User login
Mutations aid resistance, growth of malaria parasite
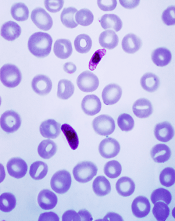
Plasmodium falciparum
Image from CDC/Mae Melvin
Some mutations that enable drug resistance in the malaria parasite Plasmodium falciparum may also help it grow, according to a study published in PLOS Pathogens.
Some strains of P falciparum have evolved to become resistant to antimalarial drugs, including chloroquine.
Often, chloroquine resistance mutations hinder P falciparum’s ability to infect the bloodstream and grow.
However, in a previous study, researchers discovered that a uniquely mutated version of the P falciparum gene pfcrt provides drug resistance while avoiding the detrimental impact of growth seen with other mutated pfcrt variants.
In the new study, the same group of researchers—Stanislaw Gabryszewski, of Columbia University Medical Center in New York, and his colleagues—investigated this version of the pfcrt gene, which is called Cam734 and has been found in certain regions in Southeast Asia.
Using zinc-finger nucleases, the team characterized the individual mutations unique to Cam734 in terms of their effects on drug resistance, metabolism, and growth rates in living parasites.
The researchers found that a mutation called A144F is required for the chloroquine resistance enabled by Cam734, and this mutation also contributes to resistance to the drugs amodiaquine and quinine.
The team identified additional mutations that contribute to resistance to chloroquine and impact the potency of other antimalarials as well.
When the researchers reversed these mutations in living parasites that had the Cam734 allele, growth slowed, indicating that these mutations also enhance infection.
Additional experiments revealed specific effects of Cam734 mutations on several metabolic pathways in P falciparum, including the digestion of human hemoglobin that parasites use to obtain amino acids for protein synthesis.
The researchers also found evidence suggesting that Cam734 helps to maintain an electrochemical gradient that allows the protein encoded by the pfcrt gene to thwart the cellular effects of chloroquine.
The team said these findings broaden our understanding of Cam734, the second most common variant of the pfcrt gene in Southeast Asia. The findings identify multiple intracellular processes and multidrug resistance phenotypes impacted by changes in pfcrt and can help inform future malaria treatment efforts. ![]()
Plasmodium falciparum
Image from CDC/Mae Melvin
Some mutations that enable drug resistance in the malaria parasite Plasmodium falciparum may also help it grow, according to a study published in PLOS Pathogens.
Some strains of P falciparum have evolved to become resistant to antimalarial drugs, including chloroquine.
Often, chloroquine resistance mutations hinder P falciparum’s ability to infect the bloodstream and grow.
However, in a previous study, researchers discovered that a uniquely mutated version of the P falciparum gene pfcrt provides drug resistance while avoiding the detrimental impact of growth seen with other mutated pfcrt variants.
In the new study, the same group of researchers—Stanislaw Gabryszewski, of Columbia University Medical Center in New York, and his colleagues—investigated this version of the pfcrt gene, which is called Cam734 and has been found in certain regions in Southeast Asia.
Using zinc-finger nucleases, the team characterized the individual mutations unique to Cam734 in terms of their effects on drug resistance, metabolism, and growth rates in living parasites.
The researchers found that a mutation called A144F is required for the chloroquine resistance enabled by Cam734, and this mutation also contributes to resistance to the drugs amodiaquine and quinine.
The team identified additional mutations that contribute to resistance to chloroquine and impact the potency of other antimalarials as well.
When the researchers reversed these mutations in living parasites that had the Cam734 allele, growth slowed, indicating that these mutations also enhance infection.
Additional experiments revealed specific effects of Cam734 mutations on several metabolic pathways in P falciparum, including the digestion of human hemoglobin that parasites use to obtain amino acids for protein synthesis.
The researchers also found evidence suggesting that Cam734 helps to maintain an electrochemical gradient that allows the protein encoded by the pfcrt gene to thwart the cellular effects of chloroquine.
The team said these findings broaden our understanding of Cam734, the second most common variant of the pfcrt gene in Southeast Asia. The findings identify multiple intracellular processes and multidrug resistance phenotypes impacted by changes in pfcrt and can help inform future malaria treatment efforts. ![]()
Plasmodium falciparum
Image from CDC/Mae Melvin
Some mutations that enable drug resistance in the malaria parasite Plasmodium falciparum may also help it grow, according to a study published in PLOS Pathogens.
Some strains of P falciparum have evolved to become resistant to antimalarial drugs, including chloroquine.
Often, chloroquine resistance mutations hinder P falciparum’s ability to infect the bloodstream and grow.
However, in a previous study, researchers discovered that a uniquely mutated version of the P falciparum gene pfcrt provides drug resistance while avoiding the detrimental impact of growth seen with other mutated pfcrt variants.
