User login
Hippocampal volume change in the Alzheimer Disease Cholesterol-Lowering Treatment trial
Alzheimer disease (AD) is a degenerative disorder characterized by a gradual deterioration in memory. In its clinically overt stages, obvious signs of neural degeneration on magnetic resonance imaging (MRI) appear as global cerebral atrophy. Even in the earliest stages of the disease, regional cell loss can be observed, particularly in the mesial temporal lobe regions, specifically the hippocampus and entorhinal cortex.1–3
MRI is used primarily as a diagnostic tool to rule out conditions other than AD. However, MRI may be useful in understanding whether the underlying processes that are associated with these cognitive changes can be attributed to general or specific effects of the disease process. Volumetric changes observed with MRI in the hippocampal region have been correlated with disease progression4,5 and predict development of AD in individuals with isolated memory impairment,6 suggesting that neuroimaging quantification may serve as a useful measure of brain integrity in patients with AD.
New treatments for AD are emerging, and assessing their efficacy is of critical importance. The rationale for testing statin drugs as a therapy for AD was bolstered by ever-mounting preclinical animal and human data suggesting that elevated circulating cholesterol exacerbates AD-like pathology and that statin treatment, in part, reverses the effect of cholesterol. This article surveys current evidence on the association between cholesterol and AD as well as between statin use and AD risk. We conclude by focusing on results from the first clinical investigation of statin therapy in patients with AD and present new results of a substudy of this trial examining the morphologic effects of statin therapy in AD patients.
LINK BETWEEN CHOLESTEROL AND AD
Early epidemiologic surveys suggested an association between a high-fat/high-cholesterol diet and increased risk of AD,7–10 and this suggestion has been supported by more recent investigations.11,12 Cholesterol levels are increased in the blood of AD patients,7,13–16 and increased cholesterol has been observed in the AD brain as a function of the apolipoprotein E allotype.8,17
Numerous clinical studies suggest a link between elevated cholesterol and increased risk of AD,17–23 with one study reporting a threefold increase in the risk of AD with elevated serum cholesterol, even after adjusting for age and presence of the apolipoprotein E4 allele.19 Another study indicates that persistently elevated cholesterol levels in midlife increase the risk of AD.23 A retrospective analysis of the Framingham Study suggested, however, that there is no relationship between total cholesterol levels and risk of incident AD.24 A more recent report indicated that language performance in elderly subjects without dementia declined faster among those individuals with higher cholesterol levels, but this effect did not remain significant after accounting for multiple comparisons.25 In contrast, the Three-City Study, a population-based cohort investigation of 9,294 subjects in France, demonstrated a significant increase in the risk of dementia among subjects who had hyperlipidemia (odds ratio [OR] = 1.43; 95% confidence interval [CI], 1.03 to 1.99).12
STATIN USE AND RISK OF AD
The preponderance of clinical data suggests that statin therapy may reduce the risk of AD later in life. Since the initial epidemiologic investigation assessing the effect of statin use on later risk of AD in the elderly, there have been 13 additional studies; all but two of these studies have reported benefit with cholesterol-lowering therapy.
In the two earliest epidemiologic studies, Wolozin et al demonstrated benefit with the use of lovastatin and pravastatin, but not with simvastatin or non-statin therapy,26 and Jick et al showed benefit associated with cholesterol-lowering therapy, but not specifically with statin use.27 Five epidemiologic studies published in 2002 suggested that prior statin use reduced the risk of dementia or AD.28–32 Meta-analysis of these first seven retrospective studies suggested a significant reduction in the risk of later cognitive impairment with statin use (relative risk = 0.43; 95% CI, 0.31 to 0.62), but the risk reduction with lipid-lowering agents collectively (not just statins) was not statistically significant.33
In 2004, Zamrini et al reported a 39% reduction in the risk of AD in statin users compared with nonusers (OR = 0.61; 95% CI, 0.42 to 0.87).34 That same year, Li et al suggested that there was no association between statin use and a reduced incidence of probable AD using a time-dependent proportional hazards model, but if the data were analyzed (inappropriately) as a case-control study, a significant protective effect was identified.35
Data from the Cache County Study cohort demonstrated no significant reduction in the risk of AD with statin use but allowed for the possibility that some benefit could be provided with longer-term statin therapy.36 In constrast, the Three-City Study of 9,294 individuals in France identified a significant reduction in the risk of AD with statin use (OR = 0.61; 95% CI, 0.41 to 0.91).12 Rea et al reported that prior statin use did not decrease the risk of dementia or AD, but when they included in their analysis individuals currently using a statin, there was a significant reduction in the hazard ratios for AD and for all-cause dementia.37 The two most recent epidemiologic studies both suggest that statin therapy slows cognitive decline in AD.38,39
COGNITIVE PERFORMANCE AND STATIN USE
A retrospective cohort study that assessed intelligence and cognition at a young age and again when subjects were in their 80s indicated that statin use had a significant beneficial effect on cognitive ability.40
In contrast, two very large prospective studies published in 2002 suggested that statins produce no positive effect on cognition in younger individuals at risk for heart disease.41,42 The Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) found that the mean Mini-Mental State Examination (MMSE) score, which was assessed only at subjects’ last on-treatment clinical visit, was comparable between the study’s pravastatin and placebo groups.41 Likewise, the Medical Research Council/British Heart Foundation (MRC/BHF) Heart Protection Study, which used the Telephone Interview for Cognitive Status questionnaire at the end of the investigation, reported that simvastatin had no positive effect on cognitive performance compared with placebo, but this finding was obtained in the absence of baseline data.42 Given the limited cognitive assessments performed in these two studies, no firm conclusions should be drawn.
A more recent prospective comparison of atorvastatin and placebo in younger subjects did include baseline and follow-up assessment of cognitive function, and it identified significantly superior performance in the statin-treated population on the MMSE and on tests of attention, psychomotor speed, mental flexibility, working memory, and memory retrieval.43
STATIN TREATMENT OF AD: THE AD CHOLESTEROL-LOWERING TREATMENT TRIAL
The initial clinical investigation of statin therapy in patients with AD—the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial—involved atorvastatin.44 Patients with mild to moderate AD were randomized to either placebo or 80 mg/day of atorvastatin for a 1-year period. Evaluable data were available for 63 patients (32 in the atorvastatin group, 31 in the placebo group). End points included the change in performance on the following measures:
MMSE
- Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-cog)
- Neuropsychiatric Inventory Caregiver Distress Scale (NPI)
- Clinical Global Impression of Change scale (CGIC)
- Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory (ADCS-ADL)
- Geriatric Depression Scale (GDS).
Cognitive results
In the setting of continued cholinesterase inhibitor use, atorvastatin provided significant benefit on the ADAS-cog at 26 weeks compared with placebo (P = .003) and marginally significant benefit at 1 year (P = .055) while producing a trend for benefit on the CGIC and NPI and a statistically significant improvement on the GDS after 1 year of active treatment.44 The observed benefit on the MMSE with atorvastatin versus placebo did not reach statistical significance, and no discernible difference was observed on the ADCS-ADL.44 In contrast, a significant difference in the slope of deterioration on the MMSE and the GDS in the atorvastatin group versus the placebo group suggested disease modification.45
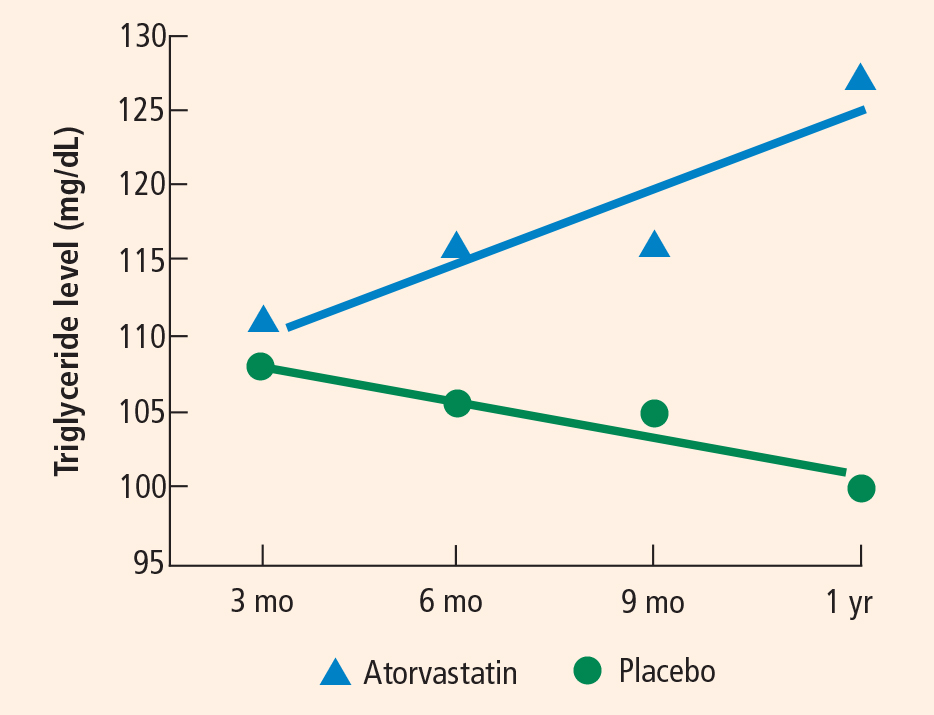
Blood test results
Secondary analysis and initial morphometric substudy
Secondary assessment indicated that the subjects who garnered the greatest benefit from atorvastatin therapy in terms of their 6-month ADAS-cog score were those who had higher cholesterol levels at trial entry, those who harbored the apolipoprotein E4 allele, and those who were less affected by AD at trial entry (ie, with higher entry MMSE scores).47
In an ADCLT substudy using new voxel-based morphometry techniques, we quantitatively assessed gray matter density in 15 ADCLT trial participants and compared it with density findings in 15 normal elderly controls.48 Regional reductions in gray matter density were observed in the AD patients compared with the controls. Large differences in gray matter concentration were observed bilaterally in the temporal lobe. The anterior cingulate, right superior temporal, left superior frontal, and posterior cingulate regions also showed significantly decreased gray matter density in the AD patients compared with the controls. A significant relationship was observed between gray matter density and ADAS-cog error scores—ie, more severe levels of cognitive impairment correlated with reduced gray matter density.48
PILOT SUBSTUDY OF ADCLT: ASSESSING MORPHOLOGIC CHANGES WITH STATIN THERAPY
Eleven of the 15 ADCLT trial participants from the above morphometric substudy returned for MRI assessment after 1 year of treatment with either atorvastatin or placebo. We report here the comparative effects of atorvastatin and placebo on hippocampal volume and the relationship with cognitive performance.
