User login
Post-ibrutinib management in MCL unclear, speaker says

NEW YORK—Despite an “unprecedented” single-agent response rate and progression-free survival (PFS) in previously treated mantle cell lymphoma (MCL) patients, those with multiple risk factors have a dismal outcome following ibrutinib failure.
So after ibrutinib, what’s next in MCL? That was the question asked at Lymphoma & Myeloma 2015.
Peter Martin, MD, of Weill Cornell Medical College in New York, New York, discussed some possibilities.
Ibrutinib (Imbruvica) was approved by the US Food and Drug Administration for MCL based on the PCYC-1104 trial, which showed an overall response rate of 68%. In the MCL2001 trial, the overall response rate was 63%.
The median PFS for MCL was 13 months in PCYC-1104 and 10.5 months in MCL2001. The median overall survival (OS) was close to 2 years in PCYC-1104 and 18 months in MCL2001.
“So this is where I think it starts to get interesting,” Dr Martin said. “People were able to live for several months after progressing on ibrutinib. [However,] our experience at Cornell was not necessarily consistent with that.”
Cornell investigators, along with colleagues from Ohio State University, compiled data on patients who had been treated in clinical trials at their institutions and reviewed their survival after progression on ibrutinib. These patients had a median OS of 4 months.
In reviewing the patients’ Mantle Cell Lymphoma International Prognostic Index (MIPI) scores, Dr Martin said they were arguably a higher-risk population.
Dr Martin collected data on 114 relapsed/refractory MCL patients from centers around the world and found they had a lower response rate (50%) with ibrutinib overall and a lower duration of ibrutinib therapy (4.7 months).
The median OS after stopping ibrutinib was 2.9 months for the entire group. For patients who did not receive any subsequent therapy after failure, it was 0.8 months. Patients who received treatment after ibrutinib failure had a median OS of 5.8 months.
“And it didn’t seem to matter what we gave,” Dr Martin said. “Those treatments were pretty short-lived.”
The median time to next treatment with the first subsequent therapy was 2.4 months. These therapies included bendamustine, cytarabine, and lenalidomide.
“There was no statistical association between survival and choice of therapy,” Dr Martin said.
What was significant, by univariate Cox regression analysis, was the patients’ MIPI prior to ibrutinib therapy (P=0.0002) and the duration of ibrutinib treatment (P=0.0465).
“So at this point in time, I think it’s fair to say that there is insufficient data to recommend any specific treatment following ibrutinib failure,” Dr Martin said.
However, he did make a few suggestions for treating high-risk patients.
Treatment suggestions after ibrutinib failure
Dr Martin’s first suggestion is to focus on symptom management rather than active therapy for the older, frailer patients. Second, consider allogeneic stem cell transplant in any high-risk patient responding to ibrutinib.
Third, consider continuing ibrutinib therapy while starting the next therapy. And fourth, consider some form of continuous therapy that does not depend on TP53.
Dr Martin admitted that what to do following ibrutinib failure remains cloudy.
“Conducting a clinical trial will be tricky,” he said, “because the median time from ibrutinib failure to the next therapy was 9 days, and we’re targeting a very high-risk patient population.”
In addition, on average 80% have expression of Ki67.
Currently, a phase 2 trial of copanlisib (NCT02455297) is the only post-ibrutinib clinical trial in MCL open. Copanlisib is a potent and reversible phosphatidylinositol-3-kinase (PI3K) inhibitor with activity against both alpha and delta isoforms of PI3K. Preliminary results of the trial demonstrated response in 5 of 7 MCL patients.
So perhaps the best approach, Dr Martin suggested, would be to improve response and prevent relapse while on ibrutinib using combination therapies.
A phase 1/1b trial of ibrutinib with bendamustine and rituximab (BR) is underway. Of 17 MCL patients treated thus far, 94% have responded, and 76% have achieved a CR. But 25% developed grade 3 rash.
Ibrutinib is also being studied in combination with rituximab in MCL. The combination has produced an overall response rate of 88%, with 40% of patients achieving a CR.
“My interpretation from all these studies is you can probably add ibrutinib to any other effective anti-MCL therapy and improve that therapy,” Dr Martin said. “But there are questions, obviously, that still arise.”
Overcoming ibrutinib resistance
Dr Martin explained that, to use combinations rationally, we need to understand mechanisms of ibrutinib resistance, “and that’s not so straightforward.”
Mutations in MCL likely have multiple mechanisms of resistance. Mutations occur predominantly in 3 groups of genes involving NF-kB, PIM/mTOR, and epigenetic modifiers.
A number of trials are underway to hit some of these pathways, Dr Martin said.
Researchers at Cornell are studying ibrutinib plus palbociclib, an inhibitor of CDK4/CDK6 approved for advanced breast cancer, in a phase 1 trial of MCL patients.
The combination “very early on, has seen a high number of complete responses, which have been exciting,” Dr Martin said.
There are many ongoing ibrutinib trials in previously treated patients, including ones with carfilzomib, palbociclib, bortezomib, venetoclax, lenalidomide, and TGR-1202. In addition, the frontline trial of BR +/- ibrutinib is expected to have results soon.
“[A]nd once that happens, my guess is that this frontline trial, once it’s read out, essentially, makes all these other trials irrelevant because the minute ibrutinib moves into the frontline setting, it makes it very difficult to evaluate in a subsequent setting,” Dr Martin said. “So within a couple of years, it will be standard in the frontline setting.”
Dr Martin is concerned that resources are insufficient—there are too many studies, too few patients, and too little time—to find another, potentially more effective agent or combination.
He said there won’t be a one-size-fits-all approach to MCL either before or after ibrutinib, and collaboration among institutions, companies, and cooperative groups will be needed.
“Management in the post-ibrutinib setting remains unclear,” he said, “and these patients should be treated in a clinical trial if possible.” ![]()
NEW YORK—Despite an “unprecedented” single-agent response rate and progression-free survival (PFS) in previously treated mantle cell lymphoma (MCL) patients, those with multiple risk factors have a dismal outcome following ibrutinib failure.
So after ibrutinib, what’s next in MCL? That was the question asked at Lymphoma & Myeloma 2015.
Peter Martin, MD, of Weill Cornell Medical College in New York, New York, discussed some possibilities.
Ibrutinib (Imbruvica) was approved by the US Food and Drug Administration for MCL based on the PCYC-1104 trial, which showed an overall response rate of 68%. In the MCL2001 trial, the overall response rate was 63%.
The median PFS for MCL was 13 months in PCYC-1104 and 10.5 months in MCL2001. The median overall survival (OS) was close to 2 years in PCYC-1104 and 18 months in MCL2001.
“So this is where I think it starts to get interesting,” Dr Martin said. “People were able to live for several months after progressing on ibrutinib. [However,] our experience at Cornell was not necessarily consistent with that.”
Cornell investigators, along with colleagues from Ohio State University, compiled data on patients who had been treated in clinical trials at their institutions and reviewed their survival after progression on ibrutinib. These patients had a median OS of 4 months.
In reviewing the patients’ Mantle Cell Lymphoma International Prognostic Index (MIPI) scores, Dr Martin said they were arguably a higher-risk population.
Dr Martin collected data on 114 relapsed/refractory MCL patients from centers around the world and found they had a lower response rate (50%) with ibrutinib overall and a lower duration of ibrutinib therapy (4.7 months).
The median OS after stopping ibrutinib was 2.9 months for the entire group. For patients who did not receive any subsequent therapy after failure, it was 0.8 months. Patients who received treatment after ibrutinib failure had a median OS of 5.8 months.
“And it didn’t seem to matter what we gave,” Dr Martin said. “Those treatments were pretty short-lived.”
The median time to next treatment with the first subsequent therapy was 2.4 months. These therapies included bendamustine, cytarabine, and lenalidomide.
“There was no statistical association between survival and choice of therapy,” Dr Martin said.
What was significant, by univariate Cox regression analysis, was the patients’ MIPI prior to ibrutinib therapy (P=0.0002) and the duration of ibrutinib treatment (P=0.0465).
“So at this point in time, I think it’s fair to say that there is insufficient data to recommend any specific treatment following ibrutinib failure,” Dr Martin said.
However, he did make a few suggestions for treating high-risk patients.
Treatment suggestions after ibrutinib failure
Dr Martin’s first suggestion is to focus on symptom management rather than active therapy for the older, frailer patients. Second, consider allogeneic stem cell transplant in any high-risk patient responding to ibrutinib.
Third, consider continuing ibrutinib therapy while starting the next therapy. And fourth, consider some form of continuous therapy that does not depend on TP53.
Dr Martin admitted that what to do following ibrutinib failure remains cloudy.
“Conducting a clinical trial will be tricky,” he said, “because the median time from ibrutinib failure to the next therapy was 9 days, and we’re targeting a very high-risk patient population.”
In addition, on average 80% have expression of Ki67.
Currently, a phase 2 trial of copanlisib (NCT02455297) is the only post-ibrutinib clinical trial in MCL open. Copanlisib is a potent and reversible phosphatidylinositol-3-kinase (PI3K) inhibitor with activity against both alpha and delta isoforms of PI3K. Preliminary results of the trial demonstrated response in 5 of 7 MCL patients.
So perhaps the best approach, Dr Martin suggested, would be to improve response and prevent relapse while on ibrutinib using combination therapies.
A phase 1/1b trial of ibrutinib with bendamustine and rituximab (BR) is underway. Of 17 MCL patients treated thus far, 94% have responded, and 76% have achieved a CR. But 25% developed grade 3 rash.
Ibrutinib is also being studied in combination with rituximab in MCL. The combination has produced an overall response rate of 88%, with 40% of patients achieving a CR.
“My interpretation from all these studies is you can probably add ibrutinib to any other effective anti-MCL therapy and improve that therapy,” Dr Martin said. “But there are questions, obviously, that still arise.”
Overcoming ibrutinib resistance
Dr Martin explained that, to use combinations rationally, we need to understand mechanisms of ibrutinib resistance, “and that’s not so straightforward.”
Mutations in MCL likely have multiple mechanisms of resistance. Mutations occur predominantly in 3 groups of genes involving NF-kB, PIM/mTOR, and epigenetic modifiers.
A number of trials are underway to hit some of these pathways, Dr Martin said.
Researchers at Cornell are studying ibrutinib plus palbociclib, an inhibitor of CDK4/CDK6 approved for advanced breast cancer, in a phase 1 trial of MCL patients.
The combination “very early on, has seen a high number of complete responses, which have been exciting,” Dr Martin said.
There are many ongoing ibrutinib trials in previously treated patients, including ones with carfilzomib, palbociclib, bortezomib, venetoclax, lenalidomide, and TGR-1202. In addition, the frontline trial of BR +/- ibrutinib is expected to have results soon.
“[A]nd once that happens, my guess is that this frontline trial, once it’s read out, essentially, makes all these other trials irrelevant because the minute ibrutinib moves into the frontline setting, it makes it very difficult to evaluate in a subsequent setting,” Dr Martin said. “So within a couple of years, it will be standard in the frontline setting.”
Dr Martin is concerned that resources are insufficient—there are too many studies, too few patients, and too little time—to find another, potentially more effective agent or combination.
He said there won’t be a one-size-fits-all approach to MCL either before or after ibrutinib, and collaboration among institutions, companies, and cooperative groups will be needed.
“Management in the post-ibrutinib setting remains unclear,” he said, “and these patients should be treated in a clinical trial if possible.” ![]()
NEW YORK—Despite an “unprecedented” single-agent response rate and progression-free survival (PFS) in previously treated mantle cell lymphoma (MCL) patients, those with multiple risk factors have a dismal outcome following ibrutinib failure.
So after ibrutinib, what’s next in MCL? That was the question asked at Lymphoma & Myeloma 2015.
Peter Martin, MD, of Weill Cornell Medical College in New York, New York, discussed some possibilities.
Ibrutinib (Imbruvica) was approved by the US Food and Drug Administration for MCL based on the PCYC-1104 trial, which showed an overall response rate of 68%. In the MCL2001 trial, the overall response rate was 63%.
The median PFS for MCL was 13 months in PCYC-1104 and 10.5 months in MCL2001. The median overall survival (OS) was close to 2 years in PCYC-1104 and 18 months in MCL2001.
“So this is where I think it starts to get interesting,” Dr Martin said. “People were able to live for several months after progressing on ibrutinib. [However,] our experience at Cornell was not necessarily consistent with that.”
Cornell investigators, along with colleagues from Ohio State University, compiled data on patients who had been treated in clinical trials at their institutions and reviewed their survival after progression on ibrutinib. These patients had a median OS of 4 months.
In reviewing the patients’ Mantle Cell Lymphoma International Prognostic Index (MIPI) scores, Dr Martin said they were arguably a higher-risk population.
Dr Martin collected data on 114 relapsed/refractory MCL patients from centers around the world and found they had a lower response rate (50%) with ibrutinib overall and a lower duration of ibrutinib therapy (4.7 months).
The median OS after stopping ibrutinib was 2.9 months for the entire group. For patients who did not receive any subsequent therapy after failure, it was 0.8 months. Patients who received treatment after ibrutinib failure had a median OS of 5.8 months.
“And it didn’t seem to matter what we gave,” Dr Martin said. “Those treatments were pretty short-lived.”
The median time to next treatment with the first subsequent therapy was 2.4 months. These therapies included bendamustine, cytarabine, and lenalidomide.
“There was no statistical association between survival and choice of therapy,” Dr Martin said.
What was significant, by univariate Cox regression analysis, was the patients’ MIPI prior to ibrutinib therapy (P=0.0002) and the duration of ibrutinib treatment (P=0.0465).
“So at this point in time, I think it’s fair to say that there is insufficient data to recommend any specific treatment following ibrutinib failure,” Dr Martin said.
However, he did make a few suggestions for treating high-risk patients.
Treatment suggestions after ibrutinib failure
Dr Martin’s first suggestion is to focus on symptom management rather than active therapy for the older, frailer patients. Second, consider allogeneic stem cell transplant in any high-risk patient responding to ibrutinib.
Third, consider continuing ibrutinib therapy while starting the next therapy. And fourth, consider some form of continuous therapy that does not depend on TP53.
Dr Martin admitted that what to do following ibrutinib failure remains cloudy.
