User login
Cytotect®CP found to be safe and effective after allo-HCT

A small retrospective study of 23 transplant patients has confirmed that CMV hyperimmune globulin (Cytotect®CP) is a safe and effective salvage therapy for patients with cytomegalovirus (CMV) infection after allogeneic hematopoietic cell transplant (allo-HCT).
Cytotect®CP used as salvage therapy resulted in a 78% overall response rate and 70% of all patients cleared CMV infection, according to investigators.
They observed no clinically significant adverse events.
CMV is a major factor contributing to high mortality rates in allo-HCT patients.
And because Cytotect®CP is less toxic than commonly used treatments for CMV infection, investigators suggested that it be used prophylactically in patients known to have a predisposition to CMV infection.
They reported their findings in the journal Bone Marrow Transplantation.
Patient characteristics and methods
All 23 patients transplanted at 8 centers in France were CMV seropositive at the time of transplant, and 70% received the transplant from a CMV serostatus negative donor.
Recipient positivity and donor negativity, the investigators indicated, is a risk factor for developing recurrent CMV infection after allo-HCT.
The patients’ median age was 53, 11 were male and 12 female.
Five patients (22%) received a haploidentical transplant and 14 (61%) from an unrelated donor, which the investigators pointed out is also a known risk factor for developing recurrent CMV infection.
Thirteen (57%) were in complete remission from their underlying disease, 4 (17%) in partial remission, 1 (4%) had stable disease, and 5 (22%) were in relapse or had progressive disease.
Most (83%) had a peripheral blood transplant, 15 (65%) had a reduced intensity conditioning regimen, and 16 (70%) had antithymocyte globulin as part of their conditioning regimen.
All patients received valacyclovir as antiviral prophylaxis prior to receiving Cytotect®CP. The investigators mentioned in the paper that valacyclovir has not been proven to be effective in treating CMV.
They noted that other CMV treatments, such as ganciclovir, foscavir, and dicofovir, cause high levels of toxicity, frequently leading to treatment discontinuation.
Seventeen patients (74%) had a history of graft-versus-host disease (GVHD), and 11 (49%) had active GVHD at the time CMV hyperimmune globulin was administered.
Investigators used quantitative polymerase chain reaction (PCR) to quantify CMV viral load in the blood.
Treatment could begin when patients’ viral load was greater than 3-3.5 log UI/mL, according to the Francophone Society of Bone Marrow Transplantation and Cellular therapy.
Three patients received Cytotect®CP at a prophylaxis dose (200 U/kg/week) to prevent CMV recurrences and 20 as preemptive therapy (400 U/kg on days 1, 4, 8 then 200 U/kg on days 12and 16).
Seven patients (30%) received Cytotect®CP as monotherapy, 5 (22%) in combination with ganciclovir, 5 (22%) in combination with foscavir, 2 (9%) in combination with ganciclovir and foscavir, and 4 (17%) with some other combination.
Investigators restricted their analysis to 100-day overall survival (OS), starting at the beginning of Cytotect®CP treatment to death within 100 days, regardless of the cause of death.
Results
Eighteen patients (78%) responded to Cytotect®CP, and 16 of the responders converted to CMV-PCR negative.
Median time to achieve CMV-PCR response was 15 days (range, 3-51).
Four patients did not respond to therapy, and 1 patient had a non-evaluable response. The latter patient died 13 days after the introduction of Cytotect®CP due to another infection.
Eight patients died within 100 days after initiation of Cytotect®CP. Two deaths were related to CMV and 6 were unrelated.
Four patients who responded to Cytotect®CP experienced CMV relapse between 9 and 49 days after their best response to therapy.
Five responders died within 100 days due to the following causes: GVHD (n = 2), other infection (n = 1), underlying disease (n = 1), and CMV-related causes (n = 1).
Two of the 4 nonresponders died of other infection (n = 1) and GVHD (n = 1).
Investigators estimated the 100-day OS from the start of Cytotect®CP to be 69.6%. They observed no statistical difference (P=0.258) between those who responded (73.7%) and those who didn’t (50.0%).
The investigators believe that Cytotect®CP is an alternative option for treatment of CMV infection because it avoids renal and bone marrow impairment and should be considered as prophylaxis in select patients.
They recommend a large prospective study be conducted to confirm safety and efficacy results of CMV hyperimmune globulin.
Cytotect®CP is authorized in more than 15 countries for the prophylaxis of CMV infection in patients receiving immunosuppressive treatment, particularly transplant recipients.
In French transplant centers, according to the study authors, use of Cytotect®CP is limited to the salvage setting for recurrent or refractory CMV infections and sometimes in combination for CMV pneumonia.
Biotest, the commercializer of Cytotect®CP, provided a grant for this study.
A small retrospective study of 23 transplant patients has confirmed that CMV hyperimmune globulin (Cytotect®CP) is a safe and effective salvage therapy for patients with cytomegalovirus (CMV) infection after allogeneic hematopoietic cell transplant (allo-HCT).
Cytotect®CP used as salvage therapy resulted in a 78% overall response rate and 70% of all patients cleared CMV infection, according to investigators.
They observed no clinically significant adverse events.
CMV is a major factor contributing to high mortality rates in allo-HCT patients.
And because Cytotect®CP is less toxic than commonly used treatments for CMV infection, investigators suggested that it be used prophylactically in patients known to have a predisposition to CMV infection.
They reported their findings in the journal Bone Marrow Transplantation.
Patient characteristics and methods
All 23 patients transplanted at 8 centers in France were CMV seropositive at the time of transplant, and 70% received the transplant from a CMV serostatus negative donor.
Recipient positivity and donor negativity, the investigators indicated, is a risk factor for developing recurrent CMV infection after allo-HCT.
The patients’ median age was 53, 11 were male and 12 female.
Five patients (22%) received a haploidentical transplant and 14 (61%) from an unrelated donor, which the investigators pointed out is also a known risk factor for developing recurrent CMV infection.
Thirteen (57%) were in complete remission from their underlying disease, 4 (17%) in partial remission, 1 (4%) had stable disease, and 5 (22%) were in relapse or had progressive disease.
Most (83%) had a peripheral blood transplant, 15 (65%) had a reduced intensity conditioning regimen, and 16 (70%) had antithymocyte globulin as part of their conditioning regimen.
All patients received valacyclovir as antiviral prophylaxis prior to receiving Cytotect®CP. The investigators mentioned in the paper that valacyclovir has not been proven to be effective in treating CMV.
They noted that other CMV treatments, such as ganciclovir, foscavir, and dicofovir, cause high levels of toxicity, frequently leading to treatment discontinuation.
Seventeen patients (74%) had a history of graft-versus-host disease (GVHD), and 11 (49%) had active GVHD at the time CMV hyperimmune globulin was administered.
Investigators used quantitative polymerase chain reaction (PCR) to quantify CMV viral load in the blood.
Treatment could begin when patients’ viral load was greater than 3-3.5 log UI/mL, according to the Francophone Society of Bone Marrow Transplantation and Cellular therapy.
Three patients received Cytotect®CP at a prophylaxis dose (200 U/kg/week) to prevent CMV recurrences and 20 as preemptive therapy (400 U/kg on days 1, 4, 8 then 200 U/kg on days 12and 16).
Seven patients (30%) received Cytotect®CP as monotherapy, 5 (22%) in combination with ganciclovir, 5 (22%) in combination with foscavir, 2 (9%) in combination with ganciclovir and foscavir, and 4 (17%) with some other combination.
Investigators restricted their analysis to 100-day overall survival (OS), starting at the beginning of Cytotect®CP treatment to death within 100 days, regardless of the cause of death.
Results
Eighteen patients (78%) responded to Cytotect®CP, and 16 of the responders converted to CMV-PCR negative.
Median time to achieve CMV-PCR response was 15 days (range, 3-51).
Four patients did not respond to therapy, and 1 patient had a non-evaluable response. The latter patient died 13 days after the introduction of Cytotect®CP due to another infection.
Eight patients died within 100 days after initiation of Cytotect®CP. Two deaths were related to CMV and 6 were unrelated.
Four patients who responded to Cytotect®CP experienced CMV relapse between 9 and 49 days after their best response to therapy.
Five responders died within 100 days due to the following causes: GVHD (n = 2), other infection (n = 1), underlying disease (n = 1), and CMV-related causes (n = 1).
Two of the 4 nonresponders died of other infection (n = 1) and GVHD (n = 1).
Investigators estimated the 100-day OS from the start of Cytotect®CP to be 69.6%. They observed no statistical difference (P=0.258) between those who responded (73.7%) and those who didn’t (50.0%).
The investigators believe that Cytotect®CP is an alternative option for treatment of CMV infection because it avoids renal and bone marrow impairment and should be considered as prophylaxis in select patients.
They recommend a large prospective study be conducted to confirm safety and efficacy results of CMV hyperimmune globulin.
Cytotect®CP is authorized in more than 15 countries for the prophylaxis of CMV infection in patients receiving immunosuppressive treatment, particularly transplant recipients.
In French transplant centers, according to the study authors, use of Cytotect®CP is limited to the salvage setting for recurrent or refractory CMV infections and sometimes in combination for CMV pneumonia.
Biotest, the commercializer of Cytotect®CP, provided a grant for this study.
A small retrospective study of 23 transplant patients has confirmed that CMV hyperimmune globulin (Cytotect®CP) is a safe and effective salvage therapy for patients with cytomegalovirus (CMV) infection after allogeneic hematopoietic cell transplant (allo-HCT).
Cytotect®CP used as salvage therapy resulted in a 78% overall response rate and 70% of all patients cleared CMV infection, according to investigators.
They observed no clinically significant adverse events.
CMV is a major factor contributing to high mortality rates in allo-HCT patients.
And because Cytotect®CP is less toxic than commonly used treatments for CMV infection, investigators suggested that it be used prophylactically in patients known to have a predisposition to CMV infection.
