User login
Reversal agent granted conditional approval in Canada

to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events. ![]()
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events. ![]()
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events. ![]()
Improving NK cell therapy

Image by Joshua Stokes
New findings published in PNAS may help scientists improve the efficacy of natural killer (NK) cell therapy for patients with leukemia.
The preclinical research revealed a tolerance mechanism that restrains the activity of NK cells, as well as a potential way to overcome this problem.
Investigators found that a transcription factor, Kruppel-like factor 2 (KFL2), is critical for NK cell expansion and survival.
Specifically, KLF2 limits immature NK cell proliferation and instructs mature NK cells to home to niches rich in interleukin 15 (IL-15), which is necessary for their continued survival.
“This is the same process likely used by cancer cells to avoid destruction by NK cells,” said study author Eric Sebzda, PhD, of Vanderbilt University Medical Center in Nashville, Tennessee.
In particular, tumors may avoid immune clearance by promoting KLF2 destruction within the NK cell population, thereby starving these cells of IL-15.
Dr Sebzda and his colleagues noted that increased expression of IL-15 can improve immune responses against tumors. Unfortunately, it’s not easy to introduce the cytokine only within a tumor microenvironment, and high systemic levels of IL-15 can be toxic.
Recruiting cells that transpresent IL-15 to the tumor microenvironment may overcome this barrier and therefore improve NK cell-mediated cancer therapy, the investigators said. However, the methodology hasn’t been worked out yet.
“Our paper should encourage this line of inquiry,” Dr Sebzda concluded. ![]()
Image by Joshua Stokes
New findings published in PNAS may help scientists improve the efficacy of natural killer (NK) cell therapy for patients with leukemia.
The preclinical research revealed a tolerance mechanism that restrains the activity of NK cells, as well as a potential way to overcome this problem.
Investigators found that a transcription factor, Kruppel-like factor 2 (KFL2), is critical for NK cell expansion and survival.
Specifically, KLF2 limits immature NK cell proliferation and instructs mature NK cells to home to niches rich in interleukin 15 (IL-15), which is necessary for their continued survival.
“This is the same process likely used by cancer cells to avoid destruction by NK cells,” said study author Eric Sebzda, PhD, of Vanderbilt University Medical Center in Nashville, Tennessee.
In particular, tumors may avoid immune clearance by promoting KLF2 destruction within the NK cell population, thereby starving these cells of IL-15.
Dr Sebzda and his colleagues noted that increased expression of IL-15 can improve immune responses against tumors. Unfortunately, it’s not easy to introduce the cytokine only within a tumor microenvironment, and high systemic levels of IL-15 can be toxic.
Recruiting cells that transpresent IL-15 to the tumor microenvironment may overcome this barrier and therefore improve NK cell-mediated cancer therapy, the investigators said. However, the methodology hasn’t been worked out yet.
“Our paper should encourage this line of inquiry,” Dr Sebzda concluded. ![]()
Image by Joshua Stokes
New findings published in PNAS may help scientists improve the efficacy of natural killer (NK) cell therapy for patients with leukemia.
The preclinical research revealed a tolerance mechanism that restrains the activity of NK cells, as well as a potential way to overcome this problem.
Investigators found that a transcription factor, Kruppel-like factor 2 (KFL2), is critical for NK cell expansion and survival.
Specifically, KLF2 limits immature NK cell proliferation and instructs mature NK cells to home to niches rich in interleukin 15 (IL-15), which is necessary for their continued survival.
“This is the same process likely used by cancer cells to avoid destruction by NK cells,” said study author Eric Sebzda, PhD, of Vanderbilt University Medical Center in Nashville, Tennessee.
In particular, tumors may avoid immune clearance by promoting KLF2 destruction within the NK cell population, thereby starving these cells of IL-15.
Dr Sebzda and his colleagues noted that increased expression of IL-15 can improve immune responses against tumors. Unfortunately, it’s not easy to introduce the cytokine only within a tumor microenvironment, and high systemic levels of IL-15 can be toxic.
Recruiting cells that transpresent IL-15 to the tumor microenvironment may overcome this barrier and therefore improve NK cell-mediated cancer therapy, the investigators said. However, the methodology hasn’t been worked out yet.
“Our paper should encourage this line of inquiry,” Dr Sebzda concluded. ![]()
CDC announces availability of funds to fight Zika

Photo courtesy of
Muhammad Mahdi Karim
The US Centers for Disease Control and Prevention (CDC) has announced that US states and territories can now apply for funds to fight Zika locally.
The agency said more than $85 million in redirected funds identified by the Department of Health and Human Services is being made available to support efforts to protect Americans from Zika infection and associated adverse health outcomes.
“These funds will allow states and territories to continue implementation of their Zika preparedness plans but are not enough to support a comprehensive Zika response and can only temporarily address what is needed,” said Stephen C. Redd, MD, director of the CDC’s Office of Public Health Preparedness and Response.
“Without the full amount of requested emergency supplemental funding, many activities that need to start now are being delayed or may have to be stopped within months.”
The more than $85 million in funds includes the more $60 million reported earlier this year (Funding opportunity number CDC-RFA-CK14-1401CONTPPHF16) as well as another $25 million that was just announced (Funding opportunity number CDC-RFA-TP16-1602).
Earlier this year, states and cities participating in the Epidemiology and Lab Capacity program became eligible for more than $60 million in funds to:
- Build laboratory capacity
- Enhance epidemiological surveillance and investigation
- Improve mosquito control and monitoring
- Keep blood supplies safe
- Contribute data to the US Zika Pregnancy registry.