Participants
Subjects were participating in the ADCLT trial, an investigator-initiated, double-blind, placebo-controlled study. Neuroimaging was performed at the Barrow Neurological Institute, Phoenix, AZ, for a subset of the participants in the trial (n = 11) as a pilot study to examine neural changes associated with atorvastatin therapy.
Each patient underwent screening, assignment to either atorvastatin 80 mg/day or placebo, and medical and cognitive assessment at Sun Health Research Institute, Sun City, AZ, prior to imaging at Barrow. All patients met Diagnostic and Statistical Manual of Mental Disorders, fourth edition, criteria for dementia as well as NINCDS-ADRDA criteria for probable AD. Each patient was free of significant psychiatric and neurological history and had a score of 4 or less on the Hachinski Modified Ischemia Scale. All MRIs were reviewed by a neuroradiologist to ensure that there was no evidence of stroke or cortical or lacunar infarcts.
Both sites’ institutional review boards approved this project, and all subjects gave written informed consent.
Cognitive assessment
A primary efficacy measure used in the parent study was the ADAS-cog,49 and the MMSE50 was a secondary measure. Change scores were determined by comparing values obtained at baseline, prior to randomization to treatment with either atorvastatin or placebo, and after 1 year of treatment. MMSE scores were obtained at the same session as the ADAS-cog scores. Cognitive assessments were obtained within 2 weeks prior to MRI.
Image acquisition
All participants underwent imaging on a single 1.5tesla GE scanner at Barrow Neurological Institute. Imaging was conducted both prior to treatment randomization and again after 1 year of treatment. Images of the whole brain were collected using a coronal SPGR (spoiled gradient) T1-weighted, three-dimensional acquisition with the following parameters:
- Number of acquisitions = 1
- Repetition time = 23 msec
- Echo time = 8 msec
- Flip angle = 35 degrees
- Bandwidth = 12.5 kHz
- Slice thickness = 1.5 mm or 1.9 mm
- 0 skip between slices
- In-plane resolution = 0.9375 x 0.9375.
Hippocampal volumetrics
All imaging analysis was performed within the Analysis of Functional Neuroimages (AFNI) package.51 We traced the outline of the hippocampus using the three-dimensional SPGR images. The hippocampi were visualized in all three planes, landmarked in the coronal and sagittal planes, and drawn in the coronal plane. We employed the guidelines of Insausti et al52 and Machulda et al53 to define the hippocampal boundaries. First we defined the anterior boundary by observing the white matter band and/or the cerebrospinal fluid space between the amygdala and hippocampus in the sagittal plane. The posterior aspect of the posterior region was initially landmarked in the sagittal plane by locating the posterior edge of the hippocampus and then checking in the coronal plane to ensure that the fornices were completely visualized. Volumes were calculated by importing the extracted hippocampi into MATLAB to measure the volumes.
Statistical analyses
Mean differences between the atorvastatin and placebo groups were evaluated using two-tailed Student t tests. Correlation between changes in cognitive measures and changes in the hippocampal volume for the total population and for the treatment groups was determined using Pearson’s r coefficient. Significance was defined as a P value less than .05; a P value between .05 and .10 was deemed a trend.
Results
In contrast to other studies,54–58 we found in this pilot study that right hippocampal volume was slightly less than left hippocampal volume (2,015 ± 141 mm3 vs 2,135 ± 183 mm3).
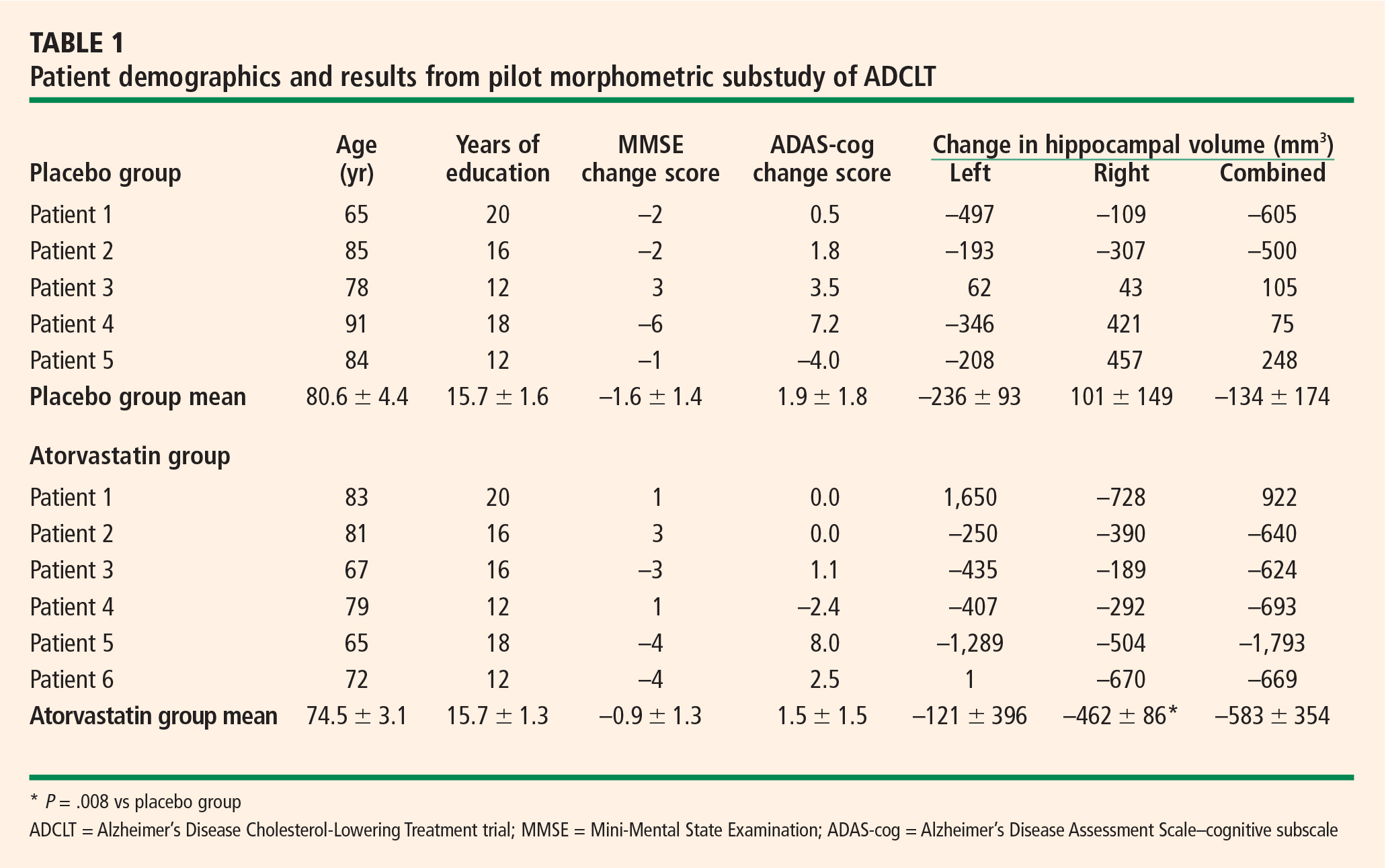
Mean changes in performance on the ADAS-cog and MMSE were less pronounced in the atorvastatin group than in the placebo group, but not significantly so (Table 1). However, there was a trend toward superiority in the atorvastatin group on performance on the free word-recall subscale of the ADAS-cog.
No significant correlations were found between change in cognitive performance and change in hip-pocampal volume.
