User login
Central Sleep Apnea in Adults: Diagnosis and Treatment
As the prevalence of obstructive sleep apnea (OSA) has steadily increased in the United States, so has the awareness of central sleep apnea (CSA). The hallmark of CSA is transient cessation of airflow during sleep due to a lack of respiratory effort triggered by the brain. This is in contrast to OSA, in which there is absence of airflow despite continued ventilatory effort due to physical airflow obstruction. The gold standard for the diagnosis and optimal treatment assessment of CSA is inlaboratory polysomnography (PSG) with esophageal manometry, but in practice, respiratory effort is generally estimated through oronasal flow and respiratory inductance plethysmography bands placed on the chest and abdomen during PSG.
Background
The literature has demonstrated a higher prevalence of moderate-to-severe OSA in the general population compared with that of CSA. While OSA is associated with worse clinical outcomes, more evidence is needed on the long-term clinical impact and optimal treatment strategies for CSA.1 CSA is overrepresented among certain clinical populations. CSA is not frequently diagnosed in the active-duty population, but is increasing in the veteran population, especially in those with heart failure (HF), stroke, neuromuscular disorders, and opioid use. It is associated with increased admissions related to comorbid cardiovascular disorders and to an increased risk of death.2-4 The clinical concerns with CSA parallel those of OSA. The absence of respiration (apneas and hypopneas due to lack of effort) results in sympathetic surge, compromise of oxygenation and ventilation, sleep fragmentation, and elevation in blood pressure. Symptoms such as excessive daytime sleepiness, morning headaches, witnessed apneas, and nocturnal arrhythmias are shared between the 2 disorders.
Ventilatory instability is the most widely accepted mechanism leading to CSA in patients. Loop gain is the concept used to explain ventilatory control. This feedback loop is influenced by controller gain (primarily represented by central and peripheral chemoreceptors causing changes in ventilation due to PaCO2 [partial pressure of CO2 in arterial blood] fluctuations), plant gain (includes lungs and respiratory muscles and their ability to eliminate CO2), and circulation time (feedback between controller and plant).5
High loop gain and narrow CO2 reserve contribute to ventilatory instability in CSA.6 Those with high loop gain have increased sensitivity to changes in CO2. These patients tend to overbreathe in response to smaller increases in PaCO2 compared with those with low loop gain. Once the PaCO2 falls below an individual’s apneic threshold (AT), an apnea will occur.7 The brainstem then pauses ventilation to allow the PaCO2 to rise back above the AT. CSAs also can occur in healthy individuals as they transition from wakefulness into non–rapid eye movement (REM) sleep in a phenomenon called sleep state oscillation, with a mechanism that is similar to hyperventilation-induced CSAs described earlier.
Primary CSA has been defined in the International Classification of Sleep Disorders 3rd edition (ICSD-3) with the following criteria: (1) diagnostic PSG with ≥ 5 events per hour of CSAs and/or central hypopneas per hour of sleep; the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; and there is no evidence of Cheyne-Stokes breathing (CSB); (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) there is no evidence of nocturnal hypoventilation; and (4) the disorder is not better explained by another medical use, substance use disorder (SUD), or other current sleep, medical, or neurologic disorder.8
A systematic clinical approach should be used to identify and treat CSA (Figure).6,7 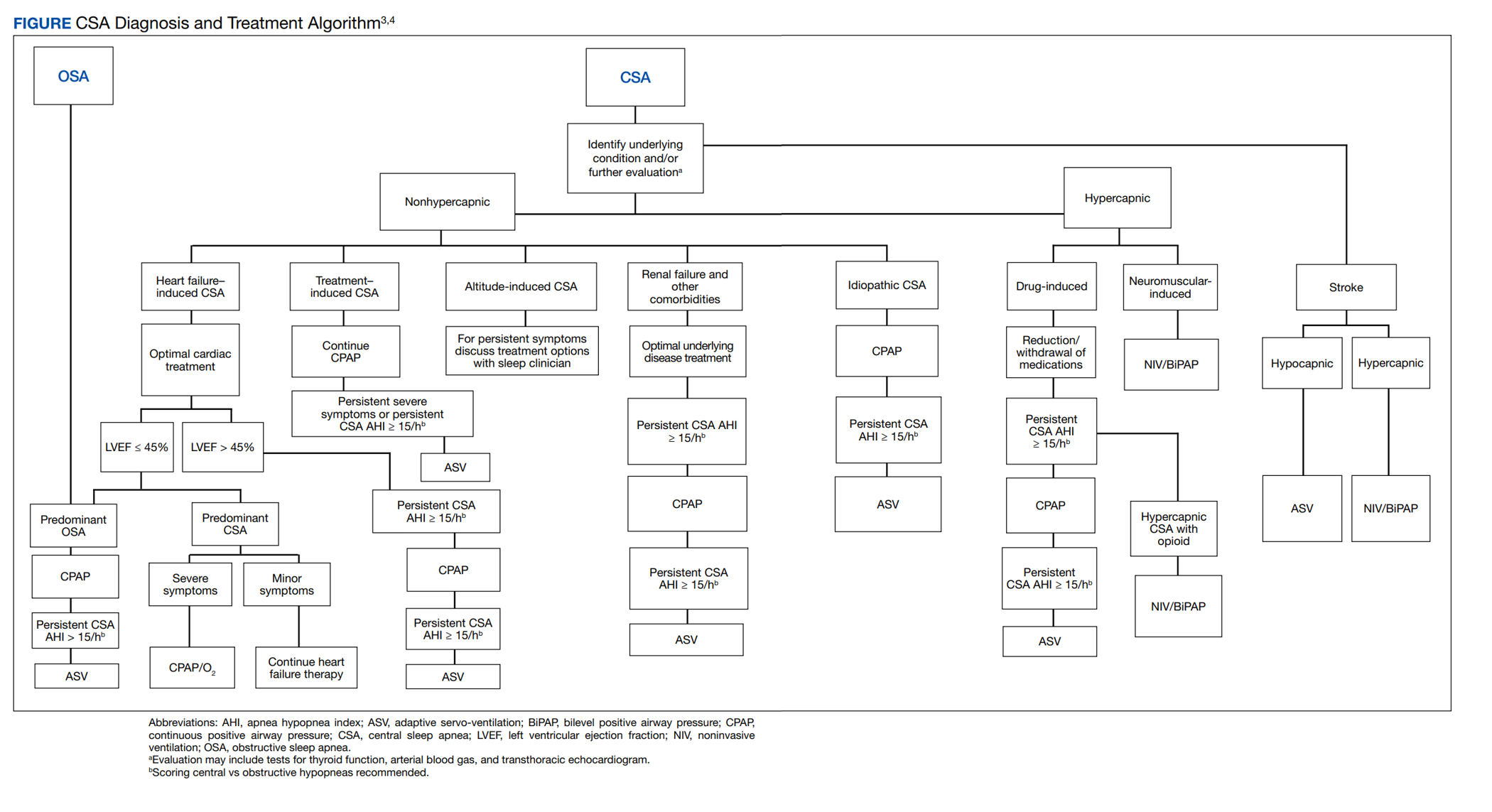
The purpose of this review is to familiarize the primary care community with CSA to aid in the identification and management of this breathing disturbance.
Nonhypercapnic CSA
Heart Failure–Induced CSA
The leading medical diagnosis causing CSA is congestive HF (CHF), and there is a correlation between HF severity and presence of CSA. In patients with stable CHF with HF reduced ejection fraction (HFrEF), CSA is highly prevalent, occurring in 25% to 40% of patients.9 In contrast to other subtypes of CSA where literature regarding prognosis is lacking, the literature is clear that patients with HFrEF with CSA have a worse prognosis, with increased risk of death independent of the severity of HF. This may be the result of CSA promoting malignant ventricular arrhythmias. The prevalence of CSA in HF with preserved ejection fraction (HFpEF) is about 18% to 30%.10,11
A significant reduction in cardiac output results in circulatory delay between the lungs and chemoreceptors that produces CSB periodic breathing, which is characteristic of the most recognized form of CSA. Per the ICSD-3, CSA with CSB requires the following 4 findings: (1) PSG reveals ≥ 5 CSAs and/or central hypopneas per hour of sleep; there are at least 3 consecutive CSAs and/or central hypopneas separated by crescendo-decrescendo breathing with a cycle length of at least 40 seconds (ie, CSB pattern), and the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) the breathing pattern is associated with atrial fibrillation/flutter, CHF, or a neurologic disorder; and (4) the disorder is not better explained by another current sleep disorder, medication use (eg, opioids), or SUD.8
Treatment of HF-induced CSA begins with guideline-based medical management with the goal of reducing pulmonary capillary wedge pressure or increasing left ventricular ejection fraction through means that may include cardiac resynchronization therapy or left ventricular assist devices, when clinically indicated. If medical optimization is not sufficient, the next step is continuous positive airway pressure (CPAP or PAP), followed by adaptive servo-ventilation (ASV) if the apnea-hypopnea index (AHI) remains > 15 events per hour and is clinically indicated.
ASV is a second-line PAP therapy modality that uses proprietary algorithms to provide variable amounts of pressure that alternate between expiratory and inspiratory phases of the respiratory cycle in combination with physician-set or automatic backup respiratory rate designed to stabilize ventilation in patients with CSA and CSB. The inability to adjust tidal volume, potentially resulting in insufficient tidal volumes or ventilation, results in the contraindication for its use in patients with CSA with comorbid conditions that may result in hypercapnic respiratory failure. These conditions include chronic hypoventilation in obesity hypoventilation syndrome (OHS), moderate-to-severe chronic obstructive pulmonary disease, chronic elevation of PaCO2 on arterial blood gas > 45 mm Hg, and restrictive thoracic or neuromuscular disease.12
Although ASV is more effective in normalizing AHI in patients with HF and CSA than is CPAP therapy, the clinical indications for ASV in the setting of HFrEF changed drastically with the publication of the landmark SERVE-HF trial, which investigated the effects of adding ASV to guideline-based medical management on survival and cardiovascular outcomes in patients with HFrEF and predominant CSA.13 The study did not show a difference between the ASV and control groups for the primary endpoint: a composite of time to first event of death from any cause, lifesaving cardiovascular intervention (transplantation, implantation of a long-term ventricular assist device, resuscitation after sudden cardiac arrest, or appropriate lifesaving shock), or unplanned hospitalization for worsening HF. However, the study showed a statistically and clinically significant increased risk of all-cause and cardiovascular mortality in the ASV group compared with the control group.13 A possible explanation for the increased all-cause and cardiovascular mortality is that CSA potentially serves a protective mechanism that when eliminated results in deleterious clinical outcomes. This resulted in significant changes in the treatment algorithm for HF-induced CSA with left ventricular ejection fraction of at least 45% becoming the cutoff for therapeutic decisions.
