User login
Preventing and treating acute gout attacks across the clinical spectrum: A roundtable discussion
Gout is the most common form of inflammatory arthritis in men, and both gout and its root cause, hyperuricemia, are increasing in prevalence in both men and women. The result is that all medical generalists and specialists undoubtedly encounter patients with acute gouty flares in the outpatient or inpatient setting.
Gout increasingly presents in patients with a number of comorbidities, including chronic kidney disease, coronary artery disease, congestive heart failure, diabetes, and hypertension. As a result, medical management of the acute attack of gout, a notably painful and debilitating condition, is growing more complicated.
I asked three of my “goutophile” rheumatology colleagues from around the country and an infectious disease specialist/hospitalist to join me earlier this year in an unscripted roundtable discussion of the management of the acute gout attack. All have extensive real-world clinical experience, and the spirit of our conversation was clinically pragmatic. We aimed to put the relevant, though limited in number, clinical trials into a very practical perspective. We focused our conversation on the diagnosis and management of the acute attack in the outpatient and inpatient settings, essentially leaving undiscussed the long-term management of hyperuricemia—the underlying cause of acute gout attacks. The roundtable and this resulting journal supplement were supported by a CME grant from URL Pharma, Inc., the manufacturer of the branded formulation of colchicine.
I believe our discussion, transcribed and published here without external editing or input, will be of value to all practitioners faced with patients enduring an acute gout attack. We’ve added references as appropriate, and I hope the result is a balanced mix of experience-based medicine from thoughtful, seasoned clinicians with quantitative supporting data provided as available from the published clinical studies. This supplement has been independently peer reviewed by a rheumatologist content expert to exclude any content that could be perceived as unjustified bias for or against any specific therapy.
Gout is a disease that has fascinated me for 30 years, and my colleagues share my sense of wonder with the dramatic nature of the acute gouty flare (and its auto-resolution). I hope our shared interest shines through here and will assist all of us in managing patients with this chronic disorder punctuated by dramatically painful attacks.
As with all issues and supplements of Cleveland Clinic Journal of Medicine, I, as the journal’s editor-in-chief, welcome your feedback and comments.
—Brian F. Mandell, MD, PhD, FACR
HOW DO WE KNOW IT’S GOUT? WHEN PRESUMPTIVE DIAGNOSIS IS (AND ISN’T) ENOUGH
Brian F. Mandell, MD, PhD: In addressing acute gouty arthritis, let’s start with diagnosis. The gold standard for diagnosis of gout is synovial fluid aspiration and analysis for the presence of monosodium urate crystals. But what’s the practicality of aspiration and the need for synovial fluid analysis in a given patient at a given time? When is fluid analysis useful, and when is it absolutely necessary?
N. Lawrence Edwards, MD: It’s important to emphasize that synovial fluid analysis remains the gold standard of diagnosis. Ultrasonography of the peripheral joints is probably specific enough that it may soon be considered on par with fluid analysis for crystals, but we’re not there yet and it is not uniformly available. Less than 10% of patients who are diagnosed with gout have ever undergone synovial fluid aspiration, however, so presumptive, or “clinical,” diagnosis is very common.
A reliable presumptive diagnosis should be based on the image of a classic gout attack. The further an episode strays from a classic attack, the more presumptive the diagnosis is. So if a patient experiences the typical rapid explosion of symptoms from no pain to maximum pain over less than half a day, and if the symptoms are in the typical joints—the midfoot, the ankle, the first metatarsophalangeal joint (great toe)—that’s very consistent with a classic gout attack. Symptoms in the knee are more problematic because the two main conditions in the differential diagnosis of gout—septic arthritis and calcium pyrophosphate dihydrate (CPPD) crystal deposition disease (pseudogout)—also commonly affect the knee.
If the patient has had acute attacks that have self-resolved or gone away with therapy over a 5- to 8-day period, that too is a profile that’s typical enough for a presumptive diagnosis of gout, especially since septic arthritis doesn’t tend to do that. Pseudogout usually is a bit slower in onset. It can look virtually identical to gout, but the affected joints often aren’t the same and the attacks tend to last for weeks rather than days, which ultimately simplifies the differential diagnosis.
The setting where synovial fluid analysis is absolutely essential is when a septic process is suspected. If there’s any nagging concern that an infection might be present, the clinician should aspirate the joint or refer the patient to someone who can. The features of a septic process may include joint swelling, rigors, and fever, but all of those can be seen in gout, so they are not distinguishing.
Dr. Mandell: John, how comfortable can you be in making the distinction between crystal-induced arthritis versus infection at the start of a given attack?
John S. Sundy, MD, PhD: Larry’s point about the features typical for gout is key, but I also consider the patient’s comorbidities and general situation. For instance, if the patient is a healthy middle-aged man with no relevant past medical history, it’s easier to feel comfortable with a presumptive diagnosis of gout. But if I’m dealing with a hospitalized patient who’s had invasive procedures and has risk factors for bacteremia, it’s imperative to get synovial fluid to rule out a septic joint. There is, of course, a continuum between these two examples. It’s important to have a construct of what typical gout is. When a case is close to that construct, it’s reasonable to work with a presumptive diagnosis; the further a case is from the construct, the more important it is to get fluid. From there it’s just a matter of overcoming the practical issues that a lot of clinicians contend with in obtaining the aspiration and fluid analysis.
Although it’s fine to think about a presumptive diagnosis and initiate treatment accordingly, it’s still incumbent upon the clinician to strive to get a firm diagnosis at some point so that the patient and physician can be 100% confident in the diagnosis.
Peter A. Simkin, MD: When we focus on the initial diagnosis we may overlook the fact that a chronically gouty joint is more susceptible to infection, so that patients who have well-established gout may still require joint aspiration to exclude acquired infection. I agree that the knee is a major site to focus on, and the olecranon bursa is another common site of infection in gouty patients.
Dr. Mandell: That’s convenient, since both of those areas are easily aspirated by rheumatologists and nonrheumatologists alike. So I’m hearing a consensus that we look at the entire clinical picture, including the historical features and the location of symptoms, to assess whether an acute attack is more likely to be gout or infection. I would add that crystal disease, especially gout, is far more common than infection, and infection is particularly unusual in the midfoot.
No objective analysis has shown that any single feature, such as leukocytosis or fever, is reliably indicative of infection versus crystal disease. Yet when we look at the entire pattern, if there isn’t significant white blood cell elevation, if there aren’t rigors, if there isn’t significant fever, and if the patient has a history of self-limited attacks, that points to a clinical diagnosis of gout that we probably can be comfortable with. That said, joint aspiration is still required if there is not a rapid response to treatment.
There are settings, as John noted, where vigilance for infection is particularly indicated, notably in hospitalized patients. Jim, as a hospitalist, you may be less apt than us rheumatologists to stick a needle into a joint. What’s your comfort level in assessing the hospitalized patient with sudden eruption of a red, swollen, painful joint?
James C. Pile, MD: I agree that the setting is very important. Let’s say I have a patient with a well-established history of gout to whom I’ve been giving aggressive diuresis for heart failure exacerbation for 2 or 3 days. If he develops a hot, swollen ankle that’s consistent with his prior episodes, I won’t typically feel obligated to stick a needle in that joint, particularly not in an ankle, for which I’d want to consult a rheumatologist in any event. If it’s a patient who underwent cardiac catheterization or other instrumentation a couple of days ago, and if this patient is febrile and has sudden eruption of a hot, swollen knee, that’s a different story. Aspiration is absolutely required in that patient, who needs to be treated as having septic arthritis until proven otherwise. But there is a lot of gray area between these two scenarios.
Dr. Mandell: Speaking of willingness to aspirate joints, there’s a concern among nonrheumatologists about putting a needle into a joint that appears to have cellulitis over it. This can stand in the way of appropriate diagnosis since the response to crystals in a joint can produce a response that looks like cellulitis. If my examination suggests joint involvement, a cellulitislike appearance does not stand in the way of aspirating the joint or bursa, which might reveal a closed-space infection.
Dr. Edwards: One population that should make us nervous any time they get an acutely swollen joint are organ transplant recipients. When these patients get gout, it’s often atypical. Not only are they usually on cyclosporine, which can cause hyperuricemia and accelerated gout, but they’re also often taking corticosteroids, which dampen the acute symptoms of gout. So whether the acute swelling is caused by gout or infection, it may not appear to be quite as profound as it actually is. Additionally, these patients may not get fever or rigors. Despite all this, they are at high risk for infection, in light of their immunosuppression. When any transplant recipient presents with acute monoarthritis, the burden of proof is on us to show that it’s not an infected joint.
Dr. Mandell: I’d add that Larry’s caveat extends to patients on any immunosuppression, including the anti–tumor necrosis factor agents, and we have seen both infection and gout in these patients.
Dr. Simkin: It’s also worth noting that joint aspiration can be therapeutic. The exquisitely painful joint often is so painful because of distention of the joint capsule. If the joint is tapped, the capsule becomes decompressed and the needle track may leave a vent through which it can remain decompressed.
Dr. Edwards: Pointing out the therapeutic benefit of joint aspiration may even be one way to get patients to allow us to put a needle into an excruciatingly painful joint. So while we advocate joint aspiration as a diagnostic procedure, often an effective approach is to explain to patients that if you tap the joint, you can treat their pain much more rapidly. Instead of having another 18 to 36 hours of pretty significant pain even on the best oral treatments, with aspiration patients might be relatively free of pain within 18 to 24 hours.
Dr. Mandell: So I’m hearing that a history of prior events consistent with attacks of crystal-induced arthritis suggests that an acute flare is likely to be another attack of crystal disease. Almost all patients with acute postoperative flares have a history of gout, though that history rarely is noted in the chart and is usually obtained later. The problem with this reliance on history is that if it’s a previously damaged joint, it may have altered microvasculature and may be more prone to infection, as Peter noted. The literature on coexistent infection in crystal disease is small, which attests to its relative infrequency, but we need to be wary of this potential for coexistence, particularly in patients at risk of infection, such as transplant recipients and those with recent infection elsewhere and bacteremia. Acute joint swelling in such patients calls for arthrocentesis.
Dr. Edwards: One last criterion for when to do a joint aspiration is when a patient with well-established gout tells you an attack is different from previous attacks. If a patient says, “This isn’t like my previous gout attacks,” that’s a red flag that usually merits joint aspiration.
Dr. Mandell: We’d be remiss if we didn’t mention the longstanding “preliminary” criteria for the diagnosis of acute gout published by the American Rheumatism Association (now the American College of Rheumatology) in 1977.1 They’ve been used for clinical research studies and also applied in principle to clinical practice. There have been a couple of recent papers examining the limitations of these criteria. How do you use these “criteria”?
Dr. Edwards: Their biggest drawback is that they’re cumbersome. They also have recently been shown to lack specificity and, to some degree, sensitivity.2
Dr. Mandell: Testing of the ACR criteria has shown that we’ve had unfounded confidence in using them in practice. As you said, their sensitivity has been reported to be 80% and their specificity even lower, at 64%.3 So even a panel of clinical indicators of gout cannot be relied on in a setting where we have concern about the possible alternative diagnosis of infection.
CAN TRIALS OF SPECIFIC THERAPIES OFFER DIAGNOSTIC UTILITY?
Dr. Mandell: What about the response to anti-inflammatory therapy? Can we use the specificity of response to a drug like colchicine to determine whether an attack is more or less likely to be attributable to crystal disease?
Dr. Simkin: It’s certainly helpful, but entities other than crystal disease can respond to anti-inflammatory drugs, of course. For instance, some people with paraneoplastic problems will have acute arthritis that goes away with anti-inflammatory therapy. So I think it’s more a matter of how fast and complete the response is rather than whether there is a response.
Dr. Mandell: Consider the cases you’ve seen that ultimately turned out to be septic arthritis. What was the initial response in those cases to either nonsteroidal antiinflammatory drugs (NSAIDs), colchicine, or corticosteroids, and what conclusions can you draw from those cases?
Dr. Sundy: In my experience, signs and symptoms are often more lingering in these infectious cases. I think it’s fair to generalize that the response is not as rapid as with a typical gout flare. This underscores the importance of follow-up. If a patient presents to the emergency room and is treated and released, the critical question is how careful the follow-up is going to be at 24, 48, and 72 hours to see if improvement and resolution are occurring as expected. If you’re going to proceed on the basis of a presumptive diagnosis, there must be planned and frequent follow-up.
Dr. Edwards: I don’t think anyone thinks that a patient with an acute septic joint is going to get much improvement at all from oral colchicine or much more than a little fever reduction from NSAIDs without parenteral antibiotics. That joint is going to progress toward destruction within days. So John’s point is crucial that any time you make a presumptive diagnosis of gout, it’s incumbent on you to make sure that the pattern thereafter is one of resolution over time—not worsening—on the medications you use.
Dr. Pile: Even with corticosteroids, which are probably most likely to mask the clinical picture, in most cases I think it’s still fairly easy to sort things out based on response in the first 24 to 48 hours after initiating therapy. I don’t think patients with bacterial septic arthritis are going to remain better on corticosteroids.
Dr. Mandell: The time frame is crucial. Therapy for acute arthritis is ideally started fast, and patients frequently will be treated initially with an anti-inflammatory—in the community, probably more often with an NSAID than with colchicine or a steroid. Even if they actually have a septic joint, they may initially get a bit better on the anti-inflammatory, perhaps from the analgesic or antipyretic effects, but the benefit will clearly plateau and the attack will worsen again. So the period for peak vigilance is probably 1 to 2 days after therapy is started; if improvement is not maintained in this period, you should be circumspect regarding diagnosis of crystal disease and you must aspirate, or reaspirate, the joint.
Let’s explore specificity a bit further. Diagnostic specificity has historically been attributed to colchicine. People have even suggested that a diagnostic colchicine trial can be useful. How does that jibe with your experience with response to colchicine therapy in patients with entities other than gout?
Dr. Edwards: The main disease in the differential diagnosis of gout other than a septic process is pseudogout, or CPPD arthropathy, and patients with pseudogout may also respond to colchicine.
Dr. Simkin: Another entity that reportedly responds to colchicine is sarcoid, particularly sarcoid of the ankle.
Dr. Edwards: In fact, colchicine has been tried for treating the acute inflammatory symptoms of many of the diseases we see as rheumatologists. Some people swear by it, while others believe it doesn’t do much for these conditions. In any case, I don’t think its specificity for gout is very strong, and we have no basis to say that response to colchicine should serve as a diagnostic test.
Dr. Mandell: I agree, and the literature over the past few years on colchicine for treatment of acute and chronic pericarditis further argues against specificity of this drug for crystal-induced inflammation.
SERUM URATE LEVEL IS UNRELIABLE FOR DIAGNOSIS
Dr. Edwards: While we’re discussing diagnosis, nothing’s been said about serum urate levels, which I think many primary care physicians rely on heavily in the diagnosis of gout. We need to underscore that a lot of people who are hyperuricemic will never develop gout. At the same time, there’s also the phenomenon during an acute attack in which an acute uricosuria accompanies the initial inflammation, causing serum urate values to fall from what would otherwise be a high baseline to a level that looks normal. These declines may be between about 1.5 to 2.5 mg/dL, so that a patient presenting to the emergency room with a serum urate of, say, 7 mg/dL might actually have a baseline chronic level of 9 mg/dL.
Relying too heavily on serum urate levels can be misleading in either direction: someone with joint pain with serum urate elevation may be diagnosed with gout inappropriately, whereas someone who comes in with a gouty attack who has a serum urate level in the normal range may be thought not to have the disease.
Dr. Mandell: The fact that urate level didn’t come up in a conversation among rheumatologists with an interest in gout testifies that none of us uses serum urate in the diagnosis of acute arthritis. Your point is well taken, though, that serum urate is used for this purpose in the community but shouldn’t be, especially not in an acute setting.
Dr. Simkin: Indeed, I’ve seen a serum urate of 3.7 mg/dL during an acute, crystal-confirmed gout attack in a patient who was not on urate-lowering therapy.
Dr. Mandell: There’s also the issue that laboratory-defined normal levels of serum urate are not the same as biologically “normal” levels in the context of urate deposition. Levels that are in the normal range in the laboratory clearly can be above the saturation point of urate in physiologic tissues, which is about 6.7 mg/dL.
Dr. Simkin: Plus, many labs report their normal range as being up to 8 mg/dL, yet most gouty arthritis in the community probably occurs in patients with urate levels below 8 mg/dL, and this misstatement of normality certainly deserves attention.
Dr. Sundy: The issue of serum urate further underscores the importance of looking at gout as a longitudinal condition and not just as an acute episodic one. We would never treat and release a patient who came in with a severe hypertensive episode without insisting that further follow-up was indicated for the hypertension. Similarly, for a person who presents in the emergency room with gout there should be a longer-term strategy to make sure the patient understands the importance of followup to address the underlying hyperuricemia.
Dr. Mandell: Yes, there is a challenge with the system of care; the providers faced with the acute attack are not the ones who will ultimately be treating the disease and its associated comorbidities.
GENERAL APPROACH TO THE ACUTE GOUT ATTACK
Dr. Mandell: Let’s return to the patient who presents with an acutely swollen, painful joint. Let’s say gout is diagnosed with confidence, supported either by synovial fluid analysis or by the overall clinical details. What are your general considerations, Jim, for initial treatment in the hospital?
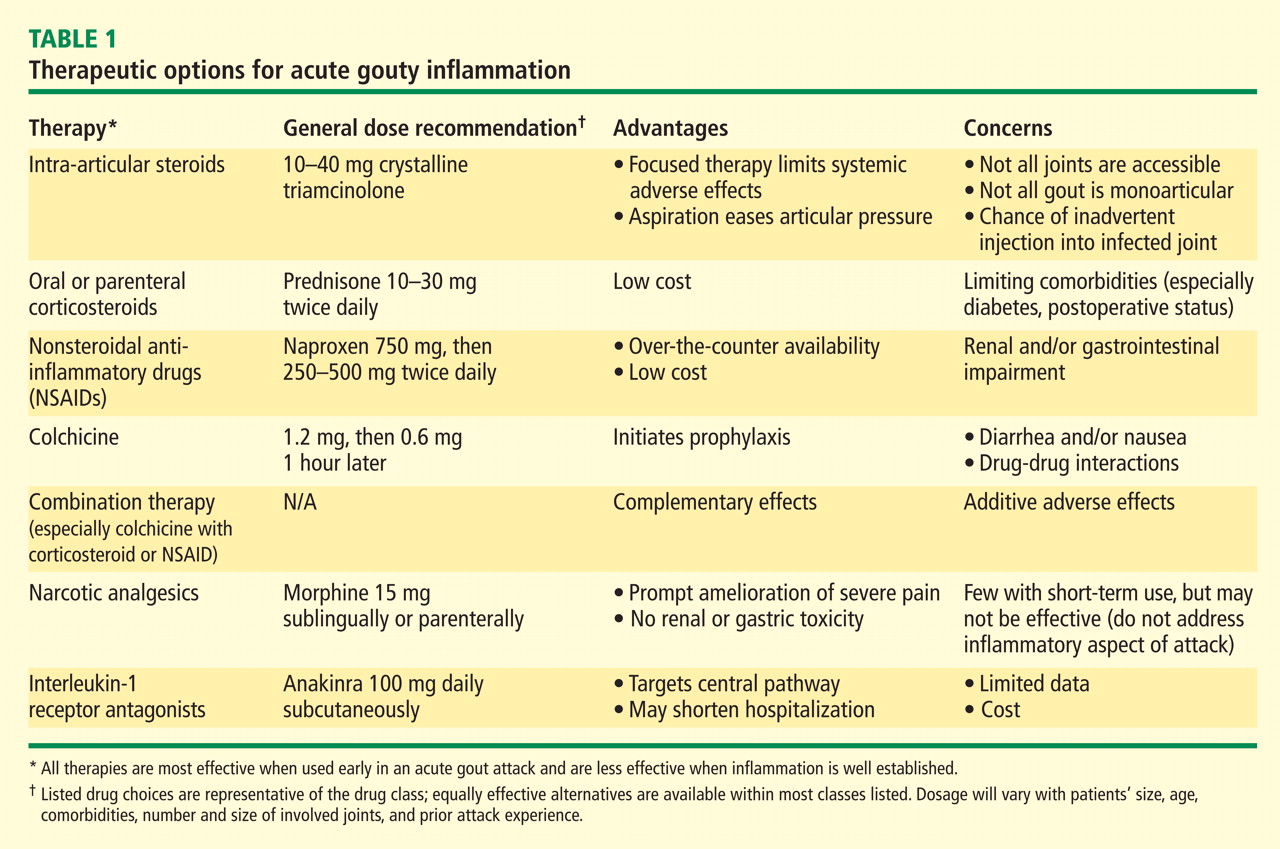
Dr. Mandell: We’ll come back and talk about each of these drug classes specifically, but what do my fellow rheumatologists tend to reach for as first-choice therapy?
Dr. Simkin: My thinking is very similar to Jim’s. As far as NSAIDs are concerned, so many of our patients have significant renal compromise that it is absolutely mandatory that we know what a patient’s renal function is before we treat acute gout with NSAIDs. I’ve seen more catastrophes from the use of NSAIDs in patients with renal compromise than from any other gout treatment scenario. So I probably wind up using steroids more often, but in the hospital setting you often have to deal with a surgeon who doesn’t want to use steroids. In such a case, if the patient also has renal problems, an agent that has entered the picture in our hospital is the interleukin-1 (IL-1) receptor antagonist anakinra, given in daily subcutaneous injections of 100 mg. When a patient is hospitalized for gout, it’s his severely painful joint that’s keeping him from going home, and in this situation anakinra in fact becomes a relatively inexpensive option, despite its absolute cost, when compared with the cost of the hospital bed.
Dr. Mandell: I think that’s the first time I’ve heard “anakinra” and “inexpensive” used in the same sentence.
Dr. Simkin: Of course biologics are terribly expensive, but so is hospitalization, and hospitals are bad places. We want to get our patients home, and in our experience this has been a very useful way to make that happen sooner.
Dr. Pile: I agree that an emphasis on hospital throughput is incredibly important. For the hospitalized patient with gout that’s preventing ambulation, that’s the issue that must be addressed before discharge is possible. Certainly the agent with the fastest onset of action is going to be very attractive.
Dr. Edwards: All of these agents for acute gout have a relatively fast onset of action if they’re given early in the disease process. Once you get out to a day and a half from the onset of symptoms or beyond, you’re fighting an uphill battle in terms of making a difference in the natural course of the attack. I believe that’s true of all three of the medication classes that are typically used.
My approach to the initial therapy choice is highly individualized, depending on what the patient’s been on before. If they’ve been on maintenance colchicine to prevent flares and then they flare, I usually won’t use colchicine for the acute attack; I will go with a steroid or an NSAID. If they’ve been on NSAIDs as preventive therapy and they flare, I might try low-dose colchicine for the flare or use steroids. In cases of a prolonged course, where the patient is in the hospital and it’s 3 or 4 days since symptom onset and a steroid taper or a trial of colchicine has failed, I’ve been very impressed with the ability of anakinra to suddenly bring the attack to a halt. Like Peter, I am on the cusp of looking at acute treatments a little differently, although there still aren’t a lot of data on IL-1 inhibition in this setting.
Dr. Sundy: There are data showing that a gout flare adds about 3 days to the hospital stay,4 so that’s a huge burden that intervention with IL-1 inhibition can really help to address.
Dr. Mandell: I think we’ve seen a trend over time toward corticosteroid therapy becoming more common, particularly in hospitalized patients. I think that’s partially related to more widespread use of appropriate deep vein thrombosis (DVT) prophylaxis, which means that more patients are on anticoagulant therapy, which is one more reason to shy away from NSAIDs, particularly the nonselective ones, in the hospital setting. Do you sense that trends in the outpatient setting have shifted, or do most clinicians still reach for NSAIDs?
Dr. Sundy: I think most people still reach for the NSAID, but it really depends on the comorbidities. I sense we’re seeing chronic kidney disease in a greater proportion of patients, and that’s probably creating a shift toward a bit more corticosteroid use. I like to reach for an NSAID as first-line therapy, but we really have to understand our patient’s overall comorbidity profile, including renal function, before doing so, as Peter said.
Dr. Edwards: I think NSAIDs are still the most commonly used acute treatment for gout. I used to calm myself when I used them by saying, “It’s only a 7- to 10-day course; how much trouble can you get the patient into?” Well, in that short a time I’ve had patients tip over into congestive heart failure because they had some renal decompensation beforehand and then had another 20% or 25% of their renal function knocked out with the NSAID, and they’d have extra sodium retention. I’ve seen GI bleeds develop in patients over that short a time. I don’t prescribe nonselective cyclooxygenase (COX) inhibitors anymore without also giving a proton pump inhibitor for stomach protection. I think that’s becoming a standard of how to use NSAIDs among rheumatologists, and it’s hard to get that stomach protection up and running in the very short time frame of an acute gout attack. So I’m personally tending away from NSAIDs; I use steroids more often and then the IL-1 inhibitor for special cases.
Dr. Mandell: Studies assessing the gastric risk of NSAIDs have shown that a GI bleed can be induced in as little as 2 or 3 days. I agree that there is a trend, at least among rheumatologists, to use a proton pump inhibitor when initiating outpatient NSAID therapy. This combination may still be cost-effective for many patients, as it can speed the return to their usual activities. But even in the outpatient setting steroids are becoming more common than they used to be.
DOSING FOR THE ACUTE ATTACK: BE AGGRESSIVE FROM THE START
Dr. Simkin: Whatever dose of steroids we use in the outpatient setting, I think it’s highly desirable to divide it. Rheumatologists are taught to use a single daily steroid dose in the morning, primarily to spare the adrenal glands. While that makes sense in patients on long-term steroid therapy, such as for lupus, it doesn’t necessarily make sense in gouty arthritis. I’ve seen patients whose gout flared up every night despite taking sufficient prednisone; when they divided the dose, their gout was controlled.
Dr. Mandell: I would generalize that further to stress the importance of using an adequate and aggressive dose of either steroids or NSAIDs for treating acute gout. A common mistake I notice in the community is the use of too low a dose of either steroids or NSAIDs. We should treat aggressively from the start with a full dose of these agents.
Dr. Edwards: Even more than the usual full dose, I would say. Many generalists have a concept of what an analgesic dose of an NSAID is, which is pretty low—in the case of ibuprofen, perhaps 400 mg three times daily. An anti-inflammatory dose is higher—perhaps 800 mg three times daily for ibuprofen. Most of us who’ve used NSAIDs for gout realize that we have to get even a little bolder than that from the start. It can be hard to convince some generalists to exceed what has been their ceiling of comfort for an NSAID, but doing so for the first 24 or 36 hours is important to getting the gout attack under control. Of course, the need for such aggressive dosing is all the more reason to make sure not to use an NSAID in a patient with significant renal disease or other comorbidities for which NSAIDs are problematic.
Dr. Mandell: And all the more reason to provide gastric protection.
Dr. Sundy: I would add that if a clinician is not comfortable using doses that high, that may be a good reason to think about choosing a different initial therapy, be it colchicine or steroids (if the reluctance is with NSAIDs) or NSAIDs (if the reluctance is with steroids).
ACUTE ANALGESIA: ANYTHING MORE THAN AN ADJUNCT?
Dr. Mandell: What about treating gout with acute analgesia alone? Consider a setting where you may want to avoid a drug with anti-inflammatory or antipyretic effects, perhaps in the postoperative period or when coexistent infection is a concern and you want to monitor the fever. Can you get by with using narcotic analgesia alone?
Dr. Sundy: I’m not impressed with that approach. Narcotics can play a role in managing a patient’s pain, but a narcotics-only approach will allow the flare to linger and not address complications of the flare or back-to-work issues. I view analgesics as cotherapy as opposed to single-agent therapy.
Dr. Mandell: Is narcotic analgesia effective even at treating the pain acutely?
Dr. Sundy: No. Gout pain is an exquisitely inflammatory pain, and the key is to tackle that inflammatory response. Analgesics are really just an adjunct.
Dr. Simkin: I totally agree, but they’re an important adjunct that we often overlook.
Dr. Edwards: I’ve been singularly unimpressed with narcotics. Gout is a cytokine-driven process that is intensely inflammatory. It’s like a lot of other types of pain that don’t respond fully to narcotics, such as herpetic neuralgia or uterine pain. Perhaps the benefit of narcotics in gout is their soporific effect—patients sleep through their attack. Yet if you touch the gouty joint or the patient moves it, the pain is just as intense as if the patient weren’t taking anything. So I don’t know that narcotics add anything unless the patient has other causes of pain. I don’t use them at all.
Dr. Sundy: I tend to make the option available to the patient. I say, “Here’s what we need to do to knock down this flare. And here’s something additional you can try for pain; we can see if it helps.” But I’ll emphasize the importance of improving the inflammatory piece.
Getting back to the postoperative setting, we should recognize that a strategy to just ride out a perioperative gout flare with an analgesic medication, perhaps because of concern about the effect of corticosteroids on wound healing or infection, can carry important risks. A patient treated that way will end up bedbound at precisely the time you want him or her to be moving around, so there’s now the risk of postoperative pneumonia and other complications that won’t get documented as complications of gout even though they actually are.
Dr. Mandell: Not to mention the untreated postoperative fever from the crystal-induced attack, which then leads to a work-up looking for DVTs, more blood cultures, and more radiographs, all the while extending the hospital stay and wasting resources. Jim, as a hospitalist, do you still see narcotics used initially as the primary treatment for gout flares in the hospital?
Dr. Pile: It depends on which hat I’m wearing. If I’m the hospitalist and the patient’s on my service, usually my antennae are up for the appearance of gout in the hospital, and the house staff may or may not recognize it if the process starts overnight. When I’m serving as the medical consultant, I sometimes encounter cases where the surgeons don’t recognize a gouty flare when it first appears, and I’ve had multiple patients in whom gout-induced postoperative fever triggers a consultation. In the latter case, it’s not uncommon to see narcotics being used in an attempt to treat gouty pain.
RELATIVE DRUG EFFICACY FOR ACUTE ATTACKS: HOW MUCH DO WE KNOW?
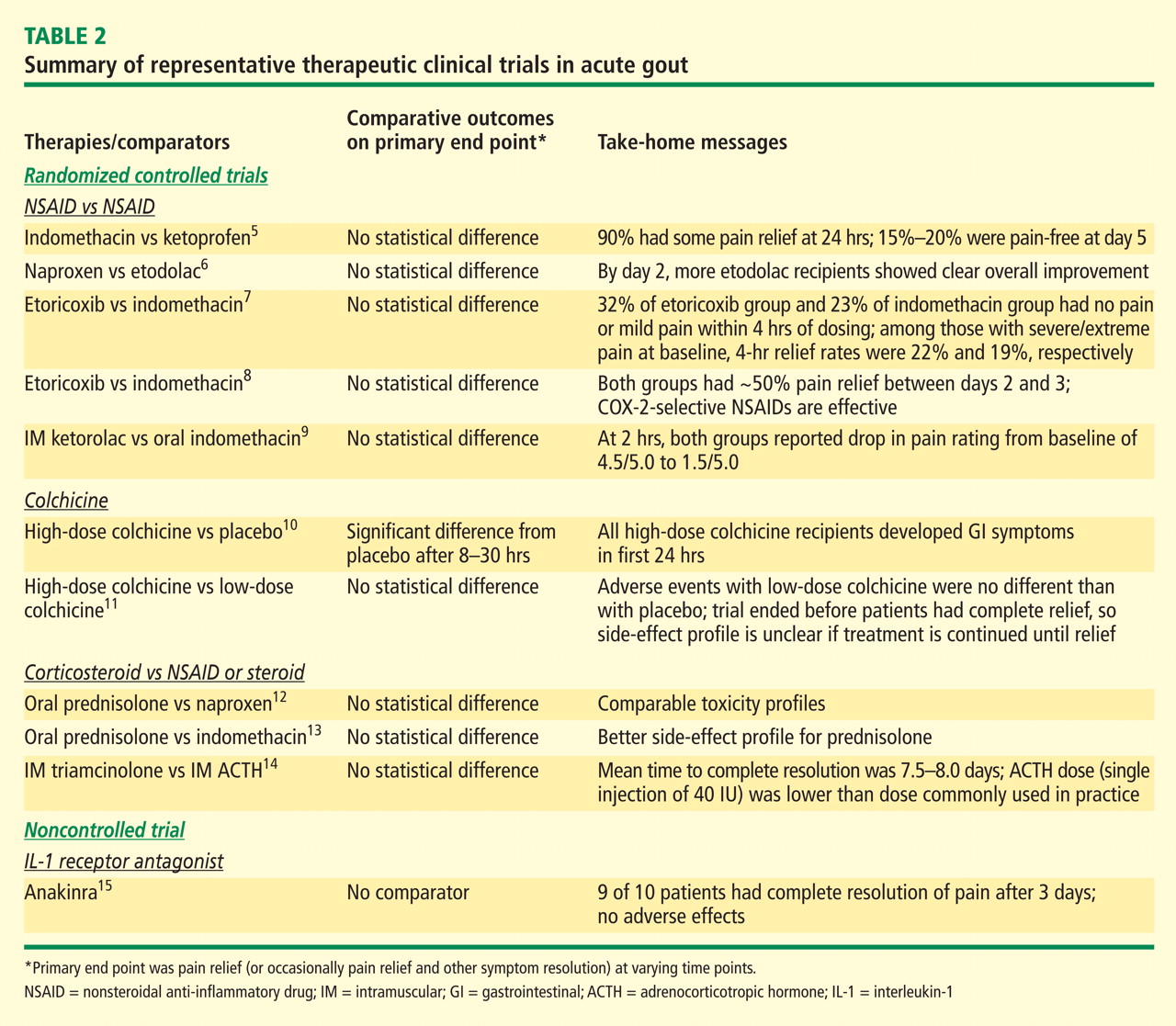
Dr. Edwards: If they’re given early, all of them have good—if not necessarily fast and ideal—effectiveness. The recent AGREE trial of colchicine (Acute Gout Flare Receiving Colchicine Evaluation) assessed pain reduction at 24 hours in patients randomized to three treatment groups: a low-dose colchicine regimen consisting of three 0.6-mg tablets (1.2 mg initially, followed by 0.6 mg 1 hour later); a more traditional high-dose colchicine regimen consisting of eight 0.6-mg tablets (1.2 mg initially, followed by 0.6 mg every hour for 6 hours), and placebo.11 Response, defined as a 50% or greater reduction in pain at 24 hours without rescue medication, was reported in 37.8% of patients in the low-dose colchicine group, 32.7% of patients in the high-dose colchicine group, and 15.5% of placebo recipients.