In the new study, the same group of researchers—Stanislaw Gabryszewski, of Columbia University Medical Center in New York, and his colleagues—investigated this version of the pfcrt gene, which is called Cam734 and has been found in certain regions in Southeast Asia.
Using zinc-finger nucleases, the team characterized the individual mutations unique to Cam734 in terms of their effects on drug resistance, metabolism, and growth rates in living parasites.
The researchers found that a mutation called A144F is required for the chloroquine resistance enabled by Cam734, and this mutation also contributes to resistance to the drugs amodiaquine and quinine.
The team identified additional mutations that contribute to resistance to chloroquine and impact the potency of other antimalarials as well.
When the researchers reversed these mutations in living parasites that had the Cam734 allele, growth slowed, indicating that these mutations also enhance infection.
Additional experiments revealed specific effects of Cam734 mutations on several metabolic pathways in P falciparum, including the digestion of human hemoglobin that parasites use to obtain amino acids for protein synthesis.
The researchers also found evidence suggesting that Cam734 helps to maintain an electrochemical gradient that allows the protein encoded by the pfcrt gene to thwart the cellular effects of chloroquine.
The team said these findings broaden our understanding of Cam734, the second most common variant of the pfcrt gene in Southeast Asia. The findings identify multiple intracellular processes and multidrug resistance phenotypes impacted by changes in pfcrt and can help inform future malaria treatment efforts. ![]()
How EBV drives lymphomagenesis

Image by Ed Uthman
Results of research published in eLIFE appear to explain how Epstein-Barr virus (EBV) controls a pair of genes to drive lymphomagenesis.
Researchers set out to determine how EBV controls MYC, which is known to drive lymphoma development when activated, and BCL2L11, a gene that normally triggers apoptosis to prevent lymphoma but can be silenced by EBV.
The team discovered that EBV controls MYC and BCL2L11 by hijacking enhancer regions of DNA, which are situated far away from the genes.
These enhancers act as “control centers” and are able to contact and control genes from long distances by the looping out of the intervening stretches of DNA.
The researchers found that EBV activates MYC by increasing contacts between a specific set of enhancers and the gene.
The team said an Epstein-Barr nuclear antigen, EBNA2, activates multiple MYC enhancers and reconfigures the MYC locus to increase upstream enhancer-promoter interactions and decrease downstream interactions.
They noted that EBNA2 recruits the BRG1 ATPase of the SWI/SNF remodeller to MYC enhancers, and BRG1 is required for enhancer-promoter interactions in EBV-infected cells.
The researchers also discovered new enhancers that control BCL2L11. In this case, though, EBV stops these control centers from contacting the gene.
Specifically, the team found a hematopoietic enhancer hub that is inactivated by the Epstein-Barr nuclear antigens EBNA3A and EBNA3C through recruitment of the H3K27 methyltransferase EZH2.
Therefore, the researchers set out to determine if an EZH1/2 inhibitor, UNC1999, could reverse this effect. They found that UNC1999 did reverse enhancer inactivation, upregulated BCL2L11, and induced apoptosis in EBV-positive Burkitt lymphoma cells.
“This is a key step towards uncovering how this common virus, which affects thousands of people every year, causes blood cancer,” said study author Michelle West, PhD, of the University of Sussex in Brighton, UK.
“It is now important to carry out further studies to determine how the Epstein-Barr virus controls other genes that are associated with lymphoma. This will tell us more about how the virus drives lymphoma development and will help to identify new ways of targeting Epstein-Barr virus-infected cancer cells with specific drugs.” ![]()
Image by Ed Uthman
Results of research published in eLIFE appear to explain how Epstein-Barr virus (EBV) controls a pair of genes to drive lymphomagenesis.
Researchers set out to determine how EBV controls MYC, which is known to drive lymphoma development when activated, and BCL2L11, a gene that normally triggers apoptosis to prevent lymphoma but can be silenced by EBV.
The team discovered that EBV controls MYC and BCL2L11 by hijacking enhancer regions of DNA, which are situated far away from the genes.
These enhancers act as “control centers” and are able to contact and control genes from long distances by the looping out of the intervening stretches of DNA.
The researchers found that EBV activates MYC by increasing contacts between a specific set of enhancers and the gene.
The team said an Epstein-Barr nuclear antigen, EBNA2, activates multiple MYC enhancers and reconfigures the MYC locus to increase upstream enhancer-promoter interactions and decrease downstream interactions.
They noted that EBNA2 recruits the BRG1 ATPase of the SWI/SNF remodeller to MYC enhancers, and BRG1 is required for enhancer-promoter interactions in EBV-infected cells.
The researchers also discovered new enhancers that control BCL2L11. In this case, though, EBV stops these control centers from contacting the gene.
Specifically, the team found a hematopoietic enhancer hub that is inactivated by the Epstein-Barr nuclear antigens EBNA3A and EBNA3C through recruitment of the H3K27 methyltransferase EZH2.
