and the other in patients younger than 60 excluding those relapsed after BMT.
The other 2 expansion arms were in untreated AML patients and in patients with any hematologic malignancy ineligible for the other arms.
Dr Stein presented results for the relapsed/refractory AML patients in the dose escalation and expansion phases of the study.
The key endpoints were safety, tolerability, maximum tolerated dose (MTD), and dose-limiting toxicities; response rates as assessed by the local investigator according to IWG criteria; and assessment of clinical activity.
Dr Stein noted the phase 2 study is now completely accrued (n=91) and the recommended enasidenib dose is 100 mg/day in relapsed/refractory AML.
The MTD was not reached at doses up to 650 mg/day.
Baseline characteristics
Median age of all 239 phase 1 patients was 70 years (range, 19-100), 57% were male, and almost all patients had intermediate- or poor-risk disease.
The investigators were also interested in the co-occurring mutations in patients on screening and whether there were differences between patients with mIDH2 at R172 and R140.
Seventy-five percent of the patients (n=179) had R140 and 24% had R172 (n=57).
There was a statistically significant difference in the number of co-occurring mutations in the R140 and R172 patients, with the R140 patients having a higher co-mutation burden compared with the R172 patients, (P=0.020).
The most frequent mutations co-occurring in R140 patients were SRSF2, followed by, in descending order of frequency, DNMT3A, RUNX1, ASXL1, and 24 others.
SFSR2 does not occur in R172 patients. DNMT3A was the most frequently co-occurring mutation in R172, followed by ASXL1, BCOR, NRAS, RUNX1, KMT2A, KRAS, and STAG2.
Safety
The most common treatment-emergent adverse events (TEAE) that occurred in 20% or more of all patients of any grade included nausea (46%), hyperbilirubinemia (45%), diarrhea and fatigue (40% each), decreased appetite (38%), vomiting (32%), dyspnea (31%), cough (29%), pyrexia and febrile neutropenia (28% each), thrombocytopenia, anemia, constipation, hypokalemia, and peripheral edema (27% each), pneumonia (21%), and hyperuricemia (20%).
The only 2 grade 3/4 TEAEs that rose above the level of 5% were hyperbilirubinemia (12%) and thrombocytopenia (6%).
“The hyperbilirubinemia, as I’ve mentioned in a number of meetings before this,” Dr Stein clarified, “is one that occurs because the enzyme is an off-target effect of inhibiting the UGT1A1 enzyme, which conjugates bilirubin.”
“So a patient who goes on this study who has a defect in bilirubin conjugation because they have Gilbert’s disease, they will have a higher level of bilirubin compared to a patient who doesn’t have Gilbert’s disease. This does not appear to have any clinical sequelae. You’ll also notice AST, ALT, alkaline phosphatase or any liver failure is not on this [TEAE] list.”
Response
The overall response rate for the patients who received enasidenib 100 mg/day was 38.5% (42/109) and for all doses 40.3% (71/176).
The true CR rate was 20.2% (100 mg/day) and 19.3% for all doses.
An additional 20% achieved a CR with incomplete hematologic recovery, CR with incomplete platelet recovery, partial response (PR), and morphologic leukemia-free state with either 100 mg enasidenib daily or all doses.
“Time to first response is not immediate,” Dr Stein pointed out. “It takes a median of 1.9 months to get there, and the time to complete remission takes even longer, a median of 3.7 months in the 100-mg experience, 3.8 months in all doses, to get to that best response.”
“I think the clinical importance of this is,” he added, “for a patient that one might have who is on this drug, it is important to keep them on the drug for a prolonged period of time so that they have the opportunity to have that response.”
Hematologic parameters also improved gradually.
Increases in platelet count, absolute neutrophil count, and hemoglobin level did not rise exponentially upon administration of study drug, but rather they slowly rose, “again getting to this point, that the drug takes time to work,” Dr Stein emphasized.
Patients in CR had very high transfusion independence rates, “which is what I would expect,” Dr Stein said. “If you are in complete remission, you should be transfusion independent.”
“What’s a little bit more interesting, though,” Dr Stein added, “is those patients who are non-CR responders. [I]n those patients who have responded but have less than a complete remission, 50% of them are independent of red cell transfusions and 50% of them are independent of platelet transfusions.”
Survival
The CR data and transfusion independence data translated into a median OS in these relapsed and refractory AML patients of 9.3 months.
And about 10% - 15% of the patients had prolonged survival up to 2 years and longer on the single agent.
Analysis of OS by best response revealed that for patients with a CR, “they really have an admirable overall survival of 19.7 months, almost 20 months,” Dr Stein said.
Patients who had a non-CR response had a median OS of 13.8 months, and non-responders had a median OS of 7.0 months.
And there was a qualitative improvement in response over time: the number of patients with CRs and PRs increased, while the number with stable disease decreased.
“Again, I think getting at the point it takes time for these responses to occur,” Dr Stein iterated.
Over the course of therapy, some responders had a differentiation of myeloblasts, so that by cycle 3, the marrow looked largely normal.
The investigators did not observe any morphological evidence of cytotoxicity or cellular aplasia.
But they did observe myeloid differentiation using FISH.
Trisomy 8 that was evident at the time of screening in responders’ myeloblasts, persisted in the promyelocytes and mature granulocyte population, and was no longer evident in the lymphoid compartment.
Baseline 2-HG levels and mIDH2 variant allele frequency were similar for responding and non-responding patients.
The investigators believe that differentiation of myeloblsts, not cytotoxicity, may drive the clinical efficacy of enasidenib.
A phase 3 trial of enasidenib monotherapy versus conventional care regimens is underway in older patients with late-stage AML, and phase 1/2 studies of enasidenib combinations are ongoing in newly diagnosed AML patients.
Enasidenib, which also has efficacy in myelodysplastic syndromes, has been granted priority review for relapsed/refractory AML by the US Food and Drug Administration. ![]()
Combo with daratumumab could be alternative to ASCT in MM

CHICAGO—Results of an open-label phase 1b study of daratumumab combined with carfilzomib, lenalidomide, and dexamethasone (KRd) in newly diagnosed multiple myeloma (MM) patients have shown the combination to be highly effective, with an overall response rate of 100%.
Ninety-one percent of patients achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR) or better.
Investigators had hypothesized that rather than using autologous stem cell transplant (ASCT) to improve results of treatment with KRd, the combination could alternatively be improved by incorporating daratumumab into a KRd regimen.
Andrzej Jakubowiak, MD, of the University of Chicago Medical Center in Illinois, presented the findings of the MMY1001 study at the 2017 ASCO Annual Meeting (abstract 8000*).
“I think what was one of the more important developments in myeloma last year,” Dr Jakubowiak said, “was data from randomized studies showing that adding daratumumab to either lenalidomide and dexamethasone in the POLLUX study or bortezomib and dexamethasone, a proteasome inhibitor, in the CASTOR study, improves responses, depth of response, and . . . dramatically improved progression-free survival.”
“[W]e have now the rationale to potentially combine daratumumab with both an IMiD and proteasome inhibitor,” he explained, “which led to the development of this phase 1b study in which we combined daratumumab with KRd and evaluated tolerability and efficacy.”
Study design
Twenty-two transplant-eligible or -ineligible newly diagnosed MM patients were enrolled on the study.
Treatment duration was planned to be 13 cycles or less and patients had the option to move to transplant after 4 cycles.
They could have no clinically significant cardiac disease and echocardiogram was required prior to transplant.
The dosing schedule was the established dosing schema for daratumumab and KRd with 2 notable differences in the 28-day cycles.
First, the daratumumab dose was a split dose. So patients received 8 mg/kg on days 1-2 of cycle 1, 16 mg/kg a week on cycle 2, 16 mg/kg every 2 weeks on cycles 3 – 6, and every 4th week thereafter.
The second difference was carfilzomib dosing was a weekly regimen with escalation from 20 mg/m2 on day 1, cycle 1 to 70 mg/m2 on day 8 of cycle 1.
Lenalidomide (25 mg on days 1-21 of each cycle) and dexamethasone (40 mg/week) were the standard regimens for these drugs.
The primary endpoint was safety and tolerability. The secondary endpoint was overall response rate (ORR), duration of response, time to response, and infusion-related reactions (IRR).
The study also had an exploratory endpoint of progression-free survival (PFS).
Baseline characteristics
Patients were a median age of 59.5 years (range 34 – 74). About two thirds were younger than 65 and one third were between 65 and 75.
A little over half were male and most (86%) were white.
A little more than half (55%) had an ECOG score of 0, 41% were ECOG 1, and 5% were ECOG 2.
Patient disposition
As of the cutoff date of March 24, 8 of the 22 patients enrolled (36%) discontinued treatment: 1 due to an adverse event (AE), 1 due to progressive disease, and 6 patients (27%) proceeded to ASCT.
Dr Jakubowiak pointed out that response was censored at this point for patients who proceeded to transplant.
The median follow-up was 10.8 months (range, 4.0 – 12.5) and the median number of treatment cycles was 11.5 (range, 1.0 – 13.0).
“What is of interest to many of us,” Dr Jakubowiak said, “is that patients were escalated to the planned dose of 70 mg/m2 by cycle 2 except for 3 patients.”
Of the 3, 1 discontinued before day 1 of the second cycle due to toxicity, 1 had a dose reduction to 56 mg/m2 at day of the second cycle, and 1 escalated to 70 mg/m2 at day 8 of cycle 3.