Applications for these funds are due May 27, 2016, and will be disbursed during the summer.
Under the latest announcement, $25 million in FY 2016 preparedness and response funding will go to 53 states, cities, and territories at risk for outbreaks of Zika virus infection.
Recipients will receive funds based on the geographic locations of the two mosquitoes known to transmit Zika virus (Aedes aegypti and Aedes albopictus), history of mosquito-borne disease outbreaks, and size of population.
The funds are intended to strengthen incident management and emergency operations coordination, information management and sharing, and community recovery and resilience.
State, local, and territorial health officials can use the funds to identify and investigate a possible outbreak of Zika virus disease in their communities, coordinate a comprehensive response across all levels of government and non-governmental partners (including the healthcare sector), and identify and connect to community services families affected by Zika virus disease.
Applications for the funds are due June 13, 2016. Funds will be disbursed during the summer and remain available through July 2017.
Zika virus disease is caused by the Zika virus, which is spread to people primarily through the bite of infected Aedes aegypti and Aedes albopictus mosquitoes, though Aedes aegypti are more likely to spread Zika. Sexual transmission and transmission via blood transfusion have been documented as well.
There is currently no vaccine or treatment for Zika. The most common symptoms of Zika are fever, rash, joint pain, and conjunctivitis. In previous outbreaks, the illness has typically been mild, with symptoms lasting for several days to a week after a person is bitten by an infected mosquito.
Zika virus infection in pregnant women is a cause of microcephaly and other severe fetal brain defects. Zika has also been linked to Guillain-Barré syndrome. ![]()
Photo courtesy of
Muhammad Mahdi Karim
The US Centers for Disease Control and Prevention (CDC) has announced that US states and territories can now apply for funds to fight Zika locally.
The agency said more than $85 million in redirected funds identified by the Department of Health and Human Services is being made available to support efforts to protect Americans from Zika infection and associated adverse health outcomes.
“These funds will allow states and territories to continue implementation of their Zika preparedness plans but are not enough to support a comprehensive Zika response and can only temporarily address what is needed,” said Stephen C. Redd, MD, director of the CDC’s Office of Public Health Preparedness and Response.
“Without the full amount of requested emergency supplemental funding, many activities that need to start now are being delayed or may have to be stopped within months.”
The more than $85 million in funds includes the more $60 million reported earlier this year (Funding opportunity number CDC-RFA-CK14-1401CONTPPHF16) as well as another $25 million that was just announced (Funding opportunity number CDC-RFA-TP16-1602).
Earlier this year, states and cities participating in the Epidemiology and Lab Capacity program became eligible for more than $60 million in funds to:
- Build laboratory capacity
- Enhance epidemiological surveillance and investigation
- Improve mosquito control and monitoring
- Keep blood supplies safe
- Contribute data to the US Zika Pregnancy registry.
Applications for these funds are due May 27, 2016, and will be disbursed during the summer.
Under the latest announcement, $25 million in FY 2016 preparedness and response funding will go to 53 states, cities, and territories at risk for outbreaks of Zika virus infection.
Recipients will receive funds based on the geographic locations of the two mosquitoes known to transmit Zika virus (Aedes aegypti and Aedes albopictus), history of mosquito-borne disease outbreaks, and size of population.
The funds are intended to strengthen incident management and emergency operations coordination, information management and sharing, and community recovery and resilience.
State, local, and territorial health officials can use the funds to identify and investigate a possible outbreak of Zika virus disease in their communities, coordinate a comprehensive response across all levels of government and non-governmental partners (including the healthcare sector), and identify and connect to community services families affected by Zika virus disease.
Applications for the funds are due June 13, 2016. Funds will be disbursed during the summer and remain available through July 2017.
Zika virus disease is caused by the Zika virus, which is spread to people primarily through the bite of infected Aedes aegypti and Aedes albopictus mosquitoes, though Aedes aegypti are more likely to spread Zika. Sexual transmission and transmission via blood transfusion have been documented as well.
There is currently no vaccine or treatment for Zika. The most common symptoms of Zika are fever, rash, joint pain, and conjunctivitis. In previous outbreaks, the illness has typically been mild, with symptoms lasting for several days to a week after a person is bitten by an infected mosquito.
Zika virus infection in pregnant women is a cause of microcephaly and other severe fetal brain defects. Zika has also been linked to Guillain-Barré syndrome. ![]()
Photo courtesy of
Muhammad Mahdi Karim
The US Centers for Disease Control and Prevention (CDC) has announced that US states and territories can now apply for funds to fight Zika locally.
The agency said more than $85 million in redirected funds identified by the Department of Health and Human Services is being made available to support efforts to protect Americans from Zika infection and associated adverse health outcomes.
“These funds will allow states and territories to continue implementation of their Zika preparedness plans but are not enough to support a comprehensive Zika response and can only temporarily address what is needed,” said Stephen C. Redd, MD, director of the CDC’s Office of Public Health Preparedness and Response.
“Without the full amount of requested emergency supplemental funding, many activities that need to start now are being delayed or may have to be stopped within months.”
The more than $85 million in funds includes the more $60 million reported earlier this year (Funding opportunity number CDC-RFA-CK14-1401CONTPPHF16) as well as another $25 million that was just announced (Funding opportunity number CDC-RFA-TP16-1602).
Earlier this year, states and cities participating in the Epidemiology and Lab Capacity program became eligible for more than $60 million in funds to:
- Build laboratory capacity
- Enhance epidemiological surveillance and investigation
- Improve mosquito control and monitoring
- Keep blood supplies safe
- Contribute data to the US Zika Pregnancy registry.
