User login
CDU is Associated with Decreased LOS
Hospitalists play a crucial role in improving hospital throughput and length of stay (LOS). The clinical decision unit (CDU) or observation unit (OU) is a strategy that was developed to facilitate both aims. CDUs and OUs are units where patients can be managed in the hospital for up to 24 hours prior to a decision being made to admit or discharge. Observation care is provided to patients who require further treatment or monitoring beyond what is accomplished in the emergency department (ED), but who do not require inpatient admission. CDUs arose in the 1990s in response to a desire to decrease inpatient costs as well as changing Medicare guidelines, which recognized observation status. Initially, CDUs and OUs were located within the ED and run by emergency medicine physicians. However, at the turn of the 21st century, hospitalists became involved in observation medicine, and the Society of Hospital Medicine issued a white paper on the OU in 2007. [1] Today, up to 50% of CDUs and OUs nationally are managed by hospitalists and located physically outside of the ED.[2, 3]
Despite the fact that nearly half of all CDUs and OUs nationally are run by hospitalists, there has been little published regarding the impact of hospitalist‐driven units. This study demonstrates the effect of observation care delivered in a hospitalist‐run geographic CDU. The primary objective was to determine the impact on LOS for patients in observation status managed in a hospitalist‐run CDU compared with LOS for observation patients with the same diagnoses cared for on medicalsurgical units prior to the existence of the CDU. The secondary objective was to determine the effect on the 30‐day ED or hospital revisit rate, as well as ED LOS. This work will guide health systems, hospitalist groups, and physicians in their decision making regarding the future structure and process of CDUs.
METHODS
Study Design
The Cooper University Hospital institutional review board approved this study. The study took place at Cooper University Hospital, a large, urban, academic safety‐net hospital providing tertiary care located in Camden, New Jersey.
We performed a retrospective observational study of all adult observation encounters at the study hospital from July 2010 to January 2011, and July 2011 through January 2012. During the second time period, patients could have been managed in the CDU or on a medicalsurgical unit. We recorded the following demographic data: age, gender, race, principal diagnosis, and payer, as well as several outcomes of interest, including: LOS (defined as the time separating the admitting physician order from discharge), ED visits within 30 days of discharge, and hospital revisits (observation or inpatient) within 30 days.
Data Sources
Data were culled by the institution's performance improvement department from the electronic medical record, as well as cost accounting and claims‐based sources.
Clinical Decision Unit
The CDU at Cooper University Hospital opened in June 2011 and is a 20‐bed geographically distinct unit adjacent to the ED. During the study period, it was staffed 24 hours a day by a hospitalist and a nurse practitioner as well as dedicated nurses and critical care technicians. Patients meeting observation status in the ED were eligible for the CDU provided that they fulfilled the CDU placement guidelines including that they were more likely than not to be discharged within a period of 24 hours of CDU care, did not meet inpatient admission criteria, did not require new placement in a rehabilitation or extended‐care facility, and did not require one‐on‐one monitoring. Additional exclusion criteria included severe vital sign or laboratory abnormalities. The overall strategy of the guidelines was to facilitate a pull culture, where the majority of observation patients were brought from the ED to the CDU once it was determined that they did not require inpatient care. The CDU had order sets and protocols in place for many of the common diagnoses. All CDU patients received priority laboratory and radiologic testing as well as priority consultation from specialty services. Medication reconciliation was performed by a pharmacy technician for higher‐risk patients, identified by Project BOOST (Better Outcomes by Optimizing Safe Transitions) criteria.[4] Structured multidisciplinary rounds occurred daily including the hospitalist, nurse practitioner, registered nurses, case manager, and pharmacy technician. A discharge planner was available to schedule follow‐up appointments.
Although chest pain was the most common CDU diagnosis, the CDU was designed to care for the majority of the hospital's observation patients rather than focus specifically on chest pain. Patients with chest pain who met observation criteria were transferred from the ED to the CDU, rather than a medicalsurgical unit, provided they did not have: positive cardiac enzymes, an electrocardiogram indicative of ischemia, known coronary artery disease presenting with pain consistent with acute coronary syndrome, need for heparin or nitroglycerin continuous infusion, symptomatic or unresolved arrhythmia, congestive heart failure meeting inpatient criteria, hypertensive urgency or emergency, pacemaker malfunction, pericarditis, or toxicity from cardiac drugs. Cardiologist consultants were involved in the care of nearly all CDU patients with chest pain.
Observation Status Determination
During the study period, observation status was recommended by a case manager in the ED based on Milliman (Milliman Care Guidelines) or McKesson InterQual (McKesson Corporation) criteria, once it was determined by the ED physician that the patient had failed usual ED care and required hospitalization. Observation status was assigned by the admitting (non‐ED) physician, who placed the order for inpatient admission or observation. Other than the implementation of the CDU, there were no significant changes to the process or criteria for assigning observation status, admission order sets, or the hospital's electronic medical record during this time period.
Statistical Analysis
Continuous data are presented as mean ( standard deviation [SD]) or median (25%75% interquartile range) as specified, and differences were assessed using one‐way analysis of variance testing and Mann‐Whitney U testing. Categorical data are presented as count (percentage) and differences evaluated using [2] analysis. P values of 0.05 or less were considered statistically significant.
To account for differences in groups with regard to outcomes, we performed a multivariate regression analysis. The following variables were entered: age (years), gender, race (African American vs other), admission diagnosis (chest pain vs other), and insurance status (Medicare vs other). All variables were entered simultaneously without forcing. Statistical analyses were done using the SPSS 20.0 Software (SPSS Inc., Chicago, IL).
RESULTS
Demographics
There were a total of 3735 patients included in the study: 1650 in the pre‐CDU group, 1469 in the post‐CDU group, and 616 in the post‐CDU group on medicalsurgical units. The post‐CDU period had a total of 2085 patients. Patients in the CDU group were younger and were more likely to have chest pain as the admission diagnosis. Patient demographics are presented in Table 1.
| Variable | Pre‐CDU, n=1,650 | Post‐CDU, n=1,469 | PostNon‐CDU, n=616 | P, CDU vs Pre‐CDU | P, Non‐CDU vs Pre‐CDU | P, CDU vs Non‐CDU |
|---|---|---|---|---|---|---|
| ||||||
| Age, y [range] | 56 [4569] | 53 [4364] | 57 [44.370] | 0.001 | 0.751 | 0.001 |
| Female gender | 918 (55.6%) | 833(56.7%) | 328 (53.2%) | 0.563 | 0.319 | 0.148 |
| African American race | 574 (34.8%) | 505 (34.4%) | 174 (28.2%) | 0.821 | 0.004 | 0.007 |
| Admission diagnosis | ||||||
| Chest pain | 462 (38%) | 528 (35.9%) | 132 (21.4%) | 0.001 | 0.002 | 0.001 |
| Syncope | 93 (5.6%) | 56 (3.8%) | 15 (2.4%) | 0.018 | 0.001 | 0.145 |
| Abdominal pain | 46 (2.8%) | 49 (3.3%) | 20(3.2%) | 0.404 | 0.575 | 1.0 |
| Other | 1,049 (63.6%) | 836 (56.9%) | 449 (72.9%) | 0.001 | 0.001 | 0.001 |
| Third‐party payer | ||||||
| Medicare | 727 (44.1%) | 491 (33.4%) | 264(43.4%) | 0.001 | 0.634 | 0.001 |
| Charity care | 187 (11.3%) | 238 (16.2%) | 73 (11.9%) | 0.001 | 0.767 | 0.010 |
| Commercial | 185 (11.1%) | 214 (14.6%) | 87 (14.1%) | 0.005 | 0.059 | 0.838 |
| Medicaid | 292 (17.7%) | 280 (19.1%) | 100 (16.2%) | 0.331 | 0.454 | 0.136 |
| Other | 153 (9.3%) | 195 (13.3%) | 60 (9.9%) | 0.001 | 0.746 | 0.028 |
| Self‐pay | 106 (6.4%) | 51(3.5%) | 32 (5.2%) | 0.001 | 0.323 | 0.085 |
Outcomes of Interest
There was a statistically significant association between LOS and CDU implementation (Table 2). Observation patients cared for in the CDU had a lower LOS than observation patients cared for on the medicalsurgical units during the same time period (17.6 vs 26.1 hours, P0.0001).
| Outcome | Pre‐CDU, n=1,650 | Post‐CDU, n=1,469 | PostNon‐CDU, n=616 | P, CDU vs Pre‐CDU | P, Non‐CDU vs Pre‐CDU | P, CDU vs Non‐CDU |
|---|---|---|---|---|---|---|
| ||||||
| All patients, n=3,735 | ||||||
| 30‐day ED or hospital revisit | 326 (19.8%) | 268 (18.2%) | 123 (17.2%) | 0.294 | 0.906 | 0.357 |
| Median LOS, h | 27.1 [17.446.4] | 17.6 [12.122.8] | 26.1 [16.941.2] | 0.001 | 0.004 | 0.001 |
| Chest‐pain patients, n=1,122 | ||||||
| 30‐day ED or hospital revisit | 69 (14.9%) | 82 (15.5%) | 23 (17.4%) | 0.859 | 0.496 | 0.596 |
| Median LOS, h | 22 [15.838.9] | 17.3 [10.922.4] | 23.2 [13.843.1] | 0.001 | 0.995 | 0.001 |
| Other diagnoses, n=2,613 | ||||||
| 30‐day ED or hospital revisit | 257 (21.6%) | 186 (19.8%) | 100 (18.4%) | 0.307 | 0.693 | 0.727 |
| Median LOS, h | 30.4 [18.649.4] | 17.8 [12.923] | 26.7 [17.231.1] | 0.001 | 0.001 | 0.001 |
In total, there were 717 total revisits including ED visits and hospital stays within 30 days of discharge (Table 2). Of all the observation encounters in the study, 19.2% were followed by a revisit within 30 days. There were no differences in the 30‐day post‐ED visit rates in between periods and between groups.
Mean ED LOS for hospitalized patients was examined for a sample of the pre‐ and post‐CDU periods, namely November 2010 to January 2011 and November 2011 to January 2012. The mean ED LOS decreased from 410 minutes (SD=61) to 393 minutes (SD=51) after implementation of the CDU (P=0.037).
To account for possible skewing of the data, we transformed LOS into ln (natural log) LOS and found the following means (SD): group 1 was 3.27 (0.94), group 2 was 2.78 (0.6), and group 3 was 3.1 (0.93). Using an independent t test, we found a significant difference between groups 1 and 2, 2 and 3, as well as 1 and 3 (P0.001 for all).
Chest‐Pain Subgroup Analysis
We analyzed the data specifically for the 1122 patients discharged with a diagnosis of chest pain. LOS was significantly lower for patients in the CDU compared to either pre‐CDU or observation on floors (Table 2).
Multivariate Regression Analysis
We performed a linear regression analysis using the following variables: age, race, gender, diagnosis, insurance status, and study period (pre‐CDU, post‐CDU, and postnon‐CDU). We performed 3 different comparisons: pre‐CDU vs post‐CDU, postnon‐CDU vs post‐CDU, and postnon‐CDU vs pre‐CDU. After adjusting for other variables, the postnon‐CDU group was significantly associated with higher LOS (P0.001). The pre‐CDU group was associated with higher LOS than both the post‐CDU and postnon‐CDU groups (P0.001 for both).
DISCUSSION
In our study of a hospitalist‐run CDU for observation patients, we observed that the care in the CDU was associated with a lower median LOS, but no increase in ED or hospital revisits within 30 days.
Previous studies have reported the impact of clinical observation or clinical diagnosis units, particularly chest‐pain units.[5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15] Studies of hospitalist‐run units suggest shorter LOS in the entire hospital,[16] or in the target unit.[17] Although one study suggested a lower 30‐day readmission rate,[18] most others did not describe this effect.[16, 17] Our study differs from previous research in that our program employed a pull‐culture aimed at accepting the majority of observation status patients without focusing on a particular diagnosis. We also implemented a structured multidisciplinary team focused on expediting care and utilized BOOST‐framed transitions, including targeted medication reconciliation and tools such as teach‐back.
The CDU in our hospital produced shorter LOS even compared to our non‐CDU units, but the revisit rate did not improve despite activities to reduce revisits. During the study period, efforts to decrease readmissions were implemented in various areas of our hospital, but not a comprehensive institution‐wide readmissions strategy. Lack of impact on revisits could be viewed as a positive finding, in that shorter LOS did not result in patients being discharged home before clinically stable. Alternatively, lack of impact could be due to the uncertain effectiveness of BOOST specifically[19, 20, 21] or inpatient‐targeted transitions interventions more generally.[22]
Our study has certain limitations. Findings in our single‐center study in an urban academic medical center may not apply to CDUs in other settings. As a prepost design, our study is subject to external trends for which our analyses may be unable to account. For example, during CDU implementation, there were hospital‐wide initiatives aimed at improving inpatient LOS, including complex case rounds, increased use of active bed management, and improved case management efforts to decrease LOS. These may have been a factor in the small decrease in observation LOS seen in the medicalsurgical patients during the post period. Additionally, though we have attempted to control for possible confounders, there could have been differences in the study groups for which we were unable to account, including code status or social variables such as homelessness, which played a role in our revisit outcomes. The decrease in LOS by 35%, or 9.5 hours, in CDU patients is clinically important, as it allows low‐risk patients to spend less time in the hospital where they may have been at risk of hospital‐acquired conditions; however, this study did not include patient satisfaction data. It would be important to measure the effect on patient experience of potentially spending 1 fewer night in the hospital. Finally, our CDU was designed with specific clinical criteria for inclusion and exclusion. Patients who were higher risk or expected to need more than 24 hours of care were not placed in the CDU. We were not able to adjust our analyses for factors that were not in our data, such as severe vital sign or laboratory abnormalities or a physician's clinical impression of a patient. It is possible, therefore, that referral bias may have occurred and influenced our results. The fact that non‐CDU chest‐pain patients in the post‐CDU period did not experience any decrease in LOS, whereas other medicalsurgical observation patients did, may be an example of this bias. Patients were excluded from the CDU by virtue of being deemed higher risk as described in Methods section. We were unable to adjust for these differences.
Implementation of CDUs may be useful for health systems seeking to improve hospital throughput and improve utilization among common but low‐acuity patient groups. Although our initial results are promising, the concept of a CDU may require enhancements. For example, at our hospital we are addressing transitions of care by looking at models that address patient risk through a systematic process, and then target individuals for specific interventions to prevent revisits. Moreover, the study of CDUs should report impact on patient and referring physician satisfaction, and whether CDUs can reduce per‐case costs.
CONCLUSION
Caring for patients in a hospitalist‐run geographic CDU was associated with a 35% decrease in observation LOS for CDU patients compared with a 3.7% decrease for observation patients cared for elsewhere in the hospital. CDU patients' LOS was significantly decreased without increasing ED or hospital revisit rates.
Acknowledgments
The authors would like to thank Ken Travis for excellent data support.
- The observation unit: an operational overview for the hospitalist. Society of Hospital Medicine website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=White_Papers18(12):1371–1379.
- , , , , , . Use of observation care in US emergency departments, 2001 to 2008. PLoS One. 2011;6(9):e24326.
- The Society of Hospital Medicine Project Boost (Better Outcomes by Optimizing Safe Transitions) Available at: http://www.hospitalmedicine.org/boost. Accessed on June 4, 2013.
- , , , , . An emergency department‐based protocol for rapidly ruling out myocardial ischemia reduces hospital time and expense: results of a randomized study (ROMIO). J Am Coll Cardiol. 1996;28(1):25–33.
- , , , , , . Implementation of the guidelines for the management of patients with chest pain through a critical pathway approach improves length of stay and patient satisfaction but not anxiety. Crit Pathw Cardiol. 2010;9(1):30–34.
- , , , et al. Costs of an emergency department‐based accelerated diagnostic protocol vs hospitalization in patients with chest pain: a randomized controlled trial. JAMA. 1997;278(20):1670–1676.
- , , , et al. Emergency‐department diagnosis of acute myocardial infarction and ischemia: a cost analysis of two diagnostic protocols. Acad Emerg Med. 1994;1(2):103–110.
- , , , et al. Impact on the care of the emergency department chest pain patient from the chest pain evaluation registry (CHEPER) study. Am J Cardiol. 1997;80(5):563–568.
- , , , et al. Cost‐effectiveness of a new short‐stay unit to “rule out” acute myocardial infarction in low risk patients. J Am Coll Cardiol. 1994;24(5):1249–1259.
- , , , et al. Emergency Department Observation Unit versus hospital inpatient care for a chronic asthmatic population: a randomized trial of health status outcome and cost. Med Care. 1998;36(4):599–609.
- , , , et al. A comparison between emergency diagnostic and treatment unit and inpatient care in the management of acute asthma. Arch Intern Med. 1997;157(18):2055–2062.
- , , , . Retrospective review of emergency department patients with non‐variceal upper gastrointestinal hemorrhage for potential outpatient management. Acad Emerg Med. 1999;6(3):196–201.
- , . Outpatient care of selected patients with acute non‐variceal upper gastrointestinal haemorrhage. Lancet. 1995;345(8942):108–111.
- , , , , . Patterns of use of an emergency department‐based observation unit. Am J Ther. 2002;9(6):499–502.
- , , . Implementation of a hospitalist‐run observation unit and impact on length of stay (LOS): a brief report. J Hosp Med. 2010;5(9):E2–E5.
- , , , , . Improving resource utilization in a teaching hospital: development of a nonteaching service for chest pain admissions. Acad Med. 2006;81(5):432–435.
- , , , . Program description: a hospitalist‐run, medical short‐stay unit in a teaching hospital. CMAJ. 2000;163(11):1477–1480.
- , , , et al. Project BOOST: effectiveness of a multihospital effort to reduce rehospitalization. J Hosp Med. 2013;8:421–427.
- , , , et al. BOOST: evidence needing a lift. J Hosp Med. 2013;8:468–469.
- . BOOST and readmissions: thinking beyond the walls of the hospital. J Hosp Med. 2013;8:470–471.
- , , , et al. Hospital‐initiated transitional care interventions as a patient safety strategy. Ann Int Med. 2013;158:433–440.
Hospitalists play a crucial role in improving hospital throughput and length of stay (LOS). The clinical decision unit (CDU) or observation unit (OU) is a strategy that was developed to facilitate both aims. CDUs and OUs are units where patients can be managed in the hospital for up to 24 hours prior to a decision being made to admit or discharge. Observation care is provided to patients who require further treatment or monitoring beyond what is accomplished in the emergency department (ED), but who do not require inpatient admission. CDUs arose in the 1990s in response to a desire to decrease inpatient costs as well as changing Medicare guidelines, which recognized observation status. Initially, CDUs and OUs were located within the ED and run by emergency medicine physicians. However, at the turn of the 21st century, hospitalists became involved in observation medicine, and the Society of Hospital Medicine issued a white paper on the OU in 2007. [1] Today, up to 50% of CDUs and OUs nationally are managed by hospitalists and located physically outside of the ED.[2, 3]
Despite the fact that nearly half of all CDUs and OUs nationally are run by hospitalists, there has been little published regarding the impact of hospitalist‐driven units. This study demonstrates the effect of observation care delivered in a hospitalist‐run geographic CDU. The primary objective was to determine the impact on LOS for patients in observation status managed in a hospitalist‐run CDU compared with LOS for observation patients with the same diagnoses cared for on medicalsurgical units prior to the existence of the CDU. The secondary objective was to determine the effect on the 30‐day ED or hospital revisit rate, as well as ED LOS. This work will guide health systems, hospitalist groups, and physicians in their decision making regarding the future structure and process of CDUs.
METHODS
Study Design
The Cooper University Hospital institutional review board approved this study. The study took place at Cooper University Hospital, a large, urban, academic safety‐net hospital providing tertiary care located in Camden, New Jersey.
We performed a retrospective observational study of all adult observation encounters at the study hospital from July 2010 to January 2011, and July 2011 through January 2012. During the second time period, patients could have been managed in the CDU or on a medicalsurgical unit. We recorded the following demographic data: age, gender, race, principal diagnosis, and payer, as well as several outcomes of interest, including: LOS (defined as the time separating the admitting physician order from discharge), ED visits within 30 days of discharge, and hospital revisits (observation or inpatient) within 30 days.
Data Sources
Data were culled by the institution's performance improvement department from the electronic medical record, as well as cost accounting and claims‐based sources.
Clinical Decision Unit
The CDU at Cooper University Hospital opened in June 2011 and is a 20‐bed geographically distinct unit adjacent to the ED. During the study period, it was staffed 24 hours a day by a hospitalist and a nurse practitioner as well as dedicated nurses and critical care technicians. Patients meeting observation status in the ED were eligible for the CDU provided that they fulfilled the CDU placement guidelines including that they were more likely than not to be discharged within a period of 24 hours of CDU care, did not meet inpatient admission criteria, did not require new placement in a rehabilitation or extended‐care facility, and did not require one‐on‐one monitoring. Additional exclusion criteria included severe vital sign or laboratory abnormalities. The overall strategy of the guidelines was to facilitate a pull culture, where the majority of observation patients were brought from the ED to the CDU once it was determined that they did not require inpatient care. The CDU had order sets and protocols in place for many of the common diagnoses. All CDU patients received priority laboratory and radiologic testing as well as priority consultation from specialty services. Medication reconciliation was performed by a pharmacy technician for higher‐risk patients, identified by Project BOOST (Better Outcomes by Optimizing Safe Transitions) criteria.[4] Structured multidisciplinary rounds occurred daily including the hospitalist, nurse practitioner, registered nurses, case manager, and pharmacy technician. A discharge planner was available to schedule follow‐up appointments.
Although chest pain was the most common CDU diagnosis, the CDU was designed to care for the majority of the hospital's observation patients rather than focus specifically on chest pain. Patients with chest pain who met observation criteria were transferred from the ED to the CDU, rather than a medicalsurgical unit, provided they did not have: positive cardiac enzymes, an electrocardiogram indicative of ischemia, known coronary artery disease presenting with pain consistent with acute coronary syndrome, need for heparin or nitroglycerin continuous infusion, symptomatic or unresolved arrhythmia, congestive heart failure meeting inpatient criteria, hypertensive urgency or emergency, pacemaker malfunction, pericarditis, or toxicity from cardiac drugs. Cardiologist consultants were involved in the care of nearly all CDU patients with chest pain.
Observation Status Determination
During the study period, observation status was recommended by a case manager in the ED based on Milliman (Milliman Care Guidelines) or McKesson InterQual (McKesson Corporation) criteria, once it was determined by the ED physician that the patient had failed usual ED care and required hospitalization. Observation status was assigned by the admitting (non‐ED) physician, who placed the order for inpatient admission or observation. Other than the implementation of the CDU, there were no significant changes to the process or criteria for assigning observation status, admission order sets, or the hospital's electronic medical record during this time period.
Statistical Analysis
Continuous data are presented as mean ( standard deviation [SD]) or median (25%75% interquartile range) as specified, and differences were assessed using one‐way analysis of variance testing and Mann‐Whitney U testing. Categorical data are presented as count (percentage) and differences evaluated using [2] analysis. P values of 0.05 or less were considered statistically significant.
To account for differences in groups with regard to outcomes, we performed a multivariate regression analysis. The following variables were entered: age (years), gender, race (African American vs other), admission diagnosis (chest pain vs other), and insurance status (Medicare vs other). All variables were entered simultaneously without forcing. Statistical analyses were done using the SPSS 20.0 Software (SPSS Inc., Chicago, IL).
RESULTS
Demographics
There were a total of 3735 patients included in the study: 1650 in the pre‐CDU group, 1469 in the post‐CDU group, and 616 in the post‐CDU group on medicalsurgical units. The post‐CDU period had a total of 2085 patients. Patients in the CDU group were younger and were more likely to have chest pain as the admission diagnosis. Patient demographics are presented in Table 1.
| Variable | Pre‐CDU, n=1,650 | Post‐CDU, n=1,469 | PostNon‐CDU, n=616 | P, CDU vs Pre‐CDU | P, Non‐CDU vs Pre‐CDU | P, CDU vs Non‐CDU |
|---|---|---|---|---|---|---|
| ||||||
| Age, y [range] | 56 [4569] | 53 [4364] | 57 [44.370] | 0.001 | 0.751 | 0.001 |
| Female gender | 918 (55.6%) | 833(56.7%) | 328 (53.2%) | 0.563 | 0.319 | 0.148 |
| African American race | 574 (34.8%) | 505 (34.4%) | 174 (28.2%) | 0.821 | 0.004 | 0.007 |
| Admission diagnosis | ||||||
| Chest pain | 462 (38%) | 528 (35.9%) | 132 (21.4%) | 0.001 | 0.002 | 0.001 |
| Syncope | 93 (5.6%) | 56 (3.8%) | 15 (2.4%) | 0.018 | 0.001 | 0.145 |
| Abdominal pain | 46 (2.8%) | 49 (3.3%) | 20(3.2%) | 0.404 | 0.575 | 1.0 |
| Other | 1,049 (63.6%) | 836 (56.9%) | 449 (72.9%) | 0.001 | 0.001 | 0.001 |
| Third‐party payer | ||||||
| Medicare | 727 (44.1%) | 491 (33.4%) | 264(43.4%) | 0.001 | 0.634 | 0.001 |
| Charity care | 187 (11.3%) | 238 (16.2%) | 73 (11.9%) | 0.001 | 0.767 | 0.010 |
| Commercial | 185 (11.1%) | 214 (14.6%) | 87 (14.1%) | 0.005 | 0.059 | 0.838 |
| Medicaid | 292 (17.7%) | 280 (19.1%) | 100 (16.2%) | 0.331 | 0.454 | 0.136 |
| Other | 153 (9.3%) | 195 (13.3%) | 60 (9.9%) | 0.001 | 0.746 | 0.028 |
| Self‐pay | 106 (6.4%) | 51(3.5%) | 32 (5.2%) | 0.001 | 0.323 | 0.085 |
Outcomes of Interest
There was a statistically significant association between LOS and CDU implementation (Table 2). Observation patients cared for in the CDU had a lower LOS than observation patients cared for on the medicalsurgical units during the same time period (17.6 vs 26.1 hours, P0.0001).
| Outcome | Pre‐CDU, n=1,650 | Post‐CDU, n=1,469 | PostNon‐CDU, n=616 | P, CDU vs Pre‐CDU | P, Non‐CDU vs Pre‐CDU | P, CDU vs Non‐CDU |
|---|---|---|---|---|---|---|
| ||||||
| All patients, n=3,735 | ||||||
| 30‐day ED or hospital revisit | 326 (19.8%) | 268 (18.2%) | 123 (17.2%) | 0.294 | 0.906 | 0.357 |
| Median LOS, h | 27.1 [17.446.4] | 17.6 [12.122.8] | 26.1 [16.941.2] | 0.001 | 0.004 | 0.001 |
| Chest‐pain patients, n=1,122 | ||||||
| 30‐day ED or hospital revisit | 69 (14.9%) | 82 (15.5%) | 23 (17.4%) | 0.859 | 0.496 | 0.596 |
| Median LOS, h | 22 [15.838.9] | 17.3 [10.922.4] | 23.2 [13.843.1] | 0.001 | 0.995 | 0.001 |
| Other diagnoses, n=2,613 | ||||||
| 30‐day ED or hospital revisit | 257 (21.6%) | 186 (19.8%) | 100 (18.4%) | 0.307 | 0.693 | 0.727 |
| Median LOS, h | 30.4 [18.649.4] | 17.8 [12.923] | 26.7 [17.231.1] | 0.001 | 0.001 | 0.001 |
In total, there were 717 total revisits including ED visits and hospital stays within 30 days of discharge (Table 2). Of all the observation encounters in the study, 19.2% were followed by a revisit within 30 days. There were no differences in the 30‐day post‐ED visit rates in between periods and between groups.
Mean ED LOS for hospitalized patients was examined for a sample of the pre‐ and post‐CDU periods, namely November 2010 to January 2011 and November 2011 to January 2012. The mean ED LOS decreased from 410 minutes (SD=61) to 393 minutes (SD=51) after implementation of the CDU (P=0.037).
To account for possible skewing of the data, we transformed LOS into ln (natural log) LOS and found the following means (SD): group 1 was 3.27 (0.94), group 2 was 2.78 (0.6), and group 3 was 3.1 (0.93). Using an independent t test, we found a significant difference between groups 1 and 2, 2 and 3, as well as 1 and 3 (P0.001 for all).
Chest‐Pain Subgroup Analysis
We analyzed the data specifically for the 1122 patients discharged with a diagnosis of chest pain. LOS was significantly lower for patients in the CDU compared to either pre‐CDU or observation on floors (Table 2).
Multivariate Regression Analysis
We performed a linear regression analysis using the following variables: age, race, gender, diagnosis, insurance status, and study period (pre‐CDU, post‐CDU, and postnon‐CDU). We performed 3 different comparisons: pre‐CDU vs post‐CDU, postnon‐CDU vs post‐CDU, and postnon‐CDU vs pre‐CDU. After adjusting for other variables, the postnon‐CDU group was significantly associated with higher LOS (P0.001). The pre‐CDU group was associated with higher LOS than both the post‐CDU and postnon‐CDU groups (P0.001 for both).