They reported their findings in the journal Bone Marrow Transplantation.
Patient characteristics and methods
All 23 patients transplanted at 8 centers in France were CMV seropositive at the time of transplant, and 70% received the transplant from a CMV serostatus negative donor.
Recipient positivity and donor negativity, the investigators indicated, is a risk factor for developing recurrent CMV infection after allo-HCT.
The patients’ median age was 53, 11 were male and 12 female.
Five patients (22%) received a haploidentical transplant and 14 (61%) from an unrelated donor, which the investigators pointed out is also a known risk factor for developing recurrent CMV infection.
Thirteen (57%) were in complete remission from their underlying disease, 4 (17%) in partial remission, 1 (4%) had stable disease, and 5 (22%) were in relapse or had progressive disease.
Most (83%) had a peripheral blood transplant, 15 (65%) had a reduced intensity conditioning regimen, and 16 (70%) had antithymocyte globulin as part of their conditioning regimen.
All patients received valacyclovir as antiviral prophylaxis prior to receiving Cytotect®CP. The investigators mentioned in the paper that valacyclovir has not been proven to be effective in treating CMV.
They noted that other CMV treatments, such as ganciclovir, foscavir, and dicofovir, cause high levels of toxicity, frequently leading to treatment discontinuation.
Seventeen patients (74%) had a history of graft-versus-host disease (GVHD), and 11 (49%) had active GVHD at the time CMV hyperimmune globulin was administered.
Investigators used quantitative polymerase chain reaction (PCR) to quantify CMV viral load in the blood.
Treatment could begin when patients’ viral load was greater than 3-3.5 log UI/mL, according to the Francophone Society of Bone Marrow Transplantation and Cellular therapy.
Three patients received Cytotect®CP at a prophylaxis dose (200 U/kg/week) to prevent CMV recurrences and 20 as preemptive therapy (400 U/kg on days 1, 4, 8 then 200 U/kg on days 12and 16).
Seven patients (30%) received Cytotect®CP as monotherapy, 5 (22%) in combination with ganciclovir, 5 (22%) in combination with foscavir, 2 (9%) in combination with ganciclovir and foscavir, and 4 (17%) with some other combination.
Investigators restricted their analysis to 100-day overall survival (OS), starting at the beginning of Cytotect®CP treatment to death within 100 days, regardless of the cause of death.
Results
Eighteen patients (78%) responded to Cytotect®CP, and 16 of the responders converted to CMV-PCR negative.
Median time to achieve CMV-PCR response was 15 days (range, 3-51).
Four patients did not respond to therapy, and 1 patient had a non-evaluable response. The latter patient died 13 days after the introduction of Cytotect®CP due to another infection.
Eight patients died within 100 days after initiation of Cytotect®CP. Two deaths were related to CMV and 6 were unrelated.
Four patients who responded to Cytotect®CP experienced CMV relapse between 9 and 49 days after their best response to therapy.
Five responders died within 100 days due to the following causes: GVHD (n = 2), other infection (n = 1), underlying disease (n = 1), and CMV-related causes (n = 1).
Two of the 4 nonresponders died of other infection (n = 1) and GVHD (n = 1).
Investigators estimated the 100-day OS from the start of Cytotect®CP to be 69.6%. They observed no statistical difference (P=0.258) between those who responded (73.7%) and those who didn’t (50.0%).
The investigators believe that Cytotect®CP is an alternative option for treatment of CMV infection because it avoids renal and bone marrow impairment and should be considered as prophylaxis in select patients.
They recommend a large prospective study be conducted to confirm safety and efficacy results of CMV hyperimmune globulin.
Cytotect®CP is authorized in more than 15 countries for the prophylaxis of CMV infection in patients receiving immunosuppressive treatment, particularly transplant recipients.
In French transplant centers, according to the study authors, use of Cytotect®CP is limited to the salvage setting for recurrent or refractory CMV infections and sometimes in combination for CMV pneumonia.
Biotest, the commercializer of Cytotect®CP, provided a grant for this study.
Liquid biopsies may be the future in managing MM patients
Scientists from the Dana-Farber Cancer Institute and the Broad Institute in Boston, Massachusetts, have shown that in multiple myeloma (MM), liquid biopsies provide similar genetic information as tumor biopsies.
Unlike tumor biopsies, liquid biopsies are noninvasive and, in the future, may offer a cost-effective way of following disease state, disease progression, and response to treatment.
The scientists used cell-free DNA (cfDNA) from 107 patients, circulating tumor cells (CTCs) from 56 patients, and compared their genomic profiling with bone marrow biopsies from 9 patients with MM.
The researchers used a two-step approach using the 2 kinds of liquid biopsies—CTCs and cfDNA. They first used “ultra-low pass” whole genome sequencing, which was a rapid and cost-effective method to identify blood samples with tumor DNA of at least 5%-10% (tumor fraction).
These samples were then subject to whole-exome sequencing (WES), which analyzes protein-coding sequence of the genome.
The researchers found genetic alterations in MM such as copy number alterations (1p, 13q deletions or 11q gain) and somatic single nucleotide variations (SSNVs), among others, which they observed in matched cfDNA, CTCs, and tumor DNA (tDNA) from tumor biopsies.
The liquid biopsies also captured the clonal heterogeneity characteristic of MM—99% of clonal mutations present in tDNA were also seen in cfDNA or CTCs, and 94% of mutations present in cfDNA or CTCs were seen in tDNA.
In addition, CTCs or cfDNA samples with higher tumor purity uncovered more mutations than were seen in tDNA.
“[The] combination of CTCs and cfDNA was able to detect almost all clonal mutations identified in the bone marrow biopsy sample and defined other subclones that were not identified in the bone marrow,” the scientists noted.
This increased sensitivity was “potentially due to the limitation of sampling site of the bone marrow,” they pointed out.
The scientists also showed that the tumor fraction in cfDNA and enriched CTCs correlated with disease progression from MGUS (monoclonal gammopathy of undetermined significance) and SMM (smoldering MM), to overt MM.
The scientists also evaluated liquid biopsies to determine response to treatment.
For example, a patient who responded to carfilzomib, lenalidomide, and dexamethasone, had a decreased cfDNA tumor fraction—from 22% to 2%.
Another patient who progressed on daratumumab over a period of 2 months showed an increase in tumor fraction—from 11% to 46%.
CTCs and cfDNA captured the mutational landscape of MM and provided a non-invasive profiling of tumor evolution. The liquid biopsy approach may be used as a novel biomarker for disease progression and response to therapy, the scientists suggest.
"Our discovery that cfDNA and CTC analyses agree with each other at the comprehensive level is an important finding,” said co-senior author Irene Ghobrial, MD, “because this means that routine genetic profiling of patient tumors from blood would be feasible."
"Our ultimate goal is to eventually use all the samples to monitor disease progression," added Jihye Park, PhD, researcher in the Ghobrial lab and co-first author on the paper.
Currently, bone marrow biopsies are the gold standard for diagnosing and monitoring the progression of MM.
However, due to their invasive nature and associated costs, they are not done at every visit. Liquid biopsies are an attractive way forward.
Nevertheless, before they can be used in the clinical management of patients with MM, these results need to be confirmed in larger prospective studies.
The investigators reported their findings in Nature Communications.
Scientists from the Dana-Farber Cancer Institute and the Broad Institute in Boston, Massachusetts, have shown that in multiple myeloma (MM), liquid biopsies provide similar genetic information as tumor biopsies.
Unlike tumor biopsies, liquid biopsies are noninvasive and, in the future, may offer a cost-effective way of following disease state, disease progression, and response to treatment.
The scientists used cell-free DNA (cfDNA) from 107 patients, circulating tumor cells (CTCs) from 56 patients, and compared their genomic profiling with bone marrow biopsies from 9 patients with MM.
The researchers used a two-step approach using the 2 kinds of liquid biopsies—CTCs and cfDNA. They first used “ultra-low pass” whole genome sequencing, which was a rapid and cost-effective method to identify blood samples with tumor DNA of at least 5%-10% (tumor fraction).
These samples were then subject to whole-exome sequencing (WES), which analyzes protein-coding sequence of the genome.
The researchers found genetic alterations in MM such as copy number alterations (1p, 13q deletions or 11q gain) and somatic single nucleotide variations (SSNVs), among others, which they observed in matched cfDNA, CTCs, and tumor DNA (tDNA) from tumor biopsies.
The liquid biopsies also captured the clonal heterogeneity characteristic of MM—99% of clonal mutations present in tDNA were also seen in cfDNA or CTCs, and 94% of mutations present in cfDNA or CTCs were seen in tDNA.
In addition, CTCs or cfDNA samples with higher tumor purity uncovered more mutations than were seen in tDNA.
“[The] combination of CTCs and cfDNA was able to detect almost all clonal mutations identified in the bone marrow biopsy sample and defined other subclones that were not identified in the bone marrow,” the scientists noted.
This increased sensitivity was “potentially due to the limitation of sampling site of the bone marrow,” they pointed out.
The scientists also showed that the tumor fraction in cfDNA and enriched CTCs correlated with disease progression from MGUS (monoclonal gammopathy of undetermined significance) and SMM (smoldering MM), to overt MM.
The scientists also evaluated liquid biopsies to determine response to treatment.
For example, a patient who responded to carfilzomib, lenalidomide, and dexamethasone, had a decreased cfDNA tumor fraction—from 22% to 2%.
Another patient who progressed on daratumumab over a period of 2 months showed an increase in tumor fraction—from 11% to 46%.
CTCs and cfDNA captured the mutational landscape of MM and provided a non-invasive profiling of tumor evolution. The liquid biopsy approach may be used as a novel biomarker for disease progression and response to therapy, the scientists suggest.