This is an excellent study that should finally take highdose colchicine for the treatment of acute gout off the table since it offered no improvement in efficacy but was significantly more toxic. However, while 50% improvement in 24 hours is a hard target to achieve, the fact that barely more than a third of subjects hit the target means we should still be looking for more effective approaches.
Dr. Simkin: Similarly, in the first controlled study of colchicine in acute gout, published by Ahern et al in 1987,10 23% of colchicine recipients noted a 50% reduction in their pain at 12 hours after starting treatment. That leaves more than three-quarters with little or no response within 12 hours, and gout is one of the most painful conditions we treat. We’d very much like to help our patients sooner than that. So while there are no head-to-head data comparing colchicine versus NSAIDs versus steroids, my impression too is that oral colchicine is a second-line choice for the acute gout attack.
Dr. Sundy: When we try to understand this literature, it’s important to look closely at how patients were ascertained. The AGREE trial evaluated patients who were enrolled when they were asymptomatic; they were given study medication and instructed on how to start it once a flare began. In contrast, most of the well-controlled NSAID trials captured patients as they came in with a flare, and they had to have had the flare no longer than 48 hours. I believe there’s a big difference between having had a flare for 12 hours versus 48 hours—and even as long as 72 hours in some corticosteroid trials. The longer the clock has been allowed to tick before treatment is started, the harder it is to achieve rapid symptom reduction.
Dr. Mandell: Obviously it’s difficult to compare between studies, but it’s interesting that in a study of the COX-2 inhibitor etoricoxib versus indomethacin,7 one of the largest studies of NSAID therapy for acute gout, approximately one-third of patients taking etoricoxib reported no pain or mild pain within 4 hours. This very rapid pain control is what we really want, since pain is the concern, along with eventual complete resolution, of course. So there really may be value in having something that has analgesic as well as anti-inflammatory effect, to Peter’s earlier point. Tackling both the pain and the inflammation that’s causing the pain certainly makes sense.
Dr. Edwards: Yes, and that etoricoxib study was designed the way people often are treated, with the unfortunate delay before the doctor is seen and the medication is started. The AGREE trial is how I hope everybody would be treated regardless of what they’re treated with. We’re probably never going to see a good head-to-head trial among the three main types of drugs we use for acute gout—the unpredictability of flares makes it extremely difficult.
Dr. Pile: I’m curious whether or not you, as rheumatologists who specialize in gout, are surprised by the results of AGREE. My experience with colchicine over time has been more favorable than what’s suggested by AGREE.
Dr. Edwards: How do you use the colchicine?
Dr. Pile: For more than a decade I’ve been using 0.6 mg three times daily. I thought that practice was unusual until I recently read the AGREE study and realized that a lot of you have been using that regimen for a while.
Dr. Mandell: Some gout patients will take a colchicine tablet when they feel the wisp of an attack coming on and it immediately will stop the attack. That gets to the lore that if you treat a flare very early on, you may be able to abort and treat an attack very quickly. Years ago, however, I was involved in an analysis of 100 patients treated with intravenous (IV) colchicine, and we found that treatment response was unaffected by whether patients were treated early or after a delay of 48 hours. I believe colchicine behaves like a different drug when given IV—and the IV form has since been withdrawn from the market for safety concerns—but this underscores that individual responses to various agents are quite unique. I think there are some patients who are exquisitely sensitive to colchicine, while others are exquisitely sensitive to an NSAID, and so on. This means we’d need a very large sample size to tease out that variation in a trial, and that’s not likely to happen.
Dr. Edwards: I too have had gout patients who tell me they get this premonition of an attack—a feeling that “something just isn’t right”—hours before they actually feel any pain from the attack. A lot of them tell me they’ve learned over time that if they take a colchicine tablet when they get this premonition—or an extra colchicine tablet, as some are on colchicine maintenance therapy (typically one or two tablets a day) plus whatever other background therapy they’re on—they don’t get the flare or the flare is diminished.
Dr. Mandell: I’d like to wrap up this portion by getting your sense of whether there’s a difference in efficacy for treating acute attacks among the drug classes we’ve discussed.
Dr. Edwards: I believe the IL-1 inhibitors are probably the most potent agents for aborting a gout attack, followed by steroids, which I think have a leg up on both colchicine and NSAIDs. The latter two options probably are equally effective in aborting an acute attack.
Dr. Sundy: Yes, I would rank them the same way.
THE ROLE OF COMORBIDITIES IN THERAPY CHOICE
Chronic kidney disease
Dr. Mandell: Let’s get more specific about comorbidities and therapy choice, starting with chronic kidney disease. What’s the threshold at which renal compromise starts to influence your choice of therapy for an acute attack of gout?
Dr. Sundy: I have a very low threshold for avoiding NSAIDs in that setting, mainly because when I want to use NSAIDs, I want to use them at a very high dose, and even short-term use of high-dose NSAIDs can reduce creatinine clearance. So I would certainly avoid NSAIDs when the patient’s creatinine clearance is less than 60 mL/min, and I’d be even more cautious if the patient had underlying hypertension or congestive heart failure.
Dr. Mandell: So even with the reversibility of almost all of the NSAIDs’ renal effects, you tend to stay away from them in that setting?
Dr. Sundy: Yes.
Dr. Edwards: I’m less guided by a single creatinine clearance level. I tend to look at patients in light of the 20% to 25% reduction in the glomerular filtration rate (GFR) that most NSAIDs will cause, even when used acutely. Though the NSAID effect is reversible, if that GFR reduction is going to make a difference to other compensated systems, primarily the heart, then the NSAID should be avoided.
Dr. Simkin: It’s my understanding that plasma flow to the kidney becomes prostaglandin dependent with renal insufficiency, and when we use antiprostaglandin agents we get into trouble on a mechanistic level. I’m not sure of the exact point at which that occurs, but I think we all recognize that serum creatinine is a fairly weak indicator of which patients have some limitation.
Dr. Mandell: So we agree on the need to be very cautious with the use of NSAIDs, recognizing that in the acute setting there is reversibly depressed renal blood flow from NSAIDs. The decreased blood flow will get better, but there’s the issue of what happens during fluid retention when there is coexistent congestive heart failure, diabetes, and chronic kidney disease—you may also occasionally bump up the potassium level. In general, most rheumatologists shy away from selective or nonselective NSAIDs in the setting of chronic kidney disease. Is that consistent with the use you see in the hospital, Jim?
Dr. Pile: It is. I would add that if patients are on certain other medications—especially angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or beta-blockers—there may be additive negative effects because of the potential for hyperkalemia if an NSAID were added to the mix.
Dr. Mandell: I would add to that list of concerning medications aminoglycosides, cyclosporine, and other nephrotoxic agents.
Diabetes
Dr. Mandell: What about diabetes? How does a patient’s diabetes enter into your choice of acute gout therapy in the outpatient and inpatient settings?
Dr. Edwards: Diabetes is especially problematic because it significantly affects two of our treatment choices—steroids and NSAIDs. The steroid doses needed to effectively treat an acute gout attack have a pretty profound effect on glucose levels in diabetic patients. That’s always of concern to me, and it’s certainly of concern to my patients with diabetes who are vigilant about monitoring their glucose levels, which may spike from 130 or 140 mg/dL to values in the high 200s. These elevations will last for the 7 to 10 days of treatment, and the longterm adverse effects of that period of hyperglycemia are not clear. If we put these patients on short courses of corticosteroids, perhaps we should be treating their diabetes more aggressively during that period, but I don’t think we have a history of doing that.
Dr. Pile: It’s very difficult to do in the short term. That’s especially the case with the diabetic patients I see as a hospitalist at a safety-net hospital. Many of my patients don’t have good glucose control at baseline: their levels of 250 mg/dL may go to 400 mg/dL or higher with acute corticosteroid therapy, so that’s clearly a problem.

Dr. Mandell: I try to avoid giving corticosteroids in the outpatient setting to a diabetic patient who is being maintained on oral hypoglycemic medications, particularly if efforts are being made to avoid more aggressive diabetes therapy. Steroids may be an option, though, for a patient who is on insulin, uses a pump, and is very comfortable measuring and managing his or her glucose at home. In the hospital setting, I’m more comfortable using corticosteroids in diabetics because I have much better control over their glucose; I can just increase the basal and premeal coverage.
Dr. Pile: I agree. With vigilance I can control almost anyone’s blood sugar in the hospital, but when patients leave the hospital on steroids, it becomes much more problematic.
Dr. Mandell: Right. You don’t know what’s going to happen when they go out and liberalize their diet on top of the steroids, because often it’s the postprandial surge that’s exacerbated by steroids.
Dr. Sundy: It can be very helpful to know at baseline what a patient’s spot blood sugar is before using a steroid, just as it’s important to know what the creatinine is before considering an NSAID. In the setting of an acute flare, the patient might already be quite hyperglycemic, and many of these patients probably don’t have the tools to manage this at home, so careful follow-up—coming back the next day for at least a blood sugar check—is critical to the proper care of these patients.
Dr. Mandell: When we talk about steroids we tend to think mostly about prednisone, but Peter mentioned injectable steroid. What about injectable adrenocorticotropic hormone (ACTH), which goes in and out of vogue as an agent to treat acute gout? What is your experience with using ACTH injections, specifically related to the issues of diabetes and heart failure?
Dr. Edwards: Before it got reformulated about 3 or 4 years ago and its price went way up, injectable ACTH was my drug of choice for treating acute gout. I had hardly any failures on it over the 10 to 15 years when it was my main means of treating acute gout. I would typically use 80 IU of ACTH gel, given subcutaneously, and the response within 12 hours was quite dramatic. About one in four patients would require a second injection at that 12-hour point, but almost everyone was symptomfree at 24 hours. This response was seen even in patients who were on fairly high-dose chronic steroids already, such as transplant patients.
This suggested that a mechanism other than just adrenocortical stimulation explained the benefit from ACTH. In fact, a lot of data have emerged suggesting that ACTH has a specific effect that is not related to adrenocortical stimulation.
Ten years ago an injection of ACTH gel would cost about $3 or $4, whereas today it costs $2,000 or more.29 I don’t think it’s justifiable at that cost.
Dr. Mandell: There’s also the issue of fluid retention with ACTH, and the higher doses, which seem to be more effective, are more likely to exacerbate congestive heart failure, which has to be a concern if you go this route.
Coronary artery disease
Dr. Mandell: Atherosclerosis, diabetes, and chronic kidney disease are increasingly recognized in the gout population, and coronary artery disease links with all of those. Does the presence of coronary artery disease enter into your choice of agents for a patient with an acute attack of gout?
Dr. Edwards: It doesn’t have as big an impact as diabetes does. To me, the priority in this setting is resolving the acute attack as quickly as possible, as I’ve seen my share of patients for whom the acute gout attack has been so painful and stressful that they’ve developed angina at the same time. So I might use steroids in patients with coronary disease because I’ll want to go with what’s likely to be most efficacious for quick resolution of the attack even though steroids may increase glucose and blood pressure and thus raise fluid retention as an issue.
Dr. Mandell: The presence of coronary disease implies the use of low-dose aspirin therapy. Despite the slight elevation in serum urate from low-dose aspirin, I recommend that my gout patients continue this for coronary protection. But even low-dose aspirin raises concerns of GI safety and even possible drug interactions with ibuprofen that could reduce the aspirin’s efficacy. Does this lessen your tendency to reach for an NSAID in these patients?
Dr. Edwards: I think the issue of cardiovascular safety and NSAIDs is still up in the air. The data that came out around the time the selective COX-2 inhibitors were released made us all think again about the role of prostaglandins and coronary blood flow and how blocking prostaglandins might have some bad effects. I don’t often use NSAIDs in patients with significant heart disease, simply because they don’t act fast enough to get patients through the attack quickly and I worry about the sodium retention and other problems putting stress on the heart.
Dr. Sundy: While coronary disease doesn’t create the acute dilemmas that diabetes does, it is a consideration in my therapy choice, and I generally avoid using NSAIDs.
Dr. Mandell: It is important to remember the additive gastric toxicity effect from low-dose aspirin and NSAIDs.
Infection
Dr. Mandell: Let’s turn to the hospitalized patient—a patient admitted to the medical service with infection. This patient is going to get fluids and drugs, and these are likely to raise or drop the serum urate level, which may precipitate a gout attack. From your perspective as an infectious disease consultant, Jim, how does the coexistence of a documented infection in the hospital—which you’re treating—factor into which agents to use or avoid when treating acute gout?
Dr. Pile: In practice, I try to avoid steroids in that situation. Theoretically, however, I’m not sure this scenario is really a contraindication to steroid use. I’m not aware of much solid data showing, for example, that patients with community-acquired pneumonia on appropriate antibiotic therapy have worse outcomes if they receive steroids than if they do not. Certainly there are a variety of serious infections for which we use steroids as a matter of course, including pneumococcal or tuberculous meningitis, tuberculous pericarditis, severe typhoid, and severe septic shock (the data for septic shock are murky, but there’s a role for at least steroid supplementation). As long as the antimicrobial therapy is appropriate, patients with serious infections are not necessarily going to do any worse with steroids.
Dr. Edwards: The biggest risk from steroids would be masking the symptoms of an unrecognized infection. If you know the infection is there and you’re treating it appropriately, I don’t think steroids are contraindicated.
Dr. Pile: A related risk is that steroids can make it more difficult to monitor the course of a known infection by blunting the overall response and bumping up the white blood cell count.
Dr. Mandell: My practice would fall in the realm of what Jim described as the theoretical course of action. If I knew what the infection was or felt comfortable in treating it, I would not shy away from systemic corticosteroids in the hospital setting, where I could monitor the patient. I think a corticosteroid may often be the safest drug to use in that setting, given the likelihood of comorbidities, at least compared with NSAIDs.
Dr. Simkin: Let’s say a patient comes in with acute knee pain and you aspirate the knee. The question arises whether you should wait for the culture results or go ahead and inject steroid while your needle is in the knee. I certainly have a much lower threshold to go ahead and inject while I’m in there, although I’d always be sure to get the culture results and revisit as needed. But if I pull out extremely inflammatory fluid during the aspiration, I’m probably not going to inject the steroid.
Dr. Edwards: And what if you actually do inject an infected joint with a corticosteroid—are you doing harm?
Dr. Simkin: I would like to think I’m not, and I may be helping.
Dr. Edwards: Yes, there are a lot of theoretical reasons why you might be helping, and there’s a small literature on steroids and infected joints that suggests you might actually slow down some bone resorptive steps and be doing various similar things.
Dr. Mandell: But in humans (a series of pediatric patients with septic arthritis30), those were systemic steroids, not intra-articular.
Dr. Edwards: In animal studies there is some amelioration of the destructive component with intra-articular steroids. The main point is that we must send off the fluid for culture when we inject steroid into a joint, and we must follow up on the cultures to make sure nothing is growing in a joint because we’re going to be calming it down. Whatever the inflammatory process is—whether infection or crystalline or rheumatoid—the patient is initially going to get better with a steroid injection. So we can’t use initial clinical improvement as the marker that we’ve done the right thing. We need to make sure the aspirate culture is negative.
Dr. Mandell: What about the use of colchicine or an IL-1 antagonist in the setting of infection?
Dr. Pile: There may be some theoretical concern, but I think low-dose colchicine would be a reasonable agent to use in the setting of infection.
Dr. Edwards: I don’t think there’s much concern since IV colchicine has been taken off the market. IV colchicine had a much more profound effect on bone marrow, and this resulted in a number of deaths from infection in immunosuppressed patients such as chemotherapy recipients. But oral colchicine in the doses we use for acute attacks and for maintenance probably doesn’t have a very profound effect on response to infectious processes or on white blood cell production (in the absence of chronic kidney disease).
Dr. Pile: Which raises the point that if the patient were on a potent inhibitor of the cytochrome P450 3A4 system, you might conceivably get into the same situation. For example, serious toxicity and even fatalities have been reported in patients taking colchicine and clarithromycin concurrently.31
Dr. Mandell: Peter, would you be comfortable giving an IL-1 antagonist to a hospitalized patient with pneumonia that’s being treated with antibiotics? Let’s say the patient has chronic kidney disease and diabetes and you’re looking for an agent to use.
Dr. Simkin: Well, I wouldn’t be comfortable, but I might feel I had to do it. If the organism were known, as you said, and the antibiotic therapy were appropriate, I might depend on that antibiotic coverage.
Dr. Sundy: Although I can envision some scenarios where I’d use the IL-1 antagonist in this situation, I’d be looking for an alternative. My concern is that people with any source of infection were excluded from clinical trials of anakinra, so we don’t have a good understanding of how it behaves in the setting of infection.
Dr. Edwards: And the fact that anakinra is not approved by the Food and Drug Administration (FDA) for treatment of acute gout should make us a bit uncomfortable.
Dr. Mandell: It’s interesting that the limited data do not show a lot of infections with IL-1 antagonist therapy other than when combined with anti–tumor necrosis factor therapy. The experience in periodic fever patients, who are on IL-1 antagonist therapy for many years, has not really shown this to be an agent predisposing to infection. I think this goes back to the point that if you document an infection and you’re treating it appropriately, some immunosuppressive or anti-inflammatory therapies may not be bad if you need to treat the attack of gout, particularly if it’s in a setting where you can monitor the patient well.
NSAIDs: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s drill down on the use of the specific classes of drugs, starting with NSAIDs. Larry made the point that NSAIDs should be used at high doses for acute gout attacks. What else is worth noting about the dosing and use of NSAIDs in this setting?
Dr. Sundy: In addition to that distinction between analgesic and anti-inflammatory dosing, it’s important that patients be told to treat at the appropriate dose intervals for whichever NSAID is chosen so that the anti-inflammatory benefit is maintained. I generally recommend that patients continue on that dosing regimen until they have substantial symptom improvement, at which point they can begin to back off, but that’s generally a 7- to 10-day treatment course, and typically closer to 10 days. Finally, I’m quick to use proton pump inhibitors or some sort of gastroprotective regimen with most patients that I start on NSAIDs in this setting.
Dr. Mandell: Your point about duration of therapy bears emphasizing. A common mistake with the use of NSAIDs is that they’re stopped too soon out of an understandable concern about their toxicity. Often they’re stopped when symptoms are still present, which means the attack hasn’t really resolved, and so it comes back. I tell patients to wait until their attack is completely gone, continue for a few days longer, and then stop the NSAID at that point.
Dr. Edwards: If we look at the natural course of disease, a patient’s first four to six gout attacks tend to be a bit shorter in duration, in the range of 5 to 7 days. As recurrent attacks occur over the decades, they tend to get more drawn out, lasting perhaps a couple or few weeks. We want treatment to at least cover the anticipated period of the natural attack since we’re not doing anything profound to the disease process itself, except perhaps with an IL-1 inhibitor. For instance, a typical treatment in emergency departments is an intramuscular injection of ketorolac. That may make patients feel good for the next 24 or 36 hours, but they’ll have a resumption of their acute attack at that point. I generally stick to 2 weeks as a reasonable treatment period. I might shorten that for a patient with diabetes in whom I have to use steroids or for a patient with congestive heart failure that I needed to put on an NSAID, but I generally try to cover patients with anti-inflammatory therapy for a couple of weeks.
Dr. Mandell: Your point about intramuscular ketorolac is important. A single shot is not likely to resolve the attack, and the intramuscular route will not ameliorate the gastric toxicity of this drug. Those are two common misperceptions.
What about the COX-2-selective NSAIDs? There’s only one that’s still marketed in the United States, celecoxib, but what’s your sense of these drugs as a class in terms of efficacy for treating gout?
Dr. Sundy: I think they’re effective. Only one COX-2 inhibitor, etoricoxib, has been studied in clinical trials for acute gout, and it’s not available in the United States, but I think the class as a whole is a reasonable choice. That’s especially true since the course of treatment is only 7 to 14 days. Even so, it’s still reasonable to use some gastric protection when giving celecoxib, as it does no harm and can add some reassurance. As with other NSAIDs, dosing of celecoxib for acute gout is higher than for other typical uses in the drug’s labeling.
Dr. Edwards: Yes, when I use celecoxib for acute gout I use 400 mg every 12 hours for the first 2 days and then take it down to 200 mg every 12 hours for the remainder of the treatment period.
Dr. Mandell: And the COX-2-selective NSAIDs do adversely affect renal function.
Dr. Simkin: One thing that troubles me when we talk about dosage for any of our agents is the large individual variation we encounter. I don’t think there’s a patient population with a wider variation of body sizes than the gout population. We see enormous patients with metabolic syndrome and several large joints, and we see frail elderly patients who have a single small joint affected. In fact, in addition to body size, the size of the joints involved can tell us a lot about the amount of inflammation we’re dealing with. Someone with two knees and an ankle affected is probably different from someone who has just a great toe affected. In light of these variables and so many others that come into play, such as age and comorbidities, I’m hesitant to recommend any particular dose because I try to make adjustments. Of course, there are no data that cover all these variables.
Dr. Mandell: But would you agree with the generality that high doses—higher than those we typically use for other musculoskeletal pains—are warranted in treating acute gout?
Dr. Simkin: Yes, but a high dose for a small elderly woman is different from a high dose for a young man who’s quite large.
Dr. Sundy: This sensitivity to individual variation can also apply to duration of therapy. As we noted, it’s not uncommon for veteran gout patients to find that starting treatment early—within 24 or 48 hours—may be sufficient to tamp down their symptoms, yet that’s an observation that is developed only over time in an individual patient. So it’s not a recommendation, but many patients will do fine with that. In those situations, I may not be so adamant about the need to continue treatment for a full 10 or 14 days.
STEROIDS: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s move to corticosteroids. A very common treatment used for acute gout is a tapered-dose regimen like the Medrol Dosepak (blister-pack) formulation of methylprednisolone. What strikes me is that this is a one-size-fits-all formulation, yet you’ve all just argued that there’s no one-size-fits-all approach. What are your thoughts?
Dr. Edwards: The Medrol Dosepak contains 21 4-mg tablets of methylprednisolone to be taken over the course of 6 days. Six tablets are taken the first day, and then one fewer tablet is taken each successive day through day 6. An obvious potential drawback is that the patient is off of medicine after 6 days, though that will be enough for some patients if they start treatment promptly.
I do use the Medrol Dosepak, and I always have patients fill a prescription ahead of time so that it’s available for quick initiation when they start to have their next attack. I instruct them to avoid storing it in a hot place, such as a car, but that it can be stored for up to 5 years at room temperature. Most of my patients like this approach, and if they do well with it they’re good about getting a new refill in advance of their next attack. This is a pretty handy way of letting patients be in charge of their attacks. If it doesn’t work and their symptoms continue, they call me and I’ll see them promptly.
A different approach is more appropriate for patients whose attacks tend to be more recalcitrant, perhaps because they’ve had gout for a long time or have bulky disease with lots of tophi. In those cases I’ll go with a steroid regimen that’s essentially double what a Medrol Dosepak would be—namely, prednisone 30 mg for the first 2 days then tapered down by 5 mg every other day. As Peter said, this approach would be overwhelming to a frail 82-year-old woman with a single tophus in her distal interphalangeal joint, but it tends to work well for a more typical 50-year-old obese male gout patient.
Dr. Sundy: I do some of the same things you describe, though my background in allergy has accustomed me to the steroid burst, so I’ll tend to use a dose of about 0.5 to 1.0 mg/kg/day for 4 or 5 days and then try to taper rapidly over another 2 to 4 days. I’m curious whether you think methylprednisolone offers advantages over prednisone.
Dr. Edwards: There are a few patients—roughly 5% to 15% of the population, according to various studies—who have trouble converting prednisone to its active form in their liver because they’re missing an enzyme. So I tend to use methylprednisolone since it has a benefit in that small segment of the population, but it otherwise doesn’t offer anything special beyond the convenient packaging that we discussed.
Dr. Mandell: I think convenience is the major factor. The blister-pack preparations are useful for patients who get confused with tapering. For patients who can make adjustments based on their symptoms, I find those personalized adjustments to be better than just giving them an absolute number of pills.
Dr. Pile: This issue of dosing duration is very important for generalists; just listening to your schedules is very instructive for me. I think a lot of nonrheumatologists, myself included, are inclined to use a one-size-fits-all approach. The mistake that I’ve been most apt to make, apparently, is using an insufficient duration of therapy. I’ve tended to use 40 mg prednisone for 5 or 6 days and then to stop rather than tapering the dose.
Dr. Mandell: From the cost perspective, steroids seem to be a very cost-effective option. The Medrol Dosepak, as a branded product, costs a bit more. Similarly, generic NSAIDs are inexpensive. There has been some feeling that nongeneric NSAIDs may offer advantages over generic NSAIDs, but I don’t know of any published evidence to support that. What are your thoughts?
Dr. Edwards: I agree with you. There has been this folklore about indomethacin and, before that, phenylbutazone being rather remarkable and specific drugs for gout. That hasn’t been borne out by the data, and virtually all of the NSAIDs work fairly comparably.
COLCHICINE: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s move to colchicine, which represents a unique class of anti-inflammatory agent. It has been available and widely used for many years, but it had not been formally licensed by the FDA until last year. Without corporate imperative and funding, data on its clinical use have been limited. As we mentioned, an IV form of colchicine was withdrawn from the market a few years ago after being associated with multisystem organ failure and death. Low-dose short-term colchicine doesn’t have those risks, and its efficacy in treating gout has been accepted for many years despite the dearth of clinical trial data until recently. How has colchicine been used in the past, and do the new data guide us on how it should be used differently, specifically for treating acute gout attacks?
Dr. Simkin: The so-called traditional regimen of starting with a pair of colchicine tablets and then giving another tablet every 1 or 2 hours until relief (or until GI toxicity developed) is one of the great embarrassments in the history of our field. The more recent trial evidence clearly indicates in the short term that there is no additional benefit beyond a very low dose. As we noted, this has been in the wind anecdotally for quite a few years, and it was confirmed by the recent AGREE trial.11 I find it interesting that the dosage used in AGREE—three 0.6-mg tablets—is very close to what we use for prophylaxis. We may well wind up concluding that any patient we’re seeing for the first time with an acute attack should be treated with that dose of colchicine, as an adjunct to another anti-inflammatory drug.
The use of colchicine as an adjunct for acute gout attacks was advanced in a review by Cronstein and Terkeltaub several years ago.32 Since we know that the mechanisms of action of the various drugs for acute gout are sufficiently different from each other, it makes sense to consider hitting the process at more than one spot. Having said that, one of the most crucial concerns in using colchicine is what other medications the patient is taking, given colchicine’s multiple drug interactions (Table 3). These include interactions with most statins, some antibiotics, and some calcium channel blockers, which share drug transporters with colchicine. So we have to be cautious about using colchicine with those other therapies.
Dr. Mandell: That’s also important in patients on chronic prophylactic colchicine therapy, as we know from recent pharmacokinetic studies and anecdotal case reports of colchicine and some of these other drugs.16–23 Do you think it’s as important in a patient on, say, chronic statin therapy who’s not on baseline colchicine, in whom you use a colchicine regimen of just two tablets followed by one tablet and then stop the colchicine right there? Is the potential for meaningful drug interactions as important in that setting?
Dr. Simkin: Probably not. One of the key issues is to educate patients about what to look for. Tell patients that if they notice numbness, weakness, or myalgia, you want to know about it. The evidence I’ve seen is that people who go into colchicine-induced rhabdomyolysis, for instance, develop it over a period of days, not hours, and if they stop the drug early, they probably will be okay. That’s the biggest concern. Neutropenia is the second concern.
Dr. Mandell: But fortunately, assuming no chronic kidney disease, those are generally patients taking colchicine chronically, not just acutely. We talked a bit before about our panel’s concerns about the efficacy of colchicine. Say you have a patient who has no history of prior colchicine use. How comfortable are you that treating such a patient with just the three-tablet colchicine regimen is likely to give the desired effect of rapid pain control and resolution of the attack?
Dr. Edwards: I’d expect that there wouldn’t be a profound or rapid improvement on that regimen, although the patient would certainly do better than if he or she were on nothing. I think that’s what the AGREE trial showed—that patients improve but still have significant morbidity days after initiating therapy.
Dr. Mandell: I think the AGREE trial did what it was intended to do: it showed that a low-dose colchicine regimen was as effective as and better tolerated than a high-dose colchicine regimen. This was something most of us believed, but it had never been demonstrated in a trial, and now there are good data to show it. And AGREE was consistent with the 1987 Ahern study10 in showing that the high-dose regimen is better than placebo. However, it in no way convinced me that colchicine in this dosing regimen is a monotherapy I’d be comfortable using to treat a severe acute attack. AGREE also didn’t give me any information as to how likely this approach would be to resolve an attack. For all the reasons we discussed, continued anti-inflammatory therapy is likely necessary beyond three pills, and I can’t be confident that a course of three colchicine pills is sufficient to maintain a drug level inside white cells to prevent other attacks from happening or a rebound attack once the regimen is over. I’m sure this approach will work for some patients; I just don’t know at the outset which ones or what percentage of patients will respond completely.
Dr. Edwards: I like the idea of combination therapy, of using colchicine as an adjunct. We’ve been talking up to now about monotherapies for acute gout because monotherapy is simple and patients and physicians alike prefer things that are fairly simple to use. But there is good theoretical reason to support using colchicine in combination with either NSAIDs or steroids, though there are probably no experimental data. I can’t imagine a reason for using corticosteroids and NSAIDs together, but pairing up colchicine with a steroid or an NSAID might allow you to keep the dose of the other agent lower and perhaps reduce the overall toxicity of your therapy. I’ve occasionally put patients on that type of combination therapy, and it’s probably a helpful way to go.
Dr. Mandell: For years my approach has been one of cotherapy, counting on either NSAIDs or corticosteroids to treat the acute attack by rapidly relieving the pain and inflammation and using colchicine in low doses—at a prophylactic dose, as Peter said—and extending it over a much longer period. I’m counting on the colchicine not to treat the acute attack but to prevent the next one and hopefully, for all the reasons we talked about, shorten the duration of therapy with the steroid or NSAID.
Dr. Edwards: Sure. The way I use colchicine is mostly for maintenance and for prophylaxis while I’m initiating urate-lowering therapy. I use it for a bit longer than most others might—I draw it out to at least 6 months after a complete resolution of symptoms, sometimes to 9 or 12 months. When a patient has a flare, I certainly have them maintain the colchicine if I’m going to treat them with something else, as I almost always do, and I believe that lets me get by with lower doses of the other agent I’m using for the acute attack.
Dr. Sundy: When I use colchicine as monotherapy for an acute flare, it’s because the patient has concluded from experience that it has worked effectively for him or her. But that’s not very common.
Dr. Mandell: There are times when the patient’s comorbidities lead me to conclude that colchicine may be the best drug to try for acute treatment. But I’m not comfortable assuming that three pills will do it all.
Dr. Pile: From the perspective of the generalist, who typically treats a simpler gout population than you do, what is the recommended treatment duration if colchicine is used as monotherapy for an acute attack? We now have this recommendation to use a single day’s worth of therapy, but you’re all expressing reservations with that.
Dr. Edwards: The recommendation in the drug’s current labeling is to take two 0.6-mg tablets at the first sign of a flare and a third 0.6-mg tablet 1 hour later. That was the low-dose regimen in AGREE, and I think it’s a good one, as only a very small percentage of patients will get the GI side effects with this regimen that occur with more prolonged therapy. The real question is what you do on day 2 and day 3 on out to day 7 or 10. If I were to use colchicine monotherapy, after that first day I’d prescribe one tablet three times daily for another 3 or 4 days and then drop down to one tablet twice daily for the rest of whatever duration of therapy I felt was justified. Of course, this is highly individualized and depends on how the drug is tolerated; probably more than 25% or 30% of patients can’t tolerate a three-times-daily regimen even for 3 or 4 days. But the total duration would be at least 7 days and up to 10 or even 14 days.
Dr. Simkin: Or months, because you’ve established a diagnosis.
Dr. Edwards: Yes—as prophylaxis, for which the dosage is one tablet once or twice daily. And then you’d leave the patient on prophylaxis and start urate-lowering therapy, if it hadn’t been started already.
Dr. Mandell: We’re now way past having trial data to support this, and this approach may negate the purported safety advantages of the low-dose regimen supported by AGREE.
Dr. Edwards: Yes, this is rank speculation.
Dr. Simkin: But there is evidence of the value of prophylaxis.
Dr. Mandell: There is certainly evidence for the value of prophylaxis, from a number of studies. We’ll get to that shortly. However, if we think just about treating the acute attack and the AGREE findings, I don’t think we can extend those results with confidence to a setting where we switch immediately from acute therapy into a prophylactic mode. Not that we shouldn’t consider doing that—it’s similar to my practice as well, though I tend to use lower doses as I extend out—but the existing trial data are limited to that very short window of acute treatment. Anything we do beyond that is an extension from our belief that the inflammatory response to crystals in a joint lasts longer. I believe it generally requires longer anti-inflammatory therapy, but we need to expect some frequency of side effects and drug interactions as we put patients on chronic colchicine therapy.
The other factor that used to be an advantage of this type of regimen was its low cost. For a long time colchicine was available from multiple manufacturers and was exceedingly inexpensive, although with less FDA guidance on its use and less oversight of its manufacturing. This enabled even patients with no insurance to go on a prophylactic therapy and remain on it. The environment is now quite different with the introduction of a single FDA-approved and -regulated branded product. So how do we deal with cost in the current environment?
Dr. Edwards: In terms of specific numbers, once-daily and twice-daily doses of unbranded colchicine used to cost about $6 and $12 a month, respectively, based on the average wholesale price. Now the cost of branded colchicine (Colcrys) is about $175 a month for the once-daily dose and $350 a month for the twice-daily dose, again based on the average wholesale price.
Dr. Simkin: I think all of us are deeply troubled by the high cost of this preparation and hope that the alternatives will still remain available to us.
Dr. Edwards: Yes, especially since it sounds like most of us consider colchicine our go-to drug for maintenance. It used to be that patients might be on maintenance colchicine therapy for 5 or 10 years without any addition of urate-lowering therapy. Today, however, when most of us put a patient on prophylactic therapy with colchicine, it’s with the intent of also addressing the hyperuricemia that’s at the heart of the disease process. In that case we still cover patients with colchicine prophylaxis during the initial months of urate-lowering therapy because of the elevated rate of gout flares—so-called mobilization flares—that occur during the process of uric acid reduction.