Applications for these funds are due May 27, 2016, and will be disbursed during the summer.
Under the latest announcement, $25 million in FY 2016 preparedness and response funding will go to 53 states, cities, and territories at risk for outbreaks of Zika virus infection.
Recipients will receive funds based on the geographic locations of the two mosquitoes known to transmit Zika virus (Aedes aegypti and Aedes albopictus), history of mosquito-borne disease outbreaks, and size of population.
The funds are intended to strengthen incident management and emergency operations coordination, information management and sharing, and community recovery and resilience.
State, local, and territorial health officials can use the funds to identify and investigate a possible outbreak of Zika virus disease in their communities, coordinate a comprehensive response across all levels of government and non-governmental partners (including the healthcare sector), and identify and connect to community services families affected by Zika virus disease.
Applications for the funds are due June 13, 2016. Funds will be disbursed during the summer and remain available through July 2017.
Zika virus disease is caused by the Zika virus, which is spread to people primarily through the bite of infected Aedes aegypti and Aedes albopictus mosquitoes, though Aedes aegypti are more likely to spread Zika. Sexual transmission and transmission via blood transfusion have been documented as well.
There is currently no vaccine or treatment for Zika. The most common symptoms of Zika are fever, rash, joint pain, and conjunctivitis. In previous outbreaks, the illness has typically been mild, with symptoms lasting for several days to a week after a person is bitten by an infected mosquito.
Zika virus infection in pregnant women is a cause of microcephaly and other severe fetal brain defects. Zika has also been linked to Guillain-Barré syndrome. ![]()
PRAC: Products may not increase risk of inhibitors

Results of a meta-analysis suggest a pair of recombinant factor VIII (FVIII) products may not increase the risk of FVIII inhibitors in previously untreated patients with severe hemophilia A.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) analyzed data from 3 observational studies and found that patients who received Kogenate or Helixate had a higher incidence of inhibitors than patients who received other recombinant FVIII products.
However, the difference was not significant, and the PRAC concluded that, overall, the evidence does not confirm an increased risk of inhibitors with Kogenate or Helixate.
This conclusion is consistent with the PRAC’s previous conclusions from a review carried out in 2013.
For the current analysis, the PRAC reviewed data from 3 studies, which were conducted by the RODIN study group, the UK Haemophilia Centre Doctors’ Organisation (UKHCDO), and the FranceCoag Network.
The analysis included 1102 previously untreated patients—481 in the RODIN study, 293 in the FranceCoag study, and 328 in the UKHCDO study—who received octocog alfa (Advate, Helixate, or Kogenate), moroctocog alfa (Refacto or Refacto AF), or other recombinant FVIII products.
Results suggested a trend toward an increase of high-titer inhibitor development and all inhibitor development with Kogenate or Helixate compared to Advate.
Overall, 37% of patients treated with Kogenate or Helixate (147/400) developed inhibitory antibodies, 22% of whom (n=88) had high-titer inhibitors. Twenty-six percent of patients who received Advate (100/385) developed inhibitors, 15% of whom (n=57) had high-titer inhibitors.
The PRAC said a similar trend was observed for other recombinant FVIII products, but the results were less pronounced due to sample size constraints.
The committee pointed out that there were several limitations with this meta-analysis, including the possibility of residual confounding. The group also said a number of parameters may have had an impact on the incidence of inhibitors in these previously untreated patients, and adjusting for all of these factors may not be possible.
In addition, the PRAC noted that there has been no signal for a similar trend of increases in inhibitor development with Kogenate in previously treated patients in other studies.
Finally, the committee recommended that companies marketing recombinant FVIII products monitor published studies on inhibitor development with the aim of keeping the product information up to date. ![]()
Results of a meta-analysis suggest a pair of recombinant factor VIII (FVIII) products may not increase the risk of FVIII inhibitors in previously untreated patients with severe hemophilia A.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) analyzed data from 3 observational studies and found that patients who received Kogenate or Helixate had a higher incidence of inhibitors than patients who received other recombinant FVIII products.
However, the difference was not significant, and the PRAC concluded that, overall, the evidence does not confirm an increased risk of inhibitors with Kogenate or Helixate.
This conclusion is consistent with the PRAC’s previous conclusions from a review carried out in 2013.
For the current analysis, the PRAC reviewed data from 3 studies, which were conducted by the RODIN study group, the UK Haemophilia Centre Doctors’ Organisation (UKHCDO), and the FranceCoag Network.
The analysis included 1102 previously untreated patients—481 in the RODIN study, 293 in the FranceCoag study, and 328 in the UKHCDO study—who received octocog alfa (Advate, Helixate, or Kogenate), moroctocog alfa (Refacto or Refacto AF), or other recombinant FVIII products.
Results suggested a trend toward an increase of high-titer inhibitor development and all inhibitor development with Kogenate or Helixate compared to Advate.
Overall, 37% of patients treated with Kogenate or Helixate (147/400) developed inhibitory antibodies, 22% of whom (n=88) had high-titer inhibitors. Twenty-six percent of patients who received Advate (100/385) developed inhibitors, 15% of whom (n=57) had high-titer inhibitors.
The PRAC said a similar trend was observed for other recombinant FVIII products, but the results were less pronounced due to sample size constraints.
The committee pointed out that there were several limitations with this meta-analysis, including the possibility of residual confounding. The group also said a number of parameters may have had an impact on the incidence of inhibitors in these previously untreated patients, and adjusting for all of these factors may not be possible.
In addition, the PRAC noted that there has been no signal for a similar trend of increases in inhibitor development with Kogenate in previously treated patients in other studies.