"Our discovery that cfDNA and CTC analyses agree with each other at the comprehensive level is an important finding,” said co-senior author Irene Ghobrial, MD, “because this means that routine genetic profiling of patient tumors from blood would be feasible."
"Our ultimate goal is to eventually use all the samples to monitor disease progression," added Jihye Park, PhD, researcher in the Ghobrial lab and co-first author on the paper.
Currently, bone marrow biopsies are the gold standard for diagnosing and monitoring the progression of MM.
However, due to their invasive nature and associated costs, they are not done at every visit. Liquid biopsies are an attractive way forward.
Nevertheless, before they can be used in the clinical management of patients with MM, these results need to be confirmed in larger prospective studies.
The investigators reported their findings in Nature Communications.
Scientists from the Dana-Farber Cancer Institute and the Broad Institute in Boston, Massachusetts, have shown that in multiple myeloma (MM), liquid biopsies provide similar genetic information as tumor biopsies.
Unlike tumor biopsies, liquid biopsies are noninvasive and, in the future, may offer a cost-effective way of following disease state, disease progression, and response to treatment.
The scientists used cell-free DNA (cfDNA) from 107 patients, circulating tumor cells (CTCs) from 56 patients, and compared their genomic profiling with bone marrow biopsies from 9 patients with MM.
The researchers used a two-step approach using the 2 kinds of liquid biopsies—CTCs and cfDNA. They first used “ultra-low pass” whole genome sequencing, which was a rapid and cost-effective method to identify blood samples with tumor DNA of at least 5%-10% (tumor fraction).
These samples were then subject to whole-exome sequencing (WES), which analyzes protein-coding sequence of the genome.
The researchers found genetic alterations in MM such as copy number alterations (1p, 13q deletions or 11q gain) and somatic single nucleotide variations (SSNVs), among others, which they observed in matched cfDNA, CTCs, and tumor DNA (tDNA) from tumor biopsies.
The liquid biopsies also captured the clonal heterogeneity characteristic of MM—99% of clonal mutations present in tDNA were also seen in cfDNA or CTCs, and 94% of mutations present in cfDNA or CTCs were seen in tDNA.
In addition, CTCs or cfDNA samples with higher tumor purity uncovered more mutations than were seen in tDNA.
“[The] combination of CTCs and cfDNA was able to detect almost all clonal mutations identified in the bone marrow biopsy sample and defined other subclones that were not identified in the bone marrow,” the scientists noted.
This increased sensitivity was “potentially due to the limitation of sampling site of the bone marrow,” they pointed out.
The scientists also showed that the tumor fraction in cfDNA and enriched CTCs correlated with disease progression from MGUS (monoclonal gammopathy of undetermined significance) and SMM (smoldering MM), to overt MM.
The scientists also evaluated liquid biopsies to determine response to treatment.
For example, a patient who responded to carfilzomib, lenalidomide, and dexamethasone, had a decreased cfDNA tumor fraction—from 22% to 2%.
Another patient who progressed on daratumumab over a period of 2 months showed an increase in tumor fraction—from 11% to 46%.
CTCs and cfDNA captured the mutational landscape of MM and provided a non-invasive profiling of tumor evolution. The liquid biopsy approach may be used as a novel biomarker for disease progression and response to therapy, the scientists suggest.
"Our discovery that cfDNA and CTC analyses agree with each other at the comprehensive level is an important finding,” said co-senior author Irene Ghobrial, MD, “because this means that routine genetic profiling of patient tumors from blood would be feasible."
"Our ultimate goal is to eventually use all the samples to monitor disease progression," added Jihye Park, PhD, researcher in the Ghobrial lab and co-first author on the paper.
Currently, bone marrow biopsies are the gold standard for diagnosing and monitoring the progression of MM.
However, due to their invasive nature and associated costs, they are not done at every visit. Liquid biopsies are an attractive way forward.
Nevertheless, before they can be used in the clinical management of patients with MM, these results need to be confirmed in larger prospective studies.
The investigators reported their findings in Nature Communications.
FDA places CTX001 for SCD on clinical hold

The US Food and Drug Administration (FDA) has placed a clinical hold on the investigational new drug application (IND) for CTX001. The agent is being developed for the treatment of sickle cell disease (SCD) and β-thalassemia.
The IND was submitted to the FDA in April to support the initiation of a phase 1/2 trial in the US in adult patients with SCD. The hold will be in place pending the resolution of questions as part of the FDA review.
The phase 1/2 trial in Europe in adult patients with transfusion-dependent β-thalassemia is expected to proceed according to schedule. Trial initiation is planned for the second half of 2018.
CTX001 is being co-developed and co-commercialized by CRISPR Therapeutics and Vertex Pharmaceuticals Incorporated.
The agent is an investigational ex vivo CRISPR gene edited therapy for patients with β-thalassemia or sickle cell disease.
The patient’s hematopoietic stem cells are engineered to produce high levels of fetal hemoglobin (HbF; hemoglobin F) in red blood cells.
The increase in HbF levels by CTX001 may potentially alleviate transfusion requirements for β-thalassemia patients and sickle crises for SCD patients.
The US Food and Drug Administration (FDA) has placed a clinical hold on the investigational new drug application (IND) for CTX001. The agent is being developed for the treatment of sickle cell disease (SCD) and β-thalassemia.
The IND was submitted to the FDA in April to support the initiation of a phase 1/2 trial in the US in adult patients with SCD. The hold will be in place pending the resolution of questions as part of the FDA review.
The phase 1/2 trial in Europe in adult patients with transfusion-dependent β-thalassemia is expected to proceed according to schedule. Trial initiation is planned for the second half of 2018.
CTX001 is being co-developed and co-commercialized by CRISPR Therapeutics and Vertex Pharmaceuticals Incorporated.
The agent is an investigational ex vivo CRISPR gene edited therapy for patients with β-thalassemia or sickle cell disease.
The patient’s hematopoietic stem cells are engineered to produce high levels of fetal hemoglobin (HbF; hemoglobin F) in red blood cells.
The increase in HbF levels by CTX001 may potentially alleviate transfusion requirements for β-thalassemia patients and sickle crises for SCD patients.
The US Food and Drug Administration (FDA) has placed a clinical hold on the investigational new drug application (IND) for CTX001. The agent is being developed for the treatment of sickle cell disease (SCD) and β-thalassemia.
The IND was submitted to the FDA in April to support the initiation of a phase 1/2 trial in the US in adult patients with SCD. The hold will be in place pending the resolution of questions as part of the FDA review.
The phase 1/2 trial in Europe in adult patients with transfusion-dependent β-thalassemia is expected to proceed according to schedule. Trial initiation is planned for the second half of 2018.
CTX001 is being co-developed and co-commercialized by CRISPR Therapeutics and Vertex Pharmaceuticals Incorporated.
The agent is an investigational ex vivo CRISPR gene edited therapy for patients with β-thalassemia or sickle cell disease.
The patient’s hematopoietic stem cells are engineered to produce high levels of fetal hemoglobin (HbF; hemoglobin F) in red blood cells.
The increase in HbF levels by CTX001 may potentially alleviate transfusion requirements for β-thalassemia patients and sickle crises for SCD patients.
VF incidence increases significantly in kids after ALL treatment
A 6-year prospective study from the Canadian STOPP investigators revealed that following treatment for acute lymphoblastic leukemia (ALL), approximately 1 out of 3 children experiences vertebral fractures (VF) and 1 out of 5 children shows non-VF.
Glucocorticoid use and vertebral fractures at diagnosis emerged as significant predictive factors.
Investigators reported a cumulative incidence of 32.5% for vertebral fractures (VF) following treatment and 23.0% for non-VF after 6 years.
Thirty-nine percent of children with VF were asymptomatic.
“The osteotoxicity of leukemia and its treatment are underscored by the observation that the non-VF incidence was more than 2.5 times higher than the general pediatric population,” the study authors wrote, “whereas the VF incidence was increased more than 2000 times.”
The STOPP investigators—the Steroid-Associated Osteoporosis in the Pediatric Population Consortium—published their findings in the Journal of Bone and Mineral Research.
To document the incidence and predictors of fractures and profile who is at greatest risk, the STOPP investigators enrolled 186 children who were diagnosed and treated at 10 children’s hospitals across Canada between 2005 and 2007.
Median age at diagnosis was 5.3 years, median duration of follow-up was 6 years, and 38.1% were boys.
Approximately 4 out of 5 children were treated with Children’s Oncology Group protocols and 1 out of 5 was treated with the Dana-Farber protocols.
In girls, glucocorticoid exposure was 7.0 g/m2 and in boys it was 8.9 g/m2.
The entire cohort received 54.3% of the total cumulative glucocorticoid dose in the first year and 83.1% in the second year.
For VF, incidence at baseline was 15.1%. Skeletal fractures at any site were reported in 36% of the children over the 6-year period, 71% occurring during the first 2 years. No VF was reported in the sixth year.
Of 38 children with VF, 32 sustained at least 1 VF in the first 2 years and 77% occurred while on therapy or within 6 months of the last glucocorticoid dose. None of the children had disease relapse.
Non-VF were reported in 1.6% of children at baseline; incidence peaked at years 2 (5.4%) and 5 (4.8%) and none occurred in the sixth year.
Thirty-one non-VFs were reported in 26 children; 18 occurred in the first 2 years and 23 occurred on therapy or within 6 months of the last glucocorticoid dose.
The STOPP investigators showed that glucocorticoid exposure, VF, and low spine bone mineral density Z-scores at baseline were significant predictors for risk of VF.
“[This] observation suggests the importance of baseline spine phenotype (both VF and low lumbar spine bone mineral density) in signaling the potential for future bone morbidity,” the STOPP investigators note.
“In revealing that vertebral fractures are frequent in children with ALL on chemotherapy, and that older children and those with more severe collapse are at risk for residual vertebral deformities, strategies to prevent vertebral fractures in those at greatest risk for permanent sequelae now merit further study,” said lead author Leanne Ward, MD, of the University of Ottawa, Canada, in a statement.