“Conducting a clinical trial will be tricky,” he said, “because the median time from ibrutinib failure to the next therapy was 9 days, and we’re targeting a very high-risk patient population.”
In addition, on average 80% have expression of Ki67.
Currently, a phase 2 trial of copanlisib (NCT02455297) is the only post-ibrutinib clinical trial in MCL open. Copanlisib is a potent and reversible phosphatidylinositol-3-kinase (PI3K) inhibitor with activity against both alpha and delta isoforms of PI3K. Preliminary results of the trial demonstrated response in 5 of 7 MCL patients.
So perhaps the best approach, Dr Martin suggested, would be to improve response and prevent relapse while on ibrutinib using combination therapies.
A phase 1/1b trial of ibrutinib with bendamustine and rituximab (BR) is underway. Of 17 MCL patients treated thus far, 94% have responded, and 76% have achieved a CR. But 25% developed grade 3 rash.
Ibrutinib is also being studied in combination with rituximab in MCL. The combination has produced an overall response rate of 88%, with 40% of patients achieving a CR.
“My interpretation from all these studies is you can probably add ibrutinib to any other effective anti-MCL therapy and improve that therapy,” Dr Martin said. “But there are questions, obviously, that still arise.”
Overcoming ibrutinib resistance
Dr Martin explained that, to use combinations rationally, we need to understand mechanisms of ibrutinib resistance, “and that’s not so straightforward.”
Mutations in MCL likely have multiple mechanisms of resistance. Mutations occur predominantly in 3 groups of genes involving NF-kB, PIM/mTOR, and epigenetic modifiers.
A number of trials are underway to hit some of these pathways, Dr Martin said.
Researchers at Cornell are studying ibrutinib plus palbociclib, an inhibitor of CDK4/CDK6 approved for advanced breast cancer, in a phase 1 trial of MCL patients.
The combination “very early on, has seen a high number of complete responses, which have been exciting,” Dr Martin said.
There are many ongoing ibrutinib trials in previously treated patients, including ones with carfilzomib, palbociclib, bortezomib, venetoclax, lenalidomide, and TGR-1202. In addition, the frontline trial of BR +/- ibrutinib is expected to have results soon.
“[A]nd once that happens, my guess is that this frontline trial, once it’s read out, essentially, makes all these other trials irrelevant because the minute ibrutinib moves into the frontline setting, it makes it very difficult to evaluate in a subsequent setting,” Dr Martin said. “So within a couple of years, it will be standard in the frontline setting.”
Dr Martin is concerned that resources are insufficient—there are too many studies, too few patients, and too little time—to find another, potentially more effective agent or combination.
He said there won’t be a one-size-fits-all approach to MCL either before or after ibrutinib, and collaboration among institutions, companies, and cooperative groups will be needed.
“Management in the post-ibrutinib setting remains unclear,” he said, “and these patients should be treated in a clinical trial if possible.” ![]()
Reprogramming the immune system

showing Hodgkin lymphoma
NEW YORK—Using a 3-pronged approach to reprogram the immune system—inhibition of critical pathways, activation of others, and depletion of malignant cells—may be the best strategy to optimize immune function in B-cell lymphomas, according to Stephen M. Ansell, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“[A]ll told, there are multiple immunological barriers to an effective immune response,” Dr Ansell said at Lymphoma & Myeloma 2015.
“So the questions are how can you use an immune checkpoint approach to try and modulate this and improve the outcome.”
Dr Ansell discussed checkpoint inhibitors, immune signal activators, and the potential of combining the approaches in Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL) in a way that enhances rather than antagonizes their effects.
Blocking CTLA-4
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) functions as an immune checkpoint that downregulates the immune system. A receptor found on the surface of inhibitor T cells, it acts as an off switch when it binds to CD80 or CD86 on the surface of antigen-presenting cells.
Ipilimumab, an antibody that targets CTLA-4, has been approved by the US Food and Drug Administration for the treatment of melanoma and is in clinical trials for lung, bladder, and prostate cancer.
Investigators wanted to see whether it also works in lymphoma, so they conducted a phase 1 study in relapsed/refractory B-cell NHL.
Eighteen patients received 3 mg/kg of ipilimumab. Two patients responded, 1 with a complete response (CR) that lasted more than 31 months, and 1 with a partial response (PR) that lasted 19 months. In 5 of 16 patients (31%), T-cell proliferation to recall antigens increased more than 2-fold.
As Dr Ansell explained, “Immune response doesn’t always correlate directly with the clinical responses. So I think we really have a lot to learn about what is really a biomarker of efficacy.”
Ipilimumab was also evaluated to treat relapse after allogeneic hematopoietic stem cell transplantation (HSCT) in 29 patients with relapsed hematologic disease. Two patients with HL achieved a CR and 1 patient with mantle cell lymphoma achieved a PR.
The investigators observed that ipilimumab did not induce or exacerbate clinical graft-versus-host disease.
Blocking PD-1
Programmed cell death protein 1 (PD-1) is a surface receptor expressed on T cells and pro-B cells. PD-1 binds 2 ligands, PD-L1 and PD-L2.
PD-1 ligands are overexpressed in inflammatory environments and attenuate the immune response through PD-1 on immune effector cells. In addition, PD-L1 expressed on malignant cells or in the tumor microenvironment suppresses tumor-infiltrating lymphocyte activity.
Pidilizumab, a humanized monoclonal antibody that binds to PD-1, weakens the apoptotic processes in lymphocytes and augments the antitumor activities of NK cells.
Investigators conducted a phase 2 trial of pidilizumab in patients with diffuse large B-cell lymphoma (DLBCL) after autologous HSCT to modulate the immune system after a transplant.
The team treated 66 patients with the antibody. At 16 months, progression-free survival (PFS) was 72%. For the 24 high-risk patients who were PET-positive after salvage chemotherapy, the 16-month PFS was 70%.
“And I think that what was most interesting,” Dr Ansell said, when focusing on the 35 patients with measurable disease after transplant, pidilizumab produced a 51% response rate “even in patients that actually had active disease.”
When pidilizumab was combined with rituximab in another trial in patients with relapsed follicular lymphoma (FL), 19 of 29 evaluable patients (66%) achieved an objective response: 15 (52%) CRs and 4 (14%) PRs.
“You might say, ‘Who cares? That’s not that great,’” Dr Ansell said. “But I think what was pretty impressive is that 52% CR rate. And most of you who treat patients with rituximab would know that that’s quite surprising, suggesting that there may be additional benefit for the use of PD-1 blockade in this subset of patients.”
Nivolumab, another monoclonal antibody that blocks the PD-1 pathway, is being investigated in a number of lymphoid malignancies, including HL, DLBCL, and T-cell lymphomas.
In a phase 1 study of nivolumab in 81 patients with relapsed or refractory lymphoid malignancies, the best preliminary overall response has been in FL and DLBCL patients, with an objective response rate of 40% and 36%, respectively, including 1 PR and 3 PRs in each subtype.
“I think what is important,” Dr Ansell said, “is that the side effects, as expected, were mainly immune-mediated, not as dramatic as have been seen with other agents, and very similar to what has been seen in solid tumor studies.”
Dr Ansell pointed out that the response rates with nivolumab varied widely by histology, suggesting that “we have a lot to learn about why patients benefit and who exactly benefits.”
There were no responses in patients with multiple myeloma or primary mediastinal B-cell lymphoma, although many patients achieved stable disease.
“Hodgkin lymphoma was completely different,” Dr Ansell said, “and there were responses in virtually every patient.”
Of 23 patients treated with nivolumab, 20 responded—4 achieved a CR and 16 a PR—including patients who had failed autologous HSCT and brentuximab vedotin treatment. Eleven patients, including 2 with CRs, have an ongoing response, some approaching 2 years. So the responses have been durable, Dr Ansell noted.
Yet another PD-1 antibody, pembrolizumab, has prompted reduction in tumor burden in HL in all but 2 of 29 evaluable patients, including 6 CRs and 13 PRs. The median duration of response has not yet been reached, and the side effect profile was similar to what has been seen with nivolumab and in solid tumors.
Activating immune stimulatory signals
Another approach to boosting the immune system is to activate immune stimulatory signals, eg, CD27 and CD40, and get a benefit that way. Varlilumab (CDX-1127) is an unconjugated monoclonal antibody that binds CD27 and activates CD27-expressing T cells.
In a phase 1 trial of varlilumab in 24 lymphoma patients, investigators found no significant depletion in absolute lymphocyte counts, T cells, or B cells. “Not quite the same success story,” Dr Ansell said, with a response—a CR—in only 1 patient.
Investigators did observe, however, evidence of increased soluble CD27, a reduction of circulating Tregs, and the induction of pro-inflammatory cytokines.
And in a phase 1 study of the anti-CD40 monoclonal antibody dacetuzumab in recurrent NHL, “the response rate was disappointingly low,” Dr Ansell said.
Investigators observed 6 objective responses, including 1 CR and 5 PRs, and a decrease in tumor size in approximately one-third of the 50 patients treated.
The investigators of the subsequent phase 2 trial did not want to take the agent forward for further study, Dr Ansell noted, “but there are antibodies now being developed in this space that will hopefully be more effective and create a greater benefit.”
Optimizing immune function
Dr Ansell suggested there are 3 main approaches to treating patients. One is going directly after the malignant cells and depleting them. A second is to inhibit critical pathways that the malignant cell is dependent upon, “starving them, if you like.” The third way is to activate the immune system and thereby create a greater benefit for patients.
“[P]robably our best strategy is to use all 3 in a reprogram approach,” he said. “Because unless you target each one of these areas, the likelihood is that the other sides of the 3-legged stool will take over.”
“This is an encouraging and exciting time for immune checkpoints therapy and an encouraging and exciting time for immune therapies in general. I think this is really the new frontier in lymphomas.” ![]()
showing Hodgkin lymphoma
NEW YORK—Using a 3-pronged approach to reprogram the immune system—inhibition of critical pathways, activation of others, and depletion of malignant cells—may be the best strategy to optimize immune function in B-cell lymphomas, according to Stephen M. Ansell, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“[A]ll told, there are multiple immunological barriers to an effective immune response,” Dr Ansell said at Lymphoma & Myeloma 2015.
“So the questions are how can you use an immune checkpoint approach to try and modulate this and improve the outcome.”
Dr Ansell discussed checkpoint inhibitors, immune signal activators, and the potential of combining the approaches in Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL) in a way that enhances rather than antagonizes their effects.
Blocking CTLA-4
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) functions as an immune checkpoint that downregulates the immune system. A receptor found on the surface of inhibitor T cells, it acts as an off switch when it binds to CD80 or CD86 on the surface of antigen-presenting cells.
Ipilimumab, an antibody that targets CTLA-4, has been approved by the US Food and Drug Administration for the treatment of melanoma and is in clinical trials for lung, bladder, and prostate cancer.
Investigators wanted to see whether it also works in lymphoma, so they conducted a phase 1 study in relapsed/refractory B-cell NHL.
Eighteen patients received 3 mg/kg of ipilimumab. Two patients responded, 1 with a complete response (CR) that lasted more than 31 months, and 1 with a partial response (PR) that lasted 19 months. In 5 of 16 patients (31%), T-cell proliferation to recall antigens increased more than 2-fold.
As Dr Ansell explained, “Immune response doesn’t always correlate directly with the clinical responses. So I think we really have a lot to learn about what is really a biomarker of efficacy.”
Ipilimumab was also evaluated to treat relapse after allogeneic hematopoietic stem cell transplantation (HSCT) in 29 patients with relapsed hematologic disease. Two patients with HL achieved a CR and 1 patient with mantle cell lymphoma achieved a PR.
The investigators observed that ipilimumab did not induce or exacerbate clinical graft-versus-host disease.
Blocking PD-1
Programmed cell death protein 1 (PD-1) is a surface receptor expressed on T cells and pro-B cells. PD-1 binds 2 ligands, PD-L1 and PD-L2.
PD-1 ligands are overexpressed in inflammatory environments and attenuate the immune response through PD-1 on immune effector cells. In addition, PD-L1 expressed on malignant cells or in the tumor microenvironment suppresses tumor-infiltrating lymphocyte activity.
Pidilizumab, a humanized monoclonal antibody that binds to PD-1, weakens the apoptotic processes in lymphocytes and augments the antitumor activities of NK cells.
Investigators conducted a phase 2 trial of pidilizumab in patients with diffuse large B-cell lymphoma (DLBCL) after autologous HSCT to modulate the immune system after a transplant.
The team treated 66 patients with the antibody. At 16 months, progression-free survival (PFS) was 72%. For the 24 high-risk patients who were PET-positive after salvage chemotherapy, the 16-month PFS was 70%.
“And I think that what was most interesting,” Dr Ansell said, when focusing on the 35 patients with measurable disease after transplant, pidilizumab produced a 51% response rate “even in patients that actually had active disease.”
When pidilizumab was combined with rituximab in another trial in patients with relapsed follicular lymphoma (FL), 19 of 29 evaluable patients (66%) achieved an objective response: 15 (52%) CRs and 4 (14%) PRs.
“You might say, ‘Who cares? That’s not that great,’” Dr Ansell said. “But I think what was pretty impressive is that 52% CR rate. And most of you who treat patients with rituximab would know that that’s quite surprising, suggesting that there may be additional benefit for the use of PD-1 blockade in this subset of patients.”
Nivolumab, another monoclonal antibody that blocks the PD-1 pathway, is being investigated in a number of lymphoid malignancies, including HL, DLBCL, and T-cell lymphomas.
In a phase 1 study of nivolumab in 81 patients with relapsed or refractory lymphoid malignancies, the best preliminary overall response has been in FL and DLBCL patients, with an objective response rate of 40% and 36%, respectively, including 1 PR and 3 PRs in each subtype.
“I think what is important,” Dr Ansell said, “is that the side effects, as expected, were mainly immune-mediated, not as dramatic as have been seen with other agents, and very similar to what has been seen in solid tumor studies.”
Dr Ansell pointed out that the response rates with nivolumab varied widely by histology, suggesting that “we have a lot to learn about why patients benefit and who exactly benefits.”
There were no responses in patients with multiple myeloma or primary mediastinal B-cell lymphoma, although many patients achieved stable disease.