DISCUSSION
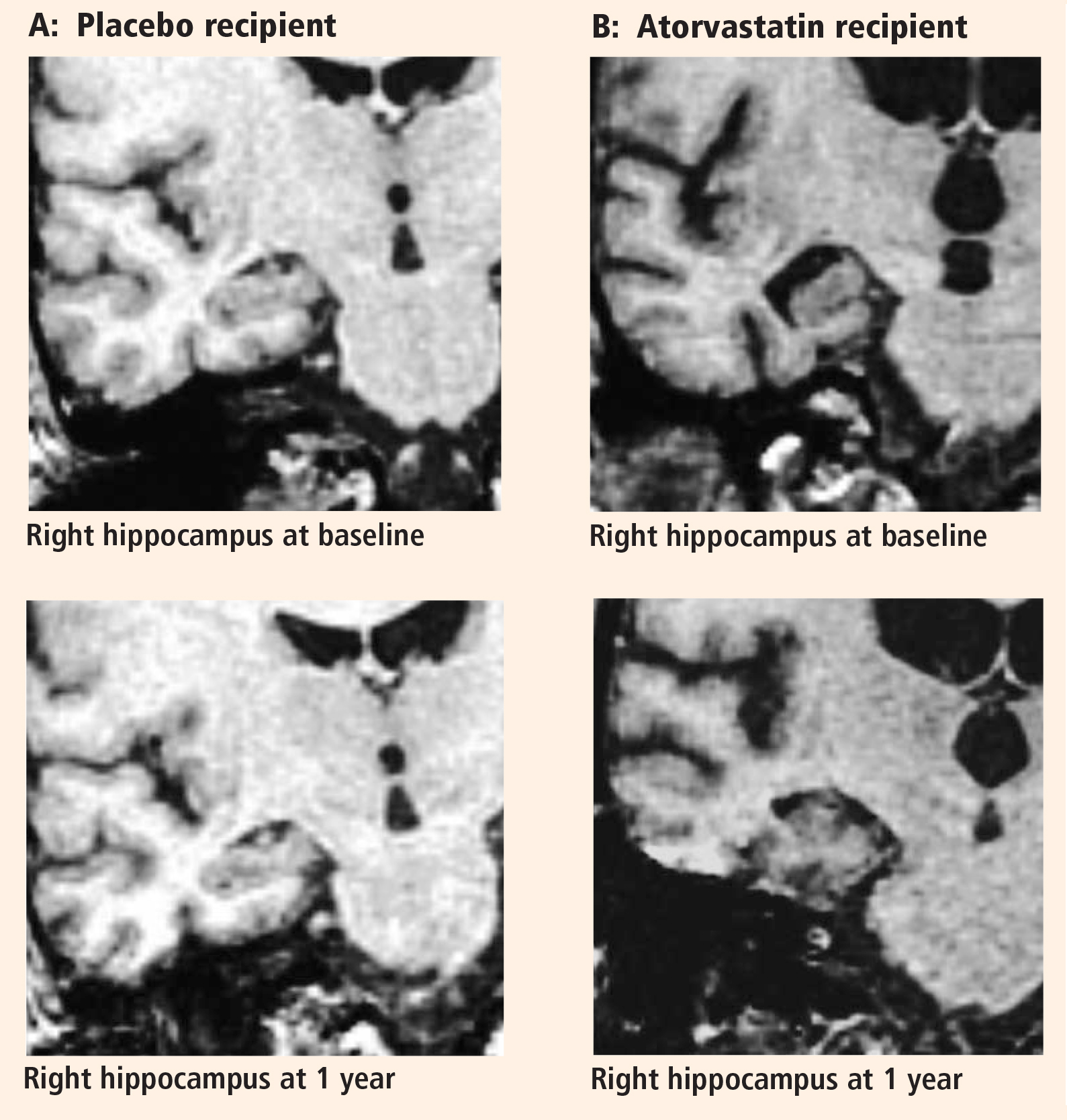
The preponderance of evidence clearly indicates that hippocampal volume is reduced in patients with AD compared with individuals with normal cognitive ability for their age. There is also evidence indicating that as cognitive performance deteriorates in AD patients, there are concurrent further reductions in hippocampal volume.54 Many studies reported that there was no significant volume difference between the right and left hippocampi, but most suggested that the left hip-pocampus was slightly smaller than the right.54–58 We identified no significant difference in volume between the sides, but we did find that the right hippocampus was smaller than the left in a very limited population of subjects with mild to moderate AD.
The major finding of this pilot study flies in the face of conventional wisdom in that there seems to be significant shrinkage of the right hippocampus with atorvastatin therapy compared with placebo in a randomized AD treatment trial that demonstrated clinical benefit with atorvastatin therapy.44 A similar finding was reported from the beta-amyloid immunization (AN1792) treatment trial in AD.59 In that study the active immunization was associated with significant clinical benefit, reduced beta-amyloid load, and reduced hippocampal volume.59 The authors suggested that removal of beta-amyloid and/or other protein constituents from the tissue might have caused a “fluid shift” out of the tissue, resulting in shrinkage.
Based on our previous finding of reduced brain tissue density in AD patients compared with age-matched normal controls,48 an alternative explanation can be proposed. Neuronal loss in the hippocampus may be accompanied by increased fluid balance (reduced density) in an attempt to retain the previous volume at the expense of function. Accordingly, as the hippocampus shrinks, it approaches a more normal density for the remaining neuronal complement, and cognitive function improves.
- Lehtovirta M, Laakso MP, Soininen H, et al. Volumes of hippocampus, amygdala and frontal lobe in Alzheimer patients with different apolipoprotein E genotypes. Neuroscience 1995; 67:65–72.
- Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996; 16:4491–4500.
- Jack CR Jr, Shiung MM, Gunter JL, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 2004; 62:591–600.
- Jack CR Jr, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999; 52:1397–1403.
- Jack CR Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology 2000; 55:484–489.
- Killiany RJ, Hyman BT, Gómez-Isla T, et al. MRI measures of entorhinal cortex vs hippocampus in preclinical AD. Neurology 2002; 58:1188–1196.
- Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer’s disease: a case-control study. Neurology 1995; 45:1092–1096.
- Sparks DL. Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann N Y Acad Sci 1997; 826:128–146.
- Grant WB. Dietary links to Alzheimer’s disease. Alzheimers Dis Rev 1997; 2:42–55.
- Desmond DW, Tatemichi TK, Paik M, Stern Y. Risk factors for cerebrovascular disease as correlates of cognitive function in a stroke-free cohort. Arch Neurol 1993; 50:162–166.
- Mainous AG III, Eschenbach SL, Wells BJ, Everett CJ, Gill JM. Cholesterol, transferrin saturation, and the development of dementia and Alzheimer’s disease: results from an 18-year population-based cohort. Fam Med 2005; 37:36–42.
- Dufouil C, Richard F, Fiévet N, et al. APOE genotype, cholesterol level, lipid-lowering treatment, and dementia: the Three-City Study. Neurology 2005; 64:1531–1538.
- Lehtonen A, Luutonen S. High-density lipoprotein cholesterol levels of very old people in the diagnosis of dementia. Age Ageing 1986; 15:267–270.
- Giubilei F, D’Antona R, Antonini R, Lenzi GL, Ricci G, Fieschi C. Serum lipoprotein pattern variations in dementia and ischemic stroke. Acta Neurol Scand 1990; 81:84–86.
- Czech C, Förstl H, Hentschel F, et al. Apolipoprotein E-4 gene dose in clinically diagnosed Alzheimer’s disease: prevalence, plasma cholesterol levels and cerebrovascular change. Eur Arch Psychiatry Clin Neurosci 1994; 243:291–292.
- Mahieux F, Couderc R, Moulignier A, Bailleul S, Podrabinek N, Laudet J. Isoform 4 of apolipoprotein E and Alzheimer disease. Specificity and clinical study [in French]. Rev Neurol (Paris) 1995; 151:231–239.
- Kuo YM, Emmerling MR, Bisgaier CL, et al. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1-42 levels. Biochem Biophys Res Commun 1998; 252:711–715.
- Chandra V, Pandav R. Gene-environment interaction in Alzheimer’s disease: a potential role for cholesterol. Neuroepidemiology 1998; 17:225–232.
- Notkola IL, Sulkava R, Pekkanen J, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 1998; 17:14–20.
- Olson RE. Discovery of the lipoproteins, their role in fat transport and their significance as risk factors. J Nutr 1998; 128(2 Suppl):439S–443S.
- Kalmijn S, Launer LJ, Ott A, Witteman JC, Hofman A, Breteler MM. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol 1997; 42:776–782.
- Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ 1997; 315:1045–1049.
- Kivipelto M, Helkala E-L, Hallikainen M, et al. Elevated systolic blood pressure and high cholesterol levels at midlife are risk factors for late-life dementia [abstract]. Neurobiol Aging 2000; 21(Suppl 1):S174. Abstract 787.
- Tan ZS, Seshadri S, Beiser A, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham Study. Arch Intern Med 2003; 163:1053–1057.
- Reitz C, Luchsinger J, Tang MX, Manly J, Mayeux R. Impact of plasma lipids and time on memory performance in healthy elderly without dementia. Neurology 2005; 64:1378–1383.
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57:1439–1443.
- Jick H, Zornberg G, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet 2000; 356:1627–1631.
- Green RC, McNagny SE, Jayakumar P, Cupples LA, Benke K, Farrer L. Statin use is associated with reduced risk of Alzheimer’s disease [abstract]. Neurology 2002; 58(Suppl 3):A81. Abstract S10.006.
- Rodriguez EG, Dodge HH, Birzescu MA, Stoehr GP, Ganguli M. Use of lipid-lowering drugs in older adults with and without dementia: a community-based epidemiological study. J Am Geriatr Soc 2002; 50:1852–1856.
- Rockwood K, Kirkland S, Hogan DB, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol 2002; 59:223–227.
- Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol 2002; 59:378–384.
- Hajjar I, Schumpert J, Hirth V, Wieland D, Eleazer GP. The impact of the use of statins on the prevalence of dementia and the progression of cognitive impairment. J Gerontol A Biol Sci Med Sci 2002; 57:M414–M418.