The study was primarily supported by an operating grant from the Canadian Institutes for Health Research (CIHR) with additional funding from a CIHR New Investigator Award, a Canadian Child Health Clinician Scientist Career Enhancement Award, a University of Ottawa Research Chair Award, a Children’s Hospital of Eastern Ontario (CHEO) Research Institute Capacity Building Award, and the CHEO Departments of Pediatrics and Surgery. This work was also supported by the University of Alberta Women and Children’s Health Research Institute.
A 6-year prospective study from the Canadian STOPP investigators revealed that following treatment for acute lymphoblastic leukemia (ALL), approximately 1 out of 3 children experiences vertebral fractures (VF) and 1 out of 5 children shows non-VF.
Glucocorticoid use and vertebral fractures at diagnosis emerged as significant predictive factors.
Investigators reported a cumulative incidence of 32.5% for vertebral fractures (VF) following treatment and 23.0% for non-VF after 6 years.
Thirty-nine percent of children with VF were asymptomatic.
“The osteotoxicity of leukemia and its treatment are underscored by the observation that the non-VF incidence was more than 2.5 times higher than the general pediatric population,” the study authors wrote, “whereas the VF incidence was increased more than 2000 times.”
The STOPP investigators—the Steroid-Associated Osteoporosis in the Pediatric Population Consortium—published their findings in the Journal of Bone and Mineral Research.
To document the incidence and predictors of fractures and profile who is at greatest risk, the STOPP investigators enrolled 186 children who were diagnosed and treated at 10 children’s hospitals across Canada between 2005 and 2007.
Median age at diagnosis was 5.3 years, median duration of follow-up was 6 years, and 38.1% were boys.
Approximately 4 out of 5 children were treated with Children’s Oncology Group protocols and 1 out of 5 was treated with the Dana-Farber protocols.
In girls, glucocorticoid exposure was 7.0 g/m2 and in boys it was 8.9 g/m2.
The entire cohort received 54.3% of the total cumulative glucocorticoid dose in the first year and 83.1% in the second year.
For VF, incidence at baseline was 15.1%. Skeletal fractures at any site were reported in 36% of the children over the 6-year period, 71% occurring during the first 2 years. No VF was reported in the sixth year.
Of 38 children with VF, 32 sustained at least 1 VF in the first 2 years and 77% occurred while on therapy or within 6 months of the last glucocorticoid dose. None of the children had disease relapse.
Non-VF were reported in 1.6% of children at baseline; incidence peaked at years 2 (5.4%) and 5 (4.8%) and none occurred in the sixth year.
Thirty-one non-VFs were reported in 26 children; 18 occurred in the first 2 years and 23 occurred on therapy or within 6 months of the last glucocorticoid dose.
The STOPP investigators showed that glucocorticoid exposure, VF, and low spine bone mineral density Z-scores at baseline were significant predictors for risk of VF.
“[This] observation suggests the importance of baseline spine phenotype (both VF and low lumbar spine bone mineral density) in signaling the potential for future bone morbidity,” the STOPP investigators note.
“In revealing that vertebral fractures are frequent in children with ALL on chemotherapy, and that older children and those with more severe collapse are at risk for residual vertebral deformities, strategies to prevent vertebral fractures in those at greatest risk for permanent sequelae now merit further study,” said lead author Leanne Ward, MD, of the University of Ottawa, Canada, in a statement.
The study was primarily supported by an operating grant from the Canadian Institutes for Health Research (CIHR) with additional funding from a CIHR New Investigator Award, a Canadian Child Health Clinician Scientist Career Enhancement Award, a University of Ottawa Research Chair Award, a Children’s Hospital of Eastern Ontario (CHEO) Research Institute Capacity Building Award, and the CHEO Departments of Pediatrics and Surgery. This work was also supported by the University of Alberta Women and Children’s Health Research Institute.
A 6-year prospective study from the Canadian STOPP investigators revealed that following treatment for acute lymphoblastic leukemia (ALL), approximately 1 out of 3 children experiences vertebral fractures (VF) and 1 out of 5 children shows non-VF.
Glucocorticoid use and vertebral fractures at diagnosis emerged as significant predictive factors.
Investigators reported a cumulative incidence of 32.5% for vertebral fractures (VF) following treatment and 23.0% for non-VF after 6 years.
Thirty-nine percent of children with VF were asymptomatic.
“The osteotoxicity of leukemia and its treatment are underscored by the observation that the non-VF incidence was more than 2.5 times higher than the general pediatric population,” the study authors wrote, “whereas the VF incidence was increased more than 2000 times.”
The STOPP investigators—the Steroid-Associated Osteoporosis in the Pediatric Population Consortium—published their findings in the Journal of Bone and Mineral Research.
To document the incidence and predictors of fractures and profile who is at greatest risk, the STOPP investigators enrolled 186 children who were diagnosed and treated at 10 children’s hospitals across Canada between 2005 and 2007.
Median age at diagnosis was 5.3 years, median duration of follow-up was 6 years, and 38.1% were boys.
Approximately 4 out of 5 children were treated with Children’s Oncology Group protocols and 1 out of 5 was treated with the Dana-Farber protocols.
In girls, glucocorticoid exposure was 7.0 g/m2 and in boys it was 8.9 g/m2.
The entire cohort received 54.3% of the total cumulative glucocorticoid dose in the first year and 83.1% in the second year.
For VF, incidence at baseline was 15.1%. Skeletal fractures at any site were reported in 36% of the children over the 6-year period, 71% occurring during the first 2 years. No VF was reported in the sixth year.
Of 38 children with VF, 32 sustained at least 1 VF in the first 2 years and 77% occurred while on therapy or within 6 months of the last glucocorticoid dose. None of the children had disease relapse.
Non-VF were reported in 1.6% of children at baseline; incidence peaked at years 2 (5.4%) and 5 (4.8%) and none occurred in the sixth year.
Thirty-one non-VFs were reported in 26 children; 18 occurred in the first 2 years and 23 occurred on therapy or within 6 months of the last glucocorticoid dose.
The STOPP investigators showed that glucocorticoid exposure, VF, and low spine bone mineral density Z-scores at baseline were significant predictors for risk of VF.
“[This] observation suggests the importance of baseline spine phenotype (both VF and low lumbar spine bone mineral density) in signaling the potential for future bone morbidity,” the STOPP investigators note.
“In revealing that vertebral fractures are frequent in children with ALL on chemotherapy, and that older children and those with more severe collapse are at risk for residual vertebral deformities, strategies to prevent vertebral fractures in those at greatest risk for permanent sequelae now merit further study,” said lead author Leanne Ward, MD, of the University of Ottawa, Canada, in a statement.
The study was primarily supported by an operating grant from the Canadian Institutes for Health Research (CIHR) with additional funding from a CIHR New Investigator Award, a Canadian Child Health Clinician Scientist Career Enhancement Award, a University of Ottawa Research Chair Award, a Children’s Hospital of Eastern Ontario (CHEO) Research Institute Capacity Building Award, and the CHEO Departments of Pediatrics and Surgery. This work was also supported by the University of Alberta Women and Children’s Health Research Institute.
Study of daratumamb with anti-PD-1 antibody in MM discontinued
Janssen is discontinuing the phase 1 MMY2036 study of daratumumab in combination with the anti PD-1 antibody JNJ-63723283 in patients with multiple myeloma (MM).
Janssen made the decision based on a Data Monitoring Committee review of Genmab’s phase 1b/2 study (LUC2001) of daratumumab plus the anti-PD-L1 antibody atezolizumab in non-small cell lung cancer (NSCLC).
Based on the DMC findings, Janssen also decided to discontinue its daratumumab-PD-1 combination study.
Janssen has an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
In the planned review, the DMC determined there was no observed benefit within the daratumumab plus atezolizumab arm compared to the atezolizumab monotherapy arm. The DMC recommended termination of the NSCLC study.
The DMC also noted an increase in mortality-related events in the combination arm.
Janssen has informed health authorities about these events and has contacted its partner companies conducting daratumumab and anti-PD-1 combination studies to discuss ceasing enrollment and dosing of the combination while the data is being further investigated.
MMY2036 study (NCT03357952)
The randomized, multicenter, multiphase study was expected to enroll up to 386 patients with relapsed or refractory MM who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory (IMiD) agent. Refractory patients had to be double refractory to both a PI and an IMiD.
The trial was to be conducted in 3 parts. Part 1 was to assess the safety of the combination of JNJ-63723283 and daratumumab. Part 2 was intended to compare the overall response rate in patients treated with the combination compared to those treated with daratumumab alone. And Part 3 was to compare progression-free survival between the 2 arms.
Daratumumab dose was planned to be 16 mg/kg weekly for 8 weeks, then once every other week for 16 weeks; then once every 4 weeks.
JNJ-63723283 dose was planned to be 240 milligrams IV fixed dose during week 1 on cycle 1 (28 days) day 2, cycle 1 day 15, then every 2 weeks thereafter.
The study was started in November 2017 and planned to be completed in December 2019.
In a news release, Genmab’s chief executive officer, Jan van de Winkel, PhD, expressed disappointment that the studies will be discontinued. He said Genmab “fully supports Janssen’s decision as patient safety is paramount in drug development. We look forward to gaining a better understanding of the data upon further analysis.”
Janssen is discontinuing the phase 1 MMY2036 study of daratumumab in combination with the anti PD-1 antibody JNJ-63723283 in patients with multiple myeloma (MM).
Janssen made the decision based on a Data Monitoring Committee review of Genmab’s phase 1b/2 study (LUC2001) of daratumumab plus the anti-PD-L1 antibody atezolizumab in non-small cell lung cancer (NSCLC).