Dr. Mandell: The manufacturer of branded colchicine has introduced a patient assistance program to ease the drug’s cost for the financially needy. We will have to see how practical it is and how it affects our patients’ use of this medication.
ACUTE FLARES IN THE SETTING OF PROPHYLAXIS AND URATE LOWERING: WHAT TO DO?
Dr. Mandell: It sounds like most of us would treat patients prophylactically with colchicine for a while after an attack, certainly after several attacks, and then continue on chronic low-dose colchicine during the introduction and adjustment of the urate-lowering therapy that constitutes comprehensive gout treatment. Yet some patients will still have flares. So how does this baseline low-dose colchicine prophylaxis—a 0.6-mg tablet once or twice daily—influence your choice of therapy for those attacks? Do you bump up the colchicine dose, or do you absolutely avoid increasing the colchicine?
Dr. Sundy: I wouldn’t absolutely avoid an increase, but I would tend not to adjust it. I would maintain the dose and add a different agent on top of it—an NSAID or corticosteroid—to manage the acute attack. In general, if a patient is on regular colchicine prophylaxis, the assumption is that they have sufficient renal function to support that use, so such a patient may do fine with an NSAID.
Dr. Simkin: I trust we all agree that it’s critically important to continue the urate-lowering therapy the patient is taking if he suffers an attack. The same thing pertains to the prophylactic regimen. Both of these components should continue through the flare, but I agree that we usually want to add something to treat a flare, and it should be something the patient has on hand and can take without needing to talk to his physician if it’s the middle of the night.
Dr. Mandell: So I’m hearing that most of us would add something else for the short term on top of the prophylactic colchicine dose when a flare developed rather than increasing the colchicine dose.
Dr. Simkin: Yes, although one exception I’d be comfortable with is the example we discussed earlier of the patient who has found that taking an extra colchicine pill or two can help abort or diminish a flare without causing diarrhea.
Dr. Mandell: We cannot extrapolate the AGREE data to support a short-course regimen for an acute attack in patients who are already on chronic colchicine for prophylaxis.
PROPHYLAXIS IN THE PERIOPERATIVE SETTING
Dr. Mandell: Admission to the hospital for acute medical or surgical reasons is not infrequently associated with gout flares. Jim, have we in the rheumatologic community made it clear that chronic colchicine prophylaxis and urate-lowering therapy should ideally be continued when patients with gout are hospitalized for any reason, or is it a reflex for these drugs be held?
Dr. Pile: I think there’s a general appreciation, at least in the hospital medicine community, that these therapies should be continued in that situation. My sense is that these prophylactic gout therapies are viewed by most hospitalists as somewhat analogous to beta-blockers for chronic heart failure, which are understood to be necessary to continue when a patient is admitted with an acute exacerbation of heart failure. I don’t have data to support this contention, however.
Dr. Edwards: My experience suggests that discontinuing urate-lowering therapy during an acute gout attack is a common mistake in general practice. I think that’s been demonstrated in surveys of primary care physicians. And when the uratelowering therapy is stopped, the attack is often prolonged and made worse. Widespread misperception about this remains—it’s a big problem.
Dr. Mandell: Based on educational lectures I’ve given to primary care audiences, I agree with Larry. If I present a case question on a scenario like this, many physicians in the audience will anonymously indicate via the audience response system that they would stop a patient’s allopurinol if an acute attack occurred.
Let’s turn to the scenario of a patient who’s admitted for surgery—elective or emergent—who has a history of gout and is on prophylaxis. What’s the general routine in managing these medications in patients who are going to surgery?
Dr. Pile: In my preoperative consultations I’ve always stressed to gout patients and their surgeons that the postoperative period is a high-risk setting for acute exacerbation of gout. My routine recommendation in this setting is that patients continue any of their goutrelated medications through the postoperative period, but that often doesn’t happen. All sorts of medications are inappropriately stopped perioperatively, including statins and beta-blockers in addition to colchicine or allopurinol. Something I haven’t done routinely is to recommend introducing prophylaxis in the postoperative period for patients who were not already receiving it. Is that something you consider doing, given the likelihood of postoperative flare?
Dr. Edwards: It’s important to appreciate the reasons why gout flares postoperatively. It has a lot to do with why uric acid levels go up postoperatively: fluid changes, starvation, ketosis for any reason, lactic acidosis—these all cause pretty remarkable changes in systemic pH, which certainly is a trigger for that. The perioperative state also changes the exchange of uric acid in the kidney so that there is greater accumulation, and anything that rapidly bumps up serum urate levels while also creating a change in the pH is a setup for flares. So any recommendations about whether or not to give prophylaxis have a lot to do with the anticipated impact of the surgery on all of those physiologic parameters.
Dr. Sundy: As rheumatologists we’re usually called after the fact, so I can’t say I’ve given the issue of prophylaxis in the perioperative setting a lot of thought. I would add that patients who are undergoing cardiac catheterization, especially with stenting procedures that may require a large dye load, are another group that tends to be at increased risk of flare, especially in the setting of acute myocardial infarction.
Dr. Edwards: They could get a subtle change in renal function related to the dye load that isn’t throwing them into acute tubular necrosis or acute renal failure but is enough to reduce elimination of urate for long enough that they get this bump of uric acid that triggers the flare.
Dr. Mandell: The retrospective analyses of perioperative gout show that patients who come in with lower urate to begin with are less likely to have flares,33 which suggests that perioperative flares are less likely in patients who have been better controlled and managed. The question of whether to introduce prophylaxis at admission, given the likelihood of postoperative flare, hasn’t been studied formally. Despite this absence of data, I would be comfortable introducing colchicine at a low dose for prophylaxis in this setting, especially in a patient who has had frequent or recent attacks. The major exception would be for patients undergoing bowel surgery or similar procedures, since colchicine’s potential to cause diarrhea and other GI side effects is a main concern with its perioperative use. But a study of prophylactic colchicine before surgery in a “high-risk” population is a trial begging to be done. The challenge is that because the event rate is low, the sample size needed would be so large as to potentially make the study impossible.
Dr. Pile: One issue I encounter from time to time as a preoperative consultant involves patients who should be on chronic urate-lowering therapy but are not. Obviously, when I’m seeing them 7 days before surgery it’s not the time to contemplate doing anything besides perhaps starting colchicine.
Dr. Mandell: Yes, that would be a time to avoid starting urate-lowering therapy since a drop in serum urate is likely to precipitate gout flares, and that’s something you don’t want to happen in a postoperative setting. So just as we shouldn’t stop urate-lowering therapy preoperatively, we shouldn’t initiate it either. When the patient is already on urate-lowering therapy, it should be continued as close as possible to the time of surgery and restarted immediately thereafter. The fluids that are given perioperatively tend to drop the patient’s urate levels, so we often can get away with the brief window of discontinuation around the time of surgery so long as we restart the allopurinol postoperatively.
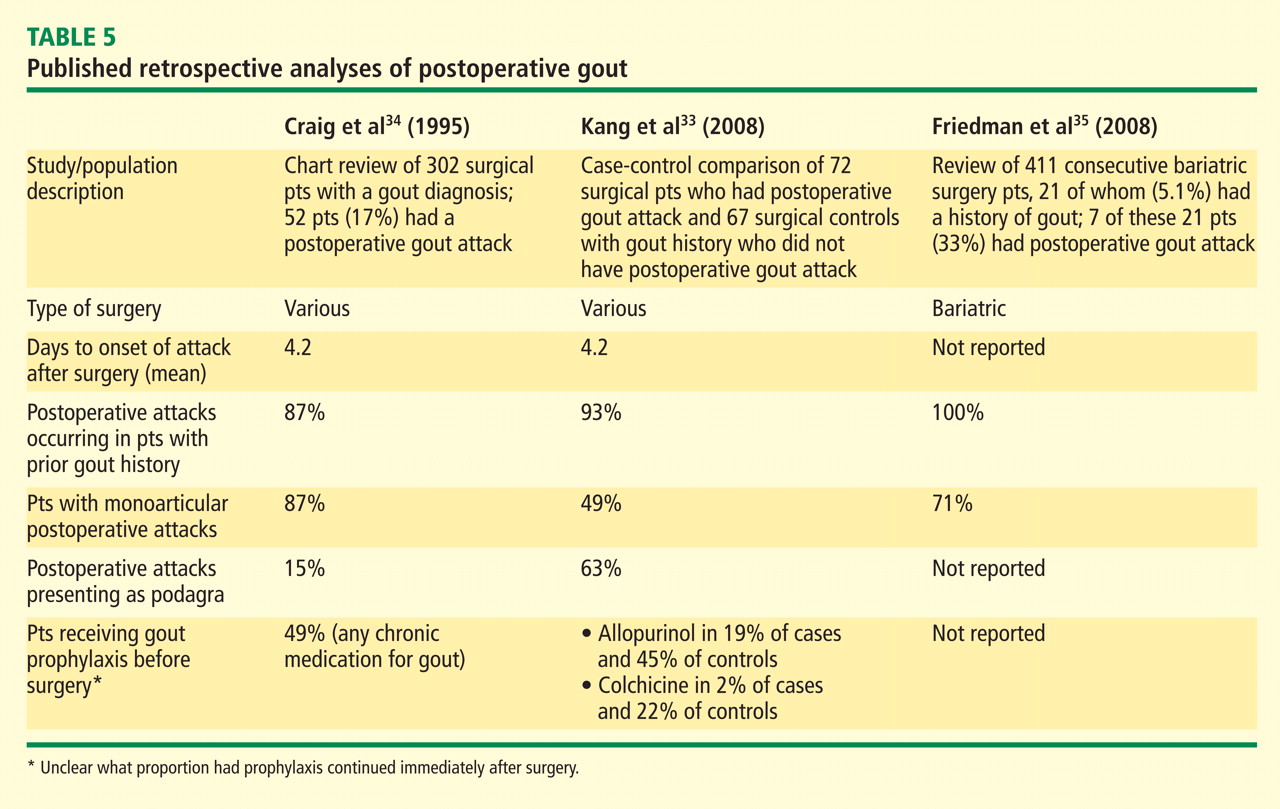
Dr. Edwards: There are certain surgeries, such as many cardiac procedures, for which the rooms are kept hypothermic, and that can be one more physiologic stimulus for an attack of gout, particularly in the body’s cooler joints. That’s a setting where podagra might be more common, whereas major abdominal surgery with a lot of bowel ischemia and lactic acidosis may render a wide range of joints more equally susceptible, although there are no data on this question.
Dr. Simkin: I’m not certain that postoperative gout is significantly different from hospitalization gout. If patients who are admitted for medical reasons—for instance, cardiac disease or pneumonia or ketoacidosis—also have an established diagnosis of gouty arthritis, they’re at elevated risk too.
Dr. Mandell: Yes, and that point—that preexisting gout is a major risk factor, whether or not it was known at the time of admission—came through from the studies Jim noted above and our experience. That piece of the history is often not obtained until an attack occurs, although more widespread use of electronic medical records may lessen this problem. It’s frustrating that attacks tend to occur about 4 days after surgery, which is typically around the time of planned discharge.
AVOIDING MOBILIZATION FLARES WHEN LOWERING URATE: START LOW, GO SLOW
Dr. Mandell: Let’s shift from this focus on the hospital setting as a precipitant for acute gout attacks and recall that the most predictable precipitant is when we pharmacologically reduce the serum urate level in an effort to decrease the total body load. As Larry noted, the down side of this very necessary intervention is that “mobilization flares” of gout are fairly predictable in this setting. While many of us have used prophylaxis to reduce the likelihood of these flares, we hadn’t had much data on duration of prophylaxis prior to some recent trials of urate-lowering therapy.36 So what are your practices now for trying to reduce the chance of acute attacks as you introduce urate-lowering therapy?
Dr. Edwards: A trigger for gouty attacks is the fluctuation of serum urate levels, be it from initiating or discontinuing urate-lowering therapy. So the approach when initiating urate-lowering therapy is to start low and go slow with your dose escalation. Most of us now buy into the notion of trying to lower the serum urate below 6.0 mg/dL for most patients, and perhaps by another 1.5 mg/dL or so for patients with bulky tophaceous disease. To get to that target, however, you should start with a fairly modest dose of whichever urate-lowering therapy you’re using—allopurinol, febuxostat, or (in rare cases) probenecid—and increase it over time. I tend to let several weeks pass between each dose escalation to ensure that the prior escalation has had time to drop the urate to its new nadir, and then I see what that level is. If it’s not at target, I will modestly escalate further.
Dr. Mandell: And we should emphasize that frequent initial monitoring of urate is appropriate to determine whether you’re reaching your target.
Dr. Edwards: That’s a critical element that isn’t done well in this country. Many studies show that regular monitoring of serum urate after initiation of urate-lowering therapy is actually a rarity. The approach to urate monitoring should be the same as for glucose monitoring with oral diabetes medications or blood pressure monitoring with antihypertensives. We should know what impact we’re having on the patient and continue to adjust the dose to find the appropriate level for the individual patient.
As with the therapies for acute gout, there is no onesize-fits-all dose regimen for urate-lowering therapy. A minority of patients will do fine on 100 mg/day of allopurinol. Most patients will still not reach their serum urate target at 300 mg/day of allopurinol, and we should consider gradual escalation to more generous doses as necessary, beyond 400 mg/day and up to the “maximum” recommended dose of 800 mg/day. Monitoring of serum urate should then continue even after we’ve found an appropriate dose to reach the target level since certain factors may change, altering concentrations of any of the urate-lowering therapies. For instance, a new drug may be added, or the patient may develop new comorbidities or a change in renal function. At this point the monitoring need not be as tight as during the initial period, but it should continue on a semiannual or yearly basis.
Dr. Simkin: I completely agree that going slow is desirable, and it’s very important for our patients to understand what we’re doing and what we’re aiming for. Every gout patient should know what his or her last serum urate level was, yet very rarely is that the case.
Dr. Edwards: Not only that, but they should know what the target level is so that they become an equal partner in their treatment plan, just as diabetics might be unsatisfied until their hemoglobin A1c is down to near 6.0%.
WHAT ROLE FOR PROPHYLAXIS DURING URATE LOWERING?
Dr. Mandell: Larry’s point that dropping the serum urate level slowly over time is less likely to induce an attack of gout is something we had suspected for a while and now have some direct trial evidence for. But no matter what we do, there’s always some likelihood of inducing an attack as we lower the urate level. So what do you do, Peter, to prevent those attacks?
Dr. Simkin: I try to get the patient on a regular dosage of colchicine prophylaxis. There’s good evidence that it’s effective. The dose is usually one or two tablets a day, largely depending on GI tolerance. It’s worth noting that more than a few of my elderly patients appreciate colchicine’s GI effects, as they provide some welcome relief from their chronic constipation. While that’s certainly not welcome by everyone, for some patients it makes colchicine prophylaxis an easier sell.
Dr. Sundy: Colchicine is my standard for prophylaxis as well. I think it’s highly effective.
Dr. Edwards: For how and when to start prophylaxis, my general recommendation is that it should precede the start of urate-lowering therapy by about 1 or 2 weeks. I think starting colchicine 2 weeks in advance is probably a good buffer for ensuring that it’s in the system. The urate-lowering therapies have very rapid onset—allopurinol will cause fluctuations in urate levels within a day or two—so the prophylaxis has to be fully on board.
Dr. Pile: Do you have a sense of how much more effective twice-daily dosing of colchicine is compared with once-daily dosing?
Dr. Edwards: I don’t have a good sense of it, but I tend to use twice-daily dosing unless renal function is a concern—specifically, if the patient’s GFR is between 50 and 60 mL/min/1.73 m2. And in such cases, you’re not really diminishing the dose with a once-daily regimen because the patient is keeping more of the drug around, in light of the renal insufficiency. That hearkens back to Peter’s earlier comments about baseline therapy with statins, some calcium channel blockers, and other drugs that will also require dose modification.
Dr. Mandell: The point is that we want to maintain a certain level of colchicine in all our patients for prophylaxis. It’s not clear that the blood level matters as much as the intracellular level, but we can’t routinely measure either. I think the answer to Jim’s question is that we don’t have data on whether once daily, twice daily, or even three times daily is the right regimen for colchicine prophylaxis; patients differ. I believe many of us start with twice-daily dosing on an empirical basis. If it’s not tolerated, typically for GI reasons, we’ll go down to once a day. If it’s tolerated but ineffective from the point of view of having flares, we may try to go to three times a day, assuming the patient’s not on other medications that would preclude that and doesn’t have chronic kidney disease or hepatobiliary disease.
WHAT ABOUT ALTERNATIVE PROPHYLAXIS OPTIONS?
Dr. Mandell: If a patient is truly intolerant of colchicine but has a reasonable GFR and no comorbidities, what about alternative prophylactic agents? The class to think about would seem to be NSAIDs, but we have no trial data to guide us with their use for prophylaxis. What’s been your experience with NSAIDs for prophylaxis of gout flares during the urate-lowering process?
Dr. Sundy: My experience is that NSAIDs are effective for this. I don’t have a notion of comparative effectiveness relative to colchicine, but I will use an NSAID in this setting when there has been GI intolerance to colchicine. The trick is the added responsibility of monitoring toxicity in terms of blood pressure, renal function, and hyperkalemia, as well as offering gastric protection, but I certainly have patients on NSAID prophylaxis.
Dr. Mandell: Do you feel compelled to use the same very high doses that we talked about for treating acute attacks of gout?
Dr. Sundy: No. I tend to use doses in more of a midrange area and make adjustments based on how the patient is doing. For example, I might increase the dose if a patient has a breakthrough flare.
Dr. Simkin: A situation where I’ve more often seen NSAIDs used for prophylaxis is in patients who have gout but are also taking NSAIDs on a regular basis for osteoarthritis. In that setting I’d be less inclined to add colchicine.
Dr. Pile: And I guess cost becomes an issue now, with the advent of branded colchicine, in a way that it hadn’t been before. At my local retail pharmacy generic naproxen costs 6 cents per 200-mg tablet, so some NSAIDs are very inexpensive.
Dr. Mandell: Colchicine used to have a similar cost invisibility as well. As Peter said before, our ability to provide cost-efficient prophylaxis is disappearing, and this may affect our ability to provide prophylaxis to some patients.
Dr. Sundy: I agree. Many times patients will stop taking drugs that are too expensive for them without telling their doctor. I sense that not using prophylaxis is a major impediment to adherence with urate-lowering drugs as well, as patients will stop those drugs because they sense appropriately that their gout has gotten worse without having been adequately educated about this risk. At the end of the day, we’re trying to eliminate gout as a medical issue for our patients, and anything that makes nonadherence more likely can be a big impediment to this goal.
Dr. Edwards: Patient education is absolutely crucial in this regard. We should step back and appreciate that these regimens can be very complicated and not terribly obvious from the patient’s perspective. First you have the disease gout, for which the treatment is allopurinol. You take that treatment and then your gout gets worse. Without good education to prepare patients for that, how can we expect them to respond, other than wanting to stop the allopurinol and probably thinking poorly of their doctor? Physicians have to educate patients on what to expect and how to stay the course. We must make clear from the start that the treatment of gout is a long-term process. We must explain that when acute flares occur the emphasis is on getting rid of the pain quickly but that the larger job of getting their disease under control by lowering their uric acid levels over time is a long haul. Both patient and physician have to be patient, go slowly, and make sure they don’t disrupt therapy just because symptoms are occurring.
Dr. Sundy: It sounds funny, but sometimes I’ve taken to congratulating patients whose gout flares after they start urate-lowering therapy. I say, “This is great—it means the drug is working, even if it doesn’t seem like it.”
SHOULD WE INTRODUCE URATE-LOWERING THERAPY AT THE TIME OF AN ACUTE ATTACK?
Dr. Mandell: Let’s consider the patient who presents with a first or second or third attack of gout but is not yet being treated for the underlying hyperuricemia. Should we introduce urate-lowering therapy at the time of the attack? After all, it’s a moment of opportunity, as the patient is “captive” and needs to be treated for his or her pain, so should we use that as a chance to also start the urate-lowering therapy that will be needed? There seem to be two schools of thought on this question. One is that if we start urate-lowering therapy immediately and drop the urate, the attack is likely to last longer. The other is that as we drop the urate by starting uratelowering therapy, we’re already treating them very aggressively for inflammation, so it may be an acceptable time to introduce urate-lowering therapy. What are your individual approaches?
Dr. Edwards: I’ve heard that argument of “we have them here now, let’s get it started.” I’d counter that when you start on this course of urate lowering, you need a very close connection to the patient during the long period of dose adjustment. Both the patient and physician need to understand that clearly. I’ve seen too many cases of a bad flare turning into a terrible flare when urate-lowering therapy is started during an acute attack. Patients are unhappy and get turned off of the idea of reintroducing allopurinol or any other urate-lowering therapy. So I don’t use that approach. Instead, I try to optimize the conditions for initiating urate-lowering therapy to minimize the chance that patients will have a bad experience. There’s no reason to rush, since most patients presenting with their first few flares have been hyperuricemic for 30 years, with urate slowly accumulating and depositing around their bodies. I’d promote an approach of letting the acute attack resolve and then getting the uratelowering therapy started about a month later.
Dr. Simkin: My approach is along the same lines as Larry’s. We all encounter gouty patients who say, “I can’t take allopurinol.” When you ask why, the most common response is, “It makes my arthritis worse.” Patients must understand that this is a hump they have to get over; we need to do everything we can to lessen that hump, and that involves good prophylaxis. But it’s also essential to educate them to expect the hump and to forewarn and forearm them in terms of how they’ll cope with it.
Dr. Sundy: A common rationale for starting urate-lowering therapy during the acute attack is that the patient may not follow up and you’ve got them on hand for intervention right now. But if follow-up is that unlikely, urate-lowering therapy is not going to be successful in any case, since frequent monitoring and dose adjustments are needed over many months. The notion that a patient can just be put on 300 mg of allopurinol and check back in 6 months is unrealistic.
Dr. Mandell: It sounds like we’re unanimous on this. One other consideration I would add is that I will avoid adding more than one drug in a patient at the same time whenever I can, to avoid confusion about which drug is causing any side effects that may develop.
REFINING THE PARADIGM: ACUTE ATTACKS AS THE ENTRY POINT TO A PROGRESSIVE DISEASE
Dr. Mandell: So our recommendation to the hospitalist who’s confronting acute gout is to treat the acute attack, start the education process, and make sure the patient understands this is a chronic disease that you’re going to make better in the hospital but which requires lifelong attention to prevent flaring up again. At that point the recommendation is to refer the patient back to the primary care physician or a rheumatologist with the understanding that the underlying hyperuricemia needs to be addressed and that the patient should make sure that happens.
Dr. Sundy: This raises some interesting handoff issues. How do we best ensure that the physician who’s going to pick up responsibility for chronic management is aware of these issues and on board with the necessary patient education efforts? There’s clearly an opportunity for doctor-to-doctor education here as well.
Dr. Pile: Yes, this is a microcosm of what we deal with all the time in the hospitalist world—safe transitions of care and trying to avoid information “voltage drop” as patients return to the care of their usual physicians.
Dr. Simkin: I think our education of both our colleagues and our patients needs to emphasize that this is not only a chronic disease but a progressive disease.
Dr. Edwards: That’s a great point, and it raises the question of the stage in the disease process where we should start urate-lowering therapy—a question without a clear-cut answer. Three decades ago, the thinking was that you should do it before the patient transitioned from acute intermittent gout to a more chronic and advanced tophaceous gout because of how terrible those joints looked and how destructive advanced gout is. Yet the destruction begins much earlier than that, and changes probably occur within the joint even before a patient has his or her first gout attack. There are soft-tissue changes around the joint as monosodium urate crystals accumulate, and the effects on bone and cartilage are occurring throughout the period of acute intermittent gout if the serum urate level hasn’t been lowered. As we’ve come to recognize this, we’ve pushed the initiation of uratelowering therapy earlier and earlier. The standard recommendation now is to start it as soon as a patient has two gout attacks within a year. Since 60% of patients have their second attack within a year of their first, that means initiating urate-lowering therapy after the second attack for about 60% of patients. An even larger majority of patients will be at that stage to qualify for urate-lowering therapy within the first few years after their initial attack.
Dr. Mandell: This is a good place to conclude, with a reminder that the acute gout attack is just one part of a chronic and potentially progressive disease. Although the acute attack is what gets the patient’s attention—and often the physician’s attention as well—it is really just the entrance into therapy for gout, a chronic disease.
- Wallace SL, Robinson H, Masi AT, et al Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum 1977; 20:895–900.
- Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol 2009; 15:22–24.
- Janssens HJEM, Janssen M, van de Lisdonk EH, et al Limited validity of the American College of Rheumatology criteria for classifying patients with gout in primary care [letter]. Ann Rheum Dis 2010; Mar 16[Epub ahead of print].
- Lin Y-H, Hsu H-L, Huang Y-C, et al Gouty arthritis in acute cerebrovascular disease. Cerebrovasc Dis 2009; 28:391–396.
- Altman RD, Honig S, Levin JM, Lightfoot RW. Ketoprofen versus indomethacin in patients with acute gouty arthritis: a multicenter, double blind comparative study. J Rheumatol 1988; 15:1422–1426.
- Maccagno A, Di Giorgio E, Romanowicz A. Effectiveness of etodolac (‘Lodine’) compared with naproxen in patients with acute gout. Curr Med Res Opin 1991; 12:423–429.
- Schumacher HR, Boice JA, Daikh DI, et al Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ 2002; 324:1488–1492.
- Rubin BR, Burton R, Navarra S, et al Efficacy and safety profile of treatment with etoricoxib 120 mg once daily compared with indomethacin 50 mg three times daily in acute gout: a randomized controlled trial. Arthritis Rheum 2004; 50:598–606.
- Shrestha M, Morgan DL, Moreden JM, et al Randomized doubleblind comparison of the analgesic efficacy of intramuscular ketorolac and oral indomethacin in the treatment of acute gouty arthritis. Ann Emerg Med 1995; 26:682–686.
- Ahern MJ, Reid C, Gordon TP, et al Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med 1987; 17:301–304.
- Terkeltaub RA, Furst DE, Bennett K, et al High vs low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum 2010; 62:1060–1068.
- Janssens HJ, Janssen M, van de Lisdonk EH, et al Use of oral prednisolone or naproxen for the treatment of gout arthritis: a doubleblind, randomised equivalence trial. Lancet 2008; 371:1854–1860.
- Man CY, Cheung IT, Cameron PA, et al Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med 2007; 49:670–677.
- Siegel LB, Alloway JA, Nashel DJ. Comparison of adrenocorticotropic hormone and triamcinolone acetonide in the treatment of acute gouty arthritis. J Rheumatol 1994; 21:1325–1327.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007; 9:R28.
- Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum 2009; 38:411–419.
- Colcrys [package insert]. Philadelphia, PA: Mutual Pharmaceutical Company; 2009.
- Simkin PA, Gardner GC. Colchicine use in cyclosporine treated transplant recipients: how little is too much? J Rheumatol 2000; 27:1334–1337.
- Dogukan A, Oymak FS, Taskapan H, et al Acute fatal colchicine intoxication in a patient on continuous ambulatory peritoneal dialysis (CAPD): possible role of clarithromycin administration. Clin Nephrol 2001; 55:181–182.
- Rollot F, Pajot O, Chauvelot-Moachon L, et al Acute colchicine intoxication during clarithromycin administration. Ann Pharmacother 2004; 38:2074–2077.
- Alayli G, Cengiz K, Canturk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother 2005; 39:1358–1361.
- Atmaca H, Sayarlioglu H, Kulah E, Demircan N, Akpolat T. Rhabdomyolysis associated with gemfibrozil-colchicine therapy. Ann Pharmacother 2002; 36:1719–1721.
- Roger U, Lins H, Scherrmann JM, et al Tetraparesis associated with colchicine is probably due to inhibition by verapamil of the P-glycoprotein efflux pump in the blood-brain barrier. BMJ 2005; 331:613.
- Levaquin [package insert]. Raritan, NJ: Ortho-McNeil; 2009.
- Phelan KM, Mosholder AD, Lu S. Lithium interaction with the cyclooxygenase 2 inhibitors rofecoxib and celecoxib and other nonsteroidal anti-inflammatory drugs. J Clin Psychiatry 2003; 64:1328–1334.
- Vistide [package insert]. Foster City, CA: Gilead Sciences; 2001.
- Hynninen V, Olkkola KT, Leino K, et al Effects of the antifungals voriconazole and fluconazole on the pharmacokinetics of S+ and R−-ibuprofen. Antimicrob Agents Chemother 2006; 50:1967–1972.
- Albengres E, Le Louet H, Tillement JP. Systemic antifungal agents. Drug interactions of clinical significance. Drug Saf 1998; 18:83–97.
- 2009 Red Book: Pharmacy’s Fundamental Reference. 113th ed. Los Angeles, CA: RAmEx Ars Medica; 2009.
- Odio CM, Ramirez T, Arias G, et al Double blind, randomized, placebo-controlled study of dexamethasone therapy for hematogenous septic arthritis in children. Pediatr Infect Dis J 2003; 22:883–888.
- Hung IF, Wu AK, Cheng VC, et al Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis 2005; 41:291–300.
- Cronstein BN, Terkeltaub R. The inflammatory process of gout and its treatment. Arthritis Res Ther 2006; 8( suppl 1):S3–S9.
- Kang EH, Lee EY, Lee YJ, et al Clinical features and risk factors of postsurgical gout. Ann Rheum Dis 2008; 67:1271–1275.
- Craig MH, Poole GV, Hauser CJ. Postsurgical gout. Am Surg 1995; 61:56–59.
- Friedman JE, Dallal RM, Lord JL. Gouty attacks occur frequently in postoperative gastric bypass patients. Surg Obes Relat Dis 2008; 4:11–15.
- Becker MA, Schumacher HR, Wortmann RL, et al Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450–2461.
Gout is the most common form of inflammatory arthritis in men, and both gout and its root cause, hyperuricemia, are increasing in prevalence in both men and women. The result is that all medical generalists and specialists undoubtedly encounter patients with acute gouty flares in the outpatient or inpatient setting.
Gout increasingly presents in patients with a number of comorbidities, including chronic kidney disease, coronary artery disease, congestive heart failure, diabetes, and hypertension. As a result, medical management of the acute attack of gout, a notably painful and debilitating condition, is growing more complicated.
I asked three of my “goutophile” rheumatology colleagues from around the country and an infectious disease specialist/hospitalist to join me earlier this year in an unscripted roundtable discussion of the management of the acute gout attack. All have extensive real-world clinical experience, and the spirit of our conversation was clinically pragmatic. We aimed to put the relevant, though limited in number, clinical trials into a very practical perspective. We focused our conversation on the diagnosis and management of the acute attack in the outpatient and inpatient settings, essentially leaving undiscussed the long-term management of hyperuricemia—the underlying cause of acute gout attacks. The roundtable and this resulting journal supplement were supported by a CME grant from URL Pharma, Inc., the manufacturer of the branded formulation of colchicine.
I believe our discussion, transcribed and published here without external editing or input, will be of value to all practitioners faced with patients enduring an acute gout attack. We’ve added references as appropriate, and I hope the result is a balanced mix of experience-based medicine from thoughtful, seasoned clinicians with quantitative supporting data provided as available from the published clinical studies. This supplement has been independently peer reviewed by a rheumatologist content expert to exclude any content that could be perceived as unjustified bias for or against any specific therapy.
Gout is a disease that has fascinated me for 30 years, and my colleagues share my sense of wonder with the dramatic nature of the acute gouty flare (and its auto-resolution). I hope our shared interest shines through here and will assist all of us in managing patients with this chronic disorder punctuated by dramatically painful attacks.
As with all issues and supplements of Cleveland Clinic Journal of Medicine, I, as the journal’s editor-in-chief, welcome your feedback and comments.
—Brian F. Mandell, MD, PhD, FACR
HOW DO WE KNOW IT’S GOUT? WHEN PRESUMPTIVE DIAGNOSIS IS (AND ISN’T) ENOUGH
Brian F. Mandell, MD, PhD: In addressing acute gouty arthritis, let’s start with diagnosis. The gold standard for diagnosis of gout is synovial fluid aspiration and analysis for the presence of monosodium urate crystals. But what’s the practicality of aspiration and the need for synovial fluid analysis in a given patient at a given time? When is fluid analysis useful, and when is it absolutely necessary?
N. Lawrence Edwards, MD: It’s important to emphasize that synovial fluid analysis remains the gold standard of diagnosis. Ultrasonography of the peripheral joints is probably specific enough that it may soon be considered on par with fluid analysis for crystals, but we’re not there yet and it is not uniformly available. Less than 10% of patients who are diagnosed with gout have ever undergone synovial fluid aspiration, however, so presumptive, or “clinical,” diagnosis is very common.
A reliable presumptive diagnosis should be based on the image of a classic gout attack. The further an episode strays from a classic attack, the more presumptive the diagnosis is. So if a patient experiences the typical rapid explosion of symptoms from no pain to maximum pain over less than half a day, and if the symptoms are in the typical joints—the midfoot, the ankle, the first metatarsophalangeal joint (great toe)—that’s very consistent with a classic gout attack. Symptoms in the knee are more problematic because the two main conditions in the differential diagnosis of gout—septic arthritis and calcium pyrophosphate dihydrate (CPPD) crystal deposition disease (pseudogout)—also commonly affect the knee.
If the patient has had acute attacks that have self-resolved or gone away with therapy over a 5- to 8-day period, that too is a profile that’s typical enough for a presumptive diagnosis of gout, especially since septic arthritis doesn’t tend to do that. Pseudogout usually is a bit slower in onset. It can look virtually identical to gout, but the affected joints often aren’t the same and the attacks tend to last for weeks rather than days, which ultimately simplifies the differential diagnosis.
The setting where synovial fluid analysis is absolutely essential is when a septic process is suspected. If there’s any nagging concern that an infection might be present, the clinician should aspirate the joint or refer the patient to someone who can. The features of a septic process may include joint swelling, rigors, and fever, but all of those can be seen in gout, so they are not distinguishing.
Dr. Mandell: John, how comfortable can you be in making the distinction between crystal-induced arthritis versus infection at the start of a given attack?
