User login
FDA grants priority review for MM drug

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted priority review for part of a supplemental biologics license application (sBLA) for daratumumab (Darzalex®).
The priority review pertains to the use of daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat patients with multiple myeloma (MM) who have received at least 1 prior therapy.
To grant an application priority review, the FDA must believe the drug would provide a significant improvement in the treatment, diagnosis, or prevention of a serious condition.
The priority review designation means the FDA’s goal is to take action on an application within 6 months, rather than the 10 months typically taken for a standard review.
The FDA has assigned a Prescription Drug User Fee Act target date of February 17, 2017, to make a decision on daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone.
Standard review
The FDA has also granted a standard review period for part of the sBLA.
The standard review pertains to the use of daratumumab in combination with pomalidomide and dexamethasone to treat patients with relapsed or refractory MM who have received at least 2 prior therapies, including a proteasome inhibitor and an immunomodulatory agent.
The Prescription Drug User Fee Act date for the combination of daratumumab with pomalidomide and dexamethasone is June 17, 2017.
Trial data
The sBLA submission included data from a pair of phase 3 studies:
- The CASTOR study, in which researchers compared the combination of daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone in patients with relapsed or refractory MM
- The POLLUX study, in which researchers compared daratumumab in combination with lenalidomide and dexamethasone to lenalidomide and dexamethasone in patients with relapsed or refractory MM.
The sBLA submission also included data from a phase 1 study of daratumumab in combination with pomalidomide and dexamethasone in patients with relapsed or refractory MM.
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to CD38, which is highly expressed on the surface of MM cells.
Daratumumab already has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
For more information on daratumumab, visit www.DARZALEX.com. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted priority review for part of a supplemental biologics license application (sBLA) for daratumumab (Darzalex®).
The priority review pertains to the use of daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat patients with multiple myeloma (MM) who have received at least 1 prior therapy.
To grant an application priority review, the FDA must believe the drug would provide a significant improvement in the treatment, diagnosis, or prevention of a serious condition.
The priority review designation means the FDA’s goal is to take action on an application within 6 months, rather than the 10 months typically taken for a standard review.
The FDA has assigned a Prescription Drug User Fee Act target date of February 17, 2017, to make a decision on daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone.
Standard review
The FDA has also granted a standard review period for part of the sBLA.
The standard review pertains to the use of daratumumab in combination with pomalidomide and dexamethasone to treat patients with relapsed or refractory MM who have received at least 2 prior therapies, including a proteasome inhibitor and an immunomodulatory agent.
The Prescription Drug User Fee Act date for the combination of daratumumab with pomalidomide and dexamethasone is June 17, 2017.
Trial data
The sBLA submission included data from a pair of phase 3 studies:
- The CASTOR study, in which researchers compared the combination of daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone in patients with relapsed or refractory MM
- The POLLUX study, in which researchers compared daratumumab in combination with lenalidomide and dexamethasone to lenalidomide and dexamethasone in patients with relapsed or refractory MM.
The sBLA submission also included data from a phase 1 study of daratumumab in combination with pomalidomide and dexamethasone in patients with relapsed or refractory MM.
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to CD38, which is highly expressed on the surface of MM cells.
Daratumumab already has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
For more information on daratumumab, visit www.DARZALEX.com. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted priority review for part of a supplemental biologics license application (sBLA) for daratumumab (Darzalex®).
The priority review pertains to the use of daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat patients with multiple myeloma (MM) who have received at least 1 prior therapy.
To grant an application priority review, the FDA must believe the drug would provide a significant improvement in the treatment, diagnosis, or prevention of a serious condition.
The priority review designation means the FDA’s goal is to take action on an application within 6 months, rather than the 10 months typically taken for a standard review.
The FDA has assigned a Prescription Drug User Fee Act target date of February 17, 2017, to make a decision on daratumumab in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone.
Standard review
The FDA has also granted a standard review period for part of the sBLA.
The standard review pertains to the use of daratumumab in combination with pomalidomide and dexamethasone to treat patients with relapsed or refractory MM who have received at least 2 prior therapies, including a proteasome inhibitor and an immunomodulatory agent.
The Prescription Drug User Fee Act date for the combination of daratumumab with pomalidomide and dexamethasone is June 17, 2017.
Trial data
The sBLA submission included data from a pair of phase 3 studies:
- The CASTOR study, in which researchers compared the combination of daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone in patients with relapsed or refractory MM
- The POLLUX study, in which researchers compared daratumumab in combination with lenalidomide and dexamethasone to lenalidomide and dexamethasone in patients with relapsed or refractory MM.
The sBLA submission also included data from a phase 1 study of daratumumab in combination with pomalidomide and dexamethasone in patients with relapsed or refractory MM.
About daratumumab
Daratumumab is a human IgG1k monoclonal antibody that binds to CD38, which is highly expressed on the surface of MM cells.
Daratumumab already has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
For more information on daratumumab, visit www.DARZALEX.com. ![]()
FDA clears use of coagulation analyzer

The US Food and Drug Administration (FDA) has granted 510(k) clearance for the Xprecia Stride Coagulation Analyzer from Siemens Healthcare Diagnostics.
This hand-held, portable coagulation analyzer is designed to deliver prothrombin time/international normalized ratio (PT/INR) testing for point-of-care (POC) monitoring and management of oral anticoagulation therapy with the vitamin K antagonist warfarin.
The Xprecia Stride Coagulation Analyzer is the first POC PT/INR device cleared by the FDA based on the new rules published in March 2016.
The analyzer uses fresh capillary whole blood, and results are expressed as INR. The Xprecia Stride Coagulation Analyzer utilizes the same Dade® Innovin® reagent used by Siemens Healthineers central lab analyzers to minimize any potential for variability.
Research has shown the performance of the Xprecia Stride Coagulation Analyzer to be equivalent to a reference laboratory hemostasis system,* with results available within minutes.
According to Siemens, the Xprecia Stride Coagulation Analyzer includes a number of innovations and features not found on most other POC analyzers.
The Xprecia Stride Coagulation Analyzer has a touchscreen interface with step-by-step instructions that help guide the user.
To further enhance usability, the analyzer features simple icons and animation in a color display more commonly found in mobile devices than medical instruments.
The Xprecia Stride Coagulation Analyzer is no bigger than a smartphone and weighs just 10.5 oz, so it can be held at virtually any angle and brought directly to the patient’s finger for blood sample collection.
The analyzer has an integrated barcode scanner intended to simplify data capture and improve patient workflow. The scanner offers patient and operator ID entry.
The Xprecia Stride Coagulation Analyzer has an operator lockout feature that restricts the analyzer’s use to trained staff only.
And the analyzer includes a first-of-its kind test strip eject button that allows the user to eject a used test strip and easily dispose of it without touching it, minimizing potential biohazard exposure.
For more information on the Xprecia Stride Coagulation Analyzer, visit www.siemens.com/xprecia. ![]()
The US Food and Drug Administration (FDA) has granted 510(k) clearance for the Xprecia Stride Coagulation Analyzer from Siemens Healthcare Diagnostics.
This hand-held, portable coagulation analyzer is designed to deliver prothrombin time/international normalized ratio (PT/INR) testing for point-of-care (POC) monitoring and management of oral anticoagulation therapy with the vitamin K antagonist warfarin.
The Xprecia Stride Coagulation Analyzer is the first POC PT/INR device cleared by the FDA based on the new rules published in March 2016.
The analyzer uses fresh capillary whole blood, and results are expressed as INR. The Xprecia Stride Coagulation Analyzer utilizes the same Dade® Innovin® reagent used by Siemens Healthineers central lab analyzers to minimize any potential for variability.
Research has shown the performance of the Xprecia Stride Coagulation Analyzer to be equivalent to a reference laboratory hemostasis system,* with results available within minutes.
According to Siemens, the Xprecia Stride Coagulation Analyzer includes a number of innovations and features not found on most other POC analyzers.
The Xprecia Stride Coagulation Analyzer has a touchscreen interface with step-by-step instructions that help guide the user.
To further enhance usability, the analyzer features simple icons and animation in a color display more commonly found in mobile devices than medical instruments.
The Xprecia Stride Coagulation Analyzer is no bigger than a smartphone and weighs just 10.5 oz, so it can be held at virtually any angle and brought directly to the patient’s finger for blood sample collection.
The analyzer has an integrated barcode scanner intended to simplify data capture and improve patient workflow. The scanner offers patient and operator ID entry.
The Xprecia Stride Coagulation Analyzer has an operator lockout feature that restricts the analyzer’s use to trained staff only.
And the analyzer includes a first-of-its kind test strip eject button that allows the user to eject a used test strip and easily dispose of it without touching it, minimizing potential biohazard exposure.
For more information on the Xprecia Stride Coagulation Analyzer, visit www.siemens.com/xprecia. ![]()
The US Food and Drug Administration (FDA) has granted 510(k) clearance for the Xprecia Stride Coagulation Analyzer from Siemens Healthcare Diagnostics.
This hand-held, portable coagulation analyzer is designed to deliver prothrombin time/international normalized ratio (PT/INR) testing for point-of-care (POC) monitoring and management of oral anticoagulation therapy with the vitamin K antagonist warfarin.
The Xprecia Stride Coagulation Analyzer is the first POC PT/INR device cleared by the FDA based on the new rules published in March 2016.
The analyzer uses fresh capillary whole blood, and results are expressed as INR. The Xprecia Stride Coagulation Analyzer utilizes the same Dade® Innovin® reagent used by Siemens Healthineers central lab analyzers to minimize any potential for variability.
Research has shown the performance of the Xprecia Stride Coagulation Analyzer to be equivalent to a reference laboratory hemostasis system,* with results available within minutes.
According to Siemens, the Xprecia Stride Coagulation Analyzer includes a number of innovations and features not found on most other POC analyzers.
The Xprecia Stride Coagulation Analyzer has a touchscreen interface with step-by-step instructions that help guide the user.
To further enhance usability, the analyzer features simple icons and animation in a color display more commonly found in mobile devices than medical instruments.
The Xprecia Stride Coagulation Analyzer is no bigger than a smartphone and weighs just 10.5 oz, so it can be held at virtually any angle and brought directly to the patient’s finger for blood sample collection.
The analyzer has an integrated barcode scanner intended to simplify data capture and improve patient workflow. The scanner offers patient and operator ID entry.