DISCUSSION
In our study of a hospitalist‐run CDU for observation patients, we observed that the care in the CDU was associated with a lower median LOS, but no increase in ED or hospital revisits within 30 days.
Previous studies have reported the impact of clinical observation or clinical diagnosis units, particularly chest‐pain units.[5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15] Studies of hospitalist‐run units suggest shorter LOS in the entire hospital,[16] or in the target unit.[17] Although one study suggested a lower 30‐day readmission rate,[18] most others did not describe this effect.[16, 17] Our study differs from previous research in that our program employed a pull‐culture aimed at accepting the majority of observation status patients without focusing on a particular diagnosis. We also implemented a structured multidisciplinary team focused on expediting care and utilized BOOST‐framed transitions, including targeted medication reconciliation and tools such as teach‐back.
The CDU in our hospital produced shorter LOS even compared to our non‐CDU units, but the revisit rate did not improve despite activities to reduce revisits. During the study period, efforts to decrease readmissions were implemented in various areas of our hospital, but not a comprehensive institution‐wide readmissions strategy. Lack of impact on revisits could be viewed as a positive finding, in that shorter LOS did not result in patients being discharged home before clinically stable. Alternatively, lack of impact could be due to the uncertain effectiveness of BOOST specifically[19, 20, 21] or inpatient‐targeted transitions interventions more generally.[22]
Our study has certain limitations. Findings in our single‐center study in an urban academic medical center may not apply to CDUs in other settings. As a prepost design, our study is subject to external trends for which our analyses may be unable to account. For example, during CDU implementation, there were hospital‐wide initiatives aimed at improving inpatient LOS, including complex case rounds, increased use of active bed management, and improved case management efforts to decrease LOS. These may have been a factor in the small decrease in observation LOS seen in the medicalsurgical patients during the post period. Additionally, though we have attempted to control for possible confounders, there could have been differences in the study groups for which we were unable to account, including code status or social variables such as homelessness, which played a role in our revisit outcomes. The decrease in LOS by 35%, or 9.5 hours, in CDU patients is clinically important, as it allows low‐risk patients to spend less time in the hospital where they may have been at risk of hospital‐acquired conditions; however, this study did not include patient satisfaction data. It would be important to measure the effect on patient experience of potentially spending 1 fewer night in the hospital. Finally, our CDU was designed with specific clinical criteria for inclusion and exclusion. Patients who were higher risk or expected to need more than 24 hours of care were not placed in the CDU. We were not able to adjust our analyses for factors that were not in our data, such as severe vital sign or laboratory abnormalities or a physician's clinical impression of a patient. It is possible, therefore, that referral bias may have occurred and influenced our results. The fact that non‐CDU chest‐pain patients in the post‐CDU period did not experience any decrease in LOS, whereas other medicalsurgical observation patients did, may be an example of this bias. Patients were excluded from the CDU by virtue of being deemed higher risk as described in Methods section. We were unable to adjust for these differences.
Implementation of CDUs may be useful for health systems seeking to improve hospital throughput and improve utilization among common but low‐acuity patient groups. Although our initial results are promising, the concept of a CDU may require enhancements. For example, at our hospital we are addressing transitions of care by looking at models that address patient risk through a systematic process, and then target individuals for specific interventions to prevent revisits. Moreover, the study of CDUs should report impact on patient and referring physician satisfaction, and whether CDUs can reduce per‐case costs.
CONCLUSION
Caring for patients in a hospitalist‐run geographic CDU was associated with a 35% decrease in observation LOS for CDU patients compared with a 3.7% decrease for observation patients cared for elsewhere in the hospital. CDU patients' LOS was significantly decreased without increasing ED or hospital revisit rates.
Acknowledgments
The authors would like to thank Ken Travis for excellent data support.
Hospitalists play a crucial role in improving hospital throughput and length of stay (LOS). The clinical decision unit (CDU) or observation unit (OU) is a strategy that was developed to facilitate both aims. CDUs and OUs are units where patients can be managed in the hospital for up to 24 hours prior to a decision being made to admit or discharge. Observation care is provided to patients who require further treatment or monitoring beyond what is accomplished in the emergency department (ED), but who do not require inpatient admission. CDUs arose in the 1990s in response to a desire to decrease inpatient costs as well as changing Medicare guidelines, which recognized observation status. Initially, CDUs and OUs were located within the ED and run by emergency medicine physicians. However, at the turn of the 21st century, hospitalists became involved in observation medicine, and the Society of Hospital Medicine issued a white paper on the OU in 2007. [1] Today, up to 50% of CDUs and OUs nationally are managed by hospitalists and located physically outside of the ED.[2, 3]
Despite the fact that nearly half of all CDUs and OUs nationally are run by hospitalists, there has been little published regarding the impact of hospitalist‐driven units. This study demonstrates the effect of observation care delivered in a hospitalist‐run geographic CDU. The primary objective was to determine the impact on LOS for patients in observation status managed in a hospitalist‐run CDU compared with LOS for observation patients with the same diagnoses cared for on medicalsurgical units prior to the existence of the CDU. The secondary objective was to determine the effect on the 30‐day ED or hospital revisit rate, as well as ED LOS. This work will guide health systems, hospitalist groups, and physicians in their decision making regarding the future structure and process of CDUs.
METHODS
Study Design
The Cooper University Hospital institutional review board approved this study. The study took place at Cooper University Hospital, a large, urban, academic safety‐net hospital providing tertiary care located in Camden, New Jersey.
We performed a retrospective observational study of all adult observation encounters at the study hospital from July 2010 to January 2011, and July 2011 through January 2012. During the second time period, patients could have been managed in the CDU or on a medicalsurgical unit. We recorded the following demographic data: age, gender, race, principal diagnosis, and payer, as well as several outcomes of interest, including: LOS (defined as the time separating the admitting physician order from discharge), ED visits within 30 days of discharge, and hospital revisits (observation or inpatient) within 30 days.
Data Sources
Data were culled by the institution's performance improvement department from the electronic medical record, as well as cost accounting and claims‐based sources.
Clinical Decision Unit
The CDU at Cooper University Hospital opened in June 2011 and is a 20‐bed geographically distinct unit adjacent to the ED. During the study period, it was staffed 24 hours a day by a hospitalist and a nurse practitioner as well as dedicated nurses and critical care technicians. Patients meeting observation status in the ED were eligible for the CDU provided that they fulfilled the CDU placement guidelines including that they were more likely than not to be discharged within a period of 24 hours of CDU care, did not meet inpatient admission criteria, did not require new placement in a rehabilitation or extended‐care facility, and did not require one‐on‐one monitoring. Additional exclusion criteria included severe vital sign or laboratory abnormalities. The overall strategy of the guidelines was to facilitate a pull culture, where the majority of observation patients were brought from the ED to the CDU once it was determined that they did not require inpatient care. The CDU had order sets and protocols in place for many of the common diagnoses. All CDU patients received priority laboratory and radiologic testing as well as priority consultation from specialty services. Medication reconciliation was performed by a pharmacy technician for higher‐risk patients, identified by Project BOOST (Better Outcomes by Optimizing Safe Transitions) criteria.[4] Structured multidisciplinary rounds occurred daily including the hospitalist, nurse practitioner, registered nurses, case manager, and pharmacy technician. A discharge planner was available to schedule follow‐up appointments.
Although chest pain was the most common CDU diagnosis, the CDU was designed to care for the majority of the hospital's observation patients rather than focus specifically on chest pain. Patients with chest pain who met observation criteria were transferred from the ED to the CDU, rather than a medicalsurgical unit, provided they did not have: positive cardiac enzymes, an electrocardiogram indicative of ischemia, known coronary artery disease presenting with pain consistent with acute coronary syndrome, need for heparin or nitroglycerin continuous infusion, symptomatic or unresolved arrhythmia, congestive heart failure meeting inpatient criteria, hypertensive urgency or emergency, pacemaker malfunction, pericarditis, or toxicity from cardiac drugs. Cardiologist consultants were involved in the care of nearly all CDU patients with chest pain.
Observation Status Determination
During the study period, observation status was recommended by a case manager in the ED based on Milliman (Milliman Care Guidelines) or McKesson InterQual (McKesson Corporation) criteria, once it was determined by the ED physician that the patient had failed usual ED care and required hospitalization. Observation status was assigned by the admitting (non‐ED) physician, who placed the order for inpatient admission or observation. Other than the implementation of the CDU, there were no significant changes to the process or criteria for assigning observation status, admission order sets, or the hospital's electronic medical record during this time period.
Statistical Analysis
Continuous data are presented as mean ( standard deviation [SD]) or median (25%75% interquartile range) as specified, and differences were assessed using one‐way analysis of variance testing and Mann‐Whitney U testing. Categorical data are presented as count (percentage) and differences evaluated using [2] analysis. P values of 0.05 or less were considered statistically significant.
To account for differences in groups with regard to outcomes, we performed a multivariate regression analysis. The following variables were entered: age (years), gender, race (African American vs other), admission diagnosis (chest pain vs other), and insurance status (Medicare vs other). All variables were entered simultaneously without forcing. Statistical analyses were done using the SPSS 20.0 Software (SPSS Inc., Chicago, IL).
RESULTS
Demographics
There were a total of 3735 patients included in the study: 1650 in the pre‐CDU group, 1469 in the post‐CDU group, and 616 in the post‐CDU group on medicalsurgical units. The post‐CDU period had a total of 2085 patients. Patients in the CDU group were younger and were more likely to have chest pain as the admission diagnosis. Patient demographics are presented in Table 1.
| Variable | Pre‐CDU, n=1,650 | Post‐CDU, n=1,469 | PostNon‐CDU, n=616 | P, CDU vs Pre‐CDU | P, Non‐CDU vs Pre‐CDU | P, CDU vs Non‐CDU |
|---|---|---|---|---|---|---|
| ||||||
| Age, y [range] | 56 [4569] | 53 [4364] | 57 [44.370] | 0.001 | 0.751 | 0.001 |
| Female gender | 918 (55.6%) | 833(56.7%) | 328 (53.2%) | 0.563 | 0.319 | 0.148 |
| African American race | 574 (34.8%) | 505 (34.4%) | 174 (28.2%) | 0.821 | 0.004 | 0.007 |
| Admission diagnosis | ||||||
| Chest pain | 462 (38%) | 528 (35.9%) | 132 (21.4%) | 0.001 | 0.002 | 0.001 |
| Syncope | 93 (5.6%) | 56 (3.8%) | 15 (2.4%) | 0.018 | 0.001 | 0.145 |
| Abdominal pain | 46 (2.8%) | 49 (3.3%) | 20(3.2%) | 0.404 | 0.575 | 1.0 |
| Other | 1,049 (63.6%) | 836 (56.9%) | 449 (72.9%) | 0.001 | 0.001 | 0.001 |
| Third‐party payer | ||||||
| Medicare | 727 (44.1%) | 491 (33.4%) | 264(43.4%) | 0.001 | 0.634 | 0.001 |
| Charity care | 187 (11.3%) | 238 (16.2%) | 73 (11.9%) | 0.001 | 0.767 | 0.010 |
| Commercial | 185 (11.1%) | 214 (14.6%) | 87 (14.1%) | 0.005 | 0.059 | 0.838 |
| Medicaid | 292 (17.7%) | 280 (19.1%) | 100 (16.2%) | 0.331 | 0.454 | 0.136 |
| Other | 153 (9.3%) | 195 (13.3%) | 60 (9.9%) | 0.001 | 0.746 | 0.028 |
| Self‐pay | 106 (6.4%) | 51(3.5%) | 32 (5.2%) | 0.001 | 0.323 | 0.085 |
Outcomes of Interest
There was a statistically significant association between LOS and CDU implementation (Table 2). Observation patients cared for in the CDU had a lower LOS than observation patients cared for on the medicalsurgical units during the same time period (17.6 vs 26.1 hours, P0.0001).
| Outcome | Pre‐CDU, n=1,650 | Post‐CDU, n=1,469 | PostNon‐CDU, n=616 | P, CDU vs Pre‐CDU | P, Non‐CDU vs Pre‐CDU | P, CDU vs Non‐CDU |
|---|---|---|---|---|---|---|
| ||||||
| All patients, n=3,735 | ||||||
| 30‐day ED or hospital revisit | 326 (19.8%) | 268 (18.2%) | 123 (17.2%) | 0.294 | 0.906 | 0.357 |
| Median LOS, h | 27.1 [17.446.4] | 17.6 [12.122.8] | 26.1 [16.941.2] | 0.001 | 0.004 | 0.001 |
| Chest‐pain patients, n=1,122 | ||||||
| 30‐day ED or hospital revisit | 69 (14.9%) | 82 (15.5%) | 23 (17.4%) | 0.859 | 0.496 | 0.596 |
| Median LOS, h | 22 [15.838.9] | 17.3 [10.922.4] | 23.2 [13.843.1] | 0.001 | 0.995 | 0.001 |
| Other diagnoses, n=2,613 | ||||||
| 30‐day ED or hospital revisit | 257 (21.6%) | 186 (19.8%) | 100 (18.4%) | 0.307 | 0.693 | 0.727 |
| Median LOS, h | 30.4 [18.649.4] | 17.8 [12.923] | 26.7 [17.231.1] | 0.001 | 0.001 | 0.001 |
In total, there were 717 total revisits including ED visits and hospital stays within 30 days of discharge (Table 2). Of all the observation encounters in the study, 19.2% were followed by a revisit within 30 days. There were no differences in the 30‐day post‐ED visit rates in between periods and between groups.
Mean ED LOS for hospitalized patients was examined for a sample of the pre‐ and post‐CDU periods, namely November 2010 to January 2011 and November 2011 to January 2012. The mean ED LOS decreased from 410 minutes (SD=61) to 393 minutes (SD=51) after implementation of the CDU (P=0.037).
To account for possible skewing of the data, we transformed LOS into ln (natural log) LOS and found the following means (SD): group 1 was 3.27 (0.94), group 2 was 2.78 (0.6), and group 3 was 3.1 (0.93). Using an independent t test, we found a significant difference between groups 1 and 2, 2 and 3, as well as 1 and 3 (P0.001 for all).
Chest‐Pain Subgroup Analysis
We analyzed the data specifically for the 1122 patients discharged with a diagnosis of chest pain. LOS was significantly lower for patients in the CDU compared to either pre‐CDU or observation on floors (Table 2).
Multivariate Regression Analysis
We performed a linear regression analysis using the following variables: age, race, gender, diagnosis, insurance status, and study period (pre‐CDU, post‐CDU, and postnon‐CDU). We performed 3 different comparisons: pre‐CDU vs post‐CDU, postnon‐CDU vs post‐CDU, and postnon‐CDU vs pre‐CDU. After adjusting for other variables, the postnon‐CDU group was significantly associated with higher LOS (P0.001). The pre‐CDU group was associated with higher LOS than both the post‐CDU and postnon‐CDU groups (P0.001 for both).
DISCUSSION
In our study of a hospitalist‐run CDU for observation patients, we observed that the care in the CDU was associated with a lower median LOS, but no increase in ED or hospital revisits within 30 days.
Previous studies have reported the impact of clinical observation or clinical diagnosis units, particularly chest‐pain units.[5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15] Studies of hospitalist‐run units suggest shorter LOS in the entire hospital,[16] or in the target unit.[17] Although one study suggested a lower 30‐day readmission rate,[18] most others did not describe this effect.[16, 17] Our study differs from previous research in that our program employed a pull‐culture aimed at accepting the majority of observation status patients without focusing on a particular diagnosis. We also implemented a structured multidisciplinary team focused on expediting care and utilized BOOST‐framed transitions, including targeted medication reconciliation and tools such as teach‐back.
The CDU in our hospital produced shorter LOS even compared to our non‐CDU units, but the revisit rate did not improve despite activities to reduce revisits. During the study period, efforts to decrease readmissions were implemented in various areas of our hospital, but not a comprehensive institution‐wide readmissions strategy. Lack of impact on revisits could be viewed as a positive finding, in that shorter LOS did not result in patients being discharged home before clinically stable. Alternatively, lack of impact could be due to the uncertain effectiveness of BOOST specifically[19, 20, 21] or inpatient‐targeted transitions interventions more generally.[22]
Our study has certain limitations. Findings in our single‐center study in an urban academic medical center may not apply to CDUs in other settings. As a prepost design, our study is subject to external trends for which our analyses may be unable to account. For example, during CDU implementation, there were hospital‐wide initiatives aimed at improving inpatient LOS, including complex case rounds, increased use of active bed management, and improved case management efforts to decrease LOS. These may have been a factor in the small decrease in observation LOS seen in the medicalsurgical patients during the post period. Additionally, though we have attempted to control for possible confounders, there could have been differences in the study groups for which we were unable to account, including code status or social variables such as homelessness, which played a role in our revisit outcomes. The decrease in LOS by 35%, or 9.5 hours, in CDU patients is clinically important, as it allows low‐risk patients to spend less time in the hospital where they may have been at risk of hospital‐acquired conditions; however, this study did not include patient satisfaction data. It would be important to measure the effect on patient experience of potentially spending 1 fewer night in the hospital. Finally, our CDU was designed with specific clinical criteria for inclusion and exclusion. Patients who were higher risk or expected to need more than 24 hours of care were not placed in the CDU. We were not able to adjust our analyses for factors that were not in our data, such as severe vital sign or laboratory abnormalities or a physician's clinical impression of a patient. It is possible, therefore, that referral bias may have occurred and influenced our results. The fact that non‐CDU chest‐pain patients in the post‐CDU period did not experience any decrease in LOS, whereas other medicalsurgical observation patients did, may be an example of this bias. Patients were excluded from the CDU by virtue of being deemed higher risk as described in Methods section. We were unable to adjust for these differences.
Implementation of CDUs may be useful for health systems seeking to improve hospital throughput and improve utilization among common but low‐acuity patient groups. Although our initial results are promising, the concept of a CDU may require enhancements. For example, at our hospital we are addressing transitions of care by looking at models that address patient risk through a systematic process, and then target individuals for specific interventions to prevent revisits. Moreover, the study of CDUs should report impact on patient and referring physician satisfaction, and whether CDUs can reduce per‐case costs.
CONCLUSION
Caring for patients in a hospitalist‐run geographic CDU was associated with a 35% decrease in observation LOS for CDU patients compared with a 3.7% decrease for observation patients cared for elsewhere in the hospital. CDU patients' LOS was significantly decreased without increasing ED or hospital revisit rates.
Acknowledgments
The authors would like to thank Ken Travis for excellent data support.
- The observation unit: an operational overview for the hospitalist. Society of Hospital Medicine website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=White_Papers18(12):1371–1379.
- , , , , , . Use of observation care in US emergency departments, 2001 to 2008. PLoS One. 2011;6(9):e24326.
- The Society of Hospital Medicine Project Boost (Better Outcomes by Optimizing Safe Transitions) Available at: http://www.hospitalmedicine.org/boost. Accessed on June 4, 2013.
- , , , , . An emergency department‐based protocol for rapidly ruling out myocardial ischemia reduces hospital time and expense: results of a randomized study (ROMIO). J Am Coll Cardiol. 1996;28(1):25–33.
- , , , , , . Implementation of the guidelines for the management of patients with chest pain through a critical pathway approach improves length of stay and patient satisfaction but not anxiety. Crit Pathw Cardiol. 2010;9(1):30–34.
- , , , et al. Costs of an emergency department‐based accelerated diagnostic protocol vs hospitalization in patients with chest pain: a randomized controlled trial. JAMA. 1997;278(20):1670–1676.
- , , , et al. Emergency‐department diagnosis of acute myocardial infarction and ischemia: a cost analysis of two diagnostic protocols. Acad Emerg Med. 1994;1(2):103–110.
- , , , et al. Impact on the care of the emergency department chest pain patient from the chest pain evaluation registry (CHEPER) study. Am J Cardiol. 1997;80(5):563–568.
- , , , et al. Cost‐effectiveness of a new short‐stay unit to “rule out” acute myocardial infarction in low risk patients. J Am Coll Cardiol. 1994;24(5):1249–1259.
- , , , et al. Emergency Department Observation Unit versus hospital inpatient care for a chronic asthmatic population: a randomized trial of health status outcome and cost. Med Care. 1998;36(4):599–609.
- , , , et al. A comparison between emergency diagnostic and treatment unit and inpatient care in the management of acute asthma. Arch Intern Med. 1997;157(18):2055–2062.
- , , , . Retrospective review of emergency department patients with non‐variceal upper gastrointestinal hemorrhage for potential outpatient management. Acad Emerg Med. 1999;6(3):196–201.
- , . Outpatient care of selected patients with acute non‐variceal upper gastrointestinal haemorrhage. Lancet. 1995;345(8942):108–111.
- , , , , . Patterns of use of an emergency department‐based observation unit. Am J Ther. 2002;9(6):499–502.
- , , . Implementation of a hospitalist‐run observation unit and impact on length of stay (LOS): a brief report. J Hosp Med. 2010;5(9):E2–E5.
- , , , , . Improving resource utilization in a teaching hospital: development of a nonteaching service for chest pain admissions. Acad Med. 2006;81(5):432–435.
- , , , . Program description: a hospitalist‐run, medical short‐stay unit in a teaching hospital. CMAJ. 2000;163(11):1477–1480.
- , , , et al. Project BOOST: effectiveness of a multihospital effort to reduce rehospitalization. J Hosp Med. 2013;8:421–427.
- , , , et al. BOOST: evidence needing a lift. J Hosp Med. 2013;8:468–469.
- . BOOST and readmissions: thinking beyond the walls of the hospital. J Hosp Med. 2013;8:470–471.
- , , , et al. Hospital‐initiated transitional care interventions as a patient safety strategy. Ann Int Med. 2013;158:433–440.
- The observation unit: an operational overview for the hospitalist. Society of Hospital Medicine website. Available at: http://www.hospitalmedicine.org/AM/Template.cfm?Section=White_Papers18(12):1371–1379.
- , , , , , . Use of observation care in US emergency departments, 2001 to 2008. PLoS One. 2011;6(9):e24326.
- The Society of Hospital Medicine Project Boost (Better Outcomes by Optimizing Safe Transitions) Available at: http://www.hospitalmedicine.org/boost. Accessed on June 4, 2013.
- , , , , . An emergency department‐based protocol for rapidly ruling out myocardial ischemia reduces hospital time and expense: results of a randomized study (ROMIO). J Am Coll Cardiol. 1996;28(1):25–33.
- , , , , , . Implementation of the guidelines for the management of patients with chest pain through a critical pathway approach improves length of stay and patient satisfaction but not anxiety. Crit Pathw Cardiol. 2010;9(1):30–34.
- , , , et al. Costs of an emergency department‐based accelerated diagnostic protocol vs hospitalization in patients with chest pain: a randomized controlled trial. JAMA. 1997;278(20):1670–1676.
- , , , et al. Emergency‐department diagnosis of acute myocardial infarction and ischemia: a cost analysis of two diagnostic protocols. Acad Emerg Med. 1994;1(2):103–110.
- , , , et al. Impact on the care of the emergency department chest pain patient from the chest pain evaluation registry (CHEPER) study. Am J Cardiol. 1997;80(5):563–568.
- , , , et al. Cost‐effectiveness of a new short‐stay unit to “rule out” acute myocardial infarction in low risk patients. J Am Coll Cardiol. 1994;24(5):1249–1259.
- , , , et al. Emergency Department Observation Unit versus hospital inpatient care for a chronic asthmatic population: a randomized trial of health status outcome and cost. Med Care. 1998;36(4):599–609.
- , , , et al. A comparison between emergency diagnostic and treatment unit and inpatient care in the management of acute asthma. Arch Intern Med. 1997;157(18):2055–2062.
- , , , . Retrospective review of emergency department patients with non‐variceal upper gastrointestinal hemorrhage for potential outpatient management. Acad Emerg Med. 1999;6(3):196–201.
- , . Outpatient care of selected patients with acute non‐variceal upper gastrointestinal haemorrhage. Lancet. 1995;345(8942):108–111.
- , , , , . Patterns of use of an emergency department‐based observation unit. Am J Ther. 2002;9(6):499–502.
- , , . Implementation of a hospitalist‐run observation unit and impact on length of stay (LOS): a brief report. J Hosp Med. 2010;5(9):E2–E5.
- , , , , . Improving resource utilization in a teaching hospital: development of a nonteaching service for chest pain admissions. Acad Med. 2006;81(5):432–435.
- , , , . Program description: a hospitalist‐run, medical short‐stay unit in a teaching hospital. CMAJ. 2000;163(11):1477–1480.
- , , , et al. Project BOOST: effectiveness of a multihospital effort to reduce rehospitalization. J Hosp Med. 2013;8:421–427.
- , , , et al. BOOST: evidence needing a lift. J Hosp Med. 2013;8:468–469.
- . BOOST and readmissions: thinking beyond the walls of the hospital. J Hosp Med. 2013;8:470–471.
- , , , et al. Hospital‐initiated transitional care interventions as a patient safety strategy. Ann Int Med. 2013;158:433–440.
Four Nephrology Myths Debunked
There are many controversial topics relating to renal disease in hospitalized patients. The aim of this review is to shed light on some important and often debated issues. We will first discuss topics related to electrolytes disorder commonly seen in hospitalized patients (hyponatremia, hyperkalemia, metabolic acidosis) then the use of diuretics in patients with allergy to sulfa containing antibiotics.
Hypothyroidism and Hyponatremia
Hyponatremia is common in hospitalized patients and is associated with worse outcomes.1 It can be seen with a variety of conditions ranging from congestive heart failure to volume depletion. Careful history and physical examination are paramount and the initial work‐up usually includes serum and urine osmolality and urine sodium concentration.
For euvolemic hyponatremia, the differential diagnosis includes the syndrome of inappropriate adenine dinucleotide (ADH) secretion (SIADH), hypoadrenalism, and beer potomania. Additionally, many authorities also include hypothyroidism.
Although the simultaneous finding of hypothyroidism and hyponatremia can occur in patients as both diseases are widely prevalent in the general population, causation has yet to be convincingly demonstrated.
ADH is released in response to effective volume depletion; consequently when hypothyroidism is encountered in the setting of complete pituitary failure there is often hyponatremia.2, 3 Alternatively, with myxedema, the ability of the kidney to handle a water load and concentrate urine can be impaired.4
However, the observation that thyroid hormone administration did not raise sodium values in newborns with congenital hypothyroidism or in adults supports the absence of causal effect.5, 6And in addition, large studies done in the hospital and outpatient setting showed no differences between the serum sodium values of hypothyroid patients and that of controls.7, 8 In the study of outpatients, among those with hypothyroidism, for every increase of 10 mU/L of thyroid‐stimulating hormone (TSH), there was a drop of only 0.14 mmol/L of Na concentration.8 Thus, the elevation of TSH required for a clinically meaningful drop in sodium to occur was considerable.
Hence in patients with hyponatremia, the hospitalist should look for etiologies other than hypothyroidism and should only consider thyroid hypofunction as a culprit in cases of myxedema, or panhypopituitarism.
Sodium Bicarbonate for Hyperkalemia
Hyperkalemia is one of the most feared electrolyte disorders encountered in hospitalized patients and can lead to dire outcomes.9, 10
Potassium (K+) homeostasis is maintained in the body by 2 complimentary systems: a short‐term system that regulates K+ variation by modifying translocation across the cellular membrane and a long‐term system that adjusts overall K+ balance. The translocation system is regulated primarily by insulin and ‐2 stimulation. Overall K+ balance is mainly controlled by the kidney (90‐95%) although the gastrointestinal (GI) tract can have a more preponderant role in anephric patients.
Hyperkalemia can ensue by either a dysregulation of the translocation system (as in diabetic Ketoacidosis secondary to insulin deficiency) or impairment of K+ elimination.
Acid‐base status was previously thought to have a prominent influence on K+ concentration, based on studies that demonstrated that. However, studies looking at metabolic acidosis revealed that contrary to the effect of mineral acidosis (excess of nonmetabolizable anions)11 where there is an inverse correlation between potassium concentration and pH; organic acidosis (excess of metabolizable anions)11 was not associated with hyperkalemia.1214 However, organic acidosis can be seen simultaneously and induced by a same underlying disease (such as organ ischemia with lactic acidosis or insulin deficiency complicated by ketoacidosis). Also, when changes in pH are induced by respiratory variations or with alkalosis, the impact on serum K+ concentration is less remarkable.15 Hence, it seems that it is the nature of the acid‐base disturbance that impacts K+ concentration more than the change in pH itself.
In the kidney, the main site for regulation of K+ balance is the collecting duct. Factors that affect elimination include urinary sodium delivery, urine flow, and aldosterone.16 In order to adequately eliminate K+ these factors must be optimized in conjunction.
Treatment of hyperkalemia includes the sequential administration of agents that stabilize the cardiac membrane (calcium gluconate), shift the potassium intracellularly (insulin, ‐2 agonists), and remove the potassium (diuretics, sodium polystyrene, or dialysis).
The use of sodium bicarbonate for treatment of hyperkalemia has been long advocated.17 It was thought to act by translocation of potassium hence could be used to quickly lower K+ concentration. However, this dogma has been challenged recently.