Treatment-Emergent CSA
Treatment-emergent CSA (TECSA, also known as complex sleep apnea) has been defined by the ICSD-3 by the following criteria: (1) diagnostic PSG with ≥ 5 events per hour of predominantly obstructive events; (2) resolution of obstructive events with PAP without a backup rate and CSA index (CAI) ≥ 5 per hour with central events ≥ 50% of the AHI; and (3) CSA not better explained by another disorder.8 Patients with TECSA can be further classified into those who have transient events that resolve within weeks to months, those with persistent events, and those with delayed events that may develop weeks to months after initiating PAP therapy.14
PAP treatment can decrease the PaCO2 below the AT due to removal of flow limitation in previously obstructed upper airways, resulting in TECSA.15,16 PAP therapy has not been the only treatment where new CSA has been identified on initiation. A 2021 systematic review identified patients who developed new-onset CSA with mandibular advancement device (MAD), hypoglossal nerve stimulator, tongue protrusion device, and nasal expiratory PAP device use, as well as after tracheostomy, maxillofacial surgery, and other surgeries, such as nasal and uvulopalatopharyngoplasty.17
The prevalence of TECSA has been noted to range between 0.6% and 20.3%, but Nigam and colleagues estimated a prevalence of 8.4% in their systematic review.11,14 The variability in prevalence between studies could be due to differences in study design (retrospective vs prospective vs cross-sectional), diagnostic and inclusion criteria, patient population, and type of study used (full-night vs split-night vs both).18,19 Risk factors for TECSA include male sex; older age; lower body mass index; higher baseline AHI, CAI, and arousal index; chronic medical issues such as CHF and coronary artery disease; opioid use; higher CPAP settings; excessive mask leak; and bilevel PAP (BiPAP) use.20 Identifying these risk factors is important, as patients with TECSA are at higher risk of discontinuing therapy and of developing PAP intolerance.15,20
Most patients with TECSA can continue CPAP therapy with resolution of events over weeks to months, but treatment of comorbid conditions should be optimized as they could be contributing factors. Zeineddine and colleagues recommend continuation of CPAP for 3 months if the patient has minor or no symptoms.19 A 2018 systematic review noted that 14.3% to 46.2% of TECSA patients will have persistent TECSA and some will develop TECSA after at least 1 month of PAP therapy.14 For these patients and those with severe symptoms in spite of therapy, a switch to BiPAP spontaneous/timed (BiPAP-S/T) or ASV should be considered, if not contraindicated based on comorbidities.21 Medications such as acetazolamide, oxygen therapy, and CO2 supplementation have also been discussed as alternative treatments, but these options should not be first-line therapies and should be used on a case-by-case basis as adjuncts to PAP therapy.16,17
Altitude-Induced CSA
Also known as CSA due to high-altitude periodic breathing (CSA-HAPB), this form of CSA occurs in nearly all lowlanders at altitudes above 3000 m, with severity increasing with altitude.15 The exact altitude at which it occurs varies based on an individual’s physiology. CSA-HAPB occurs in response to the low barometric pressure at altitude, combined with stable fraction of oxygen, resulting in decreased inspired partial pressure of oxygen and hypoxia. The normal physiologic response to hypoxia is increased ventilation, which can cause hypocapnia, suppressing respiratory drive and resulting in CSAs. The instability of the respiratory response results in cyclical CSAs followed by hyperventilation. This periodic breathing then causes arousals from sleep, driving further sleep fragmentation and exacerbation of baseline desaturation and instability in the cyclical respiratory response. There is individual variability in hypoxic chemoresponsiveness (loop gain). Compensatory mechanisms are most robust when an individual routinely dwells at high altitude, resulting in acclimatization, rather than traveling there for a brief stay. Genetics and cardiac output also contribute to the effectiveness of compensation to altitude.
CSA-HAPB is defined by the following ICSD-3 criteria: (1) Recent ascent to a high altitude (typically ≥ 2500 m, although some individuals may exhibit the disorder at altitudes as low as 1500 m); (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) symptoms are clinically attributable to HAPB, or PSG, if performed, reveals recurrent CSAs or hypopneas primarily during non-REM sleep at a frequency of ≥ 5 events per hour; (4) the disorder is not better explained by another current sleep disorder, medical or neurological disorder, medication use (eg, narcotics), or SUD.8
Treatment options to improve nocturnal oxygen saturation and reduce or eliminate CSA-HAPB in nonacclimatized individuals include oxygen-enriched air, acetazolamide, or combination treatment with acetazolamide and automatic PAP (APAP).22 A meta-analysis looking at the effectiveness of acetazolamide in 8 different randomized controlled trials demonstrated that a dose of 250 mg per day was effective in improving sleep apnea at altitude as measured by a decrease in AHI, decrease in percentage of periodic breathing, and increasing oxygenation during sleep.15 The question of superiority of combined acetazolamide with APAP to placebo with APAP in treatment of high-altitude OSA was addressed in a randomized double-blind, placebo-controlled trial. The results showed that combined APAP (5-15 cm of water pressure) and acetazolamide (250 mg morning, 500 mg evening) decreased the AHI to normal range, whereas central events persisted in the APAP and placebo group.23 In addition, Latshang and colleagues have demonstrated that ASV may not be as efficacious for controlling CSA-HAPB in nonacclimatized individuals compared with oxygen therapy and suggested that further research is warranted examining if ASV device algorithm adjustment improves efficacy of this therapeutic option.24
Comorbidity-Induced CSA
Several medical conditions may be associated with CSA, including chronic kidney disease (CKD), pulmonary hypertension, acromegaly, and hypothyroidism. The common pathophysiologic link is that these disorders may result in alteration of ventilatory responses to CO2, ultimately resulting in CSA.
As many as 10% of patients with CKD may experience CSA.25,26 The complications encountered in CKD include fluid overload with pulmonary edema, chronic metabolic acidosis, and anemia. These can provoke hyperventilation in addition to poor sleep quality, triggering arousals that further drive CSA in these patients. Additional risk factors for CSA in this population include atrial fibrillation and cardiac dysfunction. Clinical interventions that have demonstrated reduction in CSA include hemodialysis at night vs daytime and using bicarbonate buffer vs acetate for hemodialysis 22-24,26-29
Hypersecretion of growth hormone in acromegaly also results in hyperventilation contributing to CSA. While medical and surgical management of acromegaly results in a reduction in OSA, there is limited evidence on the outcome of the CSA after these interventions.
Hypothyroidism and CSA both present with similar symptoms of fatigue, daytime sleepiness, depression, and headaches. Studies suggest that respiratory muscle fatigue and decreased ventilatory response to hypercapnia and hypoxia contribute to apnea in this population. In one study, 27% of hypothyroid patients had a blunted response to hypercapnia, and 34% suffered from a blunted response to hypoxia. The same study showed universal reversal of the impairment following thyroid replacement therapy and return to euthyroid state.30 Similarly, multiple studies have shown reversal of respiratory muscle fatigue after initiation of thyroid replacement.30-32 Assessing thyroid function is an appropriate initial step during any sleep-disordered breathing workup, as it is a reversible cause of apnea. Up to 2.4% of patients presenting for PSG (and diagnosed with OSA) are found to have undiagnosed hypothyroidism.32,33 In a military population, treatment of a secondary cause of CSA, such as hypothyroidism, could remove some administrative burden as well as improve service members’ quality of life.
If CSA persists despite previous treatment strategies, then clinicians should focus on the optimization of treatment for comorbid conditions. If that does not resolve CSA, CPAP should be used when AHI remains above 15 events per hour or ASV can be used.
Idiopathic CSA
There are limited data on the pathophysiology and prevalence of idiopathic CSA. In most cases it is hypocapnic CSA, which occurs after an arousal from sleep causing hyperventilation that causes hypocapnia below the apnea threshold similar to CSA-HAPB. Therapeutic options based on addressing underlying pathophysiology include increasing CO2 by inhalation or addition of dead space. Additional therapeutic options to reduce the arousals and CSAs include hypnotics, such as zolpidem and acetazolamide, but these should be administered only with close clinical monitoring. If symptoms continue, CPAP or ASV may be trialed; however, limited clinical evidence of efficacy exists.15
For patients with moderate-to-severe CSA, an additional treatment option includes an implantable device (eg, Zoll remede¯), which stimulates the phrenic nerve to move the diaphragm and restore normal breathing. This device is not indicated for those with OSA. Based on data submitted to the US Food and Drug Administration, AHI is reduced by ≥ 50% in 51% of patients with the implanted device and by 11% in patients without the device. Five-year follow-up data show sustained improvements.34
Hypercapnic CSA
CSA due to a medication or substance requires the following criteria: (1) the patient is taking an opioid or other respiratory depressant; (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia (difficulty initiating or maintaining sleep, frequent awakenings, or nonrestorative sleep); (3) PSG reveals ≥ 5 CSAs and/or central hypopneas per hour of sleep; the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; and there is no evidence of CSB; and (4) the disorder is not better explained by another current sleep disorder.8
Drugs that affect the respiratory centers, such as opiates and opioids, γ aminobutyric acid (GABA) type A and B receptor agonists, and P2Y(12) receptor antagonists such as ticagrelor, may result in alterations in ventilatory drive in the central nervous system respiratory centers, resulting in CSA.
Opioids are prescribed either for chronic pain or to treat opiate addiction with methadone, resulting in about one-third of chronic opioid users having some form of CSA.35 CSA may be seen after opioids have been used for at least 2 months. A dose-dependent effect exists with high doses of opioids, typically resulting in hypoventilation, hypercapnia, and hypoxemia with ataxic or erratic breathing and a periodic breathing pattern similar to those described in CSA-HAPB or idiopathic CSA. About 14% to 60% of methadone patients also demonstrate CSA or ataxic breathing.35,36
Benzodiazepines (GABA-A receptor agonists) and baclofen (a GABA-B receptor agonist) depress central ventilatory drive, blunt the response to hypoxia and hypercapnia, leading to CSAs, and increase risk for OSA by increasing upper airway obstruction through reduction in tone. Use of these medications with antidepressants or opioids further exacerbates this response.
Unlike the other medications previously described, ticagrelor, a first-line dual antiplatelet therapy medication indicated for acute coronary syndrome treatment, actually increases the activity of the respiratory centers but may result in CSA.
First-line treatment, if possible, is reduction in medication dose or complete withdrawal. Additional treatment options include PAP therapies: CPAP, BiPAP, ASV, and oxygen therapy with or without PAP.37,38 The literature has demonstrated that for the treatment of opioid-associated CSA, ASV (in cases of normocapnia) and noninvasive ventilation (NIV)/BiPAP (in cases with hypercapnia or REM sleep hypoventilation) are superior treatment options when compared with conventional CPAP for elimination of respiratory events. CPAP with oxygen therapy and BiPAP with oxygen therapy are more effective than CPAP alone in reducing respiratory events. However, concerns remain that as with CSA in HF, CSA in chronic opioid users may serve as a physiologic protective mechanism to prevent further clinical injury from opioids. Similarly, as in the use of ASV in the SERVE-HF trial, focusing on elimination of respiratory events may prove detrimental. More studies are needed to determine whether reducing the number of CSA events in chronic opioid users is clinically beneficial when other health outcomes, such as cardiovascular, neurocognitive, hospital/intensive care unit admissions, and mortality risks are examined.
Neuromuscular-Induced CSA
CSA also is highly prevalent in neuromuscular conditions, such as amyotrophic lateral sclerosis, Duchenne muscular dystrophy, myotonic dystrophy, advanced multiple sclerosis, and acid maltase deficiency. There is reduced respiratory muscle strength and tone in these disorders, resulting in alveolar hypoventilation with hypercapnia. Given the hypercapnia, NIV/BiPAP is the first-line treatment to improve survival, gas exchange, symptom burden, and quality of life.
Stroke-Induced CSA
Extensive cerebrovascular events commonly precipitate sleep-related breathing disorders. The incidence increases in the acute phase of stroke and decreases 3 to 6 months poststroke; however, incidence also depends on the severity of the stroke.7,39,40 Stroke also has been shown to be a predictor of CSA (odds ratio, 1.65; 95% CI, 1.50-1.82; P < .001) in a retrospective analysis of a large cohort of US veterans.2 The location of the lesion often determines whether normocapnic or hypercapnic CSA will predominate, based on ventilatory instability resulting in normocapnia or reduced ventilatory drive resulting in hypercapnic CSA. PSG results and blood gases direct the treatment options. CSA with normocapnia is treated with ASV, and patients with hypercapnia/REM sleep hypoventilation are treated with NIV/BiPAP.
