User login
Erythrocytosis due to presumed polycythemia vera
A 40-year-old woman with hypertrophic obstructive cardiomyopathy presents to the hematology clinic for a second opinion regarding a history of headaches and fatigue for the past 10 years. She has been diagnosed with idiopathic erythrocytosis, presumed to be due to polycythemia vera. She periodically undergoes phlebotomy to keep her hematocrit below 41%, and this markedly improves her headaches. She denies shortness of breath, cough, fever, weight loss, joint pain, and visual or other neurologic symptoms. She has never reported pruritus related to bathing or exposure to water.
She does not smoke, drink alcohol, or use illicit drugs. She works as a pharmacy technician. She says her father died of cancer (no further details available) and describes a family history of gastrointestinal malignancy in her grandfather and paternal aunt. She takes aspirin, metoprolol, and spironolactone for her cardiomyopathy.
Physical examination reveals generalized plethora, more marked on her cheeks and face, and mild bilateral pitting pedal edema. No lymphadenopathy or hepatosplenomegaly can be palpated. Other systems, including the cardiac, respiratory, and nervous systems, are normal.
ERYTHROCYTOSIS AND POLYCYTHEMIA VERA
1. In patients with erythrocytosis, which of the following is not characteristic of polycythemia vera?
- Erythromelalgia and postbathing pruritus
- Splenomegaly
- History of thrombosis
- Gout
- Hematuria

Erythrocytosis—an abnormally high concentration of red blood cells in the peripheral blood—is a laboratory finding. It often reflects an increase in the total quantity or mass of red blood cells in the body (polycythemia) but can sometimes be due to decreased plasma volume (spurious polycythemia).1 Erythrocytosis can be caused by a number of diseases, hereditary and acquired, and can be classified as primary or secondary (Table 1).
Symptoms arise from an increase in the total blood volume and red blood cell mass, often leading to dilated capillaries and other blood vessels. Symptoms can occur regardless of the cause and classically include headache (often described as diffuse heaviness), dizziness, and a tendency for bleeding or thrombosis.2 Symptoms are relieved when the hematocrit is lowered.
Several features in the history and physical examination of a patient being evaluated for erythrocytosis can suggest an underlying cause. Smoking, chronic respiratory insufficiency, and congenital cyanotic heart disease point to secondary erythrocytosis and can usually be identified at the outset. A history of occupational exposure to carbon monoxide (such as engine exhaust) should be elicited carefully. A family history of erythrocytosis should raise suspicion of a heritable condition such as a hemoglobinopathy associated with increased oxygen affinity or rare forms of primary erythrocytosis associated with endogenous overproduction of erythropoietin or activating mutations of the erythropoietin receptor.3 Iatrogenic causes such as androgen supplementation, erythropoietin abuse, and postrenal-transplant erythrocytosis should also be considered.
Secretion of erythropoietin or erythropoietinlike proteins by a malignant neoplasm is a rare but important cause of erythrocytosis. For example, renal cell carcinoma may present with erythrocytosis secondary to excessive erythropoietin production, and hematuria can be an early symptom.
Polycythemia vera
Polycythemia vera, a myeloproliferative neoplasm, is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The bone marrow shows a panmyelosis that is often accompanied by leukocytosis or thrombocytosis, or both, in the peripheral blood.
Symptoms such as severe itching after exposure to hot water (aquagenic pruritus) and periodic attacks of redness, swelling, and pain in the hands or feet, or both (erythromelalgia), have been described in patients with polycythemia vera. Splenomegaly is relatively common, seen in approximately two-thirds of patients.4 Hyperuricemia (from increased cell turnover) and gout are also associated with polycythemia vera, as is a history of arterial and venous thrombosis.5
Hematuria is not commonly seen in polycythemia vera, although bleeding from the bladder, vagina, or uterus has been described.
CASE RESUMED: INITIAL LABORATORY TESTS
Results of our patient’s initial laboratory tests are:
- Hemoglobin 16.9 g/dL (reference range 11.5–15.5)
- Hematocrit 48.8% (36.0–46.0)
- Mean corpuscular volume 85.2 fL (80–100)
- Platelet count 328 × 109/L (150–400)
- White blood cell count 9.14 × 109/L (3.7–11.0)
- Absolute neutrophil count 5.95 × 109/L (1.45–7.5)
- Blood urea nitrogen 12 mg/dL (8–25)
- Creatinine 0.5 mg/dL (0.7–1.4)
- Lactate dehydrogenase 180 U/L (100–220)
- Uric acid 3.0 mg/dL (2.0–7.0)
- Thyroid-stimulating hormone 2.2 µU/mL (0.4–5.5).
The patient undergoes additional tests, including a serum erythropoietin level and hemoglobinopathy screen. Bone marrow aspiration and biopsy are performed, with cytogenetic analysis, chromosomal microarray analysis, and molecular testing for mutation of the Janus kinase 2 (JAK2) gene.
CONFIRMING SUSPECTED POLYCYTHEMIA VERA
2. In patients with suspected polycythemia vera, which of the following laboratory tests is most useful in making the diagnosis?
- Hemoglobin, hematocrit, and red blood cell mass
- Serum erythropoietin level
- Arterial blood gases with co-oximetry
- Testing for the JAK2 mutation
- Bone marrow aspiration and biopsy
The aim of the initial workup of erythrocytosis is to differentiate polycythemia vera from secondary causes of erythrocytosis.
Hemoglobin, hematocrit, red cell mass
Erythrocytosis is defined by an abnormal elevation in the hematocrit (> 48% in women or > 49% in men), hemoglobin concentration (> 16.0 g/dL in women or > 16.5 g/dL in men), or red blood cell mass. The red blood cell count should not be used as a surrogate for red blood cell mass, since some anemias (especially thalassemia minor) can be associated with an increase in the number of red blood cells but a low hemoglobin concentration.
Isotope dilution techniques to determine the red cell mass and plasma volume can differentiate true erythrocytosis from a spurious elevation due to a decrease in plasma volume.6,7 However, this is an expensive, time-consuming test that is not widely available and so is rarely performed.8
JAK2 mutation testing
The initial evaluation of a patient with erythrocytosis has changed significantly in the past 10 years with the discovery of the JAK2 gene and its role in the pathogenesis of polycythemia vera and other myeloproliferative neoplasms.
JAK2, located at 9p24, codes for a tyrosine kinase important for signal transduction in hematopoietic cells. Mutations in this gene have been shown to promote hypersensitivity to cytokines, including erythropoietin.9 The most common somatic mutation occurs within exon 14 at base pair 1849 and results in a phenylalanine-for-valine amino acid substitution in the JAK2 protein, designated V617F. Less commonly, mutations occur elsewhere in exons 12 to 15, with more than 50 different mutations described; nonpolymorphic mutations are assumed to have biologic effects similar to those of V617F.