John S. Sundy, MD, PhD: Larry’s point about the features typical for gout is key, but I also consider the patient’s comorbidities and general situation. For instance, if the patient is a healthy middle-aged man with no relevant past medical history, it’s easier to feel comfortable with a presumptive diagnosis of gout. But if I’m dealing with a hospitalized patient who’s had invasive procedures and has risk factors for bacteremia, it’s imperative to get synovial fluid to rule out a septic joint. There is, of course, a continuum between these two examples. It’s important to have a construct of what typical gout is. When a case is close to that construct, it’s reasonable to work with a presumptive diagnosis; the further a case is from the construct, the more important it is to get fluid. From there it’s just a matter of overcoming the practical issues that a lot of clinicians contend with in obtaining the aspiration and fluid analysis.
Although it’s fine to think about a presumptive diagnosis and initiate treatment accordingly, it’s still incumbent upon the clinician to strive to get a firm diagnosis at some point so that the patient and physician can be 100% confident in the diagnosis.
Peter A. Simkin, MD: When we focus on the initial diagnosis we may overlook the fact that a chronically gouty joint is more susceptible to infection, so that patients who have well-established gout may still require joint aspiration to exclude acquired infection. I agree that the knee is a major site to focus on, and the olecranon bursa is another common site of infection in gouty patients.
Dr. Mandell: That’s convenient, since both of those areas are easily aspirated by rheumatologists and nonrheumatologists alike. So I’m hearing a consensus that we look at the entire clinical picture, including the historical features and the location of symptoms, to assess whether an acute attack is more likely to be gout or infection. I would add that crystal disease, especially gout, is far more common than infection, and infection is particularly unusual in the midfoot.
No objective analysis has shown that any single feature, such as leukocytosis or fever, is reliably indicative of infection versus crystal disease. Yet when we look at the entire pattern, if there isn’t significant white blood cell elevation, if there aren’t rigors, if there isn’t significant fever, and if the patient has a history of self-limited attacks, that points to a clinical diagnosis of gout that we probably can be comfortable with. That said, joint aspiration is still required if there is not a rapid response to treatment.
There are settings, as John noted, where vigilance for infection is particularly indicated, notably in hospitalized patients. Jim, as a hospitalist, you may be less apt than us rheumatologists to stick a needle into a joint. What’s your comfort level in assessing the hospitalized patient with sudden eruption of a red, swollen, painful joint?
James C. Pile, MD: I agree that the setting is very important. Let’s say I have a patient with a well-established history of gout to whom I’ve been giving aggressive diuresis for heart failure exacerbation for 2 or 3 days. If he develops a hot, swollen ankle that’s consistent with his prior episodes, I won’t typically feel obligated to stick a needle in that joint, particularly not in an ankle, for which I’d want to consult a rheumatologist in any event. If it’s a patient who underwent cardiac catheterization or other instrumentation a couple of days ago, and if this patient is febrile and has sudden eruption of a hot, swollen knee, that’s a different story. Aspiration is absolutely required in that patient, who needs to be treated as having septic arthritis until proven otherwise. But there is a lot of gray area between these two scenarios.
Dr. Mandell: Speaking of willingness to aspirate joints, there’s a concern among nonrheumatologists about putting a needle into a joint that appears to have cellulitis over it. This can stand in the way of appropriate diagnosis since the response to crystals in a joint can produce a response that looks like cellulitis. If my examination suggests joint involvement, a cellulitislike appearance does not stand in the way of aspirating the joint or bursa, which might reveal a closed-space infection.
Dr. Edwards: One population that should make us nervous any time they get an acutely swollen joint are organ transplant recipients. When these patients get gout, it’s often atypical. Not only are they usually on cyclosporine, which can cause hyperuricemia and accelerated gout, but they’re also often taking corticosteroids, which dampen the acute symptoms of gout. So whether the acute swelling is caused by gout or infection, it may not appear to be quite as profound as it actually is. Additionally, these patients may not get fever or rigors. Despite all this, they are at high risk for infection, in light of their immunosuppression. When any transplant recipient presents with acute monoarthritis, the burden of proof is on us to show that it’s not an infected joint.
Dr. Mandell: I’d add that Larry’s caveat extends to patients on any immunosuppression, including the anti–tumor necrosis factor agents, and we have seen both infection and gout in these patients.
Dr. Simkin: It’s also worth noting that joint aspiration can be therapeutic. The exquisitely painful joint often is so painful because of distention of the joint capsule. If the joint is tapped, the capsule becomes decompressed and the needle track may leave a vent through which it can remain decompressed.
Dr. Edwards: Pointing out the therapeutic benefit of joint aspiration may even be one way to get patients to allow us to put a needle into an excruciatingly painful joint. So while we advocate joint aspiration as a diagnostic procedure, often an effective approach is to explain to patients that if you tap the joint, you can treat their pain much more rapidly. Instead of having another 18 to 36 hours of pretty significant pain even on the best oral treatments, with aspiration patients might be relatively free of pain within 18 to 24 hours.
Dr. Mandell: So I’m hearing that a history of prior events consistent with attacks of crystal-induced arthritis suggests that an acute flare is likely to be another attack of crystal disease. Almost all patients with acute postoperative flares have a history of gout, though that history rarely is noted in the chart and is usually obtained later. The problem with this reliance on history is that if it’s a previously damaged joint, it may have altered microvasculature and may be more prone to infection, as Peter noted. The literature on coexistent infection in crystal disease is small, which attests to its relative infrequency, but we need to be wary of this potential for coexistence, particularly in patients at risk of infection, such as transplant recipients and those with recent infection elsewhere and bacteremia. Acute joint swelling in such patients calls for arthrocentesis.
Dr. Edwards: One last criterion for when to do a joint aspiration is when a patient with well-established gout tells you an attack is different from previous attacks. If a patient says, “This isn’t like my previous gout attacks,” that’s a red flag that usually merits joint aspiration.
Dr. Mandell: We’d be remiss if we didn’t mention the longstanding “preliminary” criteria for the diagnosis of acute gout published by the American Rheumatism Association (now the American College of Rheumatology) in 1977.1 They’ve been used for clinical research studies and also applied in principle to clinical practice. There have been a couple of recent papers examining the limitations of these criteria. How do you use these “criteria”?
Dr. Edwards: Their biggest drawback is that they’re cumbersome. They also have recently been shown to lack specificity and, to some degree, sensitivity.2
Dr. Mandell: Testing of the ACR criteria has shown that we’ve had unfounded confidence in using them in practice. As you said, their sensitivity has been reported to be 80% and their specificity even lower, at 64%.3 So even a panel of clinical indicators of gout cannot be relied on in a setting where we have concern about the possible alternative diagnosis of infection.
CAN TRIALS OF SPECIFIC THERAPIES OFFER DIAGNOSTIC UTILITY?
Dr. Mandell: What about the response to anti-inflammatory therapy? Can we use the specificity of response to a drug like colchicine to determine whether an attack is more or less likely to be attributable to crystal disease?
Dr. Simkin: It’s certainly helpful, but entities other than crystal disease can respond to anti-inflammatory drugs, of course. For instance, some people with paraneoplastic problems will have acute arthritis that goes away with anti-inflammatory therapy. So I think it’s more a matter of how fast and complete the response is rather than whether there is a response.
Dr. Mandell: Consider the cases you’ve seen that ultimately turned out to be septic arthritis. What was the initial response in those cases to either nonsteroidal antiinflammatory drugs (NSAIDs), colchicine, or corticosteroids, and what conclusions can you draw from those cases?
Dr. Sundy: In my experience, signs and symptoms are often more lingering in these infectious cases. I think it’s fair to generalize that the response is not as rapid as with a typical gout flare. This underscores the importance of follow-up. If a patient presents to the emergency room and is treated and released, the critical question is how careful the follow-up is going to be at 24, 48, and 72 hours to see if improvement and resolution are occurring as expected. If you’re going to proceed on the basis of a presumptive diagnosis, there must be planned and frequent follow-up.
Dr. Edwards: I don’t think anyone thinks that a patient with an acute septic joint is going to get much improvement at all from oral colchicine or much more than a little fever reduction from NSAIDs without parenteral antibiotics. That joint is going to progress toward destruction within days. So John’s point is crucial that any time you make a presumptive diagnosis of gout, it’s incumbent on you to make sure that the pattern thereafter is one of resolution over time—not worsening—on the medications you use.
Dr. Pile: Even with corticosteroids, which are probably most likely to mask the clinical picture, in most cases I think it’s still fairly easy to sort things out based on response in the first 24 to 48 hours after initiating therapy. I don’t think patients with bacterial septic arthritis are going to remain better on corticosteroids.
Dr. Mandell: The time frame is crucial. Therapy for acute arthritis is ideally started fast, and patients frequently will be treated initially with an anti-inflammatory—in the community, probably more often with an NSAID than with colchicine or a steroid. Even if they actually have a septic joint, they may initially get a bit better on the anti-inflammatory, perhaps from the analgesic or antipyretic effects, but the benefit will clearly plateau and the attack will worsen again. So the period for peak vigilance is probably 1 to 2 days after therapy is started; if improvement is not maintained in this period, you should be circumspect regarding diagnosis of crystal disease and you must aspirate, or reaspirate, the joint.
Let’s explore specificity a bit further. Diagnostic specificity has historically been attributed to colchicine. People have even suggested that a diagnostic colchicine trial can be useful. How does that jibe with your experience with response to colchicine therapy in patients with entities other than gout?
Dr. Edwards: The main disease in the differential diagnosis of gout other than a septic process is pseudogout, or CPPD arthropathy, and patients with pseudogout may also respond to colchicine.
Dr. Simkin: Another entity that reportedly responds to colchicine is sarcoid, particularly sarcoid of the ankle.
Dr. Edwards: In fact, colchicine has been tried for treating the acute inflammatory symptoms of many of the diseases we see as rheumatologists. Some people swear by it, while others believe it doesn’t do much for these conditions. In any case, I don’t think its specificity for gout is very strong, and we have no basis to say that response to colchicine should serve as a diagnostic test.
Dr. Mandell: I agree, and the literature over the past few years on colchicine for treatment of acute and chronic pericarditis further argues against specificity of this drug for crystal-induced inflammation.
SERUM URATE LEVEL IS UNRELIABLE FOR DIAGNOSIS
Dr. Edwards: While we’re discussing diagnosis, nothing’s been said about serum urate levels, which I think many primary care physicians rely on heavily in the diagnosis of gout. We need to underscore that a lot of people who are hyperuricemic will never develop gout. At the same time, there’s also the phenomenon during an acute attack in which an acute uricosuria accompanies the initial inflammation, causing serum urate values to fall from what would otherwise be a high baseline to a level that looks normal. These declines may be between about 1.5 to 2.5 mg/dL, so that a patient presenting to the emergency room with a serum urate of, say, 7 mg/dL might actually have a baseline chronic level of 9 mg/dL.
Relying too heavily on serum urate levels can be misleading in either direction: someone with joint pain with serum urate elevation may be diagnosed with gout inappropriately, whereas someone who comes in with a gouty attack who has a serum urate level in the normal range may be thought not to have the disease.
Dr. Mandell: The fact that urate level didn’t come up in a conversation among rheumatologists with an interest in gout testifies that none of us uses serum urate in the diagnosis of acute arthritis. Your point is well taken, though, that serum urate is used for this purpose in the community but shouldn’t be, especially not in an acute setting.
Dr. Simkin: Indeed, I’ve seen a serum urate of 3.7 mg/dL during an acute, crystal-confirmed gout attack in a patient who was not on urate-lowering therapy.
Dr. Mandell: There’s also the issue that laboratory-defined normal levels of serum urate are not the same as biologically “normal” levels in the context of urate deposition. Levels that are in the normal range in the laboratory clearly can be above the saturation point of urate in physiologic tissues, which is about 6.7 mg/dL.
Dr. Simkin: Plus, many labs report their normal range as being up to 8 mg/dL, yet most gouty arthritis in the community probably occurs in patients with urate levels below 8 mg/dL, and this misstatement of normality certainly deserves attention.
Dr. Sundy: The issue of serum urate further underscores the importance of looking at gout as a longitudinal condition and not just as an acute episodic one. We would never treat and release a patient who came in with a severe hypertensive episode without insisting that further follow-up was indicated for the hypertension. Similarly, for a person who presents in the emergency room with gout there should be a longer-term strategy to make sure the patient understands the importance of followup to address the underlying hyperuricemia.
Dr. Mandell: Yes, there is a challenge with the system of care; the providers faced with the acute attack are not the ones who will ultimately be treating the disease and its associated comorbidities.
GENERAL APPROACH TO THE ACUTE GOUT ATTACK
Dr. Mandell: Let’s return to the patient who presents with an acutely swollen, painful joint. Let’s say gout is diagnosed with confidence, supported either by synovial fluid analysis or by the overall clinical details. What are your general considerations, Jim, for initial treatment in the hospital?
Dr. Mandell: We’ll come back and talk about each of these drug classes specifically, but what do my fellow rheumatologists tend to reach for as first-choice therapy?
Dr. Simkin: My thinking is very similar to Jim’s. As far as NSAIDs are concerned, so many of our patients have significant renal compromise that it is absolutely mandatory that we know what a patient’s renal function is before we treat acute gout with NSAIDs. I’ve seen more catastrophes from the use of NSAIDs in patients with renal compromise than from any other gout treatment scenario. So I probably wind up using steroids more often, but in the hospital setting you often have to deal with a surgeon who doesn’t want to use steroids. In such a case, if the patient also has renal problems, an agent that has entered the picture in our hospital is the interleukin-1 (IL-1) receptor antagonist anakinra, given in daily subcutaneous injections of 100 mg. When a patient is hospitalized for gout, it’s his severely painful joint that’s keeping him from going home, and in this situation anakinra in fact becomes a relatively inexpensive option, despite its absolute cost, when compared with the cost of the hospital bed.
Dr. Mandell: I think that’s the first time I’ve heard “anakinra” and “inexpensive” used in the same sentence.
Dr. Simkin: Of course biologics are terribly expensive, but so is hospitalization, and hospitals are bad places. We want to get our patients home, and in our experience this has been a very useful way to make that happen sooner.
Dr. Pile: I agree that an emphasis on hospital throughput is incredibly important. For the hospitalized patient with gout that’s preventing ambulation, that’s the issue that must be addressed before discharge is possible. Certainly the agent with the fastest onset of action is going to be very attractive.
Dr. Edwards: All of these agents for acute gout have a relatively fast onset of action if they’re given early in the disease process. Once you get out to a day and a half from the onset of symptoms or beyond, you’re fighting an uphill battle in terms of making a difference in the natural course of the attack. I believe that’s true of all three of the medication classes that are typically used.
My approach to the initial therapy choice is highly individualized, depending on what the patient’s been on before. If they’ve been on maintenance colchicine to prevent flares and then they flare, I usually won’t use colchicine for the acute attack; I will go with a steroid or an NSAID. If they’ve been on NSAIDs as preventive therapy and they flare, I might try low-dose colchicine for the flare or use steroids. In cases of a prolonged course, where the patient is in the hospital and it’s 3 or 4 days since symptom onset and a steroid taper or a trial of colchicine has failed, I’ve been very impressed with the ability of anakinra to suddenly bring the attack to a halt. Like Peter, I am on the cusp of looking at acute treatments a little differently, although there still aren’t a lot of data on IL-1 inhibition in this setting.
Dr. Sundy: There are data showing that a gout flare adds about 3 days to the hospital stay,4 so that’s a huge burden that intervention with IL-1 inhibition can really help to address.
Dr. Mandell: I think we’ve seen a trend over time toward corticosteroid therapy becoming more common, particularly in hospitalized patients. I think that’s partially related to more widespread use of appropriate deep vein thrombosis (DVT) prophylaxis, which means that more patients are on anticoagulant therapy, which is one more reason to shy away from NSAIDs, particularly the nonselective ones, in the hospital setting. Do you sense that trends in the outpatient setting have shifted, or do most clinicians still reach for NSAIDs?
Dr. Sundy: I think most people still reach for the NSAID, but it really depends on the comorbidities. I sense we’re seeing chronic kidney disease in a greater proportion of patients, and that’s probably creating a shift toward a bit more corticosteroid use. I like to reach for an NSAID as first-line therapy, but we really have to understand our patient’s overall comorbidity profile, including renal function, before doing so, as Peter said.
Dr. Edwards: I think NSAIDs are still the most commonly used acute treatment for gout. I used to calm myself when I used them by saying, “It’s only a 7- to 10-day course; how much trouble can you get the patient into?” Well, in that short a time I’ve had patients tip over into congestive heart failure because they had some renal decompensation beforehand and then had another 20% or 25% of their renal function knocked out with the NSAID, and they’d have extra sodium retention. I’ve seen GI bleeds develop in patients over that short a time. I don’t prescribe nonselective cyclooxygenase (COX) inhibitors anymore without also giving a proton pump inhibitor for stomach protection. I think that’s becoming a standard of how to use NSAIDs among rheumatologists, and it’s hard to get that stomach protection up and running in the very short time frame of an acute gout attack. So I’m personally tending away from NSAIDs; I use steroids more often and then the IL-1 inhibitor for special cases.
Dr. Mandell: Studies assessing the gastric risk of NSAIDs have shown that a GI bleed can be induced in as little as 2 or 3 days. I agree that there is a trend, at least among rheumatologists, to use a proton pump inhibitor when initiating outpatient NSAID therapy. This combination may still be cost-effective for many patients, as it can speed the return to their usual activities. But even in the outpatient setting steroids are becoming more common than they used to be.
DOSING FOR THE ACUTE ATTACK: BE AGGRESSIVE FROM THE START
Dr. Simkin: Whatever dose of steroids we use in the outpatient setting, I think it’s highly desirable to divide it. Rheumatologists are taught to use a single daily steroid dose in the morning, primarily to spare the adrenal glands. While that makes sense in patients on long-term steroid therapy, such as for lupus, it doesn’t necessarily make sense in gouty arthritis. I’ve seen patients whose gout flared up every night despite taking sufficient prednisone; when they divided the dose, their gout was controlled.
Dr. Mandell: I would generalize that further to stress the importance of using an adequate and aggressive dose of either steroids or NSAIDs for treating acute gout. A common mistake I notice in the community is the use of too low a dose of either steroids or NSAIDs. We should treat aggressively from the start with a full dose of these agents.
Dr. Edwards: Even more than the usual full dose, I would say. Many generalists have a concept of what an analgesic dose of an NSAID is, which is pretty low—in the case of ibuprofen, perhaps 400 mg three times daily. An anti-inflammatory dose is higher—perhaps 800 mg three times daily for ibuprofen. Most of us who’ve used NSAIDs for gout realize that we have to get even a little bolder than that from the start. It can be hard to convince some generalists to exceed what has been their ceiling of comfort for an NSAID, but doing so for the first 24 or 36 hours is important to getting the gout attack under control. Of course, the need for such aggressive dosing is all the more reason to make sure not to use an NSAID in a patient with significant renal disease or other comorbidities for which NSAIDs are problematic.
Dr. Mandell: And all the more reason to provide gastric protection.
Dr. Sundy: I would add that if a clinician is not comfortable using doses that high, that may be a good reason to think about choosing a different initial therapy, be it colchicine or steroids (if the reluctance is with NSAIDs) or NSAIDs (if the reluctance is with steroids).
ACUTE ANALGESIA: ANYTHING MORE THAN AN ADJUNCT?
Dr. Mandell: What about treating gout with acute analgesia alone? Consider a setting where you may want to avoid a drug with anti-inflammatory or antipyretic effects, perhaps in the postoperative period or when coexistent infection is a concern and you want to monitor the fever. Can you get by with using narcotic analgesia alone?
Dr. Sundy: I’m not impressed with that approach. Narcotics can play a role in managing a patient’s pain, but a narcotics-only approach will allow the flare to linger and not address complications of the flare or back-to-work issues. I view analgesics as cotherapy as opposed to single-agent therapy.
Dr. Mandell: Is narcotic analgesia effective even at treating the pain acutely?
Dr. Sundy: No. Gout pain is an exquisitely inflammatory pain, and the key is to tackle that inflammatory response. Analgesics are really just an adjunct.
Dr. Simkin: I totally agree, but they’re an important adjunct that we often overlook.
Dr. Edwards: I’ve been singularly unimpressed with narcotics. Gout is a cytokine-driven process that is intensely inflammatory. It’s like a lot of other types of pain that don’t respond fully to narcotics, such as herpetic neuralgia or uterine pain. Perhaps the benefit of narcotics in gout is their soporific effect—patients sleep through their attack. Yet if you touch the gouty joint or the patient moves it, the pain is just as intense as if the patient weren’t taking anything. So I don’t know that narcotics add anything unless the patient has other causes of pain. I don’t use them at all.
Dr. Sundy: I tend to make the option available to the patient. I say, “Here’s what we need to do to knock down this flare. And here’s something additional you can try for pain; we can see if it helps.” But I’ll emphasize the importance of improving the inflammatory piece.
Getting back to the postoperative setting, we should recognize that a strategy to just ride out a perioperative gout flare with an analgesic medication, perhaps because of concern about the effect of corticosteroids on wound healing or infection, can carry important risks. A patient treated that way will end up bedbound at precisely the time you want him or her to be moving around, so there’s now the risk of postoperative pneumonia and other complications that won’t get documented as complications of gout even though they actually are.
Dr. Mandell: Not to mention the untreated postoperative fever from the crystal-induced attack, which then leads to a work-up looking for DVTs, more blood cultures, and more radiographs, all the while extending the hospital stay and wasting resources. Jim, as a hospitalist, do you still see narcotics used initially as the primary treatment for gout flares in the hospital?
Dr. Pile: It depends on which hat I’m wearing. If I’m the hospitalist and the patient’s on my service, usually my antennae are up for the appearance of gout in the hospital, and the house staff may or may not recognize it if the process starts overnight. When I’m serving as the medical consultant, I sometimes encounter cases where the surgeons don’t recognize a gouty flare when it first appears, and I’ve had multiple patients in whom gout-induced postoperative fever triggers a consultation. In the latter case, it’s not uncommon to see narcotics being used in an attempt to treat gouty pain.
RELATIVE DRUG EFFICACY FOR ACUTE ATTACKS: HOW MUCH DO WE KNOW?
Dr. Edwards: If they’re given early, all of them have good—if not necessarily fast and ideal—effectiveness. The recent AGREE trial of colchicine (Acute Gout Flare Receiving Colchicine Evaluation) assessed pain reduction at 24 hours in patients randomized to three treatment groups: a low-dose colchicine regimen consisting of three 0.6-mg tablets (1.2 mg initially, followed by 0.6 mg 1 hour later); a more traditional high-dose colchicine regimen consisting of eight 0.6-mg tablets (1.2 mg initially, followed by 0.6 mg every hour for 6 hours), and placebo.11 Response, defined as a 50% or greater reduction in pain at 24 hours without rescue medication, was reported in 37.8% of patients in the low-dose colchicine group, 32.7% of patients in the high-dose colchicine group, and 15.5% of placebo recipients.
This is an excellent study that should finally take highdose colchicine for the treatment of acute gout off the table since it offered no improvement in efficacy but was significantly more toxic. However, while 50% improvement in 24 hours is a hard target to achieve, the fact that barely more than a third of subjects hit the target means we should still be looking for more effective approaches.
Dr. Simkin: Similarly, in the first controlled study of colchicine in acute gout, published by Ahern et al in 1987,10 23% of colchicine recipients noted a 50% reduction in their pain at 12 hours after starting treatment. That leaves more than three-quarters with little or no response within 12 hours, and gout is one of the most painful conditions we treat. We’d very much like to help our patients sooner than that. So while there are no head-to-head data comparing colchicine versus NSAIDs versus steroids, my impression too is that oral colchicine is a second-line choice for the acute gout attack.
Dr. Sundy: When we try to understand this literature, it’s important to look closely at how patients were ascertained. The AGREE trial evaluated patients who were enrolled when they were asymptomatic; they were given study medication and instructed on how to start it once a flare began. In contrast, most of the well-controlled NSAID trials captured patients as they came in with a flare, and they had to have had the flare no longer than 48 hours. I believe there’s a big difference between having had a flare for 12 hours versus 48 hours—and even as long as 72 hours in some corticosteroid trials. The longer the clock has been allowed to tick before treatment is started, the harder it is to achieve rapid symptom reduction.
Dr. Mandell: Obviously it’s difficult to compare between studies, but it’s interesting that in a study of the COX-2 inhibitor etoricoxib versus indomethacin,7 one of the largest studies of NSAID therapy for acute gout, approximately one-third of patients taking etoricoxib reported no pain or mild pain within 4 hours. This very rapid pain control is what we really want, since pain is the concern, along with eventual complete resolution, of course. So there really may be value in having something that has analgesic as well as anti-inflammatory effect, to Peter’s earlier point. Tackling both the pain and the inflammation that’s causing the pain certainly makes sense.
Dr. Edwards: Yes, and that etoricoxib study was designed the way people often are treated, with the unfortunate delay before the doctor is seen and the medication is started. The AGREE trial is how I hope everybody would be treated regardless of what they’re treated with. We’re probably never going to see a good head-to-head trial among the three main types of drugs we use for acute gout—the unpredictability of flares makes it extremely difficult.
Dr. Pile: I’m curious whether or not you, as rheumatologists who specialize in gout, are surprised by the results of AGREE. My experience with colchicine over time has been more favorable than what’s suggested by AGREE.
Dr. Edwards: How do you use the colchicine?
Dr. Pile: For more than a decade I’ve been using 0.6 mg three times daily. I thought that practice was unusual until I recently read the AGREE study and realized that a lot of you have been using that regimen for a while.
Dr. Mandell: Some gout patients will take a colchicine tablet when they feel the wisp of an attack coming on and it immediately will stop the attack. That gets to the lore that if you treat a flare very early on, you may be able to abort and treat an attack very quickly. Years ago, however, I was involved in an analysis of 100 patients treated with intravenous (IV) colchicine, and we found that treatment response was unaffected by whether patients were treated early or after a delay of 48 hours. I believe colchicine behaves like a different drug when given IV—and the IV form has since been withdrawn from the market for safety concerns—but this underscores that individual responses to various agents are quite unique. I think there are some patients who are exquisitely sensitive to colchicine, while others are exquisitely sensitive to an NSAID, and so on. This means we’d need a very large sample size to tease out that variation in a trial, and that’s not likely to happen.
Dr. Edwards: I too have had gout patients who tell me they get this premonition of an attack—a feeling that “something just isn’t right”—hours before they actually feel any pain from the attack. A lot of them tell me they’ve learned over time that if they take a colchicine tablet when they get this premonition—or an extra colchicine tablet, as some are on colchicine maintenance therapy (typically one or two tablets a day) plus whatever other background therapy they’re on—they don’t get the flare or the flare is diminished.
Dr. Mandell: I’d like to wrap up this portion by getting your sense of whether there’s a difference in efficacy for treating acute attacks among the drug classes we’ve discussed.
Dr. Edwards: I believe the IL-1 inhibitors are probably the most potent agents for aborting a gout attack, followed by steroids, which I think have a leg up on both colchicine and NSAIDs. The latter two options probably are equally effective in aborting an acute attack.
Dr. Sundy: Yes, I would rank them the same way.
THE ROLE OF COMORBIDITIES IN THERAPY CHOICE
Chronic kidney disease
Dr. Mandell: Let’s get more specific about comorbidities and therapy choice, starting with chronic kidney disease. What’s the threshold at which renal compromise starts to influence your choice of therapy for an acute attack of gout?
Dr. Sundy: I have a very low threshold for avoiding NSAIDs in that setting, mainly because when I want to use NSAIDs, I want to use them at a very high dose, and even short-term use of high-dose NSAIDs can reduce creatinine clearance. So I would certainly avoid NSAIDs when the patient’s creatinine clearance is less than 60 mL/min, and I’d be even more cautious if the patient had underlying hypertension or congestive heart failure.
Dr. Mandell: So even with the reversibility of almost all of the NSAIDs’ renal effects, you tend to stay away from them in that setting?
Dr. Sundy: Yes.
Dr. Edwards: I’m less guided by a single creatinine clearance level. I tend to look at patients in light of the 20% to 25% reduction in the glomerular filtration rate (GFR) that most NSAIDs will cause, even when used acutely. Though the NSAID effect is reversible, if that GFR reduction is going to make a difference to other compensated systems, primarily the heart, then the NSAID should be avoided.
Dr. Simkin: It’s my understanding that plasma flow to the kidney becomes prostaglandin dependent with renal insufficiency, and when we use antiprostaglandin agents we get into trouble on a mechanistic level. I’m not sure of the exact point at which that occurs, but I think we all recognize that serum creatinine is a fairly weak indicator of which patients have some limitation.
Dr. Mandell: So we agree on the need to be very cautious with the use of NSAIDs, recognizing that in the acute setting there is reversibly depressed renal blood flow from NSAIDs. The decreased blood flow will get better, but there’s the issue of what happens during fluid retention when there is coexistent congestive heart failure, diabetes, and chronic kidney disease—you may also occasionally bump up the potassium level. In general, most rheumatologists shy away from selective or nonselective NSAIDs in the setting of chronic kidney disease. Is that consistent with the use you see in the hospital, Jim?
Dr. Pile: It is. I would add that if patients are on certain other medications—especially angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or beta-blockers—there may be additive negative effects because of the potential for hyperkalemia if an NSAID were added to the mix.
Dr. Mandell: I would add to that list of concerning medications aminoglycosides, cyclosporine, and other nephrotoxic agents.
Diabetes
Dr. Mandell: What about diabetes? How does a patient’s diabetes enter into your choice of acute gout therapy in the outpatient and inpatient settings?
Dr. Edwards: Diabetes is especially problematic because it significantly affects two of our treatment choices—steroids and NSAIDs. The steroid doses needed to effectively treat an acute gout attack have a pretty profound effect on glucose levels in diabetic patients. That’s always of concern to me, and it’s certainly of concern to my patients with diabetes who are vigilant about monitoring their glucose levels, which may spike from 130 or 140 mg/dL to values in the high 200s. These elevations will last for the 7 to 10 days of treatment, and the longterm adverse effects of that period of hyperglycemia are not clear. If we put these patients on short courses of corticosteroids, perhaps we should be treating their diabetes more aggressively during that period, but I don’t think we have a history of doing that.
Dr. Pile: It’s very difficult to do in the short term. That’s especially the case with the diabetic patients I see as a hospitalist at a safety-net hospital. Many of my patients don’t have good glucose control at baseline: their levels of 250 mg/dL may go to 400 mg/dL or higher with acute corticosteroid therapy, so that’s clearly a problem.
Dr. Mandell: I try to avoid giving corticosteroids in the outpatient setting to a diabetic patient who is being maintained on oral hypoglycemic medications, particularly if efforts are being made to avoid more aggressive diabetes therapy. Steroids may be an option, though, for a patient who is on insulin, uses a pump, and is very comfortable measuring and managing his or her glucose at home. In the hospital setting, I’m more comfortable using corticosteroids in diabetics because I have much better control over their glucose; I can just increase the basal and premeal coverage.
Dr. Pile: I agree. With vigilance I can control almost anyone’s blood sugar in the hospital, but when patients leave the hospital on steroids, it becomes much more problematic.
Dr. Mandell: Right. You don’t know what’s going to happen when they go out and liberalize their diet on top of the steroids, because often it’s the postprandial surge that’s exacerbated by steroids.
Dr. Sundy: It can be very helpful to know at baseline what a patient’s spot blood sugar is before using a steroid, just as it’s important to know what the creatinine is before considering an NSAID. In the setting of an acute flare, the patient might already be quite hyperglycemic, and many of these patients probably don’t have the tools to manage this at home, so careful follow-up—coming back the next day for at least a blood sugar check—is critical to the proper care of these patients.
Dr. Mandell: When we talk about steroids we tend to think mostly about prednisone, but Peter mentioned injectable steroid. What about injectable adrenocorticotropic hormone (ACTH), which goes in and out of vogue as an agent to treat acute gout? What is your experience with using ACTH injections, specifically related to the issues of diabetes and heart failure?
Dr. Edwards: Before it got reformulated about 3 or 4 years ago and its price went way up, injectable ACTH was my drug of choice for treating acute gout. I had hardly any failures on it over the 10 to 15 years when it was my main means of treating acute gout. I would typically use 80 IU of ACTH gel, given subcutaneously, and the response within 12 hours was quite dramatic. About one in four patients would require a second injection at that 12-hour point, but almost everyone was symptomfree at 24 hours. This response was seen even in patients who were on fairly high-dose chronic steroids already, such as transplant patients.
This suggested that a mechanism other than just adrenocortical stimulation explained the benefit from ACTH. In fact, a lot of data have emerged suggesting that ACTH has a specific effect that is not related to adrenocortical stimulation.
Ten years ago an injection of ACTH gel would cost about $3 or $4, whereas today it costs $2,000 or more.29 I don’t think it’s justifiable at that cost.
Dr. Mandell: There’s also the issue of fluid retention with ACTH, and the higher doses, which seem to be more effective, are more likely to exacerbate congestive heart failure, which has to be a concern if you go this route.
Coronary artery disease
Dr. Mandell: Atherosclerosis, diabetes, and chronic kidney disease are increasingly recognized in the gout population, and coronary artery disease links with all of those. Does the presence of coronary artery disease enter into your choice of agents for a patient with an acute attack of gout?
Dr. Edwards: It doesn’t have as big an impact as diabetes does. To me, the priority in this setting is resolving the acute attack as quickly as possible, as I’ve seen my share of patients for whom the acute gout attack has been so painful and stressful that they’ve developed angina at the same time. So I might use steroids in patients with coronary disease because I’ll want to go with what’s likely to be most efficacious for quick resolution of the attack even though steroids may increase glucose and blood pressure and thus raise fluid retention as an issue.
Dr. Mandell: The presence of coronary disease implies the use of low-dose aspirin therapy. Despite the slight elevation in serum urate from low-dose aspirin, I recommend that my gout patients continue this for coronary protection. But even low-dose aspirin raises concerns of GI safety and even possible drug interactions with ibuprofen that could reduce the aspirin’s efficacy. Does this lessen your tendency to reach for an NSAID in these patients?
Dr. Edwards: I think the issue of cardiovascular safety and NSAIDs is still up in the air. The data that came out around the time the selective COX-2 inhibitors were released made us all think again about the role of prostaglandins and coronary blood flow and how blocking prostaglandins might have some bad effects. I don’t often use NSAIDs in patients with significant heart disease, simply because they don’t act fast enough to get patients through the attack quickly and I worry about the sodium retention and other problems putting stress on the heart.