“Hodgkin lymphoma was completely different,” Dr Ansell said, “and there were responses in virtually every patient.”
Of 23 patients treated with nivolumab, 20 responded—4 achieved a CR and 16 a PR—including patients who had failed autologous HSCT and brentuximab vedotin treatment. Eleven patients, including 2 with CRs, have an ongoing response, some approaching 2 years. So the responses have been durable, Dr Ansell noted.
Yet another PD-1 antibody, pembrolizumab, has prompted reduction in tumor burden in HL in all but 2 of 29 evaluable patients, including 6 CRs and 13 PRs. The median duration of response has not yet been reached, and the side effect profile was similar to what has been seen with nivolumab and in solid tumors.
Activating immune stimulatory signals
Another approach to boosting the immune system is to activate immune stimulatory signals, eg, CD27 and CD40, and get a benefit that way. Varlilumab (CDX-1127) is an unconjugated monoclonal antibody that binds CD27 and activates CD27-expressing T cells.
In a phase 1 trial of varlilumab in 24 lymphoma patients, investigators found no significant depletion in absolute lymphocyte counts, T cells, or B cells. “Not quite the same success story,” Dr Ansell said, with a response—a CR—in only 1 patient.
Investigators did observe, however, evidence of increased soluble CD27, a reduction of circulating Tregs, and the induction of pro-inflammatory cytokines.
And in a phase 1 study of the anti-CD40 monoclonal antibody dacetuzumab in recurrent NHL, “the response rate was disappointingly low,” Dr Ansell said.
Investigators observed 6 objective responses, including 1 CR and 5 PRs, and a decrease in tumor size in approximately one-third of the 50 patients treated.
The investigators of the subsequent phase 2 trial did not want to take the agent forward for further study, Dr Ansell noted, “but there are antibodies now being developed in this space that will hopefully be more effective and create a greater benefit.”
Optimizing immune function
Dr Ansell suggested there are 3 main approaches to treating patients. One is going directly after the malignant cells and depleting them. A second is to inhibit critical pathways that the malignant cell is dependent upon, “starving them, if you like.” The third way is to activate the immune system and thereby create a greater benefit for patients.
“[P]robably our best strategy is to use all 3 in a reprogram approach,” he said. “Because unless you target each one of these areas, the likelihood is that the other sides of the 3-legged stool will take over.”
“This is an encouraging and exciting time for immune checkpoints therapy and an encouraging and exciting time for immune therapies in general. I think this is really the new frontier in lymphomas.” ![]()
showing Hodgkin lymphoma
NEW YORK—Using a 3-pronged approach to reprogram the immune system—inhibition of critical pathways, activation of others, and depletion of malignant cells—may be the best strategy to optimize immune function in B-cell lymphomas, according to Stephen M. Ansell, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“[A]ll told, there are multiple immunological barriers to an effective immune response,” Dr Ansell said at Lymphoma & Myeloma 2015.
“So the questions are how can you use an immune checkpoint approach to try and modulate this and improve the outcome.”
Dr Ansell discussed checkpoint inhibitors, immune signal activators, and the potential of combining the approaches in Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL) in a way that enhances rather than antagonizes their effects.
Blocking CTLA-4
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) functions as an immune checkpoint that downregulates the immune system. A receptor found on the surface of inhibitor T cells, it acts as an off switch when it binds to CD80 or CD86 on the surface of antigen-presenting cells.
Ipilimumab, an antibody that targets CTLA-4, has been approved by the US Food and Drug Administration for the treatment of melanoma and is in clinical trials for lung, bladder, and prostate cancer.
Investigators wanted to see whether it also works in lymphoma, so they conducted a phase 1 study in relapsed/refractory B-cell NHL.
Eighteen patients received 3 mg/kg of ipilimumab. Two patients responded, 1 with a complete response (CR) that lasted more than 31 months, and 1 with a partial response (PR) that lasted 19 months. In 5 of 16 patients (31%), T-cell proliferation to recall antigens increased more than 2-fold.
As Dr Ansell explained, “Immune response doesn’t always correlate directly with the clinical responses. So I think we really have a lot to learn about what is really a biomarker of efficacy.”
Ipilimumab was also evaluated to treat relapse after allogeneic hematopoietic stem cell transplantation (HSCT) in 29 patients with relapsed hematologic disease. Two patients with HL achieved a CR and 1 patient with mantle cell lymphoma achieved a PR.
The investigators observed that ipilimumab did not induce or exacerbate clinical graft-versus-host disease.
Blocking PD-1
Programmed cell death protein 1 (PD-1) is a surface receptor expressed on T cells and pro-B cells. PD-1 binds 2 ligands, PD-L1 and PD-L2.
PD-1 ligands are overexpressed in inflammatory environments and attenuate the immune response through PD-1 on immune effector cells. In addition, PD-L1 expressed on malignant cells or in the tumor microenvironment suppresses tumor-infiltrating lymphocyte activity.
Pidilizumab, a humanized monoclonal antibody that binds to PD-1, weakens the apoptotic processes in lymphocytes and augments the antitumor activities of NK cells.
Investigators conducted a phase 2 trial of pidilizumab in patients with diffuse large B-cell lymphoma (DLBCL) after autologous HSCT to modulate the immune system after a transplant.
The team treated 66 patients with the antibody. At 16 months, progression-free survival (PFS) was 72%. For the 24 high-risk patients who were PET-positive after salvage chemotherapy, the 16-month PFS was 70%.
“And I think that what was most interesting,” Dr Ansell said, when focusing on the 35 patients with measurable disease after transplant, pidilizumab produced a 51% response rate “even in patients that actually had active disease.”
When pidilizumab was combined with rituximab in another trial in patients with relapsed follicular lymphoma (FL), 19 of 29 evaluable patients (66%) achieved an objective response: 15 (52%) CRs and 4 (14%) PRs.
“You might say, ‘Who cares? That’s not that great,’” Dr Ansell said. “But I think what was pretty impressive is that 52% CR rate. And most of you who treat patients with rituximab would know that that’s quite surprising, suggesting that there may be additional benefit for the use of PD-1 blockade in this subset of patients.”
Nivolumab, another monoclonal antibody that blocks the PD-1 pathway, is being investigated in a number of lymphoid malignancies, including HL, DLBCL, and T-cell lymphomas.
In a phase 1 study of nivolumab in 81 patients with relapsed or refractory lymphoid malignancies, the best preliminary overall response has been in FL and DLBCL patients, with an objective response rate of 40% and 36%, respectively, including 1 PR and 3 PRs in each subtype.
“I think what is important,” Dr Ansell said, “is that the side effects, as expected, were mainly immune-mediated, not as dramatic as have been seen with other agents, and very similar to what has been seen in solid tumor studies.”
Dr Ansell pointed out that the response rates with nivolumab varied widely by histology, suggesting that “we have a lot to learn about why patients benefit and who exactly benefits.”
There were no responses in patients with multiple myeloma or primary mediastinal B-cell lymphoma, although many patients achieved stable disease.
“Hodgkin lymphoma was completely different,” Dr Ansell said, “and there were responses in virtually every patient.”
Of 23 patients treated with nivolumab, 20 responded—4 achieved a CR and 16 a PR—including patients who had failed autologous HSCT and brentuximab vedotin treatment. Eleven patients, including 2 with CRs, have an ongoing response, some approaching 2 years. So the responses have been durable, Dr Ansell noted.
Yet another PD-1 antibody, pembrolizumab, has prompted reduction in tumor burden in HL in all but 2 of 29 evaluable patients, including 6 CRs and 13 PRs. The median duration of response has not yet been reached, and the side effect profile was similar to what has been seen with nivolumab and in solid tumors.
Activating immune stimulatory signals
Another approach to boosting the immune system is to activate immune stimulatory signals, eg, CD27 and CD40, and get a benefit that way. Varlilumab (CDX-1127) is an unconjugated monoclonal antibody that binds CD27 and activates CD27-expressing T cells.
In a phase 1 trial of varlilumab in 24 lymphoma patients, investigators found no significant depletion in absolute lymphocyte counts, T cells, or B cells. “Not quite the same success story,” Dr Ansell said, with a response—a CR—in only 1 patient.
Investigators did observe, however, evidence of increased soluble CD27, a reduction of circulating Tregs, and the induction of pro-inflammatory cytokines.
And in a phase 1 study of the anti-CD40 monoclonal antibody dacetuzumab in recurrent NHL, “the response rate was disappointingly low,” Dr Ansell said.
Investigators observed 6 objective responses, including 1 CR and 5 PRs, and a decrease in tumor size in approximately one-third of the 50 patients treated.
The investigators of the subsequent phase 2 trial did not want to take the agent forward for further study, Dr Ansell noted, “but there are antibodies now being developed in this space that will hopefully be more effective and create a greater benefit.”
Optimizing immune function
Dr Ansell suggested there are 3 main approaches to treating patients. One is going directly after the malignant cells and depleting them. A second is to inhibit critical pathways that the malignant cell is dependent upon, “starving them, if you like.” The third way is to activate the immune system and thereby create a greater benefit for patients.
“[P]robably our best strategy is to use all 3 in a reprogram approach,” he said. “Because unless you target each one of these areas, the likelihood is that the other sides of the 3-legged stool will take over.”
“This is an encouraging and exciting time for immune checkpoints therapy and an encouraging and exciting time for immune therapies in general. I think this is really the new frontier in lymphomas.” ![]()
Median DOR, PFS not yet reached for ibrutinib in CLL

Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
The search continues for additional targets in CLL
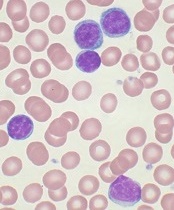
showing CLL
Image by Mary Ann Thompson
NEW YORK—Despite enormous advances in therapies for chronic lymphocytic leukemia (CLL) that target the B-cell receptor (BCR) signaling pathway, there is still room for improvement, according to investigators at the Mayo Clinic.
Bruton’s tyrosine kinase (BTK) and phosphoinositide-3 kinase delta (PI3Kδ) inhibitors are major players in mediating BCR signaling, yet both have off-target effects.
“These agents are not curative, and resistance develops,” explained Neil Kay, MD, of the Mayo Clinic in Rochester, Minnesota.
He described the Axl receptor tyrosine kinase and its inhibitor, TP-0903, at Lymphoma & Myeloma 2015, and discussed how it may be another promising target for CLL therapy.
Pros and cons of current therapies
Ibrutinib, a potent, irreversible, covalent inhibitor of BTK, inhibits interleukin 2 kinase, an essential enzyme in Th2 T cells.
“The potential benefit of this,” Dr Kay said, “is it shifts the balance between Th1 and Th2 T cells and potentially enhances antitumor immune responses.”
However, ibrutinib may cause off-target effects, including defects of platelet function, atrial fibrillation probably related to the inhibition of cardiac PI3K-AKT signaling, and decreased efficacy of anti-CD20 antibodies as mediated by natural killer cells.
In addition, long-term exposure to ibrutinib can induce signal mutations associated with resistance.
Idelalisib, a reversible inhibitor of the delta isoform of PI3K, modulates malignant B-cell signaling and inhibits the chemokines CCL3 and 4. It does not inhibit antibody-dependent cell-mediated cytotoxicity mediated by anti-CD20 antibodies, and it may stimulate antitumor responses.
However, idelalisib may also increase the incidence of diarrhea and colitis and can cause transaminitis and pneumonitis.
Axl receptor tyrosine kinase
So Dr Kay and fellow researchers explored whether CLL B cells express other active receptor tyrosine kinases (RTKs), and if so, what their functional implication is in CLL B-cell survival.
And what they detected is active Axl RTK and basic fibroblast growth factor receptor 3 (FGFR3) RTK in CLL B cells. The human Axl gene, a member of the TAM family of RTKs, is located on chromosome 19q13.2 and encodes a protein weighing 100–140 kD.
When Axl is activated, it initiates various signaling pathways, including cell survival, proliferation, apoptosis inhibition, migration, cell adhesion, and cytokine production. It does this through interactions with a number of signaling molecules, including PI3K and phospholipase C γ2 (PLCγ2), among others.
Most important, Axl is overexpressed in a number of human malignancies, including acute myeloid leukemia, and is associated with poor survivorship.
The research team immunoprecipitated Axl RTK from CLL B-cell lysates and examined them for co-localization by Western blot for the proteins PI3K, c-Src, Syk, PLCγ2, ZAP-70, and Lyn.
“What we think currently is that Axl provides a docking site for these key signaling molecules,” Dr Kay said.
The team found that, by inhibiting Axl with TP-0903, they were able to induce robust apoptosis in CLL B cells, including those from high-risk 17p– and 11q– CLL. They also observed that Axl inhibition resulted in significant reduction of the anti-apoptotic proteins Bcl-2, XIAP, and Mcl-1 and upregulation of the pro-apoptotic protein BIM in CLL B cells.
They then tested the BTK inhibitors ibrutinib and TP-4216 with and without the Axl inhibitor TP-0903 and found that concurrent administration of TP-0903 with the reversible BTK inhibitor TP-4216 had additive effects on apoptosis. This was not the case when it was added to ibrutinib.
“There was much more dramatic upregulation with the reversible BTK inhibitor,” Dr Kay emphasized.
He said Axl inhibition is a therapeutic opportunity in that these inhibitors are orally bioavailable and would be candidates for clinical testing. These inhibitors may minimize drug resistance and prevent the emergence of Richter’s transformation, he added.
The researchers received TP-0903 from Tolero Pharmaceuticals. ![]()
showing CLL
Image by Mary Ann Thompson
NEW YORK—Despite enormous advances in therapies for chronic lymphocytic leukemia (CLL) that target the B-cell receptor (BCR) signaling pathway, there is still room for improvement, according to investigators at the Mayo Clinic.
Bruton’s tyrosine kinase (BTK) and phosphoinositide-3 kinase delta (PI3Kδ) inhibitors are major players in mediating BCR signaling, yet both have off-target effects.
“These agents are not curative, and resistance develops,” explained Neil Kay, MD, of the Mayo Clinic in Rochester, Minnesota.
He described the Axl receptor tyrosine kinase and its inhibitor, TP-0903, at Lymphoma & Myeloma 2015, and discussed how it may be another promising target for CLL therapy.