- Etminan M, Gill S, Samii A. The role of lipid-lowering drugs in cognitive function: a meta-analysis of observational studies. Pharmacotherapy 2003; 23:726–730.
- Zamrini E, McGwin G, Roseman JM. Association between statin use and Alzheimer’s disease. Neuroepidemiology 2004; 23:94–98.
- Li G, Higdon R, Kukull WA, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology 2004; 63:1624–1628.
- Zandi PP, Sparks DL, Khachaturian AS, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry 2005; 62:217–224.
- Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol 2005; 62:1047–1051.
- Masse I, Bordet R, Deplanque D, et al. Lipid lowering agents are associated with a slower cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2005; 76:1624–1629.
- Bernick C, Katz R, Smith NL, et al. Statins and cognitive function in the elderly: the Cardiovascular Health Study. Neurology 2005; 65:1388–1394.
- Starr JM, McGurn B, Whiteman M, Pattie A, Whalley LJ, Deary IJ. Life long changes in cognitive ability are associated with prescribed medications in old age. Int J Geriatr Psychiatry 2004; 19:327–332.
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Parale GP, Baheti NN, Kulkarni PM, Panchal NV. Effects of atorvastatin on higher functions. Eur J Clin Pharmacol 2006; 62:259–265.
- Sparks DL, Sabbagh MN, Connor DJ, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol 2005; 62:753–757.
- Sparks DL, Sabbagh M, Connor D, et al. Statin therapy in Alzheimer’s disease. Acta Neurol Scand 2006; 114(Suppl 185):78–86.
- Stankovic G, Sparks DL. Change in circulating C-reactive protein is not associated with atorvastatin treatment in Alzheimer’s disease. Neurol Res 2006; 28:621–624.
- Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer’s disease: results of the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial. Acta Neurol Scand 2006; 114(Suppl 185):3–7.
- Baxter LC, Sparks DL, Johnson SC, et al. Relationship of cognitive measures and gray and white matter in Alzheimer’s disease. J Alzheimers Dis 2006; 9:253–260.
- Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984; 141:1356–1364.
- Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the mental state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Cox RW. AFNI: sofware for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res 1996; 29:162–173.
- Insausti R, Juottonen K, Soininen H, et al. MR volumetric analysis of the human entorhinal, perirhinal, and temporopolar cortices. AJNR Am J Neuroradiol 1998; 19:659–671.
- Machulda MM, Ward HA, Cha R, O’Brien P, Jack CR Jr. Functional inferences vary with the method of analysis in fMRI. Neuroimage 2001; 14:1122–1127.
- Jack CR Jr, Slomkowski M, Gracon S, et al. MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology 2003; 60:253–260.
- Pruessner JC, Li LM, Serles W, et al. Volumetry of hippocampus and amygdala with high-resolution MRI and three-dimensional analysis software: minimizing the discrepancies between laboratories. Cereb Cortex 2000; 10:433–442.
- Pruessner JC, Collins DL, Pruessner M, Evans AC. Age and gender predict volume decline in the anterior and posterior hippocam-pus in early adulthood. J Neurosci 2001; 21:194–200.
- Hackert VH, den Heijer T, Oudkerk M, Koudstaal PJ, Hofman A, Breteler MM. Hippocampal head size associated with verbal memory performance in nondemented elderly. Neuroimage 2002; 17:1365–1372.
- Rosen AC, Prull MW, Gabrieli JD, et al. Differential associations between entorhinal and hippocampal volumes and memory performance in older adults. Behav Neurosci 2003; 117:1150–1160.
- Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005; 64:1563–1572.
Alzheimer disease (AD) is a degenerative disorder characterized by a gradual deterioration in memory. In its clinically overt stages, obvious signs of neural degeneration on magnetic resonance imaging (MRI) appear as global cerebral atrophy. Even in the earliest stages of the disease, regional cell loss can be observed, particularly in the mesial temporal lobe regions, specifically the hippocampus and entorhinal cortex.1–3
MRI is used primarily as a diagnostic tool to rule out conditions other than AD. However, MRI may be useful in understanding whether the underlying processes that are associated with these cognitive changes can be attributed to general or specific effects of the disease process. Volumetric changes observed with MRI in the hippocampal region have been correlated with disease progression4,5 and predict development of AD in individuals with isolated memory impairment,6 suggesting that neuroimaging quantification may serve as a useful measure of brain integrity in patients with AD.
New treatments for AD are emerging, and assessing their efficacy is of critical importance. The rationale for testing statin drugs as a therapy for AD was bolstered by ever-mounting preclinical animal and human data suggesting that elevated circulating cholesterol exacerbates AD-like pathology and that statin treatment, in part, reverses the effect of cholesterol. This article surveys current evidence on the association between cholesterol and AD as well as between statin use and AD risk. We conclude by focusing on results from the first clinical investigation of statin therapy in patients with AD and present new results of a substudy of this trial examining the morphologic effects of statin therapy in AD patients.
LINK BETWEEN CHOLESTEROL AND AD
Early epidemiologic surveys suggested an association between a high-fat/high-cholesterol diet and increased risk of AD,7–10 and this suggestion has been supported by more recent investigations.11,12 Cholesterol levels are increased in the blood of AD patients,7,13–16 and increased cholesterol has been observed in the AD brain as a function of the apolipoprotein E allotype.8,17
Numerous clinical studies suggest a link between elevated cholesterol and increased risk of AD,17–23 with one study reporting a threefold increase in the risk of AD with elevated serum cholesterol, even after adjusting for age and presence of the apolipoprotein E4 allele.19 Another study indicates that persistently elevated cholesterol levels in midlife increase the risk of AD.23 A retrospective analysis of the Framingham Study suggested, however, that there is no relationship between total cholesterol levels and risk of incident AD.24 A more recent report indicated that language performance in elderly subjects without dementia declined faster among those individuals with higher cholesterol levels, but this effect did not remain significant after accounting for multiple comparisons.25 In contrast, the Three-City Study, a population-based cohort investigation of 9,294 subjects in France, demonstrated a significant increase in the risk of dementia among subjects who had hyperlipidemia (odds ratio [OR] = 1.43; 95% confidence interval [CI], 1.03 to 1.99).12
STATIN USE AND RISK OF AD
The preponderance of clinical data suggests that statin therapy may reduce the risk of AD later in life. Since the initial epidemiologic investigation assessing the effect of statin use on later risk of AD in the elderly, there have been 13 additional studies; all but two of these studies have reported benefit with cholesterol-lowering therapy.
In the two earliest epidemiologic studies, Wolozin et al demonstrated benefit with the use of lovastatin and pravastatin, but not with simvastatin or non-statin therapy,26 and Jick et al showed benefit associated with cholesterol-lowering therapy, but not specifically with statin use.27 Five epidemiologic studies published in 2002 suggested that prior statin use reduced the risk of dementia or AD.28–32 Meta-analysis of these first seven retrospective studies suggested a significant reduction in the risk of later cognitive impairment with statin use (relative risk = 0.43; 95% CI, 0.31 to 0.62), but the risk reduction with lipid-lowering agents collectively (not just statins) was not statistically significant.33
In 2004, Zamrini et al reported a 39% reduction in the risk of AD in statin users compared with nonusers (OR = 0.61; 95% CI, 0.42 to 0.87).34 That same year, Li et al suggested that there was no association between statin use and a reduced incidence of probable AD using a time-dependent proportional hazards model, but if the data were analyzed (inappropriately) as a case-control study, a significant protective effect was identified.35
Data from the Cache County Study cohort demonstrated no significant reduction in the risk of AD with statin use but allowed for the possibility that some benefit could be provided with longer-term statin therapy.36 In constrast, the Three-City Study of 9,294 individuals in France identified a significant reduction in the risk of AD with statin use (OR = 0.61; 95% CI, 0.41 to 0.91).12 Rea et al reported that prior statin use did not decrease the risk of dementia or AD, but when they included in their analysis individuals currently using a statin, there was a significant reduction in the hazard ratios for AD and for all-cause dementia.37 The two most recent epidemiologic studies both suggest that statin therapy slows cognitive decline in AD.38,39
COGNITIVE PERFORMANCE AND STATIN USE
A retrospective cohort study that assessed intelligence and cognition at a young age and again when subjects were in their 80s indicated that statin use had a significant beneficial effect on cognitive ability.40
In contrast, two very large prospective studies published in 2002 suggested that statins produce no positive effect on cognition in younger individuals at risk for heart disease.41,42 The Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) found that the mean Mini-Mental State Examination (MMSE) score, which was assessed only at subjects’ last on-treatment clinical visit, was comparable between the study’s pravastatin and placebo groups.41 Likewise, the Medical Research Council/British Heart Foundation (MRC/BHF) Heart Protection Study, which used the Telephone Interview for Cognitive Status questionnaire at the end of the investigation, reported that simvastatin had no positive effect on cognitive performance compared with placebo, but this finding was obtained in the absence of baseline data.42 Given the limited cognitive assessments performed in these two studies, no firm conclusions should be drawn.
A more recent prospective comparison of atorvastatin and placebo in younger subjects did include baseline and follow-up assessment of cognitive function, and it identified significantly superior performance in the statin-treated population on the MMSE and on tests of attention, psychomotor speed, mental flexibility, working memory, and memory retrieval.43
STATIN TREATMENT OF AD: THE AD CHOLESTEROL-LOWERING TREATMENT TRIAL
The initial clinical investigation of statin therapy in patients with AD—the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial—involved atorvastatin.44 Patients with mild to moderate AD were randomized to either placebo or 80 mg/day of atorvastatin for a 1-year period. Evaluable data were available for 63 patients (32 in the atorvastatin group, 31 in the placebo group). End points included the change in performance on the following measures:
MMSE
- Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-cog)
- Neuropsychiatric Inventory Caregiver Distress Scale (NPI)
- Clinical Global Impression of Change scale (CGIC)
- Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory (ADCS-ADL)
- Geriatric Depression Scale (GDS).