Taken together, the JAK2 V617F and non-V617F mutations have a diagnostic sensitivity of 98% to 100% for polycythemia vera. For practical purposes, this means that the presence of a JAK2 mutation can be used as a clonal marker to distinguish polycythemia vera from reactive secondary causes of erythrocytosis. A JAK2 mutation is one of three major diagnostic criteria for polycythemia vera in the 2016 revision to the 2008 World Health Organization criteria (Table 2).10 Of note, this mutation is not specific for polycythemia vera and can also be found in other myeloproliferative neoplasms, including primary myelofibrosis and essential thrombocythemia.
Absence of a JAK2 mutation makes polycythemia vera unlikely, so this test is most useful in making the diagnosis.
Serum erythropoietin
Serum erythropoietin testing can be very useful to distinguish polycythemia vera from secondary erythrocytosis. Low levels suggest polycythemia vera, while high levels are seen in secondary processes.11
This test is best used along with JAK2 V617F mutation analysis as an initial step in evaluating patients with erythrocytosis. When JAK2 V617F mutation analysis is negative, a low serum erythropoietin level should prompt further testing for non-V617F JAK2 mutations, whereas a normal or elevated erythropoietin level should be evaluated further with tests to distinguish hereditary from acquired secondary causes of erythrocytosis.
Arterial blood gas analysis and co-oximetry
Arterial blood gas analysis can reveal hypoxemia, pointing to a cardiorespiratory process driving the erythrocytosis, whereas co-oximetry can be used to identify the presence and amount of carboxyhemoglobin in the blood.
Bone marrow biopsy
An increase in pleomorphic megakaryocytes in the bone marrow without stainable iron is often described as characteristic in polycythemia vera patients, but it is not diagnostic. Panmyelosis with increased cellularity is the norm but can be seen in other myeloproliferative neoplasms. The morphologic features of bone marrow are now included as one of the major diagnostic criteria for polycythemia vera (Table 2).
OUR PATIENT’S FURTHER WORKUP
Our patient’s erythropoietin level is 34.2 mIU/mL (reference range 4.7–28.6). Her oxygen saturation is 96%, and her carboxyhemoglobin level is 1.1% (0–5).
She undergoes bone marrow biopsy. Analysis finds that the marrow is normocellular (60%) with trilineage hematopoiesis and decreased stainable iron.
Cytogenetic analysis shows a 46,XX[20] karyotype. Chromosomal microarray analysis shows no pathogenic copy-number changes. There is no detectable JAK2 V617F or exon 12-to-15 mutation.
The patient’s erythrocytosis and abnormal hemoglobin electrophoresis study raise suspicion for a variant type of hemoglobin that has a higher affinity for oxygen than normal.
3. What is the next best step to evaluate this patient?
- Red-cell oxygen equilibrium curve to calculate the P50 (the partial pressure of oxygen that is required to saturate 50% of the hemoglobin.)
- High-performance liquid chromatography
- Globin gene DNA sequencing
- Testing 2,3-bisphosphoglycerate mutase (BPGM) activity
Nearly 200 mutational variants in alpha and beta globin chains that lead to an increased affinity of hemoglobin for oxygen have been reported.12 While not all mutations are clinically significant, increased oxygen affinity variants can lead to impaired oxygen delivery to tissues, especially the kidneys, resulting in a physiologic increase in erythropoietin and erythrocytosis.
In patients being evaluated for a high-oxygen-affinity hemoglobinopathy, a two-step approach has been outlined.13 The first involves measuring the oxygen-binding properties of a freshly collected sample of blood by directly measuring the oxygen saturation of the hemoglobin and pO2 using a co-oximeter. This information is used to create a red cell oxygen equilibrium curve and to calculate the P50. A low P50 correlates with an abnormally high affinity of hemoglobin for oxygen.
The second step is to identify the abnormal hemoglobin. High-performance liquid chromatography is now widely available as a screening test but does not detect all variants. For many years, sequencing of globin chain DNA has been a gold standard for identifying specific mutations. Subsequent to analyzing a catalog of known hemoglobin variants, mass spectrometry can serve as a screening and identification technique. Mass spectroscopy can also detect known rare variants with posttranslational modifications14 that are not recognized by DNA analysis. Mass spectroscopy and DNA sequencing are complementary techniques available only in specialized reference laboratories.
Erythrocytosis due to BPGM deficiency is very rare. Clinical and laboratory features mimic those of high-oxygen-affinity hemoglobin, but patients do not have a demonstrable mutation in alpha or beta globin genes. The level of BPGM is low, and the diagnosis is established by measuring BPGM levels and sequencing the BPGM gene.15
RESULTS OF THE ADDITIONAL WORKUP
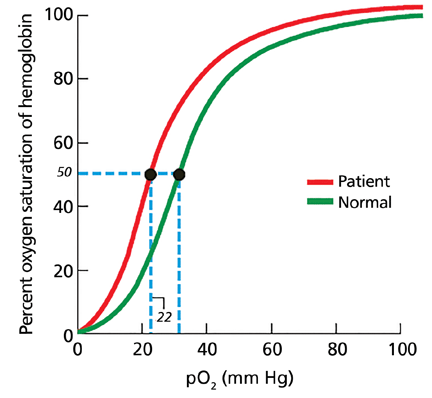
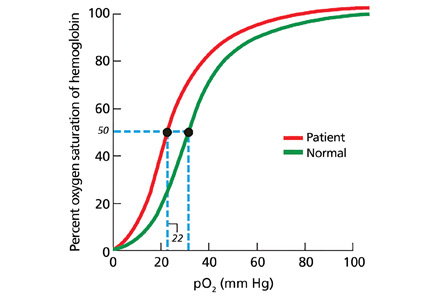
In our patient, hemoglobin electrophoresis reveals an abnormal hemoglobin variant. High-performance liquid chromatography reveals an abnormal peak that comprises approximately 23.7% of the total hemoglobin, consistent with an alpha globin variant. Further characterization (using a sample of venous blood) shows an oxygen dissociation P50 of 22 mm Hg (normal 24–30 mm Hg) (Figure 1).
Mass spectrometry identifies the variant as hemoglobin Tarrant. This variant is characterized by a substitution of asparagine for aspartic acid at position 126 of the alpha globin chain, a known site of contact between the alpha 1 and beta 1 chains.16 It has been seen in patients of Hispanic heritage and clinically correlates with mild erythrocytosis. Indeed, this woman’s mother was from Mexico.