Pros and cons of current therapies
Ibrutinib, a potent, irreversible, covalent inhibitor of BTK, inhibits interleukin 2 kinase, an essential enzyme in Th2 T cells.
“The potential benefit of this,” Dr Kay said, “is it shifts the balance between Th1 and Th2 T cells and potentially enhances antitumor immune responses.”
However, ibrutinib may cause off-target effects, including defects of platelet function, atrial fibrillation probably related to the inhibition of cardiac PI3K-AKT signaling, and decreased efficacy of anti-CD20 antibodies as mediated by natural killer cells.
In addition, long-term exposure to ibrutinib can induce signal mutations associated with resistance.
Idelalisib, a reversible inhibitor of the delta isoform of PI3K, modulates malignant B-cell signaling and inhibits the chemokines CCL3 and 4. It does not inhibit antibody-dependent cell-mediated cytotoxicity mediated by anti-CD20 antibodies, and it may stimulate antitumor responses.
However, idelalisib may also increase the incidence of diarrhea and colitis and can cause transaminitis and pneumonitis.
Axl receptor tyrosine kinase
So Dr Kay and fellow researchers explored whether CLL B cells express other active receptor tyrosine kinases (RTKs), and if so, what their functional implication is in CLL B-cell survival.
And what they detected is active Axl RTK and basic fibroblast growth factor receptor 3 (FGFR3) RTK in CLL B cells. The human Axl gene, a member of the TAM family of RTKs, is located on chromosome 19q13.2 and encodes a protein weighing 100–140 kD.
When Axl is activated, it initiates various signaling pathways, including cell survival, proliferation, apoptosis inhibition, migration, cell adhesion, and cytokine production. It does this through interactions with a number of signaling molecules, including PI3K and phospholipase C γ2 (PLCγ2), among others.
Most important, Axl is overexpressed in a number of human malignancies, including acute myeloid leukemia, and is associated with poor survivorship.
The research team immunoprecipitated Axl RTK from CLL B-cell lysates and examined them for co-localization by Western blot for the proteins PI3K, c-Src, Syk, PLCγ2, ZAP-70, and Lyn.
“What we think currently is that Axl provides a docking site for these key signaling molecules,” Dr Kay said.
The team found that, by inhibiting Axl with TP-0903, they were able to induce robust apoptosis in CLL B cells, including those from high-risk 17p– and 11q– CLL. They also observed that Axl inhibition resulted in significant reduction of the anti-apoptotic proteins Bcl-2, XIAP, and Mcl-1 and upregulation of the pro-apoptotic protein BIM in CLL B cells.
They then tested the BTK inhibitors ibrutinib and TP-4216 with and without the Axl inhibitor TP-0903 and found that concurrent administration of TP-0903 with the reversible BTK inhibitor TP-4216 had additive effects on apoptosis. This was not the case when it was added to ibrutinib.
“There was much more dramatic upregulation with the reversible BTK inhibitor,” Dr Kay emphasized.
He said Axl inhibition is a therapeutic opportunity in that these inhibitors are orally bioavailable and would be candidates for clinical testing. These inhibitors may minimize drug resistance and prevent the emergence of Richter’s transformation, he added.
The researchers received TP-0903 from Tolero Pharmaceuticals. ![]()
showing CLL
Image by Mary Ann Thompson
NEW YORK—Despite enormous advances in therapies for chronic lymphocytic leukemia (CLL) that target the B-cell receptor (BCR) signaling pathway, there is still room for improvement, according to investigators at the Mayo Clinic.
Bruton’s tyrosine kinase (BTK) and phosphoinositide-3 kinase delta (PI3Kδ) inhibitors are major players in mediating BCR signaling, yet both have off-target effects.
“These agents are not curative, and resistance develops,” explained Neil Kay, MD, of the Mayo Clinic in Rochester, Minnesota.
He described the Axl receptor tyrosine kinase and its inhibitor, TP-0903, at Lymphoma & Myeloma 2015, and discussed how it may be another promising target for CLL therapy.
Pros and cons of current therapies
Ibrutinib, a potent, irreversible, covalent inhibitor of BTK, inhibits interleukin 2 kinase, an essential enzyme in Th2 T cells.
“The potential benefit of this,” Dr Kay said, “is it shifts the balance between Th1 and Th2 T cells and potentially enhances antitumor immune responses.”
However, ibrutinib may cause off-target effects, including defects of platelet function, atrial fibrillation probably related to the inhibition of cardiac PI3K-AKT signaling, and decreased efficacy of anti-CD20 antibodies as mediated by natural killer cells.
In addition, long-term exposure to ibrutinib can induce signal mutations associated with resistance.
Idelalisib, a reversible inhibitor of the delta isoform of PI3K, modulates malignant B-cell signaling and inhibits the chemokines CCL3 and 4. It does not inhibit antibody-dependent cell-mediated cytotoxicity mediated by anti-CD20 antibodies, and it may stimulate antitumor responses.
However, idelalisib may also increase the incidence of diarrhea and colitis and can cause transaminitis and pneumonitis.
Axl receptor tyrosine kinase
So Dr Kay and fellow researchers explored whether CLL B cells express other active receptor tyrosine kinases (RTKs), and if so, what their functional implication is in CLL B-cell survival.
And what they detected is active Axl RTK and basic fibroblast growth factor receptor 3 (FGFR3) RTK in CLL B cells. The human Axl gene, a member of the TAM family of RTKs, is located on chromosome 19q13.2 and encodes a protein weighing 100–140 kD.
When Axl is activated, it initiates various signaling pathways, including cell survival, proliferation, apoptosis inhibition, migration, cell adhesion, and cytokine production. It does this through interactions with a number of signaling molecules, including PI3K and phospholipase C γ2 (PLCγ2), among others.
Most important, Axl is overexpressed in a number of human malignancies, including acute myeloid leukemia, and is associated with poor survivorship.
The research team immunoprecipitated Axl RTK from CLL B-cell lysates and examined them for co-localization by Western blot for the proteins PI3K, c-Src, Syk, PLCγ2, ZAP-70, and Lyn.
“What we think currently is that Axl provides a docking site for these key signaling molecules,” Dr Kay said.
The team found that, by inhibiting Axl with TP-0903, they were able to induce robust apoptosis in CLL B cells, including those from high-risk 17p– and 11q– CLL. They also observed that Axl inhibition resulted in significant reduction of the anti-apoptotic proteins Bcl-2, XIAP, and Mcl-1 and upregulation of the pro-apoptotic protein BIM in CLL B cells.
They then tested the BTK inhibitors ibrutinib and TP-4216 with and without the Axl inhibitor TP-0903 and found that concurrent administration of TP-0903 with the reversible BTK inhibitor TP-4216 had additive effects on apoptosis. This was not the case when it was added to ibrutinib.
“There was much more dramatic upregulation with the reversible BTK inhibitor,” Dr Kay emphasized.
He said Axl inhibition is a therapeutic opportunity in that these inhibitors are orally bioavailable and would be candidates for clinical testing. These inhibitors may minimize drug resistance and prevent the emergence of Richter’s transformation, he added.
The researchers received TP-0903 from Tolero Pharmaceuticals. ![]()
Ibrutinib may prove useful in MM, research shows

showing multiple myeloma
NEW YORK—Results of an open-label, phase 2, dose-escalation study of ibrutinib combined with low-dose dexamethasone suggest the Bruton’s tyrosine kinase (BTK) inhibitor may be useful in treating relapsed or relapsed and refractory patients with multiple myeloma (MM).
In the highest dose cohort, 23% of patients experienced a clinical benefit, which was defined as a minimal response or better by International Myeloma Working Group criteria.
“Ibrutinib is a remarkable agent and, through BTK inhibition, has been a game-changer in CLL [chronic lymphocytic leukemia],” said Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
Dr Richardson reported results with ibrutinib in MM at Lymphoma & Myeloma 2015.
The rationale for the use of ibrutinib in myeloma is the robust BTK expression in the plasma cells of MM patients as well as its regulation of myeloma “stemness” in the bone marrow.
Ibrutinib plus low-dose dexamethasone has previously demonstrated activity in myeloma at the highest dose tested in a phase 1 trial.
So investigators continued to evaluate ibrutinib in 4 dose cohorts, with and without dexamathesone, and enrolled 92 relapsed/refractory MM patients. Dr Richardson described the results of cohort 4, which met criteria for expansion as of March 2015.
The investigators enrolled 43 patients in cohort 4 to receive 840 mg daily of ibrutinib plus 40 mg of dexamethasone weekly. This dose of ibrutinib is double the dose approved by the US Food and Drug Administration (FDA) for treatment of CLL and Waldenström’s macroglobulinemia.
The patient population was “very typical” for a relapsed/refractory patient population, Dr Richardson noted. The median age was 65 years (range, 43–81), almost two-thirds were male, and they had a median of 4 (range, 2–10) prior therapies.
More than half of patients had an ECOG performance status of 0 or 1, and their median time since diagnosis was 6.5 years.
Nineteen percent had t(11;14), and other chromosomal abnormalities included del 13q14, t(4:14), and del 17p. Twenty-three patients were ISS stage I, 16 were ISS stage II, and 4 were ISS stage III.
Seventy-seven percent of patients had received an autologous stem cell transplant, 95% had prior alkylator treatment, 91% prior lenalidomide, 58% prior thalidomide, and 91% prior bortezomib treatment.
All patients were steroid-refractory. Thirty-five percent were refractory to alkylator treatment, 61% were refractory to lenalidomide, and 49% were refractory to bortezomib.
“And even a number of them had been exposed to pomalidomide and also to carfilzomib,” Dr Richardson said, “recognizing that these are the new and exciting drugs on the block, but, at the same time, a significant number of our patients already received those therapies.”
Five percent of patients achieved a partial response to ibrutinib and low-dose dexamethasone, and 18% achieved a minimal response, for a clinical benefit rate of 23%. Thirty percent had stable disease after 4 or more cycles.
“What was particularly striking,” Dr Richardson said, “was at the top dose, the 840-mg dose, the progression-free survival.”
The median time to disease progression was 5.4 months (range, 0.0–16.4), which the investigators thought was compelling in this large study.
Dr Richardson noted that there were no significant differences in safety observed across all dose cohorts, although 55% of patients experienced a grade 3 or greater treatment-emergent adverse event (AE), while 29% experienced at least 1 serious AE. And 10% of patients experienced peripheral neuropathy (PN), 8 of whom had a prior history of PN.
Treatment-emergent hematologic AEs of any grade in cohort 4 occurring in more than 20% of patients included anemia (23%) and thrombocytopenia (21%). Grade 3/4 anemia and thrombocytopenia occurred in 9% of patients in each category.
Treatment-emergent non-hematologic AEs of any grade in cohort 4 occurring in more than 20% of patients included diarrhea (63%), fatigue (49%), cough (26%), nausea (23%), and muscle spasms (21%). Two percent of patients had grade 3/4 diarrhea. There were no other grade 3/4 adverse events in the cohort.
Eighty-six percent of patients in cohort 4 discontinued treatment, 60% due to progressive disease, 12% due to an AE, and 2% at the discretion of the investigator. Twelve percent withdrew, were noncompliant, or required concomitant medication that was not permitted by the protocol.
Overall, Dr Richardson said, the safety profile was manageable, similar across the dosing cohorts, and consistent with those seen in CLL and Waldenström’s macroglobulinemia. The 840-mg dose did not increase toxicity and demonstrated activity in this heavily pretreated population.
“The real signal that struck us,” Dr Richardson emphasized, “was the progression-free survival at 5.4 months . . . for a 92-patient, multicenter experience, this is obviously, I think, an encouraging start.”
Investigators continue to explore ibrutinib in 2 ongoing combination studies, one with carfilzomib and dexamethasone (PCYC-1119) and another with pomalidomide and dexamethasone (PCYC-1138). Another trial with lenalidomide and dexamethasone is planned.
Ibrutinib now has 4 FDA-approved indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
showing multiple myeloma
NEW YORK—Results of an open-label, phase 2, dose-escalation study of ibrutinib combined with low-dose dexamethasone suggest the Bruton’s tyrosine kinase (BTK) inhibitor may be useful in treating relapsed or relapsed and refractory patients with multiple myeloma (MM).
In the highest dose cohort, 23% of patients experienced a clinical benefit, which was defined as a minimal response or better by International Myeloma Working Group criteria.
“Ibrutinib is a remarkable agent and, through BTK inhibition, has been a game-changer in CLL [chronic lymphocytic leukemia],” said Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
Dr Richardson reported results with ibrutinib in MM at Lymphoma & Myeloma 2015.
The rationale for the use of ibrutinib in myeloma is the robust BTK expression in the plasma cells of MM patients as well as its regulation of myeloma “stemness” in the bone marrow.
Ibrutinib plus low-dose dexamethasone has previously demonstrated activity in myeloma at the highest dose tested in a phase 1 trial.
So investigators continued to evaluate ibrutinib in 4 dose cohorts, with and without dexamathesone, and enrolled 92 relapsed/refractory MM patients. Dr Richardson described the results of cohort 4, which met criteria for expansion as of March 2015.
The investigators enrolled 43 patients in cohort 4 to receive 840 mg daily of ibrutinib plus 40 mg of dexamethasone weekly. This dose of ibrutinib is double the dose approved by the US Food and Drug Administration (FDA) for treatment of CLL and Waldenström’s macroglobulinemia.
The patient population was “very typical” for a relapsed/refractory patient population, Dr Richardson noted. The median age was 65 years (range, 43–81), almost two-thirds were male, and they had a median of 4 (range, 2–10) prior therapies.
More than half of patients had an ECOG performance status of 0 or 1, and their median time since diagnosis was 6.5 years.
Nineteen percent had t(11;14), and other chromosomal abnormalities included del 13q14, t(4:14), and del 17p. Twenty-three patients were ISS stage I, 16 were ISS stage II, and 4 were ISS stage III.
Seventy-seven percent of patients had received an autologous stem cell transplant, 95% had prior alkylator treatment, 91% prior lenalidomide, 58% prior thalidomide, and 91% prior bortezomib treatment.
All patients were steroid-refractory. Thirty-five percent were refractory to alkylator treatment, 61% were refractory to lenalidomide, and 49% were refractory to bortezomib.