Cognitive results
In the setting of continued cholinesterase inhibitor use, atorvastatin provided significant benefit on the ADAS-cog at 26 weeks compared with placebo (P = .003) and marginally significant benefit at 1 year (P = .055) while producing a trend for benefit on the CGIC and NPI and a statistically significant improvement on the GDS after 1 year of active treatment.44 The observed benefit on the MMSE with atorvastatin versus placebo did not reach statistical significance, and no discernible difference was observed on the ADCS-ADL.44 In contrast, a significant difference in the slope of deterioration on the MMSE and the GDS in the atorvastatin group versus the placebo group suggested disease modification.45
Blood test results
Secondary analysis and initial morphometric substudy
Secondary assessment indicated that the subjects who garnered the greatest benefit from atorvastatin therapy in terms of their 6-month ADAS-cog score were those who had higher cholesterol levels at trial entry, those who harbored the apolipoprotein E4 allele, and those who were less affected by AD at trial entry (ie, with higher entry MMSE scores).47
In an ADCLT substudy using new voxel-based morphometry techniques, we quantitatively assessed gray matter density in 15 ADCLT trial participants and compared it with density findings in 15 normal elderly controls.48 Regional reductions in gray matter density were observed in the AD patients compared with the controls. Large differences in gray matter concentration were observed bilaterally in the temporal lobe. The anterior cingulate, right superior temporal, left superior frontal, and posterior cingulate regions also showed significantly decreased gray matter density in the AD patients compared with the controls. A significant relationship was observed between gray matter density and ADAS-cog error scores—ie, more severe levels of cognitive impairment correlated with reduced gray matter density.48
PILOT SUBSTUDY OF ADCLT: ASSESSING MORPHOLOGIC CHANGES WITH STATIN THERAPY
Eleven of the 15 ADCLT trial participants from the above morphometric substudy returned for MRI assessment after 1 year of treatment with either atorvastatin or placebo. We report here the comparative effects of atorvastatin and placebo on hippocampal volume and the relationship with cognitive performance.
Participants
Subjects were participating in the ADCLT trial, an investigator-initiated, double-blind, placebo-controlled study. Neuroimaging was performed at the Barrow Neurological Institute, Phoenix, AZ, for a subset of the participants in the trial (n = 11) as a pilot study to examine neural changes associated with atorvastatin therapy.
Each patient underwent screening, assignment to either atorvastatin 80 mg/day or placebo, and medical and cognitive assessment at Sun Health Research Institute, Sun City, AZ, prior to imaging at Barrow. All patients met Diagnostic and Statistical Manual of Mental Disorders, fourth edition, criteria for dementia as well as NINCDS-ADRDA criteria for probable AD. Each patient was free of significant psychiatric and neurological history and had a score of 4 or less on the Hachinski Modified Ischemia Scale. All MRIs were reviewed by a neuroradiologist to ensure that there was no evidence of stroke or cortical or lacunar infarcts.
Both sites’ institutional review boards approved this project, and all subjects gave written informed consent.
Cognitive assessment
A primary efficacy measure used in the parent study was the ADAS-cog,49 and the MMSE50 was a secondary measure. Change scores were determined by comparing values obtained at baseline, prior to randomization to treatment with either atorvastatin or placebo, and after 1 year of treatment. MMSE scores were obtained at the same session as the ADAS-cog scores. Cognitive assessments were obtained within 2 weeks prior to MRI.
Image acquisition
All participants underwent imaging on a single 1.5tesla GE scanner at Barrow Neurological Institute. Imaging was conducted both prior to treatment randomization and again after 1 year of treatment. Images of the whole brain were collected using a coronal SPGR (spoiled gradient) T1-weighted, three-dimensional acquisition with the following parameters:
- Number of acquisitions = 1
- Repetition time = 23 msec
- Echo time = 8 msec
- Flip angle = 35 degrees
- Bandwidth = 12.5 kHz
- Slice thickness = 1.5 mm or 1.9 mm
- 0 skip between slices
- In-plane resolution = 0.9375 x 0.9375.
Hippocampal volumetrics
All imaging analysis was performed within the Analysis of Functional Neuroimages (AFNI) package.51 We traced the outline of the hippocampus using the three-dimensional SPGR images. The hippocampi were visualized in all three planes, landmarked in the coronal and sagittal planes, and drawn in the coronal plane. We employed the guidelines of Insausti et al52 and Machulda et al53 to define the hippocampal boundaries. First we defined the anterior boundary by observing the white matter band and/or the cerebrospinal fluid space between the amygdala and hippocampus in the sagittal plane. The posterior aspect of the posterior region was initially landmarked in the sagittal plane by locating the posterior edge of the hippocampus and then checking in the coronal plane to ensure that the fornices were completely visualized. Volumes were calculated by importing the extracted hippocampi into MATLAB to measure the volumes.
Statistical analyses
Mean differences between the atorvastatin and placebo groups were evaluated using two-tailed Student t tests. Correlation between changes in cognitive measures and changes in the hippocampal volume for the total population and for the treatment groups was determined using Pearson’s r coefficient. Significance was defined as a P value less than .05; a P value between .05 and .10 was deemed a trend.
Results
In contrast to other studies,54–58 we found in this pilot study that right hippocampal volume was slightly less than left hippocampal volume (2,015 ± 141 mm3 vs 2,135 ± 183 mm3).
Mean changes in performance on the ADAS-cog and MMSE were less pronounced in the atorvastatin group than in the placebo group, but not significantly so (Table 1). However, there was a trend toward superiority in the atorvastatin group on performance on the free word-recall subscale of the ADAS-cog.
No significant correlations were found between change in cognitive performance and change in hip-pocampal volume.
DISCUSSION
The preponderance of evidence clearly indicates that hippocampal volume is reduced in patients with AD compared with individuals with normal cognitive ability for their age. There is also evidence indicating that as cognitive performance deteriorates in AD patients, there are concurrent further reductions in hippocampal volume.54 Many studies reported that there was no significant volume difference between the right and left hippocampi, but most suggested that the left hip-pocampus was slightly smaller than the right.54–58 We identified no significant difference in volume between the sides, but we did find that the right hippocampus was smaller than the left in a very limited population of subjects with mild to moderate AD.
The major finding of this pilot study flies in the face of conventional wisdom in that there seems to be significant shrinkage of the right hippocampus with atorvastatin therapy compared with placebo in a randomized AD treatment trial that demonstrated clinical benefit with atorvastatin therapy.44 A similar finding was reported from the beta-amyloid immunization (AN1792) treatment trial in AD.59 In that study the active immunization was associated with significant clinical benefit, reduced beta-amyloid load, and reduced hippocampal volume.59 The authors suggested that removal of beta-amyloid and/or other protein constituents from the tissue might have caused a “fluid shift” out of the tissue, resulting in shrinkage.
Based on our previous finding of reduced brain tissue density in AD patients compared with age-matched normal controls,48 an alternative explanation can be proposed. Neuronal loss in the hippocampus may be accompanied by increased fluid balance (reduced density) in an attempt to retain the previous volume at the expense of function. Accordingly, as the hippocampus shrinks, it approaches a more normal density for the remaining neuronal complement, and cognitive function improves.
Alzheimer disease (AD) is a degenerative disorder characterized by a gradual deterioration in memory. In its clinically overt stages, obvious signs of neural degeneration on magnetic resonance imaging (MRI) appear as global cerebral atrophy. Even in the earliest stages of the disease, regional cell loss can be observed, particularly in the mesial temporal lobe regions, specifically the hippocampus and entorhinal cortex.1–3
MRI is used primarily as a diagnostic tool to rule out conditions other than AD. However, MRI may be useful in understanding whether the underlying processes that are associated with these cognitive changes can be attributed to general or specific effects of the disease process. Volumetric changes observed with MRI in the hippocampal region have been correlated with disease progression4,5 and predict development of AD in individuals with isolated memory impairment,6 suggesting that neuroimaging quantification may serve as a useful measure of brain integrity in patients with AD.
New treatments for AD are emerging, and assessing their efficacy is of critical importance. The rationale for testing statin drugs as a therapy for AD was bolstered by ever-mounting preclinical animal and human data suggesting that elevated circulating cholesterol exacerbates AD-like pathology and that statin treatment, in part, reverses the effect of cholesterol. This article surveys current evidence on the association between cholesterol and AD as well as between statin use and AD risk. We conclude by focusing on results from the first clinical investigation of statin therapy in patients with AD and present new results of a substudy of this trial examining the morphologic effects of statin therapy in AD patients.