Conclusions
While much has been learned about CSA in recent decades, more evidence needs to be gathered to determine optimal treatment strategies and the impact on patient prognosis. The identification of CSA can lead to the diagnosis of previously unrecognized medical conditions. With proper diagnosis and treatment, we can optimize clinical management and improve patients’ prognosis and quality of life.
Acknowledgments
The authors thank the librarians of the Franzello Aeromedical Library in particular Sara Craycraft, Catherine Stahl, Kristen Young and Elizabeth Irvine for their support of this publication.
1. Heinzer R, Vat S, Marques-Vidal P, et al. Prevalence of sleep-disordered breathing in the general population: the HypnoLaus study. Lancet Respir Med. 2015;3(4):310-318. Epub 2015 Feb 12. doi:10.1016/S2213-2600(15)00043-0
2. Ratz D, Wiitala W, Safwan Badr M, Burns J, Chowdhuri S. Correlates and consequences of central sleep apnea in a national sample of US veterans. Sleep. 2018;41(9):zy058. doi:10.1093/sleep/zsyn058
3. Agrawal R, Sharafkhaneneh A, Gottlief, DJ, Nowakowski S, Razjouyan J. Mortality patterns associated with central sleep apnea among veterans: a large, retrospective, longitudinal report. Ann Am Thorac Soc. 2022;10.1513/AnnalsATS.202207-648OC. doi:10.1513/annalsATS. 202207-648OC
4. Mysliwiec V, McGraw L, Pierce R, Smith, P, Trapp, B, Roth B. Sleep disorders and associated medical comorbidities in active duty military personnel. Sleep. 2013;36(2):167-174. doi:10.5665/sleep.2364
5. Badr MS, Dingell JD, Javaheri S. Central sleep apnea: a brief review. Curr Pulmonol Rep. 2019;8(1):14-21. Epub 2019 Mar 13. doi:10.1007/s13665-019-0221-z
6. Baillieul S, Revol B, Jullian-Desayes I, Joyeux-Faure M, Tamisier R, Pépin JL. Diagnosis and management of central sleep apnea syndrome. Expert Rev Respir Med. 2019;13(6):545-557.1604226. Epub 2019 Apr 24. doi:10.1080/17476348.2019
7. Randerath W, Verbraecken J, Andreas S, et al. Definition, discrimination, diagnosis and treatment of central breathing disturbances during sleep. Eur Respir J. 2017;49(1):1600959. doi:10.1183/13993003.00959-2016
8. American Academy of Sleep Medicine. International Classification of Sleep Disorders. 3rd ed. American Academy of Sleep Medicine; 2014.
9. Lévy P, Pépin J-L, Tamisier R, Neuder Y, Baguet J-P, Javaheri S. Prevalence and impact of central sleep apnea in heart failure. Sleep Med Clinics. 2007;2(4):615-621. doi:10.1016/j.jsmc.2007.08.001
10. Bitter T, Faber L, Hering D, Langer C, Horstkotte D, Oldenburg O. Sleep-disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail. 2009;11(6):602-608. doi:10.1093/eurjhf/hfp057
11. Sekizuka H, Osada N, Miyake F. Sleep disordered breathing in heart failure patients with reduced versus preserved ejection fraction. Heart Lung Circ. 2013;22(2):104-109. Epub 2012 Oct 26. doi:10.1016/j.hlc.2012.08.006
12. Iotti GA, Polito A, Belliato M, et al. Adaptive support ventilation versus conventional ventilation for total ventilatory support in acute respiratory failure. Intensive Care Med. 2010;36(8):1371-1379. Epub 2010 May 26. doi:10.1007/s00134-010-1917-2
13. Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373(12):1095-105. Epub 2015 Sep 1. doi:10.1056/NEJMoa1506459
14. Nigam G, Riaz M, Chang ET, Camacho M. Natural history of treatment-emergent central sleep apnea on positive airway pressure: a systematic review. Ann Thorac Med. 2018;13(2):86-91. doi:10.4103/atm.ATM_321_17
15. Orr JE, Malhotra A, Sands SA. Pathogenesis of central and complex sleep apnoea. Respirology. 2017;22(1):43-52. Epub 2016 Oct 31. doi:10.1111/resp.12927
16. Berger M, Solelhac G, Horvath C, Heinzer R, Brill AK. Treatment-emergent central sleep apnea associated with non-positive airway pressure therapies in obstructive sleep apnea patients: a systematic review. Sleep Med Rev. 2021; 58:101513. Epub 2021 Jun 5. doi:10.1016/j.smrv.2021.101513
17. Zhang J, Wang L, Guo HJ, Wang Y, Cao J, Chen BY. Treatment-emergent central sleep apnea: a unique sleep-disordered breathing. Chin Med J (Engl). 2020;133(22):2721-2730. doi:10.1097/CM9.0000000000001125
18. Nigam G, Pathak C, Riaz M. A systematic review on prevalence and risk factors associated with treatment- emergent central sleep apnea. Ann Thorac Med. 2016;11(3):202-210. doi:10.4103/1817-1737.185761
19. Zeineddine S, Badr MS. Treatment-emergent central apnea: physiologic mechanisms informing clinical practice. Chest. 2021;159(6):2449-2457. Epub 2021 Jan 23. doi:10.1016/j.hest.2021.01.036
20. Liu D, Armitstead J, Benjafield A. Trajectories of emergent central sleep apnea during CPAP therapy. Chest. 2017;152(4):751-760. Epub 2017 Jun 16. doi:10.1016/j.chest.2017.06.010
21. Moro M, Gannon K, Lovell K, Merlino M, Mojica J, Bianchi MT. Clinical predictors of central sleep apnea evoked by positive airway pressure titration. Nat Sci Sleep. 2016;8:259-266. doi:10.2147/NSS.S110032
22. Orr JE, Heinrich EC, Djokic M, et al. Adaptive servoventilation as treatment for central sleep apnea due to high-altitude periodic breathing in nonacclimatized healthy individuals. High Alt Med Biol. 2018;19(2):178-184. Epub 2018 Mar 13. doi:10.1089/ham.2017.0147
23. Liu HM, Chiang IJ, Kuo KN, Liou CM, Chen C. The effect of acetazolamide on sleep apnea at high altitude: a systematic review and meta-analysis. Ther Adv Respir Dis. 2017;11(1):20-29. Epub 2016 Nov 15. doi:10.1177/1753465816677006
24. Latshang TD, Nussbaumer-Ochsner Y, Henn RM, et al. Effect of acetazolamide and autoCPAP therapy on breathing disturbances among patients with obstructive sleep apnea syndrome who travel to altitude: a randomized controlled trial. JAMA. 2012;308(22):2390-8. doi:10.1001/jama.2012.94847
25. Nigam G, Pathak C, Riaz M. A systematic review of central sleep apnea in adult patients with chronic kidney disease. Sleep Breath. 2016;20(3):957-964. Epub 2016 Jan 27. doi:10.1007/s11325-016-1317-0
26. Nigam G, Riaz M. Pathophysiology of central sleep apnea in chronic kidney disease. Saudi J Kidney Dis Transpl. 2016;27(5):1068-1070. doi:10.4103/1319-2442.190907
27. Hanly PJ, Pierratos A. Improvement of sleep apnea in patients with chronic renal failure who undergo nocturnal hemodialysis. N Engl J Med. 2001;344(2):102-107. doi:10.1056/NEJM200101113440204
28. Jean G, Piperno D, François B, Charra B. Sleep apnea incidence in maintenance hemodialysis patients: influence of dialysate buffer. Nephron. 1995;71(2):138-142. doi:10.1159/000188701
29. Pressman MR, Benz RL, Schleifer CR, Peterson DD. Sleep disordered breathing in ESRD: acute beneficial effects of treatment with nasal continuous positive airway pressure. Kidney Int. 1993;43(5):1134-1139. doi:10.1038/ki.1993.159
30. Ladenson PW, Goldenheim PD, Ridgway EC. Prediction and reversal of blunted ventilatory responsiveness in patients with hypothyroidism. Am J Med. 1988;84(5):877-883. doi:10.1016/0002-9343(88)90066-6
31. Siafakas NM, Salesiotou V, Filaditaki V, Tzanakis N, Thalassinos N, Bouros D. Respiratory muscle strength in hypothyroidism. Chest. 1992;102(1):189-194. doi:10.1378/chest.102.1.189
32. Laroche CM, Cairns T, Moxham J, Green M. Hypothyroidism presenting with respiratory muscle weakness. Am Rev Respir Dis. 1988;138(2):472-474. doi:10.1164/ajrccm/138.2.472

33. Skjodt NM, Atkar R, Easton PA. Screening for hypothyroidism in sleep apnea. Am J Respir Crit Care Med. 1999;160(2):732-735. doi:10.1164/ajrccm.160.2.9802051
34. American Academy of Sleep Medicine. FDA approves Remede¯ implantable device to treat central sleep apnea. Accessed February 3, 2023. https://aasm.org/fda-approves-remede-implantable-device-treat-central-sleep-apnea
35. Wang D, Teichtahl H, Drummer O, et al. Central sleep apnea in stable methadone maintenance treatment patients. Chest. 2005;128(3):1348-1356. doi:10.1378/chest.128.3.1348
36. Sharkey KM, Kurth ME, Anderson BJ, Corso RP, Millman RP, Stein MD. Obstructive sleep apnea is more common than central sleep apnea in methadone maintenance patients with subjective sleep complaints. Drug Alcohol Depend. 2010;108(1-2):77-83. Epub 2010 Jan 15. doi:10.1016/j.drugalcdep.2009.11.019
37. Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg. 2015;120:1273-1285. doi:10.1213/ANE.0000000000000672
38. Wang, D, Yee, BJ, Gunstein RR, Chung F. Chronic opioid use and central sleep apnea, where are we now and where to go? A state of the art review. Anesth Analg. 2021;132(5):1244-1253. doi:10.1213/ANE.0000000000005378
39. Schütz SG, Lisabeth LD, Hsu CW, Kim S, Chervin RD, Brown DL. Central sleep apnea is uncommon after stroke. Sleep Med. 2021;77:304-306. Epub 2020 Aug 28. doi:10.1016/j.sleep.2020.08.025
40. Seiler A, Camilo M, Korostovtseva L, et al. Prevalence of sleep-disordered breathing after stroke and TIA: a meta-analysis. Neurology. 2019;92(7):e648-e654. Epub 2019 Jan 11. doi:10.1212/WNL.0000000000006904
As the prevalence of obstructive sleep apnea (OSA) has steadily increased in the United States, so has the awareness of central sleep apnea (CSA). The hallmark of CSA is transient cessation of airflow during sleep due to a lack of respiratory effort triggered by the brain. This is in contrast to OSA, in which there is absence of airflow despite continued ventilatory effort due to physical airflow obstruction. The gold standard for the diagnosis and optimal treatment assessment of CSA is inlaboratory polysomnography (PSG) with esophageal manometry, but in practice, respiratory effort is generally estimated through oronasal flow and respiratory inductance plethysmography bands placed on the chest and abdomen during PSG.
Background
The literature has demonstrated a higher prevalence of moderate-to-severe OSA in the general population compared with that of CSA. While OSA is associated with worse clinical outcomes, more evidence is needed on the long-term clinical impact and optimal treatment strategies for CSA.1 CSA is overrepresented among certain clinical populations. CSA is not frequently diagnosed in the active-duty population, but is increasing in the veteran population, especially in those with heart failure (HF), stroke, neuromuscular disorders, and opioid use. It is associated with increased admissions related to comorbid cardiovascular disorders and to an increased risk of death.2-4 The clinical concerns with CSA parallel those of OSA. The absence of respiration (apneas and hypopneas due to lack of effort) results in sympathetic surge, compromise of oxygenation and ventilation, sleep fragmentation, and elevation in blood pressure. Symptoms such as excessive daytime sleepiness, morning headaches, witnessed apneas, and nocturnal arrhythmias are shared between the 2 disorders.