Ultimately, all patients who remained on study were able to escalate to 70 mg/m2.
Safety
The hematologic treatment-emergent adverse events (TEAE) generally followed what has been observed in similar studies before, Dr Jakubowiak noted.
Hematologic TEAEs of all grades occurring in 30% or more of patients were lymphopenia (68%), thrombocytopenia (55%), anemia (46%), leukopenia (41%), and neutropenia (32%).
The most common non-hematologic TEAEs of all grades occurring in 30% of patients or more were diarrhea (73%), upper respiratory infection (59%) cough, constipation, and fatigue (50% each), dyspnea and insomnia (46%), nausea, rash, and back pain (41%), muscle spasm (36%), and vomiting, pain in extremity, hyperglycemia, and increased ALT (32%).
The most common grade 3/4 TEAEs were infrequent and many events had none of grade 3/4 severity.
The safety profile is consistent with what was previously reported for daratumumab or KRd, Dr Jakubowiak affirmed.
Serious TEAEs
Serious TEAEs occurred in 10 patients (46%), with many occurring in just 1 patient. Pulmonary embolism (PE) was the most frequent, occurring in 3 patients.
All patients were required to be on aspirin prophylaxis and 1 of the patients who had a PE discontinued therapy.
The number of patients with a serious TEAE reasonably related to an individual study drug were 3 (14%) for daratumumab, 5 (23%) for carfilzomib, 5 (23%) for lenalidomide, and 2 (9%) for dexamethasone.
The TEAEs of interest—tachycardia, congestive heart failure, and hypertension—occurred in a single patient each.
Overall, serious TEAEs were consistent with previous reports from KRd studies.
Echocardiogram assessment
Investigators conducted 30 systemic evaluations on the impact of this regimen on heart function. The investigators observed no change from baseline through the duration of treatment in patients’ left ventricular ejection fractions.
One patient developed congestive heart failure, possibly related to daratumumab or carfilzomib. This patient resumed treatment with a reduced carfilzomib dose, elected ASCT on study day 113, and ended treatment with a VGPR.
“In all,” Dr Jakubowiak said, “we feel that there is no apparent signal of adverse impact of the addition of daratumumab on cardiac function.”
Infusion times and reactions
Overall, IRRs occurred in 27% of the patients, “which appears lower than with previous daratumumab studies,” Dr Jakubowiak noted. And IRRs occurred more frequently during the first infusion than subsequent infusions.
The split-dose infusion time was very similar to that of second and subsequent cycles.
There were limited events related to infusions. All were grade 1 or 2 and most occurred in only a single patient.
Response rate
The median number of treatment cycles administered was 11.5 (range, 2.0 – 13.0). The best response was 100% PR or better, 91% achieved VGPR or better, 42% CR or better, and 29% a stringent CR.
The depth of response improved with duration of treatment. For example, the sCR rate increased from 5% after 4 cycles to 29% at the end of treatment.
PFS was an exploratory endpoint. One patient progressed at 10.8 months and the 12-month PFS rate was 94% with all patients alive.
Stem cell harvest and ASCT
“For many of us,” Dr Jakubowiak commented, “it’s also of interest how this regimen will impact stem cell harvest.”
Nineteen of 22 patients were deemed to be transplant eligible, and the median number of CD34+ cells collected from them was 10.4 x 106 cells/kg.
Patients had a median of 5 treatment cycles prior to stem cell harvest, and 14 (74%) had a VGPR or better prior to harvest.
The investigators believe stem cell yield was consistent with previous KRd studies.
Dr Jakubowiak commented that the deepening of response over time “is a phenomenon we think is important. . . . In all, the data from this small phase 1b study provide support for further evaluation of this regimen in newly diagnosed myeloma."
The study was funded by Janssen Research and Development, LLC. ![]()
*Data presented during the meeting differ from the abstract.
CHICAGO—Results of an open-label phase 1b study of daratumumab combined with carfilzomib, lenalidomide, and dexamethasone (KRd) in newly diagnosed multiple myeloma (MM) patients have shown the combination to be highly effective, with an overall response rate of 100%.
Ninety-one percent of patients achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR) or better.
Investigators had hypothesized that rather than using autologous stem cell transplant (ASCT) to improve results of treatment with KRd, the combination could alternatively be improved by incorporating daratumumab into a KRd regimen.
Andrzej Jakubowiak, MD, of the University of Chicago Medical Center in Illinois, presented the findings of the MMY1001 study at the 2017 ASCO Annual Meeting (abstract 8000*).
“I think what was one of the more important developments in myeloma last year,” Dr Jakubowiak said, “was data from randomized studies showing that adding daratumumab to either lenalidomide and dexamethasone in the POLLUX study or bortezomib and dexamethasone, a proteasome inhibitor, in the CASTOR study, improves responses, depth of response, and . . . dramatically improved progression-free survival.”
“[W]e have now the rationale to potentially combine daratumumab with both an IMiD and proteasome inhibitor,” he explained, “which led to the development of this phase 1b study in which we combined daratumumab with KRd and evaluated tolerability and efficacy.”
Study design
Twenty-two transplant-eligible or -ineligible newly diagnosed MM patients were enrolled on the study.
Treatment duration was planned to be 13 cycles or less and patients had the option to move to transplant after 4 cycles.
They could have no clinically significant cardiac disease and echocardiogram was required prior to transplant.
The dosing schedule was the established dosing schema for daratumumab and KRd with 2 notable differences in the 28-day cycles.
First, the daratumumab dose was a split dose. So patients received 8 mg/kg on days 1-2 of cycle 1, 16 mg/kg a week on cycle 2, 16 mg/kg every 2 weeks on cycles 3 – 6, and every 4th week thereafter.
The second difference was carfilzomib dosing was a weekly regimen with escalation from 20 mg/m2 on day 1, cycle 1 to 70 mg/m2 on day 8 of cycle 1.
Lenalidomide (25 mg on days 1-21 of each cycle) and dexamethasone (40 mg/week) were the standard regimens for these drugs.
The primary endpoint was safety and tolerability. The secondary endpoint was overall response rate (ORR), duration of response, time to response, and infusion-related reactions (IRR).
The study also had an exploratory endpoint of progression-free survival (PFS).
Baseline characteristics
Patients were a median age of 59.5 years (range 34 – 74). About two thirds were younger than 65 and one third were between 65 and 75.
A little over half were male and most (86%) were white.
A little more than half (55%) had an ECOG score of 0, 41% were ECOG 1, and 5% were ECOG 2.
Patient disposition
As of the cutoff date of March 24, 8 of the 22 patients enrolled (36%) discontinued treatment: 1 due to an adverse event (AE), 1 due to progressive disease, and 6 patients (27%) proceeded to ASCT.
Dr Jakubowiak pointed out that response was censored at this point for patients who proceeded to transplant.
The median follow-up was 10.8 months (range, 4.0 – 12.5) and the median number of treatment cycles was 11.5 (range, 1.0 – 13.0).
“What is of interest to many of us,” Dr Jakubowiak said, “is that patients were escalated to the planned dose of 70 mg/m2 by cycle 2 except for 3 patients.”
Of the 3, 1 discontinued before day 1 of the second cycle due to toxicity, 1 had a dose reduction to 56 mg/m2 at day of the second cycle, and 1 escalated to 70 mg/m2 at day 8 of cycle 3.
Ultimately, all patients who remained on study were able to escalate to 70 mg/m2.
Safety
The hematologic treatment-emergent adverse events (TEAE) generally followed what has been observed in similar studies before, Dr Jakubowiak noted.
Hematologic TEAEs of all grades occurring in 30% or more of patients were lymphopenia (68%), thrombocytopenia (55%), anemia (46%), leukopenia (41%), and neutropenia (32%).
The most common non-hematologic TEAEs of all grades occurring in 30% of patients or more were diarrhea (73%), upper respiratory infection (59%) cough, constipation, and fatigue (50% each), dyspnea and insomnia (46%), nausea, rash, and back pain (41%), muscle spasm (36%), and vomiting, pain in extremity, hyperglycemia, and increased ALT (32%).
The most common grade 3/4 TEAEs were infrequent and many events had none of grade 3/4 severity.
The safety profile is consistent with what was previously reported for daratumumab or KRd, Dr Jakubowiak affirmed.
Serious TEAEs
Serious TEAEs occurred in 10 patients (46%), with many occurring in just 1 patient. Pulmonary embolism (PE) was the most frequent, occurring in 3 patients.
All patients were required to be on aspirin prophylaxis and 1 of the patients who had a PE discontinued therapy.
The number of patients with a serious TEAE reasonably related to an individual study drug were 3 (14%) for daratumumab, 5 (23%) for carfilzomib, 5 (23%) for lenalidomide, and 2 (9%) for dexamethasone.
The TEAEs of interest—tachycardia, congestive heart failure, and hypertension—occurred in a single patient each.
Overall, serious TEAEs were consistent with previous reports from KRd studies.
Echocardiogram assessment
Investigators conducted 30 systemic evaluations on the impact of this regimen on heart function. The investigators observed no change from baseline through the duration of treatment in patients’ left ventricular ejection fractions.
One patient developed congestive heart failure, possibly related to daratumumab or carfilzomib. This patient resumed treatment with a reduced carfilzomib dose, elected ASCT on study day 113, and ended treatment with a VGPR.
“In all,” Dr Jakubowiak said, “we feel that there is no apparent signal of adverse impact of the addition of daratumumab on cardiac function.”