To assess the true impact of sodium bicarbonate on potassium translocation, studies have been conducted on anephric patients with hyperkalemia. Bicarbonate infusion failed to elicit a significant rapid change in serum K+ concentration despite increase in bicarbonate concentration, arguing against a translocation mechanism.1821 After 60 minutes of treatment, neither isotonic nor hypertonic bicarbonate infusion affected Serum K+ levels in end‐stage renal disease (ESRD) patients.19, 20 On the contrary, hypertonic sodium bicarbonate increased the K+ concentration after 180 minutes of treatment,20 and it took a prolonged infusion of 4 hours to see a significant decrease in K+ concentration (0.6 mmol/L); half of which could be accounted for solely by volume administration. Moreover, this reduction was highly variable.
Rather, sodium bicarbonate seems to enhance potassium elimination by increasing sodium delivery to the distal tubule, increasing urinary pH and negative luminal charge and potentiating the action of diuretics.23 In an elegant study on normovolemic patients, the induction of bicarbonaturia practically doubled potassium excretion.23 However, such an effect is heterogeneous and usually takes place over 4 hours to 6 hours.17
At the cellular level, 2 ion exchange pumps cooperate to handle Na/K/H movement across the cellular membrane: an Na+/H+ exchanger (NHE) and the Na+/K+ ATP‐ase pump (Figure 1). The NHE is normally inactive and is only upregulated in cases of severe intracellular acidosis.24 The infusion of sodium bicarbonate to patients with severe metabolic acidosis could possibly decrease the serum potassium concentration by translocation if the NHE was significantly upregulated. However, this treatment can be associated with a drop in the ionized calcium level, a worsening of the intracellular acidosis, and a decreased peripheral oxygen delivery.25 Thus, the benefits should be balanced with the potential adverse effects and, even in cases of severe metabolic acidosis with hyperkalemia, we would advise the clinician to restrictively administer sodium bicarbonate.
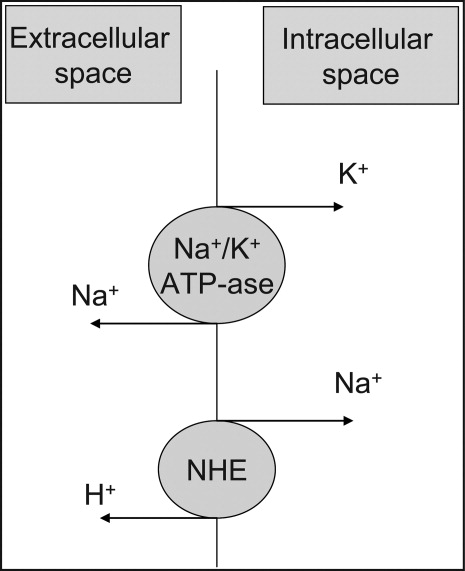
In addition, in ESRD patients, the administration of sodium bicarbonate can be problematic owing to the osmotic and volume burden it carries. It should also be avoided in patients who are volume overload or in those with decreased ability to eliminate potassium.
When treating hyperkalemic patients, hospitalists should use sodium bicarbonate to potentiate urinary elimination of potassium and should consider administering it either with acetazolamide or a loop diuretic, anticipating a lowering effect after a few hours.26 It should be avoided in patients with volume overload and anuria. Immediate translocation of potassium into cells is best achieved by insulin and ‐2 agonists.
5‐Oxoprolinuria: A Newly Recognized Cause of High Anion Gap Metabolic Acidosis
There are several causes of metabolic high anion gap acidosis in hospitalized patients. However, despite careful investigations, the cause of that disorder is not always apparent.27 Recently, 5‐oxoprolinuria (also called pyroglutamic acidosis) has become increasingly recognized as a potential etiology.2832
The metabolism of glutamate (though the ‐Glutamyl cycle) generates glutathione which provides negative feedback to the ‐Glutamyl‐cysteine synthetase enzyme. The depletion of glutathione increases 5‐oxoproline production owing to the loss of that inhibition (Figures 2 and 3). Low glutathione levels can be seen with liver disease,33 chronic alcohol intake,34 acetaminophen use,35 malnutrition,36 renal dysfunction,29 use of vigabatrim (an antiepileptic that received Food and Drug Administration [FDA] approval for use in April 2009)37 and sepsis.38 Most of the reported cases were female and had more than one risk factor.39
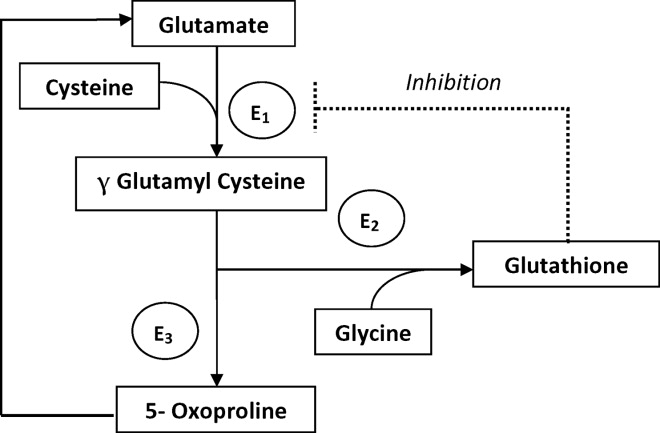
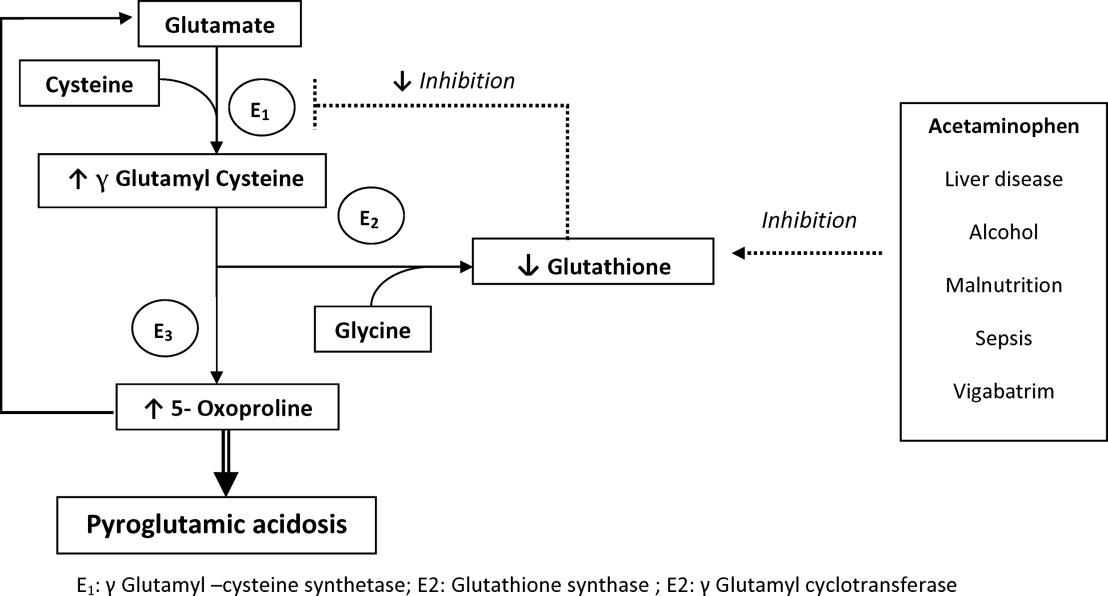
Typically, patients present with high anion gap acidosis (often more than 20)28 with normal acetaminophen levels and all usual tests being negative. A history of chronic acetaminophen use with or without other risk factors can frequently be found. The true independent impact of this type of acidosis on outcomes is difficult to determine as all of the reported cases had many confounding factors.
A urinary organic acid level is diagnostic and will reveal increased levels of pyroglutamic acid. Alternatively, the finding of a positive urinary anion gap (UNa +UK UCl) with a positive urinary osmolar gap (Uosmmeasured‐Uosmcalculated) in the appropriate clinical setting (unexplained high anion gap acidosis with negative workup and presence of risk factors for 5‐oxoproliniuria) can point towards the diagnosis.40
A study of patients with unexplained metabolic acidosis did not find any cases of 5‐oxoprolinuria.41 Although this might suggest that the incidence of this disease is low, very few of those patients were actually taking acetaminophen (therefore had a reduced propensity for developing pyroglutamic acidosis).41 Thus, the actual incidence of 5‐oxoprolinuria is hard to determine.
Once recognized, acetaminophen should be withheld and N‐acetylcysteine (NAC) can be used to replete glutathione levels although there is no convincing evidence for this use.42 It is important for hospitalists to be aware of this disorder as it can pose a diagnostic challenge (negative usual work‐up), is easy to treat by stopping acetaminophen, and can (possibly) negatively affect outcomes.
Furosemide and Patients With Sulfa Allergy
Allergic reactions are a common occurrence with sulfa‐containing antibiotics (SCA) and reports estimates the incidence to be approximately 3% to 5%.43, 44
One misbelief is that patients who are allergic to SCAs should not receive sulfa containing diuretics or other sulfa‐containing medications.45 This leads some physicians to substitute commonly used diuretics (such as furosemide or thiazides) for ethacrynic acid. The use of ethacrynic acid has several challenges: the limited supply of the intravenous form, the discontinuation of the oral form, the increased cost, and the risk of permanent ototoxicity.
The evidence for potential allergic cross‐reactivity among medications containing the sulfa moiety has been primarily derived from Case Reports.4649
The molecular structures of sulfamethoxazole and furosemide are shown below. The allergic antigen is most often the N1 component,45 and sometimes N4 but not the sulfa moiety. Both of the incriminated antigens are not present in the furosemide structure (as well as all other sulfa containing diuretics) (Figures 4 and 5).
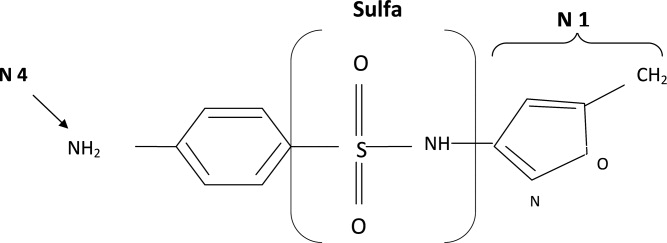
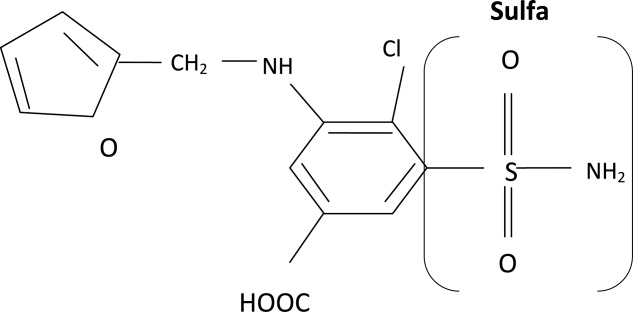
Experimental data showed that serum from patients allergic to SCAs did not bind to diuretics.50 In addition, clinical reports failed to demonstrate cross‐reactivity.5153 In a large clinical trial, Strom et al.53 showed that although there was a higher risk for allergic reaction to sulfa containing medications (SCM) in patients allergic to SCA (compared to those who were not), it was lower among patients with an allergy to sulfa antibiotic than among patients with a history of hypersensitivity to penicillins, suggesting this was due to a predisposition to allergic reactions in general rather than true cross‐reactivity. In another report, patients who were receiving ethacrynic acid for many years were successfully and uneventfully switched to furosemide.54
Taken together, these findings suggest that there is no evidence for withholding sulfa nonantibiotics in patients allergic to sulfa containing antibiotics.
Conclusion
Hypothyroidism, unlike myxedema, is not a cause of hyponatremia (although it can be sometimes seen in conjunction with the latter) and additional investigations should be done to determine its etiology. Sodium bicarbonate is effective for treatment of hyperkalemia by enhancing renal potassium elimination, rather than from shifting potassium into cells. The 5‐oxoprolinuria is a newly recognized cause of high anion‐gap metabolic acidosis and should be considered in patients who have taken acetaminophen. Furosemide (and sulfa containing diuretics) can be used safely in patients with an allergy to SCA.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122(9):857–865.
- ,.Hyponatremia as the presenting manifestation of Sheehan's syndrome in elderly patients.Aging Clin Exp Res.2006;18(6):536–539.
- ,,.Sodium and water disturbances in patients with Sheehan's syndrome.Am J Kidney Dis.2001;38(3):E14.
- ,,,,,.Effect of acute water loading on plasma levels of antidiuretic hormone AVP aldosterone, ANP fractional excretion of sodium and plasma and urine osmolalities in myxedema.Chin Med J (Engl).1990;103(9):704–708.
- ,.Sodium handling in congenitally hypothyroid neonates.Acta Paediatr.2004;93(1):22–24.
- ,,.Prevalence and severity of hyponatremia and hypercreatininemia in short‐term uncomplicated hypothyroidism.J Endocrinol Invest.1999;22(1):35–39.
- ,,,,.Absence of relation between hyponatraemia and hypothyroidism.Lancet.1997;350(9088):1402.
- ,,.The effect of newly diagnosed hypothyroidism on serum sodium concentrations: a retrospective study.Clin Endocrinol (Oxf).2006;64(5):598–599.
- .Disorders of potassium homeostasis. Hypokalemia and hyperkalemia.Crit Care Clin.2002;18(2):273–288,vi.
- ,.Hyperkalemia in hospitalized patients.Int Urol Nephrol.2000;32(2):177–180.
- ,.Treatment of metabolic acidosis.Curr Opin Crit Care.2003;9(4):260–265.
- ,,,.Natural history of lactic acidosis after grand‐mal seizures. A model for the study of an anion‐gap acidosis not associated with hyperkalemia.N Engl J Med.1977;297(15):796–799.
- ,,,,.The plasma potassium concentration in metabolic acidosis: a re‐evaluation.Am J Kidney Dis.1988;11(3):220–224.
- ,,,.Determinants of plasma potassium levels in diabetic ketoacidosis.Medicine (Baltimore).1986;65(3):163–172.
- ,.Changes in plasma potassium concentration during acute acid‐base disturbances.Am J Med.1981;71(3):456–467.
- ,,.New aspects of renal potassium transport.Pflugers Arch.2003;446(3):289–297.
- ,.Correction of hyperkalemia by bicarbonate despite constant blood pH.Kidney Int.1977;12(5):354–360.
- ,,,.Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure.Am J Med.1988;85(4):507–512.
- ,.Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol.Am J Kidney Dis.1996;28(4):508–514.
- ,,,,.Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end‐stage renal disease.Miner Electrolyte Metab.1991;17(5):297–302.
- .Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end‐stage renal disease patients.Nephron.1996;72(3):476–482.
- ,,.Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure.Kidney Int.1992;41(2):369–374.
- ,,, et al.Modulation of the secretion of potassium by accompanying anions in humans.Kidney Int.1991;39(6):1206–1212.
- ,.Controversial issues in the treatment of hyperkalaemia.Nephrol Dial Transplant.2003;18(11):2215–2218.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- .Management of severe hyperkalemia.Crit Care Med.2008;36(12):3246–3251.
- ,.Pyroglutamic acid and high anion gap: looking through the keyhole?Crit Care Med.2000;28(6):2140–2141.
- ,,,,.Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen.Clin J Am Soc Nephrol.2006;1(3):441–447.
- ,,,.Pyroglutamic acidemia: a cause of high anion gap metabolic acidosis.Crit Care Med.2000;28(6):1803–1807.
- ,,,,,.Acetaminophen‐induced anion gap metabolic acidosis and 5‐oxoprolinuria (pyroglutamic aciduria) acquired in hospital.Am J Kidney Dis.2005;46(1):143–146.
- ,.Anion gap acidosis associated with acetaminophen.Ann Intern Med.2000;133(9):752–753.
- ,,.Pyroglutamic acidosis in a renal transplant patient.Nephrol Dial Transplant.2005;20(12):2836–2838.
- ,,.Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases.Life Sci.1988;43(12):991–998.
- ,,,,,.Decreased hepatic glutathione in chronic alcoholic patients.J Hepatol.1986;3(1):1–6.
- ,.Intracellular signaling mechanisms of acetaminophen‐induced liver cell death.Toxicol Sci.2006;89(1):31–41.
- ,,.Urinary excretion of 5‐L‐oxoproline (pyroglutamic acid) is increased during recovery from severe childhood malnutrition and responds to supplemental glycine.J Nutr.1996;126(11):2823–2830.
- ,,,.Pyroglutamicaciduria from vigabatrin.Lancet.1989;1(8652):1452–1453.
- ,,, et al.Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients.Crit Care Med.2001;29(4):870–877.
- ,.Transient 5‐oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects.Clin Chem.1998;44(7):1497–1503.
- ,,,.Guilty as charged: unmeasured urinary anions in a case of pyroglutamic acidosis.Neth J Med.2008;66(8):351–353.
- ,,.Unexplained metabolic acidosis in critically ill patients: the role of pyroglutamic acid.Intensive Care Med.2004;30(3):502–505.
- ,,.A therapeutic trial with N‐acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5‐oxoprolinuria).J Inherit Metab Dis.1989;12(2):120–130.
- .Diagnosis of allergic reactions to sulfonamides.Allergy.1999;54Suppl 58:28–32.
- .Practical issues in the management of hypersensitivity reactions: sulfonamides.South Med J.2001;94(8):817–824.
- ,,.Should celecoxib be contraindicated in patients who are allergic to sulfonamides? Revisiting the meaning of ‘sulfa’ allergy.Drug Saf.2001;24(4):239–247.
- .Thrombocytopenia due to sulfonamide cross‐sensitivity.Wis Med J.1982;81(6):21–23.
- ,,.Leukocytoclastic vasculitis induced by use of glyburide: a case of possible cross‐reaction of a sulfonamide and a sulfonylurea.Cutis.2003;71(3):235–238.
- ,.Celecoxib‐induced erythema multiforme with glyburide cross‐reactivity.Pharmacotherapy.2002;22(5):637–640.
- ,.Vesiculobullous rash in a patient with systemic lupus erythematosus.Ann Allergy.1993;70(3):196–203.
- ,,, et al.Use of optical biosensor technology to study immunological cross‐reactivity between different sulfonamide drugs.Anal Biochem.2002;300(2):177–184.
- ,,,.Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis.Adverse Drug React Toxicol Rev.1996;15(1):9–50.
- ,,.Cross‐reactivity in HIV‐infected patients switched from trimethoprim‐sulfamethoxazole to dapsone.Pharmacotherapy.1998;18(4):831–835.
- ,,, et al.Absence of cross‐reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics.N Engl J Med.2003;349(17):1628–1635.
- ,,.Furosemide challenge in patients with heart failure and adverse reactions to sulfa‐containing diuretics.Ann Intern Med.2003;138(4):358–359.
There are many controversial topics relating to renal disease in hospitalized patients. The aim of this review is to shed light on some important and often debated issues. We will first discuss topics related to electrolytes disorder commonly seen in hospitalized patients (hyponatremia, hyperkalemia, metabolic acidosis) then the use of diuretics in patients with allergy to sulfa containing antibiotics.
Hypothyroidism and Hyponatremia
Hyponatremia is common in hospitalized patients and is associated with worse outcomes.1 It can be seen with a variety of conditions ranging from congestive heart failure to volume depletion. Careful history and physical examination are paramount and the initial work‐up usually includes serum and urine osmolality and urine sodium concentration.
For euvolemic hyponatremia, the differential diagnosis includes the syndrome of inappropriate adenine dinucleotide (ADH) secretion (SIADH), hypoadrenalism, and beer potomania. Additionally, many authorities also include hypothyroidism.
Although the simultaneous finding of hypothyroidism and hyponatremia can occur in patients as both diseases are widely prevalent in the general population, causation has yet to be convincingly demonstrated.
ADH is released in response to effective volume depletion; consequently when hypothyroidism is encountered in the setting of complete pituitary failure there is often hyponatremia.2, 3 Alternatively, with myxedema, the ability of the kidney to handle a water load and concentrate urine can be impaired.4
However, the observation that thyroid hormone administration did not raise sodium values in newborns with congenital hypothyroidism or in adults supports the absence of causal effect.5, 6And in addition, large studies done in the hospital and outpatient setting showed no differences between the serum sodium values of hypothyroid patients and that of controls.7, 8 In the study of outpatients, among those with hypothyroidism, for every increase of 10 mU/L of thyroid‐stimulating hormone (TSH), there was a drop of only 0.14 mmol/L of Na concentration.8 Thus, the elevation of TSH required for a clinically meaningful drop in sodium to occur was considerable.
Hence in patients with hyponatremia, the hospitalist should look for etiologies other than hypothyroidism and should only consider thyroid hypofunction as a culprit in cases of myxedema, or panhypopituitarism.
Sodium Bicarbonate for Hyperkalemia
Hyperkalemia is one of the most feared electrolyte disorders encountered in hospitalized patients and can lead to dire outcomes.9, 10
Potassium (K+) homeostasis is maintained in the body by 2 complimentary systems: a short‐term system that regulates K+ variation by modifying translocation across the cellular membrane and a long‐term system that adjusts overall K+ balance. The translocation system is regulated primarily by insulin and ‐2 stimulation. Overall K+ balance is mainly controlled by the kidney (90‐95%) although the gastrointestinal (GI) tract can have a more preponderant role in anephric patients.
Hyperkalemia can ensue by either a dysregulation of the translocation system (as in diabetic Ketoacidosis secondary to insulin deficiency) or impairment of K+ elimination.
Acid‐base status was previously thought to have a prominent influence on K+ concentration, based on studies that demonstrated that. However, studies looking at metabolic acidosis revealed that contrary to the effect of mineral acidosis (excess of nonmetabolizable anions)11 where there is an inverse correlation between potassium concentration and pH; organic acidosis (excess of metabolizable anions)11 was not associated with hyperkalemia.1214 However, organic acidosis can be seen simultaneously and induced by a same underlying disease (such as organ ischemia with lactic acidosis or insulin deficiency complicated by ketoacidosis). Also, when changes in pH are induced by respiratory variations or with alkalosis, the impact on serum K+ concentration is less remarkable.15 Hence, it seems that it is the nature of the acid‐base disturbance that impacts K+ concentration more than the change in pH itself.
In the kidney, the main site for regulation of K+ balance is the collecting duct. Factors that affect elimination include urinary sodium delivery, urine flow, and aldosterone.16 In order to adequately eliminate K+ these factors must be optimized in conjunction.
Treatment of hyperkalemia includes the sequential administration of agents that stabilize the cardiac membrane (calcium gluconate), shift the potassium intracellularly (insulin, ‐2 agonists), and remove the potassium (diuretics, sodium polystyrene, or dialysis).
The use of sodium bicarbonate for treatment of hyperkalemia has been long advocated.17 It was thought to act by translocation of potassium hence could be used to quickly lower K+ concentration. However, this dogma has been challenged recently.
To assess the true impact of sodium bicarbonate on potassium translocation, studies have been conducted on anephric patients with hyperkalemia. Bicarbonate infusion failed to elicit a significant rapid change in serum K+ concentration despite increase in bicarbonate concentration, arguing against a translocation mechanism.1821 After 60 minutes of treatment, neither isotonic nor hypertonic bicarbonate infusion affected Serum K+ levels in end‐stage renal disease (ESRD) patients.19, 20 On the contrary, hypertonic sodium bicarbonate increased the K+ concentration after 180 minutes of treatment,20 and it took a prolonged infusion of 4 hours to see a significant decrease in K+ concentration (0.6 mmol/L); half of which could be accounted for solely by volume administration. Moreover, this reduction was highly variable.
Rather, sodium bicarbonate seems to enhance potassium elimination by increasing sodium delivery to the distal tubule, increasing urinary pH and negative luminal charge and potentiating the action of diuretics.23 In an elegant study on normovolemic patients, the induction of bicarbonaturia practically doubled potassium excretion.23 However, such an effect is heterogeneous and usually takes place over 4 hours to 6 hours.17
At the cellular level, 2 ion exchange pumps cooperate to handle Na/K/H movement across the cellular membrane: an Na+/H+ exchanger (NHE) and the Na+/K+ ATP‐ase pump (Figure 1). The NHE is normally inactive and is only upregulated in cases of severe intracellular acidosis.24 The infusion of sodium bicarbonate to patients with severe metabolic acidosis could possibly decrease the serum potassium concentration by translocation if the NHE was significantly upregulated. However, this treatment can be associated with a drop in the ionized calcium level, a worsening of the intracellular acidosis, and a decreased peripheral oxygen delivery.25 Thus, the benefits should be balanced with the potential adverse effects and, even in cases of severe metabolic acidosis with hyperkalemia, we would advise the clinician to restrictively administer sodium bicarbonate.
In addition, in ESRD patients, the administration of sodium bicarbonate can be problematic owing to the osmotic and volume burden it carries. It should also be avoided in patients who are volume overload or in those with decreased ability to eliminate potassium.
When treating hyperkalemic patients, hospitalists should use sodium bicarbonate to potentiate urinary elimination of potassium and should consider administering it either with acetazolamide or a loop diuretic, anticipating a lowering effect after a few hours.26 It should be avoided in patients with volume overload and anuria. Immediate translocation of potassium into cells is best achieved by insulin and ‐2 agonists.
5‐Oxoprolinuria: A Newly Recognized Cause of High Anion Gap Metabolic Acidosis
There are several causes of metabolic high anion gap acidosis in hospitalized patients. However, despite careful investigations, the cause of that disorder is not always apparent.27 Recently, 5‐oxoprolinuria (also called pyroglutamic acidosis) has become increasingly recognized as a potential etiology.2832
The metabolism of glutamate (though the ‐Glutamyl cycle) generates glutathione which provides negative feedback to the ‐Glutamyl‐cysteine synthetase enzyme. The depletion of glutathione increases 5‐oxoproline production owing to the loss of that inhibition (Figures 2 and 3). Low glutathione levels can be seen with liver disease,33 chronic alcohol intake,34 acetaminophen use,35 malnutrition,36 renal dysfunction,29 use of vigabatrim (an antiepileptic that received Food and Drug Administration [FDA] approval for use in April 2009)37 and sepsis.38 Most of the reported cases were female and had more than one risk factor.39
Typically, patients present with high anion gap acidosis (often more than 20)28 with normal acetaminophen levels and all usual tests being negative. A history of chronic acetaminophen use with or without other risk factors can frequently be found. The true independent impact of this type of acidosis on outcomes is difficult to determine as all of the reported cases had many confounding factors.
A urinary organic acid level is diagnostic and will reveal increased levels of pyroglutamic acid. Alternatively, the finding of a positive urinary anion gap (UNa +UK UCl) with a positive urinary osmolar gap (Uosmmeasured‐Uosmcalculated) in the appropriate clinical setting (unexplained high anion gap acidosis with negative workup and presence of risk factors for 5‐oxoproliniuria) can point towards the diagnosis.40
A study of patients with unexplained metabolic acidosis did not find any cases of 5‐oxoprolinuria.41 Although this might suggest that the incidence of this disease is low, very few of those patients were actually taking acetaminophen (therefore had a reduced propensity for developing pyroglutamic acidosis).41 Thus, the actual incidence of 5‐oxoprolinuria is hard to determine.
Once recognized, acetaminophen should be withheld and N‐acetylcysteine (NAC) can be used to replete glutathione levels although there is no convincing evidence for this use.42 It is important for hospitalists to be aware of this disorder as it can pose a diagnostic challenge (negative usual work‐up), is easy to treat by stopping acetaminophen, and can (possibly) negatively affect outcomes.
Furosemide and Patients With Sulfa Allergy
Allergic reactions are a common occurrence with sulfa‐containing antibiotics (SCA) and reports estimates the incidence to be approximately 3% to 5%.43, 44
One misbelief is that patients who are allergic to SCAs should not receive sulfa containing diuretics or other sulfa‐containing medications.45 This leads some physicians to substitute commonly used diuretics (such as furosemide or thiazides) for ethacrynic acid. The use of ethacrynic acid has several challenges: the limited supply of the intravenous form, the discontinuation of the oral form, the increased cost, and the risk of permanent ototoxicity.
The evidence for potential allergic cross‐reactivity among medications containing the sulfa moiety has been primarily derived from Case Reports.4649
The molecular structures of sulfamethoxazole and furosemide are shown below. The allergic antigen is most often the N1 component,45 and sometimes N4 but not the sulfa moiety. Both of the incriminated antigens are not present in the furosemide structure (as well as all other sulfa containing diuretics) (Figures 4 and 5).
Experimental data showed that serum from patients allergic to SCAs did not bind to diuretics.50 In addition, clinical reports failed to demonstrate cross‐reactivity.5153 In a large clinical trial, Strom et al.53 showed that although there was a higher risk for allergic reaction to sulfa containing medications (SCM) in patients allergic to SCA (compared to those who were not), it was lower among patients with an allergy to sulfa antibiotic than among patients with a history of hypersensitivity to penicillins, suggesting this was due to a predisposition to allergic reactions in general rather than true cross‐reactivity. In another report, patients who were receiving ethacrynic acid for many years were successfully and uneventfully switched to furosemide.54
Taken together, these findings suggest that there is no evidence for withholding sulfa nonantibiotics in patients allergic to sulfa containing antibiotics.