Ventilatory instability is the most widely accepted mechanism leading to CSA in patients. Loop gain is the concept used to explain ventilatory control. This feedback loop is influenced by controller gain (primarily represented by central and peripheral chemoreceptors causing changes in ventilation due to PaCO2 [partial pressure of CO2 in arterial blood] fluctuations), plant gain (includes lungs and respiratory muscles and their ability to eliminate CO2), and circulation time (feedback between controller and plant).5
High loop gain and narrow CO2 reserve contribute to ventilatory instability in CSA.6 Those with high loop gain have increased sensitivity to changes in CO2. These patients tend to overbreathe in response to smaller increases in PaCO2 compared with those with low loop gain. Once the PaCO2 falls below an individual’s apneic threshold (AT), an apnea will occur.7 The brainstem then pauses ventilation to allow the PaCO2 to rise back above the AT. CSAs also can occur in healthy individuals as they transition from wakefulness into non–rapid eye movement (REM) sleep in a phenomenon called sleep state oscillation, with a mechanism that is similar to hyperventilation-induced CSAs described earlier.
Primary CSA has been defined in the International Classification of Sleep Disorders 3rd edition (ICSD-3) with the following criteria: (1) diagnostic PSG with ≥ 5 events per hour of CSAs and/or central hypopneas per hour of sleep; the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; and there is no evidence of Cheyne-Stokes breathing (CSB); (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) there is no evidence of nocturnal hypoventilation; and (4) the disorder is not better explained by another medical use, substance use disorder (SUD), or other current sleep, medical, or neurologic disorder.8
A systematic clinical approach should be used to identify and treat CSA (Figure).6,7
The purpose of this review is to familiarize the primary care community with CSA to aid in the identification and management of this breathing disturbance.
Nonhypercapnic CSA
Heart Failure–Induced CSA
The leading medical diagnosis causing CSA is congestive HF (CHF), and there is a correlation between HF severity and presence of CSA. In patients with stable CHF with HF reduced ejection fraction (HFrEF), CSA is highly prevalent, occurring in 25% to 40% of patients.9 In contrast to other subtypes of CSA where literature regarding prognosis is lacking, the literature is clear that patients with HFrEF with CSA have a worse prognosis, with increased risk of death independent of the severity of HF. This may be the result of CSA promoting malignant ventricular arrhythmias. The prevalence of CSA in HF with preserved ejection fraction (HFpEF) is about 18% to 30%.10,11
A significant reduction in cardiac output results in circulatory delay between the lungs and chemoreceptors that produces CSB periodic breathing, which is characteristic of the most recognized form of CSA. Per the ICSD-3, CSA with CSB requires the following 4 findings: (1) PSG reveals ≥ 5 CSAs and/or central hypopneas per hour of sleep; there are at least 3 consecutive CSAs and/or central hypopneas separated by crescendo-decrescendo breathing with a cycle length of at least 40 seconds (ie, CSB pattern), and the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) the breathing pattern is associated with atrial fibrillation/flutter, CHF, or a neurologic disorder; and (4) the disorder is not better explained by another current sleep disorder, medication use (eg, opioids), or SUD.8
Treatment of HF-induced CSA begins with guideline-based medical management with the goal of reducing pulmonary capillary wedge pressure or increasing left ventricular ejection fraction through means that may include cardiac resynchronization therapy or left ventricular assist devices, when clinically indicated. If medical optimization is not sufficient, the next step is continuous positive airway pressure (CPAP or PAP), followed by adaptive servo-ventilation (ASV) if the apnea-hypopnea index (AHI) remains > 15 events per hour and is clinically indicated.
ASV is a second-line PAP therapy modality that uses proprietary algorithms to provide variable amounts of pressure that alternate between expiratory and inspiratory phases of the respiratory cycle in combination with physician-set or automatic backup respiratory rate designed to stabilize ventilation in patients with CSA and CSB. The inability to adjust tidal volume, potentially resulting in insufficient tidal volumes or ventilation, results in the contraindication for its use in patients with CSA with comorbid conditions that may result in hypercapnic respiratory failure. These conditions include chronic hypoventilation in obesity hypoventilation syndrome (OHS), moderate-to-severe chronic obstructive pulmonary disease, chronic elevation of PaCO2 on arterial blood gas > 45 mm Hg, and restrictive thoracic or neuromuscular disease.12
Although ASV is more effective in normalizing AHI in patients with HF and CSA than is CPAP therapy, the clinical indications for ASV in the setting of HFrEF changed drastically with the publication of the landmark SERVE-HF trial, which investigated the effects of adding ASV to guideline-based medical management on survival and cardiovascular outcomes in patients with HFrEF and predominant CSA.13 The study did not show a difference between the ASV and control groups for the primary endpoint: a composite of time to first event of death from any cause, lifesaving cardiovascular intervention (transplantation, implantation of a long-term ventricular assist device, resuscitation after sudden cardiac arrest, or appropriate lifesaving shock), or unplanned hospitalization for worsening HF. However, the study showed a statistically and clinically significant increased risk of all-cause and cardiovascular mortality in the ASV group compared with the control group.13 A possible explanation for the increased all-cause and cardiovascular mortality is that CSA potentially serves a protective mechanism that when eliminated results in deleterious clinical outcomes. This resulted in significant changes in the treatment algorithm for HF-induced CSA with left ventricular ejection fraction of at least 45% becoming the cutoff for therapeutic decisions.
Treatment-Emergent CSA
Treatment-emergent CSA (TECSA, also known as complex sleep apnea) has been defined by the ICSD-3 by the following criteria: (1) diagnostic PSG with ≥ 5 events per hour of predominantly obstructive events; (2) resolution of obstructive events with PAP without a backup rate and CSA index (CAI) ≥ 5 per hour with central events ≥ 50% of the AHI; and (3) CSA not better explained by another disorder.8 Patients with TECSA can be further classified into those who have transient events that resolve within weeks to months, those with persistent events, and those with delayed events that may develop weeks to months after initiating PAP therapy.14
PAP treatment can decrease the PaCO2 below the AT due to removal of flow limitation in previously obstructed upper airways, resulting in TECSA.15,16 PAP therapy has not been the only treatment where new CSA has been identified on initiation. A 2021 systematic review identified patients who developed new-onset CSA with mandibular advancement device (MAD), hypoglossal nerve stimulator, tongue protrusion device, and nasal expiratory PAP device use, as well as after tracheostomy, maxillofacial surgery, and other surgeries, such as nasal and uvulopalatopharyngoplasty.17
The prevalence of TECSA has been noted to range between 0.6% and 20.3%, but Nigam and colleagues estimated a prevalence of 8.4% in their systematic review.11,14 The variability in prevalence between studies could be due to differences in study design (retrospective vs prospective vs cross-sectional), diagnostic and inclusion criteria, patient population, and type of study used (full-night vs split-night vs both).18,19 Risk factors for TECSA include male sex; older age; lower body mass index; higher baseline AHI, CAI, and arousal index; chronic medical issues such as CHF and coronary artery disease; opioid use; higher CPAP settings; excessive mask leak; and bilevel PAP (BiPAP) use.20 Identifying these risk factors is important, as patients with TECSA are at higher risk of discontinuing therapy and of developing PAP intolerance.15,20
Most patients with TECSA can continue CPAP therapy with resolution of events over weeks to months, but treatment of comorbid conditions should be optimized as they could be contributing factors. Zeineddine and colleagues recommend continuation of CPAP for 3 months if the patient has minor or no symptoms.19 A 2018 systematic review noted that 14.3% to 46.2% of TECSA patients will have persistent TECSA and some will develop TECSA after at least 1 month of PAP therapy.14 For these patients and those with severe symptoms in spite of therapy, a switch to BiPAP spontaneous/timed (BiPAP-S/T) or ASV should be considered, if not contraindicated based on comorbidities.21 Medications such as acetazolamide, oxygen therapy, and CO2 supplementation have also been discussed as alternative treatments, but these options should not be first-line therapies and should be used on a case-by-case basis as adjuncts to PAP therapy.16,17
Altitude-Induced CSA
Also known as CSA due to high-altitude periodic breathing (CSA-HAPB), this form of CSA occurs in nearly all lowlanders at altitudes above 3000 m, with severity increasing with altitude.15 The exact altitude at which it occurs varies based on an individual’s physiology. CSA-HAPB occurs in response to the low barometric pressure at altitude, combined with stable fraction of oxygen, resulting in decreased inspired partial pressure of oxygen and hypoxia. The normal physiologic response to hypoxia is increased ventilation, which can cause hypocapnia, suppressing respiratory drive and resulting in CSAs. The instability of the respiratory response results in cyclical CSAs followed by hyperventilation. This periodic breathing then causes arousals from sleep, driving further sleep fragmentation and exacerbation of baseline desaturation and instability in the cyclical respiratory response. There is individual variability in hypoxic chemoresponsiveness (loop gain). Compensatory mechanisms are most robust when an individual routinely dwells at high altitude, resulting in acclimatization, rather than traveling there for a brief stay. Genetics and cardiac output also contribute to the effectiveness of compensation to altitude.
CSA-HAPB is defined by the following ICSD-3 criteria: (1) Recent ascent to a high altitude (typically ≥ 2500 m, although some individuals may exhibit the disorder at altitudes as low as 1500 m); (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) symptoms are clinically attributable to HAPB, or PSG, if performed, reveals recurrent CSAs or hypopneas primarily during non-REM sleep at a frequency of ≥ 5 events per hour; (4) the disorder is not better explained by another current sleep disorder, medical or neurological disorder, medication use (eg, narcotics), or SUD.8
Treatment options to improve nocturnal oxygen saturation and reduce or eliminate CSA-HAPB in nonacclimatized individuals include oxygen-enriched air, acetazolamide, or combination treatment with acetazolamide and automatic PAP (APAP).22 A meta-analysis looking at the effectiveness of acetazolamide in 8 different randomized controlled trials demonstrated that a dose of 250 mg per day was effective in improving sleep apnea at altitude as measured by a decrease in AHI, decrease in percentage of periodic breathing, and increasing oxygenation during sleep.15 The question of superiority of combined acetazolamide with APAP to placebo with APAP in treatment of high-altitude OSA was addressed in a randomized double-blind, placebo-controlled trial. The results showed that combined APAP (5-15 cm of water pressure) and acetazolamide (250 mg morning, 500 mg evening) decreased the AHI to normal range, whereas central events persisted in the APAP and placebo group.23 In addition, Latshang and colleagues have demonstrated that ASV may not be as efficacious for controlling CSA-HAPB in nonacclimatized individuals compared with oxygen therapy and suggested that further research is warranted examining if ASV device algorithm adjustment improves efficacy of this therapeutic option.24
Comorbidity-Induced CSA
Several medical conditions may be associated with CSA, including chronic kidney disease (CKD), pulmonary hypertension, acromegaly, and hypothyroidism. The common pathophysiologic link is that these disorders may result in alteration of ventilatory responses to CO2, ultimately resulting in CSA.
As many as 10% of patients with CKD may experience CSA.25,26 The complications encountered in CKD include fluid overload with pulmonary edema, chronic metabolic acidosis, and anemia. These can provoke hyperventilation in addition to poor sleep quality, triggering arousals that further drive CSA in these patients. Additional risk factors for CSA in this population include atrial fibrillation and cardiac dysfunction. Clinical interventions that have demonstrated reduction in CSA include hemodialysis at night vs daytime and using bicarbonate buffer vs acetate for hemodialysis 22-24,26-29
Hypersecretion of growth hormone in acromegaly also results in hyperventilation contributing to CSA. While medical and surgical management of acromegaly results in a reduction in OSA, there is limited evidence on the outcome of the CSA after these interventions.