“And even a number of them had been exposed to pomalidomide and also to carfilzomib,” Dr Richardson said, “recognizing that these are the new and exciting drugs on the block, but, at the same time, a significant number of our patients already received those therapies.”
Five percent of patients achieved a partial response to ibrutinib and low-dose dexamethasone, and 18% achieved a minimal response, for a clinical benefit rate of 23%. Thirty percent had stable disease after 4 or more cycles.
“What was particularly striking,” Dr Richardson said, “was at the top dose, the 840-mg dose, the progression-free survival.”
The median time to disease progression was 5.4 months (range, 0.0–16.4), which the investigators thought was compelling in this large study.
Dr Richardson noted that there were no significant differences in safety observed across all dose cohorts, although 55% of patients experienced a grade 3 or greater treatment-emergent adverse event (AE), while 29% experienced at least 1 serious AE. And 10% of patients experienced peripheral neuropathy (PN), 8 of whom had a prior history of PN.
Treatment-emergent hematologic AEs of any grade in cohort 4 occurring in more than 20% of patients included anemia (23%) and thrombocytopenia (21%). Grade 3/4 anemia and thrombocytopenia occurred in 9% of patients in each category.
Treatment-emergent non-hematologic AEs of any grade in cohort 4 occurring in more than 20% of patients included diarrhea (63%), fatigue (49%), cough (26%), nausea (23%), and muscle spasms (21%). Two percent of patients had grade 3/4 diarrhea. There were no other grade 3/4 adverse events in the cohort.
Eighty-six percent of patients in cohort 4 discontinued treatment, 60% due to progressive disease, 12% due to an AE, and 2% at the discretion of the investigator. Twelve percent withdrew, were noncompliant, or required concomitant medication that was not permitted by the protocol.
Overall, Dr Richardson said, the safety profile was manageable, similar across the dosing cohorts, and consistent with those seen in CLL and Waldenström’s macroglobulinemia. The 840-mg dose did not increase toxicity and demonstrated activity in this heavily pretreated population.
“The real signal that struck us,” Dr Richardson emphasized, “was the progression-free survival at 5.4 months . . . for a 92-patient, multicenter experience, this is obviously, I think, an encouraging start.”
Investigators continue to explore ibrutinib in 2 ongoing combination studies, one with carfilzomib and dexamethasone (PCYC-1119) and another with pomalidomide and dexamethasone (PCYC-1138). Another trial with lenalidomide and dexamethasone is planned.
Ibrutinib now has 4 FDA-approved indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
showing multiple myeloma
NEW YORK—Results of an open-label, phase 2, dose-escalation study of ibrutinib combined with low-dose dexamethasone suggest the Bruton’s tyrosine kinase (BTK) inhibitor may be useful in treating relapsed or relapsed and refractory patients with multiple myeloma (MM).
In the highest dose cohort, 23% of patients experienced a clinical benefit, which was defined as a minimal response or better by International Myeloma Working Group criteria.
“Ibrutinib is a remarkable agent and, through BTK inhibition, has been a game-changer in CLL [chronic lymphocytic leukemia],” said Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
Dr Richardson reported results with ibrutinib in MM at Lymphoma & Myeloma 2015.
The rationale for the use of ibrutinib in myeloma is the robust BTK expression in the plasma cells of MM patients as well as its regulation of myeloma “stemness” in the bone marrow.
Ibrutinib plus low-dose dexamethasone has previously demonstrated activity in myeloma at the highest dose tested in a phase 1 trial.
So investigators continued to evaluate ibrutinib in 4 dose cohorts, with and without dexamathesone, and enrolled 92 relapsed/refractory MM patients. Dr Richardson described the results of cohort 4, which met criteria for expansion as of March 2015.
The investigators enrolled 43 patients in cohort 4 to receive 840 mg daily of ibrutinib plus 40 mg of dexamethasone weekly. This dose of ibrutinib is double the dose approved by the US Food and Drug Administration (FDA) for treatment of CLL and Waldenström’s macroglobulinemia.
The patient population was “very typical” for a relapsed/refractory patient population, Dr Richardson noted. The median age was 65 years (range, 43–81), almost two-thirds were male, and they had a median of 4 (range, 2–10) prior therapies.
More than half of patients had an ECOG performance status of 0 or 1, and their median time since diagnosis was 6.5 years.
Nineteen percent had t(11;14), and other chromosomal abnormalities included del 13q14, t(4:14), and del 17p. Twenty-three patients were ISS stage I, 16 were ISS stage II, and 4 were ISS stage III.
Seventy-seven percent of patients had received an autologous stem cell transplant, 95% had prior alkylator treatment, 91% prior lenalidomide, 58% prior thalidomide, and 91% prior bortezomib treatment.
All patients were steroid-refractory. Thirty-five percent were refractory to alkylator treatment, 61% were refractory to lenalidomide, and 49% were refractory to bortezomib.
“And even a number of them had been exposed to pomalidomide and also to carfilzomib,” Dr Richardson said, “recognizing that these are the new and exciting drugs on the block, but, at the same time, a significant number of our patients already received those therapies.”
Five percent of patients achieved a partial response to ibrutinib and low-dose dexamethasone, and 18% achieved a minimal response, for a clinical benefit rate of 23%. Thirty percent had stable disease after 4 or more cycles.
“What was particularly striking,” Dr Richardson said, “was at the top dose, the 840-mg dose, the progression-free survival.”
The median time to disease progression was 5.4 months (range, 0.0–16.4), which the investigators thought was compelling in this large study.
Dr Richardson noted that there were no significant differences in safety observed across all dose cohorts, although 55% of patients experienced a grade 3 or greater treatment-emergent adverse event (AE), while 29% experienced at least 1 serious AE. And 10% of patients experienced peripheral neuropathy (PN), 8 of whom had a prior history of PN.
Treatment-emergent hematologic AEs of any grade in cohort 4 occurring in more than 20% of patients included anemia (23%) and thrombocytopenia (21%). Grade 3/4 anemia and thrombocytopenia occurred in 9% of patients in each category.
Treatment-emergent non-hematologic AEs of any grade in cohort 4 occurring in more than 20% of patients included diarrhea (63%), fatigue (49%), cough (26%), nausea (23%), and muscle spasms (21%). Two percent of patients had grade 3/4 diarrhea. There were no other grade 3/4 adverse events in the cohort.
Eighty-six percent of patients in cohort 4 discontinued treatment, 60% due to progressive disease, 12% due to an AE, and 2% at the discretion of the investigator. Twelve percent withdrew, were noncompliant, or required concomitant medication that was not permitted by the protocol.
Overall, Dr Richardson said, the safety profile was manageable, similar across the dosing cohorts, and consistent with those seen in CLL and Waldenström’s macroglobulinemia. The 840-mg dose did not increase toxicity and demonstrated activity in this heavily pretreated population.
“The real signal that struck us,” Dr Richardson emphasized, “was the progression-free survival at 5.4 months . . . for a 92-patient, multicenter experience, this is obviously, I think, an encouraging start.”
Investigators continue to explore ibrutinib in 2 ongoing combination studies, one with carfilzomib and dexamethasone (PCYC-1119) and another with pomalidomide and dexamethasone (PCYC-1138). Another trial with lenalidomide and dexamethasone is planned.
Ibrutinib now has 4 FDA-approved indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Treating EBV-associated lymphomas with VSTs
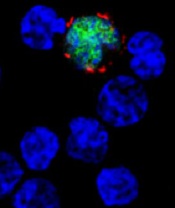
among uninfected cells (blue)
Image by Benjamin
Chaigne-Delalande
NEW YORK—Type 2 Epstein-Barr virus (EBV) tumors, such as Hodgkin and non-Hodgkin lymphomas, are challenging to treat with virus-specific T (VST) cells, according to researchers.
These lymphomas express a more restricted array of EBV antigens that are not particularly immunogenic.
Nevertheless, researchers are devising an approach using peptide mixtures to activate EBV VSTs for use in these patients.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described this work at the inaugural CRI-CIMT-EATI-AACR International Immunotherapy of Cancer Conference. She also described the researchers’ efforts to create off-the-shelf VSTs.
Dr Heslop explained that, in addition to the more restricted array of EBV antigens, EBV-associated tumors often produce inhibitory cytokines that can impede the activity of T cells.
So the researchers devised a strategy to expand these low-frequency clones by stimulating responding T cells with dendritic cells genetically modified with an ADV viral vector to overexpress LMP1 and LMP2. After multiple stimulations, they obtained an autologous product from the patient.
The team then tested the cytotoxic T cells in 21 patients with relapsed disease and in 29 patients as adjuvant therapy after stem cell transplant (n=14) or chemotherapy (n=15).
In the adjuvant arm, all patients but 1 remain in remission up to 5 years later.
In the relapsed arm, 11 had a complete response (CR), 2 had a partial response (PR), and 8 had progressive disease within 2 to 8 weeks.
“Importantly, there was no toxicity,” Dr Heslop said. “[A]ll were heavily pretreated with multiple lines of therapy for lymphoma, so I think the response rate is encouraging.”
An alternative approach: Pepmix-activated EBV VSTs
Although the antitumor effects of the above approach were encouraging, “we had a very complex manufacturing methodology that we didn’t think was sufficiently scaleable and robust for clinical studies,” Dr Heslop said.
“We also thought there would be potential regulatory issues with live EBV virus and the adenoviral vector,” she added.
And the researchers were concerned about the competition from the EBV/Ad-LMP dominant antigens.
So they devised an alternative approach using peptide mixture (pepmix)-activated EBVSTs.
This approach used autologous monocyte-derived dendritic cells as the antigen-presenting cells for the first stimulation.
The researchers pulsed them with overlapping peptides derived from 4 EBV antigens expressed in the tumors (EBV-LMP1, LMP2, EBNA1, and BARF1). They then expanded and opsonized the cells with IL-7 and IL-15.
For the second stimulation, the team used the T cells pulsed with the peptides and a K562 line pulsed with co-stimulatory molecules. And this process took 23 days, as opposed to the 2-3 months with the previous product.
The researchers have treated 9 patients with these EBVSTs as adjuvant therapy after autologous stem cell transplant. All patients remain in remission.
They also treated 6 patients with active disease. Two are in CR, 2 are in PR, and 2 have progressed.
This trial is ongoing, but the researchers believe that targeting the more challenging type 2 latency tumors with autologous cells can overcome T-cell anergy by using IL-7 and IL-15.
“Obviously, we need more numbers to know what the range of response is,” Dr Heslop said, although, at this early stage, it appears pepmix-activated T cells can produce antitumor responses. ![]()
among uninfected cells (blue)
Image by Benjamin
Chaigne-Delalande
NEW YORK—Type 2 Epstein-Barr virus (EBV) tumors, such as Hodgkin and non-Hodgkin lymphomas, are challenging to treat with virus-specific T (VST) cells, according to researchers.
These lymphomas express a more restricted array of EBV antigens that are not particularly immunogenic.
Nevertheless, researchers are devising an approach using peptide mixtures to activate EBV VSTs for use in these patients.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described this work at the inaugural CRI-CIMT-EATI-AACR International Immunotherapy of Cancer Conference. She also described the researchers’ efforts to create off-the-shelf VSTs.
Dr Heslop explained that, in addition to the more restricted array of EBV antigens, EBV-associated tumors often produce inhibitory cytokines that can impede the activity of T cells.
So the researchers devised a strategy to expand these low-frequency clones by stimulating responding T cells with dendritic cells genetically modified with an ADV viral vector to overexpress LMP1 and LMP2. After multiple stimulations, they obtained an autologous product from the patient.
The team then tested the cytotoxic T cells in 21 patients with relapsed disease and in 29 patients as adjuvant therapy after stem cell transplant (n=14) or chemotherapy (n=15).
In the adjuvant arm, all patients but 1 remain in remission up to 5 years later.
In the relapsed arm, 11 had a complete response (CR), 2 had a partial response (PR), and 8 had progressive disease within 2 to 8 weeks.
“Importantly, there was no toxicity,” Dr Heslop said. “[A]ll were heavily pretreated with multiple lines of therapy for lymphoma, so I think the response rate is encouraging.”
An alternative approach: Pepmix-activated EBV VSTs
Although the antitumor effects of the above approach were encouraging, “we had a very complex manufacturing methodology that we didn’t think was sufficiently scaleable and robust for clinical studies,” Dr Heslop said.
“We also thought there would be potential regulatory issues with live EBV virus and the adenoviral vector,” she added.
And the researchers were concerned about the competition from the EBV/Ad-LMP dominant antigens.
So they devised an alternative approach using peptide mixture (pepmix)-activated EBVSTs.
This approach used autologous monocyte-derived dendritic cells as the antigen-presenting cells for the first stimulation.
The researchers pulsed them with overlapping peptides derived from 4 EBV antigens expressed in the tumors (EBV-LMP1, LMP2, EBNA1, and BARF1). They then expanded and opsonized the cells with IL-7 and IL-15.
For the second stimulation, the team used the T cells pulsed with the peptides and a K562 line pulsed with co-stimulatory molecules. And this process took 23 days, as opposed to the 2-3 months with the previous product.
The researchers have treated 9 patients with these EBVSTs as adjuvant therapy after autologous stem cell transplant. All patients remain in remission.
They also treated 6 patients with active disease. Two are in CR, 2 are in PR, and 2 have progressed.
This trial is ongoing, but the researchers believe that targeting the more challenging type 2 latency tumors with autologous cells can overcome T-cell anergy by using IL-7 and IL-15.
“Obviously, we need more numbers to know what the range of response is,” Dr Heslop said, although, at this early stage, it appears pepmix-activated T cells can produce antitumor responses. ![]()
among uninfected cells (blue)
Image by Benjamin
Chaigne-Delalande
NEW YORK—Type 2 Epstein-Barr virus (EBV) tumors, such as Hodgkin and non-Hodgkin lymphomas, are challenging to treat with virus-specific T (VST) cells, according to researchers.
These lymphomas express a more restricted array of EBV antigens that are not particularly immunogenic.