Conclusion
Hypothyroidism, unlike myxedema, is not a cause of hyponatremia (although it can be sometimes seen in conjunction with the latter) and additional investigations should be done to determine its etiology. Sodium bicarbonate is effective for treatment of hyperkalemia by enhancing renal potassium elimination, rather than from shifting potassium into cells. The 5‐oxoprolinuria is a newly recognized cause of high anion‐gap metabolic acidosis and should be considered in patients who have taken acetaminophen. Furosemide (and sulfa containing diuretics) can be used safely in patients with an allergy to SCA.
There are many controversial topics relating to renal disease in hospitalized patients. The aim of this review is to shed light on some important and often debated issues. We will first discuss topics related to electrolytes disorder commonly seen in hospitalized patients (hyponatremia, hyperkalemia, metabolic acidosis) then the use of diuretics in patients with allergy to sulfa containing antibiotics.
Hypothyroidism and Hyponatremia
Hyponatremia is common in hospitalized patients and is associated with worse outcomes.1 It can be seen with a variety of conditions ranging from congestive heart failure to volume depletion. Careful history and physical examination are paramount and the initial work‐up usually includes serum and urine osmolality and urine sodium concentration.
For euvolemic hyponatremia, the differential diagnosis includes the syndrome of inappropriate adenine dinucleotide (ADH) secretion (SIADH), hypoadrenalism, and beer potomania. Additionally, many authorities also include hypothyroidism.
Although the simultaneous finding of hypothyroidism and hyponatremia can occur in patients as both diseases are widely prevalent in the general population, causation has yet to be convincingly demonstrated.
ADH is released in response to effective volume depletion; consequently when hypothyroidism is encountered in the setting of complete pituitary failure there is often hyponatremia.2, 3 Alternatively, with myxedema, the ability of the kidney to handle a water load and concentrate urine can be impaired.4
However, the observation that thyroid hormone administration did not raise sodium values in newborns with congenital hypothyroidism or in adults supports the absence of causal effect.5, 6And in addition, large studies done in the hospital and outpatient setting showed no differences between the serum sodium values of hypothyroid patients and that of controls.7, 8 In the study of outpatients, among those with hypothyroidism, for every increase of 10 mU/L of thyroid‐stimulating hormone (TSH), there was a drop of only 0.14 mmol/L of Na concentration.8 Thus, the elevation of TSH required for a clinically meaningful drop in sodium to occur was considerable.
Hence in patients with hyponatremia, the hospitalist should look for etiologies other than hypothyroidism and should only consider thyroid hypofunction as a culprit in cases of myxedema, or panhypopituitarism.
Sodium Bicarbonate for Hyperkalemia
Hyperkalemia is one of the most feared electrolyte disorders encountered in hospitalized patients and can lead to dire outcomes.9, 10
Potassium (K+) homeostasis is maintained in the body by 2 complimentary systems: a short‐term system that regulates K+ variation by modifying translocation across the cellular membrane and a long‐term system that adjusts overall K+ balance. The translocation system is regulated primarily by insulin and ‐2 stimulation. Overall K+ balance is mainly controlled by the kidney (90‐95%) although the gastrointestinal (GI) tract can have a more preponderant role in anephric patients.
Hyperkalemia can ensue by either a dysregulation of the translocation system (as in diabetic Ketoacidosis secondary to insulin deficiency) or impairment of K+ elimination.
Acid‐base status was previously thought to have a prominent influence on K+ concentration, based on studies that demonstrated that. However, studies looking at metabolic acidosis revealed that contrary to the effect of mineral acidosis (excess of nonmetabolizable anions)11 where there is an inverse correlation between potassium concentration and pH; organic acidosis (excess of metabolizable anions)11 was not associated with hyperkalemia.1214 However, organic acidosis can be seen simultaneously and induced by a same underlying disease (such as organ ischemia with lactic acidosis or insulin deficiency complicated by ketoacidosis). Also, when changes in pH are induced by respiratory variations or with alkalosis, the impact on serum K+ concentration is less remarkable.15 Hence, it seems that it is the nature of the acid‐base disturbance that impacts K+ concentration more than the change in pH itself.
In the kidney, the main site for regulation of K+ balance is the collecting duct. Factors that affect elimination include urinary sodium delivery, urine flow, and aldosterone.16 In order to adequately eliminate K+ these factors must be optimized in conjunction.
Treatment of hyperkalemia includes the sequential administration of agents that stabilize the cardiac membrane (calcium gluconate), shift the potassium intracellularly (insulin, ‐2 agonists), and remove the potassium (diuretics, sodium polystyrene, or dialysis).
The use of sodium bicarbonate for treatment of hyperkalemia has been long advocated.17 It was thought to act by translocation of potassium hence could be used to quickly lower K+ concentration. However, this dogma has been challenged recently.
To assess the true impact of sodium bicarbonate on potassium translocation, studies have been conducted on anephric patients with hyperkalemia. Bicarbonate infusion failed to elicit a significant rapid change in serum K+ concentration despite increase in bicarbonate concentration, arguing against a translocation mechanism.1821 After 60 minutes of treatment, neither isotonic nor hypertonic bicarbonate infusion affected Serum K+ levels in end‐stage renal disease (ESRD) patients.19, 20 On the contrary, hypertonic sodium bicarbonate increased the K+ concentration after 180 minutes of treatment,20 and it took a prolonged infusion of 4 hours to see a significant decrease in K+ concentration (0.6 mmol/L); half of which could be accounted for solely by volume administration. Moreover, this reduction was highly variable.
Rather, sodium bicarbonate seems to enhance potassium elimination by increasing sodium delivery to the distal tubule, increasing urinary pH and negative luminal charge and potentiating the action of diuretics.23 In an elegant study on normovolemic patients, the induction of bicarbonaturia practically doubled potassium excretion.23 However, such an effect is heterogeneous and usually takes place over 4 hours to 6 hours.17
At the cellular level, 2 ion exchange pumps cooperate to handle Na/K/H movement across the cellular membrane: an Na+/H+ exchanger (NHE) and the Na+/K+ ATP‐ase pump (Figure 1). The NHE is normally inactive and is only upregulated in cases of severe intracellular acidosis.24 The infusion of sodium bicarbonate to patients with severe metabolic acidosis could possibly decrease the serum potassium concentration by translocation if the NHE was significantly upregulated. However, this treatment can be associated with a drop in the ionized calcium level, a worsening of the intracellular acidosis, and a decreased peripheral oxygen delivery.25 Thus, the benefits should be balanced with the potential adverse effects and, even in cases of severe metabolic acidosis with hyperkalemia, we would advise the clinician to restrictively administer sodium bicarbonate.
In addition, in ESRD patients, the administration of sodium bicarbonate can be problematic owing to the osmotic and volume burden it carries. It should also be avoided in patients who are volume overload or in those with decreased ability to eliminate potassium.
When treating hyperkalemic patients, hospitalists should use sodium bicarbonate to potentiate urinary elimination of potassium and should consider administering it either with acetazolamide or a loop diuretic, anticipating a lowering effect after a few hours.26 It should be avoided in patients with volume overload and anuria. Immediate translocation of potassium into cells is best achieved by insulin and ‐2 agonists.
5‐Oxoprolinuria: A Newly Recognized Cause of High Anion Gap Metabolic Acidosis
There are several causes of metabolic high anion gap acidosis in hospitalized patients. However, despite careful investigations, the cause of that disorder is not always apparent.27 Recently, 5‐oxoprolinuria (also called pyroglutamic acidosis) has become increasingly recognized as a potential etiology.2832
The metabolism of glutamate (though the ‐Glutamyl cycle) generates glutathione which provides negative feedback to the ‐Glutamyl‐cysteine synthetase enzyme. The depletion of glutathione increases 5‐oxoproline production owing to the loss of that inhibition (Figures 2 and 3). Low glutathione levels can be seen with liver disease,33 chronic alcohol intake,34 acetaminophen use,35 malnutrition,36 renal dysfunction,29 use of vigabatrim (an antiepileptic that received Food and Drug Administration [FDA] approval for use in April 2009)37 and sepsis.38 Most of the reported cases were female and had more than one risk factor.39
Typically, patients present with high anion gap acidosis (often more than 20)28 with normal acetaminophen levels and all usual tests being negative. A history of chronic acetaminophen use with or without other risk factors can frequently be found. The true independent impact of this type of acidosis on outcomes is difficult to determine as all of the reported cases had many confounding factors.
A urinary organic acid level is diagnostic and will reveal increased levels of pyroglutamic acid. Alternatively, the finding of a positive urinary anion gap (UNa +UK UCl) with a positive urinary osmolar gap (Uosmmeasured‐Uosmcalculated) in the appropriate clinical setting (unexplained high anion gap acidosis with negative workup and presence of risk factors for 5‐oxoproliniuria) can point towards the diagnosis.40
A study of patients with unexplained metabolic acidosis did not find any cases of 5‐oxoprolinuria.41 Although this might suggest that the incidence of this disease is low, very few of those patients were actually taking acetaminophen (therefore had a reduced propensity for developing pyroglutamic acidosis).41 Thus, the actual incidence of 5‐oxoprolinuria is hard to determine.
Once recognized, acetaminophen should be withheld and N‐acetylcysteine (NAC) can be used to replete glutathione levels although there is no convincing evidence for this use.42 It is important for hospitalists to be aware of this disorder as it can pose a diagnostic challenge (negative usual work‐up), is easy to treat by stopping acetaminophen, and can (possibly) negatively affect outcomes.
Furosemide and Patients With Sulfa Allergy
Allergic reactions are a common occurrence with sulfa‐containing antibiotics (SCA) and reports estimates the incidence to be approximately 3% to 5%.43, 44
One misbelief is that patients who are allergic to SCAs should not receive sulfa containing diuretics or other sulfa‐containing medications.45 This leads some physicians to substitute commonly used diuretics (such as furosemide or thiazides) for ethacrynic acid. The use of ethacrynic acid has several challenges: the limited supply of the intravenous form, the discontinuation of the oral form, the increased cost, and the risk of permanent ototoxicity.
The evidence for potential allergic cross‐reactivity among medications containing the sulfa moiety has been primarily derived from Case Reports.4649
The molecular structures of sulfamethoxazole and furosemide are shown below. The allergic antigen is most often the N1 component,45 and sometimes N4 but not the sulfa moiety. Both of the incriminated antigens are not present in the furosemide structure (as well as all other sulfa containing diuretics) (Figures 4 and 5).
Experimental data showed that serum from patients allergic to SCAs did not bind to diuretics.50 In addition, clinical reports failed to demonstrate cross‐reactivity.5153 In a large clinical trial, Strom et al.53 showed that although there was a higher risk for allergic reaction to sulfa containing medications (SCM) in patients allergic to SCA (compared to those who were not), it was lower among patients with an allergy to sulfa antibiotic than among patients with a history of hypersensitivity to penicillins, suggesting this was due to a predisposition to allergic reactions in general rather than true cross‐reactivity. In another report, patients who were receiving ethacrynic acid for many years were successfully and uneventfully switched to furosemide.54
Taken together, these findings suggest that there is no evidence for withholding sulfa nonantibiotics in patients allergic to sulfa containing antibiotics.
Conclusion
Hypothyroidism, unlike myxedema, is not a cause of hyponatremia (although it can be sometimes seen in conjunction with the latter) and additional investigations should be done to determine its etiology. Sodium bicarbonate is effective for treatment of hyperkalemia by enhancing renal potassium elimination, rather than from shifting potassium into cells. The 5‐oxoprolinuria is a newly recognized cause of high anion‐gap metabolic acidosis and should be considered in patients who have taken acetaminophen. Furosemide (and sulfa containing diuretics) can be used safely in patients with an allergy to SCA.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122(9):857–865.
- ,.Hyponatremia as the presenting manifestation of Sheehan's syndrome in elderly patients.Aging Clin Exp Res.2006;18(6):536–539.
- ,,.Sodium and water disturbances in patients with Sheehan's syndrome.Am J Kidney Dis.2001;38(3):E14.
- ,,,,,.Effect of acute water loading on plasma levels of antidiuretic hormone AVP aldosterone, ANP fractional excretion of sodium and plasma and urine osmolalities in myxedema.Chin Med J (Engl).1990;103(9):704–708.
- ,.Sodium handling in congenitally hypothyroid neonates.Acta Paediatr.2004;93(1):22–24.
- ,,.Prevalence and severity of hyponatremia and hypercreatininemia in short‐term uncomplicated hypothyroidism.J Endocrinol Invest.1999;22(1):35–39.
- ,,,,.Absence of relation between hyponatraemia and hypothyroidism.Lancet.1997;350(9088):1402.
- ,,.The effect of newly diagnosed hypothyroidism on serum sodium concentrations: a retrospective study.Clin Endocrinol (Oxf).2006;64(5):598–599.
- .Disorders of potassium homeostasis. Hypokalemia and hyperkalemia.Crit Care Clin.2002;18(2):273–288,vi.
- ,.Hyperkalemia in hospitalized patients.Int Urol Nephrol.2000;32(2):177–180.
- ,.Treatment of metabolic acidosis.Curr Opin Crit Care.2003;9(4):260–265.
- ,,,.Natural history of lactic acidosis after grand‐mal seizures. A model for the study of an anion‐gap acidosis not associated with hyperkalemia.N Engl J Med.1977;297(15):796–799.
- ,,,,.The plasma potassium concentration in metabolic acidosis: a re‐evaluation.Am J Kidney Dis.1988;11(3):220–224.
- ,,,.Determinants of plasma potassium levels in diabetic ketoacidosis.Medicine (Baltimore).1986;65(3):163–172.
- ,.Changes in plasma potassium concentration during acute acid‐base disturbances.Am J Med.1981;71(3):456–467.
- ,,.New aspects of renal potassium transport.Pflugers Arch.2003;446(3):289–297.
- ,.Correction of hyperkalemia by bicarbonate despite constant blood pH.Kidney Int.1977;12(5):354–360.
- ,,,.Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure.Am J Med.1988;85(4):507–512.
- ,.Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol.Am J Kidney Dis.1996;28(4):508–514.
- ,,,,.Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end‐stage renal disease.Miner Electrolyte Metab.1991;17(5):297–302.
- .Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end‐stage renal disease patients.Nephron.1996;72(3):476–482.
- ,,.Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure.Kidney Int.1992;41(2):369–374.
- ,,, et al.Modulation of the secretion of potassium by accompanying anions in humans.Kidney Int.1991;39(6):1206–1212.
- ,.Controversial issues in the treatment of hyperkalaemia.Nephrol Dial Transplant.2003;18(11):2215–2218.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- .Management of severe hyperkalemia.Crit Care Med.2008;36(12):3246–3251.
- ,.Pyroglutamic acid and high anion gap: looking through the keyhole?Crit Care Med.2000;28(6):2140–2141.
- ,,,,.Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen.Clin J Am Soc Nephrol.2006;1(3):441–447.
- ,,,.Pyroglutamic acidemia: a cause of high anion gap metabolic acidosis.Crit Care Med.2000;28(6):1803–1807.
- ,,,,,.Acetaminophen‐induced anion gap metabolic acidosis and 5‐oxoprolinuria (pyroglutamic aciduria) acquired in hospital.Am J Kidney Dis.2005;46(1):143–146.
- ,.Anion gap acidosis associated with acetaminophen.Ann Intern Med.2000;133(9):752–753.
- ,,.Pyroglutamic acidosis in a renal transplant patient.Nephrol Dial Transplant.2005;20(12):2836–2838.
- ,,.Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases.Life Sci.1988;43(12):991–998.
- ,,,,,.Decreased hepatic glutathione in chronic alcoholic patients.J Hepatol.1986;3(1):1–6.
- ,.Intracellular signaling mechanisms of acetaminophen‐induced liver cell death.Toxicol Sci.2006;89(1):31–41.
- ,,.Urinary excretion of 5‐L‐oxoproline (pyroglutamic acid) is increased during recovery from severe childhood malnutrition and responds to supplemental glycine.J Nutr.1996;126(11):2823–2830.
- ,,,.Pyroglutamicaciduria from vigabatrin.Lancet.1989;1(8652):1452–1453.
- ,,, et al.Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients.Crit Care Med.2001;29(4):870–877.
- ,.Transient 5‐oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects.Clin Chem.1998;44(7):1497–1503.
- ,,,.Guilty as charged: unmeasured urinary anions in a case of pyroglutamic acidosis.Neth J Med.2008;66(8):351–353.
- ,,.Unexplained metabolic acidosis in critically ill patients: the role of pyroglutamic acid.Intensive Care Med.2004;30(3):502–505.
- ,,.A therapeutic trial with N‐acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5‐oxoprolinuria).J Inherit Metab Dis.1989;12(2):120–130.
- .Diagnosis of allergic reactions to sulfonamides.Allergy.1999;54Suppl 58:28–32.
- .Practical issues in the management of hypersensitivity reactions: sulfonamides.South Med J.2001;94(8):817–824.
- ,,.Should celecoxib be contraindicated in patients who are allergic to sulfonamides? Revisiting the meaning of ‘sulfa’ allergy.Drug Saf.2001;24(4):239–247.
- .Thrombocytopenia due to sulfonamide cross‐sensitivity.Wis Med J.1982;81(6):21–23.
- ,,.Leukocytoclastic vasculitis induced by use of glyburide: a case of possible cross‐reaction of a sulfonamide and a sulfonylurea.Cutis.2003;71(3):235–238.
- ,.Celecoxib‐induced erythema multiforme with glyburide cross‐reactivity.Pharmacotherapy.2002;22(5):637–640.
- ,.Vesiculobullous rash in a patient with systemic lupus erythematosus.Ann Allergy.1993;70(3):196–203.
- ,,, et al.Use of optical biosensor technology to study immunological cross‐reactivity between different sulfonamide drugs.Anal Biochem.2002;300(2):177–184.
- ,,,.Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis.Adverse Drug React Toxicol Rev.1996;15(1):9–50.
- ,,.Cross‐reactivity in HIV‐infected patients switched from trimethoprim‐sulfamethoxazole to dapsone.Pharmacotherapy.1998;18(4):831–835.
- ,,, et al.Absence of cross‐reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics.N Engl J Med.2003;349(17):1628–1635.
- ,,.Furosemide challenge in patients with heart failure and adverse reactions to sulfa‐containing diuretics.Ann Intern Med.2003;138(4):358–359.
- ,,.Mortality after hospitalization with mild, moderate, and severe hyponatremia.Am J Med.2009;122(9):857–865.
- ,.Hyponatremia as the presenting manifestation of Sheehan's syndrome in elderly patients.Aging Clin Exp Res.2006;18(6):536–539.
- ,,.Sodium and water disturbances in patients with Sheehan's syndrome.Am J Kidney Dis.2001;38(3):E14.
- ,,,,,.Effect of acute water loading on plasma levels of antidiuretic hormone AVP aldosterone, ANP fractional excretion of sodium and plasma and urine osmolalities in myxedema.Chin Med J (Engl).1990;103(9):704–708.
- ,.Sodium handling in congenitally hypothyroid neonates.Acta Paediatr.2004;93(1):22–24.
- ,,.Prevalence and severity of hyponatremia and hypercreatininemia in short‐term uncomplicated hypothyroidism.J Endocrinol Invest.1999;22(1):35–39.
- ,,,,.Absence of relation between hyponatraemia and hypothyroidism.Lancet.1997;350(9088):1402.
- ,,.The effect of newly diagnosed hypothyroidism on serum sodium concentrations: a retrospective study.Clin Endocrinol (Oxf).2006;64(5):598–599.
- .Disorders of potassium homeostasis. Hypokalemia and hyperkalemia.Crit Care Clin.2002;18(2):273–288,vi.
- ,.Hyperkalemia in hospitalized patients.Int Urol Nephrol.2000;32(2):177–180.
- ,.Treatment of metabolic acidosis.Curr Opin Crit Care.2003;9(4):260–265.
- ,,,.Natural history of lactic acidosis after grand‐mal seizures. A model for the study of an anion‐gap acidosis not associated with hyperkalemia.N Engl J Med.1977;297(15):796–799.
- ,,,,.The plasma potassium concentration in metabolic acidosis: a re‐evaluation.Am J Kidney Dis.1988;11(3):220–224.
- ,,,.Determinants of plasma potassium levels in diabetic ketoacidosis.Medicine (Baltimore).1986;65(3):163–172.
- ,.Changes in plasma potassium concentration during acute acid‐base disturbances.Am J Med.1981;71(3):456–467.
- ,,.New aspects of renal potassium transport.Pflugers Arch.2003;446(3):289–297.
- ,.Correction of hyperkalemia by bicarbonate despite constant blood pH.Kidney Int.1977;12(5):354–360.
- ,,,.Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure.Am J Med.1988;85(4):507–512.
- ,.Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol.Am J Kidney Dis.1996;28(4):508–514.
- ,,,,.Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end‐stage renal disease.Miner Electrolyte Metab.1991;17(5):297–302.
- .Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end‐stage renal disease patients.Nephron.1996;72(3):476–482.
- ,,.Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure.Kidney Int.1992;41(2):369–374.
- ,,, et al.Modulation of the secretion of potassium by accompanying anions in humans.Kidney Int.1991;39(6):1206–1212.
- ,.Controversial issues in the treatment of hyperkalaemia.Nephrol Dial Transplant.2003;18(11):2215–2218.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- .Management of severe hyperkalemia.Crit Care Med.2008;36(12):3246–3251.
- ,.Pyroglutamic acid and high anion gap: looking through the keyhole?Crit Care Med.2000;28(6):2140–2141.
- ,,,,.Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen.Clin J Am Soc Nephrol.2006;1(3):441–447.
- ,,,.Pyroglutamic acidemia: a cause of high anion gap metabolic acidosis.Crit Care Med.2000;28(6):1803–1807.
- ,,,,,.Acetaminophen‐induced anion gap metabolic acidosis and 5‐oxoprolinuria (pyroglutamic aciduria) acquired in hospital.Am J Kidney Dis.2005;46(1):143–146.
- ,.Anion gap acidosis associated with acetaminophen.Ann Intern Med.2000;133(9):752–753.
- ,,.Pyroglutamic acidosis in a renal transplant patient.Nephrol Dial Transplant.2005;20(12):2836–2838.
- ,,.Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases.Life Sci.1988;43(12):991–998.
- ,,,,,.Decreased hepatic glutathione in chronic alcoholic patients.J Hepatol.1986;3(1):1–6.
- ,.Intracellular signaling mechanisms of acetaminophen‐induced liver cell death.Toxicol Sci.2006;89(1):31–41.
- ,,.Urinary excretion of 5‐L‐oxoproline (pyroglutamic acid) is increased during recovery from severe childhood malnutrition and responds to supplemental glycine.J Nutr.1996;126(11):2823–2830.
- ,,,.Pyroglutamicaciduria from vigabatrin.Lancet.1989;1(8652):1452–1453.
- ,,, et al.Cysteine metabolism and whole blood glutathione synthesis in septic pediatric patients.Crit Care Med.2001;29(4):870–877.
- ,.Transient 5‐oxoprolinuria and high anion gap metabolic acidosis: clinical and biochemical findings in eleven subjects.Clin Chem.1998;44(7):1497–1503.
- ,,,.Guilty as charged: unmeasured urinary anions in a case of pyroglutamic acidosis.Neth J Med.2008;66(8):351–353.
- ,,.Unexplained metabolic acidosis in critically ill patients: the role of pyroglutamic acid.Intensive Care Med.2004;30(3):502–505.
- ,,.A therapeutic trial with N‐acetylcysteine in subjects with hereditary glutathione synthetase deficiency (5‐oxoprolinuria).J Inherit Metab Dis.1989;12(2):120–130.
- .Diagnosis of allergic reactions to sulfonamides.Allergy.1999;54Suppl 58:28–32.
- .Practical issues in the management of hypersensitivity reactions: sulfonamides.South Med J.2001;94(8):817–824.
- ,,.Should celecoxib be contraindicated in patients who are allergic to sulfonamides? Revisiting the meaning of ‘sulfa’ allergy.Drug Saf.2001;24(4):239–247.
- .Thrombocytopenia due to sulfonamide cross‐sensitivity.Wis Med J.1982;81(6):21–23.
- ,,.Leukocytoclastic vasculitis induced by use of glyburide: a case of possible cross‐reaction of a sulfonamide and a sulfonylurea.Cutis.2003;71(3):235–238.
- ,.Celecoxib‐induced erythema multiforme with glyburide cross‐reactivity.Pharmacotherapy.2002;22(5):637–640.
- ,.Vesiculobullous rash in a patient with systemic lupus erythematosus.Ann Allergy.1993;70(3):196–203.
- ,,, et al.Use of optical biosensor technology to study immunological cross‐reactivity between different sulfonamide drugs.Anal Biochem.2002;300(2):177–184.
- ,,,.Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis.Adverse Drug React Toxicol Rev.1996;15(1):9–50.
- ,,.Cross‐reactivity in HIV‐infected patients switched from trimethoprim‐sulfamethoxazole to dapsone.Pharmacotherapy.1998;18(4):831–835.
- ,,, et al.Absence of cross‐reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics.N Engl J Med.2003;349(17):1628–1635.
- ,,.Furosemide challenge in patients with heart failure and adverse reactions to sulfa‐containing diuretics.Ann Intern Med.2003;138(4):358–359.
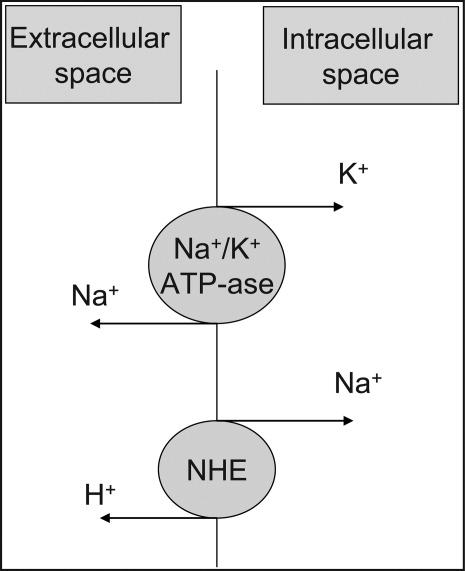
Treatment of Lactic Acidosis
Lactic acidosis (LA) is common in hospitalized patients and is associated with a high mortality.1, 2 Commonly, it is defined as a lactic acid concentration greater than 5 mmol/L with a pH less than 7.35.3 There are no evidence‐based guidelines for the treatment of LA despite progress in our understanding of its pathophysiology.36 This is not surprising, given the uncertainty regarding the impact of LA itself on clinical outcomes. In this regard, it is interesting to note that, despite its well‐recognized role as a marker of tissue hypoxia, lactate accumulation appears to have beneficial effects and may function as an adaptive mechanism. This raises the possibility that therapy directed at altering this adaptation may be detrimental. Pursuing correction of the pH in LA has been shown to have untoward physiologic effects. These and other ambiguities in the pathophysiology and treatment of LA are the focus of this review.
Lactate Metabolism
The body produces approximately 1400 mmol of lactate daily.7 Lactate is derived from the metabolism of pyruvate through an anaerobic reaction that occurs in all tissues (Figure 1). The liver is the primary site of lactate clearance and can metabolize up to 100 mmol per hour under normal conditions.8 There, lactate is converted to glucose to serve as an energy source during periods of hypoxia (Figure 2).9
Approximately 20% to 30% of the daily lactate load is metabolized by the kidneys.10, 11 Renal clearance is increased in acidosis12 and is maintained even in the presence of low renal perfusion.10, 12, 13 Renal lactate clearance is primarily through metabolism and not excretion.10, 14
LA Subtypes
Generally, lactic acid accumulation results from excess lactic acid production and not from reduced clearance.15 In cases of fulminant liver failure, it is due to a combination of decreased clearance and tissue hypoxia.16 In the setting of tissue hypoxia, an impairment of mitochondrial oxidative capacity results in the accumulation of pyruvate and generation of lactate. Lactic acid accumulation through this mechanism has historically been described as Type A LA.7 Hence, in critically ill patients lactate has traditionally been viewed as a marker of tissue hypoxia.15, 1721 Hyperlactatemia without tissue hypoxia has been referred to as type B LA. This is seen in a variety of circumstances. In sepsis, for example, several studies have shown lactic acid accumulation, despite adequate oxygen delivery.2224
Hyperlactatemia may also occur in cases of pure mitochondrial dysfunction, which can be induced by commonly prescribed medications such as the biguanides, nucleoside analog reverse‐transcriptase inhibitors (NRTIs), and linezolid.2527 Alternatively, lactate generation from metabolism of agents such as propylene glycol is possible. Finally, excessive lactate generation may occur following stress due to altered carbohydrate metabolism, or with respiratory alkalosis.2831
Lactate: A Metabolic Adaptation
Lactate was traditionally considered only as a marker of tissue hypoxia and anaerobic metabolism.17 This is certainly the case in situations of poor perfusion such as cardiogenic,15, 18 vasopressor‐resistant,19 or hypovolemic shock.20, 21
Alternative explanations for lactic acid accumulation, without tissue hypoperfusion, include catecholamine‐induced alterations in glycolysis,32, 33 mitochondrial disturbances,3436 and increased pyruvate production combined with increased glucose entry into cells.24, 37 In addition, the activity of an enzyme regulating lactate metabolism, pyruvate dehydrogenase kinase, increases in sepsis.38 This enzyme inactivates the pyruvate dehydrogenase (PDH) complex, which metabolizes pyruvate. Pyruvate and lactate may accumulate as a result. These changes partly explain the generation of LA in sepsis, independent of any effect of diminished tissue perfusion.