Hypothyroidism and CSA both present with similar symptoms of fatigue, daytime sleepiness, depression, and headaches. Studies suggest that respiratory muscle fatigue and decreased ventilatory response to hypercapnia and hypoxia contribute to apnea in this population. In one study, 27% of hypothyroid patients had a blunted response to hypercapnia, and 34% suffered from a blunted response to hypoxia. The same study showed universal reversal of the impairment following thyroid replacement therapy and return to euthyroid state.30 Similarly, multiple studies have shown reversal of respiratory muscle fatigue after initiation of thyroid replacement.30-32 Assessing thyroid function is an appropriate initial step during any sleep-disordered breathing workup, as it is a reversible cause of apnea. Up to 2.4% of patients presenting for PSG (and diagnosed with OSA) are found to have undiagnosed hypothyroidism.32,33 In a military population, treatment of a secondary cause of CSA, such as hypothyroidism, could remove some administrative burden as well as improve service members’ quality of life.
If CSA persists despite previous treatment strategies, then clinicians should focus on the optimization of treatment for comorbid conditions. If that does not resolve CSA, CPAP should be used when AHI remains above 15 events per hour or ASV can be used.
Idiopathic CSA
There are limited data on the pathophysiology and prevalence of idiopathic CSA. In most cases it is hypocapnic CSA, which occurs after an arousal from sleep causing hyperventilation that causes hypocapnia below the apnea threshold similar to CSA-HAPB. Therapeutic options based on addressing underlying pathophysiology include increasing CO2 by inhalation or addition of dead space. Additional therapeutic options to reduce the arousals and CSAs include hypnotics, such as zolpidem and acetazolamide, but these should be administered only with close clinical monitoring. If symptoms continue, CPAP or ASV may be trialed; however, limited clinical evidence of efficacy exists.15
For patients with moderate-to-severe CSA, an additional treatment option includes an implantable device (eg, Zoll remede¯), which stimulates the phrenic nerve to move the diaphragm and restore normal breathing. This device is not indicated for those with OSA. Based on data submitted to the US Food and Drug Administration, AHI is reduced by ≥ 50% in 51% of patients with the implanted device and by 11% in patients without the device. Five-year follow-up data show sustained improvements.34
Hypercapnic CSA
CSA due to a medication or substance requires the following criteria: (1) the patient is taking an opioid or other respiratory depressant; (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia (difficulty initiating or maintaining sleep, frequent awakenings, or nonrestorative sleep); (3) PSG reveals ≥ 5 CSAs and/or central hypopneas per hour of sleep; the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; and there is no evidence of CSB; and (4) the disorder is not better explained by another current sleep disorder.8
Drugs that affect the respiratory centers, such as opiates and opioids, γ aminobutyric acid (GABA) type A and B receptor agonists, and P2Y(12) receptor antagonists such as ticagrelor, may result in alterations in ventilatory drive in the central nervous system respiratory centers, resulting in CSA.
Opioids are prescribed either for chronic pain or to treat opiate addiction with methadone, resulting in about one-third of chronic opioid users having some form of CSA.35 CSA may be seen after opioids have been used for at least 2 months. A dose-dependent effect exists with high doses of opioids, typically resulting in hypoventilation, hypercapnia, and hypoxemia with ataxic or erratic breathing and a periodic breathing pattern similar to those described in CSA-HAPB or idiopathic CSA. About 14% to 60% of methadone patients also demonstrate CSA or ataxic breathing.35,36
Benzodiazepines (GABA-A receptor agonists) and baclofen (a GABA-B receptor agonist) depress central ventilatory drive, blunt the response to hypoxia and hypercapnia, leading to CSAs, and increase risk for OSA by increasing upper airway obstruction through reduction in tone. Use of these medications with antidepressants or opioids further exacerbates this response.
Unlike the other medications previously described, ticagrelor, a first-line dual antiplatelet therapy medication indicated for acute coronary syndrome treatment, actually increases the activity of the respiratory centers but may result in CSA.
First-line treatment, if possible, is reduction in medication dose or complete withdrawal. Additional treatment options include PAP therapies: CPAP, BiPAP, ASV, and oxygen therapy with or without PAP.37,38 The literature has demonstrated that for the treatment of opioid-associated CSA, ASV (in cases of normocapnia) and noninvasive ventilation (NIV)/BiPAP (in cases with hypercapnia or REM sleep hypoventilation) are superior treatment options when compared with conventional CPAP for elimination of respiratory events. CPAP with oxygen therapy and BiPAP with oxygen therapy are more effective than CPAP alone in reducing respiratory events. However, concerns remain that as with CSA in HF, CSA in chronic opioid users may serve as a physiologic protective mechanism to prevent further clinical injury from opioids. Similarly, as in the use of ASV in the SERVE-HF trial, focusing on elimination of respiratory events may prove detrimental. More studies are needed to determine whether reducing the number of CSA events in chronic opioid users is clinically beneficial when other health outcomes, such as cardiovascular, neurocognitive, hospital/intensive care unit admissions, and mortality risks are examined.
Neuromuscular-Induced CSA
CSA also is highly prevalent in neuromuscular conditions, such as amyotrophic lateral sclerosis, Duchenne muscular dystrophy, myotonic dystrophy, advanced multiple sclerosis, and acid maltase deficiency. There is reduced respiratory muscle strength and tone in these disorders, resulting in alveolar hypoventilation with hypercapnia. Given the hypercapnia, NIV/BiPAP is the first-line treatment to improve survival, gas exchange, symptom burden, and quality of life.
Stroke-Induced CSA
Extensive cerebrovascular events commonly precipitate sleep-related breathing disorders. The incidence increases in the acute phase of stroke and decreases 3 to 6 months poststroke; however, incidence also depends on the severity of the stroke.7,39,40 Stroke also has been shown to be a predictor of CSA (odds ratio, 1.65; 95% CI, 1.50-1.82; P < .001) in a retrospective analysis of a large cohort of US veterans.2 The location of the lesion often determines whether normocapnic or hypercapnic CSA will predominate, based on ventilatory instability resulting in normocapnia or reduced ventilatory drive resulting in hypercapnic CSA. PSG results and blood gases direct the treatment options. CSA with normocapnia is treated with ASV, and patients with hypercapnia/REM sleep hypoventilation are treated with NIV/BiPAP.
Conclusions
While much has been learned about CSA in recent decades, more evidence needs to be gathered to determine optimal treatment strategies and the impact on patient prognosis. The identification of CSA can lead to the diagnosis of previously unrecognized medical conditions. With proper diagnosis and treatment, we can optimize clinical management and improve patients’ prognosis and quality of life.
Acknowledgments
The authors thank the librarians of the Franzello Aeromedical Library in particular Sara Craycraft, Catherine Stahl, Kristen Young and Elizabeth Irvine for their support of this publication.
As the prevalence of obstructive sleep apnea (OSA) has steadily increased in the United States, so has the awareness of central sleep apnea (CSA). The hallmark of CSA is transient cessation of airflow during sleep due to a lack of respiratory effort triggered by the brain. This is in contrast to OSA, in which there is absence of airflow despite continued ventilatory effort due to physical airflow obstruction. The gold standard for the diagnosis and optimal treatment assessment of CSA is inlaboratory polysomnography (PSG) with esophageal manometry, but in practice, respiratory effort is generally estimated through oronasal flow and respiratory inductance plethysmography bands placed on the chest and abdomen during PSG.
Background
The literature has demonstrated a higher prevalence of moderate-to-severe OSA in the general population compared with that of CSA. While OSA is associated with worse clinical outcomes, more evidence is needed on the long-term clinical impact and optimal treatment strategies for CSA.1 CSA is overrepresented among certain clinical populations. CSA is not frequently diagnosed in the active-duty population, but is increasing in the veteran population, especially in those with heart failure (HF), stroke, neuromuscular disorders, and opioid use. It is associated with increased admissions related to comorbid cardiovascular disorders and to an increased risk of death.2-4 The clinical concerns with CSA parallel those of OSA. The absence of respiration (apneas and hypopneas due to lack of effort) results in sympathetic surge, compromise of oxygenation and ventilation, sleep fragmentation, and elevation in blood pressure. Symptoms such as excessive daytime sleepiness, morning headaches, witnessed apneas, and nocturnal arrhythmias are shared between the 2 disorders.
Ventilatory instability is the most widely accepted mechanism leading to CSA in patients. Loop gain is the concept used to explain ventilatory control. This feedback loop is influenced by controller gain (primarily represented by central and peripheral chemoreceptors causing changes in ventilation due to PaCO2 [partial pressure of CO2 in arterial blood] fluctuations), plant gain (includes lungs and respiratory muscles and their ability to eliminate CO2), and circulation time (feedback between controller and plant).5
High loop gain and narrow CO2 reserve contribute to ventilatory instability in CSA.6 Those with high loop gain have increased sensitivity to changes in CO2. These patients tend to overbreathe in response to smaller increases in PaCO2 compared with those with low loop gain. Once the PaCO2 falls below an individual’s apneic threshold (AT), an apnea will occur.7 The brainstem then pauses ventilation to allow the PaCO2 to rise back above the AT. CSAs also can occur in healthy individuals as they transition from wakefulness into non–rapid eye movement (REM) sleep in a phenomenon called sleep state oscillation, with a mechanism that is similar to hyperventilation-induced CSAs described earlier.
Primary CSA has been defined in the International Classification of Sleep Disorders 3rd edition (ICSD-3) with the following criteria: (1) diagnostic PSG with ≥ 5 events per hour of CSAs and/or central hypopneas per hour of sleep; the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; and there is no evidence of Cheyne-Stokes breathing (CSB); (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) there is no evidence of nocturnal hypoventilation; and (4) the disorder is not better explained by another medical use, substance use disorder (SUD), or other current sleep, medical, or neurologic disorder.8
A systematic clinical approach should be used to identify and treat CSA (Figure).6,7
The purpose of this review is to familiarize the primary care community with CSA to aid in the identification and management of this breathing disturbance.
Nonhypercapnic CSA
Heart Failure–Induced CSA
The leading medical diagnosis causing CSA is congestive HF (CHF), and there is a correlation between HF severity and presence of CSA. In patients with stable CHF with HF reduced ejection fraction (HFrEF), CSA is highly prevalent, occurring in 25% to 40% of patients.9 In contrast to other subtypes of CSA where literature regarding prognosis is lacking, the literature is clear that patients with HFrEF with CSA have a worse prognosis, with increased risk of death independent of the severity of HF. This may be the result of CSA promoting malignant ventricular arrhythmias. The prevalence of CSA in HF with preserved ejection fraction (HFpEF) is about 18% to 30%.10,11
A significant reduction in cardiac output results in circulatory delay between the lungs and chemoreceptors that produces CSB periodic breathing, which is characteristic of the most recognized form of CSA. Per the ICSD-3, CSA with CSB requires the following 4 findings: (1) PSG reveals ≥ 5 CSAs and/or central hypopneas per hour of sleep; there are at least 3 consecutive CSAs and/or central hypopneas separated by crescendo-decrescendo breathing with a cycle length of at least 40 seconds (ie, CSB pattern), and the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) the breathing pattern is associated with atrial fibrillation/flutter, CHF, or a neurologic disorder; and (4) the disorder is not better explained by another current sleep disorder, medication use (eg, opioids), or SUD.8
Treatment of HF-induced CSA begins with guideline-based medical management with the goal of reducing pulmonary capillary wedge pressure or increasing left ventricular ejection fraction through means that may include cardiac resynchronization therapy or left ventricular assist devices, when clinically indicated. If medical optimization is not sufficient, the next step is continuous positive airway pressure (CPAP or PAP), followed by adaptive servo-ventilation (ASV) if the apnea-hypopnea index (AHI) remains > 15 events per hour and is clinically indicated.