LINK BETWEEN CHOLESTEROL AND AD
Early epidemiologic surveys suggested an association between a high-fat/high-cholesterol diet and increased risk of AD,7–10 and this suggestion has been supported by more recent investigations.11,12 Cholesterol levels are increased in the blood of AD patients,7,13–16 and increased cholesterol has been observed in the AD brain as a function of the apolipoprotein E allotype.8,17
Numerous clinical studies suggest a link between elevated cholesterol and increased risk of AD,17–23 with one study reporting a threefold increase in the risk of AD with elevated serum cholesterol, even after adjusting for age and presence of the apolipoprotein E4 allele.19 Another study indicates that persistently elevated cholesterol levels in midlife increase the risk of AD.23 A retrospective analysis of the Framingham Study suggested, however, that there is no relationship between total cholesterol levels and risk of incident AD.24 A more recent report indicated that language performance in elderly subjects without dementia declined faster among those individuals with higher cholesterol levels, but this effect did not remain significant after accounting for multiple comparisons.25 In contrast, the Three-City Study, a population-based cohort investigation of 9,294 subjects in France, demonstrated a significant increase in the risk of dementia among subjects who had hyperlipidemia (odds ratio [OR] = 1.43; 95% confidence interval [CI], 1.03 to 1.99).12
STATIN USE AND RISK OF AD
The preponderance of clinical data suggests that statin therapy may reduce the risk of AD later in life. Since the initial epidemiologic investigation assessing the effect of statin use on later risk of AD in the elderly, there have been 13 additional studies; all but two of these studies have reported benefit with cholesterol-lowering therapy.
In the two earliest epidemiologic studies, Wolozin et al demonstrated benefit with the use of lovastatin and pravastatin, but not with simvastatin or non-statin therapy,26 and Jick et al showed benefit associated with cholesterol-lowering therapy, but not specifically with statin use.27 Five epidemiologic studies published in 2002 suggested that prior statin use reduced the risk of dementia or AD.28–32 Meta-analysis of these first seven retrospective studies suggested a significant reduction in the risk of later cognitive impairment with statin use (relative risk = 0.43; 95% CI, 0.31 to 0.62), but the risk reduction with lipid-lowering agents collectively (not just statins) was not statistically significant.33
In 2004, Zamrini et al reported a 39% reduction in the risk of AD in statin users compared with nonusers (OR = 0.61; 95% CI, 0.42 to 0.87).34 That same year, Li et al suggested that there was no association between statin use and a reduced incidence of probable AD using a time-dependent proportional hazards model, but if the data were analyzed (inappropriately) as a case-control study, a significant protective effect was identified.35
Data from the Cache County Study cohort demonstrated no significant reduction in the risk of AD with statin use but allowed for the possibility that some benefit could be provided with longer-term statin therapy.36 In constrast, the Three-City Study of 9,294 individuals in France identified a significant reduction in the risk of AD with statin use (OR = 0.61; 95% CI, 0.41 to 0.91).12 Rea et al reported that prior statin use did not decrease the risk of dementia or AD, but when they included in their analysis individuals currently using a statin, there was a significant reduction in the hazard ratios for AD and for all-cause dementia.37 The two most recent epidemiologic studies both suggest that statin therapy slows cognitive decline in AD.38,39
COGNITIVE PERFORMANCE AND STATIN USE
A retrospective cohort study that assessed intelligence and cognition at a young age and again when subjects were in their 80s indicated that statin use had a significant beneficial effect on cognitive ability.40
In contrast, two very large prospective studies published in 2002 suggested that statins produce no positive effect on cognition in younger individuals at risk for heart disease.41,42 The Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) found that the mean Mini-Mental State Examination (MMSE) score, which was assessed only at subjects’ last on-treatment clinical visit, was comparable between the study’s pravastatin and placebo groups.41 Likewise, the Medical Research Council/British Heart Foundation (MRC/BHF) Heart Protection Study, which used the Telephone Interview for Cognitive Status questionnaire at the end of the investigation, reported that simvastatin had no positive effect on cognitive performance compared with placebo, but this finding was obtained in the absence of baseline data.42 Given the limited cognitive assessments performed in these two studies, no firm conclusions should be drawn.
A more recent prospective comparison of atorvastatin and placebo in younger subjects did include baseline and follow-up assessment of cognitive function, and it identified significantly superior performance in the statin-treated population on the MMSE and on tests of attention, psychomotor speed, mental flexibility, working memory, and memory retrieval.43
STATIN TREATMENT OF AD: THE AD CHOLESTEROL-LOWERING TREATMENT TRIAL
The initial clinical investigation of statin therapy in patients with AD—the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial—involved atorvastatin.44 Patients with mild to moderate AD were randomized to either placebo or 80 mg/day of atorvastatin for a 1-year period. Evaluable data were available for 63 patients (32 in the atorvastatin group, 31 in the placebo group). End points included the change in performance on the following measures:
MMSE
- Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-cog)
- Neuropsychiatric Inventory Caregiver Distress Scale (NPI)
- Clinical Global Impression of Change scale (CGIC)
- Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory (ADCS-ADL)
- Geriatric Depression Scale (GDS).
Cognitive results
In the setting of continued cholinesterase inhibitor use, atorvastatin provided significant benefit on the ADAS-cog at 26 weeks compared with placebo (P = .003) and marginally significant benefit at 1 year (P = .055) while producing a trend for benefit on the CGIC and NPI and a statistically significant improvement on the GDS after 1 year of active treatment.44 The observed benefit on the MMSE with atorvastatin versus placebo did not reach statistical significance, and no discernible difference was observed on the ADCS-ADL.44 In contrast, a significant difference in the slope of deterioration on the MMSE and the GDS in the atorvastatin group versus the placebo group suggested disease modification.45
Blood test results
Secondary analysis and initial morphometric substudy
Secondary assessment indicated that the subjects who garnered the greatest benefit from atorvastatin therapy in terms of their 6-month ADAS-cog score were those who had higher cholesterol levels at trial entry, those who harbored the apolipoprotein E4 allele, and those who were less affected by AD at trial entry (ie, with higher entry MMSE scores).47
In an ADCLT substudy using new voxel-based morphometry techniques, we quantitatively assessed gray matter density in 15 ADCLT trial participants and compared it with density findings in 15 normal elderly controls.48 Regional reductions in gray matter density were observed in the AD patients compared with the controls. Large differences in gray matter concentration were observed bilaterally in the temporal lobe. The anterior cingulate, right superior temporal, left superior frontal, and posterior cingulate regions also showed significantly decreased gray matter density in the AD patients compared with the controls. A significant relationship was observed between gray matter density and ADAS-cog error scores—ie, more severe levels of cognitive impairment correlated with reduced gray matter density.48
PILOT SUBSTUDY OF ADCLT: ASSESSING MORPHOLOGIC CHANGES WITH STATIN THERAPY
Eleven of the 15 ADCLT trial participants from the above morphometric substudy returned for MRI assessment after 1 year of treatment with either atorvastatin or placebo. We report here the comparative effects of atorvastatin and placebo on hippocampal volume and the relationship with cognitive performance.
Participants
Subjects were participating in the ADCLT trial, an investigator-initiated, double-blind, placebo-controlled study. Neuroimaging was performed at the Barrow Neurological Institute, Phoenix, AZ, for a subset of the participants in the trial (n = 11) as a pilot study to examine neural changes associated with atorvastatin therapy.
Each patient underwent screening, assignment to either atorvastatin 80 mg/day or placebo, and medical and cognitive assessment at Sun Health Research Institute, Sun City, AZ, prior to imaging at Barrow. All patients met Diagnostic and Statistical Manual of Mental Disorders, fourth edition, criteria for dementia as well as NINCDS-ADRDA criteria for probable AD. Each patient was free of significant psychiatric and neurological history and had a score of 4 or less on the Hachinski Modified Ischemia Scale. All MRIs were reviewed by a neuroradiologist to ensure that there was no evidence of stroke or cortical or lacunar infarcts.
Both sites’ institutional review boards approved this project, and all subjects gave written informed consent.
Cognitive assessment
A primary efficacy measure used in the parent study was the ADAS-cog,49 and the MMSE50 was a secondary measure. Change scores were determined by comparing values obtained at baseline, prior to randomization to treatment with either atorvastatin or placebo, and after 1 year of treatment. MMSE scores were obtained at the same session as the ADAS-cog scores. Cognitive assessments were obtained within 2 weeks prior to MRI.
Image acquisition
All participants underwent imaging on a single 1.5tesla GE scanner at Barrow Neurological Institute. Imaging was conducted both prior to treatment randomization and again after 1 year of treatment. Images of the whole brain were collected using a coronal SPGR (spoiled gradient) T1-weighted, three-dimensional acquisition with the following parameters:
- Number of acquisitions = 1
- Repetition time = 23 msec
- Echo time = 8 msec
- Flip angle = 35 degrees
- Bandwidth = 12.5 kHz
- Slice thickness = 1.5 mm or 1.9 mm
- 0 skip between slices
- In-plane resolution = 0.9375 x 0.9375.
Hippocampal volumetrics
All imaging analysis was performed within the Analysis of Functional Neuroimages (AFNI) package.51 We traced the outline of the hippocampus using the three-dimensional SPGR images. The hippocampi were visualized in all three planes, landmarked in the coronal and sagittal planes, and drawn in the coronal plane. We employed the guidelines of Insausti et al52 and Machulda et al53 to define the hippocampal boundaries. First we defined the anterior boundary by observing the white matter band and/or the cerebrospinal fluid space between the amygdala and hippocampus in the sagittal plane. The posterior aspect of the posterior region was initially landmarked in the sagittal plane by locating the posterior edge of the hippocampus and then checking in the coronal plane to ensure that the fornices were completely visualized. Volumes were calculated by importing the extracted hippocampi into MATLAB to measure the volumes.