EDUCATING PATIENTS
4. What should patients know about their high-oxygen-affinity hemoglobinopathy?
- High altitudes and air travel can be risky
- Pregnancy may have adverse outcomes
- Systemic anticoagulation may lower the risk of venous thromboembolism
- Periodic phlebotomy may help control symptoms
Most patients with high-oxygen-affinity hemoglobin do not require specific clinical management but only counseling and education about their condition. Establishing an accurate diagnosis is important in order to avoid further inappropriate, invasive, and expensive testing.
Although exposure to high altitudes may be associated with decreased ambient oxygen levels, hypoxia is usually not a problem because of hemoglobin’s high affinity for oxygen.
Impaired delivery of oxygen across the placenta may be anticipated in a mother with high-oxygen-affinity hemoglobin, but this has not been observed clinically.17
Compared with patients with polycythemia vera, patients with high-oxygen-affinity hemoglobin have fewer complications from hyperviscosity and thrombosis, even with comparable degrees of erythrocytosis.
Although patients usually do not require treatment, phlebotomy may be helpful for symptoms that can be attributed to the higher hemoglobin concentration.
Our patient continues to be seen in clinic for periodic blood counts and phlebotomy for her headaches, as required.
HEMOGLOBIN: RELAXED OR TENSE
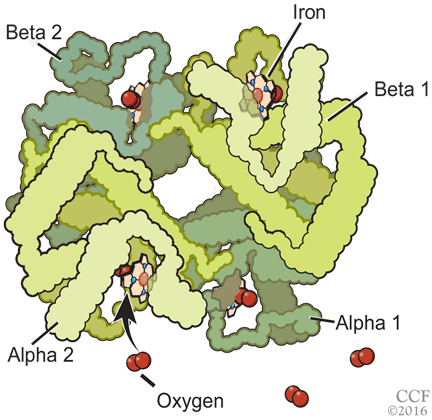
Normal adult hemoglobin is a tetramer composed of two pairs of globin polypeptide chains: alpha and beta (Figure 2). The intrinsic properties of the constituent globin chains and their allosteric conformation—as well as extrinsic factors including temperature, pH, and the binding of hydrogen ion and 2,3-BPG—play important roles in modifying the affinity of hemoglobin for oxygen. The major modulator of hemoglobin-oxygen affinity in human erythrocytes is 2,3-BPG.
The hemoglobin tetramer, consisting of two identical halves, alpha 1-beta 1 and alpha 2-beta 2, oscillates between two quaternary conformations, “relaxed” (fully oxygenated) and “tense” (fully deoxygenated).18 High-oxygen-affinity hemoglobins can result from factors that enhance the relaxed state, either by stabilizing the relaxed state or by destabilizing the tense state. Structural modifications in hemoglobin typically affect the main contacts involved in the transition from the deoxygenated to the oxygenated state, the 2,3-BPG binding sites, the heme pocket, or elongation of globin chains by various mutations. In hemoglobin Tarrant, the mutation prevents formation of noncovalent salt bridges in the alpha 1-beta 1 contact that normally stabilize the deoxygenated conformation of hemoglobin. As a result, the deoxygenated (tense) state is destabilized, shifting the allosteric equilibrium in favor of the oxygenated (relaxed) state with consequent high oxygen affinity.16
MORE ABOUT HIGH-OXYGEN-AFFINITY HEMOGLOBINS
The first case of erythrocytosis due to an abnormal hemoglobin was identified in 1966. This was an alpha chain variant with an arginine-to-leucine substitution at position 92, named hemoglobin Chesapeake.19
High-oxygen-affinity hemoglobin variants are usually transmitted as autosomal dominant traits. Patients are most often identified because of unexplained erythrocytosis detected on a routine blood cell count, as in our patient.
Not all high-oxygen-affinity hemoglobinopathies are associated with erythrocytosis. The degree of increased oxygen affinity may only be mild or the abnormal hemoglobin may be slightly unstable, thereby masking the usual clinical signs and symptoms.
Therapeutic phlebotomy should be used cautiously since it can decrease delivery of oxygen to tissues. A subset of patients whose symptoms are related to an elevated red cell mass may experience some relief, as did our patient.
- Kremyanskaya M, Mascarenhas J, Hoffman R. Why does my patient have erythrocytosis? Hematol Oncol Clin North Am 2012; 26:267–283.
- Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ 2013; 347:f6667.
- Agarwal N, Gordeuk RV, Prchal JT. Genetic mechanisms underlying regulation of hemoglobin mass. Adv Exp Med Biol 2007; 618:195–210.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87:285–293.
- Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 2008; 22:2020–2028.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151–154.
- Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169–186; discussion 186a–186g.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405.
- Messinezy M, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, Pearson TC. Serum erythropoietin values in erythrocytosis and in primary thrombocythaemia. Br J Haematol 2002; 117:47–53.
- Hardison RC, Chui DHK, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutat 2002; 19:225–233.
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009; 94:1321–1322.
- Kattamis AC, Kelly KM, Ohene-Frempong K, et al. Hb Osler [beta 145(HC2)Tyr-->Asp] results from posttranslational modification. Hemoglobin 1997; 21:109–120.
- Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol 2004; 75:205–208.
- Moo-Penn WF, Jue DL, Johnson MH, Wilson SM, Therrell B Jr, Schmidt RM. Hemoglobin Tarrant: alpha126(H9) asp leads to asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity. Biochim Biophys Acta 1977; 490:443–451.
- Bard H, Peri KG, Gagnon C. The biologic implications of a rare hemoglobin mutant that decreases oxygen affinity. Pediatr Res 2001; 49:69–73.
- Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis: new variants and concepts. Hemoglobin 2005; 29:91–106.
- Clegg JB, Naughton MA, Weatherall DJ. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol 1966; 19:91–108.
A 40-year-old woman with hypertrophic obstructive cardiomyopathy presents to the hematology clinic for a second opinion regarding a history of headaches and fatigue for the past 10 years. She has been diagnosed with idiopathic erythrocytosis, presumed to be due to polycythemia vera. She periodically undergoes phlebotomy to keep her hematocrit below 41%, and this markedly improves her headaches. She denies shortness of breath, cough, fever, weight loss, joint pain, and visual or other neurologic symptoms. She has never reported pruritus related to bathing or exposure to water.
She does not smoke, drink alcohol, or use illicit drugs. She works as a pharmacy technician. She says her father died of cancer (no further details available) and describes a family history of gastrointestinal malignancy in her grandfather and paternal aunt. She takes aspirin, metoprolol, and spironolactone for her cardiomyopathy.
Physical examination reveals generalized plethora, more marked on her cheeks and face, and mild bilateral pitting pedal edema. No lymphadenopathy or hepatosplenomegaly can be palpated. Other systems, including the cardiac, respiratory, and nervous systems, are normal.