Dr. Sundy: While coronary disease doesn’t create the acute dilemmas that diabetes does, it is a consideration in my therapy choice, and I generally avoid using NSAIDs.
Dr. Mandell: It is important to remember the additive gastric toxicity effect from low-dose aspirin and NSAIDs.
Infection
Dr. Mandell: Let’s turn to the hospitalized patient—a patient admitted to the medical service with infection. This patient is going to get fluids and drugs, and these are likely to raise or drop the serum urate level, which may precipitate a gout attack. From your perspective as an infectious disease consultant, Jim, how does the coexistence of a documented infection in the hospital—which you’re treating—factor into which agents to use or avoid when treating acute gout?
Dr. Pile: In practice, I try to avoid steroids in that situation. Theoretically, however, I’m not sure this scenario is really a contraindication to steroid use. I’m not aware of much solid data showing, for example, that patients with community-acquired pneumonia on appropriate antibiotic therapy have worse outcomes if they receive steroids than if they do not. Certainly there are a variety of serious infections for which we use steroids as a matter of course, including pneumococcal or tuberculous meningitis, tuberculous pericarditis, severe typhoid, and severe septic shock (the data for septic shock are murky, but there’s a role for at least steroid supplementation). As long as the antimicrobial therapy is appropriate, patients with serious infections are not necessarily going to do any worse with steroids.
Dr. Edwards: The biggest risk from steroids would be masking the symptoms of an unrecognized infection. If you know the infection is there and you’re treating it appropriately, I don’t think steroids are contraindicated.
Dr. Pile: A related risk is that steroids can make it more difficult to monitor the course of a known infection by blunting the overall response and bumping up the white blood cell count.
Dr. Mandell: My practice would fall in the realm of what Jim described as the theoretical course of action. If I knew what the infection was or felt comfortable in treating it, I would not shy away from systemic corticosteroids in the hospital setting, where I could monitor the patient. I think a corticosteroid may often be the safest drug to use in that setting, given the likelihood of comorbidities, at least compared with NSAIDs.
Dr. Simkin: Let’s say a patient comes in with acute knee pain and you aspirate the knee. The question arises whether you should wait for the culture results or go ahead and inject steroid while your needle is in the knee. I certainly have a much lower threshold to go ahead and inject while I’m in there, although I’d always be sure to get the culture results and revisit as needed. But if I pull out extremely inflammatory fluid during the aspiration, I’m probably not going to inject the steroid.
Dr. Edwards: And what if you actually do inject an infected joint with a corticosteroid—are you doing harm?
Dr. Simkin: I would like to think I’m not, and I may be helping.
Dr. Edwards: Yes, there are a lot of theoretical reasons why you might be helping, and there’s a small literature on steroids and infected joints that suggests you might actually slow down some bone resorptive steps and be doing various similar things.
Dr. Mandell: But in humans (a series of pediatric patients with septic arthritis30), those were systemic steroids, not intra-articular.
Dr. Edwards: In animal studies there is some amelioration of the destructive component with intra-articular steroids. The main point is that we must send off the fluid for culture when we inject steroid into a joint, and we must follow up on the cultures to make sure nothing is growing in a joint because we’re going to be calming it down. Whatever the inflammatory process is—whether infection or crystalline or rheumatoid—the patient is initially going to get better with a steroid injection. So we can’t use initial clinical improvement as the marker that we’ve done the right thing. We need to make sure the aspirate culture is negative.
Dr. Mandell: What about the use of colchicine or an IL-1 antagonist in the setting of infection?
Dr. Pile: There may be some theoretical concern, but I think low-dose colchicine would be a reasonable agent to use in the setting of infection.
Dr. Edwards: I don’t think there’s much concern since IV colchicine has been taken off the market. IV colchicine had a much more profound effect on bone marrow, and this resulted in a number of deaths from infection in immunosuppressed patients such as chemotherapy recipients. But oral colchicine in the doses we use for acute attacks and for maintenance probably doesn’t have a very profound effect on response to infectious processes or on white blood cell production (in the absence of chronic kidney disease).
Dr. Pile: Which raises the point that if the patient were on a potent inhibitor of the cytochrome P450 3A4 system, you might conceivably get into the same situation. For example, serious toxicity and even fatalities have been reported in patients taking colchicine and clarithromycin concurrently.31
Dr. Mandell: Peter, would you be comfortable giving an IL-1 antagonist to a hospitalized patient with pneumonia that’s being treated with antibiotics? Let’s say the patient has chronic kidney disease and diabetes and you’re looking for an agent to use.
Dr. Simkin: Well, I wouldn’t be comfortable, but I might feel I had to do it. If the organism were known, as you said, and the antibiotic therapy were appropriate, I might depend on that antibiotic coverage.
Dr. Sundy: Although I can envision some scenarios where I’d use the IL-1 antagonist in this situation, I’d be looking for an alternative. My concern is that people with any source of infection were excluded from clinical trials of anakinra, so we don’t have a good understanding of how it behaves in the setting of infection.
Dr. Edwards: And the fact that anakinra is not approved by the Food and Drug Administration (FDA) for treatment of acute gout should make us a bit uncomfortable.
Dr. Mandell: It’s interesting that the limited data do not show a lot of infections with IL-1 antagonist therapy other than when combined with anti–tumor necrosis factor therapy. The experience in periodic fever patients, who are on IL-1 antagonist therapy for many years, has not really shown this to be an agent predisposing to infection. I think this goes back to the point that if you document an infection and you’re treating it appropriately, some immunosuppressive or anti-inflammatory therapies may not be bad if you need to treat the attack of gout, particularly if it’s in a setting where you can monitor the patient well.
NSAIDs: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s drill down on the use of the specific classes of drugs, starting with NSAIDs. Larry made the point that NSAIDs should be used at high doses for acute gout attacks. What else is worth noting about the dosing and use of NSAIDs in this setting?
Dr. Sundy: In addition to that distinction between analgesic and anti-inflammatory dosing, it’s important that patients be told to treat at the appropriate dose intervals for whichever NSAID is chosen so that the anti-inflammatory benefit is maintained. I generally recommend that patients continue on that dosing regimen until they have substantial symptom improvement, at which point they can begin to back off, but that’s generally a 7- to 10-day treatment course, and typically closer to 10 days. Finally, I’m quick to use proton pump inhibitors or some sort of gastroprotective regimen with most patients that I start on NSAIDs in this setting.
Dr. Mandell: Your point about duration of therapy bears emphasizing. A common mistake with the use of NSAIDs is that they’re stopped too soon out of an understandable concern about their toxicity. Often they’re stopped when symptoms are still present, which means the attack hasn’t really resolved, and so it comes back. I tell patients to wait until their attack is completely gone, continue for a few days longer, and then stop the NSAID at that point.
Dr. Edwards: If we look at the natural course of disease, a patient’s first four to six gout attacks tend to be a bit shorter in duration, in the range of 5 to 7 days. As recurrent attacks occur over the decades, they tend to get more drawn out, lasting perhaps a couple or few weeks. We want treatment to at least cover the anticipated period of the natural attack since we’re not doing anything profound to the disease process itself, except perhaps with an IL-1 inhibitor. For instance, a typical treatment in emergency departments is an intramuscular injection of ketorolac. That may make patients feel good for the next 24 or 36 hours, but they’ll have a resumption of their acute attack at that point. I generally stick to 2 weeks as a reasonable treatment period. I might shorten that for a patient with diabetes in whom I have to use steroids or for a patient with congestive heart failure that I needed to put on an NSAID, but I generally try to cover patients with anti-inflammatory therapy for a couple of weeks.
Dr. Mandell: Your point about intramuscular ketorolac is important. A single shot is not likely to resolve the attack, and the intramuscular route will not ameliorate the gastric toxicity of this drug. Those are two common misperceptions.
What about the COX-2-selective NSAIDs? There’s only one that’s still marketed in the United States, celecoxib, but what’s your sense of these drugs as a class in terms of efficacy for treating gout?
Dr. Sundy: I think they’re effective. Only one COX-2 inhibitor, etoricoxib, has been studied in clinical trials for acute gout, and it’s not available in the United States, but I think the class as a whole is a reasonable choice. That’s especially true since the course of treatment is only 7 to 14 days. Even so, it’s still reasonable to use some gastric protection when giving celecoxib, as it does no harm and can add some reassurance. As with other NSAIDs, dosing of celecoxib for acute gout is higher than for other typical uses in the drug’s labeling.
Dr. Edwards: Yes, when I use celecoxib for acute gout I use 400 mg every 12 hours for the first 2 days and then take it down to 200 mg every 12 hours for the remainder of the treatment period.
Dr. Mandell: And the COX-2-selective NSAIDs do adversely affect renal function.
Dr. Simkin: One thing that troubles me when we talk about dosage for any of our agents is the large individual variation we encounter. I don’t think there’s a patient population with a wider variation of body sizes than the gout population. We see enormous patients with metabolic syndrome and several large joints, and we see frail elderly patients who have a single small joint affected. In fact, in addition to body size, the size of the joints involved can tell us a lot about the amount of inflammation we’re dealing with. Someone with two knees and an ankle affected is probably different from someone who has just a great toe affected. In light of these variables and so many others that come into play, such as age and comorbidities, I’m hesitant to recommend any particular dose because I try to make adjustments. Of course, there are no data that cover all these variables.
Dr. Mandell: But would you agree with the generality that high doses—higher than those we typically use for other musculoskeletal pains—are warranted in treating acute gout?
Dr. Simkin: Yes, but a high dose for a small elderly woman is different from a high dose for a young man who’s quite large.
Dr. Sundy: This sensitivity to individual variation can also apply to duration of therapy. As we noted, it’s not uncommon for veteran gout patients to find that starting treatment early—within 24 or 48 hours—may be sufficient to tamp down their symptoms, yet that’s an observation that is developed only over time in an individual patient. So it’s not a recommendation, but many patients will do fine with that. In those situations, I may not be so adamant about the need to continue treatment for a full 10 or 14 days.
STEROIDS: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s move to corticosteroids. A very common treatment used for acute gout is a tapered-dose regimen like the Medrol Dosepak (blister-pack) formulation of methylprednisolone. What strikes me is that this is a one-size-fits-all formulation, yet you’ve all just argued that there’s no one-size-fits-all approach. What are your thoughts?
Dr. Edwards: The Medrol Dosepak contains 21 4-mg tablets of methylprednisolone to be taken over the course of 6 days. Six tablets are taken the first day, and then one fewer tablet is taken each successive day through day 6. An obvious potential drawback is that the patient is off of medicine after 6 days, though that will be enough for some patients if they start treatment promptly.
I do use the Medrol Dosepak, and I always have patients fill a prescription ahead of time so that it’s available for quick initiation when they start to have their next attack. I instruct them to avoid storing it in a hot place, such as a car, but that it can be stored for up to 5 years at room temperature. Most of my patients like this approach, and if they do well with it they’re good about getting a new refill in advance of their next attack. This is a pretty handy way of letting patients be in charge of their attacks. If it doesn’t work and their symptoms continue, they call me and I’ll see them promptly.
A different approach is more appropriate for patients whose attacks tend to be more recalcitrant, perhaps because they’ve had gout for a long time or have bulky disease with lots of tophi. In those cases I’ll go with a steroid regimen that’s essentially double what a Medrol Dosepak would be—namely, prednisone 30 mg for the first 2 days then tapered down by 5 mg every other day. As Peter said, this approach would be overwhelming to a frail 82-year-old woman with a single tophus in her distal interphalangeal joint, but it tends to work well for a more typical 50-year-old obese male gout patient.
Dr. Sundy: I do some of the same things you describe, though my background in allergy has accustomed me to the steroid burst, so I’ll tend to use a dose of about 0.5 to 1.0 mg/kg/day for 4 or 5 days and then try to taper rapidly over another 2 to 4 days. I’m curious whether you think methylprednisolone offers advantages over prednisone.
Dr. Edwards: There are a few patients—roughly 5% to 15% of the population, according to various studies—who have trouble converting prednisone to its active form in their liver because they’re missing an enzyme. So I tend to use methylprednisolone since it has a benefit in that small segment of the population, but it otherwise doesn’t offer anything special beyond the convenient packaging that we discussed.
Dr. Mandell: I think convenience is the major factor. The blister-pack preparations are useful for patients who get confused with tapering. For patients who can make adjustments based on their symptoms, I find those personalized adjustments to be better than just giving them an absolute number of pills.
Dr. Pile: This issue of dosing duration is very important for generalists; just listening to your schedules is very instructive for me. I think a lot of nonrheumatologists, myself included, are inclined to use a one-size-fits-all approach. The mistake that I’ve been most apt to make, apparently, is using an insufficient duration of therapy. I’ve tended to use 40 mg prednisone for 5 or 6 days and then to stop rather than tapering the dose.
Dr. Mandell: From the cost perspective, steroids seem to be a very cost-effective option. The Medrol Dosepak, as a branded product, costs a bit more. Similarly, generic NSAIDs are inexpensive. There has been some feeling that nongeneric NSAIDs may offer advantages over generic NSAIDs, but I don’t know of any published evidence to support that. What are your thoughts?
Dr. Edwards: I agree with you. There has been this folklore about indomethacin and, before that, phenylbutazone being rather remarkable and specific drugs for gout. That hasn’t been borne out by the data, and virtually all of the NSAIDs work fairly comparably.
COLCHICINE: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s move to colchicine, which represents a unique class of anti-inflammatory agent. It has been available and widely used for many years, but it had not been formally licensed by the FDA until last year. Without corporate imperative and funding, data on its clinical use have been limited. As we mentioned, an IV form of colchicine was withdrawn from the market a few years ago after being associated with multisystem organ failure and death. Low-dose short-term colchicine doesn’t have those risks, and its efficacy in treating gout has been accepted for many years despite the dearth of clinical trial data until recently. How has colchicine been used in the past, and do the new data guide us on how it should be used differently, specifically for treating acute gout attacks?
Dr. Simkin: The so-called traditional regimen of starting with a pair of colchicine tablets and then giving another tablet every 1 or 2 hours until relief (or until GI toxicity developed) is one of the great embarrassments in the history of our field. The more recent trial evidence clearly indicates in the short term that there is no additional benefit beyond a very low dose. As we noted, this has been in the wind anecdotally for quite a few years, and it was confirmed by the recent AGREE trial.11 I find it interesting that the dosage used in AGREE—three 0.6-mg tablets—is very close to what we use for prophylaxis. We may well wind up concluding that any patient we’re seeing for the first time with an acute attack should be treated with that dose of colchicine, as an adjunct to another anti-inflammatory drug.
The use of colchicine as an adjunct for acute gout attacks was advanced in a review by Cronstein and Terkeltaub several years ago.32 Since we know that the mechanisms of action of the various drugs for acute gout are sufficiently different from each other, it makes sense to consider hitting the process at more than one spot. Having said that, one of the most crucial concerns in using colchicine is what other medications the patient is taking, given colchicine’s multiple drug interactions (Table 3). These include interactions with most statins, some antibiotics, and some calcium channel blockers, which share drug transporters with colchicine. So we have to be cautious about using colchicine with those other therapies.
Dr. Mandell: That’s also important in patients on chronic prophylactic colchicine therapy, as we know from recent pharmacokinetic studies and anecdotal case reports of colchicine and some of these other drugs.16–23 Do you think it’s as important in a patient on, say, chronic statin therapy who’s not on baseline colchicine, in whom you use a colchicine regimen of just two tablets followed by one tablet and then stop the colchicine right there? Is the potential for meaningful drug interactions as important in that setting?
Dr. Simkin: Probably not. One of the key issues is to educate patients about what to look for. Tell patients that if they notice numbness, weakness, or myalgia, you want to know about it. The evidence I’ve seen is that people who go into colchicine-induced rhabdomyolysis, for instance, develop it over a period of days, not hours, and if they stop the drug early, they probably will be okay. That’s the biggest concern. Neutropenia is the second concern.
Dr. Mandell: But fortunately, assuming no chronic kidney disease, those are generally patients taking colchicine chronically, not just acutely. We talked a bit before about our panel’s concerns about the efficacy of colchicine. Say you have a patient who has no history of prior colchicine use. How comfortable are you that treating such a patient with just the three-tablet colchicine regimen is likely to give the desired effect of rapid pain control and resolution of the attack?
Dr. Edwards: I’d expect that there wouldn’t be a profound or rapid improvement on that regimen, although the patient would certainly do better than if he or she were on nothing. I think that’s what the AGREE trial showed—that patients improve but still have significant morbidity days after initiating therapy.
Dr. Mandell: I think the AGREE trial did what it was intended to do: it showed that a low-dose colchicine regimen was as effective as and better tolerated than a high-dose colchicine regimen. This was something most of us believed, but it had never been demonstrated in a trial, and now there are good data to show it. And AGREE was consistent with the 1987 Ahern study10 in showing that the high-dose regimen is better than placebo. However, it in no way convinced me that colchicine in this dosing regimen is a monotherapy I’d be comfortable using to treat a severe acute attack. AGREE also didn’t give me any information as to how likely this approach would be to resolve an attack. For all the reasons we discussed, continued anti-inflammatory therapy is likely necessary beyond three pills, and I can’t be confident that a course of three colchicine pills is sufficient to maintain a drug level inside white cells to prevent other attacks from happening or a rebound attack once the regimen is over. I’m sure this approach will work for some patients; I just don’t know at the outset which ones or what percentage of patients will respond completely.
Dr. Edwards: I like the idea of combination therapy, of using colchicine as an adjunct. We’ve been talking up to now about monotherapies for acute gout because monotherapy is simple and patients and physicians alike prefer things that are fairly simple to use. But there is good theoretical reason to support using colchicine in combination with either NSAIDs or steroids, though there are probably no experimental data. I can’t imagine a reason for using corticosteroids and NSAIDs together, but pairing up colchicine with a steroid or an NSAID might allow you to keep the dose of the other agent lower and perhaps reduce the overall toxicity of your therapy. I’ve occasionally put patients on that type of combination therapy, and it’s probably a helpful way to go.
Dr. Mandell: For years my approach has been one of cotherapy, counting on either NSAIDs or corticosteroids to treat the acute attack by rapidly relieving the pain and inflammation and using colchicine in low doses—at a prophylactic dose, as Peter said—and extending it over a much longer period. I’m counting on the colchicine not to treat the acute attack but to prevent the next one and hopefully, for all the reasons we talked about, shorten the duration of therapy with the steroid or NSAID.
Dr. Edwards: Sure. The way I use colchicine is mostly for maintenance and for prophylaxis while I’m initiating urate-lowering therapy. I use it for a bit longer than most others might—I draw it out to at least 6 months after a complete resolution of symptoms, sometimes to 9 or 12 months. When a patient has a flare, I certainly have them maintain the colchicine if I’m going to treat them with something else, as I almost always do, and I believe that lets me get by with lower doses of the other agent I’m using for the acute attack.
Dr. Sundy: When I use colchicine as monotherapy for an acute flare, it’s because the patient has concluded from experience that it has worked effectively for him or her. But that’s not very common.
Dr. Mandell: There are times when the patient’s comorbidities lead me to conclude that colchicine may be the best drug to try for acute treatment. But I’m not comfortable assuming that three pills will do it all.
Dr. Pile: From the perspective of the generalist, who typically treats a simpler gout population than you do, what is the recommended treatment duration if colchicine is used as monotherapy for an acute attack? We now have this recommendation to use a single day’s worth of therapy, but you’re all expressing reservations with that.
Dr. Edwards: The recommendation in the drug’s current labeling is to take two 0.6-mg tablets at the first sign of a flare and a third 0.6-mg tablet 1 hour later. That was the low-dose regimen in AGREE, and I think it’s a good one, as only a very small percentage of patients will get the GI side effects with this regimen that occur with more prolonged therapy. The real question is what you do on day 2 and day 3 on out to day 7 or 10. If I were to use colchicine monotherapy, after that first day I’d prescribe one tablet three times daily for another 3 or 4 days and then drop down to one tablet twice daily for the rest of whatever duration of therapy I felt was justified. Of course, this is highly individualized and depends on how the drug is tolerated; probably more than 25% or 30% of patients can’t tolerate a three-times-daily regimen even for 3 or 4 days. But the total duration would be at least 7 days and up to 10 or even 14 days.
Dr. Simkin: Or months, because you’ve established a diagnosis.
Dr. Edwards: Yes—as prophylaxis, for which the dosage is one tablet once or twice daily. And then you’d leave the patient on prophylaxis and start urate-lowering therapy, if it hadn’t been started already.
Dr. Mandell: We’re now way past having trial data to support this, and this approach may negate the purported safety advantages of the low-dose regimen supported by AGREE.
Dr. Edwards: Yes, this is rank speculation.
Dr. Simkin: But there is evidence of the value of prophylaxis.
Dr. Mandell: There is certainly evidence for the value of prophylaxis, from a number of studies. We’ll get to that shortly. However, if we think just about treating the acute attack and the AGREE findings, I don’t think we can extend those results with confidence to a setting where we switch immediately from acute therapy into a prophylactic mode. Not that we shouldn’t consider doing that—it’s similar to my practice as well, though I tend to use lower doses as I extend out—but the existing trial data are limited to that very short window of acute treatment. Anything we do beyond that is an extension from our belief that the inflammatory response to crystals in a joint lasts longer. I believe it generally requires longer anti-inflammatory therapy, but we need to expect some frequency of side effects and drug interactions as we put patients on chronic colchicine therapy.
The other factor that used to be an advantage of this type of regimen was its low cost. For a long time colchicine was available from multiple manufacturers and was exceedingly inexpensive, although with less FDA guidance on its use and less oversight of its manufacturing. This enabled even patients with no insurance to go on a prophylactic therapy and remain on it. The environment is now quite different with the introduction of a single FDA-approved and -regulated branded product. So how do we deal with cost in the current environment?
Dr. Edwards: In terms of specific numbers, once-daily and twice-daily doses of unbranded colchicine used to cost about $6 and $12 a month, respectively, based on the average wholesale price. Now the cost of branded colchicine (Colcrys) is about $175 a month for the once-daily dose and $350 a month for the twice-daily dose, again based on the average wholesale price.
Dr. Simkin: I think all of us are deeply troubled by the high cost of this preparation and hope that the alternatives will still remain available to us.
Dr. Edwards: Yes, especially since it sounds like most of us consider colchicine our go-to drug for maintenance. It used to be that patients might be on maintenance colchicine therapy for 5 or 10 years without any addition of urate-lowering therapy. Today, however, when most of us put a patient on prophylactic therapy with colchicine, it’s with the intent of also addressing the hyperuricemia that’s at the heart of the disease process. In that case we still cover patients with colchicine prophylaxis during the initial months of urate-lowering therapy because of the elevated rate of gout flares—so-called mobilization flares—that occur during the process of uric acid reduction.
Dr. Mandell: The manufacturer of branded colchicine has introduced a patient assistance program to ease the drug’s cost for the financially needy. We will have to see how practical it is and how it affects our patients’ use of this medication.
ACUTE FLARES IN THE SETTING OF PROPHYLAXIS AND URATE LOWERING: WHAT TO DO?
Dr. Mandell: It sounds like most of us would treat patients prophylactically with colchicine for a while after an attack, certainly after several attacks, and then continue on chronic low-dose colchicine during the introduction and adjustment of the urate-lowering therapy that constitutes comprehensive gout treatment. Yet some patients will still have flares. So how does this baseline low-dose colchicine prophylaxis—a 0.6-mg tablet once or twice daily—influence your choice of therapy for those attacks? Do you bump up the colchicine dose, or do you absolutely avoid increasing the colchicine?
Dr. Sundy: I wouldn’t absolutely avoid an increase, but I would tend not to adjust it. I would maintain the dose and add a different agent on top of it—an NSAID or corticosteroid—to manage the acute attack. In general, if a patient is on regular colchicine prophylaxis, the assumption is that they have sufficient renal function to support that use, so such a patient may do fine with an NSAID.
Dr. Simkin: I trust we all agree that it’s critically important to continue the urate-lowering therapy the patient is taking if he suffers an attack. The same thing pertains to the prophylactic regimen. Both of these components should continue through the flare, but I agree that we usually want to add something to treat a flare, and it should be something the patient has on hand and can take without needing to talk to his physician if it’s the middle of the night.
Dr. Mandell: So I’m hearing that most of us would add something else for the short term on top of the prophylactic colchicine dose when a flare developed rather than increasing the colchicine dose.
Dr. Simkin: Yes, although one exception I’d be comfortable with is the example we discussed earlier of the patient who has found that taking an extra colchicine pill or two can help abort or diminish a flare without causing diarrhea.
Dr. Mandell: We cannot extrapolate the AGREE data to support a short-course regimen for an acute attack in patients who are already on chronic colchicine for prophylaxis.
PROPHYLAXIS IN THE PERIOPERATIVE SETTING
Dr. Mandell: Admission to the hospital for acute medical or surgical reasons is not infrequently associated with gout flares. Jim, have we in the rheumatologic community made it clear that chronic colchicine prophylaxis and urate-lowering therapy should ideally be continued when patients with gout are hospitalized for any reason, or is it a reflex for these drugs be held?
Dr. Pile: I think there’s a general appreciation, at least in the hospital medicine community, that these therapies should be continued in that situation. My sense is that these prophylactic gout therapies are viewed by most hospitalists as somewhat analogous to beta-blockers for chronic heart failure, which are understood to be necessary to continue when a patient is admitted with an acute exacerbation of heart failure. I don’t have data to support this contention, however.
Dr. Edwards: My experience suggests that discontinuing urate-lowering therapy during an acute gout attack is a common mistake in general practice. I think that’s been demonstrated in surveys of primary care physicians. And when the uratelowering therapy is stopped, the attack is often prolonged and made worse. Widespread misperception about this remains—it’s a big problem.
Dr. Mandell: Based on educational lectures I’ve given to primary care audiences, I agree with Larry. If I present a case question on a scenario like this, many physicians in the audience will anonymously indicate via the audience response system that they would stop a patient’s allopurinol if an acute attack occurred.
Let’s turn to the scenario of a patient who’s admitted for surgery—elective or emergent—who has a history of gout and is on prophylaxis. What’s the general routine in managing these medications in patients who are going to surgery?
Dr. Pile: In my preoperative consultations I’ve always stressed to gout patients and their surgeons that the postoperative period is a high-risk setting for acute exacerbation of gout. My routine recommendation in this setting is that patients continue any of their goutrelated medications through the postoperative period, but that often doesn’t happen. All sorts of medications are inappropriately stopped perioperatively, including statins and beta-blockers in addition to colchicine or allopurinol. Something I haven’t done routinely is to recommend introducing prophylaxis in the postoperative period for patients who were not already receiving it. Is that something you consider doing, given the likelihood of postoperative flare?
Dr. Edwards: It’s important to appreciate the reasons why gout flares postoperatively. It has a lot to do with why uric acid levels go up postoperatively: fluid changes, starvation, ketosis for any reason, lactic acidosis—these all cause pretty remarkable changes in systemic pH, which certainly is a trigger for that. The perioperative state also changes the exchange of uric acid in the kidney so that there is greater accumulation, and anything that rapidly bumps up serum urate levels while also creating a change in the pH is a setup for flares. So any recommendations about whether or not to give prophylaxis have a lot to do with the anticipated impact of the surgery on all of those physiologic parameters.
Dr. Sundy: As rheumatologists we’re usually called after the fact, so I can’t say I’ve given the issue of prophylaxis in the perioperative setting a lot of thought. I would add that patients who are undergoing cardiac catheterization, especially with stenting procedures that may require a large dye load, are another group that tends to be at increased risk of flare, especially in the setting of acute myocardial infarction.
Dr. Edwards: They could get a subtle change in renal function related to the dye load that isn’t throwing them into acute tubular necrosis or acute renal failure but is enough to reduce elimination of urate for long enough that they get this bump of uric acid that triggers the flare.
Dr. Mandell: The retrospective analyses of perioperative gout show that patients who come in with lower urate to begin with are less likely to have flares,33 which suggests that perioperative flares are less likely in patients who have been better controlled and managed. The question of whether to introduce prophylaxis at admission, given the likelihood of postoperative flare, hasn’t been studied formally. Despite this absence of data, I would be comfortable introducing colchicine at a low dose for prophylaxis in this setting, especially in a patient who has had frequent or recent attacks. The major exception would be for patients undergoing bowel surgery or similar procedures, since colchicine’s potential to cause diarrhea and other GI side effects is a main concern with its perioperative use. But a study of prophylactic colchicine before surgery in a “high-risk” population is a trial begging to be done. The challenge is that because the event rate is low, the sample size needed would be so large as to potentially make the study impossible.
Dr. Pile: One issue I encounter from time to time as a preoperative consultant involves patients who should be on chronic urate-lowering therapy but are not. Obviously, when I’m seeing them 7 days before surgery it’s not the time to contemplate doing anything besides perhaps starting colchicine.
Dr. Mandell: Yes, that would be a time to avoid starting urate-lowering therapy since a drop in serum urate is likely to precipitate gout flares, and that’s something you don’t want to happen in a postoperative setting. So just as we shouldn’t stop urate-lowering therapy preoperatively, we shouldn’t initiate it either. When the patient is already on urate-lowering therapy, it should be continued as close as possible to the time of surgery and restarted immediately thereafter. The fluids that are given perioperatively tend to drop the patient’s urate levels, so we often can get away with the brief window of discontinuation around the time of surgery so long as we restart the allopurinol postoperatively.
Dr. Edwards: There are certain surgeries, such as many cardiac procedures, for which the rooms are kept hypothermic, and that can be one more physiologic stimulus for an attack of gout, particularly in the body’s cooler joints. That’s a setting where podagra might be more common, whereas major abdominal surgery with a lot of bowel ischemia and lactic acidosis may render a wide range of joints more equally susceptible, although there are no data on this question.
Dr. Simkin: I’m not certain that postoperative gout is significantly different from hospitalization gout. If patients who are admitted for medical reasons—for instance, cardiac disease or pneumonia or ketoacidosis—also have an established diagnosis of gouty arthritis, they’re at elevated risk too.
Dr. Mandell: Yes, and that point—that preexisting gout is a major risk factor, whether or not it was known at the time of admission—came through from the studies Jim noted above and our experience. That piece of the history is often not obtained until an attack occurs, although more widespread use of electronic medical records may lessen this problem. It’s frustrating that attacks tend to occur about 4 days after surgery, which is typically around the time of planned discharge.
AVOIDING MOBILIZATION FLARES WHEN LOWERING URATE: START LOW, GO SLOW
Dr. Mandell: Let’s shift from this focus on the hospital setting as a precipitant for acute gout attacks and recall that the most predictable precipitant is when we pharmacologically reduce the serum urate level in an effort to decrease the total body load. As Larry noted, the down side of this very necessary intervention is that “mobilization flares” of gout are fairly predictable in this setting. While many of us have used prophylaxis to reduce the likelihood of these flares, we hadn’t had much data on duration of prophylaxis prior to some recent trials of urate-lowering therapy.36 So what are your practices now for trying to reduce the chance of acute attacks as you introduce urate-lowering therapy?
Dr. Edwards: A trigger for gouty attacks is the fluctuation of serum urate levels, be it from initiating or discontinuing urate-lowering therapy. So the approach when initiating urate-lowering therapy is to start low and go slow with your dose escalation. Most of us now buy into the notion of trying to lower the serum urate below 6.0 mg/dL for most patients, and perhaps by another 1.5 mg/dL or so for patients with bulky tophaceous disease. To get to that target, however, you should start with a fairly modest dose of whichever urate-lowering therapy you’re using—allopurinol, febuxostat, or (in rare cases) probenecid—and increase it over time. I tend to let several weeks pass between each dose escalation to ensure that the prior escalation has had time to drop the urate to its new nadir, and then I see what that level is. If it’s not at target, I will modestly escalate further.
Dr. Mandell: And we should emphasize that frequent initial monitoring of urate is appropriate to determine whether you’re reaching your target.
Dr. Edwards: That’s a critical element that isn’t done well in this country. Many studies show that regular monitoring of serum urate after initiation of urate-lowering therapy is actually a rarity. The approach to urate monitoring should be the same as for glucose monitoring with oral diabetes medications or blood pressure monitoring with antihypertensives. We should know what impact we’re having on the patient and continue to adjust the dose to find the appropriate level for the individual patient.
As with the therapies for acute gout, there is no onesize-fits-all dose regimen for urate-lowering therapy. A minority of patients will do fine on 100 mg/day of allopurinol. Most patients will still not reach their serum urate target at 300 mg/day of allopurinol, and we should consider gradual escalation to more generous doses as necessary, beyond 400 mg/day and up to the “maximum” recommended dose of 800 mg/day. Monitoring of serum urate should then continue even after we’ve found an appropriate dose to reach the target level since certain factors may change, altering concentrations of any of the urate-lowering therapies. For instance, a new drug may be added, or the patient may develop new comorbidities or a change in renal function. At this point the monitoring need not be as tight as during the initial period, but it should continue on a semiannual or yearly basis.
Dr. Simkin: I completely agree that going slow is desirable, and it’s very important for our patients to understand what we’re doing and what we’re aiming for. Every gout patient should know what his or her last serum urate level was, yet very rarely is that the case.
Dr. Edwards: Not only that, but they should know what the target level is so that they become an equal partner in their treatment plan, just as diabetics might be unsatisfied until their hemoglobin A1c is down to near 6.0%.
WHAT ROLE FOR PROPHYLAXIS DURING URATE LOWERING?
Dr. Mandell: Larry’s point that dropping the serum urate level slowly over time is less likely to induce an attack of gout is something we had suspected for a while and now have some direct trial evidence for. But no matter what we do, there’s always some likelihood of inducing an attack as we lower the urate level. So what do you do, Peter, to prevent those attacks?
Dr. Simkin: I try to get the patient on a regular dosage of colchicine prophylaxis. There’s good evidence that it’s effective. The dose is usually one or two tablets a day, largely depending on GI tolerance. It’s worth noting that more than a few of my elderly patients appreciate colchicine’s GI effects, as they provide some welcome relief from their chronic constipation. While that’s certainly not welcome by everyone, for some patients it makes colchicine prophylaxis an easier sell.
Dr. Sundy: Colchicine is my standard for prophylaxis as well. I think it’s highly effective.