Therefore, the researchers set out to determine if an EZH1/2 inhibitor, UNC1999, could reverse this effect. They found that UNC1999 did reverse enhancer inactivation, upregulated BCL2L11, and induced apoptosis in EBV-positive Burkitt lymphoma cells.
“This is a key step towards uncovering how this common virus, which affects thousands of people every year, causes blood cancer,” said study author Michelle West, PhD, of the University of Sussex in Brighton, UK.
“It is now important to carry out further studies to determine how the Epstein-Barr virus controls other genes that are associated with lymphoma. This will tell us more about how the virus drives lymphoma development and will help to identify new ways of targeting Epstein-Barr virus-infected cancer cells with specific drugs.” ![]()
Image by Ed Uthman
Results of research published in eLIFE appear to explain how Epstein-Barr virus (EBV) controls a pair of genes to drive lymphomagenesis.
Researchers set out to determine how EBV controls MYC, which is known to drive lymphoma development when activated, and BCL2L11, a gene that normally triggers apoptosis to prevent lymphoma but can be silenced by EBV.
The team discovered that EBV controls MYC and BCL2L11 by hijacking enhancer regions of DNA, which are situated far away from the genes.
These enhancers act as “control centers” and are able to contact and control genes from long distances by the looping out of the intervening stretches of DNA.
The researchers found that EBV activates MYC by increasing contacts between a specific set of enhancers and the gene.
The team said an Epstein-Barr nuclear antigen, EBNA2, activates multiple MYC enhancers and reconfigures the MYC locus to increase upstream enhancer-promoter interactions and decrease downstream interactions.
They noted that EBNA2 recruits the BRG1 ATPase of the SWI/SNF remodeller to MYC enhancers, and BRG1 is required for enhancer-promoter interactions in EBV-infected cells.
The researchers also discovered new enhancers that control BCL2L11. In this case, though, EBV stops these control centers from contacting the gene.
Specifically, the team found a hematopoietic enhancer hub that is inactivated by the Epstein-Barr nuclear antigens EBNA3A and EBNA3C through recruitment of the H3K27 methyltransferase EZH2.
Therefore, the researchers set out to determine if an EZH1/2 inhibitor, UNC1999, could reverse this effect. They found that UNC1999 did reverse enhancer inactivation, upregulated BCL2L11, and induced apoptosis in EBV-positive Burkitt lymphoma cells.
“This is a key step towards uncovering how this common virus, which affects thousands of people every year, causes blood cancer,” said study author Michelle West, PhD, of the University of Sussex in Brighton, UK.
“It is now important to carry out further studies to determine how the Epstein-Barr virus controls other genes that are associated with lymphoma. This will tell us more about how the virus drives lymphoma development and will help to identify new ways of targeting Epstein-Barr virus-infected cancer cells with specific drugs.” ![]()
CHMP recommends drug for hemophilia A

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy.
Lonoctocog alfa is intended for the treatment and prophylaxis of bleeding in hemophilia A patients of all ages.
The CHMP’s recommendation will be reviewed by the European Commission, which is expected to make a decision in the next few months.
Lonoctocog alfa is designed to provide lasting protection from bleeds with 2- to 3-times weekly dosing. The product uses a covalent bond that forms one structural entity, a single polypeptide-chain, to improve the stability of FVIII and provide longer-lasting FVIII activity.
Lonoctocog alfa is being developed by CSL Behring GmbH.
Clinical trials
The CHMP’s positive opinion of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate was 1.14 in the adults and adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy.
Lonoctocog alfa is intended for the treatment and prophylaxis of bleeding in hemophilia A patients of all ages.
The CHMP’s recommendation will be reviewed by the European Commission, which is expected to make a decision in the next few months.
Lonoctocog alfa is designed to provide lasting protection from bleeds with 2- to 3-times weekly dosing. The product uses a covalent bond that forms one structural entity, a single polypeptide-chain, to improve the stability of FVIII and provide longer-lasting FVIII activity.
Lonoctocog alfa is being developed by CSL Behring GmbH.
Clinical trials
The CHMP’s positive opinion of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate was 1.14 in the adults and adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy.
Lonoctocog alfa is intended for the treatment and prophylaxis of bleeding in hemophilia A patients of all ages.
The CHMP’s recommendation will be reviewed by the European Commission, which is expected to make a decision in the next few months.
Lonoctocog alfa is designed to provide lasting protection from bleeds with 2- to 3-times weekly dosing. The product uses a covalent bond that forms one structural entity, a single polypeptide-chain, to improve the stability of FVIII and provide longer-lasting FVIII activity.
Lonoctocog alfa is being developed by CSL Behring GmbH.