ERYTHROCYTOSIS AND POLYCYTHEMIA VERA
1. In patients with erythrocytosis, which of the following is not characteristic of polycythemia vera?
- Erythromelalgia and postbathing pruritus
- Splenomegaly
- History of thrombosis
- Gout
- Hematuria
Erythrocytosis—an abnormally high concentration of red blood cells in the peripheral blood—is a laboratory finding. It often reflects an increase in the total quantity or mass of red blood cells in the body (polycythemia) but can sometimes be due to decreased plasma volume (spurious polycythemia).1 Erythrocytosis can be caused by a number of diseases, hereditary and acquired, and can be classified as primary or secondary (Table 1).
Symptoms arise from an increase in the total blood volume and red blood cell mass, often leading to dilated capillaries and other blood vessels. Symptoms can occur regardless of the cause and classically include headache (often described as diffuse heaviness), dizziness, and a tendency for bleeding or thrombosis.2 Symptoms are relieved when the hematocrit is lowered.
Several features in the history and physical examination of a patient being evaluated for erythrocytosis can suggest an underlying cause. Smoking, chronic respiratory insufficiency, and congenital cyanotic heart disease point to secondary erythrocytosis and can usually be identified at the outset. A history of occupational exposure to carbon monoxide (such as engine exhaust) should be elicited carefully. A family history of erythrocytosis should raise suspicion of a heritable condition such as a hemoglobinopathy associated with increased oxygen affinity or rare forms of primary erythrocytosis associated with endogenous overproduction of erythropoietin or activating mutations of the erythropoietin receptor.3 Iatrogenic causes such as androgen supplementation, erythropoietin abuse, and postrenal-transplant erythrocytosis should also be considered.
Secretion of erythropoietin or erythropoietinlike proteins by a malignant neoplasm is a rare but important cause of erythrocytosis. For example, renal cell carcinoma may present with erythrocytosis secondary to excessive erythropoietin production, and hematuria can be an early symptom.
Polycythemia vera
Polycythemia vera, a myeloproliferative neoplasm, is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The bone marrow shows a panmyelosis that is often accompanied by leukocytosis or thrombocytosis, or both, in the peripheral blood.
Symptoms such as severe itching after exposure to hot water (aquagenic pruritus) and periodic attacks of redness, swelling, and pain in the hands or feet, or both (erythromelalgia), have been described in patients with polycythemia vera. Splenomegaly is relatively common, seen in approximately two-thirds of patients.4 Hyperuricemia (from increased cell turnover) and gout are also associated with polycythemia vera, as is a history of arterial and venous thrombosis.5
Hematuria is not commonly seen in polycythemia vera, although bleeding from the bladder, vagina, or uterus has been described.
CASE RESUMED: INITIAL LABORATORY TESTS
Results of our patient’s initial laboratory tests are:
- Hemoglobin 16.9 g/dL (reference range 11.5–15.5)
- Hematocrit 48.8% (36.0–46.0)
- Mean corpuscular volume 85.2 fL (80–100)
- Platelet count 328 × 109/L (150–400)
- White blood cell count 9.14 × 109/L (3.7–11.0)
- Absolute neutrophil count 5.95 × 109/L (1.45–7.5)
- Blood urea nitrogen 12 mg/dL (8–25)
- Creatinine 0.5 mg/dL (0.7–1.4)
- Lactate dehydrogenase 180 U/L (100–220)
- Uric acid 3.0 mg/dL (2.0–7.0)
- Thyroid-stimulating hormone 2.2 µU/mL (0.4–5.5).
The patient undergoes additional tests, including a serum erythropoietin level and hemoglobinopathy screen. Bone marrow aspiration and biopsy are performed, with cytogenetic analysis, chromosomal microarray analysis, and molecular testing for mutation of the Janus kinase 2 (JAK2) gene.
CONFIRMING SUSPECTED POLYCYTHEMIA VERA
2. In patients with suspected polycythemia vera, which of the following laboratory tests is most useful in making the diagnosis?
- Hemoglobin, hematocrit, and red blood cell mass
- Serum erythropoietin level
- Arterial blood gases with co-oximetry
- Testing for the JAK2 mutation
- Bone marrow aspiration and biopsy
The aim of the initial workup of erythrocytosis is to differentiate polycythemia vera from secondary causes of erythrocytosis.
Hemoglobin, hematocrit, red cell mass
Erythrocytosis is defined by an abnormal elevation in the hematocrit (> 48% in women or > 49% in men), hemoglobin concentration (> 16.0 g/dL in women or > 16.5 g/dL in men), or red blood cell mass. The red blood cell count should not be used as a surrogate for red blood cell mass, since some anemias (especially thalassemia minor) can be associated with an increase in the number of red blood cells but a low hemoglobin concentration.
Isotope dilution techniques to determine the red cell mass and plasma volume can differentiate true erythrocytosis from a spurious elevation due to a decrease in plasma volume.6,7 However, this is an expensive, time-consuming test that is not widely available and so is rarely performed.8
JAK2 mutation testing
The initial evaluation of a patient with erythrocytosis has changed significantly in the past 10 years with the discovery of the JAK2 gene and its role in the pathogenesis of polycythemia vera and other myeloproliferative neoplasms.
JAK2, located at 9p24, codes for a tyrosine kinase important for signal transduction in hematopoietic cells. Mutations in this gene have been shown to promote hypersensitivity to cytokines, including erythropoietin.9 The most common somatic mutation occurs within exon 14 at base pair 1849 and results in a phenylalanine-for-valine amino acid substitution in the JAK2 protein, designated V617F. Less commonly, mutations occur elsewhere in exons 12 to 15, with more than 50 different mutations described; nonpolymorphic mutations are assumed to have biologic effects similar to those of V617F.
Taken together, the JAK2 V617F and non-V617F mutations have a diagnostic sensitivity of 98% to 100% for polycythemia vera. For practical purposes, this means that the presence of a JAK2 mutation can be used as a clonal marker to distinguish polycythemia vera from reactive secondary causes of erythrocytosis. A JAK2 mutation is one of three major diagnostic criteria for polycythemia vera in the 2016 revision to the 2008 World Health Organization criteria (Table 2).10 Of note, this mutation is not specific for polycythemia vera and can also be found in other myeloproliferative neoplasms, including primary myelofibrosis and essential thrombocythemia.
Absence of a JAK2 mutation makes polycythemia vera unlikely, so this test is most useful in making the diagnosis.
Serum erythropoietin
Serum erythropoietin testing can be very useful to distinguish polycythemia vera from secondary erythrocytosis. Low levels suggest polycythemia vera, while high levels are seen in secondary processes.11
This test is best used along with JAK2 V617F mutation analysis as an initial step in evaluating patients with erythrocytosis. When JAK2 V617F mutation analysis is negative, a low serum erythropoietin level should prompt further testing for non-V617F JAK2 mutations, whereas a normal or elevated erythropoietin level should be evaluated further with tests to distinguish hereditary from acquired secondary causes of erythrocytosis.