Based on the DMC findings, Janssen also decided to discontinue its daratumumab-PD-1 combination study.
Janssen has an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
In the planned review, the DMC determined there was no observed benefit within the daratumumab plus atezolizumab arm compared to the atezolizumab monotherapy arm. The DMC recommended termination of the NSCLC study.
The DMC also noted an increase in mortality-related events in the combination arm.
Janssen has informed health authorities about these events and has contacted its partner companies conducting daratumumab and anti-PD-1 combination studies to discuss ceasing enrollment and dosing of the combination while the data is being further investigated.
MMY2036 study (NCT03357952)
The randomized, multicenter, multiphase study was expected to enroll up to 386 patients with relapsed or refractory MM who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory (IMiD) agent. Refractory patients had to be double refractory to both a PI and an IMiD.
The trial was to be conducted in 3 parts. Part 1 was to assess the safety of the combination of JNJ-63723283 and daratumumab. Part 2 was intended to compare the overall response rate in patients treated with the combination compared to those treated with daratumumab alone. And Part 3 was to compare progression-free survival between the 2 arms.
Daratumumab dose was planned to be 16 mg/kg weekly for 8 weeks, then once every other week for 16 weeks; then once every 4 weeks.
JNJ-63723283 dose was planned to be 240 milligrams IV fixed dose during week 1 on cycle 1 (28 days) day 2, cycle 1 day 15, then every 2 weeks thereafter.
The study was started in November 2017 and planned to be completed in December 2019.
In a news release, Genmab’s chief executive officer, Jan van de Winkel, PhD, expressed disappointment that the studies will be discontinued. He said Genmab “fully supports Janssen’s decision as patient safety is paramount in drug development. We look forward to gaining a better understanding of the data upon further analysis.”
Janssen is discontinuing the phase 1 MMY2036 study of daratumumab in combination with the anti PD-1 antibody JNJ-63723283 in patients with multiple myeloma (MM).
Janssen made the decision based on a Data Monitoring Committee review of Genmab’s phase 1b/2 study (LUC2001) of daratumumab plus the anti-PD-L1 antibody atezolizumab in non-small cell lung cancer (NSCLC).
Based on the DMC findings, Janssen also decided to discontinue its daratumumab-PD-1 combination study.
Janssen has an exclusive worldwide license from Genmab to develop, manufacture, and commercialize daratumumab.
In the planned review, the DMC determined there was no observed benefit within the daratumumab plus atezolizumab arm compared to the atezolizumab monotherapy arm. The DMC recommended termination of the NSCLC study.
The DMC also noted an increase in mortality-related events in the combination arm.
Janssen has informed health authorities about these events and has contacted its partner companies conducting daratumumab and anti-PD-1 combination studies to discuss ceasing enrollment and dosing of the combination while the data is being further investigated.
MMY2036 study (NCT03357952)
The randomized, multicenter, multiphase study was expected to enroll up to 386 patients with relapsed or refractory MM who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory (IMiD) agent. Refractory patients had to be double refractory to both a PI and an IMiD.
The trial was to be conducted in 3 parts. Part 1 was to assess the safety of the combination of JNJ-63723283 and daratumumab. Part 2 was intended to compare the overall response rate in patients treated with the combination compared to those treated with daratumumab alone. And Part 3 was to compare progression-free survival between the 2 arms.
Daratumumab dose was planned to be 16 mg/kg weekly for 8 weeks, then once every other week for 16 weeks; then once every 4 weeks.
JNJ-63723283 dose was planned to be 240 milligrams IV fixed dose during week 1 on cycle 1 (28 days) day 2, cycle 1 day 15, then every 2 weeks thereafter.
The study was started in November 2017 and planned to be completed in December 2019.
In a news release, Genmab’s chief executive officer, Jan van de Winkel, PhD, expressed disappointment that the studies will be discontinued. He said Genmab “fully supports Janssen’s decision as patient safety is paramount in drug development. We look forward to gaining a better understanding of the data upon further analysis.”
FDA grants priority review to gilteritinib for R/R AML

The US Food and Drug Administration (FDA) has granted priority review to gilteritinib for the treatment of adult patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who have a FLT3 mutation.
At present, no FLT3-targeting agents are approved for the treatment of R/R FLT3-mutation-positive AML, according to Astellas, the developer of the drug.
The FDA previously granted gilteritinib orphan drug designation and fast track designation.
The European Commission (EC) also granted gilteritinib orphan designation and the Japan Ministry of Health, Labor and Welfare (MHLW) did likewise.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT tyrosine kinase domain (TKD) mutation. These 2 FLT3 mutations are present in approximately 1/3 of AML patients.
The priority review for the new drug application is based on the ongoing phase 3 ADMIRAL trial.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The goal date for a decision by the FDA is the end of November.
This open-label multicenter randomized study compares gilteritinib with salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy.
The study has enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by a central lab.
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy.
Salvage chemotherapy could consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, cytarabine) induction chemotherapy, or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin) induction chemotherapy.
The primary endpoints of the study are overall survival and complete remission or complete remission with partial hematologic recovery.
Secondary endpoints include event-free survival, complete remission rate, leukemia-free survival, duration of remission, transplantation rate, fatigue inventory, among other outcomes.
The study is estimated to be completed in October for its primary endpoint and February 2020 for the entire study.
The US Food and Drug Administration (FDA) has granted priority review to gilteritinib for the treatment of adult patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who have a FLT3 mutation.
At present, no FLT3-targeting agents are approved for the treatment of R/R FLT3-mutation-positive AML, according to Astellas, the developer of the drug.
The FDA previously granted gilteritinib orphan drug designation and fast track designation.
The European Commission (EC) also granted gilteritinib orphan designation and the Japan Ministry of Health, Labor and Welfare (MHLW) did likewise.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT tyrosine kinase domain (TKD) mutation. These 2 FLT3 mutations are present in approximately 1/3 of AML patients.
The priority review for the new drug application is based on the ongoing phase 3 ADMIRAL trial.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The goal date for a decision by the FDA is the end of November.
This open-label multicenter randomized study compares gilteritinib with salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy.
The study has enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by a central lab.
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy.
Salvage chemotherapy could consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, cytarabine) induction chemotherapy, or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin) induction chemotherapy.
The primary endpoints of the study are overall survival and complete remission or complete remission with partial hematologic recovery.
Secondary endpoints include event-free survival, complete remission rate, leukemia-free survival, duration of remission, transplantation rate, fatigue inventory, among other outcomes.
The study is estimated to be completed in October for its primary endpoint and February 2020 for the entire study.
The US Food and Drug Administration (FDA) has granted priority review to gilteritinib for the treatment of adult patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who have a FLT3 mutation.
At present, no FLT3-targeting agents are approved for the treatment of R/R FLT3-mutation-positive AML, according to Astellas, the developer of the drug.
The FDA previously granted gilteritinib orphan drug designation and fast track designation.
The European Commission (EC) also granted gilteritinib orphan designation and the Japan Ministry of Health, Labor and Welfare (MHLW) did likewise.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT tyrosine kinase domain (TKD) mutation. These 2 FLT3 mutations are present in approximately 1/3 of AML patients.
The priority review for the new drug application is based on the ongoing phase 3 ADMIRAL trial.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The goal date for a decision by the FDA is the end of November.
This open-label multicenter randomized study compares gilteritinib with salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy.
The study has enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by a central lab.
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy.
Salvage chemotherapy could consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, cytarabine) induction chemotherapy, or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin) induction chemotherapy.
The primary endpoints of the study are overall survival and complete remission or complete remission with partial hematologic recovery.
Secondary endpoints include event-free survival, complete remission rate, leukemia-free survival, duration of remission, transplantation rate, fatigue inventory, among other outcomes.
The study is estimated to be completed in October for its primary endpoint and February 2020 for the entire study.
A closer look at the BK polyomavirus
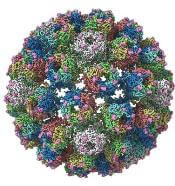
A detailed image at near-atomic levels of the BK polyomavirus (BKV), which affects kidney and bone marrow transplant patients, is now available for the first time.
This molecular-level structural visualization could allow scientists to study potential targets for antiviral therapies or drugs.
Approximately 80% to 90% of the general population are seropositive for the most prevalent BKV genotypes. Yet the virus rarely causes illness in people with healthy immune systems.
But the BKV causes nephropathy and hemorrhagic cystitis in immunosuppressed patients. Currently, no effective antiviral agents are available specifically targeting BKV.
Using high-resolution cryoelectron microscopy (cryo-EM), the research team at the University of Leeds Astbury Centre for Structural Molecular Biology determined the structure of BKV at 3.8 Å resolution.
At this resolution, they could highlight differences between human BKV and BKV pathogens that infect simian and murine hosts.
The investigators created the structures by freezing infectious BKV particles and taking thousands of images using the microscopes. The two-dimensional images were then combined computationally to produce a high-resolution, three-dimensional view of the virus.
They reported their findings in the journal Structure.
Senior investigator Neil A. Ranson, PhD, of the University of Leeds , noted that cryo-EM has been around for 30 years.
Although it’s been useful, the technology lacked the ability to routinely look at molecules at the level of detail needed, he said.
"However, the Titan Krios microscopes we have installed in Leeds are absolutely state-of-the-art and mean that these limitations have been shattered. Researchers and industry users who work with us can now image biological molecules with an incredible resolution. “
The investigators believe the quality and completeness of the cryo-EM structures they captured allows researchers to describe the various conformations of the C termini of the major capsid protein VP1, which play a fundamental role in the assembly and stability of a polyomavirus capsid.
The investigators say the images also present a more complete picture of disulfide bonding in the capsid.
Thus, the images provide insights into the structure of the human pathogen at near native conditions and give scientists a better-quality research tool to use.
“Crucially, we'll also be able to see how these molecules interact with each other," Dr Ransom said.