Clinical trials
The CHMP’s positive opinion of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate was 1.14 in the adults and adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis. ![]()
Drug dubbed ‘breakthrough’ for CTCL subtypes

Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to brentuximab vedotin (Adcetris) as a treatment for 2 subtypes of cutaneous T-cell lymphoma (CTCL).
The drug now has this designation for the treatment of patients with CD30-expressing mycosis fungoides (MF) and patients with primary cutaneous anaplastic large-cell lymphoma (pcALCL) who require systemic therapy and have received 1 prior systemic therapy.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Brentuximab vedotin in CTCL
Brentuximab vedotin is an antibody-drug conjugate directed to CD30, which is expressed on skin lesions in approximately 50% of patients with CTCL. The drug is being developed by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Brentuximab vedotin has orphan drug designation from the FDA for the treatment of MF. The drug also received orphan drug designation from the European Commission for CTCL, including subtypes pcALCL and MF.
Brentuximab vedotin has been evaluated in CD30-expressing CTCL in investigator- and corporate-sponsored clinical trials, including the phase 3 ALCANZA trial.
This trial was designed to compare single-agent brentuximab vedotin to investigator’s choice of standard therapies—methotrexate or bexarotene—in patients with CD30-expressing CTCL, including those with pcALCL or MF.
The trial has enrolled 131 patients at 50 sites globally. Patients with pcALCL must have received at least 1 prior systemic or radiation therapy, and patients with MF must have received at least 1 prior systemic therapy.
The study’s primary endpoint is objective response lasting at least 4 months (ORR4), as assessed by Global Response Score, in the brentuximab vedotin arm compared to the control arm. Key secondary endpoints are complete response rate, progression-free survival, and reduction in the burden of symptoms during treatment.
Topline results of the trial were announced in August. The data showed a significant improvement in the ORR4 for the brentuximab vedotin arm compared to the control arm. The ORR4 was 56.3% and 12.5%, respectively (P<0.0001).
The key secondary endpoints were all statistically significant in favor of the brentuximab vedotin arm. And investigators said the safety profile of brentuximab vedotin was generally consistent with the existing prescribing information.
An abstract detailing results of the ALCANZA trial was accepted for oral presentation at the upcoming ASH Annual Meeting (abstract 182). ![]()
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to brentuximab vedotin (Adcetris) as a treatment for 2 subtypes of cutaneous T-cell lymphoma (CTCL).
The drug now has this designation for the treatment of patients with CD30-expressing mycosis fungoides (MF) and patients with primary cutaneous anaplastic large-cell lymphoma (pcALCL) who require systemic therapy and have received 1 prior systemic therapy.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Brentuximab vedotin in CTCL
Brentuximab vedotin is an antibody-drug conjugate directed to CD30, which is expressed on skin lesions in approximately 50% of patients with CTCL. The drug is being developed by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Brentuximab vedotin has orphan drug designation from the FDA for the treatment of MF. The drug also received orphan drug designation from the European Commission for CTCL, including subtypes pcALCL and MF.
Brentuximab vedotin has been evaluated in CD30-expressing CTCL in investigator- and corporate-sponsored clinical trials, including the phase 3 ALCANZA trial.
This trial was designed to compare single-agent brentuximab vedotin to investigator’s choice of standard therapies—methotrexate or bexarotene—in patients with CD30-expressing CTCL, including those with pcALCL or MF.
The trial has enrolled 131 patients at 50 sites globally. Patients with pcALCL must have received at least 1 prior systemic or radiation therapy, and patients with MF must have received at least 1 prior systemic therapy.
The study’s primary endpoint is objective response lasting at least 4 months (ORR4), as assessed by Global Response Score, in the brentuximab vedotin arm compared to the control arm. Key secondary endpoints are complete response rate, progression-free survival, and reduction in the burden of symptoms during treatment.
Topline results of the trial were announced in August. The data showed a significant improvement in the ORR4 for the brentuximab vedotin arm compared to the control arm. The ORR4 was 56.3% and 12.5%, respectively (P<0.0001).
The key secondary endpoints were all statistically significant in favor of the brentuximab vedotin arm. And investigators said the safety profile of brentuximab vedotin was generally consistent with the existing prescribing information.
An abstract detailing results of the ALCANZA trial was accepted for oral presentation at the upcoming ASH Annual Meeting (abstract 182). ![]()
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to brentuximab vedotin (Adcetris) as a treatment for 2 subtypes of cutaneous T-cell lymphoma (CTCL).
The drug now has this designation for the treatment of patients with CD30-expressing mycosis fungoides (MF) and patients with primary cutaneous anaplastic large-cell lymphoma (pcALCL) who require systemic therapy and have received 1 prior systemic therapy.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Brentuximab vedotin in CTCL
Brentuximab vedotin is an antibody-drug conjugate directed to CD30, which is expressed on skin lesions in approximately 50% of patients with CTCL. The drug is being developed by Seattle Genetics and Takeda Pharmaceutical Company Limited.