The Xprecia Stride Coagulation Analyzer has an operator lockout feature that restricts the analyzer’s use to trained staff only.
And the analyzer includes a first-of-its kind test strip eject button that allows the user to eject a used test strip and easily dispose of it without touching it, minimizing potential biohazard exposure.
For more information on the Xprecia Stride Coagulation Analyzer, visit www.siemens.com/xprecia. ![]()
Immunotherapy produces CRs in kids with rel/ref ALL
The bispecific T-cell engager (BiTE®) antibody blinatumomab can produce complete responses (CRs) in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), according to a phase 1/2 study published in the Journal of Clinical Oncology.
Of the patients who received the recommended dosage of blinatumomab, 39% achieved a CR within the first 2 treatment cycles.
And 52% of these patients achieved a complete minimal residual disease (MRD) response.
“This study showed that [blinatumomab] can induce deep molecular remissions in children with highly refractory, multiply relapsed ALL,” said study author Lia Gore, MD, of University of Colorado Anschutz Medical Campus in Aurora, Colorado.
However, most of these remissions did not last. Although a few of the complete responders were still alive and in CR at the study’s 2-year follow-up, more than half had relapsed, and two-thirds had died.
This trial, known as Study ‘205, was supported by Amgen.
Study ‘205 included 93 pediatric patients with relapsed or refractory B-cell precursor ALL. Patients received blinatumomab as a continuous intravenous infusion—49 patients in the phase 1 portion of the trial and 44 in phase 2. The patients were followed for 2 years.
Toxicities and recommended dose
There were 4 dose-limiting toxicities during the phase 1 portion of the trial, and 2 of these events were fatal. One patient treated at 15 μg/m2/day developed grade 4 cytokine release syndrome (CRS), which was deemed related to grade 4 gastrointestinal hemorrhage.
Two patients treated at 30 μg/m2/day had grade 4 CRS. One case was attributed to grade 5 cardiac failure, and the other was treated successfully with tocilizumab.
One patient treated at 15 μg/m2/day had grade 5 respiratory failure with cardiac arrest after hypotonia and muscle weakness after 7 days of infusion with blinatumomab. This patient experienced febrile neutropenia and pneumonia shortly before the start of the infusion.
Based on these toxicities, the maximum-tolerated dose of blinatumomab was 15 μg/m2/day, but a step-wise dosage was recommended to reduce the risk of CRS.
So the recommended dose was 5 μg/m2/day on days 1-7 and 15 μg/m2/day on days 8-28 for cycle 1, and 15 μg/m2/day on days 1-28 for subsequent cycles.
Dose adjustment was possible in case of adverse events. Patients who responded to blinatumomab but later relapsed had the option to be retreated with blinatumomab.
Treatment at recommended dose
Seventy patients received at least 1 infusion of blinatumomab at the recommended dose. The median number of treatment cycles was 1 (range, 1 to 5).
The patients’ median age was 8 years (range, 7 months to 17 years). Forty patients (57%) had undergone allogeneic transplant prior to receiving blinatumomab, and 39 (56%) had refractory disease. Four patients had less than the 25% bone marrow blasts required for protocol entry but had more than 5% blasts.
Adverse events
The most common adverse events among the patients who received the recommended dose of blinatumomab were pyrexia (80%), anemia (41%), nausea (33%), and headache (30%).
The most frequent grade 3 or higher events were anemia (36%), thrombocytopenia (21%), febrile neutropenia (17%), hypokalemia (17%), and neutropenia (17%).
Eight patients developed CRS. Three had grade 3 and 1 had grade 4 CRS. Two of these patients had treatment interruptions, and 2 discontinued treatment permanently. All 4 patients achieved a CR.
Ten patients (14%) had treatment interruptions due to adverse events, and 4 (6%) discontinued treatment permanently because of adverse events.
Six patients had fatal adverse events. Three died after they went on to allogeneic transplant—1 of multiorgan failure, 1 of sepsis, and 1 of respiratory failure. The 3 other deaths were due to fungal infection, multiorgan failure, and thrombocytopenia.
Response and follow-up
Among the 70 patients who received the recommended dose of blinatumomab, 27 (39%) achieved a CR within the first 2 cycles. Fourteen of these patients (52%) achieved complete MRD response.
CRs were achieved across subgroups, and complete MRD response rates were similar across subgroups.
Thirteen of the 27 patients (48%) who achieved a CR went on to receive an allogeneic transplant.
At the end of the 2-year follow-up, 4 of the 27 complete responders were still in remission.
Two of the patients had relapsed but were still alive, 3 had withdrawn consent (1 in CR and 2 after relapse), 3 had died in CR after transplant, and 15 had relapsed and died.
Of the 43 patients who did not achieve a CR within the first 2 treatment cycles, 8 were still alive at the end of the 2-year follow-up. ![]()
The bispecific T-cell engager (BiTE®) antibody blinatumomab can produce complete responses (CRs) in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), according to a phase 1/2 study published in the Journal of Clinical Oncology.
Of the patients who received the recommended dosage of blinatumomab, 39% achieved a CR within the first 2 treatment cycles.
And 52% of these patients achieved a complete minimal residual disease (MRD) response.
“This study showed that [blinatumomab] can induce deep molecular remissions in children with highly refractory, multiply relapsed ALL,” said study author Lia Gore, MD, of University of Colorado Anschutz Medical Campus in Aurora, Colorado.
However, most of these remissions did not last. Although a few of the complete responders were still alive and in CR at the study’s 2-year follow-up, more than half had relapsed, and two-thirds had died.
This trial, known as Study ‘205, was supported by Amgen.
Study ‘205 included 93 pediatric patients with relapsed or refractory B-cell precursor ALL. Patients received blinatumomab as a continuous intravenous infusion—49 patients in the phase 1 portion of the trial and 44 in phase 2. The patients were followed for 2 years.
Toxicities and recommended dose
There were 4 dose-limiting toxicities during the phase 1 portion of the trial, and 2 of these events were fatal. One patient treated at 15 μg/m2/day developed grade 4 cytokine release syndrome (CRS), which was deemed related to grade 4 gastrointestinal hemorrhage.
Two patients treated at 30 μg/m2/day had grade 4 CRS. One case was attributed to grade 5 cardiac failure, and the other was treated successfully with tocilizumab.
One patient treated at 15 μg/m2/day had grade 5 respiratory failure with cardiac arrest after hypotonia and muscle weakness after 7 days of infusion with blinatumomab. This patient experienced febrile neutropenia and pneumonia shortly before the start of the infusion.
Based on these toxicities, the maximum-tolerated dose of blinatumomab was 15 μg/m2/day, but a step-wise dosage was recommended to reduce the risk of CRS.
So the recommended dose was 5 μg/m2/day on days 1-7 and 15 μg/m2/day on days 8-28 for cycle 1, and 15 μg/m2/day on days 1-28 for subsequent cycles.
Dose adjustment was possible in case of adverse events. Patients who responded to blinatumomab but later relapsed had the option to be retreated with blinatumomab.
Treatment at recommended dose
Seventy patients received at least 1 infusion of blinatumomab at the recommended dose. The median number of treatment cycles was 1 (range, 1 to 5).
The patients’ median age was 8 years (range, 7 months to 17 years). Forty patients (57%) had undergone allogeneic transplant prior to receiving blinatumomab, and 39 (56%) had refractory disease. Four patients had less than the 25% bone marrow blasts required for protocol entry but had more than 5% blasts.
Adverse events
The most common adverse events among the patients who received the recommended dose of blinatumomab were pyrexia (80%), anemia (41%), nausea (33%), and headache (30%).
The most frequent grade 3 or higher events were anemia (36%), thrombocytopenia (21%), febrile neutropenia (17%), hypokalemia (17%), and neutropenia (17%).
Eight patients developed CRS. Three had grade 3 and 1 had grade 4 CRS. Two of these patients had treatment interruptions, and 2 discontinued treatment permanently. All 4 patients achieved a CR.
Ten patients (14%) had treatment interruptions due to adverse events, and 4 (6%) discontinued treatment permanently because of adverse events.
Six patients had fatal adverse events. Three died after they went on to allogeneic transplant—1 of multiorgan failure, 1 of sepsis, and 1 of respiratory failure. The 3 other deaths were due to fungal infection, multiorgan failure, and thrombocytopenia.
Response and follow-up
Among the 70 patients who received the recommended dose of blinatumomab, 27 (39%) achieved a CR within the first 2 cycles. Fourteen of these patients (52%) achieved complete MRD response.
CRs were achieved across subgroups, and complete MRD response rates were similar across subgroups.
Thirteen of the 27 patients (48%) who achieved a CR went on to receive an allogeneic transplant.
At the end of the 2-year follow-up, 4 of the 27 complete responders were still in remission.
Two of the patients had relapsed but were still alive, 3 had withdrawn consent (1 in CR and 2 after relapse), 3 had died in CR after transplant, and 15 had relapsed and died.
Of the 43 patients who did not achieve a CR within the first 2 treatment cycles, 8 were still alive at the end of the 2-year follow-up. ![]()
The bispecific T-cell engager (BiTE®) antibody blinatumomab can produce complete responses (CRs) in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), according to a phase 1/2 study published in the Journal of Clinical Oncology.
Of the patients who received the recommended dosage of blinatumomab, 39% achieved a CR within the first 2 treatment cycles.
And 52% of these patients achieved a complete minimal residual disease (MRD) response.
“This study showed that [blinatumomab] can induce deep molecular remissions in children with highly refractory, multiply relapsed ALL,” said study author Lia Gore, MD, of University of Colorado Anschutz Medical Campus in Aurora, Colorado.
However, most of these remissions did not last. Although a few of the complete responders were still alive and in CR at the study’s 2-year follow-up, more than half had relapsed, and two-thirds had died.
This trial, known as Study ‘205, was supported by Amgen.
Study ‘205 included 93 pediatric patients with relapsed or refractory B-cell precursor ALL. Patients received blinatumomab as a continuous intravenous infusion—49 patients in the phase 1 portion of the trial and 44 in phase 2. The patients were followed for 2 years.
Toxicities and recommended dose
There were 4 dose-limiting toxicities during the phase 1 portion of the trial, and 2 of these events were fatal. One patient treated at 15 μg/m2/day developed grade 4 cytokine release syndrome (CRS), which was deemed related to grade 4 gastrointestinal hemorrhage.