The BK Polyomavirus structure research was funded by Wellcome, Kidney Research UK, and Kidney Research Yorkshire.
A detailed image at near-atomic levels of the BK polyomavirus (BKV), which affects kidney and bone marrow transplant patients, is now available for the first time.
This molecular-level structural visualization could allow scientists to study potential targets for antiviral therapies or drugs.
Approximately 80% to 90% of the general population are seropositive for the most prevalent BKV genotypes. Yet the virus rarely causes illness in people with healthy immune systems.
But the BKV causes nephropathy and hemorrhagic cystitis in immunosuppressed patients. Currently, no effective antiviral agents are available specifically targeting BKV.
Using high-resolution cryoelectron microscopy (cryo-EM), the research team at the University of Leeds Astbury Centre for Structural Molecular Biology determined the structure of BKV at 3.8 Å resolution.
At this resolution, they could highlight differences between human BKV and BKV pathogens that infect simian and murine hosts.
The investigators created the structures by freezing infectious BKV particles and taking thousands of images using the microscopes. The two-dimensional images were then combined computationally to produce a high-resolution, three-dimensional view of the virus.
They reported their findings in the journal Structure.
Senior investigator Neil A. Ranson, PhD, of the University of Leeds , noted that cryo-EM has been around for 30 years.
Although it’s been useful, the technology lacked the ability to routinely look at molecules at the level of detail needed, he said.
"However, the Titan Krios microscopes we have installed in Leeds are absolutely state-of-the-art and mean that these limitations have been shattered. Researchers and industry users who work with us can now image biological molecules with an incredible resolution. “
The investigators believe the quality and completeness of the cryo-EM structures they captured allows researchers to describe the various conformations of the C termini of the major capsid protein VP1, which play a fundamental role in the assembly and stability of a polyomavirus capsid.
The investigators say the images also present a more complete picture of disulfide bonding in the capsid.
Thus, the images provide insights into the structure of the human pathogen at near native conditions and give scientists a better-quality research tool to use.
“Crucially, we'll also be able to see how these molecules interact with each other," Dr Ransom said.
The BK Polyomavirus structure research was funded by Wellcome, Kidney Research UK, and Kidney Research Yorkshire.
A detailed image at near-atomic levels of the BK polyomavirus (BKV), which affects kidney and bone marrow transplant patients, is now available for the first time.
This molecular-level structural visualization could allow scientists to study potential targets for antiviral therapies or drugs.
Approximately 80% to 90% of the general population are seropositive for the most prevalent BKV genotypes. Yet the virus rarely causes illness in people with healthy immune systems.
But the BKV causes nephropathy and hemorrhagic cystitis in immunosuppressed patients. Currently, no effective antiviral agents are available specifically targeting BKV.
Using high-resolution cryoelectron microscopy (cryo-EM), the research team at the University of Leeds Astbury Centre for Structural Molecular Biology determined the structure of BKV at 3.8 Å resolution.
At this resolution, they could highlight differences between human BKV and BKV pathogens that infect simian and murine hosts.
The investigators created the structures by freezing infectious BKV particles and taking thousands of images using the microscopes. The two-dimensional images were then combined computationally to produce a high-resolution, three-dimensional view of the virus.
They reported their findings in the journal Structure.
Senior investigator Neil A. Ranson, PhD, of the University of Leeds , noted that cryo-EM has been around for 30 years.
Although it’s been useful, the technology lacked the ability to routinely look at molecules at the level of detail needed, he said.
"However, the Titan Krios microscopes we have installed in Leeds are absolutely state-of-the-art and mean that these limitations have been shattered. Researchers and industry users who work with us can now image biological molecules with an incredible resolution. “
The investigators believe the quality and completeness of the cryo-EM structures they captured allows researchers to describe the various conformations of the C termini of the major capsid protein VP1, which play a fundamental role in the assembly and stability of a polyomavirus capsid.
The investigators say the images also present a more complete picture of disulfide bonding in the capsid.
Thus, the images provide insights into the structure of the human pathogen at near native conditions and give scientists a better-quality research tool to use.
“Crucially, we'll also be able to see how these molecules interact with each other," Dr Ransom said.
The BK Polyomavirus structure research was funded by Wellcome, Kidney Research UK, and Kidney Research Yorkshire.
Perioperative rVWF alone sufficient for some VWD patients
GLASGOW—Recombinant von Willebrand factor (rVWF) alone can be sufficient as perioperative management for some patients with severe von Willebrand disease (VWD), according to researchers.
In a phase 3 study, 10 of 15 patients were able to achieve hemostatic efficacy ratings of “good” or “excellent” when receiving only rVWF before, during, and/or after surgery.
The remaining 5 patients also achieved favorable hemostatic efficacy ratings, but they received recombinant factor VIII (FVIII) as well.
These results were presented at the World Federation of Hemophilia (WFH) 2018 World Congress (abstract W-MP-63 [749]). The research was sponsored by Shire, the company marketing rVWF as Vonvendi.
“There is an unmet clinical need for those living with von Willebrand disease, as they face a heightened risk of bleeding during surgery,” said study investigator Flora Peyvandi, MD, PhD, of the University of Milan in Italy.
“People with von Willebrand disease lack proper function or quantity of von Willebrand factor, and some also have a secondary factor VIII deficiency. In this study, recombinant von Willebrand factor was administered to replace the insufficient or dysfunctional von Willebrand factor, allowing the body to naturally replenish FVIII in most patients. These study results demonstrate clinical promise as physicians were able to tailor treatment based on each patient’s individual need for one or both factor therapies.”
The study included 15 adults with severe VWD who were undergoing elective surgical procedures. Ten patients were undergoing major surgery, 4 minor, and 1 oral surgery.
The patients’ median age was 40 (range, 20-70), and 8 were female. Most (n=8) had type 3 VWD, 3 had type 1, 2 had type 2A, 1 had 2B, and 1 had 2M.
At baseline, the mean endogenous FVIII level (FVIII:C) was 16.4 IU/dL, and the mean VWF ristocetin cofactor (VWF:Rco) was 10.6 IU/dL.
The patients received rVWF at 40 to 60 IU/kg VWF:RCo intravenously 12 to 24 hours before surgery to allow FVIII:C levels to increase to at least 30 IU/dL for minor or oral surgery or to at least 60 IU/dL for major surgery, within 3 hours before surgery.
If the desired levels were achieved, rVWF could be given alone. If the levels were not achieved, patients would receive rFVIII as well, within 1 to 2 hours before surgery. Patients were monitored for 14 days after surgery.
Results
All 15 patients had overall/intraoperative hemostatic efficacy ratings of “excellent” (as good as or better than expected) or “good” (probably as good as expected).
The patients received a median of 6 (range, 2 to 15) rVWF infusions at a median dose of 55 IU/kg (range, 36.1 to 59.9). Most patients (n=11) did not receive rVWF every day. For some, infusions were separated by 2 to 9 days.
Ten patients received rVWF alone, 12 did not receive any preoperative FVIII, and 2 did not receive rVWF postoperatively.
Most rVWF infusions (89.4%, 93/104) were given alone, and 70% (7/10) of the major surgeries were performed with rVWF alone.
The researchers said that, with rVWF alone, patients had hemostatically effective levels of FVIII:C as early as 6 hours after surgery, and this was sustained for 72 to 96 hours.
There were 5 patients who received rVWF with rFVIII. Of the 11 infusions these patients received, 9 were given when FVIII:C levels were above 60 IU/dL.
Three patients received rVWF with rFVIII 1 hour before major surgery—total hip replacement, molar extraction, and left ankle prosthesis. However, 2 of these patients had FVIII:C levels above 60 IU/dL.
The patient undergoing a molar extraction received rVWF with rFVIII 6 times after surgery. In 5 cases, the patient’s FVIII:C levels were 110 to 152 IU/dL. In the remaining case, the FVIII:C level was 23 IU/dL.
Two patients received rVWF with rFVIII for minor surgery. One patient undergoing a tooth extraction received rVWF with rFVIII intraoperatively when the FVIII:C level was 72 IU/dL.
The other patient received rVWF with rFVIII after radioisotope synovectomy when the FVIII:C level was 73 IU/dL. This patient received a postoperative dose of rVWF alone as well.
One patient tested positive for binding antibodies to VWF, and 1 patient developed deep vein thrombosis 3 days after total hip replacement while receiving rVWF.
GLASGOW—Recombinant von Willebrand factor (rVWF) alone can be sufficient as perioperative management for some patients with severe von Willebrand disease (VWD), according to researchers.
In a phase 3 study, 10 of 15 patients were able to achieve hemostatic efficacy ratings of “good” or “excellent” when receiving only rVWF before, during, and/or after surgery.
The remaining 5 patients also achieved favorable hemostatic efficacy ratings, but they received recombinant factor VIII (FVIII) as well.
These results were presented at the World Federation of Hemophilia (WFH) 2018 World Congress (abstract W-MP-63 [749]). The research was sponsored by Shire, the company marketing rVWF as Vonvendi.
“There is an unmet clinical need for those living with von Willebrand disease, as they face a heightened risk of bleeding during surgery,” said study investigator Flora Peyvandi, MD, PhD, of the University of Milan in Italy.
“People with von Willebrand disease lack proper function or quantity of von Willebrand factor, and some also have a secondary factor VIII deficiency. In this study, recombinant von Willebrand factor was administered to replace the insufficient or dysfunctional von Willebrand factor, allowing the body to naturally replenish FVIII in most patients. These study results demonstrate clinical promise as physicians were able to tailor treatment based on each patient’s individual need for one or both factor therapies.”
The study included 15 adults with severe VWD who were undergoing elective surgical procedures. Ten patients were undergoing major surgery, 4 minor, and 1 oral surgery.