ASV is a second-line PAP therapy modality that uses proprietary algorithms to provide variable amounts of pressure that alternate between expiratory and inspiratory phases of the respiratory cycle in combination with physician-set or automatic backup respiratory rate designed to stabilize ventilation in patients with CSA and CSB. The inability to adjust tidal volume, potentially resulting in insufficient tidal volumes or ventilation, results in the contraindication for its use in patients with CSA with comorbid conditions that may result in hypercapnic respiratory failure. These conditions include chronic hypoventilation in obesity hypoventilation syndrome (OHS), moderate-to-severe chronic obstructive pulmonary disease, chronic elevation of PaCO2 on arterial blood gas > 45 mm Hg, and restrictive thoracic or neuromuscular disease.12
Although ASV is more effective in normalizing AHI in patients with HF and CSA than is CPAP therapy, the clinical indications for ASV in the setting of HFrEF changed drastically with the publication of the landmark SERVE-HF trial, which investigated the effects of adding ASV to guideline-based medical management on survival and cardiovascular outcomes in patients with HFrEF and predominant CSA.13 The study did not show a difference between the ASV and control groups for the primary endpoint: a composite of time to first event of death from any cause, lifesaving cardiovascular intervention (transplantation, implantation of a long-term ventricular assist device, resuscitation after sudden cardiac arrest, or appropriate lifesaving shock), or unplanned hospitalization for worsening HF. However, the study showed a statistically and clinically significant increased risk of all-cause and cardiovascular mortality in the ASV group compared with the control group.13 A possible explanation for the increased all-cause and cardiovascular mortality is that CSA potentially serves a protective mechanism that when eliminated results in deleterious clinical outcomes. This resulted in significant changes in the treatment algorithm for HF-induced CSA with left ventricular ejection fraction of at least 45% becoming the cutoff for therapeutic decisions.
Treatment-Emergent CSA
Treatment-emergent CSA (TECSA, also known as complex sleep apnea) has been defined by the ICSD-3 by the following criteria: (1) diagnostic PSG with ≥ 5 events per hour of predominantly obstructive events; (2) resolution of obstructive events with PAP without a backup rate and CSA index (CAI) ≥ 5 per hour with central events ≥ 50% of the AHI; and (3) CSA not better explained by another disorder.8 Patients with TECSA can be further classified into those who have transient events that resolve within weeks to months, those with persistent events, and those with delayed events that may develop weeks to months after initiating PAP therapy.14
PAP treatment can decrease the PaCO2 below the AT due to removal of flow limitation in previously obstructed upper airways, resulting in TECSA.15,16 PAP therapy has not been the only treatment where new CSA has been identified on initiation. A 2021 systematic review identified patients who developed new-onset CSA with mandibular advancement device (MAD), hypoglossal nerve stimulator, tongue protrusion device, and nasal expiratory PAP device use, as well as after tracheostomy, maxillofacial surgery, and other surgeries, such as nasal and uvulopalatopharyngoplasty.17
The prevalence of TECSA has been noted to range between 0.6% and 20.3%, but Nigam and colleagues estimated a prevalence of 8.4% in their systematic review.11,14 The variability in prevalence between studies could be due to differences in study design (retrospective vs prospective vs cross-sectional), diagnostic and inclusion criteria, patient population, and type of study used (full-night vs split-night vs both).18,19 Risk factors for TECSA include male sex; older age; lower body mass index; higher baseline AHI, CAI, and arousal index; chronic medical issues such as CHF and coronary artery disease; opioid use; higher CPAP settings; excessive mask leak; and bilevel PAP (BiPAP) use.20 Identifying these risk factors is important, as patients with TECSA are at higher risk of discontinuing therapy and of developing PAP intolerance.15,20
Most patients with TECSA can continue CPAP therapy with resolution of events over weeks to months, but treatment of comorbid conditions should be optimized as they could be contributing factors. Zeineddine and colleagues recommend continuation of CPAP for 3 months if the patient has minor or no symptoms.19 A 2018 systematic review noted that 14.3% to 46.2% of TECSA patients will have persistent TECSA and some will develop TECSA after at least 1 month of PAP therapy.14 For these patients and those with severe symptoms in spite of therapy, a switch to BiPAP spontaneous/timed (BiPAP-S/T) or ASV should be considered, if not contraindicated based on comorbidities.21 Medications such as acetazolamide, oxygen therapy, and CO2 supplementation have also been discussed as alternative treatments, but these options should not be first-line therapies and should be used on a case-by-case basis as adjuncts to PAP therapy.16,17
Altitude-Induced CSA
Also known as CSA due to high-altitude periodic breathing (CSA-HAPB), this form of CSA occurs in nearly all lowlanders at altitudes above 3000 m, with severity increasing with altitude.15 The exact altitude at which it occurs varies based on an individual’s physiology. CSA-HAPB occurs in response to the low barometric pressure at altitude, combined with stable fraction of oxygen, resulting in decreased inspired partial pressure of oxygen and hypoxia. The normal physiologic response to hypoxia is increased ventilation, which can cause hypocapnia, suppressing respiratory drive and resulting in CSAs. The instability of the respiratory response results in cyclical CSAs followed by hyperventilation. This periodic breathing then causes arousals from sleep, driving further sleep fragmentation and exacerbation of baseline desaturation and instability in the cyclical respiratory response. There is individual variability in hypoxic chemoresponsiveness (loop gain). Compensatory mechanisms are most robust when an individual routinely dwells at high altitude, resulting in acclimatization, rather than traveling there for a brief stay. Genetics and cardiac output also contribute to the effectiveness of compensation to altitude.
CSA-HAPB is defined by the following ICSD-3 criteria: (1) Recent ascent to a high altitude (typically ≥ 2500 m, although some individuals may exhibit the disorder at altitudes as low as 1500 m); (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia; (3) symptoms are clinically attributable to HAPB, or PSG, if performed, reveals recurrent CSAs or hypopneas primarily during non-REM sleep at a frequency of ≥ 5 events per hour; (4) the disorder is not better explained by another current sleep disorder, medical or neurological disorder, medication use (eg, narcotics), or SUD.8
Treatment options to improve nocturnal oxygen saturation and reduce or eliminate CSA-HAPB in nonacclimatized individuals include oxygen-enriched air, acetazolamide, or combination treatment with acetazolamide and automatic PAP (APAP).22 A meta-analysis looking at the effectiveness of acetazolamide in 8 different randomized controlled trials demonstrated that a dose of 250 mg per day was effective in improving sleep apnea at altitude as measured by a decrease in AHI, decrease in percentage of periodic breathing, and increasing oxygenation during sleep.15 The question of superiority of combined acetazolamide with APAP to placebo with APAP in treatment of high-altitude OSA was addressed in a randomized double-blind, placebo-controlled trial. The results showed that combined APAP (5-15 cm of water pressure) and acetazolamide (250 mg morning, 500 mg evening) decreased the AHI to normal range, whereas central events persisted in the APAP and placebo group.23 In addition, Latshang and colleagues have demonstrated that ASV may not be as efficacious for controlling CSA-HAPB in nonacclimatized individuals compared with oxygen therapy and suggested that further research is warranted examining if ASV device algorithm adjustment improves efficacy of this therapeutic option.24
Comorbidity-Induced CSA
Several medical conditions may be associated with CSA, including chronic kidney disease (CKD), pulmonary hypertension, acromegaly, and hypothyroidism. The common pathophysiologic link is that these disorders may result in alteration of ventilatory responses to CO2, ultimately resulting in CSA.
As many as 10% of patients with CKD may experience CSA.25,26 The complications encountered in CKD include fluid overload with pulmonary edema, chronic metabolic acidosis, and anemia. These can provoke hyperventilation in addition to poor sleep quality, triggering arousals that further drive CSA in these patients. Additional risk factors for CSA in this population include atrial fibrillation and cardiac dysfunction. Clinical interventions that have demonstrated reduction in CSA include hemodialysis at night vs daytime and using bicarbonate buffer vs acetate for hemodialysis 22-24,26-29
Hypersecretion of growth hormone in acromegaly also results in hyperventilation contributing to CSA. While medical and surgical management of acromegaly results in a reduction in OSA, there is limited evidence on the outcome of the CSA after these interventions.
Hypothyroidism and CSA both present with similar symptoms of fatigue, daytime sleepiness, depression, and headaches. Studies suggest that respiratory muscle fatigue and decreased ventilatory response to hypercapnia and hypoxia contribute to apnea in this population. In one study, 27% of hypothyroid patients had a blunted response to hypercapnia, and 34% suffered from a blunted response to hypoxia. The same study showed universal reversal of the impairment following thyroid replacement therapy and return to euthyroid state.30 Similarly, multiple studies have shown reversal of respiratory muscle fatigue after initiation of thyroid replacement.30-32 Assessing thyroid function is an appropriate initial step during any sleep-disordered breathing workup, as it is a reversible cause of apnea. Up to 2.4% of patients presenting for PSG (and diagnosed with OSA) are found to have undiagnosed hypothyroidism.32,33 In a military population, treatment of a secondary cause of CSA, such as hypothyroidism, could remove some administrative burden as well as improve service members’ quality of life.
If CSA persists despite previous treatment strategies, then clinicians should focus on the optimization of treatment for comorbid conditions. If that does not resolve CSA, CPAP should be used when AHI remains above 15 events per hour or ASV can be used.
Idiopathic CSA
There are limited data on the pathophysiology and prevalence of idiopathic CSA. In most cases it is hypocapnic CSA, which occurs after an arousal from sleep causing hyperventilation that causes hypocapnia below the apnea threshold similar to CSA-HAPB. Therapeutic options based on addressing underlying pathophysiology include increasing CO2 by inhalation or addition of dead space. Additional therapeutic options to reduce the arousals and CSAs include hypnotics, such as zolpidem and acetazolamide, but these should be administered only with close clinical monitoring. If symptoms continue, CPAP or ASV may be trialed; however, limited clinical evidence of efficacy exists.15
For patients with moderate-to-severe CSA, an additional treatment option includes an implantable device (eg, Zoll remede¯), which stimulates the phrenic nerve to move the diaphragm and restore normal breathing. This device is not indicated for those with OSA. Based on data submitted to the US Food and Drug Administration, AHI is reduced by ≥ 50% in 51% of patients with the implanted device and by 11% in patients without the device. Five-year follow-up data show sustained improvements.34
Hypercapnic CSA
CSA due to a medication or substance requires the following criteria: (1) the patient is taking an opioid or other respiratory depressant; (2) the patient reports sleepiness, awakening with shortness of breath, snoring, witnessed apneas, or insomnia (difficulty initiating or maintaining sleep, frequent awakenings, or nonrestorative sleep); (3) PSG reveals ≥ 5 CSAs and/or central hypopneas per hour of sleep; the number of CSAs and/or central hypopneas is > 50% of the total number of apneas and hypopneas; and there is no evidence of CSB; and (4) the disorder is not better explained by another current sleep disorder.8
Drugs that affect the respiratory centers, such as opiates and opioids, γ aminobutyric acid (GABA) type A and B receptor agonists, and P2Y(12) receptor antagonists such as ticagrelor, may result in alterations in ventilatory drive in the central nervous system respiratory centers, resulting in CSA.