Two patients treated at 30 μg/m2/day had grade 4 CRS. One case was attributed to grade 5 cardiac failure, and the other was treated successfully with tocilizumab.
One patient treated at 15 μg/m2/day had grade 5 respiratory failure with cardiac arrest after hypotonia and muscle weakness after 7 days of infusion with blinatumomab. This patient experienced febrile neutropenia and pneumonia shortly before the start of the infusion.
Based on these toxicities, the maximum-tolerated dose of blinatumomab was 15 μg/m2/day, but a step-wise dosage was recommended to reduce the risk of CRS.
So the recommended dose was 5 μg/m2/day on days 1-7 and 15 μg/m2/day on days 8-28 for cycle 1, and 15 μg/m2/day on days 1-28 for subsequent cycles.
Dose adjustment was possible in case of adverse events. Patients who responded to blinatumomab but later relapsed had the option to be retreated with blinatumomab.
Treatment at recommended dose
Seventy patients received at least 1 infusion of blinatumomab at the recommended dose. The median number of treatment cycles was 1 (range, 1 to 5).
The patients’ median age was 8 years (range, 7 months to 17 years). Forty patients (57%) had undergone allogeneic transplant prior to receiving blinatumomab, and 39 (56%) had refractory disease. Four patients had less than the 25% bone marrow blasts required for protocol entry but had more than 5% blasts.
Adverse events
The most common adverse events among the patients who received the recommended dose of blinatumomab were pyrexia (80%), anemia (41%), nausea (33%), and headache (30%).
The most frequent grade 3 or higher events were anemia (36%), thrombocytopenia (21%), febrile neutropenia (17%), hypokalemia (17%), and neutropenia (17%).
Eight patients developed CRS. Three had grade 3 and 1 had grade 4 CRS. Two of these patients had treatment interruptions, and 2 discontinued treatment permanently. All 4 patients achieved a CR.
Ten patients (14%) had treatment interruptions due to adverse events, and 4 (6%) discontinued treatment permanently because of adverse events.
Six patients had fatal adverse events. Three died after they went on to allogeneic transplant—1 of multiorgan failure, 1 of sepsis, and 1 of respiratory failure. The 3 other deaths were due to fungal infection, multiorgan failure, and thrombocytopenia.
Response and follow-up
Among the 70 patients who received the recommended dose of blinatumomab, 27 (39%) achieved a CR within the first 2 cycles. Fourteen of these patients (52%) achieved complete MRD response.
CRs were achieved across subgroups, and complete MRD response rates were similar across subgroups.
Thirteen of the 27 patients (48%) who achieved a CR went on to receive an allogeneic transplant.
At the end of the 2-year follow-up, 4 of the 27 complete responders were still in remission.
Two of the patients had relapsed but were still alive, 3 had withdrawn consent (1 in CR and 2 after relapse), 3 had died in CR after transplant, and 15 had relapsed and died.
Of the 43 patients who did not achieve a CR within the first 2 treatment cycles, 8 were still alive at the end of the 2-year follow-up. ![]()
Study shows more bleeding with rivaroxaban than dabigatran

A large, retrospective study comparing first-time users of rivaroxaban and dabigatran suggests that rivaroxaban poses a higher risk of bleeding in elderly patients with nonvalvular atrial fibrillation (NVAF).
Treatment with rivaroxaban was associated with significant increases in intracranial hemorrhage (ICH) and major extracranial bleeding, a nonsignificant reduction in thromboembolic stroke, and a nonsignificant increase in mortality.
David J. Graham, MD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues reported these results in JAMA Internal Medicine.
The investigators analyzed 118,891 NVAF patients older than 65 who were enrolled in Medicare. The patients had started treatment with dabigatran (150 mg twice daily) or rivaroxaban (20 mg once daily) from November 4, 2011, through June 30, 2014.
There were 52,240 patients receiving dabigatran and 66,651 receiving rivaroxaban.
The study’s primary outcomes were thromboembolic stroke, ICH, major extracranial bleeding events (including major gastrointestinal bleeding), and mortality.
The investigators adjusted for differences in baseline characteristics and calculated hazard ratios (HRs) for the primary outcomes. They also calculated adjusted incidence rate differences.
The data showed that, compared to patients who received dabigatran, those who received rivaroxaban had:
- 1.8 fewer cases of thromboembolic stroke per 1000 person-years (HR=0.81; 95% CI, 0.65-1.01; P=0.07)
- 2.3 excess cases of ICH per 1000 person-years (HR=1.65; 95% CI, 1.20-2.26; P=0.002)
- 13.0 excess cases of major extracranial bleeding per 1000 person-years (HR=1.48; 95% CI, 1.32-1.67; P<0.001)
- 9.4 excess cases of major gastrointestinal bleeding per 1000 person-years (HR=1.40; 95% CI, 1.23-1.59; P<0.001)
- 3.1 excess deaths per 1000 person-years (HR=1.15; 95% CI, 1.00-1.32; P=0.051).
The investigators noted that this study had several limitations. The mean follow-up time was 4 months, which led to a smaller sample size at longer durations of use.
In addition, the study was restricted to patients age 65 and older. Although this age group accounts for 80% of patients with NVAF, the comparative effects of treatment with dabigatran and rivaroxaban could be different in younger populations.
Because the study was observational, it may also be subject to residual confounding by unmeasured factors.
And finally, the investigators examined warfarin-naïve, first-time users of dabigatran and rivaroxaban. Results could differ in patients switching from warfarin to a novel oral anticoagulant. ![]()
A large, retrospective study comparing first-time users of rivaroxaban and dabigatran suggests that rivaroxaban poses a higher risk of bleeding in elderly patients with nonvalvular atrial fibrillation (NVAF).
Treatment with rivaroxaban was associated with significant increases in intracranial hemorrhage (ICH) and major extracranial bleeding, a nonsignificant reduction in thromboembolic stroke, and a nonsignificant increase in mortality.
David J. Graham, MD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues reported these results in JAMA Internal Medicine.
The investigators analyzed 118,891 NVAF patients older than 65 who were enrolled in Medicare. The patients had started treatment with dabigatran (150 mg twice daily) or rivaroxaban (20 mg once daily) from November 4, 2011, through June 30, 2014.
There were 52,240 patients receiving dabigatran and 66,651 receiving rivaroxaban.
The study’s primary outcomes were thromboembolic stroke, ICH, major extracranial bleeding events (including major gastrointestinal bleeding), and mortality.
The investigators adjusted for differences in baseline characteristics and calculated hazard ratios (HRs) for the primary outcomes. They also calculated adjusted incidence rate differences.
The data showed that, compared to patients who received dabigatran, those who received rivaroxaban had:
- 1.8 fewer cases of thromboembolic stroke per 1000 person-years (HR=0.81; 95% CI, 0.65-1.01; P=0.07)
- 2.3 excess cases of ICH per 1000 person-years (HR=1.65; 95% CI, 1.20-2.26; P=0.002)
- 13.0 excess cases of major extracranial bleeding per 1000 person-years (HR=1.48; 95% CI, 1.32-1.67; P<0.001)
- 9.4 excess cases of major gastrointestinal bleeding per 1000 person-years (HR=1.40; 95% CI, 1.23-1.59; P<0.001)
- 3.1 excess deaths per 1000 person-years (HR=1.15; 95% CI, 1.00-1.32; P=0.051).
The investigators noted that this study had several limitations. The mean follow-up time was 4 months, which led to a smaller sample size at longer durations of use.
In addition, the study was restricted to patients age 65 and older. Although this age group accounts for 80% of patients with NVAF, the comparative effects of treatment with dabigatran and rivaroxaban could be different in younger populations.
Because the study was observational, it may also be subject to residual confounding by unmeasured factors.
And finally, the investigators examined warfarin-naïve, first-time users of dabigatran and rivaroxaban. Results could differ in patients switching from warfarin to a novel oral anticoagulant. ![]()
A large, retrospective study comparing first-time users of rivaroxaban and dabigatran suggests that rivaroxaban poses a higher risk of bleeding in elderly patients with nonvalvular atrial fibrillation (NVAF).
Treatment with rivaroxaban was associated with significant increases in intracranial hemorrhage (ICH) and major extracranial bleeding, a nonsignificant reduction in thromboembolic stroke, and a nonsignificant increase in mortality.
David J. Graham, MD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues reported these results in JAMA Internal Medicine.
The investigators analyzed 118,891 NVAF patients older than 65 who were enrolled in Medicare. The patients had started treatment with dabigatran (150 mg twice daily) or rivaroxaban (20 mg once daily) from November 4, 2011, through June 30, 2014.
There were 52,240 patients receiving dabigatran and 66,651 receiving rivaroxaban.
The study’s primary outcomes were thromboembolic stroke, ICH, major extracranial bleeding events (including major gastrointestinal bleeding), and mortality.
The investigators adjusted for differences in baseline characteristics and calculated hazard ratios (HRs) for the primary outcomes. They also calculated adjusted incidence rate differences.
The data showed that, compared to patients who received dabigatran, those who received rivaroxaban had:
- 1.8 fewer cases of thromboembolic stroke per 1000 person-years (HR=0.81; 95% CI, 0.65-1.01; P=0.07)
- 2.3 excess cases of ICH per 1000 person-years (HR=1.65; 95% CI, 1.20-2.26; P=0.002)
- 13.0 excess cases of major extracranial bleeding per 1000 person-years (HR=1.48; 95% CI, 1.32-1.67; P<0.001)
- 9.4 excess cases of major gastrointestinal bleeding per 1000 person-years (HR=1.40; 95% CI, 1.23-1.59; P<0.001)
- 3.1 excess deaths per 1000 person-years (HR=1.15; 95% CI, 1.00-1.32; P=0.051).
The investigators noted that this study had several limitations. The mean follow-up time was 4 months, which led to a smaller sample size at longer durations of use.
In addition, the study was restricted to patients age 65 and older. Although this age group accounts for 80% of patients with NVAF, the comparative effects of treatment with dabigatran and rivaroxaban could be different in younger populations.
Because the study was observational, it may also be subject to residual confounding by unmeasured factors.
And finally, the investigators examined warfarin-naïve, first-time users of dabigatran and rivaroxaban. Results could differ in patients switching from warfarin to a novel oral anticoagulant. ![]()
Triplet improves PFS in relapsed/refractory MM

Results of the phase 3 POLLUX trial suggest that adding daratumumab to treatment with lenalidomide and dexamethasone can improve progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (MM).