The patients’ median age was 40 (range, 20-70), and 8 were female. Most (n=8) had type 3 VWD, 3 had type 1, 2 had type 2A, 1 had 2B, and 1 had 2M.
At baseline, the mean endogenous FVIII level (FVIII:C) was 16.4 IU/dL, and the mean VWF ristocetin cofactor (VWF:Rco) was 10.6 IU/dL.
The patients received rVWF at 40 to 60 IU/kg VWF:RCo intravenously 12 to 24 hours before surgery to allow FVIII:C levels to increase to at least 30 IU/dL for minor or oral surgery or to at least 60 IU/dL for major surgery, within 3 hours before surgery.
If the desired levels were achieved, rVWF could be given alone. If the levels were not achieved, patients would receive rFVIII as well, within 1 to 2 hours before surgery. Patients were monitored for 14 days after surgery.
Results
All 15 patients had overall/intraoperative hemostatic efficacy ratings of “excellent” (as good as or better than expected) or “good” (probably as good as expected).
The patients received a median of 6 (range, 2 to 15) rVWF infusions at a median dose of 55 IU/kg (range, 36.1 to 59.9). Most patients (n=11) did not receive rVWF every day. For some, infusions were separated by 2 to 9 days.
Ten patients received rVWF alone, 12 did not receive any preoperative FVIII, and 2 did not receive rVWF postoperatively.
Most rVWF infusions (89.4%, 93/104) were given alone, and 70% (7/10) of the major surgeries were performed with rVWF alone.
The researchers said that, with rVWF alone, patients had hemostatically effective levels of FVIII:C as early as 6 hours after surgery, and this was sustained for 72 to 96 hours.
There were 5 patients who received rVWF with rFVIII. Of the 11 infusions these patients received, 9 were given when FVIII:C levels were above 60 IU/dL.
Three patients received rVWF with rFVIII 1 hour before major surgery—total hip replacement, molar extraction, and left ankle prosthesis. However, 2 of these patients had FVIII:C levels above 60 IU/dL.
The patient undergoing a molar extraction received rVWF with rFVIII 6 times after surgery. In 5 cases, the patient’s FVIII:C levels were 110 to 152 IU/dL. In the remaining case, the FVIII:C level was 23 IU/dL.
Two patients received rVWF with rFVIII for minor surgery. One patient undergoing a tooth extraction received rVWF with rFVIII intraoperatively when the FVIII:C level was 72 IU/dL.
The other patient received rVWF with rFVIII after radioisotope synovectomy when the FVIII:C level was 73 IU/dL. This patient received a postoperative dose of rVWF alone as well.
One patient tested positive for binding antibodies to VWF, and 1 patient developed deep vein thrombosis 3 days after total hip replacement while receiving rVWF.
GLASGOW—Recombinant von Willebrand factor (rVWF) alone can be sufficient as perioperative management for some patients with severe von Willebrand disease (VWD), according to researchers.
In a phase 3 study, 10 of 15 patients were able to achieve hemostatic efficacy ratings of “good” or “excellent” when receiving only rVWF before, during, and/or after surgery.
The remaining 5 patients also achieved favorable hemostatic efficacy ratings, but they received recombinant factor VIII (FVIII) as well.
These results were presented at the World Federation of Hemophilia (WFH) 2018 World Congress (abstract W-MP-63 [749]). The research was sponsored by Shire, the company marketing rVWF as Vonvendi.
“There is an unmet clinical need for those living with von Willebrand disease, as they face a heightened risk of bleeding during surgery,” said study investigator Flora Peyvandi, MD, PhD, of the University of Milan in Italy.
“People with von Willebrand disease lack proper function or quantity of von Willebrand factor, and some also have a secondary factor VIII deficiency. In this study, recombinant von Willebrand factor was administered to replace the insufficient or dysfunctional von Willebrand factor, allowing the body to naturally replenish FVIII in most patients. These study results demonstrate clinical promise as physicians were able to tailor treatment based on each patient’s individual need for one or both factor therapies.”
The study included 15 adults with severe VWD who were undergoing elective surgical procedures. Ten patients were undergoing major surgery, 4 minor, and 1 oral surgery.
The patients’ median age was 40 (range, 20-70), and 8 were female. Most (n=8) had type 3 VWD, 3 had type 1, 2 had type 2A, 1 had 2B, and 1 had 2M.
At baseline, the mean endogenous FVIII level (FVIII:C) was 16.4 IU/dL, and the mean VWF ristocetin cofactor (VWF:Rco) was 10.6 IU/dL.
The patients received rVWF at 40 to 60 IU/kg VWF:RCo intravenously 12 to 24 hours before surgery to allow FVIII:C levels to increase to at least 30 IU/dL for minor or oral surgery or to at least 60 IU/dL for major surgery, within 3 hours before surgery.
If the desired levels were achieved, rVWF could be given alone. If the levels were not achieved, patients would receive rFVIII as well, within 1 to 2 hours before surgery. Patients were monitored for 14 days after surgery.
Results
All 15 patients had overall/intraoperative hemostatic efficacy ratings of “excellent” (as good as or better than expected) or “good” (probably as good as expected).
The patients received a median of 6 (range, 2 to 15) rVWF infusions at a median dose of 55 IU/kg (range, 36.1 to 59.9). Most patients (n=11) did not receive rVWF every day. For some, infusions were separated by 2 to 9 days.
Ten patients received rVWF alone, 12 did not receive any preoperative FVIII, and 2 did not receive rVWF postoperatively.
Most rVWF infusions (89.4%, 93/104) were given alone, and 70% (7/10) of the major surgeries were performed with rVWF alone.
The researchers said that, with rVWF alone, patients had hemostatically effective levels of FVIII:C as early as 6 hours after surgery, and this was sustained for 72 to 96 hours.
There were 5 patients who received rVWF with rFVIII. Of the 11 infusions these patients received, 9 were given when FVIII:C levels were above 60 IU/dL.
Three patients received rVWF with rFVIII 1 hour before major surgery—total hip replacement, molar extraction, and left ankle prosthesis. However, 2 of these patients had FVIII:C levels above 60 IU/dL.
The patient undergoing a molar extraction received rVWF with rFVIII 6 times after surgery. In 5 cases, the patient’s FVIII:C levels were 110 to 152 IU/dL. In the remaining case, the FVIII:C level was 23 IU/dL.
Two patients received rVWF with rFVIII for minor surgery. One patient undergoing a tooth extraction received rVWF with rFVIII intraoperatively when the FVIII:C level was 72 IU/dL.
The other patient received rVWF with rFVIII after radioisotope synovectomy when the FVIII:C level was 73 IU/dL. This patient received a postoperative dose of rVWF alone as well.
One patient tested positive for binding antibodies to VWF, and 1 patient developed deep vein thrombosis 3 days after total hip replacement while receiving rVWF.
FDA clears device for treatment of PE

The US Food and Drug Administration (FDA) has granted 510(k) clearance to the FlowTriever System for the treatment of pulmonary embolism (PE).
This makes the FlowTriever System the first and only thrombectomy device cleared by the FDA for the treatment of PE.
The 510(k) clearance was based on results from the FlowTriever Pulmonary Embolectomy (FLARE) study.
In this prospective, single-arm study, researchers evaluated the FlowTriever System in 106 patients with acute PE. Patients with proximal PE and right heart strain (RV/LV ratio ≥ 0.9) were eligible to participate.
Treatment with the FlowTriever System was used to non-surgically remove blood clots in the pulmonary arteries without the need for thrombolytic drugs.
The mean RV/LV ratio decreased from a baseline of 1.53 to 1.15 at 48 hours post-procedure, a difference of 0.39 (P<0.0001).
At 30 days, the rate of major adverse events was 3.8%. There were no device-related complications.
Patients had a median hospital stay of 3 days and a median stay in the intensive care unit of 1 day.
“The FlowTriever System is an exciting advancement in the treatment of acute pulmonary embolism patients,” said FLARE investigator Wissam Jaber, MD, of Emory University Hospital in Atlanta, Georgia.
“Until now, there has not been an approach to rapidly restore flow to reverse right heart strain without the use of thrombolytic drugs and their inherent risk of bleeding complications. The FlowTriever System represents a breakthrough in treatment options for this large patient population.”
The FlowTriever System is a product of Inari Medical, Inc.
The US Food and Drug Administration (FDA) has granted 510(k) clearance to the FlowTriever System for the treatment of pulmonary embolism (PE).
This makes the FlowTriever System the first and only thrombectomy device cleared by the FDA for the treatment of PE.
The 510(k) clearance was based on results from the FlowTriever Pulmonary Embolectomy (FLARE) study.
In this prospective, single-arm study, researchers evaluated the FlowTriever System in 106 patients with acute PE. Patients with proximal PE and right heart strain (RV/LV ratio ≥ 0.9) were eligible to participate.
Treatment with the FlowTriever System was used to non-surgically remove blood clots in the pulmonary arteries without the need for thrombolytic drugs.
The mean RV/LV ratio decreased from a baseline of 1.53 to 1.15 at 48 hours post-procedure, a difference of 0.39 (P<0.0001).
At 30 days, the rate of major adverse events was 3.8%. There were no device-related complications.
Patients had a median hospital stay of 3 days and a median stay in the intensive care unit of 1 day.
“The FlowTriever System is an exciting advancement in the treatment of acute pulmonary embolism patients,” said FLARE investigator Wissam Jaber, MD, of Emory University Hospital in Atlanta, Georgia.
“Until now, there has not been an approach to rapidly restore flow to reverse right heart strain without the use of thrombolytic drugs and their inherent risk of bleeding complications. The FlowTriever System represents a breakthrough in treatment options for this large patient population.”
The FlowTriever System is a product of Inari Medical, Inc.
The US Food and Drug Administration (FDA) has granted 510(k) clearance to the FlowTriever System for the treatment of pulmonary embolism (PE).