Finally, the committee recommended that companies marketing recombinant FVIII products monitor published studies on inhibitor development with the aim of keeping the product information up to date. ![]()
Results of a meta-analysis suggest a pair of recombinant factor VIII (FVIII) products may not increase the risk of FVIII inhibitors in previously untreated patients with severe hemophilia A.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) analyzed data from 3 observational studies and found that patients who received Kogenate or Helixate had a higher incidence of inhibitors than patients who received other recombinant FVIII products.
However, the difference was not significant, and the PRAC concluded that, overall, the evidence does not confirm an increased risk of inhibitors with Kogenate or Helixate.
This conclusion is consistent with the PRAC’s previous conclusions from a review carried out in 2013.
For the current analysis, the PRAC reviewed data from 3 studies, which were conducted by the RODIN study group, the UK Haemophilia Centre Doctors’ Organisation (UKHCDO), and the FranceCoag Network.
The analysis included 1102 previously untreated patients—481 in the RODIN study, 293 in the FranceCoag study, and 328 in the UKHCDO study—who received octocog alfa (Advate, Helixate, or Kogenate), moroctocog alfa (Refacto or Refacto AF), or other recombinant FVIII products.
Results suggested a trend toward an increase of high-titer inhibitor development and all inhibitor development with Kogenate or Helixate compared to Advate.
Overall, 37% of patients treated with Kogenate or Helixate (147/400) developed inhibitory antibodies, 22% of whom (n=88) had high-titer inhibitors. Twenty-six percent of patients who received Advate (100/385) developed inhibitors, 15% of whom (n=57) had high-titer inhibitors.
The PRAC said a similar trend was observed for other recombinant FVIII products, but the results were less pronounced due to sample size constraints.
The committee pointed out that there were several limitations with this meta-analysis, including the possibility of residual confounding. The group also said a number of parameters may have had an impact on the incidence of inhibitors in these previously untreated patients, and adjusting for all of these factors may not be possible.
In addition, the PRAC noted that there has been no signal for a similar trend of increases in inhibitor development with Kogenate in previously treated patients in other studies.
Finally, the committee recommended that companies marketing recombinant FVIII products monitor published studies on inhibitor development with the aim of keeping the product information up to date. ![]()
FDA warns of counterfeit BiCNU
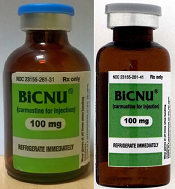
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing [email protected].
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing [email protected].
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing [email protected].
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
EC approves FC fusion protein for hemophilia B

Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study. ![]()
Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study. ![]()
Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study. ![]()
Test can monitor blood coagulation at home, team says

CINCINNATI—Researchers say they have developed a lateral flow assay device that patients can use at home to monitor their blood coagulation.
The device consists of nanofiber membranes inside a paper-based, porous test strip that is housed in a plastic cassette.
The researchers said this device can quickly reveal the level of the blood’s ability to clot using just a drop of blood from a finger prick.
“We have developed a blood-screening device for patients on medications like Coumadin (warfarin) or other blood thinners who need to monitor their blood-clotting levels on a regular basis,” said Andrew Steckl, PhD, of the University of Cincinnati in Ohio.
“Patients can soon monitor their blood coagulation characteristics from home quickly and painlessly before making needless trips to the lab or hospital.”
Hua Li, a student researcher in Dr Steckl’s lab, presented details on this device at the 8th International Conference on Porous Media and Annual Meeting of the International Society for Porous Media (abstract 1371).
Dr Steckl noted that slight changes in the level of coagulation properties will occur normally, but a major change in levels immediately shows up with his team’s device.
The researchers found the device was able to detect coagulation ability in rabbit blood and in blood from patients receiving warfarin.
“This simple test is not intended to replace the very careful and accurate measurements that get accomplished in a laboratory facility, but, at a relatively minimal cost, a patient can do this on their own between scheduled visits or when in doubt,” Dr Steckl said. “And it shouldn’t require a caregiver, as most patients can perform this test quickly on their own.”
Furthermore, the researchers said this technology can be calibrated to a specific patient’s condition. For example, a patient whose normal blood coagulation rate is significantly different from the general population because of a genetic disorder can use a tailor-made test kit that includes a different porous membrane.
The technology may also help patients who have a known inherited blood clotting disorder detect concerning levels early.
“By identifying potential blood-clotting problems early enough, we hope to prevent potential injury or death and the exorbitant associated costs,” Dr Steckl said. “By shifting from what were once mandatory expensive laboratory tests to using more in-home screening tests, patients can take more control of their lives, reduce healthcare costs, and, ultimately, save more lives.” ![]()
CINCINNATI—Researchers say they have developed a lateral flow assay device that patients can use at home to monitor their blood coagulation.
The device consists of nanofiber membranes inside a paper-based, porous test strip that is housed in a plastic cassette.
The researchers said this device can quickly reveal the level of the blood’s ability to clot using just a drop of blood from a finger prick.
“We have developed a blood-screening device for patients on medications like Coumadin (warfarin) or other blood thinners who need to monitor their blood-clotting levels on a regular basis,” said Andrew Steckl, PhD, of the University of Cincinnati in Ohio.
“Patients can soon monitor their blood coagulation characteristics from home quickly and painlessly before making needless trips to the lab or hospital.”
Hua Li, a student researcher in Dr Steckl’s lab, presented details on this device at the 8th International Conference on Porous Media and Annual Meeting of the International Society for Porous Media (abstract 1371).
Dr Steckl noted that slight changes in the level of coagulation properties will occur normally, but a major change in levels immediately shows up with his team’s device.