Statistical analyses
Mean differences between the atorvastatin and placebo groups were evaluated using two-tailed Student t tests. Correlation between changes in cognitive measures and changes in the hippocampal volume for the total population and for the treatment groups was determined using Pearson’s r coefficient. Significance was defined as a P value less than .05; a P value between .05 and .10 was deemed a trend.
Results
In contrast to other studies,54–58 we found in this pilot study that right hippocampal volume was slightly less than left hippocampal volume (2,015 ± 141 mm3 vs 2,135 ± 183 mm3).
Mean changes in performance on the ADAS-cog and MMSE were less pronounced in the atorvastatin group than in the placebo group, but not significantly so (Table 1). However, there was a trend toward superiority in the atorvastatin group on performance on the free word-recall subscale of the ADAS-cog.
No significant correlations were found between change in cognitive performance and change in hip-pocampal volume.
DISCUSSION
The preponderance of evidence clearly indicates that hippocampal volume is reduced in patients with AD compared with individuals with normal cognitive ability for their age. There is also evidence indicating that as cognitive performance deteriorates in AD patients, there are concurrent further reductions in hippocampal volume.54 Many studies reported that there was no significant volume difference between the right and left hippocampi, but most suggested that the left hip-pocampus was slightly smaller than the right.54–58 We identified no significant difference in volume between the sides, but we did find that the right hippocampus was smaller than the left in a very limited population of subjects with mild to moderate AD.
The major finding of this pilot study flies in the face of conventional wisdom in that there seems to be significant shrinkage of the right hippocampus with atorvastatin therapy compared with placebo in a randomized AD treatment trial that demonstrated clinical benefit with atorvastatin therapy.44 A similar finding was reported from the beta-amyloid immunization (AN1792) treatment trial in AD.59 In that study the active immunization was associated with significant clinical benefit, reduced beta-amyloid load, and reduced hippocampal volume.59 The authors suggested that removal of beta-amyloid and/or other protein constituents from the tissue might have caused a “fluid shift” out of the tissue, resulting in shrinkage.
Based on our previous finding of reduced brain tissue density in AD patients compared with age-matched normal controls,48 an alternative explanation can be proposed. Neuronal loss in the hippocampus may be accompanied by increased fluid balance (reduced density) in an attempt to retain the previous volume at the expense of function. Accordingly, as the hippocampus shrinks, it approaches a more normal density for the remaining neuronal complement, and cognitive function improves.
- Lehtovirta M, Laakso MP, Soininen H, et al. Volumes of hippocampus, amygdala and frontal lobe in Alzheimer patients with different apolipoprotein E genotypes. Neuroscience 1995; 67:65–72.
- Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996; 16:4491–4500.
- Jack CR Jr, Shiung MM, Gunter JL, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 2004; 62:591–600.
- Jack CR Jr, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999; 52:1397–1403.
- Jack CR Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology 2000; 55:484–489.
- Killiany RJ, Hyman BT, Gómez-Isla T, et al. MRI measures of entorhinal cortex vs hippocampus in preclinical AD. Neurology 2002; 58:1188–1196.
- Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer’s disease: a case-control study. Neurology 1995; 45:1092–1096.
- Sparks DL. Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann N Y Acad Sci 1997; 826:128–146.
- Grant WB. Dietary links to Alzheimer’s disease. Alzheimers Dis Rev 1997; 2:42–55.
- Desmond DW, Tatemichi TK, Paik M, Stern Y. Risk factors for cerebrovascular disease as correlates of cognitive function in a stroke-free cohort. Arch Neurol 1993; 50:162–166.
- Mainous AG III, Eschenbach SL, Wells BJ, Everett CJ, Gill JM. Cholesterol, transferrin saturation, and the development of dementia and Alzheimer’s disease: results from an 18-year population-based cohort. Fam Med 2005; 37:36–42.
- Dufouil C, Richard F, Fiévet N, et al. APOE genotype, cholesterol level, lipid-lowering treatment, and dementia: the Three-City Study. Neurology 2005; 64:1531–1538.
- Lehtonen A, Luutonen S. High-density lipoprotein cholesterol levels of very old people in the diagnosis of dementia. Age Ageing 1986; 15:267–270.
- Giubilei F, D’Antona R, Antonini R, Lenzi GL, Ricci G, Fieschi C. Serum lipoprotein pattern variations in dementia and ischemic stroke. Acta Neurol Scand 1990; 81:84–86.
- Czech C, Förstl H, Hentschel F, et al. Apolipoprotein E-4 gene dose in clinically diagnosed Alzheimer’s disease: prevalence, plasma cholesterol levels and cerebrovascular change. Eur Arch Psychiatry Clin Neurosci 1994; 243:291–292.
- Mahieux F, Couderc R, Moulignier A, Bailleul S, Podrabinek N, Laudet J. Isoform 4 of apolipoprotein E and Alzheimer disease. Specificity and clinical study [in French]. Rev Neurol (Paris) 1995; 151:231–239.
- Kuo YM, Emmerling MR, Bisgaier CL, et al. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1-42 levels. Biochem Biophys Res Commun 1998; 252:711–715.
- Chandra V, Pandav R. Gene-environment interaction in Alzheimer’s disease: a potential role for cholesterol. Neuroepidemiology 1998; 17:225–232.
- Notkola IL, Sulkava R, Pekkanen J, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 1998; 17:14–20.
- Olson RE. Discovery of the lipoproteins, their role in fat transport and their significance as risk factors. J Nutr 1998; 128(2 Suppl):439S–443S.
- Kalmijn S, Launer LJ, Ott A, Witteman JC, Hofman A, Breteler MM. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol 1997; 42:776–782.
- Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ 1997; 315:1045–1049.
- Kivipelto M, Helkala E-L, Hallikainen M, et al. Elevated systolic blood pressure and high cholesterol levels at midlife are risk factors for late-life dementia [abstract]. Neurobiol Aging 2000; 21(Suppl 1):S174. Abstract 787.
- Tan ZS, Seshadri S, Beiser A, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham Study. Arch Intern Med 2003; 163:1053–1057.
- Reitz C, Luchsinger J, Tang MX, Manly J, Mayeux R. Impact of plasma lipids and time on memory performance in healthy elderly without dementia. Neurology 2005; 64:1378–1383.
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57:1439–1443.
- Jick H, Zornberg G, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet 2000; 356:1627–1631.
- Green RC, McNagny SE, Jayakumar P, Cupples LA, Benke K, Farrer L. Statin use is associated with reduced risk of Alzheimer’s disease [abstract]. Neurology 2002; 58(Suppl 3):A81. Abstract S10.006.
- Rodriguez EG, Dodge HH, Birzescu MA, Stoehr GP, Ganguli M. Use of lipid-lowering drugs in older adults with and without dementia: a community-based epidemiological study. J Am Geriatr Soc 2002; 50:1852–1856.
- Rockwood K, Kirkland S, Hogan DB, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol 2002; 59:223–227.
- Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol 2002; 59:378–384.
- Hajjar I, Schumpert J, Hirth V, Wieland D, Eleazer GP. The impact of the use of statins on the prevalence of dementia and the progression of cognitive impairment. J Gerontol A Biol Sci Med Sci 2002; 57:M414–M418.
- Etminan M, Gill S, Samii A. The role of lipid-lowering drugs in cognitive function: a meta-analysis of observational studies. Pharmacotherapy 2003; 23:726–730.
- Zamrini E, McGwin G, Roseman JM. Association between statin use and Alzheimer’s disease. Neuroepidemiology 2004; 23:94–98.
- Li G, Higdon R, Kukull WA, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology 2004; 63:1624–1628.
- Zandi PP, Sparks DL, Khachaturian AS, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry 2005; 62:217–224.
- Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol 2005; 62:1047–1051.
- Masse I, Bordet R, Deplanque D, et al. Lipid lowering agents are associated with a slower cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2005; 76:1624–1629.
- Bernick C, Katz R, Smith NL, et al. Statins and cognitive function in the elderly: the Cardiovascular Health Study. Neurology 2005; 65:1388–1394.
- Starr JM, McGurn B, Whiteman M, Pattie A, Whalley LJ, Deary IJ. Life long changes in cognitive ability are associated with prescribed medications in old age. Int J Geriatr Psychiatry 2004; 19:327–332.
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Parale GP, Baheti NN, Kulkarni PM, Panchal NV. Effects of atorvastatin on higher functions. Eur J Clin Pharmacol 2006; 62:259–265.
- Sparks DL, Sabbagh MN, Connor DJ, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol 2005; 62:753–757.
- Sparks DL, Sabbagh M, Connor D, et al. Statin therapy in Alzheimer’s disease. Acta Neurol Scand 2006; 114(Suppl 185):78–86.
- Stankovic G, Sparks DL. Change in circulating C-reactive protein is not associated with atorvastatin treatment in Alzheimer’s disease. Neurol Res 2006; 28:621–624.
- Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer’s disease: results of the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial. Acta Neurol Scand 2006; 114(Suppl 185):3–7.
- Baxter LC, Sparks DL, Johnson SC, et al. Relationship of cognitive measures and gray and white matter in Alzheimer’s disease. J Alzheimers Dis 2006; 9:253–260.
- Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984; 141:1356–1364.
- Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the mental state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Cox RW. AFNI: sofware for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res 1996; 29:162–173.
- Insausti R, Juottonen K, Soininen H, et al. MR volumetric analysis of the human entorhinal, perirhinal, and temporopolar cortices. AJNR Am J Neuroradiol 1998; 19:659–671.
- Machulda MM, Ward HA, Cha R, O’Brien P, Jack CR Jr. Functional inferences vary with the method of analysis in fMRI. Neuroimage 2001; 14:1122–1127.