Dr. Edwards: For how and when to start prophylaxis, my general recommendation is that it should precede the start of urate-lowering therapy by about 1 or 2 weeks. I think starting colchicine 2 weeks in advance is probably a good buffer for ensuring that it’s in the system. The urate-lowering therapies have very rapid onset—allopurinol will cause fluctuations in urate levels within a day or two—so the prophylaxis has to be fully on board.
Dr. Pile: Do you have a sense of how much more effective twice-daily dosing of colchicine is compared with once-daily dosing?
Dr. Edwards: I don’t have a good sense of it, but I tend to use twice-daily dosing unless renal function is a concern—specifically, if the patient’s GFR is between 50 and 60 mL/min/1.73 m2. And in such cases, you’re not really diminishing the dose with a once-daily regimen because the patient is keeping more of the drug around, in light of the renal insufficiency. That hearkens back to Peter’s earlier comments about baseline therapy with statins, some calcium channel blockers, and other drugs that will also require dose modification.
Dr. Mandell: The point is that we want to maintain a certain level of colchicine in all our patients for prophylaxis. It’s not clear that the blood level matters as much as the intracellular level, but we can’t routinely measure either. I think the answer to Jim’s question is that we don’t have data on whether once daily, twice daily, or even three times daily is the right regimen for colchicine prophylaxis; patients differ. I believe many of us start with twice-daily dosing on an empirical basis. If it’s not tolerated, typically for GI reasons, we’ll go down to once a day. If it’s tolerated but ineffective from the point of view of having flares, we may try to go to three times a day, assuming the patient’s not on other medications that would preclude that and doesn’t have chronic kidney disease or hepatobiliary disease.
WHAT ABOUT ALTERNATIVE PROPHYLAXIS OPTIONS?
Dr. Mandell: If a patient is truly intolerant of colchicine but has a reasonable GFR and no comorbidities, what about alternative prophylactic agents? The class to think about would seem to be NSAIDs, but we have no trial data to guide us with their use for prophylaxis. What’s been your experience with NSAIDs for prophylaxis of gout flares during the urate-lowering process?
Dr. Sundy: My experience is that NSAIDs are effective for this. I don’t have a notion of comparative effectiveness relative to colchicine, but I will use an NSAID in this setting when there has been GI intolerance to colchicine. The trick is the added responsibility of monitoring toxicity in terms of blood pressure, renal function, and hyperkalemia, as well as offering gastric protection, but I certainly have patients on NSAID prophylaxis.
Dr. Mandell: Do you feel compelled to use the same very high doses that we talked about for treating acute attacks of gout?
Dr. Sundy: No. I tend to use doses in more of a midrange area and make adjustments based on how the patient is doing. For example, I might increase the dose if a patient has a breakthrough flare.
Dr. Simkin: A situation where I’ve more often seen NSAIDs used for prophylaxis is in patients who have gout but are also taking NSAIDs on a regular basis for osteoarthritis. In that setting I’d be less inclined to add colchicine.
Dr. Pile: And I guess cost becomes an issue now, with the advent of branded colchicine, in a way that it hadn’t been before. At my local retail pharmacy generic naproxen costs 6 cents per 200-mg tablet, so some NSAIDs are very inexpensive.
Dr. Mandell: Colchicine used to have a similar cost invisibility as well. As Peter said before, our ability to provide cost-efficient prophylaxis is disappearing, and this may affect our ability to provide prophylaxis to some patients.
Dr. Sundy: I agree. Many times patients will stop taking drugs that are too expensive for them without telling their doctor. I sense that not using prophylaxis is a major impediment to adherence with urate-lowering drugs as well, as patients will stop those drugs because they sense appropriately that their gout has gotten worse without having been adequately educated about this risk. At the end of the day, we’re trying to eliminate gout as a medical issue for our patients, and anything that makes nonadherence more likely can be a big impediment to this goal.
Dr. Edwards: Patient education is absolutely crucial in this regard. We should step back and appreciate that these regimens can be very complicated and not terribly obvious from the patient’s perspective. First you have the disease gout, for which the treatment is allopurinol. You take that treatment and then your gout gets worse. Without good education to prepare patients for that, how can we expect them to respond, other than wanting to stop the allopurinol and probably thinking poorly of their doctor? Physicians have to educate patients on what to expect and how to stay the course. We must make clear from the start that the treatment of gout is a long-term process. We must explain that when acute flares occur the emphasis is on getting rid of the pain quickly but that the larger job of getting their disease under control by lowering their uric acid levels over time is a long haul. Both patient and physician have to be patient, go slowly, and make sure they don’t disrupt therapy just because symptoms are occurring.
Dr. Sundy: It sounds funny, but sometimes I’ve taken to congratulating patients whose gout flares after they start urate-lowering therapy. I say, “This is great—it means the drug is working, even if it doesn’t seem like it.”
SHOULD WE INTRODUCE URATE-LOWERING THERAPY AT THE TIME OF AN ACUTE ATTACK?
Dr. Mandell: Let’s consider the patient who presents with a first or second or third attack of gout but is not yet being treated for the underlying hyperuricemia. Should we introduce urate-lowering therapy at the time of the attack? After all, it’s a moment of opportunity, as the patient is “captive” and needs to be treated for his or her pain, so should we use that as a chance to also start the urate-lowering therapy that will be needed? There seem to be two schools of thought on this question. One is that if we start urate-lowering therapy immediately and drop the urate, the attack is likely to last longer. The other is that as we drop the urate by starting uratelowering therapy, we’re already treating them very aggressively for inflammation, so it may be an acceptable time to introduce urate-lowering therapy. What are your individual approaches?
Dr. Edwards: I’ve heard that argument of “we have them here now, let’s get it started.” I’d counter that when you start on this course of urate lowering, you need a very close connection to the patient during the long period of dose adjustment. Both the patient and physician need to understand that clearly. I’ve seen too many cases of a bad flare turning into a terrible flare when urate-lowering therapy is started during an acute attack. Patients are unhappy and get turned off of the idea of reintroducing allopurinol or any other urate-lowering therapy. So I don’t use that approach. Instead, I try to optimize the conditions for initiating urate-lowering therapy to minimize the chance that patients will have a bad experience. There’s no reason to rush, since most patients presenting with their first few flares have been hyperuricemic for 30 years, with urate slowly accumulating and depositing around their bodies. I’d promote an approach of letting the acute attack resolve and then getting the uratelowering therapy started about a month later.
Dr. Simkin: My approach is along the same lines as Larry’s. We all encounter gouty patients who say, “I can’t take allopurinol.” When you ask why, the most common response is, “It makes my arthritis worse.” Patients must understand that this is a hump they have to get over; we need to do everything we can to lessen that hump, and that involves good prophylaxis. But it’s also essential to educate them to expect the hump and to forewarn and forearm them in terms of how they’ll cope with it.
Dr. Sundy: A common rationale for starting urate-lowering therapy during the acute attack is that the patient may not follow up and you’ve got them on hand for intervention right now. But if follow-up is that unlikely, urate-lowering therapy is not going to be successful in any case, since frequent monitoring and dose adjustments are needed over many months. The notion that a patient can just be put on 300 mg of allopurinol and check back in 6 months is unrealistic.
Dr. Mandell: It sounds like we’re unanimous on this. One other consideration I would add is that I will avoid adding more than one drug in a patient at the same time whenever I can, to avoid confusion about which drug is causing any side effects that may develop.
REFINING THE PARADIGM: ACUTE ATTACKS AS THE ENTRY POINT TO A PROGRESSIVE DISEASE
Dr. Mandell: So our recommendation to the hospitalist who’s confronting acute gout is to treat the acute attack, start the education process, and make sure the patient understands this is a chronic disease that you’re going to make better in the hospital but which requires lifelong attention to prevent flaring up again. At that point the recommendation is to refer the patient back to the primary care physician or a rheumatologist with the understanding that the underlying hyperuricemia needs to be addressed and that the patient should make sure that happens.
Dr. Sundy: This raises some interesting handoff issues. How do we best ensure that the physician who’s going to pick up responsibility for chronic management is aware of these issues and on board with the necessary patient education efforts? There’s clearly an opportunity for doctor-to-doctor education here as well.
Dr. Pile: Yes, this is a microcosm of what we deal with all the time in the hospitalist world—safe transitions of care and trying to avoid information “voltage drop” as patients return to the care of their usual physicians.
Dr. Simkin: I think our education of both our colleagues and our patients needs to emphasize that this is not only a chronic disease but a progressive disease.
Dr. Edwards: That’s a great point, and it raises the question of the stage in the disease process where we should start urate-lowering therapy—a question without a clear-cut answer. Three decades ago, the thinking was that you should do it before the patient transitioned from acute intermittent gout to a more chronic and advanced tophaceous gout because of how terrible those joints looked and how destructive advanced gout is. Yet the destruction begins much earlier than that, and changes probably occur within the joint even before a patient has his or her first gout attack. There are soft-tissue changes around the joint as monosodium urate crystals accumulate, and the effects on bone and cartilage are occurring throughout the period of acute intermittent gout if the serum urate level hasn’t been lowered. As we’ve come to recognize this, we’ve pushed the initiation of uratelowering therapy earlier and earlier. The standard recommendation now is to start it as soon as a patient has two gout attacks within a year. Since 60% of patients have their second attack within a year of their first, that means initiating urate-lowering therapy after the second attack for about 60% of patients. An even larger majority of patients will be at that stage to qualify for urate-lowering therapy within the first few years after their initial attack.
Dr. Mandell: This is a good place to conclude, with a reminder that the acute gout attack is just one part of a chronic and potentially progressive disease. Although the acute attack is what gets the patient’s attention—and often the physician’s attention as well—it is really just the entrance into therapy for gout, a chronic disease.
Gout is the most common form of inflammatory arthritis in men, and both gout and its root cause, hyperuricemia, are increasing in prevalence in both men and women. The result is that all medical generalists and specialists undoubtedly encounter patients with acute gouty flares in the outpatient or inpatient setting.
Gout increasingly presents in patients with a number of comorbidities, including chronic kidney disease, coronary artery disease, congestive heart failure, diabetes, and hypertension. As a result, medical management of the acute attack of gout, a notably painful and debilitating condition, is growing more complicated.
I asked three of my “goutophile” rheumatology colleagues from around the country and an infectious disease specialist/hospitalist to join me earlier this year in an unscripted roundtable discussion of the management of the acute gout attack. All have extensive real-world clinical experience, and the spirit of our conversation was clinically pragmatic. We aimed to put the relevant, though limited in number, clinical trials into a very practical perspective. We focused our conversation on the diagnosis and management of the acute attack in the outpatient and inpatient settings, essentially leaving undiscussed the long-term management of hyperuricemia—the underlying cause of acute gout attacks. The roundtable and this resulting journal supplement were supported by a CME grant from URL Pharma, Inc., the manufacturer of the branded formulation of colchicine.
I believe our discussion, transcribed and published here without external editing or input, will be of value to all practitioners faced with patients enduring an acute gout attack. We’ve added references as appropriate, and I hope the result is a balanced mix of experience-based medicine from thoughtful, seasoned clinicians with quantitative supporting data provided as available from the published clinical studies. This supplement has been independently peer reviewed by a rheumatologist content expert to exclude any content that could be perceived as unjustified bias for or against any specific therapy.
Gout is a disease that has fascinated me for 30 years, and my colleagues share my sense of wonder with the dramatic nature of the acute gouty flare (and its auto-resolution). I hope our shared interest shines through here and will assist all of us in managing patients with this chronic disorder punctuated by dramatically painful attacks.
As with all issues and supplements of Cleveland Clinic Journal of Medicine, I, as the journal’s editor-in-chief, welcome your feedback and comments.
—Brian F. Mandell, MD, PhD, FACR
HOW DO WE KNOW IT’S GOUT? WHEN PRESUMPTIVE DIAGNOSIS IS (AND ISN’T) ENOUGH
Brian F. Mandell, MD, PhD: In addressing acute gouty arthritis, let’s start with diagnosis. The gold standard for diagnosis of gout is synovial fluid aspiration and analysis for the presence of monosodium urate crystals. But what’s the practicality of aspiration and the need for synovial fluid analysis in a given patient at a given time? When is fluid analysis useful, and when is it absolutely necessary?
N. Lawrence Edwards, MD: It’s important to emphasize that synovial fluid analysis remains the gold standard of diagnosis. Ultrasonography of the peripheral joints is probably specific enough that it may soon be considered on par with fluid analysis for crystals, but we’re not there yet and it is not uniformly available. Less than 10% of patients who are diagnosed with gout have ever undergone synovial fluid aspiration, however, so presumptive, or “clinical,” diagnosis is very common.
A reliable presumptive diagnosis should be based on the image of a classic gout attack. The further an episode strays from a classic attack, the more presumptive the diagnosis is. So if a patient experiences the typical rapid explosion of symptoms from no pain to maximum pain over less than half a day, and if the symptoms are in the typical joints—the midfoot, the ankle, the first metatarsophalangeal joint (great toe)—that’s very consistent with a classic gout attack. Symptoms in the knee are more problematic because the two main conditions in the differential diagnosis of gout—septic arthritis and calcium pyrophosphate dihydrate (CPPD) crystal deposition disease (pseudogout)—also commonly affect the knee.
If the patient has had acute attacks that have self-resolved or gone away with therapy over a 5- to 8-day period, that too is a profile that’s typical enough for a presumptive diagnosis of gout, especially since septic arthritis doesn’t tend to do that. Pseudogout usually is a bit slower in onset. It can look virtually identical to gout, but the affected joints often aren’t the same and the attacks tend to last for weeks rather than days, which ultimately simplifies the differential diagnosis.
The setting where synovial fluid analysis is absolutely essential is when a septic process is suspected. If there’s any nagging concern that an infection might be present, the clinician should aspirate the joint or refer the patient to someone who can. The features of a septic process may include joint swelling, rigors, and fever, but all of those can be seen in gout, so they are not distinguishing.
Dr. Mandell: John, how comfortable can you be in making the distinction between crystal-induced arthritis versus infection at the start of a given attack?
John S. Sundy, MD, PhD: Larry’s point about the features typical for gout is key, but I also consider the patient’s comorbidities and general situation. For instance, if the patient is a healthy middle-aged man with no relevant past medical history, it’s easier to feel comfortable with a presumptive diagnosis of gout. But if I’m dealing with a hospitalized patient who’s had invasive procedures and has risk factors for bacteremia, it’s imperative to get synovial fluid to rule out a septic joint. There is, of course, a continuum between these two examples. It’s important to have a construct of what typical gout is. When a case is close to that construct, it’s reasonable to work with a presumptive diagnosis; the further a case is from the construct, the more important it is to get fluid. From there it’s just a matter of overcoming the practical issues that a lot of clinicians contend with in obtaining the aspiration and fluid analysis.
Although it’s fine to think about a presumptive diagnosis and initiate treatment accordingly, it’s still incumbent upon the clinician to strive to get a firm diagnosis at some point so that the patient and physician can be 100% confident in the diagnosis.
Peter A. Simkin, MD: When we focus on the initial diagnosis we may overlook the fact that a chronically gouty joint is more susceptible to infection, so that patients who have well-established gout may still require joint aspiration to exclude acquired infection. I agree that the knee is a major site to focus on, and the olecranon bursa is another common site of infection in gouty patients.
Dr. Mandell: That’s convenient, since both of those areas are easily aspirated by rheumatologists and nonrheumatologists alike. So I’m hearing a consensus that we look at the entire clinical picture, including the historical features and the location of symptoms, to assess whether an acute attack is more likely to be gout or infection. I would add that crystal disease, especially gout, is far more common than infection, and infection is particularly unusual in the midfoot.
No objective analysis has shown that any single feature, such as leukocytosis or fever, is reliably indicative of infection versus crystal disease. Yet when we look at the entire pattern, if there isn’t significant white blood cell elevation, if there aren’t rigors, if there isn’t significant fever, and if the patient has a history of self-limited attacks, that points to a clinical diagnosis of gout that we probably can be comfortable with. That said, joint aspiration is still required if there is not a rapid response to treatment.
There are settings, as John noted, where vigilance for infection is particularly indicated, notably in hospitalized patients. Jim, as a hospitalist, you may be less apt than us rheumatologists to stick a needle into a joint. What’s your comfort level in assessing the hospitalized patient with sudden eruption of a red, swollen, painful joint?
James C. Pile, MD: I agree that the setting is very important. Let’s say I have a patient with a well-established history of gout to whom I’ve been giving aggressive diuresis for heart failure exacerbation for 2 or 3 days. If he develops a hot, swollen ankle that’s consistent with his prior episodes, I won’t typically feel obligated to stick a needle in that joint, particularly not in an ankle, for which I’d want to consult a rheumatologist in any event. If it’s a patient who underwent cardiac catheterization or other instrumentation a couple of days ago, and if this patient is febrile and has sudden eruption of a hot, swollen knee, that’s a different story. Aspiration is absolutely required in that patient, who needs to be treated as having septic arthritis until proven otherwise. But there is a lot of gray area between these two scenarios.
Dr. Mandell: Speaking of willingness to aspirate joints, there’s a concern among nonrheumatologists about putting a needle into a joint that appears to have cellulitis over it. This can stand in the way of appropriate diagnosis since the response to crystals in a joint can produce a response that looks like cellulitis. If my examination suggests joint involvement, a cellulitislike appearance does not stand in the way of aspirating the joint or bursa, which might reveal a closed-space infection.
Dr. Edwards: One population that should make us nervous any time they get an acutely swollen joint are organ transplant recipients. When these patients get gout, it’s often atypical. Not only are they usually on cyclosporine, which can cause hyperuricemia and accelerated gout, but they’re also often taking corticosteroids, which dampen the acute symptoms of gout. So whether the acute swelling is caused by gout or infection, it may not appear to be quite as profound as it actually is. Additionally, these patients may not get fever or rigors. Despite all this, they are at high risk for infection, in light of their immunosuppression. When any transplant recipient presents with acute monoarthritis, the burden of proof is on us to show that it’s not an infected joint.
Dr. Mandell: I’d add that Larry’s caveat extends to patients on any immunosuppression, including the anti–tumor necrosis factor agents, and we have seen both infection and gout in these patients.
Dr. Simkin: It’s also worth noting that joint aspiration can be therapeutic. The exquisitely painful joint often is so painful because of distention of the joint capsule. If the joint is tapped, the capsule becomes decompressed and the needle track may leave a vent through which it can remain decompressed.
Dr. Edwards: Pointing out the therapeutic benefit of joint aspiration may even be one way to get patients to allow us to put a needle into an excruciatingly painful joint. So while we advocate joint aspiration as a diagnostic procedure, often an effective approach is to explain to patients that if you tap the joint, you can treat their pain much more rapidly. Instead of having another 18 to 36 hours of pretty significant pain even on the best oral treatments, with aspiration patients might be relatively free of pain within 18 to 24 hours.
Dr. Mandell: So I’m hearing that a history of prior events consistent with attacks of crystal-induced arthritis suggests that an acute flare is likely to be another attack of crystal disease. Almost all patients with acute postoperative flares have a history of gout, though that history rarely is noted in the chart and is usually obtained later. The problem with this reliance on history is that if it’s a previously damaged joint, it may have altered microvasculature and may be more prone to infection, as Peter noted. The literature on coexistent infection in crystal disease is small, which attests to its relative infrequency, but we need to be wary of this potential for coexistence, particularly in patients at risk of infection, such as transplant recipients and those with recent infection elsewhere and bacteremia. Acute joint swelling in such patients calls for arthrocentesis.
Dr. Edwards: One last criterion for when to do a joint aspiration is when a patient with well-established gout tells you an attack is different from previous attacks. If a patient says, “This isn’t like my previous gout attacks,” that’s a red flag that usually merits joint aspiration.
Dr. Mandell: We’d be remiss if we didn’t mention the longstanding “preliminary” criteria for the diagnosis of acute gout published by the American Rheumatism Association (now the American College of Rheumatology) in 1977.1 They’ve been used for clinical research studies and also applied in principle to clinical practice. There have been a couple of recent papers examining the limitations of these criteria. How do you use these “criteria”?
Dr. Edwards: Their biggest drawback is that they’re cumbersome. They also have recently been shown to lack specificity and, to some degree, sensitivity.2
Dr. Mandell: Testing of the ACR criteria has shown that we’ve had unfounded confidence in using them in practice. As you said, their sensitivity has been reported to be 80% and their specificity even lower, at 64%.3 So even a panel of clinical indicators of gout cannot be relied on in a setting where we have concern about the possible alternative diagnosis of infection.
CAN TRIALS OF SPECIFIC THERAPIES OFFER DIAGNOSTIC UTILITY?
Dr. Mandell: What about the response to anti-inflammatory therapy? Can we use the specificity of response to a drug like colchicine to determine whether an attack is more or less likely to be attributable to crystal disease?
Dr. Simkin: It’s certainly helpful, but entities other than crystal disease can respond to anti-inflammatory drugs, of course. For instance, some people with paraneoplastic problems will have acute arthritis that goes away with anti-inflammatory therapy. So I think it’s more a matter of how fast and complete the response is rather than whether there is a response.
Dr. Mandell: Consider the cases you’ve seen that ultimately turned out to be septic arthritis. What was the initial response in those cases to either nonsteroidal antiinflammatory drugs (NSAIDs), colchicine, or corticosteroids, and what conclusions can you draw from those cases?
Dr. Sundy: In my experience, signs and symptoms are often more lingering in these infectious cases. I think it’s fair to generalize that the response is not as rapid as with a typical gout flare. This underscores the importance of follow-up. If a patient presents to the emergency room and is treated and released, the critical question is how careful the follow-up is going to be at 24, 48, and 72 hours to see if improvement and resolution are occurring as expected. If you’re going to proceed on the basis of a presumptive diagnosis, there must be planned and frequent follow-up.
Dr. Edwards: I don’t think anyone thinks that a patient with an acute septic joint is going to get much improvement at all from oral colchicine or much more than a little fever reduction from NSAIDs without parenteral antibiotics. That joint is going to progress toward destruction within days. So John’s point is crucial that any time you make a presumptive diagnosis of gout, it’s incumbent on you to make sure that the pattern thereafter is one of resolution over time—not worsening—on the medications you use.
Dr. Pile: Even with corticosteroids, which are probably most likely to mask the clinical picture, in most cases I think it’s still fairly easy to sort things out based on response in the first 24 to 48 hours after initiating therapy. I don’t think patients with bacterial septic arthritis are going to remain better on corticosteroids.
Dr. Mandell: The time frame is crucial. Therapy for acute arthritis is ideally started fast, and patients frequently will be treated initially with an anti-inflammatory—in the community, probably more often with an NSAID than with colchicine or a steroid. Even if they actually have a septic joint, they may initially get a bit better on the anti-inflammatory, perhaps from the analgesic or antipyretic effects, but the benefit will clearly plateau and the attack will worsen again. So the period for peak vigilance is probably 1 to 2 days after therapy is started; if improvement is not maintained in this period, you should be circumspect regarding diagnosis of crystal disease and you must aspirate, or reaspirate, the joint.
Let’s explore specificity a bit further. Diagnostic specificity has historically been attributed to colchicine. People have even suggested that a diagnostic colchicine trial can be useful. How does that jibe with your experience with response to colchicine therapy in patients with entities other than gout?
Dr. Edwards: The main disease in the differential diagnosis of gout other than a septic process is pseudogout, or CPPD arthropathy, and patients with pseudogout may also respond to colchicine.
Dr. Simkin: Another entity that reportedly responds to colchicine is sarcoid, particularly sarcoid of the ankle.
Dr. Edwards: In fact, colchicine has been tried for treating the acute inflammatory symptoms of many of the diseases we see as rheumatologists. Some people swear by it, while others believe it doesn’t do much for these conditions. In any case, I don’t think its specificity for gout is very strong, and we have no basis to say that response to colchicine should serve as a diagnostic test.
Dr. Mandell: I agree, and the literature over the past few years on colchicine for treatment of acute and chronic pericarditis further argues against specificity of this drug for crystal-induced inflammation.
SERUM URATE LEVEL IS UNRELIABLE FOR DIAGNOSIS
Dr. Edwards: While we’re discussing diagnosis, nothing’s been said about serum urate levels, which I think many primary care physicians rely on heavily in the diagnosis of gout. We need to underscore that a lot of people who are hyperuricemic will never develop gout. At the same time, there’s also the phenomenon during an acute attack in which an acute uricosuria accompanies the initial inflammation, causing serum urate values to fall from what would otherwise be a high baseline to a level that looks normal. These declines may be between about 1.5 to 2.5 mg/dL, so that a patient presenting to the emergency room with a serum urate of, say, 7 mg/dL might actually have a baseline chronic level of 9 mg/dL.
Relying too heavily on serum urate levels can be misleading in either direction: someone with joint pain with serum urate elevation may be diagnosed with gout inappropriately, whereas someone who comes in with a gouty attack who has a serum urate level in the normal range may be thought not to have the disease.
Dr. Mandell: The fact that urate level didn’t come up in a conversation among rheumatologists with an interest in gout testifies that none of us uses serum urate in the diagnosis of acute arthritis. Your point is well taken, though, that serum urate is used for this purpose in the community but shouldn’t be, especially not in an acute setting.
Dr. Simkin: Indeed, I’ve seen a serum urate of 3.7 mg/dL during an acute, crystal-confirmed gout attack in a patient who was not on urate-lowering therapy.
Dr. Mandell: There’s also the issue that laboratory-defined normal levels of serum urate are not the same as biologically “normal” levels in the context of urate deposition. Levels that are in the normal range in the laboratory clearly can be above the saturation point of urate in physiologic tissues, which is about 6.7 mg/dL.
Dr. Simkin: Plus, many labs report their normal range as being up to 8 mg/dL, yet most gouty arthritis in the community probably occurs in patients with urate levels below 8 mg/dL, and this misstatement of normality certainly deserves attention.
Dr. Sundy: The issue of serum urate further underscores the importance of looking at gout as a longitudinal condition and not just as an acute episodic one. We would never treat and release a patient who came in with a severe hypertensive episode without insisting that further follow-up was indicated for the hypertension. Similarly, for a person who presents in the emergency room with gout there should be a longer-term strategy to make sure the patient understands the importance of followup to address the underlying hyperuricemia.
Dr. Mandell: Yes, there is a challenge with the system of care; the providers faced with the acute attack are not the ones who will ultimately be treating the disease and its associated comorbidities.
GENERAL APPROACH TO THE ACUTE GOUT ATTACK
Dr. Mandell: Let’s return to the patient who presents with an acutely swollen, painful joint. Let’s say gout is diagnosed with confidence, supported either by synovial fluid analysis or by the overall clinical details. What are your general considerations, Jim, for initial treatment in the hospital?
Dr. Mandell: We’ll come back and talk about each of these drug classes specifically, but what do my fellow rheumatologists tend to reach for as first-choice therapy?
Dr. Simkin: My thinking is very similar to Jim’s. As far as NSAIDs are concerned, so many of our patients have significant renal compromise that it is absolutely mandatory that we know what a patient’s renal function is before we treat acute gout with NSAIDs. I’ve seen more catastrophes from the use of NSAIDs in patients with renal compromise than from any other gout treatment scenario. So I probably wind up using steroids more often, but in the hospital setting you often have to deal with a surgeon who doesn’t want to use steroids. In such a case, if the patient also has renal problems, an agent that has entered the picture in our hospital is the interleukin-1 (IL-1) receptor antagonist anakinra, given in daily subcutaneous injections of 100 mg. When a patient is hospitalized for gout, it’s his severely painful joint that’s keeping him from going home, and in this situation anakinra in fact becomes a relatively inexpensive option, despite its absolute cost, when compared with the cost of the hospital bed.
Dr. Mandell: I think that’s the first time I’ve heard “anakinra” and “inexpensive” used in the same sentence.
Dr. Simkin: Of course biologics are terribly expensive, but so is hospitalization, and hospitals are bad places. We want to get our patients home, and in our experience this has been a very useful way to make that happen sooner.
Dr. Pile: I agree that an emphasis on hospital throughput is incredibly important. For the hospitalized patient with gout that’s preventing ambulation, that’s the issue that must be addressed before discharge is possible. Certainly the agent with the fastest onset of action is going to be very attractive.
Dr. Edwards: All of these agents for acute gout have a relatively fast onset of action if they’re given early in the disease process. Once you get out to a day and a half from the onset of symptoms or beyond, you’re fighting an uphill battle in terms of making a difference in the natural course of the attack. I believe that’s true of all three of the medication classes that are typically used.
My approach to the initial therapy choice is highly individualized, depending on what the patient’s been on before. If they’ve been on maintenance colchicine to prevent flares and then they flare, I usually won’t use colchicine for the acute attack; I will go with a steroid or an NSAID. If they’ve been on NSAIDs as preventive therapy and they flare, I might try low-dose colchicine for the flare or use steroids. In cases of a prolonged course, where the patient is in the hospital and it’s 3 or 4 days since symptom onset and a steroid taper or a trial of colchicine has failed, I’ve been very impressed with the ability of anakinra to suddenly bring the attack to a halt. Like Peter, I am on the cusp of looking at acute treatments a little differently, although there still aren’t a lot of data on IL-1 inhibition in this setting.
Dr. Sundy: There are data showing that a gout flare adds about 3 days to the hospital stay,4 so that’s a huge burden that intervention with IL-1 inhibition can really help to address.
Dr. Mandell: I think we’ve seen a trend over time toward corticosteroid therapy becoming more common, particularly in hospitalized patients. I think that’s partially related to more widespread use of appropriate deep vein thrombosis (DVT) prophylaxis, which means that more patients are on anticoagulant therapy, which is one more reason to shy away from NSAIDs, particularly the nonselective ones, in the hospital setting. Do you sense that trends in the outpatient setting have shifted, or do most clinicians still reach for NSAIDs?
Dr. Sundy: I think most people still reach for the NSAID, but it really depends on the comorbidities. I sense we’re seeing chronic kidney disease in a greater proportion of patients, and that’s probably creating a shift toward a bit more corticosteroid use. I like to reach for an NSAID as first-line therapy, but we really have to understand our patient’s overall comorbidity profile, including renal function, before doing so, as Peter said.
Dr. Edwards: I think NSAIDs are still the most commonly used acute treatment for gout. I used to calm myself when I used them by saying, “It’s only a 7- to 10-day course; how much trouble can you get the patient into?” Well, in that short a time I’ve had patients tip over into congestive heart failure because they had some renal decompensation beforehand and then had another 20% or 25% of their renal function knocked out with the NSAID, and they’d have extra sodium retention. I’ve seen GI bleeds develop in patients over that short a time. I don’t prescribe nonselective cyclooxygenase (COX) inhibitors anymore without also giving a proton pump inhibitor for stomach protection. I think that’s becoming a standard of how to use NSAIDs among rheumatologists, and it’s hard to get that stomach protection up and running in the very short time frame of an acute gout attack. So I’m personally tending away from NSAIDs; I use steroids more often and then the IL-1 inhibitor for special cases.
Dr. Mandell: Studies assessing the gastric risk of NSAIDs have shown that a GI bleed can be induced in as little as 2 or 3 days. I agree that there is a trend, at least among rheumatologists, to use a proton pump inhibitor when initiating outpatient NSAID therapy. This combination may still be cost-effective for many patients, as it can speed the return to their usual activities. But even in the outpatient setting steroids are becoming more common than they used to be.
DOSING FOR THE ACUTE ATTACK: BE AGGRESSIVE FROM THE START
Dr. Simkin: Whatever dose of steroids we use in the outpatient setting, I think it’s highly desirable to divide it. Rheumatologists are taught to use a single daily steroid dose in the morning, primarily to spare the adrenal glands. While that makes sense in patients on long-term steroid therapy, such as for lupus, it doesn’t necessarily make sense in gouty arthritis. I’ve seen patients whose gout flared up every night despite taking sufficient prednisone; when they divided the dose, their gout was controlled.
Dr. Mandell: I would generalize that further to stress the importance of using an adequate and aggressive dose of either steroids or NSAIDs for treating acute gout. A common mistake I notice in the community is the use of too low a dose of either steroids or NSAIDs. We should treat aggressively from the start with a full dose of these agents.
Dr. Edwards: Even more than the usual full dose, I would say. Many generalists have a concept of what an analgesic dose of an NSAID is, which is pretty low—in the case of ibuprofen, perhaps 400 mg three times daily. An anti-inflammatory dose is higher—perhaps 800 mg three times daily for ibuprofen. Most of us who’ve used NSAIDs for gout realize that we have to get even a little bolder than that from the start. It can be hard to convince some generalists to exceed what has been their ceiling of comfort for an NSAID, but doing so for the first 24 or 36 hours is important to getting the gout attack under control. Of course, the need for such aggressive dosing is all the more reason to make sure not to use an NSAID in a patient with significant renal disease or other comorbidities for which NSAIDs are problematic.
Dr. Mandell: And all the more reason to provide gastric protection.
Dr. Sundy: I would add that if a clinician is not comfortable using doses that high, that may be a good reason to think about choosing a different initial therapy, be it colchicine or steroids (if the reluctance is with NSAIDs) or NSAIDs (if the reluctance is with steroids).
ACUTE ANALGESIA: ANYTHING MORE THAN AN ADJUNCT?
Dr. Mandell: What about treating gout with acute analgesia alone? Consider a setting where you may want to avoid a drug with anti-inflammatory or antipyretic effects, perhaps in the postoperative period or when coexistent infection is a concern and you want to monitor the fever. Can you get by with using narcotic analgesia alone?
Dr. Sundy: I’m not impressed with that approach. Narcotics can play a role in managing a patient’s pain, but a narcotics-only approach will allow the flare to linger and not address complications of the flare or back-to-work issues. I view analgesics as cotherapy as opposed to single-agent therapy.
Dr. Mandell: Is narcotic analgesia effective even at treating the pain acutely?
Dr. Sundy: No. Gout pain is an exquisitely inflammatory pain, and the key is to tackle that inflammatory response. Analgesics are really just an adjunct.
Dr. Simkin: I totally agree, but they’re an important adjunct that we often overlook.
Dr. Edwards: I’ve been singularly unimpressed with narcotics. Gout is a cytokine-driven process that is intensely inflammatory. It’s like a lot of other types of pain that don’t respond fully to narcotics, such as herpetic neuralgia or uterine pain. Perhaps the benefit of narcotics in gout is their soporific effect—patients sleep through their attack. Yet if you touch the gouty joint or the patient moves it, the pain is just as intense as if the patient weren’t taking anything. So I don’t know that narcotics add anything unless the patient has other causes of pain. I don’t use them at all.