Compared to patients who received only lenalidomide and dexamethasone, those who received daratumumab as well had a significantly higher response rate and longer PFS.
Treatment with daratumumab was also associated with infusion-related reactions and a higher incidence of neutropenia.
These results were published in NEJM. They were previously presented at the 21st Congress of the European Hematology Association. The POLLUX trial was funded by Janssen Research and Development.
POLLUX enrolled 569 patients who had relapsed or refractory MM. The patients were randomized to receive either daratumumab, lenalidomide, and dexamethasone (DRd, n=286) or lenalidomide and dexamethasone alone (Rd, n=283).
Patient characteristics were similar between the treatment arms. The median age was 65 in both arms (overall range, 34-89). About half of patients in each arm had ISS stage I disease, about half had an ECOG performance score of 1 or 2, and patients were, roughly, a median of 4 years from diagnosis.
In both arms, patients had a median of 1 prior lines of therapy (overall range, 1-11).
More than 60% of patients in both arms (63%-64%) had received a prior transplant; 86% had received a proteasome inhibitor; 55% had received an immunomodulatory agent; 44% had received a proteasome inhibitor and an immunomodulatory agent; and 41%-43% had received a proteasome inhibitor, an immunomodulatory agent, and an alkylating agent.
Discontinuation
At the clinical cutoff date on March 7, 2016, 23.3% (n=66) of patients in the DRd arm and 47.0% (n=132) of those in the Rd arm discontinued treatment.
The most common reasons for discontinuation were progression (14.1% in the DRd arm and 34.2% in the Rd arm) and adverse events (6.7% and 8.2%, respectively).
Response
The overall response rate among evaluable patients (281 in the DRd arm and 276 in the Rd arm) was 92.9% in DRd arm and 76.4% in the Rd arm (P<0.001).
The rates of complete response or better were 43.1% and 19.2%, respectively. And the rates of stringent complete response were 18.1% and 7.2%, respectively.
In the DRd arm, 22.4% of patients had results below the threshold for minimal residual disease (1 tumor cell per 105 white cells). The same was true for 4.6% of patients in the Rd arm (P<0.001).
PFS and OS
At a median follow-up of 13.5 months, there were 169 events of disease progression or death. The incidence was 18.5% (53/286) in the DRd arm and 41% (116/283) in the Rd arm. The hazard ratio was 0.37 (P<0.001).
At 12 months, the rate of PFS was 83.2% in the DRd arm and 60.1% in the Rd arm. The median PFS has not been reached in the DRd arm but was 18.4 months for patients in the Rd arm.
The improvement in PFS for the DRd arm was seen across all patient subgroups, regardless of age, ISS stage, prior treatment, etc.
In addition, there was an overall survival (OS) advantage with DRd. The 12-month OS was 92.1% in the DRd arm and 86.8% in the Rd arm.
The median OS was not reached in the DRd arm and was 20.3 months in the Rd arm. The hazard ratio was 0.64 (P=0.0534). The researchers said follow-up for long-term survival is ongoing.
Safety
The most common hematologic adverse events (in the DRd and Rd arms, respectively) were neutropenia (59.4% and 43.1%), anemia (31.1% and 34.9%), thrombocytopenia (26.9% and 27.4%), lymphopenia (6.0% and 5.3%), and febrile neutropenia (5.7% and 2.5%). Deep vein thrombosis occurred in 1.8% and 3.9% of patients, respectively.
The most common non-hematologic adverse events (in the DRd and Rd arms, respectively) were diarrhea (42.8% and 24.6%), fatigue (35.3% and 27.8%), upper respiratory tract infection (31.8% and 20.6%), constipation (29.3% and 25.3%), cough (29.0% and 12.5%), muscle spasms (25.8% and 18.5%), and pneumonia (14.1% and 13.2%).
Patients in the DRd arm had a higher rate of several grade 3/4 events, including neutropenia (51.9% and 37.0%), diarrhea (5.3% and 3.2%), fatigue (6.4% and 2.5%), nausea (1.4% and 0%), dyspnea (3.2% and 0.7%), and infection (28.3% and 22.8%).
Serious adverse events occurred in 48.8% of patients in the DRd arm and 42.0% in the Rd arm. Pneumonia was the most common serious event, occurring in 8.1% and 8.5% of patients, respectively.
Daratumumab-infusion-related reactions occurred in 47.7% of patients, were mostly grade 1/2, and occurred predominantly during the first infusion.
The researchers also noted that daratumumab interferes with laboratory-based blood compatibility tests because the drug binds to CD38 on red cells.
Daratumumab development
Data from the phase 3 CASTOR trial—in which researchers compared daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone alone—were also recently published in NEJM.
“Following the publication of the phase 3 CASTOR data, we are pleased that the positive data from the phase 3 POLLUX study has now also been published in the New England Journal of Medicine,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“The data from this study formed the basis, along with data from the CASTOR study, of 2 regulatory submissions in August—the supplemental Biologics License Application submitted to the US Food and Drug Administration and the submission of the variation to the Marketing Authorization to the European Medicines Agency.”
Both submissions seek to expand the current indication for daratumumab so the drug can be used in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat MM patients who have received at least 1 prior therapy.
Daratumumab currently has conditional approval from the European Commission as monotherapy for adults with relapsed and refractory MM who progressed on their last therapy and have received treatment with a proteasome inhibitor and an immunomodulatory agent.
Daratumumab has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab. ![]()
Results of the phase 3 POLLUX trial suggest that adding daratumumab to treatment with lenalidomide and dexamethasone can improve progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (MM).
Compared to patients who received only lenalidomide and dexamethasone, those who received daratumumab as well had a significantly higher response rate and longer PFS.
Treatment with daratumumab was also associated with infusion-related reactions and a higher incidence of neutropenia.
These results were published in NEJM. They were previously presented at the 21st Congress of the European Hematology Association. The POLLUX trial was funded by Janssen Research and Development.
POLLUX enrolled 569 patients who had relapsed or refractory MM. The patients were randomized to receive either daratumumab, lenalidomide, and dexamethasone (DRd, n=286) or lenalidomide and dexamethasone alone (Rd, n=283).
Patient characteristics were similar between the treatment arms. The median age was 65 in both arms (overall range, 34-89). About half of patients in each arm had ISS stage I disease, about half had an ECOG performance score of 1 or 2, and patients were, roughly, a median of 4 years from diagnosis.
In both arms, patients had a median of 1 prior lines of therapy (overall range, 1-11).
More than 60% of patients in both arms (63%-64%) had received a prior transplant; 86% had received a proteasome inhibitor; 55% had received an immunomodulatory agent; 44% had received a proteasome inhibitor and an immunomodulatory agent; and 41%-43% had received a proteasome inhibitor, an immunomodulatory agent, and an alkylating agent.
Discontinuation
At the clinical cutoff date on March 7, 2016, 23.3% (n=66) of patients in the DRd arm and 47.0% (n=132) of those in the Rd arm discontinued treatment.
The most common reasons for discontinuation were progression (14.1% in the DRd arm and 34.2% in the Rd arm) and adverse events (6.7% and 8.2%, respectively).
Response
The overall response rate among evaluable patients (281 in the DRd arm and 276 in the Rd arm) was 92.9% in DRd arm and 76.4% in the Rd arm (P<0.001).
The rates of complete response or better were 43.1% and 19.2%, respectively. And the rates of stringent complete response were 18.1% and 7.2%, respectively.
In the DRd arm, 22.4% of patients had results below the threshold for minimal residual disease (1 tumor cell per 105 white cells). The same was true for 4.6% of patients in the Rd arm (P<0.001).
PFS and OS
At a median follow-up of 13.5 months, there were 169 events of disease progression or death. The incidence was 18.5% (53/286) in the DRd arm and 41% (116/283) in the Rd arm. The hazard ratio was 0.37 (P<0.001).
At 12 months, the rate of PFS was 83.2% in the DRd arm and 60.1% in the Rd arm. The median PFS has not been reached in the DRd arm but was 18.4 months for patients in the Rd arm.
The improvement in PFS for the DRd arm was seen across all patient subgroups, regardless of age, ISS stage, prior treatment, etc.
In addition, there was an overall survival (OS) advantage with DRd. The 12-month OS was 92.1% in the DRd arm and 86.8% in the Rd arm.
The median OS was not reached in the DRd arm and was 20.3 months in the Rd arm. The hazard ratio was 0.64 (P=0.0534). The researchers said follow-up for long-term survival is ongoing.
Safety
The most common hematologic adverse events (in the DRd and Rd arms, respectively) were neutropenia (59.4% and 43.1%), anemia (31.1% and 34.9%), thrombocytopenia (26.9% and 27.4%), lymphopenia (6.0% and 5.3%), and febrile neutropenia (5.7% and 2.5%). Deep vein thrombosis occurred in 1.8% and 3.9% of patients, respectively.
The most common non-hematologic adverse events (in the DRd and Rd arms, respectively) were diarrhea (42.8% and 24.6%), fatigue (35.3% and 27.8%), upper respiratory tract infection (31.8% and 20.6%), constipation (29.3% and 25.3%), cough (29.0% and 12.5%), muscle spasms (25.8% and 18.5%), and pneumonia (14.1% and 13.2%).
Patients in the DRd arm had a higher rate of several grade 3/4 events, including neutropenia (51.9% and 37.0%), diarrhea (5.3% and 3.2%), fatigue (6.4% and 2.5%), nausea (1.4% and 0%), dyspnea (3.2% and 0.7%), and infection (28.3% and 22.8%).
Serious adverse events occurred in 48.8% of patients in the DRd arm and 42.0% in the Rd arm. Pneumonia was the most common serious event, occurring in 8.1% and 8.5% of patients, respectively.
Daratumumab-infusion-related reactions occurred in 47.7% of patients, were mostly grade 1/2, and occurred predominantly during the first infusion.
The researchers also noted that daratumumab interferes with laboratory-based blood compatibility tests because the drug binds to CD38 on red cells.
Daratumumab development
Data from the phase 3 CASTOR trial—in which researchers compared daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone alone—were also recently published in NEJM.