Recognizing the body's tendency toward homeostasis, it is appealing to speculate that lactate accumulation is adaptive.9 A number of findings support this. For example, lactate may act to shuttle energy between organs, or between cell types in the same organ. The astrocyteneuron lactate shuttle and the spermatogenic lactate shuttle are 2 examples of lactate's valuable effects on cellular metabolism.39 In the astrocyteneuron lactate shuttle, astrocytes support the increased metabolic demands of neurons through lactic acid production.40 Specifically, the neurotransmitter glutamate is released by the neurons and taken up by the astrocytes. Astrocytes produce lactate, which then moves back to the neuron to be used as an energy source. Glutamine, also released by the astrocytes, leads to the regeneration of glutamate and the potential to restart the cycle.39
Animal and human studies have suggested that, in periods of stress, lactate is the preferential energy substrate in the brain.4144 The usefulness of increased lactate production routinely seen in sepsis may thus represent multiple adaptive processes aimed primarily at improving the delivery of energy substrates. Thus, therapeutic strategies aimed specifically at lowering lactic acid levels may prove to have deleterious effects on cellular metabolism.
Impact of LA on Morbidity and Mortality
The poor prognosis in patients with LA is well recognized.2, 4548 For example, in a study of 126 patients with various causes of LA, the median survival was 38.5 hours and 30‐day survival was 17%.2
Studies have revealed that LA with low pH is associated with adverse effects on the cardiovascular system, particularly a decrease in cardiac contractility.49, 50 This effect is particularly prominent with a pH below 7.20. In contrast, acidosis in animal models has been shown to limit myocardial infarct size after reperfusion.51, 52 Variable effects of LA on cell death have been found. A worsening of apoptosis in myocytes has been noted;53 alternatively, protection from hypoxic injury in hepatocytes and myocardium has been observed.52, 54 Thus, although LA is associated with poor outcomes in human studies,2, 4547 it is still unclear to what extent lactic acid accumulation is a marker of severe illness, an independent effector of pathology, or a mechanism with the potential to serve a protective role.
Available data indicate that lactate itself is not harmful. Studies on infusion of lactate solutions to postoperative patients was shown to be safe.55 Also, the fact that lactate generation in states of respiratory alkalosis, stress, or altered carbohydrate metabolism without sepsis is not associated with worse outcomes supports the fact that lactic acid alone may not be maladaptive.2831
Similarly, low pH is not necessarily maladaptive. In the postictal state,56 diabetic ketoacidosis,57 spontaneous respiratory acidosis,58 or permissive hypercapnia,59 low blood pH is not deleterious.
In summary, LA is associated with poor outcomes, and indirect evidence suggests that it is the underlying causative condition rather than the low pH or the lactate that is responsible for the dire outcomes.
Treatment of LA with Sodium Bicarbonate
Since excessive lactic acid generation is accompanied by consumption of plasma bicarbonate and a fall in plasma pH, sodium bicarbonate has been long proposed as a treatment for LA. While theoretically appealing, this strategy has not been validated by studies in animals or humans. Indeed, bicarbonate administration in LA often has been shown to be detrimental.60, 61 The adverse effects of bicarbonate administration in LA, while initially paradoxical, have a number of possible explanations.
First, bicarbonate administration can induce a reduction in intracellular pH.60, 62, 63 The mechanism involves bicarbonate's effect to increase carbon dioxide (CO2) generation through mass action effect. Because the cell membrane is more permeable to CO2 than to bicarbonate, intracellular pH falls.64, 65 In sepsis, this intracellular/extracellular pH discrepancy may be more pronounced due to alterations in blood flow.66 Other reports on outcomes of intracellular pH with bicarbonate therapy show variable effects.6772
Second, to the extent that bicarbonate administration raises extracellular pH, it is associated with a reduction in ionized calcium concentration, since the binding of calcium to albumin is pH dependent.73 A sodium bicarbonate load administered to patients with LA was associated with a significant fall in ionized calcium concentration, whereas a sodium chloride load was not.1 This can affect cardiac function, as the latter varies proportionally with calcium levels.74
Third, bicarbonate administration may reduce tissue oxygen delivery since the affinity of hemoglobin for oxygen increases as pH rises (Bohr effect).75 The administration of bicarbonate worsened systemic oxygen consumption in one study76 and decreased oxygen delivery in another.75
Fourth, bicarbonate administration may indirectly increase intracellular calcium concentration. Low intracellular pH (see above) stimulates proton efflux by way of proton transporters and exchangers, increasing intracellular sodium content.77 A high cell sodium content then may increase intracellular calcium, through the Na/Ca exchanger, impairing cellular function.7779 Compounding this, the reduced function of the Na/H ATPase as a regulator of intracellular sodium in sepsis may not be adequate to limit cell swelling.77
Against this background of mechanistic concerns with the use of bicarbonate treatment, it is not surprising that clinical outcomes have been inconsistent at best. In animal models of LA, the use of sodium bicarbonate has either negative effects on cardiac output60, 72 or no significant hemodynamic effect when compared to sodium chloride infusion.67, 80, 81 One animal study did show some benefit with sodium bicarbonate compared to saline, though all animals subsequently died.50
In humans, sodium bicarbonate was studied in 2 randomized trials of sepsis‐induced LA.1, 82 In a study by Cooper et al.,1 14 critically‐ill patients received sequential infusions of sodium bicarbonate or sodium chloride. Neither solution was superior to the other in terms of hemodynamic improvement. No benefit was noted even when analysis was limited to those with very low pH (7.2). Mathieu et al.82 randomized 10 critically‐ill patients to sequential infusion of either sodium bicarbonate or sodium chloride. Similarly, no significant difference in hemodynamic variables was noted.
When taken together, these studies evaluating sodium bicarbonate in LA fail to show convincing benefit and raise serious questions about its detrimental effects. Extracellular pH may be a misleading marker of success in the treatment of LA, given its direct influence by sodium bicarbonate administration.
Treatment of LA and Use of Other Buffers
Other buffers (Carbicarb, dichloroacetate, and tromethamine [THAM]) have been studied for treatment of LA. Human studies have not shown superiority of any of the buffers as far as improving pH,83, 84 hemodynamics, or survival.85
Treatment of LA by Renal Replacement Therapy
Renal replacement therapy (RRT; dialysis and its variants) has been studied for the treatment of severe acidosis. RRT has a number of theoretical advantages over purely medical therapies in the treatment of LA: it can deliver large quantities of base without contributing to volume overload; it can directly remove lactate from the plasma; and it can mitigate the effect of alkalinization on ionized calcium concentration by delivering calcium.
In critically ill patients with intact liver function, continuous venovenous hemofiltration (CVVH) appears to contribute very little (less then 3%) to overall lactate clearance.86 While outcome studies are limited, continuous dialysis modalities consistently show improved resolution of acidosis of various types when compared to intermittent modalities.87, 88 As described above, this is related to base administration and is not a surprising finding. There are no studies comparing RRT and medical therapy with respect to clinical outcomes in patients with LA.
Special Situations
Biguanides
Biguanide‐induced LA can be due to impairment of hepatic neoglucogenesis, in the case of metformin, or increasing hepatic oxidative phosphorylation, in the case of phenformin.89 This infrequent complication90, 91 is associated with a high mortality.92 Proposed therapy has included the use of sodium bicarbonate infusion.93 In this setting, it is unclear if the use of bicarbonate alone improves clinical outcomes.94
Renal replacement therapy in a wide variety of formats has been used to treat this condition.93, 95101 Metformin has a high clearance during dialysis due to its low molecular weight and lack of protein binding.97, 98, 102 Nonetheless, its high volume of distribution suggests a longer dialysis time would be more beneficial if the main goal is reducing metformin levels.97, 103 The limited prospective literature and lack of conclusive evidence about what levels of metformin induce LA makes generalized recommendations about duration of hemodialysis purely speculative.104
NRTIs
The use of NRTIs is associated with LA due to impairment of mitochondrial oxidative phosphorylation.105108 This uncommon complication, if not recognized early, is associated with a high mortality.101, 109 Investigations are ongoing into agents directed at improving mitochondrial function such as riboflavin, thiamine, and L‐carnitine.110112 As with biguanide‐associated LA, RRT decisions should be individualized based on metabolic circumstances.
Lorazepam
Many intravenous medications are formulated in the alcohol solvent, propylene glycol. Injectable lorazepam has the highest proportional amount of propylene glycol compared with other commonly used agents.113, 114 The kidney normally eliminates 12% to 50% of administered propylene glycol via proximal tubule secretion.115 The remainder is metabolized by the liver to form pyruvate and lactate.114, 116, 117
When propylene glycol accumulates, as in cases of reduced renal function, it results in hyperosmolarity, LA, and can even induce additional kidney injury (probably through proximal tubular cell necrosis).118
LA due to propylene glycol has been reported by many authors and its incidence with high dose intravenous (IV) lorazepam has been estimated to be as high as 19%.114, 116, 119, 120 This disorder can frequently go unrecognized, as many other factors that induce LA often coincide in such patients. But when identified and promptly addressed, its prognosis seems to be favorable.114
The best treatment is prevention, by avoiding the use of IV lorazepam in patients with impaired renal function. Once it is recognized, the drug should be promptly withdrawn. In addition, removal by hemodialysis can quickly lower propylene glycol levels since it is a small, highly water soluble, non‐protein‐bound molecule.121 As no rebound in the level is expected, intermittent dialysis should be an acceptable modality.117
Linezolid
Recently, Gram‐positive bacteria in general and methicillin‐resistant Staphylococcus aureus in particular have emerged as major causes of nosocomial and community‐acquired infections. Linezolid, an oxazolidinone, is increasingly used to treat such infections. Several cases of LA have been associated with linezolid.27, 122, 123 and a survey of the Infectious Diseases Society of America (IDSA) Emerging Infections Network members revealed that this complication was commonly encountered.124 Linezolid causes LA by mitochondrial toxicity125, 126 and risk factors include prolonged exposure and older age. Once the disorder is recognized, the clinician should stop the drug immediately. Chemistries should be monitored frequently in patients on long‐term therapy.
Conclusions
Many studies note the association between LA and adverse outcomes.2, 4547 Though metabolic acidosis from elevated lactate levels may negatively affect organ function, the evidence supporting therapy specifically aimed at increasing pH in these settings is consistently poor.3, 127 Limitations have included small numbers of subjects,1, 82 variable outcomes studied, and the inability to assess intracellular metabolic stability.1, 61 When taking these factors into account it is hard to justify aggressive treatment of LA with mechanisms aimed at raising pH. Literature on the treatment of patients with LA and very low pH (below 7.2) is even more limited.
Moreover, lactate elevations may not represent tissue hypoperfusion. Lactate may have an important role in improving energy metabolism. This represents 1 additional reason to be hesitant when attempting to normalize pH in LA; we may be disrupting the body's physiologic response to sepsis. A conflict for clinicians emerges, however, as lactate is often used to define tissue ischemia. Obviously, more specific markers of tissue hypoperfusion would be ideal.
Bicarbonate therapy is an understandably attractive means to improve the acidemia, but there are serious mechanistic concerns with it use. Moreover, neither animal nor human studies, limited as they may be, show a convincing benefit. LA in the setting of acute kidney injury may be best treated with renal replacement therapy with bicarbonate‐based buffers, but controlled trials are lacking.
A number of commonly used drugs can cause LA. A heightened awareness on the part of clinicians will lead to prompt recognition of these cases, and timely treatment.
- ,,,.Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study.Ann Intern Med.1990;112(7):492–498.
- ,,, et al.Natural history and course of acquired lactic acidosis in adults. DCA‐Lactic Acidosis Study Group.Am J Med.1994;97(1):47–54.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- ,.Management of life‐threatening acid‐base disorders. First of two parts.N Engl J Med.1998;338(1):26–34.
- .Indications for use of bicarbonate in patients with metabolic acidosis.Br J Anaesth.1991;67(2):165–177.
- ,.Bench‐to‐bedside review: treating acid‐base abnormalities in the intensive care unit—the role of buffers.Crit Care.2004;8(4):259–265.
- .Lactic acidosis update for critical care clinicians.J Am Soc Nephrol.2001;12(suppl 17):S15–S19.
- ,.Lactic acidosis: diagnosis and treatment.Clin Endocrinol Metab.1980;9(3):513–541.
- .Lactate and shock state: the metabolic view.Curr Opin Crit Care.2006;12(4):315–321.
- .Bench‐to‐bedside review: lactate and the kidney.Crit Care.2002;6(4):322–326.
- ,,,.The influence of renal function on lactate and glucose metabolism.Biochem J.1984;219(1):73–78.
- ,.The effect of acidosis on lactate removal by the perfused rat kidney.Clin Sci Mol Med.1976;50(3):185–194.
- ,,.Transvisceral lactate fluxes during early endotoxemia.Chest.1996;110(1):198–204.
- ,,,,.Metabolism of lactate by the intact functioning kidney of the dog.Am J Physiol.1973;224(6):1463–1467.
- ,,, et al.Lactate and glucose metabolism in severe sepsis and cardiogenic shock.Crit Care Med.2005;33(10):2235–2240.
- ,,,, et al.Lactic acidosis in fulminant hepatic failure. Some aspects of pathogenesis and prognosis.J Hepatol.1985;1(4):405–416.
- ,.Lactic acidosis in critical illness.Crit Care Med.1992;20(1):80–93.
- ,,, et al.Effects of cardiogenic shock on lactate and glucose metabolism after heart surgery.Crit Care Med.2000;28(12):3784–3791.
- ,,,,,.Evolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine‐treated septic shock.Crit Care Med.2000;28(1):114–119.
- ,,, et al.Oxygen debt and metabolic acidemia as quantitative predictors of mortality and the severity of the ischemic insult in hemorrhagic shock.Crit Care Med.1991;19(2):231–243.
- ,,, et al.Early goal‐directed therapy in the treatment of severe sepsis and septic shock.N Engl J Med.2001;345(19):1368–1377.
- ,,,.Skeletal muscle partial pressure of oxygen in patients with sepsis.Crit Care Med.1994;22(4):640–650.
- ,.Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis.JAMA.1992;267(11):1503–1510.
- ,,,.Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability.Ann Surg.1996;224(1):97–102.
- ,,,.Biguanide‐associated lactic acidosis. Case report and review of the literature.Arch Intern Med.1992;152(11):2333–2336.
- ,,,.Bench‐to‐bedside review: severe lactic acidosis in HIV patients treated with nucleoside analogue reverse transcriptase inhibitors.Crit Care.2003;7(3):226–232.
- ,,,.Lactic acidosis after treatment with linezolid.Infection.2007;35(4):278–281.
- ,,,.Isotopic evaluation of the metabolism of pyruvate and related substrates in normal adult volunteers and severely burned children: effect of dichloroacetate and glucose infusion.Surgery.1991;110(1):54–67.
- .Alterations in carbohydrate metabolism during stress: a review of the literature.Am J Med.1995;98(1):75–84.
- .,,,.Lactic acid kinetics in respiratory alkalosis.Crit Care Med.1991;19(9):1120–1124.
- .Significance of hyperlactatemia without acidosis during hypermetabolic stress.Crit Care Med.1997;25(11):1780–1781.
- ,,,.Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis.Lancet.1999;354(9177):505–508.
- ,,,,.Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study.Lancet.2005;365(9462):871–875.
- ,.Sepsis induces diaphragm electron transport chain dysfunction and protein depletion.Am J Respir Crit Care Med.2005;172(7):861–868.
- ,,, et al.Association between mitochondrial dysfunction and severity and outcome of septic shock.Lancet.2002;360(9328):219–223.
- ,.Mitochondrial dysfunction in sepsis.Biochem Soc Symp.1999;66:149–166.
- ,,, et al.Altered glucose transporter mRNA abundance in a rat model of endotoxic shock.Biochem Biophys Res Commun.1991;176(1):535–540.
- .Increased pyruvate dehydrogenase kinase activity in response to sepsis.Am J Physiol.1991;260(5 Pt 1):E669–E674.
- .Lactate metabolism: a new paradigm for the third millennium.J Physiol.2004;558(Pt 1):5–30.
- ,,, et al.Evidence supporting the existence of an activity‐dependent astrocyte‐neuron lactate shuttle.Dev Neurosci.1998;20(4–5):291–299.
- ,,,.Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: further in vitro validation.J Neurochem.1997;69(1):423–426.
- .Bench‐to‐bedside review: a possible resolution of the glucose paradox of cerebral ischemia.Crit Care.2002;6(4):330–334.
- ,,,,,.Intravenous lactate prevents cerebral dysfunction during hypoglycaemia in insulin‐dependent diabetes mellitus.Clin Sci (Lond).1998;94(2):157–163.
- ,,,.Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study.Brain Res.1997;744(1):105–111.
- ,,,,,.Clinical prognostic markers in patients with severe sepsis: a prospective analysis of 139 consecutive cases.J Infect.2003;47(4):300–306.
- ,,, et al.Serum lactate as a predictor of mortality in patients with infection.Intensive Care Med.2007;33(6):970–977.
- ,,,.Lactate versus non‐lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients.Crit Care.2006;10(1):R22.
- .Abnormal resting blood lactate. I. The significance of hyperlactatemia in hospitalized patients.Am J Med.1961;30:840–848.
- ,,,,.Effect of lactic acidosis on canine hemodynamics and left ventricular function.Am J Physiol.1990;258(4 Pt 2):H1193–H1199.
- ,,,.Alkali therapy extends the period of survival during hypoxia: studies in rats.Am J Physiol.1996;271(2 Pt 2):R381–R387.
- ,,.Acidosis during early reperfusion prevents myocardial stunning in perfused ferret hearts.J Clin Invest.1988;82(3):920–927.
- ,,, et al.Effect of acidotic blood reperfusion on reperfusion injury after coronary artery occlusion in the dog heart.J Cardiovasc Pharmacol.1998;31(2):179–186.
- ,,,.Hypoxia and acidosis activate cardiac myocyte death through the Bcl‐2 family protein BNIP3.Proc Natl Acad Sci U S A.2002;99(20):12825–12830.
- ,,,,,.Extracellular acidosis delays onset of cell death in ATP‐depleted hepatocytes.Am J Physiol.1988;255(3 Pt 1):C315–C322.
- ,.Metabolic and hemodynamic effects of hypertonic solutions: sodium‐lactate versus sodium chloride infusion in postoperative patients.Shock.2002;18(4):306–310.
- .Medical complications of status epilepticus.Adv Neurol.1983;34:395–398.
- ,,,,,.Bicarbonate therapy in severe diabetic ketoacidosis. A double blind, randomized, placebo controlled trial.Rev Invest Clin.1991;43(3):234–238.
- ,,.Supercarbia in children: clinical course and outcome.Crit Care Med.1990;18(2):166–168.
- ,,,.Low mortality rate in adult respiratory distress syndrome using low‐volume, pressure‐limited ventilation with permissive hypercapnia: a prospective study.Crit Care Med.1994;22(10):1568–1578.
- ,,,.Systemic effects of NaHCO3 in experimental lactic acidosis in dogs.Am J Physiol.1982;242(6):F586–F591.
- ,,,,.Lactic acidosis: effect of treatment on intracellular pH and energetics in living rat heart.Am J Physiol.1992;262(5 Pt 2):H1572–H1578.
- ,,,,,.Effect of sodium bicarbonate on intracellular pH under different buffering conditions.Kidney Int.1996;49(5):1262–1267.
- ,,.Hemodynamic and hepatic pH responses to sodium bicarbonate and Carbicarb during systemic acidosis.Magn Reson Med.1990;16(3):403–410.
- ,,, et al.The increase in CO2 production induced by NaHCO3 depends on blood albumin and hemoglobin concentrations.Intensive Care Med.2000;26(5):558–564.
- ,,, et al.Initial effect of sodium bicarbonate on intracellular pH depends on the extracellular nonbicarbonate buffering capacity.Crit Care Med.2001;29(5):1033–1039.
- ,,,,,.Dynamic study of the distribution of microcirculatory blood flow in multiple splanchnic organs in septic shock.Crit Care Med.2000;28(9):3233–3241.
- ,,,,,.The effects of sodium bicarbonate and a mixture of sodium bicarbonate and carbonate (“Carbicarb”) on skeletal muscle pH and hemodynamic status in rats with hypovolemic shock.Metabolism.1994;43(4):518–522.
- ,,, et al.Haemodynamic and metabolic effects in diabetic ketoacidosis in rats of treatment with sodium bicarbonate or a mixture of sodium bicarbonate and sodium carbonate.Diabetologia.1995;38(8):889–898.
- ,,,,,.Effects of sodium bicarbonate on striated muscle metabolism and intracellular pH during endotoxic shock.Shock.1994;1(3):196–200.
- ,,,,.Carbicarb, sodium bicarbonate, and sodium chloride in hypoxic lactic acidosis. Effect on arterial blood gases, lactate concentrations, hemodynamic variables, and myocardial intracellular pH.Chest.1993;104(3):913–918.
- ,.Improved hemodynamic function during hypoxia with Carbicarb, a new agent for the management of acidosis.Circulation.1988;77(1):227–233.
- ,,.Metabolic effects of sodium bicarbonate in hypoxic lactic acidosis in dogs.Am J Physiol.1985;249(5 Pt 2):F630–F635.
- .Binding of calcium to serum albumin. II. Effect of pH via competitive hydrogen and calcium ion binding to the imidazole groups of albumin.Scand J Clin Lab Invest.1972;29(1):75–83.
- ,,,,.Left ventricular contractility varies directly with blood ionized calcium.Ann Intern Med.1988;108(4):524–529.
- ,,.Regulatory mechanisms of hemoglobin oxygen affinity in acidosis and alkalosis.J Clin Invest.1971;50(3):700–706.
- ,,.Metabolic and hemodynamic consequences of sodium bicarbonate administration in patients with heart disease.Am J Med.1989;87(1):7–14.
- ,,.Sodium ion/hydrogen ion exchange inhibition: a new pharmacologic approach to myocardial ischemia and reperfusion injury.J Clin Pharmacol.1998;38(10):887–897.
- ,.SEA0400: a novel sodium‐calcium exchange inhibitor with cardioprotective properties.Cardiovasc Drug Rev.2004;22(4):334–347.
- ,,,.Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart.Circ Res.1987;60(5):700–707.
- ,.Acute haemodynamic effects of sodium bicarbonate administration in respiratory and metabolic acidosis in anaesthetized dogs.Anaesth Intensive Care.1997;25(6):615–620.
- ,,,.Bicarbonate does not increase left ventricular contractility during L‐lactic acidemia in pigs.Am Rev Respir Dis.1993;148(2):317–322.
- ,,,,.Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study.Crit Care Med.1991;19(11):1352–1356.
- ,,, et al.Safety and efficacy of intravenous Carbicarb in patients undergoing surgery: comparison with sodium bicarbonate in the treatment of mild metabolic acidosis. SPI Research Group. Study of Perioperative Ischemia.Crit Care Med.1994;22(10):1540–1549.
- ,,, et al.Sodium bicarbonate versus THAM in ICU patients with mild metabolic acidosis.J Nephrol.2005;18(3):303–307.
- ,,, et al.A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate‐Lactic Acidosis Study Group.N Engl J Med.1992;327(22):1564–1569.
- ,,,,,.Effect of continuous venovenous hemofiltration with dialysis on lactate clearance in critically ill patients.Crit Care Med.1997;25(1):58–62.
- ,,,,.A pilot randomised controlled comparison of continuous veno‐venous haemofiltration and extended daily dialysis with filtration: effect on small solutes and acid‐base balance.Intensive Care Med.2007;33(5):830–835.
- ,,.Intermittent versus continuous renal replacement therapy in the ICU: impact on electrolyte and acid‐base balance.Intensive Care Med.2001;27(6):1037–1043.
- ,,,.Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus: systematic review and meta‐analysis.Arch Intern Med.2003;163(21):2594–2602.
- ,,,,.Severe acidosis in patients taking metformin—rapid reversal and survival despite high APACHE score.Diabet Med.2006;23(4):432–435.
- ,,.Incidence of lactic acidosis in metformin users.Diabetes Care.1999;22(6):925–927.
- ,.Lactic acidosis in metformin‐treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations.Drug Saf.1999;20(4):377–384.
- ,,,.The management of metformin overdose.Anaesthesia.1998;53(7):698–701.
- ,,.Lactic acidosis in biguanide‐treated diabetics: a review of 330 cases.Diabetologia.1978;14(2):75–87.
- ,,,,.Treatment of metformin‐associated lactic acidosis with closed recirculation bicarbonate‐buffered hemodialysis.Arch Intern Med.1984;144(1):203–205.
- ,,,.High anion gap metabolic acidosis in suicide: don't forget metformin intoxication—two patients' experiences.Ren Fail.2002;24(5):671–675.
- ,,, et al.Hemodialysis in the treatment of lactic acidosis in diabetics treated by metformin: a study of metformin elimination.Int J Clin Pharmacol Ther Toxicol.1989;27(6):285–288.
- ,,.Bicarbonate haemodialysis as a treatment of metformin overdose.Nephrol Dial Transplant.1997;12(5):1046–1047.
- ,,,.Combination of intermittent haemodialysis and high‐volume continuous haemofiltration for the treatment of severe metformin‐induced lactic acidosis.Nephrol Dial Transplant.2004;19(8):2157–2158.
- ,,,.When a friend can become an enemy! Recognition and management of metformin‐associated lactic acidosis.Kidney Int.2007;72(9):1157–1160.
- .,, et al.Severe nucleoside‐associated lactic acidosis in human immunodeficiency virus‐infected patients: report of 12 cases and review of the literature.Clin Infect Dis.2002;34(6):838–846.
- ,,.Clearance of metformin by hemofiltration in overdose.J Toxicol Clin Toxicol.2002;40(2):177–180.
- .Metformin‐associated lactic acidosis.J Emerg Med.2001;20(3):267–272.
- ,.Contraindications to use of metformin. Blanket banning of metformin two days before surgery may not be a good idea.BMJ.2003;326(7392):762; author reply 762.
- ,,, et al.Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B.N Engl J Med.1995;333(17):1099–1105.
- ,,,.Zidovudine‐induced fatal lactic acidosis and hepatic failure in patients with acquired immunodeficiency syndrome: report of two patients and review of the literature.Crit Care Med.1997;25(8):1425–1430.
- ,.Mitochondrial toxicity of antiviral drugs.Nat Med.1995;1(5):417–422.
- ,.Mitochondrial toxicity of nucleoside analogue reverse transcriptase inhibitors: a looming obstacle for long‐term antiretroviral therapy?Curr Opin Infect Dis.2000;13(1):5–11.
- ,,, et al.Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society‐USA panel.JAMA.2006;296(7):827–843.
- ,,.Riboflavin to treat nucleoside analogue‐induced lactic acidosis.Lancet.1998;352(9124):291–292.
- ,,,,,.Severe lactic acidosis and thiamine administration in an HIV‐infected patient on HAART.Int J STD AIDS.2001;12(6):407–409.
- ,,, et al.Detecting life‐threatening lactic acidosis related to nucleoside‐analog treatment of human immunodeficiency virus‐infected patients, and treatment with L‐carnitine.Crit Care Med.2003;31(4):1042–1047.
- ,.Hyperosmolar metabolic acidosis and intravenous Lorazepam.N Engl J Med.2002;347(11):857–858; author reply 857–858.
- ,,,.Propylene glycol toxicity: a severe iatrogenic illness in ICU patients receiving IV benzodiazepines: a case series and prospective, observational pilot study.Chest.2005;128(3):1674–1681.
- ,,, et al.Propylene glycol pharmacokinetics and effects after intravenous infusion in humans.Ther Drug Monit.1987;9(3):255–258.
- .Short‐term lorazepam infusion and concern for propylene glycol toxicity: case report and review.Pharmacotherapy.2001;21(9):1140–1144.
- ,,.Recognition, treatment, and prevention of propylene glycol toxicity.Semin Dial.2007;20(3):217–219.
- ,,.Propylene glycol‐mediated cell injury in a primary culture of human proximal tubule cells.Toxicol Sci.1998;46(2):410–417.
- ,.Osmolar gap metabolic acidosis in a 60‐year‐old man treated for hypoxemic respiratory failure.Chest.2000;118(2):545–546.
- ,,,.Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults.Crit Care Med.2004;32(8):1709–1714.
- ,,,.Removal of propylene glycol and correction of increased osmolar gap by hemodialysis in a patient on high dose lorazepam infusion therapy.Intensive Care Med.2002;28(1):81–84.
- ,,.Linezolid use associated with lactic acidosis.Scand J Infect Dis.2005;37(2):153–154.