Nevertheless, researchers are devising an approach using peptide mixtures to activate EBV VSTs for use in these patients.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described this work at the inaugural CRI-CIMT-EATI-AACR International Immunotherapy of Cancer Conference. She also described the researchers’ efforts to create off-the-shelf VSTs.
Dr Heslop explained that, in addition to the more restricted array of EBV antigens, EBV-associated tumors often produce inhibitory cytokines that can impede the activity of T cells.
So the researchers devised a strategy to expand these low-frequency clones by stimulating responding T cells with dendritic cells genetically modified with an ADV viral vector to overexpress LMP1 and LMP2. After multiple stimulations, they obtained an autologous product from the patient.
The team then tested the cytotoxic T cells in 21 patients with relapsed disease and in 29 patients as adjuvant therapy after stem cell transplant (n=14) or chemotherapy (n=15).
In the adjuvant arm, all patients but 1 remain in remission up to 5 years later.
In the relapsed arm, 11 had a complete response (CR), 2 had a partial response (PR), and 8 had progressive disease within 2 to 8 weeks.
“Importantly, there was no toxicity,” Dr Heslop said. “[A]ll were heavily pretreated with multiple lines of therapy for lymphoma, so I think the response rate is encouraging.”
An alternative approach: Pepmix-activated EBV VSTs
Although the antitumor effects of the above approach were encouraging, “we had a very complex manufacturing methodology that we didn’t think was sufficiently scaleable and robust for clinical studies,” Dr Heslop said.
“We also thought there would be potential regulatory issues with live EBV virus and the adenoviral vector,” she added.
And the researchers were concerned about the competition from the EBV/Ad-LMP dominant antigens.
So they devised an alternative approach using peptide mixture (pepmix)-activated EBVSTs.
This approach used autologous monocyte-derived dendritic cells as the antigen-presenting cells for the first stimulation.
The researchers pulsed them with overlapping peptides derived from 4 EBV antigens expressed in the tumors (EBV-LMP1, LMP2, EBNA1, and BARF1). They then expanded and opsonized the cells with IL-7 and IL-15.
For the second stimulation, the team used the T cells pulsed with the peptides and a K562 line pulsed with co-stimulatory molecules. And this process took 23 days, as opposed to the 2-3 months with the previous product.
The researchers have treated 9 patients with these EBVSTs as adjuvant therapy after autologous stem cell transplant. All patients remain in remission.
They also treated 6 patients with active disease. Two are in CR, 2 are in PR, and 2 have progressed.
This trial is ongoing, but the researchers believe that targeting the more challenging type 2 latency tumors with autologous cells can overcome T-cell anergy by using IL-7 and IL-15.
“Obviously, we need more numbers to know what the range of response is,” Dr Heslop said, although, at this early stage, it appears pepmix-activated T cells can produce antitumor responses. ![]()
Creating off-the-shelf VSTs

Photo courtesy of Baylor
College of Medicine
NEW YORK—Researchers are creating virus-specific T cells (VST) to treat and prevent viral infections in patients who undergo hematopoietic stem cell transplant.
Thus far, the group has modified T cells with 5 viral vectors—Epstein-Barr virus (EBV), cytomegalovirus (CMV), adenovirus (ADV), BK virus (BKV), and human herpesvirus 6 (HHV6)—and are devising methods whereby these VSTs can be made readily available, off-the-shelf products.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described the efforts of the Baylor research team at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
The team’s tri-virus and 5-virus VST approaches have been described previously. Here, we focus on the team’s efforts to create an off-the-shelf product.
A companion story describes the team’s VST approach to treating type 2 EBV-associated lymphomas.
Despite promising results with these earlier methodologies both as prophylaxis and treatment after stem cell transplant, problems existed with the approaches that would impede broader implementation of VSTs.
“Most of these initial methodologies were complex,” Dr Heslop said.
And even though the production time of the 5-virus VSTs is only 10 days, it does not allow for urgent use.
To solve this problem, the researchers are developing VSTs as off-the-shelf, third-party banked cells for patients who don’t have the time to wait for donor-specific cells to be made.
“The strategy here is that we make lines that are well-characterized but are HLA-restricted and tied to specific viruses,” Dr Heslop said.
The cells are then cryopreserved so that they’re available, and patients receive VSTs according to their HLA type and the line that has suitable activity for their infections.
“We initially evaluated this approach through a multicenter study sponsored by the NHLBI [National Heart, Lung, and Blood Institute],” Dr Heslop said.
Tri-virus approach
The researchers used the original tri-virus methodology, which was lengthy, “but, in this case, because the cells were already available, the time was not a major issue,” Dr Heslop said.
The team also made some new lines for donors with common alleles. In all, they had 32 lines available while the study was running.
They treated 50 patients: 23 received VSTs for CMV, 18 for ADV, and 9 for EBV.
One patient who had CMV colitis received the VSTs and had a complete response (CR), as evidenced by a normal endoscopy with no viral inclusions.
Another patient had EBV persistent after 6 doses of rituximab. One month after receiving the VST infusion, the patient had a CR, even though the cell line had only 1 class 2 allele. And the patient has sustained the CR for several years.
The overall response rate for the study is 74.0% at day 42 after infusion.
The response rate was not significantly different for each virus, Dr Heslop pointed out. Patients with CMV had a 73.9% response rate, those with EBV had a 66.7% response rate, and those with ADV had a 77.8% response rate.
“This is a little bit lower than with the donor-specific T cells, but I think it’s still a promising approach,” she added.
5-virus peptide mix
The researchers are now evaluating the more rapid, 5-virus peptide mix method for creating off-the-shelf VSTs.
This method replaced live viruses with overlapping peptide pools and added immunogenic antigens for 5 viruses, including BKV and HHV6. The process takes only 10 days to produce VSTs.
The team has identified a line for over 90% of the patients screened.
“That’s because we will accept a line that’s only matched at 1 HLA allele, as long as we have activity against the infecting virus through the shared allele,” Dr Heslop explained.
So they’re conducting a clinical trial using this method, and thus far, they have enrolled 22 patients. Sixteen patients have had 1 infusion, and 6 patients have had multiple infusions.
The 22 patients had 25 infections: 10 CMV, 10 BKV, 2 EBV, 2 ADV, and 1 HHV6.
The overall response rate is 88%. Nine of 10 responses in patients with BKV were partial because BK is difficult to clear from urine. However, the BK patients who had hemorrhagic cystitis all had symptomatic improvement.
Dr Heslop pointed out that these results are similar to those at other centers using EBV third-party T cells.
The initial third-party studies were done in Scotland and had a response rate of about 60%, which increased to around 80% in follow-up studies, where they characterized the T cells more extensively.
Both donor-specific and third-party VSTs have low toxicity, sustained response rates, and activity confirmed by studies in multiple centers.
Dr Heslop believes the rapid manufacturing methodologies will facilitate definitive clinical trials.
She said Cell Medica provided support for some of the trials with EBV tumors. ![]()
Photo courtesy of Baylor
College of Medicine
NEW YORK—Researchers are creating virus-specific T cells (VST) to treat and prevent viral infections in patients who undergo hematopoietic stem cell transplant.
Thus far, the group has modified T cells with 5 viral vectors—Epstein-Barr virus (EBV), cytomegalovirus (CMV), adenovirus (ADV), BK virus (BKV), and human herpesvirus 6 (HHV6)—and are devising methods whereby these VSTs can be made readily available, off-the-shelf products.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described the efforts of the Baylor research team at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
The team’s tri-virus and 5-virus VST approaches have been described previously. Here, we focus on the team’s efforts to create an off-the-shelf product.
A companion story describes the team’s VST approach to treating type 2 EBV-associated lymphomas.
Despite promising results with these earlier methodologies both as prophylaxis and treatment after stem cell transplant, problems existed with the approaches that would impede broader implementation of VSTs.
“Most of these initial methodologies were complex,” Dr Heslop said.
And even though the production time of the 5-virus VSTs is only 10 days, it does not allow for urgent use.
To solve this problem, the researchers are developing VSTs as off-the-shelf, third-party banked cells for patients who don’t have the time to wait for donor-specific cells to be made.
“The strategy here is that we make lines that are well-characterized but are HLA-restricted and tied to specific viruses,” Dr Heslop said.
The cells are then cryopreserved so that they’re available, and patients receive VSTs according to their HLA type and the line that has suitable activity for their infections.
“We initially evaluated this approach through a multicenter study sponsored by the NHLBI [National Heart, Lung, and Blood Institute],” Dr Heslop said.
Tri-virus approach
The researchers used the original tri-virus methodology, which was lengthy, “but, in this case, because the cells were already available, the time was not a major issue,” Dr Heslop said.
The team also made some new lines for donors with common alleles. In all, they had 32 lines available while the study was running.
They treated 50 patients: 23 received VSTs for CMV, 18 for ADV, and 9 for EBV.
One patient who had CMV colitis received the VSTs and had a complete response (CR), as evidenced by a normal endoscopy with no viral inclusions.
Another patient had EBV persistent after 6 doses of rituximab. One month after receiving the VST infusion, the patient had a CR, even though the cell line had only 1 class 2 allele. And the patient has sustained the CR for several years.
The overall response rate for the study is 74.0% at day 42 after infusion.
The response rate was not significantly different for each virus, Dr Heslop pointed out. Patients with CMV had a 73.9% response rate, those with EBV had a 66.7% response rate, and those with ADV had a 77.8% response rate.
“This is a little bit lower than with the donor-specific T cells, but I think it’s still a promising approach,” she added.
5-virus peptide mix
The researchers are now evaluating the more rapid, 5-virus peptide mix method for creating off-the-shelf VSTs.
This method replaced live viruses with overlapping peptide pools and added immunogenic antigens for 5 viruses, including BKV and HHV6. The process takes only 10 days to produce VSTs.
The team has identified a line for over 90% of the patients screened.
“That’s because we will accept a line that’s only matched at 1 HLA allele, as long as we have activity against the infecting virus through the shared allele,” Dr Heslop explained.
So they’re conducting a clinical trial using this method, and thus far, they have enrolled 22 patients. Sixteen patients have had 1 infusion, and 6 patients have had multiple infusions.
The 22 patients had 25 infections: 10 CMV, 10 BKV, 2 EBV, 2 ADV, and 1 HHV6.
The overall response rate is 88%. Nine of 10 responses in patients with BKV were partial because BK is difficult to clear from urine. However, the BK patients who had hemorrhagic cystitis all had symptomatic improvement.
Dr Heslop pointed out that these results are similar to those at other centers using EBV third-party T cells.
The initial third-party studies were done in Scotland and had a response rate of about 60%, which increased to around 80% in follow-up studies, where they characterized the T cells more extensively.
Both donor-specific and third-party VSTs have low toxicity, sustained response rates, and activity confirmed by studies in multiple centers.
Dr Heslop believes the rapid manufacturing methodologies will facilitate definitive clinical trials.
She said Cell Medica provided support for some of the trials with EBV tumors. ![]()
Photo courtesy of Baylor
College of Medicine
NEW YORK—Researchers are creating virus-specific T cells (VST) to treat and prevent viral infections in patients who undergo hematopoietic stem cell transplant.
Thus far, the group has modified T cells with 5 viral vectors—Epstein-Barr virus (EBV), cytomegalovirus (CMV), adenovirus (ADV), BK virus (BKV), and human herpesvirus 6 (HHV6)—and are devising methods whereby these VSTs can be made readily available, off-the-shelf products.
Helen Heslop, MD, of Baylor College of Medicine in Houston, Texas, described the efforts of the Baylor research team at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
The team’s tri-virus and 5-virus VST approaches have been described previously. Here, we focus on the team’s efforts to create an off-the-shelf product.
A companion story describes the team’s VST approach to treating type 2 EBV-associated lymphomas.
Despite promising results with these earlier methodologies both as prophylaxis and treatment after stem cell transplant, problems existed with the approaches that would impede broader implementation of VSTs.
“Most of these initial methodologies were complex,” Dr Heslop said.
And even though the production time of the 5-virus VSTs is only 10 days, it does not allow for urgent use.
To solve this problem, the researchers are developing VSTs as off-the-shelf, third-party banked cells for patients who don’t have the time to wait for donor-specific cells to be made.
“The strategy here is that we make lines that are well-characterized but are HLA-restricted and tied to specific viruses,” Dr Heslop said.
The cells are then cryopreserved so that they’re available, and patients receive VSTs according to their HLA type and the line that has suitable activity for their infections.
“We initially evaluated this approach through a multicenter study sponsored by the NHLBI [National Heart, Lung, and Blood Institute],” Dr Heslop said.
Tri-virus approach
The researchers used the original tri-virus methodology, which was lengthy, “but, in this case, because the cells were already available, the time was not a major issue,” Dr Heslop said.
The team also made some new lines for donors with common alleles. In all, they had 32 lines available while the study was running.
They treated 50 patients: 23 received VSTs for CMV, 18 for ADV, and 9 for EBV.
One patient who had CMV colitis received the VSTs and had a complete response (CR), as evidenced by a normal endoscopy with no viral inclusions.
Another patient had EBV persistent after 6 doses of rituximab. One month after receiving the VST infusion, the patient had a CR, even though the cell line had only 1 class 2 allele. And the patient has sustained the CR for several years.
The overall response rate for the study is 74.0% at day 42 after infusion.
The response rate was not significantly different for each virus, Dr Heslop pointed out. Patients with CMV had a 73.9% response rate, those with EBV had a 66.7% response rate, and those with ADV had a 77.8% response rate.
“This is a little bit lower than with the donor-specific T cells, but I think it’s still a promising approach,” she added.
5-virus peptide mix
The researchers are now evaluating the more rapid, 5-virus peptide mix method for creating off-the-shelf VSTs.
This method replaced live viruses with overlapping peptide pools and added immunogenic antigens for 5 viruses, including BKV and HHV6. The process takes only 10 days to produce VSTs.
The team has identified a line for over 90% of the patients screened.
“That’s because we will accept a line that’s only matched at 1 HLA allele, as long as we have activity against the infecting virus through the shared allele,” Dr Heslop explained.
So they’re conducting a clinical trial using this method, and thus far, they have enrolled 22 patients. Sixteen patients have had 1 infusion, and 6 patients have had multiple infusions.
The 22 patients had 25 infections: 10 CMV, 10 BKV, 2 EBV, 2 ADV, and 1 HHV6.