“Following the publication of the phase 3 CASTOR data, we are pleased that the positive data from the phase 3 POLLUX study has now also been published in the New England Journal of Medicine,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“The data from this study formed the basis, along with data from the CASTOR study, of 2 regulatory submissions in August—the supplemental Biologics License Application submitted to the US Food and Drug Administration and the submission of the variation to the Marketing Authorization to the European Medicines Agency.”
Both submissions seek to expand the current indication for daratumumab so the drug can be used in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat MM patients who have received at least 1 prior therapy.
Daratumumab currently has conditional approval from the European Commission as monotherapy for adults with relapsed and refractory MM who progressed on their last therapy and have received treatment with a proteasome inhibitor and an immunomodulatory agent.
Daratumumab has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab. ![]()
Results of the phase 3 POLLUX trial suggest that adding daratumumab to treatment with lenalidomide and dexamethasone can improve progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (MM).
Compared to patients who received only lenalidomide and dexamethasone, those who received daratumumab as well had a significantly higher response rate and longer PFS.
Treatment with daratumumab was also associated with infusion-related reactions and a higher incidence of neutropenia.
These results were published in NEJM. They were previously presented at the 21st Congress of the European Hematology Association. The POLLUX trial was funded by Janssen Research and Development.
POLLUX enrolled 569 patients who had relapsed or refractory MM. The patients were randomized to receive either daratumumab, lenalidomide, and dexamethasone (DRd, n=286) or lenalidomide and dexamethasone alone (Rd, n=283).
Patient characteristics were similar between the treatment arms. The median age was 65 in both arms (overall range, 34-89). About half of patients in each arm had ISS stage I disease, about half had an ECOG performance score of 1 or 2, and patients were, roughly, a median of 4 years from diagnosis.
In both arms, patients had a median of 1 prior lines of therapy (overall range, 1-11).
More than 60% of patients in both arms (63%-64%) had received a prior transplant; 86% had received a proteasome inhibitor; 55% had received an immunomodulatory agent; 44% had received a proteasome inhibitor and an immunomodulatory agent; and 41%-43% had received a proteasome inhibitor, an immunomodulatory agent, and an alkylating agent.
Discontinuation
At the clinical cutoff date on March 7, 2016, 23.3% (n=66) of patients in the DRd arm and 47.0% (n=132) of those in the Rd arm discontinued treatment.
The most common reasons for discontinuation were progression (14.1% in the DRd arm and 34.2% in the Rd arm) and adverse events (6.7% and 8.2%, respectively).
Response
The overall response rate among evaluable patients (281 in the DRd arm and 276 in the Rd arm) was 92.9% in DRd arm and 76.4% in the Rd arm (P<0.001).
The rates of complete response or better were 43.1% and 19.2%, respectively. And the rates of stringent complete response were 18.1% and 7.2%, respectively.
In the DRd arm, 22.4% of patients had results below the threshold for minimal residual disease (1 tumor cell per 105 white cells). The same was true for 4.6% of patients in the Rd arm (P<0.001).
PFS and OS
At a median follow-up of 13.5 months, there were 169 events of disease progression or death. The incidence was 18.5% (53/286) in the DRd arm and 41% (116/283) in the Rd arm. The hazard ratio was 0.37 (P<0.001).
At 12 months, the rate of PFS was 83.2% in the DRd arm and 60.1% in the Rd arm. The median PFS has not been reached in the DRd arm but was 18.4 months for patients in the Rd arm.
The improvement in PFS for the DRd arm was seen across all patient subgroups, regardless of age, ISS stage, prior treatment, etc.
In addition, there was an overall survival (OS) advantage with DRd. The 12-month OS was 92.1% in the DRd arm and 86.8% in the Rd arm.
The median OS was not reached in the DRd arm and was 20.3 months in the Rd arm. The hazard ratio was 0.64 (P=0.0534). The researchers said follow-up for long-term survival is ongoing.
Safety
The most common hematologic adverse events (in the DRd and Rd arms, respectively) were neutropenia (59.4% and 43.1%), anemia (31.1% and 34.9%), thrombocytopenia (26.9% and 27.4%), lymphopenia (6.0% and 5.3%), and febrile neutropenia (5.7% and 2.5%). Deep vein thrombosis occurred in 1.8% and 3.9% of patients, respectively.
The most common non-hematologic adverse events (in the DRd and Rd arms, respectively) were diarrhea (42.8% and 24.6%), fatigue (35.3% and 27.8%), upper respiratory tract infection (31.8% and 20.6%), constipation (29.3% and 25.3%), cough (29.0% and 12.5%), muscle spasms (25.8% and 18.5%), and pneumonia (14.1% and 13.2%).
Patients in the DRd arm had a higher rate of several grade 3/4 events, including neutropenia (51.9% and 37.0%), diarrhea (5.3% and 3.2%), fatigue (6.4% and 2.5%), nausea (1.4% and 0%), dyspnea (3.2% and 0.7%), and infection (28.3% and 22.8%).
Serious adverse events occurred in 48.8% of patients in the DRd arm and 42.0% in the Rd arm. Pneumonia was the most common serious event, occurring in 8.1% and 8.5% of patients, respectively.
Daratumumab-infusion-related reactions occurred in 47.7% of patients, were mostly grade 1/2, and occurred predominantly during the first infusion.
The researchers also noted that daratumumab interferes with laboratory-based blood compatibility tests because the drug binds to CD38 on red cells.
Daratumumab development
Data from the phase 3 CASTOR trial—in which researchers compared daratumumab, bortezomib, and dexamethasone to bortezomib and dexamethasone alone—were also recently published in NEJM.
“Following the publication of the phase 3 CASTOR data, we are pleased that the positive data from the phase 3 POLLUX study has now also been published in the New England Journal of Medicine,” said Jan van de Winkel, PhD, chief executive officer of Genmab.
“The data from this study formed the basis, along with data from the CASTOR study, of 2 regulatory submissions in August—the supplemental Biologics License Application submitted to the US Food and Drug Administration and the submission of the variation to the Marketing Authorization to the European Medicines Agency.”
Both submissions seek to expand the current indication for daratumumab so the drug can be used in combination with lenalidomide and dexamethasone or bortezomib and dexamethasone to treat MM patients who have received at least 1 prior therapy.
Daratumumab currently has conditional approval from the European Commission as monotherapy for adults with relapsed and refractory MM who progressed on their last therapy and have received treatment with a proteasome inhibitor and an immunomodulatory agent.
Daratumumab has accelerated approval in the US as monotherapy for MM patients who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is being developed by Janssen Biotech, Inc. under an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab. ![]()
Drug granted conditional approval to treat CLL in Canada

of venetoclax (Venclexta)
Photo courtesy of AbbVie
Health Canada has issued a Notice of Compliance with Conditions (NOC/c) for the BCL-2 inhibitor venetoclax (Venclexta™).
This means venetoclax is conditionally approved for use in patients with previously treated chronic lymphocytic leukemia (CLL) who have 17p deletion or no other available treatment options.
An NOC/c is authorization to market a drug with the condition that the sponsor perform additional studies to verify a clinical benefit.
The NOC/c policy is designed to provide access to:
- Drugs that can treat serious, life-threatening, or severely debilitating diseases
- Drugs that can treat conditions for which no drug is currently marketed in Canada
- Drugs that provide a significant increase in efficacy or significant decrease in risk when compared to existing drugs marketed in Canada.
Venetoclax (previously ABT‐199) is being developed by AbbVie and Genentech, a member of the Roche Group. The drug is jointly commercialized by the companies in the US and by AbbVie outside of the US.
Venetoclax is currently under evaluation in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL.
Phase 2 trial
Results from a phase 2 trial of venetoclax in CLL (M13-982, NCT01889186) were published in The Lancet Oncology in June. The trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
At a median follow-up of 12.1 months, 85 patients had responded to treatment, for an overall response rate of 79%.
Eight patients (8%) achieved a complete response or complete response with incomplete count recovery, 3 (3%) had a near-partial response, and 74 (69%) had a partial response. Twenty-two patients (21%) did not respond.
At the time of analysis, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72%, and the overall survival estimate was 87%.
The incidence of treatment-emergent adverse was 96%. The most frequent grade 3/4 adverse events were neutropenia (40%), infection (20%), anemia (18%), and thrombocytopenia (15%).
The incidence of serious adverse events was 55%. The most common of these events were pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Grade 3 laboratory tumor lysis syndrome (TLS) was reported in 5 patients during the ramp-up period only. Three of these patients continued on venetoclax, but 2 patients required a dose interruption of 1 day each.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS. ![]()
of venetoclax (Venclexta)
Photo courtesy of AbbVie
Health Canada has issued a Notice of Compliance with Conditions (NOC/c) for the BCL-2 inhibitor venetoclax (Venclexta™).
This means venetoclax is conditionally approved for use in patients with previously treated chronic lymphocytic leukemia (CLL) who have 17p deletion or no other available treatment options.
An NOC/c is authorization to market a drug with the condition that the sponsor perform additional studies to verify a clinical benefit.
The NOC/c policy is designed to provide access to:
- Drugs that can treat serious, life-threatening, or severely debilitating diseases
- Drugs that can treat conditions for which no drug is currently marketed in Canada
- Drugs that provide a significant increase in efficacy or significant decrease in risk when compared to existing drugs marketed in Canada.
Venetoclax (previously ABT‐199) is being developed by AbbVie and Genentech, a member of the Roche Group. The drug is jointly commercialized by the companies in the US and by AbbVie outside of the US.
Venetoclax is currently under evaluation in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL.
Phase 2 trial
Results from a phase 2 trial of venetoclax in CLL (M13-982, NCT01889186) were published in The Lancet Oncology in June. The trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
At a median follow-up of 12.1 months, 85 patients had responded to treatment, for an overall response rate of 79%.
Eight patients (8%) achieved a complete response or complete response with incomplete count recovery, 3 (3%) had a near-partial response, and 74 (69%) had a partial response. Twenty-two patients (21%) did not respond.
At the time of analysis, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72%, and the overall survival estimate was 87%.
The incidence of treatment-emergent adverse was 96%. The most frequent grade 3/4 adverse events were neutropenia (40%), infection (20%), anemia (18%), and thrombocytopenia (15%).
The incidence of serious adverse events was 55%. The most common of these events were pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Grade 3 laboratory tumor lysis syndrome (TLS) was reported in 5 patients during the ramp-up period only. Three of these patients continued on venetoclax, but 2 patients required a dose interruption of 1 day each.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS. ![]()
of venetoclax (Venclexta)
Photo courtesy of AbbVie
Health Canada has issued a Notice of Compliance with Conditions (NOC/c) for the BCL-2 inhibitor venetoclax (Venclexta™).