Brentuximab vedotin has orphan drug designation from the FDA for the treatment of MF. The drug also received orphan drug designation from the European Commission for CTCL, including subtypes pcALCL and MF.
Brentuximab vedotin has been evaluated in CD30-expressing CTCL in investigator- and corporate-sponsored clinical trials, including the phase 3 ALCANZA trial.
This trial was designed to compare single-agent brentuximab vedotin to investigator’s choice of standard therapies—methotrexate or bexarotene—in patients with CD30-expressing CTCL, including those with pcALCL or MF.
The trial has enrolled 131 patients at 50 sites globally. Patients with pcALCL must have received at least 1 prior systemic or radiation therapy, and patients with MF must have received at least 1 prior systemic therapy.
The study’s primary endpoint is objective response lasting at least 4 months (ORR4), as assessed by Global Response Score, in the brentuximab vedotin arm compared to the control arm. Key secondary endpoints are complete response rate, progression-free survival, and reduction in the burden of symptoms during treatment.
Topline results of the trial were announced in August. The data showed a significant improvement in the ORR4 for the brentuximab vedotin arm compared to the control arm. The ORR4 was 56.3% and 12.5%, respectively (P<0.0001).
The key secondary endpoints were all statistically significant in favor of the brentuximab vedotin arm. And investigators said the safety profile of brentuximab vedotin was generally consistent with the existing prescribing information.
An abstract detailing results of the ALCANZA trial was accepted for oral presentation at the upcoming ASH Annual Meeting (abstract 182). ![]()
CHMP recommends expanding use of drug in CLL

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
NCCN issues challenge to ‘bag’ vincristine
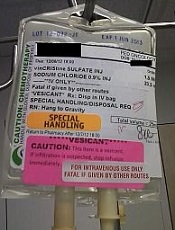
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Health Canada approves drug for patients with VTE, NVAF

Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Treating RBCs with NO may make them safer

Sheep Experiment Station
Research conducted in sheep indicates that pretreating red blood cells (RBCs) with nitric oxide (NO) may make it safer to transfuse blood nearing its expiration date.
Past studies have suggested that RBCs stored for more than 30 days are less likely than “fresher” RBCs to survive after transfusion, and receiving a transfusion of RBCs nearing their expiration date of 42 days may increase the risk of pulmonary hypertension.
However, a new study published in Anesthesiology suggests that pretreating older RBCs with NO may increase their likelihood of survival after transfusion and reduce the risk of pulmonary hypertension in the recipient.
“Extended storage of RBCs makes them rigid and decreases their ability to change shape, which is necessary as they travel through small blood vessels,” said study author Warren M. Zapol, MD, of Massachusetts General Hospital in Boston.
“We found that pretreatment with nitric oxide actually rejuvenates RBCs, making them more flexible so they can more easily travel through blood vessels. This can further reduce the risk of pulmonary hypertension.”
Dr Zapol and his colleagues performed their experiments on RBCs derived from lambs. The team treated RBCs with NO gas, a short-lived NO donor, or gas without NO (control).
The RBCs were then stored for either 2 days (hereafter referred to as “fresh” RBCs) or 40 days (referred to as “stored” RBCs) and transfused back into the original lambs.
RBC survival
The researchers found that treatment with NO gas improved the early post-transfusion survival of stored RBCs.
At 1 hour after transfusion, 75.3 ± 5.8% of the control-treated stored RBCs remained in the circulation, compared to 86.8 ± 8.1% of the NO-treated stored RBCs and 94.2 ± 4.6% of the fresh RBCs.
At 24 hours after transfusion, 73.4 ± 3.8% of the control-treated stored RBCs remained in the circulation, compared to 78.3 ± 6.3% of the NO-treated stored RBCs, 90.8 ± 4.1% of control-treated fresh RBCs, and 91.4 ± 1.4% of NO-treated fresh RBCs.
The differences between stored RBCs that were treated with NO gas and stored control RBCs was statistically significant at both 1 hour and 24 hours, with P values of 0.002 and 0.046, respectively.
Seven days after transfusion, there was no significant difference in the percentage of NO-treated and control-treated RBCs in the circulation.
Pulmonary hypertension
The researchers found that pretreating RBCs with NO prevented transfusion-associated pulmonary hypertension in the lambs.
Lambs that received control-treated stored RBCs had an increase in pulmonary arterial pressure (PAP) during and after transfusion—from 13.4 ± 0.8 mmHg at baseline to a maximum of 22.7 ± 2.2 mmHg.
However, lambs that received stored RBCs treated with NO gas did not have an increase in PAP when compared to lambs that received fresh RBCs.
At 20 minutes, PAP was 14.5 ± 1.4 mmHg for NO-treated stored RBCs, 13.9 ± 0.6 mmHg for control-treated fresh RBCs, and 14 ± 1.2 mmHg for NO-treated fresh RBCs.