Arterial blood gas analysis and co-oximetry
Arterial blood gas analysis can reveal hypoxemia, pointing to a cardiorespiratory process driving the erythrocytosis, whereas co-oximetry can be used to identify the presence and amount of carboxyhemoglobin in the blood.
Bone marrow biopsy
An increase in pleomorphic megakaryocytes in the bone marrow without stainable iron is often described as characteristic in polycythemia vera patients, but it is not diagnostic. Panmyelosis with increased cellularity is the norm but can be seen in other myeloproliferative neoplasms. The morphologic features of bone marrow are now included as one of the major diagnostic criteria for polycythemia vera (Table 2).
OUR PATIENT’S FURTHER WORKUP
Our patient’s erythropoietin level is 34.2 mIU/mL (reference range 4.7–28.6). Her oxygen saturation is 96%, and her carboxyhemoglobin level is 1.1% (0–5).
She undergoes bone marrow biopsy. Analysis finds that the marrow is normocellular (60%) with trilineage hematopoiesis and decreased stainable iron.
Cytogenetic analysis shows a 46,XX[20] karyotype. Chromosomal microarray analysis shows no pathogenic copy-number changes. There is no detectable JAK2 V617F or exon 12-to-15 mutation.
The patient’s erythrocytosis and abnormal hemoglobin electrophoresis study raise suspicion for a variant type of hemoglobin that has a higher affinity for oxygen than normal.
3. What is the next best step to evaluate this patient?
- Red-cell oxygen equilibrium curve to calculate the P50 (the partial pressure of oxygen that is required to saturate 50% of the hemoglobin.)
- High-performance liquid chromatography
- Globin gene DNA sequencing
- Testing 2,3-bisphosphoglycerate mutase (BPGM) activity
Nearly 200 mutational variants in alpha and beta globin chains that lead to an increased affinity of hemoglobin for oxygen have been reported.12 While not all mutations are clinically significant, increased oxygen affinity variants can lead to impaired oxygen delivery to tissues, especially the kidneys, resulting in a physiologic increase in erythropoietin and erythrocytosis.
In patients being evaluated for a high-oxygen-affinity hemoglobinopathy, a two-step approach has been outlined.13 The first involves measuring the oxygen-binding properties of a freshly collected sample of blood by directly measuring the oxygen saturation of the hemoglobin and pO2 using a co-oximeter. This information is used to create a red cell oxygen equilibrium curve and to calculate the P50. A low P50 correlates with an abnormally high affinity of hemoglobin for oxygen.
The second step is to identify the abnormal hemoglobin. High-performance liquid chromatography is now widely available as a screening test but does not detect all variants. For many years, sequencing of globin chain DNA has been a gold standard for identifying specific mutations. Subsequent to analyzing a catalog of known hemoglobin variants, mass spectrometry can serve as a screening and identification technique. Mass spectroscopy can also detect known rare variants with posttranslational modifications14 that are not recognized by DNA analysis. Mass spectroscopy and DNA sequencing are complementary techniques available only in specialized reference laboratories.
Erythrocytosis due to BPGM deficiency is very rare. Clinical and laboratory features mimic those of high-oxygen-affinity hemoglobin, but patients do not have a demonstrable mutation in alpha or beta globin genes. The level of BPGM is low, and the diagnosis is established by measuring BPGM levels and sequencing the BPGM gene.15
RESULTS OF THE ADDITIONAL WORKUP
In our patient, hemoglobin electrophoresis reveals an abnormal hemoglobin variant. High-performance liquid chromatography reveals an abnormal peak that comprises approximately 23.7% of the total hemoglobin, consistent with an alpha globin variant. Further characterization (using a sample of venous blood) shows an oxygen dissociation P50 of 22 mm Hg (normal 24–30 mm Hg) (Figure 1).
Mass spectrometry identifies the variant as hemoglobin Tarrant. This variant is characterized by a substitution of asparagine for aspartic acid at position 126 of the alpha globin chain, a known site of contact between the alpha 1 and beta 1 chains.16 It has been seen in patients of Hispanic heritage and clinically correlates with mild erythrocytosis. Indeed, this woman’s mother was from Mexico.
EDUCATING PATIENTS
4. What should patients know about their high-oxygen-affinity hemoglobinopathy?
- High altitudes and air travel can be risky
- Pregnancy may have adverse outcomes
- Systemic anticoagulation may lower the risk of venous thromboembolism
- Periodic phlebotomy may help control symptoms
Most patients with high-oxygen-affinity hemoglobin do not require specific clinical management but only counseling and education about their condition. Establishing an accurate diagnosis is important in order to avoid further inappropriate, invasive, and expensive testing.
Although exposure to high altitudes may be associated with decreased ambient oxygen levels, hypoxia is usually not a problem because of hemoglobin’s high affinity for oxygen.
Impaired delivery of oxygen across the placenta may be anticipated in a mother with high-oxygen-affinity hemoglobin, but this has not been observed clinically.17
Compared with patients with polycythemia vera, patients with high-oxygen-affinity hemoglobin have fewer complications from hyperviscosity and thrombosis, even with comparable degrees of erythrocytosis.
Although patients usually do not require treatment, phlebotomy may be helpful for symptoms that can be attributed to the higher hemoglobin concentration.
Our patient continues to be seen in clinic for periodic blood counts and phlebotomy for her headaches, as required.
HEMOGLOBIN: RELAXED OR TENSE
Normal adult hemoglobin is a tetramer composed of two pairs of globin polypeptide chains: alpha and beta (Figure 2). The intrinsic properties of the constituent globin chains and their allosteric conformation—as well as extrinsic factors including temperature, pH, and the binding of hydrogen ion and 2,3-BPG—play important roles in modifying the affinity of hemoglobin for oxygen. The major modulator of hemoglobin-oxygen affinity in human erythrocytes is 2,3-BPG.