The overall response rate is 88%. Nine of 10 responses in patients with BKV were partial because BK is difficult to clear from urine. However, the BK patients who had hemorrhagic cystitis all had symptomatic improvement.
Dr Heslop pointed out that these results are similar to those at other centers using EBV third-party T cells.
The initial third-party studies were done in Scotland and had a response rate of about 60%, which increased to around 80% in follow-up studies, where they characterized the T cells more extensively.
Both donor-specific and third-party VSTs have low toxicity, sustained response rates, and activity confirmed by studies in multiple centers.
Dr Heslop believes the rapid manufacturing methodologies will facilitate definitive clinical trials.
She said Cell Medica provided support for some of the trials with EBV tumors.
New chimeric CD19 antibody may reduce MRD in ALL

NEW YORK—Researchers have developed a pharmaceutical-grade, third-generation, CD19-specific antibody that reduced minimal residual disease (MRD) in pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
This chimerized, Fc-optimized antibody—4G7SDIE—was used on a compassionate-need basis in 14 patients with relapsed or refractory BCP-ALL. Nine of the patients had prior stem cell transplants.
Ursula JE Seidel, a PhD candidate at University Children’s Hospital Tubingen in Germany, discussed early results with the new antibody (poster B144) during the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Patients received 4G7SDIE infusions ranging from 5 mg/m2 to 50 mg/m2 twice a week for a year or longer.
They rarely experienced fever, nausea, or headache, according to the investigators, and all had B-cell depletion.
“The good thing about this antibody is it has a very low toxicity profile,” Seidel noted.
Upon discontinuation of therapy, B-cell counts recovered rapidly to normal levels.
The researchers followed the patients for a median of 543 days after transplant (range, 208–1137) and a median of 720 days after administration of 4G7SDIE (range, 264–1115).
Nine of the 14 patients had a reduction in MRD by 1 log or more, 2 of whom were receiving additional therapy with tyrosine kinase inhibitors.
Five patients had a reduction in MRD below the quantifiable level, and 2 patients became MRD-negative.
Six patients relapsed, and 5 of them died from relapsed disease. Two patients died of sepsis or chemotoxicity while in complete molecular remission. And 6 patients remain in complete molecular remission.
Functional characterization of 4G7SDIE
Through analysis of cells from healthy volunteers and BCP-ALL blasts of untreated and treated patients, the researchers determined that 4G7SDIE mediates enhanced antibody‑dependent cellular cytotoxicity through its improved capability to recruit FcγRIIIa-bearing effector cells.
They identified natural killer cells and γδ T cells as the main effector cells. And they determined that the FcγRIIIa-V158F polymorphism did not influence the effect of 4G7SDIE-mediated antibody‑dependent cellular cytotoxicity.
The researchers believe that the promising anti-leukemic effects of 4G7SDIE both in vitro and in vivo call for additional exploration. They are currently planning a phase 1/2 study to further assess the therapeutic activity of 4G7SDIE.
NEW YORK—Researchers have developed a pharmaceutical-grade, third-generation, CD19-specific antibody that reduced minimal residual disease (MRD) in pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
This chimerized, Fc-optimized antibody—4G7SDIE—was used on a compassionate-need basis in 14 patients with relapsed or refractory BCP-ALL. Nine of the patients had prior stem cell transplants.
Ursula JE Seidel, a PhD candidate at University Children’s Hospital Tubingen in Germany, discussed early results with the new antibody (poster B144) during the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Patients received 4G7SDIE infusions ranging from 5 mg/m2 to 50 mg/m2 twice a week for a year or longer.
They rarely experienced fever, nausea, or headache, according to the investigators, and all had B-cell depletion.
“The good thing about this antibody is it has a very low toxicity profile,” Seidel noted.
Upon discontinuation of therapy, B-cell counts recovered rapidly to normal levels.
The researchers followed the patients for a median of 543 days after transplant (range, 208–1137) and a median of 720 days after administration of 4G7SDIE (range, 264–1115).
Nine of the 14 patients had a reduction in MRD by 1 log or more, 2 of whom were receiving additional therapy with tyrosine kinase inhibitors.
Five patients had a reduction in MRD below the quantifiable level, and 2 patients became MRD-negative.
Six patients relapsed, and 5 of them died from relapsed disease. Two patients died of sepsis or chemotoxicity while in complete molecular remission. And 6 patients remain in complete molecular remission.
Functional characterization of 4G7SDIE
Through analysis of cells from healthy volunteers and BCP-ALL blasts of untreated and treated patients, the researchers determined that 4G7SDIE mediates enhanced antibody‑dependent cellular cytotoxicity through its improved capability to recruit FcγRIIIa-bearing effector cells.
They identified natural killer cells and γδ T cells as the main effector cells. And they determined that the FcγRIIIa-V158F polymorphism did not influence the effect of 4G7SDIE-mediated antibody‑dependent cellular cytotoxicity.
The researchers believe that the promising anti-leukemic effects of 4G7SDIE both in vitro and in vivo call for additional exploration. They are currently planning a phase 1/2 study to further assess the therapeutic activity of 4G7SDIE.
NEW YORK—Researchers have developed a pharmaceutical-grade, third-generation, CD19-specific antibody that reduced minimal residual disease (MRD) in pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
This chimerized, Fc-optimized antibody—4G7SDIE—was used on a compassionate-need basis in 14 patients with relapsed or refractory BCP-ALL. Nine of the patients had prior stem cell transplants.
Ursula JE Seidel, a PhD candidate at University Children’s Hospital Tubingen in Germany, discussed early results with the new antibody (poster B144) during the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Patients received 4G7SDIE infusions ranging from 5 mg/m2 to 50 mg/m2 twice a week for a year or longer.
They rarely experienced fever, nausea, or headache, according to the investigators, and all had B-cell depletion.
“The good thing about this antibody is it has a very low toxicity profile,” Seidel noted.
Upon discontinuation of therapy, B-cell counts recovered rapidly to normal levels.
The researchers followed the patients for a median of 543 days after transplant (range, 208–1137) and a median of 720 days after administration of 4G7SDIE (range, 264–1115).
Nine of the 14 patients had a reduction in MRD by 1 log or more, 2 of whom were receiving additional therapy with tyrosine kinase inhibitors.
Five patients had a reduction in MRD below the quantifiable level, and 2 patients became MRD-negative.
Six patients relapsed, and 5 of them died from relapsed disease. Two patients died of sepsis or chemotoxicity while in complete molecular remission. And 6 patients remain in complete molecular remission.
Functional characterization of 4G7SDIE
Through analysis of cells from healthy volunteers and BCP-ALL blasts of untreated and treated patients, the researchers determined that 4G7SDIE mediates enhanced antibody‑dependent cellular cytotoxicity through its improved capability to recruit FcγRIIIa-bearing effector cells.
They identified natural killer cells and γδ T cells as the main effector cells. And they determined that the FcγRIIIa-V158F polymorphism did not influence the effect of 4G7SDIE-mediated antibody‑dependent cellular cytotoxicity.
The researchers believe that the promising anti-leukemic effects of 4G7SDIE both in vitro and in vivo call for additional exploration. They are currently planning a phase 1/2 study to further assess the therapeutic activity of 4G7SDIE.
Nonviral gene transfer of CARs tested in humans

Photo courtesy of MDACC
NEW YORK—Researchers have used a nonviral approach to create chimeric antigen receptor (CAR) T cells and tested these cells in safety trials.
Patients with advanced lymphoma or leukemia were infused with the nonvirally modified CD19-directed CAR T cells after autologous or allogeneic hematopoietic stem cell transplant (HSCT).
Eighty-six percent of autologous HSCT recipients were alive 24 months after infusion, and 53% of allogeneic HSCT recipients were alive with a median follow-up of 6.5 months.
“Gratifyingly, the patients have not demonstrated any acute or late toxicity to these CAR T-cell infusions,” said Laurence Cooper, MD, PhD, formerly of MD Anderson Cancer Center (MDACC) in Houston, Texas, and now with Ziopharm Oncology.
Dr Cooper presented these results at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Some of the technology he described was conducted at MDACC. Dr Cooper is currently a visiting scientist there and will continue to supervise the development of this technology.
Dr Cooper said the appeal of this nonviral approach, which is a modified Sleeping Beauty approach, “is it essentially avoids the complexity of making a virus, a lentivirus or a retrovirus, it can be done at quite low cost, and really allows for a nimbleness to this system.”
Using a simple blood draw of 200 cc of peripheral blood—the process does not require apheresis—the T cells can be expanded on a feeder cell layer and genetically reprogrammed.
Sleeping Beauty system
The researchers reprogrammed the T cells using a 2-plasmid Sleeping Beauty system, which is a transposon/transposase system.
The transposon DNA plasmid codes for the cargo load, which, in this case, is the CAR. At the same time, the transposase DNA plasmid is electroporated, “which is really the secret sauce of the transposition event,” Dr Cooper explained.
After electroporation, the transposon/transposase are co-cultured with K562-derived artificial antigen-presenting cells (aAPC) and expanded with the integrated transposon of K562-aAPC. In this case, CD19 is on the aAPC.
CD19 is co-expressed with other co-stimulatory molecules, CD86 and 4-1BB ligand.
In addition, the researchers added a molecule of interleukin 15 that’s sewn in frame to the Fc region of an immunoglobulin that then activates the T cell in the context of these co-stimulatory molecules.
The T cells that have stable integrants of the CAR grow out over time. And those that have transient expression of the CAR die by neglect.
“By day 14, most of the T cells have the CAR sewn into the genome and are stably expressed,” Dr Cooper said.
The CAR used for these safety trials at MDACC targets CD19 and uses mouse scFv held in frame with an immunoglobulin 4 Fc (IgG4Fc) stalk.
It’s tunneled through the T-cell membrane and has 2 costimulatory molecules, signal 1 delivered by phosphorylation of the immunoreceptor tyrosine-based activation motif in CD3ζ and signal 2 by the costimulatory domain CD28.
The researchers tested the CD19 CARs in 2 clinical settings—one with T cells that were patient-derived and infused after autologous HSCT, and the second with T cells that were derived from a third party and infused after allogeneic HSCT.
Infusion after autologous HSCT
The researchers first tried the CARs in 7 non-Hodgkin lymphoma patients who had an autologous HSCT. Their median age was 52 (range, 36-61).
Five patients received a starting CAR T-cell dose of 5x108 cells/m2, and 2 received 5x109 cells/m2.
Six patients (86%) remain alive and are in complete remission (CR) at a median follow-up of 24 months.
Infusion after allogeneic HSCT
The researchers expanded the investigation to a wider cohort of 19 patients who had undergone allogeneic HSCT.
Seventeen patients had advanced CD19-positive acute lymphoblastic leukemia, and 2 had non-Hodgkin lymphoma. Their median age was 35 (range, 21-56).
All patients were on graft-versus-host disease (GVHD) prophylaxis with tacrolimus at the time of CAR infusion. A subset of these allogeneic transplant patients had haploidentical donors rather than matched sibling donors.
Five patients received a CAR T-cell dose of 106, 6 patients received 107, 5 received 5x107, and 3 received 5x108 cells/m2 based on recipient body surface area.
Fifty-eight percent of patients (11/19) achieved a CR, and 10 remain alive a median of 6.5 months after CAR T-cell infusion.
Three patients developed GVHD, 1 with steroid-refractory acute liver disease, 1 with grade 2 acute skin disease, and 1 with chronic limited skin disease. The incidence of GVHD was lower than historical controls at MDACC, Dr Cooper said.
“[G]ratifyingly, in this clinical setting of minimal disease, patients did not have any acute or late toxicity from these infusions,” he added.
And the rate of cytomegalovirus reactivation after CAR T-cell infusion was 24%, compared with 41% for patients after transplant at MDACC without CAR T-cell infusion.
Eight patients received haploidentical HSCT followed by CAR T-cell infusion, and 75% (6/8) remain in CR.
Persistence of infused T cells
The researchers used 2 forms of PCR—qPCR and droplet PCR—to map the fate of the CARs.
“Roughly speaking, for these patients, and this is in line with the literature, in terms of those T cells that are activated through CD28 in contrast to 4-1BB, these T cells are, on average, living about 28 or so days post-infusion,” Dr Cooper noted.
He said this is similar to results observed with CARs being tested at the National Cancer Institute and Memorial Sloan-Kettering Cancer Center.
Photo courtesy of MDACC
NEW YORK—Researchers have used a nonviral approach to create chimeric antigen receptor (CAR) T cells and tested these cells in safety trials.
Patients with advanced lymphoma or leukemia were infused with the nonvirally modified CD19-directed CAR T cells after autologous or allogeneic hematopoietic stem cell transplant (HSCT).
Eighty-six percent of autologous HSCT recipients were alive 24 months after infusion, and 53% of allogeneic HSCT recipients were alive with a median follow-up of 6.5 months.
“Gratifyingly, the patients have not demonstrated any acute or late toxicity to these CAR T-cell infusions,” said Laurence Cooper, MD, PhD, formerly of MD Anderson Cancer Center (MDACC) in Houston, Texas, and now with Ziopharm Oncology.
Dr Cooper presented these results at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Some of the technology he described was conducted at MDACC. Dr Cooper is currently a visiting scientist there and will continue to supervise the development of this technology.
Dr Cooper said the appeal of this nonviral approach, which is a modified Sleeping Beauty approach, “is it essentially avoids the complexity of making a virus, a lentivirus or a retrovirus, it can be done at quite low cost, and really allows for a nimbleness to this system.”
Using a simple blood draw of 200 cc of peripheral blood—the process does not require apheresis—the T cells can be expanded on a feeder cell layer and genetically reprogrammed.
Sleeping Beauty system
The researchers reprogrammed the T cells using a 2-plasmid Sleeping Beauty system, which is a transposon/transposase system.
The transposon DNA plasmid codes for the cargo load, which, in this case, is the CAR. At the same time, the transposase DNA plasmid is electroporated, “which is really the secret sauce of the transposition event,” Dr Cooper explained.
After electroporation, the transposon/transposase are co-cultured with K562-derived artificial antigen-presenting cells (aAPC) and expanded with the integrated transposon of K562-aAPC. In this case, CD19 is on the aAPC.