The researchers also found that transfusion of stored RBCs caused a transient increase in the pulmonary vascular resistance index (PVRI) from 10 minutes to 30 minutes after transfusion, but pretreatment with NO gas prevented this increase.
At 20 minutes, the PVRI was 211.1 ± 44.4 dyn·sec·cm−5·m−2 for control-treated stored RBCs and 114.6 ± 18.9 dyn·sec·cm−5·m−2 for NO-treated stored RBCs (P<0.0001).
Transfusion of fresh RBCs, with or without prior NO exposure, did not alter the PVRI.
Finally, the researchers found that treating stored RBCs with the NO donor compound MAHMA NONOate prevented transfusion-associated pulmonary hypertension and pulmonary vasoconstriction in awake lambs.
The team said studies with human RBCs are required to confirm the beneficial effects of NO exposure observed in this study.
Sheep Experiment Station
Research conducted in sheep indicates that pretreating red blood cells (RBCs) with nitric oxide (NO) may make it safer to transfuse blood nearing its expiration date.
Past studies have suggested that RBCs stored for more than 30 days are less likely than “fresher” RBCs to survive after transfusion, and receiving a transfusion of RBCs nearing their expiration date of 42 days may increase the risk of pulmonary hypertension.
However, a new study published in Anesthesiology suggests that pretreating older RBCs with NO may increase their likelihood of survival after transfusion and reduce the risk of pulmonary hypertension in the recipient.
“Extended storage of RBCs makes them rigid and decreases their ability to change shape, which is necessary as they travel through small blood vessels,” said study author Warren M. Zapol, MD, of Massachusetts General Hospital in Boston.
“We found that pretreatment with nitric oxide actually rejuvenates RBCs, making them more flexible so they can more easily travel through blood vessels. This can further reduce the risk of pulmonary hypertension.”
Dr Zapol and his colleagues performed their experiments on RBCs derived from lambs. The team treated RBCs with NO gas, a short-lived NO donor, or gas without NO (control).
The RBCs were then stored for either 2 days (hereafter referred to as “fresh” RBCs) or 40 days (referred to as “stored” RBCs) and transfused back into the original lambs.
RBC survival
The researchers found that treatment with NO gas improved the early post-transfusion survival of stored RBCs.
At 1 hour after transfusion, 75.3 ± 5.8% of the control-treated stored RBCs remained in the circulation, compared to 86.8 ± 8.1% of the NO-treated stored RBCs and 94.2 ± 4.6% of the fresh RBCs.
At 24 hours after transfusion, 73.4 ± 3.8% of the control-treated stored RBCs remained in the circulation, compared to 78.3 ± 6.3% of the NO-treated stored RBCs, 90.8 ± 4.1% of control-treated fresh RBCs, and 91.4 ± 1.4% of NO-treated fresh RBCs.
The differences between stored RBCs that were treated with NO gas and stored control RBCs was statistically significant at both 1 hour and 24 hours, with P values of 0.002 and 0.046, respectively.
Seven days after transfusion, there was no significant difference in the percentage of NO-treated and control-treated RBCs in the circulation.
Pulmonary hypertension
The researchers found that pretreating RBCs with NO prevented transfusion-associated pulmonary hypertension in the lambs.
Lambs that received control-treated stored RBCs had an increase in pulmonary arterial pressure (PAP) during and after transfusion—from 13.4 ± 0.8 mmHg at baseline to a maximum of 22.7 ± 2.2 mmHg.
However, lambs that received stored RBCs treated with NO gas did not have an increase in PAP when compared to lambs that received fresh RBCs.
At 20 minutes, PAP was 14.5 ± 1.4 mmHg for NO-treated stored RBCs, 13.9 ± 0.6 mmHg for control-treated fresh RBCs, and 14 ± 1.2 mmHg for NO-treated fresh RBCs.
The researchers also found that transfusion of stored RBCs caused a transient increase in the pulmonary vascular resistance index (PVRI) from 10 minutes to 30 minutes after transfusion, but pretreatment with NO gas prevented this increase.
At 20 minutes, the PVRI was 211.1 ± 44.4 dyn·sec·cm−5·m−2 for control-treated stored RBCs and 114.6 ± 18.9 dyn·sec·cm−5·m−2 for NO-treated stored RBCs (P<0.0001).
Transfusion of fresh RBCs, with or without prior NO exposure, did not alter the PVRI.
Finally, the researchers found that treating stored RBCs with the NO donor compound MAHMA NONOate prevented transfusion-associated pulmonary hypertension and pulmonary vasoconstriction in awake lambs.
The team said studies with human RBCs are required to confirm the beneficial effects of NO exposure observed in this study.
Sheep Experiment Station
Research conducted in sheep indicates that pretreating red blood cells (RBCs) with nitric oxide (NO) may make it safer to transfuse blood nearing its expiration date.