Dr. Sundy: I tend to make the option available to the patient. I say, “Here’s what we need to do to knock down this flare. And here’s something additional you can try for pain; we can see if it helps.” But I’ll emphasize the importance of improving the inflammatory piece.
Getting back to the postoperative setting, we should recognize that a strategy to just ride out a perioperative gout flare with an analgesic medication, perhaps because of concern about the effect of corticosteroids on wound healing or infection, can carry important risks. A patient treated that way will end up bedbound at precisely the time you want him or her to be moving around, so there’s now the risk of postoperative pneumonia and other complications that won’t get documented as complications of gout even though they actually are.
Dr. Mandell: Not to mention the untreated postoperative fever from the crystal-induced attack, which then leads to a work-up looking for DVTs, more blood cultures, and more radiographs, all the while extending the hospital stay and wasting resources. Jim, as a hospitalist, do you still see narcotics used initially as the primary treatment for gout flares in the hospital?
Dr. Pile: It depends on which hat I’m wearing. If I’m the hospitalist and the patient’s on my service, usually my antennae are up for the appearance of gout in the hospital, and the house staff may or may not recognize it if the process starts overnight. When I’m serving as the medical consultant, I sometimes encounter cases where the surgeons don’t recognize a gouty flare when it first appears, and I’ve had multiple patients in whom gout-induced postoperative fever triggers a consultation. In the latter case, it’s not uncommon to see narcotics being used in an attempt to treat gouty pain.
RELATIVE DRUG EFFICACY FOR ACUTE ATTACKS: HOW MUCH DO WE KNOW?
Dr. Edwards: If they’re given early, all of them have good—if not necessarily fast and ideal—effectiveness. The recent AGREE trial of colchicine (Acute Gout Flare Receiving Colchicine Evaluation) assessed pain reduction at 24 hours in patients randomized to three treatment groups: a low-dose colchicine regimen consisting of three 0.6-mg tablets (1.2 mg initially, followed by 0.6 mg 1 hour later); a more traditional high-dose colchicine regimen consisting of eight 0.6-mg tablets (1.2 mg initially, followed by 0.6 mg every hour for 6 hours), and placebo.11 Response, defined as a 50% or greater reduction in pain at 24 hours without rescue medication, was reported in 37.8% of patients in the low-dose colchicine group, 32.7% of patients in the high-dose colchicine group, and 15.5% of placebo recipients.
This is an excellent study that should finally take highdose colchicine for the treatment of acute gout off the table since it offered no improvement in efficacy but was significantly more toxic. However, while 50% improvement in 24 hours is a hard target to achieve, the fact that barely more than a third of subjects hit the target means we should still be looking for more effective approaches.
Dr. Simkin: Similarly, in the first controlled study of colchicine in acute gout, published by Ahern et al in 1987,10 23% of colchicine recipients noted a 50% reduction in their pain at 12 hours after starting treatment. That leaves more than three-quarters with little or no response within 12 hours, and gout is one of the most painful conditions we treat. We’d very much like to help our patients sooner than that. So while there are no head-to-head data comparing colchicine versus NSAIDs versus steroids, my impression too is that oral colchicine is a second-line choice for the acute gout attack.
Dr. Sundy: When we try to understand this literature, it’s important to look closely at how patients were ascertained. The AGREE trial evaluated patients who were enrolled when they were asymptomatic; they were given study medication and instructed on how to start it once a flare began. In contrast, most of the well-controlled NSAID trials captured patients as they came in with a flare, and they had to have had the flare no longer than 48 hours. I believe there’s a big difference between having had a flare for 12 hours versus 48 hours—and even as long as 72 hours in some corticosteroid trials. The longer the clock has been allowed to tick before treatment is started, the harder it is to achieve rapid symptom reduction.
Dr. Mandell: Obviously it’s difficult to compare between studies, but it’s interesting that in a study of the COX-2 inhibitor etoricoxib versus indomethacin,7 one of the largest studies of NSAID therapy for acute gout, approximately one-third of patients taking etoricoxib reported no pain or mild pain within 4 hours. This very rapid pain control is what we really want, since pain is the concern, along with eventual complete resolution, of course. So there really may be value in having something that has analgesic as well as anti-inflammatory effect, to Peter’s earlier point. Tackling both the pain and the inflammation that’s causing the pain certainly makes sense.
Dr. Edwards: Yes, and that etoricoxib study was designed the way people often are treated, with the unfortunate delay before the doctor is seen and the medication is started. The AGREE trial is how I hope everybody would be treated regardless of what they’re treated with. We’re probably never going to see a good head-to-head trial among the three main types of drugs we use for acute gout—the unpredictability of flares makes it extremely difficult.
Dr. Pile: I’m curious whether or not you, as rheumatologists who specialize in gout, are surprised by the results of AGREE. My experience with colchicine over time has been more favorable than what’s suggested by AGREE.
Dr. Edwards: How do you use the colchicine?
Dr. Pile: For more than a decade I’ve been using 0.6 mg three times daily. I thought that practice was unusual until I recently read the AGREE study and realized that a lot of you have been using that regimen for a while.
Dr. Mandell: Some gout patients will take a colchicine tablet when they feel the wisp of an attack coming on and it immediately will stop the attack. That gets to the lore that if you treat a flare very early on, you may be able to abort and treat an attack very quickly. Years ago, however, I was involved in an analysis of 100 patients treated with intravenous (IV) colchicine, and we found that treatment response was unaffected by whether patients were treated early or after a delay of 48 hours. I believe colchicine behaves like a different drug when given IV—and the IV form has since been withdrawn from the market for safety concerns—but this underscores that individual responses to various agents are quite unique. I think there are some patients who are exquisitely sensitive to colchicine, while others are exquisitely sensitive to an NSAID, and so on. This means we’d need a very large sample size to tease out that variation in a trial, and that’s not likely to happen.
Dr. Edwards: I too have had gout patients who tell me they get this premonition of an attack—a feeling that “something just isn’t right”—hours before they actually feel any pain from the attack. A lot of them tell me they’ve learned over time that if they take a colchicine tablet when they get this premonition—or an extra colchicine tablet, as some are on colchicine maintenance therapy (typically one or two tablets a day) plus whatever other background therapy they’re on—they don’t get the flare or the flare is diminished.
Dr. Mandell: I’d like to wrap up this portion by getting your sense of whether there’s a difference in efficacy for treating acute attacks among the drug classes we’ve discussed.
Dr. Edwards: I believe the IL-1 inhibitors are probably the most potent agents for aborting a gout attack, followed by steroids, which I think have a leg up on both colchicine and NSAIDs. The latter two options probably are equally effective in aborting an acute attack.
Dr. Sundy: Yes, I would rank them the same way.
THE ROLE OF COMORBIDITIES IN THERAPY CHOICE
Chronic kidney disease
Dr. Mandell: Let’s get more specific about comorbidities and therapy choice, starting with chronic kidney disease. What’s the threshold at which renal compromise starts to influence your choice of therapy for an acute attack of gout?
Dr. Sundy: I have a very low threshold for avoiding NSAIDs in that setting, mainly because when I want to use NSAIDs, I want to use them at a very high dose, and even short-term use of high-dose NSAIDs can reduce creatinine clearance. So I would certainly avoid NSAIDs when the patient’s creatinine clearance is less than 60 mL/min, and I’d be even more cautious if the patient had underlying hypertension or congestive heart failure.
Dr. Mandell: So even with the reversibility of almost all of the NSAIDs’ renal effects, you tend to stay away from them in that setting?
Dr. Sundy: Yes.
Dr. Edwards: I’m less guided by a single creatinine clearance level. I tend to look at patients in light of the 20% to 25% reduction in the glomerular filtration rate (GFR) that most NSAIDs will cause, even when used acutely. Though the NSAID effect is reversible, if that GFR reduction is going to make a difference to other compensated systems, primarily the heart, then the NSAID should be avoided.
Dr. Simkin: It’s my understanding that plasma flow to the kidney becomes prostaglandin dependent with renal insufficiency, and when we use antiprostaglandin agents we get into trouble on a mechanistic level. I’m not sure of the exact point at which that occurs, but I think we all recognize that serum creatinine is a fairly weak indicator of which patients have some limitation.
Dr. Mandell: So we agree on the need to be very cautious with the use of NSAIDs, recognizing that in the acute setting there is reversibly depressed renal blood flow from NSAIDs. The decreased blood flow will get better, but there’s the issue of what happens during fluid retention when there is coexistent congestive heart failure, diabetes, and chronic kidney disease—you may also occasionally bump up the potassium level. In general, most rheumatologists shy away from selective or nonselective NSAIDs in the setting of chronic kidney disease. Is that consistent with the use you see in the hospital, Jim?
Dr. Pile: It is. I would add that if patients are on certain other medications—especially angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or beta-blockers—there may be additive negative effects because of the potential for hyperkalemia if an NSAID were added to the mix.
Dr. Mandell: I would add to that list of concerning medications aminoglycosides, cyclosporine, and other nephrotoxic agents.
Diabetes
Dr. Mandell: What about diabetes? How does a patient’s diabetes enter into your choice of acute gout therapy in the outpatient and inpatient settings?
Dr. Edwards: Diabetes is especially problematic because it significantly affects two of our treatment choices—steroids and NSAIDs. The steroid doses needed to effectively treat an acute gout attack have a pretty profound effect on glucose levels in diabetic patients. That’s always of concern to me, and it’s certainly of concern to my patients with diabetes who are vigilant about monitoring their glucose levels, which may spike from 130 or 140 mg/dL to values in the high 200s. These elevations will last for the 7 to 10 days of treatment, and the longterm adverse effects of that period of hyperglycemia are not clear. If we put these patients on short courses of corticosteroids, perhaps we should be treating their diabetes more aggressively during that period, but I don’t think we have a history of doing that.
Dr. Pile: It’s very difficult to do in the short term. That’s especially the case with the diabetic patients I see as a hospitalist at a safety-net hospital. Many of my patients don’t have good glucose control at baseline: their levels of 250 mg/dL may go to 400 mg/dL or higher with acute corticosteroid therapy, so that’s clearly a problem.
Dr. Mandell: I try to avoid giving corticosteroids in the outpatient setting to a diabetic patient who is being maintained on oral hypoglycemic medications, particularly if efforts are being made to avoid more aggressive diabetes therapy. Steroids may be an option, though, for a patient who is on insulin, uses a pump, and is very comfortable measuring and managing his or her glucose at home. In the hospital setting, I’m more comfortable using corticosteroids in diabetics because I have much better control over their glucose; I can just increase the basal and premeal coverage.
Dr. Pile: I agree. With vigilance I can control almost anyone’s blood sugar in the hospital, but when patients leave the hospital on steroids, it becomes much more problematic.
Dr. Mandell: Right. You don’t know what’s going to happen when they go out and liberalize their diet on top of the steroids, because often it’s the postprandial surge that’s exacerbated by steroids.
Dr. Sundy: It can be very helpful to know at baseline what a patient’s spot blood sugar is before using a steroid, just as it’s important to know what the creatinine is before considering an NSAID. In the setting of an acute flare, the patient might already be quite hyperglycemic, and many of these patients probably don’t have the tools to manage this at home, so careful follow-up—coming back the next day for at least a blood sugar check—is critical to the proper care of these patients.
Dr. Mandell: When we talk about steroids we tend to think mostly about prednisone, but Peter mentioned injectable steroid. What about injectable adrenocorticotropic hormone (ACTH), which goes in and out of vogue as an agent to treat acute gout? What is your experience with using ACTH injections, specifically related to the issues of diabetes and heart failure?
Dr. Edwards: Before it got reformulated about 3 or 4 years ago and its price went way up, injectable ACTH was my drug of choice for treating acute gout. I had hardly any failures on it over the 10 to 15 years when it was my main means of treating acute gout. I would typically use 80 IU of ACTH gel, given subcutaneously, and the response within 12 hours was quite dramatic. About one in four patients would require a second injection at that 12-hour point, but almost everyone was symptomfree at 24 hours. This response was seen even in patients who were on fairly high-dose chronic steroids already, such as transplant patients.
This suggested that a mechanism other than just adrenocortical stimulation explained the benefit from ACTH. In fact, a lot of data have emerged suggesting that ACTH has a specific effect that is not related to adrenocortical stimulation.
Ten years ago an injection of ACTH gel would cost about $3 or $4, whereas today it costs $2,000 or more.29 I don’t think it’s justifiable at that cost.
Dr. Mandell: There’s also the issue of fluid retention with ACTH, and the higher doses, which seem to be more effective, are more likely to exacerbate congestive heart failure, which has to be a concern if you go this route.
Coronary artery disease
Dr. Mandell: Atherosclerosis, diabetes, and chronic kidney disease are increasingly recognized in the gout population, and coronary artery disease links with all of those. Does the presence of coronary artery disease enter into your choice of agents for a patient with an acute attack of gout?
Dr. Edwards: It doesn’t have as big an impact as diabetes does. To me, the priority in this setting is resolving the acute attack as quickly as possible, as I’ve seen my share of patients for whom the acute gout attack has been so painful and stressful that they’ve developed angina at the same time. So I might use steroids in patients with coronary disease because I’ll want to go with what’s likely to be most efficacious for quick resolution of the attack even though steroids may increase glucose and blood pressure and thus raise fluid retention as an issue.
Dr. Mandell: The presence of coronary disease implies the use of low-dose aspirin therapy. Despite the slight elevation in serum urate from low-dose aspirin, I recommend that my gout patients continue this for coronary protection. But even low-dose aspirin raises concerns of GI safety and even possible drug interactions with ibuprofen that could reduce the aspirin’s efficacy. Does this lessen your tendency to reach for an NSAID in these patients?
Dr. Edwards: I think the issue of cardiovascular safety and NSAIDs is still up in the air. The data that came out around the time the selective COX-2 inhibitors were released made us all think again about the role of prostaglandins and coronary blood flow and how blocking prostaglandins might have some bad effects. I don’t often use NSAIDs in patients with significant heart disease, simply because they don’t act fast enough to get patients through the attack quickly and I worry about the sodium retention and other problems putting stress on the heart.
Dr. Sundy: While coronary disease doesn’t create the acute dilemmas that diabetes does, it is a consideration in my therapy choice, and I generally avoid using NSAIDs.
Dr. Mandell: It is important to remember the additive gastric toxicity effect from low-dose aspirin and NSAIDs.
Infection
Dr. Mandell: Let’s turn to the hospitalized patient—a patient admitted to the medical service with infection. This patient is going to get fluids and drugs, and these are likely to raise or drop the serum urate level, which may precipitate a gout attack. From your perspective as an infectious disease consultant, Jim, how does the coexistence of a documented infection in the hospital—which you’re treating—factor into which agents to use or avoid when treating acute gout?
Dr. Pile: In practice, I try to avoid steroids in that situation. Theoretically, however, I’m not sure this scenario is really a contraindication to steroid use. I’m not aware of much solid data showing, for example, that patients with community-acquired pneumonia on appropriate antibiotic therapy have worse outcomes if they receive steroids than if they do not. Certainly there are a variety of serious infections for which we use steroids as a matter of course, including pneumococcal or tuberculous meningitis, tuberculous pericarditis, severe typhoid, and severe septic shock (the data for septic shock are murky, but there’s a role for at least steroid supplementation). As long as the antimicrobial therapy is appropriate, patients with serious infections are not necessarily going to do any worse with steroids.
Dr. Edwards: The biggest risk from steroids would be masking the symptoms of an unrecognized infection. If you know the infection is there and you’re treating it appropriately, I don’t think steroids are contraindicated.
Dr. Pile: A related risk is that steroids can make it more difficult to monitor the course of a known infection by blunting the overall response and bumping up the white blood cell count.
Dr. Mandell: My practice would fall in the realm of what Jim described as the theoretical course of action. If I knew what the infection was or felt comfortable in treating it, I would not shy away from systemic corticosteroids in the hospital setting, where I could monitor the patient. I think a corticosteroid may often be the safest drug to use in that setting, given the likelihood of comorbidities, at least compared with NSAIDs.
Dr. Simkin: Let’s say a patient comes in with acute knee pain and you aspirate the knee. The question arises whether you should wait for the culture results or go ahead and inject steroid while your needle is in the knee. I certainly have a much lower threshold to go ahead and inject while I’m in there, although I’d always be sure to get the culture results and revisit as needed. But if I pull out extremely inflammatory fluid during the aspiration, I’m probably not going to inject the steroid.
Dr. Edwards: And what if you actually do inject an infected joint with a corticosteroid—are you doing harm?
Dr. Simkin: I would like to think I’m not, and I may be helping.
Dr. Edwards: Yes, there are a lot of theoretical reasons why you might be helping, and there’s a small literature on steroids and infected joints that suggests you might actually slow down some bone resorptive steps and be doing various similar things.
Dr. Mandell: But in humans (a series of pediatric patients with septic arthritis30), those were systemic steroids, not intra-articular.
Dr. Edwards: In animal studies there is some amelioration of the destructive component with intra-articular steroids. The main point is that we must send off the fluid for culture when we inject steroid into a joint, and we must follow up on the cultures to make sure nothing is growing in a joint because we’re going to be calming it down. Whatever the inflammatory process is—whether infection or crystalline or rheumatoid—the patient is initially going to get better with a steroid injection. So we can’t use initial clinical improvement as the marker that we’ve done the right thing. We need to make sure the aspirate culture is negative.
Dr. Mandell: What about the use of colchicine or an IL-1 antagonist in the setting of infection?
Dr. Pile: There may be some theoretical concern, but I think low-dose colchicine would be a reasonable agent to use in the setting of infection.
Dr. Edwards: I don’t think there’s much concern since IV colchicine has been taken off the market. IV colchicine had a much more profound effect on bone marrow, and this resulted in a number of deaths from infection in immunosuppressed patients such as chemotherapy recipients. But oral colchicine in the doses we use for acute attacks and for maintenance probably doesn’t have a very profound effect on response to infectious processes or on white blood cell production (in the absence of chronic kidney disease).
Dr. Pile: Which raises the point that if the patient were on a potent inhibitor of the cytochrome P450 3A4 system, you might conceivably get into the same situation. For example, serious toxicity and even fatalities have been reported in patients taking colchicine and clarithromycin concurrently.31
Dr. Mandell: Peter, would you be comfortable giving an IL-1 antagonist to a hospitalized patient with pneumonia that’s being treated with antibiotics? Let’s say the patient has chronic kidney disease and diabetes and you’re looking for an agent to use.
Dr. Simkin: Well, I wouldn’t be comfortable, but I might feel I had to do it. If the organism were known, as you said, and the antibiotic therapy were appropriate, I might depend on that antibiotic coverage.
Dr. Sundy: Although I can envision some scenarios where I’d use the IL-1 antagonist in this situation, I’d be looking for an alternative. My concern is that people with any source of infection were excluded from clinical trials of anakinra, so we don’t have a good understanding of how it behaves in the setting of infection.
Dr. Edwards: And the fact that anakinra is not approved by the Food and Drug Administration (FDA) for treatment of acute gout should make us a bit uncomfortable.
Dr. Mandell: It’s interesting that the limited data do not show a lot of infections with IL-1 antagonist therapy other than when combined with anti–tumor necrosis factor therapy. The experience in periodic fever patients, who are on IL-1 antagonist therapy for many years, has not really shown this to be an agent predisposing to infection. I think this goes back to the point that if you document an infection and you’re treating it appropriately, some immunosuppressive or anti-inflammatory therapies may not be bad if you need to treat the attack of gout, particularly if it’s in a setting where you can monitor the patient well.
NSAIDs: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s drill down on the use of the specific classes of drugs, starting with NSAIDs. Larry made the point that NSAIDs should be used at high doses for acute gout attacks. What else is worth noting about the dosing and use of NSAIDs in this setting?
Dr. Sundy: In addition to that distinction between analgesic and anti-inflammatory dosing, it’s important that patients be told to treat at the appropriate dose intervals for whichever NSAID is chosen so that the anti-inflammatory benefit is maintained. I generally recommend that patients continue on that dosing regimen until they have substantial symptom improvement, at which point they can begin to back off, but that’s generally a 7- to 10-day treatment course, and typically closer to 10 days. Finally, I’m quick to use proton pump inhibitors or some sort of gastroprotective regimen with most patients that I start on NSAIDs in this setting.
Dr. Mandell: Your point about duration of therapy bears emphasizing. A common mistake with the use of NSAIDs is that they’re stopped too soon out of an understandable concern about their toxicity. Often they’re stopped when symptoms are still present, which means the attack hasn’t really resolved, and so it comes back. I tell patients to wait until their attack is completely gone, continue for a few days longer, and then stop the NSAID at that point.
Dr. Edwards: If we look at the natural course of disease, a patient’s first four to six gout attacks tend to be a bit shorter in duration, in the range of 5 to 7 days. As recurrent attacks occur over the decades, they tend to get more drawn out, lasting perhaps a couple or few weeks. We want treatment to at least cover the anticipated period of the natural attack since we’re not doing anything profound to the disease process itself, except perhaps with an IL-1 inhibitor. For instance, a typical treatment in emergency departments is an intramuscular injection of ketorolac. That may make patients feel good for the next 24 or 36 hours, but they’ll have a resumption of their acute attack at that point. I generally stick to 2 weeks as a reasonable treatment period. I might shorten that for a patient with diabetes in whom I have to use steroids or for a patient with congestive heart failure that I needed to put on an NSAID, but I generally try to cover patients with anti-inflammatory therapy for a couple of weeks.
Dr. Mandell: Your point about intramuscular ketorolac is important. A single shot is not likely to resolve the attack, and the intramuscular route will not ameliorate the gastric toxicity of this drug. Those are two common misperceptions.
What about the COX-2-selective NSAIDs? There’s only one that’s still marketed in the United States, celecoxib, but what’s your sense of these drugs as a class in terms of efficacy for treating gout?
Dr. Sundy: I think they’re effective. Only one COX-2 inhibitor, etoricoxib, has been studied in clinical trials for acute gout, and it’s not available in the United States, but I think the class as a whole is a reasonable choice. That’s especially true since the course of treatment is only 7 to 14 days. Even so, it’s still reasonable to use some gastric protection when giving celecoxib, as it does no harm and can add some reassurance. As with other NSAIDs, dosing of celecoxib for acute gout is higher than for other typical uses in the drug’s labeling.
Dr. Edwards: Yes, when I use celecoxib for acute gout I use 400 mg every 12 hours for the first 2 days and then take it down to 200 mg every 12 hours for the remainder of the treatment period.
Dr. Mandell: And the COX-2-selective NSAIDs do adversely affect renal function.
Dr. Simkin: One thing that troubles me when we talk about dosage for any of our agents is the large individual variation we encounter. I don’t think there’s a patient population with a wider variation of body sizes than the gout population. We see enormous patients with metabolic syndrome and several large joints, and we see frail elderly patients who have a single small joint affected. In fact, in addition to body size, the size of the joints involved can tell us a lot about the amount of inflammation we’re dealing with. Someone with two knees and an ankle affected is probably different from someone who has just a great toe affected. In light of these variables and so many others that come into play, such as age and comorbidities, I’m hesitant to recommend any particular dose because I try to make adjustments. Of course, there are no data that cover all these variables.
Dr. Mandell: But would you agree with the generality that high doses—higher than those we typically use for other musculoskeletal pains—are warranted in treating acute gout?
Dr. Simkin: Yes, but a high dose for a small elderly woman is different from a high dose for a young man who’s quite large.
Dr. Sundy: This sensitivity to individual variation can also apply to duration of therapy. As we noted, it’s not uncommon for veteran gout patients to find that starting treatment early—within 24 or 48 hours—may be sufficient to tamp down their symptoms, yet that’s an observation that is developed only over time in an individual patient. So it’s not a recommendation, but many patients will do fine with that. In those situations, I may not be so adamant about the need to continue treatment for a full 10 or 14 days.
STEROIDS: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s move to corticosteroids. A very common treatment used for acute gout is a tapered-dose regimen like the Medrol Dosepak (blister-pack) formulation of methylprednisolone. What strikes me is that this is a one-size-fits-all formulation, yet you’ve all just argued that there’s no one-size-fits-all approach. What are your thoughts?
Dr. Edwards: The Medrol Dosepak contains 21 4-mg tablets of methylprednisolone to be taken over the course of 6 days. Six tablets are taken the first day, and then one fewer tablet is taken each successive day through day 6. An obvious potential drawback is that the patient is off of medicine after 6 days, though that will be enough for some patients if they start treatment promptly.
I do use the Medrol Dosepak, and I always have patients fill a prescription ahead of time so that it’s available for quick initiation when they start to have their next attack. I instruct them to avoid storing it in a hot place, such as a car, but that it can be stored for up to 5 years at room temperature. Most of my patients like this approach, and if they do well with it they’re good about getting a new refill in advance of their next attack. This is a pretty handy way of letting patients be in charge of their attacks. If it doesn’t work and their symptoms continue, they call me and I’ll see them promptly.
A different approach is more appropriate for patients whose attacks tend to be more recalcitrant, perhaps because they’ve had gout for a long time or have bulky disease with lots of tophi. In those cases I’ll go with a steroid regimen that’s essentially double what a Medrol Dosepak would be—namely, prednisone 30 mg for the first 2 days then tapered down by 5 mg every other day. As Peter said, this approach would be overwhelming to a frail 82-year-old woman with a single tophus in her distal interphalangeal joint, but it tends to work well for a more typical 50-year-old obese male gout patient.
Dr. Sundy: I do some of the same things you describe, though my background in allergy has accustomed me to the steroid burst, so I’ll tend to use a dose of about 0.5 to 1.0 mg/kg/day for 4 or 5 days and then try to taper rapidly over another 2 to 4 days. I’m curious whether you think methylprednisolone offers advantages over prednisone.
Dr. Edwards: There are a few patients—roughly 5% to 15% of the population, according to various studies—who have trouble converting prednisone to its active form in their liver because they’re missing an enzyme. So I tend to use methylprednisolone since it has a benefit in that small segment of the population, but it otherwise doesn’t offer anything special beyond the convenient packaging that we discussed.
Dr. Mandell: I think convenience is the major factor. The blister-pack preparations are useful for patients who get confused with tapering. For patients who can make adjustments based on their symptoms, I find those personalized adjustments to be better than just giving them an absolute number of pills.
Dr. Pile: This issue of dosing duration is very important for generalists; just listening to your schedules is very instructive for me. I think a lot of nonrheumatologists, myself included, are inclined to use a one-size-fits-all approach. The mistake that I’ve been most apt to make, apparently, is using an insufficient duration of therapy. I’ve tended to use 40 mg prednisone for 5 or 6 days and then to stop rather than tapering the dose.
Dr. Mandell: From the cost perspective, steroids seem to be a very cost-effective option. The Medrol Dosepak, as a branded product, costs a bit more. Similarly, generic NSAIDs are inexpensive. There has been some feeling that nongeneric NSAIDs may offer advantages over generic NSAIDs, but I don’t know of any published evidence to support that. What are your thoughts?
Dr. Edwards: I agree with you. There has been this folklore about indomethacin and, before that, phenylbutazone being rather remarkable and specific drugs for gout. That hasn’t been borne out by the data, and virtually all of the NSAIDs work fairly comparably.
COLCHICINE: SPECIAL CONSIDERATIONS FOR USE
Dr. Mandell: Let’s move to colchicine, which represents a unique class of anti-inflammatory agent. It has been available and widely used for many years, but it had not been formally licensed by the FDA until last year. Without corporate imperative and funding, data on its clinical use have been limited. As we mentioned, an IV form of colchicine was withdrawn from the market a few years ago after being associated with multisystem organ failure and death. Low-dose short-term colchicine doesn’t have those risks, and its efficacy in treating gout has been accepted for many years despite the dearth of clinical trial data until recently. How has colchicine been used in the past, and do the new data guide us on how it should be used differently, specifically for treating acute gout attacks?
Dr. Simkin: The so-called traditional regimen of starting with a pair of colchicine tablets and then giving another tablet every 1 or 2 hours until relief (or until GI toxicity developed) is one of the great embarrassments in the history of our field. The more recent trial evidence clearly indicates in the short term that there is no additional benefit beyond a very low dose. As we noted, this has been in the wind anecdotally for quite a few years, and it was confirmed by the recent AGREE trial.11 I find it interesting that the dosage used in AGREE—three 0.6-mg tablets—is very close to what we use for prophylaxis. We may well wind up concluding that any patient we’re seeing for the first time with an acute attack should be treated with that dose of colchicine, as an adjunct to another anti-inflammatory drug.
The use of colchicine as an adjunct for acute gout attacks was advanced in a review by Cronstein and Terkeltaub several years ago.32 Since we know that the mechanisms of action of the various drugs for acute gout are sufficiently different from each other, it makes sense to consider hitting the process at more than one spot. Having said that, one of the most crucial concerns in using colchicine is what other medications the patient is taking, given colchicine’s multiple drug interactions (Table 3). These include interactions with most statins, some antibiotics, and some calcium channel blockers, which share drug transporters with colchicine. So we have to be cautious about using colchicine with those other therapies.
Dr. Mandell: That’s also important in patients on chronic prophylactic colchicine therapy, as we know from recent pharmacokinetic studies and anecdotal case reports of colchicine and some of these other drugs.16–23 Do you think it’s as important in a patient on, say, chronic statin therapy who’s not on baseline colchicine, in whom you use a colchicine regimen of just two tablets followed by one tablet and then stop the colchicine right there? Is the potential for meaningful drug interactions as important in that setting?
Dr. Simkin: Probably not. One of the key issues is to educate patients about what to look for. Tell patients that if they notice numbness, weakness, or myalgia, you want to know about it. The evidence I’ve seen is that people who go into colchicine-induced rhabdomyolysis, for instance, develop it over a period of days, not hours, and if they stop the drug early, they probably will be okay. That’s the biggest concern. Neutropenia is the second concern.
Dr. Mandell: But fortunately, assuming no chronic kidney disease, those are generally patients taking colchicine chronically, not just acutely. We talked a bit before about our panel’s concerns about the efficacy of colchicine. Say you have a patient who has no history of prior colchicine use. How comfortable are you that treating such a patient with just the three-tablet colchicine regimen is likely to give the desired effect of rapid pain control and resolution of the attack?
Dr. Edwards: I’d expect that there wouldn’t be a profound or rapid improvement on that regimen, although the patient would certainly do better than if he or she were on nothing. I think that’s what the AGREE trial showed—that patients improve but still have significant morbidity days after initiating therapy.
Dr. Mandell: I think the AGREE trial did what it was intended to do: it showed that a low-dose colchicine regimen was as effective as and better tolerated than a high-dose colchicine regimen. This was something most of us believed, but it had never been demonstrated in a trial, and now there are good data to show it. And AGREE was consistent with the 1987 Ahern study10 in showing that the high-dose regimen is better than placebo. However, it in no way convinced me that colchicine in this dosing regimen is a monotherapy I’d be comfortable using to treat a severe acute attack. AGREE also didn’t give me any information as to how likely this approach would be to resolve an attack. For all the reasons we discussed, continued anti-inflammatory therapy is likely necessary beyond three pills, and I can’t be confident that a course of three colchicine pills is sufficient to maintain a drug level inside white cells to prevent other attacks from happening or a rebound attack once the regimen is over. I’m sure this approach will work for some patients; I just don’t know at the outset which ones or what percentage of patients will respond completely.
Dr. Edwards: I like the idea of combination therapy, of using colchicine as an adjunct. We’ve been talking up to now about monotherapies for acute gout because monotherapy is simple and patients and physicians alike prefer things that are fairly simple to use. But there is good theoretical reason to support using colchicine in combination with either NSAIDs or steroids, though there are probably no experimental data. I can’t imagine a reason for using corticosteroids and NSAIDs together, but pairing up colchicine with a steroid or an NSAID might allow you to keep the dose of the other agent lower and perhaps reduce the overall toxicity of your therapy. I’ve occasionally put patients on that type of combination therapy, and it’s probably a helpful way to go.
Dr. Mandell: For years my approach has been one of cotherapy, counting on either NSAIDs or corticosteroids to treat the acute attack by rapidly relieving the pain and inflammation and using colchicine in low doses—at a prophylactic dose, as Peter said—and extending it over a much longer period. I’m counting on the colchicine not to treat the acute attack but to prevent the next one and hopefully, for all the reasons we talked about, shorten the duration of therapy with the steroid or NSAID.
Dr. Edwards: Sure. The way I use colchicine is mostly for maintenance and for prophylaxis while I’m initiating urate-lowering therapy. I use it for a bit longer than most others might—I draw it out to at least 6 months after a complete resolution of symptoms, sometimes to 9 or 12 months. When a patient has a flare, I certainly have them maintain the colchicine if I’m going to treat them with something else, as I almost always do, and I believe that lets me get by with lower doses of the other agent I’m using for the acute attack.
Dr. Sundy: When I use colchicine as monotherapy for an acute flare, it’s because the patient has concluded from experience that it has worked effectively for him or her. But that’s not very common.
Dr. Mandell: There are times when the patient’s comorbidities lead me to conclude that colchicine may be the best drug to try for acute treatment. But I’m not comfortable assuming that three pills will do it all.
Dr. Pile: From the perspective of the generalist, who typically treats a simpler gout population than you do, what is the recommended treatment duration if colchicine is used as monotherapy for an acute attack? We now have this recommendation to use a single day’s worth of therapy, but you’re all expressing reservations with that.
Dr. Edwards: The recommendation in the drug’s current labeling is to take two 0.6-mg tablets at the first sign of a flare and a third 0.6-mg tablet 1 hour later. That was the low-dose regimen in AGREE, and I think it’s a good one, as only a very small percentage of patients will get the GI side effects with this regimen that occur with more prolonged therapy. The real question is what you do on day 2 and day 3 on out to day 7 or 10. If I were to use colchicine monotherapy, after that first day I’d prescribe one tablet three times daily for another 3 or 4 days and then drop down to one tablet twice daily for the rest of whatever duration of therapy I felt was justified. Of course, this is highly individualized and depends on how the drug is tolerated; probably more than 25% or 30% of patients can’t tolerate a three-times-daily regimen even for 3 or 4 days. But the total duration would be at least 7 days and up to 10 or even 14 days.
Dr. Simkin: Or months, because you’ve established a diagnosis.
Dr. Edwards: Yes—as prophylaxis, for which the dosage is one tablet once or twice daily. And then you’d leave the patient on prophylaxis and start urate-lowering therapy, if it hadn’t been started already.