CD19 is co-expressed with other co-stimulatory molecules, CD86 and 4-1BB ligand.
In addition, the researchers added a molecule of interleukin 15 that’s sewn in frame to the Fc region of an immunoglobulin that then activates the T cell in the context of these co-stimulatory molecules.
The T cells that have stable integrants of the CAR grow out over time. And those that have transient expression of the CAR die by neglect.
“By day 14, most of the T cells have the CAR sewn into the genome and are stably expressed,” Dr Cooper said.
The CAR used for these safety trials at MDACC targets CD19 and uses mouse scFv held in frame with an immunoglobulin 4 Fc (IgG4Fc) stalk.
It’s tunneled through the T-cell membrane and has 2 costimulatory molecules, signal 1 delivered by phosphorylation of the immunoreceptor tyrosine-based activation motif in CD3ζ and signal 2 by the costimulatory domain CD28.
The researchers tested the CD19 CARs in 2 clinical settings—one with T cells that were patient-derived and infused after autologous HSCT, and the second with T cells that were derived from a third party and infused after allogeneic HSCT.
Infusion after autologous HSCT
The researchers first tried the CARs in 7 non-Hodgkin lymphoma patients who had an autologous HSCT. Their median age was 52 (range, 36-61).
Five patients received a starting CAR T-cell dose of 5x108 cells/m2, and 2 received 5x109 cells/m2.
Six patients (86%) remain alive and are in complete remission (CR) at a median follow-up of 24 months.
Infusion after allogeneic HSCT
The researchers expanded the investigation to a wider cohort of 19 patients who had undergone allogeneic HSCT.
Seventeen patients had advanced CD19-positive acute lymphoblastic leukemia, and 2 had non-Hodgkin lymphoma. Their median age was 35 (range, 21-56).
All patients were on graft-versus-host disease (GVHD) prophylaxis with tacrolimus at the time of CAR infusion. A subset of these allogeneic transplant patients had haploidentical donors rather than matched sibling donors.
Five patients received a CAR T-cell dose of 106, 6 patients received 107, 5 received 5x107, and 3 received 5x108 cells/m2 based on recipient body surface area.
Fifty-eight percent of patients (11/19) achieved a CR, and 10 remain alive a median of 6.5 months after CAR T-cell infusion.
Three patients developed GVHD, 1 with steroid-refractory acute liver disease, 1 with grade 2 acute skin disease, and 1 with chronic limited skin disease. The incidence of GVHD was lower than historical controls at MDACC, Dr Cooper said.
“[G]ratifyingly, in this clinical setting of minimal disease, patients did not have any acute or late toxicity from these infusions,” he added.
And the rate of cytomegalovirus reactivation after CAR T-cell infusion was 24%, compared with 41% for patients after transplant at MDACC without CAR T-cell infusion.
Eight patients received haploidentical HSCT followed by CAR T-cell infusion, and 75% (6/8) remain in CR.
Persistence of infused T cells
The researchers used 2 forms of PCR—qPCR and droplet PCR—to map the fate of the CARs.
“Roughly speaking, for these patients, and this is in line with the literature, in terms of those T cells that are activated through CD28 in contrast to 4-1BB, these T cells are, on average, living about 28 or so days post-infusion,” Dr Cooper noted.
He said this is similar to results observed with CARs being tested at the National Cancer Institute and Memorial Sloan-Kettering Cancer Center.
Photo courtesy of MDACC
NEW YORK—Researchers have used a nonviral approach to create chimeric antigen receptor (CAR) T cells and tested these cells in safety trials.
Patients with advanced lymphoma or leukemia were infused with the nonvirally modified CD19-directed CAR T cells after autologous or allogeneic hematopoietic stem cell transplant (HSCT).
Eighty-six percent of autologous HSCT recipients were alive 24 months after infusion, and 53% of allogeneic HSCT recipients were alive with a median follow-up of 6.5 months.
“Gratifyingly, the patients have not demonstrated any acute or late toxicity to these CAR T-cell infusions,” said Laurence Cooper, MD, PhD, formerly of MD Anderson Cancer Center (MDACC) in Houston, Texas, and now with Ziopharm Oncology.
Dr Cooper presented these results at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Some of the technology he described was conducted at MDACC. Dr Cooper is currently a visiting scientist there and will continue to supervise the development of this technology.
Dr Cooper said the appeal of this nonviral approach, which is a modified Sleeping Beauty approach, “is it essentially avoids the complexity of making a virus, a lentivirus or a retrovirus, it can be done at quite low cost, and really allows for a nimbleness to this system.”
Using a simple blood draw of 200 cc of peripheral blood—the process does not require apheresis—the T cells can be expanded on a feeder cell layer and genetically reprogrammed.
Sleeping Beauty system
The researchers reprogrammed the T cells using a 2-plasmid Sleeping Beauty system, which is a transposon/transposase system.
The transposon DNA plasmid codes for the cargo load, which, in this case, is the CAR. At the same time, the transposase DNA plasmid is electroporated, “which is really the secret sauce of the transposition event,” Dr Cooper explained.
After electroporation, the transposon/transposase are co-cultured with K562-derived artificial antigen-presenting cells (aAPC) and expanded with the integrated transposon of K562-aAPC. In this case, CD19 is on the aAPC.
CD19 is co-expressed with other co-stimulatory molecules, CD86 and 4-1BB ligand.
In addition, the researchers added a molecule of interleukin 15 that’s sewn in frame to the Fc region of an immunoglobulin that then activates the T cell in the context of these co-stimulatory molecules.
The T cells that have stable integrants of the CAR grow out over time. And those that have transient expression of the CAR die by neglect.
“By day 14, most of the T cells have the CAR sewn into the genome and are stably expressed,” Dr Cooper said.
The CAR used for these safety trials at MDACC targets CD19 and uses mouse scFv held in frame with an immunoglobulin 4 Fc (IgG4Fc) stalk.
It’s tunneled through the T-cell membrane and has 2 costimulatory molecules, signal 1 delivered by phosphorylation of the immunoreceptor tyrosine-based activation motif in CD3ζ and signal 2 by the costimulatory domain CD28.
The researchers tested the CD19 CARs in 2 clinical settings—one with T cells that were patient-derived and infused after autologous HSCT, and the second with T cells that were derived from a third party and infused after allogeneic HSCT.
Infusion after autologous HSCT
The researchers first tried the CARs in 7 non-Hodgkin lymphoma patients who had an autologous HSCT. Their median age was 52 (range, 36-61).
Five patients received a starting CAR T-cell dose of 5x108 cells/m2, and 2 received 5x109 cells/m2.
Six patients (86%) remain alive and are in complete remission (CR) at a median follow-up of 24 months.
Infusion after allogeneic HSCT
The researchers expanded the investigation to a wider cohort of 19 patients who had undergone allogeneic HSCT.
Seventeen patients had advanced CD19-positive acute lymphoblastic leukemia, and 2 had non-Hodgkin lymphoma. Their median age was 35 (range, 21-56).
All patients were on graft-versus-host disease (GVHD) prophylaxis with tacrolimus at the time of CAR infusion. A subset of these allogeneic transplant patients had haploidentical donors rather than matched sibling donors.
Five patients received a CAR T-cell dose of 106, 6 patients received 107, 5 received 5x107, and 3 received 5x108 cells/m2 based on recipient body surface area.
Fifty-eight percent of patients (11/19) achieved a CR, and 10 remain alive a median of 6.5 months after CAR T-cell infusion.
Three patients developed GVHD, 1 with steroid-refractory acute liver disease, 1 with grade 2 acute skin disease, and 1 with chronic limited skin disease. The incidence of GVHD was lower than historical controls at MDACC, Dr Cooper said.
“[G]ratifyingly, in this clinical setting of minimal disease, patients did not have any acute or late toxicity from these infusions,” he added.
And the rate of cytomegalovirus reactivation after CAR T-cell infusion was 24%, compared with 41% for patients after transplant at MDACC without CAR T-cell infusion.
Eight patients received haploidentical HSCT followed by CAR T-cell infusion, and 75% (6/8) remain in CR.
Persistence of infused T cells
The researchers used 2 forms of PCR—qPCR and droplet PCR—to map the fate of the CARs.
“Roughly speaking, for these patients, and this is in line with the literature, in terms of those T cells that are activated through CD28 in contrast to 4-1BB, these T cells are, on average, living about 28 or so days post-infusion,” Dr Cooper noted.
He said this is similar to results observed with CARs being tested at the National Cancer Institute and Memorial Sloan-Kettering Cancer Center.
Abs from transplanted AML patients enhance GvL effect in vitro

NEW YORK—Investigators have found that B cells may play a role in stimulating graft-versus-leukemia (GvL) responses in patients with acute myeloid leukemia (AML) who have undergone allogeneic hematopoietic stem cell transplant (HSCT).
The team created B cell lines from these patients, isolated AML-specific antibodies, and found that these antibodies can induce the death of AML cells through oncosis.
Oncosis is a non-apoptotic type of cell death that involves swelling and coagulation of the cytoplasm.
Mette Hazenberg, MD, PhD, from the Academic Medical Center in Amsterdam, The Netherlands, reported these findings at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference as poster B052.
The investigators cloned B cells from 3 high-risk AML patients who had a strong GvL response after HSCT.
The team transduced memory B cells from the patients’ peripheral blood using BCL-6 and BCL-xL. They then screened the B cells for those that produced antibodies that bound specifically to surface antigens on AML cell lines and blasts.
Six of the 15 AML antibodies retrieved from the patients bound specifically to snRNp200. In normal cells, snRNp200 is in the nucleus, but, in AML, it is exposed on the cell membrane.
The investigators then confirmed this by ELISA.
They found 7 of the 15 AML antibodies directly lysed AML blasts without the addition of effector cells or complement. Time-lapse images showed that cell death by the AML antibodies occurred rapidly, within minutes after incubation.
“The leukemia blasts popped like balloons,” Dr Hazenberg said.
The investigators confirmed that the antibodies induced cell death by oncosis and that oncosis occurred independently of temperature. The antibodies were cytotoxic at 4°C and 37°C.
Cytotoxicity of the antibodies could be blocked by the membrane stabilizer cytochalasin D but not by apoptosis inhibitors.
The team concluded that a potent GvL response could be mediated by these antibodies against tumor-associated antigens on AML cells.
Dr Hazenberg’s hope is that, at some point, these antibodies can be combined with chemotherapy—as is rituximab—so patients won’t need to undergo transplant.
NEW YORK—Investigators have found that B cells may play a role in stimulating graft-versus-leukemia (GvL) responses in patients with acute myeloid leukemia (AML) who have undergone allogeneic hematopoietic stem cell transplant (HSCT).
The team created B cell lines from these patients, isolated AML-specific antibodies, and found that these antibodies can induce the death of AML cells through oncosis.
Oncosis is a non-apoptotic type of cell death that involves swelling and coagulation of the cytoplasm.
Mette Hazenberg, MD, PhD, from the Academic Medical Center in Amsterdam, The Netherlands, reported these findings at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference as poster B052.
The investigators cloned B cells from 3 high-risk AML patients who had a strong GvL response after HSCT.
The team transduced memory B cells from the patients’ peripheral blood using BCL-6 and BCL-xL. They then screened the B cells for those that produced antibodies that bound specifically to surface antigens on AML cell lines and blasts.
Six of the 15 AML antibodies retrieved from the patients bound specifically to snRNp200. In normal cells, snRNp200 is in the nucleus, but, in AML, it is exposed on the cell membrane.
The investigators then confirmed this by ELISA.
They found 7 of the 15 AML antibodies directly lysed AML blasts without the addition of effector cells or complement. Time-lapse images showed that cell death by the AML antibodies occurred rapidly, within minutes after incubation.
“The leukemia blasts popped like balloons,” Dr Hazenberg said.
The investigators confirmed that the antibodies induced cell death by oncosis and that oncosis occurred independently of temperature. The antibodies were cytotoxic at 4°C and 37°C.
Cytotoxicity of the antibodies could be blocked by the membrane stabilizer cytochalasin D but not by apoptosis inhibitors.
The team concluded that a potent GvL response could be mediated by these antibodies against tumor-associated antigens on AML cells.
Dr Hazenberg’s hope is that, at some point, these antibodies can be combined with chemotherapy—as is rituximab—so patients won’t need to undergo transplant.
NEW YORK—Investigators have found that B cells may play a role in stimulating graft-versus-leukemia (GvL) responses in patients with acute myeloid leukemia (AML) who have undergone allogeneic hematopoietic stem cell transplant (HSCT).
The team created B cell lines from these patients, isolated AML-specific antibodies, and found that these antibodies can induce the death of AML cells through oncosis.
Oncosis is a non-apoptotic type of cell death that involves swelling and coagulation of the cytoplasm.
Mette Hazenberg, MD, PhD, from the Academic Medical Center in Amsterdam, The Netherlands, reported these findings at the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference as poster B052.
The investigators cloned B cells from 3 high-risk AML patients who had a strong GvL response after HSCT.
The team transduced memory B cells from the patients’ peripheral blood using BCL-6 and BCL-xL. They then screened the B cells for those that produced antibodies that bound specifically to surface antigens on AML cell lines and blasts.
Six of the 15 AML antibodies retrieved from the patients bound specifically to snRNp200. In normal cells, snRNp200 is in the nucleus, but, in AML, it is exposed on the cell membrane.
The investigators then confirmed this by ELISA.
They found 7 of the 15 AML antibodies directly lysed AML blasts without the addition of effector cells or complement. Time-lapse images showed that cell death by the AML antibodies occurred rapidly, within minutes after incubation.
“The leukemia blasts popped like balloons,” Dr Hazenberg said.
The investigators confirmed that the antibodies induced cell death by oncosis and that oncosis occurred independently of temperature. The antibodies were cytotoxic at 4°C and 37°C.
Cytotoxicity of the antibodies could be blocked by the membrane stabilizer cytochalasin D but not by apoptosis inhibitors.
The team concluded that a potent GvL response could be mediated by these antibodies against tumor-associated antigens on AML cells.
Dr Hazenberg’s hope is that, at some point, these antibodies can be combined with chemotherapy—as is rituximab—so patients won’t need to undergo transplant.