Opioids are prescribed either for chronic pain or to treat opiate addiction with methadone, resulting in about one-third of chronic opioid users having some form of CSA.35 CSA may be seen after opioids have been used for at least 2 months. A dose-dependent effect exists with high doses of opioids, typically resulting in hypoventilation, hypercapnia, and hypoxemia with ataxic or erratic breathing and a periodic breathing pattern similar to those described in CSA-HAPB or idiopathic CSA. About 14% to 60% of methadone patients also demonstrate CSA or ataxic breathing.35,36
Benzodiazepines (GABA-A receptor agonists) and baclofen (a GABA-B receptor agonist) depress central ventilatory drive, blunt the response to hypoxia and hypercapnia, leading to CSAs, and increase risk for OSA by increasing upper airway obstruction through reduction in tone. Use of these medications with antidepressants or opioids further exacerbates this response.
Unlike the other medications previously described, ticagrelor, a first-line dual antiplatelet therapy medication indicated for acute coronary syndrome treatment, actually increases the activity of the respiratory centers but may result in CSA.
First-line treatment, if possible, is reduction in medication dose or complete withdrawal. Additional treatment options include PAP therapies: CPAP, BiPAP, ASV, and oxygen therapy with or without PAP.37,38 The literature has demonstrated that for the treatment of opioid-associated CSA, ASV (in cases of normocapnia) and noninvasive ventilation (NIV)/BiPAP (in cases with hypercapnia or REM sleep hypoventilation) are superior treatment options when compared with conventional CPAP for elimination of respiratory events. CPAP with oxygen therapy and BiPAP with oxygen therapy are more effective than CPAP alone in reducing respiratory events. However, concerns remain that as with CSA in HF, CSA in chronic opioid users may serve as a physiologic protective mechanism to prevent further clinical injury from opioids. Similarly, as in the use of ASV in the SERVE-HF trial, focusing on elimination of respiratory events may prove detrimental. More studies are needed to determine whether reducing the number of CSA events in chronic opioid users is clinically beneficial when other health outcomes, such as cardiovascular, neurocognitive, hospital/intensive care unit admissions, and mortality risks are examined.
Neuromuscular-Induced CSA
CSA also is highly prevalent in neuromuscular conditions, such as amyotrophic lateral sclerosis, Duchenne muscular dystrophy, myotonic dystrophy, advanced multiple sclerosis, and acid maltase deficiency. There is reduced respiratory muscle strength and tone in these disorders, resulting in alveolar hypoventilation with hypercapnia. Given the hypercapnia, NIV/BiPAP is the first-line treatment to improve survival, gas exchange, symptom burden, and quality of life.
Stroke-Induced CSA
Extensive cerebrovascular events commonly precipitate sleep-related breathing disorders. The incidence increases in the acute phase of stroke and decreases 3 to 6 months poststroke; however, incidence also depends on the severity of the stroke.7,39,40 Stroke also has been shown to be a predictor of CSA (odds ratio, 1.65; 95% CI, 1.50-1.82; P < .001) in a retrospective analysis of a large cohort of US veterans.2 The location of the lesion often determines whether normocapnic or hypercapnic CSA will predominate, based on ventilatory instability resulting in normocapnia or reduced ventilatory drive resulting in hypercapnic CSA. PSG results and blood gases direct the treatment options. CSA with normocapnia is treated with ASV, and patients with hypercapnia/REM sleep hypoventilation are treated with NIV/BiPAP.
Conclusions
While much has been learned about CSA in recent decades, more evidence needs to be gathered to determine optimal treatment strategies and the impact on patient prognosis. The identification of CSA can lead to the diagnosis of previously unrecognized medical conditions. With proper diagnosis and treatment, we can optimize clinical management and improve patients’ prognosis and quality of life.
Acknowledgments
The authors thank the librarians of the Franzello Aeromedical Library in particular Sara Craycraft, Catherine Stahl, Kristen Young and Elizabeth Irvine for their support of this publication.
1. Heinzer R, Vat S, Marques-Vidal P, et al. Prevalence of sleep-disordered breathing in the general population: the HypnoLaus study. Lancet Respir Med. 2015;3(4):310-318. Epub 2015 Feb 12. doi:10.1016/S2213-2600(15)00043-0
2. Ratz D, Wiitala W, Safwan Badr M, Burns J, Chowdhuri S. Correlates and consequences of central sleep apnea in a national sample of US veterans. Sleep. 2018;41(9):zy058. doi:10.1093/sleep/zsyn058
3. Agrawal R, Sharafkhaneneh A, Gottlief, DJ, Nowakowski S, Razjouyan J. Mortality patterns associated with central sleep apnea among veterans: a large, retrospective, longitudinal report. Ann Am Thorac Soc. 2022;10.1513/AnnalsATS.202207-648OC. doi:10.1513/annalsATS. 202207-648OC
4. Mysliwiec V, McGraw L, Pierce R, Smith, P, Trapp, B, Roth B. Sleep disorders and associated medical comorbidities in active duty military personnel. Sleep. 2013;36(2):167-174. doi:10.5665/sleep.2364
5. Badr MS, Dingell JD, Javaheri S. Central sleep apnea: a brief review. Curr Pulmonol Rep. 2019;8(1):14-21. Epub 2019 Mar 13. doi:10.1007/s13665-019-0221-z
6. Baillieul S, Revol B, Jullian-Desayes I, Joyeux-Faure M, Tamisier R, Pépin JL. Diagnosis and management of central sleep apnea syndrome. Expert Rev Respir Med. 2019;13(6):545-557.1604226. Epub 2019 Apr 24. doi:10.1080/17476348.2019
7. Randerath W, Verbraecken J, Andreas S, et al. Definition, discrimination, diagnosis and treatment of central breathing disturbances during sleep. Eur Respir J. 2017;49(1):1600959. doi:10.1183/13993003.00959-2016
8. American Academy of Sleep Medicine. International Classification of Sleep Disorders. 3rd ed. American Academy of Sleep Medicine; 2014.
9. Lévy P, Pépin J-L, Tamisier R, Neuder Y, Baguet J-P, Javaheri S. Prevalence and impact of central sleep apnea in heart failure. Sleep Med Clinics. 2007;2(4):615-621. doi:10.1016/j.jsmc.2007.08.001
10. Bitter T, Faber L, Hering D, Langer C, Horstkotte D, Oldenburg O. Sleep-disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail. 2009;11(6):602-608. doi:10.1093/eurjhf/hfp057
11. Sekizuka H, Osada N, Miyake F. Sleep disordered breathing in heart failure patients with reduced versus preserved ejection fraction. Heart Lung Circ. 2013;22(2):104-109. Epub 2012 Oct 26. doi:10.1016/j.hlc.2012.08.006
12. Iotti GA, Polito A, Belliato M, et al. Adaptive support ventilation versus conventional ventilation for total ventilatory support in acute respiratory failure. Intensive Care Med. 2010;36(8):1371-1379. Epub 2010 May 26. doi:10.1007/s00134-010-1917-2
13. Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373(12):1095-105. Epub 2015 Sep 1. doi:10.1056/NEJMoa1506459
14. Nigam G, Riaz M, Chang ET, Camacho M. Natural history of treatment-emergent central sleep apnea on positive airway pressure: a systematic review. Ann Thorac Med. 2018;13(2):86-91. doi:10.4103/atm.ATM_321_17
15. Orr JE, Malhotra A, Sands SA. Pathogenesis of central and complex sleep apnoea. Respirology. 2017;22(1):43-52. Epub 2016 Oct 31. doi:10.1111/resp.12927
16. Berger M, Solelhac G, Horvath C, Heinzer R, Brill AK. Treatment-emergent central sleep apnea associated with non-positive airway pressure therapies in obstructive sleep apnea patients: a systematic review. Sleep Med Rev. 2021; 58:101513. Epub 2021 Jun 5. doi:10.1016/j.smrv.2021.101513
17. Zhang J, Wang L, Guo HJ, Wang Y, Cao J, Chen BY. Treatment-emergent central sleep apnea: a unique sleep-disordered breathing. Chin Med J (Engl). 2020;133(22):2721-2730. doi:10.1097/CM9.0000000000001125
18. Nigam G, Pathak C, Riaz M. A systematic review on prevalence and risk factors associated with treatment- emergent central sleep apnea. Ann Thorac Med. 2016;11(3):202-210. doi:10.4103/1817-1737.185761
19. Zeineddine S, Badr MS. Treatment-emergent central apnea: physiologic mechanisms informing clinical practice. Chest. 2021;159(6):2449-2457. Epub 2021 Jan 23. doi:10.1016/j.hest.2021.01.036
20. Liu D, Armitstead J, Benjafield A. Trajectories of emergent central sleep apnea during CPAP therapy. Chest. 2017;152(4):751-760. Epub 2017 Jun 16. doi:10.1016/j.chest.2017.06.010
21. Moro M, Gannon K, Lovell K, Merlino M, Mojica J, Bianchi MT. Clinical predictors of central sleep apnea evoked by positive airway pressure titration. Nat Sci Sleep. 2016;8:259-266. doi:10.2147/NSS.S110032
22. Orr JE, Heinrich EC, Djokic M, et al. Adaptive servoventilation as treatment for central sleep apnea due to high-altitude periodic breathing in nonacclimatized healthy individuals. High Alt Med Biol. 2018;19(2):178-184. Epub 2018 Mar 13. doi:10.1089/ham.2017.0147
23. Liu HM, Chiang IJ, Kuo KN, Liou CM, Chen C. The effect of acetazolamide on sleep apnea at high altitude: a systematic review and meta-analysis. Ther Adv Respir Dis. 2017;11(1):20-29. Epub 2016 Nov 15. doi:10.1177/1753465816677006
24. Latshang TD, Nussbaumer-Ochsner Y, Henn RM, et al. Effect of acetazolamide and autoCPAP therapy on breathing disturbances among patients with obstructive sleep apnea syndrome who travel to altitude: a randomized controlled trial. JAMA. 2012;308(22):2390-8. doi:10.1001/jama.2012.94847
25. Nigam G, Pathak C, Riaz M. A systematic review of central sleep apnea in adult patients with chronic kidney disease. Sleep Breath. 2016;20(3):957-964. Epub 2016 Jan 27. doi:10.1007/s11325-016-1317-0
26. Nigam G, Riaz M. Pathophysiology of central sleep apnea in chronic kidney disease. Saudi J Kidney Dis Transpl. 2016;27(5):1068-1070. doi:10.4103/1319-2442.190907
27. Hanly PJ, Pierratos A. Improvement of sleep apnea in patients with chronic renal failure who undergo nocturnal hemodialysis. N Engl J Med. 2001;344(2):102-107. doi:10.1056/NEJM200101113440204
28. Jean G, Piperno D, François B, Charra B. Sleep apnea incidence in maintenance hemodialysis patients: influence of dialysate buffer. Nephron. 1995;71(2):138-142. doi:10.1159/000188701
29. Pressman MR, Benz RL, Schleifer CR, Peterson DD. Sleep disordered breathing in ESRD: acute beneficial effects of treatment with nasal continuous positive airway pressure. Kidney Int. 1993;43(5):1134-1139. doi:10.1038/ki.1993.159