This means venetoclax is conditionally approved for use in patients with previously treated chronic lymphocytic leukemia (CLL) who have 17p deletion or no other available treatment options.
An NOC/c is authorization to market a drug with the condition that the sponsor perform additional studies to verify a clinical benefit.
The NOC/c policy is designed to provide access to:
- Drugs that can treat serious, life-threatening, or severely debilitating diseases
- Drugs that can treat conditions for which no drug is currently marketed in Canada
- Drugs that provide a significant increase in efficacy or significant decrease in risk when compared to existing drugs marketed in Canada.
Venetoclax (previously ABT‐199) is being developed by AbbVie and Genentech, a member of the Roche Group. The drug is jointly commercialized by the companies in the US and by AbbVie outside of the US.
Venetoclax is currently under evaluation in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL.
Phase 2 trial
Results from a phase 2 trial of venetoclax in CLL (M13-982, NCT01889186) were published in The Lancet Oncology in June. The trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
At a median follow-up of 12.1 months, 85 patients had responded to treatment, for an overall response rate of 79%.
Eight patients (8%) achieved a complete response or complete response with incomplete count recovery, 3 (3%) had a near-partial response, and 74 (69%) had a partial response. Twenty-two patients (21%) did not respond.
At the time of analysis, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72%, and the overall survival estimate was 87%.
The incidence of treatment-emergent adverse was 96%. The most frequent grade 3/4 adverse events were neutropenia (40%), infection (20%), anemia (18%), and thrombocytopenia (15%).
The incidence of serious adverse events was 55%. The most common of these events were pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Grade 3 laboratory tumor lysis syndrome (TLS) was reported in 5 patients during the ramp-up period only. Three of these patients continued on venetoclax, but 2 patients required a dose interruption of 1 day each.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS. ![]()
Findings may aid drug delivery, bioimaging

to illuminate microfluidic device
simulating a blood vessel.
Photo from Anson Ma/UConn
A study published in Biophysical Journal has revealed new information about how particles behave in the bloodstream, and investigators believe the findings have implications for bioimaging and targeted drug delivery in cancer.
The investigators used a microfluidic channel device to observe, track, and measure how individual particles behave in a simulated blood vessel.
Their goal was to learn more about the physics influencing a particle’s behavior as it travels in the blood and to determine which particle size might be the most effective for delivering drugs to their targets.
“Even before particles reach a target site, you have to worry about what is going to happen with them after they get injected into the bloodstream,” said study author Anson Ma, PhD, of the University of Connecticut in Storrs, Connecticut.
“Are they going to clump together? How are they going to move around? Are they going to get swept away and flushed out of our bodies?”
Using a high-powered fluorescence microscope, Dr Ma and his colleagues were able to observe particles being carried along in the simulated blood vessel in what could be described as a vascular “Running of the Bulls.”
Red blood cells raced through the middle of the channel, and the particles were carried along in the rush, bumping and bouncing off the blood cells until they were pushed to open spaces—called the cell-free layer—along the vessel’s walls.
The investigators found that larger particles—the optimum size appeared to be about 2 microns—were most likely to get pushed to the cell-free layer, where their chances of carrying a drug to a targeted site are greatest.
The team also determined that 2 microns was the largest size that should be used if particles are going to have any chance of going through the leaky blood vessel walls to the site.
“When it comes to using particles for the delivery of cancer drugs, size matters,” Dr Ma said. “When you have a bigger particle, the chance of it bumping into blood cells is much higher, there are a lot more collisions, and they tend to get pushed to the blood vessel walls.”
These results were somewhat surprising. The investigators had theorized that smaller particles would probably be the most effective since they would move the most in collisions with blood cells.
But the opposite proved true. The smaller particles appeared to skirt through the mass of moving blood cells and were less likely to get bounced to the cell-free layer.
Knowing how particles behave in the circulatory system should help improve targeted drug delivery, Dr Ma said. And this should further reduce the side effects caused by potent cancer drugs missing their target.
Measuring how different sized particles move in the bloodstream may also be beneficial in bioimaging, where the goal is to keep particles circulating in the bloodstream long enough for imaging to occur. In that case, smaller particles would be better, Dr Ma said.
Moving forward, Dr Ma would like to explore other aspects of particle flow in the circulatory system, such as how particles behave when they pass through a constricted area, like from a blood vessel to a capillary.
Capillaries are only about 7 microns in diameter. Dr Ma said he would like to know how that constricted space might impact particle flow or the ability of particles to accumulate near the vessel walls.
“We have all of this complex geometry in our bodies,” Dr Ma said. “Most people just assume there is no impact when a particle moves from a bigger channel to a smaller channel because they haven’t quantified it. Our plan is to do some experiments to look at this more carefully, building on the work that we just published.” ![]()
to illuminate microfluidic device
simulating a blood vessel.
Photo from Anson Ma/UConn
A study published in Biophysical Journal has revealed new information about how particles behave in the bloodstream, and investigators believe the findings have implications for bioimaging and targeted drug delivery in cancer.
The investigators used a microfluidic channel device to observe, track, and measure how individual particles behave in a simulated blood vessel.
Their goal was to learn more about the physics influencing a particle’s behavior as it travels in the blood and to determine which particle size might be the most effective for delivering drugs to their targets.
“Even before particles reach a target site, you have to worry about what is going to happen with them after they get injected into the bloodstream,” said study author Anson Ma, PhD, of the University of Connecticut in Storrs, Connecticut.
“Are they going to clump together? How are they going to move around? Are they going to get swept away and flushed out of our bodies?”
Using a high-powered fluorescence microscope, Dr Ma and his colleagues were able to observe particles being carried along in the simulated blood vessel in what could be described as a vascular “Running of the Bulls.”
Red blood cells raced through the middle of the channel, and the particles were carried along in the rush, bumping and bouncing off the blood cells until they were pushed to open spaces—called the cell-free layer—along the vessel’s walls.
The investigators found that larger particles—the optimum size appeared to be about 2 microns—were most likely to get pushed to the cell-free layer, where their chances of carrying a drug to a targeted site are greatest.
The team also determined that 2 microns was the largest size that should be used if particles are going to have any chance of going through the leaky blood vessel walls to the site.
“When it comes to using particles for the delivery of cancer drugs, size matters,” Dr Ma said. “When you have a bigger particle, the chance of it bumping into blood cells is much higher, there are a lot more collisions, and they tend to get pushed to the blood vessel walls.”
These results were somewhat surprising. The investigators had theorized that smaller particles would probably be the most effective since they would move the most in collisions with blood cells.
But the opposite proved true. The smaller particles appeared to skirt through the mass of moving blood cells and were less likely to get bounced to the cell-free layer.
Knowing how particles behave in the circulatory system should help improve targeted drug delivery, Dr Ma said. And this should further reduce the side effects caused by potent cancer drugs missing their target.
Measuring how different sized particles move in the bloodstream may also be beneficial in bioimaging, where the goal is to keep particles circulating in the bloodstream long enough for imaging to occur. In that case, smaller particles would be better, Dr Ma said.
Moving forward, Dr Ma would like to explore other aspects of particle flow in the circulatory system, such as how particles behave when they pass through a constricted area, like from a blood vessel to a capillary.
Capillaries are only about 7 microns in diameter. Dr Ma said he would like to know how that constricted space might impact particle flow or the ability of particles to accumulate near the vessel walls.
“We have all of this complex geometry in our bodies,” Dr Ma said. “Most people just assume there is no impact when a particle moves from a bigger channel to a smaller channel because they haven’t quantified it. Our plan is to do some experiments to look at this more carefully, building on the work that we just published.” ![]()
to illuminate microfluidic device
simulating a blood vessel.
Photo from Anson Ma/UConn
A study published in Biophysical Journal has revealed new information about how particles behave in the bloodstream, and investigators believe the findings have implications for bioimaging and targeted drug delivery in cancer.
The investigators used a microfluidic channel device to observe, track, and measure how individual particles behave in a simulated blood vessel.
Their goal was to learn more about the physics influencing a particle’s behavior as it travels in the blood and to determine which particle size might be the most effective for delivering drugs to their targets.
“Even before particles reach a target site, you have to worry about what is going to happen with them after they get injected into the bloodstream,” said study author Anson Ma, PhD, of the University of Connecticut in Storrs, Connecticut.
“Are they going to clump together? How are they going to move around? Are they going to get swept away and flushed out of our bodies?”
Using a high-powered fluorescence microscope, Dr Ma and his colleagues were able to observe particles being carried along in the simulated blood vessel in what could be described as a vascular “Running of the Bulls.”
Red blood cells raced through the middle of the channel, and the particles were carried along in the rush, bumping and bouncing off the blood cells until they were pushed to open spaces—called the cell-free layer—along the vessel’s walls.
The investigators found that larger particles—the optimum size appeared to be about 2 microns—were most likely to get pushed to the cell-free layer, where their chances of carrying a drug to a targeted site are greatest.
The team also determined that 2 microns was the largest size that should be used if particles are going to have any chance of going through the leaky blood vessel walls to the site.
“When it comes to using particles for the delivery of cancer drugs, size matters,” Dr Ma said. “When you have a bigger particle, the chance of it bumping into blood cells is much higher, there are a lot more collisions, and they tend to get pushed to the blood vessel walls.”
These results were somewhat surprising. The investigators had theorized that smaller particles would probably be the most effective since they would move the most in collisions with blood cells.
But the opposite proved true. The smaller particles appeared to skirt through the mass of moving blood cells and were less likely to get bounced to the cell-free layer.
Knowing how particles behave in the circulatory system should help improve targeted drug delivery, Dr Ma said. And this should further reduce the side effects caused by potent cancer drugs missing their target.
Measuring how different sized particles move in the bloodstream may also be beneficial in bioimaging, where the goal is to keep particles circulating in the bloodstream long enough for imaging to occur. In that case, smaller particles would be better, Dr Ma said.
Moving forward, Dr Ma would like to explore other aspects of particle flow in the circulatory system, such as how particles behave when they pass through a constricted area, like from a blood vessel to a capillary.
Capillaries are only about 7 microns in diameter. Dr Ma said he would like to know how that constricted space might impact particle flow or the ability of particles to accumulate near the vessel walls.