The researchers found the device was able to detect coagulation ability in rabbit blood and in blood from patients receiving warfarin.
“This simple test is not intended to replace the very careful and accurate measurements that get accomplished in a laboratory facility, but, at a relatively minimal cost, a patient can do this on their own between scheduled visits or when in doubt,” Dr Steckl said. “And it shouldn’t require a caregiver, as most patients can perform this test quickly on their own.”
Furthermore, the researchers said this technology can be calibrated to a specific patient’s condition. For example, a patient whose normal blood coagulation rate is significantly different from the general population because of a genetic disorder can use a tailor-made test kit that includes a different porous membrane.
The technology may also help patients who have a known inherited blood clotting disorder detect concerning levels early.
“By identifying potential blood-clotting problems early enough, we hope to prevent potential injury or death and the exorbitant associated costs,” Dr Steckl said. “By shifting from what were once mandatory expensive laboratory tests to using more in-home screening tests, patients can take more control of their lives, reduce healthcare costs, and, ultimately, save more lives.” ![]()
CINCINNATI—Researchers say they have developed a lateral flow assay device that patients can use at home to monitor their blood coagulation.
The device consists of nanofiber membranes inside a paper-based, porous test strip that is housed in a plastic cassette.
The researchers said this device can quickly reveal the level of the blood’s ability to clot using just a drop of blood from a finger prick.
“We have developed a blood-screening device for patients on medications like Coumadin (warfarin) or other blood thinners who need to monitor their blood-clotting levels on a regular basis,” said Andrew Steckl, PhD, of the University of Cincinnati in Ohio.
“Patients can soon monitor their blood coagulation characteristics from home quickly and painlessly before making needless trips to the lab or hospital.”
Hua Li, a student researcher in Dr Steckl’s lab, presented details on this device at the 8th International Conference on Porous Media and Annual Meeting of the International Society for Porous Media (abstract 1371).
Dr Steckl noted that slight changes in the level of coagulation properties will occur normally, but a major change in levels immediately shows up with his team’s device.
The researchers found the device was able to detect coagulation ability in rabbit blood and in blood from patients receiving warfarin.
“This simple test is not intended to replace the very careful and accurate measurements that get accomplished in a laboratory facility, but, at a relatively minimal cost, a patient can do this on their own between scheduled visits or when in doubt,” Dr Steckl said. “And it shouldn’t require a caregiver, as most patients can perform this test quickly on their own.”
Furthermore, the researchers said this technology can be calibrated to a specific patient’s condition. For example, a patient whose normal blood coagulation rate is significantly different from the general population because of a genetic disorder can use a tailor-made test kit that includes a different porous membrane.
The technology may also help patients who have a known inherited blood clotting disorder detect concerning levels early.
“By identifying potential blood-clotting problems early enough, we hope to prevent potential injury or death and the exorbitant associated costs,” Dr Steckl said. “By shifting from what were once mandatory expensive laboratory tests to using more in-home screening tests, patients can take more control of their lives, reduce healthcare costs, and, ultimately, save more lives.”
CDC issues interim guidance for Zika testing

The US Centers for Disease Control and Prevention (CDC) has released an interim guidance for testing urine for the Zika virus.
The agency noted that, in most patients, Zika virus RNA is unlikely to be detected in serum after the first week of illness.
However, recent reports have suggested that Zika virus RNA can be detected in urine for at least 2 weeks after the onset of symptoms.
Therefore, the CDC recommends that real-time reverse transcription–polymerase chain reaction (rRT-PCR) be performed on urine collected less than 14 days after the onset of symptoms in patients with suspected Zika virus.
The agency said further investigation is needed to determine the utility of rRT-PCR on urine specimens collected at 14 days or beyond.
The CDC also recommends that urine testing be performed in conjunction with serum testing for specimens collected less than 7 days after symptom onset. The agency said its recommendations for testing serum and other clinical specimens for Zika virus have not changed.
At present, the CDC’s Trioplex rRT-PCR assay is the only diagnostic tool authorized by the US Food and Drug Administration to test urine for Zika virus.
The US Centers for Disease Control and Prevention (CDC) has released an interim guidance for testing urine for the Zika virus.
The agency noted that, in most patients, Zika virus RNA is unlikely to be detected in serum after the first week of illness.
However, recent reports have suggested that Zika virus RNA can be detected in urine for at least 2 weeks after the onset of symptoms.
Therefore, the CDC recommends that real-time reverse transcription–polymerase chain reaction (rRT-PCR) be performed on urine collected less than 14 days after the onset of symptoms in patients with suspected Zika virus.
The agency said further investigation is needed to determine the utility of rRT-PCR on urine specimens collected at 14 days or beyond.
The CDC also recommends that urine testing be performed in conjunction with serum testing for specimens collected less than 7 days after symptom onset. The agency said its recommendations for testing serum and other clinical specimens for Zika virus have not changed.
At present, the CDC’s Trioplex rRT-PCR assay is the only diagnostic tool authorized by the US Food and Drug Administration to test urine for Zika virus.
The US Centers for Disease Control and Prevention (CDC) has released an interim guidance for testing urine for the Zika virus.
The agency noted that, in most patients, Zika virus RNA is unlikely to be detected in serum after the first week of illness.
However, recent reports have suggested that Zika virus RNA can be detected in urine for at least 2 weeks after the onset of symptoms.
Therefore, the CDC recommends that real-time reverse transcription–polymerase chain reaction (rRT-PCR) be performed on urine collected less than 14 days after the onset of symptoms in patients with suspected Zika virus.