This makes the FlowTriever System the first and only thrombectomy device cleared by the FDA for the treatment of PE.
The 510(k) clearance was based on results from the FlowTriever Pulmonary Embolectomy (FLARE) study.
In this prospective, single-arm study, researchers evaluated the FlowTriever System in 106 patients with acute PE. Patients with proximal PE and right heart strain (RV/LV ratio ≥ 0.9) were eligible to participate.
Treatment with the FlowTriever System was used to non-surgically remove blood clots in the pulmonary arteries without the need for thrombolytic drugs.
The mean RV/LV ratio decreased from a baseline of 1.53 to 1.15 at 48 hours post-procedure, a difference of 0.39 (P<0.0001).
At 30 days, the rate of major adverse events was 3.8%. There were no device-related complications.
Patients had a median hospital stay of 3 days and a median stay in the intensive care unit of 1 day.
“The FlowTriever System is an exciting advancement in the treatment of acute pulmonary embolism patients,” said FLARE investigator Wissam Jaber, MD, of Emory University Hospital in Atlanta, Georgia.
“Until now, there has not been an approach to rapidly restore flow to reverse right heart strain without the use of thrombolytic drugs and their inherent risk of bleeding complications. The FlowTriever System represents a breakthrough in treatment options for this large patient population.”
The FlowTriever System is a product of Inari Medical, Inc.
Emicizumab controls bleeding regardless of inhibitors

GLASGOW—Emicizumab prophylaxis provides “clinically meaningful” control of bleeding whether or not patients have factor VIII inhibitors, according to researchers.
In the phase 3 HAVEN 4 study, researchers evaluated emicizumab prophylaxis, given every 4 weeks, in hemophilia A patients with or without factor VIII inhibitors.
At a median follow-up of about 26 weeks, patients had a median annualized bleeding rate (ABR) of 0.0 for treated bleeds and 2.1 for all bleeds.
About 30% of patients had 0 bleeds, and about 56% had 0 treated bleeds.
There were no serious adverse events (AEs) related to emicizumab. The most common AE was injection-site reaction.
Steve Pipe, MD, of Mott Children’s Hospital in Ann Arbor, Michigan, presented these results at the World Federation of Hemophilia (WFH) 2018 World Congress during the late-breaking abstract session on Monday.
The trial was sponsored by Hoffmann-La Roche.
HAVEN 4 included 48 patients, age 12 and older, who had hemophilia A with or without factor VIII inhibitors. Patients were previously treated with factor VIII or bypassing agents, on-demand or as prophylaxis.
The study was conducted in 2 parts: a pharmacokinetic (PK) run-in and an expansion cohort.
All patients in the PK run-in (n=7) were previously treated on-demand and received subcutaneous emicizumab at 6 mg/kg to fully characterize the PK profile after a single dose during 4 weeks. This was followed by 6 mg/kg every 4 weeks for at least 24 weeks.
Patients in the expansion cohort (n=41) received subcutaneous emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 6 mg/kg every 4 weeks for at least 24 weeks.
Episodic treatment of breakthrough bleeds with factor VIII therapy or bypassing agents, depending on a patient’s factor VIII inhibitor status, was allowed per study protocol.
Results
The efficacy analysis included the 41 patients in the expansion cohort, 5 of whom had inhibitors at baseline.
The median efficacy period was 25.6 weeks. The median ABR was 2.1 for all bleeds and 0.0 for treated bleeds.
The percentage of patients with 0 bleeds was 29.3% for all bleeds, 56.1% for treated bleeds, 82.9% for treated spontaneous bleeds, 70.7% for treated joint bleeds, and 85.4% for treated target joint bleeds.
Most treated bleeds (74.5%, 38/51) were traumatic.
There were 148 AEs, and 73.2% of patients had at least 1 AE.
Injection-site reaction was the most common AE related to emicizumab, occurring in 22.0% of patients (n=9).
There were 2 serious AEs (grade ≥3)—hypertension and rhabdomyolysis. Both were considered unrelated to emicizumab.
There were no AEs leading to emicizumab discontinuation or withdrawal. There were no thrombotic events, cases of thrombotic microangiopathy, hypersensitivity reactions, or fatalities.
None of the patients developed de novo factor VIII inhibitors, and there were no anti-drug antibodies detected.
GLASGOW—Emicizumab prophylaxis provides “clinically meaningful” control of bleeding whether or not patients have factor VIII inhibitors, according to researchers.
In the phase 3 HAVEN 4 study, researchers evaluated emicizumab prophylaxis, given every 4 weeks, in hemophilia A patients with or without factor VIII inhibitors.
At a median follow-up of about 26 weeks, patients had a median annualized bleeding rate (ABR) of 0.0 for treated bleeds and 2.1 for all bleeds.
About 30% of patients had 0 bleeds, and about 56% had 0 treated bleeds.
There were no serious adverse events (AEs) related to emicizumab. The most common AE was injection-site reaction.
Steve Pipe, MD, of Mott Children’s Hospital in Ann Arbor, Michigan, presented these results at the World Federation of Hemophilia (WFH) 2018 World Congress during the late-breaking abstract session on Monday.
The trial was sponsored by Hoffmann-La Roche.
HAVEN 4 included 48 patients, age 12 and older, who had hemophilia A with or without factor VIII inhibitors. Patients were previously treated with factor VIII or bypassing agents, on-demand or as prophylaxis.
The study was conducted in 2 parts: a pharmacokinetic (PK) run-in and an expansion cohort.
All patients in the PK run-in (n=7) were previously treated on-demand and received subcutaneous emicizumab at 6 mg/kg to fully characterize the PK profile after a single dose during 4 weeks. This was followed by 6 mg/kg every 4 weeks for at least 24 weeks.
Patients in the expansion cohort (n=41) received subcutaneous emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 6 mg/kg every 4 weeks for at least 24 weeks.
Episodic treatment of breakthrough bleeds with factor VIII therapy or bypassing agents, depending on a patient’s factor VIII inhibitor status, was allowed per study protocol.
Results
The efficacy analysis included the 41 patients in the expansion cohort, 5 of whom had inhibitors at baseline.
The median efficacy period was 25.6 weeks. The median ABR was 2.1 for all bleeds and 0.0 for treated bleeds.
The percentage of patients with 0 bleeds was 29.3% for all bleeds, 56.1% for treated bleeds, 82.9% for treated spontaneous bleeds, 70.7% for treated joint bleeds, and 85.4% for treated target joint bleeds.
Most treated bleeds (74.5%, 38/51) were traumatic.
There were 148 AEs, and 73.2% of patients had at least 1 AE.
Injection-site reaction was the most common AE related to emicizumab, occurring in 22.0% of patients (n=9).
There were 2 serious AEs (grade ≥3)—hypertension and rhabdomyolysis. Both were considered unrelated to emicizumab.
There were no AEs leading to emicizumab discontinuation or withdrawal. There were no thrombotic events, cases of thrombotic microangiopathy, hypersensitivity reactions, or fatalities.
None of the patients developed de novo factor VIII inhibitors, and there were no anti-drug antibodies detected.
GLASGOW—Emicizumab prophylaxis provides “clinically meaningful” control of bleeding whether or not patients have factor VIII inhibitors, according to researchers.
In the phase 3 HAVEN 4 study, researchers evaluated emicizumab prophylaxis, given every 4 weeks, in hemophilia A patients with or without factor VIII inhibitors.
At a median follow-up of about 26 weeks, patients had a median annualized bleeding rate (ABR) of 0.0 for treated bleeds and 2.1 for all bleeds.
About 30% of patients had 0 bleeds, and about 56% had 0 treated bleeds.
There were no serious adverse events (AEs) related to emicizumab. The most common AE was injection-site reaction.
Steve Pipe, MD, of Mott Children’s Hospital in Ann Arbor, Michigan, presented these results at the World Federation of Hemophilia (WFH) 2018 World Congress during the late-breaking abstract session on Monday.
The trial was sponsored by Hoffmann-La Roche.
HAVEN 4 included 48 patients, age 12 and older, who had hemophilia A with or without factor VIII inhibitors. Patients were previously treated with factor VIII or bypassing agents, on-demand or as prophylaxis.
The study was conducted in 2 parts: a pharmacokinetic (PK) run-in and an expansion cohort.
All patients in the PK run-in (n=7) were previously treated on-demand and received subcutaneous emicizumab at 6 mg/kg to fully characterize the PK profile after a single dose during 4 weeks. This was followed by 6 mg/kg every 4 weeks for at least 24 weeks.
Patients in the expansion cohort (n=41) received subcutaneous emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 6 mg/kg every 4 weeks for at least 24 weeks.
Episodic treatment of breakthrough bleeds with factor VIII therapy or bypassing agents, depending on a patient’s factor VIII inhibitor status, was allowed per study protocol.
Results
The efficacy analysis included the 41 patients in the expansion cohort, 5 of whom had inhibitors at baseline.
The median efficacy period was 25.6 weeks. The median ABR was 2.1 for all bleeds and 0.0 for treated bleeds.
The percentage of patients with 0 bleeds was 29.3% for all bleeds, 56.1% for treated bleeds, 82.9% for treated spontaneous bleeds, 70.7% for treated joint bleeds, and 85.4% for treated target joint bleeds.
Most treated bleeds (74.5%, 38/51) were traumatic.
There were 148 AEs, and 73.2% of patients had at least 1 AE.
Injection-site reaction was the most common AE related to emicizumab, occurring in 22.0% of patients (n=9).
There were 2 serious AEs (grade ≥3)—hypertension and rhabdomyolysis. Both were considered unrelated to emicizumab.
There were no AEs leading to emicizumab discontinuation or withdrawal. There were no thrombotic events, cases of thrombotic microangiopathy, hypersensitivity reactions, or fatalities.
None of the patients developed de novo factor VIII inhibitors, and there were no anti-drug antibodies detected.