The hemoglobin tetramer, consisting of two identical halves, alpha 1-beta 1 and alpha 2-beta 2, oscillates between two quaternary conformations, “relaxed” (fully oxygenated) and “tense” (fully deoxygenated).18 High-oxygen-affinity hemoglobins can result from factors that enhance the relaxed state, either by stabilizing the relaxed state or by destabilizing the tense state. Structural modifications in hemoglobin typically affect the main contacts involved in the transition from the deoxygenated to the oxygenated state, the 2,3-BPG binding sites, the heme pocket, or elongation of globin chains by various mutations. In hemoglobin Tarrant, the mutation prevents formation of noncovalent salt bridges in the alpha 1-beta 1 contact that normally stabilize the deoxygenated conformation of hemoglobin. As a result, the deoxygenated (tense) state is destabilized, shifting the allosteric equilibrium in favor of the oxygenated (relaxed) state with consequent high oxygen affinity.16
MORE ABOUT HIGH-OXYGEN-AFFINITY HEMOGLOBINS
The first case of erythrocytosis due to an abnormal hemoglobin was identified in 1966. This was an alpha chain variant with an arginine-to-leucine substitution at position 92, named hemoglobin Chesapeake.19
High-oxygen-affinity hemoglobin variants are usually transmitted as autosomal dominant traits. Patients are most often identified because of unexplained erythrocytosis detected on a routine blood cell count, as in our patient.
Not all high-oxygen-affinity hemoglobinopathies are associated with erythrocytosis. The degree of increased oxygen affinity may only be mild or the abnormal hemoglobin may be slightly unstable, thereby masking the usual clinical signs and symptoms.
Therapeutic phlebotomy should be used cautiously since it can decrease delivery of oxygen to tissues. A subset of patients whose symptoms are related to an elevated red cell mass may experience some relief, as did our patient.
A 40-year-old woman with hypertrophic obstructive cardiomyopathy presents to the hematology clinic for a second opinion regarding a history of headaches and fatigue for the past 10 years. She has been diagnosed with idiopathic erythrocytosis, presumed to be due to polycythemia vera. She periodically undergoes phlebotomy to keep her hematocrit below 41%, and this markedly improves her headaches. She denies shortness of breath, cough, fever, weight loss, joint pain, and visual or other neurologic symptoms. She has never reported pruritus related to bathing or exposure to water.
She does not smoke, drink alcohol, or use illicit drugs. She works as a pharmacy technician. She says her father died of cancer (no further details available) and describes a family history of gastrointestinal malignancy in her grandfather and paternal aunt. She takes aspirin, metoprolol, and spironolactone for her cardiomyopathy.
Physical examination reveals generalized plethora, more marked on her cheeks and face, and mild bilateral pitting pedal edema. No lymphadenopathy or hepatosplenomegaly can be palpated. Other systems, including the cardiac, respiratory, and nervous systems, are normal.
ERYTHROCYTOSIS AND POLYCYTHEMIA VERA
1. In patients with erythrocytosis, which of the following is not characteristic of polycythemia vera?
- Erythromelalgia and postbathing pruritus
- Splenomegaly
- History of thrombosis
- Gout
- Hematuria
Erythrocytosis—an abnormally high concentration of red blood cells in the peripheral blood—is a laboratory finding. It often reflects an increase in the total quantity or mass of red blood cells in the body (polycythemia) but can sometimes be due to decreased plasma volume (spurious polycythemia).1 Erythrocytosis can be caused by a number of diseases, hereditary and acquired, and can be classified as primary or secondary (Table 1).
Symptoms arise from an increase in the total blood volume and red blood cell mass, often leading to dilated capillaries and other blood vessels. Symptoms can occur regardless of the cause and classically include headache (often described as diffuse heaviness), dizziness, and a tendency for bleeding or thrombosis.2 Symptoms are relieved when the hematocrit is lowered.
Several features in the history and physical examination of a patient being evaluated for erythrocytosis can suggest an underlying cause. Smoking, chronic respiratory insufficiency, and congenital cyanotic heart disease point to secondary erythrocytosis and can usually be identified at the outset. A history of occupational exposure to carbon monoxide (such as engine exhaust) should be elicited carefully. A family history of erythrocytosis should raise suspicion of a heritable condition such as a hemoglobinopathy associated with increased oxygen affinity or rare forms of primary erythrocytosis associated with endogenous overproduction of erythropoietin or activating mutations of the erythropoietin receptor.3 Iatrogenic causes such as androgen supplementation, erythropoietin abuse, and postrenal-transplant erythrocytosis should also be considered.
Secretion of erythropoietin or erythropoietinlike proteins by a malignant neoplasm is a rare but important cause of erythrocytosis. For example, renal cell carcinoma may present with erythrocytosis secondary to excessive erythropoietin production, and hematuria can be an early symptom.
Polycythemia vera
Polycythemia vera, a myeloproliferative neoplasm, is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The bone marrow shows a panmyelosis that is often accompanied by leukocytosis or thrombocytosis, or both, in the peripheral blood.
Symptoms such as severe itching after exposure to hot water (aquagenic pruritus) and periodic attacks of redness, swelling, and pain in the hands or feet, or both (erythromelalgia), have been described in patients with polycythemia vera. Splenomegaly is relatively common, seen in approximately two-thirds of patients.4 Hyperuricemia (from increased cell turnover) and gout are also associated with polycythemia vera, as is a history of arterial and venous thrombosis.5
Hematuria is not commonly seen in polycythemia vera, although bleeding from the bladder, vagina, or uterus has been described.
CASE RESUMED: INITIAL LABORATORY TESTS
Results of our patient’s initial laboratory tests are:
- Hemoglobin 16.9 g/dL (reference range 11.5–15.5)
- Hematocrit 48.8% (36.0–46.0)
- Mean corpuscular volume 85.2 fL (80–100)
- Platelet count 328 × 109/L (150–400)
- White blood cell count 9.14 × 109/L (3.7–11.0)
- Absolute neutrophil count 5.95 × 109/L (1.45–7.5)
- Blood urea nitrogen 12 mg/dL (8–25)
- Creatinine 0.5 mg/dL (0.7–1.4)
- Lactate dehydrogenase 180 U/L (100–220)
- Uric acid 3.0 mg/dL (2.0–7.0)
- Thyroid-stimulating hormone 2.2 µU/mL (0.4–5.5).
The patient undergoes additional tests, including a serum erythropoietin level and hemoglobinopathy screen. Bone marrow aspiration and biopsy are performed, with cytogenetic analysis, chromosomal microarray analysis, and molecular testing for mutation of the Janus kinase 2 (JAK2) gene.
CONFIRMING SUSPECTED POLYCYTHEMIA VERA
2. In patients with suspected polycythemia vera, which of the following laboratory tests is most useful in making the diagnosis?
- Hemoglobin, hematocrit, and red blood cell mass
- Serum erythropoietin level
- Arterial blood gases with co-oximetry
- Testing for the JAK2 mutation
- Bone marrow aspiration and biopsy
The aim of the initial workup of erythrocytosis is to differentiate polycythemia vera from secondary causes of erythrocytosis.