The agency said further investigation is needed to determine the utility of rRT-PCR on urine specimens collected at 14 days or beyond.
The CDC also recommends that urine testing be performed in conjunction with serum testing for specimens collected less than 7 days after symptom onset. The agency said its recommendations for testing serum and other clinical specimens for Zika virus have not changed.
At present, the CDC’s Trioplex rRT-PCR assay is the only diagnostic tool authorized by the US Food and Drug Administration to test urine for Zika virus.
Health Canada approves ibrutinib for WM

Photo from Janssen Biotech
Health Canada has approved the BTK inhibitor ibrutinib (Imbruvica) as a treatment for patients with Waldenström’s macroglobulinemia (WM).
Ibrutinib was first approved in Canada in November 2014 for the treatment of patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL and 17p deletion.
In July 2015, ibrutinib was granted conditional approval for the treatment of patients with relapsed or refractory mantle cell lymphoma.
Health Canada’s approval of ibrutinib for WM was based on results of a multicenter, phase 2 study in which researchers tested the drug (given at 420 mg once daily) in 63 patients with previously treated WM.
The patients’ median age was 63 (range, 44-86), and their median number of prior therapies was 2 (range, 1-11).
Initial data showed an overall response rate of 87.3% in patients who received ibrutinib for a median of 11.7 months.
Updated results from the study were published in NEJM in April 2015. After a median treatment duration of 19.1 months, the overall response rate was 91%.
At 24 months, the estimated rate of progression-free survival was 69%, and the estimated rate of overall survival was 95%.
The most common grade 2-4 adverse events were neutropenia (22%) and thrombocytopenia (14%). Ibrutinib-related neutropenia and thrombocytopenia were reversible but required a dose reduction in 3 patients and treatment discontinuation in 4 patients.
Grade 2 or higher bleeding events occurred in 4 patients, and there were 15 infections considered possibly related to ibrutinib.
Treatment-related atrial fibrillation (AFib) occurred in 3 patients, all of whom had a prior history of paroxysmal AFib. AFib resolved when treatment was withheld, and all 3 patients were able to continue on therapy per protocol without an additional event.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada.
Photo from Janssen Biotech
Health Canada has approved the BTK inhibitor ibrutinib (Imbruvica) as a treatment for patients with Waldenström’s macroglobulinemia (WM).
Ibrutinib was first approved in Canada in November 2014 for the treatment of patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL and 17p deletion.
In July 2015, ibrutinib was granted conditional approval for the treatment of patients with relapsed or refractory mantle cell lymphoma.
Health Canada’s approval of ibrutinib for WM was based on results of a multicenter, phase 2 study in which researchers tested the drug (given at 420 mg once daily) in 63 patients with previously treated WM.
The patients’ median age was 63 (range, 44-86), and their median number of prior therapies was 2 (range, 1-11).
Initial data showed an overall response rate of 87.3% in patients who received ibrutinib for a median of 11.7 months.
Updated results from the study were published in NEJM in April 2015. After a median treatment duration of 19.1 months, the overall response rate was 91%.
At 24 months, the estimated rate of progression-free survival was 69%, and the estimated rate of overall survival was 95%.
The most common grade 2-4 adverse events were neutropenia (22%) and thrombocytopenia (14%). Ibrutinib-related neutropenia and thrombocytopenia were reversible but required a dose reduction in 3 patients and treatment discontinuation in 4 patients.
Grade 2 or higher bleeding events occurred in 4 patients, and there were 15 infections considered possibly related to ibrutinib.
Treatment-related atrial fibrillation (AFib) occurred in 3 patients, all of whom had a prior history of paroxysmal AFib. AFib resolved when treatment was withheld, and all 3 patients were able to continue on therapy per protocol without an additional event.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada.
Photo from Janssen Biotech
Health Canada has approved the BTK inhibitor ibrutinib (Imbruvica) as a treatment for patients with Waldenström’s macroglobulinemia (WM).
Ibrutinib was first approved in Canada in November 2014 for the treatment of patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL and 17p deletion.
In July 2015, ibrutinib was granted conditional approval for the treatment of patients with relapsed or refractory mantle cell lymphoma.
Health Canada’s approval of ibrutinib for WM was based on results of a multicenter, phase 2 study in which researchers tested the drug (given at 420 mg once daily) in 63 patients with previously treated WM.
The patients’ median age was 63 (range, 44-86), and their median number of prior therapies was 2 (range, 1-11).
Initial data showed an overall response rate of 87.3% in patients who received ibrutinib for a median of 11.7 months.
Updated results from the study were published in NEJM in April 2015. After a median treatment duration of 19.1 months, the overall response rate was 91%.
At 24 months, the estimated rate of progression-free survival was 69%, and the estimated rate of overall survival was 95%.
The most common grade 2-4 adverse events were neutropenia (22%) and thrombocytopenia (14%). Ibrutinib-related neutropenia and thrombocytopenia were reversible but required a dose reduction in 3 patients and treatment discontinuation in 4 patients.
Grade 2 or higher bleeding events occurred in 4 patients, and there were 15 infections considered possibly related to ibrutinib.
Treatment-related atrial fibrillation (AFib) occurred in 3 patients, all of whom had a prior history of paroxysmal AFib. AFib resolved when treatment was withheld, and all 3 patients were able to continue on therapy per protocol without an additional event.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada.
Drug may reduce severity of AEs from dexamethasone

Photo by Bill Branson
Adding a physiologic dose of hydrocortisone to treatment with dexamethasone can reduce the severity of certain adverse effects (AEs) in pediatric patients with acute lymphoblastic leukemia (ALL), according to researchers.