Past studies have suggested that RBCs stored for more than 30 days are less likely than “fresher” RBCs to survive after transfusion, and receiving a transfusion of RBCs nearing their expiration date of 42 days may increase the risk of pulmonary hypertension.
However, a new study published in Anesthesiology suggests that pretreating older RBCs with NO may increase their likelihood of survival after transfusion and reduce the risk of pulmonary hypertension in the recipient.
“Extended storage of RBCs makes them rigid and decreases their ability to change shape, which is necessary as they travel through small blood vessels,” said study author Warren M. Zapol, MD, of Massachusetts General Hospital in Boston.
“We found that pretreatment with nitric oxide actually rejuvenates RBCs, making them more flexible so they can more easily travel through blood vessels. This can further reduce the risk of pulmonary hypertension.”
Dr Zapol and his colleagues performed their experiments on RBCs derived from lambs. The team treated RBCs with NO gas, a short-lived NO donor, or gas without NO (control).
The RBCs were then stored for either 2 days (hereafter referred to as “fresh” RBCs) or 40 days (referred to as “stored” RBCs) and transfused back into the original lambs.
RBC survival
The researchers found that treatment with NO gas improved the early post-transfusion survival of stored RBCs.
At 1 hour after transfusion, 75.3 ± 5.8% of the control-treated stored RBCs remained in the circulation, compared to 86.8 ± 8.1% of the NO-treated stored RBCs and 94.2 ± 4.6% of the fresh RBCs.
At 24 hours after transfusion, 73.4 ± 3.8% of the control-treated stored RBCs remained in the circulation, compared to 78.3 ± 6.3% of the NO-treated stored RBCs, 90.8 ± 4.1% of control-treated fresh RBCs, and 91.4 ± 1.4% of NO-treated fresh RBCs.
The differences between stored RBCs that were treated with NO gas and stored control RBCs was statistically significant at both 1 hour and 24 hours, with P values of 0.002 and 0.046, respectively.
Seven days after transfusion, there was no significant difference in the percentage of NO-treated and control-treated RBCs in the circulation.
Pulmonary hypertension
The researchers found that pretreating RBCs with NO prevented transfusion-associated pulmonary hypertension in the lambs.
Lambs that received control-treated stored RBCs had an increase in pulmonary arterial pressure (PAP) during and after transfusion—from 13.4 ± 0.8 mmHg at baseline to a maximum of 22.7 ± 2.2 mmHg.
However, lambs that received stored RBCs treated with NO gas did not have an increase in PAP when compared to lambs that received fresh RBCs.
At 20 minutes, PAP was 14.5 ± 1.4 mmHg for NO-treated stored RBCs, 13.9 ± 0.6 mmHg for control-treated fresh RBCs, and 14 ± 1.2 mmHg for NO-treated fresh RBCs.
The researchers also found that transfusion of stored RBCs caused a transient increase in the pulmonary vascular resistance index (PVRI) from 10 minutes to 30 minutes after transfusion, but pretreatment with NO gas prevented this increase.
At 20 minutes, the PVRI was 211.1 ± 44.4 dyn·sec·cm−5·m−2 for control-treated stored RBCs and 114.6 ± 18.9 dyn·sec·cm−5·m−2 for NO-treated stored RBCs (P<0.0001).
Transfusion of fresh RBCs, with or without prior NO exposure, did not alter the PVRI.
Finally, the researchers found that treating stored RBCs with the NO donor compound MAHMA NONOate prevented transfusion-associated pulmonary hypertension and pulmonary vasoconstriction in awake lambs.
The team said studies with human RBCs are required to confirm the beneficial effects of NO exposure observed in this study.
Tool provides info for cancer patients, survivors

receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute.
receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute.
receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute.
Blood test can predict outcomes in DLBCL, team says

Photo by Juan D. Alfonso
A blood test can reveal genetic features linked to outcomes in patients with diffuse large B-cell lymphoma (DLBCL), according to research published in Science Translational Medicine.
Investigators used targeted sequencing to analyze circulating tumor DNA (ctDNA) in blood samples from DLBCL patients.
This allowed the team to identify the cell of origin, detect minimal residual disease (MRD), and predict progression-free survival (PFS) in these patients.
Florian Scherer, MD, of Stanford University in California, and his colleagues conducted this research.
They used cancer personalized profiling by deep sequencing (CAPP-Seq) to analyze tumor biopsies and cell-free DNA samples from 92 patients with DLBCL and 24 healthy controls.
The investigators found that CAPP-Seq could effectively detect somatic mutations in DLBCL plasma samples as well as tumor biopsies. They said their results suggest ctDNA is a “robust surrogate for direct assessment of primary tumor genotypes” in most DLBCL patients.