- ,.Linezolid‐induced lactic acidosis.N Engl J Med.2003;348(1):86–87.
- ,,.Toxicity of extended courses of linezolid: results of an Infectious Diseases Society of America Emerging Infections Network survey.Diagn Microbiol Infect Dis.2008;62(4):407–410.
- ,,.Mitochondrial toxicity associated with linezolid.N Engl J Med.2005;353(21):2305–2306.
- ,,, et al.Linezolid‐induced inhibition of mitochondrial protein synthesis.Clin Infect Dis.2006;42(8):1111–1117.
- ,.Use of base in the treatment of severe acidemic states.Am J Kidney Dis.2001;38(4):703–727.
Lactic acidosis (LA) is common in hospitalized patients and is associated with a high mortality.1, 2 Commonly, it is defined as a lactic acid concentration greater than 5 mmol/L with a pH less than 7.35.3 There are no evidence‐based guidelines for the treatment of LA despite progress in our understanding of its pathophysiology.36 This is not surprising, given the uncertainty regarding the impact of LA itself on clinical outcomes. In this regard, it is interesting to note that, despite its well‐recognized role as a marker of tissue hypoxia, lactate accumulation appears to have beneficial effects and may function as an adaptive mechanism. This raises the possibility that therapy directed at altering this adaptation may be detrimental. Pursuing correction of the pH in LA has been shown to have untoward physiologic effects. These and other ambiguities in the pathophysiology and treatment of LA are the focus of this review.
Lactate Metabolism
The body produces approximately 1400 mmol of lactate daily.7 Lactate is derived from the metabolism of pyruvate through an anaerobic reaction that occurs in all tissues (Figure 1). The liver is the primary site of lactate clearance and can metabolize up to 100 mmol per hour under normal conditions.8 There, lactate is converted to glucose to serve as an energy source during periods of hypoxia (Figure 2).9
Approximately 20% to 30% of the daily lactate load is metabolized by the kidneys.10, 11 Renal clearance is increased in acidosis12 and is maintained even in the presence of low renal perfusion.10, 12, 13 Renal lactate clearance is primarily through metabolism and not excretion.10, 14
LA Subtypes
Generally, lactic acid accumulation results from excess lactic acid production and not from reduced clearance.15 In cases of fulminant liver failure, it is due to a combination of decreased clearance and tissue hypoxia.16 In the setting of tissue hypoxia, an impairment of mitochondrial oxidative capacity results in the accumulation of pyruvate and generation of lactate. Lactic acid accumulation through this mechanism has historically been described as Type A LA.7 Hence, in critically ill patients lactate has traditionally been viewed as a marker of tissue hypoxia.15, 1721 Hyperlactatemia without tissue hypoxia has been referred to as type B LA. This is seen in a variety of circumstances. In sepsis, for example, several studies have shown lactic acid accumulation, despite adequate oxygen delivery.2224
Hyperlactatemia may also occur in cases of pure mitochondrial dysfunction, which can be induced by commonly prescribed medications such as the biguanides, nucleoside analog reverse‐transcriptase inhibitors (NRTIs), and linezolid.2527 Alternatively, lactate generation from metabolism of agents such as propylene glycol is possible. Finally, excessive lactate generation may occur following stress due to altered carbohydrate metabolism, or with respiratory alkalosis.2831
Lactate: A Metabolic Adaptation
Lactate was traditionally considered only as a marker of tissue hypoxia and anaerobic metabolism.17 This is certainly the case in situations of poor perfusion such as cardiogenic,15, 18 vasopressor‐resistant,19 or hypovolemic shock.20, 21
Alternative explanations for lactic acid accumulation, without tissue hypoperfusion, include catecholamine‐induced alterations in glycolysis,32, 33 mitochondrial disturbances,3436 and increased pyruvate production combined with increased glucose entry into cells.24, 37 In addition, the activity of an enzyme regulating lactate metabolism, pyruvate dehydrogenase kinase, increases in sepsis.38 This enzyme inactivates the pyruvate dehydrogenase (PDH) complex, which metabolizes pyruvate. Pyruvate and lactate may accumulate as a result. These changes partly explain the generation of LA in sepsis, independent of any effect of diminished tissue perfusion.
Recognizing the body's tendency toward homeostasis, it is appealing to speculate that lactate accumulation is adaptive.9 A number of findings support this. For example, lactate may act to shuttle energy between organs, or between cell types in the same organ. The astrocyteneuron lactate shuttle and the spermatogenic lactate shuttle are 2 examples of lactate's valuable effects on cellular metabolism.39 In the astrocyteneuron lactate shuttle, astrocytes support the increased metabolic demands of neurons through lactic acid production.40 Specifically, the neurotransmitter glutamate is released by the neurons and taken up by the astrocytes. Astrocytes produce lactate, which then moves back to the neuron to be used as an energy source. Glutamine, also released by the astrocytes, leads to the regeneration of glutamate and the potential to restart the cycle.39
Animal and human studies have suggested that, in periods of stress, lactate is the preferential energy substrate in the brain.4144 The usefulness of increased lactate production routinely seen in sepsis may thus represent multiple adaptive processes aimed primarily at improving the delivery of energy substrates. Thus, therapeutic strategies aimed specifically at lowering lactic acid levels may prove to have deleterious effects on cellular metabolism.
Impact of LA on Morbidity and Mortality
The poor prognosis in patients with LA is well recognized.2, 4548 For example, in a study of 126 patients with various causes of LA, the median survival was 38.5 hours and 30‐day survival was 17%.2
Studies have revealed that LA with low pH is associated with adverse effects on the cardiovascular system, particularly a decrease in cardiac contractility.49, 50 This effect is particularly prominent with a pH below 7.20. In contrast, acidosis in animal models has been shown to limit myocardial infarct size after reperfusion.51, 52 Variable effects of LA on cell death have been found. A worsening of apoptosis in myocytes has been noted;53 alternatively, protection from hypoxic injury in hepatocytes and myocardium has been observed.52, 54 Thus, although LA is associated with poor outcomes in human studies,2, 4547 it is still unclear to what extent lactic acid accumulation is a marker of severe illness, an independent effector of pathology, or a mechanism with the potential to serve a protective role.
Available data indicate that lactate itself is not harmful. Studies on infusion of lactate solutions to postoperative patients was shown to be safe.55 Also, the fact that lactate generation in states of respiratory alkalosis, stress, or altered carbohydrate metabolism without sepsis is not associated with worse outcomes supports the fact that lactic acid alone may not be maladaptive.2831
Similarly, low pH is not necessarily maladaptive. In the postictal state,56 diabetic ketoacidosis,57 spontaneous respiratory acidosis,58 or permissive hypercapnia,59 low blood pH is not deleterious.
In summary, LA is associated with poor outcomes, and indirect evidence suggests that it is the underlying causative condition rather than the low pH or the lactate that is responsible for the dire outcomes.
Treatment of LA with Sodium Bicarbonate
Since excessive lactic acid generation is accompanied by consumption of plasma bicarbonate and a fall in plasma pH, sodium bicarbonate has been long proposed as a treatment for LA. While theoretically appealing, this strategy has not been validated by studies in animals or humans. Indeed, bicarbonate administration in LA often has been shown to be detrimental.60, 61 The adverse effects of bicarbonate administration in LA, while initially paradoxical, have a number of possible explanations.
First, bicarbonate administration can induce a reduction in intracellular pH.60, 62, 63 The mechanism involves bicarbonate's effect to increase carbon dioxide (CO2) generation through mass action effect. Because the cell membrane is more permeable to CO2 than to bicarbonate, intracellular pH falls.64, 65 In sepsis, this intracellular/extracellular pH discrepancy may be more pronounced due to alterations in blood flow.66 Other reports on outcomes of intracellular pH with bicarbonate therapy show variable effects.6772
Second, to the extent that bicarbonate administration raises extracellular pH, it is associated with a reduction in ionized calcium concentration, since the binding of calcium to albumin is pH dependent.73 A sodium bicarbonate load administered to patients with LA was associated with a significant fall in ionized calcium concentration, whereas a sodium chloride load was not.1 This can affect cardiac function, as the latter varies proportionally with calcium levels.74
Third, bicarbonate administration may reduce tissue oxygen delivery since the affinity of hemoglobin for oxygen increases as pH rises (Bohr effect).75 The administration of bicarbonate worsened systemic oxygen consumption in one study76 and decreased oxygen delivery in another.75
Fourth, bicarbonate administration may indirectly increase intracellular calcium concentration. Low intracellular pH (see above) stimulates proton efflux by way of proton transporters and exchangers, increasing intracellular sodium content.77 A high cell sodium content then may increase intracellular calcium, through the Na/Ca exchanger, impairing cellular function.7779 Compounding this, the reduced function of the Na/H ATPase as a regulator of intracellular sodium in sepsis may not be adequate to limit cell swelling.77
Against this background of mechanistic concerns with the use of bicarbonate treatment, it is not surprising that clinical outcomes have been inconsistent at best. In animal models of LA, the use of sodium bicarbonate has either negative effects on cardiac output60, 72 or no significant hemodynamic effect when compared to sodium chloride infusion.67, 80, 81 One animal study did show some benefit with sodium bicarbonate compared to saline, though all animals subsequently died.50
In humans, sodium bicarbonate was studied in 2 randomized trials of sepsis‐induced LA.1, 82 In a study by Cooper et al.,1 14 critically‐ill patients received sequential infusions of sodium bicarbonate or sodium chloride. Neither solution was superior to the other in terms of hemodynamic improvement. No benefit was noted even when analysis was limited to those with very low pH (7.2). Mathieu et al.82 randomized 10 critically‐ill patients to sequential infusion of either sodium bicarbonate or sodium chloride. Similarly, no significant difference in hemodynamic variables was noted.
When taken together, these studies evaluating sodium bicarbonate in LA fail to show convincing benefit and raise serious questions about its detrimental effects. Extracellular pH may be a misleading marker of success in the treatment of LA, given its direct influence by sodium bicarbonate administration.
Treatment of LA and Use of Other Buffers
Other buffers (Carbicarb, dichloroacetate, and tromethamine [THAM]) have been studied for treatment of LA. Human studies have not shown superiority of any of the buffers as far as improving pH,83, 84 hemodynamics, or survival.85
Treatment of LA by Renal Replacement Therapy
Renal replacement therapy (RRT; dialysis and its variants) has been studied for the treatment of severe acidosis. RRT has a number of theoretical advantages over purely medical therapies in the treatment of LA: it can deliver large quantities of base without contributing to volume overload; it can directly remove lactate from the plasma; and it can mitigate the effect of alkalinization on ionized calcium concentration by delivering calcium.
In critically ill patients with intact liver function, continuous venovenous hemofiltration (CVVH) appears to contribute very little (less then 3%) to overall lactate clearance.86 While outcome studies are limited, continuous dialysis modalities consistently show improved resolution of acidosis of various types when compared to intermittent modalities.87, 88 As described above, this is related to base administration and is not a surprising finding. There are no studies comparing RRT and medical therapy with respect to clinical outcomes in patients with LA.
Special Situations
Biguanides
Biguanide‐induced LA can be due to impairment of hepatic neoglucogenesis, in the case of metformin, or increasing hepatic oxidative phosphorylation, in the case of phenformin.89 This infrequent complication90, 91 is associated with a high mortality.92 Proposed therapy has included the use of sodium bicarbonate infusion.93 In this setting, it is unclear if the use of bicarbonate alone improves clinical outcomes.94
Renal replacement therapy in a wide variety of formats has been used to treat this condition.93, 95101 Metformin has a high clearance during dialysis due to its low molecular weight and lack of protein binding.97, 98, 102 Nonetheless, its high volume of distribution suggests a longer dialysis time would be more beneficial if the main goal is reducing metformin levels.97, 103 The limited prospective literature and lack of conclusive evidence about what levels of metformin induce LA makes generalized recommendations about duration of hemodialysis purely speculative.104
NRTIs
The use of NRTIs is associated with LA due to impairment of mitochondrial oxidative phosphorylation.105108 This uncommon complication, if not recognized early, is associated with a high mortality.101, 109 Investigations are ongoing into agents directed at improving mitochondrial function such as riboflavin, thiamine, and L‐carnitine.110112 As with biguanide‐associated LA, RRT decisions should be individualized based on metabolic circumstances.
Lorazepam
Many intravenous medications are formulated in the alcohol solvent, propylene glycol. Injectable lorazepam has the highest proportional amount of propylene glycol compared with other commonly used agents.113, 114 The kidney normally eliminates 12% to 50% of administered propylene glycol via proximal tubule secretion.115 The remainder is metabolized by the liver to form pyruvate and lactate.114, 116, 117
When propylene glycol accumulates, as in cases of reduced renal function, it results in hyperosmolarity, LA, and can even induce additional kidney injury (probably through proximal tubular cell necrosis).118
LA due to propylene glycol has been reported by many authors and its incidence with high dose intravenous (IV) lorazepam has been estimated to be as high as 19%.114, 116, 119, 120 This disorder can frequently go unrecognized, as many other factors that induce LA often coincide in such patients. But when identified and promptly addressed, its prognosis seems to be favorable.114
The best treatment is prevention, by avoiding the use of IV lorazepam in patients with impaired renal function. Once it is recognized, the drug should be promptly withdrawn. In addition, removal by hemodialysis can quickly lower propylene glycol levels since it is a small, highly water soluble, non‐protein‐bound molecule.121 As no rebound in the level is expected, intermittent dialysis should be an acceptable modality.117
Linezolid
Recently, Gram‐positive bacteria in general and methicillin‐resistant Staphylococcus aureus in particular have emerged as major causes of nosocomial and community‐acquired infections. Linezolid, an oxazolidinone, is increasingly used to treat such infections. Several cases of LA have been associated with linezolid.27, 122, 123 and a survey of the Infectious Diseases Society of America (IDSA) Emerging Infections Network members revealed that this complication was commonly encountered.124 Linezolid causes LA by mitochondrial toxicity125, 126 and risk factors include prolonged exposure and older age. Once the disorder is recognized, the clinician should stop the drug immediately. Chemistries should be monitored frequently in patients on long‐term therapy.
Conclusions
Many studies note the association between LA and adverse outcomes.2, 4547 Though metabolic acidosis from elevated lactate levels may negatively affect organ function, the evidence supporting therapy specifically aimed at increasing pH in these settings is consistently poor.3, 127 Limitations have included small numbers of subjects,1, 82 variable outcomes studied, and the inability to assess intracellular metabolic stability.1, 61 When taking these factors into account it is hard to justify aggressive treatment of LA with mechanisms aimed at raising pH. Literature on the treatment of patients with LA and very low pH (below 7.2) is even more limited.
Moreover, lactate elevations may not represent tissue hypoperfusion. Lactate may have an important role in improving energy metabolism. This represents 1 additional reason to be hesitant when attempting to normalize pH in LA; we may be disrupting the body's physiologic response to sepsis. A conflict for clinicians emerges, however, as lactate is often used to define tissue ischemia. Obviously, more specific markers of tissue hypoperfusion would be ideal.
Bicarbonate therapy is an understandably attractive means to improve the acidemia, but there are serious mechanistic concerns with it use. Moreover, neither animal nor human studies, limited as they may be, show a convincing benefit. LA in the setting of acute kidney injury may be best treated with renal replacement therapy with bicarbonate‐based buffers, but controlled trials are lacking.
A number of commonly used drugs can cause LA. A heightened awareness on the part of clinicians will lead to prompt recognition of these cases, and timely treatment.
Lactic acidosis (LA) is common in hospitalized patients and is associated with a high mortality.1, 2 Commonly, it is defined as a lactic acid concentration greater than 5 mmol/L with a pH less than 7.35.3 There are no evidence‐based guidelines for the treatment of LA despite progress in our understanding of its pathophysiology.36 This is not surprising, given the uncertainty regarding the impact of LA itself on clinical outcomes. In this regard, it is interesting to note that, despite its well‐recognized role as a marker of tissue hypoxia, lactate accumulation appears to have beneficial effects and may function as an adaptive mechanism. This raises the possibility that therapy directed at altering this adaptation may be detrimental. Pursuing correction of the pH in LA has been shown to have untoward physiologic effects. These and other ambiguities in the pathophysiology and treatment of LA are the focus of this review.
Lactate Metabolism
The body produces approximately 1400 mmol of lactate daily.7 Lactate is derived from the metabolism of pyruvate through an anaerobic reaction that occurs in all tissues (Figure 1). The liver is the primary site of lactate clearance and can metabolize up to 100 mmol per hour under normal conditions.8 There, lactate is converted to glucose to serve as an energy source during periods of hypoxia (Figure 2).9
Approximately 20% to 30% of the daily lactate load is metabolized by the kidneys.10, 11 Renal clearance is increased in acidosis12 and is maintained even in the presence of low renal perfusion.10, 12, 13 Renal lactate clearance is primarily through metabolism and not excretion.10, 14
LA Subtypes
Generally, lactic acid accumulation results from excess lactic acid production and not from reduced clearance.15 In cases of fulminant liver failure, it is due to a combination of decreased clearance and tissue hypoxia.16 In the setting of tissue hypoxia, an impairment of mitochondrial oxidative capacity results in the accumulation of pyruvate and generation of lactate. Lactic acid accumulation through this mechanism has historically been described as Type A LA.7 Hence, in critically ill patients lactate has traditionally been viewed as a marker of tissue hypoxia.15, 1721 Hyperlactatemia without tissue hypoxia has been referred to as type B LA. This is seen in a variety of circumstances. In sepsis, for example, several studies have shown lactic acid accumulation, despite adequate oxygen delivery.2224
Hyperlactatemia may also occur in cases of pure mitochondrial dysfunction, which can be induced by commonly prescribed medications such as the biguanides, nucleoside analog reverse‐transcriptase inhibitors (NRTIs), and linezolid.2527 Alternatively, lactate generation from metabolism of agents such as propylene glycol is possible. Finally, excessive lactate generation may occur following stress due to altered carbohydrate metabolism, or with respiratory alkalosis.2831
Lactate: A Metabolic Adaptation
Lactate was traditionally considered only as a marker of tissue hypoxia and anaerobic metabolism.17 This is certainly the case in situations of poor perfusion such as cardiogenic,15, 18 vasopressor‐resistant,19 or hypovolemic shock.20, 21
Alternative explanations for lactic acid accumulation, without tissue hypoperfusion, include catecholamine‐induced alterations in glycolysis,32, 33 mitochondrial disturbances,3436 and increased pyruvate production combined with increased glucose entry into cells.24, 37 In addition, the activity of an enzyme regulating lactate metabolism, pyruvate dehydrogenase kinase, increases in sepsis.38 This enzyme inactivates the pyruvate dehydrogenase (PDH) complex, which metabolizes pyruvate. Pyruvate and lactate may accumulate as a result. These changes partly explain the generation of LA in sepsis, independent of any effect of diminished tissue perfusion.
Recognizing the body's tendency toward homeostasis, it is appealing to speculate that lactate accumulation is adaptive.9 A number of findings support this. For example, lactate may act to shuttle energy between organs, or between cell types in the same organ. The astrocyteneuron lactate shuttle and the spermatogenic lactate shuttle are 2 examples of lactate's valuable effects on cellular metabolism.39 In the astrocyteneuron lactate shuttle, astrocytes support the increased metabolic demands of neurons through lactic acid production.40 Specifically, the neurotransmitter glutamate is released by the neurons and taken up by the astrocytes. Astrocytes produce lactate, which then moves back to the neuron to be used as an energy source. Glutamine, also released by the astrocytes, leads to the regeneration of glutamate and the potential to restart the cycle.39
Animal and human studies have suggested that, in periods of stress, lactate is the preferential energy substrate in the brain.4144 The usefulness of increased lactate production routinely seen in sepsis may thus represent multiple adaptive processes aimed primarily at improving the delivery of energy substrates. Thus, therapeutic strategies aimed specifically at lowering lactic acid levels may prove to have deleterious effects on cellular metabolism.
Impact of LA on Morbidity and Mortality
The poor prognosis in patients with LA is well recognized.2, 4548 For example, in a study of 126 patients with various causes of LA, the median survival was 38.5 hours and 30‐day survival was 17%.2
Studies have revealed that LA with low pH is associated with adverse effects on the cardiovascular system, particularly a decrease in cardiac contractility.49, 50 This effect is particularly prominent with a pH below 7.20. In contrast, acidosis in animal models has been shown to limit myocardial infarct size after reperfusion.51, 52 Variable effects of LA on cell death have been found. A worsening of apoptosis in myocytes has been noted;53 alternatively, protection from hypoxic injury in hepatocytes and myocardium has been observed.52, 54 Thus, although LA is associated with poor outcomes in human studies,2, 4547 it is still unclear to what extent lactic acid accumulation is a marker of severe illness, an independent effector of pathology, or a mechanism with the potential to serve a protective role.
Available data indicate that lactate itself is not harmful. Studies on infusion of lactate solutions to postoperative patients was shown to be safe.55 Also, the fact that lactate generation in states of respiratory alkalosis, stress, or altered carbohydrate metabolism without sepsis is not associated with worse outcomes supports the fact that lactic acid alone may not be maladaptive.2831
Similarly, low pH is not necessarily maladaptive. In the postictal state,56 diabetic ketoacidosis,57 spontaneous respiratory acidosis,58 or permissive hypercapnia,59 low blood pH is not deleterious.
In summary, LA is associated with poor outcomes, and indirect evidence suggests that it is the underlying causative condition rather than the low pH or the lactate that is responsible for the dire outcomes.
Treatment of LA with Sodium Bicarbonate
Since excessive lactic acid generation is accompanied by consumption of plasma bicarbonate and a fall in plasma pH, sodium bicarbonate has been long proposed as a treatment for LA. While theoretically appealing, this strategy has not been validated by studies in animals or humans. Indeed, bicarbonate administration in LA often has been shown to be detrimental.60, 61 The adverse effects of bicarbonate administration in LA, while initially paradoxical, have a number of possible explanations.
First, bicarbonate administration can induce a reduction in intracellular pH.60, 62, 63 The mechanism involves bicarbonate's effect to increase carbon dioxide (CO2) generation through mass action effect. Because the cell membrane is more permeable to CO2 than to bicarbonate, intracellular pH falls.64, 65 In sepsis, this intracellular/extracellular pH discrepancy may be more pronounced due to alterations in blood flow.66 Other reports on outcomes of intracellular pH with bicarbonate therapy show variable effects.6772
Second, to the extent that bicarbonate administration raises extracellular pH, it is associated with a reduction in ionized calcium concentration, since the binding of calcium to albumin is pH dependent.73 A sodium bicarbonate load administered to patients with LA was associated with a significant fall in ionized calcium concentration, whereas a sodium chloride load was not.1 This can affect cardiac function, as the latter varies proportionally with calcium levels.74
Third, bicarbonate administration may reduce tissue oxygen delivery since the affinity of hemoglobin for oxygen increases as pH rises (Bohr effect).75 The administration of bicarbonate worsened systemic oxygen consumption in one study76 and decreased oxygen delivery in another.75
Fourth, bicarbonate administration may indirectly increase intracellular calcium concentration. Low intracellular pH (see above) stimulates proton efflux by way of proton transporters and exchangers, increasing intracellular sodium content.77 A high cell sodium content then may increase intracellular calcium, through the Na/Ca exchanger, impairing cellular function.7779 Compounding this, the reduced function of the Na/H ATPase as a regulator of intracellular sodium in sepsis may not be adequate to limit cell swelling.77
Against this background of mechanistic concerns with the use of bicarbonate treatment, it is not surprising that clinical outcomes have been inconsistent at best. In animal models of LA, the use of sodium bicarbonate has either negative effects on cardiac output60, 72 or no significant hemodynamic effect when compared to sodium chloride infusion.67, 80, 81 One animal study did show some benefit with sodium bicarbonate compared to saline, though all animals subsequently died.50
In humans, sodium bicarbonate was studied in 2 randomized trials of sepsis‐induced LA.1, 82 In a study by Cooper et al.,1 14 critically‐ill patients received sequential infusions of sodium bicarbonate or sodium chloride. Neither solution was superior to the other in terms of hemodynamic improvement. No benefit was noted even when analysis was limited to those with very low pH (7.2). Mathieu et al.82 randomized 10 critically‐ill patients to sequential infusion of either sodium bicarbonate or sodium chloride. Similarly, no significant difference in hemodynamic variables was noted.
When taken together, these studies evaluating sodium bicarbonate in LA fail to show convincing benefit and raise serious questions about its detrimental effects. Extracellular pH may be a misleading marker of success in the treatment of LA, given its direct influence by sodium bicarbonate administration.
Treatment of LA and Use of Other Buffers
Other buffers (Carbicarb, dichloroacetate, and tromethamine [THAM]) have been studied for treatment of LA. Human studies have not shown superiority of any of the buffers as far as improving pH,83, 84 hemodynamics, or survival.85
Treatment of LA by Renal Replacement Therapy
Renal replacement therapy (RRT; dialysis and its variants) has been studied for the treatment of severe acidosis. RRT has a number of theoretical advantages over purely medical therapies in the treatment of LA: it can deliver large quantities of base without contributing to volume overload; it can directly remove lactate from the plasma; and it can mitigate the effect of alkalinization on ionized calcium concentration by delivering calcium.
In critically ill patients with intact liver function, continuous venovenous hemofiltration (CVVH) appears to contribute very little (less then 3%) to overall lactate clearance.86 While outcome studies are limited, continuous dialysis modalities consistently show improved resolution of acidosis of various types when compared to intermittent modalities.87, 88 As described above, this is related to base administration and is not a surprising finding. There are no studies comparing RRT and medical therapy with respect to clinical outcomes in patients with LA.
Special Situations
Biguanides
Biguanide‐induced LA can be due to impairment of hepatic neoglucogenesis, in the case of metformin, or increasing hepatic oxidative phosphorylation, in the case of phenformin.89 This infrequent complication90, 91 is associated with a high mortality.92 Proposed therapy has included the use of sodium bicarbonate infusion.93 In this setting, it is unclear if the use of bicarbonate alone improves clinical outcomes.94
Renal replacement therapy in a wide variety of formats has been used to treat this condition.93, 95101 Metformin has a high clearance during dialysis due to its low molecular weight and lack of protein binding.97, 98, 102 Nonetheless, its high volume of distribution suggests a longer dialysis time would be more beneficial if the main goal is reducing metformin levels.97, 103 The limited prospective literature and lack of conclusive evidence about what levels of metformin induce LA makes generalized recommendations about duration of hemodialysis purely speculative.104
NRTIs
The use of NRTIs is associated with LA due to impairment of mitochondrial oxidative phosphorylation.105108 This uncommon complication, if not recognized early, is associated with a high mortality.101, 109 Investigations are ongoing into agents directed at improving mitochondrial function such as riboflavin, thiamine, and L‐carnitine.110112 As with biguanide‐associated LA, RRT decisions should be individualized based on metabolic circumstances.
Lorazepam
Many intravenous medications are formulated in the alcohol solvent, propylene glycol. Injectable lorazepam has the highest proportional amount of propylene glycol compared with other commonly used agents.113, 114 The kidney normally eliminates 12% to 50% of administered propylene glycol via proximal tubule secretion.115 The remainder is metabolized by the liver to form pyruvate and lactate.114, 116, 117
When propylene glycol accumulates, as in cases of reduced renal function, it results in hyperosmolarity, LA, and can even induce additional kidney injury (probably through proximal tubular cell necrosis).118
LA due to propylene glycol has been reported by many authors and its incidence with high dose intravenous (IV) lorazepam has been estimated to be as high as 19%.114, 116, 119, 120 This disorder can frequently go unrecognized, as many other factors that induce LA often coincide in such patients. But when identified and promptly addressed, its prognosis seems to be favorable.114
The best treatment is prevention, by avoiding the use of IV lorazepam in patients with impaired renal function. Once it is recognized, the drug should be promptly withdrawn. In addition, removal by hemodialysis can quickly lower propylene glycol levels since it is a small, highly water soluble, non‐protein‐bound molecule.121 As no rebound in the level is expected, intermittent dialysis should be an acceptable modality.117
Linezolid
Recently, Gram‐positive bacteria in general and methicillin‐resistant Staphylococcus aureus in particular have emerged as major causes of nosocomial and community‐acquired infections. Linezolid, an oxazolidinone, is increasingly used to treat such infections. Several cases of LA have been associated with linezolid.27, 122, 123 and a survey of the Infectious Diseases Society of America (IDSA) Emerging Infections Network members revealed that this complication was commonly encountered.124 Linezolid causes LA by mitochondrial toxicity125, 126 and risk factors include prolonged exposure and older age. Once the disorder is recognized, the clinician should stop the drug immediately. Chemistries should be monitored frequently in patients on long‐term therapy.
Conclusions
Many studies note the association between LA and adverse outcomes.2, 4547 Though metabolic acidosis from elevated lactate levels may negatively affect organ function, the evidence supporting therapy specifically aimed at increasing pH in these settings is consistently poor.3, 127 Limitations have included small numbers of subjects,1, 82 variable outcomes studied, and the inability to assess intracellular metabolic stability.1, 61 When taking these factors into account it is hard to justify aggressive treatment of LA with mechanisms aimed at raising pH. Literature on the treatment of patients with LA and very low pH (below 7.2) is even more limited.