30. Ladenson PW, Goldenheim PD, Ridgway EC. Prediction and reversal of blunted ventilatory responsiveness in patients with hypothyroidism. Am J Med. 1988;84(5):877-883. doi:10.1016/0002-9343(88)90066-6
31. Siafakas NM, Salesiotou V, Filaditaki V, Tzanakis N, Thalassinos N, Bouros D. Respiratory muscle strength in hypothyroidism. Chest. 1992;102(1):189-194. doi:10.1378/chest.102.1.189
32. Laroche CM, Cairns T, Moxham J, Green M. Hypothyroidism presenting with respiratory muscle weakness. Am Rev Respir Dis. 1988;138(2):472-474. doi:10.1164/ajrccm/138.2.472
33. Skjodt NM, Atkar R, Easton PA. Screening for hypothyroidism in sleep apnea. Am J Respir Crit Care Med. 1999;160(2):732-735. doi:10.1164/ajrccm.160.2.9802051
34. American Academy of Sleep Medicine. FDA approves Remede¯ implantable device to treat central sleep apnea. Accessed February 3, 2023. https://aasm.org/fda-approves-remede-implantable-device-treat-central-sleep-apnea
35. Wang D, Teichtahl H, Drummer O, et al. Central sleep apnea in stable methadone maintenance treatment patients. Chest. 2005;128(3):1348-1356. doi:10.1378/chest.128.3.1348
36. Sharkey KM, Kurth ME, Anderson BJ, Corso RP, Millman RP, Stein MD. Obstructive sleep apnea is more common than central sleep apnea in methadone maintenance patients with subjective sleep complaints. Drug Alcohol Depend. 2010;108(1-2):77-83. Epub 2010 Jan 15. doi:10.1016/j.drugalcdep.2009.11.019
37. Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg. 2015;120:1273-1285. doi:10.1213/ANE.0000000000000672
38. Wang, D, Yee, BJ, Gunstein RR, Chung F. Chronic opioid use and central sleep apnea, where are we now and where to go? A state of the art review. Anesth Analg. 2021;132(5):1244-1253. doi:10.1213/ANE.0000000000005378
39. Schütz SG, Lisabeth LD, Hsu CW, Kim S, Chervin RD, Brown DL. Central sleep apnea is uncommon after stroke. Sleep Med. 2021;77:304-306. Epub 2020 Aug 28. doi:10.1016/j.sleep.2020.08.025
40. Seiler A, Camilo M, Korostovtseva L, et al. Prevalence of sleep-disordered breathing after stroke and TIA: a meta-analysis. Neurology. 2019;92(7):e648-e654. Epub 2019 Jan 11. doi:10.1212/WNL.0000000000006904
1. Heinzer R, Vat S, Marques-Vidal P, et al. Prevalence of sleep-disordered breathing in the general population: the HypnoLaus study. Lancet Respir Med. 2015;3(4):310-318. Epub 2015 Feb 12. doi:10.1016/S2213-2600(15)00043-0
2. Ratz D, Wiitala W, Safwan Badr M, Burns J, Chowdhuri S. Correlates and consequences of central sleep apnea in a national sample of US veterans. Sleep. 2018;41(9):zy058. doi:10.1093/sleep/zsyn058
3. Agrawal R, Sharafkhaneneh A, Gottlief, DJ, Nowakowski S, Razjouyan J. Mortality patterns associated with central sleep apnea among veterans: a large, retrospective, longitudinal report. Ann Am Thorac Soc. 2022;10.1513/AnnalsATS.202207-648OC. doi:10.1513/annalsATS. 202207-648OC
4. Mysliwiec V, McGraw L, Pierce R, Smith, P, Trapp, B, Roth B. Sleep disorders and associated medical comorbidities in active duty military personnel. Sleep. 2013;36(2):167-174. doi:10.5665/sleep.2364
5. Badr MS, Dingell JD, Javaheri S. Central sleep apnea: a brief review. Curr Pulmonol Rep. 2019;8(1):14-21. Epub 2019 Mar 13. doi:10.1007/s13665-019-0221-z
6. Baillieul S, Revol B, Jullian-Desayes I, Joyeux-Faure M, Tamisier R, Pépin JL. Diagnosis and management of central sleep apnea syndrome. Expert Rev Respir Med. 2019;13(6):545-557.1604226. Epub 2019 Apr 24. doi:10.1080/17476348.2019
7. Randerath W, Verbraecken J, Andreas S, et al. Definition, discrimination, diagnosis and treatment of central breathing disturbances during sleep. Eur Respir J. 2017;49(1):1600959. doi:10.1183/13993003.00959-2016
8. American Academy of Sleep Medicine. International Classification of Sleep Disorders. 3rd ed. American Academy of Sleep Medicine; 2014.
9. Lévy P, Pépin J-L, Tamisier R, Neuder Y, Baguet J-P, Javaheri S. Prevalence and impact of central sleep apnea in heart failure. Sleep Med Clinics. 2007;2(4):615-621. doi:10.1016/j.jsmc.2007.08.001
10. Bitter T, Faber L, Hering D, Langer C, Horstkotte D, Oldenburg O. Sleep-disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail. 2009;11(6):602-608. doi:10.1093/eurjhf/hfp057
11. Sekizuka H, Osada N, Miyake F. Sleep disordered breathing in heart failure patients with reduced versus preserved ejection fraction. Heart Lung Circ. 2013;22(2):104-109. Epub 2012 Oct 26. doi:10.1016/j.hlc.2012.08.006
12. Iotti GA, Polito A, Belliato M, et al. Adaptive support ventilation versus conventional ventilation for total ventilatory support in acute respiratory failure. Intensive Care Med. 2010;36(8):1371-1379. Epub 2010 May 26. doi:10.1007/s00134-010-1917-2
13. Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373(12):1095-105. Epub 2015 Sep 1. doi:10.1056/NEJMoa1506459
14. Nigam G, Riaz M, Chang ET, Camacho M. Natural history of treatment-emergent central sleep apnea on positive airway pressure: a systematic review. Ann Thorac Med. 2018;13(2):86-91. doi:10.4103/atm.ATM_321_17
15. Orr JE, Malhotra A, Sands SA. Pathogenesis of central and complex sleep apnoea. Respirology. 2017;22(1):43-52. Epub 2016 Oct 31. doi:10.1111/resp.12927
16. Berger M, Solelhac G, Horvath C, Heinzer R, Brill AK. Treatment-emergent central sleep apnea associated with non-positive airway pressure therapies in obstructive sleep apnea patients: a systematic review. Sleep Med Rev. 2021; 58:101513. Epub 2021 Jun 5. doi:10.1016/j.smrv.2021.101513
17. Zhang J, Wang L, Guo HJ, Wang Y, Cao J, Chen BY. Treatment-emergent central sleep apnea: a unique sleep-disordered breathing. Chin Med J (Engl). 2020;133(22):2721-2730. doi:10.1097/CM9.0000000000001125
18. Nigam G, Pathak C, Riaz M. A systematic review on prevalence and risk factors associated with treatment- emergent central sleep apnea. Ann Thorac Med. 2016;11(3):202-210. doi:10.4103/1817-1737.185761
19. Zeineddine S, Badr MS. Treatment-emergent central apnea: physiologic mechanisms informing clinical practice. Chest. 2021;159(6):2449-2457. Epub 2021 Jan 23. doi:10.1016/j.hest.2021.01.036
20. Liu D, Armitstead J, Benjafield A. Trajectories of emergent central sleep apnea during CPAP therapy. Chest. 2017;152(4):751-760. Epub 2017 Jun 16. doi:10.1016/j.chest.2017.06.010
21. Moro M, Gannon K, Lovell K, Merlino M, Mojica J, Bianchi MT. Clinical predictors of central sleep apnea evoked by positive airway pressure titration. Nat Sci Sleep. 2016;8:259-266. doi:10.2147/NSS.S110032
22. Orr JE, Heinrich EC, Djokic M, et al. Adaptive servoventilation as treatment for central sleep apnea due to high-altitude periodic breathing in nonacclimatized healthy individuals. High Alt Med Biol. 2018;19(2):178-184. Epub 2018 Mar 13. doi:10.1089/ham.2017.0147
23. Liu HM, Chiang IJ, Kuo KN, Liou CM, Chen C. The effect of acetazolamide on sleep apnea at high altitude: a systematic review and meta-analysis. Ther Adv Respir Dis. 2017;11(1):20-29. Epub 2016 Nov 15. doi:10.1177/1753465816677006
24. Latshang TD, Nussbaumer-Ochsner Y, Henn RM, et al. Effect of acetazolamide and autoCPAP therapy on breathing disturbances among patients with obstructive sleep apnea syndrome who travel to altitude: a randomized controlled trial. JAMA. 2012;308(22):2390-8. doi:10.1001/jama.2012.94847
25. Nigam G, Pathak C, Riaz M. A systematic review of central sleep apnea in adult patients with chronic kidney disease. Sleep Breath. 2016;20(3):957-964. Epub 2016 Jan 27. doi:10.1007/s11325-016-1317-0
26. Nigam G, Riaz M. Pathophysiology of central sleep apnea in chronic kidney disease. Saudi J Kidney Dis Transpl. 2016;27(5):1068-1070. doi:10.4103/1319-2442.190907
27. Hanly PJ, Pierratos A. Improvement of sleep apnea in patients with chronic renal failure who undergo nocturnal hemodialysis. N Engl J Med. 2001;344(2):102-107. doi:10.1056/NEJM200101113440204
28. Jean G, Piperno D, François B, Charra B. Sleep apnea incidence in maintenance hemodialysis patients: influence of dialysate buffer. Nephron. 1995;71(2):138-142. doi:10.1159/000188701
29. Pressman MR, Benz RL, Schleifer CR, Peterson DD. Sleep disordered breathing in ESRD: acute beneficial effects of treatment with nasal continuous positive airway pressure. Kidney Int. 1993;43(5):1134-1139. doi:10.1038/ki.1993.159
30. Ladenson PW, Goldenheim PD, Ridgway EC. Prediction and reversal of blunted ventilatory responsiveness in patients with hypothyroidism. Am J Med. 1988;84(5):877-883. doi:10.1016/0002-9343(88)90066-6
31. Siafakas NM, Salesiotou V, Filaditaki V, Tzanakis N, Thalassinos N, Bouros D. Respiratory muscle strength in hypothyroidism. Chest. 1992;102(1):189-194. doi:10.1378/chest.102.1.189
32. Laroche CM, Cairns T, Moxham J, Green M. Hypothyroidism presenting with respiratory muscle weakness. Am Rev Respir Dis. 1988;138(2):472-474. doi:10.1164/ajrccm/138.2.472
33. Skjodt NM, Atkar R, Easton PA. Screening for hypothyroidism in sleep apnea. Am J Respir Crit Care Med. 1999;160(2):732-735. doi:10.1164/ajrccm.160.2.9802051
34. American Academy of Sleep Medicine. FDA approves Remede¯ implantable device to treat central sleep apnea. Accessed February 3, 2023. https://aasm.org/fda-approves-remede-implantable-device-treat-central-sleep-apnea
35. Wang D, Teichtahl H, Drummer O, et al. Central sleep apnea in stable methadone maintenance treatment patients. Chest. 2005;128(3):1348-1356. doi:10.1378/chest.128.3.1348
36. Sharkey KM, Kurth ME, Anderson BJ, Corso RP, Millman RP, Stein MD. Obstructive sleep apnea is more common than central sleep apnea in methadone maintenance patients with subjective sleep complaints. Drug Alcohol Depend. 2010;108(1-2):77-83. Epub 2010 Jan 15. doi:10.1016/j.drugalcdep.2009.11.019
37. Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg. 2015;120:1273-1285. doi:10.1213/ANE.0000000000000672
38. Wang, D, Yee, BJ, Gunstein RR, Chung F. Chronic opioid use and central sleep apnea, where are we now and where to go? A state of the art review. Anesth Analg. 2021;132(5):1244-1253. doi:10.1213/ANE.0000000000005378
39. Schütz SG, Lisabeth LD, Hsu CW, Kim S, Chervin RD, Brown DL. Central sleep apnea is uncommon after stroke. Sleep Med. 2021;77:304-306. Epub 2020 Aug 28. doi:10.1016/j.sleep.2020.08.025
40. Seiler A, Camilo M, Korostovtseva L, et al. Prevalence of sleep-disordered breathing after stroke and TIA: a meta-analysis. Neurology. 2019;92(7):e648-e654. Epub 2019 Jan 11. doi:10.1212/WNL.0000000000006904