Hydrocortisone did not decrease the incidence of psychosocial problems or sleep-related issues in these patients, but the drug did reduce the severity of these problems among patients who experienced them.
Lidewij T. Warris, MD, of Erasmus MC-Sophia Children’s Hospital in Rotterdam, the Netherlands, and her colleagues reported these results in the Journal of Clinical Oncology.
The team conducted this study to determine whether a physiologic dose of hydrocortisone could reduce neuropsychologic and metabolic AEs in children with ALL who were receiving dexamethasone.
The study enrolled 50 patients (ages 3 to 16) who were set to receive 2 consecutive courses of dexamethasone in accordance with Dutch Childhood Oncology Group ALL protocols.
The patients were randomized to receive either hydrocortisone or placebo in a circadian rhythm (10 mg/m2/d) during their first dexamethasone course. During their second course, the patients were assigned to the opposite arm.
The treatment groups were similar with regard to age, type of leukemia, treatment protocol, and CNS status at diagnosis.
Psychosocial problems
The researchers assessed psychosocial problems by having parents complete the Strength and Difficulties Questionnaire (SDQ). Forty-six parents completed the questionnaire at all 4 time points tested.
The results showed that 4 days of dexamethasone treatment significantly increased patient problems, as reported by all SDQ scales and subscales. However, one-third of the population did not have any increase in SDQ total difficulties with dexamethasone.
The addition of hydrocortisone did not affect patients’ total difficulties score (mean difference, -0.8 ± 5.5; P=0.33), emotional symptoms (mean difference, -0.6 ± 2.3; P=0.08), conduct problems (mean difference, 0.0 ± 1.5; P=1.00), or other SDQ subscales.
However, hydrocortisone did have a clinically significant effect in the subset of 16 patients who had clinically relevant dexamethasone-related AEs. This was defined as an increase of ≥5 in their SDQ total difficulties score.
In these patients, hydrocortisone improved the total difficulties delta-score (median difference, -5.0; IQR, -7.8 to -3.0), emotional symptoms score (median difference, -1.5; IQR, -4.0 to -1.0), conduct problems score (median difference, -1.0; IQR, -2.0 to 0.0), and impact of stress score (median difference, -1.0; IQR, -2.0 to 0.0).
Sleep issues
The researchers used the Sleep Disturbance Scale for Children (SDSC) to assess sleep quality and sleep disturbances. Forty-seven parents completed the questionnaire at all 4 time points tested.
Results showed that dexamethasone significantly increased disorders of arousal (P=0.04), sleep-wake transition disorders (P=0.01), and disorders of excessive somnolence (P=0.01).
The addition of hydrocortisone had no significant effect on patients’ total SDSC score (P=0.84), disorders of initiating and maintaining sleep (P=0.74), disorders of excessive somnolence (P=0.29), or sleep-wake transition disorder (P=0.29).
However, hydrocortisone did have a clinically significant effect in the subset of 9 children who had clinically relevant dexamethasone-induced sleeping problems, which were defined as a change of ≥7 in SDSC total score.
Hydrocortisone reduced SDSC total scores (median difference, -11.0; IQR, -16.0 to 0.0) and disorders of initiating and maintaining sleep scores (median difference, -3.0; IQR, -7.0 to –0.5).
Other outcomes
The researchers also found that dexamethasone treatment alone did not affect patients’ attention, visual-spatial functions, memory, or processing speed.
However, the addition of hydrocortisone significantly improved patients’ long-term visual memory (P=0.01).
Hydrocortisone did not have any effect on other neuropsychological tests or on metabolic parameters.
Photo by Bill Branson
Adding a physiologic dose of hydrocortisone to treatment with dexamethasone can reduce the severity of certain adverse effects (AEs) in pediatric patients with acute lymphoblastic leukemia (ALL), according to researchers.
Hydrocortisone did not decrease the incidence of psychosocial problems or sleep-related issues in these patients, but the drug did reduce the severity of these problems among patients who experienced them.
Lidewij T. Warris, MD, of Erasmus MC-Sophia Children’s Hospital in Rotterdam, the Netherlands, and her colleagues reported these results in the Journal of Clinical Oncology.
The team conducted this study to determine whether a physiologic dose of hydrocortisone could reduce neuropsychologic and metabolic AEs in children with ALL who were receiving dexamethasone.
The study enrolled 50 patients (ages 3 to 16) who were set to receive 2 consecutive courses of dexamethasone in accordance with Dutch Childhood Oncology Group ALL protocols.
The patients were randomized to receive either hydrocortisone or placebo in a circadian rhythm (10 mg/m2/d) during their first dexamethasone course. During their second course, the patients were assigned to the opposite arm.
The treatment groups were similar with regard to age, type of leukemia, treatment protocol, and CNS status at diagnosis.
Psychosocial problems
The researchers assessed psychosocial problems by having parents complete the Strength and Difficulties Questionnaire (SDQ). Forty-six parents completed the questionnaire at all 4 time points tested.
The results showed that 4 days of dexamethasone treatment significantly increased patient problems, as reported by all SDQ scales and subscales. However, one-third of the population did not have any increase in SDQ total difficulties with dexamethasone.
The addition of hydrocortisone did not affect patients’ total difficulties score (mean difference, -0.8 ± 5.5; P=0.33), emotional symptoms (mean difference, -0.6 ± 2.3; P=0.08), conduct problems (mean difference, 0.0 ± 1.5; P=1.00), or other SDQ subscales.