In addition, ctDNA profiling with CAPP-Seq revealed mutations associated with resistance to the BTK inhibitor ibrutinib.
The investigators also said their results suggest ctDNA profiling can be used to classify DLBCL subtypes. The overall concordance in cell of origin predictions between tumor tissue and plasma genotyping was 88%.
Another key finding of this study is that the amount of ctDNA at DLBCL diagnosis was predictive of PFS. The investigators said higher ctDNA levels at diagnosis were “continuously and independently” correlated with inferior PFS.
Dr Scherer and his colleagues also discovered that ctDNA profiling could detect MRD with greater accuracy than immunoglobulin sequencing and radiographic imaging. And patients with ctDNA in their plasma had significantly worse PFS than patients with undetectable ctDNA.
Finally, the investigators found evidence to suggest that ctDNA profiling could provide early detection of disease transformation. They identified “distinct patterns of clonal evolution” by which they could distinguish indolent follicular lymphomas from follicular lymphomas that transformed into DLBCL.
Photo by Juan D. Alfonso
A blood test can reveal genetic features linked to outcomes in patients with diffuse large B-cell lymphoma (DLBCL), according to research published in Science Translational Medicine.
Investigators used targeted sequencing to analyze circulating tumor DNA (ctDNA) in blood samples from DLBCL patients.
This allowed the team to identify the cell of origin, detect minimal residual disease (MRD), and predict progression-free survival (PFS) in these patients.
Florian Scherer, MD, of Stanford University in California, and his colleagues conducted this research.
They used cancer personalized profiling by deep sequencing (CAPP-Seq) to analyze tumor biopsies and cell-free DNA samples from 92 patients with DLBCL and 24 healthy controls.
The investigators found that CAPP-Seq could effectively detect somatic mutations in DLBCL plasma samples as well as tumor biopsies. They said their results suggest ctDNA is a “robust surrogate for direct assessment of primary tumor genotypes” in most DLBCL patients.
In addition, ctDNA profiling with CAPP-Seq revealed mutations associated with resistance to the BTK inhibitor ibrutinib.
The investigators also said their results suggest ctDNA profiling can be used to classify DLBCL subtypes. The overall concordance in cell of origin predictions between tumor tissue and plasma genotyping was 88%.
Another key finding of this study is that the amount of ctDNA at DLBCL diagnosis was predictive of PFS. The investigators said higher ctDNA levels at diagnosis were “continuously and independently” correlated with inferior PFS.
Dr Scherer and his colleagues also discovered that ctDNA profiling could detect MRD with greater accuracy than immunoglobulin sequencing and radiographic imaging. And patients with ctDNA in their plasma had significantly worse PFS than patients with undetectable ctDNA.
Finally, the investigators found evidence to suggest that ctDNA profiling could provide early detection of disease transformation. They identified “distinct patterns of clonal evolution” by which they could distinguish indolent follicular lymphomas from follicular lymphomas that transformed into DLBCL.
Photo by Juan D. Alfonso
A blood test can reveal genetic features linked to outcomes in patients with diffuse large B-cell lymphoma (DLBCL), according to research published in Science Translational Medicine.
Investigators used targeted sequencing to analyze circulating tumor DNA (ctDNA) in blood samples from DLBCL patients.
This allowed the team to identify the cell of origin, detect minimal residual disease (MRD), and predict progression-free survival (PFS) in these patients.
Florian Scherer, MD, of Stanford University in California, and his colleagues conducted this research.
They used cancer personalized profiling by deep sequencing (CAPP-Seq) to analyze tumor biopsies and cell-free DNA samples from 92 patients with DLBCL and 24 healthy controls.
The investigators found that CAPP-Seq could effectively detect somatic mutations in DLBCL plasma samples as well as tumor biopsies. They said their results suggest ctDNA is a “robust surrogate for direct assessment of primary tumor genotypes” in most DLBCL patients.
In addition, ctDNA profiling with CAPP-Seq revealed mutations associated with resistance to the BTK inhibitor ibrutinib.
The investigators also said their results suggest ctDNA profiling can be used to classify DLBCL subtypes. The overall concordance in cell of origin predictions between tumor tissue and plasma genotyping was 88%.
Another key finding of this study is that the amount of ctDNA at DLBCL diagnosis was predictive of PFS. The investigators said higher ctDNA levels at diagnosis were “continuously and independently” correlated with inferior PFS.
Dr Scherer and his colleagues also discovered that ctDNA profiling could detect MRD with greater accuracy than immunoglobulin sequencing and radiographic imaging. And patients with ctDNA in their plasma had significantly worse PFS than patients with undetectable ctDNA.
Finally, the investigators found evidence to suggest that ctDNA profiling could provide early detection of disease transformation. They identified “distinct patterns of clonal evolution” by which they could distinguish indolent follicular lymphomas from follicular lymphomas that transformed into DLBCL.