Moreover, lactate elevations may not represent tissue hypoperfusion. Lactate may have an important role in improving energy metabolism. This represents 1 additional reason to be hesitant when attempting to normalize pH in LA; we may be disrupting the body's physiologic response to sepsis. A conflict for clinicians emerges, however, as lactate is often used to define tissue ischemia. Obviously, more specific markers of tissue hypoperfusion would be ideal.
Bicarbonate therapy is an understandably attractive means to improve the acidemia, but there are serious mechanistic concerns with it use. Moreover, neither animal nor human studies, limited as they may be, show a convincing benefit. LA in the setting of acute kidney injury may be best treated with renal replacement therapy with bicarbonate‐based buffers, but controlled trials are lacking.
A number of commonly used drugs can cause LA. A heightened awareness on the part of clinicians will lead to prompt recognition of these cases, and timely treatment.
- ,,,.Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study.Ann Intern Med.1990;112(7):492–498.
- ,,, et al.Natural history and course of acquired lactic acidosis in adults. DCA‐Lactic Acidosis Study Group.Am J Med.1994;97(1):47–54.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- ,.Management of life‐threatening acid‐base disorders. First of two parts.N Engl J Med.1998;338(1):26–34.
- .Indications for use of bicarbonate in patients with metabolic acidosis.Br J Anaesth.1991;67(2):165–177.
- ,.Bench‐to‐bedside review: treating acid‐base abnormalities in the intensive care unit—the role of buffers.Crit Care.2004;8(4):259–265.
- .Lactic acidosis update for critical care clinicians.J Am Soc Nephrol.2001;12(suppl 17):S15–S19.
- ,.Lactic acidosis: diagnosis and treatment.Clin Endocrinol Metab.1980;9(3):513–541.
- .Lactate and shock state: the metabolic view.Curr Opin Crit Care.2006;12(4):315–321.
- .Bench‐to‐bedside review: lactate and the kidney.Crit Care.2002;6(4):322–326.
- ,,,.The influence of renal function on lactate and glucose metabolism.Biochem J.1984;219(1):73–78.
- ,.The effect of acidosis on lactate removal by the perfused rat kidney.Clin Sci Mol Med.1976;50(3):185–194.
- ,,.Transvisceral lactate fluxes during early endotoxemia.Chest.1996;110(1):198–204.
- ,,,,.Metabolism of lactate by the intact functioning kidney of the dog.Am J Physiol.1973;224(6):1463–1467.
- ,,, et al.Lactate and glucose metabolism in severe sepsis and cardiogenic shock.Crit Care Med.2005;33(10):2235–2240.
- ,,,, et al.Lactic acidosis in fulminant hepatic failure. Some aspects of pathogenesis and prognosis.J Hepatol.1985;1(4):405–416.
- ,.Lactic acidosis in critical illness.Crit Care Med.1992;20(1):80–93.
- ,,, et al.Effects of cardiogenic shock on lactate and glucose metabolism after heart surgery.Crit Care Med.2000;28(12):3784–3791.
- ,,,,,.Evolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine‐treated septic shock.Crit Care Med.2000;28(1):114–119.
- ,,, et al.Oxygen debt and metabolic acidemia as quantitative predictors of mortality and the severity of the ischemic insult in hemorrhagic shock.Crit Care Med.1991;19(2):231–243.
- ,,, et al.Early goal‐directed therapy in the treatment of severe sepsis and septic shock.N Engl J Med.2001;345(19):1368–1377.
- ,,,.Skeletal muscle partial pressure of oxygen in patients with sepsis.Crit Care Med.1994;22(4):640–650.
- ,.Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis.JAMA.1992;267(11):1503–1510.
- ,,,.Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability.Ann Surg.1996;224(1):97–102.
- ,,,.Biguanide‐associated lactic acidosis. Case report and review of the literature.Arch Intern Med.1992;152(11):2333–2336.
- ,,,.Bench‐to‐bedside review: severe lactic acidosis in HIV patients treated with nucleoside analogue reverse transcriptase inhibitors.Crit Care.2003;7(3):226–232.
- ,,,.Lactic acidosis after treatment with linezolid.Infection.2007;35(4):278–281.
- ,,,.Isotopic evaluation of the metabolism of pyruvate and related substrates in normal adult volunteers and severely burned children: effect of dichloroacetate and glucose infusion.Surgery.1991;110(1):54–67.
- .Alterations in carbohydrate metabolism during stress: a review of the literature.Am J Med.1995;98(1):75–84.
- .,,,.Lactic acid kinetics in respiratory alkalosis.Crit Care Med.1991;19(9):1120–1124.
- .Significance of hyperlactatemia without acidosis during hypermetabolic stress.Crit Care Med.1997;25(11):1780–1781.
- ,,,.Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis.Lancet.1999;354(9177):505–508.
- ,,,,.Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study.Lancet.2005;365(9462):871–875.
- ,.Sepsis induces diaphragm electron transport chain dysfunction and protein depletion.Am J Respir Crit Care Med.2005;172(7):861–868.
- ,,, et al.Association between mitochondrial dysfunction and severity and outcome of septic shock.Lancet.2002;360(9328):219–223.
- ,.Mitochondrial dysfunction in sepsis.Biochem Soc Symp.1999;66:149–166.
- ,,, et al.Altered glucose transporter mRNA abundance in a rat model of endotoxic shock.Biochem Biophys Res Commun.1991;176(1):535–540.
- .Increased pyruvate dehydrogenase kinase activity in response to sepsis.Am J Physiol.1991;260(5 Pt 1):E669–E674.
- .Lactate metabolism: a new paradigm for the third millennium.J Physiol.2004;558(Pt 1):5–30.
- ,,, et al.Evidence supporting the existence of an activity‐dependent astrocyte‐neuron lactate shuttle.Dev Neurosci.1998;20(4–5):291–299.
- ,,,.Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: further in vitro validation.J Neurochem.1997;69(1):423–426.
- .Bench‐to‐bedside review: a possible resolution of the glucose paradox of cerebral ischemia.Crit Care.2002;6(4):330–334.
- ,,,,,.Intravenous lactate prevents cerebral dysfunction during hypoglycaemia in insulin‐dependent diabetes mellitus.Clin Sci (Lond).1998;94(2):157–163.
- ,,,.Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study.Brain Res.1997;744(1):105–111.
- ,,,,,.Clinical prognostic markers in patients with severe sepsis: a prospective analysis of 139 consecutive cases.J Infect.2003;47(4):300–306.
- ,,, et al.Serum lactate as a predictor of mortality in patients with infection.Intensive Care Med.2007;33(6):970–977.
- ,,,.Lactate versus non‐lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients.Crit Care.2006;10(1):R22.
- .Abnormal resting blood lactate. I. The significance of hyperlactatemia in hospitalized patients.Am J Med.1961;30:840–848.
- ,,,,.Effect of lactic acidosis on canine hemodynamics and left ventricular function.Am J Physiol.1990;258(4 Pt 2):H1193–H1199.
- ,,,.Alkali therapy extends the period of survival during hypoxia: studies in rats.Am J Physiol.1996;271(2 Pt 2):R381–R387.
- ,,.Acidosis during early reperfusion prevents myocardial stunning in perfused ferret hearts.J Clin Invest.1988;82(3):920–927.
- ,,, et al.Effect of acidotic blood reperfusion on reperfusion injury after coronary artery occlusion in the dog heart.J Cardiovasc Pharmacol.1998;31(2):179–186.
- ,,,.Hypoxia and acidosis activate cardiac myocyte death through the Bcl‐2 family protein BNIP3.Proc Natl Acad Sci U S A.2002;99(20):12825–12830.
- ,,,,,.Extracellular acidosis delays onset of cell death in ATP‐depleted hepatocytes.Am J Physiol.1988;255(3 Pt 1):C315–C322.
- ,.Metabolic and hemodynamic effects of hypertonic solutions: sodium‐lactate versus sodium chloride infusion in postoperative patients.Shock.2002;18(4):306–310.
- .Medical complications of status epilepticus.Adv Neurol.1983;34:395–398.
- ,,,,,.Bicarbonate therapy in severe diabetic ketoacidosis. A double blind, randomized, placebo controlled trial.Rev Invest Clin.1991;43(3):234–238.
- ,,.Supercarbia in children: clinical course and outcome.Crit Care Med.1990;18(2):166–168.
- ,,,.Low mortality rate in adult respiratory distress syndrome using low‐volume, pressure‐limited ventilation with permissive hypercapnia: a prospective study.Crit Care Med.1994;22(10):1568–1578.
- ,,,.Systemic effects of NaHCO3 in experimental lactic acidosis in dogs.Am J Physiol.1982;242(6):F586–F591.
- ,,,,.Lactic acidosis: effect of treatment on intracellular pH and energetics in living rat heart.Am J Physiol.1992;262(5 Pt 2):H1572–H1578.
- ,,,,,.Effect of sodium bicarbonate on intracellular pH under different buffering conditions.Kidney Int.1996;49(5):1262–1267.
- ,,.Hemodynamic and hepatic pH responses to sodium bicarbonate and Carbicarb during systemic acidosis.Magn Reson Med.1990;16(3):403–410.
- ,,, et al.The increase in CO2 production induced by NaHCO3 depends on blood albumin and hemoglobin concentrations.Intensive Care Med.2000;26(5):558–564.
- ,,, et al.Initial effect of sodium bicarbonate on intracellular pH depends on the extracellular nonbicarbonate buffering capacity.Crit Care Med.2001;29(5):1033–1039.
- ,,,,,.Dynamic study of the distribution of microcirculatory blood flow in multiple splanchnic organs in septic shock.Crit Care Med.2000;28(9):3233–3241.
- ,,,,,.The effects of sodium bicarbonate and a mixture of sodium bicarbonate and carbonate (“Carbicarb”) on skeletal muscle pH and hemodynamic status in rats with hypovolemic shock.Metabolism.1994;43(4):518–522.
- ,,, et al.Haemodynamic and metabolic effects in diabetic ketoacidosis in rats of treatment with sodium bicarbonate or a mixture of sodium bicarbonate and sodium carbonate.Diabetologia.1995;38(8):889–898.
- ,,,,,.Effects of sodium bicarbonate on striated muscle metabolism and intracellular pH during endotoxic shock.Shock.1994;1(3):196–200.
- ,,,,.Carbicarb, sodium bicarbonate, and sodium chloride in hypoxic lactic acidosis. Effect on arterial blood gases, lactate concentrations, hemodynamic variables, and myocardial intracellular pH.Chest.1993;104(3):913–918.
- ,.Improved hemodynamic function during hypoxia with Carbicarb, a new agent for the management of acidosis.Circulation.1988;77(1):227–233.
- ,,.Metabolic effects of sodium bicarbonate in hypoxic lactic acidosis in dogs.Am J Physiol.1985;249(5 Pt 2):F630–F635.
- .Binding of calcium to serum albumin. II. Effect of pH via competitive hydrogen and calcium ion binding to the imidazole groups of albumin.Scand J Clin Lab Invest.1972;29(1):75–83.
- ,,,,.Left ventricular contractility varies directly with blood ionized calcium.Ann Intern Med.1988;108(4):524–529.
- ,,.Regulatory mechanisms of hemoglobin oxygen affinity in acidosis and alkalosis.J Clin Invest.1971;50(3):700–706.
- ,,.Metabolic and hemodynamic consequences of sodium bicarbonate administration in patients with heart disease.Am J Med.1989;87(1):7–14.
- ,,.Sodium ion/hydrogen ion exchange inhibition: a new pharmacologic approach to myocardial ischemia and reperfusion injury.J Clin Pharmacol.1998;38(10):887–897.
- ,.SEA0400: a novel sodium‐calcium exchange inhibitor with cardioprotective properties.Cardiovasc Drug Rev.2004;22(4):334–347.
- ,,,.Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart.Circ Res.1987;60(5):700–707.
- ,.Acute haemodynamic effects of sodium bicarbonate administration in respiratory and metabolic acidosis in anaesthetized dogs.Anaesth Intensive Care.1997;25(6):615–620.
- ,,,.Bicarbonate does not increase left ventricular contractility during L‐lactic acidemia in pigs.Am Rev Respir Dis.1993;148(2):317–322.
- ,,,,.Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study.Crit Care Med.1991;19(11):1352–1356.
- ,,, et al.Safety and efficacy of intravenous Carbicarb in patients undergoing surgery: comparison with sodium bicarbonate in the treatment of mild metabolic acidosis. SPI Research Group. Study of Perioperative Ischemia.Crit Care Med.1994;22(10):1540–1549.
- ,,, et al.Sodium bicarbonate versus THAM in ICU patients with mild metabolic acidosis.J Nephrol.2005;18(3):303–307.
- ,,, et al.A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate‐Lactic Acidosis Study Group.N Engl J Med.1992;327(22):1564–1569.
- ,,,,,.Effect of continuous venovenous hemofiltration with dialysis on lactate clearance in critically ill patients.Crit Care Med.1997;25(1):58–62.
- ,,,,.A pilot randomised controlled comparison of continuous veno‐venous haemofiltration and extended daily dialysis with filtration: effect on small solutes and acid‐base balance.Intensive Care Med.2007;33(5):830–835.
- ,,.Intermittent versus continuous renal replacement therapy in the ICU: impact on electrolyte and acid‐base balance.Intensive Care Med.2001;27(6):1037–1043.
- ,,,.Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus: systematic review and meta‐analysis.Arch Intern Med.2003;163(21):2594–2602.
- ,,,,.Severe acidosis in patients taking metformin—rapid reversal and survival despite high APACHE score.Diabet Med.2006;23(4):432–435.
- ,,.Incidence of lactic acidosis in metformin users.Diabetes Care.1999;22(6):925–927.
- ,.Lactic acidosis in metformin‐treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations.Drug Saf.1999;20(4):377–384.
- ,,,.The management of metformin overdose.Anaesthesia.1998;53(7):698–701.
- ,,.Lactic acidosis in biguanide‐treated diabetics: a review of 330 cases.Diabetologia.1978;14(2):75–87.
- ,,,,.Treatment of metformin‐associated lactic acidosis with closed recirculation bicarbonate‐buffered hemodialysis.Arch Intern Med.1984;144(1):203–205.
- ,,,.High anion gap metabolic acidosis in suicide: don't forget metformin intoxication—two patients' experiences.Ren Fail.2002;24(5):671–675.
- ,,, et al.Hemodialysis in the treatment of lactic acidosis in diabetics treated by metformin: a study of metformin elimination.Int J Clin Pharmacol Ther Toxicol.1989;27(6):285–288.
- ,,.Bicarbonate haemodialysis as a treatment of metformin overdose.Nephrol Dial Transplant.1997;12(5):1046–1047.
- ,,,.Combination of intermittent haemodialysis and high‐volume continuous haemofiltration for the treatment of severe metformin‐induced lactic acidosis.Nephrol Dial Transplant.2004;19(8):2157–2158.
- ,,,.When a friend can become an enemy! Recognition and management of metformin‐associated lactic acidosis.Kidney Int.2007;72(9):1157–1160.
- .,, et al.Severe nucleoside‐associated lactic acidosis in human immunodeficiency virus‐infected patients: report of 12 cases and review of the literature.Clin Infect Dis.2002;34(6):838–846.
- ,,.Clearance of metformin by hemofiltration in overdose.J Toxicol Clin Toxicol.2002;40(2):177–180.
- .Metformin‐associated lactic acidosis.J Emerg Med.2001;20(3):267–272.
- ,.Contraindications to use of metformin. Blanket banning of metformin two days before surgery may not be a good idea.BMJ.2003;326(7392):762; author reply 762.
- ,,, et al.Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B.N Engl J Med.1995;333(17):1099–1105.
- ,,,.Zidovudine‐induced fatal lactic acidosis and hepatic failure in patients with acquired immunodeficiency syndrome: report of two patients and review of the literature.Crit Care Med.1997;25(8):1425–1430.
- ,.Mitochondrial toxicity of antiviral drugs.Nat Med.1995;1(5):417–422.
- ,.Mitochondrial toxicity of nucleoside analogue reverse transcriptase inhibitors: a looming obstacle for long‐term antiretroviral therapy?Curr Opin Infect Dis.2000;13(1):5–11.
- ,,, et al.Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society‐USA panel.JAMA.2006;296(7):827–843.
- ,,.Riboflavin to treat nucleoside analogue‐induced lactic acidosis.Lancet.1998;352(9124):291–292.
- ,,,,,.Severe lactic acidosis and thiamine administration in an HIV‐infected patient on HAART.Int J STD AIDS.2001;12(6):407–409.
- ,,, et al.Detecting life‐threatening lactic acidosis related to nucleoside‐analog treatment of human immunodeficiency virus‐infected patients, and treatment with L‐carnitine.Crit Care Med.2003;31(4):1042–1047.
- ,.Hyperosmolar metabolic acidosis and intravenous Lorazepam.N Engl J Med.2002;347(11):857–858; author reply 857–858.
- ,,,.Propylene glycol toxicity: a severe iatrogenic illness in ICU patients receiving IV benzodiazepines: a case series and prospective, observational pilot study.Chest.2005;128(3):1674–1681.
- ,,, et al.Propylene glycol pharmacokinetics and effects after intravenous infusion in humans.Ther Drug Monit.1987;9(3):255–258.
- .Short‐term lorazepam infusion and concern for propylene glycol toxicity: case report and review.Pharmacotherapy.2001;21(9):1140–1144.
- ,,.Recognition, treatment, and prevention of propylene glycol toxicity.Semin Dial.2007;20(3):217–219.
- ,,.Propylene glycol‐mediated cell injury in a primary culture of human proximal tubule cells.Toxicol Sci.1998;46(2):410–417.
- ,.Osmolar gap metabolic acidosis in a 60‐year‐old man treated for hypoxemic respiratory failure.Chest.2000;118(2):545–546.
- ,,,.Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults.Crit Care Med.2004;32(8):1709–1714.
- ,,,.Removal of propylene glycol and correction of increased osmolar gap by hemodialysis in a patient on high dose lorazepam infusion therapy.Intensive Care Med.2002;28(1):81–84.
- ,,.Linezolid use associated with lactic acidosis.Scand J Infect Dis.2005;37(2):153–154.
- ,.Linezolid‐induced lactic acidosis.N Engl J Med.2003;348(1):86–87.
- ,,.Toxicity of extended courses of linezolid: results of an Infectious Diseases Society of America Emerging Infections Network survey.Diagn Microbiol Infect Dis.2008;62(4):407–410.
- ,,.Mitochondrial toxicity associated with linezolid.N Engl J Med.2005;353(21):2305–2306.
- ,,, et al.Linezolid‐induced inhibition of mitochondrial protein synthesis.Clin Infect Dis.2006;42(8):1111–1117.
- ,.Use of base in the treatment of severe acidemic states.Am J Kidney Dis.2001;38(4):703–727.
- ,,,.Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study.Ann Intern Med.1990;112(7):492–498.
- ,,, et al.Natural history and course of acquired lactic acidosis in adults. DCA‐Lactic Acidosis Study Group.Am J Med.1994;97(1):47–54.
- ,.Sodium bicarbonate for the treatment of lactic acidosis.Chest.2000;117(1):260–267.
- ,.Management of life‐threatening acid‐base disorders. First of two parts.N Engl J Med.1998;338(1):26–34.
- .Indications for use of bicarbonate in patients with metabolic acidosis.Br J Anaesth.1991;67(2):165–177.
- ,.Bench‐to‐bedside review: treating acid‐base abnormalities in the intensive care unit—the role of buffers.Crit Care.2004;8(4):259–265.
- .Lactic acidosis update for critical care clinicians.J Am Soc Nephrol.2001;12(suppl 17):S15–S19.
- ,.Lactic acidosis: diagnosis and treatment.Clin Endocrinol Metab.1980;9(3):513–541.
- .Lactate and shock state: the metabolic view.Curr Opin Crit Care.2006;12(4):315–321.
- .Bench‐to‐bedside review: lactate and the kidney.Crit Care.2002;6(4):322–326.
- ,,,.The influence of renal function on lactate and glucose metabolism.Biochem J.1984;219(1):73–78.
- ,.The effect of acidosis on lactate removal by the perfused rat kidney.Clin Sci Mol Med.1976;50(3):185–194.
- ,,.Transvisceral lactate fluxes during early endotoxemia.Chest.1996;110(1):198–204.
- ,,,,.Metabolism of lactate by the intact functioning kidney of the dog.Am J Physiol.1973;224(6):1463–1467.
- ,,, et al.Lactate and glucose metabolism in severe sepsis and cardiogenic shock.Crit Care Med.2005;33(10):2235–2240.
- ,,,, et al.Lactic acidosis in fulminant hepatic failure. Some aspects of pathogenesis and prognosis.J Hepatol.1985;1(4):405–416.
- ,.Lactic acidosis in critical illness.Crit Care Med.1992;20(1):80–93.
- ,,, et al.Effects of cardiogenic shock on lactate and glucose metabolism after heart surgery.Crit Care Med.2000;28(12):3784–3791.
- ,,,,,.Evolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine‐treated septic shock.Crit Care Med.2000;28(1):114–119.
- ,,, et al.Oxygen debt and metabolic acidemia as quantitative predictors of mortality and the severity of the ischemic insult in hemorrhagic shock.Crit Care Med.1991;19(2):231–243.
- ,,, et al.Early goal‐directed therapy in the treatment of severe sepsis and septic shock.N Engl J Med.2001;345(19):1368–1377.
- ,,,.Skeletal muscle partial pressure of oxygen in patients with sepsis.Crit Care Med.1994;22(4):640–650.
- ,.Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis.JAMA.1992;267(11):1503–1510.
- ,,,.Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability.Ann Surg.1996;224(1):97–102.
- ,,,.Biguanide‐associated lactic acidosis. Case report and review of the literature.Arch Intern Med.1992;152(11):2333–2336.
- ,,,.Bench‐to‐bedside review: severe lactic acidosis in HIV patients treated with nucleoside analogue reverse transcriptase inhibitors.Crit Care.2003;7(3):226–232.
- ,,,.Lactic acidosis after treatment with linezolid.Infection.2007;35(4):278–281.
- ,,,.Isotopic evaluation of the metabolism of pyruvate and related substrates in normal adult volunteers and severely burned children: effect of dichloroacetate and glucose infusion.Surgery.1991;110(1):54–67.
- .Alterations in carbohydrate metabolism during stress: a review of the literature.Am J Med.1995;98(1):75–84.
- .,,,.Lactic acid kinetics in respiratory alkalosis.Crit Care Med.1991;19(9):1120–1124.
- .Significance of hyperlactatemia without acidosis during hypermetabolic stress.Crit Care Med.1997;25(11):1780–1781.
- ,,,.Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis.Lancet.1999;354(9177):505–508.
- ,,,,.Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study.Lancet.2005;365(9462):871–875.
- ,.Sepsis induces diaphragm electron transport chain dysfunction and protein depletion.Am J Respir Crit Care Med.2005;172(7):861–868.
- ,,, et al.Association between mitochondrial dysfunction and severity and outcome of septic shock.Lancet.2002;360(9328):219–223.
- ,.Mitochondrial dysfunction in sepsis.Biochem Soc Symp.1999;66:149–166.
- ,,, et al.Altered glucose transporter mRNA abundance in a rat model of endotoxic shock.Biochem Biophys Res Commun.1991;176(1):535–540.
- .Increased pyruvate dehydrogenase kinase activity in response to sepsis.Am J Physiol.1991;260(5 Pt 1):E669–E674.
- .Lactate metabolism: a new paradigm for the third millennium.J Physiol.2004;558(Pt 1):5–30.
- ,,, et al.Evidence supporting the existence of an activity‐dependent astrocyte‐neuron lactate shuttle.Dev Neurosci.1998;20(4–5):291–299.
- ,,,.Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: further in vitro validation.J Neurochem.1997;69(1):423–426.
- .Bench‐to‐bedside review: a possible resolution of the glucose paradox of cerebral ischemia.Crit Care.2002;6(4):330–334.
- ,,,,,.Intravenous lactate prevents cerebral dysfunction during hypoglycaemia in insulin‐dependent diabetes mellitus.Clin Sci (Lond).1998;94(2):157–163.
- ,,,.Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study.Brain Res.1997;744(1):105–111.
- ,,,,,.Clinical prognostic markers in patients with severe sepsis: a prospective analysis of 139 consecutive cases.J Infect.2003;47(4):300–306.
- ,,, et al.Serum lactate as a predictor of mortality in patients with infection.Intensive Care Med.2007;33(6):970–977.
- ,,,.Lactate versus non‐lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients.Crit Care.2006;10(1):R22.
- .Abnormal resting blood lactate. I. The significance of hyperlactatemia in hospitalized patients.Am J Med.1961;30:840–848.
- ,,,,.Effect of lactic acidosis on canine hemodynamics and left ventricular function.Am J Physiol.1990;258(4 Pt 2):H1193–H1199.
- ,,,.Alkali therapy extends the period of survival during hypoxia: studies in rats.Am J Physiol.1996;271(2 Pt 2):R381–R387.
- ,,.Acidosis during early reperfusion prevents myocardial stunning in perfused ferret hearts.J Clin Invest.1988;82(3):920–927.
- ,,, et al.Effect of acidotic blood reperfusion on reperfusion injury after coronary artery occlusion in the dog heart.J Cardiovasc Pharmacol.1998;31(2):179–186.
- ,,,.Hypoxia and acidosis activate cardiac myocyte death through the Bcl‐2 family protein BNIP3.Proc Natl Acad Sci U S A.2002;99(20):12825–12830.
- ,,,,,.Extracellular acidosis delays onset of cell death in ATP‐depleted hepatocytes.Am J Physiol.1988;255(3 Pt 1):C315–C322.
- ,.Metabolic and hemodynamic effects of hypertonic solutions: sodium‐lactate versus sodium chloride infusion in postoperative patients.Shock.2002;18(4):306–310.
- .Medical complications of status epilepticus.Adv Neurol.1983;34:395–398.
- ,,,,,.Bicarbonate therapy in severe diabetic ketoacidosis. A double blind, randomized, placebo controlled trial.Rev Invest Clin.1991;43(3):234–238.
- ,,.Supercarbia in children: clinical course and outcome.Crit Care Med.1990;18(2):166–168.
- ,,,.Low mortality rate in adult respiratory distress syndrome using low‐volume, pressure‐limited ventilation with permissive hypercapnia: a prospective study.Crit Care Med.1994;22(10):1568–1578.
- ,,,.Systemic effects of NaHCO3 in experimental lactic acidosis in dogs.Am J Physiol.1982;242(6):F586–F591.
- ,,,,.Lactic acidosis: effect of treatment on intracellular pH and energetics in living rat heart.Am J Physiol.1992;262(5 Pt 2):H1572–H1578.
- ,,,,,.Effect of sodium bicarbonate on intracellular pH under different buffering conditions.Kidney Int.1996;49(5):1262–1267.
- ,,.Hemodynamic and hepatic pH responses to sodium bicarbonate and Carbicarb during systemic acidosis.Magn Reson Med.1990;16(3):403–410.
- ,,, et al.The increase in CO2 production induced by NaHCO3 depends on blood albumin and hemoglobin concentrations.Intensive Care Med.2000;26(5):558–564.
- ,,, et al.Initial effect of sodium bicarbonate on intracellular pH depends on the extracellular nonbicarbonate buffering capacity.Crit Care Med.2001;29(5):1033–1039.
- ,,,,,.Dynamic study of the distribution of microcirculatory blood flow in multiple splanchnic organs in septic shock.Crit Care Med.2000;28(9):3233–3241.
- ,,,,,.The effects of sodium bicarbonate and a mixture of sodium bicarbonate and carbonate (“Carbicarb”) on skeletal muscle pH and hemodynamic status in rats with hypovolemic shock.Metabolism.1994;43(4):518–522.
- ,,, et al.Haemodynamic and metabolic effects in diabetic ketoacidosis in rats of treatment with sodium bicarbonate or a mixture of sodium bicarbonate and sodium carbonate.Diabetologia.1995;38(8):889–898.
- ,,,,,.Effects of sodium bicarbonate on striated muscle metabolism and intracellular pH during endotoxic shock.Shock.1994;1(3):196–200.
- ,,,,.Carbicarb, sodium bicarbonate, and sodium chloride in hypoxic lactic acidosis. Effect on arterial blood gases, lactate concentrations, hemodynamic variables, and myocardial intracellular pH.Chest.1993;104(3):913–918.
- ,.Improved hemodynamic function during hypoxia with Carbicarb, a new agent for the management of acidosis.Circulation.1988;77(1):227–233.
- ,,.Metabolic effects of sodium bicarbonate in hypoxic lactic acidosis in dogs.Am J Physiol.1985;249(5 Pt 2):F630–F635.
- .Binding of calcium to serum albumin. II. Effect of pH via competitive hydrogen and calcium ion binding to the imidazole groups of albumin.Scand J Clin Lab Invest.1972;29(1):75–83.
- ,,,,.Left ventricular contractility varies directly with blood ionized calcium.Ann Intern Med.1988;108(4):524–529.