However, hydrocortisone did have a clinically significant effect in the subset of 16 patients who had clinically relevant dexamethasone-related AEs. This was defined as an increase of ≥5 in their SDQ total difficulties score.
In these patients, hydrocortisone improved the total difficulties delta-score (median difference, -5.0; IQR, -7.8 to -3.0), emotional symptoms score (median difference, -1.5; IQR, -4.0 to -1.0), conduct problems score (median difference, -1.0; IQR, -2.0 to 0.0), and impact of stress score (median difference, -1.0; IQR, -2.0 to 0.0).
Sleep issues
The researchers used the Sleep Disturbance Scale for Children (SDSC) to assess sleep quality and sleep disturbances. Forty-seven parents completed the questionnaire at all 4 time points tested.
Results showed that dexamethasone significantly increased disorders of arousal (P=0.04), sleep-wake transition disorders (P=0.01), and disorders of excessive somnolence (P=0.01).
The addition of hydrocortisone had no significant effect on patients’ total SDSC score (P=0.84), disorders of initiating and maintaining sleep (P=0.74), disorders of excessive somnolence (P=0.29), or sleep-wake transition disorder (P=0.29).
However, hydrocortisone did have a clinically significant effect in the subset of 9 children who had clinically relevant dexamethasone-induced sleeping problems, which were defined as a change of ≥7 in SDSC total score.
Hydrocortisone reduced SDSC total scores (median difference, -11.0; IQR, -16.0 to 0.0) and disorders of initiating and maintaining sleep scores (median difference, -3.0; IQR, -7.0 to –0.5).
Other outcomes
The researchers also found that dexamethasone treatment alone did not affect patients’ attention, visual-spatial functions, memory, or processing speed.
However, the addition of hydrocortisone significantly improved patients’ long-term visual memory (P=0.01).
Hydrocortisone did not have any effect on other neuropsychological tests or on metabolic parameters.
Photo by Bill Branson
Adding a physiologic dose of hydrocortisone to treatment with dexamethasone can reduce the severity of certain adverse effects (AEs) in pediatric patients with acute lymphoblastic leukemia (ALL), according to researchers.
Hydrocortisone did not decrease the incidence of psychosocial problems or sleep-related issues in these patients, but the drug did reduce the severity of these problems among patients who experienced them.
Lidewij T. Warris, MD, of Erasmus MC-Sophia Children’s Hospital in Rotterdam, the Netherlands, and her colleagues reported these results in the Journal of Clinical Oncology.
The team conducted this study to determine whether a physiologic dose of hydrocortisone could reduce neuropsychologic and metabolic AEs in children with ALL who were receiving dexamethasone.
The study enrolled 50 patients (ages 3 to 16) who were set to receive 2 consecutive courses of dexamethasone in accordance with Dutch Childhood Oncology Group ALL protocols.
The patients were randomized to receive either hydrocortisone or placebo in a circadian rhythm (10 mg/m2/d) during their first dexamethasone course. During their second course, the patients were assigned to the opposite arm.
The treatment groups were similar with regard to age, type of leukemia, treatment protocol, and CNS status at diagnosis.
Psychosocial problems
The researchers assessed psychosocial problems by having parents complete the Strength and Difficulties Questionnaire (SDQ). Forty-six parents completed the questionnaire at all 4 time points tested.
The results showed that 4 days of dexamethasone treatment significantly increased patient problems, as reported by all SDQ scales and subscales. However, one-third of the population did not have any increase in SDQ total difficulties with dexamethasone.
The addition of hydrocortisone did not affect patients’ total difficulties score (mean difference, -0.8 ± 5.5; P=0.33), emotional symptoms (mean difference, -0.6 ± 2.3; P=0.08), conduct problems (mean difference, 0.0 ± 1.5; P=1.00), or other SDQ subscales.
However, hydrocortisone did have a clinically significant effect in the subset of 16 patients who had clinically relevant dexamethasone-related AEs. This was defined as an increase of ≥5 in their SDQ total difficulties score.
In these patients, hydrocortisone improved the total difficulties delta-score (median difference, -5.0; IQR, -7.8 to -3.0), emotional symptoms score (median difference, -1.5; IQR, -4.0 to -1.0), conduct problems score (median difference, -1.0; IQR, -2.0 to 0.0), and impact of stress score (median difference, -1.0; IQR, -2.0 to 0.0).
Sleep issues
The researchers used the Sleep Disturbance Scale for Children (SDSC) to assess sleep quality and sleep disturbances. Forty-seven parents completed the questionnaire at all 4 time points tested.
Results showed that dexamethasone significantly increased disorders of arousal (P=0.04), sleep-wake transition disorders (P=0.01), and disorders of excessive somnolence (P=0.01).
The addition of hydrocortisone had no significant effect on patients’ total SDSC score (P=0.84), disorders of initiating and maintaining sleep (P=0.74), disorders of excessive somnolence (P=0.29), or sleep-wake transition disorder (P=0.29).
However, hydrocortisone did have a clinically significant effect in the subset of 9 children who had clinically relevant dexamethasone-induced sleeping problems, which were defined as a change of ≥7 in SDSC total score.
Hydrocortisone reduced SDSC total scores (median difference, -11.0; IQR, -16.0 to 0.0) and disorders of initiating and maintaining sleep scores (median difference, -3.0; IQR, -7.0 to –0.5).
Other outcomes
The researchers also found that dexamethasone treatment alone did not affect patients’ attention, visual-spatial functions, memory, or processing speed.
However, the addition of hydrocortisone significantly improved patients’ long-term visual memory (P=0.01).
Hydrocortisone did not have any effect on other neuropsychological tests or on metabolic parameters.