Infusion times and reactions
Overall, IRRs occurred in 27% of the patients, “which appears lower than with previous daratumumab studies,” Dr Jakubowiak noted. And IRRs occurred more frequently during the first infusion than subsequent infusions.
The split-dose infusion time was very similar to that of second and subsequent cycles.
There were limited events related to infusions. All were grade 1 or 2 and most occurred in only a single patient.
Response rate
The median number of treatment cycles administered was 11.5 (range, 2.0 – 13.0). The best response was 100% PR or better, 91% achieved VGPR or better, 42% CR or better, and 29% a stringent CR.
The depth of response improved with duration of treatment. For example, the sCR rate increased from 5% after 4 cycles to 29% at the end of treatment.
PFS was an exploratory endpoint. One patient progressed at 10.8 months and the 12-month PFS rate was 94% with all patients alive.
Stem cell harvest and ASCT
“For many of us,” Dr Jakubowiak commented, “it’s also of interest how this regimen will impact stem cell harvest.”
Nineteen of 22 patients were deemed to be transplant eligible, and the median number of CD34+ cells collected from them was 10.4 x 106 cells/kg.
Patients had a median of 5 treatment cycles prior to stem cell harvest, and 14 (74%) had a VGPR or better prior to harvest.
The investigators believe stem cell yield was consistent with previous KRd studies.
Dr Jakubowiak commented that the deepening of response over time “is a phenomenon we think is important. . . . In all, the data from this small phase 1b study provide support for further evaluation of this regimen in newly diagnosed myeloma."
The study was funded by Janssen Research and Development, LLC. ![]()
*Data presented during the meeting differ from the abstract.
CHICAGO—Results of an open-label phase 1b study of daratumumab combined with carfilzomib, lenalidomide, and dexamethasone (KRd) in newly diagnosed multiple myeloma (MM) patients have shown the combination to be highly effective, with an overall response rate of 100%.
Ninety-one percent of patients achieved a very good partial response (VGPR) or better, and 43% achieved a complete response (CR) or better.
Investigators had hypothesized that rather than using autologous stem cell transplant (ASCT) to improve results of treatment with KRd, the combination could alternatively be improved by incorporating daratumumab into a KRd regimen.
Andrzej Jakubowiak, MD, of the University of Chicago Medical Center in Illinois, presented the findings of the MMY1001 study at the 2017 ASCO Annual Meeting (abstract 8000*).
“I think what was one of the more important developments in myeloma last year,” Dr Jakubowiak said, “was data from randomized studies showing that adding daratumumab to either lenalidomide and dexamethasone in the POLLUX study or bortezomib and dexamethasone, a proteasome inhibitor, in the CASTOR study, improves responses, depth of response, and . . . dramatically improved progression-free survival.”
“[W]e have now the rationale to potentially combine daratumumab with both an IMiD and proteasome inhibitor,” he explained, “which led to the development of this phase 1b study in which we combined daratumumab with KRd and evaluated tolerability and efficacy.”
Study design
Twenty-two transplant-eligible or -ineligible newly diagnosed MM patients were enrolled on the study.
Treatment duration was planned to be 13 cycles or less and patients had the option to move to transplant after 4 cycles.
They could have no clinically significant cardiac disease and echocardiogram was required prior to transplant.
The dosing schedule was the established dosing schema for daratumumab and KRd with 2 notable differences in the 28-day cycles.
First, the daratumumab dose was a split dose. So patients received 8 mg/kg on days 1-2 of cycle 1, 16 mg/kg a week on cycle 2, 16 mg/kg every 2 weeks on cycles 3 – 6, and every 4th week thereafter.
The second difference was carfilzomib dosing was a weekly regimen with escalation from 20 mg/m2 on day 1, cycle 1 to 70 mg/m2 on day 8 of cycle 1.
Lenalidomide (25 mg on days 1-21 of each cycle) and dexamethasone (40 mg/week) were the standard regimens for these drugs.
The primary endpoint was safety and tolerability. The secondary endpoint was overall response rate (ORR), duration of response, time to response, and infusion-related reactions (IRR).
The study also had an exploratory endpoint of progression-free survival (PFS).
Baseline characteristics
Patients were a median age of 59.5 years (range 34 – 74). About two thirds were younger than 65 and one third were between 65 and 75.
A little over half were male and most (86%) were white.
A little more than half (55%) had an ECOG score of 0, 41% were ECOG 1, and 5% were ECOG 2.
Patient disposition
As of the cutoff date of March 24, 8 of the 22 patients enrolled (36%) discontinued treatment: 1 due to an adverse event (AE), 1 due to progressive disease, and 6 patients (27%) proceeded to ASCT.
Dr Jakubowiak pointed out that response was censored at this point for patients who proceeded to transplant.
The median follow-up was 10.8 months (range, 4.0 – 12.5) and the median number of treatment cycles was 11.5 (range, 1.0 – 13.0).
“What is of interest to many of us,” Dr Jakubowiak said, “is that patients were escalated to the planned dose of 70 mg/m2 by cycle 2 except for 3 patients.”
Of the 3, 1 discontinued before day 1 of the second cycle due to toxicity, 1 had a dose reduction to 56 mg/m2 at day of the second cycle, and 1 escalated to 70 mg/m2 at day 8 of cycle 3.
Ultimately, all patients who remained on study were able to escalate to 70 mg/m2.
Safety
The hematologic treatment-emergent adverse events (TEAE) generally followed what has been observed in similar studies before, Dr Jakubowiak noted.
Hematologic TEAEs of all grades occurring in 30% or more of patients were lymphopenia (68%), thrombocytopenia (55%), anemia (46%), leukopenia (41%), and neutropenia (32%).
The most common non-hematologic TEAEs of all grades occurring in 30% of patients or more were diarrhea (73%), upper respiratory infection (59%) cough, constipation, and fatigue (50% each), dyspnea and insomnia (46%), nausea, rash, and back pain (41%), muscle spasm (36%), and vomiting, pain in extremity, hyperglycemia, and increased ALT (32%).
The most common grade 3/4 TEAEs were infrequent and many events had none of grade 3/4 severity.
The safety profile is consistent with what was previously reported for daratumumab or KRd, Dr Jakubowiak affirmed.
Serious TEAEs
Serious TEAEs occurred in 10 patients (46%), with many occurring in just 1 patient. Pulmonary embolism (PE) was the most frequent, occurring in 3 patients.
All patients were required to be on aspirin prophylaxis and 1 of the patients who had a PE discontinued therapy.
The number of patients with a serious TEAE reasonably related to an individual study drug were 3 (14%) for daratumumab, 5 (23%) for carfilzomib, 5 (23%) for lenalidomide, and 2 (9%) for dexamethasone.
The TEAEs of interest—tachycardia, congestive heart failure, and hypertension—occurred in a single patient each.
Overall, serious TEAEs were consistent with previous reports from KRd studies.
Echocardiogram assessment
Investigators conducted 30 systemic evaluations on the impact of this regimen on heart function. The investigators observed no change from baseline through the duration of treatment in patients’ left ventricular ejection fractions.
One patient developed congestive heart failure, possibly related to daratumumab or carfilzomib. This patient resumed treatment with a reduced carfilzomib dose, elected ASCT on study day 113, and ended treatment with a VGPR.
“In all,” Dr Jakubowiak said, “we feel that there is no apparent signal of adverse impact of the addition of daratumumab on cardiac function.”
Infusion times and reactions
Overall, IRRs occurred in 27% of the patients, “which appears lower than with previous daratumumab studies,” Dr Jakubowiak noted. And IRRs occurred more frequently during the first infusion than subsequent infusions.
The split-dose infusion time was very similar to that of second and subsequent cycles.
There were limited events related to infusions. All were grade 1 or 2 and most occurred in only a single patient.
Response rate
The median number of treatment cycles administered was 11.5 (range, 2.0 – 13.0). The best response was 100% PR or better, 91% achieved VGPR or better, 42% CR or better, and 29% a stringent CR.
The depth of response improved with duration of treatment. For example, the sCR rate increased from 5% after 4 cycles to 29% at the end of treatment.
PFS was an exploratory endpoint. One patient progressed at 10.8 months and the 12-month PFS rate was 94% with all patients alive.
Stem cell harvest and ASCT
“For many of us,” Dr Jakubowiak commented, “it’s also of interest how this regimen will impact stem cell harvest.”
Nineteen of 22 patients were deemed to be transplant eligible, and the median number of CD34+ cells collected from them was 10.4 x 106 cells/kg.
Patients had a median of 5 treatment cycles prior to stem cell harvest, and 14 (74%) had a VGPR or better prior to harvest.
The investigators believe stem cell yield was consistent with previous KRd studies.
Dr Jakubowiak commented that the deepening of response over time “is a phenomenon we think is important. . . . In all, the data from this small phase 1b study provide support for further evaluation of this regimen in newly diagnosed myeloma."
The study was funded by Janssen Research and Development, LLC. ![]()
*Data presented during the meeting differ from the abstract.
Deep molecular responses achievable in AML pts treated with gilteritinib

CHICAGO—Next generation sequencing (NGS) has shown that the FLT3 inhibitor gilteritinib can produce deep molecular responses in a subset of patients with acute myeloid leukemia (AML), according to new research.
Gilteritinib is a highly selective FLT3/AXL inhibitor that is active against FLT3-ITD and FLT3-D835 mutations, but minimal residual disease (MRD) had not systematically been assessed previously in AML patients treated with potent FLT3 inhibitors.