- ,,.Regulatory mechanisms of hemoglobin oxygen affinity in acidosis and alkalosis.J Clin Invest.1971;50(3):700–706.
- ,,.Metabolic and hemodynamic consequences of sodium bicarbonate administration in patients with heart disease.Am J Med.1989;87(1):7–14.
- ,,.Sodium ion/hydrogen ion exchange inhibition: a new pharmacologic approach to myocardial ischemia and reperfusion injury.J Clin Pharmacol.1998;38(10):887–897.
- ,.SEA0400: a novel sodium‐calcium exchange inhibitor with cardioprotective properties.Cardiovasc Drug Rev.2004;22(4):334–347.
- ,,,.Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart.Circ Res.1987;60(5):700–707.
- ,.Acute haemodynamic effects of sodium bicarbonate administration in respiratory and metabolic acidosis in anaesthetized dogs.Anaesth Intensive Care.1997;25(6):615–620.
- ,,,.Bicarbonate does not increase left ventricular contractility during L‐lactic acidemia in pigs.Am Rev Respir Dis.1993;148(2):317–322.
- ,,,,.Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study.Crit Care Med.1991;19(11):1352–1356.
- ,,, et al.Safety and efficacy of intravenous Carbicarb in patients undergoing surgery: comparison with sodium bicarbonate in the treatment of mild metabolic acidosis. SPI Research Group. Study of Perioperative Ischemia.Crit Care Med.1994;22(10):1540–1549.
- ,,, et al.Sodium bicarbonate versus THAM in ICU patients with mild metabolic acidosis.J Nephrol.2005;18(3):303–307.
- ,,, et al.A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate‐Lactic Acidosis Study Group.N Engl J Med.1992;327(22):1564–1569.
- ,,,,,.Effect of continuous venovenous hemofiltration with dialysis on lactate clearance in critically ill patients.Crit Care Med.1997;25(1):58–62.
- ,,,,.A pilot randomised controlled comparison of continuous veno‐venous haemofiltration and extended daily dialysis with filtration: effect on small solutes and acid‐base balance.Intensive Care Med.2007;33(5):830–835.
- ,,.Intermittent versus continuous renal replacement therapy in the ICU: impact on electrolyte and acid‐base balance.Intensive Care Med.2001;27(6):1037–1043.
- ,,,.Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus: systematic review and meta‐analysis.Arch Intern Med.2003;163(21):2594–2602.
- ,,,,.Severe acidosis in patients taking metformin—rapid reversal and survival despite high APACHE score.Diabet Med.2006;23(4):432–435.
- ,,.Incidence of lactic acidosis in metformin users.Diabetes Care.1999;22(6):925–927.
- ,.Lactic acidosis in metformin‐treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations.Drug Saf.1999;20(4):377–384.
- ,,,.The management of metformin overdose.Anaesthesia.1998;53(7):698–701.
- ,,.Lactic acidosis in biguanide‐treated diabetics: a review of 330 cases.Diabetologia.1978;14(2):75–87.
- ,,,,.Treatment of metformin‐associated lactic acidosis with closed recirculation bicarbonate‐buffered hemodialysis.Arch Intern Med.1984;144(1):203–205.
- ,,,.High anion gap metabolic acidosis in suicide: don't forget metformin intoxication—two patients' experiences.Ren Fail.2002;24(5):671–675.
- ,,, et al.Hemodialysis in the treatment of lactic acidosis in diabetics treated by metformin: a study of metformin elimination.Int J Clin Pharmacol Ther Toxicol.1989;27(6):285–288.
- ,,.Bicarbonate haemodialysis as a treatment of metformin overdose.Nephrol Dial Transplant.1997;12(5):1046–1047.
- ,,,.Combination of intermittent haemodialysis and high‐volume continuous haemofiltration for the treatment of severe metformin‐induced lactic acidosis.Nephrol Dial Transplant.2004;19(8):2157–2158.
- ,,,.When a friend can become an enemy! Recognition and management of metformin‐associated lactic acidosis.Kidney Int.2007;72(9):1157–1160.
- .,, et al.Severe nucleoside‐associated lactic acidosis in human immunodeficiency virus‐infected patients: report of 12 cases and review of the literature.Clin Infect Dis.2002;34(6):838–846.
- ,,.Clearance of metformin by hemofiltration in overdose.J Toxicol Clin Toxicol.2002;40(2):177–180.
- .Metformin‐associated lactic acidosis.J Emerg Med.2001;20(3):267–272.
- ,.Contraindications to use of metformin. Blanket banning of metformin two days before surgery may not be a good idea.BMJ.2003;326(7392):762; author reply 762.
- ,,, et al.Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B.N Engl J Med.1995;333(17):1099–1105.
- ,,,.Zidovudine‐induced fatal lactic acidosis and hepatic failure in patients with acquired immunodeficiency syndrome: report of two patients and review of the literature.Crit Care Med.1997;25(8):1425–1430.
- ,.Mitochondrial toxicity of antiviral drugs.Nat Med.1995;1(5):417–422.
- ,.Mitochondrial toxicity of nucleoside analogue reverse transcriptase inhibitors: a looming obstacle for long‐term antiretroviral therapy?Curr Opin Infect Dis.2000;13(1):5–11.
- ,,, et al.Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society‐USA panel.JAMA.2006;296(7):827–843.
- ,,.Riboflavin to treat nucleoside analogue‐induced lactic acidosis.Lancet.1998;352(9124):291–292.
- ,,,,,.Severe lactic acidosis and thiamine administration in an HIV‐infected patient on HAART.Int J STD AIDS.2001;12(6):407–409.
- ,,, et al.Detecting life‐threatening lactic acidosis related to nucleoside‐analog treatment of human immunodeficiency virus‐infected patients, and treatment with L‐carnitine.Crit Care Med.2003;31(4):1042–1047.
- ,.Hyperosmolar metabolic acidosis and intravenous Lorazepam.N Engl J Med.2002;347(11):857–858; author reply 857–858.
- ,,,.Propylene glycol toxicity: a severe iatrogenic illness in ICU patients receiving IV benzodiazepines: a case series and prospective, observational pilot study.Chest.2005;128(3):1674–1681.
- ,,, et al.Propylene glycol pharmacokinetics and effects after intravenous infusion in humans.Ther Drug Monit.1987;9(3):255–258.
- .Short‐term lorazepam infusion and concern for propylene glycol toxicity: case report and review.Pharmacotherapy.2001;21(9):1140–1144.
- ,,.Recognition, treatment, and prevention of propylene glycol toxicity.Semin Dial.2007;20(3):217–219.
- ,,.Propylene glycol‐mediated cell injury in a primary culture of human proximal tubule cells.Toxicol Sci.1998;46(2):410–417.
- ,.Osmolar gap metabolic acidosis in a 60‐year‐old man treated for hypoxemic respiratory failure.Chest.2000;118(2):545–546.
- ,,,.Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults.Crit Care Med.2004;32(8):1709–1714.
- ,,,.Removal of propylene glycol and correction of increased osmolar gap by hemodialysis in a patient on high dose lorazepam infusion therapy.Intensive Care Med.2002;28(1):81–84.
- ,,.Linezolid use associated with lactic acidosis.Scand J Infect Dis.2005;37(2):153–154.
- ,.Linezolid‐induced lactic acidosis.N Engl J Med.2003;348(1):86–87.
- ,,.Toxicity of extended courses of linezolid: results of an Infectious Diseases Society of America Emerging Infections Network survey.Diagn Microbiol Infect Dis.2008;62(4):407–410.
- ,,.Mitochondrial toxicity associated with linezolid.N Engl J Med.2005;353(21):2305–2306.
- ,,, et al.Linezolid‐induced inhibition of mitochondrial protein synthesis.Clin Infect Dis.2006;42(8):1111–1117.
- ,.Use of base in the treatment of severe acidemic states.Am J Kidney Dis.2001;38(4):703–727.
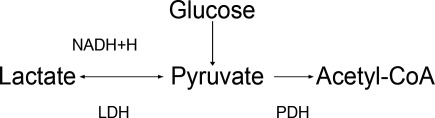
Cefepime: Underrecognized Cause of NCSE
Hospitalized patients with sepsis or severe nosocomial infections are frequently treated empirically with broad‐spectrum antibiotics. Cefepime hydrochloride, a fourth‐generation cephalosporin, is a common antibiotic of first choice. Its proconvulsant properties are well described in the literature,1, 2 but its importance as a potential cause of change in mental status is probably underestimated. We report a case of change in mental status related to nonconvulsive status epilepticus (NCSE) caused by the use of cefepime in an elderly, hospitalized patient. Our goal is to raise awareness about this uncommon and still underrecognized complication.
Case Report
A 72‐year‐old woman with stage III chronic kidney disease secondary to hypertension with a stable creatinine of 1.5 mg/dL (glomerular filtration rate (GFR) estimated by the modification of diet in renal disease (MDRD) at 36 mL/minute/1.73 m2) was admitted to the hospital for worsening of her chronic back pain. She had a past medical history significant for hyperlipidemia, asthma, and peripheral vascular disease, with breast cancer in remission since 1989. She had no history of seizures or cerebrovascular disease. Her medications were ibuprofen, oxycontin, cilastazol, acetaminophen/oxycodone, and an albuterol/empratropium inhaler. Her physical examination was remarkable only for decreased strength in the right lower extremity. Magnetic resonance imaging (MRI) of the lumbosacral spine showed signs consistent with an inflammatory process at the level of L4‐L5. A computed tomography (CT)‐guided biopsy was performed and confirmed a diagnosis of osteomyelitis on biopsy. Cultures from the biopsy grew Pseudomonas aeruginosa and treatment with intravenous cefepime at a dose of 1 g every 12 hours was initiated. Over the next 3 days, the patient had a gradual worsening of her mental status, leading to pronounced somnolence with occasional episodes of agitation during which she had no focal motor deficits. Her mental status declined to the point of unresponsiveness to simple verbal commands. She had not received any new medications other than cefepime. Her creatinine level was stable throughout this time period at 1.6 mg/dL. No other abnormalities were found on laboratory evaluation or on CT and MRI scans of the brain. An electroencephalogram (EEG) was markedly abnormal due to a generalized background slowing and disorganization with frequent bilateral paroxysmal epileptiform discharges, confirming the clinical diagnosis of subclinical generalized status epilepticus. Given that there were no other intrinsic neurological or metabolic reasons for this mental status change, and given that cefepime was the only new medication added before the patient started deteriorating, cefepime was discontinued and treatment for seizures was started with intravenous benzodiazepines. Over the next 2 days, her mental status returned to normal. She was soon discharged to a rehabilitation center.
Discussion
Beta‐lactam antibiotics have been described to induce seizures due to their direct and/or indirect inhibition of the gamma‐aminobutyric acid (GABA) system.1, 3 Previous experiments have shown a dose‐dependent effect on seizures, and suggest that the cephalosporin with the most pronounced proconvulsant effect is cefazolin.1, 3
Cefepime has been associated with neurological side effects such as headache, confusion, hallucinations, agitation, myoclonus, ataxia, seizures, and coma. Another underrecognized but critical side effect is NSCE. This is defined as seizure activity for more than 30 minutes, with cognitive and behavioral changes, but without convulsive clinical manifestations. This complication has been reported in the literature, but it is probably underrecognized.3‐7 The tendency for cefepime to produce more subclinical activity than the other cephalosporins is not well understood.
Cefepime is mainly eliminated though renal excretion (85%) and displays linear pharmacokinetic properties, thus its dose needs to be adjusted according to renal function. Consequently, in the case of renal dysfunction, accumulation of the drug is proportional to the degree of renal impairment. For NSCE, the most important risk factor is renal impairment, although cases in patients with normal kidney function have been described.36 Age, preexisting central nervous system (CNS) disease, sepsis, and cardiopulmonary bypass have also been reported as possible risk factors for NCSE.1
Cefepime can accumulate in the cerebrospinal fluid (CSF) in the setting of renal dysfunction, decreased protein‐binding capacity (as is sometimes seen in the elderly), and increased blood‐brain permeability in the setting of CNS infections. Accumulation of the drug in the CSF can lead to blockade of the GABA‐A receptor through a mechanism of competitive antagonism1, 8
The onset of NSCE varies between 1 and 16 days after initiation of cefepime therapy.3‐5, 7 It is frequently confused with delirium, since hospitalized patients treated with broad‐spectrum antibiotics such as cefepime frequently have other comorbidities and risk factors for delirium.
This can delay the diagnosis of NSCE due to a lack of awareness of this critical complication in the setting of renal dysfunction. In order to quantify the likelihood that the NSCE was related to cefepime and not to other causes, we calculated a Naranjo adverse drug events probability score, which consists of 9 questions on the relationship between the adverse event and the incriminated drug.9 Each answer is scored from 1 to +2 points. This score was designed to quantify the strength of the association between any adverse event and a pharmacological agent.
In our patient, the Naranjo score was 7 points, suggesting that the diagnosis of cefepime‐induced NCSE was probable.
The diagnosis of NCSE is made through a combination of a high index of clinical suspicion, specific findings on EEG, and improvement with withdrawal of the drug. Fatal outcomes have been reported.5, 6 Early and prompt recognition of the condition is crucial for the prevention of its morbidity and mortality.
The mainstay of treatment is prompt withdrawal of antibiotics and symptomatic treatment with benzodiazepines or barbiturates. Very severe cases with refractory seizures have been treated with hemodialysis. Phenytoin should be avoided as a treatment of this condition due to its lack of GABA‐agonist activity.
Conclusion
Cefepime can cause NCSE, predominantly in patients with renal dysfunction. Its frequency is probably underestimated in hospitalized patients with multiple comorbid conditions. Hospitalists should be aware of this unusual but critical relationship, especially in patients with renal failure. A high level of clinical suspicion and an emergency EEG are essential to obtain a prompt and accurate diagnosis.
- .Antibiotic‐induced convulsions.Crit Care Clin.1997;13:741‐762.
- ,.Cefepime neurotoxicity: case report, pharmacokinetic considerations, and literature review.Pharmacotherapy.2006;26(8):1169‐1174.
- ,,,,.Relationship between structure and convulsant properties of some β‐lactams antibiotics following intracerebroventricular microinjections in rats.Antimicrob Agents Chemother.1995;39:232‐237.
- ,,,,,.Cefepime‐ and cefixime‐induced encephalopathy in a patient with normal renal function.Neurology.2005;65(11):1840.
- ,,, et al.Cefepime induced neurotoxicity: an underestimated complication of antibiotherapy in patients with acute renal failure.Intensive Care Med.2002;28:214‐217.
- ,,,.Nonconvulsive status epilepticus due to cefepime in a patient with normal renal function.Epilepsy Behav.2006;8(1):312‐314.
- ,,, et al.Nonconvulsive status epilepticus associated with cephalosporins in patients with renal failure.Am J Med.2001;111(2):115‐119.
- ,,, et al.Evidence for the involvement of GABA(A) receptor blockade in convulsions induced by cephalosporins.Neuropharmacology.2003;45(3):304‐314.
- ,,, et al.A method for estimating the probability of adverse drug reactions.Clin Pharmacol Ther.1981;30(2):239‐245.
Hospitalized patients with sepsis or severe nosocomial infections are frequently treated empirically with broad‐spectrum antibiotics. Cefepime hydrochloride, a fourth‐generation cephalosporin, is a common antibiotic of first choice. Its proconvulsant properties are well described in the literature,1, 2 but its importance as a potential cause of change in mental status is probably underestimated. We report a case of change in mental status related to nonconvulsive status epilepticus (NCSE) caused by the use of cefepime in an elderly, hospitalized patient. Our goal is to raise awareness about this uncommon and still underrecognized complication.
Case Report
A 72‐year‐old woman with stage III chronic kidney disease secondary to hypertension with a stable creatinine of 1.5 mg/dL (glomerular filtration rate (GFR) estimated by the modification of diet in renal disease (MDRD) at 36 mL/minute/1.73 m2) was admitted to the hospital for worsening of her chronic back pain. She had a past medical history significant for hyperlipidemia, asthma, and peripheral vascular disease, with breast cancer in remission since 1989. She had no history of seizures or cerebrovascular disease. Her medications were ibuprofen, oxycontin, cilastazol, acetaminophen/oxycodone, and an albuterol/empratropium inhaler. Her physical examination was remarkable only for decreased strength in the right lower extremity. Magnetic resonance imaging (MRI) of the lumbosacral spine showed signs consistent with an inflammatory process at the level of L4‐L5. A computed tomography (CT)‐guided biopsy was performed and confirmed a diagnosis of osteomyelitis on biopsy. Cultures from the biopsy grew Pseudomonas aeruginosa and treatment with intravenous cefepime at a dose of 1 g every 12 hours was initiated. Over the next 3 days, the patient had a gradual worsening of her mental status, leading to pronounced somnolence with occasional episodes of agitation during which she had no focal motor deficits. Her mental status declined to the point of unresponsiveness to simple verbal commands. She had not received any new medications other than cefepime. Her creatinine level was stable throughout this time period at 1.6 mg/dL. No other abnormalities were found on laboratory evaluation or on CT and MRI scans of the brain. An electroencephalogram (EEG) was markedly abnormal due to a generalized background slowing and disorganization with frequent bilateral paroxysmal epileptiform discharges, confirming the clinical diagnosis of subclinical generalized status epilepticus. Given that there were no other intrinsic neurological or metabolic reasons for this mental status change, and given that cefepime was the only new medication added before the patient started deteriorating, cefepime was discontinued and treatment for seizures was started with intravenous benzodiazepines. Over the next 2 days, her mental status returned to normal. She was soon discharged to a rehabilitation center.
Discussion
Beta‐lactam antibiotics have been described to induce seizures due to their direct and/or indirect inhibition of the gamma‐aminobutyric acid (GABA) system.1, 3 Previous experiments have shown a dose‐dependent effect on seizures, and suggest that the cephalosporin with the most pronounced proconvulsant effect is cefazolin.1, 3
Cefepime has been associated with neurological side effects such as headache, confusion, hallucinations, agitation, myoclonus, ataxia, seizures, and coma. Another underrecognized but critical side effect is NSCE. This is defined as seizure activity for more than 30 minutes, with cognitive and behavioral changes, but without convulsive clinical manifestations. This complication has been reported in the literature, but it is probably underrecognized.3‐7 The tendency for cefepime to produce more subclinical activity than the other cephalosporins is not well understood.
Cefepime is mainly eliminated though renal excretion (85%) and displays linear pharmacokinetic properties, thus its dose needs to be adjusted according to renal function. Consequently, in the case of renal dysfunction, accumulation of the drug is proportional to the degree of renal impairment. For NSCE, the most important risk factor is renal impairment, although cases in patients with normal kidney function have been described.36 Age, preexisting central nervous system (CNS) disease, sepsis, and cardiopulmonary bypass have also been reported as possible risk factors for NCSE.1
Cefepime can accumulate in the cerebrospinal fluid (CSF) in the setting of renal dysfunction, decreased protein‐binding capacity (as is sometimes seen in the elderly), and increased blood‐brain permeability in the setting of CNS infections. Accumulation of the drug in the CSF can lead to blockade of the GABA‐A receptor through a mechanism of competitive antagonism1, 8
The onset of NSCE varies between 1 and 16 days after initiation of cefepime therapy.3‐5, 7 It is frequently confused with delirium, since hospitalized patients treated with broad‐spectrum antibiotics such as cefepime frequently have other comorbidities and risk factors for delirium.
This can delay the diagnosis of NSCE due to a lack of awareness of this critical complication in the setting of renal dysfunction. In order to quantify the likelihood that the NSCE was related to cefepime and not to other causes, we calculated a Naranjo adverse drug events probability score, which consists of 9 questions on the relationship between the adverse event and the incriminated drug.9 Each answer is scored from 1 to +2 points. This score was designed to quantify the strength of the association between any adverse event and a pharmacological agent.
In our patient, the Naranjo score was 7 points, suggesting that the diagnosis of cefepime‐induced NCSE was probable.
The diagnosis of NCSE is made through a combination of a high index of clinical suspicion, specific findings on EEG, and improvement with withdrawal of the drug. Fatal outcomes have been reported.5, 6 Early and prompt recognition of the condition is crucial for the prevention of its morbidity and mortality.
The mainstay of treatment is prompt withdrawal of antibiotics and symptomatic treatment with benzodiazepines or barbiturates. Very severe cases with refractory seizures have been treated with hemodialysis. Phenytoin should be avoided as a treatment of this condition due to its lack of GABA‐agonist activity.
Conclusion
Cefepime can cause NCSE, predominantly in patients with renal dysfunction. Its frequency is probably underestimated in hospitalized patients with multiple comorbid conditions. Hospitalists should be aware of this unusual but critical relationship, especially in patients with renal failure. A high level of clinical suspicion and an emergency EEG are essential to obtain a prompt and accurate diagnosis.
Hospitalized patients with sepsis or severe nosocomial infections are frequently treated empirically with broad‐spectrum antibiotics. Cefepime hydrochloride, a fourth‐generation cephalosporin, is a common antibiotic of first choice. Its proconvulsant properties are well described in the literature,1, 2 but its importance as a potential cause of change in mental status is probably underestimated. We report a case of change in mental status related to nonconvulsive status epilepticus (NCSE) caused by the use of cefepime in an elderly, hospitalized patient. Our goal is to raise awareness about this uncommon and still underrecognized complication.
Case Report
A 72‐year‐old woman with stage III chronic kidney disease secondary to hypertension with a stable creatinine of 1.5 mg/dL (glomerular filtration rate (GFR) estimated by the modification of diet in renal disease (MDRD) at 36 mL/minute/1.73 m2) was admitted to the hospital for worsening of her chronic back pain. She had a past medical history significant for hyperlipidemia, asthma, and peripheral vascular disease, with breast cancer in remission since 1989. She had no history of seizures or cerebrovascular disease. Her medications were ibuprofen, oxycontin, cilastazol, acetaminophen/oxycodone, and an albuterol/empratropium inhaler. Her physical examination was remarkable only for decreased strength in the right lower extremity. Magnetic resonance imaging (MRI) of the lumbosacral spine showed signs consistent with an inflammatory process at the level of L4‐L5. A computed tomography (CT)‐guided biopsy was performed and confirmed a diagnosis of osteomyelitis on biopsy. Cultures from the biopsy grew Pseudomonas aeruginosa and treatment with intravenous cefepime at a dose of 1 g every 12 hours was initiated. Over the next 3 days, the patient had a gradual worsening of her mental status, leading to pronounced somnolence with occasional episodes of agitation during which she had no focal motor deficits. Her mental status declined to the point of unresponsiveness to simple verbal commands. She had not received any new medications other than cefepime. Her creatinine level was stable throughout this time period at 1.6 mg/dL. No other abnormalities were found on laboratory evaluation or on CT and MRI scans of the brain. An electroencephalogram (EEG) was markedly abnormal due to a generalized background slowing and disorganization with frequent bilateral paroxysmal epileptiform discharges, confirming the clinical diagnosis of subclinical generalized status epilepticus. Given that there were no other intrinsic neurological or metabolic reasons for this mental status change, and given that cefepime was the only new medication added before the patient started deteriorating, cefepime was discontinued and treatment for seizures was started with intravenous benzodiazepines. Over the next 2 days, her mental status returned to normal. She was soon discharged to a rehabilitation center.
Discussion
Beta‐lactam antibiotics have been described to induce seizures due to their direct and/or indirect inhibition of the gamma‐aminobutyric acid (GABA) system.1, 3 Previous experiments have shown a dose‐dependent effect on seizures, and suggest that the cephalosporin with the most pronounced proconvulsant effect is cefazolin.1, 3
Cefepime has been associated with neurological side effects such as headache, confusion, hallucinations, agitation, myoclonus, ataxia, seizures, and coma. Another underrecognized but critical side effect is NSCE. This is defined as seizure activity for more than 30 minutes, with cognitive and behavioral changes, but without convulsive clinical manifestations. This complication has been reported in the literature, but it is probably underrecognized.3‐7 The tendency for cefepime to produce more subclinical activity than the other cephalosporins is not well understood.
Cefepime is mainly eliminated though renal excretion (85%) and displays linear pharmacokinetic properties, thus its dose needs to be adjusted according to renal function. Consequently, in the case of renal dysfunction, accumulation of the drug is proportional to the degree of renal impairment. For NSCE, the most important risk factor is renal impairment, although cases in patients with normal kidney function have been described.36 Age, preexisting central nervous system (CNS) disease, sepsis, and cardiopulmonary bypass have also been reported as possible risk factors for NCSE.1
Cefepime can accumulate in the cerebrospinal fluid (CSF) in the setting of renal dysfunction, decreased protein‐binding capacity (as is sometimes seen in the elderly), and increased blood‐brain permeability in the setting of CNS infections. Accumulation of the drug in the CSF can lead to blockade of the GABA‐A receptor through a mechanism of competitive antagonism1, 8
The onset of NSCE varies between 1 and 16 days after initiation of cefepime therapy.3‐5, 7 It is frequently confused with delirium, since hospitalized patients treated with broad‐spectrum antibiotics such as cefepime frequently have other comorbidities and risk factors for delirium.
This can delay the diagnosis of NSCE due to a lack of awareness of this critical complication in the setting of renal dysfunction. In order to quantify the likelihood that the NSCE was related to cefepime and not to other causes, we calculated a Naranjo adverse drug events probability score, which consists of 9 questions on the relationship between the adverse event and the incriminated drug.9 Each answer is scored from 1 to +2 points. This score was designed to quantify the strength of the association between any adverse event and a pharmacological agent.
In our patient, the Naranjo score was 7 points, suggesting that the diagnosis of cefepime‐induced NCSE was probable.
The diagnosis of NCSE is made through a combination of a high index of clinical suspicion, specific findings on EEG, and improvement with withdrawal of the drug. Fatal outcomes have been reported.5, 6 Early and prompt recognition of the condition is crucial for the prevention of its morbidity and mortality.
The mainstay of treatment is prompt withdrawal of antibiotics and symptomatic treatment with benzodiazepines or barbiturates. Very severe cases with refractory seizures have been treated with hemodialysis. Phenytoin should be avoided as a treatment of this condition due to its lack of GABA‐agonist activity.
Conclusion
Cefepime can cause NCSE, predominantly in patients with renal dysfunction. Its frequency is probably underestimated in hospitalized patients with multiple comorbid conditions. Hospitalists should be aware of this unusual but critical relationship, especially in patients with renal failure. A high level of clinical suspicion and an emergency EEG are essential to obtain a prompt and accurate diagnosis.
- .Antibiotic‐induced convulsions.Crit Care Clin.1997;13:741‐762.
- ,.Cefepime neurotoxicity: case report, pharmacokinetic considerations, and literature review.Pharmacotherapy.2006;26(8):1169‐1174.
- ,,,,.Relationship between structure and convulsant properties of some β‐lactams antibiotics following intracerebroventricular microinjections in rats.Antimicrob Agents Chemother.1995;39:232‐237.
- ,,,,,.Cefepime‐ and cefixime‐induced encephalopathy in a patient with normal renal function.Neurology.2005;65(11):1840.
- ,,, et al.Cefepime induced neurotoxicity: an underestimated complication of antibiotherapy in patients with acute renal failure.Intensive Care Med.2002;28:214‐217.
- ,,,.Nonconvulsive status epilepticus due to cefepime in a patient with normal renal function.Epilepsy Behav.2006;8(1):312‐314.
- ,,, et al.Nonconvulsive status epilepticus associated with cephalosporins in patients with renal failure.Am J Med.2001;111(2):115‐119.
- ,,, et al.Evidence for the involvement of GABA(A) receptor blockade in convulsions induced by cephalosporins.Neuropharmacology.2003;45(3):304‐314.
- ,,, et al.A method for estimating the probability of adverse drug reactions.Clin Pharmacol Ther.1981;30(2):239‐245.
- .Antibiotic‐induced convulsions.Crit Care Clin.1997;13:741‐762.
- ,.Cefepime neurotoxicity: case report, pharmacokinetic considerations, and literature review.Pharmacotherapy.2006;26(8):1169‐1174.
- ,,,,.Relationship between structure and convulsant properties of some β‐lactams antibiotics following intracerebroventricular microinjections in rats.Antimicrob Agents Chemother.1995;39:232‐237.
- ,,,,,.Cefepime‐ and cefixime‐induced encephalopathy in a patient with normal renal function.Neurology.2005;65(11):1840.
- ,,, et al.Cefepime induced neurotoxicity: an underestimated complication of antibiotherapy in patients with acute renal failure.Intensive Care Med.2002;28:214‐217.
- ,,,.Nonconvulsive status epilepticus due to cefepime in a patient with normal renal function.Epilepsy Behav.2006;8(1):312‐314.
- ,,, et al.Nonconvulsive status epilepticus associated with cephalosporins in patients with renal failure.Am J Med.2001;111(2):115‐119.
- ,,, et al.Evidence for the involvement of GABA(A) receptor blockade in convulsions induced by cephalosporins.Neuropharmacology.2003;45(3):304‐314.
- ,,, et al.A method for estimating the probability of adverse drug reactions.Clin Pharmacol Ther.1981;30(2):239‐245.