“We have all of this complex geometry in our bodies,” Dr Ma said. “Most people just assume there is no impact when a particle moves from a bigger channel to a smaller channel because they haven’t quantified it. Our plan is to do some experiments to look at this more carefully, building on the work that we just published.”
Vitamin D affects HSPC production, team says
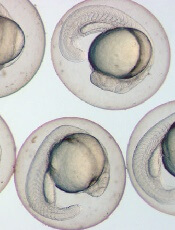
Photo by Ian Johnston
The availability of vitamin D during embryonic development can affect hematopoietic stem and progenitor cells (HSPCs), according to research published in Cell Reports.
Experiments with zebrafish embryos suggested that vitamin D acts directly on HSPCs to increase proliferation.
Similarly, in HSPCs from human umbilical cords, treatment with vitamin D enhanced hematopoietic colony numbers.
Researchers therefore theorized that vitamin D supplementation might be useful for HSPC expansion prior to transplant.
“We clearly showed that not getting enough vitamin D can alter how blood stem cells are formed,” said study author Trista North, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Vitamin D was having a direct response on the blood stem cells, and it changed what those cells did in terms of multiplying and staying alive.”
The researchers found, in both human and zebrafish tissue, that 1,25(OH)D3 (active vitamin D3) had an impact on HSPC production and function.
Investigation into the mechanism revealed that CXCL8-CXCR1/2 signaling functions downstream of 1,25(OH)D3-mediated vitamin D receptor stimulation to directly regulate HSPC production and expansion.
“What was surprising was that vitamin D is having an impact so early,” Dr North said. “We really only thought about vitamin D in terms of bone development and maintenance, but we clearly show that, whether they were zebrafish or human blood stem cells, they can respond directly to the nutrient.”
One caveat is the researchers did face difficulty testing the response in mice, as the animals don’t have the same vitamin D inflammatory targets observed in zebrafish and humans.
Additionally, the team didn’t know the vitamin D levels in the umbilical cord blood samples they tested, which may have influenced the outcome of their analysis.
As a next step, Dr North and her colleagues hope to test cord blood samples for which they know the vitamin D status to see if umbilical cords with healthy levels respond better or worse to stimulation than cords from vitamin-D-deficient donors.
Photo by Ian Johnston
The availability of vitamin D during embryonic development can affect hematopoietic stem and progenitor cells (HSPCs), according to research published in Cell Reports.
Experiments with zebrafish embryos suggested that vitamin D acts directly on HSPCs to increase proliferation.
Similarly, in HSPCs from human umbilical cords, treatment with vitamin D enhanced hematopoietic colony numbers.
Researchers therefore theorized that vitamin D supplementation might be useful for HSPC expansion prior to transplant.
“We clearly showed that not getting enough vitamin D can alter how blood stem cells are formed,” said study author Trista North, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Vitamin D was having a direct response on the blood stem cells, and it changed what those cells did in terms of multiplying and staying alive.”
The researchers found, in both human and zebrafish tissue, that 1,25(OH)D3 (active vitamin D3) had an impact on HSPC production and function.
Investigation into the mechanism revealed that CXCL8-CXCR1/2 signaling functions downstream of 1,25(OH)D3-mediated vitamin D receptor stimulation to directly regulate HSPC production and expansion.
“What was surprising was that vitamin D is having an impact so early,” Dr North said. “We really only thought about vitamin D in terms of bone development and maintenance, but we clearly show that, whether they were zebrafish or human blood stem cells, they can respond directly to the nutrient.”
One caveat is the researchers did face difficulty testing the response in mice, as the animals don’t have the same vitamin D inflammatory targets observed in zebrafish and humans.
Additionally, the team didn’t know the vitamin D levels in the umbilical cord blood samples they tested, which may have influenced the outcome of their analysis.
As a next step, Dr North and her colleagues hope to test cord blood samples for which they know the vitamin D status to see if umbilical cords with healthy levels respond better or worse to stimulation than cords from vitamin-D-deficient donors.
Photo by Ian Johnston
The availability of vitamin D during embryonic development can affect hematopoietic stem and progenitor cells (HSPCs), according to research published in Cell Reports.
Experiments with zebrafish embryos suggested that vitamin D acts directly on HSPCs to increase proliferation.
Similarly, in HSPCs from human umbilical cords, treatment with vitamin D enhanced hematopoietic colony numbers.
Researchers therefore theorized that vitamin D supplementation might be useful for HSPC expansion prior to transplant.
“We clearly showed that not getting enough vitamin D can alter how blood stem cells are formed,” said study author Trista North, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Vitamin D was having a direct response on the blood stem cells, and it changed what those cells did in terms of multiplying and staying alive.”
The researchers found, in both human and zebrafish tissue, that 1,25(OH)D3 (active vitamin D3) had an impact on HSPC production and function.
Investigation into the mechanism revealed that CXCL8-CXCR1/2 signaling functions downstream of 1,25(OH)D3-mediated vitamin D receptor stimulation to directly regulate HSPC production and expansion.
“What was surprising was that vitamin D is having an impact so early,” Dr North said. “We really only thought about vitamin D in terms of bone development and maintenance, but we clearly show that, whether they were zebrafish or human blood stem cells, they can respond directly to the nutrient.”
One caveat is the researchers did face difficulty testing the response in mice, as the animals don’t have the same vitamin D inflammatory targets observed in zebrafish and humans.
Additionally, the team didn’t know the vitamin D levels in the umbilical cord blood samples they tested, which may have influenced the outcome of their analysis.
As a next step, Dr North and her colleagues hope to test cord blood samples for which they know the vitamin D status to see if umbilical cords with healthy levels respond better or worse to stimulation than cords from vitamin-D-deficient donors.
Lifestyle may impact life expectancy in mild SCD

alongside a normal one
Image by Betty Pace
A case series published in Blood indicates that some patients with mildly symptomatic sickle cell disease (SCD) can live long lives if they
comply with treatment recommendations and lead a healthy lifestyle.
The paper includes details on 4 women with milder forms of SCD who survived beyond age 80.
“For those with mild forms of SCD, these women show that lifestyle modifications may improve disease outcomes,” said author Samir K. Ballas, MD, of Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia, Pennsylvania.
Three of the women described in this case series were treated at the Sickle Cell Center of Thomas Jefferson University, and 1 was treated in Brazil’s Instituto de Hematologia Arthur de Siqueira Cavalcanti in Rio de Janeiro.
The women had different ancestries—2 African-American, 1 Italian-American, and 1 African-Brazilian—and different diagnoses—2 with hemoglobin SC disease and 2 with sickle cell anemia. But all 4 women had what Dr Ballas called “desirable” disease states.
“These women never had a stroke, never had recurrent acute chest syndrome, had a relatively high fetal hemoglobin count, and had infrequent painful crises,” Dr Ballas said. “Patients like this usually—but not always—experience relatively mild SCD, and they live longer with better quality of life.”
In addition, all of the women took steps to maintain and improve their health and had long-term family support. Dr Ballas said these factors likely contributed to the women’s long lives and high quality of life.
“All of the women were non-smokers who consumed little to no alcohol and maintained a normal body mass index,” he said. “This was coupled with a strong compliance to their treatment regimens and excellent family support at home.”
Family support was defined as having a spouse or child who provided attentive, ongoing care. And all of the women had at least 1 such caregiver.
Treatment compliance was based on observations by healthcare providers, including study authors. According to these observations, all of the women showed “excellent” adherence when it came to medication intake, appointments, and referrals.
As the women had relatively mild disease states, none of them were qualified to receive treatment with hydroxyurea. Instead, they received hydration, vaccination (including annual flu shots), and blood transfusion and analgesics as needed.
Even with their mild disease states and healthy lifestyles, these women did not live crisis-free lives. Each experienced disease-related complications necessitating medical attention.
The women had 0 to 3 vaso-occlusive crises per year. Two women required frequent transfusions (and had iron overload), and 2 required occasional transfusions. One woman had 2 episodes of acute chest syndrome, and the second episode led to her death.
Ultimately, 3 of the women died. One died of acute chest syndrome and septicemia at age 82, and another died of cardiac complications at age 86. For a third woman, the cause of death, at age 82, was unknown. The fourth woman remains alive at age 82.
As the median life expectancy of women with SCD in the US is 47, Dr Ballas and his colleagues said these 4 women may “provide a blueprint of how to live a long life despite having a serious medical condition like SCD.”
“I would often come out to the waiting room and find these ladies talking with other SCD patients, and I could tell that they gave others hope, that just because they have SCD does not mean that they are doomed to die by their 40s . . . ,” Dr Ballas said. “[I]f they take care of themselves and live closely with those who can help keep them well, that there is hope for them to lead long, full lives.”
alongside a normal one
Image by Betty Pace
A case series published in Blood indicates that some patients with mildly symptomatic sickle cell disease (SCD) can live long lives if they
comply with treatment recommendations and lead a healthy lifestyle.
The paper includes details on 4 women with milder forms of SCD who survived beyond age 80.
“For those with mild forms of SCD, these women show that lifestyle modifications may improve disease outcomes,” said author Samir K. Ballas, MD, of Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia, Pennsylvania.
Three of the women described in this case series were treated at the Sickle Cell Center of Thomas Jefferson University, and 1 was treated in Brazil’s Instituto de Hematologia Arthur de Siqueira Cavalcanti in Rio de Janeiro.
The women had different ancestries—2 African-American, 1 Italian-American, and 1 African-Brazilian—and different diagnoses—2 with hemoglobin SC disease and 2 with sickle cell anemia. But all 4 women had what Dr Ballas called “desirable” disease states.
“These women never had a stroke, never had recurrent acute chest syndrome, had a relatively high fetal hemoglobin count, and had infrequent painful crises,” Dr Ballas said. “Patients like this usually—but not always—experience relatively mild SCD, and they live longer with better quality of life.”
In addition, all of the women took steps to maintain and improve their health and had long-term family support. Dr Ballas said these factors likely contributed to the women’s long lives and high quality of life.
“All of the women were non-smokers who consumed little to no alcohol and maintained a normal body mass index,” he said. “This was coupled with a strong compliance to their treatment regimens and excellent family support at home.”
Family support was defined as having a spouse or child who provided attentive, ongoing care. And all of the women had at least 1 such caregiver.