Investigators believed that MRD evaluation in these patients could serve as a useful marker of FLT3 inhibitor efficacy. They therefore conducted an exploratory analysis of a subset of AML patients treated with gilteritinib on the Chrysalis study.
Jessica K. Altman, MD, of the Robert H. Lurie Cancer Center of Northwestern University in Chicago, Illinois, presented the findings at the ASCO 2017 Annual Meeting (abstract 7003).
Chrysalis study: Efficacy and survival
The phase 1/2 Chrysalis study examined the tolerability and antileukemic activity of once daily gilteritinib in a FLT3-ITD-enriched relapsed/refractory AML population of approximately 250 patients.
Overall, gilteritinib was well tolerated and had consistency and potent FLT3 inhibition at doses of >80 mg/day.
The maximum tolerated dose was 300 mg/day. Dose-limiting toxicities were diarrhea and liver function abnormalities.
The greatest overall response rate was 52% and the longest median overall survival (OS) duration was 31 weeks, observed in patients at doses >80 mg/day.
The composite complete remission (CR) rate, comprised of CR, CR with incomplete count recovery (Cri), and CR with incomplete platelet recovery (CRp), was 41%.
The median OS was 31 weeks, and median duration of response 20 weeks.
“Survival probabilities demonstrated that the overall survival for patients who received 80 mg of gilteritinib was higher than those who received less than 80 mg,” Dr Altman said.
Molecular response assessment
Dr Altman then presented the molecular response assessment.
The investigators included all FLT3-ITD mutated patients enrolled in the gilteritinib 120 and 200 mg/day dose cohorts and had bone marrow aspirates available at baseline and at 1 or more additional time points.
“The group I’m reporting on,” Dr Altman explained, “comprises 51% of all FLT3-ITD mutated patients treated at these 2 dose levels.”
FLT3-ITD and total FLT3 alleles were quantified by a novel NGS assay using an Illumina® sequencing platform. Read depth of at least 100,000 reads per sample were implemented.
“Evaluation of MRD was exploratory and it was not prespecified in the study,” Dr Altman noted.
Hence, the investigators defined a molecular response as an ITD signal ratio—FLT3-ITD : FLT3 total—of <10-2.
They defined major molecular response (MMR) as an ITD signal ratio of <10-3, and negative MRD status as <10-4.
Patient characteristics
Baseline characteristics of the 80 patients in the MRD analysis group were similar to those of the entire Chrysalis study population.
Median age was 61 years (range, 23 – 86) and the patients were heavily pretreated: 35% had 3 or more prior lines of AML therapy, and 28% had received a prior FLT3 inhibitor. About a third had prior allogeneic hematopoietic stem cell transplant.
Molecular response
Median OS in this cohort was 32.6 weeks, very similar to the entire study population.
Twenty patients (25%) achieved a molecular response, 18 (23%) an MMR, and 13 (16%) were MRD negative.
The median time to achieve a minimum ITD signal ratio was 8.2 weeks (range, 3.7 – 64).
And molecular response correlated with improved OS.
“The 20 patients who achieved a molecular response had a median overall survival of 59.6 weeks,” Dr Altman said, “which is statistically significantly different and I think clinically different than those who did not attain a molecular response.”
Patients who did not achieve a molecular response had a median overall survival of 28.4 weeks.
“As you could predict,” she added, “the molecular response was greater in those who attained a complete remission than those who had a CRp or Cri.”
Investigators observed similar results in patients who achieved an MMR, using the the cutoff point of 10-3.
“When we stratified by MRD negative status,” she said, “which was an ITD signal ratio of 10-4 or better, there’s clear separation of the Kaplan Meier curves for OS in this cohort again.”
Dr Altman pointed out that this was the first clinical trial to demonstrate that patients with AML treated with a FLT3 inhibitor can attain a molecular response.
“Also, and importantly, there is now a sensitive and specific assay for the detection of minimal residual disease in FLT3-ITD mutated patients and it has the potential to be widely adopted across trials and in clinical practice.”
MRD is prospectively being evaluated in 2 gilteritinib phase 3 maintenance studies.
The trial was sponsored by Astellas Pharma Global Development, Inc. ![]()
CHICAGO—Next generation sequencing (NGS) has shown that the FLT3 inhibitor gilteritinib can produce deep molecular responses in a subset of patients with acute myeloid leukemia (AML), according to new research.
Gilteritinib is a highly selective FLT3/AXL inhibitor that is active against FLT3-ITD and FLT3-D835 mutations, but minimal residual disease (MRD) had not systematically been assessed previously in AML patients treated with potent FLT3 inhibitors.
Investigators believed that MRD evaluation in these patients could serve as a useful marker of FLT3 inhibitor efficacy. They therefore conducted an exploratory analysis of a subset of AML patients treated with gilteritinib on the Chrysalis study.
Jessica K. Altman, MD, of the Robert H. Lurie Cancer Center of Northwestern University in Chicago, Illinois, presented the findings at the ASCO 2017 Annual Meeting (abstract 7003).
Chrysalis study: Efficacy and survival
The phase 1/2 Chrysalis study examined the tolerability and antileukemic activity of once daily gilteritinib in a FLT3-ITD-enriched relapsed/refractory AML population of approximately 250 patients.
Overall, gilteritinib was well tolerated and had consistency and potent FLT3 inhibition at doses of >80 mg/day.
The maximum tolerated dose was 300 mg/day. Dose-limiting toxicities were diarrhea and liver function abnormalities.
The greatest overall response rate was 52% and the longest median overall survival (OS) duration was 31 weeks, observed in patients at doses >80 mg/day.
The composite complete remission (CR) rate, comprised of CR, CR with incomplete count recovery (Cri), and CR with incomplete platelet recovery (CRp), was 41%.
The median OS was 31 weeks, and median duration of response 20 weeks.
“Survival probabilities demonstrated that the overall survival for patients who received 80 mg of gilteritinib was higher than those who received less than 80 mg,” Dr Altman said.
Molecular response assessment
Dr Altman then presented the molecular response assessment.
The investigators included all FLT3-ITD mutated patients enrolled in the gilteritinib 120 and 200 mg/day dose cohorts and had bone marrow aspirates available at baseline and at 1 or more additional time points.
“The group I’m reporting on,” Dr Altman explained, “comprises 51% of all FLT3-ITD mutated patients treated at these 2 dose levels.”
FLT3-ITD and total FLT3 alleles were quantified by a novel NGS assay using an Illumina® sequencing platform. Read depth of at least 100,000 reads per sample were implemented.
“Evaluation of MRD was exploratory and it was not prespecified in the study,” Dr Altman noted.
Hence, the investigators defined a molecular response as an ITD signal ratio—FLT3-ITD : FLT3 total—of <10-2.
They defined major molecular response (MMR) as an ITD signal ratio of <10-3, and negative MRD status as <10-4.
Patient characteristics
Baseline characteristics of the 80 patients in the MRD analysis group were similar to those of the entire Chrysalis study population.
Median age was 61 years (range, 23 – 86) and the patients were heavily pretreated: 35% had 3 or more prior lines of AML therapy, and 28% had received a prior FLT3 inhibitor. About a third had prior allogeneic hematopoietic stem cell transplant.
Molecular response
Median OS in this cohort was 32.6 weeks, very similar to the entire study population.
Twenty patients (25%) achieved a molecular response, 18 (23%) an MMR, and 13 (16%) were MRD negative.
The median time to achieve a minimum ITD signal ratio was 8.2 weeks (range, 3.7 – 64).
And molecular response correlated with improved OS.
“The 20 patients who achieved a molecular response had a median overall survival of 59.6 weeks,” Dr Altman said, “which is statistically significantly different and I think clinically different than those who did not attain a molecular response.”
Patients who did not achieve a molecular response had a median overall survival of 28.4 weeks.
“As you could predict,” she added, “the molecular response was greater in those who attained a complete remission than those who had a CRp or Cri.”
Investigators observed similar results in patients who achieved an MMR, using the the cutoff point of 10-3.
“When we stratified by MRD negative status,” she said, “which was an ITD signal ratio of 10-4 or better, there’s clear separation of the Kaplan Meier curves for OS in this cohort again.”
Dr Altman pointed out that this was the first clinical trial to demonstrate that patients with AML treated with a FLT3 inhibitor can attain a molecular response.
“Also, and importantly, there is now a sensitive and specific assay for the detection of minimal residual disease in FLT3-ITD mutated patients and it has the potential to be widely adopted across trials and in clinical practice.”
MRD is prospectively being evaluated in 2 gilteritinib phase 3 maintenance studies.
The trial was sponsored by Astellas Pharma Global Development, Inc. ![]()
CHICAGO—Next generation sequencing (NGS) has shown that the FLT3 inhibitor gilteritinib can produce deep molecular responses in a subset of patients with acute myeloid leukemia (AML), according to new research.
Gilteritinib is a highly selective FLT3/AXL inhibitor that is active against FLT3-ITD and FLT3-D835 mutations, but minimal residual disease (MRD) had not systematically been assessed previously in AML patients treated with potent FLT3 inhibitors.
Investigators believed that MRD evaluation in these patients could serve as a useful marker of FLT3 inhibitor efficacy. They therefore conducted an exploratory analysis of a subset of AML patients treated with gilteritinib on the Chrysalis study.
Jessica K. Altman, MD, of the Robert H. Lurie Cancer Center of Northwestern University in Chicago, Illinois, presented the findings at the ASCO 2017 Annual Meeting (abstract 7003).