Dr. Mandell: We’re now way past having trial data to support this, and this approach may negate the purported safety advantages of the low-dose regimen supported by AGREE.
Dr. Edwards: Yes, this is rank speculation.
Dr. Simkin: But there is evidence of the value of prophylaxis.
Dr. Mandell: There is certainly evidence for the value of prophylaxis, from a number of studies. We’ll get to that shortly. However, if we think just about treating the acute attack and the AGREE findings, I don’t think we can extend those results with confidence to a setting where we switch immediately from acute therapy into a prophylactic mode. Not that we shouldn’t consider doing that—it’s similar to my practice as well, though I tend to use lower doses as I extend out—but the existing trial data are limited to that very short window of acute treatment. Anything we do beyond that is an extension from our belief that the inflammatory response to crystals in a joint lasts longer. I believe it generally requires longer anti-inflammatory therapy, but we need to expect some frequency of side effects and drug interactions as we put patients on chronic colchicine therapy.
The other factor that used to be an advantage of this type of regimen was its low cost. For a long time colchicine was available from multiple manufacturers and was exceedingly inexpensive, although with less FDA guidance on its use and less oversight of its manufacturing. This enabled even patients with no insurance to go on a prophylactic therapy and remain on it. The environment is now quite different with the introduction of a single FDA-approved and -regulated branded product. So how do we deal with cost in the current environment?
Dr. Edwards: In terms of specific numbers, once-daily and twice-daily doses of unbranded colchicine used to cost about $6 and $12 a month, respectively, based on the average wholesale price. Now the cost of branded colchicine (Colcrys) is about $175 a month for the once-daily dose and $350 a month for the twice-daily dose, again based on the average wholesale price.
Dr. Simkin: I think all of us are deeply troubled by the high cost of this preparation and hope that the alternatives will still remain available to us.
Dr. Edwards: Yes, especially since it sounds like most of us consider colchicine our go-to drug for maintenance. It used to be that patients might be on maintenance colchicine therapy for 5 or 10 years without any addition of urate-lowering therapy. Today, however, when most of us put a patient on prophylactic therapy with colchicine, it’s with the intent of also addressing the hyperuricemia that’s at the heart of the disease process. In that case we still cover patients with colchicine prophylaxis during the initial months of urate-lowering therapy because of the elevated rate of gout flares—so-called mobilization flares—that occur during the process of uric acid reduction.
Dr. Mandell: The manufacturer of branded colchicine has introduced a patient assistance program to ease the drug’s cost for the financially needy. We will have to see how practical it is and how it affects our patients’ use of this medication.
ACUTE FLARES IN THE SETTING OF PROPHYLAXIS AND URATE LOWERING: WHAT TO DO?
Dr. Mandell: It sounds like most of us would treat patients prophylactically with colchicine for a while after an attack, certainly after several attacks, and then continue on chronic low-dose colchicine during the introduction and adjustment of the urate-lowering therapy that constitutes comprehensive gout treatment. Yet some patients will still have flares. So how does this baseline low-dose colchicine prophylaxis—a 0.6-mg tablet once or twice daily—influence your choice of therapy for those attacks? Do you bump up the colchicine dose, or do you absolutely avoid increasing the colchicine?
Dr. Sundy: I wouldn’t absolutely avoid an increase, but I would tend not to adjust it. I would maintain the dose and add a different agent on top of it—an NSAID or corticosteroid—to manage the acute attack. In general, if a patient is on regular colchicine prophylaxis, the assumption is that they have sufficient renal function to support that use, so such a patient may do fine with an NSAID.
Dr. Simkin: I trust we all agree that it’s critically important to continue the urate-lowering therapy the patient is taking if he suffers an attack. The same thing pertains to the prophylactic regimen. Both of these components should continue through the flare, but I agree that we usually want to add something to treat a flare, and it should be something the patient has on hand and can take without needing to talk to his physician if it’s the middle of the night.
Dr. Mandell: So I’m hearing that most of us would add something else for the short term on top of the prophylactic colchicine dose when a flare developed rather than increasing the colchicine dose.
Dr. Simkin: Yes, although one exception I’d be comfortable with is the example we discussed earlier of the patient who has found that taking an extra colchicine pill or two can help abort or diminish a flare without causing diarrhea.
Dr. Mandell: We cannot extrapolate the AGREE data to support a short-course regimen for an acute attack in patients who are already on chronic colchicine for prophylaxis.
PROPHYLAXIS IN THE PERIOPERATIVE SETTING
Dr. Mandell: Admission to the hospital for acute medical or surgical reasons is not infrequently associated with gout flares. Jim, have we in the rheumatologic community made it clear that chronic colchicine prophylaxis and urate-lowering therapy should ideally be continued when patients with gout are hospitalized for any reason, or is it a reflex for these drugs be held?
Dr. Pile: I think there’s a general appreciation, at least in the hospital medicine community, that these therapies should be continued in that situation. My sense is that these prophylactic gout therapies are viewed by most hospitalists as somewhat analogous to beta-blockers for chronic heart failure, which are understood to be necessary to continue when a patient is admitted with an acute exacerbation of heart failure. I don’t have data to support this contention, however.
Dr. Edwards: My experience suggests that discontinuing urate-lowering therapy during an acute gout attack is a common mistake in general practice. I think that’s been demonstrated in surveys of primary care physicians. And when the uratelowering therapy is stopped, the attack is often prolonged and made worse. Widespread misperception about this remains—it’s a big problem.
Dr. Mandell: Based on educational lectures I’ve given to primary care audiences, I agree with Larry. If I present a case question on a scenario like this, many physicians in the audience will anonymously indicate via the audience response system that they would stop a patient’s allopurinol if an acute attack occurred.
Let’s turn to the scenario of a patient who’s admitted for surgery—elective or emergent—who has a history of gout and is on prophylaxis. What’s the general routine in managing these medications in patients who are going to surgery?
Dr. Pile: In my preoperative consultations I’ve always stressed to gout patients and their surgeons that the postoperative period is a high-risk setting for acute exacerbation of gout. My routine recommendation in this setting is that patients continue any of their goutrelated medications through the postoperative period, but that often doesn’t happen. All sorts of medications are inappropriately stopped perioperatively, including statins and beta-blockers in addition to colchicine or allopurinol. Something I haven’t done routinely is to recommend introducing prophylaxis in the postoperative period for patients who were not already receiving it. Is that something you consider doing, given the likelihood of postoperative flare?
Dr. Edwards: It’s important to appreciate the reasons why gout flares postoperatively. It has a lot to do with why uric acid levels go up postoperatively: fluid changes, starvation, ketosis for any reason, lactic acidosis—these all cause pretty remarkable changes in systemic pH, which certainly is a trigger for that. The perioperative state also changes the exchange of uric acid in the kidney so that there is greater accumulation, and anything that rapidly bumps up serum urate levels while also creating a change in the pH is a setup for flares. So any recommendations about whether or not to give prophylaxis have a lot to do with the anticipated impact of the surgery on all of those physiologic parameters.
Dr. Sundy: As rheumatologists we’re usually called after the fact, so I can’t say I’ve given the issue of prophylaxis in the perioperative setting a lot of thought. I would add that patients who are undergoing cardiac catheterization, especially with stenting procedures that may require a large dye load, are another group that tends to be at increased risk of flare, especially in the setting of acute myocardial infarction.
Dr. Edwards: They could get a subtle change in renal function related to the dye load that isn’t throwing them into acute tubular necrosis or acute renal failure but is enough to reduce elimination of urate for long enough that they get this bump of uric acid that triggers the flare.
Dr. Mandell: The retrospective analyses of perioperative gout show that patients who come in with lower urate to begin with are less likely to have flares,33 which suggests that perioperative flares are less likely in patients who have been better controlled and managed. The question of whether to introduce prophylaxis at admission, given the likelihood of postoperative flare, hasn’t been studied formally. Despite this absence of data, I would be comfortable introducing colchicine at a low dose for prophylaxis in this setting, especially in a patient who has had frequent or recent attacks. The major exception would be for patients undergoing bowel surgery or similar procedures, since colchicine’s potential to cause diarrhea and other GI side effects is a main concern with its perioperative use. But a study of prophylactic colchicine before surgery in a “high-risk” population is a trial begging to be done. The challenge is that because the event rate is low, the sample size needed would be so large as to potentially make the study impossible.
Dr. Pile: One issue I encounter from time to time as a preoperative consultant involves patients who should be on chronic urate-lowering therapy but are not. Obviously, when I’m seeing them 7 days before surgery it’s not the time to contemplate doing anything besides perhaps starting colchicine.
Dr. Mandell: Yes, that would be a time to avoid starting urate-lowering therapy since a drop in serum urate is likely to precipitate gout flares, and that’s something you don’t want to happen in a postoperative setting. So just as we shouldn’t stop urate-lowering therapy preoperatively, we shouldn’t initiate it either. When the patient is already on urate-lowering therapy, it should be continued as close as possible to the time of surgery and restarted immediately thereafter. The fluids that are given perioperatively tend to drop the patient’s urate levels, so we often can get away with the brief window of discontinuation around the time of surgery so long as we restart the allopurinol postoperatively.
Dr. Edwards: There are certain surgeries, such as many cardiac procedures, for which the rooms are kept hypothermic, and that can be one more physiologic stimulus for an attack of gout, particularly in the body’s cooler joints. That’s a setting where podagra might be more common, whereas major abdominal surgery with a lot of bowel ischemia and lactic acidosis may render a wide range of joints more equally susceptible, although there are no data on this question.
Dr. Simkin: I’m not certain that postoperative gout is significantly different from hospitalization gout. If patients who are admitted for medical reasons—for instance, cardiac disease or pneumonia or ketoacidosis—also have an established diagnosis of gouty arthritis, they’re at elevated risk too.
Dr. Mandell: Yes, and that point—that preexisting gout is a major risk factor, whether or not it was known at the time of admission—came through from the studies Jim noted above and our experience. That piece of the history is often not obtained until an attack occurs, although more widespread use of electronic medical records may lessen this problem. It’s frustrating that attacks tend to occur about 4 days after surgery, which is typically around the time of planned discharge.
AVOIDING MOBILIZATION FLARES WHEN LOWERING URATE: START LOW, GO SLOW
Dr. Mandell: Let’s shift from this focus on the hospital setting as a precipitant for acute gout attacks and recall that the most predictable precipitant is when we pharmacologically reduce the serum urate level in an effort to decrease the total body load. As Larry noted, the down side of this very necessary intervention is that “mobilization flares” of gout are fairly predictable in this setting. While many of us have used prophylaxis to reduce the likelihood of these flares, we hadn’t had much data on duration of prophylaxis prior to some recent trials of urate-lowering therapy.36 So what are your practices now for trying to reduce the chance of acute attacks as you introduce urate-lowering therapy?
Dr. Edwards: A trigger for gouty attacks is the fluctuation of serum urate levels, be it from initiating or discontinuing urate-lowering therapy. So the approach when initiating urate-lowering therapy is to start low and go slow with your dose escalation. Most of us now buy into the notion of trying to lower the serum urate below 6.0 mg/dL for most patients, and perhaps by another 1.5 mg/dL or so for patients with bulky tophaceous disease. To get to that target, however, you should start with a fairly modest dose of whichever urate-lowering therapy you’re using—allopurinol, febuxostat, or (in rare cases) probenecid—and increase it over time. I tend to let several weeks pass between each dose escalation to ensure that the prior escalation has had time to drop the urate to its new nadir, and then I see what that level is. If it’s not at target, I will modestly escalate further.
Dr. Mandell: And we should emphasize that frequent initial monitoring of urate is appropriate to determine whether you’re reaching your target.
Dr. Edwards: That’s a critical element that isn’t done well in this country. Many studies show that regular monitoring of serum urate after initiation of urate-lowering therapy is actually a rarity. The approach to urate monitoring should be the same as for glucose monitoring with oral diabetes medications or blood pressure monitoring with antihypertensives. We should know what impact we’re having on the patient and continue to adjust the dose to find the appropriate level for the individual patient.
As with the therapies for acute gout, there is no onesize-fits-all dose regimen for urate-lowering therapy. A minority of patients will do fine on 100 mg/day of allopurinol. Most patients will still not reach their serum urate target at 300 mg/day of allopurinol, and we should consider gradual escalation to more generous doses as necessary, beyond 400 mg/day and up to the “maximum” recommended dose of 800 mg/day. Monitoring of serum urate should then continue even after we’ve found an appropriate dose to reach the target level since certain factors may change, altering concentrations of any of the urate-lowering therapies. For instance, a new drug may be added, or the patient may develop new comorbidities or a change in renal function. At this point the monitoring need not be as tight as during the initial period, but it should continue on a semiannual or yearly basis.
Dr. Simkin: I completely agree that going slow is desirable, and it’s very important for our patients to understand what we’re doing and what we’re aiming for. Every gout patient should know what his or her last serum urate level was, yet very rarely is that the case.
Dr. Edwards: Not only that, but they should know what the target level is so that they become an equal partner in their treatment plan, just as diabetics might be unsatisfied until their hemoglobin A1c is down to near 6.0%.
WHAT ROLE FOR PROPHYLAXIS DURING URATE LOWERING?
Dr. Mandell: Larry’s point that dropping the serum urate level slowly over time is less likely to induce an attack of gout is something we had suspected for a while and now have some direct trial evidence for. But no matter what we do, there’s always some likelihood of inducing an attack as we lower the urate level. So what do you do, Peter, to prevent those attacks?
Dr. Simkin: I try to get the patient on a regular dosage of colchicine prophylaxis. There’s good evidence that it’s effective. The dose is usually one or two tablets a day, largely depending on GI tolerance. It’s worth noting that more than a few of my elderly patients appreciate colchicine’s GI effects, as they provide some welcome relief from their chronic constipation. While that’s certainly not welcome by everyone, for some patients it makes colchicine prophylaxis an easier sell.
Dr. Sundy: Colchicine is my standard for prophylaxis as well. I think it’s highly effective.
Dr. Edwards: For how and when to start prophylaxis, my general recommendation is that it should precede the start of urate-lowering therapy by about 1 or 2 weeks. I think starting colchicine 2 weeks in advance is probably a good buffer for ensuring that it’s in the system. The urate-lowering therapies have very rapid onset—allopurinol will cause fluctuations in urate levels within a day or two—so the prophylaxis has to be fully on board.
Dr. Pile: Do you have a sense of how much more effective twice-daily dosing of colchicine is compared with once-daily dosing?
Dr. Edwards: I don’t have a good sense of it, but I tend to use twice-daily dosing unless renal function is a concern—specifically, if the patient’s GFR is between 50 and 60 mL/min/1.73 m2. And in such cases, you’re not really diminishing the dose with a once-daily regimen because the patient is keeping more of the drug around, in light of the renal insufficiency. That hearkens back to Peter’s earlier comments about baseline therapy with statins, some calcium channel blockers, and other drugs that will also require dose modification.
Dr. Mandell: The point is that we want to maintain a certain level of colchicine in all our patients for prophylaxis. It’s not clear that the blood level matters as much as the intracellular level, but we can’t routinely measure either. I think the answer to Jim’s question is that we don’t have data on whether once daily, twice daily, or even three times daily is the right regimen for colchicine prophylaxis; patients differ. I believe many of us start with twice-daily dosing on an empirical basis. If it’s not tolerated, typically for GI reasons, we’ll go down to once a day. If it’s tolerated but ineffective from the point of view of having flares, we may try to go to three times a day, assuming the patient’s not on other medications that would preclude that and doesn’t have chronic kidney disease or hepatobiliary disease.
WHAT ABOUT ALTERNATIVE PROPHYLAXIS OPTIONS?
Dr. Mandell: If a patient is truly intolerant of colchicine but has a reasonable GFR and no comorbidities, what about alternative prophylactic agents? The class to think about would seem to be NSAIDs, but we have no trial data to guide us with their use for prophylaxis. What’s been your experience with NSAIDs for prophylaxis of gout flares during the urate-lowering process?
Dr. Sundy: My experience is that NSAIDs are effective for this. I don’t have a notion of comparative effectiveness relative to colchicine, but I will use an NSAID in this setting when there has been GI intolerance to colchicine. The trick is the added responsibility of monitoring toxicity in terms of blood pressure, renal function, and hyperkalemia, as well as offering gastric protection, but I certainly have patients on NSAID prophylaxis.
Dr. Mandell: Do you feel compelled to use the same very high doses that we talked about for treating acute attacks of gout?
Dr. Sundy: No. I tend to use doses in more of a midrange area and make adjustments based on how the patient is doing. For example, I might increase the dose if a patient has a breakthrough flare.
Dr. Simkin: A situation where I’ve more often seen NSAIDs used for prophylaxis is in patients who have gout but are also taking NSAIDs on a regular basis for osteoarthritis. In that setting I’d be less inclined to add colchicine.
Dr. Pile: And I guess cost becomes an issue now, with the advent of branded colchicine, in a way that it hadn’t been before. At my local retail pharmacy generic naproxen costs 6 cents per 200-mg tablet, so some NSAIDs are very inexpensive.
Dr. Mandell: Colchicine used to have a similar cost invisibility as well. As Peter said before, our ability to provide cost-efficient prophylaxis is disappearing, and this may affect our ability to provide prophylaxis to some patients.
Dr. Sundy: I agree. Many times patients will stop taking drugs that are too expensive for them without telling their doctor. I sense that not using prophylaxis is a major impediment to adherence with urate-lowering drugs as well, as patients will stop those drugs because they sense appropriately that their gout has gotten worse without having been adequately educated about this risk. At the end of the day, we’re trying to eliminate gout as a medical issue for our patients, and anything that makes nonadherence more likely can be a big impediment to this goal.
Dr. Edwards: Patient education is absolutely crucial in this regard. We should step back and appreciate that these regimens can be very complicated and not terribly obvious from the patient’s perspective. First you have the disease gout, for which the treatment is allopurinol. You take that treatment and then your gout gets worse. Without good education to prepare patients for that, how can we expect them to respond, other than wanting to stop the allopurinol and probably thinking poorly of their doctor? Physicians have to educate patients on what to expect and how to stay the course. We must make clear from the start that the treatment of gout is a long-term process. We must explain that when acute flares occur the emphasis is on getting rid of the pain quickly but that the larger job of getting their disease under control by lowering their uric acid levels over time is a long haul. Both patient and physician have to be patient, go slowly, and make sure they don’t disrupt therapy just because symptoms are occurring.
Dr. Sundy: It sounds funny, but sometimes I’ve taken to congratulating patients whose gout flares after they start urate-lowering therapy. I say, “This is great—it means the drug is working, even if it doesn’t seem like it.”
SHOULD WE INTRODUCE URATE-LOWERING THERAPY AT THE TIME OF AN ACUTE ATTACK?
Dr. Mandell: Let’s consider the patient who presents with a first or second or third attack of gout but is not yet being treated for the underlying hyperuricemia. Should we introduce urate-lowering therapy at the time of the attack? After all, it’s a moment of opportunity, as the patient is “captive” and needs to be treated for his or her pain, so should we use that as a chance to also start the urate-lowering therapy that will be needed? There seem to be two schools of thought on this question. One is that if we start urate-lowering therapy immediately and drop the urate, the attack is likely to last longer. The other is that as we drop the urate by starting uratelowering therapy, we’re already treating them very aggressively for inflammation, so it may be an acceptable time to introduce urate-lowering therapy. What are your individual approaches?
Dr. Edwards: I’ve heard that argument of “we have them here now, let’s get it started.” I’d counter that when you start on this course of urate lowering, you need a very close connection to the patient during the long period of dose adjustment. Both the patient and physician need to understand that clearly. I’ve seen too many cases of a bad flare turning into a terrible flare when urate-lowering therapy is started during an acute attack. Patients are unhappy and get turned off of the idea of reintroducing allopurinol or any other urate-lowering therapy. So I don’t use that approach. Instead, I try to optimize the conditions for initiating urate-lowering therapy to minimize the chance that patients will have a bad experience. There’s no reason to rush, since most patients presenting with their first few flares have been hyperuricemic for 30 years, with urate slowly accumulating and depositing around their bodies. I’d promote an approach of letting the acute attack resolve and then getting the uratelowering therapy started about a month later.
Dr. Simkin: My approach is along the same lines as Larry’s. We all encounter gouty patients who say, “I can’t take allopurinol.” When you ask why, the most common response is, “It makes my arthritis worse.” Patients must understand that this is a hump they have to get over; we need to do everything we can to lessen that hump, and that involves good prophylaxis. But it’s also essential to educate them to expect the hump and to forewarn and forearm them in terms of how they’ll cope with it.
Dr. Sundy: A common rationale for starting urate-lowering therapy during the acute attack is that the patient may not follow up and you’ve got them on hand for intervention right now. But if follow-up is that unlikely, urate-lowering therapy is not going to be successful in any case, since frequent monitoring and dose adjustments are needed over many months. The notion that a patient can just be put on 300 mg of allopurinol and check back in 6 months is unrealistic.
Dr. Mandell: It sounds like we’re unanimous on this. One other consideration I would add is that I will avoid adding more than one drug in a patient at the same time whenever I can, to avoid confusion about which drug is causing any side effects that may develop.
REFINING THE PARADIGM: ACUTE ATTACKS AS THE ENTRY POINT TO A PROGRESSIVE DISEASE
Dr. Mandell: So our recommendation to the hospitalist who’s confronting acute gout is to treat the acute attack, start the education process, and make sure the patient understands this is a chronic disease that you’re going to make better in the hospital but which requires lifelong attention to prevent flaring up again. At that point the recommendation is to refer the patient back to the primary care physician or a rheumatologist with the understanding that the underlying hyperuricemia needs to be addressed and that the patient should make sure that happens.
Dr. Sundy: This raises some interesting handoff issues. How do we best ensure that the physician who’s going to pick up responsibility for chronic management is aware of these issues and on board with the necessary patient education efforts? There’s clearly an opportunity for doctor-to-doctor education here as well.
Dr. Pile: Yes, this is a microcosm of what we deal with all the time in the hospitalist world—safe transitions of care and trying to avoid information “voltage drop” as patients return to the care of their usual physicians.
Dr. Simkin: I think our education of both our colleagues and our patients needs to emphasize that this is not only a chronic disease but a progressive disease.
Dr. Edwards: That’s a great point, and it raises the question of the stage in the disease process where we should start urate-lowering therapy—a question without a clear-cut answer. Three decades ago, the thinking was that you should do it before the patient transitioned from acute intermittent gout to a more chronic and advanced tophaceous gout because of how terrible those joints looked and how destructive advanced gout is. Yet the destruction begins much earlier than that, and changes probably occur within the joint even before a patient has his or her first gout attack. There are soft-tissue changes around the joint as monosodium urate crystals accumulate, and the effects on bone and cartilage are occurring throughout the period of acute intermittent gout if the serum urate level hasn’t been lowered. As we’ve come to recognize this, we’ve pushed the initiation of uratelowering therapy earlier and earlier. The standard recommendation now is to start it as soon as a patient has two gout attacks within a year. Since 60% of patients have their second attack within a year of their first, that means initiating urate-lowering therapy after the second attack for about 60% of patients. An even larger majority of patients will be at that stage to qualify for urate-lowering therapy within the first few years after their initial attack.
Dr. Mandell: This is a good place to conclude, with a reminder that the acute gout attack is just one part of a chronic and potentially progressive disease. Although the acute attack is what gets the patient’s attention—and often the physician’s attention as well—it is really just the entrance into therapy for gout, a chronic disease.
- Wallace SL, Robinson H, Masi AT, et al Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum 1977; 20:895–900.
- Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol 2009; 15:22–24.
- Janssens HJEM, Janssen M, van de Lisdonk EH, et al Limited validity of the American College of Rheumatology criteria for classifying patients with gout in primary care [letter]. Ann Rheum Dis 2010; Mar 16[Epub ahead of print].
- Lin Y-H, Hsu H-L, Huang Y-C, et al Gouty arthritis in acute cerebrovascular disease. Cerebrovasc Dis 2009; 28:391–396.
- Altman RD, Honig S, Levin JM, Lightfoot RW. Ketoprofen versus indomethacin in patients with acute gouty arthritis: a multicenter, double blind comparative study. J Rheumatol 1988; 15:1422–1426.
- Maccagno A, Di Giorgio E, Romanowicz A. Effectiveness of etodolac (‘Lodine’) compared with naproxen in patients with acute gout. Curr Med Res Opin 1991; 12:423–429.
- Schumacher HR, Boice JA, Daikh DI, et al Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ 2002; 324:1488–1492.
- Rubin BR, Burton R, Navarra S, et al Efficacy and safety profile of treatment with etoricoxib 120 mg once daily compared with indomethacin 50 mg three times daily in acute gout: a randomized controlled trial. Arthritis Rheum 2004; 50:598–606.
- Shrestha M, Morgan DL, Moreden JM, et al Randomized doubleblind comparison of the analgesic efficacy of intramuscular ketorolac and oral indomethacin in the treatment of acute gouty arthritis. Ann Emerg Med 1995; 26:682–686.
- Ahern MJ, Reid C, Gordon TP, et al Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med 1987; 17:301–304.
- Terkeltaub RA, Furst DE, Bennett K, et al High vs low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum 2010; 62:1060–1068.
- Janssens HJ, Janssen M, van de Lisdonk EH, et al Use of oral prednisolone or naproxen for the treatment of gout arthritis: a doubleblind, randomised equivalence trial. Lancet 2008; 371:1854–1860.
- Man CY, Cheung IT, Cameron PA, et al Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med 2007; 49:670–677.
- Siegel LB, Alloway JA, Nashel DJ. Comparison of adrenocorticotropic hormone and triamcinolone acetonide in the treatment of acute gouty arthritis. J Rheumatol 1994; 21:1325–1327.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007; 9:R28.
- Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum 2009; 38:411–419.
- Colcrys [package insert]. Philadelphia, PA: Mutual Pharmaceutical Company; 2009.
- Simkin PA, Gardner GC. Colchicine use in cyclosporine treated transplant recipients: how little is too much? J Rheumatol 2000; 27:1334–1337.
- Dogukan A, Oymak FS, Taskapan H, et al Acute fatal colchicine intoxication in a patient on continuous ambulatory peritoneal dialysis (CAPD): possible role of clarithromycin administration. Clin Nephrol 2001; 55:181–182.
- Rollot F, Pajot O, Chauvelot-Moachon L, et al Acute colchicine intoxication during clarithromycin administration. Ann Pharmacother 2004; 38:2074–2077.
- Alayli G, Cengiz K, Canturk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother 2005; 39:1358–1361.
- Atmaca H, Sayarlioglu H, Kulah E, Demircan N, Akpolat T. Rhabdomyolysis associated with gemfibrozil-colchicine therapy. Ann Pharmacother 2002; 36:1719–1721.
- Roger U, Lins H, Scherrmann JM, et al Tetraparesis associated with colchicine is probably due to inhibition by verapamil of the P-glycoprotein efflux pump in the blood-brain barrier. BMJ 2005; 331:613.
- Levaquin [package insert]. Raritan, NJ: Ortho-McNeil; 2009.
- Phelan KM, Mosholder AD, Lu S. Lithium interaction with the cyclooxygenase 2 inhibitors rofecoxib and celecoxib and other nonsteroidal anti-inflammatory drugs. J Clin Psychiatry 2003; 64:1328–1334.
- Vistide [package insert]. Foster City, CA: Gilead Sciences; 2001.
- Hynninen V, Olkkola KT, Leino K, et al Effects of the antifungals voriconazole and fluconazole on the pharmacokinetics of S+ and R−-ibuprofen. Antimicrob Agents Chemother 2006; 50:1967–1972.
- Albengres E, Le Louet H, Tillement JP. Systemic antifungal agents. Drug interactions of clinical significance. Drug Saf 1998; 18:83–97.
- 2009 Red Book: Pharmacy’s Fundamental Reference. 113th ed. Los Angeles, CA: RAmEx Ars Medica; 2009.
- Odio CM, Ramirez T, Arias G, et al Double blind, randomized, placebo-controlled study of dexamethasone therapy for hematogenous septic arthritis in children. Pediatr Infect Dis J 2003; 22:883–888.
- Hung IF, Wu AK, Cheng VC, et al Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis 2005; 41:291–300.
- Cronstein BN, Terkeltaub R. The inflammatory process of gout and its treatment. Arthritis Res Ther 2006; 8( suppl 1):S3–S9.
- Kang EH, Lee EY, Lee YJ, et al Clinical features and risk factors of postsurgical gout. Ann Rheum Dis 2008; 67:1271–1275.
- Craig MH, Poole GV, Hauser CJ. Postsurgical gout. Am Surg 1995; 61:56–59.
- Friedman JE, Dallal RM, Lord JL. Gouty attacks occur frequently in postoperative gastric bypass patients. Surg Obes Relat Dis 2008; 4:11–15.
- Becker MA, Schumacher HR, Wortmann RL, et al Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450–2461.
- Wallace SL, Robinson H, Masi AT, et al Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum 1977; 20:895–900.
- Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol 2009; 15:22–24.
- Janssens HJEM, Janssen M, van de Lisdonk EH, et al Limited validity of the American College of Rheumatology criteria for classifying patients with gout in primary care [letter]. Ann Rheum Dis 2010; Mar 16[Epub ahead of print].
- Lin Y-H, Hsu H-L, Huang Y-C, et al Gouty arthritis in acute cerebrovascular disease. Cerebrovasc Dis 2009; 28:391–396.
- Altman RD, Honig S, Levin JM, Lightfoot RW. Ketoprofen versus indomethacin in patients with acute gouty arthritis: a multicenter, double blind comparative study. J Rheumatol 1988; 15:1422–1426.
- Maccagno A, Di Giorgio E, Romanowicz A. Effectiveness of etodolac (‘Lodine’) compared with naproxen in patients with acute gout. Curr Med Res Opin 1991; 12:423–429.
- Schumacher HR, Boice JA, Daikh DI, et al Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ 2002; 324:1488–1492.
- Rubin BR, Burton R, Navarra S, et al Efficacy and safety profile of treatment with etoricoxib 120 mg once daily compared with indomethacin 50 mg three times daily in acute gout: a randomized controlled trial. Arthritis Rheum 2004; 50:598–606.
- Shrestha M, Morgan DL, Moreden JM, et al Randomized doubleblind comparison of the analgesic efficacy of intramuscular ketorolac and oral indomethacin in the treatment of acute gouty arthritis. Ann Emerg Med 1995; 26:682–686.
- Ahern MJ, Reid C, Gordon TP, et al Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med 1987; 17:301–304.
- Terkeltaub RA, Furst DE, Bennett K, et al High vs low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum 2010; 62:1060–1068.
- Janssens HJ, Janssen M, van de Lisdonk EH, et al Use of oral prednisolone or naproxen for the treatment of gout arthritis: a doubleblind, randomised equivalence trial. Lancet 2008; 371:1854–1860.
- Man CY, Cheung IT, Cameron PA, et al Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med 2007; 49:670–677.
- Siegel LB, Alloway JA, Nashel DJ. Comparison of adrenocorticotropic hormone and triamcinolone acetonide in the treatment of acute gouty arthritis. J Rheumatol 1994; 21:1325–1327.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007; 9:R28.
- Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum 2009; 38:411–419.
- Colcrys [package insert]. Philadelphia, PA: Mutual Pharmaceutical Company; 2009.
- Simkin PA, Gardner GC. Colchicine use in cyclosporine treated transplant recipients: how little is too much? J Rheumatol 2000; 27:1334–1337.
- Dogukan A, Oymak FS, Taskapan H, et al Acute fatal colchicine intoxication in a patient on continuous ambulatory peritoneal dialysis (CAPD): possible role of clarithromycin administration. Clin Nephrol 2001; 55:181–182.
- Rollot F, Pajot O, Chauvelot-Moachon L, et al Acute colchicine intoxication during clarithromycin administration. Ann Pharmacother 2004; 38:2074–2077.
- Alayli G, Cengiz K, Canturk F, Durmus D, Akyol Y, Menekse EB. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother 2005; 39:1358–1361.
- Atmaca H, Sayarlioglu H, Kulah E, Demircan N, Akpolat T. Rhabdomyolysis associated with gemfibrozil-colchicine therapy. Ann Pharmacother 2002; 36:1719–1721.
- Roger U, Lins H, Scherrmann JM, et al Tetraparesis associated with colchicine is probably due to inhibition by verapamil of the P-glycoprotein efflux pump in the blood-brain barrier. BMJ 2005; 331:613.
- Levaquin [package insert]. Raritan, NJ: Ortho-McNeil; 2009.
- Phelan KM, Mosholder AD, Lu S. Lithium interaction with the cyclooxygenase 2 inhibitors rofecoxib and celecoxib and other nonsteroidal anti-inflammatory drugs. J Clin Psychiatry 2003; 64:1328–1334.
- Vistide [package insert]. Foster City, CA: Gilead Sciences; 2001.
- Hynninen V, Olkkola KT, Leino K, et al Effects of the antifungals voriconazole and fluconazole on the pharmacokinetics of S+ and R−-ibuprofen. Antimicrob Agents Chemother 2006; 50:1967–1972.
- Albengres E, Le Louet H, Tillement JP. Systemic antifungal agents. Drug interactions of clinical significance. Drug Saf 1998; 18:83–97.
- 2009 Red Book: Pharmacy’s Fundamental Reference. 113th ed. Los Angeles, CA: RAmEx Ars Medica; 2009.
- Odio CM, Ramirez T, Arias G, et al Double blind, randomized, placebo-controlled study of dexamethasone therapy for hematogenous septic arthritis in children. Pediatr Infect Dis J 2003; 22:883–888.
- Hung IF, Wu AK, Cheng VC, et al Fatal interaction between clarithromycin and colchicine in patients with renal insufficiency: a retrospective study. Clin Infect Dis 2005; 41:291–300.
- Cronstein BN, Terkeltaub R. The inflammatory process of gout and its treatment. Arthritis Res Ther 2006; 8( suppl 1):S3–S9.
- Kang EH, Lee EY, Lee YJ, et al Clinical features and risk factors of postsurgical gout. Ann Rheum Dis 2008; 67:1271–1275.
- Craig MH, Poole GV, Hauser CJ. Postsurgical gout. Am Surg 1995; 61:56–59.
- Friedman JE, Dallal RM, Lord JL. Gouty attacks occur frequently in postoperative gastric bypass patients. Surg Obes Relat Dis 2008; 4:11–15.
- Becker MA, Schumacher HR, Wortmann RL, et al Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450–2461.