- Jack CR Jr, Slomkowski M, Gracon S, et al. MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology 2003; 60:253–260.
- Pruessner JC, Li LM, Serles W, et al. Volumetry of hippocampus and amygdala with high-resolution MRI and three-dimensional analysis software: minimizing the discrepancies between laboratories. Cereb Cortex 2000; 10:433–442.
- Pruessner JC, Collins DL, Pruessner M, Evans AC. Age and gender predict volume decline in the anterior and posterior hippocam-pus in early adulthood. J Neurosci 2001; 21:194–200.
- Hackert VH, den Heijer T, Oudkerk M, Koudstaal PJ, Hofman A, Breteler MM. Hippocampal head size associated with verbal memory performance in nondemented elderly. Neuroimage 2002; 17:1365–1372.
- Rosen AC, Prull MW, Gabrieli JD, et al. Differential associations between entorhinal and hippocampal volumes and memory performance in older adults. Behav Neurosci 2003; 117:1150–1160.
- Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005; 64:1563–1572.
- Lehtovirta M, Laakso MP, Soininen H, et al. Volumes of hippocampus, amygdala and frontal lobe in Alzheimer patients with different apolipoprotein E genotypes. Neuroscience 1995; 67:65–72.
- Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 1996; 16:4491–4500.
- Jack CR Jr, Shiung MM, Gunter JL, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 2004; 62:591–600.
- Jack CR Jr, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999; 52:1397–1403.
- Jack CR Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology 2000; 55:484–489.
- Killiany RJ, Hyman BT, Gómez-Isla T, et al. MRI measures of entorhinal cortex vs hippocampus in preclinical AD. Neurology 2002; 58:1188–1196.
- Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer’s disease: a case-control study. Neurology 1995; 45:1092–1096.
- Sparks DL. Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann N Y Acad Sci 1997; 826:128–146.
- Grant WB. Dietary links to Alzheimer’s disease. Alzheimers Dis Rev 1997; 2:42–55.
- Desmond DW, Tatemichi TK, Paik M, Stern Y. Risk factors for cerebrovascular disease as correlates of cognitive function in a stroke-free cohort. Arch Neurol 1993; 50:162–166.
- Mainous AG III, Eschenbach SL, Wells BJ, Everett CJ, Gill JM. Cholesterol, transferrin saturation, and the development of dementia and Alzheimer’s disease: results from an 18-year population-based cohort. Fam Med 2005; 37:36–42.
- Dufouil C, Richard F, Fiévet N, et al. APOE genotype, cholesterol level, lipid-lowering treatment, and dementia: the Three-City Study. Neurology 2005; 64:1531–1538.
- Lehtonen A, Luutonen S. High-density lipoprotein cholesterol levels of very old people in the diagnosis of dementia. Age Ageing 1986; 15:267–270.
- Giubilei F, D’Antona R, Antonini R, Lenzi GL, Ricci G, Fieschi C. Serum lipoprotein pattern variations in dementia and ischemic stroke. Acta Neurol Scand 1990; 81:84–86.
- Czech C, Förstl H, Hentschel F, et al. Apolipoprotein E-4 gene dose in clinically diagnosed Alzheimer’s disease: prevalence, plasma cholesterol levels and cerebrovascular change. Eur Arch Psychiatry Clin Neurosci 1994; 243:291–292.
- Mahieux F, Couderc R, Moulignier A, Bailleul S, Podrabinek N, Laudet J. Isoform 4 of apolipoprotein E and Alzheimer disease. Specificity and clinical study [in French]. Rev Neurol (Paris) 1995; 151:231–239.
- Kuo YM, Emmerling MR, Bisgaier CL, et al. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1-42 levels. Biochem Biophys Res Commun 1998; 252:711–715.
- Chandra V, Pandav R. Gene-environment interaction in Alzheimer’s disease: a potential role for cholesterol. Neuroepidemiology 1998; 17:225–232.
- Notkola IL, Sulkava R, Pekkanen J, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 1998; 17:14–20.
- Olson RE. Discovery of the lipoproteins, their role in fat transport and their significance as risk factors. J Nutr 1998; 128(2 Suppl):439S–443S.
- Kalmijn S, Launer LJ, Ott A, Witteman JC, Hofman A, Breteler MM. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol 1997; 42:776–782.
- Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ 1997; 315:1045–1049.
- Kivipelto M, Helkala E-L, Hallikainen M, et al. Elevated systolic blood pressure and high cholesterol levels at midlife are risk factors for late-life dementia [abstract]. Neurobiol Aging 2000; 21(Suppl 1):S174. Abstract 787.
- Tan ZS, Seshadri S, Beiser A, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham Study. Arch Intern Med 2003; 163:1053–1057.
- Reitz C, Luchsinger J, Tang MX, Manly J, Mayeux R. Impact of plasma lipids and time on memory performance in healthy elderly without dementia. Neurology 2005; 64:1378–1383.
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57:1439–1443.
- Jick H, Zornberg G, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet 2000; 356:1627–1631.
- Green RC, McNagny SE, Jayakumar P, Cupples LA, Benke K, Farrer L. Statin use is associated with reduced risk of Alzheimer’s disease [abstract]. Neurology 2002; 58(Suppl 3):A81. Abstract S10.006.
- Rodriguez EG, Dodge HH, Birzescu MA, Stoehr GP, Ganguli M. Use of lipid-lowering drugs in older adults with and without dementia: a community-based epidemiological study. J Am Geriatr Soc 2002; 50:1852–1856.
- Rockwood K, Kirkland S, Hogan DB, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol 2002; 59:223–227.
- Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol 2002; 59:378–384.
- Hajjar I, Schumpert J, Hirth V, Wieland D, Eleazer GP. The impact of the use of statins on the prevalence of dementia and the progression of cognitive impairment. J Gerontol A Biol Sci Med Sci 2002; 57:M414–M418.
- Etminan M, Gill S, Samii A. The role of lipid-lowering drugs in cognitive function: a meta-analysis of observational studies. Pharmacotherapy 2003; 23:726–730.
- Zamrini E, McGwin G, Roseman JM. Association between statin use and Alzheimer’s disease. Neuroepidemiology 2004; 23:94–98.
- Li G, Higdon R, Kukull WA, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology 2004; 63:1624–1628.
- Zandi PP, Sparks DL, Khachaturian AS, et al. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry 2005; 62:217–224.
- Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol 2005; 62:1047–1051.
- Masse I, Bordet R, Deplanque D, et al. Lipid lowering agents are associated with a slower cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2005; 76:1624–1629.
- Bernick C, Katz R, Smith NL, et al. Statins and cognitive function in the elderly: the Cardiovascular Health Study. Neurology 2005; 65:1388–1394.
- Starr JM, McGurn B, Whiteman M, Pattie A, Whalley LJ, Deary IJ. Life long changes in cognitive ability are associated with prescribed medications in old age. Int J Geriatr Psychiatry 2004; 19:327–332.
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Parale GP, Baheti NN, Kulkarni PM, Panchal NV. Effects of atorvastatin on higher functions. Eur J Clin Pharmacol 2006; 62:259–265.
- Sparks DL, Sabbagh MN, Connor DJ, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol 2005; 62:753–757.
- Sparks DL, Sabbagh M, Connor D, et al. Statin therapy in Alzheimer’s disease. Acta Neurol Scand 2006; 114(Suppl 185):78–86.
- Stankovic G, Sparks DL. Change in circulating C-reactive protein is not associated with atorvastatin treatment in Alzheimer’s disease. Neurol Res 2006; 28:621–624.
- Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer’s disease: results of the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial. Acta Neurol Scand 2006; 114(Suppl 185):3–7.
- Baxter LC, Sparks DL, Johnson SC, et al. Relationship of cognitive measures and gray and white matter in Alzheimer’s disease. J Alzheimers Dis 2006; 9:253–260.
- Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984; 141:1356–1364.
- Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the mental state of patients for the clinician. J Psychiatr Res 1975; 12:189–198.
- Cox RW. AFNI: sofware for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res 1996; 29:162–173.
- Insausti R, Juottonen K, Soininen H, et al. MR volumetric analysis of the human entorhinal, perirhinal, and temporopolar cortices. AJNR Am J Neuroradiol 1998; 19:659–671.
- Machulda MM, Ward HA, Cha R, O’Brien P, Jack CR Jr. Functional inferences vary with the method of analysis in fMRI. Neuroimage 2001; 14:1122–1127.
- Jack CR Jr, Slomkowski M, Gracon S, et al. MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology 2003; 60:253–260.
- Pruessner JC, Li LM, Serles W, et al. Volumetry of hippocampus and amygdala with high-resolution MRI and three-dimensional analysis software: minimizing the discrepancies between laboratories. Cereb Cortex 2000; 10:433–442.
- Pruessner JC, Collins DL, Pruessner M, Evans AC. Age and gender predict volume decline in the anterior and posterior hippocam-pus in early adulthood. J Neurosci 2001; 21:194–200.
- Hackert VH, den Heijer T, Oudkerk M, Koudstaal PJ, Hofman A, Breteler MM. Hippocampal head size associated with verbal memory performance in nondemented elderly. Neuroimage 2002; 17:1365–1372.
- Rosen AC, Prull MW, Gabrieli JD, et al. Differential associations between entorhinal and hippocampal volumes and memory performance in older adults. Behav Neurosci 2003; 117:1150–1160.
- Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005; 64:1563–1572.