Chrysalis study: Efficacy and survival
The phase 1/2 Chrysalis study examined the tolerability and antileukemic activity of once daily gilteritinib in a FLT3-ITD-enriched relapsed/refractory AML population of approximately 250 patients.
Overall, gilteritinib was well tolerated and had consistency and potent FLT3 inhibition at doses of >80 mg/day.
The maximum tolerated dose was 300 mg/day. Dose-limiting toxicities were diarrhea and liver function abnormalities.
The greatest overall response rate was 52% and the longest median overall survival (OS) duration was 31 weeks, observed in patients at doses >80 mg/day.
The composite complete remission (CR) rate, comprised of CR, CR with incomplete count recovery (Cri), and CR with incomplete platelet recovery (CRp), was 41%.
The median OS was 31 weeks, and median duration of response 20 weeks.
“Survival probabilities demonstrated that the overall survival for patients who received 80 mg of gilteritinib was higher than those who received less than 80 mg,” Dr Altman said.
Molecular response assessment
Dr Altman then presented the molecular response assessment.
The investigators included all FLT3-ITD mutated patients enrolled in the gilteritinib 120 and 200 mg/day dose cohorts and had bone marrow aspirates available at baseline and at 1 or more additional time points.
“The group I’m reporting on,” Dr Altman explained, “comprises 51% of all FLT3-ITD mutated patients treated at these 2 dose levels.”
FLT3-ITD and total FLT3 alleles were quantified by a novel NGS assay using an Illumina® sequencing platform. Read depth of at least 100,000 reads per sample were implemented.
“Evaluation of MRD was exploratory and it was not prespecified in the study,” Dr Altman noted.
Hence, the investigators defined a molecular response as an ITD signal ratio—FLT3-ITD : FLT3 total—of <10-2.
They defined major molecular response (MMR) as an ITD signal ratio of <10-3, and negative MRD status as <10-4.
Patient characteristics
Baseline characteristics of the 80 patients in the MRD analysis group were similar to those of the entire Chrysalis study population.
Median age was 61 years (range, 23 – 86) and the patients were heavily pretreated: 35% had 3 or more prior lines of AML therapy, and 28% had received a prior FLT3 inhibitor. About a third had prior allogeneic hematopoietic stem cell transplant.
Molecular response
Median OS in this cohort was 32.6 weeks, very similar to the entire study population.
Twenty patients (25%) achieved a molecular response, 18 (23%) an MMR, and 13 (16%) were MRD negative.
The median time to achieve a minimum ITD signal ratio was 8.2 weeks (range, 3.7 – 64).
And molecular response correlated with improved OS.
“The 20 patients who achieved a molecular response had a median overall survival of 59.6 weeks,” Dr Altman said, “which is statistically significantly different and I think clinically different than those who did not attain a molecular response.”
Patients who did not achieve a molecular response had a median overall survival of 28.4 weeks.
“As you could predict,” she added, “the molecular response was greater in those who attained a complete remission than those who had a CRp or Cri.”
Investigators observed similar results in patients who achieved an MMR, using the the cutoff point of 10-3.
“When we stratified by MRD negative status,” she said, “which was an ITD signal ratio of 10-4 or better, there’s clear separation of the Kaplan Meier curves for OS in this cohort again.”
Dr Altman pointed out that this was the first clinical trial to demonstrate that patients with AML treated with a FLT3 inhibitor can attain a molecular response.
“Also, and importantly, there is now a sensitive and specific assay for the detection of minimal residual disease in FLT3-ITD mutated patients and it has the potential to be widely adopted across trials and in clinical practice.”
MRD is prospectively being evaluated in 2 gilteritinib phase 3 maintenance studies.
The trial was sponsored by Astellas Pharma Global Development, Inc. ![]()
Dasatinib potentially a new SOC for children with CML-CP

CHICAGO—The largest ongoing and prospective trial of pediatric patients with chronic myeloid leukemia in chronic phase (CML-CP), according to the best knowledge of the investigators, has found dasatinib to be safe and effective as first- or second-line therapy for these children.
Patients refractory to or intolerant of imatinib had a major cytogenetic response (MCyR) by 3 months and responses at 12 and 24 months exceeded 90%.
Newly diagnosed patients had a complete cytogenetic response (CCyR) by 6 months.
“We believe our data suggests that dasatinib could be considered as a new standard of care (SOC) for children with CML in chronic phase,” said study author Lia Gore, MD, of the University of Colorado School of Medicine/Children’s Hospital Colorado in Aurora.
She presented the findings of the study at the ASCO 2017 Annual Meeting (abstract 10511).
Study design
CA 180-226 is a phase 2, open-label, nonrandomized, prospective study conducted in 18 countries. Patients younger than 18 years with newly diagnosed CML-CP, or imatinib-resistant/intolerant (R/I) CML-CP, or CML in accelerated phase, or Ph+ acute lymphoblastic leukemia (ALL) were enrolled on the study between March 2009 and September 2014.
Dr Gore’s presentation focused on the CML-CP patients in the study, both the newly diagnosed and the imatinib-R/I patients.
The imatinib-R/I patients received dasatinib 60 mg/m2 tablets once daily, and the newly diagnosed patients received the same tablet dosage daily or a powdered formulation for oral suspension (PFOS) of dasatinib at 72 mg/m2 daily.
Dr Gore noted the different dosage in the oral suspension formulation is based on bioavailability studies performed in adults, which was determined to be equivalent to the 60 mg/m2 tablet formulation.
Once accrual was reached in the tablet cohort, newly diagnosed patients were accrued to the PFOS cohort. The patients on PFOS could switch to tablets after a year or more on the oral suspension.
Patients remained on treatment until disease progression, unacceptable toxicity occurred, or the patient/physician preference.
All patients had a minimum follow-up of 2 years. The longest follow-up was more than 90 months.
Primary objectives of the study were MCyR greater than 30% for imatinib-R/I patients and complete CCyR greater than 55% for newly diagnosed patients.
Secondary objectives included time to and duration of response, major molecular response (MMR), progression-free survival (PFS), overall survival (OS), and safety.
Baseline patient characteristics
One hundred thirteen patients were treated across the 3 cohorts—29 in the imatinib-R/I receiving tablets, 51 newly diagnosed patients receiving tablets, and 33 newly diagnosed patients in the PFOS arm. A total of 84 patients had newly diagnosed disease.
Of the 29 imatinib-R/I patients, 25 were resistant, 2 intolerant, and 2 undetermined. And 6 of the 25 resistant patients had defined imatinib-resistance mutations.
Median age was 13.8 years in the imatinib-R/I cohort, 12.9 years in the newly diagnosed on tablets, and 11.7 years in the PFOS group. Other baseline characteristics were similar among the cohorts.
“Importantly, there were 3 patients less than 2 years of age, and a substantial proportion of patients were actually less than 12 years of age,” Dr Gore pointed out.
Dasatinib exposure
The median duration of therapy was 50 months and 42 months in the R/I and newly diagnosed cohorts, respectively.
Forty-eight percent of the imatinib-R/I patients and 73% of the newly diagnosed patients are still on treatment. A relatively small number of patients discontinued therapy.
The median duration of therapy was shorter in the PFOS cohort because they were enrolled only after accrual to the tablet cohort. However, they were also followed up for more than 2 years.
Of the 33 patients on PFOS, 22 eventually switched to tablet formulation.
Results
The primary endpoint for imatinib-R/I patients—MCyR greater than 30%—was reached by 3 months, and MCyR at 12 and 24 months exceeded 90%. The median time to response was 3.1 months (range, 2.8 – 4.1), and median duration of response was not yet reached (range, 54.9 – not estimable).
For newly diagnosed patients, the preset defined rate of interest of 55% for CCyR was reached as early as 6 months, and exceeded 90% by 12 and 24 months.
Dr Gore pointed out that intolerant patients also reached CCyR relatively quickly, although it was not a specified endpoint.
Data indicate that responses occurred relatively quickly and continued to increase over time of follow-up.
MMR also continued to increase over time and showed no difference between formulation and response rate.
Median PFS has not been reached, as only 7 patients in each cohort had disease progression.
One imatinib-R/I patient died 1 year after stopping treatment. The patient, who had a GI bleed unrelated to dasatinib, had discontinued therapy for progressive disease with loss of MCyR.
Safety
Overall safety was very similar to the dasatinib exposure and experience in adults, Dr Gore said, and there were no differences in events between PFOS and tablets.
One patient in the PFOS cohort had a dasatinib-related grade 3 hypersensitivity reaction, which resolved after discontinuation of dasatinib.
“What’s important here,” she said, “is that there were almost no adverse events of severity in either cohort, only 1 in the imatinib refractory and intolerant and 1 in the newly diagnosed cohorts.”
“Most importantly for those of us with a lot of experience in this field,” she added, “there were no occurrences of pleural effusion, pericardial effusion, pulmonary edema, pulmonary hypertension, or any vascular occlusive events in patients noted on this trial.”
“Additionally, for pediatricians, we care a lot about what happens to growth in these patients and prospectively we collected a lot of data related to growth parameters in bone growth and development."
Of the dasatinib-related adverse events occurring in 10% or more of patients, there were only 5 growth and development events noted out of the 113 patients treated and all were grade 1 or 2 events, Dr Gore pointed out.
In the R/I cohort, one patient had osteopenia and gynecomastia. At the time of data analysis, this event had resolved even though the patient continued on dasatinib.
“We believe our data suggests that dasatinib could be considered as a new standard of care for children with CML in chronic phase,” she said.