Hemoglobin, hematocrit, red cell mass
Erythrocytosis is defined by an abnormal elevation in the hematocrit (> 48% in women or > 49% in men), hemoglobin concentration (> 16.0 g/dL in women or > 16.5 g/dL in men), or red blood cell mass. The red blood cell count should not be used as a surrogate for red blood cell mass, since some anemias (especially thalassemia minor) can be associated with an increase in the number of red blood cells but a low hemoglobin concentration.
Isotope dilution techniques to determine the red cell mass and plasma volume can differentiate true erythrocytosis from a spurious elevation due to a decrease in plasma volume.6,7 However, this is an expensive, time-consuming test that is not widely available and so is rarely performed.8
JAK2 mutation testing
The initial evaluation of a patient with erythrocytosis has changed significantly in the past 10 years with the discovery of the JAK2 gene and its role in the pathogenesis of polycythemia vera and other myeloproliferative neoplasms.
JAK2, located at 9p24, codes for a tyrosine kinase important for signal transduction in hematopoietic cells. Mutations in this gene have been shown to promote hypersensitivity to cytokines, including erythropoietin.9 The most common somatic mutation occurs within exon 14 at base pair 1849 and results in a phenylalanine-for-valine amino acid substitution in the JAK2 protein, designated V617F. Less commonly, mutations occur elsewhere in exons 12 to 15, with more than 50 different mutations described; nonpolymorphic mutations are assumed to have biologic effects similar to those of V617F.
Taken together, the JAK2 V617F and non-V617F mutations have a diagnostic sensitivity of 98% to 100% for polycythemia vera. For practical purposes, this means that the presence of a JAK2 mutation can be used as a clonal marker to distinguish polycythemia vera from reactive secondary causes of erythrocytosis. A JAK2 mutation is one of three major diagnostic criteria for polycythemia vera in the 2016 revision to the 2008 World Health Organization criteria (Table 2).10 Of note, this mutation is not specific for polycythemia vera and can also be found in other myeloproliferative neoplasms, including primary myelofibrosis and essential thrombocythemia.
Absence of a JAK2 mutation makes polycythemia vera unlikely, so this test is most useful in making the diagnosis.
Serum erythropoietin
Serum erythropoietin testing can be very useful to distinguish polycythemia vera from secondary erythrocytosis. Low levels suggest polycythemia vera, while high levels are seen in secondary processes.11
This test is best used along with JAK2 V617F mutation analysis as an initial step in evaluating patients with erythrocytosis. When JAK2 V617F mutation analysis is negative, a low serum erythropoietin level should prompt further testing for non-V617F JAK2 mutations, whereas a normal or elevated erythropoietin level should be evaluated further with tests to distinguish hereditary from acquired secondary causes of erythrocytosis.
Arterial blood gas analysis and co-oximetry
Arterial blood gas analysis can reveal hypoxemia, pointing to a cardiorespiratory process driving the erythrocytosis, whereas co-oximetry can be used to identify the presence and amount of carboxyhemoglobin in the blood.
Bone marrow biopsy
An increase in pleomorphic megakaryocytes in the bone marrow without stainable iron is often described as characteristic in polycythemia vera patients, but it is not diagnostic. Panmyelosis with increased cellularity is the norm but can be seen in other myeloproliferative neoplasms. The morphologic features of bone marrow are now included as one of the major diagnostic criteria for polycythemia vera (Table 2).
OUR PATIENT’S FURTHER WORKUP
Our patient’s erythropoietin level is 34.2 mIU/mL (reference range 4.7–28.6). Her oxygen saturation is 96%, and her carboxyhemoglobin level is 1.1% (0–5).
She undergoes bone marrow biopsy. Analysis finds that the marrow is normocellular (60%) with trilineage hematopoiesis and decreased stainable iron.
Cytogenetic analysis shows a 46,XX[20] karyotype. Chromosomal microarray analysis shows no pathogenic copy-number changes. There is no detectable JAK2 V617F or exon 12-to-15 mutation.
The patient’s erythrocytosis and abnormal hemoglobin electrophoresis study raise suspicion for a variant type of hemoglobin that has a higher affinity for oxygen than normal.
3. What is the next best step to evaluate this patient?
- Red-cell oxygen equilibrium curve to calculate the P50 (the partial pressure of oxygen that is required to saturate 50% of the hemoglobin.)
- High-performance liquid chromatography
- Globin gene DNA sequencing
- Testing 2,3-bisphosphoglycerate mutase (BPGM) activity
Nearly 200 mutational variants in alpha and beta globin chains that lead to an increased affinity of hemoglobin for oxygen have been reported.12 While not all mutations are clinically significant, increased oxygen affinity variants can lead to impaired oxygen delivery to tissues, especially the kidneys, resulting in a physiologic increase in erythropoietin and erythrocytosis.
In patients being evaluated for a high-oxygen-affinity hemoglobinopathy, a two-step approach has been outlined.13 The first involves measuring the oxygen-binding properties of a freshly collected sample of blood by directly measuring the oxygen saturation of the hemoglobin and pO2 using a co-oximeter. This information is used to create a red cell oxygen equilibrium curve and to calculate the P50. A low P50 correlates with an abnormally high affinity of hemoglobin for oxygen.
The second step is to identify the abnormal hemoglobin. High-performance liquid chromatography is now widely available as a screening test but does not detect all variants. For many years, sequencing of globin chain DNA has been a gold standard for identifying specific mutations. Subsequent to analyzing a catalog of known hemoglobin variants, mass spectrometry can serve as a screening and identification technique. Mass spectroscopy can also detect known rare variants with posttranslational modifications14 that are not recognized by DNA analysis. Mass spectroscopy and DNA sequencing are complementary techniques available only in specialized reference laboratories.
Erythrocytosis due to BPGM deficiency is very rare. Clinical and laboratory features mimic those of high-oxygen-affinity hemoglobin, but patients do not have a demonstrable mutation in alpha or beta globin genes. The level of BPGM is low, and the diagnosis is established by measuring BPGM levels and sequencing the BPGM gene.15
RESULTS OF THE ADDITIONAL WORKUP
In our patient, hemoglobin electrophoresis reveals an abnormal hemoglobin variant. High-performance liquid chromatography reveals an abnormal peak that comprises approximately 23.7% of the total hemoglobin, consistent with an alpha globin variant. Further characterization (using a sample of venous blood) shows an oxygen dissociation P50 of 22 mm Hg (normal 24–30 mm Hg) (Figure 1).
Mass spectrometry identifies the variant as hemoglobin Tarrant. This variant is characterized by a substitution of asparagine for aspartic acid at position 126 of the alpha globin chain, a known site of contact between the alpha 1 and beta 1 chains.16 It has been seen in patients of Hispanic heritage and clinically correlates with mild erythrocytosis. Indeed, this woman’s mother was from Mexico.