Treatment compliance was based on observations by healthcare providers, including study authors. According to these observations, all of the women showed “excellent” adherence when it came to medication intake, appointments, and referrals.
As the women had relatively mild disease states, none of them were qualified to receive treatment with hydroxyurea. Instead, they received hydration, vaccination (including annual flu shots), and blood transfusion and analgesics as needed.
Even with their mild disease states and healthy lifestyles, these women did not live crisis-free lives. Each experienced disease-related complications necessitating medical attention.
The women had 0 to 3 vaso-occlusive crises per year. Two women required frequent transfusions (and had iron overload), and 2 required occasional transfusions. One woman had 2 episodes of acute chest syndrome, and the second episode led to her death.
Ultimately, 3 of the women died. One died of acute chest syndrome and septicemia at age 82, and another died of cardiac complications at age 86. For a third woman, the cause of death, at age 82, was unknown. The fourth woman remains alive at age 82.
As the median life expectancy of women with SCD in the US is 47, Dr Ballas and his colleagues said these 4 women may “provide a blueprint of how to live a long life despite having a serious medical condition like SCD.”
“I would often come out to the waiting room and find these ladies talking with other SCD patients, and I could tell that they gave others hope, that just because they have SCD does not mean that they are doomed to die by their 40s . . . ,” Dr Ballas said. “[I]f they take care of themselves and live closely with those who can help keep them well, that there is hope for them to lead long, full lives.”
alongside a normal one
Image by Betty Pace
A case series published in Blood indicates that some patients with mildly symptomatic sickle cell disease (SCD) can live long lives if they
comply with treatment recommendations and lead a healthy lifestyle.
The paper includes details on 4 women with milder forms of SCD who survived beyond age 80.
“For those with mild forms of SCD, these women show that lifestyle modifications may improve disease outcomes,” said author Samir K. Ballas, MD, of Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia, Pennsylvania.
Three of the women described in this case series were treated at the Sickle Cell Center of Thomas Jefferson University, and 1 was treated in Brazil’s Instituto de Hematologia Arthur de Siqueira Cavalcanti in Rio de Janeiro.
The women had different ancestries—2 African-American, 1 Italian-American, and 1 African-Brazilian—and different diagnoses—2 with hemoglobin SC disease and 2 with sickle cell anemia. But all 4 women had what Dr Ballas called “desirable” disease states.
“These women never had a stroke, never had recurrent acute chest syndrome, had a relatively high fetal hemoglobin count, and had infrequent painful crises,” Dr Ballas said. “Patients like this usually—but not always—experience relatively mild SCD, and they live longer with better quality of life.”
In addition, all of the women took steps to maintain and improve their health and had long-term family support. Dr Ballas said these factors likely contributed to the women’s long lives and high quality of life.
“All of the women were non-smokers who consumed little to no alcohol and maintained a normal body mass index,” he said. “This was coupled with a strong compliance to their treatment regimens and excellent family support at home.”
Family support was defined as having a spouse or child who provided attentive, ongoing care. And all of the women had at least 1 such caregiver.
Treatment compliance was based on observations by healthcare providers, including study authors. According to these observations, all of the women showed “excellent” adherence when it came to medication intake, appointments, and referrals.
As the women had relatively mild disease states, none of them were qualified to receive treatment with hydroxyurea. Instead, they received hydration, vaccination (including annual flu shots), and blood transfusion and analgesics as needed.
Even with their mild disease states and healthy lifestyles, these women did not live crisis-free lives. Each experienced disease-related complications necessitating medical attention.
The women had 0 to 3 vaso-occlusive crises per year. Two women required frequent transfusions (and had iron overload), and 2 required occasional transfusions. One woman had 2 episodes of acute chest syndrome, and the second episode led to her death.
Ultimately, 3 of the women died. One died of acute chest syndrome and septicemia at age 82, and another died of cardiac complications at age 86. For a third woman, the cause of death, at age 82, was unknown. The fourth woman remains alive at age 82.
As the median life expectancy of women with SCD in the US is 47, Dr Ballas and his colleagues said these 4 women may “provide a blueprint of how to live a long life despite having a serious medical condition like SCD.”
“I would often come out to the waiting room and find these ladies talking with other SCD patients, and I could tell that they gave others hope, that just because they have SCD does not mean that they are doomed to die by their 40s . . . ,” Dr Ballas said. “[I]f they take care of themselves and live closely with those who can help keep them well, that there is hope for them to lead long, full lives.”
Factor IX therapy approved in Australia

The Australian Therapeutic Goods Administration has approved albutrepenonacog alfa (Idelvion) to treat hemophilia B patients of all ages.
Albutrepenonacog alfa is a fusion protein linking recombinant coagulation factor IX with recombinant albumin.
The product is now approved in Australia for use as routine prophylaxis to prevent and reduce the frequency of bleeding, for on-demand control of bleeding, and for perioperative management of
bleeding.
Albutrepenonacog alfa has also been approved in Canada, the European Union, Japan, Switzerland, and the US.
Albutrepenonacog alfa is being developed by CSL Behring.
The company says albutrepenonacog alfa is the first and only Australian-registered factor IX therapy that delivers high-level protection from bleeding with up to 14-day dosing for appropriate patients.
According to CSL Behring, albutrepenonacog alfa can deliver high-level protection by maintaining factor IX activity levels at an average of 20% in patients treated prophylactically every 7 days and an average of 12% in patients treated prophylactically every 14 days.
“The Australian Haemophilia Centre Directors’ Organisation (AHCDO) views the development of effective long-acting clotting factor concentrates as a major step forward in the management of our patients with hemophilia,” said Simon McRae, MMBS, consultant hematologist and chairman of AHCDO.
“The ability to maintain clotting factor levels above a level that prevent the vast majority of bleeding events with less frequent infusions has the potential to improve long-term outcomes in individuals with hemophilia.”
Phase 3 trial
The Therapeutic Goods Administration approved albutrepenonacog alfa based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were published in Blood. The study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks, followed by a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache.
“I have seen first-hand the benefits Idelvion has had on children with hemophilia B,” said PROLONG-9FP investigator Julie Curtin, MBBS, PhD, of The Children’s Hospital at Westmead in New South Wales.
“Idelvion has enabled children on regular treatment with factor IX to reduce the frequency of infusions without increasing their risk of bleeding. For a child to only need an injection every 1-2 weeks is a good step forward in the management of hemophilia B, which I welcome, and I am sure my patients will welcome this improvement too.”
The Australian Therapeutic Goods Administration has approved albutrepenonacog alfa (Idelvion) to treat hemophilia B patients of all ages.
Albutrepenonacog alfa is a fusion protein linking recombinant coagulation factor IX with recombinant albumin.
The product is now approved in Australia for use as routine prophylaxis to prevent and reduce the frequency of bleeding, for on-demand control of bleeding, and for perioperative management of
bleeding.
Albutrepenonacog alfa has also been approved in Canada, the European Union, Japan, Switzerland, and the US.
Albutrepenonacog alfa is being developed by CSL Behring.
The company says albutrepenonacog alfa is the first and only Australian-registered factor IX therapy that delivers high-level protection from bleeding with up to 14-day dosing for appropriate patients.
According to CSL Behring, albutrepenonacog alfa can deliver high-level protection by maintaining factor IX activity levels at an average of 20% in patients treated prophylactically every 7 days and an average of 12% in patients treated prophylactically every 14 days.
“The Australian Haemophilia Centre Directors’ Organisation (AHCDO) views the development of effective long-acting clotting factor concentrates as a major step forward in the management of our patients with hemophilia,” said Simon McRae, MMBS, consultant hematologist and chairman of AHCDO.
“The ability to maintain clotting factor levels above a level that prevent the vast majority of bleeding events with less frequent infusions has the potential to improve long-term outcomes in individuals with hemophilia.”
Phase 3 trial
The Therapeutic Goods Administration approved albutrepenonacog alfa based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were published in Blood. The study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks, followed by a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache.
“I have seen first-hand the benefits Idelvion has had on children with hemophilia B,” said PROLONG-9FP investigator Julie Curtin, MBBS, PhD, of The Children’s Hospital at Westmead in New South Wales.
“Idelvion has enabled children on regular treatment with factor IX to reduce the frequency of infusions without increasing their risk of bleeding. For a child to only need an injection every 1-2 weeks is a good step forward in the management of hemophilia B, which I welcome, and I am sure my patients will welcome this improvement too.”
The Australian Therapeutic Goods Administration has approved albutrepenonacog alfa (Idelvion) to treat hemophilia B patients of all ages.
Albutrepenonacog alfa is a fusion protein linking recombinant coagulation factor IX with recombinant albumin.
The product is now approved in Australia for use as routine prophylaxis to prevent and reduce the frequency of bleeding, for on-demand control of bleeding, and for perioperative management of
bleeding.
Albutrepenonacog alfa has also been approved in Canada, the European Union, Japan, Switzerland, and the US.
Albutrepenonacog alfa is being developed by CSL Behring.
The company says albutrepenonacog alfa is the first and only Australian-registered factor IX therapy that delivers high-level protection from bleeding with up to 14-day dosing for appropriate patients.
According to CSL Behring, albutrepenonacog alfa can deliver high-level protection by maintaining factor IX activity levels at an average of 20% in patients treated prophylactically every 7 days and an average of 12% in patients treated prophylactically every 14 days.
“The Australian Haemophilia Centre Directors’ Organisation (AHCDO) views the development of effective long-acting clotting factor concentrates as a major step forward in the management of our patients with hemophilia,” said Simon McRae, MMBS, consultant hematologist and chairman of AHCDO.
“The ability to maintain clotting factor levels above a level that prevent the vast majority of bleeding events with less frequent infusions has the potential to improve long-term outcomes in individuals with hemophilia.”
Phase 3 trial
The Therapeutic Goods Administration approved albutrepenonacog alfa based on results of the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were published in Blood. The study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks, followed by a 7-, 10-, or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache.
“I have seen first-hand the benefits Idelvion has had on children with hemophilia B,” said PROLONG-9FP investigator Julie Curtin, MBBS, PhD, of The Children’s Hospital at Westmead in New South Wales.
“Idelvion has enabled children on regular treatment with factor IX to reduce the frequency of infusions without increasing their risk of bleeding. For a child to only need an injection every 1-2 weeks is a good step forward in the management of hemophilia B, which I welcome, and I am sure my patients will welcome this improvement too.”