“It includes the advantage of a liquid formulation as well as the advantages of once daily dosing and administration without regard to fed or fasting state,” she added, “which for all of us who treat children know could be quite important.”
The study was funded by Bristol-Myers Squibb. ![]()
CHICAGO—The largest ongoing and prospective trial of pediatric patients with chronic myeloid leukemia in chronic phase (CML-CP), according to the best knowledge of the investigators, has found dasatinib to be safe and effective as first- or second-line therapy for these children.
Patients refractory to or intolerant of imatinib had a major cytogenetic response (MCyR) by 3 months and responses at 12 and 24 months exceeded 90%.
Newly diagnosed patients had a complete cytogenetic response (CCyR) by 6 months.
“We believe our data suggests that dasatinib could be considered as a new standard of care (SOC) for children with CML in chronic phase,” said study author Lia Gore, MD, of the University of Colorado School of Medicine/Children’s Hospital Colorado in Aurora.
She presented the findings of the study at the ASCO 2017 Annual Meeting (abstract 10511).
Study design
CA 180-226 is a phase 2, open-label, nonrandomized, prospective study conducted in 18 countries. Patients younger than 18 years with newly diagnosed CML-CP, or imatinib-resistant/intolerant (R/I) CML-CP, or CML in accelerated phase, or Ph+ acute lymphoblastic leukemia (ALL) were enrolled on the study between March 2009 and September 2014.
Dr Gore’s presentation focused on the CML-CP patients in the study, both the newly diagnosed and the imatinib-R/I patients.
The imatinib-R/I patients received dasatinib 60 mg/m2 tablets once daily, and the newly diagnosed patients received the same tablet dosage daily or a powdered formulation for oral suspension (PFOS) of dasatinib at 72 mg/m2 daily.
Dr Gore noted the different dosage in the oral suspension formulation is based on bioavailability studies performed in adults, which was determined to be equivalent to the 60 mg/m2 tablet formulation.
Once accrual was reached in the tablet cohort, newly diagnosed patients were accrued to the PFOS cohort. The patients on PFOS could switch to tablets after a year or more on the oral suspension.
Patients remained on treatment until disease progression, unacceptable toxicity occurred, or the patient/physician preference.
All patients had a minimum follow-up of 2 years. The longest follow-up was more than 90 months.
Primary objectives of the study were MCyR greater than 30% for imatinib-R/I patients and complete CCyR greater than 55% for newly diagnosed patients.
Secondary objectives included time to and duration of response, major molecular response (MMR), progression-free survival (PFS), overall survival (OS), and safety.
Baseline patient characteristics
One hundred thirteen patients were treated across the 3 cohorts—29 in the imatinib-R/I receiving tablets, 51 newly diagnosed patients receiving tablets, and 33 newly diagnosed patients in the PFOS arm. A total of 84 patients had newly diagnosed disease.
Of the 29 imatinib-R/I patients, 25 were resistant, 2 intolerant, and 2 undetermined. And 6 of the 25 resistant patients had defined imatinib-resistance mutations.
Median age was 13.8 years in the imatinib-R/I cohort, 12.9 years in the newly diagnosed on tablets, and 11.7 years in the PFOS group. Other baseline characteristics were similar among the cohorts.
“Importantly, there were 3 patients less than 2 years of age, and a substantial proportion of patients were actually less than 12 years of age,” Dr Gore pointed out.
Dasatinib exposure
The median duration of therapy was 50 months and 42 months in the R/I and newly diagnosed cohorts, respectively.
Forty-eight percent of the imatinib-R/I patients and 73% of the newly diagnosed patients are still on treatment. A relatively small number of patients discontinued therapy.
The median duration of therapy was shorter in the PFOS cohort because they were enrolled only after accrual to the tablet cohort. However, they were also followed up for more than 2 years.
Of the 33 patients on PFOS, 22 eventually switched to tablet formulation.
Results
The primary endpoint for imatinib-R/I patients—MCyR greater than 30%—was reached by 3 months, and MCyR at 12 and 24 months exceeded 90%. The median time to response was 3.1 months (range, 2.8 – 4.1), and median duration of response was not yet reached (range, 54.9 – not estimable).
For newly diagnosed patients, the preset defined rate of interest of 55% for CCyR was reached as early as 6 months, and exceeded 90% by 12 and 24 months.
Dr Gore pointed out that intolerant patients also reached CCyR relatively quickly, although it was not a specified endpoint.
Data indicate that responses occurred relatively quickly and continued to increase over time of follow-up.
MMR also continued to increase over time and showed no difference between formulation and response rate.
Median PFS has not been reached, as only 7 patients in each cohort had disease progression.
One imatinib-R/I patient died 1 year after stopping treatment. The patient, who had a GI bleed unrelated to dasatinib, had discontinued therapy for progressive disease with loss of MCyR.
Safety
Overall safety was very similar to the dasatinib exposure and experience in adults, Dr Gore said, and there were no differences in events between PFOS and tablets.
One patient in the PFOS cohort had a dasatinib-related grade 3 hypersensitivity reaction, which resolved after discontinuation of dasatinib.
“What’s important here,” she said, “is that there were almost no adverse events of severity in either cohort, only 1 in the imatinib refractory and intolerant and 1 in the newly diagnosed cohorts.”
“Most importantly for those of us with a lot of experience in this field,” she added, “there were no occurrences of pleural effusion, pericardial effusion, pulmonary edema, pulmonary hypertension, or any vascular occlusive events in patients noted on this trial.”
“Additionally, for pediatricians, we care a lot about what happens to growth in these patients and prospectively we collected a lot of data related to growth parameters in bone growth and development."
Of the dasatinib-related adverse events occurring in 10% or more of patients, there were only 5 growth and development events noted out of the 113 patients treated and all were grade 1 or 2 events, Dr Gore pointed out.
In the R/I cohort, one patient had osteopenia and gynecomastia. At the time of data analysis, this event had resolved even though the patient continued on dasatinib.
“We believe our data suggests that dasatinib could be considered as a new standard of care for children with CML in chronic phase,” she said.
“It includes the advantage of a liquid formulation as well as the advantages of once daily dosing and administration without regard to fed or fasting state,” she added, “which for all of us who treat children know could be quite important.”
The study was funded by Bristol-Myers Squibb. ![]()
CHICAGO—The largest ongoing and prospective trial of pediatric patients with chronic myeloid leukemia in chronic phase (CML-CP), according to the best knowledge of the investigators, has found dasatinib to be safe and effective as first- or second-line therapy for these children.
Patients refractory to or intolerant of imatinib had a major cytogenetic response (MCyR) by 3 months and responses at 12 and 24 months exceeded 90%.
Newly diagnosed patients had a complete cytogenetic response (CCyR) by 6 months.
“We believe our data suggests that dasatinib could be considered as a new standard of care (SOC) for children with CML in chronic phase,” said study author Lia Gore, MD, of the University of Colorado School of Medicine/Children’s Hospital Colorado in Aurora.
She presented the findings of the study at the ASCO 2017 Annual Meeting (abstract 10511).
Study design
CA 180-226 is a phase 2, open-label, nonrandomized, prospective study conducted in 18 countries. Patients younger than 18 years with newly diagnosed CML-CP, or imatinib-resistant/intolerant (R/I) CML-CP, or CML in accelerated phase, or Ph+ acute lymphoblastic leukemia (ALL) were enrolled on the study between March 2009 and September 2014.
Dr Gore’s presentation focused on the CML-CP patients in the study, both the newly diagnosed and the imatinib-R/I patients.
The imatinib-R/I patients received dasatinib 60 mg/m2 tablets once daily, and the newly diagnosed patients received the same tablet dosage daily or a powdered formulation for oral suspension (PFOS) of dasatinib at 72 mg/m2 daily.
Dr Gore noted the different dosage in the oral suspension formulation is based on bioavailability studies performed in adults, which was determined to be equivalent to the 60 mg/m2 tablet formulation.
Once accrual was reached in the tablet cohort, newly diagnosed patients were accrued to the PFOS cohort. The patients on PFOS could switch to tablets after a year or more on the oral suspension.
Patients remained on treatment until disease progression, unacceptable toxicity occurred, or the patient/physician preference.
All patients had a minimum follow-up of 2 years. The longest follow-up was more than 90 months.
Primary objectives of the study were MCyR greater than 30% for imatinib-R/I patients and complete CCyR greater than 55% for newly diagnosed patients.
Secondary objectives included time to and duration of response, major molecular response (MMR), progression-free survival (PFS), overall survival (OS), and safety.
Baseline patient characteristics
One hundred thirteen patients were treated across the 3 cohorts—29 in the imatinib-R/I receiving tablets, 51 newly diagnosed patients receiving tablets, and 33 newly diagnosed patients in the PFOS arm. A total of 84 patients had newly diagnosed disease.
Of the 29 imatinib-R/I patients, 25 were resistant, 2 intolerant, and 2 undetermined. And 6 of the 25 resistant patients had defined imatinib-resistance mutations.
Median age was 13.8 years in the imatinib-R/I cohort, 12.9 years in the newly diagnosed on tablets, and 11.7 years in the PFOS group. Other baseline characteristics were similar among the cohorts.
“Importantly, there were 3 patients less than 2 years of age, and a substantial proportion of patients were actually less than 12 years of age,” Dr Gore pointed out.
Dasatinib exposure
The median duration of therapy was 50 months and 42 months in the R/I and newly diagnosed cohorts, respectively.