EDUCATING PATIENTS
4. What should patients know about their high-oxygen-affinity hemoglobinopathy?
- High altitudes and air travel can be risky
- Pregnancy may have adverse outcomes
- Systemic anticoagulation may lower the risk of venous thromboembolism
- Periodic phlebotomy may help control symptoms
Most patients with high-oxygen-affinity hemoglobin do not require specific clinical management but only counseling and education about their condition. Establishing an accurate diagnosis is important in order to avoid further inappropriate, invasive, and expensive testing.
Although exposure to high altitudes may be associated with decreased ambient oxygen levels, hypoxia is usually not a problem because of hemoglobin’s high affinity for oxygen.
Impaired delivery of oxygen across the placenta may be anticipated in a mother with high-oxygen-affinity hemoglobin, but this has not been observed clinically.17
Compared with patients with polycythemia vera, patients with high-oxygen-affinity hemoglobin have fewer complications from hyperviscosity and thrombosis, even with comparable degrees of erythrocytosis.
Although patients usually do not require treatment, phlebotomy may be helpful for symptoms that can be attributed to the higher hemoglobin concentration.
Our patient continues to be seen in clinic for periodic blood counts and phlebotomy for her headaches, as required.
HEMOGLOBIN: RELAXED OR TENSE
Normal adult hemoglobin is a tetramer composed of two pairs of globin polypeptide chains: alpha and beta (Figure 2). The intrinsic properties of the constituent globin chains and their allosteric conformation—as well as extrinsic factors including temperature, pH, and the binding of hydrogen ion and 2,3-BPG—play important roles in modifying the affinity of hemoglobin for oxygen. The major modulator of hemoglobin-oxygen affinity in human erythrocytes is 2,3-BPG.
The hemoglobin tetramer, consisting of two identical halves, alpha 1-beta 1 and alpha 2-beta 2, oscillates between two quaternary conformations, “relaxed” (fully oxygenated) and “tense” (fully deoxygenated).18 High-oxygen-affinity hemoglobins can result from factors that enhance the relaxed state, either by stabilizing the relaxed state or by destabilizing the tense state. Structural modifications in hemoglobin typically affect the main contacts involved in the transition from the deoxygenated to the oxygenated state, the 2,3-BPG binding sites, the heme pocket, or elongation of globin chains by various mutations. In hemoglobin Tarrant, the mutation prevents formation of noncovalent salt bridges in the alpha 1-beta 1 contact that normally stabilize the deoxygenated conformation of hemoglobin. As a result, the deoxygenated (tense) state is destabilized, shifting the allosteric equilibrium in favor of the oxygenated (relaxed) state with consequent high oxygen affinity.16
MORE ABOUT HIGH-OXYGEN-AFFINITY HEMOGLOBINS
The first case of erythrocytosis due to an abnormal hemoglobin was identified in 1966. This was an alpha chain variant with an arginine-to-leucine substitution at position 92, named hemoglobin Chesapeake.19
High-oxygen-affinity hemoglobin variants are usually transmitted as autosomal dominant traits. Patients are most often identified because of unexplained erythrocytosis detected on a routine blood cell count, as in our patient.
Not all high-oxygen-affinity hemoglobinopathies are associated with erythrocytosis. The degree of increased oxygen affinity may only be mild or the abnormal hemoglobin may be slightly unstable, thereby masking the usual clinical signs and symptoms.
Therapeutic phlebotomy should be used cautiously since it can decrease delivery of oxygen to tissues. A subset of patients whose symptoms are related to an elevated red cell mass may experience some relief, as did our patient.
- Kremyanskaya M, Mascarenhas J, Hoffman R. Why does my patient have erythrocytosis? Hematol Oncol Clin North Am 2012; 26:267–283.
- Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ 2013; 347:f6667.
- Agarwal N, Gordeuk RV, Prchal JT. Genetic mechanisms underlying regulation of hemoglobin mass. Adv Exp Med Biol 2007; 618:195–210.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87:285–293.
- Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 2008; 22:2020–2028.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151–154.
- Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169–186; discussion 186a–186g.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405.
- Messinezy M, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, Pearson TC. Serum erythropoietin values in erythrocytosis and in primary thrombocythaemia. Br J Haematol 2002; 117:47–53.
- Hardison RC, Chui DHK, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutat 2002; 19:225–233.
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009; 94:1321–1322.
- Kattamis AC, Kelly KM, Ohene-Frempong K, et al. Hb Osler [beta 145(HC2)Tyr-->Asp] results from posttranslational modification. Hemoglobin 1997; 21:109–120.
- Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol 2004; 75:205–208.
- Moo-Penn WF, Jue DL, Johnson MH, Wilson SM, Therrell B Jr, Schmidt RM. Hemoglobin Tarrant: alpha126(H9) asp leads to asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity. Biochim Biophys Acta 1977; 490:443–451.
- Bard H, Peri KG, Gagnon C. The biologic implications of a rare hemoglobin mutant that decreases oxygen affinity. Pediatr Res 2001; 49:69–73.
- Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis: new variants and concepts. Hemoglobin 2005; 29:91–106.
- Clegg JB, Naughton MA, Weatherall DJ. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol 1966; 19:91–108.
- Kremyanskaya M, Mascarenhas J, Hoffman R. Why does my patient have erythrocytosis? Hematol Oncol Clin North Am 2012; 26:267–283.
- Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ 2013; 347:f6667.
- Agarwal N, Gordeuk RV, Prchal JT. Genetic mechanisms underlying regulation of hemoglobin mass. Adv Exp Med Biol 2007; 618:195–210.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87:285–293.
- Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 2008; 22:2020–2028.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151–154.
- Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169–186; discussion 186a–186g.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405.
- Messinezy M, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, Pearson TC. Serum erythropoietin values in erythrocytosis and in primary thrombocythaemia. Br J Haematol 2002; 117:47–53.
- Hardison RC, Chui DHK, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutat 2002; 19:225–233.
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009; 94:1321–1322.
- Kattamis AC, Kelly KM, Ohene-Frempong K, et al. Hb Osler [beta 145(HC2)Tyr-->Asp] results from posttranslational modification. Hemoglobin 1997; 21:109–120.
- Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol 2004; 75:205–208.
- Moo-Penn WF, Jue DL, Johnson MH, Wilson SM, Therrell B Jr, Schmidt RM. Hemoglobin Tarrant: alpha126(H9) asp leads to asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity. Biochim Biophys Acta 1977; 490:443–451.
- Bard H, Peri KG, Gagnon C. The biologic implications of a rare hemoglobin mutant that decreases oxygen affinity. Pediatr Res 2001; 49:69–73.
- Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis: new variants and concepts. Hemoglobin 2005; 29:91–106.
- Clegg JB, Naughton MA, Weatherall DJ. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol 1966; 19:91–108.