Forty-eight percent of the imatinib-R/I patients and 73% of the newly diagnosed patients are still on treatment. A relatively small number of patients discontinued therapy.
The median duration of therapy was shorter in the PFOS cohort because they were enrolled only after accrual to the tablet cohort. However, they were also followed up for more than 2 years.
Of the 33 patients on PFOS, 22 eventually switched to tablet formulation.
Results
The primary endpoint for imatinib-R/I patients—MCyR greater than 30%—was reached by 3 months, and MCyR at 12 and 24 months exceeded 90%. The median time to response was 3.1 months (range, 2.8 – 4.1), and median duration of response was not yet reached (range, 54.9 – not estimable).
For newly diagnosed patients, the preset defined rate of interest of 55% for CCyR was reached as early as 6 months, and exceeded 90% by 12 and 24 months.
Dr Gore pointed out that intolerant patients also reached CCyR relatively quickly, although it was not a specified endpoint.
Data indicate that responses occurred relatively quickly and continued to increase over time of follow-up.
MMR also continued to increase over time and showed no difference between formulation and response rate.
Median PFS has not been reached, as only 7 patients in each cohort had disease progression.
One imatinib-R/I patient died 1 year after stopping treatment. The patient, who had a GI bleed unrelated to dasatinib, had discontinued therapy for progressive disease with loss of MCyR.
Safety
Overall safety was very similar to the dasatinib exposure and experience in adults, Dr Gore said, and there were no differences in events between PFOS and tablets.
One patient in the PFOS cohort had a dasatinib-related grade 3 hypersensitivity reaction, which resolved after discontinuation of dasatinib.
“What’s important here,” she said, “is that there were almost no adverse events of severity in either cohort, only 1 in the imatinib refractory and intolerant and 1 in the newly diagnosed cohorts.”
“Most importantly for those of us with a lot of experience in this field,” she added, “there were no occurrences of pleural effusion, pericardial effusion, pulmonary edema, pulmonary hypertension, or any vascular occlusive events in patients noted on this trial.”
“Additionally, for pediatricians, we care a lot about what happens to growth in these patients and prospectively we collected a lot of data related to growth parameters in bone growth and development."
Of the dasatinib-related adverse events occurring in 10% or more of patients, there were only 5 growth and development events noted out of the 113 patients treated and all were grade 1 or 2 events, Dr Gore pointed out.
In the R/I cohort, one patient had osteopenia and gynecomastia. At the time of data analysis, this event had resolved even though the patient continued on dasatinib.
“We believe our data suggests that dasatinib could be considered as a new standard of care for children with CML in chronic phase,” she said.
“It includes the advantage of a liquid formulation as well as the advantages of once daily dosing and administration without regard to fed or fasting state,” she added, “which for all of us who treat children know could be quite important.”
The study was funded by Bristol-Myers Squibb.
Pembrolizumab enhances CAR T-cell persistence in relapsed ALL
CHICAGO—Three of 6 pediatric patients with relapsed or refractory acute lymphoblastic leukemia (ALL) whose CD19 chimeric antigen receptor (CAR) T cells did not persist even after reinfusion demonstrated persistence when the PD-1 checkpoint inhibitor pembrolizumab was added to the regimen.
CAR T cells can persist for months or even years and have the potential to mediate long-term disease control.
But some patients recover their normal B cells, which is a marker of loss of functional CAR T cells. These patients are at a higher risk of disease relapse.
Investigators, therefore, undertook a pilot study to determine whether PD-1 checkpoint pathway inhibition can improve CAR T persistence in these patients.
Shannon L. Maude, MD, of the Children’s Hospital of Philadelphia and the University of Pennsylvania Perelman School of Medicine, shared some of the patient cases in this pilot study at the ASCO 2017 Annual Meeting (Abstract 103*).
The investigators hypothesized that possible anti-murine immunogenicity could be causing poor CAR T-cell persistence, since the first CAR T developed used scFv domains of murine origin.
If T-cell exhaustion caused poor CAR T-cell persistence, immune checkpoints might play a role. In this case, combination with PD-1 checkpoint blockade could improve persistence, they hypothesized.
The investigators proposed to administer a repeat CAR T-cell infusion for relapsed or refractory ALL patients with poor persistence and add pembrolizumab after retreatment if the patients still had poor persistence. Patients were offered the option of another reinfusion prior to treatment with pembrolizumab if their CAR T-cell persistence continued to be poor.
Investigators added pembrolizumab no earlier than 14 days after infusion and only after patients recovered from cytokine release syndrome (CRS).
The infusions in the pilot study were humanized CART19 (huCART19, CTL119), unless otherwise specified.
Patient 1 – Pembrolizumab for partial response
This patient had no response to the prior murine CD19 CAR infusion, but responded well to infusion with huCART19 with good CAR T-cell proliferation.
By day 28, the patient had achieved a complete response (CR) in bone marrow but had a minimal residual disease (MRD) level of 1.2%.
At 7 weeks, the patient had a CD19+ relapse with low levels of huCART19.
Investigators added pembrolizumb on day 52 after infusion, and the patient had a modest increase in huCART19.
The patient had a temporary clearance of peripheral blasts followed by disease progression.
Patient 2 – Pembrolizumab for no response
This patient had a CD19+ relapse at 12 months after prior murine CD19 CAR infusion.
The patient was treated with huCART19, had good proliferation, but a rapid drop of CART19, and no response of the disease with a CD19+ relapse.
The patient had a reinfusion of huCART19 at 6 weeks and investigators added pembrolizumab on day 14 after reinfusion.
The patient experienced good huCART19 proliferation and prolonged persistence, but by day 28 had persistent disease, this time with decreased expression of CD19+ cells.
Patient 3 – Pembrolizumab for poor persistence
This patient had a CR with a prior murine CD19 CAR, but had poor persistence and early B cell recovery at 2 months.
The patient had a CD19+ relapse, was treated with huCART19, and had good proliferation again.
The patient achieved an MRD-negative CR, but because of short persistence and early B-cell recovery at 2 months, relapsed at 15 months.
The patient was reinfused at 17 months and pembrolizumab was added on day 14. The patient entered CR with prolonged persistence, but ultimately B-cell recovery occurred.
The patient was reinfused a third time, and pembrolizumab was added to the regimen every 3 weeks. The patient is experiencing prolonged persistence and continued B-cell aplasia.
Patient 4 – Pembrolizumab for poor persistence
This patient had achieved a CR with murine CAR19, but had a CD19+ relapse at 9 months. The patient then received huCART19, had good proliferation and achieved a CR, but had short persistence and B-cell recovery at 2 months.
The patient relapsed at 12 months and was reinfused with huCART19 at 14 months.
Pembrolizumab was added on day 14 after reinfusion, but the patient had no huCART19 proliferation, no response, and CD19+ MRD.
Patient 5 – Pembrolizumab for poor persistence
The patient responded to a prior murine CART19, but had a CD19+ relapse at 12 months.
The patient was infused with huCART19 and had good proliferation but short persistence. The patient was reinfused at 6 months, but again had short persistence.
The patient was reinfused again at 8 months because of B-cell recovery, and pembrolizumab was added on day 14 and administered every 3 weeks thereafter.
The patient is experiencing prolonged persistence and continued B-cell aplasia.
Patient 6 – Pembrolizumab for lymphomatous disease
This patient, who had widespread lymphadenopathy and M3 bone marrow, had not received prior CAR T cells and was treated for the first time with a murine CART19 for r/r ALL.
The patient had good expansion of CART19, but by day 28, PET scan showed widespread lymph node disease despite CR in the bone marrow.
The patient was given pembrolizumab on day 32 after infusion and every 2-3 weeks thereafter. After the addition of pembrolizumab, the patient had increased CART19 cells in blood and a significant decrease in PET-avid disease.
Summary
Three of six patients achieved objective clinical responses with pembrolizumab: 2 had prolonged B-cell aplasia, and another had a decrease in PET-avid lymphomatous disease.
The addition of pembrolizumab was also well tolerated by the patients, with minimal side effects of fever in 2 patients, cytopenias in 2 patients, and no instances of severe CRS.
The investigators believe the addition of checkpoint pathway inhibitors has the potential to prolong CAR T-cell persistence and warrants further investigation.
CHICAGO—Three of 6 pediatric patients with relapsed or refractory acute lymphoblastic leukemia (ALL) whose CD19 chimeric antigen receptor (CAR) T cells did not persist even after reinfusion demonstrated persistence when the PD-1 checkpoint inhibitor pembrolizumab was added to the regimen.
CAR T cells can persist for months or even years and have the potential to mediate long-term disease control.
But some patients recover their normal B cells, which is a marker of loss of functional CAR T cells. These patients are at a higher risk of disease relapse.
Investigators, therefore, undertook a pilot study to determine whether PD-1 checkpoint pathway inhibition can improve CAR T persistence in these patients.
Shannon L. Maude, MD, of the Children’s Hospital of Philadelphia and the University of Pennsylvania Perelman School of Medicine, shared some of the patient cases in this pilot study at the ASCO 2017 Annual Meeting (Abstract 103*).
The investigators hypothesized that possible anti-murine immunogenicity could be causing poor CAR T-cell persistence, since the first CAR T developed used scFv domains of murine origin.
If T-cell exhaustion caused poor CAR T-cell persistence, immune checkpoints might play a role. In this case, combination with PD-1 checkpoint blockade could improve persistence, they hypothesized.
The investigators proposed to administer a repeat CAR T-cell infusion for relapsed or refractory ALL patients with poor persistence and add pembrolizumab after retreatment if the patients still had poor persistence. Patients were offered the option of another reinfusion prior to treatment with pembrolizumab if their CAR T-cell persistence continued to be poor.
Investigators added pembrolizumab no earlier than 14 days after infusion and only after patients recovered from cytokine release syndrome (CRS).
The infusions in the pilot study were humanized CART19 (huCART19, CTL119), unless otherwise specified.
Patient 1 – Pembrolizumab for partial response
This patient had no response to the prior murine CD19 CAR infusion, but responded well to infusion with huCART19 with good CAR T-cell proliferation.
By day 28, the patient had achieved a complete response (CR) in bone marrow but had a minimal residual disease (MRD) level of 1.2%.
At 7 weeks, the patient had a CD19+ relapse with low levels of huCART19.
Investigators added pembrolizumb on day 52 after infusion, and the patient had a modest increase in huCART19.
The patient had a temporary clearance of peripheral blasts followed by disease progression.
Patient 2 – Pembrolizumab for no response
This patient had a CD19+ relapse at 12 months after prior murine CD19 CAR infusion.
The patient was treated with huCART19, had good proliferation, but a rapid drop of CART19, and no response of the disease with a CD19+ relapse.
The patient had a reinfusion of huCART19 at 6 weeks and investigators added pembrolizumab on day 14 after reinfusion.
The patient experienced good huCART19 proliferation and prolonged persistence, but by day 28 had persistent disease, this time with decreased expression of CD19+ cells.
Patient 3 – Pembrolizumab for poor persistence
This patient had a CR with a prior murine CD19 CAR, but had poor persistence and early B cell recovery at 2 months.
The patient had a CD19+ relapse, was treated with huCART19, and had good proliferation again.
The patient achieved an MRD-negative CR, but because of short persistence and early B-cell recovery at 2 months, relapsed at 15 months.
The patient was reinfused at 17 months and pembrolizumab was added on day 14. The patient entered CR with prolonged persistence, but ultimately B-cell recovery occurred.
The patient was reinfused a third time, and pembrolizumab was added to the regimen every 3 weeks. The patient is experiencing prolonged persistence and continued B-cell aplasia.
Patient 4 – Pembrolizumab for poor persistence
This patient had achieved a CR with murine CAR19, but had a CD19+ relapse at 9 months. The patient then received huCART19, had good proliferation and achieved a CR, but had short persistence and B-cell recovery at 2 months.
The patient relapsed at 12 months and was reinfused with huCART19 at 14 months.
Pembrolizumab was added on day 14 after reinfusion, but the patient had no huCART19 proliferation, no response, and CD19+ MRD.
Patient 5 – Pembrolizumab for poor persistence
The patient responded to a prior murine CART19, but had a CD19+ relapse at 12 months.
The patient was infused with huCART19 and had good proliferation but short persistence. The patient was reinfused at 6 months, but again had short persistence.
The patient was reinfused again at 8 months because of B-cell recovery, and pembrolizumab was added on day 14 and administered every 3 weeks thereafter.
The patient is experiencing prolonged persistence and continued B-cell aplasia.
Patient 6 – Pembrolizumab for lymphomatous disease
This patient, who had widespread lymphadenopathy and M3 bone marrow, had not received prior CAR T cells and was treated for the first time with a murine CART19 for r/r ALL.
The patient had good expansion of CART19, but by day 28, PET scan showed widespread lymph node disease despite CR in the bone marrow.
The patient was given pembrolizumab on day 32 after infusion and every 2-3 weeks thereafter. After the addition of pembrolizumab, the patient had increased CART19 cells in blood and a significant decrease in PET-avid disease.
Summary
Three of six patients achieved objective clinical responses with pembrolizumab: 2 had prolonged B-cell aplasia, and another had a decrease in PET-avid lymphomatous disease.
The addition of pembrolizumab was also well tolerated by the patients, with minimal side effects of fever in 2 patients, cytopenias in 2 patients, and no instances of severe CRS.
The investigators believe the addition of checkpoint pathway inhibitors has the potential to prolong CAR T-cell persistence and warrants further investigation.
CHICAGO—Three of 6 pediatric patients with relapsed or refractory acute lymphoblastic leukemia (ALL) whose CD19 chimeric antigen receptor (CAR) T cells did not persist even after reinfusion demonstrated persistence when the PD-1 checkpoint inhibitor pembrolizumab was added to the regimen.
CAR T cells can persist for months or even years and have the potential to mediate long-term disease control.
But some patients recover their normal B cells, which is a marker of loss of functional CAR T cells. These patients are at a higher risk of disease relapse.
Investigators, therefore, undertook a pilot study to determine whether PD-1 checkpoint pathway inhibition can improve CAR T persistence in these patients.
Shannon L. Maude, MD, of the Children’s Hospital of Philadelphia and the University of Pennsylvania Perelman School of Medicine, shared some of the patient cases in this pilot study at the ASCO 2017 Annual Meeting (Abstract 103*).
The investigators hypothesized that possible anti-murine immunogenicity could be causing poor CAR T-cell persistence, since the first CAR T developed used scFv domains of murine origin.
If T-cell exhaustion caused poor CAR T-cell persistence, immune checkpoints might play a role. In this case, combination with PD-1 checkpoint blockade could improve persistence, they hypothesized.
The investigators proposed to administer a repeat CAR T-cell infusion for relapsed or refractory ALL patients with poor persistence and add pembrolizumab after retreatment if the patients still had poor persistence. Patients were offered the option of another reinfusion prior to treatment with pembrolizumab if their CAR T-cell persistence continued to be poor.
Investigators added pembrolizumab no earlier than 14 days after infusion and only after patients recovered from cytokine release syndrome (CRS).
The infusions in the pilot study were humanized CART19 (huCART19, CTL119), unless otherwise specified.
Patient 1 – Pembrolizumab for partial response
This patient had no response to the prior murine CD19 CAR infusion, but responded well to infusion with huCART19 with good CAR T-cell proliferation.
By day 28, the patient had achieved a complete response (CR) in bone marrow but had a minimal residual disease (MRD) level of 1.2%.
At 7 weeks, the patient had a CD19+ relapse with low levels of huCART19.
Investigators added pembrolizumb on day 52 after infusion, and the patient had a modest increase in huCART19.
The patient had a temporary clearance of peripheral blasts followed by disease progression.
Patient 2 – Pembrolizumab for no response
This patient had a CD19+ relapse at 12 months after prior murine CD19 CAR infusion.
The patient was treated with huCART19, had good proliferation, but a rapid drop of CART19, and no response of the disease with a CD19+ relapse.
The patient had a reinfusion of huCART19 at 6 weeks and investigators added pembrolizumab on day 14 after reinfusion.
The patient experienced good huCART19 proliferation and prolonged persistence, but by day 28 had persistent disease, this time with decreased expression of CD19+ cells.
Patient 3 – Pembrolizumab for poor persistence
This patient had a CR with a prior murine CD19 CAR, but had poor persistence and early B cell recovery at 2 months.
The patient had a CD19+ relapse, was treated with huCART19, and had good proliferation again.
The patient achieved an MRD-negative CR, but because of short persistence and early B-cell recovery at 2 months, relapsed at 15 months.
The patient was reinfused at 17 months and pembrolizumab was added on day 14. The patient entered CR with prolonged persistence, but ultimately B-cell recovery occurred.
The patient was reinfused a third time, and pembrolizumab was added to the regimen every 3 weeks. The patient is experiencing prolonged persistence and continued B-cell aplasia.
Patient 4 – Pembrolizumab for poor persistence
This patient had achieved a CR with murine CAR19, but had a CD19+ relapse at 9 months. The patient then received huCART19, had good proliferation and achieved a CR, but had short persistence and B-cell recovery at 2 months.
The patient relapsed at 12 months and was reinfused with huCART19 at 14 months.
Pembrolizumab was added on day 14 after reinfusion, but the patient had no huCART19 proliferation, no response, and CD19+ MRD.
Patient 5 – Pembrolizumab for poor persistence
The patient responded to a prior murine CART19, but had a CD19+ relapse at 12 months.
The patient was infused with huCART19 and had good proliferation but short persistence. The patient was reinfused at 6 months, but again had short persistence.
The patient was reinfused again at 8 months because of B-cell recovery, and pembrolizumab was added on day 14 and administered every 3 weeks thereafter.
The patient is experiencing prolonged persistence and continued B-cell aplasia.
Patient 6 – Pembrolizumab for lymphomatous disease
This patient, who had widespread lymphadenopathy and M3 bone marrow, had not received prior CAR T cells and was treated for the first time with a murine CART19 for r/r ALL.
The patient had good expansion of CART19, but by day 28, PET scan showed widespread lymph node disease despite CR in the bone marrow.
The patient was given pembrolizumab on day 32 after infusion and every 2-3 weeks thereafter. After the addition of pembrolizumab, the patient had increased CART19 cells in blood and a significant decrease in PET-avid disease.
Summary
Three of six patients achieved objective clinical responses with pembrolizumab: 2 had prolonged B-cell aplasia, and another had a decrease in PET-avid lymphomatous disease.
The addition of pembrolizumab was also well tolerated by the patients, with minimal side effects of fever in 2 patients, cytopenias in 2 patients, and no instances of severe CRS.
The investigators believe the addition of checkpoint pathway inhibitors has the potential to prolong CAR T-cell persistence and warrants further investigation.